Изобретение относится к способам получения соединений платины (IV), а именно транс-1,2-диаминциклогексантетрахлороплатины (IV) (ормаплатин, тетраплатин), которое является противоопухолевым препаратом II поколения.
Существует несколько способов получения соединения транс-1,2-диаминциклогексантетрахлороплатины (IV), которые основаны на реакциях аминирования комплекса платины (II) и дальнейшем окислении его в среде N,N-диметилформамида-HCl-Cl2 при температуре 100°C (Eastland G.J. // Drugs Fut. 1987. V.12. i.2. p.139-141; Anderson W.K., Quagliato D.A., Haugwitz R.D., et al. // Cancer Treat. Rep. 1986. V.70. p.997-1002). Синтез осуществляется по реакции (1) (где C6H10(NH2)2-транс-d,l-1,2-диаминциклогексан):
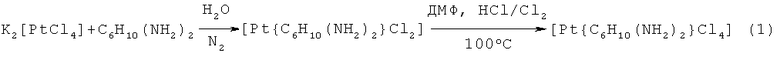
Наиболее близким к заявляемому является способ получения транс-1,2-диаминциклогексантетрахлороплатины (IV), основанный на взаимодействии гексахлороплатината (IV) калия с дигидрохлоридом транс-1,2-диаминциклогексана (Wyrig S.D., Chaney S.G. // J. Label. Compounds Radiopharm. 1990. V.28. i.7. p.753-756; Upjohn. Natl Canser Inst. // Drugs Fut. 1991. V.16. i.2. p.187). Синтез осуществляется по реакции (2):
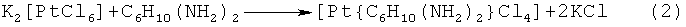
Раствор гексахлороплатината (IV) калия (6.2 мг, 0.0128 ммоль) в 2.9 мл 1.0 N соляной кислоты выпарили почти досуха под вакуумом до образования оранжевого осадка, и затем прибавили раствор 2.4 мг (0.013 ммоль) транс-1,2-диаминоциклогексана дигидрохлорида в 2 мл воды. Этот раствор, защищенный от света, перемешивали в течение 10 часов. После этого раствор выпарили под вакуумом, а твердый продукт отделили от исходного материала высачиванием в 2 мл ацетона. Ацетоновый раствор отфильтровали, концентрировали до 0.2 мл и разбавляли 2.0 мл диэтилового эфира. Полученную суспензию центрифугировали и верхнюю часть раствора отсасывали пипеткой. Добавляли эфир, центрифугировали и удаляли всплывшее на поверхность вещество. Процедуру повторяли дважды. Образовавшийся желтый осадок высушивали до постоянного веса. Выход целевого продукта составил 3.1 мг (54%).
Недостатком этого способа, как и предыдущего, является низкий выход основного продукта, использование дополнительных реагентов (ацетон, диэтиловый эфир, диметилформамид), а также сложность и длительность процесса выделения этого вещества с применением упаривания, высаливания, центрифугирования.
Поставленная цель достигается:
- созданием оптимального соотношения концентрации комплекса в растворе, концентрации транс-1,2-диаминциклогексана дигидрохлорида и pH среды;
- использованием гидроксида лития для повышения выхода основного продукта;
- уменьшением времени синтеза.
Сущность заявляемого способа состоит в следующем.
К платинохлористоводородной кислоте в соляной кислоте при комнатной температуре добавляют транс-1,2-диаминциклогексан дигидрохлорид. Образовавшийся осадок отфильтровывают, промывают ледяной уксусной кислотой и сушат. Затем осадок растворяют в воде, добавляя гироксид лития. Реакцию ведут при комнатной температуре. Процесс заканчивают, когда раствор над осадком становится бледно-желтым. Соединение транс-1,2-диаминциклогексантетрахлороплатины (IV) отфильтровывают, промывают спиртом и сушат.
В основу предлагаемого способа получения транс-1,2-диаминциклогексантетрахлороплатины (IV) положены следующие новые данные:
- найдено максимальное накопление целевого продукта от pH среды;
- минимизированы процессы гидролиза комплексного соединения платины (IV), которые понижают выход и чистоту продукта.
Из сравнения с прототипом видно, что предлагаемый способ имеет следующие преимущества:
- упрощается синтез целевого продукта;
- не используются дополнительные реагенты;
- исключаются процессы упаривания, высаливания и центрифугирования;
- сокращается время получения целевого комплекса платины (IV);
В результате получаем повышение выхода и чистоты целевого продукта - транс-1,2-диаминциклогексантетрахлороплатины (IV), до 80%.
Примеры осуществления способа
Пример 1
К 4 г Н2[PtCl6]*6H2O (7.7·10-3 моль) добавляют 1.48 г транс-1,2-C6Н10(NH3)2*2Cl (7.9·10-3 моль) в 10 мл 3 М HCl при комнатной температуре. Через 20 мин образовавшийся осадок отфильтровывают, промывают ледяной уксусной кислотой и сушат в вакууме. К полученному промежуточному продукту - 3.84 г {транс-1,2-C6Н10(NH3)2}[PtCl6] (7.3·10-3 моль) прибавляют 0.63 г LiOH*H2O (1.5·10-2 моль) в 30 мл H2O. Реакцию ведут при комнатной температуре при перемешивании в течение 2 часов до изменения цвета раствора над осадком с красного в бледно-желтый. При этом процесс прекращают, а осадок отфильтровывают, промывают спиртом и сушат. Полученное соединение кристаллизуется в виде кристаллогидрата - [Pt{транс-1,2-C6Н10(NH2)2}Cl4]*1/3H2O, согласно данным рентгеноструктурного анализа (Khokhar A.R., Xu Q., Al-Baker S. // J. Inorg. Biochem. 1993. V.52. i.1. p.51-58). Количество целевого продукта 2.82 г, что составляет 80% от теоретического. Целевой продукт охарактеризован следующими методами: элементным анализом, РФА, ИК-спектроскопией и УФ-спектрофотометрией. Примеси других комплексных соединений платины (IV-II) не обнаружены.
Пример 2.
К 2 г Н2[PtCl6]*6H2O (3.85·10-3 моль) добавляют 0.74 г транс-1,2-C6Н10(NH3)2*2Cl (3.95·10-3 моль) в 5 мл 3 М HCl при комнатной температуре. Через 20 мин образовавшийся осадок отфильтровывают, промывают ледяной уксусной кислотой и сушат в вакууме. К полученному продукту - 1.92 г {транс-1,2-C6Н10(NH3)2}[PtCl6] (3.7·10-3 моль) прибавляют 15 мл H2O. Реакцию ведут при комнатной температуре при перемешивании. Через 2 часа процесс прекращают, а осадок отфильтровывают, промывают спиртом и сушат. Выход целевого продукта - 48% от теоретического (0.85 г). Целевой продукт выделен в виде кристаллической фазы [Pt{транс-1,2-C6Н10(NH2)2}Cl4]*1/3H2O, которая охарактеризована элементным анализом, РФА, ИК-спектроскопией и УФ-спектрофотометрией. Примеси других комплексных соединений платины (IV-II) не обнаружены.
Пример 3.
К 6 г Н2[PtCl6]*6H2O (1.15·10-2 моль) добавляют 2.22 г транс-1,2-C6Н10(NH3)2*2Cl (1.2·10-2 моль) в 15 мл 3 М HCl при комнатной температуре. Через 20 мин образовавшийся осадок отфильтровывают, промывают ледяной уксусной кислотой и сушат в вакууме. К полученному продукту - 5.76 г {транс-1,2-C6Н10(NH3)2}[PtCl6] (1.1·10-2 моль) прибавляют 1.26 г LiOH*H2O (3.0·10-2 моль) в 45 мл H2O. Реакцию ведут при комнатной температуре при перемешивании. Через 2 часа, когда раствор над осадком становится бледно-желтым, процесс прекращают, а осадок отфильтровывают, промывают спиртом и сушат. Выход целевого продукта 58% от теоретического (3.05 г). Продукт - [Pt{транс-1,2-C6H10(NH2)2}Cl4]*1/3H2O охарактеризован следующими методами: элементным анализом, РФА, ИК-спектроскопией и УФ-спектрофотометрией. Примеси других комплексных соединений платины (IV-II) не обнаружены.
В примере 1 приведены оптимальные условия получения комплексного соединения транс-1,2-диаминциклогексантетрахлороплатины (IV), как по концентрации платины в растворе, так и по чистоте основного продукта. Добавление соляной кислоты предотвращает гидролиз {транс-1,2-C6H10(NH3)2}[PtCl6] и не дает протекания реакции аминирования при получении промежуточного продукта на первой стадии. Гидроксид лития необходим для нейтрализации соляной кислоты, выделяющейся при гидролизе {транс-1,2-C6H10(NH3)2}[PtCl6], для того, чтобы процесс аминирования проходил полностью и выход целевого продукта повышался. Если нейтрализацию гидроксидом лития не проводить, то в результате протекания реакции аминирования pH раствора будет понижаться, что приведет к протонированию транс-1,2-диаминциклогексана и скорость получения целевого продукта будет падать, а выход будет снижаться (пример 2). При увеличении концентрации гидроксида лития реализуется процесс щелочного гидролиза, и образуются гидролизованные формы комплексов платины (IV), а количество основного продукта уменьшается (пример 3).
Таким образом, данный способ позволяет получить соединение транс-1,2-диаминциклогексантетрахлороплатины (IV) с большим выходом и без примесей (пример 1).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ЦИКЛОГЕКСАН-ТРАНС-1,2-d,l-ДИАМИНОТЕТРАХЛОРИДА ПЛАТИНЫ (IV) | 2014 |
|
RU2568438C1 |
| СПОСОБ ПОЛУЧЕНИЯ ТРАНС-ДИХЛОРОМЕТИЛАМИНЭТИЛАМИНПЛАТИНЫ (II) | 2011 |
|
RU2464232C1 |
| СПОСОБ ПОЛУЧЕНИЯ ТРАНС-ДИХЛОРОМЕТИЛАМИНИЗОПРОПИЛАМИНПЛАТИНЫ (II) | 2011 |
|
RU2464231C2 |
| СПОСОБ ПОЛУЧЕНИЯ ТРИХЛОРОАММИНПЛАТИНАТА (II) КАЛИЯ ИЛИ АММОНИЯ ИЗ ТЕТРАХЛОРОПЛАТИНАТА (II) КАЛИЯ | 2006 |
|
RU2323886C2 |
| СПОСОБ ПОЛУЧЕНИЯ 2,7-БИС[2-(ДИЭТИЛАМИНО)ЭТОКСИ]ФЛОУРЕНОНА ДИГИДРОХЛОРИДА | 1994 |
|
RU2076097C1 |
| СПОСОБ ПОЛУЧЕНИЯ ЦИС-ДИХЛОРОДИИЗОПРОПИЛАМИНПЛАТИНЫ(II) | 2007 |
|
RU2360866C1 |
| СПОСОБ ПОЛУЧЕНИЯ ДИГИДРОХЛОРИДА 1-(2,3,4-ТРИМЕТОКСИБЕНЗИЛ)ПИПЕРАЗИНА | 2007 |
|
RU2340606C1 |
| ИНГИБИТОРЫ ТРОМБИНА И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ИХ ПОЛУЧЕНИЯ | 1995 |
|
RU2172741C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЦИС-ДИХЛОРОАММИНИЗОПРОПИЛАМИНПЛАТИНЫ (II) | 2006 |
|
RU2309158C1 |
| СПОСОБ ОЧИСТКИ ЦИС-ДИХЛОРОАММИНИЗОПРОПИЛАМИНПЛАТИНЫ (II) | 1999 |
|
RU2186068C2 |
Изобретение относится к области органической химии и фармацевтики и касается способа получения противоопухолевого препарата II поколения, а именно транс-1,2-диаминциклогексантетрахлороплатины (IV), заключающегося в том, что платинохлористоводородную кислоту подвергают взаимодействию с транс-1,2-диаминциклогексаном дигидрохлоридом в водном растворе соляной кислоты с последующим фильтрованием, отделением осадка, растворением его в воде и добавлением гироксида лития. Выход составляет 80% от теоретического. 3 пр.
Способ получения транс-1,2-диаминциклогексантетрахлороплатины (IV) из водного раствора с использованием транс-1,2-диаминциклогексана дигидрохлорида, отличающийся тем, что платинохлористоводородную кислоту подвергают взаимодействию с транс-1,2-диаминциклогексаном дигидрохлоридом в водном растворе соляной кислоты при комнатной температуре, образующийся осадок отфильтровывают, промывают ледяной уксусной кислотой, сушат и растворяют в воде, добавляют 2 эквивалента гидрохлорида лития по отношению к исходному комплексу платины и процесс ведут при комнатной температуре с последующим выделением целевого продукта фильтрованием.
| ТРЕХПЛАТИНОВЫЕ КОМПЛЕКСЫ PT(II), СПОСОБ ИХ ПОЛУЧЕНИЯ | 1994 |
|
RU2140422C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОТИВООПУХОЛЕВОГО КОМПЛЕКСА ПЛАТИНЫ | 1988 |
|
RU2007413C1 |
| КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ ПЛАТИНЫ (II), СПОСОБ ИХ ПОЛУЧЕНИЯ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 1991 |
|
RU2067099C1 |

