Настоящее изобретение относится к способу аминирования орто-бициклопропил- или орто-C6-C7алкилзамещенных галогенбензолов, 5-галогенбензонорборненов или 5-галогенбензонорборнадиенов.
Орто-бициклопропил- или орто-C6-C7алкилзамещенные первичные анилины, такие как, например, 2-бициклопропил-2-илфениламин и 2-(1,3-диметилбутил)фениламин являются ценными промежуточными соединениями при получении фунгицидов, таких как описанные, например, в патентных документах WO 03/074491 и WO 03/010149.
5-Аминобензонорборнены и 5-аминобензонорборнадиены, такие как, например, 9-изопропил-1,2,3,4-тетрагидро-1,4-метано-нафталин-5-иламин, являются ценными промежуточными соединениями при получении фунгицидов, таких как описанные, например, в патентном документе WO 04/035589.
Химические препараты для сельского хозяйства обычно производят в больших количествах. Например, производство фунгицида хлорталонила составило в 2005 году более 23000 метрических тонн.
В общих чертах, анилины со стерически менее жесткими орто-заместителями, такие как орто-толиламин, могут быть получены взаимодействием галогенбензолов с аммиаком при помощи катализируемых палладием реакций кросс-сочетания, описанных в публикации Journal of the American Chemical Society, 128, 10028-10029, 2006. Но успешное применение палладийсодержащих катализаторов при одностадийном аминировании более стерически затрудненных галогенбензолов, таких как орто-бициклопропилзамещенных галогенбензолов, 5-галогенбензонорборненов или 5-галогенбензонорборнадиенов, описано не было.
Согласно патентному документу WO 03/074491 орто-бициклопропилзамещенные первичные анилины могут быть получены взаимодействием соответствующих орто-бициклопропилзамещенных галогенбензолов в результате двухстадийного химического превращения сначала по катализируемой палладием(II) реакции с бензофенонимином и затем взаимодействием продуктов реакции с гидрохлоридом гидроксиламина и ацетатом натрия или с кислотами, например хлористоводородной кислотой. Однако такая методика получения первичных анилинов является неудобной при крупномасштабном производстве орто-бициклопропилзамещенных первичных анилинов вследствие необходимости проведения второй стадии и относительно высокой цены бензофенонимина. Кроме того, в патентном документе WO 03/074491 методика описана исключительно для бром- или йодбензолов, но не для хлорбензолов. Было обнаружено, что методика, описанная в патентном документе WO 03/074491, мало подходит для иминирования с высокими выходами менее реакционноспособных, но более дешевых 2-(2-хлор-фенил)бициклопропанов.
В патентном документе WO 06/061226 раскрыто и описано успешное одностадийное аминирование стерически затрудненных орто-бициклопропилзамещенных галогенбензолов при использовании медьсодержащих катализаторов. Такая методика получения первичных анилинов не является приемлемой для крупномасштабного производства орто-алкилзамещенных первичных анилинов вследствие высокой стоимости удаления и переработки отходов соли меди. Кроме того, было обнаружено, что описанная в патентном документе WO 06/061226 методика плохо подходит для аминирования с высокими выходами менее реакционноспособных, но более дешевых 2-(2-хлор-фенил)бициклопропанов.
В патентном документе WO 04/035589 описаны различные 5-амино-бензонорборнены или 5-амино-бензонорборнадиены, способы их получения и их применение в качестве промежуточных соединений при получении микробиоцидов. Согласно патентному документу WO 04/035589 эти амины могут быть получены, как показано на схеме 1 ниже.
Схема 1
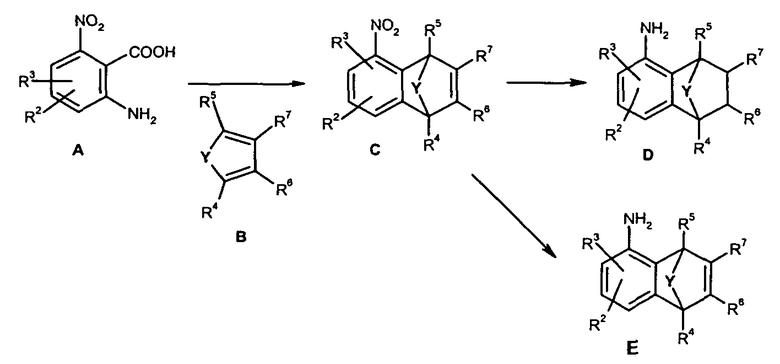
В синтезе, показанном на схеме 1, 3-нитробензол, образующийся из 6-нитроантраниловой кислоты (A), взаимодействует с циклическим 1,4-диеном (B), таким как 5-изопропил-циклопентадиеном с образованием 5-нитробензонорборнадиена (C) по реакции Дильса-Альдера. При стандартных условиях каталитического восстановления (например, использование никеля Ренея или палладия на угле в растворителе, таком как метанол), восстанавливаются и 5-нитрогруппа, и 2,3-двойная связь 5-нитробензонорборнадиена (C) с образованием 5-амино-бензонорбонена (D). При мягких условиях каталитического восстановления (например, использование металлического цинка в присутствии хлористого аммония или амальгамы алюминия), образуются аминобензонорборнадиены (E). Примером соединения (D) является 5-амино-9-изопропилбензонорборнен, который является предшественником амида, например амида 3-дифторметил-1-метил-1H-пиразол-4-карбоновой кислоты.
Недостатком синтеза, показанного на схеме 1, является образование ряда нежелательных изомерных примесей. Например, при получении по реакции Дильса-Альдера 5-нитробензонорборнадиена (C), где R4, R5, R6 и R7 все являются Н, и Y является CH-изопропилом, образуются следующие региоизомеры:
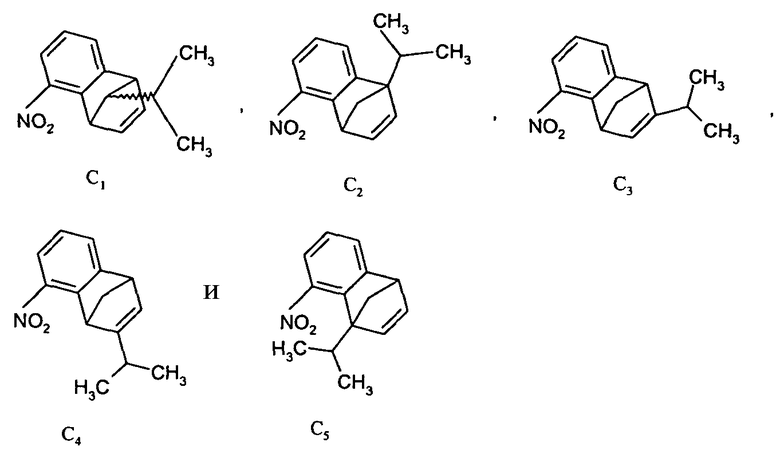
К сожалению, требуемый изомер C1 образуется с относительно низким выходом. Хотя нежелательные изомеры могут быть удалены либо в конце реакции Дильса-Альдера, либо на более поздней стадии при помощи традиционных методов, таких как фракционная кристаллизация или фракционная дистилляция, или при помощи хроматографических методов, этот синтетический подход не подходит в должной мере для крупномасштабного производства.
Соответственно, задачей настоящего изобретения является разработка нового способа получения орто-бициклопропил- или орто-C6-C7алкилзамещенных первичных анилинов, 5-аминобензонорборненов и 5-аминобензонорборнадиенов, который избавлен от упомянутых выше недостатков известного способа, и позволяет получать эти соединения при экономически целесообразных затратах и легко управляемым методом с высокими выходами и с высоким качеством.
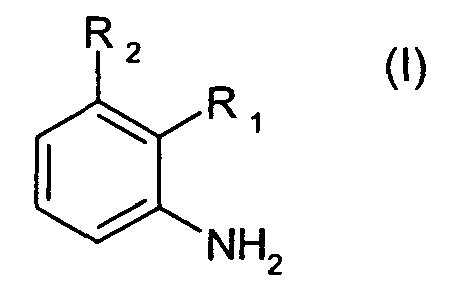
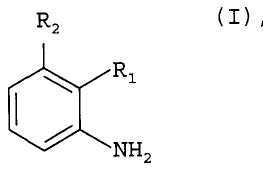
Соответственно, настоящее изобретение относится к способу получения соединений формулы I
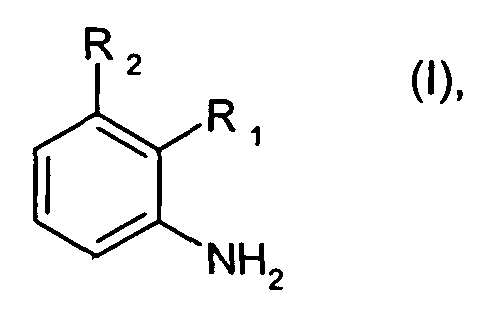
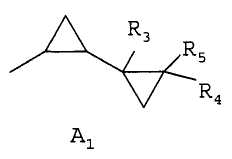
где R1 является 1,3-диметилбутилом, 1,3,3-триметилбутилом или группой A1
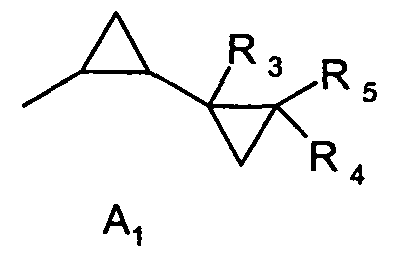
где R3, R4 и R5 каждая независимо друг от друга является водородом или C1-C4алкилом; и
R2 является водородом; или
R1 и R2 вместе образуют группу A2
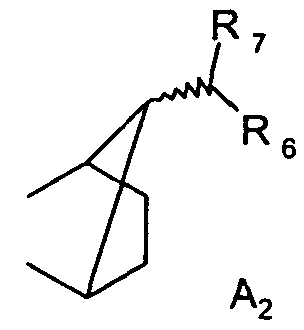
где R6 и R7 каждая независимо друг от друга является водородом или C1-C4алкилом; или R1 и R2 вместе образуют группу A3
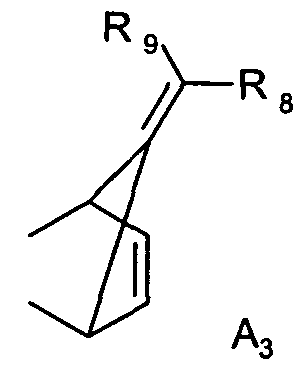
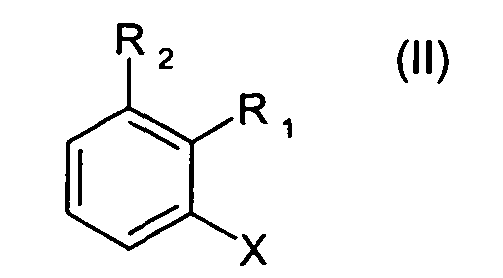
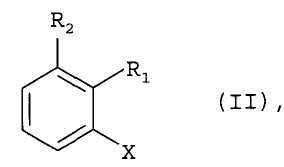
где R8 и R9 каждая независимо друг от друга является водородом или C1-C4алкилом; где соединение формулы II
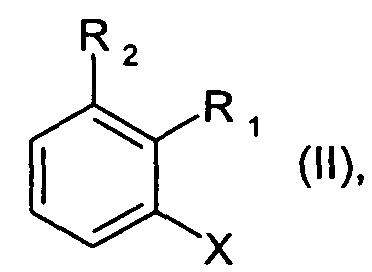
где R1 и R2 определены для формулы I, и X является бромом или хлором, взаимодействует с аммиаком в присутствии основания и каталитического количества, по меньшей мере, одного комплексного соединения палладия, где комплексное соединение палладия включает, по меньшей мере, один ферроценилбифосфиновый лиганд.
Соединения формулы I находятся в различных стереоизомерных формах. Способ согласно изобретению включает получение указанных индивидуальных стереоизомерных форм и получение смесей указанных стереоизомерных форм в любом соотношении.
Предпочтительно, чтобы способ согласно изобретению применялся для получения соединений формулы I, где R1 является группой A1, где R3, R4 и R5 каждая независимо друг от друга является водородом или C1-C4алкилом; и R2 является водородом; или R1 и R2 вместе образуют группу A2, где R6 и R7 каждая независимо друг от друга является водородом или C1-C4алкилом; или R1 и R2 вместе образуют группу A3, где R8 и R9 каждая независимо друг от друга является водородом или C1-C4алкилом.
Предпочтительно, чтобы способ согласно изобретению применялся для получения соединений формулы I, где R1 является A1, R3 является водородом или C1-C4алкилом и R2, R4 и R5 являются водородом.
Предпочтительно, чтобы способ согласно изобретению применялся для получения соединений формулы I, где R1 является A1, R3 является водородом или метилом и R2, R4 и R5 являются водородом.
Предпочтительно, чтобы способ согласно изобретению применялся для получения соединений формулы IA
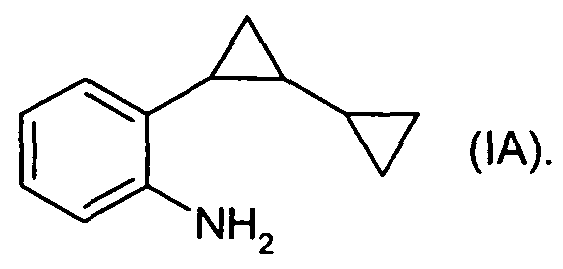
Предпочтительно, чтобы способ согласно изобретению применялся для получения соединений формулы I, где R1 является 1,3-диметилбутилом и R2 является водородом.
Предпочтительно, чтобы способ согласно изобретению применялся для получения соединений формулы I, где R1 является 1,3,3-триметил-бутилом и R2 является водородом.
Предпочтительно, чтобы способ согласно изобретению применялся для получения соединений формулы I, где R1 и R2 вместе образуют группу A2, где R6 и R7 каждая независимо друг от друга является водородом или C1-C4алкилом.
Предпочтительно, чтобы способ согласно изобретению применялся для получения соединений формулы I, где R1 и R2 вместе образуют группу A2, где R6 и R7 каждая является метилом.
Предпочтительно, чтобы способ согласно изобретению применялся для получения соединений формулы I, где R1 и R2 вместе образуют группу A3, где R8 и R9 каждая независимо друг от друга является водородом или C1-C4алкилом.
Предпочтительно, чтобы способ согласно изобретению применялся для получения соединений формулы I, где R1 и R2 вместе образуют группу A3, где R8 и R9 каждая является метилом.
Предпочтительно, чтобы в способе согласно изобретению применялись соединения формулы II, где X является бромом.
Предпочтительно, чтобы в способе согласно изобретению применялись соединения формулы II, где X является хлором.
В способе согласно изобретению соединения формулы II обычно могут быть использованы в концентрациях от 0,01 M до 5 M. Более предпочтительно, чтобы соединения формулы II использовались в концентрациях от 0,1 M до 5 M. Еще более предпочтительно, чтобы соединения формулы I использовались в концентрациях от 0,1 M до 2 M. Возможность использования высоких концентраций соединения формулы II является важным преимуществом способа согласно изобретению, так как при высоких концентрациях выделяемого вещества требуется меньше растворителя, что делает способ согласно изобретению особенно подходящим для крупномасштабного производства.
Комплексные соединения палладия, которые используют в способе согласно изобретению, образуются из предшественника палладия и, по меньшей мере, одного ферроценилбифосфинового лиганда. Предпочтительно, чтобы в способе согласно изобретению комплексные соединения палладия присутствовали в растворенной форме в виде комплексов палладий-лиганд.
Комплексные соединения палладия в способе согласно изобретению могут быть использованы в виде уже образованных комплексных соединений палладия, или они образуются in situ в способе согласно изобретению.
При образовании комплексных соединений палладия предшественник палладия взаимодействует, по меньшей мере, с одним ферроценилбифосфиновым лигандом. В случае неполного взаимодействия может иметь место ситуация, когда незначительные количества предшественника палладия или лиганда не растворяются в реакционной смеси.
Подходящими предшественниками палладия являются ацетат палладия, дихлорид палладия, раствор дихлорида палладия, палладий2(дибензилиден-ацетон)3 или палладий(дибензилиден-ацетон)2, палладий тетракис(трифенилфосфин), палладий-на-угле, палладий дихлорбис(бензонитрил), палладий(трис-трет-бутилфосфин)2 или смесь палладий2(дибензилиден-ацетон)3 и палладий (трис-трет-бутилфосфин)2.
Ферроценилбифосфиновые лиганды являются бидентатными третичными фосфиновыми лигандами, обычно используемыми в реакциях, катализируемых палладием. Такие бидентатные лиганды занимают два координационных места и поэтому способны образовывать хелаты с соединениями палладия.
Подходящими ферроценилбифосфиновыми лигандами являются:
(R)-(-)-1-[(S)-2-(дициклогексилфосфино)ферроценил]этилди-трет-бутилфосфин
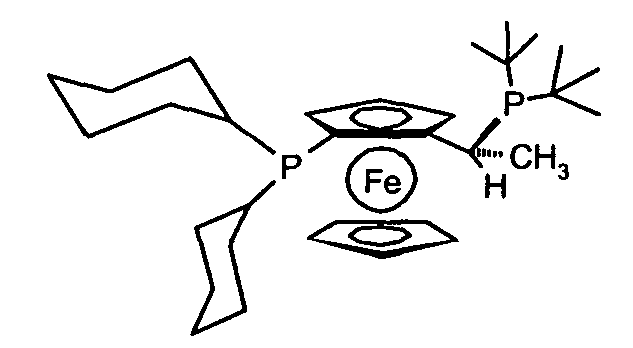
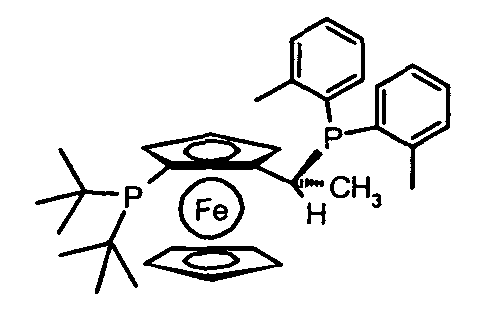
1,1'-бис(дифенилфосфино)ферроцен (dppf), 1,1'-бис(ди-трет-бутилфосфино)ферроцен, (R)-(-)-1-[(S)-2(бис(4-трифторметил-фенил)фосфино)ферроценил]этилди-трет-бутилфосфин, (R)-(-)-1-[(S)-2-(ди(3,5-бис-трифторметилфенил)фосфино)ферроценил]этилдициклогексилфосфин, (R)-(-)-1-[(S)-2-(ди(3,5-бис-трифторметил-фенил)фосфино)ферроценил]этилди(3,5-диметилфенил)фосфин, (R)-(-)-1-[(S)-2-(дициклогексилфосфино)ферроценил]этилдициклогексилфосфин, (S)-(+)-1-[(R)-2-(дициклогексилфосфино)ферроценил]этилдициклогексилфосфин, (S)-(+)-1-[(R)-2-(дициклогексилфосфино)ферроценил]этилдифенилфосфин, (R)-(-)-1-[(S)-2-(бис(3,5-диметил-4-метоксифенил)фосфино)ферроценил]этилдициклогексилфосфин, (S)-(+)-1-[(R)-2-(ди-фурилфосфино)ферроценил]этилди-3,5-ксилилфосфин, (R)-(-)-1-[(S)-2-(дифенилфосфино)ферроценил]-этилди-трет-бутилфосфин, (S)-(+)-1-[(R)-2-(дифенилфосфино)ферроценил]этилди-трет-бутилфосфин, (R)-(-)-1-[(S)-2-(дифенилфосфино)ферроценил]этилдициклогексилфосфин, (R)-(+)-1-[(R)-2-(дифенилфосфино)ферроценил]этилдициклогексилфосфин, (S)-(+)-1-[(R)-2-(дифенилфосфино)ферроценил]этилдициклогексилфосфин, (R)-(-)-1-[(S)-2-(дициклогексилфосфино)ферроценил]этилдифенилфосфин, (R)-(-)-1-[(S)-2-(дифенил)фосфино)ферроценил]этилди(3,5-диметил- фенил)фосфин, (R)-(-)-1-[(S)-2-(ди-трет-бутилфосфино)ферроценил]этилди-o-толилфосфин
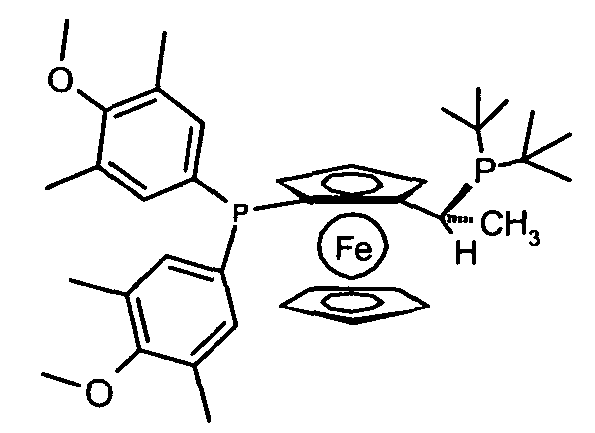
(R)-(-)-1-[(S)-2-(бис(3,5-диметил-4-метоксифенил)фосфино)-ферроценил]этилди-трет-бутилфосфин
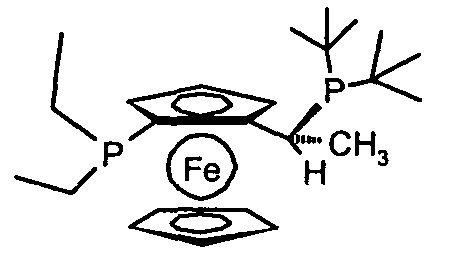
(R)-(-)-1-[(S)-2-(диэтилфосфино)ферроценил]этилди-трет-бутилфосфин
(R)-(-)-1-[(S)-2-(P-метил-P-изопропилфосфино)-ферроценил]этилдициклогексилфосфин
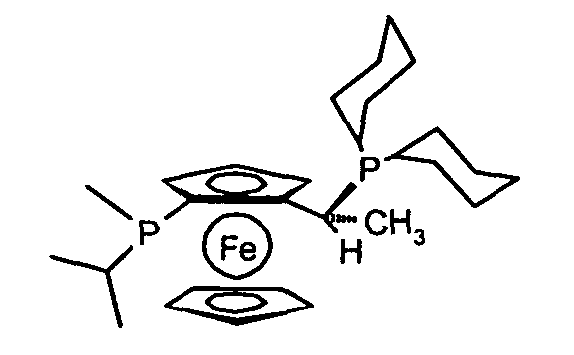
(R)-(-)-1-[(S)-2-(P-метил-P-фенилфосфино)ферроценил]этил-ди-трет-бутилфосфин
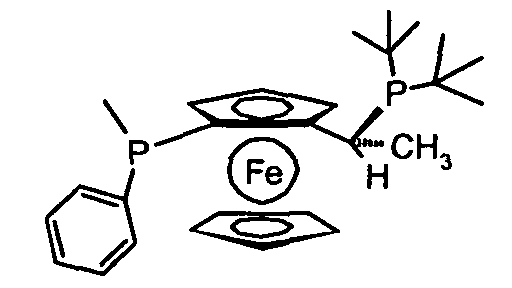
и их рацемические смеси, в частности, рацемические смеси 1-[2-(ди-трет-бутилфосфино)ферроценил]этилди-o-толилфосфина, 1-[2-(дициклогексилфосфино)ферроценил]этилди-трет-бутилфосфина и 1-[2-(дифенилфосфино)ферроценил]этилдициклогексилфосфина.
В способе согласно изобретению может быть использовано одно комплексное соединение палладия или смесь комплексных соединений палладия.
При получении комплексного соединения палладия предпочтение отдается использованию в качестве предшественника палладия ацетата палладия, палладий2(дибензилиден-ацетон)3, палладий(дибензилиден-ацетон)2, раствора дихлорида палладия, дихлорида палладия или смеси палладий2(дибензилиден-ацетон)3 и палладий(трис-трет-бутилфосфин)2. Особое предпочтение отдается использованию ацетата палладия или дихлорида палладия.
Для образования комплексного соединения палладия используют, по меньшей мере, один лиганд.
Предпочтение отдается использованию комплексных соединений палладия, которые включают, по меньшей мере, один лиганд, выбранный из (R)-(-)-1-[(S)-2-(дициклогексилфосфино)ферроценил]-этилди-трет-бутилфосфина и рацемического 1-[2-(дициклогексилфосфино)ферроценил]этилди-трет-бутилфосфина. Предпочтение отдается использованию комплексных соединений палладия, которые включают рацемический 1-[2-(дициклогексилфосфино)ферроценил]-этилди-трет-бутилфосфин.
В способе согласно изобретению комплексные соединения палладия, предшественники палладия и/или лиганды используют в каталитических количествах.
Предпочтительно, чтобы комплексные соединения палладия использовались при соотношении от 1:10 до 1:10000 относительно соединений формулы II, особенно, при соотношении от 1:100 до 1:1000.
Предпочтительно, чтобы предшественники палладия использовались при соотношении от 1:10 до 1:10000 относительно соединений формулы II, особенно, при соотношении от 1:100 до 1:1000.
Предпочтительно, чтобы лиганды использовались при соотношении от 1:10 до 1:10000 относительно соединений формулы II, особенно, при соотношении от 1:100 до 1:1000.
Подходящими основаниями являются, например, алкоголяты, например, трет-бутилат натрия, трет-бутилат калия, метилaт натрия или этилат натрия, или неорганические основания, такие как карбонаты, например K2CO3, Na2CO3 или Cs2CO3, гидроксиды, например NaOH или KOH, фосфаты, например K3PO4, или амиды, например LiNH2, NaNH2 или KNH2; в одном варианте осуществления, предпочтение отдается алкоголятам, и особое предпочтение отдается трет-бутилату натрия; в другом варианте осуществления, предпочтение отдается амидам, и особое предпочтение отдается NaNH2, KNH2 или их смеси.
Когда в качестве основания используют NaOH или KOH, может быть использован катализатор межфазного переноса, такой как, например, цетилтриметиламмоний бромид.
Подходящими количествами основания для этой реакции являются, например, от 1 до 3 эквивалентов, особенно от 1 до 2 эквивалентов.
Реакция согласно изобретению может быть проведена в инертном растворителе.
В одном варианте осуществления изобретения, реакцию согласно изобретению проводят в инертном растворителе. Подходящими растворителями являются, например, соединения формулы V
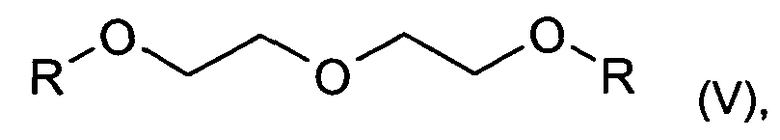
где R является C1-C6алкилом, предпочтительно, метилом; диметоксиэтан; трет-бутилметиловый эфир; тетрагидрофуран; диоксан; трет-бутанол; толуол; ксилол; анизол или триметилбензолы, такие как, например, мезитилен; и также их смеси; предпочтительными растворителями являются диметоксиэтан, тетрагидрофуран или диглим. В таком варианте осуществления предпочтительно, чтобы инертный растворитель являлся безводным.
Реакцию согласно изобретению проводят при комнатной температуре или при повышенной температуре, предпочтительно в температурном интервале от 50°C до 180°C, в частности - в температурном интервале от 50°C до 120°C.
Реакцию согласно изобретению обычно проводят при повышенном давлении. В одном варианте осуществления реакцию согласно изобретению проводят при давлении в интервале 1-100 бар, предпочтительно - в интервале 5-80 бар.
Время проведения реакции согласно изобретению обычно составляет от 1 до 48 часов, предпочтительно - от 4 до 30 часов, в частности - от 4 до 18 часов.
Реакция согласно изобретению может быть проведена в атмосфере инертного газа. Например, в качестве инертного газа используют азот или аргон.
В одном варианте осуществления реакции согласно изобретению реакцию проводят в атмосфере азота.
В реакциях согласно изобретению аммиак используют в эквимолярных количествах или в избытке относительно соединений формулы II, предпочтительно - вплоть до 500-кратного избытка, в частности - вплоть до 200-кратного избытка, более конкретно - в интервале от 80-кратного до 120-кратного избытка. В одном варианте осуществления изобретения аммиак используют в интервале от 10-кратного до 30-кратного избытка.
В способе согласно изобретению аммиак может быть введен в реактор в жидкой форме или в газовой форме.
Соединения формулы II, где X является бромом, R1 является группой A1 и R2 является водородом, хорошо известны и могут быть получены в соответствии со способами, описанными в патентном документе WO 03/074491. Соединения формулы II, где X является хлором, R1 является группой A1, и R2 является водородом, могут быть получены способом, аналогичным способам, описанным в патентном документе WO 03/074491 для соответствующих соединений формулы II, где X является бромом, R1 является группой A1 и R2 является водородом. Например, соединение формулы II, где X является хлором, R1 является группой A1 и R2, R3, R4 и R5 являются водородом (соединение номер B1), может быть получено, как показано на схеме реакции 1 и как объяснено в примерах A1-A3, которые приведены далее:
Схема 2:
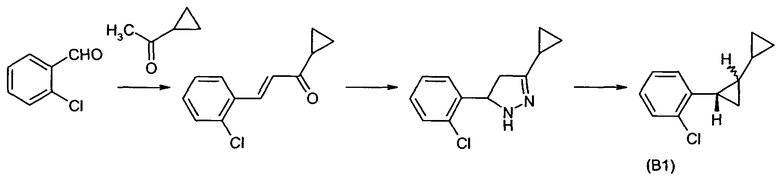
Пример синтеза A1: Получение 3-(2-хлорфенил)-1-циклопропил-пропенона:
67 г 30% раствора гидроксида натрия смешивают с 350 мл воды и 97,5 г (1,1 моль) циклопропилметилкетона и нагревают до 90°C при перемешивании. К полученной смеси добавляют по каплям 143,5 г (1 моль) 2-хлорбензальдегида и проводят перемешивание в течение 5 часов. В процессе перемешивания, после 2 часов и после дополнительных 3 часов, в каждом случае добавляют 2 мл циклопропилметилкетона. После суммарного времени реакции 6 часов проводят охлаждение до 50°C. Реакционную смесь фильтруют и фазы разделяют. Органическую фазу концентрируют. Получают 188,6 г 3-(2-хлорфенил)-1-циклопропилпропенона в виде желтого масла.
1H ЯМР (CDCl3): 0,95-1,04 (м, 2H); 1,16-1,23 (м, 2H); 2,29-2,37 (м, 1H); 6,83 (д, J=15 Гц); 7,27-7,35 (м, 2H); 7,40-7,47 (м, 1H); 8,03 (д, J=15 Гц).
Пример синтеза A2: Получение 5-(2-хлорфенил)-3-циклопропил-4,5-дигидро-1H-пиразола:
250 г этанола добавляют к 188,6 г 3-(2-хлорфенил)-1-циклопропилпропенона (1 моль), полученного в соответствии с примером A1. Добавляют по каплям 53 г (1,05 моль) гидрата гидразина при 20°C при перемешивании. Реакционную смесь перемешивают при 70°C в течение 2 часов. Реакционную смесь затем охлаждают до 50°C. Добавляют смесь 5,5 г дигидрата щавелевой кислоты (0,044 моль) и 20 г этанола, в результате чего осаждается твердое вещество. Реакционную смесь охлаждают до 25°C и фильтруют через вакуум-фильтр из пористого стекла и промывают 50 г этанола. Получают желтый фильтрат, который концентрируют путем испарения в роторном испарителе при 60°C и давлении до 20 миллибар с получением желтого масла. Получают 201,5 г изомерной смеси, содержащей в качестве основного компонента 5-(2-хлорфенил)-3-циклопропил-4,5-дигидро-1H-пиразол в виде желтого масла.
Пример синтеза A3: Синтез 2-(2-хлорфенил)бициклопропила:
К раствору 50 г (0,36 моль) карбоната калия в 600 г этиленгликоля добавляют при 190°C в течение 2 часов 201,5 г 5-(2-хлорфенил)-3-циклопропил-4,5-дигидро-1H-пиразола, полученного как описано в примере A2. Затем проводят перемешивание в течение 2 часов при 190°C. Признаком окончания реакции является прекращение выделения газа. Реакционную смесь затем охлаждают до 100°C, в результате чего происходит расслоение фаз, и верхнюю фазу продукта отделяют. Получают 158 г 2-(2-хлорфенил)бициклопропила в виде неочищенного продукта, который может быть дополнительно очищен, например, ректификацией.
1H ЯМР (CDCl3): 0,0-1,13 (м, 8H); 1,95-2,02 (м, 0,63H, транс-изомер) и 2,14-2,22 (м, 0,37H, цис-изомер); 6,88-6,94 (м); 7,05-7,24 (м); 7,31-7,42 (м).
Соединения формулы II, где X является бромом или хлором, и R1 и R2 вместе образуют группу A2 или A3, могут быть получены согласно способам, описанным в патентном документе WO 07/068417.
Комплексные соединения палладия, предшественники палладия и лиганды, используемые в способе согласно изобретению, хорошо известны и, большей частью, выпускаются в промышленном масштабе.
Далее настоящее изобретение объясняется более подробно с помощью следующих примеров:
Пример P1: Получение 2-бисциклопропиланилина (отношение субстрат/катализатор = 20:1)
Смесь 385 мг 2-(2-хлорфенил)бициклопропила (2 ммоль, отношение транс/цис около 3:2), 288 мг трет-бутоксида (трет-бутилата) натрия (3 ммоль), 22,4 мг ацетата палладия (0,1 ммоль), 61 мг R(-)-ди- трет-бутил-[1-[(S)-2-(дициклогексилфосфанил)-1-ферроценил]этил]-фосфина (0,11 ммоль), 4 г газообразного аммиака (0,235 моль) и 1,5 мл диглима перемешивали при повышенном давлении в резервуаре высокого давления при 160°C в течение 18 часов (атмосфера аргона). Затем смесь разбавляли 20 мл этилацетата и фильтровали. Оставшуюся жидкую фазу концентрировали при пониженном давлении, и неочищенный материал очищали колонной хроматографией на силикагеле (элюент:этилацетат/гептан 1:5). 0,26 г (75% от теории) чистого 2-бисциклопропиланилина получали в виде слегка коричневатой жидкости (отношение транс/цис около 1:1).
Пример P2: Получение 2-бисциклопропиланилина (отношение субстрат/катализатор = 100:1)
Смесь 385 мг 2-(2-хлорфенил)бициклопропила (2 ммоль, отношение транс/цис около 3:2), 288 мг трет-бутоксида натрия (3 ммоль), 4,5 мг ацетата палладия (0,02 ммоль), 12,2 мг R(-)-ди- трет-бутил[1-[(S)-2-(дициклогексилфосфанил)-1-ферроценил]этил]-фосфина (0,022 ммоль), 4 г газообразного аммиака (0,235 моль) и 1,5 мл тетрагидрофурана перемешивали при повышенном давлении в резервуаре высокого давления при 120°C в течение 17 часов (атмосфера аргона). Выход 2-бисциклопропиланилина определяли с помощью газовой хроматографии: 86% (сумма изомеров).
Пример P3: Получение 2-бисциклопропиланилина (отношение субстрат/катализатор = 100:1)
Смесь 385 мг 2-(2-хлорфенил)бициклопропила (2 ммоль, отношение транс/цис около 3:2), 288 мг трет-бутоксида натрия (3 ммоль), 4,5 мг ацетата палладия (0,02 ммоль), 12,2 мг R(-)-ди- трет-бутил-[1-[(S)-2-(дициклогексилфосфанил)-1-ферроценил]этил]-фосфина (0,022 ммоль), 4 г газообразного аммиака (0,235 моль) и 1,5 мл диметоксиэтана перемешивали при повышенном давлении в резервуаре высокого давления при 120°C в течение 16 часов (атмосфера аргона). Выход 2-бисциклопропиланилина определяли с помощью газовой хроматографии: 80% (сумма изомеров).
Пример P3: Получение 2-бисциклопропиланилина (отношение субстрат/катализатор = 500:1)
Смесь 385 мг 2-(2-хлорфенил)бициклопропила (2 ммоль, отношение транс/цис около 3:2), 288 мг трет-бутоксида натрия (3 ммоль), 0,9 мг ацетата палладия (0,004 ммоль), 2,44 мг R(-)-ди- трет-бутил-[1-[(S)-2-(дициклогексилфосфанил)-1-ферроценил]этил]-фосфина (0,0044 ммоль), 4 г газообразного аммиака (0,235 моль) и 1,5 мл диметоксиэтана перемешивали при повышенном давлении в резервуаре высокого давления при 120°C в течение 16 часов (атмосфера аргона). Выход 2-бисциклопропиланилина определяли с помощью газовой хроматографии: 86% (сумма изомеров).
Пример P4: Получение 2-бисциклопропиланилина (отношение субстрат/катализатор = 100:1)
В атмосфере аргона 599 мг (1,1 ммоль) (R)-(-)-1-[(S)-2-(дициклогексилфосфино)ферроценил]этилди-трет-бутилфосфина и 160 мг (0,24 ммоль) ацетата палладия (тример) в 2 мл диметоксиэтана перемешивали в течение 30 минут при комнатной температуре и в течение 1 минуты при 50°C. В атмосфере аргона добавляли каталитическую систему и 2 мл диметоксиэтана к 20,8 г (95%, 0,11 моль) 2-(2-хлорфенил)бициклопропила и 10,5 г (0,11 моль) трет-бутилата натрия в 30 мл диметоксиэтана в автоклаве. Затем добавляли 36 г (2,11 моль) аммиака (жидкого) и суспензию нагревали до 119°C при давлении 61 бар. После 18 часов реакционную массу охлаждали до комнатной температуры, продували дважды азотом и быстро охлаждали с помощью 30 мл воды. Реакционную массу фильтровали через hyflow, фильтр споласкивали ксилолом и водой и водную фазу экстрагировали три раза ксилолом. Органические растворители удаляли под вакуумом. Содержание 2-бисциклопропиланилина определяли с помощью газовой хроматографии: 78% (площадь пика на хроматограмме), оставшийся исходный материал 4,97% (площадь пика на хроматограмме). Кроме того, обнаруживаются 3,57% (площадь пика на хроматограмме) димерного побочного продукта и 3,55% (площадь пика на хроматограмме) дегалогенированного побочного продукта.
Пример P5: Получение 5-амино-9-изопропилбензонорборнена, обогащенного син-изомером (отношение субстрат/катализатор = 100:1)
Смесь 221 мг 5-хлор-9-изопропилбензонорборнена (1 ммоль, >98% син-изомера), 192 мг трет-бутоксида натрия (2 ммоль), 2,25 мг ацетата палладия (0,01 ммоль), 6,1 мг R(-)-ди- трет-бутил-[1-[(S)-2-(дициклогексилфосфанил)-1-ферроценил]этил]фосфина (0,011 ммоль), 4 г газообразного аммиака (0,235 моль) и 5 мл диметоксиэтана перемешивали при повышенном давлении в резервуаре высокого давления при 100°C в течение 21 часа (атмосфера аргона). Выход 5-амино-9-изопропилбензонорборнена определяли с помощью газовой хроматографии: 90% (>98% син-изомера).
При осуществлении вышеприведенных примеров могут быть получены следующие соединения формулы I:
Таблица 1: Соединения формулы I
№
Следующие соединения формулы II являются подходящими для использования в способе согласно изобретению:
Таблица 2: Соединения формулы II
№
В результате создания настоящего изобретения можно аминировать орто-бициклопропилзамещенные галогенбензолы, 5-галогенбензонорборнены и 5-галогенбензонорборнадиены с высокими выходами и с низкими затратами.
Исходные соединения способа настоящего изобретения отличаются тем, что они легко доступны и легко транспортируются и, кроме того, они являются недорогими.
В предпочтительном варианте осуществления способа согласно изобретению палладий и/или комплексное соединение палладия, используемое в способе, подвергают рециркуляции. Этот вариант осуществления является вариантом способа согласно изобретению, который представляет особый интерес с экономической точки зрения.
В предпочтительном варианте осуществления изобретения используют соединения формулы II, где X является хлором. Исходные соединения этого предпочтительного варианта осуществления способа изобретения признаются особенно легко доступными и дешевыми. Однако известно, что при условиях катализируемого палладием кросс-сочетания этот класс стерически затрудненных нереакционноспособных исходных соединений, по меньшей мере, субстратов орто-замещенных хлорбензолов, особенно трудно аминировать вследствие чрезвычайно низкой реакционной способности уходящей группы хлора, по сравнению с субстратами бромбензола. Так как настоящий вариант осуществления изобретения делает данные исходные соединения доступными для катализируемого палладием кросс-сочетания, этот вариант осуществления, соответственно, является вариантом способа согласно изобретению, который представляет особый интерес с экономической точки зрения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ БИАРИЛЗАМЕЩЕННОЙ 4-АМИНОМАСЛЯНОЙ КИСЛОТЫ ИЛИ ЕЕ ПРОИЗВОДНЫХ И ИХ ПРИМЕНЕНИЕ В ИЗГОТОВЛЕНИИ ИНГИБИТОРОВ НЭП | 2007 |
|
RU2469019C2 |
| НОВЫЙ СПОСОБ ПОЛУЧЕНИЯ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ, ИСПОЛЬЗУЕМЫХПРИ ПОЛУЧЕНИИИНГИБИТОРОВ NEP | 2011 |
|
RU2573824C2 |
| СПОСОБ ПОЛУЧЕНИЯ ВОРИКОНАЗОЛА И ЕГО АНАЛОГОВ | 2013 |
|
RU2619928C2 |
| ФУНГИЦИДНЫЕ ФЕНИЛПИРИМИДИНИЛАМИНО ПРОИЗВОДНЫЕ | 2008 |
|
RU2459819C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ПРОПИОНОВОЙ КИСЛОТЫ | 2010 |
|
RU2544989C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ПРОПИОНОВОЙ КИСЛОТЫ | 2011 |
|
RU2575345C2 |
| СПОСОБ | 2014 |
|
RU2671972C1 |
| ФУНГИЦИД НА ОСНОВЕ ГЕТЕРОЦИКЛИЛ-ПИРИМИДИНИЛ-АМИНОПРОИЗВОДНЫХ | 2008 |
|
RU2471793C2 |
| ПИРРОЛО[2,3-d]ПИРИМИДИНОВЫЕ ПРОИЗВОДНЫЕ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И СИНТЕЗ | 2006 |
|
RU2384583C2 |
| НОВЫЕ 2-ПИРИДИНИЛЭТИЛБЕНЗАМИДНЫЕ СОЕДИНЕНИЯ | 2004 |
|
RU2352562C2 |
Изобретение относится к улучшенному способу получения соединений формулы I
где группа R1 является группой A1, в которой группы R2, R3, R4 и R5 являются каждая водородом. Указанные соединения являются промежуточными продуктами для синтеза фунгицидов. Способ заключается в том, что соединение формулы II
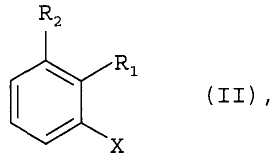
где группы R1 и R2 определены как указано для формулы 1, и X является хлором, взаимодействует с аммиаком в присутствии основания и каталитического количества, по меньшей мере, одного комплексного соединения палладия, где комплексное соединение палладия включает, по меньшей мере, один ферроценилбифосфиновый лиганд, выбранный из R(-)-ди-тpeт-бyтил[1-[(S)-2-(дициклогексилфосфинил)ферроценил]этил]-фосфина и рацемического ди-трет-бутил[1-[2-(дициклогексилфосфинил)ферроценил]этил]-фосфина. Реакцию проводят в инертном растворителе. При этом комплексное соединение палладия предпочтительно используют при соотношении от 1:10000 до 1:10 по отношению к соединению формулы II, а соединение формулы II используют при концентрации от 0,01 М до 5 М. Способ позволяет получать продукты с высоким выходом и хорошего качества по упрощенной технологии. Предлагаемый способ может быть использован при промышленном производстве целевого продукта. 3 з.п. ф-лы, 2 табл.
1. Способ получения соединений формулы I
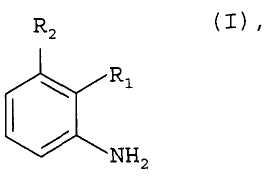
где группа R1 является группой А1
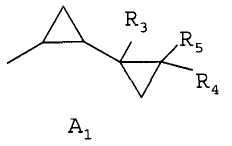
где группы R3, R4 и R5 являются каждая водородом; и группа R2 является водородом; в котором соединение формулы II
где группы R1 и R2 определены как указано для формулы I, и X является хлором, взаимодействует с аммиаком в присутствии основания и каталитического количества, по меньшей мере, одного комплексного соединения палладия, где комплексное соединение палладия включает, по меньшей мере, один ферроценилбифосфиновый лиганд, выбранный из R(-)-ди-трет-бутил[1-[(S)-2-(дициклогексилфосфинил)ферроценил]этил]-фосфина и рацемического ди-трет-бутил[1-[2-(дициклогексилфосфинил)ферроценил]этил]-фосфина, и где данную реакцию проводят в инертном растворителе.
2. Способ по п.1, где комплексное соединение палладия включает рацемический ди-трет-бутил-[1-[2-(дициклогексилфосфинил)ферроценил]этил]фосфин.
3. Способ по п.1, где комплексное соединение палладия используют при соотношении от 1:10000 до 1:10 по отношению к соединению формулы II.
4. Способ по п.1, где соединение формулы II используют при концентрации от 0,01 М до 5 М.
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| SHEN HARTWIG, Palladium-catalyzed coupling of ammonia and lithium amide with aryl halides; Journal of the American Chemical Siciety, 2006, v.128, p.1028-1029 | |||
| RU 2004128580 A, 10.04.2005 | |||
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Компонентный магнитометр | 1979 |
|
SU824099A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |

