Область изобретения
Настоящее изобретение относится к способам визуализации и подавления нежелательных, быстро пролиферирующих клеток в многоклеточных организмах.
Предпосылки создания изобретения
Аномальная пролиферация клеток, прежде всего гиперпролиферация, является источником множества заболеваний, из которых наиболее тяжелым является рак. Только в США каждый год диагноз рака зарегистрирован приблизительно у 1,5 млн человек и летальный исход от рака у 0,5 млн человек. В настоящее время, несмотря на значительный прогресс в лечении рака, способы лечения характеризуются множеством недостатков. Основные проблемы связаны с тяжелыми побочными действиями противоопухолевых средств и с развитием поколения устойчивых раковых клеток, а также ранняя локализация, точная локализация опухоли и метастазирование.
Гиперпролиферативные клетки, как многие типы раковых клеток, зависят от повышенного притока питательных веществ, ростовых факторов, энергии и витаминов. В связи с этим существует возможность доставлять лекарственные средства в такие нежелательные клетки с использованием пути транспорта витаминов, которые являются незаменимыми для клеточного роста и в большинстве случаев запас которых ограничен.
Кобаламин (Cbl), известный также как витамин В12, присутствующий в виде цианокобаламина (CN-Cbl), гидроксикобаламина (OH-Cbl) или аквакобаламина (Н2О-Cbl), является жизненно необходимым, а его концентрация в организме чрезвычайно мала. Высшие организмы, включая человека, вынуждены получать витамин из пищевых продуктов. Биосинтез кобаламина осуществляется только у некоторых прокариотов, таких как анаэробные бактерии. Кобаламин незаменим для нормальной функции нервной системы и необходим для нормального метаболизма углеводов, белков и жиров. Кобаламин утилизируется в нескольких жизненно важных внутриклеточных метаболических путях. В виде метилкобаламина (Ме-Cbl) он используется в качестве кофактора для метионинсинтазы. В виде 5'-дезоксиаденозилкобаламина (Ado-Cbl) он действует совместно с метилмалонил-СоА-мутазой при превращении метилмалонил-СоА в сукцинил-СоА. Недостаток кобаламина может привести к пернициозной анемии. Кобаламин включен также в процесс восстановительного превращения рибонуклеотидов в дезоксирибонуклеотиды при образовании ДНК.
В организме млекопитающих в большинстве случаев поглощение клетками кобаламина регулируется сывороточными транспортными белками и клеточными мембранными рецепторами. Существует два типа кобаламин-связывающих белков в плазме: негликозилированный белок транскобаламин II (ТСII) и гликозилированные белки транскобаламин I и III (TCI и TCIII), также называемые R-связывающими белками или гаптокорринами. TCI и TCIII характеризуются иммуноперекрестной реакцией и возможно отличаются только углеводным составом. TCI является первичным R-связывающим белком, найденным в кровотоке. Для простоты термин TCI используют для обозначения обоих типов R-связывающих белков TCI и TCIII. Оба типа транспортных белков (векторов) TCI и TCII циркулируют в кровотоке млекопитающих в виде частично насыщенных (голобелки) или частично ненасыщенных (апобелки) кобаламином. В клетках млекопитающих присутствует также безвекторная система поглощениеа кобаламина, которая в нормальных клетках характеризуется достаточной низкой эффективностью (см. статью Sennet С. и Rosenberg L.E., Ann. Rev. Biochem. 50, 1053-86 (1981)).
Функция TCII заключается в доставке плазматического кобаламина во все метаболически активные клетки по механизму опосредованного рецептором эндоцитоза. Известно, что ускоренная клеточная пролиферация при возникновении и развитии опухоли (неоплазия) напрямую вызывает повышенное потребление связанного с кобаламином TCII из кровотока по опосредованному рецептором механизму эндоцитозного поглощения. Во многих работах установлено, что повышенная регуляция ряда рецепторов TCII наблюдается в линиях злокачественных клеток для удовлетворения возрастающей метаболической потребности в продуцировании тимидина и метионина, в реакциях метилирования при синтезе ДНК и снабжения энергией клеток по механизму митохондриального метаболизма.
Основной рецептор TCII присутствует во всех типах тканей, в то время как второй рецептор TCII с более выраженной специфичностью к органам, так называемый мегалин, в основном экспрессируется в проксимальных канальцах почек и в некоторых других видах абсорбционного эпителия. После эндоцитозной интернализации TCII разлагается в лизосомах и свободный кобаламин транспортируется в цитоплазму и в ядерную мембрану, где он превращается в Me-Cbl и Ado-Cbl. Эти две формы проявляют функцию активных коферментов витамина В12. О важной роли TCII свидетельствует тот факт, что наследственный врожденный дефицит TCII вызывает мегалобластную анемию, тяжелые неврологические нарушения и летальный исход, если не проводить лечение избытком кобаламина.
Практически все клетки способны продуцировать TCII. Множество клеток, таких как гепатоциты, фибробласты, нервные клетки, энтероциты и макрофаги, синтезируют повышенные количества TCII. Предполагается, что первичным источником TCII является эндотелий кровеносных сосудов. Приблизительно 20-30% циркулирующего кобаламина связано с TCII в форме голо-TCII, который является метаболически эффективной формой, обеспечивающей интернализацию кобаламина во всех тканях (см. статью Rothenberg E. и др., в книге: Chemistry and Biochemistry of В12, ред. R.Banerjee, New York, NY, cc.441-473 (1999)).
TCI присутствует в крови и плазме, а также в большинстве внешнесекреторных выделений и других типов жидкостей. Этот белок в основном продуцируется в тканях передней кишки, слизистой оболочке желудка, слюнной и слезной железах и в секреторном эпителии внутреннего уха. TCI, в отличии от TCII, не поставляет связанный с ним кобаламин непосредственно для поглощения клетками, характеризуется продолжительным периодом полураспада в крови и таким образом в любой данный момент удерживает более 75% циркулирующего кобаламина (и коррина). Практически весь TCI циркулирует в форме голо-TCI. Его роль еще недостаточно изучена. Предполагается, что он функционирует в качестве бактериостатического агента, который предотвращает доставку всех типов кобаламинов и корринов в микроорганизмы. Этот белок стабилизирует также аденозилкобаламин и защищает его от фотолиза. В отличии от TCI, концентрация которого при циркуляции выше по сравнению с TCII, уровень TCII может быстро возрастать за счет синтеза de novo апо-TCII в ответ на поступающий кобаламин. TCI накапливается достаточно медленно и в значительной степени не стимулируется в ответ на любой инициирующий импульс (см. в указанной выше книге Alpers D. и Russell G., Chemistry and Biochemistry of B12, cc.411-441).
До настоящего времени безвекторная доставка кобаламина в клетки млекопитающих не рассматривалась как альтернативный путь поглощения производных кобаламина гиперпролиферативными клетками. Не вызывает сомнения тот факт, что физиологически важные механизмы поглощения кобаламина доброкачественными клетками млеокпитающих включают векторы TCII и TCI (а также собственный фактор в пищеварительном тракте). Однако по данным исследований in vivo и in vitro установлено, что свободный кобаламин также способен проникать через плазматическую мембрану без участия векторных белков. Прямое доказательство дополнительной способности поглощения свободного кобаламина получено при исследовании детей с врожденным и полным дефицитом TCII, у которых при парентеральном введении свободного кобаламина наблюдалась заметная ремиссия клинических и химических признаков внутриклеточного дефицита кобаламина (см. статью Hall С.Е. и др., Blood, 53, 251-263 (1979)). При исследовании in vitro наблюдалось поглощение свободного кобаламина клетками HeLa и фибробластами. В клетках HeLa поглощение свободного кобаламина составляет от 1% до 2% от поглощения связанного с TCII кобаламина. В фибробластах человека накопление свободного кобаламина с интервалом в 2 ч составляет приблизительно 20% от связанного с TCII витамина. Система поглощения свободного витамина фибробластами человека достаточно подробно изучена, как описано в статье Berliner N. и Rosenberg L.E., Metabolism, 30, 230-236 (1981)). Поглощение свободного CN-[57Co]-Cbl исследовали в двухстадийной системе: начальное поглощение происходит с высокой скоростью по механизму насыщения и специфически ингибируется избытком немеченых CN-Cbl и OH-Cbl, процесс поглощения завершается через 30 мин. Вторая стадия поглощения происходит с более низкой скоростью в линейной зависимости от времени, не ингибируется избытком немеченного кобаламина и не выходит на плато даже через 8 ч, что свидетельствует о характерном неспецифическом процессе. Первая стадия поглощения является высокоспецифичным процессом проникновения через мембрану, который опосредуется белком и чувствителен к сульфгидрильным реагентам, кроме того этот процесс значительно ингибируется циклогексимидом (см. статью Sennet С.и Rosenberg L.E., Ann. Rev. Biochem. 50, 1053-86 (1981)). Эти свойства согласуются с присутствием опосредованной белком системы быстрого поглощения свободного кобаламина у млекопитающих.
Установлено, что многие бактерии и все эукариотные протисты являются ауксотрофными к витамину В12 и связывают его с более высокой аффинностью по сравнению с собственным фактором млекопитающих, TCI и TCII. Связывающие В12 белки из бактерий и простейших являются белками, взаимодействующими по безвекторному механизму с клеточной поверхностью и способными связывать множество корринов (включая сам кобаламин) с высокой аффинностью. Следовательно, существует возможность детектирования бактериальных инфекций при визуализации всего организма после введения радиоактивно меченного производного кобаламина (см. статью Collins D.A. и др., Mayo Clin. Proc. 75, 568-580 (2000)). Развитие гиперпролиферативных форм клеток млекопитающих может привести к развитию в ходе многостадийного канцерогенеза более эффективных форм уже существующих безвекторных систем поглощения кобаламина.
В настоящее время опубликованы и запатентованы подходы к применению кобаламина в качестве носителя для широкого спектра биологически активных агентов, включая радиоактивные изотопы металлов (см. патент Collins D.A., US Pat. Appl. No.2003/0144198). При испытании на животных и пациентах с использованием радиоактивно меченных производных кобаламина наблюдалось включение метки в опухолевые ткани, но при этом отмечено также значительное накопление радиоактивности в здоровых тканях, таких как почки и печень. Следовательно, визуализация и радиотерапия пока еще не являются оптимальными. В настоящий момент применение таких опубликованных методов ограничено возможным повреждением некоторых здоровых отделов организма.
Таким образом, существует необходимость в соединениях, композициях и способах для введения диагностических и терапевтических производных кобаламина в быстро пролифелирующие клетки в более высокой концентрации по сравнению с нормальными клетками. Объектом настоящего изобретения является разработка новых способов идентификации, синтеза, характеристики и введения производных кобаламина с высокой специфичностью в отношении клеток, характеризующихся аномально высокой пролиферацией, и при этом исключить развитие поколения устойчивых клеток.
Краткое описание сущности изобретения
Настоящее изобретение основано на том факте, что в отличие от самого кобаламина производные кобаламина, которые не связываются или связываются в значительно меньшей степени с транспортным белком, транскобаламином II (TCII), при соответствующем способе введения накапливаются в значительно меньшей степени в крови и доброкачественных органах, таких как почки и печень, по сравнению со скоростью накопления в неопластических тканях и, кроме того, быстро удаляются из крови. При использовании производных кобаламина, действующих в качестве заменителей витамина В12, значительно снижается риск формирования поколения устойчивых неопластических тканей.
Изобретение относится к производным кобаламина,
(а) не обладающим связывающей аффинностью или обладающим низкой связывающей аффинностью в отношении транскобаламина II и
(б) сохраняющим активность заменителя витамина В12.
Прежде всего, изобретение относится к производным кобаламина,
(а) обладающим менее 20%, предпочтительно менее 5%, связывающей аффинности в отношении транскобаламина II по сравнению со связывающей активностью немодифицированного кобаламина, которую определяют методом анализа связывания, и
(б) сохраняющим более 2% активности заменителя витамина В12, которую определяют методом анализа роста клеток.
Примеры соединений по настоящему изобретению, обладающих низкой связывающей активностью в отношении TCII или не обладающие такой активностью, включают специфические производные кобаламина, содержащие терапевтический и/или диагностический агент, такой как радиоактивный металл. Соединения по настоящему изобретению выбирают по результатам определения связывающей активности с использованием очищенного TCII и анализа роста клеток с использованием в качестве исследуемого организма бактерий Lactobacillus delbrueckii.
Настоящее изобретение относится также к способу диагностики неопластического заболевания или инфекции микроорганизма у млекопитающих, включающему
(а) соблюдение диеты, не содержащей витамин В12, для млекопитающего, который подвержен неопластическому заболеванию или инфекции, и
(б) последующее введение производного кобаламина по изобретению, содержащего диагностический агент.
Аналогичным образом настоящее изобретение относится к способу лечения млекопитающего, страдающего от неопластического заболевания или от инфекции микроорганизма, включающему
(а) соблюдение диеты, не содержащей витамин В12, для млекопитающего, нуждающегося в лечении, и
(б) последующее введение производного кобаламина по изобретению, содержащего терапевтический агент.
Настоящее изобретение относится также к применению производного кобаламина в способе диагностики неопластического заболевания или инфекции микроорганизма или при лечении млекопитающего, страдающего от неопластического заболевания или инфекции микроорганизма.
Кроме того, настоящее изобретение относится к фармацевтическим композициям, содержащим производные кобаламина по настоящему изобретению, прежде всего к композициям, пригодным для диагностики, и к фармацевтическим композициям, пригодным для лечения, а также к применению таких фармацевтических композиций в способе диагностики и в способе лечения соответственно.
Настоящее изобретение относится также к промежуточным соединениям, которые используются для получения соединений, пригодных для диагностики или лечения по настоящему изобретению, прежде всего к соединениям, содержащим хелатные заместители для связывания радиоактивных металлов, но не содержащим связанный с хелатным заместителем металл или нерадиоактивный металл.
Производные кобаламина по настоящему изобретению являются, прежде всего, ценными для диагностики и/или лечения агрессивных, быстро прогрессирующих неопластических заболеваний, таких как рак, и/или диагностики и/или лечения локальных инфекций патогенных микроорганизмов.
Краткое описание чертежей
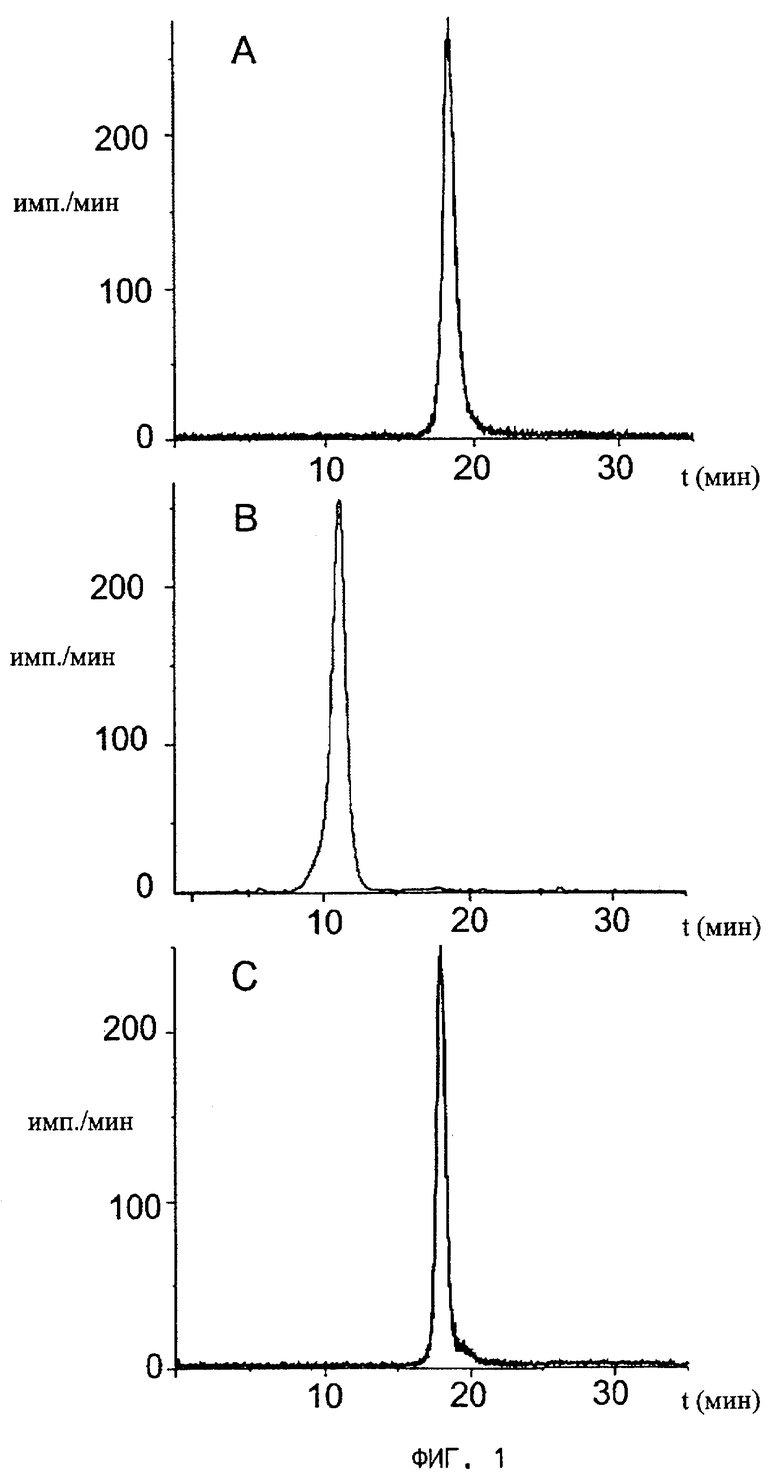
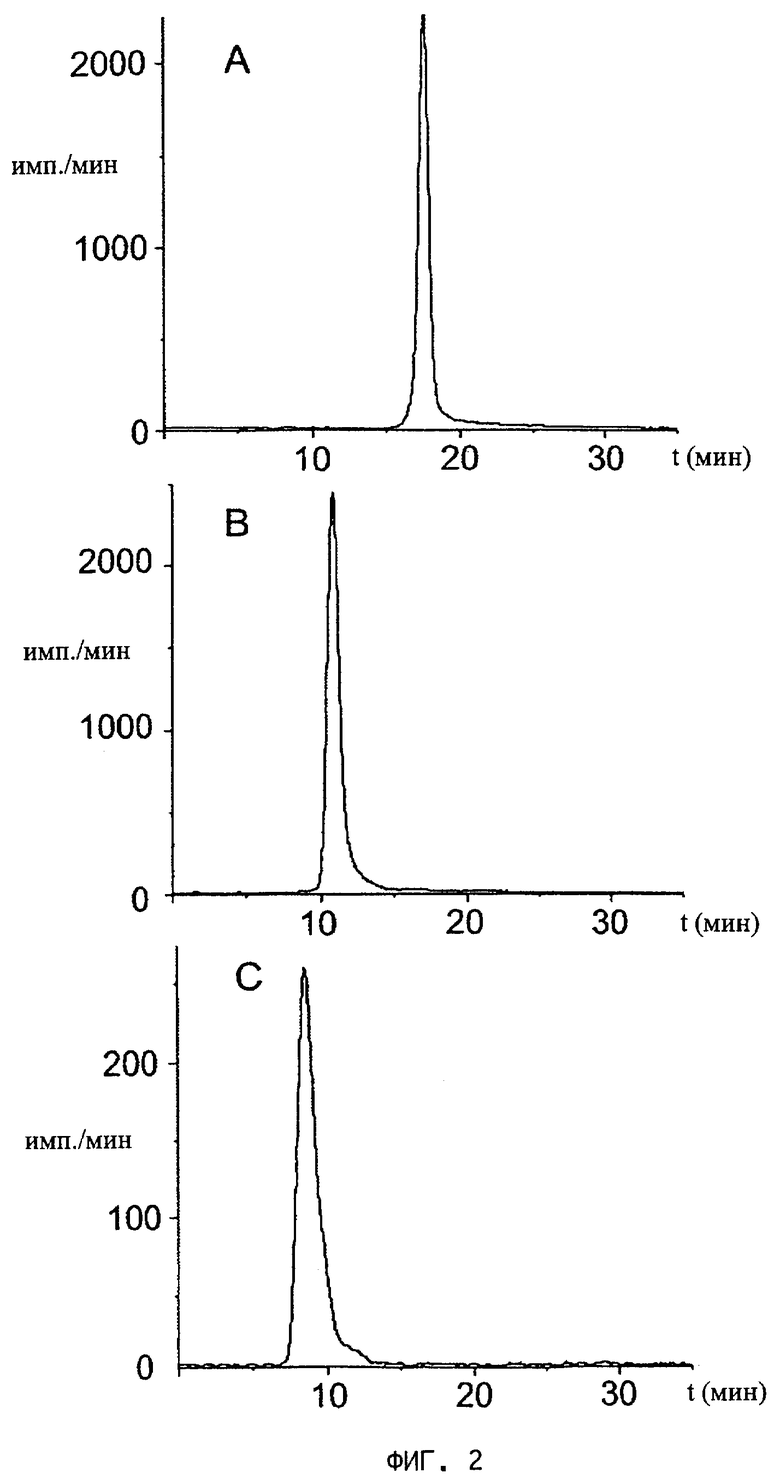
На фиг.1 представлены графики анализа взаимодействия радиоактивно меченного цианокобаламин-b-пропил-PAMA-OEt (пример 11), не связывающего TCII, с транспортными белками методом гель-фильтрации по сдвигу молекулярной массы (на оси ординат - время, на оси абсцисс - количество импульсов в мин (имп./мин)),
A) гель-фильтрация радиоактивно меченного производного на колонке Superdex 75 (пик соответствует молекулярной массе 1,5 кДа),
B) гель-фильтрация производного в смеси с TCI (сдвиг молекулярной массы от 1,5 кДа до 44 кДа),
C) гель-фильтрация производного в смеси с TCII (пик соответствует молекулярной массе 1,5 кДа, то есть цианокобаламин-b-пропил-PAMA-OEt практически не связывается с TCII).
На фиг.2 представлены графики анализа взаимодействия радиоактивно меченного цианокобаламин-b-бутил-PAPAcet (пример 5), связывающего TCII, с транспортными белками методом гель-фильтрации по сдвигу молекулярной массы (на оси ординат - время, на оси абсцисс - количество импульсов в мин (имп./мин)),
A) гель-фильтрация радиоактивно меченного производного на колонке Superdex 75 (пик соответствует молекулярной массе 1,5 кДа),
B) гель-фильтрация производного в смеси с TCI (сдвиг молекулярной массы от 1,5 кДа до 44 кДа),
C) гель-фильтрация производного в смеси с TCII (сдвиг молекулярной массы от 1,5 кДа до 60 кДа, то есть цианокобаламин-b-бутил-PAPAcet связывается с TCII).
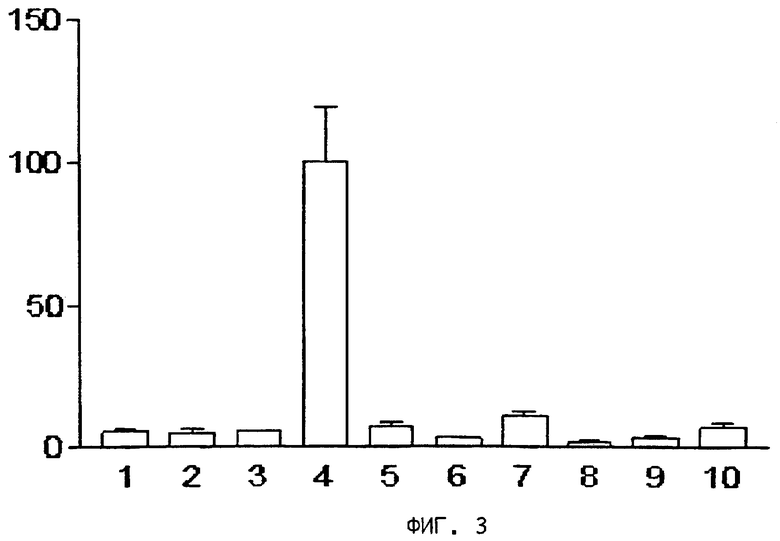
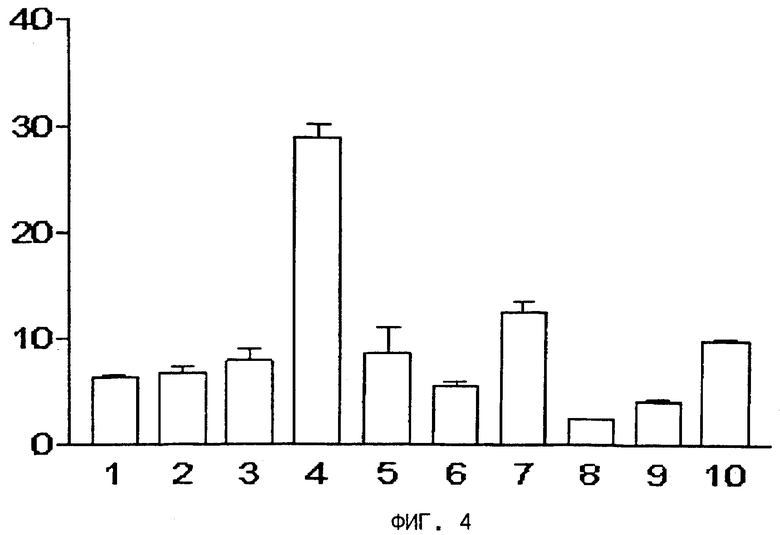
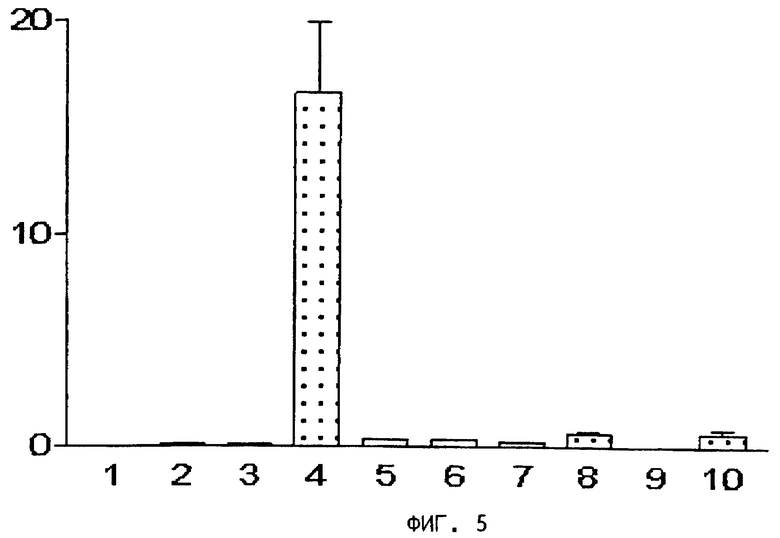
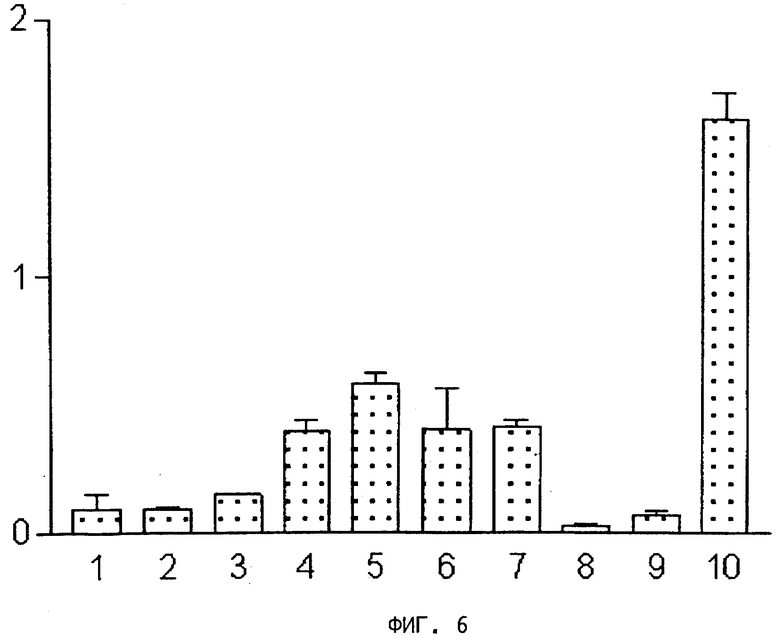
На фиг.3, 4, 5 и 6 представлены диаграммы распределения в тканях,
на оси ординат - процент введенной радиоактивности на грамм ткани,
на оси абсцисс - органы: 1) кровь, 2) сердце, 3) селезенка, 4) почки, 5) желудок, 6) кишечник, 7) печень, 8) мышцы, 9) кость, 10) опухоль.
На фиг.3 представлена диаграмма распределения после внутривенной инъекции радиоактивного цианокобаламина (57Co-CN-Cbl) в тканях мышей, получавших нормальный корм.
На фиг.4 представлена диаграмма распределения после внутривенной инъекции радиоактивного цианокобаламина (57Co-CN-Cbl) в тканях мышей, получавших корм без витамина В12.
На фиг.5 представлена диаграмма распределения после внутривенной инъекции радиоактивного цианокобаламин-β-пропил-PAMA-OEt (пример 11) в тканях мышей, получавших нормальный корм.
На фиг.6 представлена диаграмма распределения после внутривенной инъекции радиоактивного цианокобаламин-β-пропил-PAMA-OEt (пример 11) в тканях мышей, получавших корм без витамина В12.
Подробное описание вариантов осуществления настоящего изобретения
При введении млекопитающим, соблюдавшим не содержащую витамин В12 диету, производных кобаламина, проявляющих чрезвычайно низкую связывающую аффинность в отношении кобаламин-векторного белка (или транспортного белка) или не обладающих такой активностью, наблюдается значительное снижение накопления в крови и в важных органах, таких как почки и печень, и при этом поддерживается высокий уровень поглощения в гиперпролиферативных клетках, что позволяет использовать такие производные для более точной диагностики и лечения неопластических заболеваний и локальных инфекций микроорганизмов.
К соединениям по настоящему изобретению, проявляющим низкую связывающую аффинность в отношении TCII и сохраняющим активность витамина В12, относятся, например, соединения формулы (I)
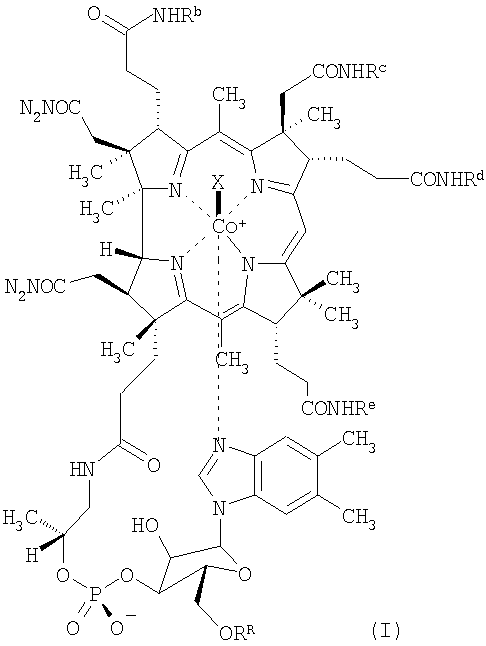
где Rb, Rc, Rd и Re независимо друг от друга означают спейсерную хелатную группу, антибиотик или антипролиферативный терапевтический агент, органическую группу с определенной конфигурацией, содержащую от 4 до 20 атомов углерода, или водород,
RR означает спейсерную хелатную группу, антибиотик или антипролиферативный терапевтический агент, каждый из которых связан через мостиковую группу Z, или водород,
при условии, что по крайней мере три остатка из Rb, Rc, Rd, Re и RR означают водород и по крайней мере один из Rb, Rc, Rd и Re не означает водород,
Х означает монодентатный лиганд, а
центральный атом кобальта (Со) необязательно находится в форме радиоактивного изотопа.
В предпочтительном варианте осуществления изобретения Re означает водород.
Монодентатным лигандом Х является, например, циано,
галоген, такой как фтор, хлор, бром или иод, цианато, изоцианато, тиоцианато или изотиоцианато,
алкил с прямой или разветвленной цепью, содержащий от 1 до 25 атомов углерода, предпочтительно от 1 до 4 атомов углерода, такой как метил, этил, н-пропил, н-бутил или изобутил или также н-гексил или н-децил, и необязательно замещенный группами: гидрокси, метокси или амино, например гидроксиметил, метоксиметил, аминометил, гидроксиэтил или метоксиэтил,
нитрил R-CN, изонитрил R-NC, карбоксилат R-COO- или тиолят R-S-, где R означает алкил с прямой или разветвленной цепью, включающий от 1 до 15 атомов углерода, предпочтительно от 1 до 6 атомов углерода, или арил, например фенил или нафтил, такой как ацетонитрил, пропионитрил, бензонитрил, метилизоцианат, фенилизоцианат, ацетат, пропионат, бензоат, метилтиолят, этилтиолят, н-гексилтиолят или тиофенолят,
фосфит (RO)3Р, где заместители R являются одинаковыми или различными и означают алкил, содержащий от 1 до 6 атомов углерода, или арил, например необязательно замещенный фенил или нафтил, такой как триметилфосфит, метилдифенилфосфит, трифенилфосфит или три-орто-толилфосфит,
гидрокси или остаток аквокислоты, или
остаток 5'-дезоксиаденозина или родственного нуклеозида.
Х предпочтительно означает циано, метил, гидрокси, остаток аквокислоты или 5'-дезоксиаденозина, наиболее предпочтительно циано.
Спейсерная хелатная группа в качестве заместителей Rb, Rc, Rd, Re или RR означает хелатную группу для атомов металла, присоединенную к NH- или O-группе кобаламина через спейсерную группу, и необязательно содержит атом металла, прежде всего атом радиоактивного металла.
Соединения формулы (I), в которых спейсерная хелатная группа не содержит атом металла, используются в качестве промежуточных производных для получения соединений, предназначенных для применения в способах диагностики и/или лечения по настоящему изобретению.
Если в качестве заместителя Rb, Rc, Rd, Re или RR используют антибиотик или антипролиферативный терапевтический агент, то антибиотик выбирают из группы, включающей аминогликозидные антибиотики, такие как гентамицин, тетрациклины, антиметаболиты, такие как селенометионин, макролиды, такие как эритромицин и триметоприм, а антипролиферативный агент выбирают из группы, включающей антиметаболиты, такие как 5-фторурацил, алкилирующий агент, такой как оксалиплатин, дакарбазин, циклофосфамид или карбоплатин, ингибитор клеточного цикла, такой как винбластин или доцетаксел, деградирующий ДНК агент (ингибитор топоизомеразы, интеркалятор, расщепляющий цепи агент), такой как доксорубицин, блеомицин или топотекан, соединение, влияющее на путь передачи сигнала, такое как модификатор активности каспазы, агонист или антагонист рецепторов гибели клеток, и модификатор нуклеаз, фосфостаз и киназ, такой как мезилат иматиниба, дексаметазон, миристат-ацетат форбола, циклоспорин А, кверцетин или тамоксифен, причем указанные соединения присоединены к NH- или O-группе кобаламина ковалентной связью напрямую или через спейсер.
Спейсер означает алифатическую цепь, содержащую от 2 до 10 атомов углерода, предпочтительно от 2 до 6 атомов углерода, например от 2 до 5 атомов углерода, причем один или два атома углерода заменены на атомы азота и/или кислорода, а алифатическая цепь замещена группами гидрокси, оксо или амино. Прежде всего, два соседних атома углерода заменены на амидогруппу -NH-CO- или сложноэфирную группу -O-СО-.
Примеры спейсеров, соединяющих NH- или O-группы кобаламина с хелатной группой, включают этилен, пропилен, бутилен или пентилен, или группы, в которых один атом углерода заменен на атом кислорода или азота, или один атом углерода заменен на атом кислорода или азота, или соседний атом углерода замещен на оксо.
Хелатное соединение содержит два, три или более донорных атомов, выбранных из азота, кислорода и серы, которые расположены на достаточном расстоянии для связывания с атомом металла. Прежде всего, хелатные соединения включают тридентатные хелатные соединения, содержащие три участка связывания с металлом, включающие донорные атомы азота, кислорода и/или серы, причем расстояние между ними является достаточным для связывания с атомами металла. Атомы азота в качестве донорных атомов входят в состав, например, алифатического амина, ароматического амина или азотсодержащего ароматического гетероцикла. Атомы кислорода в качестве донорных атомов входят в состав, например, спиртов, простых и сложных эфиров или карбоксильной группы. Атомы серы в качестве донорных атомов входят в состав, например, тиоэфиров или тиолов. Доноры присоединены, например, через алифатические углеводородные цепи или цепи, включающие амидные связи, и являются производными аминокислот, полиэфиров и т.п.
Предпочтительными хелатными соединениями являются хелатные соединения формул (II)-(IX). Карбоксильные группы могут присутствовать в виде сложных эфиров, которые отщепляются одновременно с образованием комплекса с атомом металла, при этом образуются координационные карбоксилатные группы. Такие этерифицированные хелатные соединения, то есть сложные эфиры, могут включать алкильные эфиры, в которых алкильная группа является прямой или разветвленной и включает от 1 до 25 атомов углерода, причем необязательно от одного до пяти атомов углерода заменены на атомы азота или кислорода, или один или два атома углерода заменены на атомы серы или фосфора, и указанные атомы необязательно замещены группами: фенил, пиридил, гидрокси, галоген, циано, оксо или амино. Сложный эфир включает также арильный или гетероарильный эфир, в котором арил или гетероарил включает от 3 до 10 атомов углерода или ноль, один или два атома кислорода, ноль, один, два или три атома азота или ноль или один атом серы. Такие сложноэфирные остатки также соответствующим образом замещены, чтобы обеспечить отщепление эфирных групп в определенных условиях реакции, например, как описано для сложноэфирных групп при их использовании в качестве защитных групп для карбоксильных групп, см. книгу Green T.W. и Wuts P.G.M., Protective groups in organic synthesis, Wiley (1999).
К этерифицированным хелатным соединениям предпочтительно относятся соединения в форме метилового, этилового или цианометилового эфиров.
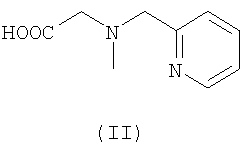
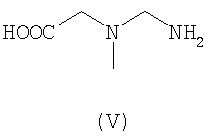
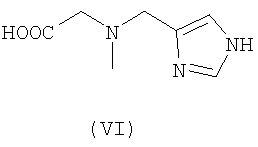
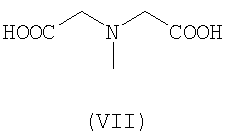
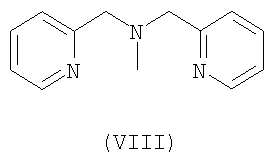
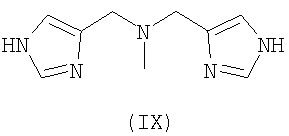
В качестве радиоактивных металлов используют радиоактивные изотопы, такие как 94mTc, 99mTc, 188Re, 186Re, 111In, 90Y, 64Cu, 67Cu и 177Lu, прежде всего 99mTc, 188Re, 186Re, и 111In.
К радиоактивным изотопам Со относятся, например, 57Со и 60Co.
К органическим группам с определенной конформацией, содержащим от 4 до 20 атомов углерода, относятся, например, алкил, циклоалкил, арил арилалкил, гетероарил или гетероарилалкил, необязательно замещенные группами: гидрокси, алкокси, оксо, амино, карбокси, карбамоил или алкоксикарбонил. Примеры арильных групп включают фенил, метилфенил, диметилфенил, гидроксифенил или нафтил. Примеры гетероарильных групп включают пиридил, пирролил, имидазолил или бензимидазолил. В алкильной цепи атомы углерода заменены на атомы азота или кислорода. Например, в алкильной цепи один атом углерода заменен на атом азота (или кислорода), а соседний атом углерода замещен оксогруппой с образованием карбоксамида (или сложноэфирной группы соответственно). Типичные примеры органических групп с определенной конформацией включают изобутил, трет-бутил, трет-пентил, орто-толил, орто-метилбензил или 2,6-диметилбензил.
Связующая группа Z, соединяющая RR со спейсерной хелатной группой или с антибиотиком или антипролиферативным агентом, означает связь или связующую группу, которую выбирают из группы, включающей фосфаты, фосфонаты, сложные эфиры карбоновых кислот или алкилены, содержащие от 1 до 10 атомов углерода или их комбинации. Такие связующие группы соединяют спейсерную хелатную группу или терапевтический агент, необязательно содержащий спейсерную группу, как определено выше, с атомом кислорода в составе кобаламина.
Соединения, которые модифицированы по группе RR, но в которых Rb, Rc, Rd и Re все означают водород, и которые связываются с транспортными белками ТС и в то же время проявляют ферментативную активность, исключены из области настоящего изобретения.
Выбор соединения по настоящему изобретению основан на следующих критериях:
(а) отсутствие связывающей аффинности или значительно сниженная связывающая аффинность в отношении TCII, например менее 20%, прежде всего менее 10%, предпочтительно менее 5%, более предпочтительно менее 2%, по сравнению с немодифицированным кобаламином, и
(б) активность в качестве заменителя витамина В12, определенная с использованием роста бактерий или клеточных линий млекопитающих, зависимых от витамина В12, составляет, например, более 2% активности, прежде всего более 10%, предпочтительно более 20% активности по сравнению с активностью витамина В12, которую проявляет немодифицированный кобаламин.
Для определения связывающей аффинности производных кобаламина (Cbl) с TCII анализ in vitro проводят с использованием частично очищенного TCII, выделенного из крови кролика. В качестве субстрата можно также использовать рекомбинантный TCII, который продуцируется в экспрессионной системе Е.coli.
Производные кобаламина по настоящему изобретению должны проявлять активность в качестве заменителей витамина В12. В связи с этим значительно снижается риск развития устойчивости, которая приводит к образованию клеток с высоким уровнем пролиферации. По всей вероятности мутантные клетки, в которых уже не происходит поглощения производных кобаламина с низкой связывающей активностью в отношении TCII или не обладающих такой активностью, теряют способность своих клеток-предшественников поддерживать высокий уровень пролиферации по механизму высокоэффективного поглощения витамина В12, независимого от TCII.
Для определения активности производных кобаламина в отношении витамина В12 использовали анализ в присутствии штамма Lactobacillus delbrueckii, который рекомендован во всем мире в качестве стандартного штамма для идентификации цианокобаламина (CN-Cbl). Добавление цианокобаламина в среду, не содержащую цианокобаламин, в ответ вызывает рост цианокобаламин-ауксотропного бактериального штамма, который можно определять методом количественного твердофазного метода диффузии на пластине. Этот метод анализа использовали для определения способности производного кобаламина (в %) замещать цианокобаламин в качестве жизненно необходимого катализатора.
Настоящее изобретение относится также к способу диагностики и к способу лечения неопластических заболеваний и локальных инфекций микроорганизмов у млекопитающих, включающий
(а) соблюдение диеты млекопитающим, не содержащей витамин В12,
(б) последующее ведение производного кобаламина по настоящему изобретению, содержащего диагностический или терапевтический агент,
и к применению производных кобаламина по изобретению в указанном способе.
Положительное влияние введения не связывающих TCII производных кобаламина на их биораспределение в организме млекопитающих после соблюдения диеты, не содержащей витамин, описано в таблице 1.
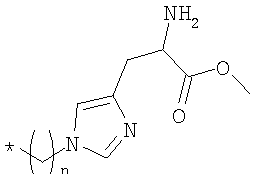
Пример 5: цианокобаламин-b-бутил-PAPAcet,
пример 6: цианокобаламин-b-бутиламинокарбоксиметил-His-OMe,
пример 8: цианокобаламин-с-бутил-PAPAcet,
пример 10: цианокобаламин-b-этил-PAMA-OEt,
пример 11: цианокобаламин-b-пропил-PAMA-OEt,
пример 12: цианокобаламин-b-бутил-PAMA-OEt,
пример 14: цианокобаламин-b-гексил-PAMA-OEt,
пример 18: цианокобаламин-d-пропил-РАМА-OEt,
пример 20: цианокобаламин-b-пропил-His-OMe,
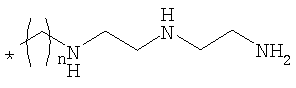
пример 22: цианокобаламин-b-этилтриамин,
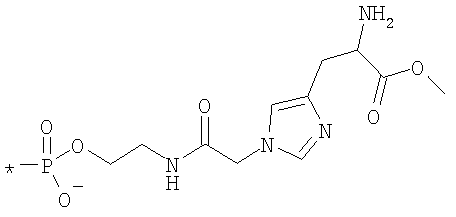
пример 25: цианокобаламин-5'-фосфоколамин-His-ОМе.
Результаты анализа биораспределения, приведенные в таблице 1, свидетельствуют о том, что не сязывающиеся с TCII соединения, например соединения по настоящему изобретению, описанные в примерах 10, 11, 12, 18 и 22, в значительной степени накапливаются в опухоли, в 5 раз или более по сравнению с кровью и по крайней мере больше, чем на половину количества, определенного в жизненно важных органах, почках и печени. Соединения, полученные в примерах 5, 6, 8, 14, 20 и 25, не относятся к соединениям по изобретению, так как они связываются с TCII и описаны только для сравнения.
Производные кобаламина по настоящему изобретению, прежде всего, являются ценными для диагностики и/или лечения агрессивных, быстро прогрессирующих опухолевых заболеваний, таких как рак. Соединения по изобретению можно использовать для обработки клеток с высокой степенью пролиферации в организме человека, включенных в процесс развития злокачественной опухоли, такой как, например, меланома, фибросаркома, карцинома яичников, карцинома поджелудочной железы, остеокарцинома и острый лейкоз, так как эти соединения не чувствительны к опосредованному TCII эндоцитозу. Способ по изобретению позволяет селективным образом защитить доброкачественные органы от опосредованного TCII разрушительного поглощения производных кобаламина, содержащих радиоактивный изотоп и/или нерадиоактивный агент, разрушающий клетки.
Соединения по настоящему изобретению можно использовать не только для получения визуализации опухолей и лечения рака, но и для визуализации и возможного лечения локальных инфекций микроорганизмов, зависящих от прямого поглощения кобаламинов в высокой степени.
Соединения по настоящему изобретению, содержащие антипролиферативный агент, можно использовать для транспорта агента в неактивной форме в гиперпролиферативные клетки, где он проявляет свое действие после внутриклеточного амидолиза.
Способ лечения опухолевого и/или инфекционного заболевания заключается в введении соединения по изобретению, содержащего пригодный терапевтический агент, в отдельности или в комбинации с одним или более других терапевтических агентов, причем предполагаемое комбинационное лечение проводят с использованием фиксированных комбинаций или соединение по изобретению и один или более других терапевтических агентов вводят поочередно или вводят независимо друг от друга или совместно вводят фиксированную комбинацию и один или более других терапевтических агентов. Соединение по изобретению можно вводить, кроме того или дополнительно, специально для лечения опухоли в комбинации с химиотерапией, иммунотерапией, хирургическим вмешательством или использовать комбинацию указанных способов лечения. В равной степени возможна долгосрочная терапия наряду с адьювантной терапией совместно с другими стратегиями лечения.
Кроме того, настоящее изобретение относится к фармацевтическим композициям, включающим производные кобаламина по изобретению, прежде всего к фармацевтическим композициям, пригодным для диагностики, и к фармацевтическим композициям, предназначенным для лечения.
Предпочтительны фармацевтические композиции для парентерального введения, такого как внутривенное, внутримышечное или подкожное введение. Композиции включают активный ингредиент в отдельности или в смеси с терапевтически примемлемым носителем. Доза активного ингредиента зависит от заболевания, которое требуется вылечить, и от субъекта, его возраста, массы тела и индивидуального состояния, от конкретных фармакокинетических данных и от способа введения.
Способы получения
Соединения по изобретению получают стандартными методами, известными в данной области химии.
Предпочтительно цианокобаламин, то есть соединение формулы (I), где Rb, Rc, Rd, Re и RR означают водород, а Х означает циано, гидролизуют в контролируемых условиях, например, разбавленной неорганической кислотой, и выделяют полученную смесь монокислот, в которых одна из карбамоильных групп CONH2 превращается в СООН. Бис-кислоты получают аналогичным способом.
Затем цианокобаламин-b-, с-, d- или е-кислота, то есть соединение формулы (I), где CONHRb, CONHRc, CONHRd или CONHRe заменены на СООН соответственно, а Х означает циано, взаимодействует с соответствующим амином Rb-NH2, Rc-NH2, Rd-NH2 и Re-NH2 соответственно, в стандартных условиях с образованием амида, например, как описано для синтеза пептидов. Функциональные группы в остатках Rb, Rc, Rd и Re, которые могут вступать в реакцию образования амидной связи, предпочтительно защищают, а затем после образования амидной связи защитные группы удаляют стандартным методом. Для получения соединений, в которых спейсер включает амидную группу, используют реакцию цианокобаламин-b-, с-, d- или е-кислоты с диамином H2N(CH2)nNH2 в стандартных условиях образования амидной связи, а затем конденсируют полученный H2N(CH2)n-цианокобаламин с соответствующей карбоновой кислотой в стандартных условиях образования амидной связи и получают соединение с заместителями Rb, Rc, Rd и Re соответственно.
Для получения соединений, в которых Rc не означает водород, предпочтительный способ включает образование о-лактона с последующим восстановительным раскрытием лактонового цикла по реакции, описанной в статье Brown и др., Inorg. Chem. 3038 (1995).
Для получения соединений, в которых RR не означает водород, цианокобаламин (или производное кобаламина, в котором Rb, Rc, Rd или Re не означают водород) взаимодействует с соединением RR-L, где L означает пригодную активирующую уходящую группу, с образованием сложноэфирной связи, например галоген, остаток смешанного ангидрида или другой активирующий остаток для образования эфиров карбоновой, фосфорной или фосфоновой кислот, которые обычно используются при синтезе пептидов и нуклеиновых кислот.
Следующие примеры приведены только для иллюстрации настоящего изобретения и не ограничивают его объем.
Примеры
В работе использовали реактивы фирмы Merck, Dietikon (CH), Aldrich или Fluka, Buchs (CH) марки х.ч. без дополнительной очистки.
ВОР - гексафторфосфат 1-бензотриазолилокситрис(диметиламино)фосфония
DCC - дициклогексилкарбодиимид
DIPEA - диизопропилэтиламин
EDC - 1-этил-3-(3-диметиламинопропил)карбодиимид
Fmoc - (9Н-флуорен-9-илметокси)карбонил
HOSu - N-гидроксисукцинимид
MES - 2-(4-морфолинил)этансульфоновая кислота
КТ - комнатная температура
TBTU - тетрафторборат бензотриазол-1-ил-N-тетраметилурония
ТЕАР - фосфат триэтиламмония
Теос - 2-триметилсилилэтоксикарбонил
ТФУ - трифторуксусная кислота
Анализ методом ЖХВР проводили в системе Merck-Hitachi L-7000, с радиометрическим детектором EG&G Berthold LB 508, с использованием колонок Waters Xterra RP8 (размер частиц 5 мкм, 1×100 мм), скорость потока 1 мл/мин, детектирование при 250 и 360 нм. Растворитель а означает водные буферные растворы. Натрий-ацетатный буферный раствор а получали при перемешивании 2,9 мл уксусной кислоты и 4,55 мл 2 М гидроксида натрия в 900 мл воды и 100 мл метанола. Трис-буферный раствор а получали при растворении трис(гидроксиметил)аминометана (605 мг) в воде, добавлении 2 М HCl до рН 8,2, доведения объема до 1000 мл и добавлении ацетонитрила (10 мл). Растворитель b всегда означает метанол.
Препаративную ЖХВР проводили в системе Varian Prostar, оборудованной двумя насосами Prostar 215 и детектором Prostar 320 УФ/вид., с использованием колонок Waters Xterra Prep RP8 (размер частиц 5 мкм, 3×100 мм и 30×100 мм). Скорость потока 4 мл/мин для колонки 3×100 мм и 30 мл/мин для колонки 30×100 мм.
Спектры УФ/вид. снимали на спектрометре Varian Cary 50, ИК-спектры снимали на спектрометре Bio-Rad FTS-45 образцы в виде таблеток KBr. Масс-спектры (MC(ESI)-ионизация электроспреем) снимали на спектрометре фирмы Merck Hitachi M-8000. При анализе соединений рения определяли массу для изотопа 187Re. ЯМР-спектры снимали на спектрометре Bruker DRX 500 МГц. Химические сдвиги определяли относительно остаточных протонов растворителя, которые использовали в качестве стандарта.
Небольшие количества производных кобаламина (несколько мг) обессоливали в водном растворе соединения с использованием картриджа Chromafix RP18ce, затем картридж тщательно промывали водой. Обессоленный продукт затем элюировали метанолом, растворитель удаляли в вакууме и продукт сушили в высоком вакууме. Большие количества (более 50 мг) обессоливали при экстракции фенолом, как описано в Meth. Enzymol. 18(3), стр.43, 1971.
Этиловый эфир (N-3-аминопропил-N-пиридин-2-илметиламино)уксусной кислоты (пропил-PAMA-Oet) получали, как описано для пентиланалога в статье Schibli и др. (Nucl. Med. Biol. 30, 465, 2003). Соединение циклизировали в щелочных условиях. Затем получали Вос-защищенное промежуточное соединение и хранили. Защитную Вос-группу удаляли непосредственно перед дальнейшей модификацией при перемешивании в разбавленной водной HCl. Этил- и гексилпроизводные получали аналогичным методом.
Карбонат Re[N-3-аминопропил-N-пиридин-2-илметиламино]уксусной кислоты ((CO)3) получали при взаимодействии (N-3-аминопропил-N-пиридин-2-илметиламино)уксусной кислоты (с удаленной защитной группой) с (Net4)2[Re(OH2)3(CO)3].
Трифторацетат метилового эфира 1-карбоксиметил-N-Fmoc-гистидина получали, как описано в статье Pak и др. (Chem. Eur. J., 9, 2053-2061, 2003). Противоион заменяли на хлорид при перемешивании соединения в 0,05 М HCl и последующем упаривании в вакууме при комнатной температуре.
Метиловый эфир 3-аминопропил-N-Теос-гистидин получали, как описано в статье Staveren и др. (Organic & Molecular Chemistry 2, 2593, 2004).
4-Нитрофениловый эфир 3-(N-2-цианоэтоксикарбонилметил)-N-пиридин-2-илметиламино)пропионовой кислоты получали, как описано в работе Kunze (Dissertation, University of Zurich, 2004).
Пример 1
Монокарбоновые кислоты цианокобаламина (b, d и е)
Витамин В12 (1,88 г, 1,39 ммоля) гидролизовали в HCl (0,1 М, 190 мл), как описано в статье Pathare и др. (Bioconjugate Chem. 217, 1996). Очистку проводили по модифицированной методике: после обессоливания при экстракции фенолом проводили хроматографию на колонке Dowex и получали три фракции. Первая фракция содержала в основном d-кислоту, вторая фракция содержала в основном b-кислоту и d-кислоту, а третья фракция содержала в основном b-кислоту и е-кислоту. Смесь b-кислоты и d-кислоты разделяли препаративной ЖХВР (колонка Waters Xterra Prep RP8, 5 мкм, 30×100 мм, градиент a/b 0,5% мин-1, в качестве начального буферного раствора использовали 100% ацетатный буферный раствор а). Смесь b-кислоты и е-кислоты разделяли в аналогичной системе, но в качестве растворителя а использовали Tris-буферный раствор. При этом получали цианокобаламин-b-кислоту (выход 280,6 мг, 14,9%), цианокобаламин-d-кислоту (выход 131,5 мг, 7,0%) и цианокобаламин-е-кислоту (выход 94,26 мг, 5,0%).
Пример 2
Цианокобаламин-b-(2-аминоэтил)амид(цианокобаламин-b-этиламин)
Цианокобаламин-b-(2-аминоэтил)амид получали, как описано в статье Pathare и др. (Bioconjugate Chem. 217, 1996) для синтеза додеканпроизводного. Этилендиамин (132 мг, 0,147 мл, 2,2 ммоля) растворяли в смеси ДМФА/Н2О (10 мл, 1:1 об./об.). Раствор подкисляли добавлением 1 М HCl до рН 5, в полученный раствор добавляли цианокобаламин-b-кислоту (60,0 мг, 44,4 мкмоля) и KCN (57 мг, 0,87 ммоля) и величину рН доводили до 5,5. Затем добавляли EDC (84,2 мг, 0,43 ммоля) и HOSu (50,6 мг, 0,44 ммоля). Смесь перемешивали при КТ в течение 3 сут и дополнительные порции EDC и HOSu добавляли с интервалом 24 ч. Смесь упаривали досуха в вакууме и очищали препаративной ЖХВР (натрий-ацетатный буферный раствор, градиент: 0,5% мин-1, в качестве начального буферного раствора использовали 100% ацетатный буферный раствор а), при этом получали 34 мг (выход 55%) цианокобаламин-b-(2-аминоэтил)амида. МС (МеОН, ESI-положит.): m/z 1398,8 [М+Н]+, 1420,1 [M+Na]+, 699,4 [M+H]2+, 711,1 [M+H+Na]2+.
Пример 3
Цианокобаламин-b-(4-аминобутил)амид[цианокобаламин-b-бутиламин]
Указанное в заголовке соединение получали, как описано выше для синтеза этиланалога. МС (МеОН, ESI-положит.): m/z 1427,1 [M+1]+, 714,5 [М+3]2+.
Пример 4
Цианокобаламин-b-этил-РАРАцет
Цианокобаламин-b-этиламин (пример 2, 24 мг, 17,2 мкмоля) растворяли в смеси ДМФА/ДМСО (5 мл, 4:1, об./об.). В полученную смесь добавляли 4-нитрофениловый эфир 3-[N-2-цианоэтоксикарбонилметил-N-пиридин-2-илметиламино]пропионовой кислоты (14 мг, 34,1 мкмоля) и DIPEA (5 мкл, 29 мкмолей). Смесь перемешивали при КТ в течение 24 ч, упаривали досуха в вакууме и очищали препаративной ЖХВР (натрий-ацетатный буферный раствор, градиент: 0,5% мин-1, в качестве начального буферного раствора использовали 100% ацетатный буферный раствор а), при этом получали 20 мг (выход 70%) цианокобаламин-b-этил-РАРАцет в виде твердого вещества красного цвета. МС (МеОН, ESI-положит.): m/z 1672,1 [М+Н]+, 836,9 [М+Н]2+.
Пример 5
Цианокобаламин-b-бутил-РАРАцет
Цианокобаламин-b-бутиламин (пример 3, 5,5 мг, 3,9 мкмоля) и 4-нитрофениловый эфир 3-[N-2-цианоэтоксикарбонилметил-N-пиридин-2-илметиламино]пропионовой кислоты (2,5 мг, 6,1 мкмоля) растворяли в сухом ДМСО (0,5 мл) и ДМФА (0,5 мл), добавляли DIPEA (5 мкл, 29 мкмолей) для рН от 8 до 9 и смесь перемешивали при комнатной температуре. Через 5 ч по данным анализа ЖХВР реакция полностью завершена. Растворитель частично упаривали в вакууме, при добавлении этилового эфира продукт выпадал в осадок. Суспензию центрифугировали и три раза декантировали, при этом получали тонкодисперсный порошок. После очистки препаративной ЖХВР (натрий-ацетатный буферный раствор, градиент: 0,5% мин-1, в качестве начального буферного раствора использовали 100% ацетатный буферный раствор а) получали чистый продукт (выход 2,7 мг, 41%). МС (ESI): m/z 850,1 [М+2]2+. УФ/вид.: λ/нм (ε/моль л-1 см-1)=279,1 (17300), 361,0 (31200), 519,9 (8700), 552,0 (9700).
Пример 6
Цианокобаламин-b-бутиламинокарбоксиметил-His-OMe
Раствор цианокобаламин-b-бутиламина (49,6 мг, 34,8 мкмоля) в сухом ДМСО (2 мл) добавляли в гидрохлорид метилового эфира 1-карбоксиметил-N-Fmoc-гистидина (35,5 мкмоля) и ВОР (46,2 мг, 104,4 мкмоля), затем добавляли DIPEA (12 мкл, 70,0 мкмоля) и раствор перемешивали при КТ в течение 16 ч. По данным анализа ЖХВР наблюдалось полное превращение исходного кобаламина в промежуточное Fmoc-защищенное соединение, которое при добавлении диэтилового эфира выпадало в осадок. Образовавшуюся суспензию центрифугировали и три раза декантировали, при этом получали тонкодисперсный порошок. Промежуточное соединение растворяли в ДМФА (5 мл) и добавляли пиперидин (225 мкл). После перемешивания при КТ в течение 1,5 ч продукт при добавлении диэтилового эфира выпадал в осадок. Полученную суспензию центрифугировали и три раза декантировали, при этом получали тонкодисперсный порошок. После очистки препаративной ЖХВР (натрий-ацетатный буферный раствор, градиент: 1% мин-1, в качестве начального буферного раствора использовали 100% ацетатный буферный раствор а) получали чистый продукт (выход 17,1 мг, 32,1%). УФ/вид.: λ/нм (ε/моль л-1 см-1)=279,1 (19200), 361,0 (24700), 521,0 (9600), 551,1 (10700).
Пример 7
Цианокобаламин-с-(4-аминобутил)амид(цианокобаламин-с-бутиламин)
Цианокобаламин-с-кислоту получали, как описано в статье Brown и др. (Inorg. Chem. 3038, 1995). 1,4-Диаминобутан (0,059 мл, 0,59 ммоля) растворяли в смеси ДМФА/Н2О (10 мл, 1:1 об./об.). Раствор подкисляли добавлением 1 М HCl до рН 5,2, в полученный раствор добавляли цианокобаламин-с-кислоту (16,0 мг, 11,8 мкмоля), KCN (15,3 мг, 0,236 ммоля), EDC (9,0 мг, 47,2 мкмоля) и HOSu (5,4 мг, 47,2 мкмоля). Смесь перемешивали при КТ в течение 4 сут. и добавляли дополнительные порции EDC и HOSu. Через 1 сут. снова добавляли дополнительные порции EDC и HOSu. Через 6 сут по данным анализа ЖХВР наблюдалось полное превращение кобаламин-производного. Смесь упаривали досуха в вакууме и очищали препаративной ЖХВР (колонка RP С18, в качестве буферного раствора а использовали 1 мМ HCl, градиент: от 20% метанола до 50% метанола в течение 30 мин), при этом получали 9,8 мг (выход 58%) цианокобаламин-с-бутиламина. МС (МеОН, ESI-положит.): m/z 1427,7 [М+2]+, 713,5 [M+1]2+.
Пример 8
Цианокобаламин-с-бутил-РАРАцет
Цианокобаламин-с-бутиламин (7,0 мг, 4,9 мкмоля) и 4-нитрофениловый эфир 3-[N-2-цианоэтоксикарбонилметил-N-пиридин-2-илметиламино]пропионовой кислоты (3,8 мг, 9,2 мкмоля) вводили в реакцию и очищали, как описано для синтеза цианокобаламин-b-бутил-РАРАцет (пример 5), при этом получали чистый продукт (выход 3,8 мг, 78%). МС (ESI): m/z 1701,0 [M+1]+, 850,1 [М+1]2+. УФ/вид.: λ/нм (ε/моль л-1 см-1)=278,1 (14500), 362,1 (25400), 550,0 (7900).
Пример 9
Цианокобаламин-b-бутил-РАРА-Re(СО)3
Цианокобаламин-b-бутиламин (пример 3, 24,6 мг, 17,2 мкмоля) и Re(СО)3(3-[N-карбоксиметил-N-пиридин-2-илметиламино]пропионовую кислоту) (9,1 мг, 17,2 мкмоля) растворяли в ДМСО, добавляли ВОР (22,9 мг, 51,7 мкмоля) и DIPEA (2,94 мкл, 17,2 мкмоля), смесь перемешивали при комнатной температуре. DIPEA и ВОР добавляли ежедневно в течение 4 сут. По данным анализа ЖХВР наблюдалось образование двух продуктов, которые при добавлении этилового эфира выпадали в осадок. Полученную суспензию центрифугировали и три раза декантировали, при этом получали тонкодисперсный порошок. После очистки препаративной ЖХВР (натрий-ацетатный буферный раствор, градиент: 1% мин-1, в качестве начального буферного раствора использовали 100% ацетатный буферный раствор а) получали фракцию основного продукта (выход 2,3 мг, 7,0%). МС (ESI): m/z 1917,5 [M+2]+, 959,9 [M+4]4+.
Пример 10
Цианокобаламин-b-этил-PAMA-OEt
Цианокобаламин-b-кислоту (20,0 мг, 14,8 мкмоля) растворяли в ДМСО (0,8 мл), затем последовательно добавляли ДМФА (2 мл) и NEt3 (0,1 мл). В другом сосуде приблизительно 5 экв. гидрохлорида этилового эфира (N-2-аминоэтил-N-пиридин-2-илметиламино)уксусной кислоты (этил-PAMA-OEt) (который получали расщеплением Вос-защищенного производного при перемешивании в смеси абсолютного EtOH/2 M HCl (7,5 мл, 4:1 об./об.) в течение ночи и удалением летучих компонентов в вакууме) растворяли в смеси ДМФА/NEt3 (4,5 мл, 8:1 об./об.). Оба раствора смешивали, добавляли TBTU (32,1 мг, 0,1 ммоля), перемешивали при КТ в течение 45 мин, растворитель удаляли в вакууме. Остаток очищали препаративной ЖХВР (натрий-ацетатный буферный раствор, градиент: 1% мин-1, в качестве начального буферного раствора использовали 100% ацетатный буферный раствор а), при этом получали 12 мг (выход 51%) цианокобаламин-b-этил-PAMA-OEt в виде твердого вещества красного цвета. МС (МеОН, ESI-положит.): m/z 1575,8 [M+H]+, 788,7 [М+Н]2+, 799,3 [M+H+Na]2+.
Пример 11
Цианокобаламин-b-пропил-PAMA-OEt
В цианокобаламин-b-кислоту (65,0 мг, 48,1 мкмоля) добавляли свежеприготовленный раствор этилового эфира (N-3-аминопропил-N-пиридин-2-илметиламино)уксусной кислоты (361 мкмоль) в воде (1 мл), затем добавляли EDC (46,1 мг, 240 мкмолей) и подщелачивали 0,1 M NaOH до рН 5,5. После перемешивания при КТ в течение 15 ч по данным анализа ЖХВР (натрий-ацетатный буферный раствор) наблюдалось образование продукта приблизительно 50%. Затем добавляли еще одну порцию EDC
(46,1 мг, 240 мкмолей), но при дальнейшем перемешивании при комнатной температуре дополнительного образования продукта не наблюдалось. Растворитель удаляли в вакууме и остаток очищали препаративной ЖХВР (градиент: а/b 0,5% мин-1, в качестве начального буферного раствора использовали 100% ацетатный буферный раствор а). Основную фракцию собирали, растворитель удаляли в вакууме и продукт обессоливали, при этом получали цианокобаламин-b-пропил-PAMA-OEt (выход 25,8 мг, 16,2 мкмоля, 33,3%). МС (ESI): m/z 806,5 [M+1+Na]2+, 795,6 [М+2]2+. УФ/вид.: λ/нм (ε/моль л-1 см-1)=278,0 (8500), 361,1 (26500), 549,1 (8000).
Пример 12
Цианокобаламин-b-бутил-PAMA-OEt
Цианокобаламин-b-кислоту (20,0 мг, 14,8 мкмоля) растворяли в ДМСО (0,8 мл), затем последовательно добавляли ДМФА (2 мл) и NEt3 (0,1 мл). В другом сосуде приблизительно 5 экв. гидрохлорида этилового эфира (N-4-аминобутил-N-пиридин-2-илметиламино)уксусной кислоты (бутил-PAMA-OEt) (который получали расщеплением Вос-защищенного производного при перемешивании в смеси абсолютного EtOH/2 M HCl (7,5 мл, 4:1 об./об.) в течение ночи и удалением летучих компонентов в вакууме) растворяли в смеси ДМФА/NEt3 (4,5 мл, 8:1 об./об.). Оба раствора смешивали, добавляли TBTU (32,1 мг, 0,1 ммоля), перемешивали при КТ в течение 45 мин, растворитель удаляли в вакууме. Остаток очищали препаративной ЖХВР (натрий-ацетатный буферный раствор, градиент: 1% мин-1, в качестве начального буферного раствора использовали 100% ацетатный буферный раствор а), при этом получали 15 мг (выход 63%) цианокобаламин-b-бутил-PAMA-OEt в виде твердого вещества красного цвета.
Пример 13
Цианокобаламин-b-бутил-РАМА-ОН
9Н-Флуорен-9-илметиловый эфир бромуксусной кислоты получали из бромацетилбромида и 9Н-флуоренилметанола в сухом ТГФ при 0°С. Вос-Бутил-PAMA-OFm (9Н-флуорен-9-илметиловый эфир [(4-трет-бутоксикарбониламинобутил)пиридин-2-илметиламино]уксусной кислоты) получали из Boc-NH-(CH2)4NH2, пиридин-2-альдегида и 9Н-флуорен-9-илметилового эфира бромуксусной кислоты по методике, описанной в статье Schibli и др. (Nucl. Med. Biol. 30, 465, 2003) для синтеза Вос-пентил-РАМА-ОМе.
Цианокобаламин-b-кислоту (20,0 мг, 14,8 мкмоля) растворяли в ДМСО (0,8 мл), затем последовательно добавляли ДМФА (2 мл) и NEt3 (0,1 мл). В другом сосуде приблизительно 5 экв. 9Н-флуорен-9-илметилового эфира [(4-аминобутил)пиридин-2-илметиламино] уксусной кислоты (бутил-PAMA-OFm) (который получали расщеплением Вос-защищенного производного при перемешивании в смеси трифторуксусная кислота/CH2Cl2 (4 мл, 1:2 об./об.) в течение 1 ч и удалением летучих компонентов в вакууме) растворяли в смеси ДМФА/NEt3 (4,5 мл, 8:1 об./об.). Оба раствора смешивали, добавляли TBTU (32,1 мг, 0,1 ммоля), перемешивали при КТ в течение 45 мин, растворитель удаляли в вакууме. Остаток очищали препаративной ЖХВР (натрий-ацетатный буферный раствор, градиент: 1,5% мин-1, в качестве начального буферного раствора использовали 100% ацетатный буферный раствор а), при этом получали 15 мг цианокобаламин-b-бутил-PAMA-OFm в виде твердого вещества красного цвета.
Цианокобаламин-b-бутил-PAMA-OFm (15 мг) растворяли в 3 мл смеси HNEt2/ДМФА (2:1 об./об.) и перемешивали при КТ в течение 2 ч. Растворитель удаляли в вакууме, остаток очищали препаративной ЖХВР (натрий-ацетатный буферный раствор, градиент: 1,0% мин-1, в качестве начального буферного раствора использовали 100% ацетатный буферный раствор а), при этом получали 9 мг цианокобаламин-b-бутил-РАМА-ОН в виде твердого вещества красного цвета.
Пример 14
Цианокобаламин-b-гексил-PAMA-OEt
Цианокобаламин-b-кислоту (20,0 мг, 14,8 мкмоля) растворяли в ДМСО (0,8 мл), затем последовательно добавляли ДМФА (2 мл) и NEt3 (0,1 мл). В другом сосуде приблизительно 5 экв. гидрохлорида этилового эфира (N-6-аминогексил-N-пиридин-2-илметиламино)уксусной кислоты (гексил-PAMA-OEt) (который получали расщеплением Вос-защищенного производного при перемешивании в смеси абсолютного EtOH/2 M HCl (7,5 мл 4:1 об./об.) в течение ночи и удалением летучих компонентов в вакууме) растворяли в смеси ДМФА/NEt3 (4,5 мл, 8:1 об./об.). Оба раствора смешивали, добавляли TBTU (32,1 мг, 0,1 ммоля), перемешивали при КТ в течение 45 мин, растворитель удаляли в вакууме. Остаток очищали препаративной ЖХВР (натрий-ацетатный буферный раствор, градиент: 1,0% мин-1, в качестве начального буферного раствора использовали 100% ацетатный буферный раствор а), при этом получали 10 мг (выход 41%) цианокобаламин-b-гексил-PAMA-OEt в виде твердого вещества красного цвета. МС (МеОН, ESI-положит.): m/z 816,9 [M+2H]+, 1632 [M+H]+.
Пример 15
Цианокобаламин-b-этил-РАМА-Re(СО)3
Цианокобаламин-b-этил-PAMA-OEt (пример 10, 11 мг, 7,0 мкмоля) растворяли в 4 мл 2 М раствора NaHCO3, добавляли раствор (NEt4)2[ReBr3(СО)3] (11 мг, 14,2 мкмоля) в 1,5 мл МеОН. Смесь нагревали при 85°С в течение 1 ч, охлаждали до КТ и очищали препаративной ЖХВР (натрий-ацетатный буферный раствор, градиент: 2,0% мин-1, в качестве начального буферного раствора использовали буферный раствор а), выход 11 мг, 86%.
Пример 16
Цианокобаламин-b-пропил-РАМА-Re(СО)3
Цианокобаламин-b-кислоту (26,7 мг, 19,8 мкмоля), карбонат Re[N-3-аминопропил-N-пиридин-2-илметиламино]уксусной кислоты(СО)3 (29,2 мг, 60 мкмолей), EDC (11,5 мг, 60 мкмолей) и HOSu (6,9 мг, 60 мкмолей) растворяли в смеси воды (5 мл) и ДМСО (0,5 мл), рН доводили разбавленной HCl и NaOH до 5,5. После перемешивания при КТ в течение 5 ч по данным анализа ЖХВР (ацетатный буферный раствор) наблюдалось образование продукта приблизительно 33%. В смесь снова добавляли EDC и HOSu. Смесь перемешивали при комнатной температуре в течение 3 сут, при этом дополнительные порции EDC и HOSu добавляли с интервалом 24 ч. Воду удаляли в вакууме, продукт осаждали добавлением диэтилового эфира. Масляную суспензию центрифугировали и декантировали, осадок дважды промывали диэтиловым эфиром до образования тонкодисперсного осадка. Неочищенный продукт сушили в высоком вакууме, очищали препаративной ЖХВР (градиент: а/b, 1% мин, в качестве начального буферного раствора использовали 100% ацетатный буферный раствор а) и продукт обессоливали, при этом получали цианокобаламин-b-пропил-РАМА-Re(СО)3 (выход 9,1 мг, 23%). МС (ESI): m/z 1831,7 [M+1]+, 916,1 [М+1]2+. УФ/вид.: λ/нм (ε/моль л-1 см-1)=278,0, 361,1, 519,9, 551,1.
Пример 17
Цианокобаламин-b-гексил-PAMA-Re(CO)3
Цианокобаламин-b-кислоту (20,0 мг, 14,8 мкмоля) растворяли в ДМСО (0,8 мл), затем последовательно добавляли ДМФА (2 мл) и NEt3 (0,1 мл). В другом сосуде приблизительно 5 экв. карбоната Re([N-3-аминопропил-N-пиридин-2-илметиламино)уксусной кислоты(СО)3·CF3COOH (который получали отщеплением Вос-защитной группы в составе комплекса в смеси CH2Cl2 и ТФУ (2:1 об./об.) в течение 1 ч при 0°С и удалением летучих компонентов при КТ в вакууме) растворяли в смеси ДМФА/NEt3 (4,5 мл, 8:1 об./об.). Оба раствора смешивали, добавляли TBTU (32,1 мг, 0,1 ммоля), перемешивали при КТ в течение 45 мин, растворитель удаляли в вакууме. Остаток очищали препаративной ЖХВР (натрий-ацетатный буферный раствор, градиент: 2,0% мин-1, в качестве начального буферного раствора использовали 100% ацетатный буферный раствор а), при этом получали 11 мг (выход 40%) цианокобаламин-b-гексил-РАМА-Re(СО)3. МС (МеОН, ESI-положит.): m/z 936,5 [М+2H]2+, 948,3 [M+H+Na]2+, 1873,8 [M+H]+.
Пример 18
Цианокобаламин-d-пропил-РАМА-OEt
Цианокобаламин-d-кислоту (9,3 мг, 6,9 мкмоля) вводили в реакцию с этиловым эфиром (N-3-аминопропил-N-пиридин-2-илметиламино)уксусной кислоты (7 мкмолей) и EDC (6,6 мг, 34 мкмоля), как описано для синтеза цианокобаламин-b-пропил-PAMA-OEt (пример 11), при этом получали указанный в заголовке продукт (выход 3,6 мг, 33%). МС (ESI): m/z 1612 [M+Na]+, 1590 [M+1]+, 806 [M+1+Na]2+, 795,1 [M+2]2+. УФ/вид.: λ/нм (ε/моль л-1 см-1)=279,0 (13400), 361,1 (23300), 549,1 (7200).
Пример 19
Цианокобаламин-d-пропил-РАМА-Re(СО)3
Цианокобаламин-d-кислоту (20,0 мг, 14,8 мкмоля) растворяли в ДМСО (1,5 мл), затем последовательно добавляли ДМФА (2 мл) и NEt3 (0,1 мл). В другом сосуде приблизительно 5 экв. карбоната Re([N-3-аминопропил-N-пиридин-2-илметиламино)уксусной кислоты(СО)3 растворяли в смеси ДМФА/NEt3 (4,5 мл, 8:1 об./об.). Оба раствора смешивали, добавляли TBTU (32,1 мг, 0,1 ммоля), перемешивали при КТ в течение 45 мин, растворитель удаляли в вакууме. Остаток очищали препаративной ЖХВР (натрий-ацетатный буферный раствор, градиент: 2,0% мин-1, в качестве начального буферного раствора использовали 100% ацетатный буферный раствор а), при этом получали 20 мг (выход 73%) цианокобаламин-d-пропил-РАМА-Re(СО)3.
Пример 20
Цианокобаламин-b-пропил-His-OMe
Цианокобаламин-b-кислоту (20,0 мг, 14,8 мкмоля) растворяли в ДМСО (0,8 мл), затем последовательно добавляли ДМФА (2 мл) и NEt3 (1 мл). В другом сосуде приблизительно 4 экв. метилового эфира 3-аминопропил-N-Teoc-гистидина растворяли в ДМФА. Оба раствора смешивали, добавляли TBTU (32,1 мг, 0,1 ммоля), перемешивали в течение 45 мин, затем упаривали досуха в вакууме. Остаток очищали препаративной ЖХВР (натрий-ацетатный буферный раствор, градиент: 2,0% мин-1, в качестве начального буферного раствора использовали буферный раствор а), при этом получали 16 мг твердого вещества красного цвета (выход 67%). МС (МеОН, ESI-положит.): m/z 1710,4 [М+Н]+, 855,0 [M+2H]+, 866,7 [M+Na+H]2+.
Образец указанного Теос-защищенного соединения (19 мг) растворяли в смеси ТФУ/CH2Cl2 (4:1, об./об.) при 0°С, перемешивали при этой температуре в течение 4 ч, при этом по данным анализа ЖХВР наблюдалось полное превращение исходного материала. Растворитель удаляли в вакууме при КТ, в остаток добавляли Et2O, затем растворитель удаляли в вакууме. Указанную стадию повторяли три раза для удаления следов ТФУ. Остаток очищали препаративной ЖХВР (натрий-ацетатный буферный раствор, градиент: 0,5% мин-1, в качестве начального буферного раствора использовали 100% буферный раствор а), при этом получали 11 мг указанного в заголовке соединения. МС (МеОН, ESI-положит.): m/z 1565,2 [M+H]+, 1587,2 [M+Na]+, 783,4 [M+2H]2+, 794,1 [M+Na+H]2+.
Пример 21
Цианокобаламин-b-пропил-His-Re(СО)3
Цианокобаламин-b-кислоту (20,0 мг, 14,8 мкмоля) растворяли в ДМСО (0,8 мл), затем последовательно добавляли ДМФА (2 мл) и NEt3 (0,1 мл). В другом сосуде приблизительно 5 экв. карбоната Re (метилового эфира 3-аминопропил-N-Теос-гистидина)(СО)з]·CF3COOH (который получали отщеплением Вос-группы в составе защищенного комплекса в CH2Cl2 и ТФУ (2:1 об./об.) в течение 1 ч при 0°С и удалением летучих компонентов при КТ в вакууме) растворяли в смеси ДМФА/NEt3 (4,5 мл, 8:1 об./об.). Оба раствора смешивали, добавляли TBTU (32,1 мг, 0,1 ммоля), перемешивали при КТ в течение 45 мин, растворитель удаляли в вакууме. Остаток очищали препаративной ЖХВР (натрий-ацетатный буферный раствор, градиент: 2,0% мин-1, в качестве начального буферного раствора использовали 100% буферный раствор а), при этом получали 7 мг (выход 73%) цианокобаламин-b-пропил-His-Re(СО)3. МС (МеОН, ESI-положит.): m/z 911.6 [M+2H]2+, 923,2 [M+H+Na]2+, 933,9 [M+2Na]2+, 1822,1 [M+H]+, 1845,6 [M+Na]+.
Пример 22
Цианокобаламин-b-этилтриамин
Триэтилентетрамин (55,4 мкл, 369 мкмолей) растворяли в смеси ДМФА (2,5 мл) и воды (2,5 мл), добавляли KCN (9,6 мг, 147 мкмолей) и подкисляли раствор добавлением водной HCl до рН 6. В полученный раствор добавляли цианокобаламин-b-кислоту (10,0 мг, 7,4 мкмоля), EDC (5,7 мг, 29 мкмолей) и HOSu (3,4 мг, 29 мкмолей). Аналогичное количество EDC и HOSu добавляли через 6, 24, 48 и 120 ч. По данным анализа ЖХВР (натрий-ацетатный буферный раствор) наблюдалось медленное образование продукта, т.е. через 48 ч получали 75%, но при дальнейшем перемешивании дополнительного образования продукта не наблюдалось. После перемешивания в течение 144 ч растворитель удаляли в вакууме и продукт очищали препаративной ЖХВР, элюент: водный раствор 0,1% ТФУ в качестве буферного раствора а, и метанол в качестве растворителя b, (градиент: 1% мин-1, в качестве начального буферного раствора использовали 80% буферный раствор а), при этом получали 7,5 мг (выход 55%) в виде трифторацетата цианокобаламин-b-этил-триамина (ЗТФУ). МС (ESI): m/z 743,1 [М+2]2+. УФ/вид.: λ/нм (ε/моль л-1 см-1)=278,0 (13000), 316,0 (23100), 519,0 (6500), 549,0 (7200).
Пример 23
Цианокобаламин-b-этилтриамин-Re(СО)3
Цианокобаламин-b-этилтриамин (5 мг, 2,7 мкмоля) и (Et4N)2[ReBr3(СО)3] (2,2 мг, 2,9 мкмоля) перемешивали в фосфатном буферном растворе, рН 7,4 (0,1 М, 0,33 мл) при 50°С. Через 1 ч по данным анализа ЖХВР наблюдалось полное превращение исходных материалов в продукт. Через 4 ч реакционную смесь обессоливали, при этом получали продукт, который по данным анализа ЖХВР содержал смесь двух стереоизомеров в соотношении приблизительно 2:1. Аналогичный состав стереоизомеров установлен при использовании меченного цианокобаламин-b-этилтриамина (99mTc). МС (ESI): m/z 1755,9 [М+1], 878,5 [М+2]2+.
Пример 24
Цианокобаламин-5'-фосфоколамин
Раствор цианокобаламина (30 мг, 22,14 мкмоля), DCC (457 мг, 2,214 ммоля) и N-Fmoc-фосфоколамина (78,9 мг, 217,2 мкмоля) в сухом ДМФА (2 мл) и сухом пиридине (1 мл) перемешивали в атмосфере N2 при комнатной температуре в течение 24 ч. После добавления 2 мл воды выпавшую в осадок дициклогексилмочевину отфильтровывали, а воду и пиридин упаривали при 60°С при пониженном давлении. Полученный раствор разбавляли до объема 8 мл раствором пиперидина в ДМФА (5%) и перемешивали при комнатной температуре в течение 2,5 ч. Продукт высаживали в диэтиловом эфире, центрифугировали и несколько раз промывали. Неочищенный продукт очищали препаративной ЖХВР (градиент: 100%→20% а, 0%→80% МеОН в течение 30 мин, a=0,1% АсОН, 10% ацетонитрил в воде, скорость потока 10 мл/мин, колонка M&N VP 250/21 Nucleosil 100-7 С18). При этом получали продукт в виде твердого вещества красного цвета (82%). МС (ESI+, МеОН): m/z 1478 [М+1]+, 762 [M+2+2Na]2+.
31Р-ЯМР (500, CD3OD): δ 0,00 (s, 1P), 0,53 (s, 1P).
Пример 25
Цианокобаламин-5'-фосфоколамин-His-OMe
Цианокобаламин-5'-фосфоколамин (50 мг, 33,8 мкмоля) и гидрохлорид метилового эфира 1-(карбоксиметил)-Н-Fmoc-гистидина (25 мг, 50,7 мкмоля) растворяли в сухом ДМСО (4 мл) и рН доводили до 6-7 добавлением 24 мкл DIPEA. В раствор добавляли ВОР (45 мг, 101,5 мкмоля) в виде твердого вещества и перемешивали при КТ. Через 1 ч образовывался кислотный раствор, который нейтрализовали. Через 5 ч по данным аналитической ЖХВР наблюдалось полное потребление исходного материала. После высаживания осадка в диэтиловом эфире, в неочищенном продукте удаляли защитные группы в смеси ДМФА и пиперидина (10 мл, 1:1) в течение 1,5 ч. После повторного высаживания и очистки препаративной ЖХВР, как описано для цианокобаламин-5'-фосфоколамина (пример 24), получали требуемый продукт (выход 46%). МС (ESI+, МеОН): m/z 1690 (M+1)+, 845,6 (M+2)+.
31Р-ЯМР (500, D2O): δ -2,16, -0,37.
Пример 26
Цианокобаламин-5'-фосфоколамин-His-Re(СО)3
Указанное соединение получали аналогично тому, как описано для цианокобаламин-5'-фосфоколамин-His-ОМе (пример 25), но при замене гидрохлорида метилового эфира 1-(карбоксиметил)-N-Fmoc-гистидина на комплекс Re(СО)3 и 1-(карбоксиметил)гистидина. При этом получали очищенный продукт (37%). МС (ESI+, МеОН): m/z 1945, 929 [M+1]+.
31Р-ЯМР (500, D2O, 333 K): δ 0,97, 2,23.
ИК (KBr, см-1): 3400, 2128, 2020, 1901, 1902, 1664, 1499, 1399, 1219, 1073.
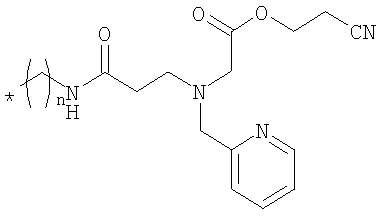
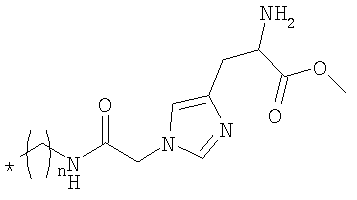
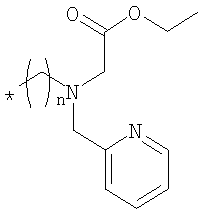
Пример 27
Общая методика введения метки
Синтез предшественника [99mTc(OH2)3(CO)3]+ получали из [99mTcO4]- с использованием набора боранокарбонатных реагентов, как описано в статье Alberto и др., J. Am. Chem. Soc. 123, 3135-3136. Стеклянный флакон объемом 10 мл с резиновой пробкой продували азотом, затем добавляли 20 мкл раствора производного цианокобаламина (0,01 М в воде), 20 мкл буферного раствора MES (1,0 М) и 200 мкл раствора [99mTc(ОН2)3(CO)3]+, реакционную смесь выдерживали при 75°С в течение от 1 до 2 ч. Завершение превращения атомов 99mTc контролировали методом ЖХВР с использованием γ-детектора. В этих условиях наблюдалось отщепление сложноэфирных защитных групп в составе хелатной группы и образование карбоксилатных комплексов.
Для испытаний in vivo и для определения связывания с транспортными векторами требуется чрезвычайно высокая удельная активность. В связи с этим 100 мкл меченого раствора вкалывали в систему ЖХВР для отделения горячего (меченого) производного кобаламина от холодного (немеченого) производного. Перед внутривенной инъекцией животному фракцию с наибольшей γ-радиоактивностью (приблизительно 300 мкл) разбавляли физиологическим раствором до концентрации 10 мкКи на одно животное. При хроматографии использовали следующие условия разделения: ацетатный буферный раствор, колонка XTerra RP8, градиент: 0% метанол (0 мин), 30% метанол (15 мин), 100% метанол (25 мин) для элюции b- и d-производных, и система ТЕАР, как описано в статье Schibli и др., Bioconjugate Chem. 343-351 (2000).
Пример 38
Выделение транскобаламина II (TCII) из цельной крови кролика
TCII очищали аффинной хроматографией с использованием цианокобаламин-агарозы (фирмы Sigma). Агарозу (5 мл) промывали сначала 200 мл 50 мМ трис/1 M NaCl, рН 8,0, затем 200 мл 0,1 М глицин/0,1 М глюкоза/1 М NaCl, рН 10, и снова 200 мл 50 мМ трис/1 M NaCl. 200 мл дважды центрифугированной цельной крови (сначала при 5000 об/мин в течение 15 мин, затем при 20000 об/мин в течение 20 мин при 4°С) наносили на аффинную колонку и колонку промывали последовательно, как описано выше. Связанный TCII элюировали 20 мл 4,0 М гидрохлоридом гуанидина/50 мМ трис, рН 8,0, затем 7,5 М гидрохлоридом гуанидина/50 мМ трис, рН 8,0. Основное количество TCII элюируется в 4 М гидрохлориде гуанидина. Образцы диализовали против избыточного количества воды в течение 2 сут при 4°С. Типичный выход составляет 5-30 нмоль/л или 7,5-10 мкг ТСII (ММ 50 кДа) из одного кролика.
Пример 29
Получение транскобаламина II из бактерий (рекомбинантный TCII)
кДНК транскобаламина II экспрессировали в Е.coli, штамм FA113, модификация К12 с двойным отсутствием trxB-гена и gor-гена, в котором цитоплазма содержит окислители и образуются дисульфидные связи. Белок, транскобаламин II, содержит участок расщепления протеазой PreScission и Т-концевой гистидиновый фрагмент. Белок выделяли из растворимой фракции экстрактов Е.Coli с использованием никель-хелатной хроматографии на сефарозе. Цианокобаламин отделяли от транскобаламина II, связанного с хелатной колонкой, при элюции 8 М мочевиной, а затем элюировали транкобаламин II имидазолом. В некоторых препаратах гистидиновый фрагмент отщепляли специфической протеазой.
Пример 30
Получение транскобаламина I (TCI, гаптокоррин)
В качестве источника транскобаламина I использовали слюну вегетарианцев-добровольцев. Слюну центрифугировали при 20000 об/мин в течение 20 мин при 4°С, смешивали с ФСБ 1:1 и стерилизовали фильтрованием. Связывающая активность транскобаламина I обычно составляет 10 нг/мл.
Пример 31
Взаимодействие производных цианокобаламина с транспортными белками TCI и TCII (фиг.1 и фиг.2)
Взаимодействие радиоактивно меченных производных цианокобаламина (57Со, 99mTc, 188Re, 111In) определяли методом сдвига пиков на колонке для гель-фильтрации. Радиоактивно меченный цианокобаламин (от 0,05 до 1 нг) инкубировали в присутствии избытка транспортных белков в течение 15 мин при комнатной температуре. Полученную смесь наносили на колонку для гель-фильтрации (супердекс 75, фирмы Amersham Biosciencies) и промывали рабочим буферным раствором ФСБ, содержащим 0,1% твин 20. Молекулярная масса биологически активного цианокобаламина, связывающегося с транспортными белками, изменяется (наблюдается сдвиг пика с ММ приблизительно 1,4 кДа до 40-70 кДа в зависимости от транспортного белка). Титрование связывающей активности транспортных белков проводили с использованием 57Со-цианокобаламина (фирмы ICN Biomedicals GmbH, Германия; 10 мкКи/50 нг).
Пример 32
Введение метки в производные цианокобаламина с использованием 188Re-трикарбонила
Получение 188Re-трикарбонила и меченых производных цианокобаламина проводили в одну стадию в одном сосуде. 7,5 мг BH3NH3 смешивали с 20 мг аскорбата натрия, 100 мкл производного цианокобаламина (10-3 М), 900 мкл элюата из источника [188ReO4]- (0,9% солевой раствор, от 40 мКи до 270 мКи), 20 мг H3PO4 (85%) и через смесь пропускали монооксид углерода (СО) в течение 20 мин. Смесь нагревали в течение от 30 мин до 2 ч при 90°С. Меченый цианокобаламин отделяли от немеченого на колонке для обращенно-фазовой ЖХВР (Waters Xterra RP8) в фосфатном буферном растворе в линейном градиенте метанола. Активную фракцию разбавляли нормальным солевым раствором перед внутривенной инъекцией до концентрации 10 мкКи на одно животное для визуализации и вплоть до 2 мКи для лечения.
Пример 33
Чувствительность шаровидных опухолевых клеток к ионизирующей радиации
Принятой в радиобиологии альтернативной моделью in vivo при исследовании облучения являются шаровидные клетки, так как эти клетки и ксенотрансплантаты, привитые в ткань мыши, проявляют аналогичную ответную реакцию на радиацию. Многоклеточные опухолевые шаровидные клетки выращивали во вращающихся колбах при непрерывном перемешивании при 37°С до среднего диаметра 400 мкм. Шаровидные клетки собирали, промывали свежей средой и затем инкубировали в течение 1 ч в присутствии холодного или 188Re-меченного производного цианокобаламина в 24-луночных культуральных планшетах с плоским дном. Диапазон радиоактивности составлял от 1 мкКи до 20 мкКи в лунке. Цитотоксичность оценивали с использованием флуоресцентных маркеров жизнеспособности клеток по диаметру шаровидных клеток и методом клоногенного анализа диспергированных шаровидных клеток в полутвердом агаре.
Пример 34
Биораспределение радиоактивно меченных производных цианокобаламина в тканях мыши (фиг.3, 4, 5, 6)
Для исследования биораспределения использовали 57Со-цианокобаламин, который смешивали в количестве 0,2 мкКи/1 нг с 180 мкл нормального солевого раствора и вводили внутривенно мышам balb/c с привитой опухолью (меланома B16-F10 у сингенных мышей). Через определенный период времени (от 5 мин до 24 ч) животных забивали, органы взвешивали и определяли радиоактивность на счетчике γ-излучения. Для исследования биораспределения использовали также 99mTc-меченный цианокобаламин, который смешивали в количестве 10 мкКи/0,5 нг с нормальным солевым раствором и анализировали, как описано выше. Для исследования биораспределения использовали также 111In-меченный цианокобаламин, который смешивали в количестве 2 мкКи/5 нг с нормальным солевым раствором и анализировали, как описано выше. Для исследования влияния не содержащего витамина В12 корма биораспределение меченого цианокобаламина сравнивали с мышами, которые получали нормальный корм, и с биораспределением у мышей, которые получали корм, не содержащий витамина В12, в течение 2 недель.
Пример 35
Лечение мышей с привитой опухолью с использованием 88Re-меченных производных цианокобаламина
Для испытаний при лечении меланому у сингенных мышей balb/c выращивали до размера приблизительно 200 мг (измеренную штангенциркулем). Затем животным вводили внутривенно возрастающие дозы (от 0,1 до 2 мКи) композиций радиоактивно меченного цианокобаламина и холодного цианокобаламина. Размер опухоли измеряли штангенциркулем. Если размер опухоли достигал 800 мг, животных забивали. В серии экспериментов животных лечили по фракционному курсу: меченый цианокобаламин вводили три раза с интервалом 1 неделя. Животных обследовали в течение 60 сут и регистрировали возобновление роста опухолей.
| название | год | авторы | номер документа |
|---|---|---|---|
| МЕЧЕНЫЕ РАДИОАКТИВНЫМ ИЗОТОПОМ ПЕПТИДЫ ДЛЯ ДИАГНОСТИКИ И ТЕРАПИИ | 1996 |
|
RU2171117C2 |
| СА-IX СПЕЦИФИЧЕСКИЕ РАДИОФАРМПРЕПАРАТЫ ДЛЯ ЛЕЧЕНИЯ И ВИЗУАЛАЗИЦИИ ЗЛОКАЧЕСТВЕННЫХ ОПУХОЛЕЙ | 2009 |
|
RU2539565C2 |
| ТЕХНЕЦИЙ- И РЕНИЙ-БИС(ГЕТЕРОАРИЛЬНЫЕ) КОМПЛЕКСЫ И МЕТОДЫ ИХ ПРИМЕНЕНИЯ ДЛЯ ИНГИБИРОВАНИЯ PSMA | 2009 |
|
RU2532912C2 |
| ПРОИЗВОДНЫЕ ХИНОЛИНПИРРОЛИДИН-2-ОНА И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2771314C1 |
| ПРОИЗВОДНЫЕ КОБАЛАМИНА И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, ВЫЗВАННЫХ ОТСУТСТВИЕМ ПОСТУПЛЕНИЯ ВИТАМИНА B | 2018 |
|
RU2777536C2 |
| КОМПОЗИЦИИ ДЛЯ НАРУЖНОГО ПРИМЕНЕНИЯ, СОДЕРЖАЩИЕ АДЕНОЗИЛКОБАЛАМИН, ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ КОЖИ | 2006 |
|
RU2379039C2 |
| ПРОИЗВОДНЫЕ ПИРАЗИН-2-КАРБОКСАМИДА В КАЧЕСТВЕ АНТАГОНИСТОВ mGluR5 | 2006 |
|
RU2407739C2 |
| ИНДОЛИЛМАЛЕИМИДНЫЕ ПРОИЗВОДНЫЕ | 2005 |
|
RU2373201C2 |
| АМПЛИФИКАЦИЯ СИСТЕМЫ ПОГЛОЩЕНИЯ ВИТАМИНА B ПРИ ПОМОЩИ ПОЛИМЕРОВ | 1994 |
|
RU2139732C1 |
| КАТИОННЫЙ ЛИПИД | 2019 |
|
RU2784335C2 |
Настоящее изобретение относится к производным кобаламина формулы (I), где Rb означает спейсерную хелатную группу формулы 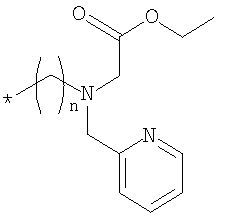 , где n равно 2, 3 или 4; Rc, Rd, Re и RR означают водород и X означает циано или где Rd означает спейсерную хелатную группу формулы , где n равно 3; Rb, Rc, Re и RR означают водород и Х означает циано или где Rb означает спейсерную хелатную группу формулы
, где n равно 2, 3 или 4; Rc, Rd, Re и RR означают водород и X означает циано или где Rd означает спейсерную хелатную группу формулы , где n равно 3; Rb, Rc, Re и RR означают водород и Х означает циано или где Rb означает спейсерную хелатную группу формулы 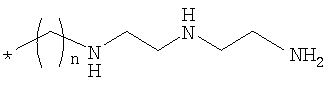 , где n равно 2; Rc, Rd, Re и RR означают водород и Х означает циано. Указанные соединения накапливаются в значительно меньшей степени в крови и доброкачественных органах, таких как почки и печень, по сравнению со скоростью накопления в неопластических тканях и, кроме того, быстро удаляются из крови. Изобретение относится также к инъецируемой фармацевтической композиции для диагностики опухолевых заболеваний, включающей производное кобаламина, несущее радиоактивный атом металла. Кроме того, изобретение относится к способу диагностики опухолевых заболеваний у млекопитающих, включающему (а) соблюдение не содержащей витамин В12 диеты млекопитающим, предрасположенным к опухолевому заболеванию, (б) последующее введение производного кобаламина, несущего радиоактивный атом металла. Изобретение относится также к применению производного кобаламина, несущего радиоактивный атом металла, в способе диагностики опухолевых заболеваний млекопитающего и для получения фармацевтической композиции, предназначенной для применения в способе диагностики опухолевого заболевания млекопитающего. 5 н. и 8 з.п. ф-лы, 2 табл., 6 ил.
, где n равно 2; Rc, Rd, Re и RR означают водород и Х означает циано. Указанные соединения накапливаются в значительно меньшей степени в крови и доброкачественных органах, таких как почки и печень, по сравнению со скоростью накопления в неопластических тканях и, кроме того, быстро удаляются из крови. Изобретение относится также к инъецируемой фармацевтической композиции для диагностики опухолевых заболеваний, включающей производное кобаламина, несущее радиоактивный атом металла. Кроме того, изобретение относится к способу диагностики опухолевых заболеваний у млекопитающих, включающему (а) соблюдение не содержащей витамин В12 диеты млекопитающим, предрасположенным к опухолевому заболеванию, (б) последующее введение производного кобаламина, несущего радиоактивный атом металла. Изобретение относится также к применению производного кобаламина, несущего радиоактивный атом металла, в способе диагностики опухолевых заболеваний млекопитающего и для получения фармацевтической композиции, предназначенной для применения в способе диагностики опухолевого заболевания млекопитающего. 5 н. и 8 з.п. ф-лы, 2 табл., 6 ил.
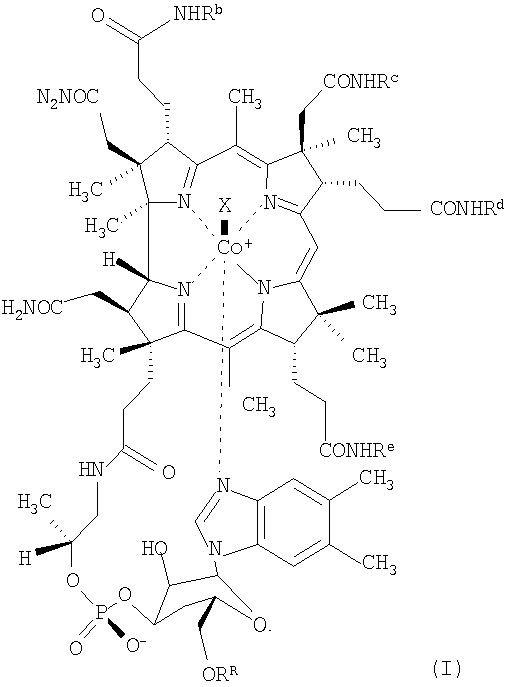
1. Производное кобаламина формулы (I)
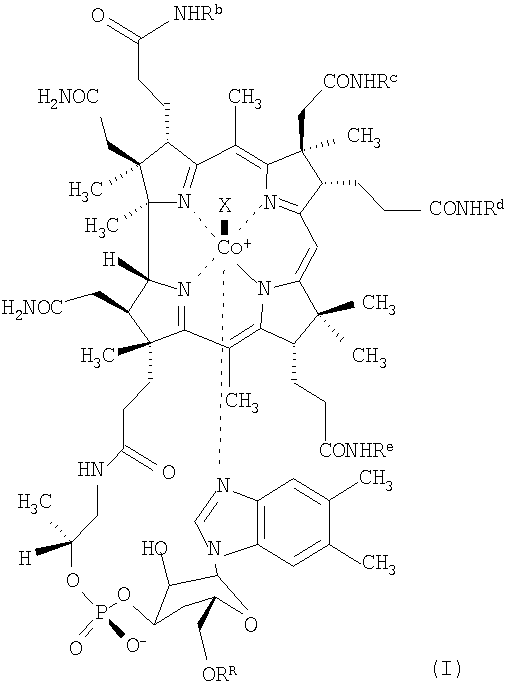
где Rb означает спейсерную хелатную группу формулы
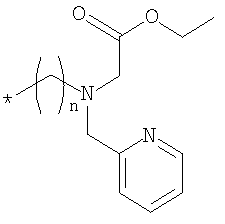 ,
,
где n равно 2, 3 или 4;
Rc, Rd, Re и RR означают водород; и X означает циано; или
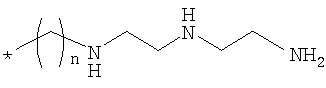
где Rd означает спейсерную хелатную группу формулы
,
где n равно 3;
Rb, Rc, Re и RR означают водород; и Х означает циано; или
где Rb означает спейсерную хелатную группу формулы
 ,
,
где n равно 2;
Rc, Rd, Re и RR означают водород; и Х означает циано.
2. Производное кобаламина по п.1 формулы (I), где
Rb означает спейсерную хелатную группу формулы
,
где n равно 2, 3 или 4;
Rc, Rd, Re и RR означают водород; и X означает циано.
3. Производное кобаламина по п.2, где n равно 4.
4. Производное кобаламина по п.2, где n равно 3.
5. Производное кобаламина по п.2, где n равно 2.
6. Производное кобаламина по любому из пп.1-5 в комплексе с радиоактивным металлом, выбранным из группы, включающей 57Со, 99mTc, 188Re и 111In, где этилэфирная функция спейсера-хелатора гидролизована.
7. Производное кобаламина по п.6, скомплексованное посредством реакции с [99mTc(ОН2)3(СО)3]+ или [188ReO4]-.
8. Инъецируемая фармацевтическая композиция для диагностики опухолевых заболеваний, включающая производное кобаламина, несущее радиоактивный атом металла по п.6 или 7.
9. Способ диагностики опухолевых заболеваний у млекопитающих, включающий
(а) соблюдение не содержащей витамин В12 диеты млекопитающим, предрасположенным к опухолевому заболеванию,
(б) последующее введение производного кобаламина по любому из пп.6 или 7, несущего радиоактивный атом металла.
10. Применение производного кобаламина по п.6 или 7, несущего радиоактивный атом металла, в способе диагностики опухолевых заболеваний млекопитающего.
11. Применение по п.10 для визуализации рака.
12. Применение производного кобаламина по п.6 или 7, несущего радиоактивный атом металла, для получения фармацевтической композиции, предназначенной для применения в способе диагностики опухолевого заболевания млекопитающего.
13. Применение по п.12 производного кобаламина, несущего радиоактивный атом металла, для получения фармацевтической композиции, предназначенной для визуализации рака.
| WO 00/62808 А2, 26.10.2000 | |||
| Кипятильник для воды | 1921 |
|
SU5A1 |
| Способ и приспособление для нагревания хлебопекарных камер | 1923 |
|
SU2003A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| Кипятильник для воды | 1921 |
|
SU5A1 |
| Способ получения витамина в 12, меченного кобальтом-57 или кобальтом-58 | 1975 |
|
SU667145A3 |

