Настоящее изобретение относится к новой полиморфной модификации N-{5-[3-(тиофен-2-карбонил)-пиразоло[1,5-а]пиримидин-7-ил]-2-фтор-фенил}-N-метил-ацетамида, способам ее получения, применению ее в качестве терапевтически активного вещества и к фармацевтическим композициям, включающим новую полиморфную модификацию.
Уровень техники
N-{5-[3-(тиофен-2-карбонил)-пиразоло[1,5-а]пиримидин-7-ил]-2-фтор-фенил}-N-метил-ацетамид представляет собой активный лиганд рецепторов γ-аминомасляной кислоты А (ГАМКА), который может применяться для лечения или профилактики тревожности, эпилепсии, расстройств сна и бессоницы, для индуцирования седативно-гипнотического эффекта, анестезии и мышечной релаксации и для модулирования времени, необходимого для наступления сна и его продолжительности так, как описано в РСТ/ЕР 2006/063243 и US 60/692866.
В тексте настоящей заявки термин "соединение (I)" относится к N-{5-[3-(тиофен-2-карбонил)-пиразоло[1,5-а]пиримидин-7-ил]-2-фтор-фенил}-N-метил-ацетамиду.
Соединение (I) является структурно-родственным N-{3-[3-(тиофен-2-карбонил)-пиразоло[1,5-а]пиримидин-7-ил]-фенил}-N-метил-ацетамиду, известному также как индиплон. Это соединение и его применение в качестве седативного или снотворного средства описано в US 6399621. В отличие от соединения (I), это соединение является только монозамещенным по фенильному циклу.
К настоящему времени опубликована единственная кристаллическая форма соединения (I), которая раскрыта в вышеупомянутом описании изобретения и показывает температуру плавления 165-167°С. В настоящем исследовании эта форма показывает кривую ДСК (дифференциальной сканирующей калориметрии) с острым пиком плавления между 166,2 и 167,4°С. Небольшое расхождение с опубликованной ранее температурой плавления является допустимым и лежит в пределах ошибки эксперимента. Эта форма обозначается в данной заявке как Полиморфная модификация А.
Важно, чтобы лекарственное вещество находилось в форме, которую было бы удобно обрабатывать и с которой было бы удобно обращаться. Это важно не только с точки зрения получения коммерчески осуществимого способа получения, но также с точки зрения последующего производства фармацевтической композиции, включающей активное соединение. Лекарственное вещество и композиция, включающая его, должны иметь достаточно большой срок хранения без проявления существенных изменений в физико-химических характеристиках активного компонента. Более того, также важно, чтобы лекарство легко получалось в наиболее чистом виде. Специалист поймет, что если лекарство может быть легко получено в стабильной кристаллической форме, вышеупомянутые проблемы можно решить. Следовательно, при производстве коммерчески жизнеспособной и фармацевтически приемлемой лекарственной композиции желательно, когда это возможно, получить лекарство в практически кристаллической и стабильной форме. Таким образом, существует потребность в стабильной кристаллической форме соединения (I), которую можно было бы легко обрабатывать и с которой было бы легко обращаться.
Раскрытие изобретения
Авторы настоящего изобретения обнаружили новую кристаллическую форму соединения (I). Эта новая форма обозначается как полиморфная модификация В.
Полиморфная модификация В соединения (I) демонстрирует кривую порошковой рентгеновской дифрактометрии, имеющую наиболее интенсивные пики при 2θ=7,1°(±0,1°) и 21,4°(±0,1°); Фурье-спектры комбинационного рассеяния (FT-Raman Spectrum) с характеристическими сигналами 3107 см-1, 1605 см-1, 1593 см-1, 1538 см-1, 1336 см-1 и 102 см-1 и кривую дифференциальной сканирующей калориметрии (ДСК) с пиком плавления при примерно 158°С.
Подобно полиморфной модификации А, полиморфная модификация В представляет собой активный лиганд ГАМКА и может применяться для лечении или профилактики тревожности, эпилепсии, расстройств сна и бессоницы, для индуцирования седативно-гипнотического эффекта, анестезии и мышечной релаксации и для модулирования времени, необходимого для наступления сна и его продолжительности.
Полиморфная модификация В соединения (I) отличается от индиплона тем, что параположение в фенильном цикле замещено атомом фтора. Полиморфная модификация В проявляет неожиданно высокую эффективность и неожиданно улучшенный предел безопасности по сравнению с соединением прототипа индиплоном, что подтверждается данными, приведенными в нижеследующем детальном описании, что, таким образом, делает соединение по изобретению неожиданно улучшенным терапевтическим лекарственным средством для достижения седативно/гипнотического эффекта.
Краткое описание чертежей
Изобретение описывается с помощью чертежей, на которых:
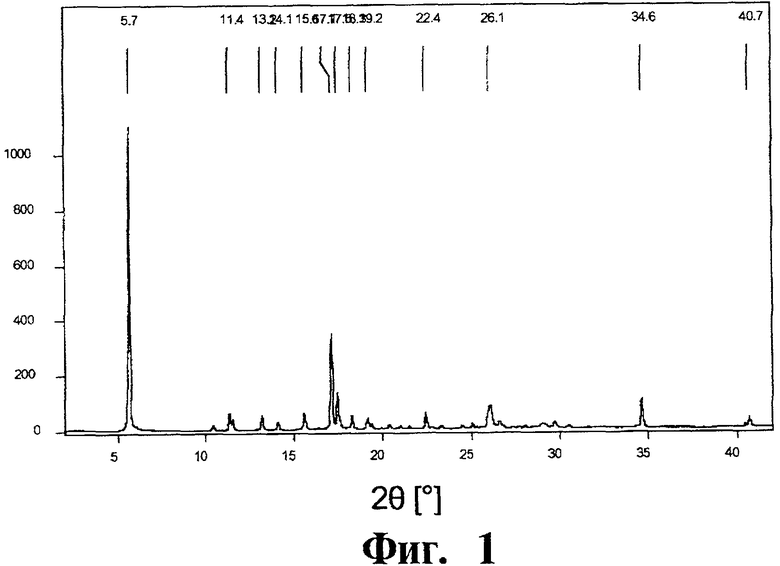
Фиг.1 представляет собой кривую порошковой рентгеновской дифрактометрии для полиморфной модификации А. Интенсивность по оси ординат представлена в импульсах в секунду (cps).
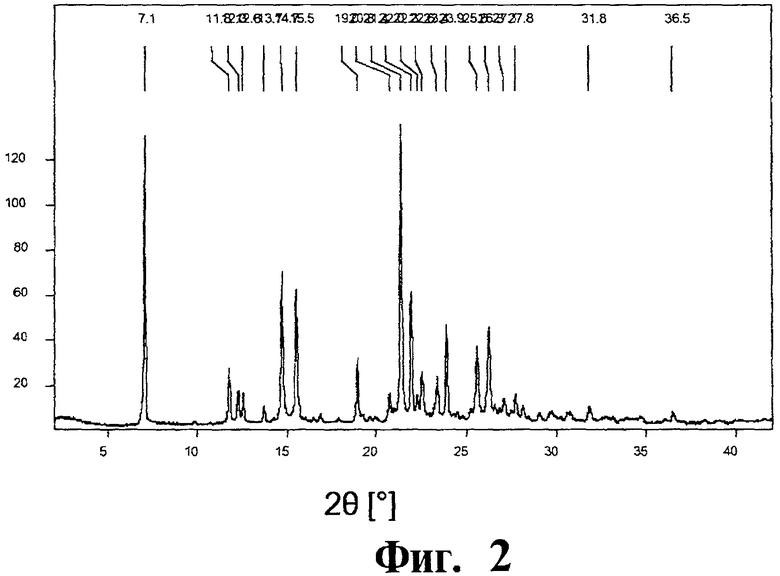
Фиг.2 представляет собой кривую порошковой рентгеновской дифрактометрии для полиморфной модификации В. Интенсивность по оси ординат представлена в импульсах в секунду (cps).
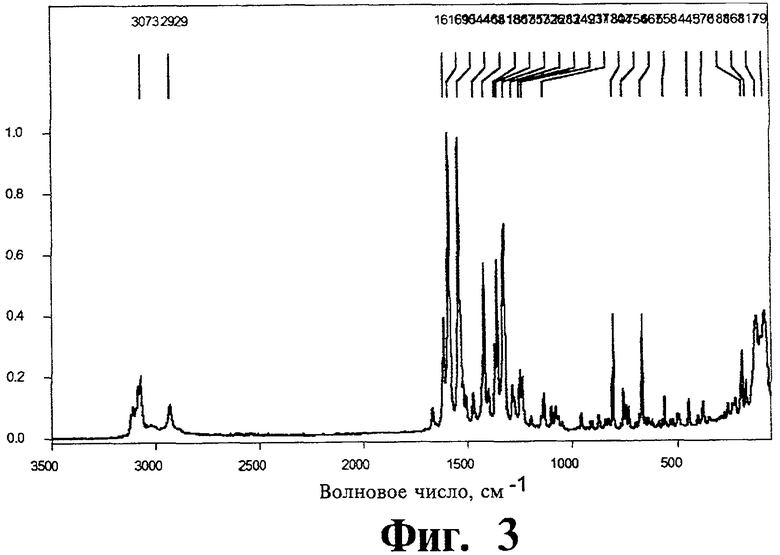
Фиг.3 представляет Фурье-спектр комбинационного рассеяния для полиморфной модификации А.
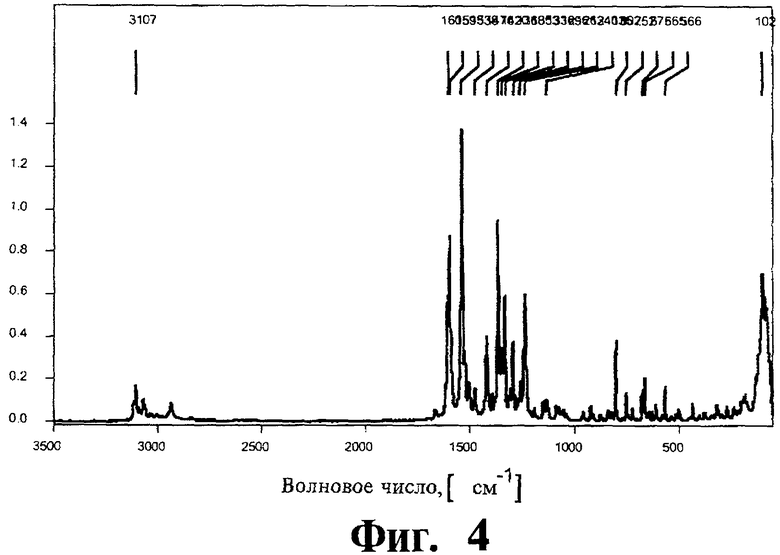
Фиг.4 представляет Фурье-спектр комбинационного рассеяния для полиморфной модификации В.
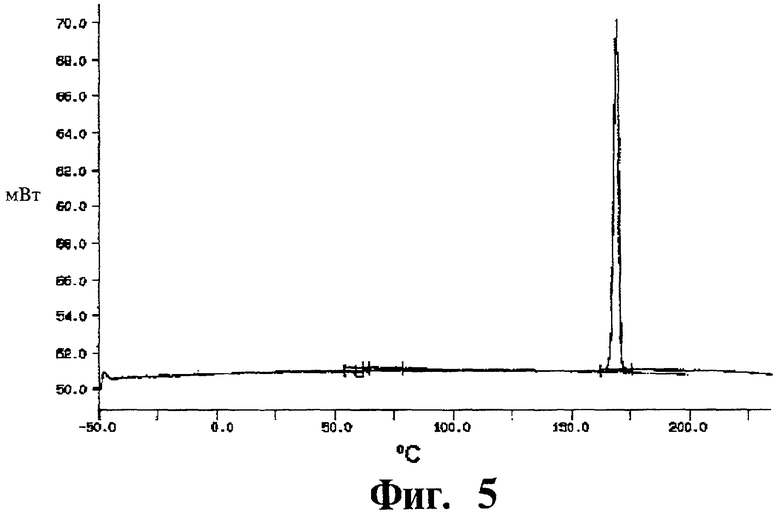
Фиг.5 представляет собой кривую дифференциальной сканирующей калориметрии (DSC) для полиморфной модификации А.
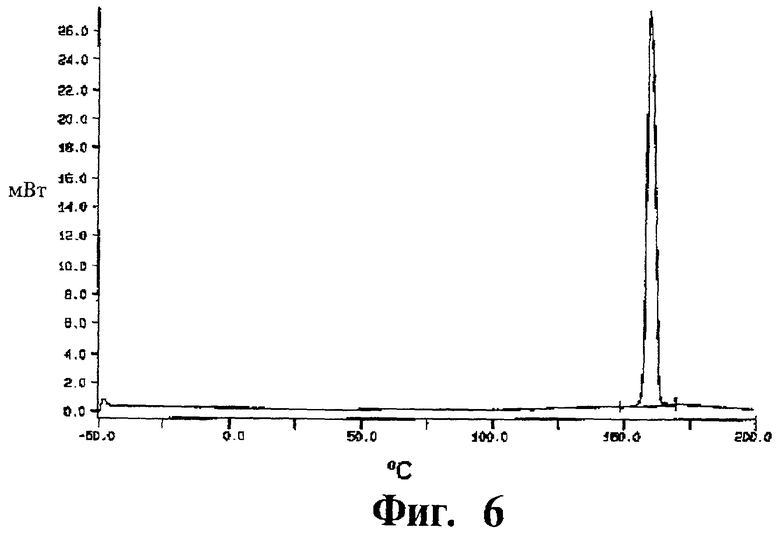
Фиг.6 представляет собой кривую дифференциальной сканирующей калориметрии (DSC) для полиморфной модификации В.
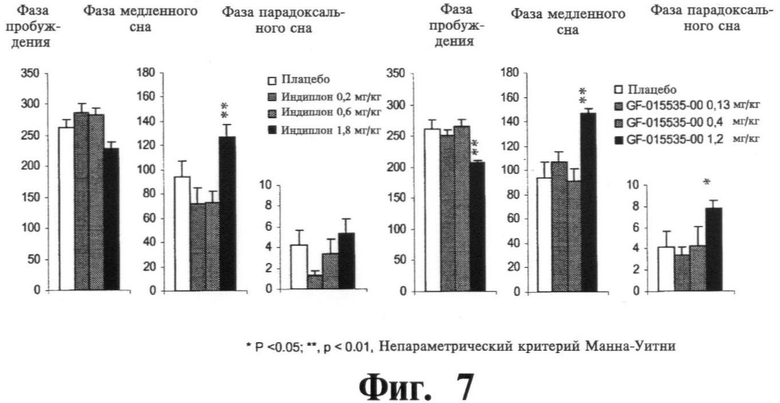
Фиг.7 показывает результаты оценки следующих параметров: время пробуждения, фазы медленного сна и фазы парадоксального сна как для соединения настоящего изобретения, так и для индиплона (US 6399621).
Осуществление изобретения
Первый аспект настоящего изобретения заключается в новой полиморфной модификации В соединения (I).
Полиморфная модификация В соединения (I) демонстрирует кривую порошковой рентгеновской дифрактометрии, имеющую наиболее интенсивные пики при 2θ=7,1°(±0,1°) и 21,4°(±0,1°); указанная полиморфная модификация характеризуется кривой порошковой рентгеновской дифрактометрии, имеющей специфические пики при 2θ=7,1°(±0,1°), 11,8°(±0,1°), 12,3°(±0,1°), 12,6°(±0,1°), 13,7°(±0,1°), 14,7°(±0,1°), 15,5°(±0,1°), 19,0°(±0,1°), 20,8°(±0,1°), 21,4°(±0,1°), 22,0°(±0,1°), 22,3°(±0,1°), 22,6°(±0,1°), 23,4°(±0,1°), 23,9°(±0,1°), 25,6°(±0,1°), 26,3°(±0,1°), 27,1°(±0,1°), 27,8°(+0,1°), 31,8°(±0,1°) и 36,5°(±0,1°). Полиморфная модификация В соединения (I) также показывает Фурье-преобразованный спектр комбинационного рассеяния с характеристическими сигналами при 3107 см-1, 1605 см-1, 1593 см-1, 1538 см-1, 1336 см-1 и 102 см-1 и кривую дифференциальной сканирующей калориметрии с пиком плавления примерно 158°С.
Вторым аспектом настоящего изобретения является предложение способа получения полиморфной модификации В соединения (I) путем суспендирования полиморфной модификации А соединения (I) при комнатной температуре (20-25°С) в растворителе, выбранном из группы, включающей С1-С6алифатические спирты, С1-С6алифатические кетоны, С1-С4алкильные эфиры С1-С4алифатические кислоты, С4-С5насыщенные циклические эфиры, С1-С6алифатические нитрилы, ароматические углеводороды и воду, и смеси, выбранные из группы, включающей С1-С6алифатический спирт и С1-С6алифатическую органическую кислоту, воду и С1-С6алифатический спирт, и воду и С4-С5насыщенный циклический эфир и последующего выделения полученных кристаллов.
Предпочтительно растворитель выбирается из группы, включающей метанол, этанол, 1-метокси-2-пропанол, метилэтилкетон, этилацетат, диоксан, ацетонитрил, толуол, воду, смесь этанола и уксусной кислоты, смесь воды и этанола, и смесь воды и тетрагидрофурана. Когда применяется смесь этанола и уксусной кислоты, объемное соотношение этанола и уксусной кислоты предпочтительно лежит в диапазоне от 90:10 до 98:2, соответственно. Более предпочтительным является соотношение 95:5. Альтернативно, когда применяется смесь воды и этанола, объемное соотношение воды и этанола предпочтительно лежит в диапазоне от 5:95 до 95:5, соответственно. Более предпочтительно соотношение лежит в пределах от 10:90 до 90:10. Если применяется смесь воды и тетрагидрофурана, то объемное соотношение воды и тетрагидрофурана лежит в пределах от 85:15 до 95:5, соответственно. Более предпочтительным является соотношение 90:10. Полученные кристаллы могут выделяться с помощью обычных процедур, таких как обычное фильтрование, фильтрование под уменьшенным давлением или фильтрование на центрифуге, с последующим промыванием, если это необходимо, и высушиванием, получая полиморфную модификацию В соединения (I) настоящего изобретения.
В рамках второго аспекта изобретения раскрывается вариант предыдущего способа, в котором смесь полиморфной модификации А соединения (I) и полиморфной модификации В соединения (I) суспендируется в ароматическом растворителе при температуре от 80°С до температуры кипения, а затем полученные кристаллы выделяются. Весовое соотношение в смеси полиморфной модификации А и полиморфной модификации В лежит в диапазоне от 25:75 до 75:25, предпочтительно 50:50. Предпочтительный ароматический ратворитель представляет собой толуол, а предпочтительный температурный диапазон - от 95 до 105°С.
Другой аспект настоящего изобретения заключается в предложении способа получения полиморфной модификации В соединения (I) путем растворения полиморфной модификации А соединения (I) в подходящем растворителе; отфильтровыванием и выдерживанием до тех пор, пока растворитель полностью не испарится. Предпочтительные растворители. представляют собой ацетон и тетрагидрофуран.
Другой аспект настоящего изобретения заключается в предложении способа получения полиморфной модификации В соединения (I) путем растворения полиморфной модификации А соединения (I) в смеси воды и тетрагидрофурана при комнатной температуре; и выделением полученного кристаллического осадка. Объемное соотношение воды и тетрагидрофурана предпочтительно лежит в границах от 5:95 до 15:85, соответственно. Более предпочтительное соотношение представляет собой 10:90. Полученный кристаллический осадок может быть выделен, как описано выше.
Другой аспект настоящего изобретения заключается в предложении способа получения полиморфной модификации В соединения (I) путем растворения полиморфной модификации А соединения (I) в растворителе, выбранном из группы, включающей С1-С6алифатические сульфоксиды, ароматические амины, С1-С6алифатические органические кислоты и смеси С1-С2 галогенированного алифатического углеводорода и C1-C6алифатического спирта; фильтрования раствора; добавления раствора к антирастворителю, выбранному из группы, включающей С1-С6алифатические спирты и С1-С4алкильные эфиры С1-C4алифатических кислот; и выделения полученных кристаллов.
Предпочтительно, чтобы растворитель выбирался из группы, включающей диметилсульфоксид, пиридин, уксусную кислоту и смесь дихлорметана и 2-пропанола. Объемное соотношение дихлорметана и 2-пропанола предпочтительно лежит в границах от 0,5:10 до 2:10, соответственно, когда применяется смесь дихлорметана и 2-пропанола. Более предпочтительное соотношение представляет собой 1:10. Антирастворитель выбирается из группы, включающей этанол, 2-пропанол и этилацетат.
Для гарантии того, что контролируемо получается полиморфная модификация В, настоятельно рекомендуется применять затравку. Это может быть затравка из равновесной суспензии, при осаждении или кристаллизации из горячего раствора. Соответственно полимерная модификация В соединения (I) может с успехом быть получена добавлением кристаллов указанной полиморфной модификации для затравки к раствору соединения (I) в подходящем растворителе для начала кристаллизации и выделением полученных кристаллов с использованием известных в химии процедур.
Другой аспект настоящего изобретения заключается в предложении использования полиморфной модификации В соединения (I) в качестве лекарственного средства.
Другой аспект настоящего изобретения заключается в предложении фармацевтической композиции, включающей полиморфную модификацию В соединения (I) в смеси с одним или более фармацевтически приемлемым носителем, наполнителем, разбавителем или вспомогательным веществом.
Другой аспект настоящего изобретения заключается в предложении фармацевтической композиции, включающей полиморфную модификацию В соединения (I), для применения при лечении или профилактике тревожности, эпилепсии, расстройств сна и бессоницы, для индуцирования седативно-снотворного действия, анестезии и мышечной релаксации и для модуляции времени, необходимого для наступления сна и его продолжительности.
Изобретение также относится к способу лечения и/или профилактики млекопитающих, включая человека, страдающих от или подверженных беспокойству, эпилепсии, расстройствам сна, для индуцирования седативно-гипнотического эффекта, анестезии и мышечной релаксации и для модулирования времени, необходимого для наступления сна и его продолжительности, указанный способ включает введение упомянутому пациенту терапевтически эффективного количества полиморфной модификации В соединения формулы (I) вместе с фармацевтически приемлемым разбавителем или наполнителем.
Фармацевтические композиции включают композиции для перорального, ректального и парентерального (включая подкожное, внутримышечное и внутривенное) введения, хотя наиболее приемлемый путь введения будет зависеть от природы и тяжести заболевания, которое предстоит лечить. Наиболее предпочтительный путь введения настоящего изобретения представляет собой пероральный путь. Композиции могут быть удобно представлены в виде единичной лекарственной дозированной формы и получены любым из способов, хорошо известных в фармацевтической отрасли.
Активное соединение может комбинироваться с фармацевтическим носителем в соответствии с обычными методиками фармацевтического смешивания. Носитель может принимать широкое разнообразие форм в зависимости от формы желаемого препарата для введения, например перорального или парентерального (включая внутривенные инъекции или инфузии). При приготовлении композиций для пероральной лекарственной формы может применяться любой из обычных лекарственных носителей. Обычный фармацевтический носитель включает, например, воду, гликоли, масла, спирты, ароматические вещества, стабилизаторы, красители и т.п. в случае жидких пероральных препаратов (таких как, например, суспензии, растворы, эмульсии и эликсиры); аэрозолей; или наполнители, такие как крахмалы, сахара, микрокристаллическая целлюлоза, разбавители, гранулирующие вещества, смазочные вещества, связующие вещества, дезинтегрирующие вещества и т.п. в случае твердых пероральных препаратов (таких как, например, порошки, капсулы и таблетки), при этом твердые пероральные препараты являются предпочтительными по сравнению с жидкими пероральными препаратами.
Из-за простоты введения таблетки и капсулы представляют собой наиболее предпочтительную пероральную лекарственную форму, в таких случаях используются твердые фармацевтические носители. При желании, таблетки могут быть покрыты оболочкой с помощью стандартных водных или неводных методик.
Границы приемлемой дозировки для использования представляют собой от примерно 0,01 мг до примерно 100,00 мг для общей суточной дозы, которая дается либо один раз в день, либо, если необходимо, разбивается на несколько приемов.
Другой аспект настоящего изобретения заключается в предложении применения полиморфной модификации В соединения (I) для приготовления лекарственного средства, предназначенного для использования при лечении или профилактике тревожности, эпилепсии, расстройств сна и бессоницы, для индуцирования седативно-гипнотического эффекта, анестезии и мышечной релаксации и для модулирования времени, необходимого для наступления сна и его продолжительности.
Прогнозируемый седативно-гипнотический эффект полиморфной модификации В соединения (I) был определен, как показано ниже, и сравнивался с эффектом, производимым индиплоном (ближайшим соединением прототипа, описанным в US 6399621).
Влияние как индиплона, так и соединения настоящего изобретения, применяемых перорально, на спонтанную двигательную активность у мышей представляет собой признанную модель для оценки индуцирования седативного эффекта, этот эксперимент считается предсказательным для эффективности гипнотического действия. В этом эксперименте ED50, т.е. доза, которая индуцирует седативное действие у 50% животных, была вычислена. Соединение прототипа (индиплон), описанное в US 6399621, показало ED50=0,2 мг/кг, в то время как соединение настоящего изобретения показало ED50=0,13 мг/кг, т.е. было на 35% более активным.
Такое усиление седативно-гипнотического эффекта было подтверждено электроэнцефалографическим (EEG) исследованием на мышах, при котором оценивалась запись цикла сна-пробуждения. Дозировки были выбраны на основании предшествующих экспериментов, и оба соединения тестировались в фармацевтических дозировках, соответствующих одинаковой активности (ED50, 3-кратная и 6-кратная в обоих случаях). Соединение настоящего изобретения продемонстрировало статистически достоверное улучшение по всем трем оцениваемым параметрам (время пробуждения, фазы медленного сна и фазы парадоксального сна), как показано на графике на Фиг.7, в то время как соединение прототипа индиплон был эффективен только по одному параметру (фаза медленного сна).
На Фиг.7, результаты представлены как среднее время (мин ± SEM), проведенное в каждом из состояний за исследуемый период в 6 ч одними и теми же животными (n=9). Наибольшая дозировка соединения настоящего изобретения (GF-015535-00) привела к увеличению фазы медленного сна (SWS, вплоть до 140 мин) и парадоксального сна (PS) и уменьшению пробуждения (W), в то время как соединения прототипа (индиплон) увеличивал только медленный сон SWS на более короткий промежуток времени по сравнению с нашим соединением (менее 140 мин), что ясно демонстрировало лучшее воздействие на сон соединения настоящего изобретения.
Наконец, был выполнен третий эксперимент для того, чтобы оценить побочные эффекты. Моделью служил двухдневный тест активного избегания, который представляет собой поведенческий тест, применяемый для оценки процессов обучения и памяти у мышей. В данном случае был получен индекс подверженности амнезии. Поскольку было опубликовано, что лекарства, подобные бенздиазепину, вызывают амнезию, этот индекс позволяет определить границу между преклинически эффективными дозировками, которые вызывают седативное состояние, и минимальной эффективной дозировкой, которая вызывает статистически достоверное ухудшение памяти у мышей (MED амнезия/ED50 седативного действия). Следовательно, индекс подверженности амнезии вычислялся для обоих соединений. Полученные результаты включены в Таблицу 1.
В результате соединение настоящего изобретения демонстрирует в 25 раз больший предел между индуцированном седативного действия и амнезией, чем соединение прототипа индиплон.
В заключение соединение настоящего изобретения явно показывает неожиданно более высокую эффективность и улучшенные границы безопасности по сравнению с соединением прототипа индиплоном.
Полиморфная модификация настоящего изобретения получается в соответствии со следующими примерами, которые приведены только в целях иллюстрации.
Препаративный пример 1: Исходный материал, полиморфная модификация А, был получен в соответствии с Примером 2 описания изобретения РСТ/ЕР 2006/063243 и US 60/692866.
Препаративный пример 2: Получение полиморфной модификации В из полиморфной модификации А в метаноле
Полиморфную модификацию А (151,8 мг) суспендировали в метаноле (2 мл) и перемешивали при комнатной температуре в течение 3 дней. Твердый осадок отфильтровали центрифугированием (фильтр 0,22 мкм) и высушили в вакууме при комнатной температуре в течение 15 мин. Была получена полиморфная модификация В (95 мг).
Препаративный пример 3: Получение полиморфной модификации В из полиморфной модификации А в ацетонитриле
Полиморфную модификацию А (151,8 мг) суспендировали в ацетонитриле (2 мл) и перемешивали при комнатной температуре в течение 3 дней. Твердый осадок отфильтровали центрифугированием (фильтр 0,22 мкм) и высушили в вакууме при комнатной температуре в течение 15 мин. Была получена полиморфная модификация В (90 мг).
Препаративный пример 4: Получение полиморфной модификации В из полиморфной модификации A в этаноле
Полиморфную модификацию А (153,3 мг) суспендировали в этаноле (2 мл) и перемешивали при комнатной температуре в течение 3 дней. Твердый осадок отфильтровали центрифугированием (фильтр 0,22 мкм) и высушили в вакууме при комнатной температуре в течение 15 мин. Была получена полиморфная модификация В (110 мг).
Препаративный пример 5: Получение полиморфной модификации В из полиморфной модификации А в 1-метокси-2-пропаноле
Полиморфную модификацию А (152,4 мг) суспендировали в 1-метокси-2-пропаноле (2 мл) и перемешивали при комнатной температуре в течение 3 дней. Твердый осадок отфильтровали центрифугированием (фильтр 0,22 мкм) и высушили в вакууме при комнатной температуре в течение 15 мин. Была получена полиморфная модификация В (90 мг).
Препаративный пример 6: Получение полиморфной модификации В из полиморфной модификации А в метилэтилкетоне
Полиморфную модификацию А (150,6 мг) суспендировали в метилэтилкетоне (2 мл) и перемешивали при комнатной температуре в течение 3 дней. Твердый осадок отфильтровали центрифугированием (фильтр 0,22 мкм) и высушили в вакууме при комнатной температуре в течение 15 мин. Была получена полиморфная модификация В (100 мг).
Препаративный пример 7: Получение полиморфной модификации В из полиморфной модификации А в этилацетате
Полиморфную модификацию А (150,0 мг) суспендировали в этилацетате (2 мл) и перемешивали при комнатной температуре в течение 3 дней. Твердый осадок отфильтровали центрифугированием (фильтр 0,22 мкм) и высушили в вакууме при комнатной температуре в течение 15 мин. Была получена полиморфная модификация В (105 мг).
Препаративный пример 8: Получение полиморфной модификации В из полиморфной модификации А в толуоле
Полиморфную модификацию А (150,0 мг) суспендировали в толуоле (2 мл) и перемешивали при комнатной температуре в течение 3 дней. Твердый осадок отфильтровали центрифугированием (фильтр 0,22 мкм) и высушили в вакууме при комнатной температуре в течение 15 мин. Была получена полиморфная модификация В (90 мг).
Препаративный пример 9: Получение полиморфной модификации В из полиморфной модификации А в смеси этанол/уксусная кислота 95:5
Полиморфную модификацию А (156,0 мг) перемешивали со смесью этанол/уксусная кислота 95:5 (2 мл) в течение 8 дней при комнатной температуре. Образец отфильтровали и высушили под вакуумом в течение 10 мин. Была получена полиморфная модификация В (100 мг).
Препаративный пример 10: Получение полиморфной модификации В из полиморфной модификации А в ацетоне
Полиморфную модификацию А (157,9 мг) растворили в ацетоне (8 мл). Раствор отфильтровали и оставили испаряться при комнатной температуре. После полного испарения растворителя за несколько дней образовались желтые кристаллы, соответствующие полиморфной модификации В.
Препаративный пример 11: Получение полиморфной модификации В из полиморфной модификации А в тетрагидрофуране
Полиморфную модификацию А (157,8 мг) растворили в тетрагидрофуране (5 мл). Раствор отфильтровали и оставили испаряться при комнатной температуре. После полного испарения растворителя за несколько дней образовались желтые кристаллы, соответствующие полиморфной модификации В.
Препаративный пример 12: Получение полиморфной модификации В из полиморфной модификации А в смеси вода/этанол 10:90
Полиморфную модификацию А (148 мг) суспендировали в H2O (0,2 мл) и этаноле (1,8 мл) и перемешивали при комнатной температуре в течение 3 дней. Твердый осадок отфильтровали и высушили в вакууме в течение 10 мин. Была получена полиморфная модификация В (110 мг).
Препаративный пример 13: Получение полиморфной модификации В из полиморфной модификации А в смеси вода/этанол 90:10
Полиморфную модификацию А (146 мг) суспендировали в Н2О (1,8 мл) и этаноле (0,2 мл) и перемешивали при комнатной температуре в течение 3 дней. Твердый осадок отфильтровали и высушили в вакууме в течение 10 мин. Была получена полиморфная модификация В (160 мг, влажный).
Препаративный пример 14: Получение полиморфной модификации В из полиморфной модификации А в смеси вода/тетрагидрофуран 10:90
Полиморфную модификацию А (154 мг) растворили в Н2О (0,2 мл) и тетрагидрофуране (1,8 мл) и перемешивали при комнатной температуре в течение 3 дней. Твердый осадок, образовавшийся за это время, отфильтровали и высушили в вакууме в течение 10 мин. Была получена полиморфная модификация В (40 мг).
Препаративный пример 15: Получение полиморфной модификации В из полиморфной модификации А в смеси вода/тетрагидрофуран 90:10
Полиморфную модификацию А (151 мг) суспендировали в H2O (1,8 мл) и тетрагидрофуране (0,2 мл) и перемешивали при комнатной температуре в течение 3 дней. Твердый осадок отфильтровали и высушили в вакууме в течение 10 мин. Была получена полиморфная модификация В (165 мг, влажный).
Препаративный пример 16: Получение полиморфной модификации В из полиморфной модификации А в диоксане
Полиморфную модификацию А (151 мг) суспендировали в диоксане (1 мл). Суспензию перемешивали при комнатной температуре в течение 6 дней. Затем твердое вещество отфильтровали и высушили в вакууме в течение нескольких минут, вещество идентифицировали как полиморфную модификацию В.
Препаративный пример 17: Получение полиморфной модификации В из смеси полиморфной модификации А/полиморфной модификации В 50:50 в толуоле
Смесь полиморфной модификации А (75,7 мг) и полиморфной модификации В (75,3 мг) суспендировали в толуоле (1 мл) и перемешивали при 99°С в течение 1 дня. Из горячего раствора был взят образец, который был немедленно идентифицирован как полиморфная модификация В.
Препаративный пример 18: Получение полиморфной модификации В из полиморфной модификации А в смеси диметилсульфоксид/2-пропанол 1:10
Полиморфную модификацию А (180,0 мг) растворили в диметилсульфоксиде (1,5 мл). Профильтрованный раствор по каплям добавили к 2-пропанолу (15 мл). Через несколько минут после окончания прибавления раствора началось образование осадка. Через 10 мин при перемешивании осадок отфильтровали, промыли 2-пропанолом и высушили в вакууме в течение 15 мин, получая 75 мг полиморфной модификации В.
Препаративный пример 19: Получение полиморфной модификации В из полиморфной модификации А в смеси пиридин/2-пропанол 1:10
Полиморфную модификацию А (182,8 мг) растворили в пиридине (1 мл). Раствор профильтровали и по каплям добавили к 2-пропанолу (10 мл). Ближе к окончанию прибавления раствора началось образование осадка. Суспензию перемешивали 5 мин, твердый осадок отфильтровали, промыли 2-пропанолом и высушили в вакууме в течение 15 мин, получая 90 мг полиморфной модификации В.
Препаративный пример 20: Получение полиморфной модификации В из полиморфной модификации А в смеси уксусная кислота/2-пропанол 1:10
Полиморфную модификацию А (180,9 мг) растворили в уксусной кислоте (1 мл). Раствор профильтровали и по каплям добавили к 2-пропанолу (10 мл). Через 1-2 мин после окончания прибавления раствора началось образование осадка. Суспензию перемешивали 5 мин, твердый осадок отфильтровали, промыли 2-пропанолом и высушили в вакууме в течение 15 мин, получая 95 мг полиморфной модификации В.
Препаративный пример 21: Получение полиморфной модификации В из полиморфной модификации А в смеси уксусная кислота/этанол 1:10
Полиморфную модификацию А (155 мг) растворили в уксусной кислоте (1 мл). Профильтрованный раствор по каплям добавили к этанолу (10 мл). Через 5 мин после окончания прибавления раствора началась кристаллизация. Суспензию перемешивали 1 ч, кристаллы отфильтровали и высушили в вакууме, получив 75 мг полиморфной модификации В.
Препаративный пример 22: Получение полиморфной модификации В из полиморфной модификации А в смеси уксусная кислота/этилацетат 1:10
Полиморфную модификацию А (158 мг) растворили в уксусной кислоте (1 мл). Профильтрованный раствор по каплям добавили к этилацетату (10 мл). Через 2 мин после окончания прибавления раствора началась кристаллизация. Суспензию перемешивали в течение 2 ч, кристаллы отфильтровали и высушили в вакууме, получив 45 мг полиморфной модификации В.
Препаративный пример 23: Получение полиморфной модификации В из полиморфной модификации А в смеси дихлорметан/2-пропанол 1:10
Полиморфную модификацию А (177,4 мг) растворили в дихлорметане (1,5 мл). Раствор профильтровали и по каплям добавили к 2-пропанолу (15 мл). Примерно через 3 мин после окончания прибавления раствора началось выпадение осадка, количество которого со временем медленно увеличивалось. Суспензию перемешивали еще в течение 30 мин, твердый осадок отфильтровали и высушили в вакууме в течение 15 мин, получив 80 мг полиморфной модификации В.
Препаративный пример 24: Получение полиморфной модификации В из полиморфной модификации А в воде
Полиморфную модификацию А (149 мг) суспендировали в воде и перемешивали при комнатной температуре в течение 5 дней. Полученные кристаллы были идентифицированы как полиморфная модификация В.
Пример композиции 1: таблетки по 5 мг
Пример композиции 2: капсулы по 10 мг
Пример композиции 3: пероральные капли
Пример композиции 4: таблетки по 2,5 мг
Пример композиции 5: капсулы по 5 мг
Пример композиции 6: пероральные капли
Характеристика полиморфных модификаций
Полиморфные модификации соединения (I) были охарактеризованы с применением следующих процедур.
Инструментальные и экспериментальные условия
Порошковая рентгеновская дифрактометрия: Bruker D8 Advance. Cu Кα-излучение; мощность рентгеновской трубки 35 кВт/45 мА; детектор VANTЕС1; 0,017° 2θ размер шага, 105±5 с на шаг, 2°-50° 2θ диапазон сканирования (записанный диапазон может быть другим). Применялся держатель для образцов с монокристаллом кремния, диаметр образца 12 мм, глубина 0,1 мм.
Спектроскопия комбинационного рассеяния с Фурье-преобразованием: Bruker RFS100. Nd:YAG 1064 нм возбуждение, мощность лазера 100 мВт, Ge-детектор, 64 сканирования, диапазон 50-3500 см-1, разрешение 2 см-1, алюминиевый держатель для образца.
Дифференциальная сканирующая калориметрия: Perkin Elmer DSC 7. Золотые тигли, скорость нагревания 2°С мин-1 или 10°С мин-1, переменная начальная и конечная температуры.
Рентгеноструктурный анализ монокристаллов: Кристалл измерялся на дифрактометре Nonius Kappa CCD при 173°К с использованием графито-монохромированного Мо Кα-излучения с λ=0,71073 Å. Набор COLLECT применялся для сбора данных и их интегрирования. Структура определялась прямыми методами с использованием программы SIR92. На всех неводородных атомах выполнялась обработка методом наименьших квадратов относительно F c использованием программы CRYSTALS. Sheldrick веса применялись для окончательной обработки. Графики получали с использованием ORTEP III для Windows.
Характеристики полиморфной модификации А
Порошковая рентгеновская дифрактометрия: Рентгеновская дифрактограмма характеризуется исключительно интенсивным пиком при 2θ=5,7°. Принимая во внимание высокоанизотропичную форму кристаллов, можно ожидать, что такая высокая интенсивность наблюдается из-за преимущественной ориентации кристаллов. Рентгеновская дифрактограмма показана на Фиг.1.
Спектроскопия комбинационного рассеяния с Фурье-преобразованием. Характеристические сигналы спектры комбинационного рассеяния представляют собой наиболее интенсивный пик в области С-Н при 3073 см-1, пики при 1616 см-1, 1590 см-1, 1544 см-1, 1326 см-1 и двойной пик при 117 см-1/79 см-1. Фурье-спектр комбинационного рассеяния показан на Фиг.3.
Дифференциальная сканирующая калориметрия: Дифференциальная сканирующая калориметрия ДСК показала острый пик плавления между 166,2 и 167,4°С (небольшие различия, зависящие от скорости сканирования) с ΔfusH=85 Дж/г. Вещество не кристаллизовалось вновь при охлаждении, даже при скорости охлаждения только 2°С/мин, а вместо этого стекловалось при 61,3°С. Кривая ДСК показана на Фиг.5.
Характеристики полиморфной модификации В
Порошковая рентгеновская дифрактометрия: Наиболее интенсивные пики на рентгеновской дифрактограмме расположены при 2θ=7,1° и 21,4°. Рентгеновская дифрактограмма показана на Фиг.2.
Спектроскопия комбинационного рассеяния с Фурье-преобразованием: Характеристические сигналы спектры комбинационного рассеяния полиморфной модификации В найдены при 3107 см-1 (наиболее интенсивный пик в области С-Н), 1605 см-1, 1593 см-1, 1538 см-1, 1336 см-1 и 102 см-1. Фурье-спектр комбинационного рассеяния показан на Фиг.4.
Дифференциальная сканирующая калориметрия: Измерения ДСК показали острый пик плавления при примерно 158°С с энтальпией плавления ΔfusH=104 Дж/г. Кривая ДСК показана на Фиг.6.
Структура монокристалла: Соединение кристаллизуется в центральносимметричную пространственную группу Р-1. Структура показывает две молекулы в асимметрической единице, которые не связаны в пространственную группу симметрии. Эти две молекулы могут почти идеально накладываться при вращении вокруг оси "а", но элементарную ячейку невозможно преобразовать так, чтобы получилась кристаллическая решетка более высокой симметрии.
Структуру можно интерпретировать как соединение, основанное на димерах. Движущая сила образования этих димеров, наиболее вероятно, представляет собой π-π взаимодействие между фенильным циклом и тиофеновым циклом, с одной стороны, и N-гетероциклами, с другой стороны. Два разных типа молекул в элементарной ячейке образуют два разных типа димеров с несколько отличающимися короткими расстояниями между конденсированными N-гетероциклами (3,348 Å и 3,308 Å для самого короткого расстояния, соответственно). Димеры располагаются в слоях наподобие структуры рыбьей кости. Области двух типов димеров всегда чередуются в структуре рыбьей кости, так же как они чередуются от одного слоя к другому. Кристаллическая структура описывается в Таблице 2.
Настоящее изобретение описывает новую полиморфную модификацию N-{5-[3-(тиофен-2-карбонил)-пиразоло[1,5-а]пиримидин-7-ил]-2-фтор-фенил}-N-метил-ацетамида, способы ее получения, применение ее в качестве лекарственного средства, применение ее для приготовления лекарственного средства и фармацевтические композиции, включающие новую полиморфную модификацию. 12 н. и 18 з.п. ф-лы, 2 табл., 7 ил.
1. Полиморфная модификация В N-{5-[3-(тиофен-2-карбонил)-пиразоло[1,5-а]пиримидин-7-ил]-2-фтор-фенил}-N-метил-ацетамида, характеризующаяся кривой порошковой рентгеновской дифрактометрии, имеющей специфические пики при 2θ=7,1°(±0,1°) и 21,4°(±0,1°).
2. Полиморфная модификация по п.1, характеризующаяся кривой порошковой рентгеновской дифрактометрии, имеющей специфические пики при 2θ=7,1°(±0,1°), 11,8°(+0,1°), 12,3°(±0,1°), 12,6°(±0,1°), 13,7°(±0,1°), 14,7°(±0,1°), 15,5°(±0,1°), 19,0°(±0,1°), 20,8°(±0,1°), 21,4°(±0,1°), 22,0°(±0,1°), 22,3°(±0,1°), 22,6°(±0,1°), 23,4°(±0,1°), 23,9°(±0,1°), 25,6°(±0,1°), 26,3°(±0,1°), 27,1°(±0,1°), 27,8°(±0,1°), 31,8°(±0,1°) и 36,5°(±0,1°).
3. Полиморфная модификация В N-{5-[3-(тиофен-2-карбонил)-пиразоло[1,5-а]пиримидин-7-ил]-2-фтор-фенил}-N-метил-ацетамида, характеризующаяся Фурье-спектром комбинационного рассеяния, имеющим характеристические сигналы при 3107 см-1, 1605 см-1, 1593 см-1, 1538 см-1, 1336 см-1, и 102 см-1.
4. Полиморфная модификация В N-{5-[3-(тиофен-2-карбонил)-пиразоло[1,5-а]пиримидин-7-ил]-2-фтор-фенил}-N-метил-ацетамида, характеризующаяся дифференциальной сканирующей калориметрией, имеющей пик плавления примерно 158°С.
5. Способ получения полиморфной модификации по любому из пп.1-4, в котором суспендируют полиморфную модификацию А N-{5-[3-(тиофен-2-карбонил)-пиразоло[1,5-а]пиримидин-7-ил]-2-фтор-фенил}-N-метил-ацетамида при комнатной температуре в растворителе, выбранном из группы, включающей C1-С6алифатические спирты, C1-С6алифатические кетоны, C1-C4алкильные эфиры С1-С4алифатических кислот, С4-С5 насыщенные циклические эфиры, C1-С6алифатические нитрилы, ароматические углеводороды и воду, и смеси, выбранные из группы, включающей C1-С6алифатический спирт и C1-С6алифатическую органическую кислоту, воду и C1-С6алифатический спирт, и воду и С4-С5 насыщенный циклический эфир, после чего выделяют образовавшиеся кристаллы.
6. Способ по п.5, в котором растворитель выбран из группы, включающей метанол, этанол, 1-метокси-2-пропанол, метилэтилкетон, этилацетат, диоксан, ацетонитрил, толуол, воду, смесь этанола и уксусной кислоты, смесь воды и этанола и смесь воды и тетрагидрофурана.
7. Способ по п.6, в котором используют смесь этанола и уксусной кислоты при следующем соотношении компонентов, об.%:
8. Способ по п.7, в котором содержание этанола и уксусной кислоты в смеси составляет 95 и 5 об.% соответственно.
9. Способ по п.6, в котором используют смесь воды и этанола при следующем соотношении компонентов, об.%:
10. Способ по п.9, в котором используют смесь воды и этанола при следующем соотношении компонентов, об.%:
11. Способ по п.6, в котором используют смесь воды и тетрагидрофурана при следующем соотношении компонентов, об.%:
12. Способ по п.11, в котором соотношение воды и тетрагидрофурана в смеси составляет 90 и 10 об.% соответственно.
13. Способ получения полиморфной модификации по любому из пп.1-4, в котором суспендируют смесь полиморфной модификации А N-{5-[3-(тиофен-2-карбонил)-пиразоло[1,5-а]пиримидин-7-ил]-2-фтор-фенил}-N-метил-ацетамида и полиморфной модификации В N-{5-[3-(тиофен-2-карбонил)-пиразоло[1,5-а]пиримидин-7-ил]-2-фтор-фенил)-N-метил-ацетамида в ароматическом растворителе при температуре от 80°С до температуры кипения, после чего выделяют образовавшиеся кристаллы.
14. Способ по п.13, в котором используют смесь полиморфной модификации А и полиморфной модификации В при следующем соотношении компонентов, мас.%:
15. Способ по п.14, в котором содержание полиморфной модификации А и полиморфной модификации В в смеси составляет 50 и 50 мас.% соответственно.
16. Способ по п.13, в котором ароматический растворитель представляет собой толуол.
17. Способ по п.13, в котором смесь суспендируют при температуре от 95°С до 105°С.
18. Способ получения полиморфной модификации по любому из пп.1-4, в котором полиморфную модификацию А N-{5-[3-(тиофен-2-карбонил)-пиразоло[1,5-а]пиримидин-7-ил]-2-фтор-фенил}-N-метил-ацетамида растворяют в растворителе, выбранном из группы, включающей ацетон и тетрагидрофуран, затем отфильтровывают ее и выдерживают до полного испарения растворителя.
19. Способ получения полиморфной модификации по любому из пп.1-4, в котором полиморфную модификации А N-{5-[3-(тиофен-2-карбонил)-пиразоло[1,5-а]пиримидин-7-ил]-2-фтор-фенил}-N-метил-ацетамида растворяют в смеси воды и тетрагидрофурана при комнатной температуре, а затем выделяют образовавшийся кристалический осадок.
20. Способ по п.19, в котором используют смесь воды и тетрагидрофурана при следующем соотношении компонентов, об.%:
21. Способ по п.20, в котором содержание воды и тетрагидрофурана в смеси составляет 10 и 90 об.% соответственно.
22. Способ получения полиморфной модификации по любому из пп.1-4, в котором полиморфную модификацию А N-{5-[3-(тиофен-2-карбонил)-пиразоло[1,5-а]пиримидин-7-ил]-2-фтор-фенил}-N-метил-ацетамида растворяют в растворителе, выбранном из группы, включающей С1-С6алифатические сульфоксиды, ароматические амины, C1-С6алифатические органические кислоты и смесь C1-C6галогенированного алифатического углеводорода и C1-С6алифатического спирта, затем раствор фильтруют, добавляют полученный раствор к антирастворителю, выбранному из группы, включающей C1-С6алифатические спирты и С1-С4алкильные эфиры C1-С4алифатических кислот, после чего выделяют образовавшиеся кристаллы.
23. Способ по п.22, в котором растворитель выбран из группы, включающей диметилсульфоксид, пиридин, уксусную кислоту и смесь дихлорметана и 2-пропанола.
24. Способ по п.23, в котором используют смесь дихлорметана и 2-пропанола при объемном соотношении компонентов (0,5-2):10.
25. Способ по п.24, в котором объемное соотношение дихлорметана и 2-пропанола в смеси составляет 1:10.
26. Способ по п.22, в котором антирастворитель выбран из группы, включающей этанол, 2-пропанол и этилацетат.
27. Применение полиморфной модификации по любому из пп.1-4, в качестве лекарственного средства при лечении или профилактике тревожности, эпилепсии, расстройств сна и бессонницы, для индуцирования седативно-гипнотического эффекта, анестезии и мышечной релаксации и для модулирования времени, необходимого для наступления сна, и его продолжительности.
28. Фармацевтическая композиция для лечения или профилактики тревожности, эпилепсии, расстройств сна и бессонницы, для индуцирования седативно-гипнотического эффекта, анестезии и мышечной релаксации и для модулирования времени, необходимого для наступления сна, и его продолжительности, включающая полиморфную модификацию по любому из пп.1-4, совместно с по меньшей мере одним фармацевтически приемлемым носителем, наполнителем, разбавителем или вспомогательным веществом.
29. Фармацевтическая композиция, для лечения или профилактики тревожности, эпилепсии, расстройств сна и бессоницы, для индуцирования седативно-гипнотического эффекта, анестезии и мышечной релаксации и для модулирования времени, необходимого для наступления сна, и его продолжительности, включающая полиморфную модификацию по любому из пп.1-4.
30. Применение полиморфной модификации по любому из пп.1-4 для приготовления лекарственного средства, предназначенного для лечения или профилактики тревожности, эпилепсии, расстройств сна и бессоницы, для индуцирования седативно-гипнотического эффекта, анестезии и мышечной релаксации и для модулирования времени, необходимого для наступления сна, и его продолжительности.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |
| Способ получения производных @ -пиразоло /1,5- @ /пиримидина или их солей (его варианты) | 1983 |
|
SU1301315A3 |

