Настоящая заявка претендует на приоритет, заявленный в предварительной заявке на патент США №60/713149, поданной 31 августа 2005 г., и в предварительной заявке на патент США №60/747762, поданной 19 мая 2006 г.
УРОВЕНЬ ТЕХНИКИ
Цизаприд является одним из представителей класса соединений, известных как производные бензамида, исходным соединением которого является метоклопрамид. Патентами США №4962115 и 5057525 (в совокупности «Van Daele», описание которых полностью включено в текст настоящей заявки в качестве ссылки) предлагаются N-(3-гидрокси-4-пипериденил)бензамиды цизаприда. В патенте Van Daele описано, что указанные соединения, их фармацевтически приемлемые кислотно-аддитивные соли и стереоизомерные формы стимулируют моторику желудочно-кишечного тракта.
Как класс, указанные производные бензамида обладают несколькими заметными фармакологическими эффектами. Заметные фармакологические эффекты производных бензамида обусловлены их воздействием на нейронные системы, которые регулируются нейромедиатором серотонином. Давно известно, что серотонин играет важную роль в ряде заболеваний, в связи с которыми изучены фармакологические свойства производных бензамида. Поэтому исследования были направлены на определение местонахождения областей выработки и накопления серотонина и серотониновых рецепторов в организме человека для того, чтобы установить связь между этими областями и различными болезненными состояниями.
В этом отношении было обнаружено, что главным местом выработки и накопления серотонина является энтерохромаффинная клетка слизистой оболочки желудочно-кишечного тракта. Также было выяснено, что серотонин является мощным стимулятором моторики желудочно-кишечного тракта, усиливающим работу гладких мышц, ускоряющим пассаж кишечного содержимого и сокращающим время всасывания, как при диарее. Указанный стимулирующий эффект также связан с тошнотой и рвотой.
Благодаря своему влиянию на серотониновую нейронную систему желудочно-кишечного тракта многие производные бензамида являются эффективными противорвотными средствами и широко используются для борьбы с рвотой при проведении химиотерапии и лучевой терапии злокачественных опухолей, особенно при применении обладающих сильным рвотным действием соединений, таких как цисплатин. Указанный эффект почти несомненно является результатом способности указанных соединений блокировать взаимодействие серотонина (5HT) со специфическими участками его действия, называемыми 5HT3-рецепторами, которые традиционно обозначаются в научной литературе, как серотониновые M-рецепторы. Химиотерапия и лучевая терапия может вызывать тошноту и рвоту вследствие высвобождения серотонина из поврежденных энтерохромаффинных клеток в желудочно-кишечном тракте. Высвободившийся нейромедиатор серотонин возбуждает как вагусные афферентные нервные волокна (вызывая, таким образом, рвотный рефлекс), так и серотониновые рецепторы в хеморецепторной триггерной зоне заднего поля головного мозга. Нерешенными остаются вопросы об анатомической области, в которой реализуется эффект производных бензамида, а также имеет ли этот эффект центральный (на центральную нервную систему - ЦНС), периферический или комбинированный характер (Barnes et al., J. Pharm. Pharmacol. 40: 586-588, 1988). Цизаприд, как и другие производные бензамида, может быть эффективным противорвотным средством благодаря способности модулировать взаимодействие серотонина с 5HT3-рецептором.
Второй заметный эффект производных бензамида заключается в усилении сократительной активности гладких мышц желудочно-кишечного тракта от пищевода до проксимальных отделов тонкого кишечника, за счет чего ускоряется пассаж содержимого через пищевод и кишечник, облегчается опорожнение желудка и повышается тонус нижнего пищеводного сфинктера (Decktor et al., Eur. J. Pharmacol. 147: 313-316, 1988). Хотя производные бензамида сами по себе не являются агонистами холинергических рецепторов, указанный выше эффект в отношении гладких мышц может блокироваться блокаторами мускариновых рецепторов, такими как атропин, или ингибиторами нейронной трансмиссии тетродотоксинового типа, воздействующими на натриевые каналы. Подобное блокирующее действие описано в отношении сократительного эффекта серотонина в тонком кишечнике. В настоящее время считается, что основные эффекты производных бензамида в отношении гладких мышц являются результатом их агонистического действия на новый класс серотониновых рецепторов, называемых 5HT4, которые расположены на вставочных нейронах межмышечного сплетения кишечной стенки. Возбуждение этих рецепторов способствует высвобождению ацетилхолина из парасимпатических нервных окончаний, расположенных приблизительно гладкомышечных волокон, и именно за счет взаимодействия ацетилхолина с его рецепторами, расположенными на мембранах гладкомышечных клеток, реализуется механизм запуска сокращения мышц.
Обсуждение различных 5HT-рецепторов, включая 5HT4-рецептор, можно найти, например, в патентах США №6331401 и 6632827, описания которых полностью включены в текст настоящей заявки в качестве ссылки.
Цизаприд, в основном, применяется для лечения гастроэзофагеальной рефлюксной болезни (ГЭРБ). Для этой болезни характерен заброс содержимого желудка в пищевод. Одним из наиболее важных факторов в патогенезе гастроэзофагеальной рефлюксной болезни является ослабление нижнего пищеводного сфинктера вследствие его недостаточности. Недостаточность нижнего пищеводного сфинктера может быть следствием низкого базального давления, расслабления сфинктера или же некомпенсированного повышения внутрижелудочного давления. Другие факторы патогенеза заболевания: задержка опорожнения желудка, недостаточная очистка пищевода из-за нарушения перистальтики, а также агрессивные свойства рефлюксного материала, который может повредить слизистую оболочку пищевода. Предполагается, что цизаприд укрепляет противорефлюксный барьер и улучшает очистку пищевода за счет повышения давления в нижнем пищеводном сфинктере и усиления перистальтических сокращений.
Поскольку цизаприд действует как прокинетический агент, он также может быть пригоден для лечения диспепсии, гастропареза, запоров, послеоперационного пареза кишечника и псевдонепроходимости кишечника. Диспепсия представляет собой состояние, характеризующееся нарушением пищеварения, которое может являться симптомом первичной дисфункции желудочно-кишечного тракта или осложнением других заболеваний, таких как аппендицит, нарушение функции желчного пузыря или нарушения питания. Гастропарез представляет собой паралич желудка, вызванный нарушением моторики желудка или являющийся осложнением таких заболеваний, как диабет, прогрессирующий системный склероз, нервная анорексия или миотоническая дистрофия. Запор представляет собой состояние, характеризующееся уреженной или затрудненной дефекацией в результате таких состояний, как недостаточность мышечного тонуса или спастичность кишечника. Послеоперационный парез кишечника представляет собой непроходимость кишечника вследствие нарушения мышечного тонуса после оперативного вмешательства. Псевдонепроходимость кишечника представляет собой состояние, характеризующееся запором, коликами и рвотой, но без признаков физической обструкции.
При лечении людей и животных большое значение имеет токсичность лекарств. Токсические побочные эффекты (неблагоприятные эффекты), возникающие вследствие применения лекарственных средств, включают в себя ряд состояний от невысокой лихорадки до смерти. Медикаментозное лечение оправдано только в том случае, если польза от применения схемы лечения превышает связанный с лечением потенциальный риск. Факторы, которые сопоставляет лечащий врач, включают в себя качественный и количественный эффект применяемого лекарственного средства, а также исход в случае, если лекарственное средство не вводится пациенту. Другие факторы, которые принимают во внимание, включают в себя физическое состояние пациента, стадию болезни и историю ее развития, а также все известные неблагоприятные эффекты, связанные с использованием лекарственного средства.
Элиминация лекарственного средства, как правило, является результатом метаболической активности в отношении лекарственного средства и его последующего выведения из организма. Метаболические процессы могут происходить в сосудистом русле и/или в клеточных органеллах или в органах. Печень является главным органом, в котором происходит метаболизм лекарственных средств. Различают синтетические и несинтетические реакции метаболического процесса. В ходе несинтетических реакций происходит химическое превращение лекарственного средства путем окисления, восстановления, гидролиза или любого сочетания упомянутых процессов. В совокупности указанные процессы называют реакциями фазы I.
В ходе реакций фазы II, известных также как синтетические реакции или реакции конъюгации, исходное лекарственное средство или его промежуточные метаболиты соединяются с эндогенными субстратами с образованием продукта присоединения или конъюгации. Метаболиты, образующиеся в ходе синтетических реакций, как правило, более полярны и лишены биологической активности. В результате указанные метаболиты легче выводятся через почки (с мочой) или печень (с желчью). Синтетические реакции включают в себя глюкуронидирование, конъюгацию с аминокислотами, ацетилирование, сульфоконъюгацию и метилирование.
Более 90% введенной дозы цизаприда метаболизируется путем окислительного N-дезалкилирования пиперидинового атома азота или путем ароматического гидроксилирования на 4-фторфенокси или бензамидных кольцах.
Было установлено, что введение цизаприда человеку вызывает тяжелые неблагоприятные эффекты, включая расстройства ЦНС, повышенное систолическое артериальное давление, взаимодействие с другими лекарственными средствами, диарею и спазмы в брюшной полости. Кроме того, отмечено, что внутривенное введение цизаприда сопровождается дополнительными неблагоприятными эффектами, которые не отмечаются после перорального введения цизаприда (Stacher et al. [1987] Digestive Diseases and Sciences 32(11):1223-1230). Считается, что указанные неблагоприятные эффекты вызываются метаболитами, которые образуются вследствие окислительного дезалкилирования или ароматического гидроксилирования соединения, которое происходит в детоксикационной системе цитохрома P450. Имеет место также ряд нежелательных взаимодействий между цизапридом и другими лекарственными средствами, которые также обусловлены метаболизмом в системе цитохрома P450.
В период с июля 1993 г. по декабрь 1999 г. зарегистрирован, по меньшей мере, 341 случай развития тяжелой аритмии сердца, связанной с приемом цизаприда (PROPULSID, Janssen Pharmaceutica Products, L.P.). Указанные аритмии включали в себя желудочковую тахикардию, фибрилляцию желудочков, трепетание-мерцание желудочков и удлинение интервала QT. Отмечено восемьдесят (80) смертельных случаев. В связи с указанными неблагоприятными эффектами производитель добровольно изъял продукт из свободной продажи в Соединенных Штатах; однако, лекарственное средство можно получить через исследовательскую программу с ограниченным доступом.
Безопасность агонистов 5HT4-рецепторов с прокинетической активностью в отношении желудочно-кишечного тракта (ЖКТ) ограничена в связи с их воздействием на сердце (удлинение интервалов QTc, тахикардия, трепетание-мерцание желудочков) и в связи с неблагоприятными лекарственными взаимодействиями, которые обусловлены метаболизмом препарата посредством печеночного цитохрома P-450. Прокинетический агент для ЖКТ данного класса, не имеющий указанных ограничений, был бы весьма ценным для ряда терапевтических областей, включая ГЭРБ и нарушения опорожнения желудка. Определенные производные цизарпида описаны в патентах США №6552046 и WO 01/093849 (описания которых полностью включены в текст данной заявки в качестве ссылки); однако, были бы желательны соединения с еще более выраженными полезными свойствами.
В настоящее время установлено, что некоторые стереоизомеры одного подобного этерифицированного структурного и/или функционального аналога цизаприда обладают явными и особо благоприятными свойствами.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к способам и процессам получения соединений и композиций формулы (Х), а также к промежуточным соединениям, пригодным для получения соединений формулы (Х), для безопасного и эффективного лечения различных желудочно-кишечных расстройств, включая, без ограничения, гастропарез, гастроэзофагеальный рефлюкс и связанные с ними состояния. Соединения по настоящему изобретению также являются пригодными для лечения ряда состояний, затрагивающих центральную нервную систему.
Соединения по настоящему изобретению включают в себя соединения формулы X:
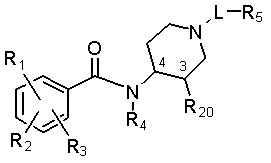 (X)
(X)
и их фармацевтически приемлемые соли, где
связи в положениях 3 и 4 находятся в цис-положении относительно друг друга;
L представляет собой -(C1-C6алкил)- (в одном варианте, -(C3-C5алкил)-), -(C1-C6алкил)-C(O)- или -C(O)-(C1-C6алкил)-, где каждая из алкильных групп необязательно замещена 1 или 2 группами, которые независимо представляют собой галоген, C1-C4алкокси или OH, и где один атом углерода в алкильной части группы L может быть заменен на -N(R9)-;
R1 представляет собой галоген;
R2 представляет собой амино, NH(C1-C4алкил) или N(C1-C4алкил)(C1-C4алкил);
R3 представляет собой OH или C1-C4алкокси;
R4 представляет собой H или метил; и
R5 представляет собой -O-C3-C8 циклоалкил, -О-гетероциклоалкил, гетероциклоалкил, арил, -О-арил, - N(R9)-(C0-C6алкил)-C(O)-арил или -N(R9)-C0-C6алкил-арил, -О-гетероарил, -N(R9)-C1-C6(O)-гетероарил или -N(R9)-C0-C6алкилгетероарил, где каждая из циклических групп может быть незамещенной или замещенной в одном более замещаемых положений следующими заместителями: C1-C6алкил, C1-C6алкокси, галоген, C1-C6галогеналкил, C1-C6галогеналкокси, гидроксил, гидрокси-C1-C4-алкил, амино, -NH(C1-C6алкил), -N(C1-C6алкил)(C1-C6алкил), -(C0-C6алкил)-C(O)R11 или -O-(C0-C6алкил)-C(O)R11, метилсульфон, C0-C6-сульфонамид или NO2; где
R9 в каждом случае независимо представляет собой H или C1-C4алкил;
R11 представляет собой C1-C6алкил, OH или
R11 представляет собой C1-C6алкокси, необязательно замещенную 1 или 2 группами, которые независимо представляют собой C1-C4алкокси, амино, -NH(C1-C6алкил), -N(C1-C6алкил)(C1-C6алкил), -(C0-C6алкил)-C(O)N(R9)-гетероциклоалкил, -О-гетероциклоалкил, -C1-C6(O)N(R9)-гетероарил или гетероарил, где
гетероциклоалкильные группы необязательно замещены 1, 2 или 3 группами, которые независимо представляют собой галоген, C1-C6алкил, C1-C6алкокси, гидрокси, гидрокси C1-C6алкил, C1-C6алкоксикарбонил, -CO2H, CF3 или OCF3,
гетероарильная группа необязательно замещена 1, 2 или 3 группами, которые независимо представляют собой галоген, C1-C6алкил, C1-C6алкокси, гидрокси, гидрокси C1-C6алкил, C1-C6алкоксикарбонил, -CO2H, CF3 или OCF3; или
R11 представляет собой -О-гетероциклоалкил, где гетероциклоалкил необязательно замещен 1, 2 или 3 группами, которые независимо представляют собой галоген, C1-C6алкил, C1-C6алкокси, гидрокси, гидрокси C1-C6алкил, C1-C6алкоксикарбонил, -CO2H, CF3 или OCF3; и
R20 представляет собой C1-C6алкокси (предпочтительно C1-C4алкокси, более предпочтительно метокси) или OH.
Изобретение относится также к композициям, включающим по меньшей мере одно соединение формулы (X), полученное с использованием способов и/или процессов по настоящему изобретению, и по меньшей мере один фармацевтически приемлемый наполнитель, адъювант, носитель или растворитель.
Соединения формулы (X), которые получены с использованием способов и/или процессов по настоящему изобретению, являются пригодными для лечения или профилактики гастроэзофагеальной рефлюксной болезни и значительно уменьшают неблагоприятные эффекты, связанные с введением цизаприда. Указанные неблагоприятные эффекты включают, без ограничения, диарею, спазмы в брюшной полости и повышение артериального давления и частоты сердечных сокращений.
Кроме того, соединения и композиции, полученные с использованием способов и/или процессов по настоящему изобретению, являются пригодными для лечения рвоты и других состояний, включая, без ограничения, диспепсию, гастропарез, запор, послеоперационный парез кишечника и псевдонепроходимость кишечника. Дополнительным преимуществом является то, что при указанных способах лечения неблагоприятные эффекты, связанные с введением цизаприда, также уменьшаются.
Соединения, полученные с использованием способов и/или процессов по настоящему изобретению, представляют собой лиганды 5HT4-рецепторов и, соответственно, могут использоваться для лечения состояний, опосредованных указанными рецепторами. Указанные рецепторы расположены в нескольких областях центральной нервной системы, и модуляцию указанных рецепторов можно использовать для достижения желательных воздействий на ЦНС.
Преимущество изобретения заключается в том, что соединения, полученные с использованием способов и/или процессов по настоящему изобретению для получения стереоизомерных соединений, обычно содержат сложноэфирную часть, которая не уменьшает способность указанных соединений оказывать терапевтическое воздействие, но которая делает их более чувствительными к расщеплению сывороточными и/или цитозольными эстеразами, в то же время позволяя избежать воздействия детоксикационной системы цитохрома P450, которая ассоциируется с неблагоприятными эффектами цизаприда, и снижая частоту возникновения указанных неблагоприятных эффектов.
Настоящее изобретение относится также к способам лечения, включающим в себя введение соединений формулы (X), полученных с использованием способов и/или процессов по настоящему изобретению, в терапевтически эффективных количествах индивидуумам, которые нуждаются в лечении гастроэзофагеальной рефлюксной болезни, диспепсии, гастропареза, запора, послеоперационного пареза кишечника и псевдонепроходимости кишечника, а также связанных с ними состояний.
Преимущество изобретения заключается в том, что терапевтические соединения, полученные с использованием способов и/или процессов по настоящему изобретению, являются стабильными при хранении и предусматривают более безопасный метаболизм по сравнению с другими лекарственными средствами; таким образом, соединения по настоящему изобретению можно использовать с меньшей частотой побочных эффектов и меньшей токсичностью.
В дополнительном аспекте настоящее изобретение относится к продуктам распада (предпочтительно к продуктам метаболического распада), которые образуются при воздействии эстераз на терапевтические соединения, полученные с использованием способов и/или процессов по настоящему изобретению. Указанные продукты распада могут применяться, как описывается в настоящем документе, для наблюдения за выведением терапевтических соединений из организма пациента.
КРАТКОЕ ОПИСАНИЕ ФИГУР
Фигура 1 представляет собой график, на котором показаны кривые зависимости эффекта от концентрации в отношении агонистического воздействия на 5-HT4-рецепторы ATI-7505, серотонина, цизаприда и ATI-7500.
Фигура 2 представляет собой график, на котором отображено опорожнение желудка у накормленных собак. Представленные данные приведены к усредненному времени возвращения ММК к исходному уровню для носителя, являющегося контролем. Величины представляют собой «среднее+SEM стандартная ошибка среднего» для 5 собак. *p<0,05 по сравнению с носителем, являющимся контролем.
Фигура 3 представляет собой график, на котором показан метаболизм ATI-7505 и ATI-7500 с CYP450-зависимым кофактором, NADPH и без такового. Показаны средние величины и стандартное отклонение концентраций ATI-7505 и ATI-7500 в мкM. ATI-7505 (2 мкM) инкубировали с человеческим микросомальным белком (1 мг) в присутствии и в отсутствии регенерирующей системы NADPH (кофактора).
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В дополнительном аспекте изобретение относится к способам и/или процессам получения соединений формулы (Х), а также к промежуточным соединениям, пригодным для получения соединений формулы (Х), где
R5 представляет собой -O-C3-C8циклоалкил, -О-гетероциклоалкил, гетероциклоалкил, где гетероциклоалкильная группа выбрана из пиперидинила, пиперазинила, пирролидинила, азабициклооктила, в некоторых вариантах азабицикло[2.2.2]октила, азабицикло[3.2.1]октила, азабициклононила, азабициклодецила, индолинила, морфолинила, тиоморфолинила, S,S-диоксотиоморфолинила и имидазолидинила, -О-арил, -N(R9)-C(O)-арил или -N(R9)-C0-C6алкиларил, где каждая из циклических групп является незамещенной или замещенной в одном или более замещаемых положений C1-C6алкилом, C1-C6алкокси, галогеном, C1-C6галогеналкилом, C1-C6галогеналкокси, гидроксилом, гидрокси-C1-C4алкилом, амино, -NH(C1-C6алкилом), -N(C1-C6алкилом)(C1-C6алкилом), -C(O)R11 или NO2; где
R9 в каждом случае независимо представляет собой H или C1-C4алкил; и
R11 представляет собой C1-C6алкил, OH, или
R11 представляет собой C1-C6алкокси, необязательно замещенный 1 или 2 группами, которые независимо представляют собой C1-C4алкокси, амино, -NH(C1-C6алкил), -N(C1-C6алкил)(C1-C6алкил), -C(O)N(R9)-гетероциклоалкил, гетероциклоалкил или гетероарил, где
гетероциклоалкильная группа выбрана из пирролидинила, пиперидинила, пиперазинила, морфолинила, азабициклооктила, в некоторых вариантах азабицикло[2.2.2]октила, азабицикло[3.2.1]октила, азабициклононила и азабициклодецила, где гетероциклоалкильные группы необязательно замещены 1, 2 или 3 группами, которые независимо представляют собой галоген, C1-C6алкил, C1-C6алкокси, гидрокси, гидрокси C1-C6алкил, C1-C6алкоксикарбонил, -CO2H, CF3 или OCF3,
гетероарильная группа выбрана из пиридила, пиримидила, хинолинила, изохинолинила и индолила, где гетероарильные группы необязательно замещены 1, 2 или 3 группами, которые независимо представляют собой галоген, C1-C6алкил, C1-C6алкокси, гидрокси, гидрокси C1-C6алкил, C1-C6алкоксикарбонил, -CO2H, CF3 или OCF3; или
R11 представляет собой -О-гетероциклоалкил, где гетероциклоалкил выбран из пиперидинила, пирролидинила, имидазолидинила, морфолинила, азабициклооктила, в некоторых вариантах азабицикло[2.2.2]октила, азабицикло[3.2.1]октила, азабициклононила, азабициклодецила и тетрагидрофуранила, и где каждая гетероциклоалкильная группа необязательно замещена 1, 2 или 3 группами, которые независимо представляют собой галоген, C1-C6алкил, C1-C6алкокси, гидрокси, гидрокси C1-C6алкил, C1-C6алкоксикарбонил, -CO2H, CF3 или OCF3.
В дополнительном аспекте настоящее изобретение относится к способам и/или процессам получения соединений формулы (Х), а также к промежуточным соединениям, пригодным для получения соединений формулы (Х), где R1 представляет собой хлор.
Еще в одном дополнительном аспекте настоящее изобретение относится к способам и/или процессам получения соединений формулы (Х), а также к промежуточным соединениям, пригодным для получения соединений формулы (Х), где R2 представляет собой амино.
В дополнительном аспекте настоящее изобретение относится к способам и/или процессам получения соединений формулы (Х), а также к промежуточным соединениям, пригодным для получения соединений формулы (Х), где R3 представляет собой метокси.
В еще одном дополнительном аспекте настоящее изобретение относится к способам и/или процессам получения соединений формулы (Х), а также к промежуточным соединениям, пригодным для получения соединений формулы (Х), где R4 представляет собой Н или метил.
В дополнительном аспекте настоящее изобретение относится к способам и/или процессам получения соединений формулы (Х), а также к промежуточным соединениям, пригодным для получения соединений формулы (Х), где R1 представляет собой хлор, R2 представляет собой амино, R3 представляет собой метокси и R4 представляет собой Н или метил.
Еще одна особенность настоящего изобретения относится к способам и/или процессам получения соединений формулы (Х), а также к промежуточным соединениям, пригодным для получения соединений формулы (Х), где R1 представляет собой хлор, R2 представляет собой амино, R3 представляет собой метокси; R4 представляет собой Н и L представляет собой -(С4-С6алкил)-С(О)-.
Еще одна особенность настоящего изобретения относится к способам и/или процессам получения соединений формулы (Х), а также к промежуточным соединениям, пригодным для получения соединений формулы (Х), где две и более описанных ранее особенностей объединены.
Еще одна особенность настоящего изобретения относится к способам и/или процессам получения соединений формулы (ХI), которые представляют собой соединения формулы (Х), где L представляет собой -(СН2)5-С(О)-:
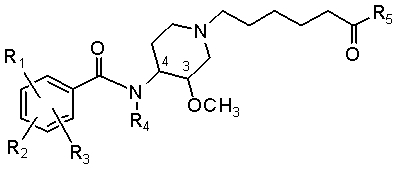 (XI).
(XI).
Еще одна особенность настоящего изобретения относится к способам и/или процессам получения соединений формулы (ХI), а также к промежуточным соединениям, пригодным для получения соединений формулы (ХI), где R1 представляет собой хлор, R2 представляет собой амино, R3 представляет собой метокси и R4 представляет собой Н или метил.
Еще одна особенность настоящего изобретения относится к способам и/или процессам получения соединений формулы (ХI), а также к промежуточным соединениям, пригодным для получения соединений формулы (ХI), где R5 представляет собой -O-гетероциклоалкил, где гетероциклоалкильная группа выбрана из азабициклооктила, в некоторых вариантах 1-азабицикло[2.2.2]окт-3-ила или 8-азабицикло[3.2.1]окт-3-ила, азабициклононила, азабициклодецила, где азот аза необязательно замещен метилом или этилом; и R4 представляет собой H или метил.
В дополнительном аспекте настоящее изобретение относится к способам и/или процессам получения соединений формулы (ХI), а также к промежуточным соединениям, пригодным для получения соединений формулы (ХI), где R5 представляет собой -O-гетероциклоалкил, где гетероциклоалкильная группа выбрана из пиперидинила, пиперазинила или пирролидинила, каждый из которых является незамещенным или замещенным в одном или двух положениях группами, которые независимо представляют собой C1-C4алкил, C1-C4алкокси, галоген, C1-C4галогеналкил (в одном варианте, CF3), C1-C4галогеналкокси (в одном варианте OCF3), гидроксил, гидрокси C1-C4алкил, амино, -NH(C1-C4алкил), -N(C1-C4алкил)(C1-C4алкил), -(C1-C6алкил)-C(O)R11 или NO2; и R4 представляет собой H или метил.
В дополнительном аспекте настоящее изобретение относится к способам и/или процессам получения соединений формулы (ХI), а также к промежуточным соединениям, пригодным для получения соединений формулы (ХI), где R5 представляет собой -O-гетероциклоалкил, где гетероциклоалкильная группа выбрана из индолинила, морфолинила, тиоморфолинила, S,S-диоксотиоморфолинила и имидазолинила, каждый из которых является незамещенным или замещенным в одном или двух положениях группами, которые независимо представляют собой C1-C4алкил, C1-C4алкокси, галоген, C1-C4галогеналкил (в одном варианте, CF3), C1-C4галогеналкокси (в одном варианте OCF3), гидроксил, гидрокси C1-C4алкил, амино, -NH(C1-C4алкил), -N(C1-C4алкил)(C1-C4алкил), -(C0-C6алкил)-C(O)R11 или NO2; и R4 представляет собой H или метил.
В дополнительном аспекте настоящее изобретение относится к способам и/или процессам получения соединений формулы (ХI), а также к промежуточным соединениям, пригодным для получения соединений формулы (ХI), где R5 представляет собой О-фенил, N(R9)-(C0-C6алкил)-C(O)-фенил или -N(R9)-C0-C4алкил-фенил, где фенильная группа замещена одной или двумя группами, которые независимо представляют собой C1-C4алкил, C1-C4алкокси, галоген, C1-C4галогеналкил (в одном варианте, CF3), C1-C4галогеналкокси (в одном варианте OCF3), гидроксил, гидрокси C1-C4алкил, амино, -NH(C1-C4алкил), -N(C1-C4алкил)(C1-C4алкил), -(C0-C6алкил)-C(O)R11 или NO2; и R4 и R9 независимо представляют собой H или метил.
В дополнительном аспекте настоящее изобретение относится к способам и/или процессам получения соединений формулы (ХI), а также к промежуточным соединениям, пригодным для получения соединений формулы (ХI), где R4 представляет собой Н.
В дополнительном аспекте настоящее изобретение относится к способам и/или процессам получения соединений формулы (ХI), а также к промежуточным соединениям, пригодным для получения соединений формулы (Х), где R11 представляет собой C1-C6алкокси, необязательно замещенный 1 или 2 группами, которые независимо представляют собой C1-C4алкокси, амино, -NH(C1-C6алкил), -N(C1-C6алкил)(C1-C6алкил), -(C0-C6алкил)-C(O)N(R9)-гетероциклоалкил или гетероциклоалкил, гетероциклоалкильная группа выбрана из пирролидинила, пиперидинила, пиперазинила и морфолинила, где гетероциклоалкильные группы необязательно замещены 1, 2 или 3 группами, которые независимо представляют собой галоген, C1-C6алкил, C1-C6алкокси, гидрокси, гидрокси C1-C6алкил, C1-C6алкоксикарбонил, -CO2H, CF3 или OCF3.
В дополнительном аспекте настоящее изобретение относится к способам и/или процессам получения соединений формулы (ХI), а также к промежуточным соединениям, пригодным для получения соединений формулы (Х), где две и более описанных ранее особенностей объединены.
В дополнительном аспекте настоящее изобретение относится к способам и/или процессам получения соединений формулы (ХII), т.е. соединений формулы (Х) следующей формулы, а также к промежуточным соединениям, пригодным для получения соединений формулы (ХII):
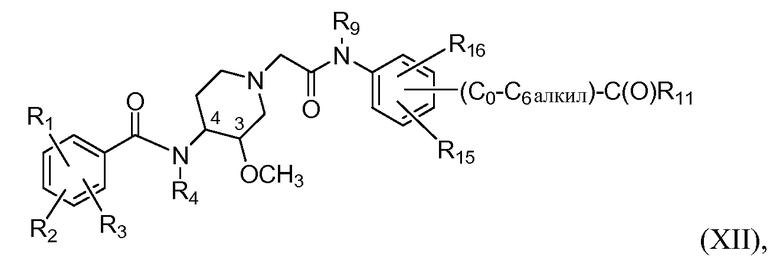
где R15 представляет собой H, C1-C6алкил, C1-C6алкокси, галоген, C1-C6галогеналкил (в одном варианте CF3), C1-C6галогеналкокси (в одном варианте OCF3), гидроксил, гидрокси C1-C4алкил, амино, -NH(C1-C6алкил), -N(C1-C6алкил)(C1-C6алкил), метилсульфон, C0-C6сульфонамид или NO2, и R16 представляет собой H или -O-(C0-C6алкил)-C(O)R11. В другом варианте R15 представляет собой H.
В еще дополнительном аспекте настоящее изобретение относится к способам и/или процессам получения соединений формулы (ХII), а также к промежуточным соединениям, пригодным для получения соединений формулы (ХII), где R4 и R9 независимо представляют собой H или метил, а R11 представляет собой OH.
В дополнительном аспекте настоящее изобретение относится к способам и/или процессам получения соединений формулы (ХII), а также к промежуточным соединениям, пригодным для получения соединений формулы (ХII), где R4 и R9 независимо представляют собой H или метил, и R11 представляет собой C1-C6алкокси, необязательно замещенный 1 или 2 группами, которые независимо представляют собой C1-C4алкокси, амино, -NH(C1-C6алкил), -N(C1-C6алкил)(C1-C6алкил), -(C0-C6алкил)-C(O)N(R9)-гетероциклоалкил или гетероциклоалкил группой, где гетероциклоалкильная группа выбрана из азабициклооктила, в некоторых вариантах 1-азабицикло[2.2.2]окт-3-ила или 8-азабицикло[3.2.1]окт-3-ила, азабициклононила, азабициклодецила, в которых азот аза необязательно замещен метилом или этилом, пирролидинила, пиперидинила, пиперазинила и морфолинила, где гетероциклоалкильные группы необязательно замещены 1, 2 или 3 группами, которые независимо представляют собой галоген, C1-C6алкил, C1-C6алкокси, гидрокси, гидрокси C1-C6алкил, C1-C6алкоксикарбонил, -CO2H, CF3 или OCF3, и R4 а R9 независимо представляют собой H или метил. В другом варианте R4, R9 и R11 определены выше, а R15 представляет собой H, R1 собой представляет хлор; R2 представляет собой амино; и R3 представляет собой метокси.
В еще дополнительном аспекте настоящее изобретение относится к способам и/или процессам получения соединений формулы (ХII), а также к промежуточным соединениям, пригодным для получения соединений формулы (ХII), где R4 и R9 независимо представляют собой H или метил, а R11 представляет собой C1-C6алкокси, необязательно замещенный 1 или 2 группами, которые независимо представляют собой C1-C4алкокси, амино, -NH(C1-C6алкил), -N(C1-C6алкил)(C1-C6алкил) или гетероарил, где гетероарильная группа выбрана из пиридила, пиримидила, хинолинила, изохинолинила и индолила, где гетероарильные группы необязательно замещены 1, 2 или 3 группами, которые независимо представляют собой галоген, C1-C6алкил, C1-C6алкокси, гидрокси, гидрокси C1-C6алкил, C1-C6алкоксикарбонил, -CO2H, CF3 или OCF3; и R4 и R9 независимо представляют собой H или метил. В другом варианте R4, R9 и R11 определены выше, R15 представляет собой H, R1 представляет собой хлор; R2 представляет собой амино; и R3 представляет собой метокси.
В дополнительном аспекте настоящее изобретение относится к способам и/или процессам получения соединений формулы (ХII), а также к промежуточным соединениям, пригодным для получения соединений формулы (ХII), где по меньшей мере один из R4 и R9 представляет собой H.
В дополнительном аспекте настоящее изобретение относится к способам и/или процессам получения соединений формулы (ХII), а также к промежуточным соединениям, пригодным для получения соединений формулы (ХII), где две или более из описанных ранее особенностей объединены.
В дополнительном аспекте настоящее изобретение относится к способам и/или процессам получения соединений формулы (ХIII), т.е. соединений формулы (Х) следующей формулы, а также к промежуточным соединениям, пригодным для получения соединений формулы (ХIII):
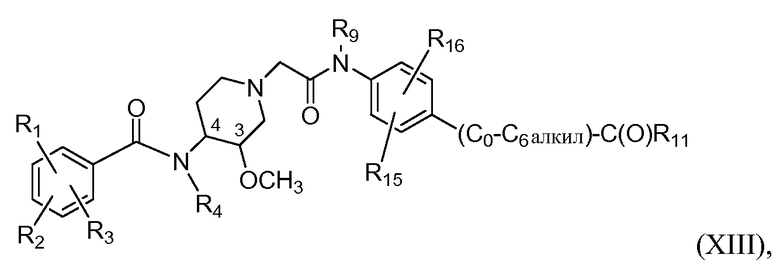
где R15 представляет собой H, C1-C6алкил, C1-C6алкокси, галоген, C1-C6галогеналкил (в одном варианте CF3), C1-C6галогеналкокси (в одном варианте OCF3), гидроксил, гидрокси C1-C4алкил, амино, -NH(C1-C6алкил), -N(C1-C6алкил)(C1-C6алкил) или метилсульфон, C0-C6-сульфонамид, NO2, и R16 представляет собой H или -O-(C0-C6алкил)-C(O)R11. В другом варианте R15 представляет собой H.
В дополнительном аспекте настоящее изобретение относится к способам и/или процессам получения соединений формулы (ХIII), а также к промежуточным соединениям, пригодным для получения соединений формулы (ХIII), где
R4 и R9 независимо представляют собой H или метил, а R11 представляет собой OH, С1-С4алкокси (в другом варианте С1-С3алкокси) или С1-С2алкокси-С1-С3алкокси. В другом варианте R4, R9 и R11 определены выше, и R1 представляет собой хлор; R2 представляет собой амино и R3 представляет собой метокси.
В еще дополнительном аспекте настоящее изобретение относится к способам и/или процессам получения соединений формулы (ХIII), а также к промежуточным соединениям, пригодным для получения соединений формулы (ХIII), где R4 и R9 независимо представляют собой H или метил, и R11 представляет C1-C4алкокси, замещенный амино, -NH(C1-C6алкилом), -N(C1-C6алкил)(C1-C6алкилом), азабициклооктилом, в некоторых вариантах 1-азабицикло[2.2.2]окт-3-илом или 8-азабицикло[3.2.1]окт-3-илом, азабициклононилом, азабициклодецилом, где азот аза необязательно замещен метилом или этилом; и R4 представляет собой H или метил, пирролидинил, пиперидинил, морфолинил, пиридил или -(C0-C6алкил)-C(O)NH-пирид-4-ил. В другом варианте R4, R9 и R11 определены выше, а R1 представляет собой хлор; R2 представляет собой амино; и R3 представляет собой метокси.
В дополнительном аспекте настоящее изобретение относится к способам и/или процессам получения соединений формулы (ХIII), а также к промежуточным соединениям, пригодным для получения соединений формулы (ХIII), где R4 и R9 независимо представляют собой H или метил, и R11 представляет C1-C4алкокси, замещенный амино, -NH(C1-C6алкилом) или -N(C1-C6алкил)(C1-C6алкилом). В другом варианте R4, R9 и R11 определены выше, а R1 представляет собой хлор; R2 представляет собой амино; и R3 представляет собой метокси.
В дополнительном аспекте настоящее изобретение относится к способам и/или процессам получения соединений формулы (ХIII), а также к промежуточным соединениям, пригодным для получения соединений формулы (ХIII), где R4 и R9 независимо представляют собой H или метил, и R11 представляет C1-C4алкокси, замещенный пирролидинилом, пиперидинилом, морфолинилом, пиридилом или -(C0-C6алкил)-C(O)NH-пирид-4-илом. В другом варианте R4, R9 и R11 определены выше, а R1 представляет собой хлор; R2 представляет собой амино; R3 представляет собой метокси.
В дополнительном аспекте настоящее изобретение относится к способам и/или процессам получения соединений формулы (ХIII), а также к промежуточным соединениям, пригодным для получения соединений формулы (ХIII), где по меньшей мере один из R4 и R9 представляет собой H.
В дополнительном аспекте настоящее изобретение относится к способам и/или процессам получения соединений формулы (ХIII), а также к промежуточным соединениям, пригодным для получения соединений формулы (ХIII), где две или более из описанных ранее особенностей объединены.
В дополнительном аспекте настоящее изобретение относится к способам и/или процессам получения соединений формулы (ХIV), т.е. соединений формулы (Х) следующей формулы, а также к промежуточным соединениям, пригодным для получения соединений формулы (ХIV):
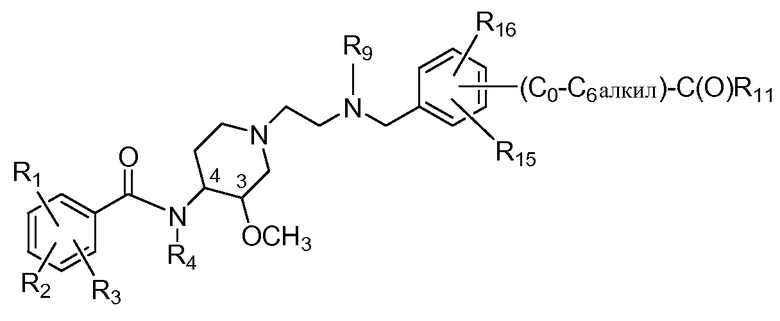 (XIV)
(XIV)
где R15 представляет собой H, C1-C6алкил, C1-C6алкокси, галоген, C1-C6галогеналкил (в одном аспекте CF3), C1-C6галогеналкокси (в одном аспекте OCF3), гидроксил, гидроксиC1-C4алкил, амино, -NH(C1-C6алкил), -N(C1-C6алкил)(C1-C6алкил), метилсульфон, C0-C6сульфонамид или NO2, а R16 представляет собой H или -O-(C0-C6алкил)-C(O)R11. В другом аспекте R15 представляет собой H.
В дополнительном аспекте настоящее изобретение относится к способам и/или процессам получения соединений формулы (ХIV), а также к промежуточным соединениям, пригодным для получения соединений формулы (ХIV), где R4 и R9 независимо представляют собой H или метил, а R11 представляет собой OH, C1-C4алкокси (в другом варианте C1-C3алкокси) или C1-C2алкокси-C1-C3алкокси-. В другом варианте R4, R9 и R11 определены выше, а R1 представляет собой хлор; R2 представляет собой амино; и R3 представляет собой метокси. В еще одном варианте по меньшей мере один из R4 и R9 представляет собой H.
В еще одном аспекте настоящее изобретение относится к способам и/или процессам получения соединений формулы (ХIV), а также к промежуточным соединениям, пригодным для получения соединений формулы (ХIV), где R4 и R9 независимо представляют собой H или метил, а R11 представляет C1-C4алкокси, замещенный амино, -NH(C1-C6алкилом), -N(C1-C6алкил)(C1-C6алкилом), азабициклооктилом, в некоторых вариантах 1-азабицикло[2.2.2]окт-3-илом или 8-азабицикло[3.2.1]окт-3-илом, азабициклононилом, азабициклодецилом, где азот аза необязательно замещен метилом или этилом; и R4 представляет собой H или метил, пирролидинил, пиперидинил, морфолинил, пиридил или -(C0-C6алкил)-C(O)NH-пирид-4-ил. В другом варианте R4, R9 и R11 определены выше, а R1 представляет собой хлор; R2 представляет собой амино; и R3 представляет собой метокси.
В еще дополнительном аспекте настоящее изобретение относится к способам и/или процессам получения соединений формулы (ХIV), а также к промежуточным соединениям, пригодным для получения соединений формулы (ХIV), где R4 и R9 независимо представляют собой H или метил, а R11 представляет C1-C4алкокси, замещенный амино, -NH(C1-C6алкилом) или -N(C1-C6алкил)(C1-C6алкилом). В другом варианте R4, R9 и R11 определены выше, а R1 представляет собой хлор; R2 представляет собой амино; и R3 представляет собой метокси.
В дополнительном аспекте настоящее изобретение относится к способам и/или процессам получения соединений формулы (ХIV), а также к промежуточным соединениям, пригодным для получения соединений формулы (ХIV), где R4 и R9 независимо представляют собой H или метил, а R11 представляет C1-C4алкокси, замещенный пирролидинилом, пиперидинилом, морфолинилом, пиридилом или -(C0-C6алкил)-C(O)NH-пирид-4-илом. В другом варианте R4, R9 и R11 определены выше, а R1 представляет собой хлор; R2 представляет собой амино; и R3 представляет собой метокси.
В дополнительном аспекте настоящее изобретение относится к способам и/или процессам получения соединений формулы (ХIV), а также к промежуточным соединениям, пригодным для получения соединений формулы (ХIV), где по меньшей мере один из R4 и R9 представляет собой H.
В другом аспекте настоящее изобретение относится к способам и/или процессам получения соединений формулы (ХIV), а также к промежуточным соединениям, пригодным для получения соединений формулы (ХIV), где две или более из описанных ранее особенностей объединены.
Еще одна особенность настоящего изобретения относится к способам и/или процессам получения соединений формулы (ХV), т.е. соединений формулы (Х) следующей формулы, а также к промежуточным соединениям, пригодным для получения соединений формулы (ХV):
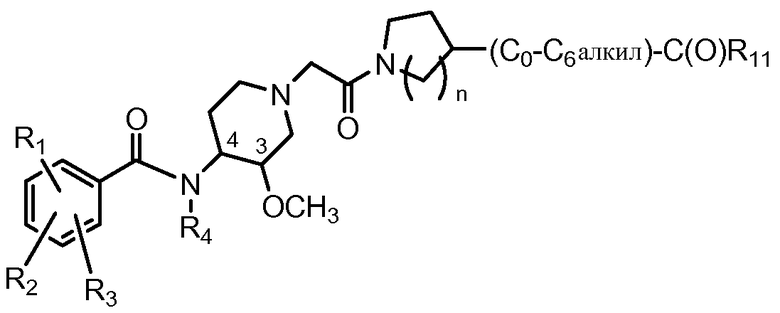 (XV)
(XV)
где n равно 1 или 2.
В дополнительном аспекте настоящее изобретение относится к способам и/или процессам получения соединений формулы (ХV), а также к промежуточным соединениям, пригодным для получения соединений формулы (ХV), где R4 представляет собой H или метил, а R11 представляет собой OH, C1-C4алкокси (в другом варианте C1-C3алкокси) или C1-C2алкокси-C1-C3алкокси-. В другом варианте R4 и R11 определены выше, а R1 представляет собой хлор; R2 представляет собой амино; и R3 представляет собой метокси. В другом варианте изобретения по меньшей мере один из R4 и R9 представляет собой H.
В еще дополнительном аспекте настоящее изобретение относится к способам и/или процессам получения соединений формулы (ХV), а также к промежуточным соединениям, пригодным для получения соединений формулы (ХV), где R4 и R9 независимо представляют собой H или метил, а R11 представляет C1-C4алкокси, замещенный амино, -NH(C1-C6алкилом), -N(C1-C6алкил)(C1-C6алкилом), азабициклооктилом, в некоторых вариантах 1-азабицикло[2.2.2]окт-3-илом или 8-азабицикло[3.2.1]окт-3-илом, азабициклононилом, азабициклодецилом, где азот аза необязательно замещен метилом или этилом; а R4 представляет собой H или метил, пирролидинил, пиперидинил, морфолинил, пиридил или C(O)NH-пирид-4-ил. В другом варианте R4, R9 и R11 определены выше, а R1 представляет собой хлор; R2 представляет собой амино; и R3 представляет собой метокси.
В дополнительном аспекте настоящее изобретение относится к способам и/или процессам получения соединений формулы (ХV), а также к промежуточным соединениям, пригодным для получения соединений формулы (ХV), где R4 и R9 независимо представляют собой H или метил, а R11 представляет C1-C4алкокси, замещенный амино, -NH(C1-C6алкилом) или -N(C1-C6алкил)(C1-C6алкилом). В другом варианте R4, R9 и R11 определены выше, а R1 представляет собой хлор; R2 представляет собой амино; и R3 представляет собой метокси.
В еще дополнительном аспекте настоящее изобретение относится к способам и/или процессам получения соединений формулы (ХV), а также к промежуточным соединениям, пригодным для получения соединений формулы (ХV), где R4 представляет собой H или метил, а R11 представляет C1-C4алкокси, замещенный азабициклооктилом, в некоторых вариантах 1-азабицикло[2.2.2]окт-3-илом или 8-азабицикло[3.2.1]окт-3-илом, азабициклононилом, азабициклодецилом, где азот аза необязательно замещен метилом или этилом; а R4 представляет собой H или метил, пирролидинил, пиперидинил, морфолинил, пиридил или -(C0-C6алкил)-C(O)NH-пирид-4-ил. В другом варианте R4, R9 и R11 определены выше, а R1 представляет собой хлор; R2 представляет собой амино; и R3 представляет собой метокси.
В другом аспекте настоящее изобретение относится к способам и/или процессам получения соединений формулы (ХV), а также к промежуточным соединениям, пригодным для получения соединений формулы (ХV), где две и более описанных ранее особенностей объединены.
В другом аспекте настоящее изобретение относится к способам и/или процессам получения соединений, а также к промежуточным соединениям, пригодным для получения соединений любой из формул (X), (XI), (XII), (XIII), (XIV) или (XV), где R1 R2 и R3 ориентированы в фенильном кольце следующим образом:
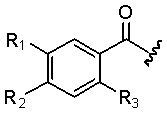 .
.
В другом аспекте настоящее изобретение относится к способам и/или процессам получения соединений, а также к промежуточным соединениям, пригодным для получения соединений любой из формул (X), (XI), (XII), (XIII), (XIV) или (XV), где связь 3 имеет «S»-конфигурацию, а связь 4 имеет «R»-конфигурацию.
В другом дополнительном аспекте настоящее изобретение относится к способам и/или процессам получения соединений, а также к промежуточным соединениям, пригодным для получения соединений любой из формул (X), (XI), (XII), (XIII), (XIV) или (XV), где R1, R2 и R3 ориентированы в фенильном кольце следующим образом:
,
и связь 3 имеет «S»-конфигурацию, а связь 4 имеет «R»-конфигурацию.
В другом аспекте настоящее изобретение относится к способам и/или процессам получения соединений, а также к промежуточным соединениям, пригодным для получения соединений любой из формул (X), (XI), (XII), (XIII), (XIV) или (XV), где связь 3 имеет «R»-конфигурацию, а связь 4 имеет «S»-конфигурацию.
В другом аспекте настоящее изобретение относится к способам и/или процессам получения соединений, а также к промежуточным соединениям, пригодным для получения соединений любой из формул (X), (XI), (XII), (XIII), (XIV) или (XV), где R1, R2 и R3 ориентированы в фенильном кольце следующим образом:
,
и связь 3 имеет «R»-конфигурацию, а связь 4 имеет «S»-конфигурацию.
В еще другом аспекте настоящее изобретение относится к способам и/или процессам получения соединений, а также к промежуточным соединениям, пригодным для получения соединений формулы (X), где R1 представляет собой хлор; R2 представляет собой амино; R3 представляет собой метокси; R4 представляет собой H, а R1, R2 и R3 ориентированы в фенильном кольце следующим образом:
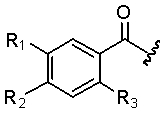 ,
,
и
L представляет собой -(C3-C5алкил)-, где один атом углерода может быть заменен на -N(R9)- или -(C2-C6алкил)-C(O)-. В другом аспекте R1, R2 и R3 определены выше и ориентированы в фенильном кольце так, как описано выше, R4 определен выше, R5 представляет собой O-гетероциклоалкил, где гетероциклоалкильная группа выбрана из азабициклооктила, в некоторых вариантах 1-азабицикло[2.2.2]окт-3-ила или 8-азабицикло[3.2.1]окт-3-ила, азабициклононила, азабициклодецила, где аза азот необязательно замещен метилом или этилом, пиперидинила, пиперазинила и пирролидинила, где пиперидинил, пиперазинил и пирролидинил являются незамещенными или замещенными в одном или двух положениях группами, которые независимо представляют собой C1-C4алкил, C1-C4алкокси, галоген, C1-C4галогеналкил, C1-C4галогеналкокси, гидроксил, гидрокси-C1-C4алкил, амино, -NH(C1-C4алкил), -N(C1-C4алкил)(C1-C4алкил), -(C0-C6алкил)-C(O)R11 или NO2, где
R11 представляет собой C1-C6алкокси, необязательно замещенный 1 или 2 группами, которые независимо представляют собой C1-C4алкокси, амино, -NH(C1-C6алкил), -N(C1-C6алкил)(C1-C6алкил), -(C0-C6алкил)-C(O)N(R9)-гетероциклоалкил или гетероциклоалкил, где гетероциклоалкильная группа выбрана из азабициклооктила, в некоторых вариантах 1-азабицикло[2.2.2]окт-3-ила или 8-азабицикло[3.2.1]окт-3-ила, азабициклононила, азабициклодецила, где азот аза необязательно замещен метилом или этилом; и R4 представляет собой H или метил, пирролидинила, пиперидинила, пиперазинила и морфолинила, где гетероциклоалкильные группы необязательно замещены 1, 2 или 3 группами, которые независимо представляют собой C1-C6алкил, C1-C6алкокси, гидрокси, гидрокси C1-C6алкил, C1-C6алкоксикарбонил, -CO2H, CF3 или OCF3.
В еще другом аспекте настоящее изобретение относится к способам и/или процессам получения соединений, а также к промежуточным соединениям, пригодным для получения соединений формулы (X), где
R1 представляет собой хлор; R2 представляет собой амино; R3 представляет собой метокси; R4 представляет собой H, а R1, R2 и R3 ориентированы в фенильном кольце следующим образом:
, и
L представляет собой -(C3-C5алкил)-, где один атом углерода может быть заменен на -N(R9)- или -(C2-C6алкил)-C(O)-. В другом варианте R1, R2 и R3 определены выше и ориентированы в фенильном кольце так, как описано выше, R4 определен выше, а R5 представляет собой гетероциклоалкил, который выбран из азабициклооктила, в некоторых вариантах 1-азабицикло[2.2.2]окт-3-ила или 8-азабицикло[3.2.1]окт-3-ила, азабициклононила, азабициклодецила, где азот аза необязательно замещен метилом или этилом.
Еще одна особенность настоящего изобретения относится к способам и/или процессам получения соединений формулы (Х), а также к промежуточным соединениям, пригодным для получения соединений формулы (X), где
R1 представляет собой хлор; R2 представляет собой амино; R3 представляет собой метокси; R4 представляет собой H, а R1, R2 и R3 ориентированы в фенильном кольце следующим образом:
, и
L представляет -(C3-C5алкил)-, где один атом углерода может быть заменен на -N(R9)- или -(C2-C6алкил)-C(O)-. В другом варианте R1, R2 и R3 определены выше и ориентированные в фенильном кольце так, как описано выше, R4 определен выше, а R5 представляет -N(R9)-C0-C4алкил-арил или -N(R9)-(C0-C6алкил)-C(O)-арил, где арильная группа является незамещенной или замещенной в одном или более замещаемых положений C1-C6алкилом, C1-C6алкокси, галогеном, C1-C6 галогеналкилом, C1-C6 галогеналкокси, гидроксилом, гидроксиалкилом, амино, -NH(C1-C6алкилом), -N(C1-C6алкил)(C1-C6алкилом), -(C0-C6алкил)-C(O)R11 или NO2. В другом варианте арильная группа представляет собой фенил, замещенный -(C0-C6алкил)-C(O)R11, и необязательно замещенный 1 или 2 группами, независимо выбранными из C1-C6алкила, C1-C6алкокси, галогена, CF3, OCF3, гидроксила, гидроксиалкила, амино, -NH(C1-C4алкила), -N(C1-C4алкил)(C1-C4алкила) или NO2, где
R11 представляет собой C1-C6алкокси, необязательно замещенный 1 или 2 группами, которые независимо представляют собой C1-C4алкокси, амино, -NH(C1-C6алкил), -N(C1-C6алкил)(C1-C6алкил), -(C0-C6алкил)-C(O)N(R9)-гетероциклоалкил или гетероциклоалкил, где гетероциклоалкильная группа выбрана из пирролидинила, пиперидинила, пиперазинила и морфолинила, где гетероциклоалкильные группы необязательно замещены 1, 2 или 3 группами, которые независимо представляют собой галоген, C1-C6алкил, C1-C6алкокси, гидрокси, гидрокси C1-C6алкил, C1-C6алкоксикарбонил, -CO2H, CF3 или OCF3. В предпочтительном варианте группа -(C0-C6алкил)-C(O)R1 присоединена к фенильному кольцу в положении 4.
В другом аспекте осуществления настоящего изобретения ориентация связей 3 и 4 соединения, полученного с использованием способов и/или процессов по настоящему изобретению, является следующей:
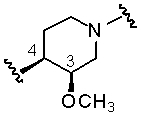 .
.
В предпочтительном аспекте осуществления настоящего изобретения ориентация связей 3 и 4 соединения, полученного с использованием способов и/или процессов по настоящему изобретению, является следующей:
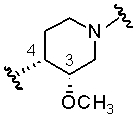 .
.
Изобретение относится также к способам лечения рвоты, диспепсии, гастропареза, запора, псевдонепроходимости кишечника, гастроэзофагеального рефлюкса или послеоперационного пареза кишечника, включающим в себя введение терапевтически эффективного количества соединения или соли соединения формулы (X), полученных с использованием способов и/или процессов по настоящему изобретению, пациентам, нуждающимся в указанном лечении.
Настоящее изобретение относится к способам и/или процессам получения соединений, которые более подвержены расщеплению под действием сывороточных и/или цитозольных эстераз, чем цизаприд, что позволяет избежать неблагоприятных эффектов, связанных с метаболизмом посредством цитохрома P450.
Преимуществом терапевтических соединений, полученных с использованием способов и/или процессов по настоящему изобретению, является их стабильность при хранении и относительно короткий полупериод существования в физиологических средах; таким образом, соединения по настоящему изобретению могут применяться с меньшей частотой побочных эффектов и меньшей токсичностью.
Предпочтительная особенность настоящего изобретения относится к терапевтическим стереоизомерным соединениям, полученным с использованием способов и/или процессов по настоящему изобретению, которые являются пригодными для лечения гастроэзофагеальной рефлюксной болезни и которые содержат сложноэфирную группу, которая является чувствительной к расщеплению эстеразами, в результате чего соединение разрушается и облегчается его эффективное выведение из организма пациента. В предпочтительном варианте осуществлялиия настоящего изобретения терапевтические стереоизомерные соединения метаболизируются с помощью системы детоксикации лекарственного средства фазы I.
Еще одна особенность настоящего изобретения относится к продуктам распада (предпочтительно к продуктам метаболического распада, т.е. метаболитам, в основном, кислотам исходных сложных эфиров), которые образуются при воздействии эстераз на терапевтические соединения, полученные с использованием способов и/или процессов по настоящему изобретению. Присутствие указанных продуктов распада в моче или сыворотке крови можно использовать для наблюдения за скоростью выведения терапевтических соединений из организма пациента.
Распад соединений, полученных с использованием способов и/или процессов по настоящему изобретению, под воздействием эстераз представляет собой особое преимущество в том, что касается метаболизма лекарственных средств, поскольку указанные ферменты повсеместно распространены в организме, и их активность не зависит от возраста, пола или болезненного состояния в той степени, в какой от указанных факторов зависит окислительный метаболизм лекарственных средств в печени.
Настоящее изобретение относится также к способам лечения расстройств, таких как гастроэзофагеальная рефлюксная болезнь, включающим в себя введение терапевтически эффективного количества по меньшей мере одного стереоизомерного структурного и/или функционального аналога цизаприда индивидууму, который нуждается в указанном лечении. Характерная особенность настоящего изобретения относится к стереоизомерным структурным и/или функциональным аналогам цизаприда и к фармацевтическим композициям указанных этерифицированных соединений.
Настоящее изобретение относится также к материалам и способам лечения рвоты и других состояний, включая, без ограничения, диспепсию, гастропарез, запор и псевдонепроходимость кишечника, которые характеризуются значительным сокращением неблагоприятных эффектов, связанных с введением цизаприда.
Предпочтительный вариант осуществления настоящего изобретения относится к способам и/или процессам получения терапевтических стереоизомерных соединений, которые являются пригодными для лечения гастроэзофагеального рефлюкса, диспепсии, гастропареза, запора, послеоперационного пареза кишечника и псевдонепроходимости кишечника и которые содержат сложноэфирную группу, на которую воздействуют эстеразы, в результате чего соединение распадается и облегчается его эффективное выведение из организма пациента.
Настоящее изобретение относится также к способам синтеза полезных соединений по настоящему изобретению. В частности, предлагаются способы изготовления и очистки указанных стереоизомерных соединений. Способы добавления указанных сложноэфирных частей и изготовления и очистки стереоизомеров хорошо известны специалистам и могут быть легко осуществлены в соответствии с представленными в настоящем документе указаниями.
Предпочтительные соединения
В предпочтительном варианте осуществления настоящее изобретение относится к способам и/или процессам получения изолированных стереоизомеров соединения I, а также к промежуточным соединениям, пригодным для получения указанных стереоизомеров, которое содержит три хиральных центра.
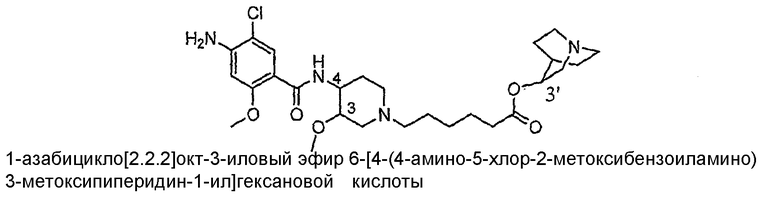
Соединение I
Два из хиральных центров находятся в цизаприде и норцизаприде и в активных лекарственных средствах имеют цис-конфигурацию:
(±)-цизаприд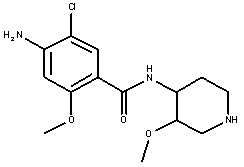
(±)-норцизаприд
Так, например, фармацевтически активный норцизаприд представляет собой рацемическую смесь двух цис-энантиомеров:
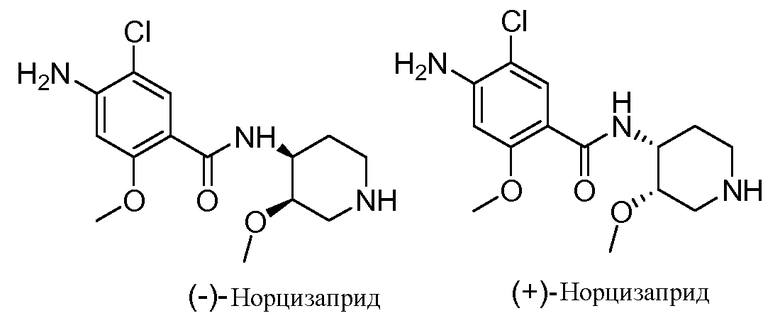
В одном аспекте настоящее изобретение относится, в частности, к способам и/или процессам получения соединений с определенной конфигурацией третьего хирального центра, в хинуклидиноловой части, а также к промежуточным соединениям, пригодным для получения указанных соединений. Указанная группа устраняется при превращении в кислотный метаболит, обозначенный в настоящем документе как ± соединение II:
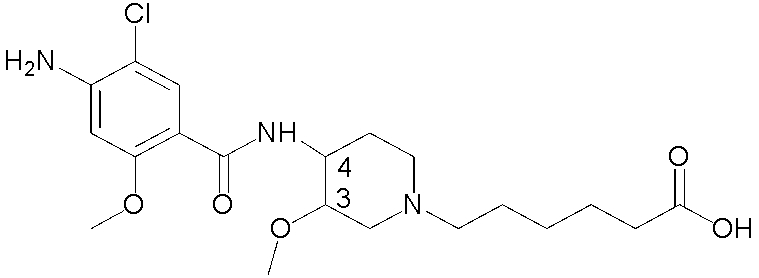
Соединение II
Несмотря на то, что стереоизомеры Соединения I можно получать конъюгацией R- или S-хинуклидинола с (+)- или (-)-норцизапридом, в результате чего образуются соединения III, IV, V и VI, предпочтительные способы описаны ниже, в которых ядро цизаприда не используется.
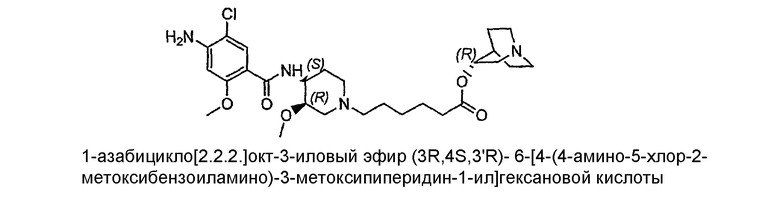
Соединение III: (-)(R)-соединение I
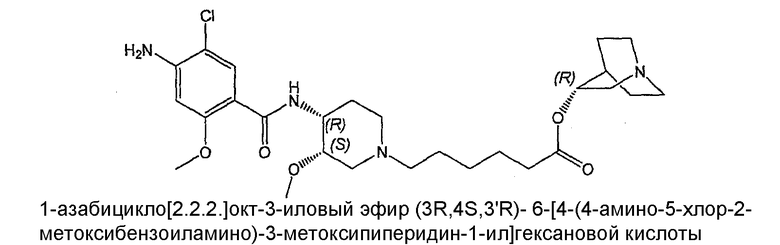
Соединение IV: (+)(R)-соединение I
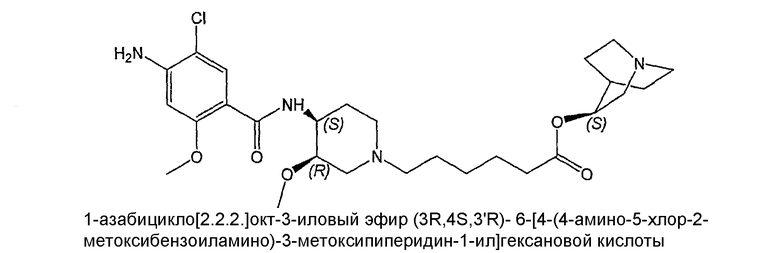
соединение V: (-)(S)-соединение
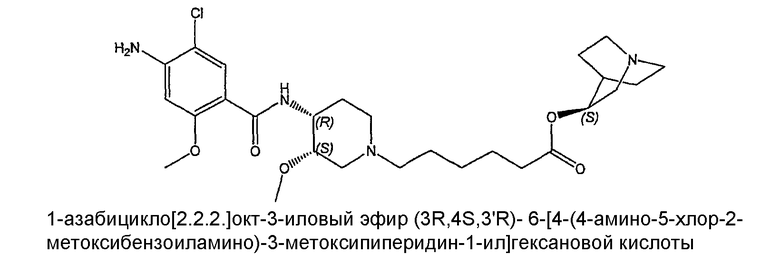
соединение VI: (+)(S)-соединение I
Предпочтительная особенность настоящего изобретения относится к способам и/или процессам получения стереоизомерно изолированных соединений, а также к промежуточным соединениям, пригодным для получения указанных соединений, и к композициям, включающим в себя указанные соединения. Изолированные стереоизомерные формы соединений, полученные с использованием способов и/или процессов по настоящему изобретению, практически не содержат другого стереоизомера (т.е. находятся в стереоизомерном избытке). Иными словами, «R»-формы соединений практически не содержат «S»-формы соединений и, таким образом, находятся в стереоизомерном избытке по отношению к «S»-формам. И наоборот, «S»-формы соединений практически не содержат «R»-формы соединений и, таким образом, находятся в стереоизомерном избытке по отношению к «R»-формам. Одна особенность настоящего изобретения относится к изолированным стереоизомерным соединениям, полученным с использованием способов и/или процессов по настоящему изобретению, которые находятся в стереоизомерном избытке, по меньшей мере, приблизительно 80%. Предпочтительная особенность настоящего изобретения относится к соединениям, полученным с использованием способов и/или процессов по настоящему изобретению, которые находятся в стереоизомерном избытке, по меньшей мере, приблизительно 90%. Более предпочтительная особенность настоящего изобретения относится к соединениям, полученным с использованием способов и/или процессов по настоящему изобретению, которые находятся в стереоизомерном избытке, по меньшей мере, приблизительно 95%. Еще более предпочтительная особенность настоящего изобретения относится к соединениям, полученным с использованием способов и/или процессов по настоящему изобретению, которые находятся в стереоизомерном избытке, по меньшей мере, приблизительно 97,5%. Наиболее предпочтительная особенность настоящего изобретения относится к соединениям, полученным с использованием способов и/или процессов по настоящему изобретению, которые находятся в стереоизомерном избытке, по меньшей мере, приблизительно 99%. Подобным образом, «(+)» и «(-)» формы соединений также получали в стереоизомерном избытке.
Как описывается в настоящем документе, различные стереоизомеры, полученные с использованием способов и/или процессов по настоящему изобретению, обладают особыми неожиданными свойствами, которые могут быть выгодно использованы для подбора лечения в определенных случаях. Так, например, соединения, содержащие (3'R)-изомер в хинуклидинильной сложноэфирной части, т.е. соединения III и IV, быстро метаболизируются эстеразами плазмы человека, тогда как метаболизм соединений, содержащих (3'S)-изомер хинуклидинола, т.е. соединений V и VI, происходит гораздо медленнее.
Соответственно, (3'R)-изомеры соединения I могут быть использованы в случаях, когда предпочтительной является короткая продолжительность действия, например, для стимуляции желудочной моторики в остром эпизоде, таких как ударное введение пациентам с острым гастропарезом или с острым гастроэзофагеальным рефлюксом. Другим преимуществом быстрого метаболизма эстеразами до значительно менее активных метаболитов, т.е. до соединения II, является очень малая вероятность лекарственных взаимодействий и токсических эффектов. Следовательно, указанные (R)-изомеры короткого действия можно успешно использовать в качестве внутривенной композиции для лечения гастроэзофагеального рефлюкса у недоношенных новорожденных, у которых, как известно, метаболизм лекарств происходит менее активно, чем у взрослых, в связи с недостаточным развитием системы CYP450. В случае указанных новорожденных, лекарственное средство, подвергающееся быстрому метаболизму без участия системы CYP450, например, эстеразами, является значительным преимуществом. С другой стороны, (3'S)-изомеры соединения I, полученные с использованием способов и/или процессов по настоящему изобретению, лучше всего использовать для лечения хронических форм тех же заболеваний, например, гастропареза у больных диабетом или онкологических больных, принимающих опиаты, а также хронического гастроэзофагеального рефлюкса у пациентов, которым требуется круглосуточная коррекция.
Помимо различий в метаболизме, указанные отдельные изомеры также обладают различным сродством к 5-HT4-рецептору, за счет чего различаются их эффекты и, соответственно, сферы терапевтического применения. Так, в порядке снижения сродства к 5-HT4-рецептору изомеры могут быть размещены следующим образом (в скобках приведены значения константы связывания Ki); соединение IV (1,4 нM), соединение VI (3,4 нM), соединение III (28 нM) и соединение V (72 нM). Указанные эксперименты по определению сродства проводились с использованием радиоизотопного метода, описанного в общедоступных руководствах, и могут быть легко воспроизведены специалистами в области молекулярной биологии.
Исходя из рассмотренных положений, можно сделать следующий вывод: когда связи 3 и 4 находятся в цис-положении относительно друг друга, соединение I представляет собой смесь 4 изомеров, состоящих из 2 пар энантиомеров. Первая пара энантиомеров представляет собой (+)(R)-соединение I и (-)(S)-соединение I (соединения IV и V, соответственно), вторая пара энантиомеров представляет собой (-)(R)-соединение I и (+)(S)-соединение I (соединения III и VI, соответственно). В каждой из пар свойства энантиомеров отличаются друг от друга в том, что касается как скорости их гидролиза эстеразами, так и их сродства к 5-HT4-рецептору. За счет указанных отличий каждое соединение обладает индивидуальными терапевтическими преимуществами, которые не являются взаимозаменяемыми, т.е. присущи определенному изомеру, и не присущи рацемической смеси. Указанные различия в сродстве к рецептору и в скорости метаболизма не являются предсказуемыми, и указанные свойства невозможно проанализировать при исследовании рацемической смеси.
Другая особенность настоящего изобретения относится к способу получения соединения формулы II'
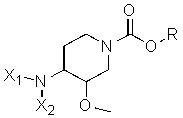 (II')
(II')
включающему в себя превращение соединения формулы (I')
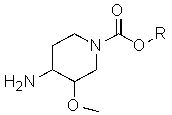 (I')
(I')
или его соли в соединение формулы (II') или его соль, соответственно, где X1 представляет собой защитную группу азота, а X2 выбран из группы, состоящей из водорода и защитной группы азота (в качестве таковой можно использовать одну из общеизвестных и широко используемых N-защитных групп, например, N-бензил; N-нитробензил; N-BoC; N-оксид; N-параметоксибензил; N-бензилсульфонил) (предпочтительно и X1, и X2 представляют собой бензил), и R представляет собой (C1-C8)алкил, предпочтительно (C1-C4)алкил, более предпочтительно этил. В другом варианте осуществления настоящего изобретения и X1, и X2 не являются бензилом.
Изобретение относится также к соединениям формулы II'
(II')
и их солям, где X1 представляет собой защитную группу азота, а X2 выбран из группы, состоящей из водорода и защитной группы азота (в качестве таковой можно использовать одну из общеизвестных и широко используемых N-защитных групп, например, N-бензил; N-нитробензил; N-BoC; N-оксид; N-параметоксибензил; N-бензилсульфонил) (предпочтительно и X1, и X2 представляют собой бензил), и R представляет собой (C1-C8)алкил, предпочтительно (C1-C4)алкил, более предпочтительно этил. В другом варианте осуществления настоящего изобретения и X1, и X2 не являются бензилом.
Другая особенность настоящего изобретения относится к способу получения соединения формулы III'
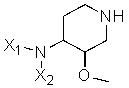 (III');
(III');
включающему в себя воздействие на соединение формулы (II')
(II')
гидроксидом или гидридом щелочного металла (например, NaOH, KOH, гидридом калия или натрия, гидридом лития-алюминия и т.п.), с получением соединения формулы (III'), где X1 представляет собой защитную группу азота, а X2 выбран из группы, состоящей из водорода и защитной группы азота (в качестве таковой можно использовать одну из общеизвестных и широко используемых N-защитных групп, например, N-бензил; N-нитробензил; N-BoC; N-оксид; N-параметоксибензил; N-бензилсульфонил) (предпочтительно и X1, и X2 представляют собой бензил), и R представляет собой (C1-C8)алкил, предпочтительно (C1-C4)алкил, более предпочтительно этил. В другом варианте осуществления настоящего изобретения и X1, и X2 не являются бензилом.
В ходе экспериментов с соединением
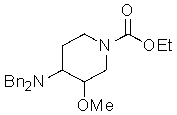
неожиданно было установлено, что при использовании по меньшей мере 12 эквивалентов KOH и 8-кратного избытка изопропилового спирта с использованием обратного холодильника реакция гидролиза дает практически 100% превращение с практически полным отсутствием примесей. При использовании меньших количеств KOH (например, 5 и 10 экв.) превращение происходило приблизительно на 83-98%, с примесями в пределах от 1,9% до 7,3%.
Изобретение относится также к соединениям формулы III'
(III');
и их солям, где X1 представляет собой защитную группу азота, а X2 выбран из группы, состоящей из водорода и защитной группы азота (в качестве таковой можно использовать одну из общеизвестных и широко используемых N-защитных групп, например, N-бензил; N-нитробензил; N-BoC; N-оксид; N-параметоксибензил; N-бензилсульфонил) (предпочтительно и X1, и X2 представляют собой бензил). В другом варианте осуществления настоящего изобретения и X1, и X2 не являются бензилом.
Другая особенность настоящего изобретения относится к способу получения соединения формулы III''
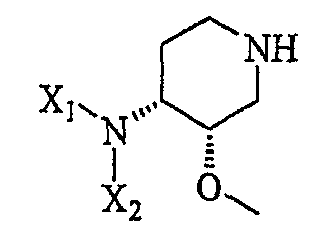 (III'');
(III'');
включающему в себя а) контактирование соединения формулы III'
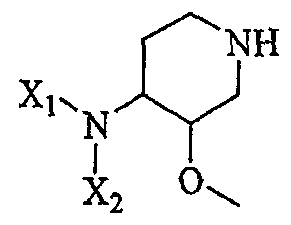 (III')
(III')
с хиральным разделяющим агентом (например, винной кислотой, миндальной кислотой, кислотой Мошера, камфарсульфоновой кислотой и т.п.), с получением хиральной соли III'' и выделением хиральной соли III'';
b) необязательно, перекристаллизацию продукта a); и
c) контактирование a) или b) с основанием, с получением соединения формулы III'';
где X1 представляет собой защитную группу азота, а X2 выбран из группы, состоящей из водорода и защитной группы азота (в качестве таковой можно использовать одну из общеизвестных и широко используемых N-защитных групп, например, N-бензил; N-нитробензил; N-BoC; N-оксид; N-параметоксибензил; N-бензилсульфонил) (предпочтительно и X1, и X2 представляют собой бензил). В другом варианте осуществления настоящего изобретения и X1, и X2 не являются бензилом.
Предпочтительно, в указанном варианте осуществления настоящего изобретения хиральным разделяющим агентом является (+)-2,3-дибензоил-D-винная кислота, а хиральной солью III' является (3S,4R)-энантиомер (+)-2,3-дибензоил-D-тартрата.
Неожиданно было установлено, что использование хиральной соли (+)-2,3-дибензоил-D-винной кислоты, предпочтительно, в количестве одного эквивалента на два эквивалента соединения III', обеспечивает повышенный выход по сравнению с другими хиральными разделяющими агентами. Взаимодействие одного эквивалента (+)-2,3-дибензоил-D-винной кислоты с двумя эквивалентами III' обеспечивает выход, более чем в три раза превышающий выход от взаимодействия одного эквивалента (+)-2,3-дибензоил-D-винной кислоты с двумя эквивалентами III'. Таким образом, в предпочтительном варианте получения соединения формулы III'' используется один эквивалент хирального разделяющего агента (предпочтительно, (+)-2,3-дибензоил-D-винной кислоты) и два эквивалента соединения формулы III'.
Другая особенность настоящего изобретения относится к соединению формулы III''
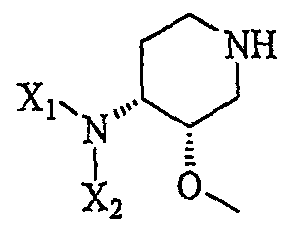 (III'')
(III'')
и его солям, где X1 представляет собой защитную группу азота, а X2 выбран из группы, состоящей из водорода и защитной группы азота (в качестве таковой можно использовать одну из общеизвестных и широко используемых N-защитных групп, например, N-бензил; N-нитробензил; N-BoC; N-оксид; N-параметоксибензил; N-бензилсульфонил) (предпочтительно и X1, и X2 представляют собой бензил). В другом варианте осуществления настоящего изобретения и X1, и X2 не являются бензилом.
Другая особенность настоящего изобретения относится к способу получения соединения формулы IV'
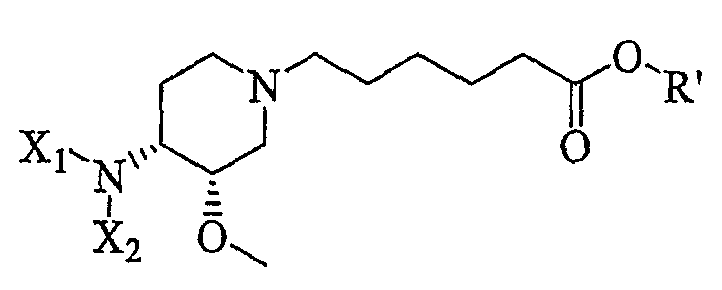 (IV');
(IV');
включающему в себя контактирование соединения формулы
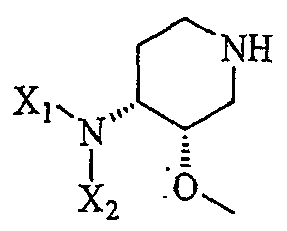 (III'');
(III'');
с (C1-C8)алкил 6-галогенгексаноатом (где галоген предпочтительно представляет собой бром), с получением соединения формулы (IV'), где R' представляет собой (C1-C8)алкил (предпочтительно этил), и X1 представляет собой защитную группу азота, а X2 выбран из группы, состоящей из водорода и защитной группы азота (в качестве таковой можно использовать одну из общеизвестных и широко используемых N-защитных групп, например, N-бензил; N-нитробензил; N-BoC; N-оксид; N-параметоксибензил; N-бензилсульфонил) (предпочтительно и X1, и X2 представляют собой бензил). В другом варианте осуществления настоящего изобретения и X1, и X2 не являются бензилом.
Другая особенность настоящего изобретения относится к соединению формулы IV'
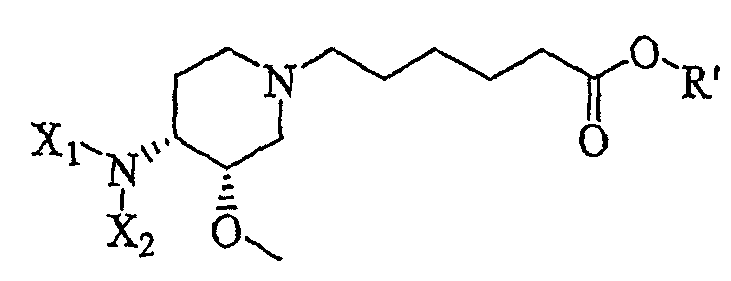 (IV').
(IV').
и его солям, где R' представляет собой (C1-C8)алкил (предпочтительно, этил), и X1 представляет собой защитную группу азота, а X2 выбран из группы, состоящей из водорода и защитной группы азота (в качестве таковой можно использовать одну из общеизвестных и широко используемых N-защитных групп, например, N-бензил; N-нитробензил; N-BoC; N-оксид; N-параметоксибензил; N-бензилсульфонил) (предпочтительно и X1, и X2 представляют собой бензил). В другом варианте осуществления настоящего изобретения и X1, и X2 не являются бензилом.
В другом аспекте настоящее изобретение относится к способу получения соединения формулы V'
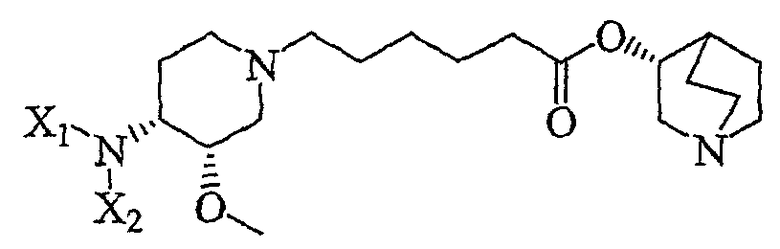 (V');
(V');
включающему в себя контактирование соединения формулы IV'
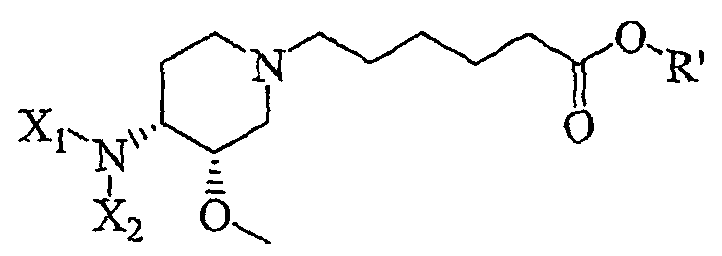 (IV').
(IV').
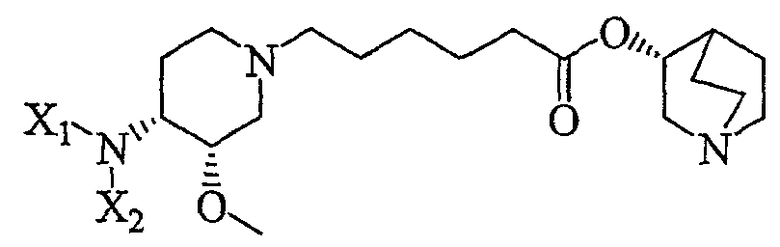
с (R)-хинуклидин-3-олом и кислотой Льюиса (например, тетраалкоксидом титана (например, Ti(OiPr)4 (тетраизопропоксидом титана) и Ti(OEt)4 (тетраэтоксидом титана)), TsOH (паратолуолсульфоновой кислотой), K2CO3 и кат. DMAP (катализатором 4-диметиламинопиридином)) в органическом растворителе (например, толуоле), где R' представляет собой (C1-C8)алкил (предпочтительно, этил), и X1 представляет собой защитную группу азота, а X2 выбран из группы, состоящей из водорода и защитной группы азота (в качестве таковой можно использовать одну из общеизвестных и широко используемых N-защитных групп, например, N-бензил; N-нитробензил; N-BoC; N-оксид; N-параметоксибензил; N-бензилсульфонил) (предпочтительно и X1, и X2 представляют собой бензил). В другом варианте осуществления настоящего изобретения и X1, и X2 не являются бензилом.
Другая особенность настоящего изобретения относится к соединению формулы V'
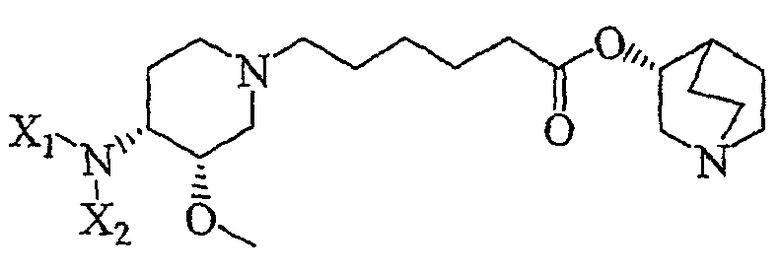 (V');
(V');
и его солям, где X1 представляет собой защитную группу азота, а X2 выбран из группы, состоящей из водорода и защитной группы азота (в качестве таковой можно использовать одну из общеизвестных и широко используемых N-защитных групп, например, N-бензил; N-нитробензил; N-BoC; N-оксид; N-параметоксибензил; N-бензилсульфонил) (предпочтительно и X1, и X2 представляют собой бензил). В другом варианте осуществления настоящего изобретения и X1, и X2 не являются бензилом.
Другая особенность настоящего изобретения относится к способу получения соединения формулы VI'
 (VI')
(VI')
включающему в себя удаление групп X1 и X2 из соединения формулы V'
 (V'),
(V'),
где X1 представляет собой защитную группу азота, а X2 выбран из группы, состоящей из водорода и защитной группы азота (в качестве таковой можно использовать одну из общеизвестных и широко используемых N-защитных групп, например, N-бензил; N-нитробензил; N-BoC; N-оксид; N-параметоксибензил; N-бензилсульфонил) (предпочтительно и X1, и X2 представляют собой бензил). В другом варианте осуществления настоящего изобретения и X1, и X2 не являются бензилом.
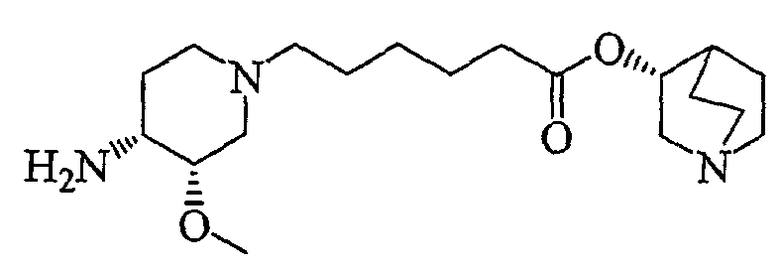
В предпочтительном варианте указанного аспекта настоящего изобретения X1 и X2 представляют собой бензил и удаляются воздействием на V' H2/Pd/C или формиатом аммония /Pd (примеры), с получением (R)-хинуклидин-3-ил 6-[(3S,4R)-4-амино-3-метоксипиперидин-1-ил]гексаноата. Заявители неожиданно установили, благодаря гидрированию с использованием H2/Pd/C указанный способ обладает значительными преимуществами по сравнению со способами дебензилирования с использованием, например, формиата аммония. Указанные способы часто являются грязными и требуют использования очистки на колонках с силикагелем для удаления реагентов (например, формиата аммония), что нецелесообразно при промышленном производстве. Гидрирование с использованием H2/Pd/C является чрезвычайно чистым процессом и не требует очистки на колонках.
В другом аспекте настоящее изобретение относится к способу получения (R)-хинуклидин-3-ил 6-((3S,4R)-4-(4-амино-5-хлор-2-метоксибензамид)-3-метоксипиперидин-1-ил)гексаноата (VII'), включающему в себя контактирование соединения формулы VI'
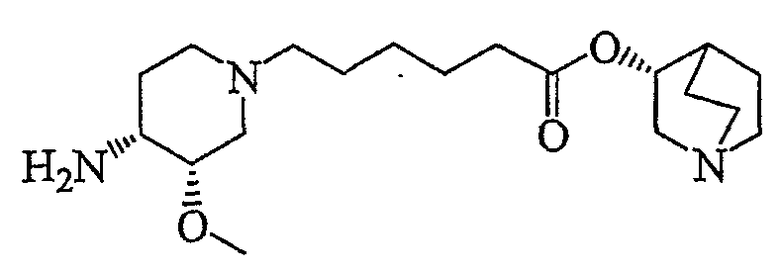 (VI')
(VI')
с 4-амино-5-хлор-2-метоксибензойной кислотой. Предпочтительно, указанное контактирование осуществляют с использованием EDCI (1-этил-3-(3'-диметиламинопропил)карбодиимида), HOBt (l-гидроксибензотриазола), HOSU (N-гидроксисукцинимида), HONB (N-гидрокси-5-норбен-эндо-2,3-дикарбоксамида), изобутилхлорформиата, пивалоилхлорида или DCC (дициклогексилкарбодиимида). Наиболее предпочтительно, указанное контактирование осуществляют с использованием пивалоилгалогенида (предпочтительно, пивалоилхлорида). Неожиданно было установлено, что при использовании пивалоилхлорида получали значительно меньше примесей, и продукт намного легче поддается очистке, чем при использовании других ацилирующих агентов. В результате обеспечивается больший выход реакции и значительно большая чистота соединения, чем при использовании других ацилирующих агентов.
Соединения формул I', II', III', III'', IV', V' и VI' представляют собой промежуточные соединения, пригодные для получения (R)-хинуклидин-3-ил 6-((3S,4R)-4-(4-амино-5-хлор-2-метоксибензамид)-3-метоксипиперидин-1-ил)гексаноата.
В другом аспекте настоящее изобретение относится к комбинациям указанных выше методов. Используемый в настоящем изобретении способ, описанный как способ X-Y, представляет собой способ, включающий комбинацию получения соединения формулы “X” и способа получения соединения формулы “Y”; и, подобным образом, способ X-Y-Z представляет собой способ X-Y с последующим применением способа получения соединения формулы “Z” и т.д. Соответственно, указанная особенность изобретения включает в себя, без ограничения, способы I'-II', II'-III', III'-III'', III''-IV', IV'-V', V'-VI', VI'-VII', I'-II'-III', II'-III'-III'', III'-III''-IV', III''-IV'-V', IV'-V'-VI', V'-VI'-VII', I'-II'-III'-III'', II'-III'-III''-IV', III'-III''-IV'-V', III''-IV'-V'-VI' и IV'-V'-VI'-VII'.
Дополнительный аспект настоящего изобретения относится к способу получения (R)-хинуклидин-3-ил 6-((3S,4R)-4-(4-амино-5-хлор-2-метоксибензамид)-3-метоксипиперидин-1-ил)гексаноата или его соли, включающему в себя:
1) необязательно, превращение соединения формулы (I')
(I')
в соль, где R представляет собой (C1-C8)алкил (предпочтительно, (C1-C6)алкил, (C1-C4)алкил, или этил);
2) превращение соединения формулы (I') или его соли в соединение формулы (II')
(II')
или его соль, соответственно, где X1 представляет собой защитную группу азота, а X2 выбран из группы, состоящей из водорода и защитной группы азота (в качестве таковой можно использовать одну из общеизвестных и широко используемых N-защитных групп, например, N-бензил; N-нитробензил; N-BoC; N-оксид; N-параметоксибензил; N-бензилсульфонил) (предпочтительно и X1, и X2 представляют собой бензил).
3) воздействие на соединение формулы (II') гидроксидом или гидридом щелочного металла (например, NaOH, KOH, гидридом натрия или калия, гидридом лития- алюминия и т.п.), с получением соединения формулы (III')
(III');
4) получение хиральной соли соединения III' воздействием на соединение формулы (III') хиральным разделяющим агентом (например, винной кислотой, миндальной кислотой, кислотой Мошера, камфарсульфокислотой и т.п., или, предпочтительно, (+)-2,3-дибензоил-D-винной кислотой, с получением хиральной соли (например, (3S,4R)-энантиомера (+)-2,3-дибензоил-D-тартрата, если хиральным разделяющим агентом является (+)-2,3-дибензоил-D-винная кислота)) и выделением цис-изомера полученного таким образом соединения III';
5) необязательно, перекристаллизацию продукта 4;
6) превращение продукта 4 или 5 в основание, с получением продукта 4 или 5 в форме свободного основания;
7) контактирование продукта 6 с (C1-C8)алкил 6-галогенгексаноатом (где галоген предпочтительно представляет собой бром), с получением соединения формулы (IV')
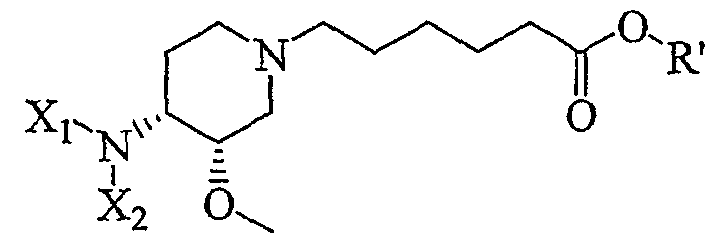 (IV');
(IV');
где R' представляет собой (C1-C8)алкил (предпочтительно этил);
8) воздействие на продукт 7 (R)-хинуклидин-3-олом и кислотой Льюиса (например, тетраалкоксидом титана (например, Ti(OiPr)4 (тетраизопропоксидом титана) и Ti(OEt)4 (тетраэтоксидом титана)), TsOH (паратолуолсульфоновой кислотой), K2CO3, и кат. DMAP (катализатором 4-диметиламинопиридином)) в органическом растворителе (например, толуоле), с получением соединения формулы (V')
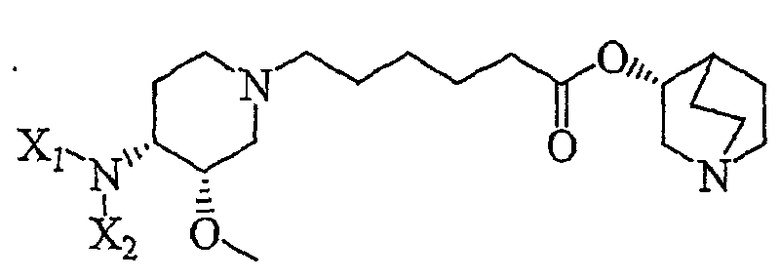 (V');
(V');
9) снятие защиты с 4-аминогруппы продукта 8 (например, с помощью H2/Pd/C или формиата аммония /Pd в качестве примеров), с получением (R)-хинуклидин-3-ил 6-[(3S,4R)-4-амино-3-метоксипиперидин-1-ил]гексаноата;
10) ацилирование продукта 9 4-амино-5-хлор-2-метоксибензойной кислотой (например, EDCI (1-этил-3-(3'-диметиламинопропил)карбодиимидом), HOBt (l-гидроксибензотриазолом), HOSU (N-гидроксисукцинимид), HONB (N-гидрокси-5-норбен-эндо-2,3-дикарбоксамидом), изобутилхлорформиатом, пивалоилхлоридом или DCC (дициклогексилкарбодиимидом), с получением (R)-хинуклидин-3-ил 6-((3S,4R)-4-(4-амино-5-хлор-2-метоксибензамид)-3-метоксипиперидин-1-ил)гексаноата;
11) необязательно, превращение продукта 10 в соль.
Дополнительный аспект изобретения относится к способу получения (R)-хинуклидин-3-ил 6-((3S,4R)-4-(4-амино-5-хлор-2-метоксибензамид)-3-метоксипиперидин-1-ил)гексаноата или его соли, включающему:
1) превращение соединения, которое представляет собой этил 4-амино-3-метоксипиперидин-1-карбоксилат
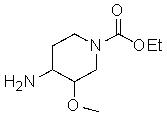
в соль;
2) превращение соли этил 4-амино-3-метоксипиперидин-1-карбоксилата в этил 4-(дифениламин)-3-метоксипиперидин-1-карбоксилат
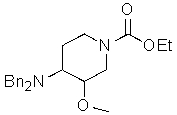 ;
;
3) воздействие на этил 4-(дифениламино)-3-метоксипиперидин-1-карбоксилат гидроксидом или гидридом щелочного металла, с получением 3-метокси-N,N-дифенилпиперидин-4-амина
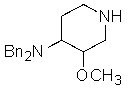 ;
;
4) Получение хиральной соли 3-метокси-N,N-дифенилпиперидин-4-амина воздействием на 3-метокси-N,N-дифенилпиперидин-4-амин с хиральным разделяющим агентом и выделением цис-изомера полученной таким способом соли 3-метокси-N,N-дифенилпиперидин-4-амина;
5) необязательно, перекристаллизацию продукта 4;
6) превращение продукта 4 или 5 в основание, с получением продукта 4 или 5 в форме свободного основания;
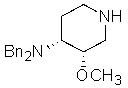 ;
;
7) алкилирование продукта 6 этил 6-бромгексаноатом, с получением этил 6-((3S,4R)-4-(дифениламин)-3-метоксипиперидин-1-ил)гексаноата
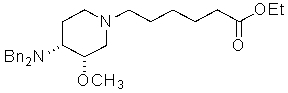 ;
;
8) этерификацию этил 6-((3S,4R)-4-(дифениламин)-3-метоксипиперидин-1-ил)гексаноата с использованием (R)-хинуклидин-3-ола, с получением (R)-хинуклидин-3-ил 6-((3S,4R)-4-(дифениламин)-3-метоксипиперидин-1-ил)гексаноата
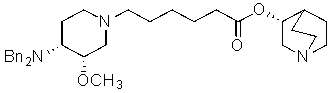 ;
;
9) снятие защиты с 4-аминогруппы продукта 8, с получением (R)-хинуклидин-3-ил 6-[(3S,4R)-4-амино-3-метоксипиперидин-1-ил]гексаноата;
10) ацилирование продукта 9 4-амино-5-хлор-2-метоксибензойной кислотой, с получением (R)-хинуклидин-3-ил 6-((3S,4R)-4-(4-амино-5-хлор-2-метоксибензамид)-3-метоксипиперидин-1-ил)гексаноата;
11) необязательно, превращение продукта 10 в соль.
Предпочтительно в указанном варианте реализации изобретения соль на стадии 1 представляет собой HCl.
Предпочтительно в указанном варианте осуществления настоящего изобретения гидроксид щелочного металла на стадии 3 представляет собой KOH.
Предпочтительно в данном варианте выполнения изобретения хиральная соль на стадии 4 представляет собой (+)-2,3-дибензоил-D-винную кислоту, после взаимодействия которой с IIIґ получали соль (3S,4R)-энантиомер (+)-2,3-дибензоил-D-тартрата.
Предпочтительно в указанном варианте осуществления настоящего изобретения для реакций на стадии 8 используется Ti(OiP)4 (изопропоксид титана (IV)).
Предпочтительно в указанном варианте осуществления настоящего изобретения для реакций на стадии 9 используется H2/Pd/C.
Предпочтительно в указанном варианте осуществления настоящего изобретения для реакций на стадии 10 используется пивалоилхлорид.
Определения
Используемый в настоящем документе термин «алкил» включает алкильные группы с заданным числом атомов углерода. Алкильные группы могут быть линейными или разветвленными. Примеры «алкила» включают в себя метил, этил, пропил, изопропил, бутил, изо-, втор- и трет-бутил, пентил, гексил, гептил, 3-этилбутил и т.п. Если число атомов углерода не определено, то обозначенная «алкильная» часть содержит от 1 до 6 атомов углерода.
Термин «алкокси» представляет алкильную группу с указанным количеством атомов углерода, которая связана с родственной молекулярной частью посредством кислородного мостика. Примеры алкоксигрупп включают в себя, без ограничения, метокси, этокси, пропокси и изопропокси.
Термин «арил» означает ароматическую карбоциклическую группу, имеющую одно кольцо (например, фенил), которое необязательно конденсировано или иным способом присоединено к другим ароматическим углеводородным кольцам или неароматическим углеводородным кольцам. Термин «арил» включает конденсированные полициклические соединения, в которых по меньшей мере одно кольцо является ароматическим (например, 1,2,3,4-тетрагидронафтил, нафтил), где каждое кольцо необязательно замещено одной, двумя или тремя группами, указанными ниже, а также неконденсированные полициклические соединения, такие как, например, бифенил или бинафтил. Предпочтительными арильными группами по настоящему изобретению являются фенил, 1-нафтил, 2-нафтил, инданил, инденил, дигидронафтил, флуоренил, тетралинил или 6,7,8,9-тетрагидро-5H-бензо[a]циклогептенил. Более предпочтительными являются фенил, бифенил и нафтил. Наиболее предпочтительным является фенил. Арильные группы в данном случае являются незамещенными или, если указано, замещенными в одном или более замещаемых положений различными группами. Например, указанные арильные группы могут быть, необязательно замещены, например, C1-C6алкилом, C1-C6алкокси, галогеном, гидрокси, циано, нитро, амино, моно(C1-C6)алкиламино, ди(C1-C6)алкиламино, C2-C6алкенилом, C2-C6алкинилом, C1-C6 галогеналкилом, C1-C6 галогеналкокси, амино(C1-C6)алкилом, моно(Ci-C6)алкиламино(CI-C6)алкилом или ди(C1-C6)алкиламино(CI-C6)алкилом.
Термин «галогеналкокси» относится к алкоксигруппе, которая замещена по меньшей мере одним атомом галогена и, необязательно, дополнительно замещена по меньшей мере одним дополнительным атомом галогена, где каждый атом галогена независимо представляют собой F, Cl, Br или I. Предпочтительными галогенами являются F или Cl. Предпочтительные галогеналкоксигруппы содержат 1-6 атома углерода, более предпочтительно, 1-4 атома углерода, и еще более предпочтительно, 1-2 атома углерода. Термин «галогеналкокси» включает в себя пергалогеналкоксигруппы, такие как OCF3 или OCF2CF3.
Термин «гетероарил» относится к ароматической циклической системе, содержащей по меньшей мере один гетероатом, выбранный из азота, кислорода и серы. Гетероарильное кольцо может быть конденсировано или иным способом присоединено одному или более гетероарильным кольцам, ароматическим или неароматическим углеводородным кольцам или гетероциклоалкильным кольцам. Примеры гетероарильных групп включают, без ограничения, пиридил, пиримидил, хинолинил, бензотиенил, индолил, индолинил, пиридазинил, пиразинил, изоиндолил, изохинолил, хиназолинил, хиноксалинил, фталазинил, имидазолил, изоксазолил, пиразолил, оксазолил, тиазолил, индолизинил, индазолил, бензотиазолил, бензимидазолил, бензофуранил, фуранил, тиенил, пирролил, оксадиазолил, тиадиазолил, бензо[1,4]оксазинил, триазолил, тетразолил, изотиазолил, нафтиридинил, изохроманил, хроманил, тетрагидроизохинолинил, изоиндолинил, изобензотетрагидрофуранил, изобензотетрагидротиенил, изобензотиенил, бензоксазолил, пиридопиридинил, бензотетрагидрофуранил, бензотетрагидротиенил, пуринил, бензодиоксолил, триазинил, птеридинил, бензотиазолил, имидазопиридинил, имидазотиазолил, дигидробензизоксазинил, бензизоксазинил, бензоксазинил, дигидробензизотиазинил, бензопиранил, бензотиопиранил, хромонил, хроманонил, пиридинил-N-оксид, тетрагидрохинолинил, дигидрохинолинил, дигидрохинолинонил, дигидроизохинолинонил, дигидрокумаринил, дигидроизокумаринил, изоиндолинонил, бензодиоксанил, бензооксазолинонил, пирролил N-оксид, пиримидинил N-оксид, пиридазинил N-оксид, пиразинил N-оксид, хинолинил N-оксид, индолил N-оксид, индолинил N-оксид, изохинолил N-оксид, хиназолинил N-оксид, хиноксалинил N-оксид, фталазинил N-оксид, имидазолил N-оксид, изоксазолил N-оксид, оксазолил N-оксид, тиазолил N-оксид, индолизинил N-оксид, индазолил N-оксид, бензотиазолил N-оксид, бензимидазолил N-оксид, пирролил N-оксид, оксадиазолил N-оксид, тиадиазолил N-оксид, триазолил N-оксид, тетразолил N-оксид, бензотиопиранил S-оксид, бензотиопиранил S,S-диоксид. Предпочтительные гетероарильные группы включают пиридил, пиримидил, хинолинил, индолил, пирролил, фуранил, тиенил и имидазолил. Более предпочтительные гетероарильные группы включают пиридил, пирролил и индолил. Гетероарильные группы в данном случае являются незамещенными или, если указано, замещенными в одном или более замещаемых положений различными группами. Например, указанные гетероарильные группы могут быть необязательно замещены, например, C1-C6алкилом, C1-C6алкокси, галогеном, гидрокси, циано, нитро, амино, моно(C1-C6)алкиламино, ди(C1-C6)алкиламино, C2-C6алкенилом, C2-C6алкинилом, C1-C6галогеналкилом, C1-C6галогеналкокси, амино(C1-C6)алкилом, моно(C1-C6)алкиламино(C1-C6)алкилом или ди(C1-C6)алкиламино(C1-C6)алкилом.
Термин «гетероциклоалкил» относится к кольцу или кольцевой системе, содержащей по меньшей мере один гетероатом, который предпочтительно выбран из азота, кислорода и серы, где указанный гетероатом находится в неароматическом кольце. Гетероциклоалкильное кольцо может быть слито или иным способом присоединено к другим гетероциклоалкильным кольцам и/или неароматическим углеводородным кольцам и/или фенильным кольцам. Предпочтительные гетероциклоалкильные группы имеют от 3 до 7 членов. Более предпочтительные гетероциклоалкильные группы имеют 5 или 6 членов. Примеры гетероциклоалкильных групп включают в себя, например, азабицикло[2.2.2]октил, азабицикло[3.2.1]октил, морфолинил, тиоморфолинил, тиоморфолинил S-оксид, тиоморфолинил S,S-диоксид, пиперазинил, гомопиперазинил, пирролидинил, пирролинил, тетрагидропиранил, пиперидинил, тетрагидрофуранил, тетрагидротиенил, гомопиперидинил, гомоморфолинил, гомотиоморфолинил, гомотиоморфолинил S,S-диоксид, оксазолидинонил, дигидропиразолил, дигидропирролил, дигидропиразинил, дигидропиридинил, дигидропиримидинил, дигидрофурил, дигидропиранил, тетрагидротиенил S-оксид, тетрагидротиенил S,S-диоксид и гомотиоморфолинил S-оксид. Предпочтительные гетероциклоалкильные группы включают азабицикло[2.2.2]октил, азабицикло[3.2.1]октил, пиперидинил, пиперазинил, пирролидинил, тиоморфолинил, S,S-диоксотиоморфолинил, морфолинил и имидалолидинил. Более предпочтительными являются азабицикло[2.2.2]октил, азабицикло[3.2.1]октил, пиперидинил, пиперазинил, пирролидинил, имидазолидинил и морфолинил. Гетероциклические группы в данном случае являются незамещенными или, если указано, замещенными в одном или более замещаемых положений различными группами. Например, указанные гетероциклические группы могут быть, необязательно замещены, например, C1-C6алкилом, C1-C6алкокси, галогеном, гидрокси, циано, нитро, амино, моно(C1-C6)алкиламино, ди(C1-C6)алкиламино, C2-C6алкенилом, C2-C6алкинилом, C1-C6галогеналкилом, C1-C6галогеналкокси, амино(C1-C6)алкилом, моно(C1-C6)алкиламино(C1-C6)алкилом, ди(C1-C6)алкиламино(C1-C6)алкилом или =O.
Термин «фармацевтически приемлемые соли» или «их фармацевтически приемлемые соли» относится к солям, полученным из фармацевтически приемлемых нетоксичных кислот или оснований, включая неорганические кислоты и основания и органические кислоты и основания. Поскольку соединение по настоящему изобретению является основным, соли могут быть получены с использованием фармацевтически приемлемых нетоксичных кислот. Подходящие фармацевтически приемлемые кислотно-аддитивные соли соединений по настоящему изобретению включают соли уксусной, бензолсульфоновой (бесилаты), бензойной, камфарсульфоновой, лимонной, этиленсульфоновой, фумаровой, глюконовой, глутаминовой, бромистоводородной, хлористоводородной, изетионовой, молочной, малеиновой, яблочной, миндальной, метансульфоновой, муциновой, азотной, памоевой, пантотеновой, фосфорной, янтарной, серной, винной, p-толуолсульфоновой и т.п. кислот. Предпочтительными кислотно-аддитивными солями являются хлориды и сульфаты. В наиболее предпочтительном варианте осуществления настоящего изобретения структурные и/или функциональные аналоги цизаприда вводят в форме свободного основания или в форме моно- или дигидрохлорида.
Используемые в настоящем документе термины «лечение» или «проведение курса лечения» относятся к профилактическому введению соединения или фармацевтической композиции, включающей в себя соединение («профилактика»), а также лечебную терапию, направленную на уменьшение или устранение заболевания или расстройства, упомянутого в настоящем документе. Профилактическое введение предназначено для предотвращения возникновения расстройств и может быть использовано для лечения пациента, входящего в группу риска или страдающего одним или более описанных выше расстройств. Таким образом, используемый в настоящем документе термин «лечение» или его производное предполагает частичное или полное ингибирование указанного болезненного состояния посредством введения активного ингредиента по настоящему изобретению с профилактической целью или после возникновения болезненного состояния, для лечения которого вводят указанный активный ингредиент. «Профилактика» относится к введению активного ингредиента(ингредиентов) млекопитающему для его защиты от любого из расстройств, описанных в настоящем документе, а также от других расстройств.
Термин «терапевтически эффективное количество» относится к количеству, необходимому для достижения терапевтического эффекта, такому как: 1) количество, достаточное для облегчения рефлюксной болезни, 2) количество, достаточное для облегчения тошноты и рвоты, или 3) количество, достаточное для облегчения состояния, вызванного нарушением моторики желудочно-кишечного тракта. терапевтически эффективные количества структурных и/или функциональных аналогов цизаприда определяются описанными выше дозировками частотой введения.
«Млекопитающим» может быть, например, мышь, крыса, свинья, лошадь, кролик, коза, корова, кошка, собака или человек. Предпочтительно, млекопитающим является человек.
Термин «индивидуум(ы)» определяется как отдельное млекопитающее, которому вводят соединение по настоящему изобретению. Млекопитающим может быть, например, мышь, крыса, свинья, лошадь, кролик, коза, корова, кошка, собака или человек. Предпочтительно, млекопитающим является человек.
Термин «этерифицированный цизаприд» относится к терапевтическим соединениям по настоящему изобретению, которые являются структурными и/или функциональными аналогами цизаприда и содержат способную гидролизоваться группу, обычно сложноэфирную, которая не уменьшает способности указанных соединений оказывать благоприятное терапевтическое действие, но которая делает указанные соединения более подверженными расщеплению гидролазами, особенно, сывороточными и/или цитозольными эстеразами, и которая уменьшает взаимодействия системы детоксикации лекарственных средств цитохрома P450 и производных цизаприда. Опосредуемый эстеразами метаболизм этерифицированных соединений цизаприда уменьшает роль системы детоксикации лекарственных средств цитохрома P450 в метаболизме цизаприда и уменьшает или устраняет неблагоприятные эффекты, вызываемые цизапридом.
Термин «структурный аналог», используемый в настоящем документе, означает, что описанное соединение имеет те же структурные характеристики, что и родоначальное соединение. Например, структурный аналог цизаприда может иметь одну или более структурных характеристик, подобных характеристикам исходного соединения цизаприда, таких как замещенное арильное кольцо, связанное с пиперидиновым кольцом посредством амидной связи, но в остальном структурно отличаться, например, включением или исключением одного или более других химических частей.
Термин «функциональный аналог», используемый в настоящем документе, означает, что описанное соединение имеет те же функциональные свойства, что и родоначальное соединение. Например, функциональный аналог цизаприда может характеризоваться незначительным структурным сходством с цизапридом или не проявлять такого сходства вообще, но выполнять похожую функцию, например, проявлять агонизм в отношении 5-HT4.
Термин «неблагоприятные эффекты» включает, без ограничения, расстройства желудочно-кишечного тракта, такие как диарея, колики и урчание в животе; усталость; головную боль; повышение систолического давления; смерть; желудочковую тахикардию; фибрилляцию желудочков; трепетание-мерцание желудочков; удлинение интервала QT; повышение частоты сердечных сокращений; неврологические расстройства и расстройства ЦНС; а также взаимодействие цизаприда с другими лекарственными средствами при одновременном применении, с такими как дигоксин, диазепам, этанол, аценокумарол, циметидин, ранитидин, парацетамол и пропранолол.
Термин «гастроэзофагеальная рефлюксная болезнь», используемый в настоящем документе, означает наличие и симптомы таких состояний, которые вызывают заброс содержимого желудка в пищевод.
Термины «оказывающий противорвотное действие» и «противорвотная терапия», используемые в настоящем документе, означают облегчение или предупреждение симптомов тошноты и рвоты, возникших спонтанно или вследствие вызывающей рвоту химиотерапии или лучевой терапии злокачественных опухолей.
Термин «лечение состояния, вызванного нарушением моторики желудочно-кишечного тракта», используемый в настоящем документе, означает лечение симптомов и состояний, связанных с указанным расстройством, которые включают, без ограничения, гастроэзофагеальную рефлюксную болезнь, диспепсию, гастропарез, запор, послеоперационный парез кишечника и псевдонепроходимость кишечника.
Термин «прокинетический», используемый в настоящем документе, означает усиление перистальтики и, следовательно, продвижения содержимого по желудочно-кишечному тракту.
Термин «диспепсия», используемый в настоящем документе, означает состояние, характеризующееся нарушением пищеварения, которое может являться симптомом первичной дисфункции желудочно-кишечного тракта или осложнением других заболеваний, таких как аппендицит, нарушение функции желчного пузыря или нарушения питания.
Термин «гастропарез», используемый в настоящем документе, означает паралич желудка, вызванный нарушением моторики желудка или являющийся осложнением таких заболеваний, как диабет, прогрессирующий системный склероз, нервная анорексия или миотоническая дистрофия.
Термин «запор», используемый в настоящем документе, означает состояние, характеризующееся уреженной или затрудненной дефекацией в результате таких состояний, как недостаточность мышечного тонуса или спастичность кишечника.
Термин «послеоперационный парез кишечника», используемый в настоящем документе, означает непроходимость кишечника вследствие нарушения мышечного тонуса после оперативного вмешательства.
Термин «псевдонепроходимость кишечника», используемый в настоящем документе, означает состояние, характеризующееся запором, коликами и рвотой, но без признаков физической обструкции.
Получение соединений
Несмотря на то, что химический синтез различных аналогов цизаприда может быть осуществлен с использованием способов, описанных в европейской патентной заявке №0076530 A2, опубликованной 13 апреля 1983 года, WO 01/093849, в патентных заявках США №4962115 и 5057525, а также в Van Daele et al., Drug Development Res. 8: 225-232 (1986), описания которых полностью включены в настоящий документ в качестве ссылок, соединения по настоящему изобретению предпочтительно получать в соответствии с описанием способов 3-6.
Изобретение проиллюстрировано далее несколькими примерами, которые приведены после способов. Способы и примеры ни в коей мере не ограничивают объем или суть изобретения конкретными приведенными в них процедурами. Специалистам будет понятно, что исходные материалы можно варьировать и использовать дополнительные стадии для получения соединений по настоящему изобретению, как показано в следующих примерах. Специалистам также будет понятно, что для осуществления некоторых из описанных выше преобразований может быть необходимо использовать различные растворители или реагенты. В некоторых случаях для осуществления описанных выше преобразований может быть необходима защита реакционноспособных функциональных групп. В целом, указанная необходимость использования защитных групп, а также условий, необходимых для присоединения или удаления указанных групп, будет очевидна для специалистов в области органического синтеза. При использовании защитной группы может потребоваться стадия снятия защиты. Подходящие защитные группы и способы защиты и снятия защиты, такие как описанные в T. Greene, Protecting Groups in Organic Synthesis, хорошо известны и приняты специалистами.
Если не указано иное, все реактивы и растворители имеют стандартное коммерческое качество и используются без дополнительной очистки. Подходящая для протекания реакции атмосфера, например, воздух, азот, водород, аргон и т.п., будет очевидна для специалистов.
Способ 1. Получение (R)-хинуклидин-3-ил 6-((3S,4R)-4-(4-амино-2-хлор-6-метоксибензамид)-3-метоксипиперидин-1-ил)гексаноата
Этап 1. Синтез этил 4-(дибензиламин)-3-метоксипиперидин-1-карбоксилата (1):
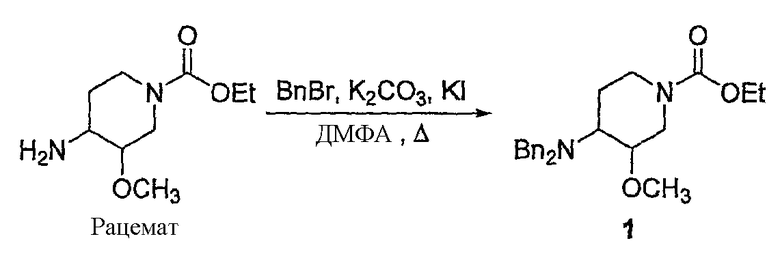
К раствору рацемического этил 4-амино-3-метоксипиперидин-1-карбоксилата (1 молярная часть) в ДМФА добавляли бензилбромид (приблизительно 2,2 молярной части), карбонат калия (приблизительно 2,4 молярной части) и йодид калия (приблизительно 0,2 молярной части), соответственно. Реакционную смесь нагревали приблизительно до 80°C (в описании дельта или “Δ” соответствует нагреванию). Приблизительно через 6 часов реакционную смесь медленно разбавляли водой (приблизительно 12 объемных частей) и экстрагировали, например, этилацетатом. Органический слой промывали солевым раствором, а затем сушили над безводным Na2SO4. После фильтрования и концентрирования растворителя получали 1 в виде масла желто-оранжевого цвета (1 молярная часть).
Этап 2. Синтез N,N-дибензил-3-метоксипиперидин-4 амина (2):
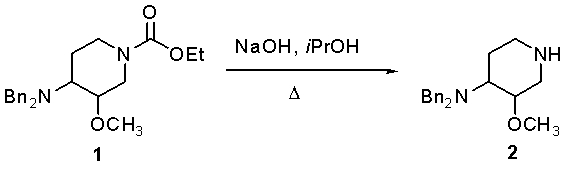
К раствору 1 добавляли NaOH (приблизительно 10 молярных частей) в изопропаноле и смесь перемешивали и нагревали до температуры кипения с обратным холодильником. По истечении от 3 до 5 часов реакционную смесь охлаждали до комнатной температуры и спиртовой растворитель удалили путем ротационного выпаривания. Смесь разбавляли водой и экстрагировали этилацетатом. Органический слой промывали солевым раствором и сушили над безводным Na2SO4. После фильтрования и концентрирования растворителя получали неочищенное масло, которое очищали через SiO2 (CH2Cl2:MeOH:NH4OH; (приблизительно) 15:1:0,01), с получением 2.
Этап 3. Синтез (3S,4R)-N,N-дибензил-3-метоксипиперидин-4 амина (3):
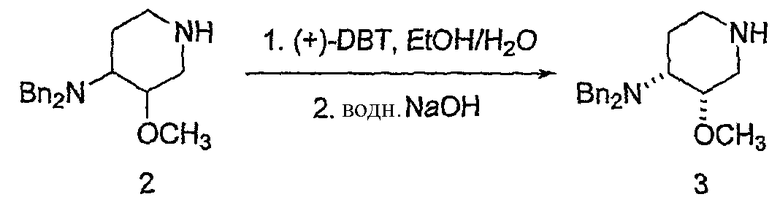
(+)-2,3-дибензоил-D-винную кислоту (приблизительно 1,2 массовой части) растворяли в этаноле, перед медленным добавлением к раствору 2 (приблизительно 1 массовая часть). Раствор осторожно нагревали, а затем позволяли ему остывать до комнатной температуры для кристаллизации соли. Соль фильтровали и промывали с использованием EtOH/H2О, перед суспендированием в воде и преобразованием в основание добавлением водн. NaOH (7%, мас./мас.), до тех пор, пока величина pH не достигала приблизительно 12. Суспензию интенсивно перемешивали при комнатной температуре и твердое вещество отфильтровывали, промывали водой и сушили в условиях вакуума, с получением цис-изомера 3.
Этап 4. Синтез этил 6-((3S,4R)-4-(дибензиламин)-3-метоксипиперидин-1-ил)гексаноата (4):
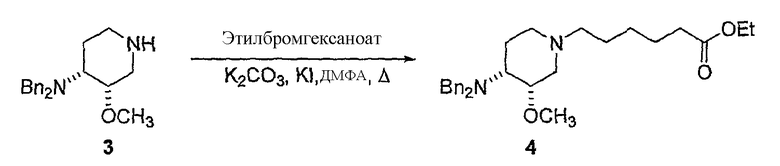
К раствору 3 (1 молярная часть) в ДМФА добавляли этил бромгексаноат (приблизительно 1,2 молярной части), карбонат калия (приблизительно 1,4 молярной части) и йодид калия (приблизительно 0,2 молярной части), соответственно. Затем реакционную смесь нагревали до 80°C. Приблизительно через 8 ч реакционную смесь медленно разбавляли водой (приблизительно 12 объемных частей) и экстрагировали этилацетатом. Органический слой промывали солевым раствором и затем сушили над безводным Na2SO4. После фильтрования и концентрирования растворителя получали неочищенный материал. После очистки через SiO2 получали алкилированный материал 4.
Этап 5. Синтез (R)-хинуклидин-3-ил 6-((3S,4R)-4-(дибензиламин)-3-метоксипиперидин-1-ил)гексаноата (5):

Тетраэтоксид титана добавляли к смеси 4 (1 молярная часть) и (R)-(-)-3-хинуклидинола (1 молярная часть) в толуоле. Реакционную смесь снабжали аппаратом Дина-Старка перед нагреванием приблизительно до 90°C, а затем использовали частичный вакуум (для поддержания требуемого уровня растворителя дополнительно добавляли толуол). Смесь затем охлаждали до комнатной температуры и реакционную смесь разбавляли этилацетатом, после чего в полученную смесь добавляли воду. Органический слой отделяли, промывали солевым раствором, сушили над безводным Na2SO4, фильтровали и концентрировали. В результате очистки через SiO2 получали энантиомерно обогащенный 5.
Этап 6. Синтез (R)-хинуклидин-3-ил 6-((3S,4R)-4-амино-3-метоксипиперидин-1-ил)гексаноата (6):
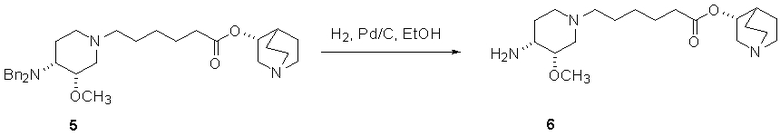
Раствор 5 (1 молярная часть) в EtOH добавляли в реакционную колбу, содержащую палладий на угле (приблизительно 0,2 молярной части). Затем из смеси откачивали воздух и подвергали ее гидрогенолизу с использованием атмосферного H2. После завершения реакции палладий отфильтровывали через целитный фильтр с последующей промывкой EtOH. Фильтрат концентрировали посредством ротационного испарения, с получением 6.
Этап 7. Синтез (R)-хинуклидин-3-ил 6-((3S,4R)-4-(4-амино-2-хлор-6-метоксибензамид)-3 -метоксипиперидин-1-ил)гексаноата (7):
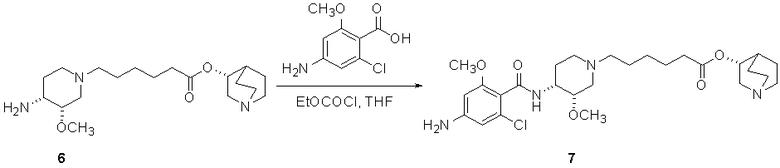
К раствору, например, этилхлорформиата (1 молярная часть) в ТГФ при температуре приблизительно 0°C по частям добавляли бензойную кислоту (1 молярная часть). Смесь нагревали до комнатной температуры в течение приблизительно 1 ч, затем охлаждали приблизительно до 0°C и по капле добавляли раствор 6 (1 молярная часть). Затем реакционную смесь нагревали до комнатной температуры. После завершения реакции ее гасили добавлением насыщенного раствора NaHCO3 и экстрагировали с использованием EA. Органический слой промывали солевым раствором, сушили над безводным Na2SO4, фильтровали и концентрировали, с получением требуемого продукта 7.
Способ 2
Синтез (R)-хинуклидин-3-ил 6-((3S,4R)-4-(4-амино-5-хлор-2-метоксибензамид)-3-метоксипиперидин-1-ил)гексаноата (или 1-азабицикло[2.2.2]окт-3'R-илового эфира 6-[4R-(4-амино-5-хлор-2-метоксибензоиламин)-3S-метоксипиперидин-1-ил]гексановой кислоты; ATI-7505):
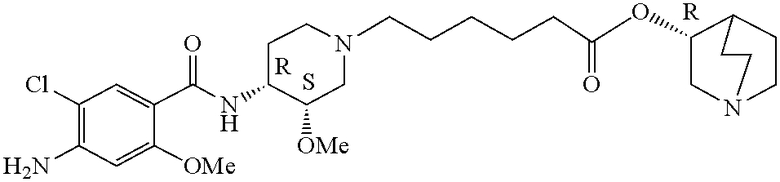
В кислой среде 1-бензилпиперидин-4-он (1) и бромистоводородная кислота взаимодействовали в присутствии уксусной кислоты с образованием N-бензил-3-бромпиперидин-4-она (2). Действие на 2 метоксида натрия и раствора метанола давало 1-бензил-4,4-диметоксипиперидин-3-ол (3). [Присутствие бета-аминогруппы сводит к нулю вероятность протекания реакции по типу Фаворского]. Осуществляли метилирование гидроксильной группы с использованием гидридного основания, с последующим воздействием иодометаном в присутствии ДМФА в качестве растворителя, с получением соединения 4.
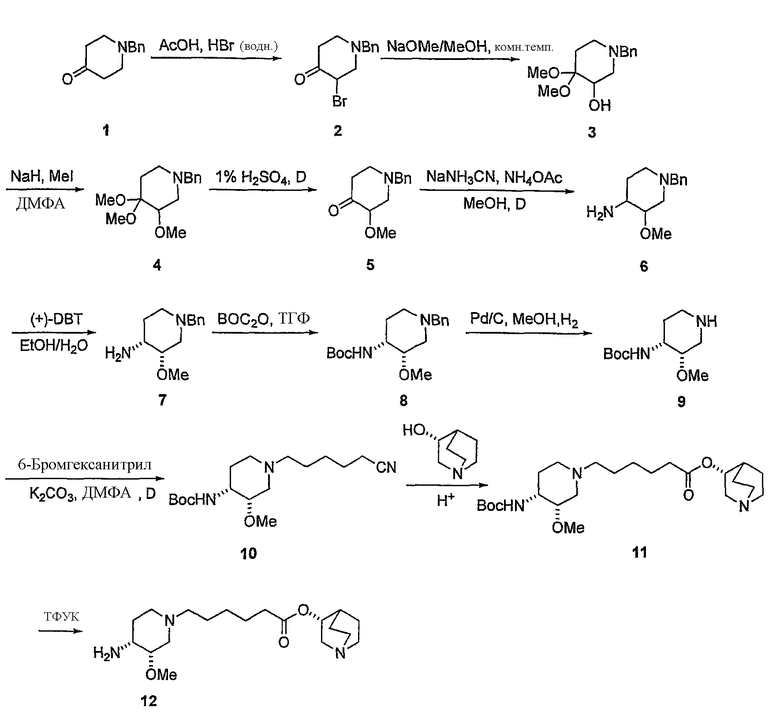
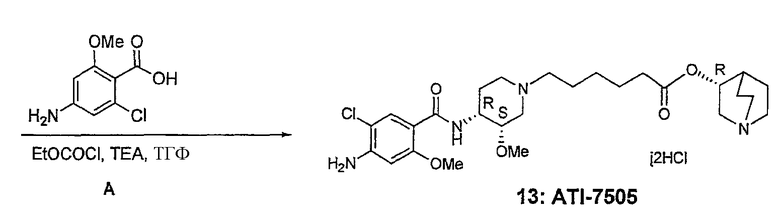
В результате последующего гидролиза ацеталя с использованием 1% серной кислоты при нагревании получали пиперидин 5, который затем подвергали восстановительному аминированию с использованием, например, цианоборогидрида натрия и ацетата аммония в метаноле, с получением 1-бензил-3-метоксипиперидин-4-амина (6). На данной стадии 6 может быть подвергнуто хиральному разделению. Для этого может быть использован (+)-DBT или другой вариант винной кислоты в присутствии подходящего растворителя для получения исключительно асимметрично чистого соединения 7. Для защиты первичного амина в 7 Boc-группой можно использовать Boc-ангидрид в присутствии растворителя ТГФ, с получением 8. Реакция дебензилирования путем гидрогенолиза с использованием Pd/C в метаноле в присутствии атмосферного газообразного водорода обеспечивала переход к этапу алкилирования. В результате воздействия 6-бромгексаннитрилом в присутствии слабого основания и ДМФА получали соединение 10. В результате превращения нитрила в сложный эфир с использованием (R)-хинуклидинола в присутствии разбавленной кислоты получали 11. В результате последующего удаления Boc-группы с использованием TFA получали свободный амин, который может участвовать в реакции сочетания с требуемой бензойной кислотой в присутствии сопрягающего реагента, такого как этилхлорформиат (и более предпочтительно, изобутилхлорформат), с получением ATI-7505 в энантиомерно чистом виде.
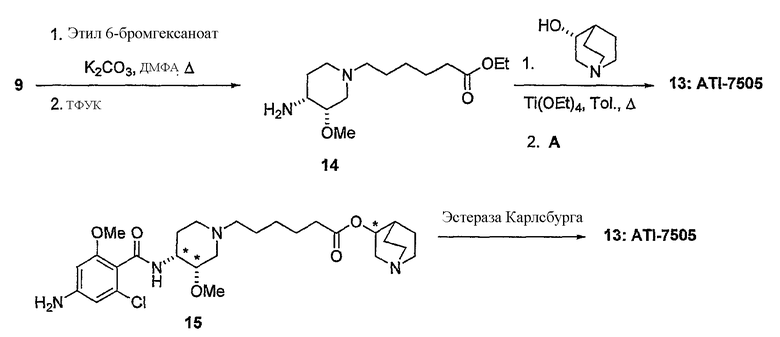
Альтернативно, соединение 9 можно алкилировать с использованием 6-бромгексаноата в присутствии слабого основания. После удаления защитной Boc-группы получали соединение 14. Путем трансэтерификации 14 посредством титана с использованием (R)-хинуклидинола и тетраэтоксида титана в растворителе толуоле получали ATI-7505. Эстераза Карлсбурга гидролизует сложные эфиры, которые имеют S-конфигурацию, поэтому сложные эфиры R-конфигурации остаются интактными. Следовательно, ATI-7505 можно также получать, воздействуя на диастереоизомерные смеси 15 эстеразой Карлсбурга.
Способ 3
Альтернативный синтез дигидрохлоридной соли (R)-хинуклидин-3-ил 6-((3S,4R)-4-(4-амино-5-хлор-2-метоксибензамид)-3-метоксипиперидин-1-ил)гексаноата - дигидрохлоридной соли ATI-7505:
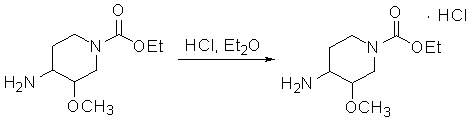
При интенсивном перемешивании хлорид водорода в диэтиловом эфире (приблизительно 1,4 молярной части) медленно добавляли к раствору карбамата пиперидина (приблизительно 1,0 молярная часть). Смесь перемешивали в течение приблизительно 8 часов, после чего фильтровали и промывали диэтиловым эфиром. Твердое вещество белого цвета дополнительно промывали дихлорметаном и диэтиловым эфиром (в объемном соотношении приблизительно 1:1) с целью удаления примесей, а затем сушили в условиях вакуума, с получением рацемической гидрохлоридной соли карбамата пиперидина в виде твердого вещества белого цвета.
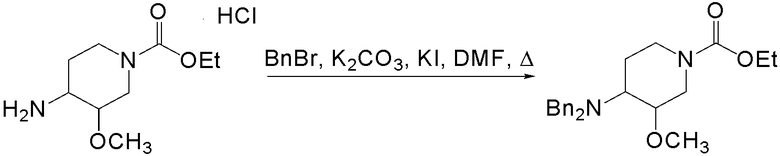
К смеси гидрохлорида пиперидина (приблизительно 1,0 молярная часть), карбоната калия (K2CO3, приблизительно 2,4 молярной части) и иодида калия (KI, приблизительно 0,1 молярной части) в диметилформамиде при комнатной температуре (к/т) добавляли бензилбромид (приблизительно 2,2 молярной части). Реакционную смесь нагревали приблизительно до 75°C. Приблизительно через 18 часов реакционную смесь охлаждали до комнатной температуры, разбавляли водой и экстрагировали этилацетатом (ЕА). Органический слой промывали солевым раствором, а затем сушили над безводн. (безводным) сульфатом натрия (Na2SO4). Затем путем фильтрования в условиях вакуума и концентрирования получали неочищенный масляный продукт. Продукт осаждали добавлением смеси изопропанола и воды (в объемном соотношении приблизительно 1:1) и перемешиванием. После фильтрования в условиях вакуума получали дибензиламинопиперидин в виде твердого вещества белого цвета.
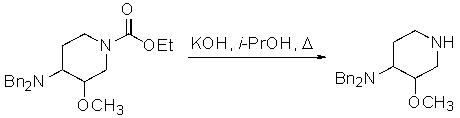
Гидроксид калия (приблизительно 10 молярных частей) по частям добавляли к перемешанному раствору дибензиламинокарбамата (приблизительно 1,0 молярная часть) в изопропаноле при комнатной температуре и смесь перемешивали и нагревали до температуры кипения с обратным холодильником. По истечении приблизительно 5 часов реакционную смесь охлаждали до комнатной температуры, а растворитель удаляли в условиях вакуума приблизительно до половины объема. Реакционную смесь разбавляли водой и экстрагировали этилацетатом. После промывания солевым раствором продукт сушили над безводным Na2SO4. Затем путем фильтрования в условиях вакуума получали пиперидин в виде полутвердого вещества.
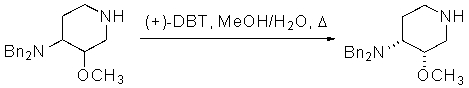
Хиральное разделение 3,4-двузамещенного пиперидина:
(+)-2,3-дибензоил-D-винную кислоту [(+)-DBT; приблизительно 1,0 молярная часть] растворяли в метаноле и медленно добавляли в нагретый раствор (приблизительно 70°C) двузамещенного пиперидина (приблизительно 1,0 молярная часть) в метаноле и воде (объемное соотношение приблизительно 1:1). Смесь перемешивали при указанной температуре в течение приблизительно 1 часа, после чего прекращали нагрев и продолжали перемешивание при комнатной температуре в течение нескольких часов, например, в одном случае в течение приблизительно 16 часов. Полученную соль собирали вакуумной фильтрацией и промывали метанолом и водой (в объемном соотношении приблизительно 1:1). Влажный осадок собирали и перекристаллизовывали еще два раза с использованием описанной процедуры.
Влажный осадок суспендировали в воде и добавляли 1 н. гидроксид натрия (до достижения pH приблизительно 12). Полученную суспензию перемешивали в течение приблизительно 3 часов при комнатной температура, после чего экстрагировали этилацетатом. Органический слой промывали солевым раствором, фильтровали и концентрировали, с получением энантиомерно обогащенного 3,4-двузамещенного пиперидина в виде твердого вещества белого цвета.
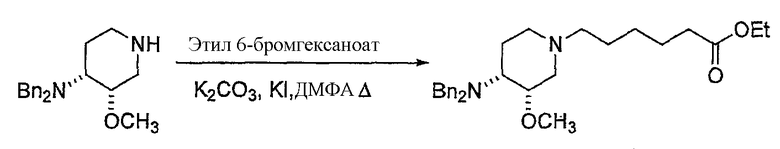
К смеси пиперидина (приблизительно 1,0 молярная часть), K2CO3 (приблизительно 1,2 молярной части) и KI (приблизительно 0,1 молярной части) в растворителе ДМФА медленно добавляли этил 6-бромгексаноат (приблизительно 1,1 молярной части). Реакционную смесь нагревали приблизительно до 70°C при перемешивании в течение приблизительно 10 часов, после чего охлаждали до комнатной температуры и разбавляли водой и экстрагировали этилацетатом. Органический слой отделяли, а затем промывали солевым раствором и, наконец, сушили над безводным Na2SO4. Затем путем фильтрования и концентрирования получали неочищенное масло. Неочищенный продукт очищали с использованием колоночной флэш-хроматографии (например, с объемным соотношением гексаны:этилацетат 1:1), с получением продукта в виде легкого масла коричневатого цвета.
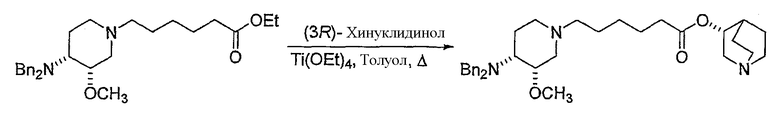
К смеси указанного выше сложного эфира пиперидина (приблизительно 1,0 молярная часть) и (3R)-хинуклидинола (приблизительно 4,0 молярные части) добавляли тетраэтоксид титана (IV) (приблизительно 1,0 молярную часть) при комнатной температуре. Реакционную смесь нагревали приблизительно до 85°C при пониженном давлении для удаления образующегося этанола. Приблизительно через 18 часов реакционную смесь охлаждали до комнатной температуры, а затем разбавляли этилацетатом и гасили водой. Органические слои промывали солевым раствором и сушили над безводным Na2SO4. После концентрирования неочищенное масло очищали с использованием колоночной флэш-хроматографии (например, приблизительно 100:10:1; CH2Cl2:MeOH:NH4OH), с получением продукта в виде прозрачного масла.
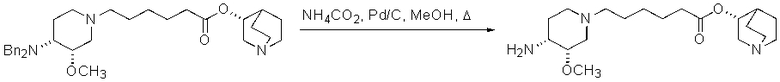
В реакционную колбу, содержащую палладий на угле, добавляли раствор указанного выше дибензилпиперидинового эфира (приблизительно 1,0 молярная часть) в метаноле и к указанной смеси добавляли формиат аммония (приблизительно 4 молярные части). Реакционную смесь нагревали до температуры кипения с обратным холодильником и по истечении приблизительно 10 часов реакционную колбу охлаждали до комнатной температуры и отфильтровывали палладий на угле через целитный фильтр. Фильтрат концентрировали до масла, которое очищали с использованием колоночной флэш-хроматографии (например, SiO2; приблизительно 150:10:1; CH2Cl2:CH3OH:NH4OH), с получением продукта аминопиперидинового эфира в виде масла желтого цвета.
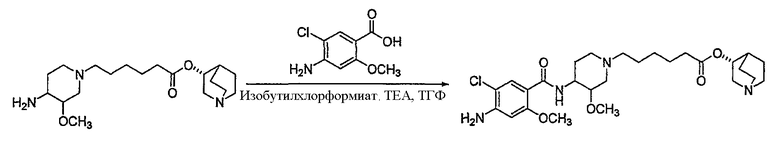
К перемешанному раствору 4-амино-5-хлор-2-метоксибензойной кислоты (приблизительно 1,2 молярной части) и триэтиламина (приблизительно 2,2 молярной части) в тетрагидрофуране (ТГФ) медленно добавляли изобутилхлорформиат (приблизительно 1,2 молярной части) при комнатной температуре. Приблизительно через 30 минут раствор пиперидинового эфира (приблизительно 1,0 молярная часть) в ТГФ добавляли к предварительно изготовленному, смешанному ангидриду. Реакционную смесь перемешивали при комнатной температуре в течение приблизительно 14 часов, после чего разбавляли насыщенным раствором бикарбоната натрия. Продукт экстрагировали с использованием, например, этилацетата и отделенный органический слой затем промывали солевым раствором и сушили над безводным сульфатом натрия. После фильтрования и концентрирования получали свободное основание ATI-7505.
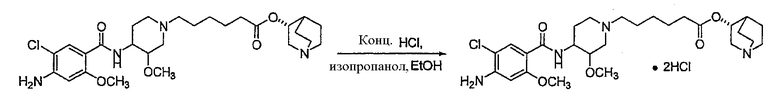
Свободное основание ATI-7505 растворяли в этаноле и изопропаноле (объемное соотношение приблизительно 1:1) и охлаждали на ледяной бане. К ледяному раствору медленно добавляли концентрированную хлористоводородную кислоту, а затем нагревали до комнатной температуры. После перемешивания в течение приблизительно 7 часов при комнатной температуре твердое вещество отфильтровывали и промывали этанолом и изопропанолом (объемное соотношение приблизительно 1:1), с получением влажного осадка. Влажный осадок ресуспендировали в этаноле, а затем нагревали до температуры кипения с обратным холодильником. Перемешиваемый раствор нагревали до комнатной температуры и оставляли для перекристаллизации. Продукт фильтровали в условиях вакуума, промывали этанолом, а затем сушили в условиях вакуума, с получением дигидрохлоридной соли ATI-7505 в виде твердого вещества белого цвета.
Способ 4:
Альтернативный способ синтеза дигидрохлоридной соли (R)-хинуклидин-3-ил 6-((3S,4R)-4-(4-амино-5-хлор-2-метоксибензамид)-3-метоксипиперидин-1-ил)гексаноата дигидрохлоридной соли - ATI-7505:
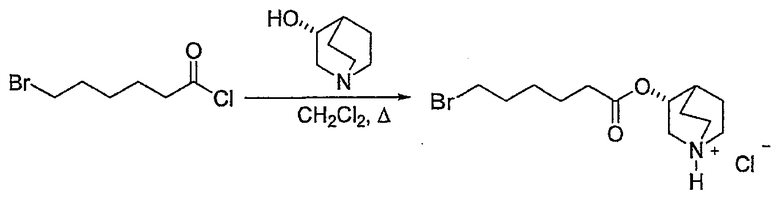
К смеси (3R)-хинуклидинола в дихлорметане по капле добавляли 6-бромгексаноилхлорид. Реакционную смесь нагревали до температуры кипения с обратным холодильником и приблизительно через 18 часов охлаждали до комнатной температуры. Непрореагировавший (3R)-хинуклидинол отфильтровывали и к фильтрату добавляли диэтиловый эфир для осаждения требуемого продукта. Продукт фильтровали в условиях вакуума и промывали CH2Cl2 и диэтиловым эфиром (объемное соотношение приблизительно 1:1), с получением продукта в виде твердого вещества белого цвета.
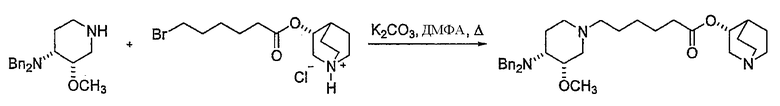
Дибензиламинопиперидин (приблизительно 1,0 молярную часть) добавляли к смеси 6-бромалканоилового эфира (приблизительно 1,0 молярной части) и карбоната калия (приблизительно 2,2 молярной части) в растворителе ДМФА. Реакционную смесь перемешивали при температуре приблизительно 70°C в течение приблизительно 11 часов, после чего охлаждали до комнатной температуры и разбавляли насыщенным раствором бикарбоната натрия. Продукт экстрагировали этилацетатом. Затем продукт промывали солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали, с получением продукта в виде бесцветного масла.
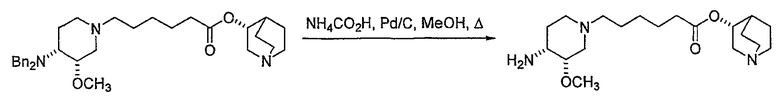
В реакционную колбу, содержащую палладий на угле, добавляли раствор указанного выше дибензилпиперидинового эфира (приблизительно 1,0 молярная часть) в метаноле и к указанной смеси добавляли формиат аммония (приблизительно 4 молярных части). Реакционную смесь нагревали до температуры кипения с обратным холодильником и по истечении приблизительно 10 часов реакционную колбу охлаждали до комнатной температуры и отфильтровывали палладий на угле, например, через целитный фильтр. Фильтрат концентрировали до масла, которое очищали с использованием колоночной флэш-хроматографии (например, SiO2: приблизительно 150:10:1; CH2Cl2:CH3OH:NH4OH), с получением продукта аминопиперидинового эфира в виде масла желтого цвета.
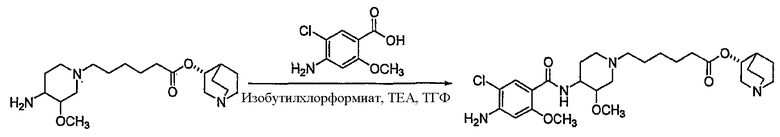
К перемешанному раствору 4-амино-5-хлор-2-метоксибензойной кислоты (приблизительно 1,2 молярной части) и триэтиламина (приблизительно 2,2 молярной части) в тетрагидрофуране (ТГФ) медленно добавляли изобутилхлорформиат (приблизительно 1,2 молярной части) при комнатной температуре. Приблизительно через 30 минут раствор пиперидинового эфира (приблизительно 1,0 молярная часть) в ТГФ добавляли к предварительно изготовленному, смешанному ангидриду. Реакционную смесь перемешивали при комнатной температуре в течение приблизительно 14 часов, после чего разбавляли насыщенным раствором бикарбоната натрия. Продукт экстрагировали с использованием, например, этилацетата, и отделенный органический слой затем промывали солевым раствором и сушили над безводным сульфатом натрия. После фильтрования и концентрирования получали свободное основание ATI-7505.
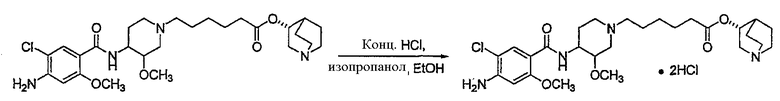
Свободное основание ATI-7505 растворяли в этаноле и изопропаноле (объемное соотношение приблизительно 1:1) и охлаждали на ледяной бане. К ледяному раствору медленно добавляли концентрированную хлористоводородную кислоту, а затем нагревали до комнатной температуры. После перемешивания в течение приблизительно 7 часов при комнатной температуре твердое вещество отфильтровывали и промывали этанолом и изопропанолом (объемное соотношение приблизительно 1:1), с получением влажного осадка. Влажный осадок ресуспендировали в этаноле, а затем нагревали до температуры кипения с обратным холодильником. Перемешиваемый раствор нагревали до комнатной температуры и оставляли для перекристаллизации. Продукт фильтровали в условиях вакуума, промывали этанолом, а затем сушили в условиях вакуума, с получением дигидрохлоридной соли ATI-7505 в виде твердого вещества белого цвета.
Способ 5:
Альтернативный способ синтеза дигидрохлоридной соли (R)-хинуклидин-3-ил 6-((3S,4R)-4-(4-амино-5-хлор-2-метоксибензамид)-3-метоксипиперидин-1-ил)гексаноата дигидрохлоридной соли - ATI-7505:
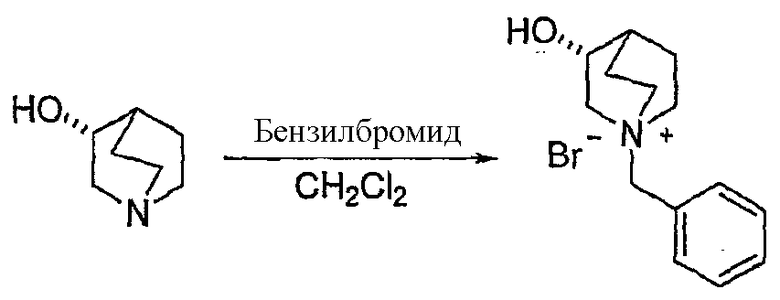
Бензилбромид (приблизительно 1,2 молярной части) добавляли к раствору (3R)-хинуклидинола (приблизительно 1,0 молярная часть) в дихлорметане. Реакционную смесь перемешивали при комнатной температуре в течение приблизительно 4 часов, затем фильтровали и промывали дихлорметаном, с получением продукта в виде твердого вещества белого цвета.
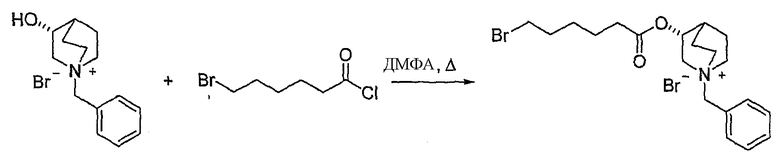
6-бромгексаноилхлорид (приблизительно 1,1 молярной части) добавляли к раствору бензилзащищенного (3R)-хинуклидинола (приблизительно 1,0 молярная часть) и реакционную смесь нагревали приблизительно до 60°C. По истечении приблизительно 12 часов реакционную смесь охлаждали до комнатной температуры и продукт осаждали добавлением диэтилового эфира. После вакуумной фильтрации, промывания эфиром и сушки получали продукт в виде аморфного твердого вещества.
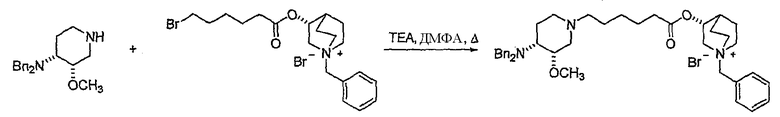
Смесь пиперидина (приблизительно 1,0 молярная часть), алканоилгалогенидного эфира (приблизительно 1,0 молярная часть) и триэтиламина (приблизительно 2,0 молярной части) В растворителе ДМФА нагревали приблизительно до 60°C в течение приблизительно 6 часов. Затем реакционную смесь охлаждали до комнатной температуры, разбавляли насыщенным раствором бикарбоната натрия и экстрагировали этилацетатом. После промывания солевым раствором и сушки над безводным сульфатом натрия органический слой концентрировали, с получением продукта в виде бесцветного масла.

В реакционную колбу, содержащую палладий на угле, добавляли раствор дибензилпиперидинового эфира (приблизительно 1,0 молярная часть) в метаноле и к указанной смеси добавляли формиат аммония (приблизительно 4 молярных части). Реакционную смесь нагревали до температуры кипения с обратным холодильником и приблизительно через 10 часов реакционную колбу охлаждали до комнатной температуры и отфильтровывали палладий на угле, например, через целитный фильтр. Фильтрат концентрировали до масла и очищали с использованием колоночной флэш-хроматографии (например, SiO2; приблизительно 150:10:1; CH2Cl2:CH3OH:NH4OH), с получением продукта аминопиперидинового эфира в виде масла желтого цвета.
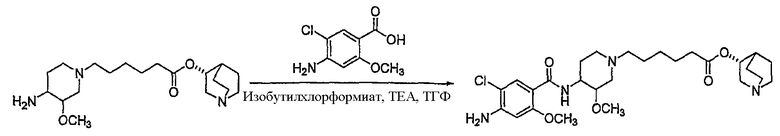
К перемешанному раствору 4-амино-5-хлор-2-метоксибензойной кислоты (приблизительно 1,2 молярной части) и триэтиламина (приблизительно 2,2 молярной части) в тетрагидрофуране (ТГФ) медленно добавляли изобутилхлорформиат (приблизительно 1,2 молярной части) при комнатной температуре. Приблизительно через 30 минут раствор пиперидинового эфира (приблизительно 1,0 молярная часть) в ТГФ добавляли к предварительно изготовленному, смешанному ангидриду. Реакционную смесь перемешивали при комнатной температуре в течение приблизительно 14 часов, после чего разбавляли насыщенным раствором бикарбоната натрия. Продукт экстрагировали, с использованием, например, этилацетата и отделенный органический слой затем промывали солевым раствором и сушили над безводным сульфатом натрия. После фильтрования и концентрирования получали свободное основание ATI-7505.
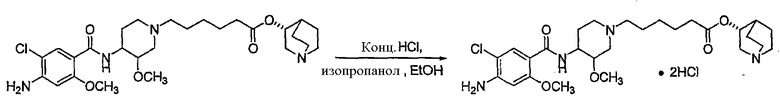
Свободное основание ATI-7505 растворяли в этаноле и изопропаноле (объемное соотношение приблизительно 1:1) и охлаждали на ледяной бане. К ледяному раствору медленно добавляли концентрированную хлористоводородную кислоту, а затем смесь нагревали до комнатной температуры. После перемешивания в течение приблизительно 7 часов при комнатной температуре твердое вещество отфильтровывали и промывали этанолом и изопропанолом (объемное соотношение приблизительно 1:1), с получением влажного осадка. Влажный осадок ресуспендировали в этаноле, а затем нагревали до температуры кипения с обратным холодильником. Перемешиваемый раствор нагревали до комнатной температуры и оставляли для перекристаллизации. Продукт фильтровали в условиях вакуума, промывали этанолом, а затем сушили в условиях вакуума, с получением дигидрохлоридной соли ATI-7505 в виде твердого вещества белого цвета.
Способ 6
Альтернативный способ синтеза дигидрохлоридной соли (R)-хинуклидин-3-ил 6-((3S,4R)-4-(4-амино-5-хлор-2-метоксибензамид)-3-метоксипиперидин-1-ил)гексаноата дигидрохлоридной соли - ATI-7505. Хотя ниже приведены конкретные условия реакций для примера синтеза способом 6, указанные условия не должны истолковываться как ограничение объема способа. Специалистам будет понятно, что в пределах указанного способа возможны изменения условий реакций, включая, без ограничения, продолжительность, температуры и растворители, используемые для реакций. Выходы реакций, где они указаны, также приводятся в качестве примера и, следовательно, могут меняться для каждого осуществления и комбинации условий реакций.
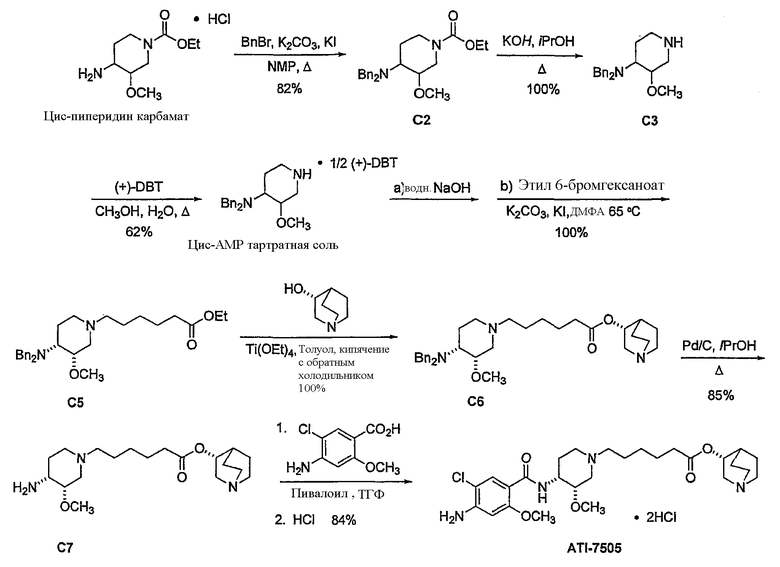
Синтез ATI-7505 из цис-APM тартрата осуществляли на основе процедуры лабораторной серии с использованием 9,7 г реагента.
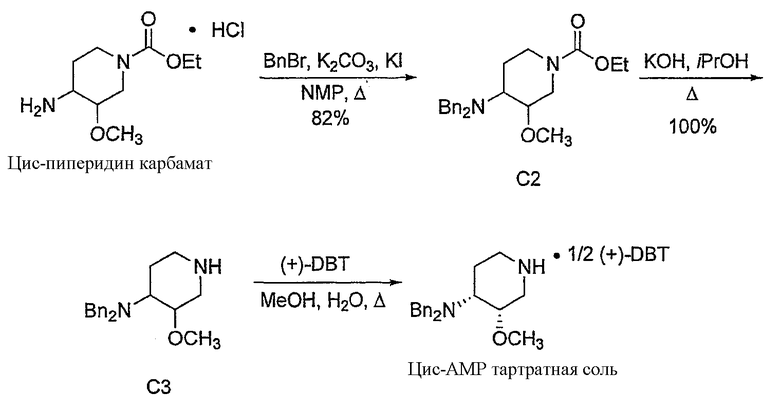
a. Синтез C2
Исходные материалы
цис-пиперидин карбамат, 24 кг
бензилбромид, 37,8 кг
KI, 1,67 кг
K2CO3, 48,7 кг
N-метилпирролидон (NMP), 200 кг
EA (этилацетат), 360 кг
вода, 600 кг
изопропиловый спирт (IPA)/вода (1:1 мас./мас.), 250 кг
Процедура
Цис-пиперидин карбамат (24 кг, 1 экв.) и K2CO3 (48,7 кг, 6 экв.) загружали в реактор, затем добавляли в реактор NMP (200 кг). В течение 15 минут перемешивали смесь при комнатной температуре. Добавляли в реактор KI (1,67 кг, 0,1 экв.), затем добавляли бензилбромид (37,8 кг, 2,2 экв.) и повышали температуру до 75°C в течение 60 минут. Отбирали образец реакционной смеси приблизительно через 4 часа; ожидаемая продолжительность реакции составляла приблизительно 7-9 часов.
После предположительного завершения реакции добавляли в реактор воду (350 кг) и экстрагировали с использованием EA (120 кг; 3 раза). Собирали слой EA и промывали слой EA водой (200 кг; 3 раза). Концентрировали слой EA до твердого вещества при 70°C. Добавляли в реактор IPA/воду (1:1, 200 кг) и нагревали реактор приблизительно до 75-80°C. Добавляли IPA/воду (1:1) частями по 25 кг при температуре 75-80°C до получения прозрачного раствора. Медленно охлаждали реакционную смесь до 5°C. Собирали твердое вещество фильтрованием и сушили влажный осадок при температуре приблизительно 60°C, с получением C2 (31,7 кг; выход 82%). Чистота полученной партии, определенная с использованием ВЭЖХ, составляла 99,3%.
b. Синтез C3
Исходные материалы
KOH, 56,3 кг
IPA, 200 кг
DCM (дихлорметан), 550 кг
Вода, 1300 кг
C2, 32 кг
Процедура
Добавляли в реактор C2 (32 кг, 1 экв.), KOH (56,3 кг, 12 экв.) и IPA (200 кг). Нагревали реакционную смесь до температуры кипения с обратным холодильником (приблизительно 82°C). Отбирали образец реакционной смеси приблизительно через 4 часа; ожидаемая продолжительность реакции составляла приблизительно 4-5 часов. После завершения реакции удаляли IPA путем дистилляции при температуре 50°C. Добавляли в реактор DCM (230 кг) и воду (700 кг) и собирали оба слоя. Осуществляли экстракцию водного слоя с использованием DCM (160 кг; 2 раза) и объединяли слои DCM. Промывали слой DCM водой (200 кг; 3 раза) и концентрировали слой DCM при температуре 70°C, с получением C3 в виде масла и переходили к следующему этапу, не выделяя продукт (предполагая 100% выход).
c. Синтез цис-АМФ тартратной соли
Исходные материалы
Добавляли в реактор, в котором содержалось полученное на предыдущем этапе соединение С3, метанол (260 кг) и воду (130 кг). Добавляли в реактор (+)-DBT (15,2 кг), растворенный в 130 кг метанола, при температуре 70°C, предпочтительно в течение 60 минут. Добавляли часть метанола (70 кг), чтобы убедиться в получении прозрачного раствора, затем охлаждали реакционную смесь до 50°C. Продукт получали приблизительно при 50°C; перед фильтрованием медленно охлаждали смесь приблизительно до 10°C. Собирали твердое вещество фильтрованием и, предпочтительно, проверяли величину энантиомерного избытка (е/е) и содержание твердого вещества.
Помещали твердое вещество в реактор. Добавляли в реактор MeOH/воду (5:1, 600 кг) и нагревали смесь до 70°C. Можно добавлять большее количество MeOH/воды (5:1), чтобы получить прозрачный раствор перед охлаждением реакционной смеси до 50°C. Медленно охлаждали смесь до 10°C, затем собирали твердое вещество фильтрованием. Сушили влажный осадок приблизительно при 60°C. В данном примере чистота полученного цис-AMP Ѕ (+)-DBT (12,6 кг, выход по массе 31% и теоретический выход 62%), определенная с использованием ВЭЖХ, составляла 99,8%, а е/е составлял 97,9%.
d. Синтез свободного основания цис-AMP
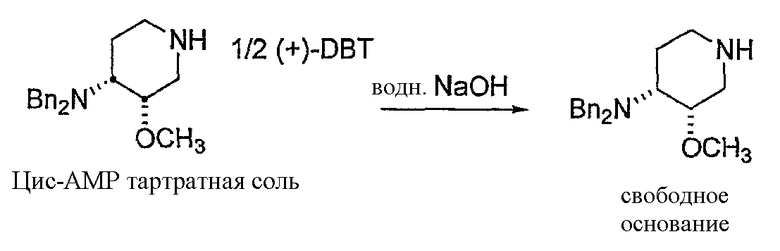
Исходные материалы
цис AMP 1/2 DBT, 10 г, 0,0279 моль
изопропиловый эфир (IPE), 50 мл
вода, 50 мл
45% NaOH, 7,2 г, 0,18 моль
Процедура
Пиперидин(+)-дибензоилтартратную соль (10,00 г; 0,0279 моль) суспендировали в 30 мл воды и 50 мл IPE при интенсивном перемешивании. 45% гидроксид натрия (7,2 г; 0,18 моль) по капле добавляли до растворения твердого вещества. Слои IPE промывали водой (10 мл; 2 раза) и после концентрирования получали твердое вещество белого цвета, представляющее собой неочищенное свободное основание (7,20 г).
e. Синтез C5
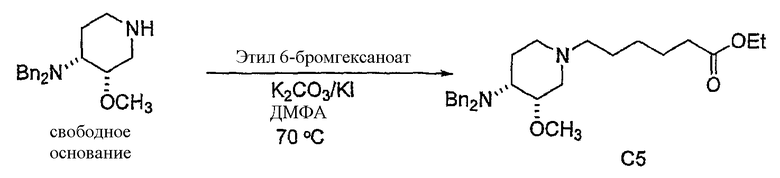
Исходные материалы
свободное основание, 7,2 г, 0,0232 моль
этил 6-бромгексаноат,4,76 г, 0,0214 моль
карбонат калия, 5,77 г, 0,0418 моль
иодид калия, 1,39 г, 8,37 ммоль
ДМФА (диметилформамид), 30 мл
изопропиловый эфир (IPE), 50 мл
вода, 50 мл
Процедура
Свободное основание (7,2 г), этил 6-бромгексаноат (4,75 г; 0,0214 моль), карбонат калия (5,77 г; 0,0418 моль), иодид калия (1,39 г; 8,37 ммоль) и ДМФА (30 мл) загружали в реактор. Реакционную смесь нагревали до 70°C в течение 1 часа и завершение реакции контролировали с использованием ВЭЖХ. Через 1 час реакционную смесь охлаждали до комнатной температуры и гасили добавлением 30 мл воды и 50 мл IPE. Слои IPE промывали водой (10 мл; 2 раза). После концентрирования получали неочищенное соединение С5 в виде масла желтого цвета (9,10 г).
f. Синтез C6
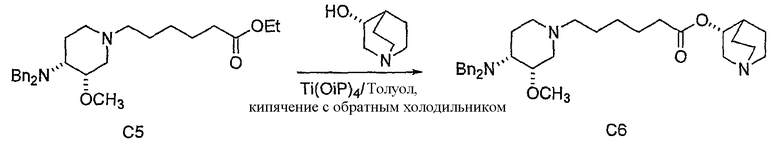
Исходные материалы
C5, 9,10 г, 0,0201 моль
(R)-3-хинуклидинол, 5,20 г, 0,0409 моль
Ti(OiP)4 (изопропоксид титана (IV)), 1,16 г, 4,08 ммоль
толуол, 120 мл
изопропиловый эфир, 60 мл
вода, 80 мл
Процедура
C5 (9,10 г; 0,0201 моль), (R)-3-хинуклидинол (5,20 г; 0,0409 моль), Ti(OiP)4 (1,16 г; 4,08 ммоль) и толуол (120 мл) загружали в реактор при перемешивании. Реактор оборудовали насадочной колонной (24/40; длина 15 см) и коротким каналом (24/40), который нагревали для отгонки EtOH, IPA и толуола (температура масляной бани 160°C). Контроль завершения реакции осуществляли путем ВЭЖХ. В данном примере синтеза ВЭЖХ продемонстрировала, что исходный материал был израсходован полностью. Для облегчения удаления толуола давление уменьшали. Реакционную смесь охлаждали до комнатной температуры и гасили добавлением 40 мл воды и 60 мл IPE, затем промывали слои IPE водой (20 мл; 2 раза) и концентрировали, с получением неочищенного C6 в виде масла желтого цвета (11,70 г).
g. Синтез C7
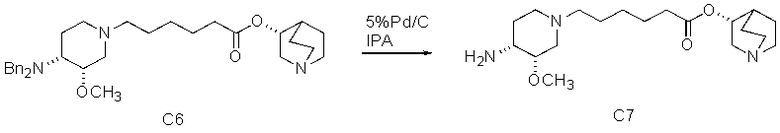
Исходные материалы
C6, 11,70 г, 0,0220 моль
5% Pd/C, 1,0 г
IPA, 30 мл
Процедура
C7 (11,70 г), 5% Pd/C (1,0 г) и IPA (30 мл) загружали в гидрогенизационный реактор (N2 инертный; H2 под давлением 5 атмосфер). Смесь перемешивали и нагревали на водяной бане до 70°C в течение 7 часов. Контроль завершения реакции осуществляли с использованием ВЭЖХ и ТСХ, которые показали, что исходные материалы были использованы полностью. Реакционную смесь охлаждали до комнатной температуры и фильтровали через целитный фильтр с промыванием IPA. После концентрирования фильтрата получали 6,66 г С7 в виде неочищенного масла.
h. Синтез основания ATI-7505
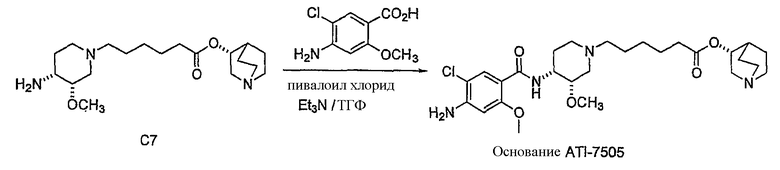
пивалоилхлорид
основание ATI-7505
Исходные материалы
4-амино-5-хлор-2-метоксибензойная кислота, 5,0 г, 0,0249 моль
ТГФ, 30 г
триэтиламин, 4,7 г, 0,0465 моль
пивалоилхлорид, 2,7 г, 0,0225 моль
C7, 66,6 г, 0,0189 моль
диэтилкетон (DEK), 100 мл
32% HCL
вода
45% NaOH
Процедура
Пивалоилхлорид (2,7 г; 0,0225 моль) по капле добавляли к раствору 4-амино-5-хлор-2-метоксибензойной кислоты (5,0 г; 0,0249 моль) и триэтиламина (4,7 г; 0,0465 ммоль) в ТГФ (20 г) при комнатной температуре. После добавления реакционная смесь помутнела и спустя 60 минут к данному предварительно изготовленному смешанному ангидриду добавляли раствор C7 (6,66 г; 0,0189 моль) в ТГФ (10 г) и оставляли перемешиваться при комнатной температуре. ВЭЖХ и ТСХ продемонстрировали, что исходный материал был израсходован полностью.
Реакцию гасили добавлением воды (40 мл) и DEK (40 мл) и добавляли 32% HCl до pH 4,0. Объединенные органические слои промывали водой (10 мл; 2 раза) и водный слой собирали. Добавляли DEK (60 мл) к водному слою и добавляли 45% NaOH до pH 12. Водный слой экстрагировали, отделяли и сливали. Объединенные органические слои промывали водой (10 мл; 2 раза) и концентрировали, с получением 12,01 г основания ATI-7505 в виде масла желтого цвета.
i. Синтез ATI-7505
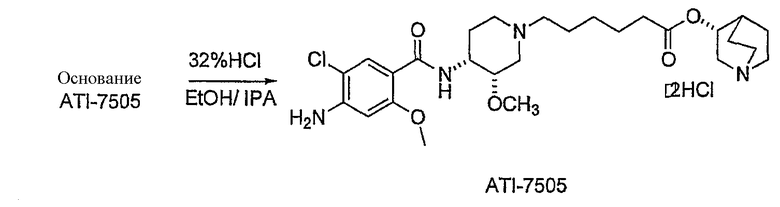
основание ATI-7505
Исходные материалы
основание ATI-7505, 12,01 г
этанол (EtOH), 50 мл
IPA, 70 мл
32% HCL
Процедура
12,01 г неочищенного основания ATI-7505 растворяли в 50 мл EtOH. При перемешивании медленно добавляли концентрированную 32% HCl до pH 4,1. После перемешивания в течение приблизительно 16 часов добавляли 50 мл IPA и реакционную смесь перемешивали в течение 2 часов. Твердое вещество отфильтровывали и промывали 20 мл IPA. Твердое вещество сушили до постоянного веса, с получением 9,71 г ATI-7505 в виде твердого вещества белого цвета. Чистота по данным ВЭЖХ составляла 98,65 %.
Пример 1
Получение дигидрохлоридной соли 1-азабицикло[2.2.2]окт-3'R-илового эфира 6-[4R-(4-амино-5-хлор-2-метоксибензоиламин)-3S-метоксипиперидин-1-ил]гексановой кислоты
Этап 1: Разделение рацемического норцизаприда
(-)-2,3-дибензоил-L-винную кислоту ((-)-DBT, приблизительно 1 массовая часть) растворяли в этаноле и фильтровали для удаления остаточных частиц. Отдельно растворяли рацемический норцизаприд (приблизительно 0,8 массовой части) в смеси этанола и воды, а затем фильтровали. Фильтрат нагревали приблизительно до 75°C, затем добавляли раствор (-)-DBT. После перемешивания при указанной температуре в течение приблизительно 30 минут смесь медленно охлаждали в течение нескольких часов приблизительно до 5°C и полученную соль собирали вакуумной фильтрацией и промывали смесью EtOH/H2О. Влажный осадок перекристаллизовывали из EtOH/H2О нагреванием приблизительно до 79°C и медленным охлаждением приблизительно до 5°C, как и ранее. Продукт собирали на вакуумном фильтре и промывали EtOH/H2О, с получением влажного осадка.
Влажный осадок суспендировали в воде и рН доводили приблизительно до 12 с использованием 7% (мас./мас.) водн. NaOH. Полученную суспензию перемешивали в течение 3 часов при комнатной температуре, после чего фильтровали в условиях вакуума, промывали твердый материал водой и сушили в условиях вакуума. Затем, чтобы получить соль, на продукт повторно воздействовали (-)-DBT, с использованием указанной выше общей процедуры. Выделенную соль затем нейтрализовали водн. NaOH, как описано выше. Продукт выделяли на фильтре и сушили, как описано ранее, с получением основания (+)-норцизаприда (приблизительно 0,25 массовой части). По данным хиральной ВЭЖХ е/е составлял приблизительно 100% (+)-норцизаприда. Оптическое вращение составляло приблизительно +5° (метанол; 25°C и 589 нм), подтверждая наличие положительного изомера норцизаприда.
Этап 2: Связывание с этил 6-бромгексаноатом
(+)-Норцизаприд (приблизительно 1 массовая часть), карбонат калия (приблизительно 0,48 массовой части) и йодид калия (приблизительно 0,063 массовой части) суспендировали в безводном этаноле USP. Этил 6-бромгексаноат (приблизительно 0,76 массовой части) медленно добавляли в суспензию при комнатной температуре. Смесь нагревали до температуры кипения с обратным холодильником до завершения реакции. После охлаждения до комнатной температуры реакционную смесь фильтровали для удаления, например, неорганических твердых веществ, и фильтрат концентрировали при пониженном давлении приблизительно до половины объема. Продукт осаждали медленным добавлением неочищенного материала в холодную воду (приблизительно 13 массовых частей) при быстром перемешивании. Осадок фильтровали в условиях вакуума и промывали водой, а затем осаждали еще два раза растворением в безводном этаноле и медленным добавлением в холодную воду, как описано выше. Полученный влажный осадок промывали н-гептаном и ресуспендировали в этилацетате и н-гептане (1:9; об./об.) и перемешивали в течение приблизительно 1 часа, фильтровали и сушили в условиях вакуума, с получением 0,73 массовой части продукта связывания в виде твердого вещества белого цвета.
Этап 3: Связывание с (R)-3-хинуклидинолом и образование дигидрохлоридной соли
Сложный эфир (1 массовая часть) и (R)-3-хинуклидинол (приблизительно 1,12 массовой части) суспендировали в толуоле, после чего медленно добавляли этоксид титана (IV) (приблизительно 0,5 массовой части) в перемешанную суспензию. Под потоком азота смесь нагревали приблизительно до 91°C и к колбе применяли частичный вакуум через дистиллятор с целью азеотропного удаления этанола. Для поддержания минимального объема растворителя в колбу дополнительно добавляли толуол. Реакцию считали завершенной по истечении приблизительно 33 часов.
Смесь охлаждали приблизительно до комнатной температуры и пятикратно экстрагировали водой. Органический слой концентрировали при пониженном давлении и полученный остаток повторно растворяли в EtOH/iPrOH (приблизительно 1:1 об./об.), а затем фильтровали через фильтр с диаметром пор мембраны 0,45 микрон для удаления всех частиц. Концентрированную хлористоводородную кислоту медленно добавляли в перемешанный фильтрат для осаждения требуемого продукта в виде дигидрохлоридной соли. Полученную суспензию перемешивали в течение нескольких часов при комнатной температуре, собирали с использованием вакуумной фильтрации и промывали EtOH/iPrOH (1:1; об./об.), с получением 0,53 массовой части неочищенной соли.
Неочищенную дигидрохлоридную соль ресуспендировали в этаноле, нагревали до температуры кипения с обратным холодильником, после чего охлаждали до комнатной температуры в течение приблизительно 1 часа. Продукт собирали вакуумной фильтрацией и промывали этанолом, а затем сушили на воздухе. Твердые вещества ресуспендировали в этаноле и нагревали до температуры приблизительно 55°C, с получением для прозрачного раствора, затем добавляли теплый изопропанол и продукт осаждали медленным охлаждением до комнатной температуры. Полученную суспензию перемешивали в течение нескольких часов с дальнейшей вакуумной фильтрацией и промывкой, например, изопропилом. Продукт сушили в условиях вакуума, сначала при комнатной температуре в течение нескольких часов, а затем при температуре приблизительно 55°C до достижения постоянной массы.
Пример 2
(+) и (-)-норцизаприд можно получать из его рацемической смеси разделением энантиомеров с использованием общепринятых средств, таких как оптически разделяющие кислоты, в соответствии со способом, описанным в патенте США №6147093 или в "Enantiomers, Racemates and Resolutions", J.Jacques, A.Collet, and S.H.Wilen (Wiley-Interscience, New York, NY) или в S.H. Wilen et al., Tetrahedron (1977) 33:2725.
4 изомера можно получать в малых количествах с использованием препаративной колоночной хроматографии, с последующим выпариванием растворителя. Указанный способ является пригодным для получения небольших количеств вещества для аналитических и исследовательских целей. Он представляет собой стандартный способ разделения, обычно применяющийся в аналитических лабораториях для выделения и изучения свойств метаболитов.
Возможные пути синтеза соединения IV, соединения VI и (+)-соединения II с использованием (+)-норцизаприда в качестве исходного материала описаны ниже. Пути синтеза соединения III, соединения V и (-)-соединения II являются идентичными, за исключением того, что исходным материалом является (-)-норцизаприд. Однако предпочтительно используются способы и процессы, описанные в способах 2-5, для получения соединений II-VI и других соединений по настоящему изобретению. Более предпочтительно используются способы 3-5 для получения соединений по настоящему изобретению.
Пример 3
Получение этилового эфира (+)-соединения II
Эквимолярную смесь (+)-норцизаприда и этил 6-бромгексаноата (по 1 эквиваленту), каталитическое количество KI и K2CO3 (2 эквивалента) в ДМФА нагревали приблизительно до 60°C в течение нескольких часов или пока анализ ТСХ не показывал завершение реакции. После охлаждения до комнатной температуры добавляли воду и смесь экстрагировали с использованием EtOAc. Объединенные органические экстракты последовательно промывали водой, 10% водным раствором LiCl и солевым раствором, затем сушили над Na2SO4. После концентрирования получали этиловый эфир (+)-соединения II.
Получение (+)-соединения II
Смесь неочищенного этилового эфира (+)-соединения II, полученного выше (1 экв.), KOH (2 М, 5 экв.) в MeOH и ТГФ (в достаточном для растворения количестве) перемешивали при комнатной температуре в течение приблизительно 1-2 часов. MeOH и ТГФ удаляли в условиях вакуума, а остаток разбавляли водой. Промывали органическим растворителем, таким как EtOAc. Водный слой подкисляли до pH~5 добавлением HCl. Осадок отфильтровывали и сушили, с получением (+)-соединения II.
Получение соединения IV и соединения VI
Смесь (+)-соединения II (1 экв.), HCl соли (R)-(-)-3-хинуклидинола (1 экв.), EDAC (1экв.) и DMAP (1 экв.) в ДМФА нагревали приблизительно до 50°C в течение ночи. После охлаждения и разбавления водой смесь очищали с использованием хроматографии или кристаллизации получали соединение IV. Подобным образом с использованием (S)-(+)-хинуклидинола получали соединение VI.
Следующие соединения получали, главным образом, в соответствии с описанными выше способами и процессами, в частности, в соответствии со способами 2-5, и, более предпочтительно, в соответствии со способами 3-5. Названия соединений составляли с использованием ChemDraw Ultra версии 8.03, которую можно получить от Cambridgesoft Corporation, или с использованием обеспечения ACD Namepro версии 6.0.
Изготовление композиций, введение и применение
Дозы и пути введения описанных соединений являются аналогичными уже используемым и известным специалистам (см., например, Physicians' Desk Reference, 54-е изд., Medical Economics Company, Montvale, NJ, 2000).
Величина профилактической и/или терапевтической дозы структурного и/или функционального аналога цизаприда для экстренного или постоянного лечения описанных в настоящем документе расстройств будет варьировать в зависимости от тяжести состояния и пути введения. Дозировка и, возможно, частота введения также будут варьировать в зависимости от возраста, массы тела и реакции отдельного пациента. В большинстве случаев общая суточная доза структурных и/или функциональных аналогов цизаприда для лечения описанных выше состояний составляет приблизительно от 1 мг до 200 мг, однократной или разделенными дозами. Предпочтительно, суточная доза должна составлять приблизительно от 5 мг до 100 мг, однократной или разделенными дозами; наиболее предпочтительно, суточная доза должна составлять приблизительно от 5 мг до 75 мг, однократной или разделенными дозами. Предпочтительно вводить дозы от 1 до 4 раз в день. В процессе лечения пациента терапию следует начинать с более низкой дозы, возможно, приблизительно от 5 мг до 10 мг, и повышать приблизительно до 50 мг и выше в зависимости от общей реакции пациента на лечение. Лечение детей и пациентов старше 65 лет, а также пациентов с нарушенной почечной или печеночной функцией рекомендуется начинать с малых доз, а затем дозы следует титровать в соответствии с индивидуальной реакцией (реакциями) и уровнем (уровнями) лекарственного средства в крови. В некоторых случаях специалистам будет понятно, что может возникнуть необходимость использования доз, выходящих за указанные пределы. Следует заметить, что клиническому или лечащему врачу будет понятно, как и когда прерывать, изменять или прекращать терапию в связи с реакцией конкретного пациента.
Соединения по настоящему изобретению можно изготавливать в различных композициях, в соответствии с известными способами изготовления фармацевтически пригодных композиций. Композиции подробно описаны во многих хорошо известных и доступных специалистам источниках. Например, в Remington's Pharmaceutical Science by E.W.Martin описаны композиции, которые можно использовать при осуществлении настоящего изобретения. В целом, композиции по настоящему изобретению изготавливают таким образом, что эффективное количество биологически активного соединения (соединений) объединяют с подходящим носителем, чтобы облегчить эффективное введение композиции.
Композиции по настоящему изобретению включают такие композиции, как суспензии, растворы и эликсиры; аэрозоли; или носители, такие как крахмалы, сахара, микрокристаллическая целлюлоза, разбавители, гранулирующие агенты, смазывающие агенты, связующие агенты, разрыхлители и т.п., в случае твердых лекарственных форм для перорального введения (таких как порошки, капсулы и таблетки); причем твердые лекарственные формы для перорального введения являются более предпочтительными, чем жидкие лекарственные формы для перорального введения. Предпочтительной твердой лекарственной формой для перорального введения являются капсулы. Наиболее предпочтительной твердой лекарственной формой для перорального введения являются таблетки. Предпочтительные количества активного ингредиента (т.е., структурного и/или функционального аналога цизаприда) в твердой дозированной форме составляет приблизительно 5 мг, 10 мг и 25 мг.
Кроме того, приемлемые носители могут быть твердыми или жидкими. Твердые лекарственные формы включают в себя порошки, таблетки, пилюли, капсулы, крахмальные капсулы, суппозитории и диспергируемые гранулы. Твердым носителем может быть одно или более веществ, которые могут действовать как разбавители, корригенты, солюбилизаторы, смазывающие агенты, суспендирующие агенты, связующие агенты, консерванты, разрыхлители для таблеток или инкапсулирующие материалы.
Описанные фармацевтические композиции могут быть разделены на стандартные дозы, содержащие подходящее количество активного компонента. Дозированная лекарственная форма может представлять собой упакованный препарат, такой как таблетки, капсулы и порошки, упакованные в бумажные или пластиковые контейнеры или во флаконы или ампулы. Кроме того, дозированная лекарственная форма может представлять собой препарат на жидкой основе или может изготавливаться для включения в твердые пищевые продукты, жевательную резинку или пастилку.
Помимо указанных выше основных лекарственных форм, соединения по настоящему изобретению можно вводить с использованием средств для контролируемого высвобождения и/или устройств для доставки, таких как, устройства, которые описаны в патентах США №3845770, 3916899, 3536809, 3598123 и 4008719, описания которых целиком включены в настоящий документ в качестве ссылок.
Любой подходящий путь введения можно использовать для введения пациенту эффективной дозы структурного и/или функционального аналога цизаприда. Например, можно использовать пероральный, ректальный, парентеральный (подкожный, внутримышечный, внутривенный), чрескожный и т.п. способы введения. Дозированные формы включают в себя таблетки, пастилки, дисперсии, суспензии, растворы, капсулы, пластыри и т.п.
Один аспект настоящего изобретения относится к способам и/или процессам получения соединений и композиций по настоящему изобретению.
Один аспект настоящего изобретения относится к способу лечения гастроэзофагеальной рефлюксной болезни у млекопитающего, при котором значительно уменьшаются сопутствующие неблагоприятные эффекты, связанные с введением цизаприда, и который включает в себя введение человеку, который нуждается в подобном лечении, терапевтически эффективного количества структурного и/или функционального аналога цизаприда или его фармацевтически приемлемой соли. Предпочтительным аспектом является лечение гастроэзофагеальной рефлюксной болезни у людей.
Другой аспект настоящего изобретения относится к композиции для лечения человека, страдающего гастроэзофагеальной рефлюксной болезнью, которая включает терапевтически эффективное количество структурного и/или функционального аналога цизаприда или его фармацевтически приемлемой соли.
Дополнительный аспект настоящего изобретения относится к способу достижения противорвотного эффекта у млекопитающего, при котором значительно уменьшаются неблагоприятные эффекты, связанные с введением цизаприда, и который включает введение млекопитающему, нуждающемуся в подобной противорвотной терапии, терапевтически эффективного количества структурного и/или функционального аналога цизаприда или его фармацевтически приемлемой соли. Предпочтительно, млекопитающим является человек.
В дополнительном аспекте настоящее изобретение относится к противорвотной композиции для лечения млекопитающего, которое нуждается в противорвотной терапии, которая включает терапевтически эффективное количество структурного и/или функционального аналога цизаприда или его фармацевтически приемлемой соли.
Дополнительный аспект настоящего изобретения относится к способу лечения состояния, вызванного нарушением моторики желудочно-кишечного тракта у млекопитающего, который включает введение млекопитающему, нуждающемуся в лечении нарушения моторики желудочно-кишечного тракта, терапевтически эффективного количества структурного и/или функционального аналога цизаприда или его фармацевтически приемлемой соли. Состояния, вызванные нарушением моторики желудочно-кишечного тракта, включают без ограничения, диспепсию, гастропарез, запор, послеоперационный парез кишечника и псевдонепроходимость кишечника. Предпочтительно, млекопитающим является человек.
Данные о том, что цизаприд проникает в центральную нервную систему и связывается с рецепторами 5HT4, указывает на то, что цизаприд может обладать центральными эффектами. Цизаприд является мощным лигандом 5HT4-рецепторов, которые расположены в нескольких областях центральной нервной системы. Модуляция серотонинергических систем вызывает ряд поведенческих эффектов. Соответственно, соединения по настоящему изобретению можно использовать для лечения: 1) когнитивных расстройств, включая, без ограничения, болезнь Альцгеймера; 2) расстройств поведения, включая, без ограничения, шизофрению, манию, обсессивно-компульсивное расстройство и расстройства, связанные с применением психотропных лекарственных средств; 3) расстройств настроения, включая, без ограничения, депрессию и тревожное состояние; и 4) расстройств контроля вегетативной функции, включая, без ограничения, эссенциальную гипертензию и расстройства сна.
Соответственно, настоящее изобретение относится также к способам лечения когнитивных, поведенческих расстройств, расстройств настроения или расстройств контроля вегетативных функций у млекопитающего, включающим введение терапевтически эффективного количества структурного и/или функционального аналога цизаприда или его фармацевтически приемлемой соли. Предпочтительно, млекопитающим является человек.
ATI-7505 обладает высоким сродством к 5-HT4-рецепторам
Известно, что 5-HT4-рецептор представляет собой основной подтип рецепторов, участвующих в прокинетической активности цизаприда в кишечнике. ATI-7505 обладает высоким сродством связывания с 5-HT4-рецептором при низких наномолярных значениях IC50. Как показано в таблице 1, сродство ATI-7505 к 5-HT4-рецептору было в 18 раз больше, чем у цизаприда, и, по крайней мере, в 360 раз больше, чем у основного метаболита ATI-7505, карбоновой кислоты.
Стандартный прототип антагониста 5-HT4-рецептора [3H]GR113808 (0,70 нM)
ATI-7505 представляет собой мощный частичный агонист 5-HT4-рецептора человека
ARYx осуществила исследования, основанные на стимуляции аденилатциклазой клеток, в которых методами генной инженерии обеспечена устойчивая экспрессия рецептора 5-HT4 человека. Было доказано, что ATI-7505 представляет собой мощный агонист 5-HT4-рецептора, в то время как его основной метаболит, ATI-7500, оказался относительно слабым агонистом (фигура 1 и таблица 2). Расчетный показатель EC50 для ATI-7505 (4 нM) был приблизительно в 10 раз меньше, чем для цизаприда (49 нM), и приблизительно в 100 раз меньше, чем для ATI-7500 (395 нM). Исходя из расчетного значения Emax, эффективность ATI-7505 по отношению к 5-HT (серотонину) составляла 85% (таблица 2); это означает, что ATI-7505 представляет собой частичный агонист 5-HT4-рецепторов.
pEC50 - отрицательный логарифм EC50
ATI-7505 ускоряет опорожнение желудка у накормленных собак
Для того чтобы оценить воздействие ATI-7505 на опорожнение желудка, были поставлены эксперименты на находящихся в сознании собаках после кормежки, с использованием тензометрических датчиков, размещенных на желудке и на тонкой кишке. Целью указанных экспериментов было измерение времени, которое требуется для возвращается мигрирующих моторных сокращений (ММС) к исходному уровню после употребления твердой пищи. Сокращение времени возвращения ММК к исходному уровню под воздействием лекарственного средства означало раннее завершение периода пищеварения за счет ускорения опорожнения желудка. Сразу же после прекращения ММК в средних отделах тонкой кишки, в течение 20 минут внутривенной (в/в) инфузией вводили различные дозы испытуемых лекарственных средств (растворитель, ATI-7505 или цизаприд). После окончания инфузии лекарственного средства собакам давали пищу. Регистрацию сокращений кишечника производили по меньшей мере в течение 60 минут до начала инфузии лекарственного средства для определения голодного состояния и для идентификации начала ММК в двенадцатиперстной кишке, и по крайней мере в течение 30 минут после возобновления ММК в двенадцатиперстной кишке. Количественное сравнение эффекта лекарственных препаратов проводили на основании времени возврата ММК к исходному уровню как показателя опорожнения желудка после употребления твердой пищи. Как представлено на фигуре 2, ATI-7505 значительно ускоряет время возврата ММК к исходному уровню, что указывает на ускорение опорожнения желудка у здоровых накормленных собак. Цизаприд демонстрировал схожее действие.
ATI-7505 усиливает моторику желудка и тонкой кишки, незначительно воздействуя на активность толстой кишки
Эксперименты проводились на голодных собаках, находящихся в сознании, чтобы оценить влияние ATI-7505 на моторику желудка, тонкой и толстой кишки, по сравнению с цизапридом. Конкретная цель исследования состояла в том, чтобы определить величины доз ATI-7505 (в/в и п/о), при введении которых характер и сила сократительной активности у собак наиболее приближены к таковым под действием цизаприда при использовании обычных терапевтических доз (0,5 мг/кг в/в; 1 мг/кг п/о).
При в/в и п/о введении как ATI-7505, так и цизаприд оказывали прокинетическое действие на кишечник собак. Начало действия обычно наблюдалось в течение 1-2 минут и 25-30 минут после в/в и п/о введения, соответственно. Воздействие ATI-7505 на моторику желудка и тонкой кишки соответствовало воздействию цизаприда. Оказалось, что подобно цизаприду, ATI-7505 вызывал дозозависимую стимуляцию сокращений антрального отдела желудка и тонкой кишки, при этом относительно незначительно воздействуя на моторику толстой кишки. Прокинетические эффекты, вызванные ATI-7505 в верхних отделах ЖК тракта, сопровождались небольшим, но значимым (p<0,05) увеличением частоты гигантских мигрирующих сокращений (GMC).
Введение ATI-7505 не было связано с развитием ретроградных гигантских мигрирующих сокращений (RGC). Подобно цизаприду, ATI-7505 оказывал минимальное воздействие на характеристики мигрирующего моторного комплекса (MMC) в антральном отделе желудка, а также в проксимальном, среднем и дистальном отделах тонкой кишки. В том, что касается частоты ММК и продолжительности Фазы III, было отмечено только одно значимое различие: п/о введение ATI-7505 увеличивало частоту ММК в проксимальном отделе тонкой кишки по сравнению с контролем. Собаки переносили в/в и п/о дозы ATI-7505 хорошо, и побочные эффекты, такие как диарея, анорексия или снижение массы тела, не наблюдались.
В целом, результаты показали, что при расчете доз в мг/кг ATI-7505 приблизительно в 2 раза сильнее цизаприда. Кроме того, эффекты ATI-7505, подобно эффектам цизаприда, соответствуют механизму, в основе которого лежит облегчение высвобождения ацетилхолина из нейронов кишечника, а не прямое воздействие на гладкие мышцы. Таким образом, ATI-7505 улучшает моторику желудка и тонкой кишки, подобно цизаприду, при минимальном воздействии или отсутствии воздействия на толстую кишку.
Метаболизм ATI-7505 происходит независимо от CYP450
На основе данных, полученных в ходе опытов с пулом микросом человека, ATI-7505 претерпевает биотрансформацию в единственный метаболит, ATI-7500, который, как оказалось, не подвергается дальнейшему метаболизму. Превращение ATI-7505 в ATI-7500 не зависит от присутствия NADPH. Таким образом, основной путь биотрансформации ATI-7505 не зависит от ферментов CYP450.
ATI-7505 не ингибирует ферменты CYP450
Для того, чтобы изучить способность ATI-7505 и/или его главного метаболита, ATI-7500, ингибировать CYP450 осуществляли скрининг указанных двух молекул с использованием теста Gentest Supersomes™. В соответствии с опубликованными сообщениями, цизаприд обладает значительной ингибирующей активностью по отношению к изоформам ферментов CYP450, CYP3A4, 2D6 и, в меньшей степени, 2C9. Ни соединение ATI-7505, ни его главный метаболит, ATI-7500, не показали значительной ингибирующей активности ни по отношению к указанным трем изоформам CYP450, ни к ряду других изоформ, которые, как известно, принимают участие в метаболизме лекарственных средств.
ATI-7505 обладает ничтожно малым сродством к каналам кардиомиоцитов IKr
Быстро активируемый калиевый (K+) ток задержанного выпрямления у человека (человеческий IKr) представляет собой K+ канал, который кодируется человеческим геном, родственным ether-a-go-go (hERG). Как известно, под действием цизаприда происходит удлинение интервала QT за счет блокады IKr, поэтому интерес представляла задача определить, оказывают ли АTI-7505 и ATI-7500 важное ингибирующее действие на IKrчеловека. В качестве тест-системы использовали HEK-293 клетки млекопитающего, экспрессирующие K+ каналы hERG, в которых калиевый ток измерялся методом фиксации потенциала с интактной клетки. Были определены величины IC50 в порядке возрастания: цизаприд (9,5 нM)>ATI-7505 (24,521 нM)>ATI-7500 (204,080 нM) (таблица 3). В целом, полученные данные показывают, что ATI-7505 имеет значительно более низкий проаритмический потенциал, чем цизаприд, и предполагают, что как ATI-7505, так и ATI-7500 обладает ничтожно малым сродством к человеческим IKr каналам.
ATI-7505 не вызывают значительных электрофизиологических изменений в сердечной мышце морской свинки
Электрофизиологическое воздействие ATI-7505 на сердечную мышцу изучали на изолированных перфузируемых сердцах морских свинок. В ходе опытов изучали ATI-7505, ATI-7500 и цизаприд в концентрациях вплоть до 10 000 нM. Уровень, при котором эффект не наблюдается (NOEL), определяли как самую высокую концентрацию испытуемого соединения, при которой не наблюдается ответ, значимо отличающийся от исходного уровня (p<0,05). Были рассмотрены следующие 6 параметров сердечной мышцы: (1) интервал QT; (2) MAPD90 (продолжительность монофазного потенциала действия); (3) интервал SA; (4) интервал QRS; (5) интервал AH и (6) HV. В то время как ATI-7505 оказался очень слабым модулятором электрофизиологических параметров сердечной мышцы, его метаболит, ATI-7500, вообще не обладал электрофизиологической активностью (таблица 4). Уровень NOEL для ATI-7500 составлял >10000 нM для всего ряд из 6 сердечно-сосудистых параметров. Поскольку цизаприд имел NOEL 10 нМ в отношении объединенного ряда из 6 изучавшихся параметров сердечной мышцы, а ATI-7505 имел объединенный NOEL 1000 нM, сделано заключение, что ATI-7505 не обладает свойством цизаприда модулировать электрофизиологические параметры сердечной мышцы. В целом, полученные данные показывают, что ATI-7505 является значительно более безопасным, чем цизаприд, в том, что касается возможности индуцировать электрофизиологические изменения в сердечной мышце.
Значимая разница (p<0,05) по сравнению с исходным состоянием наблюдалась при повышении концентрации молекулы в 10 раз, за исключением величин, указанных как >10000 нМ.
Метаболизм в микросомальных препаратах человека
Метаболизм указанных соединений изучали на пуле микросом человека в присутствии и в отсутствии кофактора NADPH системы цитохрома P450 и мониторировали время исчезновения родоначального соединения и появления соответствующего кислотного метаболита, т.е. соответствующего изомера соединения II.
Как показано в таблице 5, соединения III и IV были быстро гидролизованы эстеразой с образованием соответствующих метаболитов - (+) и (-) соединения II. Метаболизм не зависел от CYP450, поскольку скорость гидролиза не зависела от присутствия NADPH, который является необходимым кофактором для функционирования CYP450. Напротив, (±)-S соединения V и VI оказались довольно стабильными с течением времени при тех же условиях. В указанном эксперименте количество субстрата (соединений III, IV, V, и VI), остававшегося в реакции спустя 5, 60 и 90 минут, оценивали двойным методом ВЭЖХ-МС. Указанное остаточное количество коррелировало с появлением метаболита соединения II. В совокупности остаточный субстрат и соединение II составляли неизменное с течением времени количество, равное количеству исходного материала в нулевой момент времени, указывая, таким образом, что гидролиз являлся единственной реакцией метаболизма, которая имела место.
Метаболизм в свежей крови человека.
Испытуемые соединения растворяли в ДМСО для получения 12,5 мМ маточного раствора и разбавляли водой до конечной концентрации 2,5 мМ (DMSO/H2O=20/80). Свежую кровь получали от 3 доноров-людей и помещали в гепаринизированные пробирки, кровь хранили на льду вплоть до инкубации. Отдельные аликвоты крови от каждого из доноров отмеряли пипеткой в 1,5 мл центрифужные пробирки и пробирки предварительно инкубировали в качалке с водяной баней при 37°C в течение 5 минут. Реакцию инициировали добавлением 10 мкл маточного раствора соответствующего испытуемого соединения в каждую пробирку (конечная концентрация 100 мкМ). Инкубацию гасили через 0, 5, 15, 30 и 60 минут добавлением ацетонитрила (750 мл), после чего пробирки центрифугировали на 12000 об./мин в течение 2 минут, а надосадочную жидкость исследовали с использованием системы Agilent 1100 для ВЭЖХ. Разделение осуществляли на колонке Keystone Intersil ODS2, 250×4,6 мм, 5 м. Водная подвижная фаза состояла из 20 мM буферного раствора ацетата аммония (pH 5,7) и органической фазы ацетонитрила. Использовали градиент: исходное условие состояло из 20% ацетонитрила в течение 1 минуты. Концентрация ацетонитрила линейно снижалась до 90% в течение следующих 8 минут и удерживалась на указанном уровне в течение 1 минуты. Затем систему возвращали к исходным условиям в течение минуты и оставляли в указанном состоянии в течение 4 минут до следующего введения. Площадь пика, соответствовавшего родоначальному соединению, определяли по поглощению на 240, 254 и 290 нм. Результаты выражали в виде остаточного количества исходного соединения и данные подвергали кинетическому анализу с использованием программного пакета WinNonLin. Полупериоды существования отдельных соединений представлены ниже в таблице 6.
Следует понимать, что примеры и особенности, описанные в настоящей заявке, служат лишь иллюстративным целям и что их различные модификации или изменения в их интерпретации могут быть предложены специалистам и должны быть включены в идею и объем настоящей заявки и объем прилагаемой формулы изобретения. Кроме того, все патенты, патентные заявки, временные заявки и публикации, на которые ссылается или которым противопоставляется настоящий документ, целиком включены в настоящий документ, в той мере, в которой они явным образом не противоречат идеям данного описания.
Изобретение, а также способы и процессы его осуществления и использования описаны в настоящем документе полными, ясными, лаконичными и точными терминами, для того, чтобы любой специалист в данной области мог осуществлять и использовать настоящее изобретение. Следует понимать, что выше описаны предпочтительные особенности изобретения и что можно осуществлять модификации без отступления от идеи или объема изобретения, в том виде, в котором оно обозначено в формуле изобретения. Для того чтобы конкретно отметить и четко заявить предмет изобретения как изобретение, следующая формула изобретения подводит итог данного описания.
| название | год | авторы | номер документа |
|---|---|---|---|
| СТЕРЕОИЗОМЕРНЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ЛЕЧЕНИЯ ЖЕЛУДОЧНО-КИШЕЧНЫХ РАССТРОЙСТВ И РАССТРОЙСТВ ЦЕНТРАЛЬНОЙ НЕРВНОЙ СИСТЕМЫ | 2005 |
|
RU2374244C2 |
| НОВЫЕ СОЕДИНЕНИЯ, КОТОРЫЕ ЯВЛЯЮТСЯ ИНГИБИТОРАМИ ERK | 2013 |
|
RU2660429C2 |
| АРИЛЬНЫЕ, ГЕТЕРОАРИЛЬНЫЕ И ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ ОПОСРЕДОВАННЫХ КОМПЛЕМЕНТОМ РАССТРОЙСТВ | 2015 |
|
RU2707749C2 |
| АНТИБАКТЕРИАЛЬНЫЕ ПРОИЗВОДНЫЕ ПИПЕРИДИНА | 2006 |
|
RU2424240C2 |
| СОЕДИНЕНИЯ, ПРЕДСТАВЛЯЮЩИЕ СОБОЙ СТИРОЛИЛЬНЫЕ ПРОИЗВОДНЫЕ, ДЛЯ ЛЕЧЕНИЯ ОФТАЛЬМИЧЕСКИХ ЗАБОЛЕВАНИЙ И РАССТРОЙСТВ | 2008 |
|
RU2494089C2 |
| БИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ АЛЛОСТЕРИЧЕСКИХ ИНГИБИТОРОВ SHP2 | 2018 |
|
RU2776846C2 |
| СПОСОБ ПОЛУЧЕНИЯ 1-(АЛКОКСИМЕТИЛ)ПИРРОЛЬНЫХ СОЕДИНЕНИЙ | 1994 |
|
RU2137758C1 |
| СПОСОБ СИНТЕЗА ПЕПТИДОВ, СОДЕРЖАЩИХ ПОДСТРУКТУРУ 4-ГИДРОКСИПРОЛИНА | 2003 |
|
RU2392265C2 |
| СПОСОБ СИНТЕЗА ПОЛИГИДРОКСИСТИЛЬБЕНОВЫХ СОЕДИНЕНИЙ | 2008 |
|
RU2474570C2 |
| СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ ОПОСРЕДОВАННЫХ КОМПЛЕМЕНТОМ НАРУШЕНИЙ | 2015 |
|
RU2703995C2 |
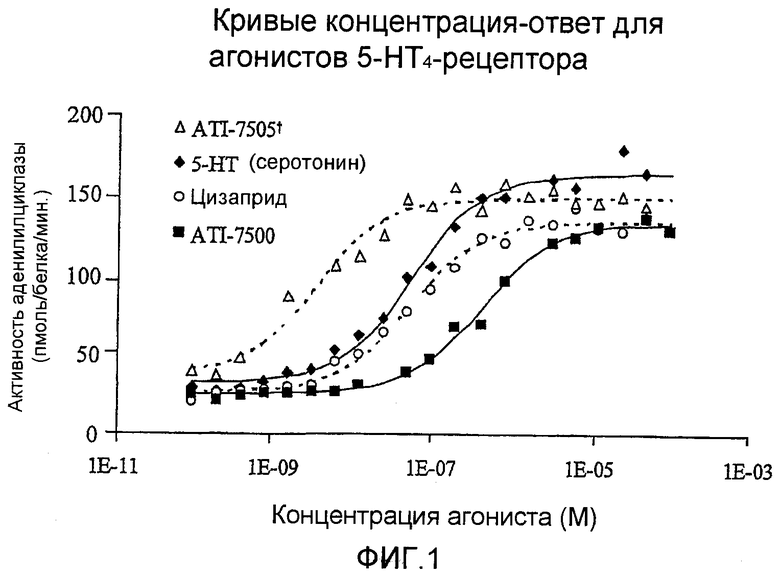
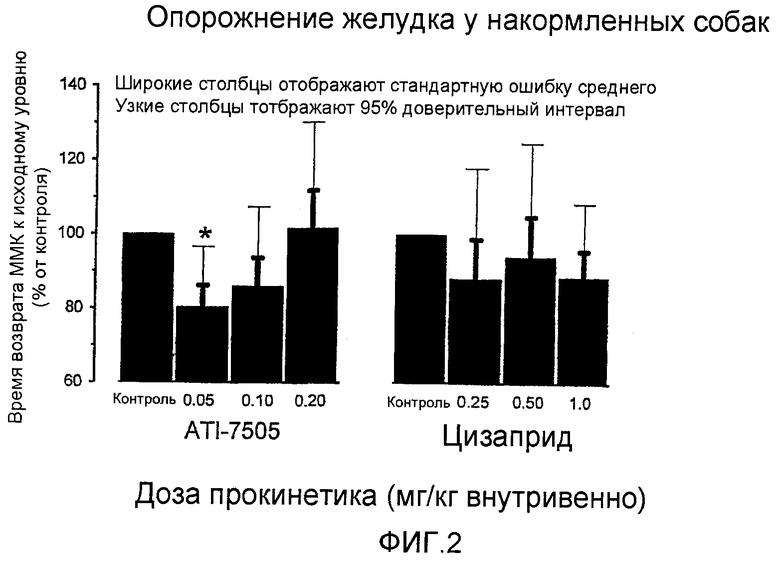
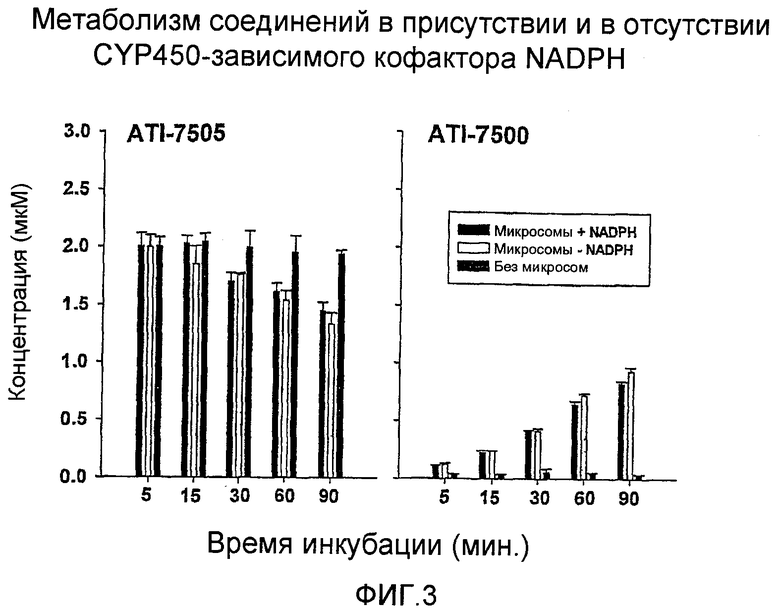
Изобретение относится к способу получения (R)-хинуклидин-3-ил 6-((3S,4R)-4-(4-амино-5-хлор-2-метоксибензамид)-3-метоксипиперидин-1-ил)гексаноата или его соли, включающий:
1) превращение соединения, которое представляет собой этил 4-амино-3-метоксипиперидин-1-карбоксилат
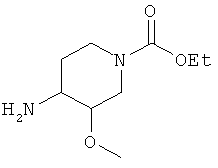
в соль;
2) превращение соли этил 4-амино-3-метоксипиперидин-1-карбоксилата в этил 4-(дифениламин)-3-метоксипиперидин-1-карбоксилат
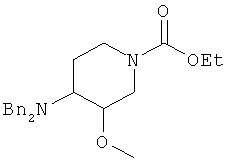 ;
;
3) обработку этил 4-(дифениламино)-3-метоксипиперидин-1-карбоксилата гидроксидом или гидридом щелочного металла, с получением 3-метокси-N,N-дифенилпиперидин-4-амина
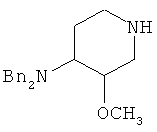 ;
;
4) получение хиральной соли цис-изомера 3-метокси-N,N-дифенилпиперидин-4-амина контактированием 3-метокси-N,N-дифенилпиперидин-4-амина с хиральным разделяющим агентом и выделением полученной указанным способом хиральной соли цис-изомера 3-метокси-N,N-дифенилпиперидин-4-амина;
5) необязательно, перекристаллизацию продукта 4;
6) превращение продукта 4 или 5 в основание, с получением продукта 4 или 5 в форме свободного основания;
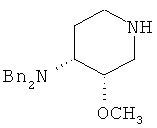 ;
;
7) контактирование продукта 6 с этил 6-бромгексаноатом, с получением этил 6-((3S,4R)-4-(дифениламин)-3-метоксипиперидин-1-ил)гексаноата
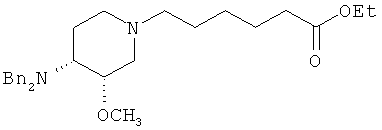 ;
;
8) этерификацию этил 6-((3S,4R)-4-(дифениламин)-3 -метоксипиперидин-1-ил)гексаноата с использованием (R)-хинуклидин-3-ола и кислоты Льюиса, с получением (R)-хинуклидин-3-ил 6-((3S,4R)-4-(дифениламин)-3-метоксипиперидин-1-ил)гексаноата
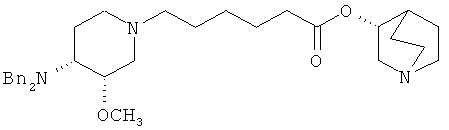 ;
;
9) снятие защиты с 4-аминогруппы продукта 8, с получением (R)-хинуклидин-3-ил 6-[(3S,4R)-4-амино-3-метоксипиперидин-1-ил]гексаноата;
10) ацилирование продукта 9 4-амино-5-хлор-2-метоксибензойной кислотой, с получением (R)-хинуклидин-3-ил 6-((3S,4R)-4-(4-амино-5-хлор-2-метоксибензамид)-3-метоксипиперидин-1-ил)гексаноата;
11) необязательно, превращение продукта 10 в соль.
Технический результат - способ позволяет увеличить выход целевого продукта и уменьшить содержание примесей. 6 з.п. ф-лы, 6 табл., 3 ил.
1. Способ получения (R)-хинуклидин-3-ил 6-((3S,4R)-4-(4-амино-5-хлор-2-метоксибензамид)-3-метоксипиперидин-1-ил)гексаноата или его соли, включающий
1) превращение соединения, которое представляет собой этил 4-амино-3-метоксипиперидин-1-карбоксилат,
в соль;
2) превращение соли этил 4-амино-3-метоксипиперидин-1-карбоксилата в этил 4-(дифениламин)-3-метоксипиперидин-1-карбоксилат
3) обработку этил 4-(дифениламино)-3-метоксипиперидин-1-карбоксилата гидроксидом или гидридом щелочного металла, с получением 3-метокси-N,N-дифенилпиперидин-4-амина
4) получение хиральной соли цис-изомера 3-метокси-N,N-дифенилпиперидин-4-амина контактированием 3-метокси-N,N-дифенилпиперидин-4-амина с хиральным разделяющим агентом и выделением полученной указанным способом хиральной соли цис-изомера 3-метокси-N,N-дифенилпиперидин-4-амина;
5) необязательно, перекристаллизацию продукта 4;
6) превращение продукта 4 или 5 в основание, с получением продукта 4 или 5 в форме свободного основания;
7) контактирование продукта 6 с этил 6-бромгексаноатом, с получением этил 6-((3S,4R)-4-(дифениламин)-3-метоксипиперидин-1-ил)гексаноата
;
8) этерификацию этил 6-((3S,4R)-4-(дифениламин)-3-метоксипиперидин-1-ил)гексаноата с использованием (R)-хинуклидин-3-ола и кислоты Льюиса, с получением (R)-хинуклидин-3-ил 6-((3S,4R)-4-(дифениламин)-3-метоксипиперидин-1-ил)гексаноата
;
9) снятие защиты с 4-аминогруппы продукта 8 с получением (R)-хинуклидин-3-ил 6-[(3S,4R)-4-амино-3-метоксипиперидин-1-ил]гексаноата;
10) ацилирование продукта 9 4-амино-5-хлор-2-метоксибензойной кислотой с получением (R)-хинуклидин-3-ил 6-((3S,4R)-4-(4-амино-5-хлор-2-метоксибензамид)-3-метоксипиперидин-1-ил)гексаноата;
11) необязательно, превращение продукта 10 в соль.
2. Способ по п.1, где соль со стадии 1 представляет собой HCl.
3. Способ по п.1, где гидроксид щелочного металла со стадии 3 представляет собой КОН.
4. Способ по п.1, где хиральная соль со стадии 4 представляет собой (+)-2,3-дибензоил-D-винную кислоту.
5. Способ по п.1, где кислота Льюиса представляет собой Ti(OiP)4 (изопропоксид титана (IV)).
6. Способ по п.1, где для снятия защиты с 4-аминогруппы используется H2/Pd/C.
7. Способ по п.1, где для ацилирования продукта 9 используется пивалоилхлорид.
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| US 5057525 A, 15.10.1991 | |||
| EP 0640601 A1, 01.03.1995 | |||
| SU 1593569 A3, 15.09.1990. | |||

