Область техники
Настоящее изобретение относится к новому производному фосфорамидита, в которое введена новая защитная группа по 2'-гидроксильной группе, и к реагенту для введения этой защитной группы.
Уровень техники
Олигорибонуклеиновые кислоты (олиго-РНК) являются полезными в качестве РНК-зондов для генного анализа, фармацевтических веществ на основе РНК (антисмысловые РНК, рибозимы, РНК для РНК-опосредуемого регулирования экспрессии генов), искусственных ферментов, и аптамеров (олигонуклеотидов, которые проявляют специфическое связывание с молекулой-мишенью). Способ твердофазного синтеза для получения олиго-РНК создали в конце восьмидесятых годов. В первом сообщении, касающемся этого способа, использовали производные фосфорамидитов с трет-бутилдиметилсилильной группой (TBDMS) или триизопропилсилильной группой (TIPS) в качестве защитной группы для 2'-гидроксильной группы (непатентный документ 1).
В химическом синтезе олиго-РНК присутствует намного больше проблем, чем в химическом синтезе олигодезоксирибонуклеиновых кислот (олиго-ДНК), состоящих только из дезоксирибонуклеотидов.
Например, использование группы TBDMS в качестве защитной группы для 2'-гидроксильной группы может вызывать побочную реакцию, в которой группа TBDMS, защищающая 2'-гидроксильную группу, мигрирует к 3'-гидроксильной группе во время фосфитилирования 3'-гидроксильной группы. Кроме того, использование объемного заместителя, такого как группа TBDMS, в качестве защитной группы для 2'-гидроксильной группы, может снизить скорость реакции конденсации при образовании межнуклеотидной связи вследствие пространственного затруднения около атома фосфора в 3'-положении, возможно приводя к разрыву или перегруппировке межнуклеотидной связи во время удаления защитной группы для 2'-гидроксильной группы после олигомеризации.
Для того чтобы преодолеть вышеприведенные проблемы, в настоящий момент изучаются более эффективные способы синтеза олиго-РНК.
В качестве защитной группы для 2'-гидроксильной группы известна 1-(2-цианоэтокси)этильная (CEE) группа, которую удаляют вместе с 3'- и 5'-защитной бис-силильной группой в нейтральной среде, обеспечивающей удаление бис-силильной защитной группы (непатентный документ 2).
На основе этой информации Wada разработал производное фосфорамидита для получения олиго-РНК, в котором группа CEE, которая может быть удалена в нейтральной среде, является введенной в 2'-гидроксигруппу (непатентный документ 3 и непатентный документ 4). Однако, поскольку введение группы СЕЕ в положение 2'-гидроксильной группы приводит к образованию нового асимметричного центра, олиго-РНК, в которых 2'-гидроксильные группы являются защищенными группой СЕЕ, представляют собой диастереоизомерную смесь. Следовательно, очищение и выделение желательной олиго-РНК являются более сложными. К тому же, поскольку олиго-РНК, в которые была введена группа СЕЕ, имеют метильную группу на углероде, присоединенном к атому кислорода в положении 2', следует ожидать возникновения некоторого пространственного затруднения вокруг атома фосфора, присоединенного к 3'-гидроксильной группе, вызывающего опасения относительно снижения эффективности конденсации и скорости реакции конденсации.
Непатентный документ 1: N.A. Usman et al., Journal of the American Chemical Society, Vol. 109, 7845 (1987).
Непатентный документ 2: Wolfgang Pfleiderer et al., Helvetica Chimica Acta, Vol. 81, 1545 (1998).
Непатентный документ 3: Takeshi Wada, Bioindustry, Vol. 21, No. 1, 17 (2004).
Непатентный документ 4: T. Umemoto et al., Tetrahedron Letters, Vol. 45, 9529 (2004).
РАСКРЫТИЕ ИЗОБРЕТЕНИЯ
Задача, которая должна быть решена с помощью изобретения
Основная цель настоящего изобретения заключается в том, чтобы обеспечить полезное и новое производное фосфорамидита для простого и высокопродуктивного способа синтеза олиго-РНК. Другая цель настоящего изобретения заключается в том, чтобы обеспечить новое эфирное соединение, которое может быть использовано для присоединения защитной группы к 2'-гидроксильной группе рибозы, при этом защитная группа может быть удалена в нейтральной cреде.
СРЕДСТВА ДЛЯ РЕШЕНИЯ ЗАДАЧ
После интенсивных и настойчивых исследований авторы настоящего изобретения нашли соединение, посредством которого можно достигнуть вышеприведенных целей, и таким образом осуществить настоящее изобретение.
I. Производное фосфорамидита настоящего изобретения
Настоящее изобретение может включать производное фосфорамидита, представленное следующей общей формулой (1) (в дальнейшем в этом документе именуется как производное фосфорамидита настоящего изобретения).
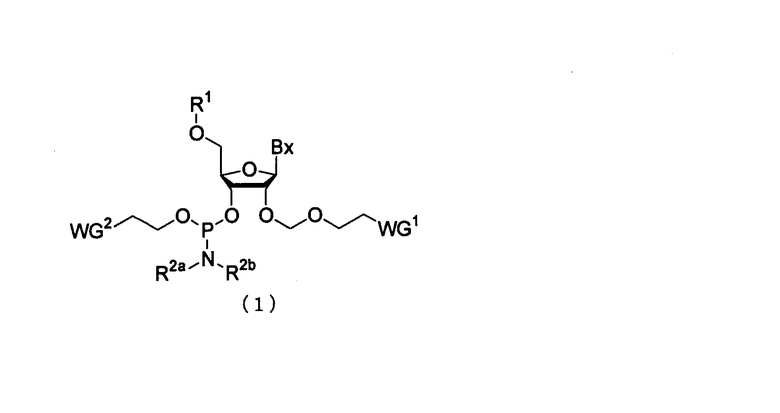
в которой
Bx представляет собой нуклеиновое основание, которое может иметь защитную группу;
R1 представляет собой заместитель, представленный следующей общей формулой (2)
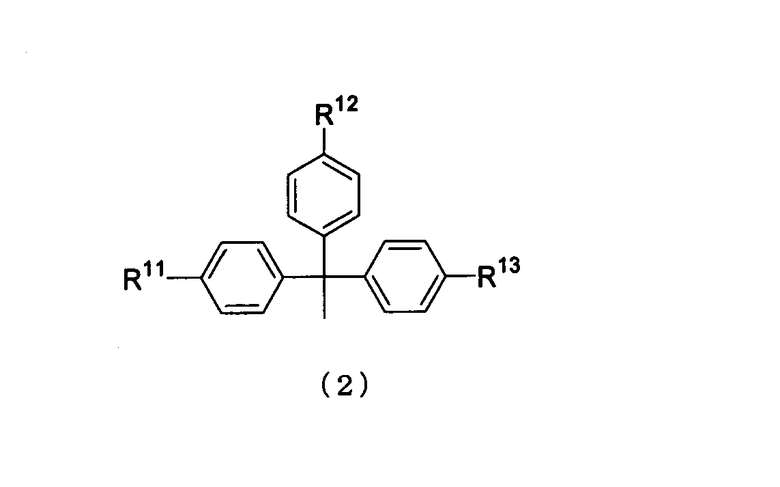
в которой
R11, R12 и R13 являются одинаковыми или различными, и каждый представляет собой водород или алкоксигруппу.
R2a и R2b являются одинаковыми или различными, и каждый представляет собой алкильную группу, или R2a и R2b, взятые вместе со смежным атомом азота, могут образовывать 5-6-членную насыщенную циклическую аминогруппу, при этом циклическая аминогруппа необязательно имеет атом кислорода или серы в составе цикла в дополнение к смежному атому азота; и WG1 и WG2 являются одинаковыми или различными, и каждый представляет собой электроноакцепторную группу.
Примеры нуклеинового основания Вх специально не ограничивают до тех пор, пока оно представляет собой нуклеиновое основание, используемое в синтезе нуклеиновой кислоты, и могут включать, например, аденин, гуанин, цитозин, урацил или их модифицированную форму.
Модифицированная форма нуклеинового основания означает соединение, в котором нуклеиновое основание несет один или более произвольных заместителей.
Примеры заместителя для модифицированной формы Вх могут включать галоген, ацильную группу, алкильную группу, арилалкильную группу, алкоксигруппу, алкоксиалкильную группу, гидроксигруппу, аминогруппу, моноалкиламиногруппу, диалкиламиногруппу, карбоксигруппу, цианогруппу и нитрогруппу. Модифицированная форма Вх может быть замещена одним-тремя такими заместителями.
Нуклеиновое основание Вх может быть защищенным. Особенно предпочтительно, чтобы была защищенной аминогруппа нуклеинового основания, имеющего аминогруппу, такого как аденин, гуанин и цитозин.
Защитную группу аминогруппы особо не ограничивают до тех пор, пока она представляет собой защитную группу, используемую в качестве защитной группы нуклеиновой кислоты, и она может включать, например, бензоильную группу, 4-метоксибензоильную группу, ацетильную группу, пропионильную группу, бутирильную группу, изобутирильную группу, фенилацетильную группу, феноксиацетильную группу, 4-трет-бутилфеноксиацетильную группу, 4-изопропилфеноксиацетильную группу и (диметиламино)метиленовую группу.
Примеры насыщенных циклических аминогрупп R2 могут включать пирролидин-1-ильную группу, пиперидин-1-ильную группу, морфолин-1-ильную группу и тиоморфолин-1-ильную группу.
Электроноакцепторные группы WG1 и WG2 могут включать цианогруппу, нитрогруппу, алкилсульфогруппу и галоген. Среди них цианогруппа является предпочтительной.
Примеры галогена производного фосфорамидита настоящего изобретения могут включать фтор, хлор, бром и йод.
Примеры ацильной группы производного фосфорамидита настоящего изобретения могут включать линейную или разветвленную алканоильную группу, имеющую 1-6 атомов углерода, и ароильную группу, имеющую 7-13 атомов углерода. Более конкретно, ацильная группа может включать, например, формильную группу, ацетильную группу, н-пропионильную группу, изопропионильную группу, н-бутироильную группу, изобутироильную группу, трет-бутироильную группу, валероильную группу, гексаноильную группу, бензоильную группу, нафтоильную группу и левулиноильную (ß-ацетилпропионильную) группу.
Примеры алкильной группы производного фосфорамидита настоящего изобретения могут включать линейную или разветвленную алкильную группу, имеющую 1-5 атомов углерода. Более конкретно, алкильная группа может включать, например, метильную группу, этильную группу, н-пропильную группу, изопропильную группу, н-бутильную группу, изобутильную группу, втор-бутильную группу, трет-бутильную группу, н-пентильную группу, изопентильную группу, неопентильную группу и трет-пентильную группу. Алкильная группа может быть замещена, и примеры заместителя могут включать галоген, алкильную группу, алкоксигруппу, цианогруппу и нитрогруппу. Алкильная группа может быть замещена одним-тремя такими заместителями.
Примеры алкильной части арилалкильной группы, алкоксиалкильной группы, моноалкиламиногруппы, диалкиламиногруппы и алкилсульфогруппы в производном фосфорамидита настоящего изобретения могут включать алкильные группы, идентичные приведенным выше.
Примеры алкоксигруппы производного фосфорамидита настоящего изобретения могут включать линейную или разветвленную алкоксигруппу, имеющую 1-4 атома углерода. Более конкретно, алкоксигруппа может включать, например, метоксигруппу, этоксигруппу, н-пропоксигруппу, изопропоксигруппу, н-бутоксигруппу, изобутоксигруппу, втор-бутоксигруппу и трет-бутоксигруппу. Среди них алкоксигруппы, имеющие 1-3 атома углерода, являются предпочтительными, а метоксигруппа является более предпочтительной.
Примеры алкоксичасти алкоксиалкильной группы производного фосфорамидита настоящего изобретения могут включать алкоксигруппы, идентичные приведенным выше.
Примеры арильной части арилалкильной группы производного фосфорамидита настоящего изобретения могут включать арильные группы, имеющие 6-12 атомов углерода.
Конкретно, арильная группа может включать, например, фенильную группу, 1-нафтильную группу, 2-нафтильную группу и бифенильную группу. Арильная группа может быть замещена, и примеры заместителя могут включать галоген, алкильную группу, алкоксигруппу, цианогруппу и нитрогруппу. Арильная группа может быть замещена одним-тремя такими заместителями. Примеры галогена, алкильной группы и алкоксигруппы, которые являются заместителями алкильной или арильной группы производного фосфорамидита настоящего изобретения, могут включать, соответственно, группы, идентичные приведенным выше.
Производное фосфорамидита настоящего изобретения может быть использовано в качестве реагента для получения олиго-РНК. Производное фосфорамидита настоящего изобретения представляет собой производное фосфорамидита, имеющее защитную группу эфирного типа в положении 2'-гидроксильной группы, которая может быть удалена в нейтральной среде. Кроме того, производное фосфорамидита настоящего изобретения отличается тем, что реакция конденсации протекает за более короткое время и приводит к лучшему выходу во время синтеза олиго-РНК по сравнению с традиционным производным фосфорамидита. Так происходит потому, что защитная группа эфирного типа, введенная в 2'-гидроксильную группу, является линейной защитной группой и, следовательно, не создает стерических затруднений в пространстве вокруг атома фосфора, присоединенного к 3'-гидроксильной группе. Производное фосфорамидита настоящего изобретения делает возможным получение олиго-РНК высокой степени чистоты способом, по существу идентичным способу, используемому в производстве олиго-ДНК.
В настоящем документе термин «олиго-ДНК» означает олигонуклеиновую кислоту, имеющую только дезоксирибонуклеотиды. К тому же, в настоящем документе термин «олиго-РНК» означает олигонуклеиновую кислоту, содержащую, по меньшей мере, один рибонуклеотид, и которая также может иметь один или более дезоксирибонуклеотидов.
Конкретные примеры производного фосфорамидита настоящего изобретения могут включать следующие соединения 1-5:
1.
N6-ацетил-5'-О-(4,4'-диметокситритил)-2'-О-(2-цианоэтоксиметил)аденозин 3'-О-(2-цианоэтил N,N-диизопропилфосфорамидит)
2.
N2-ацетил-5'-О-(4,4'-диметокситритил)-2'-О-(2-цианоэтоксиметил)гуанозин 3'-О-(2-цианоэтил N,N-диизопропилфосфорамидит)
3.
N2-феноксиацетил-5'-О-(4,4'-диметокситритил)-2'-О-(2-цианоэтоксиметил)гуанозин 3'-О-(2-цианоэтил N,N-диизопропилфосфорамидит)
4.
N4-ацетил-5'-О-(4,4'-диметокситритил)-2'-О-(2-цианоэтоксиметил)цитидин 3'-О-(2-цианоэтил N,N-диизопропилфосфорамидит)
5.
5'-О-(4,4'-диметокситритил)-2'-О-(2-цианоэтоксиметил)уридин 3'-О-(2-цианоэтил N,N-диизопропилфосфорамидит)
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Чертеж показывает хроматограмму, полученную методом высокоэффективной жидкостной хроматографии (ВЭЖХ) с обращенной фазой.
На чертеже вертикальная ось указывает время (мин) и горизонтальная ось указывает оптическое поглощение.
ЛУЧШИЙ ВАРИАНТ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
II. Способ получения производного фосфорамидита настоящего изобретения
Производное фосфорамидита настоящего изобретения может быть получено следующим образом.
В следующем способе получения в тех случаях, когда исходные вещества имеют заместитель, который оказывает влияние на реакцию (например, гидроксильная группа, аминогруппа и карбоксигруппа), эти исходные вещества традиционно используют в реакции, защищая их соответствующей защитной группой в соответствии с известным способом.
После завершения реакции защитная группа может быть удалена известным способом, таким как каталитическое восстановление, щелочная обработка, кислотная обработка или т.п. Производное фосфорамидита настоящего изобретения может быть получено из известного соединения или промежуточного соединения, которое может быть легко получено, например, посредством следующих стадий а-h.
Способ получения производного фосфорамидита настоящего изобретения описан подробно ниже.
(1) Стадия а:
Процесс получения производного нуклеозида, представленного следующими общими формулами (15) и (15'), где защитную группу эфирного типа, которая может быть удалена в нейтральной среде, вводят по 2'-гидроксильной группе путем воздействия алкилирующего реагента на производное нуклеозида, представленное следующей общей формулой (14)
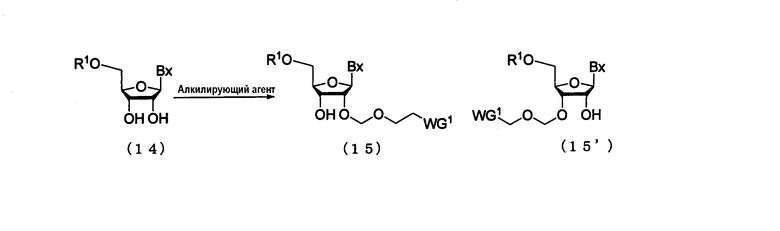
где
Вх, R1 и WG1 такие, как определено выше.
Примеры алкилирующего реагента могут включать эфирное соединение, представленное следующей общей формулой (13)
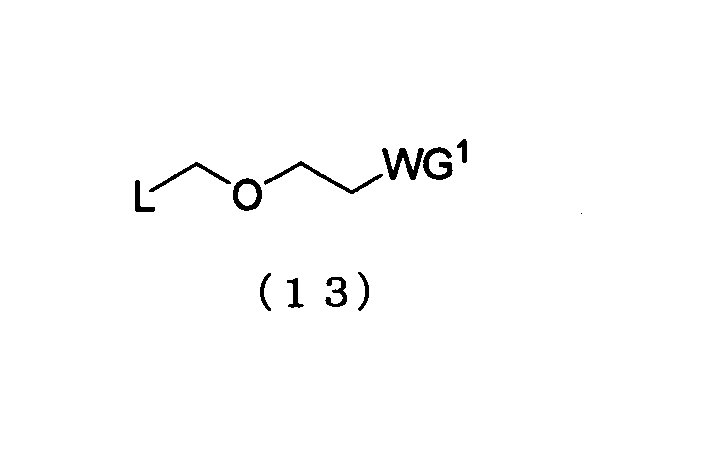
в которой
L представляет собой галоген, арилтиогруппу, алкилсульфоксидную группу или алкилтиогруппу; и WG1 такая, как определено выше.
Примеры галогена, арильной части арилтиогруппы, и алкильных частей алкилсульфоксидной группы и алкилтиогруппы L могут включать соответственно такие же галоген, арильную и алкильную группы, как галоген, арильная и алкильная группы производного фосфорамидита настоящего изобретения.
Конкретные примеры эфирного соединения (13) могут включать следующие соединения 1 и 2:
1. Хлорметил 2-цианоэтиловый эфир
2. 2-Цианоэтилметилтиометиловый эфир
Эфирное соединение (13) представляет собой новый алкилирующий реагент, который может вводить заместитель эфирного типа, который является удаляемым в нейтральной среде, в положение 2'-гидроксильной группы в щелочной среде, и который является полезным в качестве реагента для получения производного фосфорамидита настоящего изобретения.
Эфирное соединение (13) может быть получено посредством следующих стадий 1-4.
Стадия 1:
Процесс получения соединения, представленного следующей общей формулой (24), путем алкилтиометилирования спиртового соединения, представленного следующей общей формулой (20)
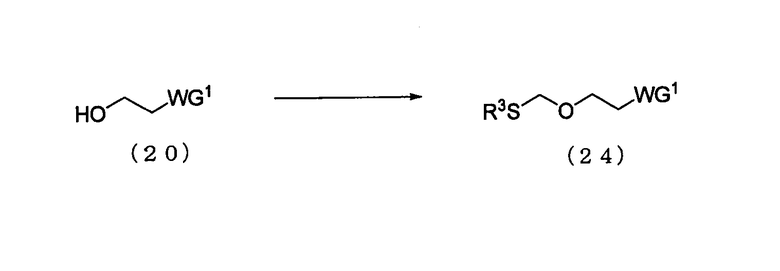
в которой
WG1 такая, как определено выше; и
R3 представляет собой алкильную или арильную группу.
Соединение (24) представляет собой эфирное соединение (13), где L является алкилтиогруппой.
Примеры алкильной группы для R3 могут включать такую же алкильную группу, как алкильная группа производного фосфорамидита настоящего изобретения.
В тех случаях, когда R3 представляет собой метильную группу, примеры алкилтиометилирующего реагента могут включать смешанный растворитель, содержащий диметилсульфоксид, уксусный ангидрид и уксусную кислоту. Количество диметилсульфоксида, которое должно быть использовано, может колебаться в пределах диапазона 10-200 моль на моль соединения (20) и предпочтительно 20-100 моль на моль соединения. Количество уксусной кислоты, которое должно быть использовано, может колебаться в пределах диапазона 10-150 моль на моль соединения (20) и предпочтительно 20-100 моль на моль соединения. Количество уксусного ангидрида, которое должно быть использовано, может находиться в диапазоне 10-150 моль на моль соединения (20) и предпочтительно 20-100 моль на моль соединения. Температура реакции предпочтительно колеблется в пределах диапазона 0-100ºС. Время реакции варьируется в зависимости от типа исходных веществ и температуры реакции и предпочтительно составляет от 1 до 48 час.
Стадия 2:
Процесс получения соединения, представленного следующей общей формулой (25), путем галогенирования соединения (24).
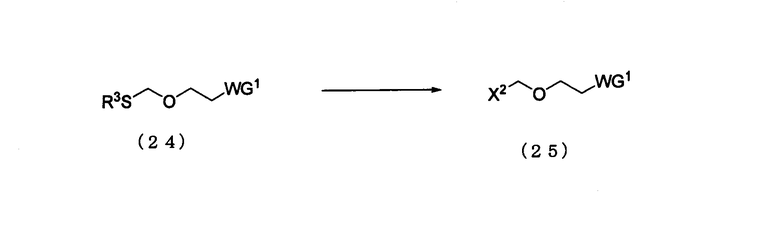
в которой
WG1 и R3 такие, как определено выше; и
X2 представляет собой галоген.
Соединение (25) представляет собой соединение, где L эфирного соединения (13) является галогеном.
Примеры галогена в Х2 могут включать такой же галоген, как галоген производного фосфорамидита настоящего изобретения.
Эта стадия может быть выполнена известным способом (например, T. Benneche et al., Synthesis 762 (1983)).
Примеры растворителя, который должен быть использован, особо не ограничивают, исключая те случаи, когда он является вовлеченным в реакцию, и могут включать, например, галогенированный углеводород, такой как дихлорметан, хлороформ, тетрахлорид углерода и 1,2-дихлорэтан.
Примеры галогенирующего агента могут включать сульфурилхлорид и оксихлорид фосфора.
Количество галогенирующего агента, которое должно быть использовано, соответственно может колебаться в пределах диапазона 1-20 моль на моль соединения (24) и предпочтительно 1-10 моль на моль соединения. Температура реакции предпочтительно колеблется в пределах диапазона 0-100ºС. Время реакции варьируется в зависимости от типа исходных веществ и температуры реакции и предпочтительно составляет от 30 мин до 24 час.
Стадия 3:
Процесс получения соединения, представленного следующей общей формулой (25а), путем арилтиолирования соединения (25).
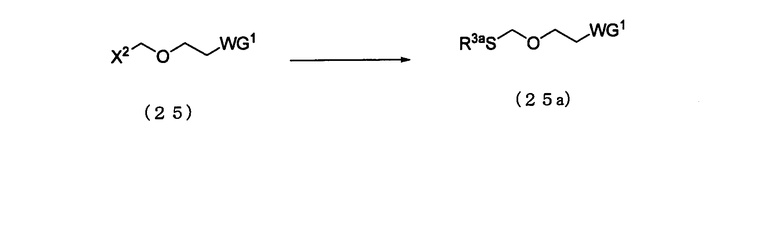
в которой
WG1 и Х2 такие, как определено выше; и
R3а представляет собой арильную группу.
Соединение (25а) представляет собой соединение класса эфирных соединений (13), где L является арилтиогруппой. Примеры арильной группы для R3а могут включать арильную группу, идентичную арильной группе производного фосфорамидита настоящего изобретения. Эта стадия может быть выполнена известным способом. Примеры растворителя, который должен быть использован, особенным образом не ограничивают, исключая те случаи, когда он является вовлеченным в реакцию, и могут включать, например, дихлорметан и ацетонитрил. Примеры арилтиолирующего реагента могут включать тиофенол и 4-метилтиофенол. Количество арилтиолирующего реагента, которое должно быть использовано, может колебаться в пределах диапазона 1-20 моль на моль соединения (25) и предпочтительно 1-5 моль на моль соединения. Температура реакции предпочтительно находится в диапазоне 0-100ºС. Время реакции варьируется в зависимости от типа исходных веществ и температуры реакции и предпочтительно составляет от 1 до 48 час.
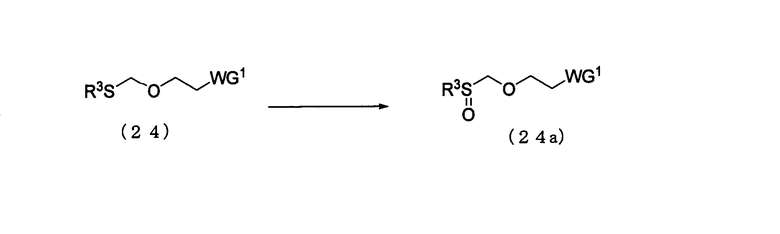
Стадия 4:
Процесс получения соединения, представленного следующей общей формулой (24а), путем окисления соединения (24).
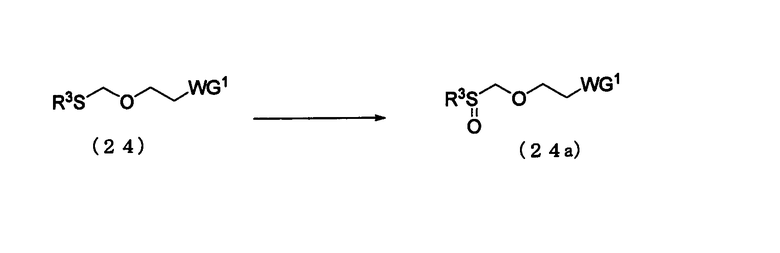
в которой
WG1 и R3 такие, как определено выше.
Соединение (24а) представляет собой соединение класса эфирных соединений (13), где L является алкилсульфоксидной группой. Примеры алкильной группы R3 могут включать алкильную группу, идентичную алкильной группе производного фосфорамидита настоящего изобретения.
Эта стадия может быть выполнена известным способом. Примеры растворителя, который должен быть использован, особо не ограничивают, исключая те случаи, когда он является вовлеченным в реакцию, и могут включать, например, дихлорметан, хлороформ и метанол. Примеры окислителя могут включать метахлорпербензойную кислоту, метапериодатную соль и пероксид водорода. Количество окислителя, которое должно быть использовано, может колебаться в пределах диапазона 1-10 моль на моль соединения (24) и предпочтительно 1-2 моля на моль соединения. Температура реакции предпочтительно находится в диапазоне 0-100ºС. Время реакции варьируется в зависимости от типа исходных веществ и температуры реакции и предпочтительно составляет от 1 до 48 час.
В тех случаях, когда в качестве алкилирующего реагента используют соединение (25), эта стадия может быть выполнена следующим образом.
Стадия может быть выполнена путем взаимодействия алкилирующего реагента и основания с соединением (14), которое имеется в продаже или является синтезированным в соответствии с известным способом.
Примеры растворителя, который должен быть использован, особенным образом не ограничивают, исключая те случаи, когда он является вовлеченным в реакцию, и могут включать, например, галогенированный углеводород, такой как дихлорметан, хлороформ, тетрахлорид углерода и 1,2-дихлорэтан. Количество алкилирующего реагента, которое должно быть использовано, может колебаться в пределах диапазона 1-20 моль на моль соединения (14) и предпочтительно 1-10 моль на моль соединения. На этой стадии при необходимости алкилирующий реагент может быть подвергнут реакции посредством промежуточного соединения, получаемого взаимодействием металлсодержащего реагента и основания с соединением (14). Примеры металлсодержащего реагента могут включать дибутилоловодихлорид. Количество металлсодержащего реагента, которое должно быть использовано, может колебаться в пределах диапазона 1-20 моль на моль соединения (14) и предпочтительно 1-10 моль на моль этого соединения. Примеры основания могут включать органическое основание, такое как пиридин, 2,6-диметилпиридин, 2,4,6-триметилпиридин, N-метилимидазол, триэтиламин, трибутиламин, N,N-диизопропилэтиламин и 1,8-диазабицикло[5.4.0]-7-ундецен. Количество основания, которое должно быть использовано, может находиться в пределах диапазона 1-20 моль на моль соединения (14) и предпочтительно 1-10 моль на моль соединения. Температура реакции предпочтительно колеблется в пределах диапазона 0-120ºС. Время реакции варьируется в зависимости от типа исходных веществ и температуры реакции и предпочтительно составляет от 30 мин до 24 час.
В тех случаях, когда в качестве алкилирующего реагента используют соединение (24) или (25а), эта стадия может быть выполнена следующим образом.
Стадия может быть выполнена путем взаимодействия алкилирующего реагента, кислоты и реагента для галогенирования атома серы с соединением (14), которое имеется в продаже или является синтезированным в соответствии с известным способом (например, M. Matteucci, Tetrahedron Letters, Vol. 31, 2385 (1990)). Количество алкилирующего реагента, которое должно быть использовано, может колебаться в пределах диапазона 1-5 моль на моль соединения (14) и предпочтительно 1,05-3 моля на моль соединения. Примеры кислоты могут включать трифторметансульфокислоту, трифторметансульфонат серебра и триметилсилилтрифторметансульфонат. Количество кислоты, которое должно быть использовано, может колебаться в пределах диапазона 0,01-20 моль на моль соединения (14) и предпочтительно 0,02-10 моль на моль соединения. Примеры растворителя, который должен быть использован, особо не ограничивают, исключая те случаи, когда он является вовлеченным в реакцию, и могут включать, например, дихлорметан, хлороформ, тетрахлорид углерода, 1,2-дихлорэтан, бензол, толуол, ксилол, тетрагидрофуран, ацетонитрил и их смеси. Примеры реагента для галогенирования атома серы, который должен быть использован на этой стадии, могут включать N-бромсукцинимид (NBS) и N-йодсукцинимид (NIS). Количество реагента для галогенирования атома серы, которое должно быть использовано, может колебаться в пределах диапазона 1-10 моль на моль соединения (14) и предпочтительно 1,05-5 моль на моль соединения. Температура реакции предпочтительно находится в пределах диапазона -78-30ºС. Время реакции варьируется в зависимости от типа исходных веществ и температуры реакции и предпочтительно составляет от 5 мин до 5 час.
В тех случаях, когда в качестве алкилирующего реагента используют соединение (24а), эта стадия может быть выполнена следующим образом.
Стадия может быть выполнена путем взаимодействия алкилирующего реагента, ангидрида кислоты и основания с соединением (14), которое имеется в продаже или является синтезированным в соответствии с известным способом. Количество алкилирующего реагента, которое должно быть использовано, может находиться в пределах диапазона 1-5 моль на моль соединения (14) и предпочтительно 1,05-3 моля на моль соединения. Примеры ангидрида кислоты могут включать ангидрид трифторметансульфокислоты и уксусный ангидрид. Количество ангидрида кислоты, которое должно быть использовано, может находиться в пределах диапазона 0,01-20 моль на моль соединения (14) и предпочтительно 0,02-10 моль на моль соединения. Примеры основания могут включать тетраметилмочевину и коллидин. Количество основания, которое должно быть использовано, может находиться в пределах диапазона 0,01-20 моль на моль соединения (14) и предпочтительно 0,02-10 моль на моль соединения. Примеры растворителя, который должен быть использован, особо не ограничивают, исключая те случаи, когда он является вовлеченным в реакцию, и могут включать, например, дихлорметан, хлороформ, тетрахлорид углерода, 1,2-дихлорэтан и их смеси. Температура реакции предпочтительно колеблется в пределах диапазона -78-30ºС. Время реакции варьируется в зависимости от типа исходных веществ и температуры реакции и предпочтительно составляет от 5 мин до 24 час.
(2) Стадия b:
Процесс выделения и очистки производного нуклеозида (15), полученного посредством стадии (а)
На этой стадии производное нуклеозида может быть выделено и очищено из смеси, полученной посредством стадии (а), путем использования стандартного метода выделения и очистки, такого как тонкослойная хроматография, колоночная хроматография на силикагеле или т.п.
(3) Стадия с:
Процесс, который осуществляют отдельно от стадии b, для получения соединения рибонуклеиновой кислоты, представленного следующей общей формулой (17), где защитную группу эфирного типа, которая может быть удалена в нейтральной среде, вводят по 2'-гидроксильной группе путем воздействия алкилирующего реагента на соединение рибонуклеиновой кислоты, представленное следующей общей формулой (16)
в которой
Вх и WG1 такие, как определено выше; и
А представляет собой кремнесодержащий заместитель, представленный следующей общей формулой (18а) или (18b)
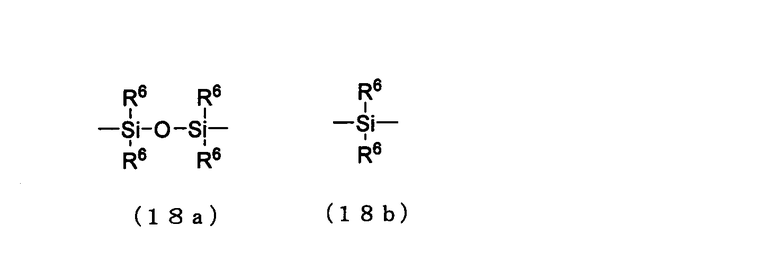
в которой
R6 представляет собой алкильную группу.
Примеры алкильной группы для R6 могут включать алкильную группу, идентичную алкильной группе производного фосфорамидита настоящего изобретения.
Примеры алкилирующего реагента могут включать алкилирующие реагенты, идентичные приведенным выше.
В тех случаях, когда в качестве алкилирующего реагента используют соединение (25), эта стадия может быть выполнена следующим образом.
Стадия может быть выполнена путем взаимодействия алкилирующего реагента и основания с соединением (16), которое имеется в продаже или является синтезированным в соответствии с известным способом.
Примеры растворителя, который должен быть использован, особо не ограничивают, исключая те случаи, когда он является вовлеченным в реакцию, и могут включать, например, галогенированный углеводород, такой как дихлорметан, хлороформ, тетрахлорид углерода и 1,2-дихлорэтан. Количество алкилирующего реагента, которое должно быть использовано, может находиться в пределах диапазона 1-20 моль на моль соединения (14) и предпочтительно 1-10 моль на моль соединения. На этой стадии при необходимости алкилирующий реагент может быть подвергнут реакции посредством промежуточного соединения, получаемого взаимодействием металлсодержащего реагента и основания с соединением (16). Примеры металлсодержащего реагента могут включать дибутилоловодихлорид и трет-бутилмагнийхлорид. Количество металлсодержащего реагента, которое должно быть использовано, может находиться в пределах диапазона 1-20 моль на моль соединения (16) и предпочтительно 1-10 моль на моль этого соединения. Примеры основания могут включать органическое основание, такое как пиридин, 2,6-диметилпиридин, 2,4,6-триметилпиридин, N-метилимидазол, триэтиламин, трибутиламин, N,N-диизопропилэтиламин и 1,8-диазабицикло[5.4.0]-7-ундецен. Количество основания, которое должно быть использовано, может находиться в пределах диапазона 1-20 моль на моль соединения (16) и предпочтительно 1-10 моль на моль соединения. Температура реакции предпочтительно находится в пределах диапазона 0-120ºС. Время реакции варьируется в зависимости от типа исходных веществ и температуры реакции и предпочтительно составляет от 30 мин до 24 час.
В тех случаях, когда в качестве алкилирующего реагента используют соединение (24) или (25а), эта стадия может быть выполнена следующим образом.
Стадия может быть выполнена путем взаимодействия алкилирующего реагента, кислоты и реагента для галогенирования атома серы с соединением (16), которое имеется в продаже или является синтезированным в соответствии с известным способом (например, M. Matteucci, Tetrahedron Letters, Vol. 31, 2385 (1990)). Количество алкилирующего реагента, которое должно быть использовано, может колебаться в пределах диапазона 1-5 моль на моль соединения (16) и предпочтительно 1,05-3 моля на моль соединения. Примеры кислоты могут включать трифторметансульфокислоту, трифторметансульфонат серебра и триметилсилил-трифторметансульфонат. Количество кислоты, которое должно быть использовано, может колебаться в пределах диапазона 0,01-20 моль на моль соединения (16) и предпочтительно 0,02-10 моль на моль соединения. Примеры растворителя, который должен быть использован, особенно не ограничивают, исключая те случаи, когда он является вовлеченным в реакцию, и могут включать, например, дихлорметан, хлороформ, тетрахлорид углерода, 1,2-дихлорэтан, бензол, толуол, ксилол, тетрагидрофуран, ацетонитрил и их смеси. Примеры реагента для галогенирования атома серы, который должен быть использован на этой стадии, могут включать N-бромсукцинимид (NBS) и N-йодсукцинимид (NIS). Количество реагента для галогенирования атома серы, которое должно быть использовано, может колебаться в пределах диапазона 1-10 моль на моль соединения (16) и предпочтительно 1,05-5 моль на моль соединения. Температура реакции предпочтительно находится в пределах диапазона -78-30ºС. Время реакции варьируется в зависимости от типа исходных веществ и температуры реакции и предпочтительно составляет от 5 мин до 5 час.
В тех случаях, когда в качестве алкилирующего реагента используют соединение (24а), эта стадия может быть выполнена следующим образом.
Стадия может быть выполнена путем взаимодействия алкилирующего реагента, ангидрида кислоты и основания с соединением (16), которое имеется в продаже или является синтезированным в соответствии с известным способом. Количество алкилирующего реагента, которое должно быть использовано, может колебаться в пределах диапазона 1-5 моль на моль соединения (16) и предпочтительно 1,05-3 моля на моль соединения. Примеры ангидрида кислоты могут включать ангидрид трифторметансульфокислоты и уксусный ангидрид. Количество ангидрида кислоты, которое должно быть использовано, может колебаться в пределах диапазона 0,01-20 моль на моль соединения (16) и предпочтительно 0,02-10 моль на моль соединения. Примеры основания могут включать тетраметилмочевину и коллидин. Количество основания, которое должно быть использовано, может колебаться в пределах диапазона 0,01-20 моль на моль соединения (16) и предпочтительно 0,02-10 моль на моль соединения. Примеры растворителя, который должен быть использован, особенно не ограничивают, исключая те случаи, когда он является вовлеченным в реакцию, и могут включать, например, дихлорметан, хлороформ, тетрахлорид углерода, 1,2-дихлорэтан и их смеси. Температура реакции предпочтительно находится в пределах диапазона -78-30ºС. Время реакции варьируется в зависимости от типа исходных веществ и температуры реакции и предпочтительно составляет от 5 мин до 24 час.
(4) Стадия d:
Процесс, который осуществляют отдельно от стадий а-с, для получения соединения рибонуклеиновой кислоты, представленного следующей общей формулой (19), путем воздействия диметилсульфоксида, уксусной кислоты и уксусного ангидрида на соединение рибонуклеиновой кислоты (16)
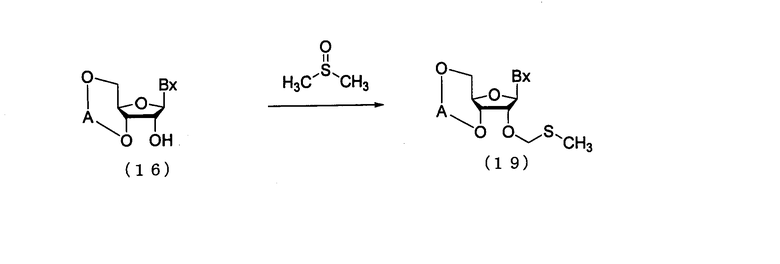
в которых
А и Вх такие, как определено выше.
Эта стадия может быть осуществлена путем взаимодействия диметилсульфоксида, уксусной кислоты и уксусного ангидрида с соединением (14), которое имеется в продаже или является синтезированным в соответствии с известным способом.
Эта стадия может быть осуществлена путем взаимодействия диметилсульфоксида, уксусной кислоты и уксусного ангидрида с соединением (14), которое имеется в продаже или является синтезированным в соответствии с известным способом.
Количество диметилсульфоксида, которое должно быть использовано, может колебаться в пределах диапазона от 10 до 200 моль на моль соединения (16) и предпочтительно от 20- до 100-кратного количества моль на моль соединения.
Количество уксусной кислоты, которое должно быть использовано, может колебаться в пределах диапазона 10-150 моль на моль соединения (16) и предпочтительно 20-100 моль на моль соединения. Количество уксусного ангидрида, которое должно быть использовано, может колебаться в пределах диапазона 10-150 моль на моль соединения (16) и предпочтительно 20-100 моль на моль соединения. Температура реакции предпочтительно находится в пределах диапазона 10-50ºС. Время реакции варьируется в зависимости от типа исходных веществ и температуры реакции и предпочтительно составляет от 30 мин до 24 час.
(5) Стадия е:
Процесс получения соединения рибонуклеиновой кислоты, представленного следующей общей формулой (17), где защитную группу эфирного типа, которая может быть удалена в нейтральной среде, вводят по 2'-гидроксильной группе путем воздействия спиртового соединения, представленного следующей общей формулой (20), кислоты и реагента для галогенирования атома серы на производное нуклеозида (19), полученное посредством стадии d.
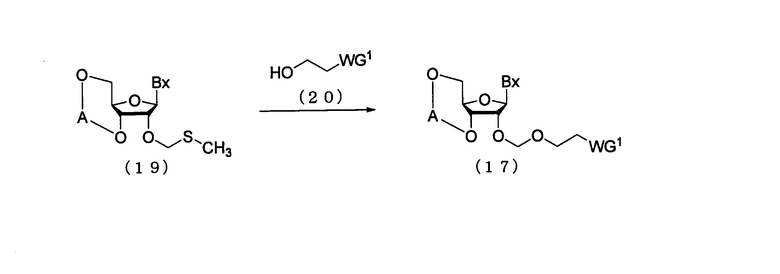
где
А, Вх и WG1 такие, как определено выше.
Стадия может быть осуществлена путем взаимодействия спиртового соединения (20), кислоты и реагента для галогенирования атома серы с соединением рибонуклеиновой кислоты (19) в соответствии с известным способом. Примеры растворителя, который должен быть использован, особо не ограничивают, исключая те случаи, когда он является вовлеченным в реакцию, и могут включать, например, дихлорметан, хлороформ, тетрахлорид углерода, 1,2-дихлорэтан, бензол, толуол, ксилол, тетрагидрофуран, ацетонитрил и их смеси. Количество спиртового соединения (20), которое должно быть использовано, может находиться в пределах диапазона 1-20 моль на моль соединения (19) и предпочтительно 1-10 моль на моль соединения. Примеры кислоты могут включать трифторметансульфокислоту, трифторметансульфонат серебра и триметилсилил-трифторметансульфонат. Примеры реагента для галогенирования атома серы могут включать N-бромсукцинимид (NBS) и N-йодсукцинимид (NIS). Количество реагента для галогенирования атома серы, которое должно быть использовано, может колебаться в пределах диапазона 0,1-20 моль на моль соединения (19) и предпочтительно 0,2-10 моль на моль соединения. Температура реакции предпочтительно находится в пределах диапазона -100-20ºС. Время реакции варьируется в зависимости от типа исходных веществ и температуры реакции и предпочтительно составляет от 5 мин до 12 час.
(6) Стадия f:
Процесс получения соединения рибонуклеиновой кислоты, представленного следующей общей формулой (21), путем удаления защитных групп для 3'- и 5'-гидроксильных групп соединения рибонуклеиновой кислоты (17), полученного на стадии с или стадии е.
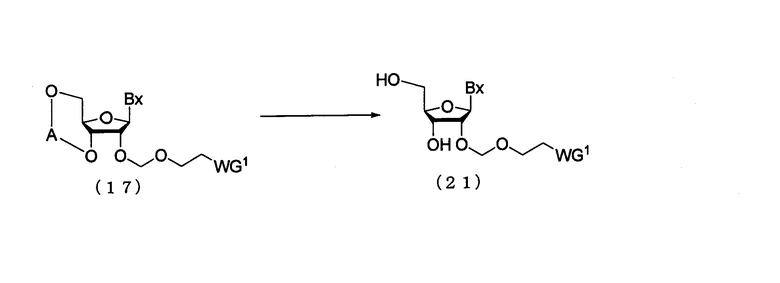
в которых
А, Вх и WG1 такие, как определено выше.
Эта стадия может быть осуществлена путем растворения соединения (17) в органическом растворителе и взаимодействия с фторирующим агентом и кислотой в виде смеси с произвольным соотношением смешения. Примеры фторирующего агента, который должен быть использован на этой стадии, могут включать фторид аммония, фторид тетра-н-бутиламмония (TBAF), триэтиламин-тригидрофторид, пиридингидрофторид. Количество фторирующего агента, которое должно быть использовано, может колебаться в пределах диапазона 0,1-20 моль на моль соединения (17), и предпочтительно 0,2-10 моль на моль соединения. Температура реакции предпочтительно находится в пределах диапазона 0-120ºС. Время реакции варьируется в зависимости от типа исходных веществ и температуры реакции и предпочтительно составляет от 30 мин до 24 час.
(7) Стадия g:
Процесс получения соединения рибонуклеиновой кислоты (15) путем введения защитной группы (R1), которая может быть удалена в кислой среде, в 5'-гидроксильную группу соединения рибонуклеиновой кислоты (21), полученного посредством стадии f.
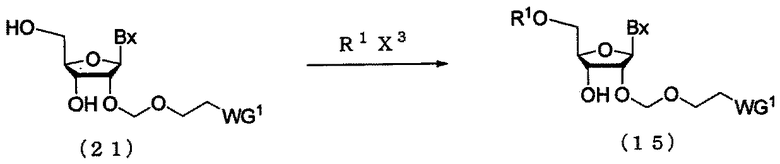
в которых
А, Вх, R1 и WG1 такие, как определено выше; и
Х3 представляет собой галоген.
Примеры галогена для Х3 могут включать галоген, идентичный галогенам производного фосфорамидита настоящего изобретения. Стадия может быть выполнена путем взаимодействия R1X3 с соединением (21) в соответствии с известным способом. Количество R1X3, которое должно быть использовано, может находиться в пределах диапазона 1-20 моль на моль соединения (21) и предпочтительно 1-10 моль на моль соединения. Примеры растворителя, который должен быть использован, особо не ограничивают, исключая те случаи, когда он является вовлеченным в реакцию, и могут включать, например, ацетонитрил и тетрагидрофуран. Примеры основания могут включать органическое основание, такое как пиридин, 2,6-диметилпиридин, 2,4,6-триметилпиридин, N-метилимидазол, триэтиламин, трибутиламин, N,N-диизопропилэтиламин и 1,8-диазабицикло[5.4.0]-7-ундецен. Количество основания, которое должно быть использовано, может колебаться в пределах диапазона 1-20 моль на моль соединения (21) и предпочтительно 1-10 моль на моль соединения. Температура реакции предпочтительно находится в пределах диапазона 0-120ºС. Время реакции варьируется в зависимости от типа исходных веществ и температуры реакции и предпочтительно составляет от 30 мин до 24 час.
(8) Стадия h:
Процесс получения производного фосфорамидита настоящего изобретения фосфтилированием 3'-гидроксильной группы путем воздействия фосфитилирующего реагента и, при необходимости, активирующего агента на производное нуклеозида (15), полученное на стадии b или стадии f.
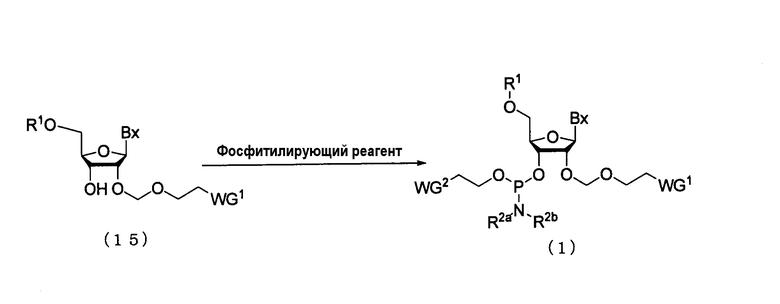
где
Вх, R1, R2a, R2b, WG1 и WG2 такие, как определено выше.
Примеры фосфитилирующего реагента могут включать соединения, представленные следующими общими формулами (22) и (23)
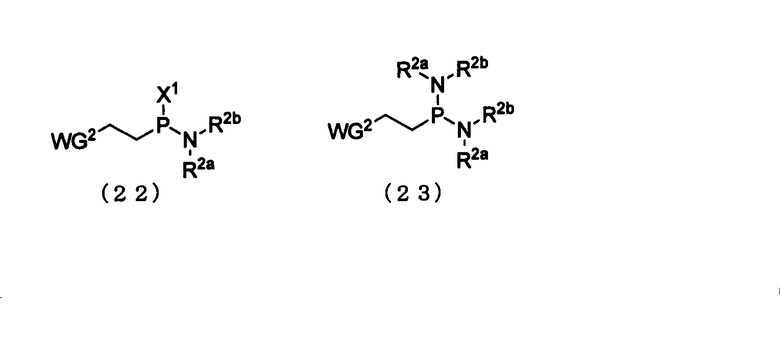
в которых
R2a, R2b и WG2 такие, как определено выше; и
Х1 представляет собой галоген.
Примеры галогена для Х1 могут включать галоген, идентичный галогенам производного фосфорамидита настоящего изобретения. Эта стадия представляет собой реакцию фосфитилирования 3'-гидроксильной группы путем взаимодействия фосфитилирующего реагента с соединением (15) и может быть осуществлена в соответствии с известным способом. При необходимости может быть использован активирующий агент. Примеры растворителя, который должен быть использован, особо не ограничивают, исключая те случаи, когда он является вовлеченным в реакцию, и могут включать, например, ацетонитрил и тетрагидрофуран.
Количество фосфитилирующего реагента, которое должно быть использовано, может колебаться в пределах диапазона 1-20 моль на моль соединения (15) и предпочтительно 1-10 моль на моль соединения. Примеры активирующего агента могут включать 1Н-тетразол, 5-этилтиотетразол, 4,5-дихлоримидазол, 4,5-дицианоимидазол, бензотриазолтрифлат, имидазолтрифлат, пиридинийтрифлат, N,N-диизопропилэтиламин и 2,4,6-коллидин/N-метилимидазол. Количество активирующего агента, которое должно быть использовано, может находиться в пределах диапазона 1-20 моль на моль соединения (15) и предпочтительно 1-10 моль на моль соединения. Температура реакции предпочтительно находится в пределах диапазона 0-120ºС. Время реакции варьируется в зависимости от типа исходных веществ и температуры реакции и предпочтительно составляет от 30 мин до 24 час. Производное фосфорамидита настоящего изобретения, полученное таким образом, может быть выделено и очищено способом, известным по своей сути, таким как концентрирование, превращение в жидкой фазе, разделение, экстракция растворителем, кристаллизация, перекристаллизация, фракционная перегонка или хроматография.
III. Способ получения олиго-РНК
Настоящее изобретение может включать способ получения олиго-РНК, представленных следующей общей формулой (3), включающий применение производного фосфорамидита настоящего изобретения.
Подробности описаны ниже.
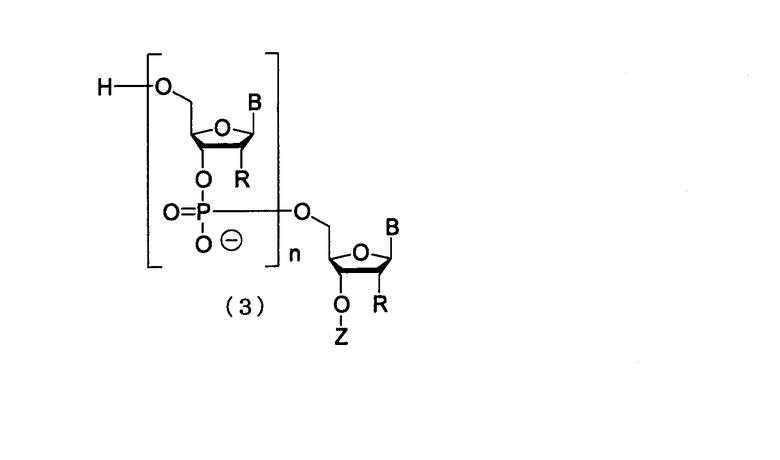
в которой
каждый В независимо представляет собой аденин, гуанин, цитозин, урацил, тимин или их модифицированную форму;
каждый R независимо представляет собой Н или гидроксильную группу, и, по меньшей мере, один из R является гидроксильной группой;
Z представляет собой Н или фосфатную группу; и
n представляет собой целое число от 1 до 100.
Предпочтительно n представляет собой целое число от 10 до 50, и более предпочтительно целое число от 15 до 30.
Примеры заместителя для модифицированной формы В могут включать галоген, ацильную группу, алкильную группу, арилалкильную группу, алкоксигруппу, гидроксильную группу, аминогруппу, моноалкиламиногруппу, диалкиламиногруппу, карбоксигруппу, цианогруппу и нитрогруппу; и модифицированная форма В может быть замещена 1-3 такими заместителями. Примеры галогена, арильной группы, алкильной группы, арилалкильной группы, алкоксигруппы, алкоксиалкильной группы, аминогруппы, моноалкиламиногруппы и диалкиламиногруппы для модифицированной формы В могут включать группы, идентичные соответствующим группам производного фосфорамидита настоящего изобретения.
Способ получения олиго-РНК (3) с применением производного фосфорамидита настоящего изобретения может быть осуществлен известным способом и, например, может быть осуществлен путем конденсации соединения мономера нуклеиновой кислоты в направлении от 3' к 5' шаг за шагом в соответствии со следующими стадиями А-G.
Соединения и реагенты, которые должны быть использованы в следующей стадии, за исключением производного фосфорамидита настоящего изобретения, особо не ограничивают, поскольку их, как правило, используют в синтезе олиго-РНК или олиго-ДНК. Кроме того, все стадии могут быть выполнены с использованием автоматического синтезатора для ДНК или вручную как в случае применения традиционных веществ для синтеза нуклеиновой кислоты. Использование автоматизированного синтезатора является желательным с точки зрения простоты и удобства способа и точности синтеза. Соединения и реагенты, описанные в следующих стадиях А-G, за исключением соединения мономера нуклеиновой кислоты, особо не ограничивают, поскольку их, как правило, используют в синтезе олиго-ДНК или олиго-РНК.
(1) Стадия А:
Процесс получения соединения, представленного следующей общей формулой (5), путем удаления 5'-гидроксильной группы из соединения, представленного следующей общей формулой (4), под действием кислоты.
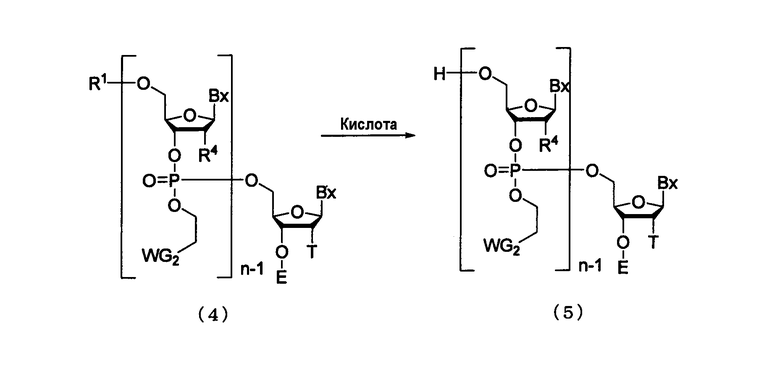
в которой
n, R1 и WG2 такие, как определено выше;
каждый В независимо представляет собой аденин, гуанин, цитозин, урацил, тимин или их модифицированную форму; и каждый R4 независимо представляет собой Н, ацилоксигруппу или заместитель, представленный следующей общей формулой (6)
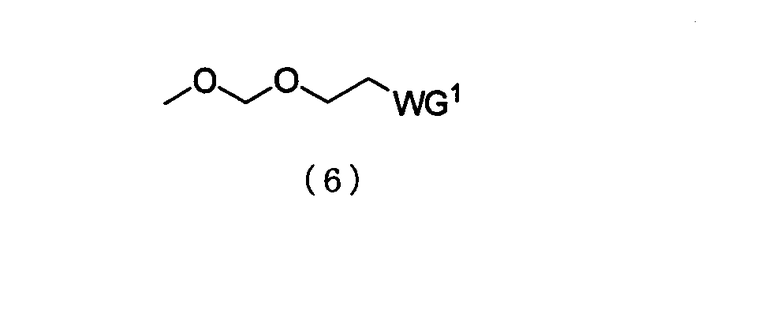
в которой
WG1 такая, как определено выше; и
Е представляет собой ацильную группу или заместитель, представленный следующей общей формулой (7)
-Q-линкер-твердый носитель (7),
в которой
Q представляет собой одинарную связь или заместитель, представленный следующей общей формулой (8)
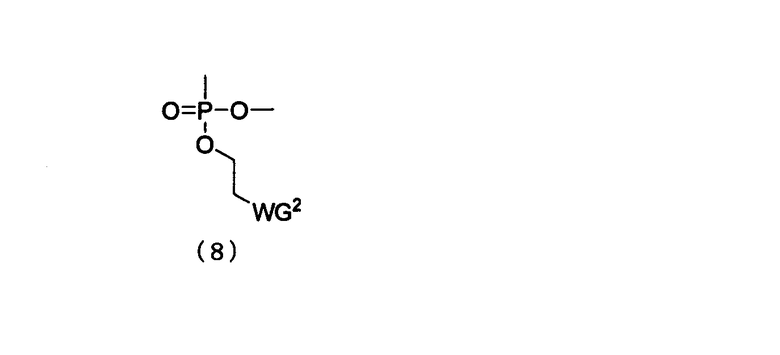
в которой
WG2 такая, как определено выше; и
Т представляет собой Н, ацилоксигруппу, заместитель, представленный вышеприведенной общей формулой (6) или (7), с условием, что либо E, либо Т является заместителем (7).
Стадию осуществляют посредством действия кислоты на соединение, представленное следующей общей формулой (26а), (26b) [соединение (4), где n является 1], которое является присоединенным к твердому носителю, или на олиго-РНК или олиго-ДНК, полученную путем выполнения процессов стадии А - стадии D [соединение (4), где n является 2-100], которая является присоединенной к твердому носителю (в дальнейшем в этом документе именуется как соединение, присоединенное к твердому носителю).
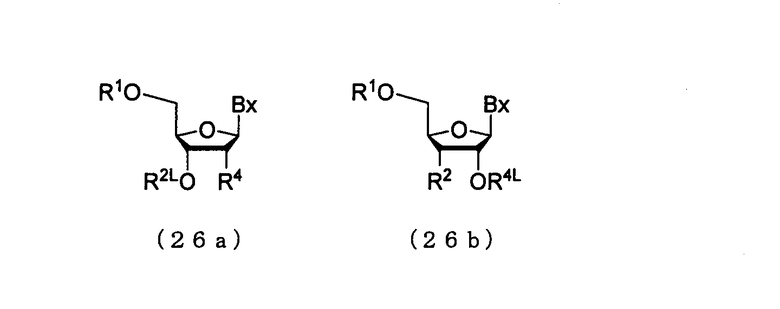
в которой
Вх и R1 такие, как определено выше;
R2L и R4L представляют собой заместитель (7);
R2 представляет собой ацилоксигруппу; и
R4 представляет собой Н, ацилоксигруппу или заместитель (6).
Примеры ацильной части ацилоксигруппы R2 и R4 могут включать ацетильную группу, пропионильную группу, бутироильную группу, изобутироильную группу, бензоильную группу, 4-метоксибензоильную группу, фенилацетильную группу, феноксиацетильную группу, 4-трет-бутилфеноксиацетильную группу и 4-изопропилфеноксиацетильную группу. Примеры твердого носителя могут включать стекло с заданным размером пор (CPG), оксалильное стекло с заданным размером пор (oxalyl-controlled pore glass) (см., например, Alul et al., Nucleic Acids Research, Vol.19, 1527 (1991)), носитель TentaGel - носитель на основе производного аминополиэтиленгликоля (см., например, Wright et al., Tetrahedron Letters, Vol.34, 3373 (1993)) и сополимер пористого полистирола и дивинилбензола. Примеры линкера могут включать 3-аминопропильную группу, сукцинильную группу, 2,2'-диэтаносульфогруппу и алкиламиногруппу с длинной цепью (LCAA). Соединения (26а) и (26b), которые являются присоединенными к твердому носителю, получают в соответствии с известным способом или приобретают в продаже, и примерами предпочтительного варианта осуществления являются соединения, представленные следующими общими формулами (27) или (28)
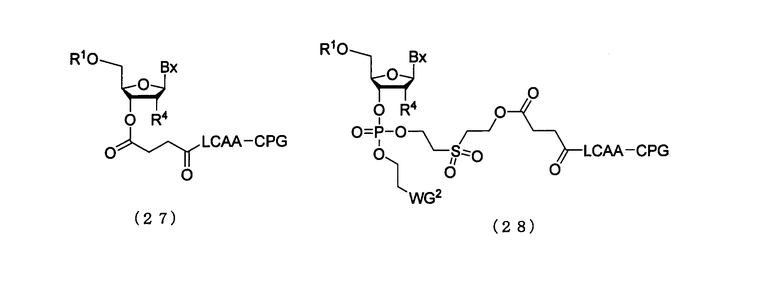
в которых
Вх, R1, R4 и WG2 такие, как определено выше.
Соединения (27) и (28), где R4 является заместителем (6), могут быть получены из производного фосфорамидита настоящего изобретения в соответствии с известным способом. Примеры кислоты, которая должна быть использована на этой стадии, могут включать трифторуксусную кислоту, дихлоруксусную кислоту, трихлоруксусную кислоту. Кислота, которая должна быть использована на этой стадии, может быть разбавлена в подходящем растворителе так, чтобы иметь концентрацию 1-5%. Примеры растворителя особым образом не ограничивают, исключая те случаи, когда он является вовлеченным в реакцию, и могут включать, например, дихлорметан, ацетонитрил, воду и их смеси. Температура реакции предпочтительно находится в пределах диапазона 20-50ºС. Время реакции варьируется в зависимости от типа кислоты и температуры реакции, и предпочтительно составляет от 1 мин до 1 час.
Количество реагента, которое должно быть использовано, предпочтительно колеблется в пределах диапазона 1-100 моль на моль соединения, присоединенного к твердому носителю, и более предпочтительно 1-10 моль на моль соединения, присоединенного к твердому носителю.
(2) Стадия В:
Процесс получения соединения, представленного следующей общей формулой (9), путем конденсации мономерного соединения нуклеиновой кислоты с соединением, полученным посредством стадии А, с использованием активирующего агента.
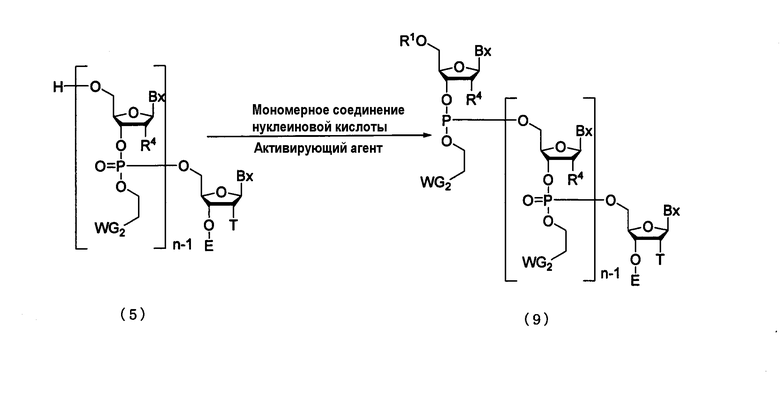
где
Вх, Е, n, R1, R4, T и WG2 такие, как определено выше.
Эта стадия может быть осуществлена путем взаимодействия соединения мономера нуклеиновой кислоты и активирующего агента с соединением, присоединенным к твердому носителю. Примеры мономерного соединения нуклеиновой кислоты могут включать производное фосфорамидита настоящего изобретения и соединение, представленное следующей общей формулой (29), которое имеется в продаже.
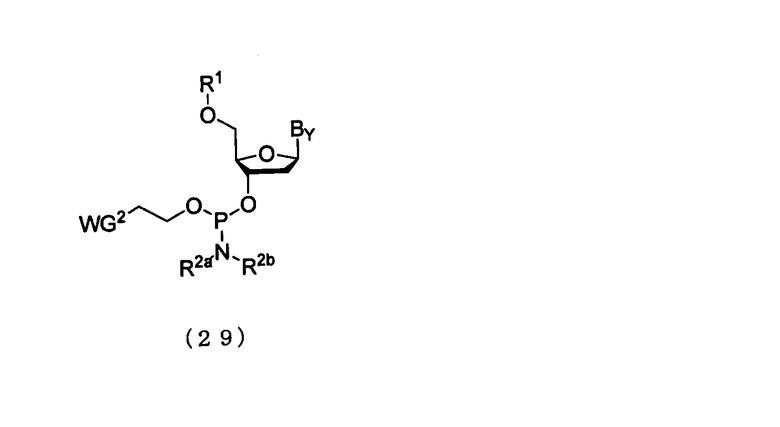
в которой
R1, R2а, R2b и WG2 такие, как определено выше; и
Вy представляет собой нуклеиновое основание, которое может иметь защитную группу.
Примеры нуклеинового основания Вy особо не ограничивают, поскольку оно представляет собой нуклеиновое основание, используемое для синтеза нуклеиновой кислоты, и могут включать, например, аденин, гуанин, цитозин, тимин и их модифицированную форму. Модифицированная форма является идентичной модифицированной форме, определенной выше для Вх. Примеры заместителя для модифицированной формы By могут включать галоген, алкильную группу, арилалкильную группу, алкоксигруппу, гидроксильную группу, аминогруппу, моноалкиламиногруппу, диалкиламиногруппу, карбоксигруппу, цианогруппу и нитрогруппу; и модифицированная форма Ву может быть замещенной одним-тремя такими заместителями.
Примеры галогена, арильной группы, алкильной группы, арилалкильной группы, алкоксигруппы, алкоксиалкильной группы, аминогруппы, моноалкиламиногруппы и диалкиламиногруппы для модифицированной формы Ву могут включать группы, идентичные соответствующим группам производного фосфорамидита настоящего изобретения.
Нуклеиновое основание Вх может быть защищено, и, в частности, нуклеиновое основание, имеющее аминогруппу (например, аденин, гуанин, цитозин) предпочтительно может быть защищено по аминогруппе. Защитная группа аминогруппы в Ву может включать группы, идентичные защитным группам аминогруппы в Вх.
Примеры активирующего агента могут включать активирующие агенты, идентичные приведенным выше.
Примеры растворителя для этой реакции особо не ограничивают, исключая только те случаи, когда он является вовлеченным в реакцию, и могут включать, например, ацетонитрил и тетрагидрофуран. Температура реакции предпочтительно находится в пределах диапазона 20-50ºС.
Время реакции варьируется в зависимости от типа активирующего агента и температуры реакции и предпочтительно составляет от 1 мин до 1 час. Количество агента, которое должно быть использовано, предпочтительно колеблется в пределах диапазона 1-100 моль на моль соединения, присоединенного к твердому носителю, и более предпочтительно 1-10 моль на моль соединения, присоединенного к твердому носителю.
(3) Стадия С:
Процесс кэппирования 5'-гидроксильной группы непрореагировавшего соединения (5) на стадии В.
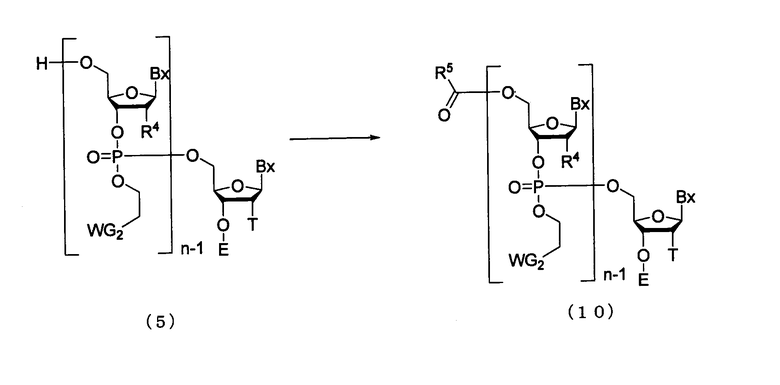
где
Bx, E, n, R4, T и WG2 такие, как определено выше; и
R5 представляет собой метильную или феноксиметильную группу.
Эта стадия представляет собой реакцию для введения защиты для 5'-гидроксильной группы, не прореагировавшей на стадии (В), и может быть осуществлена путем взаимодействия кэппирующего агента с соединением, присоединенным к твердому носителю. Примеры кэппирующего агента могут включать уксусный ангидрид и феноксиуксусный ангидрид. Кэппирующий агент, который должен быть использован, может быть разбавлен в подходящем растворителе так, чтобы иметь концентрацию 0,05-1 М. Примеры растворителя особо не ограничивают, исключая те случаи, когда он является вовлеченным в реакцию, и могут включать, например, пиридин, дихлорметан, ацетонитрил, тетрагидрофуран и их смеси. Кроме того, на этой стадии, при необходимости, могут быть использованы, например, 4-диметиламинопиридин, N-метилимидазол в качестве ускорителя реакции. Температура реакции предпочтительно находится в пределах диапазона 20-50ºС. Время реакции варьируется в зависимости от типа кэппирующего агента и температуры реакции и предпочтительно составляет от 1 до 30 мин. Количество агента, которое должно быть использовано, предпочтительно колеблется в пределах диапазона 1-100 моль на моль соединения, присоединенного к твердому носителю, и более предпочтительно 1-10 моль на моль соединения, присоединенного к твердому носителю.
(4) Стадия D:
Процесс превращения фосфорильной группы в фосфатную группу путем взаимодействия окислителя с соединением (9), полученным на стадии В.
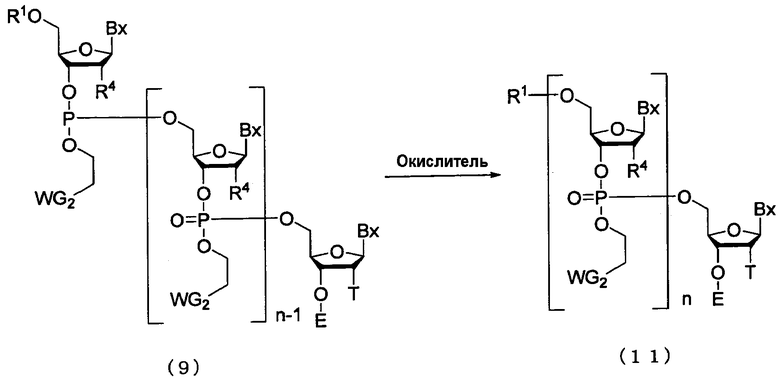
где
Bx, E, n, R1, R4, T и WG2 такие, как определено выше.
Эта стадия представляет собой реакцию для превращения трехвалентного фосфора в пятивалентный фосфор посредством окислителя и может быть осуществлена путем взаимодействия окислителя с соединением, присоединенным к твердому носителю. Примеры окислителя могут включать йод и трет-бутилгидропероксид.
Кроме того, окислитель, который должен быть использован, может быть разбавлен в подходящем растворителе так, чтобы иметь концентрацию 0,05-1 М. Примеры растворителя особо не ограничивают, исключая те случаи, когда он является вовлеченным в реакцию, и могут включать, например, пиридин, тетрагидрофуран, воду и их смеси. Например, могут быть использованы йод/вода/пиридин-тетрагидрофуран, йод/пиридин-уксусная кислота и агент для перекисного окисления (трет-бутилгидропероксид/метиленхлорид и т.п.). Температура реакции предпочтительно находится в пределах диапазона 20-50ºС. Время реакции варьируется в зависимости от типа окислителя и температуры реакции и предпочтительно составляет от 1 до 30 мин. Количество агента, которое должно быть использовано, предпочтительно колеблется в пределах диапазона 1-100 моль на моль соединения, присоединенного к твердому носителю, и более предпочтительно 1-50 моль на моль соединения.
(5) Стадия Е:
Процесс отщепления соединения (11), полученного на стадии D, от твердого носителя, и затем удаления защитных групп каждого нуклеинового основания и каждой 2'-гидроксильной группы.
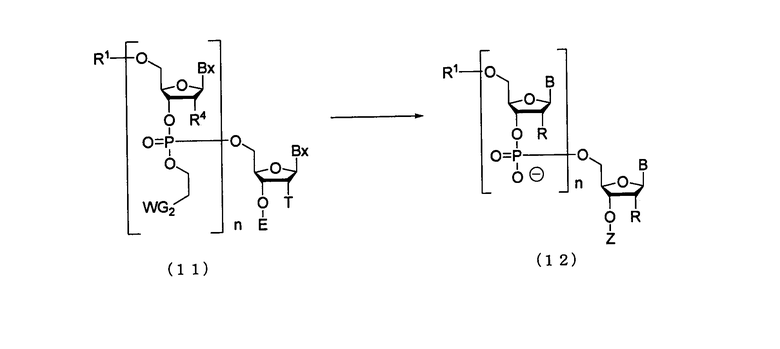
где
B, Bx, E, R, R1, R4, n, T, WG2 и Z такие, как определено выше.
Cтадия отщепления представляет собой реакцию отщепления олиго-РНК, имеющей желательную длину цепи, от твердого носителя и линкера посредством отщепляющего агента и которую осуществляют путем добавления отщепляющего агента к твердому носителю, который содержит олиго-РНК, имеющую желательную длину цепи.
На этой стадии может быть удалена защитная группа нуклеинового основания. Примеры отщепляющего агента могут включать концентрированный водный раствор аммиака и метиламин. Отщепляющий агент, который должен быть использован на этой стадии, может быть разбавлен, например, метанолом, этанолом, изопропиловым спиртом, ацетонитрилом, тетрагидрофураном и их смесями. Среди них этанол является предпочтительным. Температура реакции может быть в пределах диапазона 15-75°С, предпочтительно 15-30°С и более предпочтительно 18-25°С. Время реакции для снятия защитных групп может колебаться в пределах диапазона 1-30 час, предпочтительно 1-24 час и более предпочтительно 1-4 час. Концентрация гидроксида аммония в растворе, который должен быть использован для снятия защитных групп, может составлять 20-30% по массе, предпочтительно 25-30% по массе, более предпочтительно 28-30% по массе. Количество агента, которое должно быть использовано, может колебаться в пределах от 1 до 100 моль на моль соединения, присоединенного к твердому носителю, и предпочтительно от 10- до 50-кратного количества моль на моль соединения. Стадию удаления защитной группы для 2'-гидроксильной группы осуществляют посредством действия агента для удаления защитной группы для 2'-гидроксильной группы, такого как фторид тетрабутиламмония, тригидрофторид/соль триэтиламина. Примеры растворителя особо не ограничивают, исключая те случаи, когда он является вовлеченным в реакцию, и могут включать, например, тетрагидрофуран, N-метилпирролидон, пиридин, диметилсульфоксид и их смеси. При необходимости, в качестве дезактивирующего агента для акрилонитрила, который является побочным продуктом на этой стадии, могут быть добавлены алкиламин, амидин, тиол, производные тиола или их смеси. Примеры алкиламина могут включать неразветвленный алкиламин, имеющий 1-6 атомов углерода. В частности, алкиламин может включать, например, метиламин, этиламин, н-пропиламин, н-бутиламин, н-пентиламин и н-гексиламин. Примеры амидина могут включать бензамидин и формамидин. Примеры тиола могут включать неразветвленный тиол, имеющий 1-6 атомов углерода. В частности, тиол может включать, например, метантиол, этантиол, 1-пропантиол, 1-бутантиол, 1-пентантиол и 1-гексантиол. Примеры производного тиола могут включать одинаковые или различные спирт и эфир, содержащие неразветвленный алкилтиол, имеющий 1-6 атомов углерода. В частности, производное тиола может включать, например, 2-меркаптоэтанол, 4-меркапто-1-бутанол, 6-меркапто-1-гексанол, меркаптометиловый эфир, 2-меркаптоэтиловый эфир, 3-меркаптопропиловый эфир, 4-меркаптобутиловый эфир, 5-меркаптопентиловый эфир и 6-меркаптогексиловый эфир. Температура реакции предпочтительно может быть в пределах диапазона 20-80ºС. Время реакции варьируется в зависимости от типа агента для удаления защитной группы, который должен быть использован, и температуры реакции и предпочтительно колеблется в пределах диапазона 1-100 час. Количество агента, которое должно быть использовано, предпочтительно колеблется в пределах диапазона 50-500 моль на моль удаляемой защитной группы и более предпочтительно 50-100 моль на моль удаляемой защитной группы. Олиго-РНК, защищенная по 5'-гидроксильной группе, может быть выделена и очищена из вышеприведенной реакционной смеси посредством стандартных методов выделения и очистки, таких как экстракция, концентрирование, нейтрализация, фильтрация, центрифугирование, перекристаллизация, колоночная хроматография на силикагеле, тонкослойная хроматография, гидрофобная колоночная хроматография, ионообменная колоночная хроматография, гель-фильтрационная колоночная хроматография, диализ, ультрафильтрация и т.п.
(6) Стадия F:
Процесс удаления 5'-гидроксильной группы в соединении (12), полученном на стадии Е.
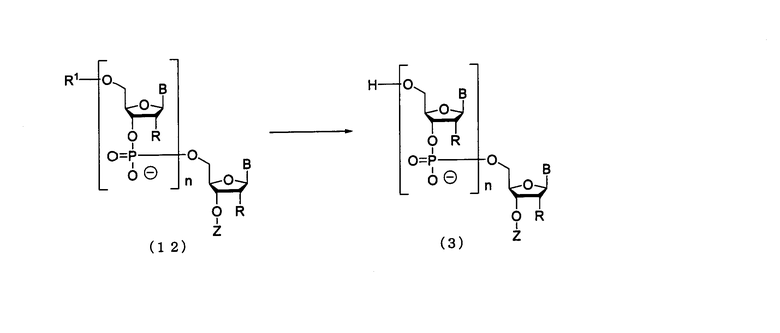
где
B, n, R, R1 и Z такие, как заместители, определенные выше.
Эта стадия представляет собой реакцию для заключительного удаления защитной группы для 5'-гидроксильной группы в олигорибонуклеотиде и может быть осуществлена посредством действия кислоты на олиго-РНК, отщепленную от твердого носителя. Примеры кислоты, которая должна быть использована на этой стадии, могут включать трихлоруксусную кислоту, дихлоруксусную кислоту и уксусную кислоту. На этой стадии может быть использована кислота, разбавленная в подходящем растворителе. Примеры растворителя особо не ограничивают, исключая те случаи, когда он является вовлеченным в реакцию, и могут включать, например, дихлорметан, ацетонитрил, воду, буферный раствор, рН которого колеблется в пределах от 2 до 5, и их смеси. Примеры буферного раствора могут включать ацетатный буферный раствор. Температура реакции предпочтительно колеблется в пределах диапазона 20-50ºС. Время реакции варьируется в зависимости от типа кислоты и температуры реакции и предпочтительно составляет от 1 мин до 1 час. Количество агента, которое должно быть использовано, предпочтительно колеблется в пределах диапазона 1-100 моль на моль соединения, присоединенного к твердому носителю, и более предпочтительно 1-10 моль на моль соединения.
(7) Стадия G:
Процесс выделения и очистки соединения (3), полученного на стадии F.
Стадия выделения и очистки представляет собой стадию выделения и очистки желательной олиго-РНК из приведенной выше реакционной смеси посредством известного способа выделения и очистки, который может включать, например, экстракцию, концентрирование, нейтрализацию, фильтрацию, разделение центрифугированием, перекристаллизацию, колоночную хроматографию с обращенной фазой (С8-С18), картридж-колонку с обращенной фазой (С8-С18), катионообменную колоночную хроматографию, анионообменную колоночную хроматографию, гель-фильтрационную колоночную хроматографию, высокоэффективную жидкостную хроматографию, диализ, ультрафильтрацию и их комбинации. Примеры элюента могут включать ацетонитрил, метанол, этанол, изопропиловый спирт, воду и растворитель, полученный смешением в произвольном соотношении. В этом случае, например, рН раствора, который должен быть в пределах диапазона рН 1-9, может быть установлен путем добавления фосфата натрия, фосфата калия, хлорида натрия, хлорида калия, ацетата аммония, ацетата триэтиламмония, ацетата натрия, ацетата калия, трис-хлористоводородной кислоты или этилендиаминтетрауксусной кислоты в качестве добавки в концентрации 1 мМ-2 М.
Олиго-РНК, имеющая желательную длину цепи, может быть получена путем повторения стадий А-D.
Кроме того, в этом способе, применяют соединение (26а), где R4 представляет собой заместитель (6), соединение (26а), где R4 представляет собой Н или ацилоксигруппу, или соединение (26b), где R2 представляет собой алкилоксигруппу и т.д.
При использовании соединения (26а), где R4 представляет собой Н или ацилоксигруппу, или соединения (26b), где R2 представляет собой алкилоксигруппу в качестве исходного вещества, необходимо применять одно или более производных фосфорамидита настоящего изобретения в качестве соединения мономера нуклеиновой кислоты.
К тому же, в этом способе выделение и очистку олиго-РНК также осуществляют путем проведения процессов стадии F перед проведением процессов стадии E, и затем процессов стадии G.
ПРИМЕРЫ
Теперь настоящее изобретение будет описано более подробно, ссылаясь на примеры, которыми, однако, не ограничено настоящее изобретение.
Пример 1
Хлорметил-2-цианоэтиловый эфир
Стадия 1
Получение метилтиометил-2-цианоэтилового эфира
3-Гидроксипропионитрил (32 г, 450 ммоль) растворяют в 450 мл диметилсульфоксида, и к этому добавляют 324 мл уксусного ангидрида и 231 мл уксусной кислоты, и реакционный раствор перемешивают при комнатной температуре в течение 24 час.
Бикарбонат натрия (990 г) растворяют в 4,5 л воды и реакционный раствор добавляют по каплям к водному раствору бикарбоната натрия в течение 1 час. Реакционный раствор перемешивают в течение 1 час и подвергают экстракции этилацетатом, и экстракт сушат над безводным сульфатом магния, и растворитель отгоняют. Полученный маслянистый продукт очищают колоночной хроматографией на силикагеле с получением 41 г метилтиометил-2-цианоэтилового эфира в виде бесцветного маслянистого продукта (выход 70%).
1Н-ЯМР (CDCl3): 2,18 (с, 3Н); 2,66 (т, 2Н, J=6,3 Гц); 3,77 (т, 2Н, J=6,3 Гц); 4,69 (с, 2Н)
Стадия 2
Получение хлорметил-2-цианоэтилового эфира
Метилтиометил-2-цианоэтиловый эфир (3,3 г, 25 ммоль) растворяют в 70 мл метиленхлорида, и добавляют по каплям 2 мл сульфурилхлорида (25 ммоль), и далее реакцию проводят при комнатной температуре в течение 1 час.
После завершения реакции растворитель отгоняют при пониженном давлении с получением 2,5 г целевого соединения в виде бесцветного маслянистого продукта (выход 85%).
Температура кипения: 84-85ºС (0,3 Торр)
1Н-ЯМР (CDCl3): 2,72 (т, 2Н, J=6,3 Гц); 3,92 (т, 2Н, J=6,3 Гц); 5,52 (с, 2Н)
Пример 2
5'-О-(4,4'-диметокситритил)-2'-О-(2-цианоэтоксиметил)уридин 3'-О-(2-цианоэтил N,N-диизопропилфосфорамидит)
Стадия 1
Получение 5'-О-(4,4'-диметокситритил)-2'-О-(2-цианоэтоксиметил)уридина
5'-О-(4,4'-диметокситритил)уридин (546 мг, 1 ммоль) растворяют в 4 мл 1,2-дихлорэтана, и к этому добавляют 452 мг диизопропилэтиламина (3,5 ммоль), и далее к этому добавляют 365 мг дибутилоловодихлорида (1,2 ммоль). Реакцию проводят при комнатной температуре в течение 1 час.
Потом реакцию проводят при 80ºС и добавляют по каплям 155,4 мг хлорметил-2-цианоэтилового эфира (1,3 ммоль), реакционный раствор перемешивают в течение 30 мин.
После завершения реакции реакционный раствор добавляют в насыщенный водный раствор бикарбоната натрия, и подвергают экстракции метиленхлоридом, и экстракт сушат над безводным сульфатом магния, и растворитель отгоняют. Полученную смесь очищают колоночной хроматографией на 30 г силикагеля с получением 5'-О-(4,4'-диметокситритил)-2'-О-(2-цианоэтоксиметил)уридина (197 мг; выход 34%).
1Н-ЯМР (CDCl3): 2,47 (д, 1Н, J=7,8 Гц); 2,69 (т, 2Н, J=6,3 Гц); 3,55 (дд, 1Н, J=11,3, 2,2 Гц); 3,62 (дд, 1Н, J=11,3, 2,2 Гц); 3,83 (с, 6Н); 3,87 (т, 2Н, J=6,3 Гц); 4,07-4,08 (м, 1Н); 4,32 (дд, 1Н, J=5,3, 1,9 Гц); 4,54 (кв., 1Н, J=5,3 Гц); 4,94, 5,11 (2д, 2Н, J=6,9 Гц); 5,32 (д, 1Н, J=8,2 Гц); 6,00 (д, 1Н, J=1,9 Гц); 6,85-6,88 (м, 4Н); 7,29-7,41 (м, 9Н); 8,02 (д, 1Н, J=8,2 Гц); 8,53 (ушир.с, 1Н)
Масс-спектрометрия с ионизацией в искровом или тлеющем разряде (ESI-Mass): 652 [M+Na]+
Стадия 2
Получение 5'-О-(4,4'-диметокситритил)-2'-О-(2-цианоэтоксиметил)уридин 3'-О-(2-цианоэтил N,N-диизопропилфосфорамидит)
5'-О-(4,4'-диметокситритил)-2'-О-(2-цианоэтоксиметил)уридин (209 г, 0,332 ммоль), полученный на стадии 1, растворяют в 2 мл ацетонитрила, и добавляют по каплям 23 мг тетразола (0,332 ммоль) и 150 мг 2-цианоэтил N,N,N',N'-тетраизопропилфосфордиамидита (0,498 ммоль), и реакцию проводят при 45ºС в течение 1,5 час.
После завершения реакции реакционный раствор смешивают с насыщенным водным раствором бикарбоната натрия, и подвергают экстракции этилацетатом, и экстракт сушат над безводным сульфатом магния, и растворитель отгоняют. Полученную смесь очищают колоночной хроматографией на 20 г силикагеля с получением целевого соединения (200 мг; выход 73%).
Масс-спектрометрия с ионизацией в искровом или тлеющем разряде (ESI-Mass): 852 [M+Na]+
Пример 3
2'-О-(2-цианоэтоксиметил)уридин
Стадия 1
Получение 3',5'-О-(тетраизопропилдисилоксан-1,3-диил)-2'-О-(2-цианоэтоксиметил)уридина
3',5'-О-(тетраизопропилдисилоксан-1,3-диил)уридин (150 мг, 0,3 ммоль) растворяют в 7 мл тетрагидрофурана в атмосфере аргона, и добавляют 54 мг метилтиометил-2-цианоэтилового эфира (0,4 ммоль) и 100 мг молекулярных сит 4Å, и реакционный раствор перемешивают в течение 10 мин.
Реакцию проводят при 0ºС и добавляют 2 мл раствора трифторметансульфокислоты (10 мг, 0,06 ммоль) в тетрагидрофуране. Затем добавляют 92 мг N-йодсукцинимида (0,4 ммоль) и реакционный раствор перемешивают в течение 1 час.
После завершения реакции реакционный раствор фильтруют через целит и промывают метиленхлоридом и полученный органический слой промывают 1 М водным раствором гидротиосульфата натрия. Органический слой промывают насыщенным водным раствором бикарбоната натрия, и сушат над безводным сульфатом магния, и растворитель отгоняют.
Полученный твердый остаток очищают тонкослойной хроматографией с получением 3',5'-О-(тетраизопропилдисилоксан-1,3-диил)-2'-О-(2-цианоэтоксиметил)уридина (150 мг; выход 85%).
1Н-ЯМР (CDCl3): 0,97-1,12 (м, 28Н); 2,68-2,73 (м, 2Н); 3,78-3,86 (м, 1Н); 3,96-4,05 (м, 2Н); 4,12-4,30 (м, 4Н); 5,0-5,04 (м, 2Н); 5,70 (д, 1Н, J=8,2 Гц); 5,75 (с, 1Н); 7,90 (д, 1Н, J=8,2 Гц); 9,62 (ушир.с, 1Н)
Масс-спектрометрия с ионизацией в искровом или тлеющем разряде (ESI-Mass): 570 [M+Н]+
Стадия 2
Получение 2'-О-(2-цианоэтоксиметил)уридина
3',5'-О-(тетраизопропилдисилоксан-1,3-диил)-2'-О-(2-цианоэтоксиметил)уридин (200 мг, 0,35 ммоль), полученный на стадии 1, растворяют в 2 мл метанола, и к этому добавляют 65 мг фторида аммония (1,76 ммоль), и реакционный раствор перемешивают при нагревании при 50ºС в течение 5 час.
После охлаждения воздухом к реакционному раствору добавляют ацетонитрил. Раствор перемешивают, и фильтруют, и концентрируют.
Полученный твердый остаток очищают колоночной хроматографией на силикагеле с получением целевого соединения (108 мг; выход 94%).
1H-ЯМР (CD3OD): 2,72-2,76 (т, 2Н, J=6,2 Гц); 3,68-3,92 (м, 4Н); 4,00-4,03 (м, 1Н); 4,26-4,32 (м, 2Н); 4,81-4,95 (м, 2Н); 5,71 (д, 1Н, J=8,1 Гц); 6,00 (д, 1Н, J=3,3 Гц); 8,10 (д, 1Н, J=8,1 Гц)
Масс-спектрометрия с ионизацией в искровом или тлеющем разряде (ESI-Mass): 350 [M+Na]+
Пример 4
Получение 5'-О-(4,4'-диметокситритил)-2'-О-(2-цианоэтоксиметил)уридина
2'-О-(2-цианоэтоксиметил)уридин (14 г, 43 ммоль) подвергают азеотропной перегонке с пиридином и затем сушат вакуумным насосом в течение 30 мин.
Твердый остаток растворяют в 300 мл тетрагидрофурана, и добавляют в атмосфере аргона 68 г пиридина (856 ммоль) и 20 г молекулярных сит 4Å, и смесь перемешивают в течение 10 мин.
К реакционному раствору добавляют тремя порциями каждый 1 час 19,6 г 4,4'-диметокситритилхлорида (57,8 ммоль) и смесь дополнительно перемешивают в течение 1 час.
После добавления 10 мл метанола и перемешивания реакционного раствора в течение 2 мин реакционный раствор фильтруют через целит и промывают этилацетатом.
После концентрирования фильтрата твердый остаток растворяют в этилацетате и промывают насыщенным водным раствором бикарбоната натрия.
После промывания органического слоя насыщенным рассолом (насыщенным раствором NaCl) и сушки над безводным сульфатом магния растворитель отгоняют.
Полученный твердый остаток очищают хроматографией на силикагеле с получением целевого соединения (26,5 г, выход 98%).
Пример 5
N 4 -ацетил-5'-О-(4,4'-диметокситритил)-2'-О-(2-цианоэтоксиметил)цитидин 3'-О-(2-цианоэтил N,N-диизопропилфосфорамидит)
Стадия 1
Получение N 4 -ацетил-5'-О-(4,4'-диметокситритил)-2'-О-(2-цианоэтоксиметил)цитидина
N4-ацетил-5'-О-(4,4'-диметокситритил)цитидин (588 мг, 1 ммоль) растворяют в 4 мл 1,2-дихлорэтана, и к этому добавляют 452 мг диизопропилэтиламина (3,5 ммоль), и затем еще добавляют 365 мг дибутилоловодихлорида (1,2 ммоль). Реакцию проводят при комнатной температуре в течение 1 час.
Затем реакцию проводят при 80ºС, и добавляют по каплям 155,4 мг хлорметил-2-цианоэтилового эфира (1,3 ммоль), и реакционный раствор перемешивают в течение 60 мин.
После завершения реакции реакционный раствор добавляют в насыщенный водный раствор бикарбоната натрия и экстрагируют метиленхлоридом. Экстракт сушат над безводным сульфатом магния и растворитель отгоняют. Полученную смесь очищают колоночной хроматографией на 30 г силикагеля с получением N4-ацетил-5'-О-(4,4'-диметокситритил)-2'-О-(2-цианоэтоксиметил)цитидина (219 мг; выход 35%).
1Н-ЯМР (CDCl3): 2,19 (с, 3Н); 2,56 (д, 1Н, J=8,8 Гц); 2,65 (т, 2Н, J=6,2 Гц); 3,55 (дд, 1Н, J=10,5, 2,5 Гц); 3,63 (дд, 1Н, J=10,5, 2,5 Гц); 3,82 (с, 6Н); 3,86 (т, 2Н, J=6,2 Гц); 4,09-4,14 (м, 1Н); 4,28 (д, 1Н, J=5,1 Гц); 4,44-4,49 (м, 1Н); 4,97, 5,24 (2д, 2Н, J=6,9 Гц); 5,96 (с, 1Н); 6,86-6,88 (м, 4Н); 7,09 (д, 1Н, J=6,9 Гц); 7,26-7,42 (м, 9Н); 8,48 (д, 1Н, J=6,9 Гц); 8,59 (ушир.с, 1Н)
Масс-спектрометрия с ионизацией в искровом или тлеющем разряде (ESI-Mass): 693 [M+Na]+
Стадия 2
Получение N 4 -ацетил-5'-О-(4,4'-диметокситритил)-2'-О-(2-цианоэтоксиметил)цитидин 3'-О-(2-цианоэтил N,N-диизопропилфосфорамидит)
N4-ацетил-5'-О-(4,4'-диметокситритил)-2'-О-(2-цианоэтоксиметил)цитидин (205 мг, 0,306 ммоль), полученный на стадии 1, растворяют в 2 мл метиленхлорида, и добавляют 105 мг диизопропилэтиламина (0,812 ммоль), и добавляют по каплям 116 мг 2-цианоэтил N,N-диизопропилхлорфосфорамидита (0,49 ммоль). Реакционный раствор подвергают реакции при комнатной температуре в течение 1 час.
После завершения реакции растворитель отгоняют и полученную смесь очищают колоночной хроматографией на 20 г силикагеля с получением целевого соединения (242 мг; выход 91%).
Масс-спектрометрия с ионизацией в искровом или тлеющем разряде (ESI-Mass): 871 [M+Н]+
Пример 6
N 4 -ацетил-2'-О-(2-цианоэтоксиметил)цитидин
Стадия 1
Получение N 4 -ацетил-3',5'-О-(тетраизопропилдисилоксан-1,3-диил)-2'-О-(2-цианоэтоксиметил)цитидина
N4-ацетил-3',5'-О-(тетраизопропилдисилоксан-1,3-диил)цитидин (1,00 г, 1,89 ммоль) и метилтиометил-2-цианоэтиловый эфир (500 мг, 3,79 ммоль) смешивают и смесь растворяют в растворителе, смешанном из 10 мл толуола и 10 мл тетрагидрофурана.
Потом добавляют 975 мг трифторметансульфоната серебра и сушат при добавлении молекулярных сит 4Å.
При охлаждении льдом добавляют 370 мг N-бромсукцинимида (2,08 ммоль) и раствор перемешивают в течение 10 мин в реакционном сосуде, защищенном от света. Кроме того, добавляют 70 мг N-бромсукцинимида (0,39 ммоль) и перемешивают в течение 25 мин.
После завершения реакции реакционный раствор разбавляют метиленхлоридом и промывают насыщенным водным раствором бикарбоната натрия. Экстракт сушат над безводным сульфатом натрия и растворитель отгоняют. Полученную смесь очищают колоночной хроматографией на силикагеле с получением N4-ацетил-3',5'-О-(тетраизопропилдисилоксан-1,3-диил)-2'-О-(2-цианоэтоксиметил)цитидина (936 мг; выход 81%).
1Н-ЯМР (CDCl3): 0,90-1,11 (м, 28Н); 2,28 (с, 3Н); 2,62-2,79 (м, 2Н); 3,78-3,89 (м, 1Н); 3,96-4,04 (м, 2Н); 4,19-4,23 (м, 3Н); 4,30 (д, 1Н, J=13,6 Гц); 5,00 (д, 1Н, J=6,8 Гц); 5,09 (д, 1Н, J=6,8 Гц); 5,77 (с, 1Н); 7,44 (д, 1Н, J=7,5 Гц); 8,30 (д, 1Н, J=7,5 Гц); 10,13 (с, 1Н)
Масс-спектрометрия с ионизацией в искровом или тлеющем разряде (ESI-Mass): 611 [M+Н]+
Стадия 2
Получение N 4 -ацетил-2'-О-(2-цианоэтоксиметил)цитидина
N4-ацетил-3',5'-О-(тетраизопропилдисилоксан-1,3-диил)-2'-О-(2-цианоэтоксиметил)цитидин (500 мг, 0,819 ммоль), полученный на стадии 1, растворяют в растворителе, смешанном из 2,5 мл тетрагидрофурана и 2,5 мл метанола, и добавляют 150 мг фторида аммония (4,10 ммоль), и затем реакционный раствор подвергают реакции при 50ºС в течение 4 час.
После завершения реакции реакционный раствор разбавляют ацетонитрилом, и фильтруют, и растворитель отгоняют. Полученную смесь очищают колоночной хроматографией на силикагеле с получением целевого соединения (210 мг; выход 70%).
1Н-ЯМР (D2О): 2,13 (с, 3Н); 2,66-2,71 (м, 2Н); 3,72-3,78 (м, 3Н); 3,90 (дд, 1Н, J=13,0, 2,6 Гц); 4,06-4,11 (м, 1Н); 4,20 (дд, 1Н, J=7,1, 5,2 Гц); 4,29 (дд, 1Н, J=5,1, 2,9 Гц); 4,83 (д, 1Н, J=7,2 Гц); 4,94 (д, 1Н, J=7,2 Гц); 5,95 (д, 1Н, J=2,9 Гц); 7,25 (д, 1Н, J=7,6 Гц); 8,25 (д, 1Н, J=7,6 Гц)
Масс-спектрометрия с ионизацией в искровом или тлеющем разряде (ESI-Mass): 391 [M+Na]+
Пример 7
Получение N 4 -ацетил-5'-О-(4,4'-диметокситритил)-2'-О-(2-цианоэтоксиметил)цитидина
2'-О-(2-цианоэтоксиметил)цитидин (9,9 г, 26,8 ммоль) подвергают азеотропной перегонке с пиридином и затем сушат вакуумным насосом в течение 30 мин. Твердый остаток растворяют в 190 мл тетрагидрофурана, и добавляют в атмосфере аргона 43 г пиридина (538 ммоль) и 20 г молекулярных сит 4Å, и смесь перемешивают в течение 10 мин.
К реакционному раствору добавляют тремя порциями каждый 1 час 11,8 г 4,4'-диметокситритилхлорида (34,9 ммоль) и смесь дополнительно перемешивают в течение 1 час.
После добавления 2 мл метанола и перемешивания реакционного раствора в течение 2 мин реакционный раствор фильтруют через целит и промывают этилацетатом.
После концентрирования фильтрата путем выпаривания твердый остаток растворяют в этилацетате и промывают насыщенным водным раствором бикарбоната натрия.
После промывания органического слоя насыщенным рассолом (насыщенным раствором NaCl) и сушки над безводным сульфатом магния растворитель отгоняют.
Полученный твердый остаток очищают хроматографией на силикагеле с получением целевого соединения (15 г, выход 83%).
Пример 8
N 2 -ацетил-5'-О-(4,4'-диметокситритил)-2'-О-(2-цианоэтоксиметил)гуанозин 3'-О-(2-цианоэтил N,N-диизопропилфосфорамидит)
Стадия 1
Получение N 2 -ацетил-5'-О-(4,4'-диметокситритил)-2'-О-(2-цианоэтоксиметил)гуанозина
N2-ацетил-5'-О-(4,4'-диметокситритил)гуанозин (627 мг, 1 ммоль) растворяют в 4 мл 1,2-дихлорэтана, и добавляют 452 мг диизопропилэтиламина (3,5 ммоль), и затем добавляют 365 мг дибутилоловодихлорида (1,2 ммоль). И затем реакционный раствор подвергают реакции при комнатной температуре в течение 1 час.
Затем реакционный раствор нагревают вплоть до 80ºС, и добавляют по каплям 155,4 мг хлорметил-2-цианоэтилового эфира (1,3 ммоль), и раствор перемешивают в течение 60 мин.
После завершения реакции реакционный раствор смешивают с насыщенным водным раствором бикарбоната натрия и подвергают экстракции метиленхлоридом. Экстракт сушат над безводным сульфатом магния и растворитель отгоняют. Полученную смесь очищают колоночной хроматографией на 30 г силикагеля с получением N2-ацетил-5'-О-(4,4'-диметокситритил)-2'-О-(2-цианоэтоксиметил)гуанозина (450 мг; выход 63%).
1Н-ЯМР (CDCl3): 1,92 (с, 3Н); 2,47-2,51 (м, 2Н); 2,68 (ушир.с, 1Н); 3,30 (дд, 1Н, J=10,7, 3,8 Гц); 3,47 (дд, 1Н, J=10,7, 3,8 Гц); 3,55-3,60 (м, 1Н); 3,65-3,70 (м, 1Н); 3,74, 3,75 (2с, 6Н); 4,22-4,23 (м, 1Н); 4,55-4,58 (м, 1Н); 4,78, 4,83 (2д, 2Н, J=7,0 Гц); 5,01 (т, 1Н, J=5,1 Гц); 5,99 (д, 1Н, J=5,1 Гц); 6,76-6,79 (м, 4Н); 7,17-7,44 (м, 9Н); 7,88 (с, 1Н); 8,36 (ушир.с, 1Н); 12,06 (ушир.с, 1Н)
Стадия 2
Получение N 2 -ацетил-5'-О-(4,4'-диметокситритил)-2'-О-(2-цианоэтоксиметил)гуанозин 3'-О-(2-цианоэтил-N,N-диизопропилфосфорамидит)
N2-ацетил-5'-О-(4,4'-диметокситритил)-2'-О-(2-цианоэтоксиметил)гуанозин (400 мг, 0,563 ммоль), полученный на стадии 1, растворяют в 2 мл метиленхлорида, и добавляют 181 мг диизопропилэтиламина (1,4 ммоль), и добавляют по каплям 161 мг 2-цианоэтил N,N-диизопропилхлорфосфорамидита (0,68 ммоль). Затем реакцию проводят при комнатной температуре в течение 1 час.
После завершения реакции растворитель отгоняют и полученную смесь очищают колоночной хроматографией на 20 г силикагеля с получением целевого соединения (471 мг; выход 92%).
Пример 9
N 6 -ацетил-5'-О-(4,4'-диметокситритил)-2'-О-(2-цианоэтоксиметил)аденозин 3'-О-(2-цианоэтил N,N-диизопропилфосфорамидит)
Стадия 1
Получение N 6 -ацетил-5'-О-(4,4'-диметокситритил)-2'-О-(2-цианоэтоксиметил)аденозина
N6-ацетил-5'-О-(4,4'-диметокситритил)аденозин (22,0 г, 36,0 ммоль) растворяют в 170 мл 1,2-дихлорэтана, и добавляют 16,3 г диизопропилэтиламина (126 ммоль), и потом добавляют 12,1 г дибутилоловодихлорида (39,7 ммоль). Затем реакцию проводят при комнатной температуре в течение 1 час.
Затем реакционный раствор нагревают вплоть до 80ºС, и добавляют по каплям 4,30 г хлорметил-2-цианоэтилового эфира (36,0 ммоль), и раствор перемешивают в течение 30 мин.
После завершения реакции реакционный раствор добавляют к насыщенному водному раствору бикарбоната натрия и подвергают экстракции метиленхлоридом. Экстракт сушат над безводным сульфатом магния и растворитель отгоняют. Полученную смесь очищают колоночной хроматографией на силикагеле с получением N6-ацетил-5'-О-(4,4'-диметокситритил)-2'-О-(2-цианоэтоксиметил)аденозина (7,47 г; выход 33%).
1Н-ЯМР (CDCl3): 2,51 (т, 2Н, J=6,2 Гц); 2,58 (д, 1Н, J=5,5 Гц); 2,61 (с, 3Н); 3,45 (дд, 1Н, J=10,7, 4,0 Гц); 3,54 (дд, 1Н, J=10,7, 3,2 Гц); 3,62-3,79 (м, 2Н); 3,79 (с, 6Н); 4,25 (ушир.кв., 1Н, J=4,6 Гц); 4,59 (кв., 1Н, J=5,2 Гц); 4,87-4,94 (м, 3Н); 6,23 (д, 1Н, J=4,4 Гц); 6,80-6,83 (м, 4Н); 7,22-7,32 (м, 7Н); 7,40-7,43 (м, 2Н); 8,20 (с, 1Н); 8,61 (ушир.с, 1Н); 8,62 (с, 1Н)
Масс-спектрометрия с ионизацией в искровом или тлеющем разряде (ESI-Mass): 695 [M+Н]+
Стадия 2
Получение N 6 -ацетил-5'-О-(4,4'-диметокситритил)-2'-О-(2-цианоэтоксиметил)аденозин 3'-О-(2-цианоэтил N,N-диизопропилфосфорамидит)
N6-ацетил-5'-О-(4,4'-диметокситритил)-2'-О-(2-цианоэтоксиметил)аденозин (10,0 г, 14,4 ммоль), полученный на стадии 1, растворяют в 75 мл метиленхлорида, и добавляют 4,7 г диизопропилэтиламина (36 ммоль), и добавляют по каплям 4,82 г 2-цианоэтил N,N-диизопропилхлорфосфорамидита (20,3 ммоль). Затем реакцию проводят при комнатной температуре в течение 1 час.
После завершения реакции растворитель отгоняют и полученную смесь, в которой остается приблизительно 30 мл растворителя, очищают колоночной хроматографией на силикагеле с получением целевого соединения (12,0 г; выход 93%).
Масс-спектрометрия с ионизацией в искровом или тлеющем разряде (ESI-Mass): 895 [M+Н]+
Пример 10
N 6 -ацетил-2'-О-(2-цианоэтоксиметил)аденозин
Стадия 1
Получение N 6 -ацетил-3',5'-О-(тетраизопропилдисилоксан-1,3-диил)-2'-О-(2-цианоэтоксиметил)аденозина
В 8 мл метиленхлорида суспендируют 245 мг N-йодсукцинимида (1,09 ммоль) и 280 мг трифторметансульфоната серебра (1,09 ммоль) и раствор сушат путем добавления молекулярных сит 4Å.
К реакционному раствору добавляют раствор N6-ацетил-3',5'-О-(тетраизопропилдисилоксан-1,3-диил)аденозина (400 мг, 0,73 ммоль) и 145 мг метилтиометил-2-цианоэтилового эфира (1,11 ммоль) в 4 мл метиленхлорида при охлаждении льдом и реакционную смесь перемешивают в течение 3 час.
После завершения реакции реакционную смесь разбавляют метиленхлоридом и промывают водным раствором тиосульфата натрия и насыщенным водным раствором бикарбоната натрия. Экстракт сушат над безводным сульфатом магния и растворитель отгоняют. Полученную смесь очищают колоночной хроматографией на силикагеле с получением N6-ацетил-3',5'-О-(тетраизопропилдисилоксан-1,3-диил)-2'-О-(2-цианоэтоксиметил)аденозина (201 мг; выход 45%).
1Н-ЯМР (CDCl3): 0,98-1,11 (м, 28Н); 2,62 (с, 3Н); 2,69 (тд, 2Н, J=6,5, 1,5 Гц); 3,81-3,89 (м, 1Н); 4,02-4,09 (м, 2Н); 4,17 (д, 1Н, J=9,4 Гц); 4,28 (д, 1Н, J=13,4 Гц); 4,50 (д, 1Н, J=4,5 Гц); 4,67 (дд, 1Н, J=8,8, 4,5 Гц); 5,02 (д, 1Н, J=7,0 Гц); 5,08 (д, 1Н, J=7,0 Гц); 6,10 (с, 1Н); 8,34 (с, 1Н); 8,66 (с, 1Н); 8,67 (с, 1Н)
Масс-спектрометрия с ионизацией в искровом или тлеющем разряде (ESI-Mass): 636 [M+Н]+
Стадия 2
Получение N 6 -ацетил-2'-О-(2-цианоэтоксиметил)аденозина
N6-ацетил-3',5'-О-(тетраизопропилдисилоксан-1,3-диил)-2'-О-(2-цианоэтоксиметил)аденозин (300 мг, 0,47 ммоль), полученный на стадии 1, растворяют в растворителе, смешанном из 0,1 мл уксусной кислоты и 2 мл 0,5 М раствора фторида тетрабутиламмония, и реакционный раствор перемешивают при комнатной температуре в течение 2 час.
После завершения реакции полученную реакционную смесь очищают колоночной хроматографией на силикагеле с получением целевого соединения (160 мг; выход 86%).
1Н-ЯМР (DMSO-d6): 2,25 (с, 3Н); 2,53-2,68 (м, 2Н); 3,41-3,46 (м, 1Н); 3,56-3,64 (м, 2Н); 3,69-3,73 (м, 1Н); 4,00-4,01 (м, 1Н); 4,36-4,37 (м, 1Н); 4,72-4,78 (м, 3Н); 5,20 (ушир.т, 2Н); 5,41 (д, 1Н, J=5,2 Гц); 6,17 (д, 1Н, J=5,7 Гц); 8,66 (с, 1Н); 8,72 (с, 1Н); 10,72 (с, 1Н)
Масс-спектрометрия с ионизацией в искровом или тлеющем разряде (ESI-Mass): 415 [M+Na]+
Пример 11
Получение N 6 -ацетил-5'-О-(4,4'-диметокситритил)-2'-О-(2-цианоэтоксиметил)аденозина
N6-ацетил-2'-О-(2-цианоэтоксиметил)аденозин (9,50 г, 24,2 ммоль) растворяют в 100 мл безводного пиридина и затем сушат путем концентрирования. Затем твердый остаток растворяют в 100 мл дегидратированного пиридина в атмосфере аргона.
При охлаждении льдом добавляют 10,7 г 4,4'-диметокситритилхлорида (31,2 ммоль) и реакцию проводят при комнатной температуре в течение 1 час 20 мин.
После завершения реакции реакционный раствор разбавляют метиленхлоридом и промывают водой. Экстракт сушат над безводным сульфатом натрия и растворитель отгоняют. Полученную смесь очищают колоночной хроматографией на силикагеле с получением целевого соединения (13,8 г; выход 82%).
Пример 12
N 2 -Феноксиацетил-5'-О-(4,4'-диметокситритил)-2'-О-(2-цианоэтоксиметил)гуанозин 3'-О-(2-цианоэтил N,N-диизопропилфосфорамидит)
Стадия 1
Получение N 2 -феноксиацетил-5'-О-(4,4'-диметокситритил)-2'-О-(2-цианоэтоксиметил)гуанозина
N2-феноксиацетил-5'-О-(4,4'-диметокситритил)гуанозин (720 мг, 1 ммоль) растворяют в 4 мл 1,2-дихлорэтана, и добавляют 452 мг диизопропилэтиламина (3,5 ммоль), и потом добавляют 365 мг дибутилоловодихлорида (1,2 ммоль). Затем реакцию проводят при комнатной температуре в течение 1 час.
Затем реакцию проводят при 80ºС, и добавляют по каплям 155,4 мг хлорметил-2-цианоэтилового эфира (1,3 ммоль), и раствор перемешивают в течение 60 мин.
После завершения реакции реакционный раствор смешивают с насыщенным водным раствором бикарбоната натрия и подвергают экстракции метиленхлоридом. Экстракт сушат над безводным сульфатом магния и растворитель отгоняют. Полученную смесь очищают колоночной хроматографией на 30 г силикагеля с получением N2-феноксиацетил-5'-О-(4,4'-диметокситритил)-2'-О-(2-цианоэтоксиметил)гуанозина (384 мг; выход 48%).
1Н-ЯМР (CDCl3): 2,47-2,51 (м, 2Н); 2,58 (ушир.с, 1Н); 3,42 (дд, 1Н, J=10,1, 3,8 Гц); 3,46 (дд, 1Н, J=10,1, 3,8 Гц); 3,53-3,57 (м, 1Н); 3,69-3,73 (м, 1Н); 3,77 (с, 6Н); 4,24-4,26 (м, 1Н); 4,48-4,50 (м, 1Н); 4,61-4,65 (м, 2Н); 4,83, 4,87 (2д, 2Н, J=7,0 Гц); 4,88 (т, 1Н, J=5,7 Гц); 6,05 (д, 1Н, J=5,7 Гц); 6,80-6,82 (м, 4Н); 6,92-6,96 (м, 3Н); 7,07-7,11 (м, 2Н); 7,20-7,42 (м, 9Н); 7,84 (с, 1Н); 8,99 (с, 1Н); 11,81 (ушир.с, 1Н)
Масс-спектрометрия с ионизацией в искровом или тлеющем разряде (ESI-Mass): 825 [M+Na]+
Стадия 2
Получение N 2 -феноксиацетил-5'-О-(4,4'-диметокситритил)-2'-О-(2-цианоэтоксиметил)гуанозин 3'-О-(2-цианоэтил N,N-диизопропилфосфорамидит)
N2-феноксиацетил-5'-О-(4,4'-диметокситритил)-2'-О-(2-цианоэтоксиметил)гуанозин (320 мг, 0,399 ммоль), полученный на стадии 1, растворяют в 4 мл метиленхлорида, и добавляют 128,8 мг диизопропилэтиламина (0,996 ммоль), и добавляют по каплям 141,5 мг 2-цианоэтил N,N-диизопропилхлорфосфорамидита (0,598 ммоль). Затем реакцию проводят при комнатной температуре в течение 1 час.
После завершения реакции растворитель отгоняют и полученную смесь очищают колоночной хроматографией на 30 г силикагеля с получением целевого соединения (316 мг; выход 79%).
Масс-спектрометрия с ионизацией в искровом или тлеющем разряде (ESI-Mass): 1003 [M+Н]+
Пример 13
N 2 -феноксиацетил-2'-О-(2-цианоэтоксиметил)гуанозин
Стадия 1
Получение N 2 -феноксиацетил-3',5'-О-(тетраизопропилдисилоксан-1,3-диил)-2'-О-(2-цианоэтоксиметил)гуанозина
N2-феноксиацетил-3',5'-О-(тетраизопропилдисилоксан-1,3-диил)гуанозин (2,0 г, 3,0 ммоль) растворяют в 16 мл тетрагидрофурана, и добавляют 0,99 г метилтиометил-2-цианоэтилового эфира (7,6 ммоль) и 1,0 г молекулярных сит 4Å, и реакционный раствор перемешивают при -45ºС в течение 10 мин в атмосфере аргона.
После добавления раствора 0,68 г трифторметансульфокислоты (4,5 ммоль) в 5 мл тетрагидрофурана и перемешивания реакционного раствора добавляют 1,02 г N-йодсукцинимида (4,5 ммоль) и реакционный раствор перемешивают в течение 15 мин.
После добавления насыщенного водного раствора бикарбоната натрия к реакционному раствору и затем фильтрации реакционного раствора органический слой промывают 1 М водным раствором гидротиосульфата натрия.
Далее, реакционный раствор последовательно промывают водой и насыщенным рассолом (насыщенный раствор NaCl), и экстракт сушат над безводным сульфатом магния, и растворитель отгоняют.
Полученный твердый остаток очищают колоночной хроматографией на силикагеле с получением N2-феноксиацетил-3',5'-О-(тетраизопропилдисилоксан-1,3-диил)-2'-О-(2-цианоэтоксиметил)гуанозина (2,0 г; выход 89%).
1Н-ЯМР (CDCl3): 0,99-1,11 (м, 28Н); 2,59-2,77 (м, 2Н); 3,82-4,05 (м, 3Н); 4,15 (д, 1Н, J=9,3 Гц); 4,25-4,35 (м, 2Н); 4,52-4,56 (дд, 1Н, J=9,3, 4,3 Гц); 5,00-5,07 (2д, 2Н, J=7,2 Гц); 5,95 (с, 1Н); 6,99-7,12 (м, 3Н); 7,35-7,40 (м, 2Н); 8,09 (с, 1Н); 9,38 (ушир.с, 1Н); 11,85 (ушир.с, 1Н)
Масс-спектрометрия с ионизацией в искровом или тлеющем разряде (ESI-Mass): 766 [M+Na]+
Стадия 2
Получение N 2 -феноксиацетил-2'-О-(2-цианоэтоксиметил)гуанозина
Готовят раствор, состоящий из 0,14 мл уксусной кислоты (0,14 ммоль) и 2,83 мл 1 М раствора фторида тетрабутиламмония в тетрагидрофуране (2,83 ммоль).
N2-феноксиацетил-3',5'-О-(тетраизопропилдисилоксан-1,3-диил)-2'-О-(2-цианоэтоксиметил)гуанозин (1,0 г, 1,35 ммоль), полученный на стадии 1, растворяют в 2,83 мл тетрагидрофурана, и добавляют раствор, приготовленный выше, и реакцию проводят при комнатной температуре в течение 1 час в атмосфере аргона.
Реакционный раствор концентрируют при пониженном давлении, и остаток растворяют в метиленхлориде, и очищают колоночной хроматографией на силикагеле с получением целевого соединения (0,67 г; выход 99%).
1Н-ЯМР (DMSO-d6): 2,59-2,66 (м, 2Н); 3,41-3,63 (м, 4Н); 3,98 (м, 1Н); 4,32 (м, 1Н); 4,58-4,62 (т, 1Н, J=5,3 Гц); 4,71-4,78 (дд, 2Н, J=13,1, 6,8 Гц); 4,87 (с, 2Н); 5,12 (с, 1Н); 5,37 (с, 1Н); 5,97 (д, 1Н, J=6,1 Гц); 6,96-6,99 (м, 3Н); 7,28-7,34 (м, 2Н); 8,30 (с, 1Н); 11,78 (ушир.с, 2Н)
Масс-спектрометрия с ионизацией в искровом или тлеющем разряде (ESI-Mass): 500 [M-Н]-
Пример 14
N 2 -феноксиацетил-5'-О-(4,4'-диметокситритил)-2'-О-(2-цианоэтоксиметил)гуанозин
N2-феноксиацетил-2'-О-(2-цианоэтоксиметил)гуанозин (660 мг, 1,32 ммоль) подвергают азеотропной перегонке с пиридином и затем сушат вакуумным насосом в течение 30 мин.
Твердый остаток растворяют в 9 мл тетрагидрофурана, и добавляют в атмосфере аргона 2,1 г пиридина (26,4 ммоль) и 600 мг молекулярных сит 4Å, и реакционный раствор перемешивают в течение 10 мин.
К реакционному раствору добавляют тремя порциями каждый 1 час 540 мг 4,4'-диметокситритилхлорида (1,58 ммоль) и реакционный раствор дополнительно перемешивают в течение 1 час.
После добавления 2 мл метанола и перемешивания реакционного раствора в течение 2 мин реакционный раствор фильтруют через целит и промывают этилацетатом.
После концентрирования фильтрата путем выпаривания остаток растворяют в этилацетате и разделяют посредством насыщенного водного раствора бикарбоната натрия.
После промывания органического слоя насыщенным рассолом (насыщенный раствор NaCl) и сушки над безводным сульфатом магния растворитель отгоняют.
Полученный твердый остаток очищают колоночной хроматографией на силикагеле с получением целевого соединения (800 мг, выход 75%).
Пример 15
N 6 -ацетил-3',5'-О-(тетраизопропилдисилоксан-1,3-диил)-2'-О-(2-цианоэтоксиметил)аденозин
Стадия 1
Получение N 6 -ацетил-3',5'-О-(тетраизопропилдисилоксан-1,3-диил)-2'-О-метилтиометиладенозина
N6-ацетил-3',5'-О-(тетраизопропилдисилоксан-1,3-диил)аденозин (2,00 г, 3,62 ммоль) растворяют в 25 мл диметилсульфоксида, и добавляют 17,5 мл уксусного ангидрида и 12,5 мл уксусной кислоты, и реакционный раствор перемешивают при комнатной температуре в течение 14 час.
После завершения реакции реакционный раствор добавляют к 200 мл воды, экстрагируют этилацетатом и промывают насыщенным водным раствором бикарбоната натрия. Экстракт сушат над безводным сульфатом натрия и растворитель отгоняют. Полученную смесь очищают колоночной хроматографией на силикагеле с получением N6-ацетил-3',5'-О-(тетраизопропилдисилоксан-1,3-диил)-2'-О-метилтиометиладенозина (1,36 г; выход 61%).
1Н-ЯМР (CDCl3): 0,96-1,11 (м, 28Н); 2,20 (с, 3Н); 2,61 (с, 3Н); 4,03 (дд, 1Н, J=13,4, 2,4 Гц); 4,18 (д, 1Н, J=9,1 Гц); 4,27 (д, 1Н, J=13,4 Гц); 4,63-4,71 (м, 2Н); 5,00 (д, 1Н, J=11,5 Гц); 5,07 (д, 1Н, J=11,5 Гц); 6,09 (с, 1Н); 8,31 (с, 1Н); 8,65 (с, 1Н); 8,69 (с, 1Н)
Масс-спектрометрия с ионизацией в искровом или тлеющем разряде (ESI-Mass): 635 [M+Na]+
Стадия 2
Получение N 6 -ацетил-3',5'-О-(тетраизопропилдисилоксан-1,3-диил)-2'-О-(2-цианоэтоксиметил)аденозина
N6-ацетил-3',5'-О-(тетраизопропилдисилоксан-1,3-диил)-2'-О-метилтиометиладенозин (1,00 г, 1,63 ммоль), полученный на стадии 1, растворяют в 25 мл тетрагидрофурана.
К реакционному раствору добавляют 5,88 г 3-гидроксипропионитрила (82,7 ммоль), и этот раствор сушат путем добавления молекулярных сит 4Å, и охлаждают до -45ºС.
К реакционному раствору добавляют 440 мг N-йодсукцинимида (1,96 ммоль) и затем 490 мг трифторметансульфокислоты (3,26 ммоль) и реакционный раствор перемешивают при -45ºС в течение 15 мин.
После завершения реакции реакционный раствор нейтрализуют путем добавления триэтиламина при охлаждении и разбавляют метиленхлоридом. Реакционный раствор промывают водным раствором тиосульфата натрия и насыщенным водным раствором бикарбоната натрия, экстракт сушат над безводным сульфатом натрия и растворитель отгоняют. Полученную смесь очищают колоночной хроматографией на силикагеле с получением целевого соединения (722 мг; выход 71%).
Пример 16
Уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридин
Олиго-РНК соединения, указанного в заглавии, синтезируют путем ввода имеющегося в продаже твердого носителя СPG (стекло с заданным размером пор) (37 мг, 1 мкмоль), содержащего 2'/3'-О-бензоил-5'-О-(4,4'-диметокситритил)уридин, в колонку со стеклянным фильтром и путем использования автоматического синтезатора нуклеиновой кислоты (ExpediteTM: Applied Biosystems (Прикладные Биосистемы)).
Используют 5'-О-(4,4'-диметокситритил)-2'-О-(2-цианоэтоксиметил)уридин 3'-О-(2-цианоэтил N,N-диизопропилфосфорамидит) в качестве соединения мономера нуклеиновой кислоты, тетразол в качестве катализатора конденсации, раствор йода в качестве окислителя, раствор уксусного ангидрида и N-метилимидазола в качестве кэппирующего раствора.
После 20-кратной конденсации соединений мономера нуклеиновой кислоты олиго-РНК отщепляют посредством действия 10 М водно-этанольного раствора метиламина в качестве отщепляющего агента при комнатной температуре в течение 1-2 час и защитные группы каждой фосфатной части удаляют.
После концентрирования реакционной смеси при пониженном давлении и удаления ненужных пиков при помощи колонки с обращенной фазой (ODS) реакционный раствор очищают элюентом (50 мМ триэтиламин - ацетатный буферный раствор в ацетонитриле).
После концентрирования остатка при пониженном давлении остаток подвергают реакции с 1 М раствором фторида тетрабутиламмония в тетрагидрофуране при комнатной температуре в течение 1 час для удаления защитной группы для 2'-гидроксильной группы.
После обессоливания реакционного раствора защитную группу для 5'-концевой группы удаляют посредством 80% раствора уксусной кислоты (обработка при комнатной температуре в течение 10 мин).
После концентрирования при пониженном давлении водный слой промывают эфиром и получают целевое соединение высокой степени чистоты без очистки.
Времяпролетная масс-спектрометрия с матричной лазерной десорбционной ионизацией (Matrix-Assisted Laser Desorption Ionization Time of Flight Mass-Spectroscopy (MALDI-TOF-MS)):
Вычисленное значение 6367,52 [М+Н]+
Найденное значение 6366,50 [М+Н]+
Из аналитических данных ВЭЖХ с обращенной фазой, представленных на чертеже, ясно, что полученное соединение имеет высокую степень чистоты.
Условия измерений являются следующими:
Прибор для Высокоэффективной Жидкостной Хроматографии (ВЭЖХ)
Устройство для аспирации: LC -6A (SHIMADZU CORPORATION)
Детектор: SPD-6A (SHIMADZU CORPORATION)
Колонка ВЭЖХ с обращенной фазой: Mightysil RP-18GP <4,6 мм ⌀×15 см> (KANTO KAGAKU)
Температура колонки: 35ºС
Градиент подвижной фазы: линейный градиент, 20 мин (раствор В: 0-70%)
Раствор А: 50 мМ триэтиламин-ацетатный буферный раствор, содержащий 5% ацетонитрила
Раствор В: 50 мМ триэтиламин-ацетатный буферный раствор, содержащий 90% ацетонитрила
Скорость потока подвижной фазы: 1 мл/мин
Длина волны для детектирования спектрофотометром в ультрафиолетовой видимой области: 260 нм
Пример 17
Цитидилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-аденилил-[3'→5']-цитидилил-[3'→5']-гуанилил-[3'→5']-цитидилил-[3'→5']-уридилил-[3'→5']-гуанилил-[3'→5']-аденилил-[3'→5']-гуанилил-[3'→5']-уридилил-[3'→5']-аденилил-[3'→5']-цитидилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-цитидилил-[3'→5']-гуанилил-[3'→5']-аденилил-[3'→5']-уридин
Олиго-РНК соединения, указанного в заглавии, синтезируют путем ввода имеющегося в продаже твердого носителя СPG (стекло с заданным размером пор) (37 мг, 1 мкмоль), содержащего 2'/3'-О-бензоил-5'-О-(4,4'-диметокситритил)уридин, в колонку со стеклянным фильтром и путем использования автоматического синтезатора нуклеиновой кислоты (ExpediteTM: Applied Biosystems (Прикладные Биосистемы)).
Используют 5'-О-(4,4'-диметокситритил)-2'-О-(2-цианоэтоксиметил)уридин 3'-О-(2-цианоэтил N,N-диизопропилфосфорамидит), N4-ацетил-5'-О-(4,4'-диметокситритил)-2'-О-(2-цианоэтоксиметил)цитидин 3'-О-(2-цианоэтил N,N-диизопропилфосфорамидит), N4-ацетил-5'-О-(4,4'-диметокситритил)-2'-О-(2-цианоэтоксиметил)аденозин 3'-О-(2-цианоэтил N,N-диизопропилфосфорамидит) и N2-феноксиацетил-5'-О-(4,4'-диметокситритил)-2'-О-(2-цианоэтоксиметил)гуанозин 3'-О-(2-цианоэтил N,N-диизопропилфосфорамидит) в качестве соединения мономера нуклеиновой кислоты; 5-этилтиотетразол в качестве катализатора конденсации; раствор йода в качестве окислителя; раствор феноксиуксусного ангидрида и N-метилимидазола в качестве кэппирующего раствора.
После 19-кратной конденсации соединений мономеров нуклеиновой кислоты защитную группу для 5'-концевой гидроксильной группы удаляют на твердой фазе. Затем олиго-РНК отщепляют посредством действия смеси концентрированного водного раствора аммиака и этанола (3:1) в качестве отщепляющего агента при 40ºС в течение 4 час и защитные группы каждой фосфатной части и основания удаляют.
После концентрирования реакционной смеси при пониженном давлении остаток подвергают реакции с 1 М раствором фторида тетрабутиламмония в тетрагидрофуране, содержащим 10% н-пропиламина и 0,6% 2-меркаптоэтилового эфира, при комнатной температуре в течение 1 час для удаления защитной группы для 2'-гидроксильной группы.
После обессоливания реакционного раствора реакционный раствор очищают посредством ДЭАЭ-ионообменной смолы (TOYOPEARLDEAE-650) для получения целевого соединения высокой степени чистоты (112 OD260; выход 58%).
Здесь поглощение ультрафиолетового излучения с длиной волны 260 нм (OD260) показывает выход целевого соединения.
В дальнейшем в этом документе поглощение (OD260) означает выход целевого соединения.
Масс-спектрометрия с матричной лазерной десорбционной ионизацией, времяпролетная (Matrix-Assisted Laser Desorption Ionization Time of Flight Mass-Spectroscopy (MALDI-TOF-MS)):
Вычисленное значение 6305,9 [М+Н]+
Найденное значение 6304,8 [М+Н]+
Пример 18
Аденилил-[3'→5']-цитидилил-[3'→5']-аденилил-[3'→5']-уридилил-[3'→5']-цитидилил-[3'→5']-аденилил-[3'→5']-цитидилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-аденилил-[3'→5']-цитидилил-[3'→5']-гуанилил-[3'→5']-цитидилил-[3'→5']-уридилил-[3'→5']-гуанилил-[3'→5']-аденилил-[3'→5']-гуанилил-[3'→5']-уридилил-[3'→5']-аденилил-[3'→5']-цитидилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-цитидилил-[3'→5']-гуанилил-[3'→5']-аденилил-[3'→5']-аденилил-[3'→5']-аденилил-[3'→5']-уридилил-[3'→5']-гуанилил-[3'→5']-уридин
Целевое соединение синтезируют аналогично тому, как это делают в примере 17 (92 OD260; выход 31%).
MALDI-TOF-MS:
Вычисленное значение 9519,8 [М+Н]+
Найденное значение 9520,4 [М+Н]+
Пример 19
Уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридин
Целевое соединение синтезируют аналогично тому, как это делают в примере 17 (254 OD260; выход 65%).
MALDI-TOF-MS:
Вычисленное значение 12185,8 [М+Н]+
Найденное значение 12183,3 [М+Н]+
Пример 20
Аденилил-[3'→5']-аденилил-[3'→5']-уридилил-[3'→5']-цитидилил-[3'→5']-аденилил-[3'→5']-цитидилил-[3'→5']-аденилил-[3'→5']-гуанилил-[3'→5']-аденилил-[3'→5']-аденилил-[3'→5']-уридилил-[3'→5']-цитидилил-[3'→5']-гуанилил-[3'→5']-уридилил-[3'→5']-цитидилил-[3'→5']-гуанилил-[3'→5']-уридилил-[3'→5']-аденилил-[3'→5']-уридилил-[3'→5']-гуанилил-[3'→5']-цитидилил-[3'→5']-аденилил-[3'→5']-гуанилил-[3'→5']-уридилил-[3'→5']-гуанилил-[3'→5']-аденилил-[3'→5']-аденилил-[3'→5']-аденилил-[3'→5']-аденилил-[3'→5']-цитидилил-[3'→5']-уридилил-[3'→5']-цитидилил-[3'→5']-уридилил-[3'→5']-цитидилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-цитидилил-[3'→5']-аденилил-[3'→5']-аденилил-[3'→5']-тимидин
Целевое соединение синтезируют аналогично тому, как это делают в примере 17 (75 OD260; выход 19%).
MALDI-TOF-MS:
Вычисленное значение 12731,8 [М+Н]+
Найденное значение 12731,7 [М+Н]+
Пример 21
Уридилил-[3'→5']-гуанилил-[3'→→5']-аденилил-[3'→5']-аденилил-[3'→5']-уридилил-[3'→5']-аденилил-[3'→5']-цитидилил-[3'→5']-аденилил-[3'→5']-аденилил-[3'→5']-аденилил-[3'→5']-уридилил-[3'→5']-цитидилил-[3'→5']-аденилил-[3'→5']-цитидилил-[3'→5']-аденилил-[3'→5']-гуанилил-[3'→5']-аденилил-[3'→5']-аденилил-[3'→5']-уридилил-[3'→5']-цитидилил-[3'→5']-гуанилил-[3'→5']-уридилил-[3'→5']-цитидилил-[3'→5']-гуанилил-[3'→5']-уридилил-[3'→5']-аденилил-[3'→5']-уридилил-[3'→5']-гуанилил-[3'→5']-цитидилил-[3'→5']-аденилил-[3'→5']-гуанилил-[3'→5']-уридилил-[3'→5']-гуанилил-[3'→5']-аденилил-[3'→5']-аденилил-[3'→5']-аденилил-[3'→5']-аденилил-[3'→5']-цитидилил-[3'→5']-уридилил-[3'→5']-цитидилил-[3'→5']-уридилил-[3'→5']-цитидилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-цитидилил-[3'→5']-аденилил-[3'→5']-аденилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-цитидилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-уридилил-[3'→5']-аденилил-[3'→5']-тимидин
Целевое соединение синтезируют аналогично тому, как это делают в примере 17 (83 OD260; выход 15%).
MALDI-TOF-MS:
Вычисленное значение 17476,6 [М+Н]+
Найденное значение 17474,6 [М+Н]+
ПРОМЫШЛЕННАЯ ПРИМЕНИМОСТЬ
Производное фосфорамидита настоящего изобретения имеет защитную группу эфирного типа, которую вводят по 2'-гидроксильной группе. Защитная группа эфирного типа представляет собой линейную защитную группу, и пространственная структура вокруг атома фосфора, присоединенного к 3'-гидроксильной группе, не является затрудненной, и, следовательно, производное фосфорамидита настоящего изобретения делает возможным протекание реакции конденсации за более короткое время и получение лучшего выхода в реакции конденсации в процессе синтезирования олиго-РНК по сравнению с традиционным производным фосфорамидита.
Применение производного фосфорамидита настоящего изобретения делает возможным получение олиго-РНК высокой степени чистоты с использованием способа, по существу идентичного способу, применяемому в производстве олиго-ДНК.
| название | год | авторы | номер документа |
|---|---|---|---|
| СИНТЕЗ ФОСФИТИЛИРОВАННЫХ СОЕДИНЕНИЙ С ИСПОЛЬЗОВАНИЕМ ЧЕТВЕРТИЧНОГО ГЕТЕРОЦИКЛИЧЕСКОГО АКТИВАТОРА | 2005 |
|
RU2440364C2 |
| НОВЫЕ НУКЛЕОЗИДНЫЕ И ОЛИГОНУКЛЕОТИДНЫЕ АНАЛОГИ | 2000 |
|
RU2233844C2 |
| 3' И/ИЛИ 2'-АМИНО- ИЛИ ТИОЛМОДИФИЦИРОВАННЫЕ НУКЛЕОЗИДЫ, НУКЛЕОТИДЫ ИЛИ ОЛИГОНУКЛЕОТИДЫ, СПОСОБ ПОЛУЧЕНИЯ СОЕДИНЕНИЙ | 1991 |
|
RU2073682C1 |
| СПОСОБ СОЕДИНЕНИЯ НУКЛЕОЗИДОВ 3'-5'-МЕЖНУКЛЕОТИДНЫМ СИЛИЛЬНЫМ ЗВЕНОМ | 1991 |
|
RU2079508C1 |
| СИНТЕЗ ОЛИГОНУКЛЕОТИДОВ | 2006 |
|
RU2465280C2 |
| НОВЫЕ АНАЛОГИ 2`, 5`-ОЛИГОАДЕНИЛАТА ИЛИ ИХ ФАРМАКОЛОГИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И ИХ ПРИМЕНЕНИЕ | 2003 |
|
RU2311422C2 |
| УЛУЧШЕННАЯ ОЛИГОНУКЛЕОТИДНАЯ КОНСТРУКЦИЯ ТИПА НАНОЧАСТИЦЫ, ОБЛАДАЮЩАЯ ВЫСОКОЙ ЭФФЕКТИВНОСТЬЮ, И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2014 |
|
RU2670164C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ХИМИЧЕСКОГО РАСЩЕПЛЕНИЯ И СНЯТИЯ ЗАЩИТЫ ДЛЯ СВЯЗАННЫХ С ПОВЕРХНОСТЬЮ ОЛИГОНУКЛЕОТИДОВ | 2019 |
|
RU2766688C2 |
| СИНТЕЗ β-L-2'-ДЕЗОКСИНУКЛЕОЗИДОВ | 2004 |
|
RU2361875C2 |
| РОДСТВЕННОЕ ВИТАМИНУ A СОЕДИНЕНИЕ И СПОСОБЫ ЕГО ПОЛУЧЕНИЯ | 1998 |
|
RU2188193C2 |
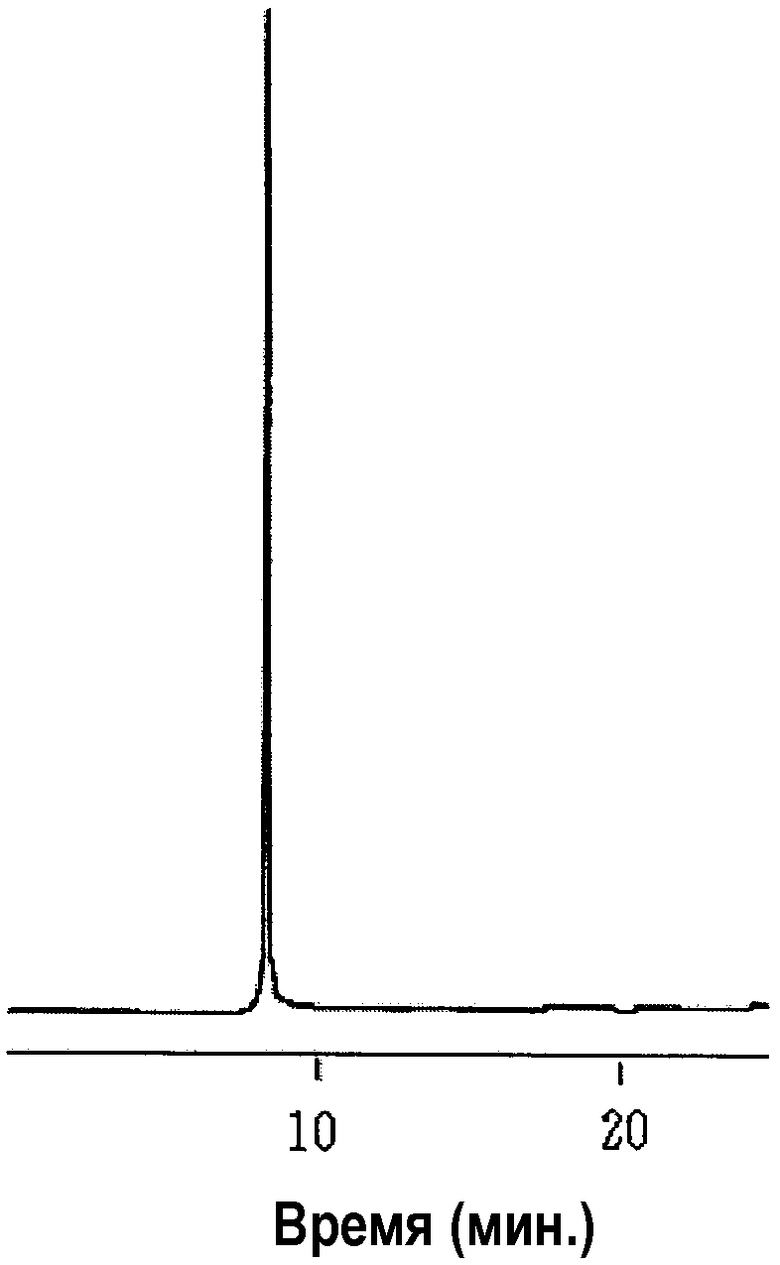
Настоящее изобретение относится к производным фосфорамидита общей формулы (1), где Вх представляет собой аденин, гуанин, цитозин, тимин или урацил, причем аминогруппа аденина, гуанина и цитозина может быть необязательно защищена защитной группой, выбранной из ацетила и феноксиацетила; R1 - заместитель общей формулы (2), в которой R11, R12 и R13 одинаковы или различны, и каждый представляет собой водород или алкокси; R2a и R2b одинаковы или различны, и каждый представляет собой алкил; и WG1, WG2 представляют собой цианогруппу. Изобретение относится также к многостадийному способу получения указанных соединений. Кроме того, изобретение относится к промежуточным соединениям названного способа, а именно к промежуточному эфирному соединению общей формулы (13), где L представляет собой галоген или С1-С5алкилтиогруппу; WG1 - цианогруппу; к промежуточному соединению общей формулы (21), где Вх представляет собой аденин, гуанин, цитозин, тимин или урацил, причем аминогруппа аденина, гуанина и цитозина может быть необязательно защищена защитной группой, выбранной из ацетильной группы и феноксиацетильной группы; и WG1 представляет собой цианогруппу; к промежуточному соединению общей формулы (15), где Вх имеет названные выше значения; R1 представляет собой заместитель общей формулы (2); к промежуточному соединению общей формулы (17), где Вх имеет названные выше значения; А представляет собой кремнесодержащий заместитель общей формулы (18а) или (18b)
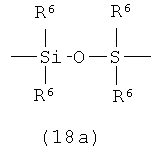
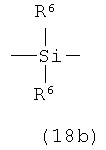 ,
,
где R6 - алкил, а WG1 - цианогруппа. Изобретение относится также к способу получения олигонуклеотида общей формулы (3), где каждый В независимо представляет собой аденин, гуанин, цитозин, урацил или тимин; каждый R независимо представляет собой Н или гидроксил, и, по меньшей мере, один из R представляет собой гидроксил; Z представляет собой Н или фосфатную группу; и n представляет собой целое число от 1 до 100, включающий стадии A-G, отличающийся использованием указанных выше производных фосфорамидита в качестве мономерного соединения нуклеиновой кислоты на стадии В. 7 н.п. ф-лы, 1 ил.
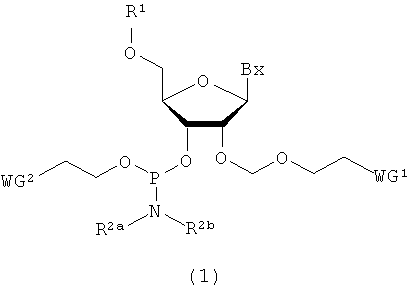
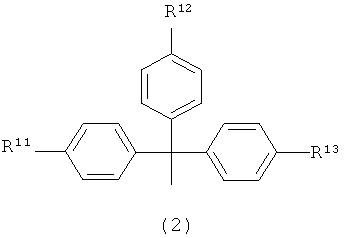
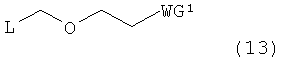
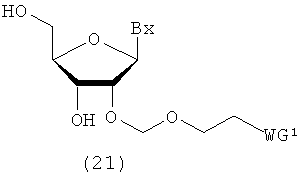
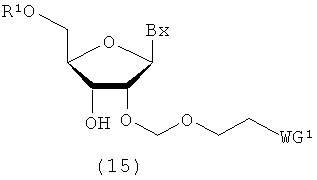
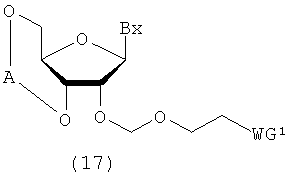
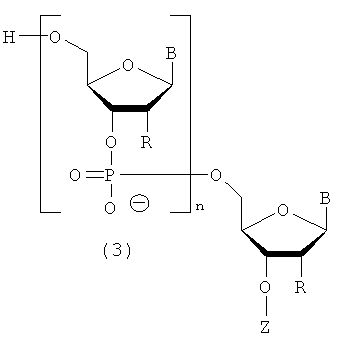
1. Производное фосфорамидита, представленное следующей общей формулой (1)
,
в которой
Вx представляет собой аденин, гуанин, цитозин, тимин или урацил, где аминогруппа аденина, гуанина и цитозина может быть необязательно защищена защитной группой, выбранной из ацетильной группы и феноксиацетильной группы;
R1 представляет собой заместитель, представленный следующей общей формулой (2)
,
в которой
R11, R12 и R13 являются одинаковыми или различными, и каждый представляет собой водород или С1-С4алкоксигруппу;
R2a и R2b являются одинаковыми или различными, и каждый представляет собой С1-С5алкильную группу; и
каждый из WG1 и WG2 представляет собой цианогруппу.
2. Способ получения олигонуклеотида, представленного следующей общей формулой (3), включающий следующие стадии A-G, отличающийся использованием производного фосфорамидита по п.1 в качестве мономерного соединения нуклеиновой кислоты на следующей стадии В
в которой
каждый В независимо представляет собой аденин, гуанин, цитозин, урацил или тимин;
каждый R независимо представляет собой Н или гидроксильную группу, и, по меньшей мере, один из R представляет собой гидроксильную группу;
Z представляет собой Н или фосфатную группу; и
n представляет собой целое число в пределах от 1 до 100,
стадия А: получение соединения, представленного следующей общей формулой (5), путем удаления 5'-гидроксильной группы взаимодействием кислоты с соединением, представленным следующей общей формулой (4)
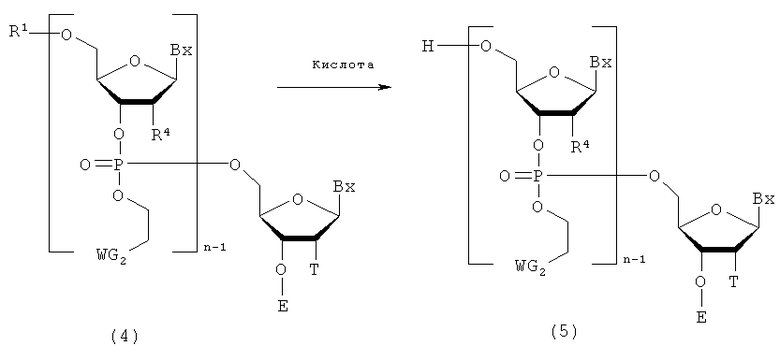
в которой
n такое, как определено выше;
каждый Вх независимо представляет собой аденин, гуанин, цитозин, тимин или урацил, где аминогруппа аденина, гуанина и цитозина может быть необязательно защищена защитной группой, выбранной из ацетильной группы и феноксиацетильной группы; и
R1 представляет собой заместитель, представленный следующей общей формулой (2)
,
в которой
R11, R12 и R13 являются одинаковыми или различными, и каждый представляет собой водород или С1-С4алкоксигруппу;
каждый WG2 представляет собой цианогруппу;
каждый R4 независимо представляет собой Н, ацилоксигруппу, где ацильный фрагмент представляет собой C1-С6алканоильную группу или С7-С13ароильную группу, или заместитель, представленный следующей общей формулой (6)
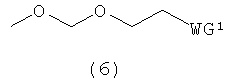 ,
,
в которой
WG1 представляет собой цианогруппу; и
Е представляет собой C1-6алканоильную группу или С7-С13ароильную группу или заместитель, представленный следующей общей формулой (7)
 ,
,
в которой
Q представляет собой одинарную связь или заместитель, представленный следующей общей формулой (8)
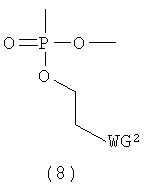 ,
,
в которой
WG2 такой как WG2, определенный выше; и
Т представляет собой Н или ацилоксигруппу, где ацильный фрагмент представляет собой C1-С6алканоильную группу или С7-С13ароильную группу, при условии, что либо Е, либо Т является заместителем (7),
причем кислота представляет собой трифторуксусную кислоту, дихлоруксусную кислоту или трихлоруксусную кислоту,
стадия В: получение соединения, представленного следующей общей формулой (9), путем конденсации мономерного соединения нуклеиновой кислоты с соединением, полученным на стадии А, с использованием активирующего агента
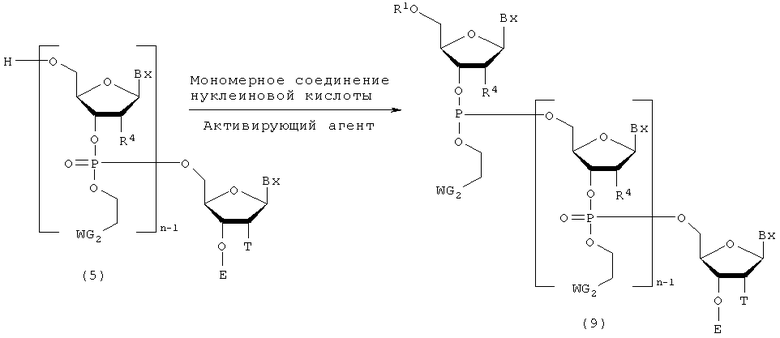
в которой
Вх, Е, n, R1, R4, Т и WG2 такие, как определено выше; и
активирующий агент представляет собой 1H-тетразол, 5-этилтиотетразол, 5-бензилмеркапто-1Н-тетразол, 4,5-дихлоримидазол, 4,5-дицианоимидазол, бензотриазолтрифлат, имидазолтрифлат, пиридинийтрифлат, N,N-диизопропилэтиламин или 2,4,6-коллидин/N-метилимидазол;
стадия С: кэппирование 5'-гидроксильной группы непрореагировавшего соединения (5) на стадии В с использованием кэппирующего агента
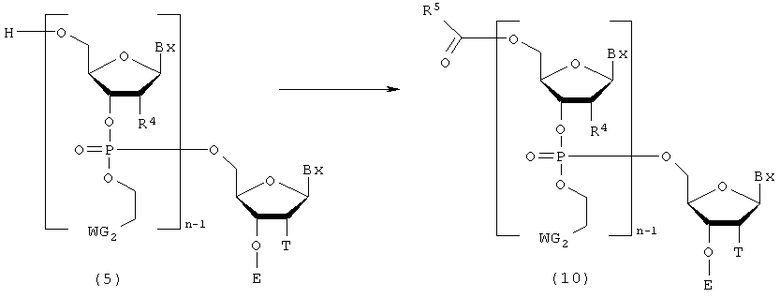
где Вх, Е, n, R4, Т и WG2 такие, как определено выше; и
R5 представляет собой метильную или феноксиметильную группу; и кэппирующий агент представляет собой уксусный ангидрид или феноксиуксусный ангидрид,
стадия D: превращение фосфорильной группы в фосфатную группу путем взаимодействия окислителя с соединением (9), которое получают на стадии В
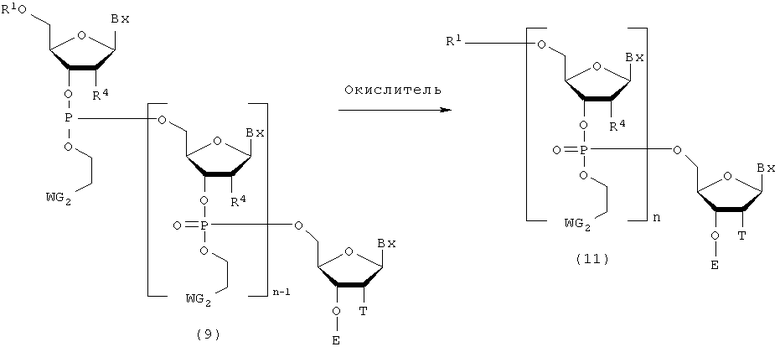
где Bx, E, n, R1, R4, Т и WG2 такие, как определено выше; и
окислитель представляет собой йод или трет-бутилгидропероксид;
стадия E: повторение стадий A-D для получения олигонуклеотида, имеющего требуемую длину цепи,
стадия F: отщепление соединения (11), полученного на стадии D, от твердого носителя и затем удаление защитных групп каждого нуклеинового основания и каждой 2'-гидроксильной группы с использованием отщепляющего агента, агента для удаления защитной группы 2'-гидроксильной группы и дезактивирующего агента
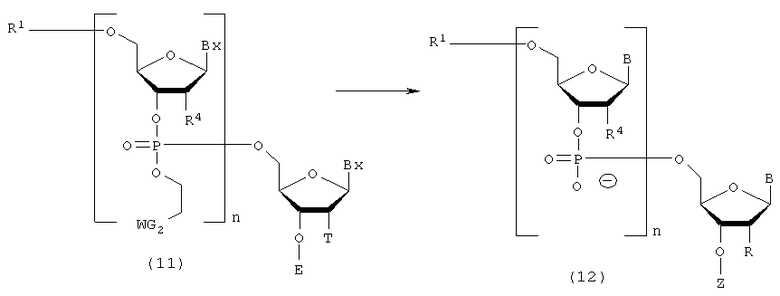
где В, Bx, E, n, R, R1, R4, Т, WG2 и Z такие, как определено выше,
отщепляющий агент представляет собой концентрированный водный раствор аммиака или метиламин;
агент для удаления защитной группы 2'-гидроксильной группы представляет собой фторид тетрабутиламмония или тригидрофторид/соль триэтиламина; и дезактивирующий агент представляет собой алкиламин, амидин, тиол, производные тиола или их смеси,
стадия G: удаление 5'-гидроксильной группы соединения (12), полученного на стадии F
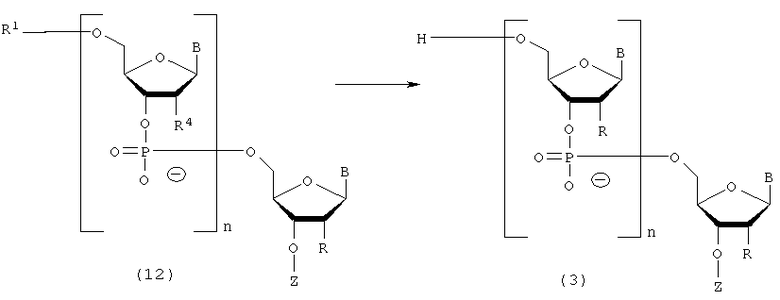
где В, n, R, R1 и Z такие, как определено выше,
стадия H: выделение и очистка олигонуклеотида (3), полученного на стадии G.
3. Эфирное соединение, представленное следующей общей формулой (13)
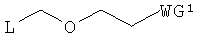 ,
,
в которой
L представляет собой галоген или С1-С5алкилтиогруппу;
WG1 представляет собой цианогруппу.
4. Способ получения производного фосфорамидита по п.1, включающий следующие стадии a-h,
стадия а: получение нуклеозида, представленного следующими общими формулами (15) и (15'), где защитную группу эфирного типа, которая может быть удалена в нейтральной среде, вводят по 2'-гидроксильной группе путем воздействия алкилирующего реагента на нуклеозид, представленный следующей общей формулой (14)
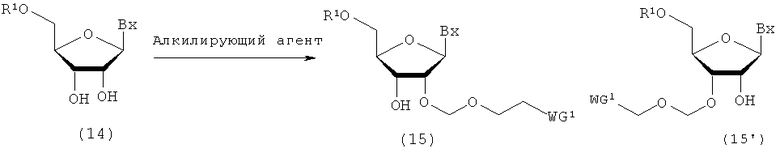 ,
,
где Вх представляет собой аденин, гуанин, цитозин, тимин или урацил, где аминогруппа аденина, гуанина и цитозина может быть необязательно защищена защитной группой, выбранной из ацетильной группы и феноксиацетильной группы;
R1 и WG1 такие, как определено в п.1; и
алкилирующий реагент представляет собой эфирное соединение, представленное следующей общей формулой (13)
в которой
L представляет собой галоген или С1-С5алкилтиогруппу; и WG1 представляет собой цианогруппу,
стадия b: выделение и очистка производного нуклеозида (15), полученного на стадии (а),
стадия с: осуществляемое отдельно от стадии b получение нуклеозида, представленного следующей общей формулой (17), где защитную группу эфирного типа, которая может быть удалена в нейтральной среде, вводят по 2'-гидроксильной группе путем воздействия алкилирующего реагента на нуклеозид, представленный следующей общей формулой (16)
 ,
,
в которой
Bx и WG1 такие, как определено выше; и
А представляет собой кремнесодержащий заместитель, представленный следующей общей формулой (18а) или (18b)
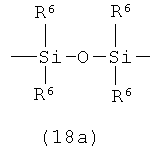 ,
,
в которой
R6 представляет собой С1-С5алкильную группу; и алкилирующий реагент такой, как определено выше,
стадия d: осуществляемое отдельно от стадий а-с получение нуклеозида, представленного следующей общей формулой (19), путем воздействия диметилсульфоксида, уксусной кислоты и уксусного ангидрида на соединение рибонуклеиновой кислоты (16)
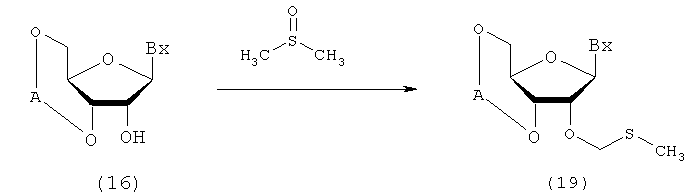 ,
,
где А и Вх такие, как определено выше,
стадия е: получение нуклеозида, представленного следующей общей формулой (17), где защитную группу эфирного типа, которая может быть удалена в нейтральной среде, вводят по 2'-гидроксильной группе, путем воздействия спиртового соединения, представленного следующей общей формулой (20), кислоты и реагента для галогенирования атома серы на нуклеозид (19), полученный на стадии d
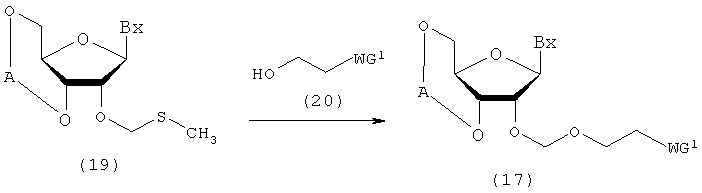
где А, Вх и WG1 такие, как определено выше;
кислота представляет собой трифторметансульфоновую кислоту, трифторметансульфонат серебра или триметилсилил трифторметансульфонат; и реагент для галогенирования атома серы представляет собой N-бромсукцинимид (NBS) или N-йодсукцинимид (NIS),
стадия f: получение нуклеозида, представленного следующей общей формулой (21), путем удаления защитных групп для 3'- и 5'-гидроксильных групп нуклеозида (17), полученного на стадии с или на стадии е
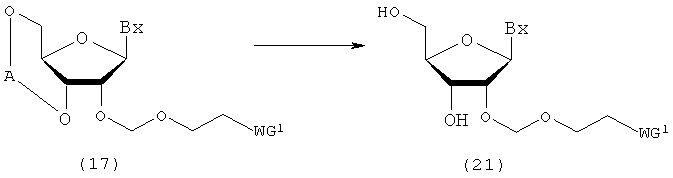 ,
,
где А, Вх и WG1 такие, как определено выше,
стадия g: получение нуклеозида (15) путем введения защитной группы (R1), которая может быть удалена в кислой среде, по 5'-гидроксильной группе нуклеозида (21), полученного на стадии f
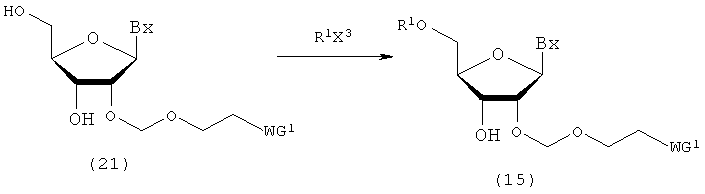 ,
,
где Вх, R1 и WG1 такие, как определено выше; и
Х3 представляет собой галоген,
стадия h: получение производного фосфорамидита, представленного следующей общей формулой (1), фосфитилированием 3'-гидроксильной группы путем воздействия фосфитилирующего реагента и, при необходимости, активирующего агента на нуклеозид (15), полученный на стадии b или стадии g
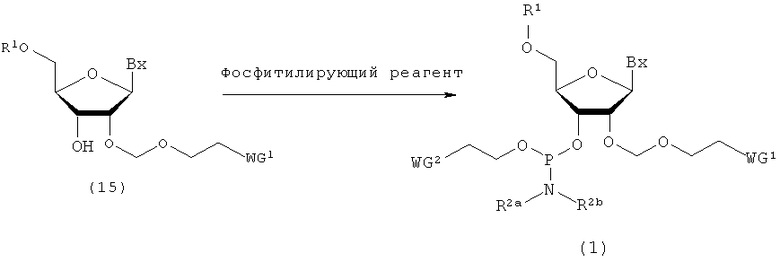
где Bx, R1 и WG1 такие, как определено выше;
каждый из R2a и R2b представляет собой С1-С5алкильную группу;
WG2 представляет собой цианогруппу;
активирующий агент представляет собой 1H-тетразол, 5-этилтиотетразол, 5-бензилмеркапто-1Н-тетразол, 4,5-дихлоримидазол, 4,5-дицианоимидазол, бензотриазолтрифлат, имидазолтрифлат, пиридинийтрифлат, N,N-диизопропилэтиламин или 2,4,6-кoллидин/N-мeтилимидaзoл;
фосфитилирующий реагент представляет собой соединение, представленное следующей общей формулой (22) или (23)
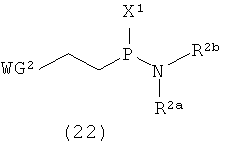
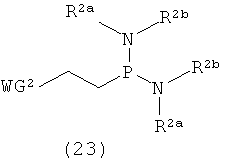 ,
,
в которых
каждый из R2a и R2b представляет собой С1-С5алкильную группу;
WG2 представляет собой цианогруппу; и
X1 представляет собой галоген.
5. Нуклеозид, представленный следующей общей формулой (21)
,
где Bx представляет собой аденин, гуанин, цитозин, тимин или урацил, где аминогруппа аденина, гуанина и цитозина может быть необязательно защищена защитной группой, выбранной из ацетильной группы и феноксиацетильной группы; и
WG1 представляет собой цианогруппу.
6. Нуклеозид, представленный следующей общей формулой (15)
,
где Вх представляет собой аденин, гуанин, цитозин, тимин или урацил, где аминогруппа аденина, гуанина и цитозина может быть необязательно защищена защитной группой, выбранной из ацетильной группы и феноксиацетильной группы;
R1 представляет собой заместитель, представленный следующей общей формулой (2)
,
где R11, R12 и R13, одинаковые или различные, каждый представляет собой водород или С1-С4алкоксигруппу; и
WG1 представляет собой цианогруппу.
7. Нуклеозид, представленный следующей общей формулой (17)
,
где Вх представляет собой аденин, гуанин, цитозин, тимин или урацил, где аминогруппа аденина, гуанина и цитозина может быть необязательно защищена защитной группой, выбранной из ацетильной группы и феноксиацетильной группы;
А представляет собой кремнесодержащий заместитель, представленный следующей общей формулой (18а) или (18b)
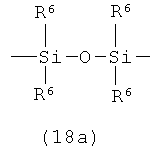 ,
,
где R6 представляет собой С1-С5алкильную группу и
WG1 представляет собой цианогруппу.
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| Кипятильник для воды | 1921 |
|
SU5A1 |
| Tadaaki Ohgi et al.//Org | |||
| Lett., 7(16), 3477-3480, 2005 | |||
| George A | |||
| Olah et al//The journal of organic chemistry | |||
| Способ изготовления звездочек для французской бороны-катка | 1922 |
|
SU46A1 |
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1923 |
|
SU571A1 |
| Домовый номерной фонарь, служащий одновременно для указания названия улицы и номера дома и для освещения прилежащего участка улицы | 1917 |
|
SU93A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| J.F.Normant et al//Tetrahedron Letters | |||
| Прибор для получения стереоскопических впечатлений от двух изображений различного масштаба | 1917 |
|
SU26A1 |
| US | |||

