Область изобретения
В настоящем изобретении раскрыты новые производные 5-замещенного 7-амино-[1,3]тиазоло[4,5-d]пиримидина, а также способы их получения, фармацевтические композиции, содержащие их, и их применение в терапии.
Предшествующий уровень техники
Хемокины играют важную роль в иммунных и воспалительных ответах при различных заболеваниях и расстройствах, включая астму, атеросклероз и аллергические заболевания, а также аутоиммунные патологии, такие как ревматоидный артрит и рассеянный склероз. Эти небольшие, секретируемые молекулы представляют собой растущее суперсемейство белков с массой 8-14 кДа, которые характеризуются консервативным цистеиновым мотивом. В настоящее время суперсемейство хемокинов включает в себя 4 группы, демонстрирующие характерные структурные мотивы, С-Х-С, С-С и С-Х3-С и ХС семейства. Семейства С-Х-С и С-С имеют сходство в последовательностях и различаются друг от друга по единственной аминокислотной вставке между NH-проксимальной парой цистеиновых остатков. Семейство С-Х3-С отличается от других двух семейств тем, что имеет тройную аминокислотную вставку между NH-проксимальной парой цистеиновых остатков. Наоборот, у членов семейства ХС отсутствует один из первых двух цистеиновых остатков.
Хемокины С-Х-С включают несколько сильных хемоаттрактантов и активаторов нейтрофилов, таких как интерлейкин-8 (IL-8) и нейтрофил-активирующий пептид 2 (NAP-2).
Хемокины С-С включают сильные хемоаттрактанты моноцитов, лимфоцитов и нейтрофилов. Примеры включают человеческие моноцитарные хемотаксические белки 1-3 (МСР-1, МСР-2 и МСР-3), RANTES (регулируемый при активации, экспрессируемый и секретируемый нормальными Т-клетками), эотаксин и воспалительные белки макрофагов 1α и 1β (MIP-1α и MIP-1β).
Хемокин С-Х3-С (также известный как фракталкин) представляет собой сильный хемоаттрактант и активатор микроглии в центральной нервной системе (ЦНС), а также моноцитов, Т-клеток, NK клеток и тучных клеток.
Исследования показали, что действия хемокинов опосредованы подсемействами рецепторов, связанных с G-белком, среди которых рецепторы, обозначенные CCR1, CCR2, CCR2A, CCR2B, CCR3, CCR4, CCR5, CCR6, CCR7, CCR8, CCR9, CCR10 и CCR11 (для семейства С-С); CXCR1, CXCR2, CXCR3, CXCR4 и CXCR5 (для семейства С-Х-С) и CX3CR1 для семейства С-Х3-С. Эти рецепторы представляют собой хорошие мишени для разработки лекарств, так как агенты, модулирующие эти рецепторы, были бы полезны при лечении нарушений и заболеваний, таких как те, которые упомянуты выше.
В WO 00/09511 раскрыты некоторые производные 2-замещенного 4-амино-тиазолопиримидина, которые полезны в качестве антагонистов рецепторов, связанных с семействами хемокинов С-Х-С и С-С, особенно в качестве антагонистов рецептора CXCR2.
Настоящее изобретение относится к группе соединений, которые частично находятся в пределах общего объема WO 00/09511, но обладают структурой, для которой в указанном документе не приведено конкретных примеров. Соединения по настоящему изобретению при сравнении с Примерами, раскрытыми в WO 00/09511, неожиданно проявляют полезные свойства в качестве антагонистов рецептора CX3CR1.
Описание изобретения
В настоящем изобретении предложены соединения формулы (I)
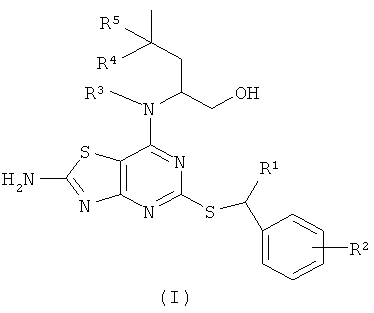
где
R1 представляет собой СН3 или СН3СН2;
R2 представляет собой Н, 2-F, 2-Cl, 3-F, 3-ОСН3, 3-CN, 3-CF3, 3-CONH2 или 3-SO2CH3;
R3 представляет собой Н или СН3;
R4 представляет собой Н или СН3; и
R5 представляет собой Н; или, когда R4 представляет собой СН3, R5 представляет собой Н или F;
и их оптические изомеры и фармацевтически приемлемые соли.
Соединения формулы (I) могут существовать в стереомерных и/или таутомерных формах. Следует понимать, что все энантиомеры, диастереомеры, рацематы, таутомеры и их смеси включены в объем настоящего изобретения.
В одном воплощении R1 представляет собой СН3. В другом воплощении R1 представляет собой СН3СН2.
В одном воплощении R2 представляет собой Н, 2-F, 3-F, 2-Cl, 3-ОСН3, 3-CN или 3-CF3. В другом воплощении R2 представляет собой Н, 2-F или 3-CN. В еще одном воплощении R2 представляет собой Н. В еще одном воплощении R2 представляет собой 2-F. В еще одном воплощении R2 представляет собой 3-CN.
В одном воплощении R3 представляет собой Н.
В одном воплощении R4 представляет собой СН3. В другом воплощении R4 представляет собой Н.
В одном воплощении R5 представляет собой Н.
В одном воплощении R4 представляет собой СН3, а R5 представляет собой F.
В одном воплощении R4 представляет собой СН3, а R5 представляет собой Н.
В одном воплощении R1 представляет собой СН3; R2 представляет собой Н, 2-F, 3-F, 2-Cl, 3-ОСН3, 3-CN или 3-CF3; R3 представляет собой Н; R4 представляет собой Н или СН3; и R5 представляет собой Н.
В еще одном воплощении R1 представляет собой СН3; R2 представляет собой Н, 2-F или 3-CN; R3 представляет собой Н; R4 представляет собой Н или СН3; и R5 представляет собой Н.
В еще одном воплощении R1 представляет собой СН3; R2 представляет собой Н, 2-F или 3-CN; R3 представляет собой Н; R4 представляет собой Н; и R5 представляет собой Н.
В еще одном воплощении R1 представляет собой СН3; R2 представляет собой Н, 2-F или 3-CN; R3 представляет собой Н; R4 представляет собой СН3; и R5 представляет собой Н или F.
В еще одном воплощении R представляет собой СН3; R представляет собой Н; R3 представляет собой Н; R4 представляет собой СН3; и R5 представляет собой Н.
Конкретные соединения формулы (I) включают:
(2R)-2-[(2-амино-5-{[(1S)-1-(2-фторфенил)этил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]пентан-1-ол;
(2R)-2-[(2-амино-5-{[(1S)-1-(2-фторфенил)этил]тио}[1,3]тиазоло[4,5-пиримидин-7-ил)амино]-4-метилпентан-1-ол;
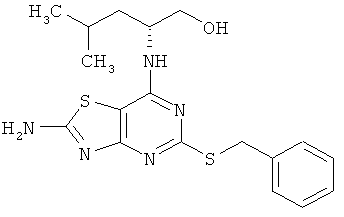
(2R)-2-({2-амино-5-[(1-фенилэтил)тио][1,3]тиазоло[4,5-d]пиримидин-7-ил}амино)-4-метилпентан-1-ол;
(2R)-2-[(2-амино-5-{[(1S)-1-фенилэтил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ол;
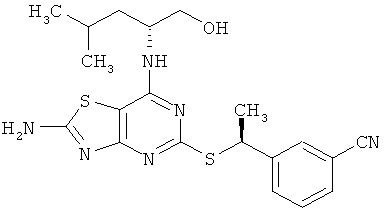
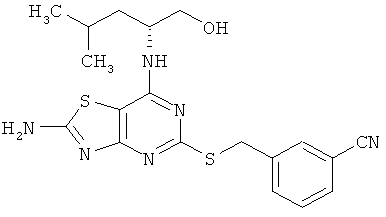
3-{(1S)-1-[(2-амино-7-{[(1R)-1-(гидроксиметил)бутил]амино}[1,3]тиазоло-[4,5-d]пиримидин-5-ил)тио]этил}бензонитрил;
(2R)-2-{[2-амино-5-({(1S)-1-[3-(метилсульфонил)фенил]этил}тио)-[1,3]тиазоло[4,5-d]пиримидин-7-ил]амино}-4-метилпентан-1-ол;
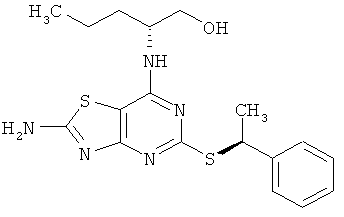
(2R)-2-[(2-амино-5-{[(1S)-1-фенилэтил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]пентан-1-ол;
3-{(1S)-1-[(2-амино-7-{[(1R)-1-(гидроксиметил)-3-метилбутил]амино}-[1,3]тиазоло[4,5-d]пиримидин-5-ил)тио]этил}бензонитрил;
(2R)-2-({2-амино-5-[(1-фенилпропил)тио][1,3]тиазоло[4,5-d]пиримидин-7-ил}амино)-4-метилпентан-1-ол;
3-{(1R)-[(2-амино-7-{[(1R)-1-(гидроксиметил)-3-метилбутил]амино}-[1,3]тиазоло[4,5-d]пиримидин-5-ил)тио]этил}бензамид;
(2R)-2-{[2-амино-5-({1-[3-(трифторметил)фенил]этил}тио)[1,3]тиазоло[4,5-d]пиримидин-7-ил]амино}-4-метилпентан-1-ол;
(2R)-2-[{2-амино-5-[(1-фенилэтил)тио][1,3]тиазоло[4,5-d]пиримидин-7-ил}(метил)амино]-4-метилпентан-1-ол;
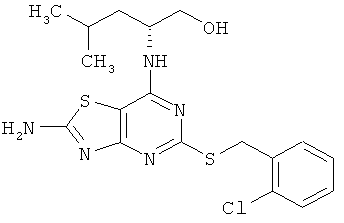
(2R)-2-[(2-амино-5-{[1-(2-хлорфенил)этил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ол;
(2R)-2-[(2-амино-5-{[1-(3-метоксифенил)этил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ол;
(2R)-2-[(2-амино-5-{[(1S)-1-фенилэтил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ол;
(2R)-2-[(2-амино-5-{[(1S)-1-фенилэтил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-фтор-4-метилпентан-1-ол;
(2R)-2-[(2-амино-5-{[(1S)-1-(2-фторфенил)этил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-фтор-4-метилпентан-1-ол;
(2R)-2-[(2-амино-5-{[(1S)-1-(3-фторфенил)этил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ол;
и их фармацевтически приемлемые соли.
По сравнению с соединениями, раскрытыми в WO 00/09511, соединения по настоящему изобретению характеризуются наличием разветвленной тиобензильной группы в положении 5 тиазолопиримидиновой кольцевой системы. То есть соединения по настоящему изобретению включают группу R1, которая не является водородом.
Согласно изобретению далее предложен способ получения соединения формулы (I) или его фармацевтически приемлемой соли, включающий:
взаимодействие соединения формулы (II)
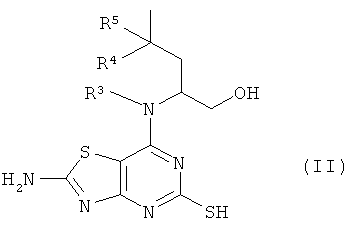
где R3, R4 и R5 являются такими, как определено в формуле (I);
с соединением формулы (III)
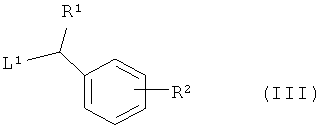
где R1 и R2 являются такими, как определено в формуле (I), и L1 представляет собой уходящую группу;
и, когда это необходимо, превращение полученного соединения формулы (I) в его фармацевтически приемлемую соль; и, если требуется, превращение полученного соединения формулы (I) в его оптический изомер.
Согласно изобретению предложен еще один способ получения соединения формулы (I) или его фармацевтически приемлемой соли, включающий взаимодействие соединения формулы (IV)
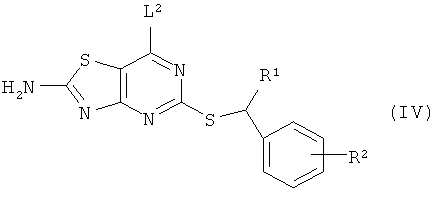
где R1 и R2 являются такими, как определено в формуле (I), и L2 представляет собой уходящую группу;
с соединением формулы (V)
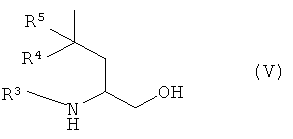
где R3, R4 и R5 являются такими, как определено в формуле (I);
и, когда это необходимо, превращение полученного соединения формулы (I) в его фармацевтически приемлемую соль; и, если требуется, превращение полученного соединения формулы (I) в его оптический изомер.
В способе (а) реагенты (II) и (III) соединяют вместе в подходящем органическом растворителе, таком как диметилсульфоксид (ДМСО), ацетонитрил или 1-метил-2-пирролидинон (NMP). Реакцию возможно осуществляют в присутствии дополнительного органического или неорганического основания, такого как триэтиламин, N,N-диизопропилэтиламин (ДИПЭА) или гидрид натрия. Реакцию возможно осуществляют в присутствии мягкого восстановителя, такого как боргидрид натрия. Реакцию проводят при подходящей температуре, обычно между комнатной температурой и температурой кипения растворителя. Реакцию обычно проводят в течение периода времени от примерно одного часа до одной недели или до тех пор, пока анализ не покажет, что образование целевого продукта завершено.
В способе (б) реагенты (IV) и (V) соединяют вместе в подходящем органическом растворителе, таком как тетрагидрофуран, ацетонитрил, диметилсульфоксид или 1-метил-2-пирролидинон. Реакцию возможно проводят в присутствии дополнительного основания. Это основание может быть органическим основанием, таким как триэтиламин или N,N-диизопропилэтиламин, или неорганическим основанием, таким как карбонат калия. Реакцию проводят при подходящей температуре, обычно между комнатной температурой и температурой кипения растворителя, но возможно при более высокой температуре, если используют герметичный реакционный сосуд. Реакцию обычно проводят в течение периода времени от примерно одного часа до одной недели или до тех пор, пока анализ не покажет, что образование целевого продукта завершено.
Подходящие уходящие группы L1 и L2 представляют собой галоген, в частности хлоро или бромо. В одном воплощении каждый из L1 и L2 представляет собой хлоро.
Специалисту в данной области техники будет понятно, что в указанных выше способах может быть желательной или необходимой защита аминной, гидроксильной или другой потенциально реакционноспособной группы. Подходящие защитные группы и подробности способов введения и удаления таких групп, в общем, хорошо известны из уровня техники. Смотри, например, "Protective Groups in Organic Synthesis", 3rd Edition (1999) by Greene and Wuts.
Настоящее изобретение включает соединения формулы (I) в форме солей. Подходящие соли включают соли, образованные с органическими или неорганическими кислотами, или органическими или неорганическими основаниями. Такие соли обычно будут фармацевтически приемлемыми; хотя соли фармацевтически неприемлемых кислот или оснований могут быть полезны в получении и очистке рассматриваемого соединения.
Соли соединений формулы (I) могут быть образованы путем взаимодействия свободного соединения или его соли, энантиомера или рацемата с одним или более эквивалентами подходящей кислоты или основания. Реакцию можно проводить в растворителе или среде, в котором(й) соль нерастворима, или в растворителе, в котором соль растворима, например воде, диоксане, этаноле, тетрагидрофуране или диэтиловом эфире, или в смеси растворителей, которые могу быть удалены под вакуумом или посредством сублимационной сушки. Реакция также может представлять собой обменный процесс или может быть проведена на ионообменной смоле.
Соединения формулы (II) или известны из WO 00/09511, или могут быть получены известными способами, которые будут очевидны специалисту в данной области техники.
Соединения формулы (IV) могут быть получены с использованием способов, аналогичным тем, которые раскрыты в WO 00/09511, или с использованием известных способов, которые будут очевидны специалисту в данной области техники.
Соединения формул (III) и (V) или имеются в продаже, или известны из литературы, или могут быть получены известными способами, которые будут очевидны специалисту в данной области техники.
Подходящие конкретные способы получения соединений формул (II), (III), (IV) и (V) подробно описаны в разделе Примеры настоящей заявки, и такие способы представляют собой конкретные воплощения способов по изобретению.
Например, соединения формулы (II) и, следовательно, соединения формулы (I), могут быть получены, как показано на схеме 1
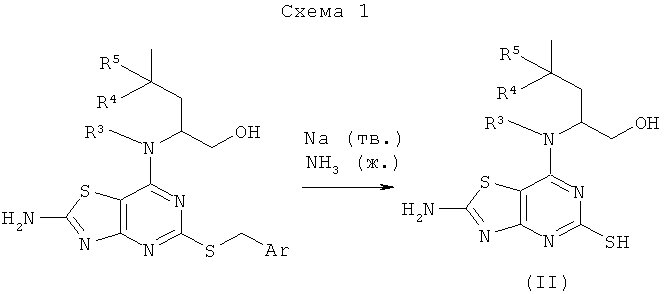
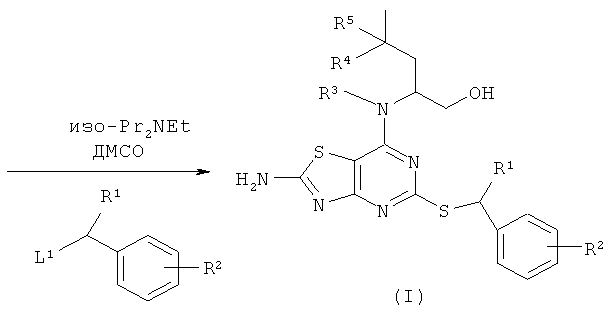
Промежуточные соединения могут быть использованы как таковые или в защищенной форме. Подходящие защитные группы и подробности способов введения и удаления таких групп, в общем, хорошо известны из уровня техники. Смотри, например, "Protective Groups in Organic Synthesis", 3rd Edition (1999) by Greene and Wuts.
Соединения по изобретению и промежуточные соединения (для их получения) могут быть выделены из реакционных смесей и, при необходимости, далее очищены с использованием стандартных методик.
Соединения формулы (I) могут существовать в стереоизомерных формах. Поэтому все энантиомеры, диастереомеры, рацематы и их смеси включены в объем настоящего изобретения. Различные оптические изомеры могут быть выделены путем разделения стереоизомерной смеси соединений с использованием обычных методик, например фракционной кристаллизации или ВЭЖХ. Альтернативно различные оптические изомеры могут быть получены непосредственно с использованием оптически активных исходных материалов.
Соединения формулы (I) содержат два стереогенных центра и, таким образом, могут существовать в четырех обособленных стереоизомерных формах, как показано в формулах (Ia)-(Id)
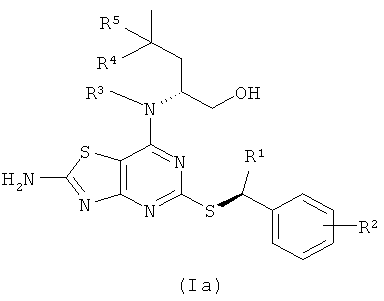
Все эти четыре стереоизомера и любые их смеси включены в объем настоящего изобретения. В одном из воплощений соединения формулы (I) имеют стереохимию согласно формуле (Ia). В другом воплощении соединения формулы (I) имеют стереохимию согласно формуле (Ib).
Промежуточные соединения также могут существовать в стереоизомерных формах и могут быть использованы в виде очищенных энантиомеров, диастереомеров, рацематов или смесей.
Соединения формулы (I) и их фармацевтически приемлемые соли являются полезными, так как они обладают фармакологической активностью в качестве антагонистов рецептора CX3CR1. В частности, по сравнению с соединениями, конкретно проиллюстрированными примерами в WO 00/09511, соединения формулы (I) по настоящему изобретению обладают значительно улучшенной эффективностью в отношении ингибирования рецептора CX3CR1 и/или пониженной эффективностью в отношении ингибирования рецептора CXCR2. Предпочтительные соединения по настоящему изобретению проявляют как повышенную эффективность в отношении ингибирования CX3CR1, так и пониженную эффективность в отношении ингибирования CXCR2.
В одном аспекте настоящего изобретения предложено соединение формулы (I) или его фармацевтически приемлемая соль для применения в качестве лекарственного средства.
В другом аспекте настоящего изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли в изготовлении лекарственного средства для лечения или профилактики заболеваний или состояний, при которых полезен антагонизм рецептора CX3CR1.
В еще одном аспекте настоящего изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли в изготовлении лекарственного средства для лечения или профилактики нейродегенеративных расстройств, демиелинизирующего заболевания, кардио- и церебро-васкулярных атеросклеротических расстройств, заболевания периферических артерий, ревматоидного артрита, легочных заболеваний, таких как ХОБЛ (хроническое обструктивное заболевание легких), астмы или боли.
В еще одном аспекте настоящего изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли в изготовлении лекарственного средства для лечения или профилактики рассеянного склероза (PC).
В еще одном аспекте настоящего изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли в изготовлении лекарственного средства для лечения или профилактики атеросклероза путем предупреждения и/или уменьшения образования новых атеросклеротических повреждений или бляшек и/или путем предупреждения или замедления развития существующих повреждений и бляшек.
В еще одном аспекте настоящего изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли в изготовлении лекарственного средства для лечения или профилактики атеросклероза путем изменения состава бляшек для снижения риска разрыва бляшек и возникновения атеротромботических событий.
Согласно изобретению также предложен способ лечения или снижения риска возникновения заболеваний или состояний человека, при которых полезен антагонизм рецептора CX3CR1, включающий введение субъекту, страдающему от указанного заболевания или состояния, или с риском возникновения указанного заболевания или состояния, терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
Также предложен способ лечения или снижения риска возникновения нейродегенеративных расстройств, демиелинизирующего заболевания, кардио- и цереброваскулярных атеросклеротических расстройств, заболевания периферических артерий, ревматоидного артрита, легочных заболеваний, таких как ХОБЛ, астмы или боли у субъекта, страдающего от указанного заболевания или состояния, или с риском возникновения указанного заболевания или состояния, включающий введение субъекту терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
Также предложен способ лечения или снижения риска возникновения рассеянного склероза (PC) у субъекта, страдающего от указанного заболевания или состояния, или с риском возникновения указанного заболевания или состояния, включающий введение субъекту терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
Также предложен способ лечения или снижения риска возникновения атеросклероза путем предупреждения и/или уменьшения образования новых атеросклеротических повреждений или бляшек и/или путем предупреждения или замедления развития существующих повреждений и бляшек у субъекта, страдающего от указанного заболевания или состояния, или с риском возникновения указанного заболевания или состояния, включающий введение субъекту терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
Также предложен способ лечения или снижения риска возникновения атеросклероза путем изменения состава бляшек для снижения риска разрыва бляшек и возникновения атеротромботических событий у субъекта, страдающего от указанного заболевания или состояния, или с риском возникновения указанного заболевания или состояния, включающий введение субъекту терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
В еще одном аспекте настоящего изобретения предложена фармацевтическая композиция, содержащая терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли в смеси с фармацевтически приемлемым адъювантом, разбавителем или носителем, для применения в лечении или профилактике заболеваний или состояний, при которых полезен антагонизм рецептора CX3CR1.
В еще одном аспекте настоящего изобретения предложена фармацевтическая композиция, содержащая терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли в смеси с фармацевтически приемлемым адъювантом, разбавителем или носителем, для применения в лечении или профилактике нейродегенеративных расстройств, демиелинизирующего заболевания, кардио- и цереброваскулярных атеросклеротических расстройств, заболевания периферических артерий, ревматоидного артрита, ХОБЛ, астмы или боли.
В еще одном аспекте настоящего изобретения предложена фармацевтическая композиция, содержащая терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли в смеси с фармацевтически приемлемым адъювантом, разбавителем или носителем, для применения в лечении или профилактике рассеянного склероза.
В еще одном аспекте настоящего изобретения предложена фармацевтическая композиция, содержащая терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли в смеси с фармацевтически приемлемым адъювантом, разбавителем или носителем, для применения в лечении или профилактике атеросклероза путем предупреждения и/или уменьшения образования новых атеросклеротических повреждений и/или бляшек и/или путем предупреждения или замедления развития существующих повреждений и бляшек.
В еще одном аспекте настоящего изобретения предложена фармацевтическая композиция, содержащая терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли в смеси с фармацевтически приемлемым адъювантом, разбавителем или носителем, для применения в лечении или профилактике атеросклероза путем изменения состава бляшек для снижения риска разрыва бляшек и возникновения атеротромботических событий.
Соединения формулы (I) и их фармацевтически приемлемые соли показаны для применения в лечении или профилактике заболеваний или состояний, при которых желательна модуляция активности рецептора CX3CR1. В частности, соединения показаны для применения в лечении нейродегенеративных расстройств или демиелинизирующего заболевания у млекопитающих, включая человека. Более конкретно, соединения показаны для применения в лечении рассеянного склероза. Соединения также полезны в лечении боли, ревматоидного артрита, остеоартрита, кардио- и церебро-васкулярных атеросклеротических расстройств, заболевания периферических артерий и легочной артериальной гипертензии.
Состояниями, которые могут быть конкретно упомянуты, являются: нейродегенеративные заболевания и различные виды деменций, например болезнь Альцгеймера, боковой амиотрофический склероз и другие заболевания двигательных нейронов, болезнь Крейтцфельдта-Якоба и другие прионовые заболевания, ВИЧ энцефалопатия, болезнь Хантингтона, лобно-височная деменция, деменция с тельцами Леви и сосудистая деменция; полиневропатии, например синдром Гийена-Барре, хроническая воспалительная демиелинизирующая полирадикулоневропатия, многоочаговая моторная невропатия и плексопатии; ЦНС демиелинизация, например острый рассеянный/геморрагический энцефаломиелит и подострый склерозирующий лейкоэнцефалит; нейромышечные нарушения, например тяжелая миастения и синдром Ламберта-Итона; позвоночные нарушения, например тропический спастический парапарез и синдром негнущегося человека; паранеопластические синдромы, например мозжечковая дегенерация и энцефаломиелит; травматическое повреждение головного мозга; мигрень; рак; отторжение аллотрансплантата; системный склероз; вирусные инфекции; заболевания, переносимые паразитами, например малярия; заболевания периодонта; инфаркт миокарда; удар; коронарная болезнь сердца; ишемическая болезнь сердца; рестеноз; ревматоидный артрит; легочные заболевания, такие как ХОБЛ; астма или боль.
Соединения по изобретению также предназначены для применения в лечении атеросклероза путем предупреждения и/или уменьшения образования новых атеросклеротических повреждений или бляшек и/или путем предупреждения или замедления развития существующих повреждений и бляшек.
Соединения по изобретению также показаны для применения в лечении атеросклероза путем изменения состава бляшек для снижения риска разрыва бляшек и возникновения атеротромботических событий.
Соединения по изобретению также показаны для применения в лечении воспалительного заболевания кишечника (IBD), например болезни Крона и неспецифического язвенного колита, путем индуцирования ремиссии и/или поддержания ремиссии IBD.
Полагают, что профилактика является особенно уместной для лечения субъектов, у которых уже наблюдался эпизод, или которые иначе рассматриваются как имеющие повышенный риск возникновения рассматриваемого заболевания или состояния. К субъектам с риском развития конкретного заболевания или состояния обычно относят тех, у кого в семейном анамнезе имеются случаи заболевания или состояния, или тех, кто признан особенно чувствительным к развитию заболевания или состояния после генетических тестов или скрининга.
Для упомянутых выше терапевтических показаний вводимую дозу конечно же будут варьировать в зависимости от используемого соединения, пути введения и желаемого лечения. Однако, в общем, удовлетворительные результаты получают, когда соединения вводят в твердой форме в дозировке от 1 мг до 2000 мг в день.
Соединения формулы (I) и их фармацевтически приемлемые производные можно использовать сами по себе или в форме подходящих фармацевтических композиций, в которых соединение или производное находится в смеси с фармацевтически приемлемым адъювантом, разбавителем или носителем. Введение может осуществляться энтеральным (включая пероральный, подъязычный или ректальный), интраназальным, внутривенным, местным или другими парентеральными путями, но не ограничивается ими. Обычные процедуры для выбора и приготовления подходящих фармацевтических препаратов описаны, например, в "Pharmaceuticals - The Science of Dosage Form Designs", M.E.Aulton, Churchill Livingstone, 1988. Фармацевтическая композиция предпочтительно содержит менее чем 80% и более предпочтительно менее чем 50% соединения формулы (I) или его фармацевтически приемлемой соли.
Также предложен способ приготовления такой фармацевтической композиции, включающий смешивание ингредиентов.
Кроме того, изобретение относится к комбинированным терапиям, где соединение формулы (I) или его фармацевтически приемлемую соль, или фармацевтическую композицию, или препарат, содержащие соединение формулы (I), вводят одновременно или последовательно с терапией и/или агентом для лечения любого из кардио- и цереброваскулярных атеросклеротических расстройств и заболевания периферических артерий.
В частности, соединение формулы (I) или его фармацевтически приемлемую соль можно вводить в комбинации с соединениями из одной или более чем одной из следующих групп;
1) противовоспалительные агенты, например,
а) НСПВП (нестероидные противовоспалительные препараты) (например, ацетилсалициловая кислота, ибупрофен, напроксен, флурбипрофен, диклофенак, индометацин),
б) ингибиторы синтеза лейкотриенов (ингибиторы 5-LO (5-липооксигеназы), например AZD4407, Зилеутон, ликофелон, CJ13610, CJ13454; ингибиторы FLAP (белка, активирующего 5-липооксигеназу), например BAY-Y-1015, DG-031, MK591, МК886, А81834; ингибиторы LTA4 (лейкотриен А4) гидролазы, например SC56938, SC57461A),
в) антагонисты рецепторов лейкотриенов (например, СР195543, амелубант, LY293111, акколат, МК571);
2) гипотензивные агенты, например,
а) бета-блокаторы (например, метопролол, атенолол, соталол),
б) ингибиторы ангиотензин-превращающего фермента (например, каптоприл, рамиприл, хинаприл, эналаприл),
в) блокаторы кальциевых каналов (например, верапамил, дилтиазем, фелодипин, амлодипин),
г) антагонисты ангиотензин II рецептора (например, ирбезартан, кандезартан, телемизартан, лозартан);
3) антикоагулянты, например,
а) ингибиторы тромбина (например, ксимелагатран), гепарины, ингибиторы фактора Ха,
б) ингибиторы агрегации тромбоцитов (например, клопидогрел, тиклопидин, прасугель, AZ4160);
4) модуляторы метаболизма липидов, например,
а) сенсибилизаторы инсулина, такие как агонисты рецепторов, активируемых пролифератором пероксисом (PPAR) (например, пиоглитазон, розиглитазон, Галида, мураглитазаар, гефемрозил, фенофибрат),
б) ингибиторы HMG-CoA (3-гидрокси-3-метилглутарил-коэнзим А) редуктазы, статины (например, симвастатин, правастатин, аторвастатин, розувастатин, флувастатин, питавастатин),
в) ингибиторы абсорбции холестерина (например, эзетимиб),
г) ингибиторы транспорта желчных кислот в подвздошной кишке (IBAT) (например, AZD-7806),
д) агонисты LXR (например, GW-683965A, Т-0901317),
е) модуляторы рецептора FXR,
ж) ингибиторы фосфолипазы;
5) агенты против стенокардии, например нитраты и нитриты;
6) модуляторы окислительного стресса, например антиоксиданты (пробукол), ингибиторы миелопероксидазы.
Изобретение проиллюстрировано следующими примерами, но не ограничено ими.
Общие способы
Все используемые растворители были аналитической степени чистоты; для реакций, как обычно, использовали коммерчески доступные безводные растворители. Реакции обычно проводили в инертной атмосфере азота или аргона.
1Н и 13С ЯМР спектры регистрировали при 400 МГц для протона и при 100 МГц для углерода-13 либо на ЯМР спектрометре Varian Unity+ 400, оснащенном 5 мм ВВО датчиком с Z-градиентами, либо на ЯМР спектрометре Bruker Avance 400, оснащенном 60 мкл датчиком двойного обратного потока с Z-градиентами, либо на ЯМР спектрометре Bruker DPX400, оснащенном 4-ядерным датчиком с Z-градиентами. 1H ЯМР спектры при 600 МГц регистрировали на ЯМР спектрометре Bruker av600, оснащенном 5 мм ВВI измерительной головкой с Z-градиентами. 1H ЯМР спектры при 300 МГц регистровали на ЯМР приборе Varian Gemini 300, оснащенном 5 мм ВВI измерительной головкой. Если в примерах не указано особо, спектры регистрировали при 400 МГц для протона и при 100 МГц для углерода-13. Использовали следующие базовые сигналы: осевая линия ДМСО-d6 δ 2.50 (1Н), δ 39.51 (13С); осевая линия CD3OD δ 3.31 (1Н) или δ 49.15 (13С); ацетон-d6 2.04 (1H), 206.5 (13С); и CDCl3 δ 7.26 (1Н), осевая линия CDCl3 δ 77.16 (13С) (если не указано иначе).
Энантиомерный избыток определяли ГХ (газовой хроматографией) на колонке Cyclodex В (изотермическое элюирование при 100°С).
Масс-спектры регистрировали на приборе жидкостной хроматографии с масс-спектрометрией (ЖХ-МС) Waters, состоящем из Alliance 2795 (ЖХ) и ZQ одноквадрупольного масс-спектрометра. Масс-спектрометр был оснащен источником ионизации электрораспылением (ИЭР), работающем в режиме положительных или отрицательных ионов. Капиллярное напряжение составляло 3 кВ; масс-спектрометр сканировал m/z 100-700 со временем считывания 0,3 или 0,8 с. Разделения проводили либо на Waters X-Terra MS, С8-колонках (3,5 мкм, 50 или 100 мм × 2,1 мм i.d. (внутренний диаметр)), либо на ScantecLab's АСЕ 3 AQ колонке (100 мм × 2,1 мм i.d.). Температура колонки была установлена 40°С. Применяли линейный градиент с использованием системы с нейтральной или кислотной подвижной фазой с изменением органической фазы от 0% до 100% за 4-5 минут, скорость потока 0,3 мл/мин. Система с нейтральной подвижной фазой: ацетонитрил/[10 мМ NH4OAc (водн.)/MeCN (95:5)], или [10 мМ NH4OAc (водн.)/MeCN (1/9)]/[10 мМ NH4Oac (водн.)/MeCN (9/1)]. Система с кислотной подвижной фазой: [133 мМ НСООН (водн.)/MeCN (5/95)]/[8 мМ НСООН (водн.)/MeCN (98/2)].
Альтернативно масс-спектры регистрировали на приборе ГХ-МС (GC 6890, 5973N MCD, Agilent Technologies) с использованием колонки VF-5 MS (i.d. 0,25 мм × 30 м, 0,25 мкм (Varian Inc.)). Применяли линейный температурный градиент (40°С - 300°С), 25°С/мин. МС прибор был оснащен источником химической ионизации (ХИ) с газом-реагентом, представляющим собой метан. МС сканировал m/z 50-500, скорость сканирования составляла 3,25 считываний/с. Анализы ВЭЖХ осуществляли на системе Agilent НР1000, состоящей из вакуумного микродегазатора G1379A, сдвоенного насоса G1312A, автосемплера для планшетов с лунками G1367A, термостатируемого блока с колонкой G1316A и детектора на диодной матрице G1315B. Колонка: Х-Terra MS, Waters, 4,6×50 мм, 3,5 мкм. Температура колонки была установлена 40°С, скорость потока составляла 1,5 мл/мин. Детектор на диодной матрице производил считывания при 210-300 нм, шаг и ширина пика были установлены 2 нм и 0,05 мин соответственно. Использовали линейный градиент, поток от 0% до 100% ацетонитрила, за 4 мин. Подвижная фаза: ацетонитрил/10 мМ ацетат аммония в 5% ацетонитриле в MilliQ воде.
Обычная методика обработки после реакции состояла в экстракции продукта растворителем, таким как этилацетат, промывки водой с последующей сушкой органической фазы над MgSO4 или Na2SO4 и концентрированием раствора в вакууме.
Тонкослойную хроматографию (ТСХ) проводили на ТСХ-пластинах Merck (силикагель 60 F254); для визуализации пятен использовали УФ. Флэш-хроматографию проводили на Combi Flash® Companion™ с использованием RediSep™ флэш-колонок с нормальной фазой или на силикагеле 60 Merck (0,040-0,063 мм). Обычные растворители, использованные для флэш-хроматографии, представляли собой смеси хлороформ/метанол, толуол/этилацетат и этилацетат/гексаны.
Препаративную хроматографию осуществляли на автопрепаративном приборе ВЭЖХ Gilson с детектором на диодной матрице. Колонка: XTerra MS С8, 19×300 мм, 7 мкм. Градиент: ацетонитрил/0,1М ацетат аммония в 5% ацетонитриле в MilliQ воде, поток от 20% до 60% ацетонитрила за 13 мин. Скорость потока: 20 мл/мин. Альтернативно очистку проводили на полупрепаративной Shimadzu LC-8A ВЭЖХ с УФ-виз.-детектором Shimadzu SPD-10А, оснащенным колонкой Waters Symmetry® (C18, 5 мкм, 100 мм × 19 мм). Градиент: ацетонитрил/0,1% трифторуксусная кислота в MilliQ воде, поток от 35% до 60% ацетонитрила за 20 мин. Скорость потока: 10 мл/мин.
Перекристаллизацию обычно проводили в растворителях или смесях растворителей, таких как эфир, этилацетат/гептаны и метанол/вода.
Использовали следующие сокращения: ДХМ - дихлорметан; DIPCI - β-хлордиизопинокамфенилборан (DIP-Хлорид™); ДИПЭА - N,N-диизопропилэтил-амин; ДМФ - N,N-диметилформамид; ДМСО - диметилсульфоксид; NCS - N-хлорсукцинимид; NMP - 1-метил-2-пирролидинон; ТГФ - тетрагидрофуран; водн. - водный; конц. - концентрированный.
Используемые исходные материалы были или имеющимися в продаже, или были получены согласно методикам, приведенным в литературных источниках, и имели экспериментальные данные, аналогичные описанным. Ниже представлены примеры исходных веществ, которые были получены:
(1S)-1-(2-фторфенил)этанол: Garrett, С.Е. Tetrahedron: Asymmetry 2002, 13, 1347-1349; Doucet, H. Chem. Eur. J. 1999, 5, 1320-1330;
(R)-N-метиллейцинол: Aitali, M.; Allaoud, S.; Karim, A.; Meliet, C.; Mortreux, A. Tetrahedron: Asymmetry 2000, 11, 1367-1374;
(2R)-2-[(2-амино-5-меркапто[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ол: WO 02/076990;
5-(бензилтио)-7-хлор[1,3]тиазоло[4,5-d]пиримидин-2-амин: WO 00/09511;
3-(1-гидроксиэтил)бензамид: Watson, C.Y; Whish, W.J.D; Threadgill, M.D. Bioorg. Med. Chem. 1998 6(6) 721-34;
1-[3-(метилсульфонил)фенил]этанон: Т.Fujita, J.Iwasa и С.Hansch, Journal of the American Chemical Society 1964, 86, 5175-5180;
(1-хлорпропил)бензол: Desai, V.R.; Nechvatal, A.; Tedder, J.M. J. Chem. Soc. (B) 1969, 30-32;
3-[(1S)-1-гидроксиэтил]бензонитрил: Belley, M. Bioorg. Med. Chem., 1999, 7, 2697-2704;
1-(3-метоксифенил)этанол: Handa, S.J. Chem. Soc. Perkin Trans. 1 1995, 1623-1633;
(2R)-2-амино-4-фтор-4-метилпентан-1-ол: Truong, V.L; Gauthier, J.Y; Boyd, M; Roy, B; Scheigetz, J. Synlett 2005, 8, 1279-1280; ниже следует путь для S энантиомера:
(1S)-1-(3-фторфенил)этанол: Pastor, I.M. Chem. Eur. J. 2003, 9, 4031-4045.
Общий способ А
Боргидрид натрия (0,1 эквив.), ДИПЭА (1,5 эквив.) и соединение общей формулы (III) (1,2 эквив.) добавляли к соединению общей формулы (II) (1,0 эквив.) в ДМСО в атмосфере азота. Полученную реакционную смесь перемешивали при 40°С до тех пор, пока реакция не завершилась (наблюдали с помощью ЖХ-МС, ВЭЖХ или ТСХ). Смесь выливали в ледяную воду, и продукт экстрагировали ДХМ или EtOAc. Объединенные органические фазы сушили и концентрировали в вакууме. Сырой продукт при необходимости очищали с использованием препаративной ВЭЖХ или колоночной флэш-хроматографией.
Общий способ Б
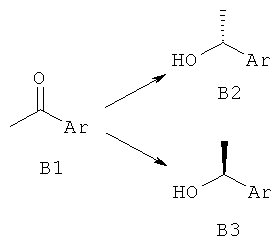
В1 (1,0 эквив.) в ТГФ добавляли при 0°С к (+)-DIPCI (с получением В2) или (-)-DIPCI (с получением В3) (1,5 эквив.) в ТГФ в атмосфере аргона. Реакционную смесь оставляли медленно нагреваться до комнатной температуры в течение ночи. Растворитель выпаривали, а затем добавляли Et2O и диэтаноламин (2,2 эквив.). Смесь перемешивали до тех пор, пока реакция не завершилась (наблюдали с помощью ЖХ-МС, ВЭЖХ или ТСХ). Образовавшийся осадок отфильтровывали, промывали Et2O, и в вакууме концентрировали фильтрат. Сырой продукт при необходимости очищали с использованием препаративной ВЭЖХ или колоночной флэш-хроматографией.
Общий способ В
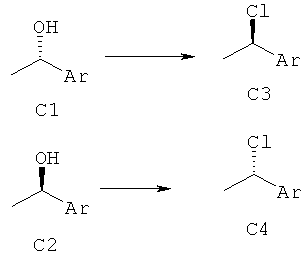
Трифенилфосфин (1,3 эквив.) в ТГФ добавляли при 0°С к NCS (1,3 эквив.) в ТГФ в атмосфере аргона. Полученную в результате смесь перемешивали при температуре окружающей среды в течение 30 мин. С1 или С2 (1 эквив.) добавляли при 0°С, и реакционную смесь перемешивали при температуре окружающей среды до тех пор, пока реакция не завершилась (наблюдали с помощью ЖХ-МС, ВЭЖХ или ТСХ). Растворитель выпаривали, а затем добавляли гексан и фильтрованием удаляли осадок. Фильтрат концентрировали в вакууме и сырой продукт при необходимости очищали с использованием препаративной ВЭЖХ или колоночной флэш-хроматографией.
Пример 1
(2R)-2-[(2-Амино-5-{[(1S)-1-(2-фторфенил)этил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]пентан-1-ол
а) 1-[(1R)-1-Хлорэтил]-2-фторбензол
Указанное в заголовке соединение получали с выходом 65% и энантиомерным избытком 93%, используя общий способ В и начиная с (1S)-1-(2-фторфенил)этанола (3,56 г, 25 ммоль).
1H ЯМР (CDCl3) δ 7.53 (td, 1H), 7.28 (m, 1H), 7.16 (m, 1H), 7.04 (m, 1H), 5.42 (q, 1H), 1.84 (d, 3H);
MC (ИЭР+) m/z 158 [M+Н]+.
б) (2R)-2-{[2-Амино-5-(бензилтио)[1,3]тиазоло[4,5-d]пиримидин-7-ил]амино}пентан-1-ол
5-(Бензилтио)-7-хлор[1,3]тиазоло[4,5-d]пиримидин-2-амин (6,0 г, 19,4 ммоль) растворяли в NMP (30 мл). Добавляли ДИПЭА (8,4 мл, 48,5 ммоль) и 2-амино-(2R)-1-пентанол (3,5 г, 33,9 ммоль), и смесь нагревали до 110°С в течение 4 дней. После охлаждения до температуры окружающей среды смесь вливали в воду (200 мл). Выпавший в осадок продукт собирали фильтрованием, промывали водой и использовали на следующей стадии без дополнительной очистки (7,0 г, выход 97%).
МС (ИЭР+) m/z 376 [М+Н]+.
в) (2R)-2-[(2-Амино-5-меркапто[1,3]тиазоло[4.5-d]пиримидин-7-ил)амино]пентан-1-ол
Круглодонную колбу снабжали конденсатором для смеси сухой лед-этанол и опускали в охлаждающую баню с использованием смеси сухой лед-этанол. В колбе конденсировали аммиак (250 мл), а затем добавляли (2R)-2-{[2-амино-5-(бензилтио)[1,3]тиазоло[4,5-d]пиримидин-7-ил]амино}пентан-1-ол (6,8 г, 18,1 ммоль). Полученную в результате смесь оставляли нагреваться до -33°С, и маленькими кусочками добавляли металлический натрий до тех пор, пока не появлялось голубое окрашивание, которое сохранялось в течение 30 секунд. Затем реакцию гасили добавлением столовой ложки твердого хлорида аммония. Аммиак выпаривали и к остатку добавляли воду (250 мл). Полученную в результате смесь нейтрализовывали 1 М HCl (водн.). Выпавший в осадок продукт собирали фильтрованием, промывали водой и сушили в вакууме с получением 4,15 г (выход 80%) указанного в заголовке соединения.
МС (ИЭР+) m/z 286 [M+H]+.
g) (2R)-2-[(2-Амино-5-{[(1S)-1-(2-фторфенил)этил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]пентан-1-ол
Указанное в заголовке соединение получали с выходом 96%, используя общий способ А и начиная с (2R)-2-[(2-амино-5-меркапто[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]пентан-1-ола (703 мг, 2,46 ммоль) и 1-[(1R)-1-хлорэтил]-2-фторбензола (469 мг, 2,96 ммоль).
1H ЯМР (ДМСО-d6) δ 8.38 (br s, 2H), 7.55 (td, 1H), 7.32 (m, 1H), 7.20 (m, 1H), 7.18 (d, 1H), 5.26 (q, 1H), 4.19 (br s, 1H), 3.43 (dd, 5.6 Гц, 1H), 3.35 (dd, 1H), 1.69 (d, 3Н), 1.66-1.42 (m, 2H), 1.39-1.21 (m, 2H), 0.86 (t, 3Н);
МС (ИЭР+) m/z 408 [M+H]+.
Пример 2
(2R)-2-[(2-Амино-5-{[(1S)-1-(2-фторфенил)этил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ол
Указанное в заголовке соединение получали с выходом 41%, используя общий способ А и начиная с (2R)-2-[(2-амино-5-меркапто[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ола (800 мг, 2,67 ммоль) и 1-[(1R)-1-хлорэтил]-2-фторбензола (509 мг, 3,21 ммоль).
1H ЯМР (ДМСО-d6) δ 7.97 (s, 2Н), 7.53 (td, 1H), 7.29 (m, 1H), 7.17 (m, 1H), 7.15 (d, 1H), 6.89 (d, 1H), 5.22 (q, 1H), 4.61 (t, 1H), 4.24 (br s, 1H), 3.38 (dt, 1H), 3.28 (m, 1H), 1.65 (d, 3Н), 1.59 (m, 1H), 1.49-1.32 (m, 2Н), 0.87 (d, 3Н), 0.84 (d, 3Н);
MC (ИЭР+) m/z 422 [M+H]+.
Пример 3
(2R)-2-({2-Амино-5-[(1-фенилэтил)тио][1,3]тиазоло[4,5-d]пиримидин-7-ил}амино)-4-метилпентан-1-ол
Указанное в заголовке соединение получали в виде смеси двух диастереомеров с выходом 67% согласно общему способу А, начиная с (2R)-2-[(2-амино-5-меркапто[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ола (320 мг, 1,01 ммоль) и (1-бромэтил)-бензола (245 мг, 1,21 ммоль).
1H ЯМР (ДМСО-d6) δ 7.94 (s, 2Н), 7.16 (m, 2Н), 7.14 (m, 2Н), 6.97 (m, 1H), 6.77 (d, 1H), 4.74 (m, 1H), 4.49 (m, 1H), 4.03 (m, 1H), 3.87 (m, 2Н), 3.22 (m, 2Н), 1.72 (dd, 1H), 1.61 (m, 1H), 1.42 (m, 2Н), 0.87 (d, 3Н), 0.85 (m, 3Н);
MC (ИЭР+) m/z 405 [М+Н]+.
Пример 4
(2R)-2-[(2-Амино-5-{[(1R)-1-фенилэтил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ол
Указанный в заголовке индивидуальный диастереомер (150 мг) получали ВЭЖХ очисткой (2R)-2-({2-амино-5-[(1-фенилэтил)тио][1,3]тиазоло[4,5-d]пиримидин-7-ил}амино)-4-метилпентан-1-ола (Пример 3) (500 мг).
1H ЯМР (ДМСО-d6) δ 7.98 (s, 2Н), 7.45 (m, 2Н), 7.32 (m, 2Н), 7.24 (m, 1H), 6.87 (d, 1H), 4.95 (q, 1H), 4.24 (br s, 1H), 3.45 (m, 1H), 3.35 (m, 1H), 1.68 (d, 3Н), 1.62 (m, 1H), 1.42 (m, 2Н), 0.88 (d, 3Н), 0.82 (d, 3Н);
MC (ИЭР+) m/z 405 [M+H]+.
Пример 5
3-{(1S)-1-[(2-Амино-7-{[(1R)-1-(гидроксиметил)бутил]амино}[1,3]тиазоло-[4,5-d]пиримидин-5-ил)тио]этил}бензонитрил
а) 3-[(1R}-1-Хлорэтил]бензонитрил
Указанное в заголовке соединение получали с выходом 79% согласно общему способу В, начиная с 3-[(1S)-1-гидроксиэтил]бензонитрила (3,35 г, 22,8 ммоль).
1H ЯМР (ДМСО-d6): δ 7.97 (s, 1Н), 7.82 (m, 2H), 7.60 (t, 1H), 5.40 (q, 1H), 1.80 (d, 3H);
13C ЯМР (ДМСО-d6): δ 144.1, 131.2, 131.6, 130.3, 129.9, 118.4, 111.6, 57.41, 25.5;
МС (ИЭР+) m/z 166 [М+Н]+.
б) 3-{(1S)-1-[(2-Амино-7-{[(1R)-1-(гидроксиметил]бутил]амино}-[1,3]тиазоло[4,5-d]пиримидин-5-ил)тио]этил}бензонитрил
Указанное в заголовке соединение получали с выходом 75% согласно общему способу А, начиная с 2-амино-7-{[(1R)-1-(гидроксиметил)бутил]амино}[1,3]тиазоло[4,5-d]пиримидин-5(6Н)-тиона (2,87 г, 10,0 ммоль) и 3-[(1R)-1-хлорэтил]бензонитрила (2,31 г, 13,9 ммоль).
1H ЯМР (ДМСО-d6): δ 8.00 (s, 2H), 7.91 (s, 1H), 7.82 (m, 1H), 7.69 (m, 1H), 7.52 (t, 1H), 6.90 (d, 1H), 5.00 (q, 1H), 4.63 (t, 1H), 4.13 (br s, 1H), 3.41 (m, 1H), 3.30 (m, 1H), 1.66 (d, 3Н), 1.57 (m, 1H), 1.43 (m, 1H), 1.29 (m, 2H), 0.86 (t, 3Н);
13С ЯМР (ДМСО-d6): δ 170.8, 168.7, 165.1, 155.7, 145.9, 132.3, 130.8, 130.6, 129.5, 118.7, 111.2, 63.3, 59.7, 51.8, 42.3, 33.0, 21.8, 18.8, 14.0;
МС (ИЭР+) m/z 415 [М+Н]+.
Пример 6
(2R)-2-{[2-Амино-5-({(1S)-1-[3-(метилсульфонил)фенил]этил}тио)[1,3]-тиазоло[4,5-d]пиримидин-7-ил]амино}-4-метилпентан-1-ол
а) (1S)- 1-[3-(Метилсульфонил)фенил]этанол
Указанное в заголовке соединение получали с выходом 58% из 1-[3-(метилсульфонил)фенил]этанона (2,00 г, 10,1 ммоль) согласно общему способу Б.
МС (ИЭР+) m/z 201 [M+H]+.
б) 1-[(1R)-1-Хлорэтил]-3-(метилсульфонил)бензол
Указанное в заголовке соединение получали с выходом 21% из (1S)-1-[3-(метилсульфонил)фенил]этанола (100 мг, 0,50 ммоль) согласно общему способу В.
МС (ИЭР+) m/z 219 [М+Н]+.
в) (2R)-2-{[2-Амино-5-({(1S)-1-[3-(метилсульфонил)фенил]этил}тио)[1,3]-тиазоло[4,5-d]пиримидин-7-ил]амино}-4-метилпентан-1-ол
Указанное в заголовке соединение получали из (2R)-2-[(2-амино-5-меркапто[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ола (16,5 г, 55,3 ммоль) и 1-[(1R)-1-хлорэтил]-3-(метилсульфонил)бензола (12,1 г, 55,3 ммоль) согласно общему способу А.
1H ЯМР (600 МГц, ДМСО-d6): δ 8.00 (m, 3Н), 7.81 (m, 2H), 7.60 (t, 1H), 6.91 (d, 1H), 5.06 (q, 1H), 4.66 (t, 1H), 4.24 (br s, 1H), 3.38 (m, 1H), 3.28 (m, 1H), 3.23 (s, 3Н), 1.69 (d, 3Н), 1.59 (m, 1H), 1.34-1.46 (m, 2H), 0.86 (m, 6H);
MC (ИЭР+) m/z 482 [M+H]+.
Пример 7
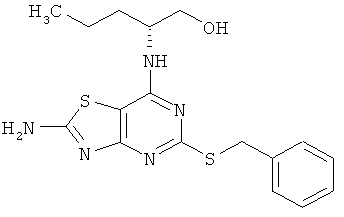
(2R)-2-[(2-Амино-5-{[(1S)-1-фенилэтил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино1пентан-1-ол
а) [(1R)-1-Хлорэтил]бензол
Указанное в заголовке соединение получали с выходом 67%, используя общий способ В и начиная с (1S)-1-фенилэтанола (25 г, 0,20 мол.).
1H ЯМР (CDCl3) δ 7.42 (m, 2H), 7.36 (m, 2H), 7.30 (m, 1H), 5.09 (q, 1H), 1.85 (d, 3Н);
МС (ХИ) m/z 141 [М+H]+.
б) (2R)-2-[(2-Амино-5-{[(1S)-1-фенилэтил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]пентан-1-ол
Указанное в заголовке соединение получали с выходом 23%, используя общий способ А и начиная с (2R)-2-[(2-амино-5-меркапто[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]пентан-1-ола (100 мг, 0,35 ммоль) и [(1R)-1-хлорэтил]бензола (54 мг, 0,38 ммоль).
1H ЯМР (ДМСО-d6) δ млн.-1 7.96 (br s, 1H), 7.43 (d, 1H), 7.31 (m, 2H), 7.22 (m, 1H), 6.86 (d, 1H), 4,95 (m, 1H), 4.64 (t, 1H), 4.17 (br s, 1H), 3.44 (m, 1H), 3.35 (m, 1H), 1.66 (d, 3Н), 1.58 (m, 1H), 1.44 (m, 1H), 1.36-1,21 (m, 2H), 0.85 (t, 3Н);
МС (ИЭР+) m/z 390 [М+Н]+.
Пример 8
3-{(1S)-1-[(2-Амино-7-{[(1R)-1-(гидроксиметил)-3-метилбутил]амино}-[1,3]тиазоло[4,5-d]пиримидин-5-ил)тио]этил}бензонитрил
Указанное в заголовке соединение получали с выходом 31% из (2R)-2-[(2-амино-5-меркапто[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ола (200 мг, 0,67 ммоль) и 3-[(1R)-1-хлорэтил]бензонитрила (166 мг, 1,0 ммоль) согласно общему способу А.
1H ЯМР (CD3OD) δ 7.89-7.76 (m, 2H), 7.57 (d, 1H), 7.49 (m, 1H), 5.12 (q, 1H), 4.42 (br s, 1H), 3.53 (m, 1H), 3.44 (m, 1H), 1.63-1.76 (m, 4H), 1.41-1.60 (m, 2H), 0.96 (t, 6H);
MC (ИЭР+) m/z 429 [M+H]+.
Пример 9
(2R)-2-({2-Амино-5-[(1-фенилпропил)тио][1,3]тиазоло[4,5-d]пиримидин-7-ил}амино)-4-метилпентан-1-ол
Указанное в заголовке соединение синтезировали в виде смеси двух диастереомеров общим способом А, начиная с реакции (2R)-2-[(2-амино-5-меркапто[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ола (30 мг, 100 мкмоль) и (1-хлорпропил)бензола (15,5 мкл, 100 мкмоль), с получением 13 мг (выход 31%) в виде масла.
1H ЯМР (CD3OD) δ 7.39 (t, 2H), 7.28 (m, 2H), 7.20 (t, 1H), 4.85 (dd, 1H), 4.57-4.40 (m, 1H), 3.62 (m, 1H), 3.59-3.48 (m, 1H), 2.25-2.11 (m, 1H), 2.01 (m, 1H), 1.79-1.65(m,1H), 1.63-1.53 (m, 1H), 1.53-1.42 (m, 1H), 1.01-0.87 (m, 9H);
МС (ИЭР+) m/z 418 [M+H]+.
Пример 10
3-{(1R)-1-[(2-Амино-7-{[(1R)-1-(гидроксиметил)-3-метилбутил]амино}-[1,3]тиазоло[4,5-d]пиримидин-5-ил)тио]этил}бензамид
а) 3-(1-Хлорэтил)бензамид
Диэтиланилин (390 мкл, 2,45 ммоль) добавляли к 3-(1-гидроксиэтил)бензамиду (400 мг, 2,45 ммоль), суспендированному в ДХМ (20 мл), и реакционную смесь охлаждали на ледяной бане. Добавляли по каплям тионилхлорид (255 мкл, 2,47 ммоль), и реакционную смесь помещали в холодильник на ночь. Добавляли воду, реакционную смесь экстрагировали дважды ДХМ, промывали 10%-ным раствором HCl, нейтрализовывали насыщенным раствором бикарбоната, обрабатывали рассолом, сушили (MgSO4), фильтровали и упаривали до сухого состояния. Сырой продукт перекристаллизовывали из смеси диэтиловый эфир/гексан с получением 335 мг (75%) указанного в заголовке соединения в виде белого твердого вещества.
1H ЯМР (хлороформ-d) δ 7.90 (s, 1Н), 7.73 (d, 1H), 7.62 (d, 1H), 7.46 (t, 1H), 5.14 (q, 1H), 1.88 (d, 3H), 1.60 (s, 2H);
МС (ИЭР+) m/z 183, 185 [M+H]+.
б) 3-{(1R)-1-[(2-Амино-7-{[(1R)-1-(гидроксиметил]-3-метилбутил]амино}-[1,3]тиазоло[4,5-d]пиримидин-5-ил)тио]этил}бензамид
Указанное в заголовке соединение синтезировали в виде смеси двух диастереомеров общим способом А, начиная с реакции (2R)-2-[(2-амино-5-меркапто[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ола (30 мг, 100 мкмоль) и 3-(1-хлорэтил)бензамида (20 мг, 100 мкмоль). Разделением смеси с помощью препаративной ВЭЖХ получали индивидуальный диастереомер (6 мг, выход 13%) в виде масла.
1H ЯМР (CD3OD) δ 7.99 (s, 1H), 7.73 (d, 1H), 7.69 (d, 1H), 7.41 (t, 1H), 5.15 (q, 1H), 4.43-4.52 (m, 1H), 3.54 (m, 1H), 3.47 (m, 1H), 1.74 (d, 3Н), 1.70 (m, 1H), 1.59-1.42 (m, 2H), 0.96 (t, 6H);
МС (ИЭР+) m/z 447 [M+H]+.
Пример 11
(2R)-2-{[2-Амино-5-({1-[3-(трифторметил)фенил]этил}тио)[1,3]тиазоло[4,5-d]пиримидин-7-ил]амино}-4-метилпентан-1-ол
Указанное в заголовке соединение синтезировали в виде смеси двух диастереомеров общим способом А, начиная с реакции (2R)-2-[(2-амино-5-меркапто[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ола (30 мг, 0,1 ммоль) и 3-(1-бромэтил)трифторметилбензола (15,5 мкл, 0,1 ммоль), с получением 38 мг (выход 81%) в виде масла.
1H ЯМР (600 МГц, CD3OD) δ 7.81-7.71 (m, 2H), 7.57-7.44 (m, 2H), 5.16 (q, 1H), 4.43 (s, 0.5H), 4.31 (s, 0.5H), 3.59 (m, 1H), 3.55-3.40 (m, 1H), 1.74 (t, 3Н), 1.69 (m, 0.5H), 1.63 (m, 0.5H), 1.54 (m, 1H), 1.49-1.37 (m, 1H), 0.96 (dd, 3Н), 0.86 (dd, 1.5H);
МС (ИЭР+) m/z 472 [M+H]+.
Пример 12
(2R)-2-[{2-Амино-5-[(1-фенилэтил)тио][1,3]тиазоло[4,5-d]пиримидин-7-ил}(метил)амино]-4-метилпентан-1-ол
а) (2R)-2-[[2-Амино-5-(бензилтио)[1,3]тиазоло[4,5-d]пиримидин-7-ил](метил)амино]-4-метилпентан-1-ол
5-(Бензилтио)-7-хлор[1,3]тиазоло[4,5-d]пиримидин-2-амин (1,5 г, 4,86 ммоль), ДИПЭА (691 мг, 5,35 ммоль) и (R)-N-метиллейцинол (956 мг, 7,29 ммоль) смешивали в NMP (7,5 мл). Полученный раствор перемешивали при 110°С в атмосфере азота в течение 2 дней. После охлаждения до комнатной температуры реакционную смесь выливали на лед. Полученный желтый осадок собирали фильтрованием, промывали водой и сушили в вакууме. Сырой продукт очищали колоночной флэш-хроматографией на диоксиде кремния (ДХМ: EtOAc, от 50:50 до 0:100) с получением 1,42 г (выход 72%) указанного в заголовке соединения в виде желтого твердого вещества.
1H ЯМР (ДМСО-d6) δ 7.97 (br s, 2H), 7.40 (m, 2H), 7.28 (m, 2H), 7.21 (m, 1H), 4.73 (dd, 1H), 4.64 (br s, 1H), 4.32 (br s, 2H), 3.52-3.37 (m, 2H), 3.00 (s, 3Н), 1.55-1.35 (m, 2H), 1.27 (m, 1H), 0.88 (d, 3Н), 0.80 (d, 3Н);
MC (ИЭР+) m/z 404 [М+Н]+.
б) (2R)-2-[(2-Амино-5-меркапто[1,3]тиазоло[4,5-d]пиримидин-7-ил)(метил)амино]-4-метилпентан-1-ол
Трехгорлую круглодонную колбу опускали в охлаждающую баню с использованием смеси сухой лед/этанол и снабжали конденсатором для смеси сухой лед/этанол. Систему продували азотом, и в колбе конденсировали аммиак (приблизительно 50 мл). (2R)-2-[[2-Амино-5-(бензилтио)[1,3]тиазоло[4,5-d]пиримидин-7-ил](метил)амино]-4-метилпентан-1-ол (1 г, 2,5 ммоль) добавляли в колбу, что привело к получению прозрачного желтого раствора. Маленькие кусочки металлического натрия (размером 2-3 мм) добавляли один за другим к реакционной смеси. Когда появлялось устойчивое синее окрашивание (>20 с), добавляли столовую ложку твердого NH4Cl для гашения реакции. Аммиак выпаривали. Добавляли воду (50 мл) и смесь нейтрализовывали 1 М HCl (водн.) до pH 7. Выпавшее в осадок желтое твердое вещество собирали фильтрованием, промывали водой и сушили в вакууме с получением 630 мг указанного в заголовке соединения (выход 80%).
1H ЯМР (ДМСО-d6) δ 12.78 (br s, 1 Н), 8.43 (br s, 2H), 4.84 (br, 2H), 3.52-3.38 (m, 2H), 3.01 (s, 3Н), 1.55-1.33 (m, 2H), 1.32-1.20 (m, 1H), 0.87 (m, 6H);
MC (ИЭР+) m/z 314 [M+H]+.
в) (2R)-2-[{2-Амино-5-[(1-фенилэтил)тио][1,3]тиазоло[4,5-d]пиримидин-7-ил}метил)амино]-4-метилпентан-1-ол
Указанное в заголовке соединение синтезировали в виде смеси двух диастереомеров общим способом А, начиная с реакции (2R)-2-[(2-амино-5-меркапто[1,3]тиазоло[4,5-d]пиримидин-7-ил)(метил)амино]-4-метилпентан-1-ола (31,4 мг, 0,1 ммоль) и 1-бромэтилбензола (13,5 мкл, 0,1 ммоль), с получением 26 мг (выход 62%).
1H ЯМР (ДМСО-d6) δ 7.96 (s, 2Н), 7.43 (t, 2H), 7.32 (m, 2H), 7.23 (t, 1H), 5.02-4.92 (m, 1H), 4.83-4.71 (m, 1H), 4.64 (s, 1H), 3.53-3.38 (m, 2H), 3.01 (d, 3H), 1.68 (dd, 3H), 1.52 (m, 1H), 1.43 (m, 1H), 1.35-1.24 (m, 1H), 0.88 (d, 3H), 0.83 (d, 3H);
МС (ИЭР+) m/z 418 [M+H]+.
Пример 13
(2R)-2-[(2-Амино-5-{[1-(2-хлорфенил)этил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино1-4-метилпентан-1-ол
а) 1-Хлор-2-(1-хлорэтил)бензол
Тионилхлорид (1,49 г, 12,6 ммоль) добавляли к 1-(2-хлорфенил)этанолу (1,0 г, 6,3 ммоль) в толуоле (50 мл) и смесь оставляли перемешиваться при комнатной температуре в течение 2 ч. К реакционной смеси добавляли 10%-ный водн. раствор HCl (20 мл). Органическую фазу отделяли и промывали еще одной порцией 10%-ного водн. раствора HCl, рассолом (20 мл), а затем отделяли, сушили и упаривали с получением 1-хлор-2-(1-хлорэтил)бензола с выходом 72%.
1H ЯМР (ДМСО-d6) δ 7.72 (1H, d), 7.48 (1H, m), 7.39 (2H, m), 5.59 (1H, q), 1.84 (3H, d).
б) (2R)-2-[(2-Амино-5-{[1-(2-хлорфенил)этил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ол
Указанное в заголовке соединение получали в виде смеси двух диастереомеров с выходом 24% согласно общему способу А, начиная с (2R)-2-[(2-амино-5-меркапто[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ола (20 мг, 0,047 ммоль) и 1-хлор-2-(1-хлорэтил)бензола (10 мг, 0,057 ммоль).
1H ЯМР (ДМСО-d6) δ 7.83 (2H, m), 7.48 (1H, d), 7.32 (1H, m), 7.15 (1H, m), 7.14 (1H, m), 6.79 (1H, d), 5.22 (1H, m), 4.13 (1H, m), 3.29-3.17 (2H, m), 1.52 (3H, dd), 1.47 (1 H, m), 1.31 (1 H, m), 1.25 (1 H, m), 0.76 (3H, dd), 0.73 (3H, dd);
13С ЯМР (ДМСО-d6) δ 171.18, 169.14, 165.75, 165.64, 155.97, 140.63, 132.62, 129.91, 129.04, 128.96, 127.94, 64.08, 63.88, 50.59, 24.70, 23.67, 22.33, 22.22, 22.01;
МС (ИЭР+) m/z 438, 440 [M+H]+.
Пример 14
(2R)-2-[(2-Амино-5-{[1-(3-метоксифенил)этил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ол
а) 1-(1-Хлорэтил)-3-метоксибензол
Указанное в заголовке соединение получали с выходом 59%, начиная с 1-(3-метоксифенил)этанола (0,5 г, 3 ммоль) и используя способ Примера 13 (а).
1H ЯМР (CDCl3) δ 7.22 (1Н, m), 6.76 (2H, m), 6.62 (1H, dd), 4.81 (1H, q), 3.57 (3H, s), 1.62 (3H, d).
б) (2R)-2-[(2-Амино-5-{[1-(3-метоксифенил)этил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпенпан-1-ол
Указанное в заголовке соединение получали в виде смеси двух диастереомеров с выходом 25% согласно общему способу А, начиная с (2R)-2-[(2-амино-5-меркапто[1,3]тиазоло[4,5-фиримидин-7-ил)амино]-4-метилпентан-1-ола (20 мг, 0,07 ммоль) и 1-(1-хлорэтил)-3-метоксибензола (9,7 мг, 0,06 ммоль).
1H ЯМР (ДМСО-d6) δ 7.89 (2H, s), 7.13 (1H, m), 6.87 (2H, m), 6.80 (1H, m), 6.71 (1H, m), 4.86 (1H, m), 4.55 (1H, br s), 4.17 (1H, m), 3.64 (3H, s), 1.51 (3H, d), 1.48 (1 H, m), 1.31 (2H, m), 0.76 (6H, m);
МС (ИЭР+) m/z 434 [M+H]+.
Пример 15
(2R)-2-[(2-амино-5-{[(1S)-1-фенилэтил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино1-4-метилпентан-1-ол
1) Использование способа (а)
(2R)-2-[(2-Амино-5-{[(1S)-1-фенилэтил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ол
Указанное в заголовке соединение получали с выходом 42%, используя общий способ А и начиная с (2R)-2-[(2-амино-5-меркапто[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ола (27 г, 90 ммоль) и [(1R)-1-хлорэтил]бензола (19 г, 135 ммоль).
1H ЯМР (ДМСО-d6) δ 7.95 (br s, 2H), 7.43 (m, 2H), 7.31 (m, 2H), 7.22 (m, 1H), 6.85 (d, 1H), 4.96 (q, 1H), 4.64 (t, 1H), 4.27 (br s, 1H), 3.44-3.30 (m, 2H), 1.66 (d, 3Н), 1.59 (m, 1H), 1.41 (m, 2H), 0.87 (d, 3Н), 0.84 (d, 3Н);
MC (ИЭР+) m/z 404 [M+H]+.
2)Использование способа (б)
а) 6-Амино-2-{[(1S)-1-фенилэтил]тио}пиримидин-4-ол
К моногидрату 6-амино-2-меркаптопиримидин-4-ола (1,6 г, 10 ммоль) в ДМФ (20 мл) добавляли NaH (60% в масле) (0,5 г, 12,5 ммоль), а затем NaBH4 (40 мг, 1 ммоль). Через 30 мин добавляли [(1R)-1-хлорэтил]бензол (1,4 г, 10 ммоль) и реакционную смесь перемешивали в течение 16 ч. Реакционную смесь концентрировали под вакуумом до объема примерно 10 мл, а затем выливали в воду (примерно 50 мл). Выпавший в осадок твердый материал отфильтровывали и промывали водой и эфиром с получением указанного в заголовке соединения (1,15 г, выход 46%).
1H ЯМР (ДМСО-d6) δ 7.46-7.22 (m, 5Н), 6.53 (br s, 2H), 4.99 (q, 1H), 4.92 (s, 1H), 1.67 (d, 3H).
б) 2-Амино-5-{[(1S)-1-фенилэтил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ол
Пиридин (0,6 г, 7,6 ммоль) добавляли к 6-амино-2-{[(1S)-1-фенилэтил]тио}пиримидин-4-олу (1 г, 4 ммоль) и KSCN (1,7 г, 16 ммоль) в ДМФ (20 мл). Реакционную смесь охлаждали до 0°С, и затем одной порцией добавляли бром (0,65 г, 4,0 ммоль). Через 2 часа реакционную смесь выливали в воду, и оранжевый осадок отфильтровывали и промывали. Твердое вещество суспендировали в смеси ДМФ (6 мл) и воды (2 мл) и нагревали до 110°С. Через 30 ч реакционную смесь выливали в воду, отфильтровывали желтоватый осадок и промывали водой и эфиром. Твердое вещество сушили в вакууме при 40°С с получением указанного в заголовке соединения (0,8 г, выход 65%).
1H ЯМР (ДМСО-d6) δ 8.16 (br s, 1H), 7.47-7.24 (m, 5Н), 5.05 (q, 1H), 1.71 (d, 3Н).
в) 7-Хлор-5-{[(1S)-1-фенилэтил]тио}[1,3]тиазоло[4,5-d]пиримидин-2-амин
POCl3 (1 мл) добавляли к ДМФ (1 мл) в диоксане (6 мл). К реакционной смеси одной порцией добавляли 2-амино-5-{[(1S)-1-фенилэтил]тио}-[1,3]тиазоло[4,5-d]пиримидин-7-ол (1 г, 3,3 ммоль). Добавляли POCl3 (1 мл) и реакционную смесь нагревали до 80°С в течение примерно 30 мин. Реакционную смесь охлаждали до комнатной температуры и выливали на лед. Полученную смесь нагревали до температуры дефлегмации в течение примерно 5 ч. Смеси давали достичь комнатной температуры, а затем экстрагировали EtOAc. Органический слой пропускали через слой силикагеля и концентрировали до сухого состояния с получением указанного в заголовке соединения (1 г, выход 95%).
1H ЯМР (ДМСО-d6) δ 8.91 (br s, 2H), 7.47-7.20 (m, 5H), 4.95 (q, 1H), 1.69 (d, 3Н).
г) (2R)-2-[(2-амино-5-{[(1S)-1-фенилэтил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ол
ДИПЭА (400 мг, 3 ммоль) и D-лейцинол (400 мг, 3,4 ммоль) добавляли к 7-хлор-5-{[(1S)-1-фенилэтил]тио}[1,3]тиазоло[4,5-d]пиримидин-2-амину (300 мг, 0,9 ммоль) в NMP (6 мл) и смесь нагревали до 120°С в течение 24 ч. Смесь выливали в воду и экстрагировали этилацетатом. Органическую фазу сушили (MgSO4), упаривали, и остаток очищали колоночной флэш-хроматографией (EtOAc) с получением указанного в заголовке соединения (200 мг, выход 54%).
1H ЯМР (ДМСО-d6) δ 7.95 (br s, 2H), 7.43 (m, 2H), 7.31 (m, 2H), 7.22 (m, 1H), 6.85 (d, 1H), 4.96 (q, 1H), 4.64 (t, 1H), 4.27 (br s, 1H), 3.44-3.30 (m, 2H), 1.66 (d, 3Н), 1.59 (m, 1H), 1.41 (m, 2H), 0.87 (d, 3Н), 0.84 (d, 3Н);
MC (ИЭР+) m/z 404 [M+H]+.
Пример 16
(2R)-2-[(2-Амино-5-{[(1S)-1-фенилэтил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-фтор-4-метилпентан-1-ол
ДИПЭА (0,83 мл, 4.75 ммоль) добавляли к раствору 7-хлор-5-{[(1S)-1-фенилэтил]тио}[1,3]тиазоло[4,5-d]пиримидин-2-амина (0,49 г, 1,52 ммоль) и (2R)-2-амино-4-фтор-4-метилпентан-1-ола (2 ммоль) в NMP (2 мл) и реакционную смесь перемешивали при 120°С в течение 22 ч. Очисткой с помощью ВЭЖХ получали указанное в заголовке соединение (0,22 г, выход 17%).
1Н ЯМР (400 МГц, CD3OD): δ 7.48 (m, 2H), 7.32 (m, 2H), 7.23 (m, 1H), 5.09 (q, 1H), 4.66 (br s, 1H), 3.61-3.47 (m, 2H), 2.12-1.89 (m, 2H), 1.75 (d, 3Н), 1.40 (m, 6Н);
МС (ИЭР+) m/z 422 [M+H]+.
Пример 17
(2R)-2-[(2-амино-5-{[(1S)-1-(2-фторфенил)этил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-фтор-4-метилпентан-1-ол
а) 6-Амино-2-{[(1S}-1-(2-фторфенил)этил]тио}пиримидин-4-ол
К моногидрату 6-амино-2-меркаптопиримидин-4-ола (4,23 г, 26,3 ммоль) в ДМФ (40 мл) порциями добавляли NaH (60% в масле, 1,05 г, 26,3 ммоль), а затем NaBH4 (0,099 г, 2,7 ммоль). Через 30 минут добавляли 1-[(1R)-1-хлорэтил]-2-фторбензол (5,0 г, 31,5 ммоль) в ДМФ (10 мл), и реакционную смесь перемешивали в течение 24 ч. Реакционную смесь концентрировали и распределяли между водой и ДХМ, органическую фазу сушили (MgSO4) и упаривали. Остаток очищали колоночной флэш-хроматографией (ступенчатый градиент: 5-10% МеОН в CHCl3) с получением указанного в заголовке соединения (5,20 г, выход 75%).
1H ЯМР (400 МГц, ДМСО-d6): δ 7.35 (m, 1H), 7.13 (m, 1H), 6.99 (m, 2H), 6.29 (s, 2H), 5.00 (q, 1H), 4.76 (br s, 1H), 1.49 (d, 3H);
МС (ИЭР+) m/z 266 [М+Н]+.
б) 2-Амино-5-{[(1S)-1-(2-фторфенил)этил]тио}[1,3]тиазоло[4,5-d]-пиримидин-7-ол
KSCN (10,76 г, 110,7 ммоль) и пиридин (3,9 мл, 49,2 ммоль) добавляли к 6-амино-2-{[(1S)-1-(2-фторфенил)этил]тио}пиримидин-4-олу (6,53 г, 24,6 ммоль) в ДМФ (70 мл). Смесь охлаждали до 0°С и по каплям добавляли Br2. Через 3,5 ч реакционную смесь выливали в воду, и образовавшийся осадок собирали фильтрованием. Твердое вещество суспендировали в смеси ДМФ (75 мл) и воды (15 мл) и нагревали до 120°С в течение 8 ч. Реакционную смесь выливали в воду, и твердое вещество собирали фильтрованием и сушили в вакууме при 40°С с получением указанного в заголовке соединения (6,42 г, выход 81%).
МС (ИЭР+) m/z 323 [М+Н]+.
в) 7-Хлор-5-{[(1S)-1-(2-фторфенил)этил]тио}[1,3]тиазоло[4,5-d]-пиримидин-2-амин
POCl3 (2,77 мл, 29,7 ммоль) добавляли к ДМФ (3,07 мл, 39,6 ммоль) в диоксане (30 мл). Через 30 минут эту смесь добавляли к раствору 2-амино-5-{[(1S)-1-(2-фторфенил)этил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ола (6,38 г, 19,8 ммоль) в диоксане (100 мл). Через 30 минут добавляли POCl3 (2,77 мл, 29,7 ммоль) и реакционную смесь нагревали до 80°С в течение 2 ч. После охлаждения до комнатной температуры осторожно добавляли воду (20 мл), и полученную в результате смесь перемешивали при 80°С в течение 30 минут и при комнатной температуре в течение 2 ч. Реакционную смесь выливали в воду и собирали образовавшийся осадок. Твердое вещество очищали колоночной флэш-хроматографией (5% МеОН в CHCl3) с получением указанного в заголовке соединения (5,91 г, выход 88%).
1H ЯМР (400 МГц, ДМСО-d6): δ 8.94 (s, 2Н), 7.58 (m, 1H), 7.32 (m, 1H), 7.20 (m, 2H), 5.22 (q, 1H), 1.71 (d, 3H);
MC (ИЭР+) m/z 341 [М+Н]+.
г) (2R)-2-[(2-Амино-5-{[(1S)-1-(2-фторфенил)этил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-фтор-4-метилпентан-1-ол
ДИПЭА (2,09 мл, 12,0 ммоль) добавляли к 7-хлор-5-{[(1S)-1-(2-фторфенил)этил]тио}[1,3]тиазоло[4,5-d]пиримидин-2-амину (1,36 г, 4,0 ммоль) и (2R)-2-амино-4-фтор-4-метилпентан-1-олу (4 ммоль) в NMP (3 мл). После перемешивания реакционной смеси при 120°С в течение 22 ч, ее выливали в воду и осадок собирали фильтрованием. Твердое вещество очищали колоночной флэш-хроматографией (ступенчатый градиент: 5%-10% МеОН в CHCl3) и препаративной ВЭЖХ с получением указанного в заголовке соединения (0,22 г, выход 13%).
1H ЯМР (400 МГц, CD3OD): δ 7.35 (m, 1H), 7.03 (m, 1H), 6.91 (m, 2Н), 5.15 (q, 1H), 4.40 (m, 1H), 3.35-3.21 (m, 2Н), 1.82-1.72 (m, 2Н), 1.50 (d, 3Н), 1.17 (m, 6H);
МС (ИЭР+) m/z 440 [М+НГ.
Пример 18
(2R)-2-[(2-Амино-5-{[(1S)-1-(3-фторфенил)этил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ол
a) 1-[(1R)- 1-Хлорэтил]-3-фторбензол
Указанное в заголовке соединение получали с выходом 49% и энантиомерным избытком 94,5%, используя общий способ В и начиная с (1S)-1-(3-фторфенил)этанола (4,20 г, 30 ммоль).
1H ЯМР (300 МГц, ДМСО-d6) δ 7.47-7.30 (m, 3Н); 7.16 (t, 1H); 5.36 (q, 1H); 1.78 (d, 3H).
б) (2R)-2-[(2-Амино-5-{[(1S)-1-(3-фторфенил)этил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ол
Указанное в заголовке соединение получали с выходом 61%, используя общий способ А и начиная с (2R)-2-[(2-амино-5-меркапто[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ола (0,30 г, 1,0 ммоль), 1-[(1R)-1-хлорэтил]-3-фторбензола (0,17 г, 1,1 ммоль) и NaBH4 (0,019 г, 0,5 ммоль).
1H ЯМР (400 МГц, CD3OD): δ 7.23 (m, 2Н), 7.13 (m, 1H), 6.86 (m, 1H), 5.01 (q, 1H), 4.38 (m, 1H), 3.43 (m, 2Н), 1.63 (d, 3Н), 1.44 (m, 2Н), 0.88 (m, 6H);
МС (ИЭР+) m/z 422 [М+Н]+.
Фармакологические исследования
Материалы
Рекомбинантный человеческий фракталкин (hCX3CL1) и рекомбинантный человеческий интерлейкин-8 (IL-8 или hCXCL8) приобретали у PeproTech Inc., UK. Рекомбинантный [125I]-фракталкин (человека) и [125I] hIL-8 с удельной активностью 2200 Ки/ммоль приобретали у NEN® Life Science Products, Inc., UK. Fluo4-AM приобретали у Molecular Probes, US. Все остальные химикаты были аналитической степени чистоты.
Клетки
Полную человеческую CX3CR1 кДНК (GenBank инвентарный номер U20350) экстрагировали из мРНК мозга человека (Superscript, Life Technologies) и лигировали в pCR-Blunt II ТОРО вектор (InVitrogen). Соответствующую вставку hCX3CR1 изолировали и далее субклонировали в pcDNA3.1zeo. С использованием набора для плазмид (Plasmid Midi Kit (Qiagen)) изготавливали ДНК плазмиду. Затем, используя трансфекционный реагент Superfect (Qiagen) согласно протоколу производителя, ввели экспрессионную плазмиду для hCX3CR1 в суспензию клеток почек человеческого эмбриона (HEKS) линии 293, содержащую вектор для стабильной экспрессии химерного G-белка Gαqi5. Создавали стабильный клон, используя селекцию на зеоцине (500 мкг/мл) и гигромицине (100 мкг/мл). Для дальнейшего применения клетки содержали в среде Игла, модифицированной Дульбекко/питательной смеси Ham's F12 (DMEM/F12), содержащей пиридоксин и обогащенной 10% (об./об.) фетальной телячьей сывороткой, 2 мМ L-глутамина, 100 ед/мл пенициллина и 100 мг/мл стрептомицина, 250 мкг/мл зеоцина и 100 мкг/мл гигромицина.
Клетки, экспрессирующие CXCR2 человека, полученные от AstraZeneca Charnwood, культивировали в ЕМЕМ, содержащей Глутамакс (Glutamax) и обогащенной 10% FBS (фетальной телячьей сывороткой) (от РАА, Austria), 1% заменимыми аминокислотами (NEAA), 100 ед/мл пенициллина и 100 мкг/мл стрептомицина (PEST) и 500 мкг/мл генетицин/G418.
Приготовление мембраны
Клетки выращивают при 37°С и 5% CO2 и собирают при слиянии 60-80% в буфере, содержащем 10 мМ Tris-HCl pH 7,4, 5 мМ EDTA, 0,1 мг/мл бацитрацина. Клетки центрифугируют при 300хg в течение 10 мин и осадок после центрифугирования ресуспендируют в буфере для сбора (10 мМ Tris-HCl, pH 7,4, 5 мМ этилендиаминтетрауксусная кислота (EDTA) и 0,1 мг/мл бацитрацина, объединяют и гомогенизируют, используя гомогенизатор Dounce. Гомогенат центрифугируют при 48000×g в течение 10 мин и ресуспендируют в буфере для сбора, используя Ultra-Turrax T8. Мембранные аликвоты хранят при -80°С. Концентрацию белка определяли на планшетах для микротитрования, как описано у Harrington (1990, Anal. Biochem. 186, 285-287).
Опыт на связывание рецептора in vitro
Исследования на конкурентное связывание [125I]фракталкина проводили в 2 мл 96-луночных планшетах (Beckman, Germany) с общим объемом 1000 мкл/лунка. Каждая лунка содержала 10 пМ [125I]-фракталкина и мембрану, эквивалентную концентрации рецептора 1 пМ в буфере для анализа (50 мМ Hepes-KOH, pH 7,4, 10 мМ MgCl2, 1 мМ EDTA, 0,1% (масс./об.) желатин). Десять концентраций (2 точки/логарифмическая единица (log unit)) тестируемых соединений предварительно растворяли в ДМСО и добавляли для достижения конечной концентрации 1% (об./об.) ДМСО. Опыт начинали добавлением мембран и инкубированием при 25°С в течение 24 ч. Реакции останавливали быстрым фильтрованием через фильтры из стекловолокна Whatman GF/B, предварительно обработанные 0,3% полиэтилимином, и последующей промывкой ледяным буфером (10 мМ Hepes-KOH pH 7,4, 500 мМ NaCl), используя харвестер для связывания рецепторов Brandel. Добавляли сцинтилляционный коктейль и с использованием жидкостного сцинтилляционного счетчика Packard 2500TR (Perkin Elmer, USA) определяли радиоактивность.
Исследования на конкурентное связывание [125I]-hIL-8 проводят однократно в 96-луночных планшетах с белым прозрачным дном isoplate с общим объемом 200 мкл; каждая лунка содержит 150 пМ [125I]-hIL-8 (удельная активность 2200 Ки/ммоль), препарат мембрана-SPA, эквивалентный 20 пМ рецепторов, и 1,5 мг SPA-гранул в буфере для анализа [50 мМ HEPES-KOH pH 7,4, 10 мМ MgCl2, 1 мМ EDTA, 0,5% (масс./об.) желатин]. Тестируемые соединения обрабатывали, как описано выше. Неспецифическое связывание определяют в присутствии 500 нМ немеченного hIL-8. Агонист hIL-8 (кривая концентрация-ответ от 3 пМ до 30 нМ) используют в качестве соединения сравнения в каждом опыте. Кривая пептида не содержит ДМСО. Реакцию связывания начинают добавлением 140 мкл препарата мембрана-SPA, и образцы инкубируют в темноте при комнатной температуре в течение 4 ч. Планшеты для анализа обсчитывают с использованием жидкостного сцинтилляционного счетчика (Wallac MicroBeta® TriLux 1450 от PerkinElmer, США).
[35S]GTPγS binding
Исследования на связывание [35S]GTPγS проводили в планшетах для микротитрования с прозрачным дном в двух экземплярах с 10 концентрациями ингибитора (2 конц./log units), разбавленного ДМСО (конечная конц. 1%) и при комнатной температуре. Мембраны, экспрессирующие рецептор hCX3CR1 (конечная концентрация 20 мкг белка/лунка), добавляли вместе с гранулами SPA (конечная концентрация 1 мг/лунка), все суспендированы в буфере для связывания GTPγS (50 мМ Tris-HCl, 100 мМ NaCl, 0,1% желатин, 15 мкг сапонин/мл и 3 мкМ GDP, pH 7,4 при комнатной температуре). Мембраны, гранулы SPA и лекарства предварительно инкубировали 30 мин перед добавлением 310 пМ фракталкина для максимальной стимуляции. Основную активность определяли как активность, обнаруженную без фракталкиновой стимуляции (буфер для связывания GTPγS). Еще через 30 мин реакцию начинали добавлением [35S]GTPγS до конечной концентрации 0,1 нМ и конечного объема опыта 0,2 мл. Эксперимент останавливали через 30 минут центрифугированием при 2000 об/мин в течение 2×5 минут (разные направления), и с использованием жидкостного сцинтилляционного счетчика (Wallac MicroBeta® TriLux 1450) определяли радиоактивность.
Результаты
Данные по связыванию рецептора для выбранных соединений по настоящему изобретению приведены в Таблице 1. Соответствующие данные для соединений сравнения приведены в Таблице 2.
Сравнение данных, приведенных в Таблицах 1 и 2, ясно показывает, что соединения по настоящему изобретению, где R1 представляет собой Me или Et, являются как более сильными антагонистами CX3CR1 рецептора, так и менее сильными антагонистами CXCR2 рецептора, чем соответствующие соединения сравнения, где R1 представляет собой Н. Полагают, что такая улучшенная селективность в отношении антагонизма CX3CR1 рецептора приводит к значительному терапевтическому преимуществу.
Изобретение относится к производному 5-замещенного 7-амино-[1,3]тиазоло[4,5-d]пиримидина формулы (I) и его оптическим изомерам и фармацевтически приемлемым солям, где R1 представляет собой СН3 или СН3СН2; R2 представляет собой Н, 2-F, 2-Cl, 3-F, 3-ОСН3, 3-CN, 3-CF3, 3-CONH2 или 3-SO2CH3; R3 представляет собой Н или СН3; R4 представляет собой Н или СН3; и R5 представляет собой Н; или, когда R4 представляет собой СН3, R5 представляет собой Н или F. Также изобретение относится к способам получения соединений формулы (I) и фармацевтическим композициям, обладающим свойствами антагониста рецептора CX3CR1, содержащим соединения формулы (I). Технический результат - производные 5-замещенного 7-амино-[1,3]тиазола[4,5-d]пиримидина в качестве селективных антагонистов рецептора CX3CR1. 5 н. и 10 з.п. ф-лы, 2 табл.
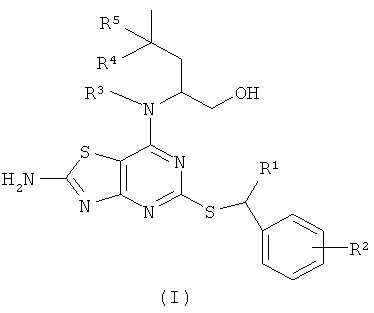
1. Соединение формулы (I)
R1 представляет собой СН3 или СН3СН2;
R2 представляет собой Н, 2-F, 2-Cl, 3-F, 3-ОСН3, 3-CN, 3-CF3, 3-CONH2 или 3-SO2CH3;
R3 представляет собой Н или CH3;
R4 представляет собой Н или CH3; и
R5 представляет собой Н; или, когда R4 представляет собой CH3, R5 представляет собой Н или F;
и его оптические изомеры и фармацевтически приемлемые соли.
2. Соединение по п.1, где R3 представляет собой H.
3. Соединение по п.1, где R1 представляет собой CH3.
4. Соединение по п.1, где R2 представляет собой Н, 2-F или 3-CN.
5. Соединение по п.1, где R4 представляет собой Н.
6. Соединение по п.1, где R4 представляет собой CH3.
7. Соединение по п.6, где R5 представляет собой F.
8. Соединение по п.6, где R5 представляет собой Н.
9. Соединение по п.1, где R1 представляет собой CH3; R2 представляет собой Н, 2-F или 3-CN; R3 представляет собой H; R4 представляет собой Н или СН3; и R5 представляет собой Н.
10. Соединение формулы (I) по п.1, представляющее собой:
(2R)-2-[(2-амино-5-{[(1S)-1-(2-фторфенил)этил]тио}[1,3]тиазоло[4,5-а]пиримидин-7-ил)амино]пентан-1-ол;
(2R)-2-[(2-амино-5-{[(1S)-1-(2-фторфенил)этил]тио}[1,3]тиазоло[4,5-а]пиримидин-7-ил)амино]-4-метилпентан-1-ол;
(2R)-2-({2-амино-5-[(1-фенилэтил)тио][1,3]тиазоло[4,5-а]пиримидин-7-ил}амино)-4-метилпентан-1-ол;
(2R)-2-[(2-амино-5-{[(1R)-1-фенилэтил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ол;
3-{(1S)-1-[(2-амино-7-{[(1R)-1-(гидроксиметил)бутил]амино}[1,3]тиазоло-[4,5-d]пиримидин-5-ил)тио]этил}бензонитрил;
(2R)-2-{[2-амино-5-({(1S)-1-[3-(метилсульфонил)фенил]этил}тио)-[1,3]тиазоло[4,5-d]пиримидин-7-ил]амино}-4-метилпентан-1-ол;
(2R)-2-[(2-амино-5-{[(1S)-1-фенилэтил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]пентан-1-ол;
3-{(1S)-1-[(2-амино-7-{[(1R)-1-(гидроксиметил)-3-метилбутил]амино}-[1,3]тиазоло[4,5-d]пиримидин-5-ил)тио]этил}бензонитрил;
(2R)-2-({2-амино-5-[(1-фенилпропил)тио][1,3]тиазоло[4,5-d]пиримидин-7-ил}амино)-4-метилпентан-1-ол;
3-{(1R)-[(2-амино-7-{[(1R)-1-(гидроксиметил)-3-метилбутил]амино}-[1,3]тиазоло[4,5-d]пиримидин-5-ил)тио]этил}бензамид;
(2R)-2-{[2-амино-5-({1-[3-(трифторметил)фенил]этил}тио)[1,3]тиазоло[4,5-d]пиримидин-7-ил]амино}-4-метилпентан-1-ол;
(2R)-2-[{2-амино-5-[(1-фенилэтил)тио][1,3]тиазоло[4,5-d]пиримидин-7-ил}(метил)амино]-4-метилпентан-1-ол;
(2R)-2-[(2-амино-5-{[1-(2-хлорфенил)этил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ол;
(2R)-2-[(2-амино-5-{[1-(3-метоксифенил)этил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ол;
(2R)-2-[(2-амино-5-{[(1S)-1-фенилэтил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ол;
(2R)-2-[(2-амино-5-{[(1S)-1-фенилэтил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-фтор-4-метилпентан-1-ол;
(2R)-2-[(2-амино-5-{[(1S)-1-(2-фторфенил)этил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-фтор-4-метилпентан-1-ол;
(2R)-2-[(2-амино-5-{[(1S)-1-(3-фторфенил)этил]тио}[1,3]тиазоло[4,5-d]пиримидин-7-ил)амино]-4-метилпентан-1-ол;
или его фармацевтически приемлемая соль.
11. Соединение формулы (I) по любому из пп.1-10 или его фармацевтически приемлемая соль для применения в качестве лекарственного средства, обладающего свойством антагониста рецептора CX3CR1.
12. Фармацевтическая композиция, обладающая свойством антагониста рецептора CX3CR1, содержащая соединение формулы (I), как оно определено в любом из пп.1-10, или его фармацевтически приемлемую соль в смеси с фармацевтически приемлемым разбавителем или носителем.
13. Применение соединения формулы (I), как оно определено в любом из пп.1-10, или его фармацевтически приемлемой соли в изготовлении лекарственного средства для лечения или профилактики заболеваний или состояний, при которых полезен антагонизм рецептора CX3CR1.
14. Способ получения соединения формулы (I), как оно определено в любом из пп.1-10, или его фармацевтически приемлемой соли, включающий:
взаимодействие соединения формулы (II):
где R3, R4 и R5 являются такими, как определено в формуле (I);
с соединением формулы (III):
где R1 и R2 являются такими, как определено в формуле (I), и L1 представляет собой уходящую группу;
и, когда это необходимо, превращение полученного соединения формулы (I) в его фармацевтически приемлемую соль; и, если требуется, превращение полученного соединения формулы (I) в его оптический изомер.
15. Способ получения соединения формулы (I), как оно определено в любом из пп.1-10, или его фармацевтически приемлемой соли, включающий:
взаимодействие соединения формулы (IV):
где R1 и R2 являются такими, как определено в формуле (I), и L2 представляет собой уходящую группу;
с соединением формулы (V):
где R3, R4 и R5 являются такими, как определено в формуле (I);
и, когда это необходимо, превращение полученного соединения формулы (I) в его фармацевтически приемлемую соль; и, если требуется, превращение полученного соединения формулы (I) в его оптический изомер.
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| WO 00/09511 A1, 24.02.00 | |||
| ПРОИЗВОДНЫЕ 5H-ТИАЗОЛ[3,2-А]ПИРИМИДИНА, ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ И ЛЕКАРСТВЕННОЕ СРЕДСТВО | 1998 |
|
RU2197493C2 |
| RU 2003110575 A, 27.01.2005. | |||

