Изобретение относится к металлургии, в частности, к способам получения из среднепрочных волокон на основе полиакрилонитрильных жгутиков высокомодульных углеродных волокон, которые могут затем использоваться для производства высококачественных композитов.
Высокое качество достигается за счет компактной формы получаемых углеродных жгутов, обеспечивающей высокое содержание углеродных волокон в композите и максимальную реализацию механических свойств композиционного материала. Получение таких углеродных волокон традиционным способом невозможно.
Известен способ получения углеродного волокна с повышенным модулем упругости, описанный в Трудах НИИГрафита "Директивный технологический процесс производства жгута углеродного марки ВПР-19С", инв. №256, 1987.
Известный способ заключается в том, что процесс проводят в статических условиях с использованием высокотемпературной обработки. Окисленное полиакрилонитрильное волокно в виде пасм длиной до 1800 мм загружают в графитовые тигли, которые помещают в электрические печи сопротивления и подвергают волокно высокотемпературной обработке в диапазоне температур 2800-3000°С.
Недостатком известного способа является то, что волокно по этому способу получают только в дискретном виде.
Известен способ получения высокомодульного углеродного волокна, описанный в патенте ВБР №1295289 по кл. С1А, опубл. 1972 г.
Известный способ заключается в том, что окисляют в среде воздуха полиакрилонитрильное волокно до плотности 1,0-1,4 г/ куб. см, затем окисленное волокно карбонизируют при температуре 675-725°С в течение 60-150 с в нейтральной среде, пропитывают 10-15%-ным раствором борной кислоты при температуре пропитки 40-70°С, затем сушат тепловым ударом при 200-300°С и подвергают высокотемпературной обработке в интервале температур 2000-2100°С не менее 20 с и с вытяжкой 0,-1,5%.
В результате получают углеродное волокно с плотностью 1,85-2,05 г/куб.см и модулем упругости 435-470 ГПа.
Недостатком известного способа является нестабильность процесса и соответственно недостаточная воспроизводимость результатов.
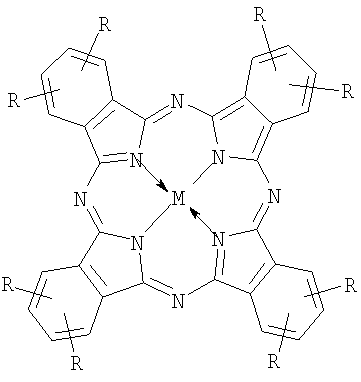
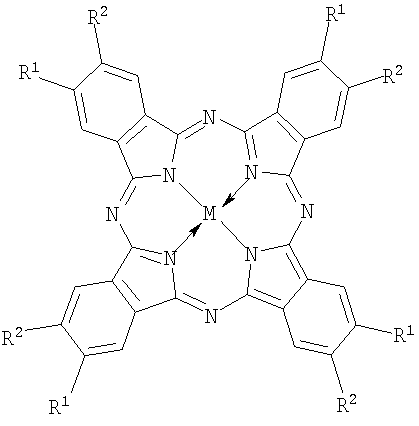
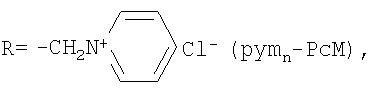
n=2÷8, M=Co, Fe;
Поставленная задача также решается тем, что мольное соотношение компонентов в ассоциате 1:1÷2.
Известен способ окисления сульфида натрия [1] с использованием катализатора, представляющего собой N-(4′-гидроксифенил)-N-(карбоксиметил)сульфамоил фталоцианин кобальта, как описано выше. Он отличается невысокой активностью.
Задача настоящего изобретения состоит в разработке способа каталитического окисления сульфида натрия, который бы обеспечивал высокую конверсию.
Поставленная задача решается тем, что процесс окисления сульфида натрия проводят в водном растворе с использованием кислорода воздуха с указанным выше катализатором при комнатной температуре.
Около 20 лет назад на примере порфиринов, а затем - смесей порфиринов и фталоцианинов было установлено, что при смешивании растворов комплексов с четырьмя противоположными по знаку заряда заместителями (R+ или R-) образуются прочные ассоциаты (в литературе используется и другие названия - супрамолекулярные агрегаты, комплексы, ионные пары с переносом заряда и проч.) строго определенного состава - димеры и триммеры [Т.Shimidzu and Т.Iyoda, Chem. Phys. Lett. 1981. p.853.; H.Segawa, H.Nishino, Т.Kamikawa, K.Honda and Т.Shimidzu, Chem. Lett. 1989. p.1917; S.Gaspard, C.R., Acad. Sci. Paris. 1984. 298. p.379]. Использование нами термина «ассоциаты» обусловлено следующими причинами. Тетрапиррольные макроциклы в растворах склонны к взаимодействию друг с другом посредством нековалентного связывания. В результате образуются агрегаты, состоящие из нескольких одинаковых молекул. Для того чтобы отличать такие агрегаты от изучаемых нами, мы используем термин «ассоциаты», так как наши образования состоят из разных молекул. Использовать термин «комплексы» также неудобно, так как исходные молекулы являются фталоцианиновыми комплексами. Таким образом, выражение «надмолекулярный ионный ассоциат» обозначает систему, состоящую из нескольких молекул (надмолекулярный), различных по своим свойствам, связанных между собой преимущественно ионными связями, но не только.
Фталоцианины с катионными аммониометильными заместителями choln-PcM и pymn-PcM получали хлорметилированием фталоцианина кобальта или фталоцианина железа α,α′-дихлорметиловым эфиром и последующим взаимодействием хлорметилзамещенного производного с 2-(диметиламино)этанолом или пиридином соответственно. Среднюю степень замещения регулировали, изменяя время реакции хлорметилирования.
Аналогично получали и анионный фосфонатометилзамещенный фталоцианин кобальта phosn-PcCo, однако при взаимодействии с хлорметилзамещенным производным использовали триметилфосфит или триэтилфосфит с последующим гидролизом диалкилфосфонатных групп.
Натриевую соль тетракарбоксифталоцианина кобальта carb4-PcCo получали нейтрализацией известного 2,9,16,23-тетракарбоксифталоцианина кобальта [С.А.Михаленко, Л.И.Соловьева, Е.А.Лукьянец // ЖОХ. 2004. Т.74. Вып.3. С.496-505].
Натриевую соль октакарбоксифталоцианина кобальта carb8-PcCo получали по способу, описанному в работе [Патент РФ 2304582, 2007, БИ №23].
Натриевую соль октакарбоксифталоцианина железа carb8-PcFe получали по методике, аналогичной carb8-PcCo.
Натриевую соль тетрасульфофталоцианина кобальта sul4-PcCo получают как в работе [Rollman L.D. Ivamoto R.T. J. Amer. Chem. Soc. 1968. V.90. №5. P.1455].
Ассоциаты получали путем смешивания водных растворов фталоцианинов с разноименно заряженными заместителями. Состав ассоциатов регулировали, изменяя стехиометрическое соотношение компонентов (R-)nPcM:(R+)nPcM. Состав катализаторов в соответствии с изобретением приведен в таблице 1.
Каталитическая активность катализаторов, выраженная в молях окисленного сульфида натрия на моль катализатора в минуту (моль(Na2S)ок/моль(катализатора)мин), приведена в таблице 2.
Как видно из таблицы 2, некоторые катализаторы показали каталитическую активность на один - три порядка большую, в сравнении с прототипом в таких же условиях: при окислении кислородом воздуха при комнатной температуре.
Таким образом, получен активный катализатор, который позволяет эффективно проводить процесс окисления сульфида натрия при комнатной температуре в атмосфере воздуха.
Нижеприведенные примеры иллюстрируют предлагаемое изобретение.
Пример 1. Получение октакис(N-(2-гидроксиэтил)-N,N-диметиламмо- ниометил)фталоцианина кобальта (chol8-PcCo).
К 11 г (0,082 моль) хлористого алюминия добавляют при перемешивании 3 мл триэтиламина. После охлаждения массы до температуры 70-80°С к смеси приливают 6 мл (0, 0075 моль) α,α′-дихлорметилового эфира, а затем загружают 3 г (0,0052 моль) фталоцианина кобальта. Смесь нагревают в течение 3 часов при перемешивании и температуре 90-93°С, после чего выгружают на лед. Осадок отфильтровывают, промывают водой, метанолом и сушат. Выход октакис(хлорметил)фталоцианина кобальта 5,65 г (78,6%).). Электронный спектр поглощения, λmax=673 нм (ДМФА).
Найдено, %: Cl 29,11.
Вычислено % Cl 29,56.
К 0,7 г (0,00073 моль) окстакис(хлорметил)фталоцианина кобальта добавляют 5 мл диметилформамида и 1,5 мл 2-(диметиламино)этанола, после чего смесь нагревают при перемешивании на кипящей водяной бане в течение 2 часов. Осадок отфильтровывают, промывают ацетоном, переосаждают из метанола с ацетоном и сушат. Выход 1,0 г (83,3%) комплекса (I). Электронный спектр поглощения, λmax=672 нм (Н2О).
Найдено, %: Cl 16,51; N 13,02.
Вычислено для C80H134N18O10C18Co, %: Cl 16,95; N 13,4.
Пример 2. Получение октакис(пиридиниометил)фталоцианина кобальта октахлорид (pym8-PcCo).
К 0,49 г (0,00051 моль) окстакис(хлорметил)фталоцианина кобальта (примеры 1-2) добавляют 5,0 мл пиридина, после чего смесь нагревают при перемешивании на кипящей водяной бане в течение 2 часов. Осадок отфильтровывают, промывают ацетоном, переосаждают из метанола с ацетоном и сушат. Выход 0,43 г (83,3%) комплекса (II). Электронный спектр поглощения, λmax=672 нм (Н2О).
Найдено, %: Cl 16,51; N 13,02.
Вычислено для C90H74N18C18Co, %: Cl 16,95; N 13,4.
Пример 3. Получение октакис(фосфонометил)фталоцианина кобальта (phos8-PcCo).
К 2,0 г (0,00208 моль) окстакис(хлорметил)фталоцианина кобальта, полученного как в примере 1, добавляют 5 мл триэтилфосфита и смесь нагревают при 150°С в течение 2 ч. Избыток триэтилфосфита удаляют в вакууме, продукт переосаждают из бензола гексаном. Выход октакис[(диэтоксифосфонил)метил]фталоцианина кобальта 3,0 г (81,3%) комплекса (I). Электронный спектр поглощения, λmax=685 нм (H2O).
Найдено, %: Р 13,31; Со 3,51.
Вычислено для C72H104CoN8O24P8, %: P 13.98; Со 3.33.
Смесь 0,50 г (0,000282 моль) выше полученного эфира и 1 мл концентрированной бромистоводородной кислоты нагревают при 110°С в течение 2 ч. Избыток бромистоводородной кислоты отгоняют в вакууме, остаток промывают водой, спиртом и сушат. Выход продукта 0,27 г (72,3%). Электронный спектр поглощения, λmax=684 нм (водный раствор NaOH, рН 10).
Найдено, %: Р 18,1; Со 4,2.
Вычислено для C40H40CoN8O24P8, %: P 18,72; Со 4,45.
Пример 4. Получение окта-4,5-карбоксифталоцианина железа (carb8-PcFe).
Данный фталоцианин железа получали взаимодействием пиромеллитового диангидрида с безводным бромидом железа (II) в присутствии мочевины, сульфата натрия и молибдата аммония (в молярном соотношении 4:1:10:10:0,1) при 205-210°С в течение трех часов с последующим омылением полученного технического тетраимида октакарбоксифталоцианина железа 25% раствором гидроокиси калия (кипячением в течение 25 часов) до калиевой соли октакарбоксифталоцианина железа. Затем подкисляли ее 10% водным раствором соляной кислоты до свободной кислоты с последующей обработкой последней водным раствором гидроокиси натрия. Выход натриевой соли технического октакарбонатного фталоцианина железа составил 35%. Полученную соль очищают от примесей, в том числе и от олигомерных продуктов методом колоночной хроматографии на оксиде алюминия. Элюент - фосфатный буфер рН 8. Из фракции с Rf 0,9 (на пластинке silufol) выделяют с выходом 10% (в расчете на диангидрид пиромеллитовой кислоты) carb8-PcFe. Готовый продукт сушат при 105-110°С в вакууме над P2O5 до постоянной массы.
Найдено, %: С 42,98; Н 1,10; N 9,81. C40H8FeN8Na8O16.
Вычислено, %: С 43,82; Н 0,74; N 10,22.
Пример 5. Получение катализатора - ассоциата pym8-PcCo:carb8-PcCo=2:1.
К 1 мл 3,30·10-5 М водного раствора carb8-PcCo прибавляют 1 мл 6,60·10-5 М водного раствора pym8-PcCo, перемешивают 15 мин. Концентрация раствора полученного ассоциата составляет 1,65·10-5 М.
Пример 6. Получение катализатора - ассоциата pym8-PcCo:carb8-РсСо=1:1.
К 1 мл 3,30·10-5 М водного раствора carb8-PcCo прибавляют 1 мл 3,30·10-5 М водного раствора pym8-PcCo и перемешивают 15 мин. Концентрация раствора полученного ассоциата составляет 1,65·10-5 М.
Пример 7. Получение катализатора, включающего ассоциат pym8-РсСо:carb8-PcCo=1:2.
К 1 мл 6,60·10-5 М водного раствора carb8-PcCo прибавляют 1 мл 3,30·10-5 М водного раствора pym8-PcCo и перемешивают 15 мин. Концентрация раствора полученного ассоциата составляет 1,65·10-5 М.
Примеры 8-24.
Остальные катализаторы согласно таблице 1 получали аналогичным образом.
Пример 25. Каталитическое окисление сульфида натрия.
Смешивают в реакторе 10 мл (0,024 М) сульфида натрия и 0,37 мл водного раствора катализатора, содержащего (1,65·10-5 М) pym8PcCo:carb8PcCo=2:1 (пример 5). Конечная концентрация катализатора составила 5,0·10-7 М. Реакцию проводили при комнатной температуре в атмосфере воздуха при интенсивном перемешивании в течение 15 минут. Анализы на остаточный сульфид натрия после опыта проводили методом потенциометрического титрования на универсальном иономере И-500 по стандартной методике раствором азотнокислого аммиаката серебра. Измерительный электрод - сульфид-серебряный, электрод сравнения - хлор-серебряный.
По результатам анализа конверсия сульфида натрия составила 100% или 3,1·103 моль(Na2S)/моль(катализатора)мин.
Примеры 26-44.
Процесс проводили по примеру 25, но с использованием катализаторов по примерам 6-24. Результаты приведены в таблице 2.
Таким образом, как видно из таблицы 2, предложенный катализатор обладает активностью, большей, чем у прототипа в 4 и более раз: самый активный катализатор показал активность в 62 раза выше, чем у прототипа.
| название | год | авторы | номер документа |
|---|---|---|---|
| КАТАЛИЗАТОР И СПОСОБ ОКИСЛЕНИЯ СУЛЬФИДА НАТРИЯ | 2008 |
|
RU2381066C1 |
| КАТАЛИЗАТОР И СПОСОБ ОКИСЛИТЕЛЬНОЙ ДЕМЕРКАПТАНИЗАЦИИ НЕФТИ И НЕФТЕПРОДУКТОВ | 2008 |
|
RU2381065C1 |
| КАТАЛИЗАТОР И СПОСОБ ГОМОГЕННОЙ ОКИСЛИТЕЛЬНОЙ ДЕМЕРКАПТАНИЗАЦИИ НЕФТИ И НЕФТЕПРОДУКТОВ | 2008 |
|
RU2381067C1 |
| ЗАМЕЩЕННЫЕ ФТАЛОЦИАНИНЫ ЖЕЛЕЗА И СПОСОБ ПОЛУЧЕНИЯ ХЛОРПРОИЗВОДНЫХ АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ | 2007 |
|
RU2340589C1 |
| СПОСОБ ПОЛУЧЕНИЯ ФОСФОНОМЕТИЛЗАМЕЩЕННЫХ ФТАЛОЦИАНИНОВ | 2011 |
|
RU2465908C1 |
| ВОЛЬТАМПЕРОМЕТРИЧЕСКИЙ СПОСОБ ОПРЕДЕЛЕНИЯ ХЛОРИНДИЯ ФТАЛОЦИАНИНА | 1996 |
|
RU2094797C1 |
| КАТАЛИЗАТОР И СПОСОБ ВАЛЕНТНОЙ ИЗОМЕРИЗАЦИИ КВАДРИЦИКЛАНА В НОРБОРНАДИЕН | 2011 |
|
RU2470030C1 |
| СПОСОБ ИЗГОТОВЛЕНИЯ ДЛИННЫХ ОРИЕНТИРОВАННЫХ ЖГУТОВ УГЛЕРОДНЫХ НАНОВОЛОКОН | 2009 |
|
RU2393276C1 |
| Катализатор для окисления сернистых соединений | 1987 |
|
SU1497830A1 |
| 4,5-БИС-(1',2':3',4'-ДИ-О-ИЗОПРОПИЛИДЕН-α-D-ГАЛАКТОПИРАНОЗО-6'-ИЛ)ФТАЛОНИТРИЛ | 2009 |
|
RU2409585C1 |
Изобретение относится к технологии получения высокомодульных углеродных волокон из среднепрочных волокон на основе полиакрилонитрильных жгутиков и может быть использовано для производства высококачественных композитов. В качестве исходного сырья используют среднепрочное углеродное волокно с линейной плотностью 200-1600 текс и модулем упругости 200-250 ГПа. Это волокно подвергают крутке до величины 30-60 круток/м при содержании в нем аппрета более 1%. Дополнительно аппретируют волокно при содержании аппрета менее 1%. Затем подкрученный жгут подвергают первичной термообработке при 2300-2500°С в течение 1-10 мин до мод значения модуля упругости углеродного жгута не менее 300 ГПа. Затем проводят вторую термообработку при температуре не ниже 3000°С в течение 1-20 сек при вытяжке жгута до 10% до возрастания модуля упругости углеродного жгута до величины не менее 450 ГПа. Высокое качество достигается за счет компактной формы получаемых углеродных жгутов, обеспечивающей высокое содержание углеродных волокон в композите и максимальную реализацию механических свойств композиционного материала. 2 табл.
Способ получения высокомодульного волокна из среднепрочных низкомодульных углеродных волокон, включающем использование углеродного волокна на основе ПАН-сырья, изготовление из него жгута, его первую термообработку при температуре не ниже 2200°С, и вторую высокоскоростную термообработку при температуре не ниже 3000°С, отличающийся тем, что в качестве исходного сырья используют среднепрочное углеродное волокно с линейной плотностью 200-1600 текс и модулем упругости 200-250 ГПа, которое подвергают крутке до величины 30-60 круток/м при содержании в нем аппрета более 1% и дополнительно аппретируют при содержании аппрета менее 1%, затем подкрученный жгут подвергают первичной термообработке при температуре 2300-2500°С в течение 1-10 мин до возрастания модуля упругости углеродного жгута до значения не менее 300 ГПа, затем проводят вторую термообработку в течение нескольких - 1-20 с при вытяжке жгута до 10% до возрастания модуля упругости углеродного жгута до величины не менее 450 ГПа.
| СПОСОБ ПОЛУЧЕНИЯ ВЫСОКОМОДУЛЬНОГО УГЛЕРОДНОГО ВОЛОКНА | 1991 |
|
RU2016147C1 |
| СПОСОБ ПОЛУЧЕНИЯ УГЛЕРОДНОГО ВОЛОКНА | 1996 |
|
RU2130516C1 |
| УГЛЕРОДНАЯ КРУЧЕНАЯ НИТЬ ДЛЯ КОМПОЗИЦИОННЫХ МАТЕРИАЛОВ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2001 |
|
RU2220235C2 |
| JP 2006307407 A, 09.11.20 | |||
| Способ варки варенья из высококислотных ягод | 1954 |
|
SU100410A1 |
| US 4869856 А, 26.09.1989 | |||
| US 5256344 А, 26.10.1993. | |||

