Настоящее изобретение относится к способам получения инсулинотропных пептидов, прежде всего глюкагонподобного пептида-1 (GLP-1) и его аналогов, с помощью методов твердофазного и жидкофазного синтеза. Настоящее изобретение относится также к пептидным фрагментам, представляющим собой промежуточные продукты, которые можно применять в указанных способах.
В литературе описаны многочисленные методы пептидного синтеза (см., например, US 6015881; Mergler и др., Tetrahedron Letters 29, 1988, cc.4005-4008; Mergler и др., Tetrahedron Letters 29, 1988, cc.4009-4012; Peptides, Chemistry and Biology, под ред. Kamber и др., изд-во ESCOM, Leiden, 1992, cc.525-526; Riniker и др., Tetrahedron Letters 49, 1993, cc.9307-9320; Lloyd-Williams и др., Tetrahedron Letters 49, 1993, cc.11065-11133, и Andersson и др., Biopolymers 55, 2000, cc.227-250). Различные методы синтеза отличаются физическим состоянием фазы, в которой протекает синтез, а именно тем, представляет ли она собой жидкую фазу или твердую фазу.
При твердофазном пептидном синтезе (SPPS) аминокислоту или пептидную группу связывают с твердой подложкой, представляющей собой смолу. Затем к связанному с подложкой пептиду присоединяют следующие аминокислоты или пептидные группы до получения представляющего интерес пептидного продукта. После этого связанный с подложкой пептид, как правило, отщепляют от подложки и подвергают дополнительному процессированию и/или очистке. В некоторых случаях твердофазный синтез позволяет получать зрелый пептидный продукт; в других случаях пептид, отщепленный от подложки (т.е. пептидный фрагмент, представляющий собой промежуточный продукт («пептидный промежуточный фрагмент»)) применяют для получения более крупного зрелого пептидного продукта.
Пептидные промежуточные фрагменты, полученные в результате твердофазных процессов, можно соединять друг с другом методом твердофазного синтеза или синтеза в растворе (далее в контексте настоящего описания обозначен как «жидкофазный синтез»). Жидкофазный синтез наиболее целесообразно применять в случаях, когда синтез требуемого зрелого пептида на твердой фазе либо невозможен, либо его трудно осуществить на практике. Например, при твердофазном синтезе более длинные пептиды могут случайно принимать неправильную конформацию, оставаясь при этом связанными с твердой подложкой, что затрудняет присоединение дополнительных аминокислот или пептидного продукта к удлиняющейся цепи. По мере удлинения пептидой цепи на смоле, представляющей собой подложку, эффективность стадий процесса, таких как сочетание и удаление защитных групп, может снижаться. Это в свою очередь может приводить помимо возрастающей потери исходных продуктов, таких как способные к активации аминокислоты, кореагенты и растворители, к удлинению времени процессирования для компенсации указанных проблем. Указанные проблемы могут возрастать по мере удлинения пептида.
Таким образом, довольно трудно найти зрелые пептиды, состоящие более чем из 30 аминокислот, синтезированные в виде одного фрагмента с использованием только метода твердофазного синтеза. Вместо этого на твердой фазе можно синтезировать по отдельности индивидуальные фрагменты, а затем осуществлять их сочетание с помощью твердофазного и/или жидкофазного синтеза с получением требуемого пептидного продукта. При использовании этого подхода требуется тщательный выбор фрагментов-кандидатов. Хотя при выборе фрагментов можно пользоваться некоторыми общими принципами, довольно часто требуется эмпирическая оценка фрагментов-кандидатов. Стратегии выбора фрагментов, которые работают в одних условиях, могут не работать в других. Даже когда выявлены приемлемые фрагменты-кандидаты, могут еще потребоваться инновации процесса для выбора стратегии синтеза, которая может работать в приемлемых с позиций стоимости условиях. Таким образом, пептидный синтез, основанный на использовании гибридных схем, часто вызывает вопросы и во многих случаях трудно предсказать, какие проблемы связаны с конкретной схемой синтеза, до фактического осуществления синтеза.
При сочетании в жидкой фазе два пептидных промежуточных фрагмента или пептидный промежуточных фрагмент и реактивную аминокислоту связывают в соответствующем растворителе, как правило, в присутствии дополнительных реагентов, которые повышают эффективность и качество реакции сочетания. Пептидные промежуточные фрагменты в процессе реакции располагаются таким образом, что N-конец одного фрагмента оказывается связанным с C-концом другого фрагмента или наоборот. Кроме того, защитные группы боковых цепей, которые присутствуют при твердофазном синтезе, как правило, сохраняют на фрагментах при сочетании в жидкой фазе для гарантии специфической реактивности концов фрагментов. Эти защитные группы боковых цепей, как правило, не удаляют до поучения зрелого пептида.
Небольшие усовершенствования одной или нескольких стадий в общей схеме синтеза могут приводить к существенным улучшениям получения зрелого пептида. Такие улучшения могут приводить к существенному общему снижению времени получения и потребления реагентов, а также могут существенно повышать чистоту и выход конечного продукта.
Хотя обсуждение важности усовершенствований гибридного синтеза можно проводить для любого типа пептида, получаемого с помощью указанных процедур, это особенно важно в контексте пептидов, которые находят терапевтическое применение и которые производятся в количестве, пригодном для коммерческого применения в медицине. Синтез более крупных биомолекулярных фармацевтических агентов, таких как терапевтические пептиды, может быть очень дорогостоящим. Вследствие значительной стоимости реагентов, продолжительности синтеза, наличия большого количества стадий синтеза, а также других факторов очень незначительные усовершенствования процесса синтеза этих крупных биомолекулярных фармацевтических агентов могут оказать существенное влияние даже на то, будет ли экономически возможно производить такое фармацевтическое средство. Указанные усовершенствования необходимы из-за высокой стоимости производства более крупных биомолекулярных фармацевтических агентов, потребность в которых подтверждается тем фактом, что во многих случаях существует немного, если они вообще существуют, приемлемых терапевтических альтернатив указанным типам более крупных биомолекулярных фармацевтических агентов.
Это, очевидно, имеет место в случае глюкагонподобного пептида-1 (GLP-1) и его аналогов. Указанные пептиды предложены в качестве возможных терапевтических агентов для лечения инсулиннезависимого сахарного диабета типа 2, а также родственных метаболических нарушений, таких как ожирение (Gutniak М.K. и др., Diabetes Care, 17, 1994, cc.1039-1044).
Lopez с соавторами и установили, что нативный GLP-1 состоит из 37 аминокислотных остатков (Lopez L.С. и др., Proc. Natl. Acad. Sci. USA, 80, 1983, cc.5485-5489). Эти данные были подтверждены исследованиями Uttenthal L.O. и др., J. Clin. Endocrinal. Metabol., 61, 1985, cc.472-479. Нативный GLP-1 можно обозначать как GLP-1(1-37). Это обозначение указывает на то, что в пептид входят все аминокислоты, начиная с 1 (N-конец) до 37 (С-конец). Нативный GLP-1 имеет аминокислотную последовательность, представленную в SEQ ID NO.1:
HDEFERHAEGTFTSDVSSYLEGQAAKEFIAWLVKGRG.
Установлено, что нативный GLP-1 (1-37), как правило, не может опосредовать биосинтез инсулина, но биологически важные фрагменты этого пептида обладают инсулинотропными свойствами. Например, нативный состоящий из 31 аминокислоты пептид GLP-1(7-37), последовательность которого представлена в SEQ ID NO.2:
HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRG,
является инсулинотропным и состоит из аминокислот, начиная с положения 7 (N-конец) до положения 37 (C-конец) нативного GLP-1. GLP-1(7-37) имеет концевой остаток глицина. Когда этот остаток глицина отсутствует, то образовавшийся пептид все еще обладает инсулинотропной активностью, и его обозначают как GLP-1(7-36), его последовательность представлена в SEQ ID NO.3:
HAEGTFTSDVSSYLEGQAAKEFIAWLVKGR.
В GLP-1 (7-36) часто C-концевой остаток аргинина находится в амидированной форме, и эту форму можно обозначать как GLP-1(7-36)-NH2.
GLP-1(1-37), как правило, превращается in vivo в свой обладающий инсулинотропной активностью аналог. Например, GLP-1(1-3 7) в естественных условиях превращается в GLP-1(7-37) in vivo. Этот пептид в свою очередь может также подвергаться дополнительному процессингу в результате протеолитического удаления C-концевого остатка глицина с образованием GLP-1(7-36), который часто существует в амидированной форме, т.е. GLP-1(7-36)-NH2. Таким образом, терапевтические применения могут включать введение GLP-1(1-37) или его аналога с расчетом на то, что обладающее инсулинотропной активностью производное образуется in vivo. Однако более часто в проходящих исследование терапевтических применениях используют введение самих обладающих инсулинотропной активностью фрагментов GLP-1.
Согласно US 6887849 инсулинотропная активность GLP-1(7-37), GLP-1(7-36) и GLP-1(7-36)-NH2, вероятно, является специфической в отношении панкреатических бета-клеток, в который эти пептиды, по-видимому, индуцируют биосинтез инсулина. Это делает указанные пептиды и их фармацевтически приемлемые аналоги пригодными для изучения патогенеза начинающегося во взрослом возрасте сахарного диабета, т.е. состояния, которое характеризуется гипергликемией, при которой динамика секреции инсулина является аномальной. Кроме того, указанные глюкагонподобные пептиды можно применять для терапии и лечения этого заболевания и для терапии и лечения гипергликемии. Согласно ЕР 1137667 В1 указанные пептиды или их фармацевтически приемлемые аналоги можно применять для лечения других типов диабета, ожирения, глюкагоном, секреторных нарушений дыхательных путей, метаболического нарушения, артрита, остеопороза, заболевания центральной нервной системы, рестеноза, нейродегенеративного заболевания, почечной недостаточности, застойной сердечной недостаточности, нефротического синдрома, цирроза, отека легких, гипертензии и/или нарушений, при которых желательно снижение потребления пищи.
Нативный GLP-1(1-37) и его нативные обладающие инсулинотропной активностью аналоги, последовательности которых представлены в SEQ ID NO.1-3, являются метаболически нестабильными, имеют время полужизни в плазме, составляющее только 1-2 мин in vivo. Экзогенно введенный GLP-1 также быстро расщепляется. Эта метаболическая нестабильность ограничивает терапевтический потенциал нативного GLP-1 и его нативных фрагментов.
Созданы синтетические аналоги GLP-1-пептидов с улучшенной стабильностью. Например, в ЕР 1137667 В1 описан пептид, последовательность которого представлена в SEQ ID NO.4:
HAibEGTFTSDVSSYLEGQAAKEFIAWLVKAibR.
Этот пептид является аналогом нативного GLP-1(7-36), за исключением того, что ахиральный остаток альфа-аминоизомасляной кислоты (сокращенно обозначен на схеме как Aib) присутствует в положениях 8 и 35 вместо соответствующих встречающихся в естественных условиях аминокислот в указанных положениях. Ахиральная альфа-аминоизомасляная кислота известна также под названием метилаланин. Указанный пептид можно обозначать формулой (Aib8,35)GLP-1(7-36)-NH2.
В ЕР 1137667 указано, что пептид, имеющий последовательность, представленную в SEQ ID NO.4, и его аналоги можно получать в виде индивидуального фрагмента с помощью твердофазного синтеза. Предложенный в ЕР 1137667 подход, предусматривающий синтез индивидуального фрагмента, является проблематичным. С одной стороны, этот подход может приводить к высоким уровням эпимеризации при сочетании конечной аминокислоты, например гистидина, в случае, например, (Aib8,35) GLP-1(7-36). Кроме того, может оказаться сложным удалять примеси в процессе хроматографической очистки, и выход может иметь тенденцию к значительному снижению. Следовательно, существует потребность в усовершенствованных стратегиях синтеза пептидов, последовательность которых представлена в SEQ ID NO.4, с целью получения возможности осуществлять производство этого пептида и его аналогов с приемлемыми с коммерческой точки зрения выходом, чистотой и в приемлемом количестве.
Помимо этих соображений большое влияние на возможность применения конкретной схемы синтеза пептидов могут оказывать показатели, связанные с выходом продукта и чистотой продукта при крупномасштабном производстве пептидов, а также с обработкой реагентов, хранением и распределением. Таким образом, продолжает сохраняться потребность в методах пептидного синтеза, обеспечивающих эффективное получение пептидных продуктов, представляющих коммерческий интерес, в виде крупных партий с повышенным выходом.
Настоящее изобретение относится к получению инсулинотропных пептидов, которые синтезируют с использованием подхода, основанного на применении твердофазного и жидкофазного («гибридного») синтеза. В целом, указанный подход предусматривает синтез трех различных пептидных промежуточных фрагментов с помощью твердофазной химии. Затем используют жидкофазную химию для добавления дополнительного аминокислотного продукта к одному из фрагментов. После этого осуществляют сочетание фрагментов друг с другом с использованием твердой и жидкой фаз. Применение псевдопролина в одном из фрагментов облегчает твердофазный синтез этого фрагмента и облегчает также последующее сочетание этого фрагмента с другими фрагментами в жидкой фазе.
Настоящее изобретение можно применять, прежде всего, для получения инсулинотропных пептидов, таких как GLP-1, GLP-1(7-36) и их встречающиеся в естественных условиях и не встречающиеся в естественных условиях аналогов, в частности GLP-1(7-36) и его встречающихся в естественных условиях и не встречающихся в естественных условиях аналогов.
Одним из объектов настоящего изобретения является способ получения инсулинотропного пептида, заключающийся в том, что:
а) получают пептидный фрагмент, который включает аминокислотную последовательность HX8EX10 (SEQ ID NO. 6), в которой X8 и X10 каждый обозначает остатки ахиральной аминокислоты, или фрагмент, являющийся его аналогом, который включает остатки Х8 и Х10, где Н, Е, Х8 и Х10 каждый необязательно несет защитную группу боковой цепи, и
б) встраивают пептидный фрагмент в инсулинотропный пептид.
X8 обозначает аминокислотный остаток, предпочтительно соответствующий метилаланину (Aib). X10 обозначает аминокислотный остаток, предпочтительно соответствующий глицину.
Следующим объектом изобретения является пептидный фрагмент, который имеет аминокислотную последовательность HX8EX10 (SEQ ID NO.6), в которой X8 и X10 каждый обозначает остаток ахиральной аминокислоты, H, Е, X8 и X10 каждый необязательно несет защитную группу боковой цепи. Предпочтительно X8 обозначает аминокислотный остаток, соответствующий Aib, a X10 обозначает аминокислотный остаток, соответствующий глицину.
Следующим объектом настоящего изобретения является способ получения инсулинотропного пептида, заключающийся в том, что:
а) получают пептидный фрагмент или его аналог, включающие аминокислотную последовательность TFTSDVX17-18YLEG (SEQ. ID No.8), в которой остаток, обозначенный символом X17-18, представляет собой остаток псевдопролина, и
б) встраивают пептидный фрагмент в инсулинотропный пептид.
Еще одним объектом настоящего изобретения является пептид или его аналог, включающие аминокислотную последовательность TFTSDVX17-18YLEG (SEQ. ID No. 8), в которой остаток, обозначенный символом X17-18, представляет собой остаток псевдопролина, где указанные аминокислотные остатки необязательно несут защитную группу боковой цепи.
Следующим объектом настоящего изобретения является способ получения инсулинотропного пептида по п.1 формулы изобретения, заключающийся в том, что:
а) осуществляют сочетание первого пептидного фрагмента, включающего аминокислотную последовательность HX8EX10 (SEQ ID NO.6), в которой X8 и X10 каждый обозначает остатки ахиральной аминокислоты, Н и Е каждый необязательно несет защитную группу боковой цепи, со вторым пептидным фрагментом, включающим аминокислотную последовательность TFTSDVX17-18YLEG (SEQ ID NO.8), в которой остаток, обозначенный символом X17-18, представляет собой дипептидный остаток псевдопролина, где указанные остатки аминокислотной последовательности необязательно несут защитную группу боковой цепи, с получением третьего пептидного фрагмента, включающего аминокислотную последовательность HX8EX10 TFTSDVX17-18YLEG (SEQ ID NO.11), где указанные аминокислотные остатки последовательности необязательно несут защитную группу боковой цепи, и
б) встраивают пептидный фрагмент в инсулинотропный пептид.
Еще одним объектом настоящего изобретения является способ получения инсулинотропного пептида, заключающийся в том, что:
а) получают пептидный фрагмент или его аналог, включающие аминокислотную последовательность QAAKEFIAWLVKX35 (SEQ ID NO.9), в которой X35 обозначает остаток ахиральной аминокислоты, где указанные остатки аминокислотной последовательности необязательно несут защитную группу боковой цепи, и
б) встраивают пептидный фрагмент в инсулинотропный пептид.
Еще одним объектом настоящего изобретения является способ получения инсулинотропного пептида, заключающийся в том, что осуществляют одну или несколько стадий, на которых:
а) получают первый пептидный фрагмент, включающий аминокислотную последовательность HX8EX10 (SEQ ID NO. 6), в которой X8 и X10 каждый обозначает остатки ахиральной аминокислоты, Н и Е каждый необязательно несет защитную группу боковой цепи;
б) получают второй пептидный фрагмент, включающий аминокислотную последовательность TFTSDVX17-18YLEG (SEQ ID NO. 8), в которой остаток, обозначенный символом X17-18, представляет собой дипептидный остаток псевдопролина, где указанные аминокислотные остатки последовательности необязательно несут защитную группу боковой цепи;
в) осуществляют сочетание первого фрагмента и второго фрагмента с получением третьего пептидного фрагмента, включающего аминокислотную последовательность HX8EX10TFTSDVX17-18YLEG (SEQ ID NO.11), где указанные аминокислотные остатки последовательности необязательно несут защитную группу боковой цепи;
г) получают четвертый пептидный фрагмент, включающий аминокислотную последовательность QAAKEFIAWLVKX35 (SEQ ID NO.9), в которой X35 обозначает остаток ахиральной аминокислоты, где указанные аминокислотные остатки последовательности необязательно несут защитную группу боковой цепи;
д) осуществляют сочетание четвертого пептидного фрагмента с аргинином с получением пятого пептидного фрагмента, включающего аминокислотную последовательность QAAKEFIAWLVK X35R (SEQ ID NO.12), где указанные аминокислотные остатки последовательности необязательно несут защитную группу боковой цепи, и
е) осуществляют сочетание пятого фрагмента с третьим фрагментом с получением инсулинотропного пептида, включающего аминокислотную последовательность HX8EX10TFTSDVX17-18YLEGQAAKEFIAWLVK X35R (SEQ ID NO.13), где указанные аминокислотные остатки последовательности необязательно несут защитную группу боковой цепи.
Предпочтительно настоящее изобретение относится к способу получения инсулинотропного пептида, заключающегося в том, что:
а) получают первый пептидный фрагмент, включающий аминокислотную последовательность HX8EX10 (SEQ ID NO.6), в которой X8 и X10 каждый обозначает остатки ахиральной аминокислоты, Н и Е каждый необязательно несет защитную группу боковой цепи;
б) получают второй пептидный фрагмент, включающий аминокислотную последовательность TFTSDVX17-18YLEG (SEQ ID NO.8), в которой остаток, обозначенный символом Х17-18, представляет собой дипептидный остаток псевдопролина, указанные аминокислотные остатки последовательности необязательно несут защитную группу боковой цепи;
в) осуществляют сочетание первого фрагмента и второго фрагмента с получением третьего пептидного фрагмента, включающего аминокислотную последовательность HX8EX10TFTSDVX17-18YLEG (SEQ ID NO.11), где указанные аминокислотные остатки последовательности необязательно несут защитную группу боковой цепи;
г) получают четвертый пептидный фрагмент, включающий аминокислотную последовательность QAAKEFIAWLVKX35 (SEQ ID NO.9), в которой X35 обозначает остаток ахиральной аминокислоты, где указанные аминокислотные остатки последовательности необязательно несут защитную группу боковой цепи;
д) осуществляют сочетание четвертого пептидного фрагмента с аргинином с получением пятого пептидного фрагмента, включающего аминокислотную последовательность QAAKEFIAWLVK X35R (SEQ ID NO.12), где указанные аминокислотные остатки последовательности необязательно несут защитную группу боковой цепи, и
е) осуществляют сочетание пятого фрагмента с третьим фрагментом с получением инсулинотропного пептида, включающего аминокислотную последовательность HX8EX10TFTSDVX17-18YLEGQAAKEFIAWLVK X35R (SEQ ID NO.13), где указанные аминокислотные остатки последовательности необязательно несут защитную группу боковой цепи.
Следующим объектом настоящего изобретения является описанный выше способ, заключающийся в том, что осуществляют следующую дополнительную стадию, на которой:
ж) удаляют защитные группы боковых цепей с получением инсулинотропного пептида, включающего аминокислотную последовательность HX8EX10TFTSDVSSYLEGQAAKEFIAWLVKX35R (SEQ ID NO.5), и его аналогов, в которых каждый из символов Х в положениях 8, 10 и 35 независимо друг от друга обозначает ахиральный, необязательно имеющий стерическую помеху аминокислотный остаток.
Удаление защитных групп боковых цепей предпочтительно осуществляют с помощью раствора для удаления защитных групп, который содержит по меньшей мере один ацидолитический агент и по меньшей мере один акцептор катионов. Ацидолитические реагенты для полного удаления защитных групп предпочтительно выбирают из группы, включающей трифторуксусную кислоту (ТФК), HCl, кислоты Льюиса, такие как BF3Et2O или Me3SiBr, жидкую плавиковую кислоту (HF), бромистый водород (HBr), трифторметансульфоновую кислоту и их комбинации. Приемлемые акцепторы катионов предпочтительно выбирают из динитротреитола (ДТТ), анизола, пара-крезола, этандитиола и диметилсульфида. Раствор для удаления защитных групп может также включать воду.
Следующим объектом настоящего изобретения является способ получения инсулинотропного пептида, заключающийся в том, что:
а) получают первый пептидный фрагмент, включающий аминокислотную последовательность HX8EX10 (SEQ ID NO.6), в которой X8 и X10 каждый обозначает остатки ахиральной аминокислоты, Н и Е каждый необязательно несет защитную группу боковой цепи;
б) получают второй пептидный фрагмент, включающий аминокислотную последовательность TFTSDVX17-18YLEG (SEQ ID NO.8), в которой остаток, обозначенный символом X17-18, представляет собой дипептидный остаток псевдопролина, где указанные аминокислотные остатки последовательности необязательно несут защитную группу боковой цепи;
в) осуществляют сочетание первого фрагмента и второго фрагмента с получением третьего пептидного фрагмента, включающего аминокислотную последовательность HX8EX10TFTSDVX17-18YLEG (SEQ ID NO.11), где указанные аминокислотные остатки последовательности необязательно несут защитную группу боковой цепи;
г) получают четвертый пептидный фрагмент, включающий аминокислотную последовательность QAAKEFIAWLVKX35 (SEQ ID NO.9), в которой X35 обозначает остаток ахиральной аминокислоты, где указанные аминокислотные остатки последовательности необязательно несут защитную группу боковой цепи;
д) осуществляют сочетание четвертого пептидного фрагмента с аргинином с получением пятого пептидного фрагмента, включающего аминокислотную последовательность QAAKEFIAWLVK X35R (SEQ ID NO.12), где указанные аминокислотные остатки последовательности необязательно несут защитную группу боковой цепи, и
е) осуществляют сочетание пятого фрагмента с третьим фрагментом с получением инсулинотропного пептида формулы (SEQ. ID NO.5) HX8EX10TFTSDVSSYLEGQAAKEFIAWLVKX35R и его аналогов, в которых каждый из символов Х в положениях 8, 10 и 35 независимо друг от друга обозначает ахиральный, необязательно имеющий стерическую помеху аминокислотный остаток и в которых один или несколько аминокислотных остатков необязательно несут защитную группу боковой цепи.
На чертеже показана схематическая диаграмма схемы синтеза, предложенной в настоящем изобретении.
Фрагмент 12 представляет собой пептидный фрагмент, включающий аминокислотную последовательность HX8EX10 (SEQ ID NO.6). Фрагмент 14 представляет собой пептидный фрагмент, включающий аминокислотную последовательность T11FTSD15VX17-18YL20EG (SEQ ID NO.8). Фрагмент 16 представляет собой пептидный фрагмент, включающий аминокислотную последовательность Q23AA25KEFIA30WLVKX35 (SEQ ID NO.9). Промежуточный фрагмент 18 представляет собой пептидный фрагмент, включающий аминокислотную последовательность H7X8EX10TFTSD15VX17-18YL20EG (SEQ ID NO.11). Промежуточный фрагмент 20 представляет собой пептидный фрагмент, включающий аминокислотную последовательность Q23AA25KEFIA30WLVKX35R (SEQ ID NO.12). Продукт 11 представляет собой требуемый защищенный пептид H7X8EX10TFTSD15VX17-18YL20EG QAA25KEFIA30WLVKX35R (SEQ ID NO.13), в котором Ser-Ser в положениях 17 и 18 еще находятся в форме защищенного псевдопролина. Ниже диаграмма будет описана более подробно.
Приведенные ниже варианты осуществления настоящего изобретения в виде конкретных форм, подробно описанным ниже, не являются исчерпывающими и не направлены на ограничение изобретения. Скорее варианты осуществления изобретения выбраны и описаны таким образом, чтобы другие специалисты в данной области смогли оценить и понять принципы и варианты воплощения на практике настоящего изобретения.
Настоящее изобретение относится к способам синтеза для получения пептидов, таких как глюкагонподобный пептид-1 (GLP-1) и его встречающиеся в естественных условиях и не встречающиеся в естественных условиях обладающие инсулинотропной активностью аналоги, с использованием методов твердофазного и/или жидкофазного синтеза. Пептидные молекулы, предлагаемые в изобретении, могут быть защищенными, незащищенными или частично защищенными. Защита может представлять собой N-концевую защиту, защиту боковой цепи и/или C-концевую защиту. Хотя изобретение относится прежде всего к синтезу глюкагонподобных пептидов, их аналогов, фрагментов и их аналогов и слитых продуктов и их аналогов, предложенные в изобретении способы можно применять также для синтеза других пептидов, в частности пептидов, которые синтезируют с использованием комбинации твердофазных и жидкофазных подходов. Настоящее изобретение относится также к синтезу пептидных промежуточных фрагментов, ассоциированных с примесями, прежде всего пироглутаматными примесями. Предпочтительные молекулы GLP-1, которые можно применять при воплощении на практике настоящего изобретения, представляют собой встречающиеся в естественных условиях и не встречающиеся в естественных условиях GLP-1(7-36) и их аналоги.
В контексте настоящего описания понятие «включающий аминокислотную последовательность» предпочтительно обозначает «имеющий аминокислотную последовательность».
В контексте настоящего описания понятие «аналог» относится к встречающимся в естественных условиях и не встречающимся в естественных условиях аналогам, производным, слитым соединениям, солям или т.п. пептида. В контексте настоящего описания понятие «пептидный аналог», как правило, относится к пептиду, имеющему модифицированную аминокислотную последовательность, например, в результате одной или нескольких аминокислотных замен, делеций, инверсий и/или добавлений относительно другого пептида или пептидного аналога. Замены могут включать одну или несколько встречающихся в естественных условиях и не встречающихся в естественных условиях аминокислот. Замены предпочтительно могут быть консервативными или высококонсервативными. Понятие «консервативная замена» относится к замене аминокислоты на другую аминокислоту, которая имеет в целом такой же чистый электронный заряд и в целом такой же размер и форму. Например, аминокислоты с алифатическими боковыми цепями или аминокислоты с замещенными алифатическими боковыми цепями имеют приблизительно одинаковый размер, когда общее количество атомов углерода и гетероатомов в их боковых цепях отличается не более чем примерно на четыре. Они имеют приблизительно одинаковую форму, когда количество ветвей в их боковых цепях отличается не более чем примерно на одну или две. Считается, что аминокислоты с фенильными или замещенными фенильными группами в их боковых цепях имеют примерно одинаковый размер и форму. Ниже перечислено пять групп аминокислот. Замена аминокислоты в соединении на другую аминокислоту из этой же группы, как правило, приводит к консервативной замене.
Группа I: глицин, аланин, валин, лейцин, изолейцин, серин, треонин, цистеин, метионин и не встречающиеся в естественных условиях аминокислоты с С1-С4алифатическими или с замещенными гидроксилом С1-С4алифатическими боковыми цепями (прямоцепочечные или одноразветвленные).
Группа II: глутаминовая кислота, аспарагиновая кислота и не встречающиеся в естественных условиях аминокислоты с замещенными карбоновой кислотой С1-С4алифатическими боковыми цепями (неразветвленные или имеющие одну точку разветвления).
Группа III: лизин, орнитин, аргинин и не встречающиеся в естественных условиях аминокислоты с замещенными амином или гуанидиногруппой С1-С4алифатическими боковыми цепями (неразветвленные или имеющие одну точку разветвления).
Группа IV: глутамин, аспаргин и не встречающиеся в естественных условиях аминокислоты с замещенными амидом С1-С4алифатическими боковыми цепями (неразветвленные или имеющие одну точку разветвления).
Группа V: фенилаланин, фенилглицин, тирозин и триптофан.
В контексте настоящего описания понятие «аналог» более предпочтительно относится к солям пептидов или к их производным, амидированным на С-конце.
«Высококонсервативная замена» представляет собой замену аминокислоты другой аминокислотой, которая имеет такую же функциональную группу в боковой цепи и приблизительно такой же размер и форму. Аминокислоты с алифатическими или аминокислоты с замещенными алифатическими боковыми цепями имеют приблизительно одинаковый размер, когда общее количество атомов углерода и гетероатомов в их боковых цепях отличается не более чем примерно на два. Они имеют приблизительно одинаковую форму, когда они имеют одинаковое количество ветвей в их боковых цепях. Примерами высококонсервативных замен являются замены валина на лейцин, треонина на серии, аспарагиновой кислоты на глутаминовую кислоту и фенилглицина на фенилаланин.
Понятие «пептидное производное», как правило, относится к пептиду, пептидному аналогу или другой пептидной копии, которые имеют химическую модификацию в одной или нескольких их боковых группах, альфа-атомах углерода, концевой аминогруппе и/или концевой карбоксильной группе. Например, химическая модификация включает (но, не ограничивается только ими) добавление химических остатков, создание новых связей и/или удаление химических остатков. Модификации боковых групп аминокислот включают (но, не ограничиваются только ими) ацилирование ε-аминогрупп лизина, N-алкилирование аргинина, гистидина или лизина, алкилирование карбоксильных групп глутаминовой или аспарагиновой кислоты и деамидирование глутамина или аспарагина. Модификации концевых аминогрупп включают (но, не ограничиваются только ими) модификации дез-амино-типа, с использованием N-(низш.)алкила, N-ди(низш.)алкила и N-ацила (например, -СО-(низш.)алкила). Модификации концевой карбоксильной группы включают (но, не ограничиваются только ими) модификации с использованием амида, (низш.)алкиламида, диалкиламида и (низш.)алкилового сложного эфира. Предпочтительными производными являются производные, амидированные на концевой карбоксильной группе, например амид, (низш.)алкиламид или диалкиламид пептида. Таким образом, частично или полностью защищенные пептиды относятся к пептидным производным.
При воплощении настоящего изобретения на практике считается, что соединение обладает «инсулинотропной» активностью, если оно может стимулировать или способствовать стимуляции синтеза или экспрессии гормона инсулина. В предпочтительных вариантах воплощения на практике инсулинотропную активность можно продемонстрировать с помощью анализов, описанных в US 6887849 и 6703365.
Предпочтительными вариантами осуществления настоящего изобретения являются пути синтеза синтетических (X8, X10, X35) GLP-1(7-36)-пептидов, имеющих следующую формулу (SEQ. ID NO.5):
HX8EX10TFTSDVSSYLEGQAAKEFIAWLVKX35R,
и их аналогов, в которых каждый из символов Х в положениях 8, 10 и 35 независимо друг от друга обозначает ахиральный, необязательно имеющий стерическую помеху аминокислотный остаток. Любой из остатков X8, X10 и/или X35 необязательно может нести защитную(ые) группу(ы) боковых цепей. Пептиды, имеющие указанную формулу, отличаются от нативного GLP-1(7-36) по меньшей мере тем, что нативные аминокислотные остатки в положениях 8 и 35 заменены на хиральные, необязательно имеющие стерическую помеху аминокислотные остатки X8 и X35. Остаток X10 может являться производным нативной ахиральной аминокислоты глицина или другой ахиральной аминокислоты. Применение ахиральных аминокислот в положениях X8, X10 и Х35 не только способствует стабилизации образовавшегося пептида, но так же, как установлено при создании настоящего изобретения, применение указанных аминокислот в качестве конструктивных элементов облегчает также предлагаемый в настоящем изобретении путь синтеза, который представлен на чертеже и дополнительно описан ниже.
Наиболее предпочтительным вариантом (X8, X10, X35) GLP-11(7-36)-пептида, который можно синтезировать согласно принципам, предлагаемым в настоящем изобретении, является пептид формулы (SEQ ID NO. 4):
HAibEGTFTSDVSSYLEGQAAKEFIAWLVKAibR
и его аналоги, предпочтительно амидированные на С-конце. В этом пептиде применяют ахиральный остаток альфа-аминоизомасляной кислоты (или метилаланин, сокращенно обозначен на схеме как Aib) в качестве как Х8, так и X35, он предпочтительно содержит амид на С-конце, остаток нативного G в положении 10, и он может быть обозначен формулой (Aib8,35)GLP-1(7-36)-NH2. Это условное изображение означает, что аминокислотный остаток, соответствующий аминокислоте «Aib», присутствует в положениях 8 и 35 вместо нативного аланина. Ахиральную альфа-аминоизомасляную кислоту называют также метилаланином. Пептид, имеющий последовательность, представленную в SEQ ID NO. 4, описан в ЕР 1137667 В1. Присутствие остатков Aib в положениях 8 и 35 замедляет метаболическое расщепление в организме, делая этот пептид более стабильным в организме по сравнению с нативным пептидом GLP-1(7-36).
Настоящее изобретение относится к усовершенствованной методологии получения пептидов GLP-1(7-36), таких как (Aib8,35)GLP-1 (7-36)-NH2. Например, на чертеже схематически проиллюстрирована одна схема 10 синтеза пептидов GLP-1(7-36) и их аналогов. Схему 10, представленную на чертеже, по-видимому, наиболее целесообразно применять для крупномасштабного синтеза пептидов GLP-1(7-36). Крупномасштабные процессы, как правило, осуществляют для получения пептида в количестве, которое можно применять для продажи. Например, количество пептида при крупномасштабном процессе может составлять 500 г или 1 кг на партию и более часто от десятков до сотен килограммов на партию или более. В предпочтительных вариантах осуществления изобретения способы, предлагаемые в изобретении, могут обеспечивать усовершенствования, которые приводят к снижению времени процессинга (синтеза), повышению выхода продуктов, улучшению чистоты продукта и/или снижению количества требуемых реагентов и исходных продуктов.
Схема синтеза 10, представленная на чертеже, основана на сочетании твердофазных и жидкофазных методов для получения пептидного продукта 11.
Как видно из чертежа, согласно схеме 10 синтезируют пептидные промежуточные фрагменты 12, 14 и 16 на твердой фазе. Фрагмент 12 представляет собой пептидный фрагмент, включающий аминокислотные остатки, представленные в SEQ ID NO. 6:
HX8EX10,
где X8 и X10 имеют указанные выше значения, или представляет собой его аналог, включающую остатки X8 и X10. Один или несколько аминокислотных остатков могут нести общепринятые защитные группы боковых цепей. В некоторых вариантах осуществления изобретения пептидный фрагмент 12 может быть связан со смолой через С-конец. Этот фрагмент необязательно может нести защитные группы на N-конце и/или на С-конце. Установлено, что Fmoc является наиболее предпочтительной защитной группой для N-конца для твердофазного синтеза пептидного фрагмента.
Фрагмент 12 включает 4 аминокислотных остатка, соответствующих аминокислотам в положениях 7-10 нативного пептида GLP-1(7-36), и поэтому его можно обозначать как (X8, Х10) GLP-1(7-10). В предпочтительных вариантах осуществления изобретения X8 обозначает Aib, a X10 обозначает глицин согласно SEQ ID NO.7:
H7AibEG10
или обозначает его аналог, содержащий остаток Aib в положении 10. Пептидный фрагмент, представленный в SEQ ID NO. 7, может быть обозначен как (Aib8) GLP-1(7-10), в этом обозначении проиллюстрирована замена на Aib нативного аланина в положении 8 нативного GLP-1(7-10).
Твердофазный синтез, как правило, осуществляют в направлении от С-конца к N-концу фрагмента 12. Так, аминокислота X10, которая присутствует в C-концевой области фрагмента, представляет собой первый аминокислотный остаток, который сочетают со смолой-подложкой для твердофазного синтеза. Затем осуществляют твердофазный синтез путем последовательного добавления аминокислотных остатков в порядке, соответствующем требуемой последовательности. Синтез пептидного промежуточного фрагмента завершают после достижения N-концевого остатка (например, N-концевой остаток гистидина (Н) добавляли к образующейся пептидной цепи).
Выбор и применение пептидного фрагмента, имеющего последовательность, представленную в SEQ ID NO.6 и 7, обеспечивает значительное преимущество для синтеза, представленного на схеме 10. Во-первых, Н может представлять собой аминокислотный остаток, который трудно включать в растущую пептидную цепь, по меньшей мере частично из-за эпимеризации. Однако фрагмент 12 является в достаточной степени небольшим, что значительно облегчает эти проблемы. При этом фрагмент 12 является достаточно длинным, что позволяет иметь два хиральных центра. В результате фрагмент можно очищать с помощью простой кристаллизации. Если фрагмент 12 заканчивается Aib, то фрагмент будет иметь только один хиральный центр и в результате его будет сложнее очищать рацемически. Позиционирование ахирального G на С-конце позволяет избегать также связанных с рацемизацией проблем, которые могут возникать, если фрагмент 12 имеет на С-конце хиральный Е. Короче говоря, выбор фрагмента 12 в качестве конструктивного элемента пептида облегчает создание фрагмента, его очистку и сочетание с другим пептидным продуктом. Выбор фрагмента обусловлен также низкой рацемизацией Н. При создании изобретения неожиданно было установлено, что добавление Н к указанному фрагменту приводит к очень незначительному увеличению степени эпимеризации, например примерно на 3 мас.% в некоторых вариантах осуществления изобретения на практике.
Фрагмент 14 представляет собой пептидный фрагмент, включающий аминокислотные остатки, представленные в SEQ ID NO.8:
T11FTSD15VX17-18YL20EG,
где остаток, обозначенный символом X17-18, представляет собой дипептидный остаток псевдопролина, который дополнительно описан ниже, или его аналог, который содержит Х17-18 в положениях 17 и 18. Фрагмент 14 включает аминокислотные остатки, которые в целом соответствуют аминокислотным остаткам в положениях 11-22 нативного пептида GLP-1(7-36), за исключением того, что дипептидный остаток псевдопролина Х17-18 применяют вместо остатков SS (Ser-Ser), которые находятся в соответствующих положениях 17 и 18 нативного GLP-1(7-36).
Один или несколько аминокислотных остатков фрагмента 14 могут нести общепринятые защитные группы. В некоторых вариантах осуществления изобретения пептидный фрагмент 14 может связываться со смолой через C-конец. Этот фрагмент необязательно может нести защитные группы на N-конце и/или C-конце. Установлено, что Fmoc является наиболее предпочтительной для твердофазного синтеза пептидного фрагмента защитной группой для N-конца. Пептидный фрагмент, который имеет последовательность, представленную в SEQ ID NO.8, можно условно обозначать как (X17-18)GLP-1(11-22), в этом обозначении проиллюстрирована замена Х17-18 на остаток псевдопролина остатка Ser-Ser в положениях 17 и 18.
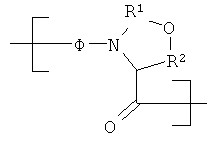
При воплощении настоящего изобретения на практике понятие «псевдопролин» относится к дипептиду, который включает остаток, содержащий функциональный гидроксил аминокислоты, такой как Ser или Thr, в котором функциональный гидроксил боковой цепи защищен с образованием напоминающего пролин, неустойчивого в присутствии ТФК оксазолидинового кольца между альфа-аминогруппой и гидроксилом боковой цепи. Из-за наличия оксазолидинового кольца дипептид функционирует в качестве обратимого миметика пролина.
В целом, типичный остаток псевдопролина, включенный в пептид, может быть представлен формулой
 ,
,
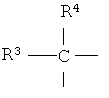
в которой Ф обозначает остаток любой аминокислоты, a R1 и R2 каждый независимо друг от друга обозначает приемлемый двухвалентный связующий радикал. Часто R1 обозначает двухвалентный радикал формулы
 ,
,
в которой R3 и R4 каждый независимо друг от друга обозначает одновалентный радикал, такой как Н или (низш.)алкил, например метил. R3 и R4 могут вместе являться членами кольцевой структуры. Предпочтительно R3 и R4 каждый обозначает метил. В случае защищенного оксазолидининовым кольцом Ser, R2 обозначает двухвалентный радикал CH2, а в случае Thr, R2 обозначает двухвалентный радикал (CH3)CH.
Понятие «(низш.)алкил» относится к разветвленному или прямоцепочечному одновалентному алкильному радикалу, состоящему из 1 -6 атомов углерода, предпочтительно 1-4 атомов углерода. Дополнительными примерами радикалов, подпадающих под указанное понятие, являются метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, н-пентил, 3-метилбутил, н-гексил, 2-этилбутил и т.п. Предпочтительными (низш.)алкильными радикалами являются метил и этил, особенно предпочтительно метил.
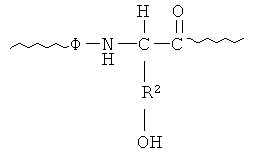
В процессе удаления защитной группы радикал R1 отщепляется с образованием дипептидного остатка следующей формулы:
 ,
,
в которой Ф и R2 имеют указанные выше значения. Во фрагменте 14 остаток псевдопролина предпочтительно соответствует остатку Ser-Ser, в котором Ser, наиболее близко расположенный относительно С-конца, защищают оксазолидиновым кольцом, и он имеет следующую структуру:
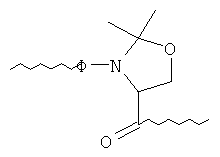 .
.
Несущую гидроксил боковую цепь Ser, ближайшего к N-концу, защищают, например, с помощью защитной группы трет-Bu. Когда защитную оксазолидиновую структуру и трет-Bu отщепляют, то образуется остаток Ser-Ser.
Применение указанного пролинового миметика в качестве конструктивного элемента при синтезе фрагмента 14 обеспечивает существенные преимущества в контексте настоящего изобретения. Во-первых, значительно облегчается твердофазный синтез фрагмента 14. Когда не применяют псевдопролин в процессе твердофазного синтеза фрагмента 14, то могут возникать существенные проблемы с удалением Fmoc-группы с остатков 13-11. Вероятно, что эта трудность может быть следствием бета-складчатой конформации. Применение псевдопролина очень упрощает удаление указанных Fmoc-групп, вероятно, в результате снижения уровня бета-складчатой конформации. Во-вторых, значительно облегчается описанное ниже последующее твердофазное сочетание фрагмента 14 с фрагментом 12 с образованием фрагмента 18. В отсутствие остатка псевдопролина растворимость фрагмента 18 в типичных растворителях, применяемых для жидкофазного сочетания, является очень плохой. Псевдопролин повышает характеристики растворимости фрагмента 18, облегчая жидкофазное сочетание этого фрагмента с фрагментом 20 (см. ниже обсуждение чертежа).
Твердофазный синтез, как правило, осуществляют в направлении от С-конца к N-концу фрагмента 14. Так, аминокислота G, которая присутствует в C-концевой области фрагмента, представляет собой первый аминокислотный остаток, который сочетают со смолой, используемой в качестве подложки для твердофазного синтеза. Затем осуществляют твердофазный синтез путем последовательного добавления аминокислотных остатков в порядке, соответствующем требуемой последовательности. Однако в качестве X17-18 добавляют дипептид псевдопролин к растущей цепи в положении 17 и 18 GLP-1(7-36) вместо последовательного добавления пары нативных остатков Ser в положениях 17 и 18. Синтез пептидного промежуточного фрагмента завершают после N-концевого остатка ((например, N-концевой остаток треонина (T) добавляли к образующейся пептидной цепи).
Фрагмент 16 представляет собой пептидный фрагмент или его аналог, включающий аминокислотные остатки, представленные в SEQ ID NO.9:
Q23AA25KEFIA30WLVKX35,
в которой X35 имеет указанные выше значения или обозначает его аналог, включающий остаток Х35. Один или несколько аминокислотных остатков могут нести общепринятые защитные группы. Фрагмент 16 включает аминокислотные остатки, соответствующие аминокислотам в положениях 23-35 нативного пептида GLP-1(7-36) за исключением того, что X35 в положении 35 заменяет нативную аминокислоту в этом положении. Фрагмент 16 можно условно обозначать как (X35)GLP-1(23-35).
В некоторых вариантах осуществления изобретения пептидный фрагмент 16 можно связывать со смолой через С-конец. Этот фрагмент необязательно может нести защитные группы боковой цепи, N-конца и/или С-конца. Установлено, что Fmoc является наиболее предпочтительной защитной группой для N-конца для твердофазного синтеза пептидного фрагмента.
В предпочтительных вариантах осуществления изобретения Х35 обозначает Aib в последовательности, представленной в SEQ ID NO.10:
Q23AA25KEFIA30WLVKAib35
или ее аналоге, включающем Aib в положении 35. Пептидный фрагмент, последовательность которого представлена в SEQ ID NO. 10, можно обозначать как (Aib35) GLP-1(23-35), в этом обозначении проиллюстрирована замена на Aib нативной аминокислоты в положении 35 нативного GLP-1(7-36).
Следует отметить, что фрагмент 16, последовательность которого представлена в SEQ ID NO. 9 и 10, еще не включает остаток аргинина R (arg) в положении 36 на С-конце. Затем осуществляют сочетание Arg с С-концом фрагмента 16 в жидкой фазе, предпочтительно используя Arg без защиты боковой цепи. Эта стратегия обеспечивает значительные преимущества при использовании схемы 10, представленной на чертеже, поскольку позволяет избегать нежелательных реакцией с участием боковой цепи, которые имеют тенденцию происходить при использовании защищенного Arg. Например, после удаления защитной группы защищенного Arg побочные продукты, возникающие при удалении защитной группы, могут иметь тенденцию к взаимодействию с другими компонентами пептида, например с триптофаном. Это снижает количество требуемого пептида, доступное в неочищенном предназначенном для очистки продукте.
Твердофазный синтез, как правило, осуществляют в направлении от С-конца к N-концу фрагмента 16. Так, аминокислота X35, которая присутствует в C-концевой области фрагмента, представляет собой первый аминокислотный остаток, который сочетают со смолой, используемой в качестве подложки для твердофазного синтеза. Затем осуществляют твердофазный синтез путем последовательного добавления аминокислотных остатков в порядке, соответствующем требуемой последовательности. Синтез пептидного промежуточного фрагмента завершают после достижения N-концевого остатка (например, N-концевой остаток глутамина (Q) добавляли к образующейся пептидной цепи). Любая аминокислота, применяемая для синтеза фрагмента 16, может нести общепринятые защитные группы боковых цепей.
Из-за стерической помехи, ближайшей к остатку X35, осуществляющему связь со смолой-подложкой, сочетание лизина (34) и валина (33) с растущей пептидой цепью может оказаться проблематичным. Даже при избытке аминокислоты трудно довести эти реакции сочетания до их завершения. Выбор растворителя и/или концевого кэпирования может способствовать решению этой проблемы. Было установлено, что природа применяемого для сочетания растворителя может влиять на уровень сочетания, достигаемый при завершении реакции. Например, в одной серии экспериментов реакции сочетания осуществляли в смеси 3:1 NМП/ДХМ, 1:1 NMP/ДХМ, 1:1 ДМФ/ДХМ и 3:1 ДМФ/ДХМ. Соотношения указанных комбинаций растворителей даны в виде объемных соотношений. NМП обозначает N-метилпирролидон, ДХМ обозначает дихлорметан, а ДМФ обозначает диметилформамид. Было установлено, что реакции сочетания в большей степени достигают завершения при использовании 1:1 ДМФ/ДХМ.
Концевое кэпирование после каждого сочетания лизина и валина можно использовать также для предупреждения участия в дополнительных реакциях сочетания непрореагировавшей смолы, представляющей собой материал подложки. Материал с концевым кэпированием легче удалять при необходимости в процессе очистки. Можно использовать общепринятые методы концевого кэпирования.
Продолжая ссылку на схему, приведенную на чертеже, фрагменты 12, 14 и 16, а также Arg, собирают для завершения сборки требуемого пептида 11. Для этого фрагмент 12 добавляют к фрагменту 14 на твердой фазе с получением более крупного промежуточного фрагмента 18, включающего аминокислотные остатки, представленные в SEQ ID NO.11:
H7X8EX10TFTSD15VX17-18EG,
где X8, X10 и X17-18 имеют указанные выше значения. В предпочтительном варианте осуществления изобретения X8 обозначает Aib, X10 обозначает нативный G, a X17-18 обозначает дипептидный остаток псевдопролина, описанный выше. Этот промежуточный пептидный фрагмент можно обозначать как (X8, X10, X17-18) GLP-1(7-22).
На чертеже показано также, что Arg добавляют к С-концу фрагмента 16 в жидкой фазе с получением более крупного промежуточного пептидного фрагмента 20, включающего аминокислотные остатки, представленные в SEQ ID NO.12:
Q23AA25KEFIA30WLVKX35R,
в которой X35 имеет указанные выше значения и предпочтительно обозначает Aib. Предпочтительно Arg, добавленный таким образом к пептидному фрагменту, не имеет защитных групп боковой цепи. Этот промежуточный пептидный фрагмент 20 может быть обозначен как (X35) GLP-1(23-36). Затем осуществляют сочетание пептидных фрагментов 18 и 20 в жидкой фазе с получением требуемого защищенного пептида 11, последовательность которого представлена в SEQ ID NO. 13, где Ser-Ser в положениях 17 и 18 все еще представляют собой псевдопролин в защищенной форме:
H7X8EX10TFTSD15VX17-18YL20EGQAA25KEFIA30WLVKX35R.
Пептид 11 можно обозначить как (X8, X10, X17-18, X35)GLP-1(7-36). В процессе осуществления этой стадии желательно сохранять имеющуюся степень защиты боковых цепей других аминокислот.
При осуществлении реакций, представленных на реакционной схеме на чертеже, твердофазный и жидкофазный синтез можно проводить с помощью стандартных методов, известных в промышленности. В репрезентативных вариантах осуществления изобретения на практике пептиды синтезируют на твердой фазе с использованием химических методов, согласно которым аминокислоты добавляют, начиная с С-конца по направлению к N-концу. Таким образом, аминокислота или пептидная группа, ближайшая к С-концу конкретного фрагмента, представляет собой первую группу, которую следует связывать со смолой. Это осуществляют путем взаимодействия C-концевой функциональной группы аминокислоты или пептидой группы с комплементарной функциональной группой смолы-подложки. N-Концевую область аминокислоты или пептидной группы маскируют с целью предупреждения нежелательных побочных реакций. Желательно также, чтобы боковые группы аминокислоты или пептидной группы были защищены. Затем к связанному с подложкой пептидному продукту присоединяют последующие аминокислоты или пептидные группы до образования представляющего интерес пептида. Большинство из них имеет также защищенные боковые цепи согласно общепринятой практике. При каждом последующем сочетании маскирующую группу на N-конце связанного со смолой пептидного продукта удаляют. Затем ее подвергают взаимодействию с С-концом следующей аминокислоты, N-конец которой замаскирован. Таким образом, продукт твердофазного синтеза представляет собой пептид, связанный со смолой-подложкой.
Можно использовать любой тип подложки, который применяют на практике при осуществлении твердофазного синтеза пептидов. В предпочтительных вариантах осуществления изобретения подложка содержит смолу, которая может состоять из одного или нескольких полимеров, сополимеров или комбинаций полимеров, таких как полиамид, полисульфамид, замещенные полиэтилены, полиэтиленгликоль, фенольные смолы, полисахариды или полистирол. Полимерная подложка может также иметь любую твердость, достаточную для того, чтобы быть нерастворимой и инертной по отношению к растворителям, применяемым в пептидном синтезе. Твердая подложка, как правило, включает связующий радикал, с которым в процессе синтеза сочетают растущий пептид и который можно расщеплять в требуемых условиях для высвобождения пептида из подложки. Приемлемые твердые подложки могут содержать линкеры, которые могут расщепляться под действием света, расщепляться в присутствии ТФК, расщепляться в присутствии HF, расщепляться в присутствии иона фтора, расщепляться в присутствии восстановителя; расщепляться в присутствии Pd(O); расщепляться в присутствии нуклеофила или расщепляться в присутствии радикалов. Предпочтительные связующие радикалы могут расщеплять в условиях, при которых боковые группы отщепляемого пептида все еще в целом остаются защищенными.
В одном из предпочтительных способов синтеза пептидные промежуточные фрагменты синтезируют на чувствительной к кислоте твердой подложке, которая содержит тритильные группы, и более предпочтительно на смоле, которая включает тритильные группы с «подвешенными» атомами хлора, например 2-хлортритилхлоридная (2-СТС) смола (Barlos и др., Tetrahedron Letters 30(30), 1989, cc.3943-3946). Примерами являются также тритилхлоридная смола, 4-метилтритилхлоридная смола, 4-метокситритилхлоридная смола, 4-аминобутан-1-ол-2-хлортритильная смола, 4-аминометилбензоил-2-хлортритильная смола, 3-аминопропан-1-ол-2-хлортритильная смола, бромуксусная кислота-2-хлортритильная смола, циануксусная кислота-2-хлортритильная смола, 4-цианбензойная кислота-2-хлортритильная смола, глицинол-2-хлортритильная смола, пропионовая-2-хлортритильная смола, этиленгликоль-2-хлортритильная смола, N-Fmoc-гидроксиламин-2-хлортритильная смола, гидразин-2-хлортритильная смола. Некоторые предпочтительные твердые подложки включают полистирол, который можно подвергать сополимеризации с дивинилбензолом с получением материала подложки, в который можно «заякоривать» реактивные группы.
Другие смолы, которые можно использовать в твердофазном синтезе, включают смолы «Ванга», которые содержат сополимер стирола и дивинилбензола с 4-гидроксиметилфенилоксиметильными заякоривающими группами (Wang S.S., J. Am. Chem. Soc., 1973), и смолу на основе 4-гидроксиметил-3-метоксифеноксимасляной кислоты (Richter и др., Tetrahedron Letters 35(27), 1994, cc.4705-4706). Смолу Ванга, 2-хлортритилхлоридную смолу и смолу на основе 4-гидроксиметил-3-метоксифеноксимасляной кислоты можно покупать, например, у фирмы Calbiochem-Novabiochem Corp., Сан-Диего, шт.Калифорния.
Для подготовки смолы для твердофазного синтеза смолу можно предварительно отмывать в приемлемом(ых) растворителе(ях). Например, используемую в качестве твердой фазы смолу, такую как смола 2-СТС, добавляют в камеру для синтеза пептидов и предварительно отмывают приемлемым растворителем. Растворитель для предварительной отмывки можно выбирать из растворителей разных типов (или смеси растворителей), которые применяют в реакции сочетания или наоборот. Пригодные для отмывки, а также для последующей реакции сочетания растворители представляют собой дихлорметан (ДХМ), дихлорэтан (ДХЭ), диметилформамид (ДМФ) и т.п. а также смеси указанных реагентов. Другие приемлемые растворители представляют собой ДМСО, пиридин, хлороформ, диоксан, тетрагидрофуран, этилацетат, N-метилпирролидон и их смеси. В некоторых случаях сочетание можно проводить в бинарной системе растворителей, такой как смесь ДМФ и ДХМ в объемном соотношении от 9:1 до 1:9, более предпочтительно от 4:1 до 1:4.
Синтез, предлагаемый в настоящем изобретении, предпочтительно осуществляют в присутствии соответствующих защитных групп, если не указано иное. Природа защитных групп и их применение широко известны. Как правило, приемлемой защитной группой является группа любого типа, которая может способствовать предупреждению участия атома или радикала, к которому она присоединена, например кислорода или азота, в нежелательных реакциях во время процессирования и синтеза. Защитные группы включают защитные группы боковых цепей и амино- или N-концевые защитные группы. Защитные группы могут предупреждать также взаимодействие или связывание карбоновых кислот, тиолов и т.п.
Понятие «защитная группа боковой цепи» относится к химическому радикалу, сцепленному с боковой цепью (т.е. R-группа в общей формуле аминокислот H2N-C(R)(H)-COOH) аминокислоты, который способствует предупреждению взаимодействия части боковой цепи с химическими веществами, применяемыми на стадиях пептидного синтеза, процессинга и т.д. Выбор защитной группы боковой цепи может зависеть от различных факторов, например типа осуществляемого синтеза, процессинга, которому должен подвергаться пептид, и требуемого промежуточного продукта или конечного продукта. Природа защитной группы боковой цепи зависит также на природы самой аминокислоты. Как правило, защитную группу боковой цепи выбирают так, чтобы в процессе удаления защитных групп она сохранялась на α-аминогруппе при твердофазном синтезе. Поэтому защитная группа α-аминогруппы и боковой цепи, как правило, не являются одинаковыми.
В некоторых случаях и в зависимости от типа реагентов, применяемых для твердофазного синтеза и другого процессинга пептидов, для аминокислоты не требуется присутствие защитной группы боковой цепи. Такие аминогруппы, как правило, не содержат реактивный кислород, азот или другой реактивный радикал в боковой цепи.
Примерами защитных групп боковых цепей являются ацетил (Ас), бензоил (Bz), трет-бутил, трифенилметил (тритил), тетрагидропиранил, простой бензиловый эфир (Bzl) и 2,6-дихлорбензил (ДХБ), трет-бутоксикарбонил (Воc), нитрогруппа, пара-толуолсульфонил (Tos), адамантилоксикарбонил, ксантил (Xan), бензил, 2,6-дихлорбензил, сложный метиловый, этиловый и трет-бутиловый эфир, бензилоксикарбонил (Z), 2-хлорбензилоксикарбонил (2-Cl-Z), трет-амилоксикарбонил (Аос) и ароматические или алифатические защитные группы уретанового типа, фотолабильные группы, такие как нитровератрилоксикарбонил (NVOC), и неустойчивые в присутствии фтористых соединений группы, такие как триметилсилилоксикарбонил (ТЕОС).
Предпочтительные защитные группы боковых цепей аминокислот, которые обычно применяют для синтеза пептидов GLP-1 при воплощении на практике настоящего изобретения, приведены ниже в таблице А:
Аминоконцевая защитная группа представляет собой химический радикал, сцепленный с альфа-аминогруппой аминокислоты. Как правило, аминоконцевую защитную группу удаляют с помощью реакции по удалению защитных групп перед добавлением следующей аминокислоты, которая должна быть интродуцирована в растущую пептидную цепь, но ее можно сохранять, когда пептид отщепляют от подложки. Выбор аминоконцевой защитной группы может зависеть от различных факторов, таких, например, как тип осуществляемого синтеза и требуемый промежуточный продукт или конечный продукт.
Примерами аминоконцевых защитных групп являются: (1) защитные группы ацильного типа, такие как формил, акрилил (Acr), бензоил (Bz) и ацетил (Ac); (2) ароматические защитные группы уретанового типа, такие как бензилоксикарбонил (Z) и замещенный Z, такой как пара-хлорбензилоксикарбонил, пара-нитробензилоксикарбонил, пара-бромбензилоксикарбонил, пара-метоксибензилоксикарбонил; (3) алифатические уретановые защитные группы, такие как трет-бутилоксикарбонил (Boc), диизопропилметоксикарбонил, изопропилоксикарбонил, этоксикарбонил, аллилоксикарбонил; (4) циклоалкильные защитные группы уретанового типа, такие как 9-флуоренилметилоксикарбонил (Fmoc), циклопентилоксикарбонил, адамантилоксикарбонил и циклогексилоксикарбонил; (5) защитные группы тиоуретанового типа, такие как фенилтиокарбонил. Предпочтительными защитными группами являются 9-флуоренилметилоксикарбонил (Fmoc), 2-(4-бифенилил)пропил(2)оксикарбонил (Bpoc), 2-фенилпропил(2)оксикарбонил (Рос) и трет-бутилоксикарбонил (Boc).
Химический процесс с использованием Fmoc или Fmoc-подобных агентов является наиболее предпочтительным для твердофазного пептидного синтеза, в виду того, что отщепление образовавшегося пептида в защищенном состоянии относительно просто осуществлять с использованием слабой кислоты в качестве агента для отщепления. Этот тип реакции расщепления является относительно чистым с позиций образующихся побочных продуктов, примесей и т.д., что делает его технически и экономически приемлемым для крупномасштабного получения пептида с использованием отмывок как для разбухания, так и сжатия смолы, с повышенным выходом. В контексте настоящего описания понятие «крупномасштабный» касательно пептидного синтеза, как правило, включает синтез партии пептида, составляющей по меньшей мере 500 г, более предпочтительно по меньшей мере 2 кг. Крупномасштабный синтез, как правило, осуществляют в крупных реакционных сосудах, таких как стальные реакционные сосуды, размер которых позволяет вносить реагенты, такие как смолы, растворители, аминокислоты, химические вещества для сочетания и для реакций по удалению защитных групп, в таких количествах, которые позволяют производить пептиды в диапазоне от килограмма до метрической тонны.
Кроме того, защитную группу Fmoc можно избирательно удалять из пептида вне зависимости от защитных групп боковой цепи, так что защита боковых цепей сохраняется, когда отщепляют Fmoc. Такой тип избирательного действия является важным при сочетании аминокислот, минимизируя реакции с участием боковых цепей. Кроме того, защитные группы боковых цепей можно избирательно отщеплять для удаления их вне зависимости от Fmoc, оставляя Fmoc на месте. Последнее указанное избирательное действие обеспечивает очень большое преимущество при схемах очистки, которые дополнительно описаны ниже.
Твердофазную реакцию сочетания можно осуществлять в присутствии одного или нескольких соединений, которые усиливают или повышают эффективность реакции сочетания. Соединения, которые могут повышать скорость реакции и снижают скорость побочных реакций, представляют собой соли фосфония и урония, которые могут в присутствии третичного основания, например диизопропилэтиламина (ДИЭА) и триэтиламина (ТЭА), превращать защищенные аминокислоты в активированные виды (например, ВОР, РуВОРО, ГБТУ и ТБТУ, которые все образуют сложные ГОБТ-эфиры). Другие реагенты способствуют предупреждению рацемизации, связанной с защитным реагентом. Эти реагенты включают карбодиимиды (например, ДЦК или ВРКДИ) с добавленным вспомогательным нуклеофилом (например, 1-гидроксибензотриазол (ГОБТ), 1-гидроксиазабензотриазол (ГОАТ) или HOSu). Наряду с азидным методом можно применять также метод, основанный на применении смешанных ангидридов, с использованием изобутилхлорформиата с добавленным вспомогательным нуклеофилом или без него благодаря низкой рацемизации, связанной с ним. Эти типы соединений могут повышать также скорость опосредуемых карбодиимидом сочетаний, а также для предупреждения дегидратации остатков Asn и Gln.
После определения того, что сочетание завершено, реакционную смесь для сочетания отмывают с помощью растворителя и цикл сочетания повторяют для каждого последующего аминокислотного остатка пептидного продукта. Для сочетания следующей аминокислоты удаление N-концевой защитной группы (например, Fmoc-группы) из связанного со смолой продукта, как правило, осуществляют обработкой реагентом, который включает 20-50 мас.% пиперидина в качестве растворителя, например N-метилпирролидона (NMП) или диметилформамида (ДМФ). После удаления защитной Fmoc-группы, как правило, осуществляют несколько отмывок для удаления оставшегося пиперидина и побочных продуктов Fmoc (таких как дибензофулвен и его пиперидиновый аддукт).
Последующие аминокислоты можно использовать в стехиометрическом избытке аминокислот относительно фактора загрузки пептидного продукта на подложке из смолы. Как правило, количество аминокислот, применяемых на стадии сочетания, по меньшей мере эквивалентно фактору загрузки первой аминокислоты в смоле (1 эквивалент или более). Предпочтительно количество аминокислот, применяемых на стадии сочетания, составляет по меньшей мере 1,3 эквивалента (избыток 0,3) или более и наиболее предпочтительно по меньшей мере примерно 1,5 эквивалента (избыток 0,5) или более. В некоторых случаях на стадии сочетания используют количество эквивалентов аминокислот в диапазоне от 1 до 3.
После последнего цикла сочетания смолу отмывают растворителем, таким как NМП, а затем отмывают инертным вторым растворителем, таким как ДХМ. Для выделения синтезированного пептидного продукта из смолы осуществляют расщепление в таком режиме, при котором отщепленный пептидный продукт все еще имел достаточное количество защитных групп боковых цепей и концевых защитных групп. Сохранение защитных групп способствует предупреждению нежелательного сочетания или других нежелательных реакций пептидных фрагментов в процесс или после отщепления от смолы. В случае применения Fmoc или аналогичной химии для синтеза пептида защиту от расщепления можно осуществлять любым требуемым образом, например путем использовании относительно слабого кислотного реагента, такого как уксусная кислота или разбавленная ТФК в растворителе, таком как ДХМ. Как правило, применяют от 0,5 до 10 мас.%, предпочтительно от 1 до 3 мас.%, ТФК в ДХМ (см., например, US 6281335).
Стадии отщепления пептидного промежуточного фрагмента от смолы, представляющей собой твердую фазу, можно осуществлять с помощью процесса, приведенного ниже в качестве примера. Однако можно применять любой пригодный процесс, при использовании которого происходит эффективное отщепление пептидного промежуточного фрагмента от смолы. Например, в сосуд, содержащий связанный со смолой пептидный продукт, добавляют примерно 5-20, предпочтительно примерно 10, объемов растворителя, содержащего кислый расщепляющий агент. Затем в реагент погружают смолу, как правило, в форме гранул. Реакция расщепления происходит при перемешивании жидкого содержимого при приемлемой температуре в течение требуемого периода времени. Перемешивание позволяет предупреждать комкование гранул. Требуемое время и температурные условия должны зависеть от таких факторов, как применяемый кислотный реагент, природа пептида, природа смолы и т.п. В целом, можно осуществлять перемешивание при температуре от примерно -15°C до примерно 5°C, предпочтительно от примерно -10°C до примерно 0°C, в течение промежутка времени, составляющего от 5 мин до 2 ч, предпочтительно от примерно 25 мин до примерно 45 мин. Продолжительность расщепления может составлять от примерно 10 мин до примерно 2 ч или даже до одного дня. Расщепление желательно осуществлять при охлаждении при таком диапазоне температур, чтобы скомпенсировать экзотермическую реакцию, которая, как правило, может иметь место. Кроме того, низкая температура реакции расщепления предупреждает удаление на этой стадии чувствительных защитных групп боковых цепей, таких как тритильные группы.
После расщепления реакцию прекращают. Для этой цели можно применять, например, комбинацию расщепляющего реагента с приемлемым основанием, таким как пиридин или т.п., и продолжать взбалтывание и перемешивание в течение дополнительного периода времени, например дополнительно в течение от 5 мин до 2 ч, предпочтительно от примерно 20 мин до примерно 40 мин. Добавление основания и продолжение перемешивания приводит к повышению температуры в сосуде. В конце перемешивания содержимое сосуда может иметь температуру, составляющую от примерно 0°C до примерно 15°C, предпочтительно от примерно 5°C до примерно 10°C.
Для повышения выхода пептида в общий процесс синтеза необязательно можно включать такие факторы, как разбухание и сжатие смолы. Эти методы, описаны, например, в опубликованной заявке на патент США 2005/0164912 А1.
Согласно некоторым объектам изобретения отщепленные пептидные фрагменты можно подготавливать для сочетания в жидкой фазе с другими пептидными фрагментами и/или аминокислотами. Сведения о реакциях пептидного сочетания в жидкой фазе обобщены, например, в: New Trends in Peptide Coupling Reagents; Albericio Fernando; Chinchilla Rafeal; Dodsworth David J.; и Najera Armen; Organic Preparations and Procedures International, 33(3), 2003, cc.203-303.
Сочетание пептидных промежуточных фрагментов с другими фрагментами или аминокислотой(ами) в жидкой фазе можно осуществлять in situ с использованием реагентов для сочетания, таких, например, как гексафторфосфат 2-(1Н-бензотриазол-1-ил)трис(диметиламино)фосфония (БОП), гексафторфосфат орто-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония (ГБТУ), гексафторфосфат орто-(7-азобензотриазол-1-ил)-1,1,3,3-тетраметилурония (ГАТУ), дициклокарбодиимид (ДЦК), водорастворимый карбодиимид (ВРКДИ) или тетрафторборат орто-(бензотриазол-1-ил)-N,N,N′,N′-гетраметилурония (ТБТУ). В других методах сочетания применяют предварительно полученные активные сложные эфиры, такие как гидроксисукцинимидные (HOSu) и пара-нитрофеноловые (HONp) сложные эфиры; предварительно полученные симметричные ангидриды; несимметричные ангидриды, такие как N-карбоксиангидриды (NCA); или галогенангидриды, такие как фтористый ацил, а также хлористый ацил.
При осуществлении реакции сочетания в жидкой фазе можно использовать приемлемый растворитель для сочетания. Очевидно, что применяемый(ые) для сочетания растворитель(и) может(ут) влиять на степень рацемизации образованной пептидной связи; растворимость пептида и/или пептидных фрагментов и скорость реакции сочетания. В некоторых вариантах осуществления изобретения растворитель для сочетания включает один или несколько смешивающихся с водой реагентов. Примерами смешивающихся с водой реагентов являются, например, ДМСО, пиридин, хлороформ, диоксан, тетрагидрофуран, этилацетат, N-метилпирролидон, диметилформамид или их смеси.
В других вариантах осуществления изобретения в реакции сочетания можно применять один или несколько не смешивающихся с водой реагентов. Примером не смешивающегося с водой растворителя является метиленхлорид. В этих вариантах осуществления изобретения не смешивающийся с водой растворитель предпочтительно совместим с реакцией по удалению защитных групп; например, если предпочтительно применяют не смешивающийся с водой растворитель, то он не оказывает вредного воздействия на реакцию по удавлению защитных групп.
После получения пептида 11 этот продукт можно подвергать при необходимости удалению защитных групп, очистке, лиофилизации, дополнительному процессингу (например, взаимодействию с другим пептидом с получением слитого белка); их комбинациям и/или т.п.
Например, согласно изобретению защитные группы боковых цепей, как правило, сохраняются в пептидных промежуточных фрагментах в процессе твердофазного синтеза, а также при осуществлении реакций сочетания в процессе жидкофазного синтеза. Как правило, после завершения стадии сочетания в жидкой фазе можно осуществлять одну или несколько стадий по удалению защитных групп для удаления одной или нескольких защитных групп у пептида.
Для удаления защитных групп боковых цепей в процессе полного удаления защитных групп, как правило, используют раствор для удаления защитных групп, который включает ацидолитический агент для отщепления защитных групп боковых цепей. Общепринятые ацидолитические реагенты для полного удаления защитных групп включают неразбавленную трифторуксусную кислоту (ТФК), HCl, кислоты Льюиса, такие как BF3Et2O или Me3SiBr, жидкую плавиковую кислоту (HF), бромистый водород (HBr), трифторметансульфоновую кислоту и их комбинации. Раствор для удаления защитных групп может включать также один или несколько пригодных акцепторов катионов, например динитротреитол (ДТТ), анизол, пора-крезол, этандитиол или диметилсульфид. Раствор для удаления защитных групп может также включать воду. В контексте настоящего описания количество реагентов, присутствующих в композиции для удаления защитных групп, как правило, выражают в виде соотношения, в котором количество индивидуального компонента выражают в числителе в «частях», таких как «массовые части» или «объемные части», а знаменатель обозначает общее количество частей в композиции. Например, раствор для удаления защитных групп, содержащий ТФК:H2O:ДТТ в соотношении 90:5:5 (мас./мас./мас.), содержит 90/100 мас. частей ТФК, 5/100 мас. частей H2O, 5/100 мас. частей ДТТ.
Согласно некоторым вариантам осуществления изобретения можно осуществлять реакцию по удалению защитных групп с использованием такого количества ацидолитического агента, предпочтительно ТФК, в композиции для удаления защитных групп, на долю которого приходится более 90/100 мас. частей. Предпочтительно использовать также количество 93/100 мас. частей или более или в диапазоне, составляющем от 93/100 мас. частей до 95/100 мас. частей.
Осаждение, как правило, осуществляют с помощью простого эфира, например диэтилового эфира или МТБЭ (метил-трет-бутиловый эфир). После осаждения пептид желательно выделять и сушить, а затем объединять с другими ингредиентами, лиофилизировать, упаковывать, хранить, дополнительно процессировать и/или иным образом обрабатывать. Это можно осуществлять любым приемлемым путем. Согласно одному приемлемому подходу пептид собирают фильтрацией, отмывают, используя достаточное количество отмывок МТБЭ для снижения конечного содержания солей до приемлемого уровня, и затем сушат.
Настоящее изобретение относится также к методам, применяемым для очистки широкого разнообразия пептидов, включая GLP-1-пептиды и их аналоги.
Наиболее предпочтительный процесс очистки включает по меньшей мере две стадии очистки через хроматографические среды, при этом по меньшей мере первую стадию осуществляют при первом значении рН, а по меньшей мере одну вторую стадию осуществляют при втором значении рН. Более предпочтительно первую стадию осуществляют при кислом значении рН, а вторую стадию при щелочном значении рН. В предпочтительных вариантах осуществления изобретения по меньшей мере первую стадию в кислых условиях осуществляют перед проведением стадии в основных условиях. В качестве иллюстрации применяемого на практике варианта этот подход очистки может предусматривать очистку полностью защищенного пептида 11, полученного согласно схеме 10, приведенной на чертеже. Первоначально осуществляют полное удаление защитных групп пептида. Отщепляют как N-концевые защитные группы, так и защитные группы боковых цепей. Первую хроматографическую стадию осуществляют в градиенте вода/АЦН (ацетонитрил), используя ТФК в количестве, достаточном для достижения значения рН примерно 1-5, предпочтительно примерно 2. Затем осуществляют вторую стадию в градиенте вода/АЦН, используя небольшое количество аммиака и/или ацетата аммония или т.п. для достижения значения рН примерно 8-9, предпочтительно 8,5-8,9.
Значения рН, как кислые, так и щелочные, повышают однородность, проявляющуюся в том, что в каждом случае присутствуют однородные ионные виды. Таким образом, кислое значение рН должно быть достаточно низким для того, чтобы протонировать практически все аминокислотные остатки в пептидном продукте. Щелочное значение рН должно быть достаточно высоким для того, чтобы депротонировать практически все аминокислотные остатки в пептидном продукте. Хроматографию в кислых и щелочных условиях можно осуществлять в любом порядке. Целесообразно, чтобы хроматография в щелочных условиях была последней, когда требуемым продуктом является ацетат пептида, поскольку ацетат может быть продуктом хроматографии.
Принципы настоящего изобретения дополнительно проиллюстрированы с помощью приведенных ниже примеров. Все указанные ниже проценты и соотношения являются объемными, если специально не указано иное.
Пример 1. Твердофазный синтез фрагмента 12 с Fmoc-защитой на N-конце и защитой боковых групп His и Glu
А. Получение загруженной Fmoc-Gly смолы 2СТС
Сначала получали загруженную Fmoc-Gly смолу 2СТС. Ниже в таблице показано количество применяемых реагентов:
Смолу 2-СТС вносили в 500-миллилитровый пептидный реактор и давали набухать в 400 мл ДХМ в течение 30 мин при 25°C. Слой осушали и добавляли раствор Fmoc-Gly-OH и ДИЭА в 8 объемах ДМФ:ДХМ (87,5:12,5). Смесь перемешивали в атмосфере азота в течение 2 ч при температуре 25°C.
Слой осушали и однократно промывали 350 мл ДМФ и однократно 175 мл ДМФ. Затем осуществляли концевое кэпирование оставшихся активных сайтов на смоле 2-СТС, используя 350 мл раствора МеОН:ДИЭА (9:1) в течение 1 ч. Слой осушали и промывали дважды 250 мл ДМФ и затем четыре раза 350 мл ДХМ. Смоле повторно давали набухать путем промывки 3×350 мл ИПС. Смолу сушили до постоянной массы с получением 38,20 г загруженной смолы. Анализ показал, что фактор загрузки составлял 0,18 ммоля/г.
Б. Твердофазный синтез
Твердофазный синтез осуществляли, начиная с 20,0 г загруженной Fmoc-Gly-2-СТС-смолы с фактором загрузки 0,18 ммоля/г, которую получали согласно методу, описанному в разделе А этого примера 1. Смоле давали набухать в ДХМ (200 мл) в течение 30 мин при 25°C. Растворитель ДХМ сливали и смолу промывали трижды NМП (для каждой промывки использовали 5 объемов).
Затем смолу обрабатывали дважды 20 об.%-ным раствором пиперидина в NМП (для каждой обработки использовали 5 объемов) для удаления защитных групп Fmoc. После второй обработки 20% пиперидином/NМП смолу промывали пять раз NМП (для каждой промывки использовали 5 объемов) до достижения отрицательной пробы на хлоранил.
Для приготовления раствора для сочетания взвешивали аминокислоту (2,85 экв.) и 6-хлор-1-гидроксибензотриазол (6-Cl-ГОБТ, 2,85 экв.), растворяли в 2,55-кратном (2,55×) объеме NМП, затем объединяли с ДИЭА (3,25 экв.) при температуре от 5 до 10°C. ТБТУ (2,85 экв.) растворяли в 1,3× объеме NMH при температуре от 5 до 10°C. Затем два раствора объединяли. Образовавшийся раствор добавляли в реакционный сосуд. Колбу отмывали 1,3× объемом ДХМ, добавляли в реактор, содержимое которого затем перемешивали в течение 2-3 ч при температуре от 25 до 27°C. Отбирали образец для осуществления теста Кайзера для оценки завершения реакции. Если реакция сочетания через 3 ч не заканчивалась (положительный тест Кайзера), то реакционный сосуд осушали и осуществляли повторное сочетание с использованием свежего раствора активированной аминокислоты. После завершения реакции сочетания раствор для сочетания сливали и смолу промывали четыре раза NМП (для каждой промывки использовали 5 объемов). Затем цикл, включающий удаление защитной группы Fmoc и реакцию сочетания, повторяли для оставшихся аминокислот фрагмента (т.е. в порядке Glu(OtBu)→Aib→His(trt)).
Из-за сложности проведения реакции сочетания между активированной Fmoc-His(trt)-OH и H-Aib-Glu(OtBu)-Gly-2-CTC и нестабильности активированной Fmoc-His(trt)-OH реакцию сочетания завершали путем слива реакционного буфера через 1 ч и немедленного осуществления повторной реакции сочетания с использованием второго раствора со свежеактивированной Fmoc-His(trt)-OH.
Все реагенты, применяемые в разделе Б этого примера, перечислены ниже в таблице:
Связанный со смолой пептидный фрагмент 4 раза промывали NМП (5 об.), 5 раз ДХМ (6 об.), 3 раза ИПС (5 об.). Повторно набухшую смолу затем сушили при 35°C в вакууме, получая 22,58 г смолы и связанного со смолой пептида.
В. Отщепление от смолы фрагмента с Fmoc-группой и с защищенными боковыми цепями
Смоле, созданной согласно методу, описанному выше в разделе Б, давали набухать в ДХМ (12,5 объемов относительно массы применяемой смолы; 12,5 мл ДХМ на 1 г смолы или 12,5 л на 1 кг) в течение 30 мин при 25°C, а затем 2 раза промывали ДХМ (для каждой промывки использовали 6,25 объема) для удаления всего остаточного NМП. Смолу охлаждали при последней промывке ДХМ до -5°C. ДХМ сливали и добавляли холодный раствор 1% ТФК/ДХМ (10 об. при температуре от -5 до -10°C) и перемешивали в течение 30 мин при 0°C. В реактор добавляли пиридин (1,3 экв. относительно ТФК) для нейтрализации ТФК. Раствор для отщепления фильтровали и собирали в колбу. После нагревания сосуда до 25°C смолу промывали 7 раз ДХМ (7,5 об.). Смывы объединяли с раствором для расщепления. Содержащий ДХМ раствор для расщепления объединяли с водой (7,5 об.). Полученную смесь перегоняли при пониженном давлении для удаления ДХМ (350 торр при 28°C). Пептидный фрагмент осаждался из воды при удалении ДХМ. Фрагмент промывали водой и сушили при 30-35°C в вакууме. Всего получили 4,73 г Fmoc-(Aib8) GLP-1(7-10)-ОН.
Пример 2
А. Получение загруженной Fmoc-Gly смолы 2СТС
Получали загруженную Fmoc-Gly смолу 2СТС. Ниже в таблице показано количество применяемых реагентов:
Смолу 2-СТС вносили в 500-миллилитровый пептидный реактор и давали набухать в 400 мл ДХМ в течение 30 мин. Смолу осушали и добавляли раствор Fmoc-Gly-OH и ДИЭА в 8 объемах ДМФ:ДХМ (87,5:12,5). Смесь перемешивали в атмосфере азота в течение 2 ч при температуре 25°C.
Слой смолы осушали и однократно промывали 400 мл ДМФ и однократно 200 мл ДМФ. Затем осуществляли концевое кэпирование оставшихся активных сайтов на смоле 2-СТС, используя 390 мл раствора МеОН:ДИЭА (9:1) в течение 1 ч. Слой вновь осушали, промывали дважды 350 мл ДМФ и затем четыре раза 350 мл ДХМ. Смоле повторно давали набухать путем промывки 3×350 мл ИПС. Смолу сушили при 35°C в вакууме до постоянной массы с получением 48,51 г загруженной смолы. Анализ показал, что фактор загрузки составлял 0,54 ммоля/г.
Б. Твердофазный синтез
Твердофазный синтез осуществляли, начиная с 27,59 г загруженной Fmoc-Gly-2-СТС-смолы с фактором загрузки 0,54 ммоля/г. Смоле давали набухать в ДХМ (300 мл) в течение 30 мин при 25°C. Растворитель ДХМ сливали и смолу промывали трижды NМП (для каждой промывки использовали 5 объемов).
Затем смолу обрабатывали дважды 20 об.%-ным раствором пиперидина в NМП (для каждой обработки использовали по 5 объемов) для удаления защитных групп Fmoc. После второй обработки 20% пиперидином/NМП смолу промывали шесть раз NМП (для каждой промывки использовали по 5 объемов) до достижения отрицательной пробы на хлоранил.
Для приготовления раствора для сочетания взвешивали аминокислоту (1,7 экв.) и 6-хлор-1-гидроксибензотриазол (6-Cl-ГОБТ, 1,7 экв.), растворяли в 4,6× объеме NМП, затем объединяли с ДИЭА (1,9 экв.) при температуре от 5 до 10°C. ТБТУ (1,7 экв.) растворяли в 2,28× объеме NМП при температуре от 5 до 10°C. Затем два раствора объединяли. Образовавшийся раствор добавляли в реакционный сосуд. Колбу отмывали 2,28× объемом ДХМ, добавляли в реактор, содержимое которого затем перемешивали в течение 2-3 ч при температуре от 25 до 27°C. Отбирали образец для теста Кайзера с целью оценки завершения реакции. После завершения реакции сочетания раствор для сочетания сливали и смолу промывали четыре раза NМП (для каждой промывки использовали по 5 объемов). Затем цикл, включающий удаление защитной группы Fmoc и реакцию сочетания, повторяли для оставшихся аминокислот фрагмента (т.е. в порядке Glu(OtBu)→Aib→His(trt)).
Все реагенты, применяемые в этом примере, перечислены ниже в таблице:
В. Отщепление фрагмента от смолы
Созданную смолу промывали 6 раз NМП (5 об.), а затем 8 раз ДХМ (6 об.) для удаления остатка NМП. Смолу охлаждали путем последней промывки ДХМ до -5°C. ДХМ сливали и после этого добавляли холодный (от -5 до -10°C) раствор 1% ТФК/ДХМ (10 об.) и образовавшуюся смесь перемешивали в течение 30 мин при 0°C. В реактор добавляли пиридин (1,3 экв. относительно ТФК) для нейтрализации ТФК. Раствор для отщепления собирали в колбу. После нагревания сосуда до 25°C смолу промывали 11 раз ДХМ (7,5 об.) и сливали в раствор для расщепления. Содержащий ДХМ раствор объединяли с водой (10 об.). Полученную смесь перегоняли при пониженном давлении для удаления ДХМ (350 торр при 28°C). Пептидный фрагмент осаждался из воды при удалении ДХМ. Фрагмент промывали водой и сушили при 30-35°C в вакууме. Всего получили 11,12 г Fmoc-(Aib8) GLP-1 (7-10)-OH (выход 78,8%).
Пример 3
А. Получение загруженной Fmoc-Gly смолы 2СТС
Получали загруженную Fmoc-Gly смолу 2СТС. Ниже в таблице показано количество применяемых реагентов:
Смолу 2-СТС вносили в 500-миллилитровый пептидный реактор и давали набухать в 400 мл ДХМ в течение 30 мин. Слой осушали и добавляли раствор Fmoc-Gly-OH и ДИЭА в 8 объемах ДМФ:ДХМ (87,5:12,5). Смесь перемешивали в атмосфере азота в течение 2 ч при температуре 25°C.
Слой осушали и однократно промывали 400 мл ДМФ и однократно 200 мл ДМФ. Затем осуществляли концевое кэпирование всех оставшихся активных сайтов на смоле 2-СТС, используя 390 мл раствора МеОН:ДИЭА (9:1) в течение 1 ч. Слой осушали, промывали дважды 350 мл ДМФ и затем четыре раза 350 мл ДХМ. Смоле повторно давали набухать путем промывки 4×250 мл ИПС. Смолу сушили при 35°C в вакууме до постоянной массы с получением 52,02 г загруженной смолы. Анализ показал, что фактор загрузки составлял 0,72 ммоля/г.
Б. Твердофазный синтез
Твердофазный синтез осуществляли, начиная с 24,43 г загруженной Fmoc-Gly-2-СТС-смолы с фактором загрузки 0,72 ммоля/г. Смоле давали набухать в ДХМ (250 мл) в течение 30 мин при 25°C.Растворитель ДХМ сливали и смолу промывали трижды NМП (для каждой промывки использовали по 5 объемов).
Затем смолу обрабатывали дважды 20 об.%-ным раствором пиперидина в NМП (для каждой обработки использовали 5 объемов) для удаления защитных групп Fmoc. После второй обработки 20% пиперидином/NМП смолу промывали шесть раз NМП (для каждой промывки использовали по 5 объемов) до достижения отрицательной пробы на хлоранил.
Для приготовления раствора для сочетания взвешивали аминокислоту и 6-хлор-1-гидроксибензотриазол (6-Cl-ГОБТ), растворяли в NМП, затем объединяли с ДИЭА при температуре 10-5°C. ТБТУ растворяли в NМП при температуре 10-5°C. Затем два раствора объединяли. Образовавшийся раствор добавляли в реакционный сосуд. Колбу отмывали ДХМ (данные о количестве см. ниже в таблице), добавляли в реактор, содержимое которого затем перемешивали в течение 2-3 ч при температуре от 25 до 27°C. Отбирали образец для теста Кайзера с целью оценки завершения реакции. Если реакция сочетания через 3 ч не заканчивалась (положительный тест Кайзера), то реакционный сосуд осушали и осуществляли повторное сочетание с использованием свежего раствора активированной аминокислоты. После завершения реакции сочетания раствор для сочетания сливали и смолу промывали четыре раза NМП (для каждой промывки использовали 5 объемов). Затем цикл, включающий удаление защитной группы Fmoc и реакцию сочетания, повторяли для оставшихся аминокислот фрагмента (т.е. в порядке Glu(OtBu)→Aib→His(trt)).
Все реагенты, применяемые в этом примере, перечислены ниже в таблице:
В. Отщепление фрагмента Fmoc-AA(7-10)-OH от смолы
Созданную смолу промывали 6 раз NМП (5 об.), а затем 7 раз ДХМ (6 об.) для удаления остатка NМП. Смолу охлаждали путем последней промывки ДХМ до -5°C. ДХМ сливали и слой смолы промывали холодным (от -5 до -10°C) раствором 1% ТФК/ДХМ (11,26 об.) в течение 5 мин при 0°C. Раствор для отщепления собирали в колбу, в которую был внесен пиридин (1,3 экв. относительно ТФК) для нейтрализации ТФК. Затем в реактор добавляли вторую часть холодного раствора 1% ТФК/ДХМ (6,14 об.) и перемешивали в течение 2 мин. Второй раствор для расщепления вновь сливали в колбу для сбора. После нагревания сосуда до 25°C смолу промывали 9 раз ДХМ (8,2 об.) и сливали в раствор для расщепления. Содержащий ДХМ раствор объединяли с водой (8,2 об.). Полученную смесь перегоняли при пониженном давлении для удаления ДХМ (350 торр при 28°C). Пептидный фрагмент осаждался из воды при удалении ДХМ. Фрагмент промывали водой и сушили при 30-35°C в вакууме. Всего получили 14,02 г Fmoc-(Aib8) GLP-1 (7-10)-OH (выход 86,6%), который имел последовательность, представленную в SEQ ID NO. 7. Анализ показал, что чистота продукта составляла 94,3% AN.
Пример 4. Твердофазный синтез Fmoc-(Aib35) GLP-1 (23-35)-OH с защищенной боковой цепью
А. Получение загруженной Fmoc-Aib смолы 2СТС
Получали загруженную Fmoc-Aib смолу 2СТС. Ниже в таблице показано количество применяемых реагентов:
Смолу 2-СТС вносили в 500-миллилитровый пептидный реактор и давали набухать в 400 мл ДХМ в течение 30 мин. Слой осушали и добавляли раствор Fmoc-Aib-OH и ДИЭА в 8 объемах ДМФ:ДХМ (87,5:12,5). Смесь перемешивали в атмосфере азота в течение 2 ч при температуре 25°C.
Слой осушали и однократно промывали 400 мл ДМФ первый раз и 200 мл второй раз. Затем осуществляли концевое кэпирование всех оставшихся активных сайтов на смоле 2-СТС, используя 400 мл раствора МеОН:ДИЭА (9:1) в течение 1 ч. Слой осушали, промывали однократно 450 мл ДМФ/МеОН/ДИЭА (4:0,9:0,1), однократно 200 мл ДМФ и четыре раза 350 мл ДХМ. Смоле повторно давали набухать путем промывки 3×350 мл ИПС. Смолу сушили до постоянной массы с получением 45,15 г загруженной смолы. Анализ показал, что фактор загрузки составлял 0,24 ммоля/г.
Б. Твердофазный синтез
10,01 г Fmoc-Aib-2-СТС-смолы с фактором загрузки 0,24 ммоля/г вносили в реакционный сосуд и давали набухать в ДХМ (120 мл) в течение 30 мин при 25°C. Растворитель ДХМ сливали и смолу промывали трижды NМП (для каждой промывки использовали 6 объемов).
Затем смолу обрабатывали дважды 5 об.%-ным раствором пиперидина в NMn (для каждой обработки использовали по 6 объемов) для удаления защитных групп Fmoc. После второй обработки 5% пиперидином/NМП смолу промывали четыре раза NМП (для каждой промывки использовали по 6 объемов).
Для приготовления раствора для сочетания аминокислоту (1,875 экв.) и моногидрат 1-гидроксибензотриазола (гидрат ГОБТ, 2,07 экв.) растворяли в 3,5× объеме NМП при температуре от 5 до 10°C и затем объединяли с 16,1 мл раствора ГБТУ (2,0 экв.) в NМП (1,5× об.). Затем 2,2 мл ДИЭА (2,63 экв.) добавляли в сосуд для активации при температуре 10-5°C. Образовавшийся раствор переносили в реакционный сосуд. Содержимое сосуда для активации смывали 1,5×объемом ДХМ в реактор, затем перемешивали в течение 2 ч при 25°C. Реакционный сосуд осушали. Реакцию сочетания повторяли еще один раз, используя свежий раствор активированной аминокислоты (1,875 экв.) После завершения второй реакции сочетания раствор для сочетания сливали и смолу промывали 4 раза NМП (по 6 об. для каждой промывки). Затем цикл, включающий удаление защитной группы Fmoc и реакцию сочетания, повторяли для оставшихся аминокислот фрагмента (т.е. в порядке Lys(Boc)→Val→Leu→Trp(Boc)→Ala→Ile→Phe→Glu(OtBu)→Lys(Boc)→Ala→Ala→Gln(trt)).
Все реагенты, применяемые в этом примере, перечислены ниже в таблице:
Созданную смолу выделяли путем промывки 4 раза NМП (6 об.), 4 раза ДХМ (6 об.) и 3 раза изопропанолом (ИПС, 6 об.). Созданную смолу сушили при 35°C в вакууме. Получили 14,3 г созданной смолы.
В. Отщепление промежуточного фрагмента от созданной смолы
6,6 г созданной согласно описанному выше методу смолы давали набухать в 10× объеме ДХМ в течение 30 мин и охлаждали до -10°C. ДХМ сливали и добавляли холодный раствор 1% ТФК/ДХМ (12 об., при температуре от -5 до -10°C) и перемешивали в течение 30 мин при 0°C. Раствор для отщепления собирали в колбу, содержащую пиридин (2-3 экв. относительно ТФК). После нагревания до 25°C смолу перемешивали с 1% ТФК/ДХМ (10× об.) в течение 5 мин и добавляли пиридин (2-3 экв.). Еще через 5 мин раствор собирали. Смолу промывали 4 раза ДХМ (10 об.). Все ДХМ-смывы объединяли с водой (вода/ДХМ=1/4). Полученную смесь перегоняли при пониженном давлении для удаления ДХМ (350 торр при 28°C). Фрагмент осаждался из воды при удалении ДХМ. Фрагмент промывали водой и сушили при 30-35°C в вакууме. Процедуру расщепления повторяли еще один раз. Всего получили 2,36 г Fmoc-(Aib35) GLP-1 (23-35)-ОН (выход 92%).
Пример 5
А. Получение загруженной Fmoc-Aib смолы 2СТС
Получали загруженную Fmoc-Aib смолу 2СТС. Ниже в таблице показано количество применяемых реагентов:
Смолу 2-СТС вносили в 500-миллилитровый пептидный реактор и давали набухать в 400 мл ДХМ в течение 30 мин. Слой осушали и добавляли раствор Fmoc-Aib-OH и ДИЭА в 8 объемах ДМФ:ДХМ (87,5:12,5). Смесь перемешивали в атмосфере азота в течение 2 ч при температуре 25°C.
Слой осушали и однократно промывали 400 мл ДМФ. Затем осуществляли концевое кэпирование всех оставшихся активных сайтов на смоле 2-СТС, используя 400 мл раствора МеОН:ДИЭА (9:1) в течение 1 ч. Слой осушали, промывали однократно 450 мл ДМФ, однократно 200 мл ДМФ и четыре раза 350 мл ДХМ. Смоле повторно давали набухать путем промывки 3×350 мл ИПС. Смолу сушили до постоянной массы с получением 45,32 г загруженной смолы. Анализ показал, что фактор загрузки составлял 0,30 ммоля/г.
Б. Твердофазный синтез
Твердофазный синтез осуществляли, начиная с 15,0 г загруженной Fmoc-Aib-2-СТС-смолы с фактором загрузки 0,30 ммоля/г. Смоле давали набухать в ДХМ (150 мл) в течение 30 мин при 25°C. Растворитель ДХМ сливали и смолу промывали дважды ДХМ (по 6 об. для каждой промывки) и трижды NМП (по 6 об. для каждой промывки).
Затем смолу обрабатывали дважды 20%-ным раствором пиперидина в NМП (по 6 об. для каждой обработки) для удаления защитных групп Fmoc. После второй обработки 20% пиперидином/NМП смолу промывали шесть раз NМП (по 6 об. для каждой промывки) до достижения отрицательной пробы на хлоранил.
Для приготовления раствора для сочетания взвешивали аминокислоту (1,7 экв.) и 6-хлор-1-гидроксибензотриазол (6-Cl-ГОБТ, 1,7 экв.), растворяли в 2,6× объеме NМП при температуре 10-5°C, затем объединяли с ДИЭА (1,9-3,0 экв.). ТБТУ или ГБТУ (1,7 экв.) растворяли в 1,33× объеме NМП при температуре 10-5°C. Затем два раствора объединяли. Образовавшийся раствор добавляли в реакционный сосуд. Колбу, в которой осуществляли смешение, промывали 1,33× объемом ДХМ, смывы добавляли в реактор, содержимое которого затем перемешивали в течение 2-3 ч при температуре от 25 до 27°C. Отбирали образец для теста Кайзера с целью оценки завершения реакции. Если реакция сочетания через 3 ч не заканчивалась (положительный тест Кайзера), то реакционный сосуд осушали и осуществляли повторное сочетание с использованием свежего раствора активированной аминокислоты. После завершения реакции сочетания раствор для сочетания сливали и смолу промывали четыре раза NМП (6 об. для каждой промывки). Затем цикл, включающий удаление защитной группы Fmoc и реакцию сочетания, повторяли для оставшихся аминокислот фрагмента (т.е. в порядке
Lys(Boc)→Val→Leu→Trp(Boc)→Ala→Ile→Phe→Glu(OtBu)→Lys(Boc)→Ala→Ala→Gln(trt)).
Из-за возможного поддерживающего взаимодействия между 2-метилаланином (Aib) и смолой 2-СТС могут возникать значительные трудности в осуществлении завершения реакций сочетаний первых двух аминокислот (Lys(Boc)-34 и Val-33). Поэтому обе реакции сочетания (Lys(Boc)-34, Val-33) осуществляли трижды (т.е. после сочетания осуществляли еще два повторных сочетания). Кроме того, уксусный ангидрид применяли для концевого кэпирования не прореагировавшего связанного со смолой продукта после реакций сочетания Lys(Boc)-34 и Val-33. Это повышало эффективность последующей очистки в результате удаления примесей из требуемого продукта в процессе хроматографической очистки.
Все реагенты, применяемые в этом примере, перечислены ниже в таблице:
В. Отщепление фрагмента от созданной смолы
Созданную согласно описанному выше методу смолу 7 раз промывали ДХМ (по 6 об. для каждой промывки) для удаления остаточных количеств NМП и смолу охлаждали с помощью последней промывки ДХМ до -5°C. ДХМ сливали и добавляли холодный раствор 1% ТФК/ДХМ (12 об., при температуре от -5 до -10°C) и перемешивали в течение 30 мин при 0°C. Раствор для отщепления собирали в колбу, содержащую пиридин (1,3 экв. относительно ТФК). После нагревания сосуда до 25°C смолу промывали 9 раз ДХМ (10 об.) и сливали в раствор для расщепления. Раствор ДХМ объединяли с водой (6 об.). Полученную смесь перегоняли при пониженном давлении для удаления ДХМ (350 торр при 28°C). Фрагмент осаждался из воды при удалении ДХМ. Фрагмент промывали и сушили при 30-35°C в вакууме. В этом примере процедуру расщепления повторяли еще один раз. Всего получили 6,78 г Fmoc-(Aib35) GLP-1 (23-35)-ОН (выход 68,1%) с чистотой 87,3% AN.
Пример 6
А. Получение загруженной Fmoc-Aib смолы 2СТС
Получали загруженную Fmoc-Aib смолу 2СТС. Ниже в таблице показаны количества применяемых реагентов:
Смолу 2-СТС вносили в 500-миллилитровый пептидный реактор и давали набухать в 400 мл ДХМ в течение 30 мин. Слой осушали и добавляли раствор Fmoc-Aib-OH и ДИЭА в 8 объемах ДМФ:ДХМ (87,5:12,5). Смесь перемешивали в атмосфере азота в течение 2 ч при температуре 25°C.
Слой осушали и промывали 400 мл ДМФ. Затем осуществляли концевое кэпирование всех оставшихся активных сайтов на смоле 2-СТС, используя 400 мл раствора МеОН:ДИЭА (9:1) в течение 1 ч. Слой осушали, промывали однократно 400 мл ДМФ, промывали однократно 200 мл ДМФ и промывали четыре раза 350 мл ДХМ. Смоле повторно давали набухать путем промывки 3×350 мл ИПС. Смолу сушили до постоянной массы с получением 47,56 г загруженной смолы. Анализ показал, что фактор загрузки составлял 0,37 ммоля/г.
Б. Твердофазный синтез
Твердофазный синтез осуществляли, начиная с 25,0 г загруженной Fmoc-Aib-2-СТС-смолы с фактором загрузки 0,37 ммоля/г. Смоле давали набухать в ДХМ (250 мл) в течение 30 мин при 25°C. Растворитель ДХМ сливали и смолу промывали дважды ДХМ (по 6 об. для каждой промывки) и трижды NMП (по 6 об. для каждой промывки).
Затем смолу обрабатывали дважды 20 об.%-ным раствором пиперидина в NМП (6 об. для каждой обработки) для удаления защитных групп Fmoc. После второй обработки 20% пиперидином/NМП смолу промывали шесть раз NMП (6 об. для каждой промывки) до достижения отрицательной пробы на хлоранил.
Для приготовления раствора для сочетания взвешивали аминокислоту и 6-хлор-1-гидроксибензотриазол (6-Cl-ГОБТ), растворяли в 3,2× NМП (или ДМФ для Lys-34, Val-33 и Gln-23), затем объединяли с ДИЭА при температуре от 10 до 5°C. ТБТУ растворяли в 1,6× объеме NМП (или ДМФ для Lys-34, Val-33 и Gln-23) при температуре от 10 до 5°C. Затем два раствора объединяли. Образовавшийся раствор добавляли с реакционный сосуд и колбу промывали 1,6× объемом ДХМ, смывы вносили в реактор, перемешивали со смолой в течение 2-3 ч при температуре от 25 до 27°C. Отбирали образец для теста Кайзера с целью оценки завершения реакции. Если реакция сочетания через 3 ч не заканчивалась (положительный тест Кайзера), то реакционный сосуд осушали и осуществляли повторное сочетание с использованием свежего раствора активированной аминокислоты. После завершения реакции сочетания раствор для сочетания сливали и смолу промывали четыре раза NМП (по 6 об. для каждой промывки). Затем цикл, включающий удаление защитной группы Fmoc и реакцию сочетания, повторяли для оставшихся аминокислот фрагмента (т.е. в порядке
Lys(Boc)→Val→Leu→Trp(Boc)→Ala→Ile→Phe→Glu(OtBu)→Lys(Boc)→Ala→Ala→Gln(trt)).
Из-за возможного поддерживающего взаимодействия между 2-метилаланином (Aib) и смолой 2-СТС могут возникать значительные трудности в осуществлении завершения реакций сочетаний первых двух аминокислот (Lys(Boc)-34 и Val-33). Условия сочетания Lys(Boc)-34, Val-33 и Gln(trt)-23 модифицировали, повышая применяемые количества как аминокислот, так и 6-Cl-ГОБТ с 1,7 экв. до 2,5 экв., а ДИЭА с 1,9 экв. до 3,0 экв. Растворитель для реакции сочетания также изменяли, используя вместо NМП ДМФ, для завершения реакции сочетания. Кроме того, в этом примере применяли уксусный ангидрид для концевого кэпирования не прореагировавшего связанного со смолой продукта после реакций сочетания Lys(Boc)-34 и Val-33. Это повышало эффективность последующей очистки в результате удаления примесей из требуемого продукта в процессе хроматографической очистки.
Все реагенты, применяемые в этом примере, перечислены ниже в таблице:
В. Отщепление фрагмента от созданной смолы
Созданную согласно описанному выше методу смолу 6 раз промывали ДХМ (по 6 об. для каждой промывки) для удаления NМП и смолу охлаждали с помощью последней промывки ДХМ до -5°C. ДХМ сливали, добавляли холодный раствор 1% ТФК/ДХМ (10 об., при температуре от -5 до -10°C) и перемешивали в течение 30 мин при 0°C. Раствор для отщепления собирали в колбу, содержащую пиридин (1,3 экв. относительно ТФК). После нагревания сосуда до 25°C смолу промывали 7 раз ДХМ (6 об.) и сливали в раствор для расщепления. Раствор ДХМ объединяли с водой (10 об.). Полученную смесь перегоняли при пониженном давлении для удаления ДХМ (350 торр при 28°C). Фрагмент осаждался из воды при удалении ДХМ. Фрагмент промывали и сушили при 30-35°C в вакууме. В этом примере процедуру расщепления повторяли еще один раз. Всего получили 12,36 г Fmoc-(Aib35) GLP-1 (23-35)-ОН (выход 59,35%) с чистотой 84,3% AN.
Пример 7
1. Промывка смолы
Процесс начинали с предварительно загруженной Fmoc-Gly-O-2-CTC-смолы (загрузка от 0,18 до 0,65 ммоля/г) или Fmoc-Aib-O-2-CTC (загрузка от 0,25 до 0,65 ммоля/г), применяя стандартную Fmoc-химию. Смоле сначала давали набухать в 10× объеме ДХМ в течение 30-60 мин. Затем ДХМ сливали и смолу промывали 10× объемом NМП в течение 3-5 мин (по 5 мин каждый раз).
2. Общий цикл синтеза
Используя смолу, промытую согласно методу, описанному в разделе А примера 7, для удаления Fmoc применяли две обработки ~10× раствором 20% пиперидина в NМП (об./об.). Каждую обработку осуществляли в течение 15-30 мин. После каждой обработки раствор пиперидина/NМП сливали. Затем смолу промывали NМП 4-5 раз (10× объем, по 5 мин каждый раз).
Для приготовления раствора для сочетания взвешивали защищенную с помощью Fmoc аминокислоту (АА) и ГОБТ (из расчета 1,5-2,0 экв.), растворяли в 4× объеме NМП при 10°C и объединяли с ДИЭА (1,5-2,0 экв.). ГБТУ (1,5-2,0 экв.) растворяли в 3× объеме NМП при 10°C. Затем два раствора объединяли и смешивали с 3× объемом ДХМ в течение 1-2 мин. Образовавшийся раствор добавляли в реакционный сосуд и смешивали со смолой при перемешивании в течение 1,5-5 ч. Отбирали образец для теста Кайзера с целью оценки завершения реакции. Продукты незавершенной реакции сочетания подвергали повторному сочетанию. После завершения реакции сочетания раствор для сочетания сливали и смолу промывали 5 раз NМП (10× объем, по 5 мин каждый раз).
А. После осуществления общей процедуры синтеза фрагмент Fmoc-GLP-1(11-22)-2-СТС, имеющий нативную последовательность, создавали с помощью постадийного процесса, используя такую же процедуру синтеза, фрагмент, последовательность которого представлена в SEQ ID NO. 7, сочетали с образовавшимся фрагментом Fmoc-GLP-1(11-22)-2-CTC, полученным согласно описанному в этом разделе А методом, используя общую процедуру синтеза.
Б. После осуществления общей процедуры синтеза фрагмент Fmoc-GLP-1(11-22)-2-СТС создавали с помощью постадийного процесса. Однако условия, в которых осуществляли удаление Fmoc, заменяли, используя раствор, содержащий 10% пиперидина и 2% ДБУ в NМП (об./об.) вместо 20% пиперидина в NМП (об./об.). Кроме того, время обработки в положении АА14 [Fmoc-Ser(tBu)-] и в положении АА13 [Fmoc-Thr(tBu)-] удлиняли до 1,5 ч с целью завершения реакции. Используя такую же общую процедуру синтеза фрагмент, последовательность которого представлена в SEQ ID NO. 7, сочетали с полученным фрагментом Fmoc-GLP-1 (11-22)-2-CTC с помощью общей процедуры синтеза.
В. Следуя общей процедуре синтеза с использованием раствора для удаления Fmoc, содержащего 10% пиперидина и 2% ДБУ в NМП (об./об.), создавали фрагмент Fmoc-(X17-18) GLP-1(11-22)-2-CTC с помощью постадийного процесса. Затем фрагмент, последовательность которого представлена в SEQ ID NO. 7, сочетали с полученным фрагментом Fmoc-(X17-18) GLP-1(11-22)-2-CTC с помощью общей процедуры синтеза.
Г. Следуя общей процедуре синтеза, создавали фрагмент Fmoc-(X17-18) GLP-1(11-22)-2-СТС с помощью постадийного процесса. Затем фрагмент, последовательность которого представлена в SEQ ID NO. 7, сочетали с полученным фрагментом Fmoc-(X17-18) GLP-1(11-22)-2-CTC с помощью общей процедуры синтеза.
Д. Следуя общей процедуре синтеза, изложенной выше в разделе Г, и за исключением различий, которые указаны в приведенной ниже таблице, создавали фрагмент Fmoc-(X17-18) GLP-1(11-22)-2-CTC с помощью постадийного процесса. Затем фрагмент, последовательность которого представлена в SEQ ID NO. 7, сочетали с полученным фрагментом Fmoc-(X17-18) GLP-1(11-22)-2-CTC с помощью общей процедуры синтеза.
Ниже в таблице обобщены детали процедур, изложенных в разделах А-Д данного примера:
Е. Отщепление фрагмента
Для осуществления отщепления любого из пептидных фрагментов, синтезированных согласно процедуре, описанной в этом примере, смоле с созданным фрагментом давали набухать в 10× объеме ДХМ в течение 30 мин и охлаждали до -10°C. ДХМ сливали и добавляли раствор 1% ТФК/ДХМ (10× объем) и перемешивали в течение 30 мин. Раствор для расщепления собирали в колбу, содержащую пиридин (2-3 экв. относительно ТФК). После нагревания до 25°C смолу обрабатывали 1% ТФК/ДХМ (10× объем) в течение 5 мин, а затем добавляли пиридин (2-3 экв. относительно ТФК). После перемешивания в течение еще 5 мин раствор собирали. Затем смолу промывали 4 раза 10×объемом ДХМ (каждый раз 5 мин). Растворы, полученные после всех промывок, и раствор для расщепления объединяли и смешивали с водой (объемное соотношение вода/ДХМ ~1/4). Полученную смесь перегоняли при пониженном давлении для удаления ДХМ (350 торр/28°C). Пептидный фрагмент осаждался из воды при удалении ДХМ, и его фильтровали. Пептидный фрагмент промывали водой и сушили при 30°C в вакууме.
Пример 8. Синтез в растворе: добавление Arg
Фрагмент (Aib35) GLP-1(23-35) (несущий защищенную боковую цепь (см. таблицу А) и несущий защитную группу Fmoc на N-конце (9,11 г, 3,94 ммоля) растворяли в ДМСО (90 мл). К этому раствору добавляли ГОБТ (2,42 г, 4 экв.), ГБТУ (5,98 г, 4 экв.), ДИЭА (3,44 мл, 5 экв.) и H-Arg (2HCl)-NH2 (3,88 г, 4 экв.) в 10 мл ДМСО. Реакционную смесь перемешивали и осуществляли мониторинг с помощью ЖХВР. Через 4 ч реакция не была завершена, поэтому добавляли 2 мл ДИЭА. Реакцию проводили в течение ночи. Затем в реакционную смесь добавляли пиперидин (5 мл). Удаление Fmoc осуществляли в течение 2 ч. Для прекращения реакции добавляли ледяную воду (800 мл) и перемешивали в течение 40 мин. Образовавшийся твердый продукт белого цвета фильтровали, промывали водой (400 мл) и сушили в течение ночи с получением фрагмента (Aib35) GLP-1(23-36) (9,65 г, массовый выход 109%).
Пример 9
Фрагмент (Aib8, X17-18) GLP-1(7-22) (7,73 г, 2,88 ммоля), несущий защитные группы боковых цепей (см. таблицу А) и защитную группу Fmoc на N-конце, растворяли в ДМСО (65 мл).
К этому раствору добавляли ГОБТ (0,73 г, 4,77 ммоля), ГБТУ (1,46 г, 3,85 ммоля), ДИЭА (0,71 мл, 7,40 ммоля) и фрагмент (Aib35) GLP-1(23-36) (8,5 г, 3,45 ммоля) в 20 мл ДМСО. Реакционную смесь перемешивали и осуществляли мониторинг с помощью ЖХВР. Через 3 ч реакция была завершена. Затем в реакционную смесь добавляли пиперидин (5 мл). Удаление Fmoc осуществляли в течение 2 ч. Для прекращения реакции добавляли ледяную воду (800 мл) и перемешивали в течение 30 мин. Образовавшийся твердый продукт белого цвета фильтровали, промывали МТБЭ (400 мл) и сушили в течение ночи с получением защищенного пептида (Aib8, X17-18, Aib35) GLP-1(7-36).
Пример 10. Полное удаление защитных групп
Пептид, полученный согласно методу, описанному в примере 9 (16,24 г), обрабатывали раствором ТФК/ДТТ/вода (100 мл/5 г/2,0 мл) в течение 2 ч и образовавшийся раствор сливали в МТБЭ (800 мл) в ледяной бане. После перемешивания в течение 30 мин образовавшееся твердое вещество белого цвета фильтровали, промывали МТБЭ (400 мл) и сушили с получением неочищенного пептидного продукта (16,0 г, массовый выход 138%). Образовавшийся пептид с удаленными защитными группами далее в контексте настоящего описания обозначен как «неочищенный» пептид.
Пример 11. Очистка
Очистку неочищенного пептида осуществляли на колонке Kromasil® C4, 10 мкм, 2,0×25 см, с получением очищенного пептида с чистотой не менее 98% (98+%) и выходом ~60% в пересчете на общее содержание. Очистка предусматривали 1-ю стадию хроматографической очистки при рН 2 с последующей 2-ой стадией при рН 9. После такой очистки очищенный пептид можно дополнительно обрабатывать или процессировать различными путями. Например, полученный очищенный пул после второй стадии можно лиофилизировать или пропускать через концентрирующую колонку и выделять осаждением. После обоих путей выделения получают требуемый пептид (ацетат).
При осуществлении обеих стадий процесса неочищенный пептид растворяли из расчета 5 мг/мл (в пересчете на общее содержание) в смеси 10% ацетонитрила/вода (0,2 М уксусная кислота). Инъецировали с дублированием по 1000 мг смеси (в пересчете на общее содержание), используя градиент ацетонитрил/ТГФ/вода/ТГФ, и объединяли фракции, имеющие чистоту ~95%. Также объединяли «рециркуляционную фракцию», имеющую чистоту ~70%, на долю которой приходится ~34% выхода. Рециркуляционную фракцию повторно инъецировали, используя такой же градиент ацетонитрил/ТГФ/вода/ТГФ, и объединяли фракцию, имеющую чистоту ~85%, с основным пулом. После 1-ой стадии хроматографической очистки выход в пересчете на общее содержание составлял 70%.
Объединенный пул после 1-ой стадии (рН 2) затем очищали дополнительно на второй стадии на такой же колонке Kromasil® C4, но используя градиент ацетонитрил/ТГФ/вода/ацетат аммония (рН 8,8). Объединяли фракции, имеющие чистоту 98+%, на долю которых приходилось ~85% выхода после 2-ой стадии. Общий выход обеих стадий очистки составлял 60% в пересчете на общее содержание.
Объединенный пул после 2-ой стадии (рН 8,8) затем концентрировали с помощью такой же колонки Kromasil® C4, но используя ступенчатый градиент метанол/вода/ацетат аммония. Фракции объединяли, и они были готовы к выделению.
А. Приготовление раствора неочищенного пептида
8 г неочищенного пептида растворяли в 500 мл раствора, содержащего 90% 0,2 н. уксусной кислоты/10% ацетонитрила. Раствор фильтровали сначала через фильтр с размером отверстий 0,45 мкм типа Durapore (диаметром 47 мм) (фирма Millipore), а затем через дисковый микронный патронный фильтр типа Supor EKV (0,65/диаметр 0,2 мм) (фирма Pall Filters). Неочищенный инъецируемый раствор анализировали с помощью ЖХВР, и было обнаружено, что он содержит 5,02 мг (мас.%) входящего в его состав пептида на 1 мл (500 мл × 5,02 мг/мл = 2512 мг входящего в состав пептида).
Б. Хроматография - 1-я стадия при рН ~2
Инъецировали в четырех повторностях неочищенный раствор в следующих условиях.
Колонка: изготовлена фирмой Kromasil, упакована С4, 10 мкм, и имеет размеры 2,0×25 см.
Детектор: ультрафиолетовый детектор, установленный на 280 нм (8 нм ширина полосы, 350/20 нм станд.).
Температура колонки: температура окружающей среды.
Скорость потока: 13,0 мл/мин (~50 бар противодавление).
Подвижная фаза:
А = смесь, содержащая 0,1% трифторуксусной кислоты/15% ацетонитрила/85% воды.
Б = смесь, содержащая 0,1% трифторуксусной кислоты/15% тетрагидрофурана/70% ацетонитрила/15% воды.
Градиент: градиент начинали со 100% подвижной фазы и поддерживали в течение 0,1 мин. Затем образец загружали вручную в колонку с помощью насоса С (см. ниже описание насоса С). После внесения образца осуществляли погон с использованием линейного градиента от 100% А до 83% А в течение 1 мин. Затем поддерживали подвижную фазу, содержащую 83% А, в течение следующих 11 мин. Затем осуществляли погон с использованием линейного градиента, содержащего 73% А, в течение 10 мин. Затем поддерживали подвижную фазу, содержащую 73% А, в течение следующих 15 мин. Затем погон завершали и осуществляли 10-минутный погон с использованием 100% Б с последующим повторным уравновешиванием колонки с помощью 100% А.
Образец вносили с помощью отдельного изократического насоса HP 1100 со скоростью 9,0 мл/мин (насос С). Примеси, которые элюировались в начала процесса, отделяли от основного пика путем начального 11-минутного изократического поддерживания 83% А с последующим линейным градиентом до 73% А в течение следующих 10 мин. Затем элюировали основной пептидный пик в течение 15 мин, поддерживая 73% А. Фракции собирали в течение 15 мин, поддерживая 73% А.
В. 1-я стадия рецикла при рН ~2
Пул для рецикла получали из фракцией, для которых было установлено, что их чистота ниже той, с которой можно добавлять фракции в объединенный пул. Эти фракции объединяли по отдельности и разводили равным объемом воды. Продукт для рецикла снова инъецировали в колонку, из которой был получен этот конкретный объединенный пул. Использовали такие же условия хроматографирования и фракции, имеющие приемлемую чистоту, объединяли, разводили равным объемом воды и добавляли в объединенный пул 1-ой стадии.
Инъецируемый объем продукта для рецикла существенно ниже по сравнению с инъекцией неочищенного продукта из-за увеличенного количества примесей, которые элюируются вблизи пика пептида. В среднем осуществляли 1 инъекцию продукта для рецикла на каждые 2 инъекции неочищенного продукта.
Г. 2-я стадия препаративной хроматографии при рН 8.8
Конечный объединенный пул после 1-ой стадии дополнительно очищали с помощью повторной хроматографии при рН 8,8. Применение значения рН 8,8 для 2-ой стадии существенно изменяло уровень элюции примесей, что позволяло получать более очищенный продукт с боле высоким выходом. Для этой 2-ой хроматографической стадии использовали следующие условия.
Колонка: изготовлена фирмой Kromasil, упакована С4, 10 мкм, и имеет размеры 2,0×25 см.
Детектор: ультрафиолетовый детектор, установленный на 280 нм (8 нм ширина полосы, 350/20 нм станд.).
Температура колонки: температура окружающей среды.
Скорость потока: 13,0 мл/мин (~50 бар противодавление).
Подвижная фаза:
А = смесь, содержащая 15% ацетонитрила/85% воды, включающая 2 г на 1 л ацетата аммония и 1 мл на 1 л концентрированного гидроксида аммония.
Б = смесь, содержащая 15% тетрагидрофурана/60% ацетонитрила/25% воды, включающая 2 г/л ацетата аммония и 1 мл/л концентрированного гидроксида аммония.
Градиент: градиент начинали со 100% подвижной фазы и поддерживали в течение 0,1 мин. Затем образец загружали вручную в колонку с помощью насоса С. После внесения образца осуществляли погон с использованием линейного градиента от 100% А до 67% А в течение 2 мин. Затем поддерживали подвижную фазу, содержащую 67% А, в течение следующих 33 мин. Затем погон завершали и осуществляли 5-минутный погон с использованием 100% Б с последующем повторным уравновешиванием колонки с помощью 100% А.
Образец вносили с помощью отдельного изократического насоса HP 1100 со скоростью 9,0 мл/мин (насос С). После 30-минутной стадии загрузки осуществляли кратковременный погон с использованием градиента от 100% А до 67% А в момент времени от 30,2 до 32 мин. Основной пептидный пик элюировался после поддержания в течение 33 мин 67% А. Фракции собирали, начиная через 20 мин после загрузки колонки, и заканчивали сбор после завершения элюции основного пика. Фракции собирали в течение примерно 15 мин. Приемлемые фракции объединяли и разводили равным объемом воды. На этой стадии очистки возможно использование рецикла, но отработанные фракции, как правило, не содержали достаточное количество пептида для того, чтобы осуществлять рециркуляционную инъекцию. Это может становиться целесообразным, если осуществляют большое количество инъекций и отработанные фракции объединяют для рецикла.
Д. Концентрирующий погон
Конечный объединенный полученный после 2-ой стадии пул вносили на ту же колонку и быстро элюировали, используя другую подвижную фазу для концентрирования пептида перед выделением. На этой стадии использовали следующие условия.
Колонка: изготовлена фирмой Kromasil, упакована С4, 10 мкм, и имеет размеры 2,0×25 см.
Детектор: ультрафиолетовый детектор, установленный на 280 нм (8 нм ширина полосы, 350/20 нм станд.).
Температура колонки: температура окружающей среды.
Скорость потока: 13,0 мл/мин (~50 бар противодавление).
Подвижная фаза:
А = смесь, содержащая 10% метанола/90% 20 мМ ацетата аммония.
Б = смесь, содержащая 90% метанола/10% 20 мМ ацетата аммония.
Градиент: градиент начинали со 100% подвижной фазы и поддерживали в течение 0,1 мин. Затем образец загружали вручную в колонку с помощью насоса C. После загрузки образца подвижную фазу поддерживали в виде 100% А в течение 5 мин. Затем подвижную фазу немедленно ступенчато заменяли на 100% Б и поддерживали в течение следующих 20 мин. Затем погон завершали и осуществляли повторное уравновешивание колонки в течение 15 мин с использованием 100% А.
Образец вносили с помощью отдельного изократического насоса HP 1100 со скоростью 9,0 мл/мин (насос С). После 45-минутной стадии загрузки в течение непродолжительного периода времени (5 мин) поддерживали 100% А, затем осуществляли погон с использованием ступенчатого градиента до 100% Б в течение 20 мин для элюции пептида. Основной пик начинал элюироваться через 2 мин после ступенчатого градиента до 100% Б. Следующие 12 мин собирали фракции, содержащие пептид, и объединяли для выделения.
Пример 12. Лиофилизация
Определяли вес 1-литрового широкогорлого флакона типа FLPE. В этот флакон добавляли очищенный пул (210 мл) ацетата (Aib8, X17-18, Aib35) GLP-1(7-36) в водном ацетонитриле/ацетате аммония. Контейнер смывали деионизированной водой 3×5 мл и смывы добавляли к пулу. Этот раствор интенсивно перемешивали до гомогенизации, затем закрывали и сосуд замораживали в жидком азоте. Замороженный флакон открывали и горло закрывали двойной безволоконной салфеткой типа KimWipe, для удержания которой на месте использовали резиновую полоску. Флакон помещали в вакутайнере в лиофилизатор и лиофилизировали. Температура конденсатора составляла -89°C, давление 19 мкм.
Через 24 ч вакутайнер открывали для оценки развития процесса; в нем еще присутствовал заметный толстый кусок льда. Лиофилизацию начинали вновь.
Еще через 18 ч флакон удаляли из лиофилизатора и взвешивали. Масса оказалось нестабильной, она быстро возрастала из-за гигроскопичной природы продукта. Липкий продукт быстро переносили во взвешенный сцинтилляционный флакон и взвешивали, установлено, что получили 0,822 г пептида.
Пример 13. Осаждение очищенного пептида
В колбе раствор, содержащий 100,5 мг чистого ацетата (Aib8, X17-18, Aib35) GLP-1(7-36) в 2 мл 20 мМ ацетата аммония в смеси МеОН/вода (9:1, об./об), разводили 1 мл 20 мМ ацетата аммония в смеси МеОН/вода (9:1, об./об.). При встряхивании медленно вводили в колбу 20 мл изопропанола (ИПС) при температуре от 20 до 25°C. Смесь в сосуде становилась мутной после добавления 15 мл ИПС. Перемешивание продолжали в течение ночи при температуре от 20 до 25°C. Осадившийся продукт фильтровали и промывали 5 мл ИПС, а затем сушили при 25°C в вакууме до постоянной массы. Получили 90,9 мг пептида (выход 90,42%).
Пример 14. Выделение очищенного пептида
В колбе раствор, содержащий 497,7 мг чистого ацетата (Aib8, Х17-18, Aib35) GLP-1(7-36) в 10 мл 20 мМ ацетата аммония в смеси МеОН/вода (9:1), разводили 6 мл 20 мМ ацетат аммония в смеси МеОН/вода (9:1). При встряхивании медленно вводили в колбу 40 мл изопропанола (ИПС) при температуре от 20 до 25°C в течение 35 мин. Смесь в сосуде становилась мутной после добавления 2 мл ИПС. Перемешивание продолжали в течение 1 ч при температуре от 20 до 25°C. Осадившийся продукт фильтровали и промывали 5 мл ИПС, а затем сушили при 25°C в вакууме до постоянной массы. Получили 458,4 мг пептида (выход 92%).
Пример 15. Выделение очищенного пептида
В колбу медленно добавляли при перемешивании 900 мл ИПС в раствор, содержащий ~1000 мг очищенного ацетата (Aib8, X17-18, Aib35) GLP-1(7-36) в 150 мл 20 мМ ацетата аммония в смеси МеОН/вода (9:1), через концентрирующую колонку при температуре от 20 до 25°C. Это добавление завершали через 45 мин. Смесь в сосуде становилась мутной после добавления 260 мл ИПС. Перемешивание продолжали в течение 40 мин при температуре от 20 до 25°C. Осадившийся продукт фильтровали и промывали 5 мл ИПС, а затем сушили при температуре от 20 до 25°C в вакууме до постоянной массы. Получили 746 мг пептида (выход 74,6%).
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНОЕ АНАЛОГА GLP-1 ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И ИХ ПРИМЕНЕНИЕ | 2010 |
|
RU2565536C2 |
| ПЕПТИДНЫЕ ПРОЛЕКАРСТВА, ПРИНАДЛЕЖАЩИЕ К СУПЕРСЕМЕЙСТВУ АМИД-СОДЕРЖАЩИХ ГЛЮКАГОНОВ | 2011 |
|
RU2580317C2 |
| ГЛИКОЗИЛИРОВАННЫЙ ПЕПТИД GLP-1 | 2009 |
|
RU2543157C2 |
| ДВОЙНЫЕ АГОНИСТЫ GLP1/GIP ИЛИ ТРОЙНЫЕ АГОНИСТЫ GLP1/GIP/ГЛЮКАГОНА | 2013 |
|
RU2652783C2 |
| ПОЛИПЕПТИДЫ, СЕЛЕКТИВНЫЕ К РЕЦЕПТОРАМ ГЛЮКАГОНА, И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2017 |
|
RU2760007C2 |
| СПОСОБ ВЫСОКОЭФФЕКТИВНОГО ПОЛУЧЕНИЯ ПОЛИПЕПТИДНОГО ФРАГМЕНТА, ПОДХОДЯЩЕГО ДЛЯ NCL | 2012 |
|
RU2605411C2 |
| ЛИНЕЙНЫЕ ЖИДКОФАЗНЫЕ ПУТИ ДЛЯ ГЕКСАПЕПТИДОВ WNT | 2019 |
|
RU2799031C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОНСТРУКЦИИ С ПОВЫШЕННОЙ АФФИННОСТЬЮ СВЯЗЫВАНИЯ С АЛЬБУМИНОМ | 2018 |
|
RU2753880C1 |
| ПЕПТИДЫ, ОБЛАДАЮЩИЕ АГОНИСТИЧЕСКОЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ РЕЦЕПТОРА НЕЙРОПЕПТИДА-2-(Y2R) | 2006 |
|
RU2383553C2 |
| ДИПЕПТИД, СОДЕРЖАЩИЙ НЕПРОТЕИНОГЕННУЮ АМИНОКИСЛОТУ | 2012 |
|
RU2643515C2 |








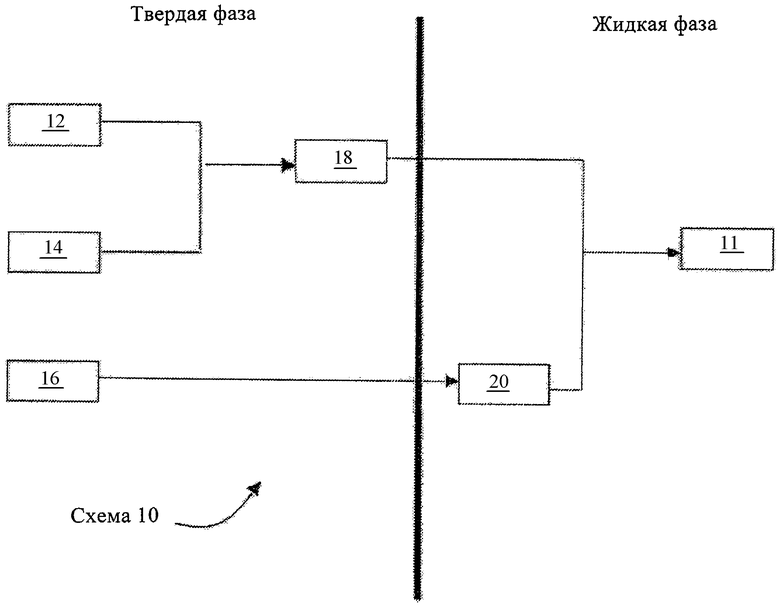
Изобретение относится к получению инсулинотропных пептидов, которые синтезируют с использованием подхода, основанного на применении твердофазного и жидкофазного («гибридного») синтеза. В целом, указанный подход предусматривает синтез трех различных пептидных промежуточных фрагментов с помощью твердофазной химии. Затем используют жидкофазную химию для добавления аминокислотного продукта к одному из фрагментов. Затем осуществляют сочетание фрагментов друг с другом с использованием твердой и жидкой фаз. Применение псевдопролина в одном из фрагментов облегчает твердофазный синтез этого фрагмента и облегчает также последующее сочетание этого фрагмента с другими фрагментами в жидкой фазе. Настоящее изобретение можно применять прежде всего для получения инсулинотропных пептидов, таких как GLP-1(7-36), и их встречающихся в естественных условиях и не встречающихся в естественных условиях аналогов. 3 н. и 16 з.п. ф-лы, 1 ил., 14 табл., 15 пр.
1. Способ получения иксулинотропного пептида, включающий этапы а)-е):
а) получения первого пептидного фрагмента, включающего аминокислотную последовательность НХ8ЕХ10 (SEQ ID NO.6), в которой X8 и X10 каждый обозначает остатки ахиральной аминокислоты, Н и Е каждый необязательно несет защитную группу боковой цепи;
б) получения второго пептидного фрагмента, включающего аминокислотную последовательность TFTSDVX17-18YLEG (SEQ ID NO.8), в которой остаток, обозначенный символом X17-18, представляет собой дипептидный остаток псевдопролина, где указанные аминокислотные остатки последовательности необязательно несут защитную группу боковой цепи;
в) сочетания первого фрагмента co вторым фрагментом с получением третьего пептидного фрагмента, который включает аминокислотную последовательность НХ8ЕХ10 TFTSDVX17-18YLEG (SEQ ID NO.11), где указанные аминокислотные остатки последовательности необязательно несут защитную группу боковой цепи;
г) получения четвертого пептидного фрагмента, включающего аминокислотную последовательность QAAKEFIAWLVKX35 (SEQ ID NO.9), в которой X35 обозначает остаток ахиральной аминокислотной кислоты, где указанные аминокислотные остатки последовательности необязательно несут защитную группу боковой цепи;
д) сочетания четвертого пептидного фрагмента с аргинином с получением пятого пептидного фрагмента, включающего аминокислотную последовательность QAAKEFIAWLVK X35R (SEQ ID NO.12), где указанные остатки последовательности необязательно несут защитную группу боковой цепи; и
е) сочетания пятого фрагмента с третьим фрагментом с получением инсулинотропного пептида, который включает аминокислотную последовательность HX8EX10TFTSDVX17-18YLEGQAAKEFIAWLVK X35R (SEQ ID N0.13), где указанные остатки последовательности необязательно несут защитную группу боковой цепи.
2. Способ по п.1, включающий дополнительную стадию, на которой:
ж) удаляют защитные группы боковых цепей с получением инсулинотропного пептида, включающего аминокислотную последовательность HX8EX10TFTSDVSSYLEGQAAKEFIAWLVKX35R (SEQ ID NO.5), где каждый из символов X в положениях 8, 10 и 35 независимо друг от друга обозначает ахиральный, необязательно имеющий стерическую помеху аминокислотный остаток.
3. Способ получения инсулинотропного пептида по п.1, включающий этапы:
а) получения первого пептидного фрагмента, включающего аминокислотную последовательность НХ8ЕХ10 (SEQ ID NO.6), в которой X8 обозначает аминокислотный остаток, соответствующий Aib, a X10 обозначает аминокислотный остаток, соответствующий глицину, Н и Е каждый необязательно несет защитную группу роковой цепи;
б) получения второго пептидного фрагмента, включающего аминокислотную последовательность TFTSDVX17-18YLEG (SEQ ID NO.8), в которой остаток, обозначенный символом X17-18, представляет собой дипептидный остаток псевдопролина, где указанные аминокислотные остатки последовательности необязательно несут защитную группу боковой цепи;
в) сочетания первого фрагмента со вторым фрагментом с получением третьего пептидного фрагмента, который включает аминокислотную последовательность HX8EX10TFTSDV X17-18YLEG (SEQ ID NO.11), где указанные аминокислотные остатки последовательности необязательно несут защитную группу боковой цепи;
г) получения четвертого пептидного фрагмента, включающего аминокислотную последовательность QAAKEFIAWLVKX35 (SEQ ID NO.9), в которой X35 обозначает аминокислотный остаток, соответствующий Aib, где указанные аминокислотные остатки последовательности необязательно несут защитную группу боковой цепи;
д) сочетания четвертого пептидного фрагмента с аргинином с получением пятого пептидного фрагмента, который включает аминокислотную последовательность QAAKEFIAWLVK X35R (SEQ ID NO.12), где указанные остатки последовательности необязательно несут защитную группу боковой цепи; и
е) сочетания пятого фрагмента с третьим фрагментом с последующим удалением защитных групп боковых цепей с получением инсулинотропного пептида формулы HX8EX1OTFTSDVSSYLEGQAAKEFIAWLVKX35R (SEQ ID NO.5), где X8 и X35 обозначают аминокислотные остатки, соответствующие Aib, a X10 обозначает аминокислотный остаток, соответствующий глицину.
4. Способ по п.1 или 3, в котором X8 обозначает аминокислотный остаток, соответствующий метилаланину.
5. Способ по п.1 или 3, в котором X10 обозначает аминокислотный остаток, соответствующий глицину.
6. Пептидный фрагмент, имеющий аминокислотную последовательность HX8EX10 (SEQ ID NO.6), в которой X8 и X10 каждый обозначает остатки ахиральной аминокислоты, где Н, Е, X8 и X10 каждый необязательно несет защитную группу боковой цепи, выбранную из групп, включающих ацетил (Ас), бензоил (Bz), трет-бутил, трифенилметил (тритил), тетрагидропиранил, простой эфир бензила (Bzl) и 2,6-дихлорбензил (DCB), t-бутоксикарбонил (Boc), нитро, р-толуолсульфонил (Tos), адамантилоксикарбонил, ксантил (Xan), бензил, 2,6-дихлорбензил, метил, этил и сложный эфир t-бутила, бензилоксикарбонил (Z), 2-хлорбензилоксикарбонил (2-Cl-Z), t-амилокикарбонил (Аос), и ароматические или алифатические защитные группы уретанового типа, фотолабильные группы, такие как нитровератрилоксикарбонил (NVOC); и неустойчивые в присутствии фтористых соединений группы, такие как триметилсилилоксикарбонил (ТЕОС).
7. Пептидный фрагмент по п.6, в котором X8 обозначает аминокислотный остаток, соответствующий Aib, a X10 обозначает аминокислотный остаток, соответствующий глицину.
8. Способ по п.5, в котором инсулинотропный пептид включает аминокислотную последовательность (SEQ ID NO.5) HX8EX10TFTSDVSSYLEGQAAKEFIAWLVKX35R, где каждый из символов X в положениях 8, 10 и 35 независимо друг от друга обозначает ахиральный, необязательно имеющий стерическую помеху аминокислотный остаток; и где один или несколько аминокислотных остатков необязательно несут защитную группу боковой цепи.
9. Способ по п.8, в котором по меньшей мере один из символов X8 и X35 обозначает остаток Aib.
10. Способ по п.8, в котором X10 обозначает остаток глицина.
11. Способ по п.1, в котором X17-18 имеет формулу
,
в которой Ф обозначает остаток любой аминокислоты, необязательно несущий защитную группу боковой цепи, и R1 и R2 каждый независимо друг от друга обозначает приемлемый двухвалентный связующий радикал.
12. Способ по п.11, в котором Ф обозначает остаток Ser, необязательно несущий защитную группу боковой цепи.
13. Способ по п.11, в котором R2 обозначает -СН2-.
14. Способ по п.11, в котором R1 обозначает
где R3 и R4 каждый независимо друг от друга обозначают одновалентный радикал, выбранный из Н или (низш.)алкила; или R3 и R4 могут также быть членами кольцевой структуры.
15. Способ по п.14, в котором R3 и R4 каждый обозначают метил.
16. Пептид с аминокислотной последовательностью TFTSDVX17-18YLEG (SEQ ID NO.8), в которой остаток, обозначенный символом X17-18, представляет собой дипептидный остаток псевдопролина, где аминокислотные остатки необязательно несут защитную группу боковой цепи, включающую трет-бутил и трет-бутиловый эфир.
17. Способ по п.1, в котором X35 обозначает аминокислотный остаток метилаланина.
18. Способ по пп.1 и 2, дополнительно включающий стадию, на которой:
д) осуществляют сочетание пептидного фрагмента, который включает аминокислотную последовательность QAAKEFIAWLVKX35 (SEQ ID NO.9), с аргинином с получением пептидного фрагмента, включающего аминокислотную последовательность QAAKEFIAWLVKX35R (SEQ ID NO.12), где указанные остатки последовательности необязательно несут защитную группу боковой цепи.
19. Способ по п.18, в котором инсулинотропный пептид представляет собой пептид, имеющий аминокислотную последовательность (SEQ ID NO.4)
HAibEGTFTSDVSSYLEGQAAKEFIAWLVKAibR.
| АНАЛОГИ GLP-1 | 1999 |
|
RU2288232C9 |
| US 6514500 B1, 04.02.2003 | |||
| WO 2004094461 A2, 04.11.2004 | |||
| Прибор для наполнения сосудов огнеопасными жидкостями до определенного уровня | 1933 |
|
SU34331A1 |
| ANDERSSON L et al | |||
| Large-scale synthesis of peptides | |||
| - BIOPOLYMERS, 2000, vol.55, no.3, 2000, pages 227-250 | |||
| SAMPSON W R et al | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |

