Область изобретения
Настоящее изобретение относится к фармацевтическому средству, в частности к новому полипептидному соединению, обладающему противогрибковой активностью, или его фармацевтически приемлемой соли.
Предпосылки изобретения
Инфекции, вызываемые грибами, подразделяются на поверхностные дерматологические грибковые заболевания (Trichophyton и подобные), вызываемые кожной инфекцией, и глубокий микоз (Candida и Aspergillus и подобные), вызываемые респираторной или оральной инфекцией. Глубокий микоз развивается у пациентов, имеющих различные факторы риска, такие как иммунодефицит или введение противораковых лекарственных средств, и в частых случаях было обнаружено, что если он развивается, он не поддается лечению или имеет плохой прогноз. В частности, когда развивается инвазивный легочный аспергиллез, смертность составляет 58% от общего количества случаев заболевания, в частности 87% у пациентов с трансплантацией костного мозга. Однако существует несколько эффективных терапевтических подходов или лекарственных средств, при этом показатель эффективности вориконазола или AmBisome, которые являются стандартными лекарственными средствами, является низким на уровне 60% или ниже. Поэтому необходимо новое противогрибковое средство, обладающее сильной активностью.
Некоторые полипептидные соединения, обладающие противогрибковой активностью, являются известными (например, Патентные документы 1-7), и Патентный документ 1 раскрывает следующее соединение.

(В представленной формуле, R1 представляет собой арил, который может быть замещен.)
Патентный документ 1; Печатное издание Международной Публикации WO2004/113368
Патентный документ 2; Печатное издание Международной Публикации WO01/60846
Патентный документ 3; Печатное издание Международной Публикации WO00/64927
Патентный документ 4; Печатное издание Международной Публикации WO99/40108
Патентный документ 5; Печатное издание Международной Публикации WO03/068807
Патентный документ 6; Патент США № US5569646
Патентный документ 7; Печатное издание Международной Публикации WO2005/005463
Раскрытие изобретения
Задача, решаемая настоящим изобретением
Задача настоящего изобретения состоит в обеспечении нового соединения, обладающего антибактериальной активностью (в частности противогрибковой активностью).
Средства для решения задачи
В результате глубоких исследований, направленных на получение нового полипептидного соединения, обладающего противогрибковой активностью, авторами настоящего изобретения было обнаружено, что соединение формулы (I), где специфическая кольцевая группа является связанной через атом углерода с атомом азота участка связывания боковой цепи в 9-положении полипептидого кольца, демонстрирует отличную противогрибковую активность, что составило настоящее изобретение.
Таким образом, настоящее изобретение относится к соединению представленной ниже формулы (I) или его фармацевтически приемлемой соли.

где символы в формуле имеют следующие значения:
R1 представляет собой -CO-NR5R6, -R00-NR5R6, -CO-(азотсодержащий гетероцикл) или -NH-R00-NR5R6,
R5 и R6 являются одинаковыми или отличными друг от друга и представляют собой H, R0, RA или RB,
RA представляет собой низший алкил, который может быть замещен одной-тремя гидроксильными группами,
RB представляет собой низший алкил, который может быть замещен одной-тремя группами, выбранными из -NH2, -NHR0 и -N(R0)2,
R2 представляет собой H, -S(O)2-OH, R0, RA, RB или -R00-NH-R00-OH,
R4 представляет собой H или -OH,
R7 в каждом случае имеет одинаковое или отличное друг от друга значение и представляет собой H, R0 или -R00-OR0,
R9 представляет собой простую связь,
R13 и R14 являются одинаковыми или отличными друг от друга и представляют собой H или R0,
кольцо Е представляет собой группу, представленную следующими формулами (II), (III), (IV), (V) или (XVIII):

где X представляет собой CH или N,
R8 в каждом случае имеет одинаковое или отличное друг от друга значение и представляет собой H, R0, -NH2, -N(R0)2, -NHR0, -OH или -R00-OR0,
R10 представляет собой H, R0, -NH2, -N(R0)2, -NHR0, -OH или -R00-OR0,
кольцо А представляет собой азотсодержащий гетероцикл или фенил,
R3 представляет собой группу, представленную следующими формулами (VI), (VII), (VIII) или (XIX):

где кольцо В и кольцо D являются одинаковыми или отличными друг от друга и представляют собой арил, который может быть замещен галогеном,
кольцо С представляет собой азотсодержащий гетероцикл,
R11 и R12 являются одинаковыми или отличными друг от друга и представляют собой группу, выбранную из H, R0, циклоалкила, -(циклоалкил)-R0, -R00-(циклоалкил), -OR0, -O-(циклоалкил), -O-(C2-10 алкилен)-OR0, -O-R00-(циклоалкил), -(азотсодержащий гетероцикл)-R00-(циклоалкил), -(азотсодержащий гетероцикл)-O-(циклоалкил) и -(азотсодержащий гетероцикл)-O-R00-(циклоалкил),
R0 представляет собой низший алкил, и
R00 представляет собой низший алкилен.
Настоящее изобретение, кроме того, относится к фармацевтической композиции, включающей соединение формулы (I) или его фармацевтически приемлемую соль и фармацевтически приемлемый эксципиент, и к фармацевтической композиции для предупреждения или лечения грибковой инфекции, включающей соединение формулы (I) или его фармацевтически приемлемую соль. Кроме того, настоящее изобретение относится к способу предупреждения и/или лечения грибковой инфекции у человека или животного (в частности, млекопитающего).
Кроме того, настоящее изобретение относится к применению соединения формулы (I) или его фармацевтически приемлемой соли для получения средства для предупреждения или лечения грибковой инфекции, и к способу предупреждения или лечения грибковой инфекции, включающему введение пациенту эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
Эффект настоящего изобретения
Соединение формулы (I) обладает противогрибковой активностью и поэтому является полезным в качестве средства для предупреждения и/или лечения грибковых инфекций.
Лучший способ осуществления изобретения
Изобретение описано подробно ниже.
В настоящем описании, “низший алкил" представляет собой линейный или разветвленный алкил, содержащий от 1 до 6 атомов углерода (далее сокращенно указан как C1-6), в частности метильные, этильные, н-пропильные, изопропильные, н-бутильные, изобутильные, втор-бутильные, трет-бутильные, н-пентильные, н-гексильные группы или подобные. В одном варианте воплощения он представляет собой C1-3 алкил, и в другом варианте воплощения он представляет собой метил, этил или изопропил.
“Низший алкилен" означает двухвалентную группу (C1-6 алкилен), образованную путем удаления произвольного атома водорода из “низшего алкила", описанного выше. В одном варианте воплощения он представляет собой C1-4 алкилен, а в другом варианте воплощения он представляет собой метилен или этилен.
“C2-10 алкилен" представляет собой линейный или разветвленный алкилен, содержащий от 2 до 10 атомов углерода. В одном варианте воплощения он представляет собой C5-6 алкилен, а в другом варианте воплощения он представляет собой пентаметилен или гексаметилен.
“Циклоалкил" означает C3-8 циклический алкил, который может быть связан мостиковой связью. В одном варианте воплощения он представляет собой C3-6 циклический алкил, а в другом варианте воплощения он представляет собой циклопропил, циклобутил, циклопентил или циклогексил.
“Галоген" означает F, Cl, Br или I.
“Арил" представляет собой C6-14 моноциклическую-трициклическую ароматическую углеводородную кольцевую группу и включает частично гидрированную кольцевую группу. В частности, он представляет собой фенильные, нафтильные, 5-тетрагидронафтильные, 1-инданильные группы или подобные. В одном варианте воплощения он представляет собой фенил и нафтил, а в другом варианте воплощения он представляет собой фенил.
“Гетероциклическая" группа означает кольцевую группу, включающую i) моноциклическое 3-8-членное, предпочтительно 5-7-членное гетероциклическое кольцо, содержащее от 1 до 4 гетероатомов, выбранных из кислорода, серы и азота, и ii) бициклическое или трициклическое гетероциклическое кольцо, содержащее от 1 до 5 гетероатомов, выбранных из кислорода, серы и азота, где указанное моноциклическое гетероциклическое кольцо является конденсированным с одним или двумя кольцами, выбранными из группы, включающей моноциклическое гетероциклическое кольцо, бензольное кольцо, C5-8 циклоалкан и C5-8 циклоалкен. Кольцевой атом серы или азота может быть окислен с образованием оксида или диоксида.
Предпочтительными примерами "гетероциклической" группы являются следующие группы.
(1) Моноциклическая насыщенная гетероциклическая группа
i) содержащая от 1 до 4 атомов азота, в частности азепанильные, диазепанильные, азиридинильные, азетидинильные, пирролидинильные, имидазолидинильные, пиперидинильные, пиразолидинильные, пиперазинильные группы или подобные;
ii) содержащая от 1 до 3 атомов азота и от 1 до 2 атомов серы и/или от 1 до 2 атомов кислорода, в частности тиоморфолинильные, тиазолидинильные, изотиазолидинильные, оксазолидинильные, морфолинильные группы или подобные;
iii) содержащие от 1 до 2 атомов серы, в частности тетрагидротинильные группы или подобные;
iv) содержащие от 1 до 2 атомов серы и от 1 до 2 атомов кислорода, в частности оксатиолановые группы или подобные;
v) содержащие от 1 до 2 атомов кислорода, в частности оксиранильные диоксоланильные, оксоланильные, тетрагидропиранильные, 1,4-диоксанильные группы или подобные.
(2) Моноциклическая ненасыщенная гетероциклическая группа
i) содержащая от 1 до 4 атомов азота, в частности пирролильные, имидазолильные, пиразолильные, пиридильные, дигидропиридильные, пиримидинильные, пиразинильные, пиридазинильные, триазолильные, тетразолильные, дигидротриазинильные, азепинильные группы или подобные;
ii) содержащая от 1 до 3 атомов азота и от 1 до 2 атомов серы и/или от 1 до 2 атомов кислорода, в частности тиазолильные, изотиазолильные, тиадиазолильные, дигидротиазинильные, оксазолильные, изоксазолильные, оксадиазолильные, оксазинильные группы или подобные;
iii) содержащая от 1 до 2 атомов серы, в частности тиенильные, тиепинильные, дигидродитиинильные, дигидродитионильные группы или подобные;
iv) содержащая от 1 до 2 атомов серы и от 1 до 2 атомов кислорода, в частности дигидрооксатиинильные группы или подобные;
v) содержащая от 1 до 2 атомов кислорода, в частности фурильные, пиранильные, оксепинильные, диоксолильные группы или подобные.
(3) Конденсированные полициклические насыщенные гетероциклические группы
i) содержащие от 1 до 5 атомов азота, в частности хинуклидиновые, 7-азабицикло[2.2.1]гептильные, 3-азабицикло[3.2.2]нонанильные группы или подобные;
ii) содержащие от 1 до 4 атомов азота и от 1 до 3 атомов серы и/или от 1 до 3 атомов кислорода, в частности тритиадиазаинденильные диоксолоимидазолидинильные группы или подобные;
iii) содержащие от 1 до 3 атомов серы и/или от 1 до 3 атомов кислорода, в частности 2,6-диоксабицикло[3.2.2]окт-7-ильные группы или подобные.
(4) Конденсированные полициклические ненасыщенные гетероциклические группы
i) содержащие от 1 до 5 атомов азота, в частности индолильные, изоиндолильные, индолинильные, индолидинильные, бензимидазолильные, хинолильные, тетрагидрохинолильные, изохинолильные, тетрагидроизохинолильные, индазолильные, имидазопиридильные, бензотриазолильные, тетразолопиридазинильные, карбазолильные, хиноксалинильные, дигидроиндазолильные, бензопиримидинильные, нафтилидинильные, хиназолинильные, циннолинильные группы или подобные;
ii) содержащие от 1 до 4 атомов азота и от 1 до 3 атомов серы и/или от 1 до 3 атомов кислорода, в частности бензотиазолильные, дигидробензотиазолильные, бензотиадиазолильные, имидазотиазолильные, имидазотиадиазолильные, бензоксазолильные, бензоксадиазолильные группы или подобные;
iii) содержащие от 1 до 3 атомов серы, в частности бензотиенильные, бензодитиинильные группы или подобные;
iv) содержащие от 1 до 3 атомов серы и от 1 до 3 атомов кислорода, в частности бензоксатиинильные, феноксазинильные группы или подобные;
v) содержащие от 1 до 3 атомов кислорода, в частности бензодиоксолильные, бензофуранильные, изобензофуранильные, хроменильные, бензодигидрофуранильные группы или подобные.
Эти гетероциклы представлены как одновалентные группы, но могут представлять собой двухвалентные или имеющие более высокую валентность группы в соответствии с положением связывания или количеством заместителей.
“Азотсодержащий гетероцикл" означает кольцо, содержащее один или несколько атомов азота в качестве атомов, составляющих кольцо, выбранное из указанных выше “гетероциклов", и, например, включает пиридил, пиримидинил, имидазотиазолил, тиадиазолил, пиперидинил, пиперазинил и подобные.
“Моноциклическое азотсодержащее гетероциклическое кольцо" означает моноциклическое кольцо, выбранное из указанных выше "азотсодержащих гетероциклов", и включает, например, пиридил, пиримидинил, тиадиазолил, пиперидил и подобные.
“Может быть замещен" означает “незамещенный" или “содержащий от 1 до 5 заместителей, которые являются одинаковыми или отличными друг от друга". Кроме того, когда содержится несколько заместителей, эти заместители могут быть одинаковыми или отличными друг от друга. Например, два R0 в -N(R0)2 могут быть отличными друг от друга.
Варианты воплощения настоящего изобретения представлены ниже.
(1) В одном варианте воплощения R1 представляет собой -CO-NR5R6 или -R00-NR5R6, в другом варианте воплощения он представляет собой -CO-NH2, -CH2-NH2, -CH2-NHRA или -CH2-NHRB, в следующем варианте воплощения он представляет собой -CO-NH2, -CO-NH-R00-NH2, -CH2-NH2 или -CH2-NH-CH(CH2OH)2, и в еще в одном варианте воплощения он представляет собой -CO-NH2, -CH2-NH2 или -CH2-NH-CH(CH2OH)2.
(2) В одном варианте воплощения R2 представляет собой -S(O)2-OH или R0, а в другом варианте воплощения он представляет собой -S(O)2-OH или метил.
(3) В одном варианте воплощения R4 представляет собой H.
(4) В одном варианте воплощения R7 представляет собой H или R0, в другом варианте воплощения он представляет собой H или метил, и в следующем варианте воплощения он представляет собой H.
(5) В одном варианте воплощения кольцо Е представляет собой группу, представленную следующей формулой (II).

В формуле (II), в одном варианте воплощения R8 представляет собой H, R0, -OH или -OR0, R10 представляет собой H, а в другом варианте воплощения R8 представляет собой H или R0, R10 представляет собой H, в следующем варианте воплощения R8 представляет собой H или метил и R10 представляет собой H. Кроме того, в одном варианте воплощения кольцо Е представляет собой циклогександиил, а в другом варианте воплощения оно представляет собой пиперидин-1,4-диил.
(6) В одном варианте воплощения X представляет собой CH, а в другом варианте воплощения он представляет собой N.
(7) В одном варианте воплощения кольцо А представляет собой моноциклическое азотсодержащее гетероциклическое кольцо, в другом варианте воплощения оно представляет собой пиридил, пиримидинил, тиадиазолил или имидазотиазолил, и в следующем варианте воплощения оно представляет собой тиадиазолил или имидазотиазолил.
(8) В одном варианте воплощения R3 представляет собой группу, представленную следующими формулами (VII) или (VIII).

в другом варианте воплощения он представляет собой группу, представленную формулой (VII), и в следующем варианте воплощения он представляет собой группу, представленную формулой (VIII).
(9) кольцо В и кольцо D являются одинаковыми или отличными друг от друга, в одном варианте воплощения представляют собой моноциклический арил, который может быть замещен галогеном, в другом варианте воплощения представляют собой моноциклический арил, и в следующем варианте воплощения представляют собой фенил.
(10) В одном варианте воплощения кольцо С представляет собой моноциклическое азотсодержащее гетероциклическое кольцо, в другом варианте воплощения оно представляет собой пиридил, пиримидинил или пиперидинил, в следующем варианте воплощения оно представляет собой пиримидинил или пиперидинил, и еще в одном варианте воплощения, вместе с R11 и R12, представляет собой группу, представленную следующими формулами (XX) или (XXI).

(11) В одном варианте воплощения любой из R11 и R12 представляет собой H или -OR0, а другой представляет собой группу, выбранную из циклоалкила, -R00-(циклоалкил), -O-(циклоалкил), -O-R00-(циклоалкил), -(азотсодержащий гетероцикл)-R00-(циклоалкил), -(азотсодержащий гетероцикл)-O-(циклоалкил) и -(азотсодержащий гетероцикл)-O-R00-(циклоалкил), в другом варианте воплощения любой из R11 и R12 представляет собой H или -OR0, а другой представляет собой группу, выбранную из циклоалкила, -O-(циклоалкил), -O-R00-(циклоалкил) и -(азотсодержащий гетероцикл)-O-(циклоалкил), и в следующем варианте воплощения любой из R11 и R12 представляет собой H или -O-(метил), а другой представляет собой группу, выбранную из циклогексила, -O-(циклогексил), -O-CH2-(циклобутил) и 4-(циклогексилокси)пиперидин-1-ила. Кроме того, в другом варианте воплощения R11 и R12 являются одинаковыми или отличными друг от друга и каждый представляет собой H, циклоалкил, -O-R0, -O-(циклоалкил), -(азотсодержащий гетероцикл)-O-(циклоалкил) или -(азотсодержащий гетероцикл)-O-R00-(циклоалкил).
Другой предпочтительный вариант воплощения представляет собой сочетание любых двух или более групп, описанных выше в (1)-(11), в частности включает соединения, представленные ниже в (12)-(16).
(12) Соединение, где R1 представляет собой -CO-NR5R6 или -R00-NR5R6, R2 представляет собой -S(O)2-OH или R0, R4 представляет собой H, R7 представляет собой H или R0, R9 представляет собой простую связь, кольцо Е представляет собой группу, представленную формулой (II), кольцо А представляет собой моноциклическое азотсодержащее гетероциклическое кольцо, R8 представляет собой H или R0, R3 представляет собой группу, представленную формулой (VII) или (VIII), кольцо В и кольцо D являются одинаковыми или отличными друг от друга, и каждый представляет собой моноциклический арил, кольцо С представляет собой моноциклическое азотсодержащее гетероциклическое кольцо, R11 и R12 являются одинаковыми или отличными друг от друга, и каждый представляет собой H, циклоалкил, -O-R0, -O-(циклоалкил), -(азотсодержащий гетероцикл)-O-(циклоалкил) или -(азотсодержащий гетероцикл)-O-R00-(циклоалкил).
(13) Соединение, где R1 представляет собой -CO-NR5R6 или -R00-NR5R6, R2 представляет собой -S(O)2-OH или R0, R4 представляет собой H, R7 представляет собой H или R0, кольцо Е представляет собой группу, представленную формулой (II), R8 представляет собой H или R0, R10 представляет собой H, кольцо А представляет собой моноциклическое азотсодержащее гетероциклическое кольцо, R3 представляет собой группу, представленную формулой (VII) или (VIII), кольцо В и кольцо D являются одинаковыми или отличными друг от друга, и каждый представляет собой моноциклический арил, который может быть замещен галогеном, кольцо С представляет собой моноциклическое азотсодержащее гетероциклическое кольцо, любой из R11 и R12 представляет собой H или -OR0, а другой представляет собой группу, выбранную из циклоалкила, -R00-(циклоалкил), -O-(циклоалкил), -O-R00-(циклоалкил), -(азотсодержащий гетероцикл)-R00-(циклоалкил), -(азотсодержащий гетероцикл)-O-(циклоалкил) и -(азотсодержащий гетероцикл)-O-R00-(циклоалкил).
(14) Соединение, где R1 представляет собой -CO-NH2, -CH2-NH2, -CH2-NHRA или -CH2-NHRB, R2 представляет собой -S(O)2-OH или метил, R4 представляет собой Н, R7 представляет собой Н или метил, кольцо Е представляет собой группу, представленную формулой (II), Х представляет собой СН, R8 представляет собой Н или метил, R10 представляет собой Н, кольцо А представляет собой пиридил, пиримидинил, тиадиазолил или имидазотиазолил, R3 представляет собой группу, представленную формулой (VII), кольцо В и кольцо D представляют собой моноциклический арил, кольцо С представляет собой пиридил, пиримидинил или пиперидинил, любой из R11 и R12 представляет собой Н или -OR0, а другой представляет собой группу, выбранную из циклоалкила, -О-(циклоалкил), -O-R00-(циклоалкил) и -(азотсодержащий гетероцикл)-О-(циклоалкил).
(15) Соединение, где R1 представляет собой -CO-NH2, -CH2-NH2 или -CH2-NH-CH(CH2OH)2, R2 представляет собой -S(O)2-OH или метил, R4 представляет собой Н, R7 представляет собой Н, кольцо Е представляет собой циклогександиил, кольцо А представляет собой тиадиазолил или имидазотиазолил, R3 представляет собой группу, представленную формулой (VII), кольцо В и кольцо D представляют собой фенил, кольцо С, вместе с R11 и R12, представляет собой группу, представленную формулой (XX) или (XXI), любой из R11 и R12 представляет собой Н или -О-(метил), а другой представляет собой группу, выбранную из циклогексила, -О-(циклогексил), -О-СН2-(циклобутил) и 4-(циклогексилокси)пиперидин-1-ила.
(16) Соединение, выбранное из следующих:
(3R)-3-{(2R,6S,11R,14aS,15S,16S,20S,23S,25aS)-9-[({транс-4-[5-(4-{2-[4-(циклопентилметил)пиперазин-1-ил]пиримидин-5-ил}фенил)-1,3,4-тиадиазол-2-ил]циклогексил}метил)амино]-2,11,15-тригидрокси-6-[(1R)-1-гидроксиэтил]-23-[(1R)-1-гидрокси-2-(4-гидрокси-3-метоксифенил)этил]-16-метил-5,8,14,19,22,25-гексаоксотетракозагидро-1H-дипирроло[2,1-c:2',1'-1][1,4,7,10,13,16]гексаазациклогеникозин-20-ил}-3-гидроксипропанамид,
(2R,6S,9S,11R,14aS,15S,16S,20S,23S,25aS)-9-{[(транс-4-{5-[4-(4-циклогексил-4-метоксипиперидин-1-ил)фенил]-1,3,4-тиадиазол-2-ил}циклогексил)метил]амино}-20-{(1R)-3-[(1,3-дигидроксиприпан-2-ил)амино]-1-гидроксипропил}-2,11,15-тригидрокси-6-[(1R)-1-гидроксиэтил]-23-[(1R)-1-гидрокси-2-(4-гидрокси-3-метоксифенил)этил]-16-метилгексадекагидро-1Н-дипирроло[2,1-с:2',1'-1][1,4,7,10,13,16]-гексаазациклогеникозин-5,8,14,19,22,25(9Н,25аН)-гексон, 2-гидрокси-5-{(2R)-2-гидрокси-2-[(2R,11R,14aS,15S,16S,20S,23S,25aS)-2,11,15-тригидрокси-6-[(1R)-1-гидроксиэтил]-20-[(1R)-1-гидрокси-3-{[2-гидрокси-1-(гидроксиметил)этил]амино}пропил]-9-({[транс-4-(2-{4-[(6-метоксигексил)окси]фенил}имидазо[2,1-b][I,3,4]тиадиазол-6-ил)циклогексил]метил}амино)-16-метил-5,8,14,19,22,25-гексаоксотетракозагидро-1Н-дипирроло[2,1-c:2',1'-1][1,4,7,10,13,16]гексаазациклогеникозин-23-ил]этил}фенил гидросульфат,
(2R,6S,11R,14aS,15S,16S,20S,23S,25aS)-20-[(1R)-3-амино-1-гидроксипропил]-9-{[(транс-4-{5-[4-(4-циклогексил-4-метоксипиперидин-1-ил)фенил]-1,3,4-тиадиазол-2-ил}циклогексил)метил]амино}-2,11,15-тригидрокси-6-[(1R)-1-гидроксиэтил]-23-[(1R)-1-гидрокси-2-(4-гидрокси-3-метоксифенил)этил]-16-метилгексадекагидро-1Н-дипирроло[2,1-с:2',1'-1][1,4,7,10,13,16]гексаазациклогеникозин-5,8,14,19,22,25(9Н,25аН)-гексон,
5-[(2R)-2-{(2R,6S,9S,11R,14aS,15S,16S,20S,23S,25aS)-20-[(1R)-3-амино-1-гидрокси-3-оксопропил]-9-[({транс-4-[5-(4-{2-[4-(циклогексилокси)пиперидин-1-ил]пиримидин-5-ил}фенил)-1,3,4-тиадиазол-2-ил]циклогексил}метил)амино]-2,11,15-тригидрокси-6-[(1R)-1-гидроксиэтил]-16-метил-5,8,14,19,22,25-гексаоксотетракозагидро-1Н-дипирроло[2,1-с:2',1'-1][1,4,7,10,13,16]гексаазациклогеникозин-23-ил}-2-гидроксиэтил]-2-гидроксифенил гидросульфат,
5-[(2R)-2-{(2R,6S,9S,11R,14aS,15S,16S,20S,23S,25aS)-20-{(1R)-3-[(2-аминоэтил)амино]-1-гидрокси-3-оксопропил}-9-[({1-[5-(4-{2-[4-(циклогексилокси)пиперидин-1-ил]пиримидин-5-ил}фенил)-1,3,4-тиадиазол-2-ил]пиперидин-4-ил}метил)амино]-2,11,15-тригидрокси-6-[(1R)-1-гидроксиэтил]-16-метил-5,8,14,19,22,25-гексаоксотетракозагидро-1Н-дипирроло[2,1-c:2',1'-1][1,4,7,10,13,16]гексаазациклогеникозин-23-ил}-2-гидроксиэтил]-2-гидроксифенил гидросульфат и
5-[(2R)-2-{(2R,6S,11R,14aS,15S,16S,20S,23S,25aS)-9-({[транс-4-(5-{4-[4-(циклогексилокси)пиперидин-1-ил]фенил}-1,3,4-тиадиазол-2-ил)циклогексил]метил}амино)-2,11,15-тригидрокси-6-[(1R)-1-гидроксиэтил]-20-[(1R)-1-гидрокси-3-{[2-гидрокси-1-(гидроксиметил)этил]амино}пропил]-16-метил-5,8,14,19,22,25-гексаоксотетракозагидро-1Н-дипирроло[2,1-c:2',1'-1][1,4,7,10,13,16]гексаазациклогеникозин-23-ил-2-гидроксиэтил]-2-гидроксифенил гидросульфат или их фармацевтически приемлемая соль.
Соединение формулы (I) может в некоторых случаях существовать в форме других таутомеров или геометрических изомеров, в зависимости от типа заместителей. В настоящем описании соединение может быть описано только в одной изомерной форме, но настоящее изобретение включает такие изомеры, выделенные формы изомеров или их смесь.
Кроме того, соединение формулы (I) может в некоторых случаях иметь асимметрические атомы углерода или аксиальную асимметрию и, соответственно, может существовать в форме оптических изомеров, такой как (R)-форма, (S)-форма и подобные. Настоящее изобретение включает смесь и выделенную форму этих оптических изомеров.
Кроме того, фармацевтически приемлемые пролекарства соединения формулы (I) также включены в настоящее изобретение. Фармацевтически приемлемое пролекарство относится к соединению, содержащему группу, которая может быть преобразована в аминогруппу, -ОН, -СO2Н или подобные группы путем сольволиза или в физиологических условиях. Примеры группы для образования пролекарства включают группы, описанные в Prog. Med., 5, 2157-2161 (1985) или "Pharmaceutical Research and Development" (Hirokawa Publishing Company, 1990), vol.7, Drug Design, 163-198.
Кроме того, соединение формулы (I) может образовывать кислотно-аддитивную соль или соль с основанием, в зависимости типа заместителей, и соль включена в настоящее изобретение при условии, что она представляет собой фармацевтически приемлемую соль. В частности, примеры таких солей включают кислотно-аддитивные соли с неорганическими кислотами, такими как хлористоводородная кислота, бромистоводородная кислота, иодистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и подобные, и с органическими кислотами, такими как муравьиная кислота, уксусная кислота, пропионовая кислота, щавелевая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, малеиновая кислота, молочная кислота, яблочная кислота, винная кислота, лимонная кислота, метансульфоновая кислота, этансульфоновая кислота, п-толуолсульфоновая кислота, аспарагиновая кислота, глутаминовая кислота и подобные, и соли с неорганическими основаниями, такие как соли натрия, калия, магния, кальция, алюминия и подобные, и органическими основаниями, такими как метиламин, этиламин, этаноламин, лизин, орнитин и подобные, соли аммония и другие.
Кроме того, настоящее изобретение также включает различные гидраты или сольваты и полиморфизм соединения формулы (I) и его фармацевтически приемлемой соли. Кроме того, настоящее изобретение также включает соединение формулы (I), меченное различными радиоактивными изотопами или нерадиоактивными изотопами.
(Способы получения)
Соединение формулы (I) и его фармацевтически приемлемую соль можно получить путем применения различных известных способов синтеза, с использованием характеристик, основанных на их основных скелетах или типе заместителей. В настоящее время, в зависимости от типа функциональных групп, в некоторых случаях является эффективным, с точки зрения технологии получения, замещение функциональной группы подходящей защитной группой (группой, которая способна легко преобразовываться в функциональную группу), на стадиях от исходных веществ до промежуточных соединений. Примеры такой функциональной группы включают аминогруппу, гидроксильную группу, карбоксильную группу и подобные, и примеры защитной группы для таких групп включают защитные группы, описанные в “Protective Groups in Organic Synthesis (4th edition, 2007)”, edited by Greene and Wuts, и подобные, которые могут быть подходящим образом выбраны и использованы в зависимости от реакционных условий. В этих способах, желаемое соединение может быть получено путем введения защитной группы для осуществления реакции и затем, если это желательно, удаления защитной группы.
Кроме того, пролекарство соединения формулы (I) можно получить путем введения определенной группы на стадиях от исходных веществ до промежуточных соединений таким же образом, как для описанных выше защитных групп, или путем осуществления реакции с использованием полученного соединения формулы (I). Реакцию можно осуществить путем применения способа, известного специалистам в данной области, такого как общий способ этерификации, амидирования, дегидратации и подобные.
Ниже объясняются репрезентативные способы получения соединения формулы (I). Каждый из способов получения также можно осуществить со ссылкой на ссылочные документы, прилагаемые в настоящем описании. Кроме того, способы получения по настоящему изобретению не ограничиваются примерами, представленными ниже.
Способ получения 1

Этот способ получения представляет собой способ для получения соединения формулы (I) путем восстановительного аминирования соединения (IX) и соединения (X).
Реакцию осуществляют с использованием соединения (IX) и соединения (X) в эквивалентных количествах или любого из них в избыточном количестве, в растворителе, инертном к реакции, в присутствии восстановителя, при температуре в пределах от -45°C до нагревания при температуре кипения с обратным холодильником, предпочтительно от 0°C до комнатной температуры, обычно при перемешивании, в течение времени от 0,1 часа до 5 дней. Растворители конкретно не ограничены, но, например, включают спирты, такие как метанол и этанол, простые эфиры, такие как диэтиловый эфир, тетрагидрофуран (ТГФ), диоксан и диметоксиэтан (DME) или смеси из таких растворителей. Восстановители включают цианоборогидрид натрия, триацетоксиборогидрид натрия, борогидрид натрия и подобные. В некоторых случаях предпочтительным является осуществление реакции с использованием агентов дегидратации, таких как молекулярные сита, или в присутствии кислот, таких как уксусная кислота, хлористоводородная кислота, комплекс изопропоксида титана (IV). В зависимости от реакции, в случае, когда форму имина, которая образуется как промежуточное соединение в реакционной системе, можно выделять стабильным образом, реакцию восстановления можно осуществить отдельно, после получения формы имина. Кроме того, реакцию можно осуществить с использованием катализатора восстановления (например, палладия на углероде, никеля Ренея и подобных) в присутствии или в отсутствие кислоты, такой как уксусная кислота и хлористоводородная кислота, в растворителе, таком как метанол, этанол и этилацетат, вместо обработки восстановителем. В этом случае, реакцию предпочтительно осуществляют в атмосфере водорода, при давлении от нормального до 50 атм, при температуре в пределах от 0°C до нагревания. (Ссылка: "Comprehensive Organic Functional Group Transformations II", vol.2, Elsevier Pergamon, 2005, edited by A. R. Katritzky and R. J. K. Taylor; "Jikken Kagaku Koza (Courses in Experimental Chemistry) (5th edition)", edited by The Chemical Society of Japan, vol. 14(2005) (Maruzen)).
Кроме того, в случае, когда защитная группа необходима в R1 соединения (IX) при осуществлении реакции, подходящую защитную группу вводят заранее, и защитную группу можно удалить после завершения реакции.
(Синтез исходных веществ)
Способ получения исходных веществ 1:

Соединение (X-a) можно получить, подвергая соединение (XI) восстановлению до спирта с получением соединения (XII), с последующим окислением до альдегида. Альтернативно, его можно получить с использованием карбоновой кислоты (XIII) и амида Вайнреба (XIV).
Способ получения исходных веществ 2:

(В описанной формуле, L представляет собой удаляемую группу, например галоген. То же применимо к описанному ниже).
Соединение (X-b) можно получить путем окисления соединения (XVII), которое получают путем нуклеофильного замещения соединения (XV) и соединения (XVI).
Исходные соединения, используемые в получении соединений Примеров настоящей заявки, которые представлены общей формулой (IX), можно получить таким же способом, как описано в WO2005/005463, WO03/068807, WO00/064927, WO99/040108 и EP0431350.
Соединение формулы (I) выделяют и очищают в виде свободного соединения, его фармацевтически приемлемых солей, гидратов, сольватов или полиморфизма. Фармацевтически приемлемую соль соединения формулы (I) также можно получить в соответствии с традиционным способом осуществления реакции солеобразования.
Выделение и очистку осуществляют путем осуществления общих химических процедур, таких как экстракция, фракционированная кристаллизация, различные типы фракционной хроматографии и подобные.
Различные изомеры можно разделить путем выбора подходящего исходного соединения или с использованием различия в физико-химических свойствах между изомерами. Например, оптический изомер может быть преобразован в стереохимически чистый изомер при помощи общих способов оптического разделения (например, фракционированной кристаллизации для индукции диастереомерных солей с использованием оптически активных оснований или кислот, хроматографии с использованием хиральной колонки, и т.д. и т.п.). Кроме того, изомеры также можно получить из подходящего оптически активного исходного соединения.
Фармакологическую активность соединения формулы (I) подтверждали при помощи Примера испытания 1 и Примера испытания 2, описанных ниже.
Пример испытания 1: Определение подверженности инфекциям C. albicans (Candida albicans) и A. fumigatus (Aspergillus fumigatus)
С использованием способа микроразведения, где использовали человеческую сыворотку, забуференную буфером 20 мМ-HEPES (pH 7,3) в качестве среды для испытания, определяли MIC (минимальная ингибирующая концентрация) в человеческой сыворотке. Суспензию посевной культуры 106 клеток/мл получали с использованием гемоцитометрической процедуры с получением посевной культуры размером от 5×103 до 2×104 клеток/мл. Микропланшеты инкубировали при 37°C в течение 24 часов в атмосфере 5% CO2. Значения MIC для C. albicans определяли как самую низкую концентрацию, при которой имело место полное ингибирование роста, и значения MIC для A. fumigatus определяли как самую низкую концентрацию, при которой имело место существенное ингибирование роста по сравнению с контролем роста. Значения представлены в виде среднего геометрического значения MIC для трех из клинических клеточных линий.
Пример испытания 2: Эффект на мышиную модель респираторной инфекции
4-недельных самцов мышей ICR использовали таким образом, чтобы в группе было по восемь мышей. 200 мг/кг циклофосфамида интраперитонеально вводили мышам за 4 дня до инфицирования и через 1 день после инфицирования. Жидкий препарат конидий (0,05 мл: приблизительно 2,2×106) A. fumigatus 20030, суспендированный в физиологическом солевом растворе, инокулировали из дыхательных путей мышей, чтобы вызвать респираторную инфекцию. Соединение вводили внутривенно в течение периода от 2 часов до 4 дней после инфицирования. Случаи выживания и гибели мышей наблюдали в течение 10-дневного периода начиная со следующего дня.
В соответствии с Примером испытания 1 и Примером испытания 2 измеряли значения MIC в человеческой сыворотке для репрезентативных соединений по настоящему изобретению и значения ED50 для мышиной модели респираторной инфекции, соответственно. Пр. означает номер примера соединения, Ср. означает сравнительные соединения, представленные Примером 3 (продукт) WO2004/113368 (то же применимо к описанному ниже).
В результате каждого из описанных выше испытаний было показано, что соединение формулы (I) обладает антибактериальной активностью (в частности противогрибковой активностью), и было подтверждено, что этот эффект превосходит эффект традиционно используемых соединений, представленных сравнительными соединениями. Учитывая вышесказанное, соединение формулы (I) можно использовать в качестве средства для лечения различных грибковых инфекций, в частности инфекционных заболеваний, вызываемых следующими грибами:
Acremonium;
Absidia (например, Absidia corymbifera и подобные);
Aspergillus (например, Aspergillus clavatus, Aspergillus flavus, Aspergillus fumigatus, Aspergillus nidulans, Aspergillus niger, Aspergillus terreus, Aspergillus versicolor и подобные);
Blastomyces (например, Blastomyces dermatitidis и подобные);
Candida (например, Candida albicans, Candida glabrata, Candida guilliermondii, Candida kefyr, Candida krusei, Candida parapsilosis, Candida stellatoidea, Candida tropicalis, Candida utilis и подобные);
Cladosporium (например, Cladosporium tricoides и подобные);
Coccidioides (например, Coccidioides immitis и подобные);
Cryptococcus (например, Cryptococcus neoformans и подобные);
Cunninghamella (например, Cunninghamella elegans и подобные);
Dermatophyte;
Exophiala (например, Exophiala dermatitidis, Exophiala spinifera и подобные);
Epidermophyton (например, Epidermophyton floccosum и подобные);
Fonsecaea (например, Fonsecaea pedrosoi и подобные);
Fusarium (например, Fusarium solani и подобные);
Geotrichum (например, Geotrichum candidum и подобные);
Histoplasma (например, Histoplasma capsulatum var. capsulatum и подобные);
Malassezia (например, Malassezia furfur и подобные);
Microsporum (например, Microsporum canis, Microsporum gypseum и подобные);
Mucor;
Paracoccidioides (например, Paracoccidioides brasiliensis и подобные);
Penicillium (например, Penicillium marneffei и подобные);
Phialophora;
Pneumocystis (например, Pneumocystis jiroveci и подобные);
Pseudallescheria (например, Pseudallescheria boydii и подобные);
Rhizopus (например, Rhizopus microsporus var. rhizopodiformis, Rhizopus oryzae и подобные);
Saccharomyces (например, Saccharomyces cerevisiae и подобные);
Scopulariopsis;
Sporothrix (например, Sporothrix schenckii и подобные);
Trichophyton (например, Trichophyton mentagrophytes, Trichophyton rubrum и подобные);
Trichosporon (например, Trichosporon asahii, Trichosporon cutaneum и подобные).
Хорошо известно, что перечисленные выше грибы вызывают различные инфекции кожи, глаз, волос, ногтей, слизистой оболочки рта, желудочно-кишечного тракта, бронхов, легких, эндокарда, головного мозга, оболочки головного мозга, мочеполовых органов, влагалища, полости рта, глазной области, инфекции, поражающие различные органы организма, почки, сердце, внешний слуховой проход, кости, носовую полость, параназальную полость, селезенку, печень, гиподерму, лимфатические каналы, желудочно-кишечный тракт, суставы, мышцы, сухожилия, интерстициальные плазменные клетки в легких, сосуды и т.п.
Поэтому соединение формулы (I) можно использовать для предупреждения и/или лечения различных инфекций, например дерматофитоза (например, трихофитоза и подобных), отрубевидного лишая, кандидоза, криптококкоза, геотрихоза, трихоспории, аспергиллеза, пенициллиоза, фузариоза, зигомикоза, споротрихоза, хромомикоза, кокцидиоидомикоза, гистоплазмоза, бластомикоза, паракокцидиоидомикоза, псевдоаллэшериаза, мицетомы, грибкового кератита, отомикоза, пневмоцистной пневмонии, гематогенной грибковой инфекции и подобных.
Препарат, включающий один, или два, или более типов соединения формулы (I) или его фармацевтически приемлемой соли в качестве активного ингредиента, можно получить в соответствии с общепринятым способом с использованием фармацевтического носителя, эксципиента или подобных веществ, которые обычно используют в фармацевтике.
Введение можно осуществить любыми средствами перорального введения через таблетки, пилюли, капсулы, гранулы, порошки, жидкие препараты или подобные, или парентерального введения через инъекции, такие как внутрисуставная, внутривенная, внутримышечная или другие, суппозитории, глазные капли, глазные мази, чрескожные жидкие препараты, мази, чрескожные пластыри, жидкие препараты для введения через слизистую оболочку, пластыри для введения через слизистую оболочку, препараты для ингаляций и подобные.
Что касается твердой композиции для перорального введения в соответствии с настоящим изобретением, используют таблетки, порошки, гранулы или подобные препараты. В такой твердой композиции один, или два, или более типов активных ингредиентов смешивают с, по меньшей мере, одним инертным эксципиентом, например лактозой, маннитом, глюкозой, гидроксипропилцеллюлозой, микрокристаллической целлюлозой, крахмалом, поливинилпирролидоном и/или алюмометасиликатом магния или подобными. В соответствии с традиционным способом, композиция может содержать инертные добавки, например смазывающее вещество, такое как стеарат магния, разрыхлитель, такой как натрий карбоксиметилкрахмал, стабилизатор и вещество, способствующее солюбилизации. Если это необходимо, на таблетки или пилюли может быть нанесено сахарное покрытие или пленочное гастро- или энетеросолюбильное покрытие.
Жидкая композиция для перорального введения включает фармацевтически приемлемые эмульсии, растворимые жидкие препараты, суспензии, сиропы, эликсиры или т.п., и содержит традиционно используемый инертный разбавитель, такой как очищенная вода или этанол. Помимо инертного разбавителя такая жидкая композиция может содержать адъювант, такой как солюбилизирующее вещество, смачивающее вещество и суспендирующее вещество, подсластитель, отдушку, ароматизатор и антисептик.
Инъекции для парентерального введения содержат стерильные водные или неводные растворимые жидкие препараты, суспензии или эмульсии. Водный растворитель включает, например, дистиллированную воду для инъекций или физиологический солевой раствор. Примеры неводного растворителя включают, например, пропиленгликоль, полиэтиленгликоль, растительные масла, такие как оливковое масло, спирты, такие как этанол, Полисорбат 80 (Japanese Pharmacopeia) и подобные. Такая композиция может дополнительно содержать агент тоничности, антисептик, увлажнитель, эмульгатор, диспергирующее вещество, стабилизатор или солюбилизирующее вещество. Их стерилизуют, например, путем фильтрования через удерживающий бактерии фильтр, смешивания с бактерицидом или облучения. Кроме того, такие препараты также можно использовать путем получения стерильной твердой композиции и ее растворения или суспендирования в стерильной воде или стерильном растворителе для инъекций перед использованием.
Средство для наружного применения включает мази, пластыри, кремы, желе, припарки, спреи, лосьоны, глазные капли, глазные мази и подобные. Такие средства содержат традиционно используемые основы для мазей, основы для лосьонов, водные или неводные жидкие препараты, суспензии, эмульсии и подобные. Примеры основы для мазей или основы для лосьонов включают полиэтиленгликоль, пропиленгликоль, белый вазелин, отбеленный пчелиный воск, полиоксиэтиленгидрированное касторовое масло, глицерилмоностеарат, стеариловый спирт, цетиловый спирт, лауромакроголь, сорбитансесквиолеат и подобные.
Что касается средств для введения через слизистую оболочку, таких как препарат для ингаляций, для введения через нос и подобные, используют средства в форме твердого, жидкого или полутвердого препарата, и их можно получить в соответствии с общеизвестным способом. Например, могут быть подходящим образом добавлены известный эксципиент, а также агент регулирования pH, антисептик, поверхностно-активное вещество, смазывающее вещество, стабилизатор, загуститель или подобные агенты. Для введения таких средств можно использовать подходящее устройство для ингаляции или вдувания. Например, соединение можно вводить отдельно или в виде сформулированной в порошок смеси, или в виде раствора или суспензии в сочетании с фармацевтически приемлемым носителем, с использованием общеизвестного устройства или распылителя, такого как ингалятор для введения отмеренных доз и подобные. Ингалятор сухого порошка или подобные устройства могут быть предназначены для однократного или многократного введения, и можно использовать сухой порошок или содержащую порошок капсулу. Альтернативно, такое средство может быть представлено в виде находящегося под давлением аэрозольного спрея, для которого используют подходящий препеллент, например подходящий газ, такой как хлорфторалкан, гидрофторалкан, диоксид углерода и подобные, или в других формах.
В частности, в случае лечения и/или предупреждения грибковых инфекций, указанные выше инъекции для парентерального введения или средства для введения через слизистую оболочку, такие как препарат для ингаляций, средство для введения через нос и подобные, являются предпочтительными.
Как правило, в случае перорального введения, суточная доза составляет от около 0,001 до 100 мг/кг, предпочтительно от 0,1 до 30 мг/кг, и более предпочтительно от 0,1 до 0 мг/кг массы тела, которую вводят как единую дозу или разделенную на 2-4 части. В случае внутривенного введения, подходящей для введения является суточная доза от около 0,0001 до 10 мг/кг массы тела, которую вводят раз в день или два или более раз в день. Кроме того, средство для введения через слизистую оболочку вводят при дозе от около 0,001 до 100 мг/кг массы тела, раз в день или два или более раз в день. Дозу подходящим образом выбирают в соответствии с конкретным случаем, принимая во внимание симптомы, возраст, пол и подобные факторы.
Соединение формулы (I) можно использовать в сочетании с различными средствами для лечения или предупреждения заболеваний, при которых соединение формулы (I), описанное выше, считается эффективным. Такие средства для лечения или предупреждения, например, включают азолы, такие как флуконазол, вориконазол, итраконазол, кетоконазол, миконазол, ER30346 и SCH56592; полиены, такие как амфотерицин B, нистатин, их липосомные и липидные формы, например Abelcet, AmBisome и Amphocil; пуриновые или пиримидиновые нуклеотидные ингибиторы, такие как флуцитозин; поликсины, такие как никкомицины, в частности никкомицин Z или никкомицин X; другие ингибиторы хитина; ингибиторы элонгации, такие как сордарин и его аналог; ингибиторы маннанов, такие как предамицин, бактерицидные/индуцирующие проницаемость (BPI) белковые продукты, такие как XMP.97 или XMP.127; или комплексные углеводные противогрибковые средства, такие как CAN-296, или иммуносупрессанты, такие как такролимус, и они являются эффективными в отношении инфекций, перечисленных выше. Комбинированное применение осуществляют путем одновременного введения или отдельного введения, и непрерывно или с желаемыми временными интервалами. Средство для одновременного введения может представлять собой комбинированное лекарственное средство или может быть отдельно получено.
Примеры
Далее способы получения соединений формулы (I) будут описаны более подробно со ссылкой на Примеры. Соединение формулы (I) не ограничивается соединениями, описанными в представленных далее Примерах. Кроме того, способы получения исходных соединений будут описаны в Примерах получения.
Daiso-гель, SP-120-40/60-ODS-B (Торговая марка, изготовитель Daiso Co., Ltd.) использовали для ODS колоночной хроматографии, если не указано иное, при получении соединений Примеров и соединений Примеров получения по настоящему изобретению. Кроме того, при описании проявляющего растворителя для колоночной хроматографии “-” означает градацию, и "→" означает, что система растворителя постепенно изменялась.
Пример получения 1
Смесь 720 мг метил 4-{2-[4-(циклопентилметил)пиперазин-1-ил]пиримидин-5-ил}бензоата, 7 мл этанола и 2,02 мл гидразинмоногидрата нагревали при температуре кипения с обратным холодильником в течение ночи. Реакционную смесь оставляли охлаждаться до комнатной температуры и осажденное твердое вещество собирали фильтрованием и сушили, с получением 610 мг 4-{2-[4-(циклопентилметил)пиперазин-1-ил]пиримидин-5-ил}бензогидразида.
Пример получения 2
В атмосфере азота, к смеси 500 мг метил транс-4-[5-(4-бромфенил)-1,3,4-тиадиазол-2-ил]циклогексанкарбоксилата, 20 мл DME, 356 мг 4-изопропоксифенилбороновой кислоты и 2 мл 2,0М водного раствора карбоната натрия добавляли 76 мг тетракис(трифенилфосфин)палладия(0), с последующим перемешиванием при температуре бани 85°С в течение ночи. Реакционную смесь оставляли охлаждаться, затем ее выливали в ледяную воду и перемешивали, осажденное твердое вещество собирали фильтрованием и промывали водой. Затем порошок, собранный фильтрованием, суспендировали в ацетонитриле с последующим перемешиванием при комнатной температуре. Твердое вещество собирали фильтрованием, промывали ацетонитрилом и сушили при 50°С при пониженном давлении, с получением 510,6 мг метил транс-4-[5-(4'-изопропоксибифенил-4-ил)-1,3,4-тиадиазол-2-ил]циклогексанкарбоксилата.
Пример получения 3
К смеси 100 мг метил транс-4-[5-(4'-изопропоксибифенил-4-ил)-1,3,4-тиадиазол-2-ил]циклогексанкарбоксилата, 1 мл ТГФ и 0,5 мл этанола добавляли 92 мкл 5М водного раствора гидроксида калия, с последующим перемешиванием при комнатной температуре в течение ночи. Затем к смеси добавляли 46 мкл 5М водного раствора гидроксида калия, с последующим перемешиванием в течение 5 часов при комнатной температуре и в течение 1 часа при температуре бани 70°С. Реакционную смесь оставляли охлаждаться, затем ее разбавляли водой, а затем подкисляли при помощи 1М хлористоводородной кислоты. Осажденное твердое вещество собирали фильтрованием, промывали последовательно водой и ацетонитрилом и сушили при 60°C при пониженном давлении, с получением 91,7 мг транс-4-[5-(4'-изопропоксибифенил-4-ил)-1,3,4-тиадиазол-2-ил]циклогексанкарбоновой кислоты.
Пример получения 4
Под потоком газообразного азота, к смеси 200 мг транс-4-{5-[4-(4-циклогексил-4-метоксипиперидин-1-ил)фенил]-1,3,4-тиадиазол-2-ил}-N-метокси-N-метилциклогексанкарбоксамида и 2 мл DME добавляли по каплям 393 мкл раствора 1,16M метиллития-диэтилового эфира при 0°C, с последующим перемешиванием в течение 2 часов. К реакционной смеси добавляли воду и осажденное твердое вещество собирали фильтрованием и очищали колоночной хроматографией на силикагеле (хлороформ:метанол=100:0-97:3), с получением 73 мг 1-(транс-4-{5-[4-(4-циклогексил-4-метоксипиперидин-1-ил)фенил]-1,3,4-тиадиазол-2-ил}циклогексил)этанона.
Пример получения 5
В атмосфере азота, 1,05 г транс-4-[5-(4'-изопропоксибифенил-4-ил)-1,3,4-тиадиазол-2-ил]-N-метокси-N-метилциклогексанкарбоксамида растворяли в 21 мл толуола и 21 мл ТГФ, добавляли по каплям 3,08 мл 0,99M раствора диизобутилалюминийгидрид-толуол при -78°C, с последующим перемешиванием в течение 2 часов при указанной температуре. Затем к смеси добавляли воду и 1M хлористоводородной кислоты и осажденное твердое вещество собирали фильтрованием, промывали последовательно 1M хлористоводородной кислоты и водой и сушили, с получением 931 мг транс-4-[5-(4'-изопропоксибифенил-4-ил)-1,3,4-тиадиазол-2-ил]циклогексанкарбоальдегида в виде твердого вещества.
Пример получения 6
К суспензии 15,86 г монометил транс-1,4 циклогександикарбоксилата, 18,32 г бромбензгидразида, 13,81 г 1-гидроксибензотриазола, 17,81 мл триэтиламина и 220 мл N,N-диметилформамида (ДМФА) добавляли 24,5 г гидрохлорида 1-(3-диметиламинопропил)-3-этилкарбодиимида, с последующим перемешиванием в течение 3 часов при комнатной температуре. Затем реакционную смесь добавляли к 1 л воды, с последующим перемешиванием в течение 30 минут. Осажденное твердое вещество собирали фильтрованием, промывали водой и затем сушили при пониженном давлении, с получением 31,58 г метил транс-4-{[2-(4-бромбензоил)гидразино]карбонил}циклогексанкарбоксилата.
Пример получения 7
Суспензию 1,5 г 5-{4-[(6-метоксигексил)окси]фенил}-1,3,4-тиадиазол-2-амина, метил транс-4-(бромацетил)циклогексанкарбоксилата в 15 мл этанола и 7,5 мл ТГФ нагревали при температуре кипения с обратным холодильником в течение 6 часов. Суспензию оставляли охлаждаться, затем к ней добавляли 1,5 трифторуксусной кислоты, с последующим нагреванием снова при температуре кипения с обратным холодильником. Смесь оставляли охлаждаться, затем добавляли диизопропиловый эфир с последующим перемешиванием и осажденное твердое вещество собирали фильтрованием. После промывки этим же растворителем и сушки получали 2,0 г метил транс-4-(2-{4-[(6-метоксигексил)окси]фенил}имидазо[2,1-b][1,3,4]тиазол-6-ил)циклогексанкарбоксилата в виде светло-коричневого порошка.
Пример получения 8
К суспензии 300 мг транс-4-(2-{4-[(6-метоксигексил)окси]фенил}имидазо[2,1-b][1,3,4]тиадиазол-6-ил)циклогексанкарбоновой кислоты и 274 мг гексафторфосфата 2-(1H-бензотриазол-1-ил)-1,1,3,3-тетраметилурония в 3 мл ДМФА добавляли 0,29 мл диизопропилэтиламина, с последующим перемешиванием в течение 1 часа при комнатной температуре. Затем к смеси добавляли 64 мг N,O-диметилгидроксиламина и 0,11 мл диизопропилэтиламина с последующим перемешиванием в течение 14 часов при комнатной температуре. К реакционной смеси добавляли 10 мл воды с последующим перемешиванием и осажденные вещества собирали фильтрованием, промывали водой и диизопропиловым эфиром и сушили, с получением 312 мг транс-N-метокси-4-(2-{4-[(6-метоксигексил)окси]фенил}имидазо[2,1-b][1,3,4]тиадиазол-6-ил)-N-метилциклогексанкарбоксамида в виде белого твердого вещества.
Пример получения 9
Раствор 10,6 г 4-{2-[4-(циклогексилокси)пиперидин-1-ил]пиримидин-5-ил}бензонитрила, 3,7 г семитиокарбазида, 106 мл толуола и 56 мл трифторуксусной кислоты перемешивали при 85°C в течение 7 часов. Реакционную смесь оставляли охлаждаться, затем концентрировали при пониженном давлении, к смеси добавляли 50 мл ТГФ и полученную смесь добавляли к 500 мл ледяной воды. Затем pH доводили до около 7 при помощи водного раствора гидроксида натрия, и осажденное твердое вещество собирали фильтрованием. Полученное твердое вещество промывали диизопропиловым эфиром и сушили в вакууме, с получением 11,4 г 5-(4-{2-[4-(циклогексилокси)пиперидин-1-ил]пиримидин-5-ил}фенил)-1,3,4-тиадиазол-2-амина.
Пример получения 10
К раствору 7,4 г 4-[(6-бромгексил)окси]бензонитрила в 74 мл метанола добавляли 25,3 мл 28% раствор метоксида натрия в метаноле с последующим нагреванием при температуре кипения с обратным холодильником в течение 3 часов. Смесь оставляли охлаждаться, затем около половины количества метанола выпаривали при пониженном давлении, добавляли воду и этилацетат для осуществления процедуры разделения. Полученный органический слой промывали водой и насыщенным солевым раствором и сушили над безводным сульфатом магния и растворитель выпаривали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии среднего давления на силикагеле (гексан:этилацетат=90:10-66:34, градиент), с получением 5,5 г 4-[(6-метоксигексил)окси]бензонитрила.
Пример получения 11
К смеси 1,07 г этил 1-{2-[4-(тетрагидро-2H-пиран-2-илокси)фенил]пиримидин-5-ил}пиперидин-4-карбоксилата и 10 мл ТГФ добавляли 1,3 мл 6M хлористоводородной кислоты с последующим перемешиванием в течение 2 часов при комнатной температуре. Затем, при охлаждении льдом, реакционный раствор нейтрализовали при помощи 1M водного раствора гидроксида натрия и осажденное твердое вещество собирали фильтрованием, с получением 0,76 г этил 1-[2-(4-гидроксифенил)пиримидин-5-ил]пиперидин-4-карбоксилата.
Пример получения 12
К раствору 0,35 г этил 1-[2-(4-гидроксифенил)пиримидин-5-ил]пиперидин-4-карбоксилата в 4,5 мл ДМФА добавляли 193 мг калия карбонат и 351 мг 6-метокси-6-метилгептил-4-метилбензолсульфоната с последующим перемешиванием при 90°C в течение ночи. Реакционную смесь оставляли охлаждаться, затем ее выливали в 100 мл ледяной воды и осажденное твердое вещество собирали фильтрованием и сушили, с получением 0,33 г этил 1-(2-{4-[(6-метокси-6-метилгептил)окси]фенил}пиримидин-5-ил)пиперидин-4-карбоксилата.
Пример получения 13
К смеси 40 г 5-бром-2-хлорпиримидина, 43,6 г 4-(циклогексилокси)пиперидина и 1 л ДМФА добавляли 62,8 г карбоната калия с последующим перемешиванием в течение 3 часов при комнатной температуре. Затем к смеси добавляли 74 мл концентрированной хлористоводородной кислоты до доведения pH до около 5,5, при охлаждении льдом, с последующим перемешиванием в течение 30 минут. Осажденное твердое вещество собирали фильтрованием и сушили, с получением 69,5 г 5-бром-2-[4-(циклогексилокси)пиперидин-1-ил]пиримидина.
Пример получения 14
Под потоком газообразного азота, к смеси 63 г 5-бром-2-[4-(циклогексилокси)пиперидин-1-ил]пиримидина, 690 мл DME и 25,8 г (4-цианофенил)бороновой кислоты добавляли последовательно 185 мл 2M водного раствора карбоната натрия и 10,6 г тетракис(трифенилфосфин)палладия(0) с последующим перемешиванием при 85°C в течение ночи. После того как смеси давали охладиться до комнатной температуры, к смеси добавляли 800 мл воды и осажденное твердое вещество собирали фильтрованием. Полученное твердое вещество суспендировали в 100 мл ацетонитрила с последующим перемешиванием в течение 30 минут при 80°C. Осажденные вещества собирали фильтрованием и сушили, с получением 42,6 г 4-{2-[4-(циклогексилокси)пиперидин-1-ил]пиримидин-5-ил}бензонитрила.
Пример получения 15
При охлаждении льдом, к раствору 9,5 г 1-{4-[4-(циклобутилметокси)пиперидин-1-ил]фенил}этанона в 76 мл уксусной кислоты добавляли 6,6 мл 30% раствор бромистого водорода в уксусной кислоте с последующим перемешиванием в течение 5 минут при указанной температуре. Затем к смеси добавляли 11,6 г пербромида пиридинийбромида при комнатной температуре с последующим перемешиванием в течение 1 часа. При охлаждении льдом, к реакционной смеси добавляли 300 мл воды и осажденное твердое вещество собирали фильтрованием, промывали водой и 500 мл охлажденного гексана и сушили, с получением 8,9 г 2-бром-1-{4-[4-(циклобутилметокси)пиперидин-1-ил]фенил}этанона в виде зеленоватого твердого вещества.
Пример получения 16
К раствору 11,2 г 5-(4-{2-[4-(циклогексилокси)пиперидин-1-ил]пиримидин-5-ил}фенил)-1,3,4-тиадиазол-2-амина в 110 мл ацетонитрила, при охлаждении льдом, добавляли 7,4 г бромида меди(II). Затем медленно по каплям добавляли 5,1 мл 3-метилбутилнитрита с последующим перемешиванием в течение 2 часов при комнатной температуре. Реакционную смесь выливали в 400 мл воды и pH доводили до около 7 при помощи водного раствора гидроксида натрия. Смесь экстрагировали при помощи 800 мл этилацетата с последующей промывкой последовательно 15% водным раствором аммиака и насыщенным солевым раствором. Органический слой сушили над безводным сульфатом магния, и растворитель выпаривали при пониженном давлении. К полученному неочищенному продукту добавляли диизопропиловый эфир и осажденные вещества собирали фильтрованием и сушили, с получением 8,6 г 5-[4-(5-бром-1,3,4-тиадиазол-2-ил)фенил]-2-[4-(циклогексилокси)пиперидин-1-ил]пиримидина.
Пример получения 17
К раствору 420 мг {1-[5-(4-{2-[4-(циклогексилокси)пиперидин-1-ил]пиримидин-5-ил}фенил)-1,3,4-тиадиазол-2-ил]пиперидин-4-ил}метанола в 11 мл дихлорметана (ДХМ), при охлаждении льдом, добавляли 399 мг периодинана Десс-Мартина при перемешивании, с последующим перемешиванием в течение 1 часа при комнатной температуре. К реакционной смеси добавляли 10% водный раствор тиосульфата натрия с последующей экстракцией при помощи 50 мл ДХМ. Органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом магния и растворитель выпаривали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (хлороформ:метанол=9:1), с получением 385 мг 1-[5-(4-{2-[4-(циклогексилокси)пиперидин-1-ил]пиримидин-5-ил}фенил)-1,3,4-тиадиазол-2-ил]пиперидин-4-карбоальдегида.
Пример получения 18
К смеси 600 мг 5-[4-(5-бром-1,3,4-тиадиазол-2-ил)фенил]-2-[4-(циклогексилокси)пиперидин-1-ил]пиримидина, 207 мг пиперидин-4-илметанола и 9 мл N,N-диметилацетамид (ДМА) добавляли 0,3 мл триэтиламина с последующим перемешиванием при 90°C в течение ночи. Смесь оставляли охлаждаться, затем осажденное твердое вещество собирали фильтрованием, промывали при помощи 20 мл воды и 20 мл 50% водного раствора ацетонитрила и сушили, с получением 0,42 г {1-[5-(4-{2-[4-(циклогексилокси)пиперидин-1-ил]пиримидин-5-ил}фенил)-1,3,4-тиадиазол-2-ил]пиперидин-4-ил}метанола.
Пример получения 19
К смеси 4,2 г карбоната цезия, 181 мг ацетата палладия(II), 336 мг 2,2'-бис(дифенилфосфино)-1,1'-бинафтила и 54 мл толуола, под потоком газообразного азота, добавляли последовательно 1,81 г 5-бром-2-[4-(тетрагидро-2H-пиран-2-илокси)фенил]пиримидина и 1 г этил пиперидин-4-карбоксилата с последующим перемешиванием в течение 20 часов при 110°C. Смесь оставляли охлаждаться, затем к смеси добавляли воду и осажденное твердое вещество собирали фильтрованием, промывали водой и ацетонитрилом и сушили, с получением 1,07 г этил 1-{2-[4-(тетрагидро-2H-пиран-2-илокси)фенил]пиримидин-5-ил}пиперидин-4-карбоксилата.
Пример получения 20
Под потоком газообразного азота, суспензию 31,5 г метил транс-4-{[2-(4-бромбензоил)гидразино]карбонил}циклогексанкарбоксилата, 850 мл ТГФ и 21,9 г пентасульфида фосфора перемешивали в течение 48 часов при комнатной температуре. Реакционную смесь выливали в 3 л воды, к смеси добавляли 100 мл 1M водного раствора гидроксида натрия с последующим перемешиванием в течение 1 часа. Осажденное твердое вещество собирали фильтрованием, промывали водой и сушили. Полученное твердое вещество и 20 г безводного сульфата магния, 15 г силикагеля и активированный уголь суспендировали в 500 мл хлороформа с последующим перемешиванием в течение 5 минут. Нерастворимое вещество отделяли фильтрованием и фильтрат концентрировали при пониженном давлении, с получением 30,2 г метил транс-4-[5-(4-бромфенил)-1,3,4-тиадиазол-2-ил]циклогексанкарбоксилата в виде белого твердого вещества.
Пример получения 21
В атмосфере азота, раствор 1,3 г метил транс-4-[5-(4-{2-[4-(циклопентилметил)пиперазин-1-ил]пиримидин-5-ил}фенил)-1,3,4-тиадиазол-2-ил]циклогексанкарбоксилата в безводном ТГФ охлаждали на ледяной бане, к смеси добавляли 135 мг литийалюминийгидрида, поддерживая при этом температуру около 0°C. После перемешивания в течение 1 часа при комнатной температуре и охлаждения снова на ледяной бане к смеси последовательно добавляли 20 мл хлороформа, 600 мг фторида натрия и 2 мл воды. Реакционную смесь фильтровали через слой целита, растворитель выпаривали при пониженном давлении и полученное твердое вещество промывали ацетонитрилом, с получением 652 мг {транс-4-[5-(4-{2-[4-(циклопентилметил)пиперазин-1-ил]пиримидин-5-ил}фенил)-1,3,4-тиадиазол-2-ил]циклогексил}метанола.
Пример получения 22
В атмосфере азота, к раствору 0,88 г транс-4-[5-(4-{2-[4-(циклогексилокси)пиперидин-1-ил]пиримидин-5-ил}фенил)-1,3,4-тиадиазол-2-ил]-N-метокси-N-метилциклогексанкарбоксамида в 20 мл ТГФ, при охлаждении льдом, добавляли 62 мг литийалюминийгидрид с последующим перемешиванием в течение 30 минут. Затем к смеси добавляли 252 мг фторида цезия и 3 мл воды с последующим фильтрованием через слой целита. Фильтрат концентрировали при пониженном давлении, к смеси добавляли диизопропиловый эфир и полученное твердое вещество собирали фильтрованием и сушили в вакууме, с получением 0,69 г {транс-4-[5-(4-{2-[4-(циклогексилокси)пиперидин-1-ил]пиримидин-5-ил}фенил)-1,3,4-тиадиазол-2-ил]циклогексил}метанола.
Пример получения 23
К смеси 474 мг соединения (продукт) Примера 138 WO00/64927, 120 мг 1,4-диоксан-2,5-диола, 4,8 мл метанола и 4,8 мл ДМФА, при комнатной температуре, добавляли 0,082 мл уксусной кислоты и 63 мг цианоборогидрида натрия с последующим перемешиванием в течение ночи. К реакционной смеси добавляли 100 мл этилацетата и осажденное твердое вещество собирали фильтрованием и сушили в вакууме. Твердое вещество растворяли в воде и очищали при помощи ODS колоночной хроматографии (20% водный раствор ацетонитрила). Фракции, содержащие целевое соединение, объединяли, ацетонитрил выпаривали при пониженном давлении, и остаток подвергали сушке замораживанием, с получением 433 мг соединения Примера получения 23.
Пример получения 24
Суспензию 630 мг соединения Примера получения 29, 9,4 мл метанола и 235 мг 10% палладия на углероде (содержание воды 50%) перемешивали при комнатной температуре в течение 4 часов в атмосфере водорода при 1 атм. Катализатор удаляли фильтрованием с последующей промывкой при помощи 50 мл метанола. Фильтрат упаривали при пониженном давлении, и остаток сушили с получением 0,24 г соединения Примера получения 24.
Пример получения 25
Суспензию 630 мг соединения Примера получения 23 в 13 мл ДХМ охлаждали до 5°C, добавляли по каплям 1,4 трифторуксусную кислоту с последующим перемешиванием в течение 2 часов при комнатной температуре. Реакционную смесь выливали в смесь 100 мл насыщенного водного раствора бикарбоната натрия и 100 мл pH 6,86 стандартного буфера. Органический растворитель выпаривали при пониженном давлении, оставшийся водный раствор очищали при помощи ODS колоночной хроматографии (5→10→15→20% ацетонитрила/вода). Фракции, содержащие целевое соединение, объединяли, ацетонитрил выпаривали при пониженном давлении, и остаток подвергали сушке замораживанием, с получением 480 мг соединения Примера получения 25.
Пример получения 26
К раствору 30 мг соединения (продукт) Примера 43 WO01/60846 в 300 мкл ДМФА добавляли 25 мкл пиперидина с последующим перемешиванием в течение 1 часа при комнатной температуре. Реакционную смесь разбавляли этилацетатом, и осажденное твердое вещество собирали фильтрованием и промывали этилацетатом. Твердое вещество сушили при пониженном давлении с получением 22,4 мг соединения Примера получения 26.
Пример получения 27
К смеси 1,04 г соединения Примера получения 1 WO00/64927 в 14 мл ДМФА и 14 мл метанола добавляли 14 мл (диметоксиметил) диметиламина с последующим перемешиванием в течение 48 часов. Затем к смеси добавляли 6 мл 1M водного раствора гидроксида натрия с последующим перемешиванием в течение ночи. Реакционную смесь нейтрализовали при помощи 1M хлористоводородной кислоты и разбавляли при помощи 500 мл воды и полученный раствор очищали при помощи 180 мл ODS колоночной хроматографии. Фракции, содержащие целевое соединение, объединяли, и растворитель выпаривали при пониженном давлении. Остаток подвергали сушке замораживанием с получением 350 мг соединения Примера получения 27.
Пример получения 28
К смеси 1,85 г соединения (продукт) Примера получения 25 WO03/068807 и 18 мл ДМФА добавляли 0,3 мл 3-бром-1-пропен и 360 мг карбоната калия с последующим перемешиванием в течение ночи. К реакционной смеси добавляли 50 мл этилацетата и осажденное твердое вещество собирали фильтрованием. Полученный порошок очищали при помощи 180 мл ODS колоночной хроматографии. Фракции, содержащие целевое соединение, объединяли, растворитель выпаривали при пониженном давлении, и остаток подвергали сушке замораживанием, с получением 1,15 г соединения Примера получения 28.
Пример получения 29
К смеси 1 г соединения Примера получения 28, 20 мл трет-бутанола и 20 мл воды добавляли 1,28 г AD-MIX-BETA (изготовитель Aldrich corporation) и 135 мг метансульфонамида с последующим перемешиванием в течение ночи. К реакционной смеси добавляли 50 мл этилацетата и осажденное твердое вещество собирали фильтрованием. Полученное твердое вещество очищали при помощи 80 мл ODS колоночной хроматографии. Фракции, содержащие целевое соединение, объединяли, ацетонитрил выпаривали при пониженном давлении, и остаток подвергали сушке замораживанием, с получением 512 мг соединения Примера получения 29.
Пример получения 30
К смеси 328 мг соединения Примера получения 27, 46,7 мг гексафторфосфата O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония и 1,4 мл ДМФА добавляли 0,35 мл N,N-диметилэтан-1,2-диамина и 83 мкл N,N-диизопропилэтиламина с последующим перемешиванием в течение ночи. Затем к смеси добавляли 50 мл этилацетата и осажденное твердое вещество собирали фильтрованием и очищали при помощи 30 мл ODS колоночной хроматографии. Фракции, содержащие целевое соединение, объединяли, растворитель выпаривали при пониженном давлении, и остаток подвергали сушке замораживанием, с получением 131 мг соединения Примера получения 30.
Пример получения 31
В атмосфере азота, 168 мг соединения Примера получения 103 растворяли в 1,68 мл метанола, к смеси добавляли 357 мкл раствора 2M триметилсилилдиазометана в гексане с последующим перемешиванием в течение 4 часов при комнатной температуре. Реакционную смесь концентрировали при пониженном давлении, и полученное твердое вещество растворяли в 2 мл ДМФА и очищали при помощи ODS колоночной хроматографии (10→20→25→30→35% водный раствор ацетонитрила). Фракции, содержащие целевое соединение объединяли, и растворитель выпаривали при пониженном давлении и подвергали сушке замораживанием, с получением 141,7 мг соединения Примера получения 31 в виде белого твердого вещества.
Пример получения 32
В атмосфере азота, смесь 80 г соединения Примера получения 26, 320 мл ДМФА, 30,69 мл диизопропилэтиламина и 46,1 г ди-трет-бутилдикарбоната перемешивали при комнатной температуре в течение ночи. Затем к смеси добавляли 20 мл толуола и растворитель выпаривали при пониженном давлении. Под потоком газообразного азота, реакционную смесь добавляли к 4 л этилацетата при перемешивании, и осажденное твердое вещество собирали фильтрованием. Твердое вещество промывали при помощи 500 мл этилацетата и сушили при пониженном давлении при 45°C в течение 8 часов, затем при комнатной температуре в течение ночи, с получением 118,36 г соединения Примера получения 32 в виде бесцветного твердого вещества.
Пример получения 33
Под потоком газообразного азота, к смеси 1,0 г метил транс-4-[5-(4-бромфенил)-1,3,4-тиадиазол-2-ил]циклогексанкарбоксилата, 10 мл толуола, 593,8 мг 4,4-диметилпиперидина, 206,4 мг 2'-(дициклогексилфосфино)-N,N-диметилбифенил-2-амина и 779,4 мг фосфата калия добавляли 150,8 мг трис(дибензилиденацетон)дипалладия с последующим перемешиванием в течение 15 часов при 100°C. К реакционной смеси добавляли 100 мл ДХМ и 30 мл 1M хлористоводородной кислоты и органический слой отделяли. Органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом магния и растворитель выпаривали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (хлороформ:метанол=100:0-97:3), с получением 350 мг метил 4-{5-[4-(4,4-диметилпиперидин-1-ил)фенил]-1,3,4-тиадиазол-2-ил}циклогексанкарбоксилата.
Пример получения 34
К смеси 800 мг метил 4-(2-пиперазин-1-илпиримидин-5-ил)бензоата дигидрохлорида, 12 мл ДХМ, 270 мкл циклопентанкарбоальдегида и 600 мкл триэтиламина добавляли 639 мг триацетоксиборогидрида натрия и 123 мкл уксусной кислоты с последующим перемешиванием при комнатной температуре в течение ночи. К реакционной смеси добавляли ДХМ и воду и органический слой отделяли. Органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом магния и растворитель концентрировали при пониженном давлении, с получением 720 мг метил 4-{2-[4-(циклопентилметил)пиперазин-1-ил]пиримидин-5-ил}бензоата.
Пример получения 35
К раствору 2,5 г трет-бутил 4-{5-[4-(метоксикарбонил)фенил]пиримидин-2-ил}пиперазин-1-карбоксилата в 15 мл 1,4-диоксана добавляли 15 мл раствора 4M хлористого водорода в 1,4-диоксане с последующим перемешиванием при комнатной температуре в течение ночи. Осажденное твердое вещество собирали фильтрованием и сушили, с получением 2,2 г метил 4-(2-пиперазин-1-илпиримидин-5-ил)бензоата дигидрохлорида.
Пример получения 36
В атмосфере азота, к раствору 1,75 г 5-бром-2-[4-(циклогексилокси)пиперидин-1-ил]пиримидина в 17,5 мл ТГФ добавляли по каплям 3,85 мл раствора 1,67M н-бутиллития в гексане в течение 10 минут при охлаждении на бане сухой лед-ацетон с последующим перемешиванием в течение 20 минут. Затем добавляли по каплям 1,42 мл триизопропилбората в течение 10 минут с последующим перемешиванием в течение 10 минут. Затем к смеси добавляли 0,4 мл метанола, охлаждающую баню удаляли, температуре давали повыситься до комнатной температуры, к смеси добавляли 50 мл воды и затем раствор подкисляли при помощи 1M хлористоводородной кислоты. К смешанному раствору добавляли этилацетат для осуществления процедуры разделения. Органический слой промывали последовательно водой и насыщенным солевым раствором, сушили над безводным сульфатом магния и концентрировали при пониженном давлении, с получением 1,47 г {2-[4-(циклогексилокси)пиперидин-1-ил]пиримидин-5-ил}бороновой кислоты.
Пример получения 125
500 мг соединения Примера получения 186 растворяли в 10 мл ДХМ, под потоком газообразного азота и при охлаждении на бане сухой лед-ацетон, добавляли по каплям 1,354 мл раствора 1M диизобутилалюминийгидрида в толуоле с последующим перемешиванием в течение 2 часов при указанной температуре. К реакционной смеси медленно добавляли 1 мл воды, затем к смеси добавляли 10 мл 1M хлористоводородной кислоты и 50 мл хлороформа для осуществления процедуры разделения. Органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом магния и растворитель выпаривали при пониженном давлении. Получали 369,1 мг соединения Примера получения 125 в виде неочищенного продукта и использовали в последующей реакции без дополнительной очистки.
Пример получения 126
При охлаждении льдом, к раствору 0,154 г соединения Примера получения 187 в 2 мл ТГФ добавляли последовательно 21 мг борогидрида лития и 0,7 мл метанола с последующим перемешиванием в течение 4 часов. Затем последовательно добавляли 1M хлористоводородной кислоты и воду, и осажденное твердое вещество собирали фильтрованием и сушили в вакууме, с получением 0,12 г соединения Примера получения 126.
Пример получения 127
К суспензии 627 мг (метоксиметил)триметилфосфонийхлорида и 5 мл ТГФ добавляли по каплям 731 мкл раствора 2,6M н-бутиллития в н-гексане, поддерживая при этом внутреннюю температуру 5°C или ниже, при охлаждении на бане лед/насыщенный солевой раствор, с последующим перемешиванием в течение 30 минут при указанной температуре. Затем к смеси добавляли по каплям раствор 300 мг трет-бутил 4-оксоазепин-1-карбоксилата в 3 мл ТГФ при указанной температуре, с последующим перемешиванием в течение 30 минут при этой же температуре. К смеси добавляли насыщенный водный раствор хлорида аммония и этилацетат для осуществления процедуры разделения. Органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом магния и растворитель выпаривали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (гексан:этилацетат=100:0-90:10), с получением 192 мг соединения Примера получения 127.
Пример получения 128
К 711 мг соединения Примера получения 192 добавляли последовательно 7 мл ДМФА, 332 мг 1-гидроксибензотриазола (HOBt), 64 мг гидрохлорида 1-этил-3-(3-диметиламинопропил)карбодиимида (WSC) и 0,46 мл триэтиламина с последующим перемешиванием в течение 1 часа при комнатной температуре. Затем к смеси добавляли 470 мг соединения Примера получения 130 с последующим перемешиванием в течение 15 часов при комнатной температуре. К реакционной смеси добавляли воду и осажденное твердое вещество собирали фильтрованием и сушили при пониженном давлении, с получением 890 мг соединения Примера получения 128.
Пример получения 129
Раствор 2,3 г соединения Примера получения 128 в 23 мл ДМА перемешивали при 130°C в течение ночи. После того как смеси давали охладиться до комнатной температуры, добавляли воду и этилацетат для осуществления процедуры разделения. Органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом магния и растворитель выпаривали при пониженном давлении, с получением 1,25 г соединения Примера получения 129.
Пример получения 130
Смесь 820 мг метил транс-4-цианоциклогексанкарбоксилата, 10 мл метанола, 1,02 мл триэтиламина и 511 мг гидроксилхлорида аммония нагревали при температуре кипения с обратным холодильником в течение 6 часов. После того как смеси давали охладиться до комнатной температуры, добавляли воду и этилацетат для осуществления процедуры разделения. Органический слой промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом магния и растворитель выпаривали при пониженном давлении, с получением 0,48 г соединения Примера получения 130.
Пример получения 131
2,380 г соединения Примера получения 141 растворяли в 200 мл воды с последующей очисткой при помощи 150 мл ODS колоночной хроматографии (0%→10%→20%→35% ацетонитрила/вода). Фракции, содержащие целевой продукт, объединяли, ацетонитрил выпаривали при пониженном давлении, и остаток подвергали сушке замораживанием, с получением 0,441 г соединения Примера получения 126.
Пример получения 132
К раствору 100 мг соединения Примера получения 127 в 1 мл ДХМ добавляли последовательно 0,22 мл анизола, 0,04 мл воды и 0,16 трифторуксусной кислоты с последующим перемешиванием в течение 1 часа при комнатной температуре. Затем к смеси добавляли 0,16 трифторуксусной кислоты с последующим перемешиванием в течение 1 часа при комнатной температуре. Реакционную смесь концентрировали при пониженном давлении и сушили при пониженном давлении, с получением 102 мг 4-формилазепинтрифторацетата.
Пример получения 133
В атмосфере водорода, смесь 570 мг соединения Примера получения 205, 11,4 мл метанола и 285 мг 10% палладия/углерод (содержание воды 50%) перемешивали в течение 4 часов при комнатной температуре. Катализатор удаляли фильтрованием с последующей промывкой при помощи 10 мл метанола, и фильтрат концентрировали при пониженном давлении. К остатку добавляли воду с последующей сушкой замораживанием, с получением 420 мг соединения Примера получения 133.
Пример получения 134
К 5,50 г соединения Примера получения 140 добавляли 100 мл ДМФА, при охлаждении льдом, при внутренней температуре 4°C, к смеси добавляли 2,30 мл DIPEA и 1,37 г 9-флуоренилметилхлороформиата (внутренняя температура 6°C) с последующим перемешиванием в течение 2 часов при указанной температуре. Реакционную смесь выливали в 1000 мл воды и добавляли 20 мл ацетонитрила. Полученную смесь очищали при помощи 120 мл ODS колоночной хроматографии (0%→20%→60% ацетонитрила/вода). Ацетонитрил из фракции, содержащей целевой продукт, выпаривали при пониженном давлении, и остаток подвергали сушке замораживанием, с получением 5,95 г соединения Примера получения 134.
Пример получения 135
К суспензии 60 г соединения (продукт) Примера 2 WO99/40108, 300 мл воды и 300 мл 1,4-диоксана добавляли по каплям 132 мл 1M водного раствора гидроксида натрия и раствор 20 г ди-трет-бутилдикарбоната в 150 мл 1,4-диоксана с последующим перемешиванием в течение 3,5 часов при комнатной температуре. 1, 4-диоксан выпаривали при пониженном давлении и остаток очищали при помощи 300 мл ODS колоночной хроматографии (0%→50% ацетонитрила/вода). Фракцию, которая просачивалась в процессе загрузки в этой процедуре, разбавляли при помощи 1500 мл воды и очищали при помощи 300 мл ODS колоночной хроматографии (после загрузки насыщенный водный раствор бикарбоната натрия и насыщенный солевой раствор вытекали последовательно, с последующим элюированием 0%→50% ацетонитрила/вода). Фракцию, которая просачивалась в процессе загрузки в этой процедуре, разбавляли при помощи 3000 мл воды и очищали при помощи ODS колоночной хроматографии, как описано выше. Ацетонитрил из фракции, содержащей целевой продукт, полученной в каждой из процедур очистки, выпаривали при пониженном давлении с последующей сушкой замораживанием, с получением 58,3 г соединения Примера получения 135.
Пример получения 136
К 58,3 г соединения Примера получения 135 добавляли последовательно 525 мл ДМФА, 13,5 мл бензилбромида и 3,57 г моногидрата гидроксида лития с последующим перемешиванием в течение 30 часов при комнатной температуре. Реакционную смесь выливали в 3500 мл этилацетата с последующим интенсивным перемешиванием. Полученной смеси давали выстояться до осаждения образовавшегося твердого осадка, надосадочную жидкость удаляли, и твердое вещество собирали фильтрованием и сушили при пониженном давлении. Полученное твердое вещество суспендировали в 1000 мл воды, к смеси добавляли 14,4 г гидрокарбоната натрия, с последующим перемешиванием, и затем нерастворимое вещество удаляли фильтрованием. Фильтрат очищали при помощи 700 мл ODS колоночной хроматографии (0%→10%→50% ацетонитрила/вода). К 2000 мл фракции, которая просачивалась, когда смесь 10% ацетонитрила/вода использовали в качестве элюента в этой очистке, добавляли 1000 мл воды и раствор очищали при помощи ODS колоночной хроматографии (после загрузки происходило вытекание, по объему в два раза превышающее объем смолы, насыщенного водного раствора бикарбоната натрия и насыщенного солевого раствора, с последующим элюированием 0%→10%→50% ацетонитрила/вода). К 1500 мл фракции, которая просачивалась в процессе загрузки в этой процедуре очистки, добавляли 1000 мл воды и раствор разделяли на три части, затем каждую часть очищали при помощи ODS колоночной хроматографии, как описано выше. Ацетонитрил из фракции, содержащей целевой продукт, полученной в каждой из процедур очистки, выпаривали при пониженном давлении с последующей сушкой замораживанием, с получением 55,4 г соединения Примера получения 136.
Пример получения 137
К 28,4 г соединения Примера получения 136 добавляли 350 мл ДМФА, добавляли по каплям 180 мл раствора 2,74M хлористого водорода в метаноле с последующим перемешиванием в течение 6 часов при охлаждении льдом. Реакционную смесь выливали в 5000 мл воды, добавляли 1M водный раствор гидроксида натрия для доведения pH до около 7. Смесь очищали при помощи 700 мл ODS колоночной хроматографии (0%→10%→20%→50% ацетонитрила/вода). Ацетонитрил из 500 мл фракции, содержащей целевой продукт, выпаривали при пониженном давлении с последующей сушкой замораживанием, с получением 25,0 г соединения Примера получения 137.
Пример получения 138
Смесь 20,0 г соединения Примера получения 137, 200 мл ДМФА, 12,4 г моногидрата гидроксида лития и 50 мл 1,2-дибромэтана перемешивали в течение 27 часов при комнатной температуре. Реакционную смесь выливали в смешанный растворитель, состоящий из 3500 мл воды и 900 мл ацетонитрила, с последующей очисткой при помощи 700 мл ODS колоночной хроматографии (0%→10%→20%→30%→40% ацетонитрила/вода). 1500 мл фракции, содержащей целевой продукт, разбавляли при помощи 1000 мл воды, добавляли ацетонитрил до растворения осажденных веществ с последующей очисткой при помощи 300 мл ODS колоночной хроматографии (смесь ацетонитрил/вода использовали в качестве растворителя для элюирования, после того как происходило вытекание объема элюента, превышающего в три раза объем смолы, при концентрации ацетонитрила 0%, элюирование осуществляли при концентрации ацетонитрила 70%). Ацетонитрил из фракции, содержащей целевой продукт, выпаривали при пониженном давлении с последующей сушкой замораживанием, с получением 14,6 г соединения Примера получения 138.
Пример получения 139
Смесь 6,50 г соединения Примера получения 138, 65 мл ДМФА и 0,753 г азида натрия перемешивали при 70°C в течение 4 часов. Реакционную смесь разбавляли при помощи 1600 мл воды с последующей очисткой при помощи 120 мл ODS колоночной хроматографии (0%→60% ацетонитрила/вода). Фракции, содержащие целевой продукт, объединяли, ацетонитрил выпаривали при пониженном давлении, и остаток подвергали сушке замораживанием, с получением 5,74 г соединения Примера получения 139.
Пример получения 140
В атмосфере водорода, смесь 5,74 г соединения Примера получения 139, 110 мл метанола и 1,70 г 10% палладия на углероде (содержание воды 50%) перемешивали в течение 12 часов при комнатной температуре. После замещения азотом, полученной смеси давали выстояться в течение ночи с последующим перемешиванием снова в течение 4 часов при комнатной температуре в атмосфере водорода. Катализатор удаляли фильтрованием через слой целита и фильтрат концентрировали при пониженном давлении. К остатку добавляли 500 мл воды с последующей очисткой при помощи 120 мл ODS колоночной хроматографии (0%→30%→70% ацетонитрила/вода). Фракции, содержащие целевой продукт, объединяли, ацетонитрил выпаривали при пониженном давлении, и остаток подвергали сушке замораживанием, с получением 5,50 г соединения Примера получения 140.
Пример получения 141
При охлаждении льдом, к смеси 2,00 г соединения Примера получения 134 и 1,00 мл воды добавляли 20,0 мл трифторуксусной кислоты с последующим перемешиванием в течение 3 часов при комнатной температуре. Реакционную смесь концентрировали при пониженном давлении и к остатку добавляли воду. Смесь подвергали сушке замораживанием, с получением 2,38 г соединения Примера получения 141.
Соединения Примеров получения, описанные в следующих далее Таблицах, получали с использованием соответствующих исходных веществ, соответственно, аналогично способам Примеров получения. Структуры, способы получения и физико-химические данные соединений Примера получения от 1 по Пример 203 представлены в Таблицах 2-23 и Таблицах 40-53.
Пример 1 (Способ получения A)
К смеси 134 мг транс-4-{5-[4-(4-циклогексил-4-метоксипиперидин-1-ил)фенил]-1,3,4-тиадиазол-2-ил}циклогексанкарбоальдегида, 2 мл метанола, 2 мл хлороформа, 2 мл ДМФА и 200 мг соединения Примера 22 (продукт) WO2005/005463 добавляли 26,4 мг цианоборогидрида натрия и 35 мкл уксусной кислоты с последующим перемешиванием при комнатной температуре в течение ночи. К реакционной смеси добавляли 567 мкл пиперидина с последующим перемешиванием в течение 1 часа. К смеси добавляли этилацетат и осажденное твердое вещество собирали фильтрованием с последующей очисткой при помощи ODS колоночной хроматографии (ацетонитрил:pH 3 водный раствор хлористоводородной кислоты =0:100→10:90→20:80→25:75→30:70). Ацетонитрил из фракции, содержащей целевой продукт, выпаривали при пониженном давлении и подвергали сушке замораживанием, с получением 214 мг соединения Примера 1 в виде желтого аморфного вещества.
Пример 2 (Способ получения B)
К смеси 385 мг 1-[5-(4-{2-[4-(циклогексилокси)пиперидин-1-ил]пиримидин-5-ил}фенил)-1,3,4-тиадиазол-2-ил]пиперидин-4-карбоальдегида, 4,8 мл метанола, 7,7 мл ДМФА, 4,8 мл ДХМ и 1,2 г соединения Примера получения 26 добавляли 0,12 мл уксусной кислоты и 91 мг цианоборогидрида натрия с последующим перемешиванием при комнатной температуре в течение ночи. К реакционной смеси добавляли 100 мл этилацетата, осажденное твердое вещество собирали фильтрованием и сушили в вакууме. Твердое вещество растворяли в водном растворе гидроксида натрия с последующей очисткой при помощи 80 мл ODS колоночной хроматографии (40% водный раствор ацетонитрила). Фракции, содержащие целевое соединение, объединяли, ацетонитрил выпаривали при пониженном давлении, и остаток подвергали сушке замораживанием, с получением 610 мг соединения Примера 2.
Пример 3 (Способ получения C)
К раствору 40 мг соединения Примера 16 в 1,2 мл метанола добавляли по каплям 1,24 мл 10% раствор хлористого водорода в метаноле при комнатной температуре с последующим перемешиванием в течение 1 часа при комнатной температуре. К реакционной смеси добавляли 200 мл воды с последующей очисткой при помощи 20 мл ODS колоночной хроматографии (ацетонитрил:вода=0:100→10:90→20:80→30:70→40:60→45:55→50:50). Фракцию, содержащую целевой продукт, разбавляли двукратно с последующей очисткой снова при помощи 20 мл ODS колоночной хроматографии (ацетонитрил:вода=80:20). Растворитель выпаривали при пониженном давлении, и остаток подвергали сушке замораживанием, с получением 33 мг соединения Примера 3.
Пример 43 (Способ получения D)
Суспензию 111 мг соединения Примера получения 204 в 2 мл ДХМ перемешивали при 5°C и добавляли по каплям 150 мкл трифторуксусной кислоты. После перемешивания в течение 2 часов при комнатной температуре суспензию выливали в смесь 10 мл насыщенного водного раствора бикарбоната натрия и 10 мл pH 6,86 стандартного буфера. Органический растворитель выпаривали при пониженном давлении, оставшийся водный раствор очищали при помощи 20 мл ODS колоночной хроматографии (5→20% ацетонитрила/вода). Фракции, содержащие целевой продукт, объединяли и ацетонитрил выпаривали при пониженном давлении. Остаток подвергали сушке замораживанием, с получением 63 мг соединения Примера 43.
Пример 44 (Способ получения E)
К смеси 30 мг соединения Примера 51, 0,5 мл ДМФА, 0,5 мл хлороформа, 4 мг формальдегида и 0,5 мл метанола добавляли 4 мкл уксусной кислоты и 2,9 мг цианоборогидрида натрия с последующим перемешиванием при комнатной температуре в течение ночи. К реакционной смеси добавляли 10 мл этилацетата и осажденное твердое вещество собирали фильтрованием и сушили при пониженном давлении. Твердое вещество растворяли в воде с последующей очисткой при помощи 20 мл ODS колоночной хроматографии (40% ацетонитрила/вода). Фракции, содержащие целевой продукт, объединяли и ацетонитрил выпаривали при пониженном давлении. Остаток подвергали сушке замораживанием, с получением 29 мг соединения Примера 44.
Пример 45 (Способ получения F)
100 мг соединения Примера 22, 32 мг гидрата глиоксиловой кислоты и 300 мг молекулярных сит 3Å суспендировали в ДМФА с последующим перемешиванием при комнатной температуре в течение ночи. Реакционную смесь разбавляли метанолом, молекулярные сита отделяли фильтрованием с последующей промывкой метанолом. Фильтрат и промывочные жидкости объединяли с последующим концентрированием при пониженном давлении, к остатку добавляли этилацетат и осажденное твердое вещество собирали фильтрованием с последующей промывкой этилацетатом. Твердое вещество сушили и растворяли в воде с последующим доведением pH до около 7,5 при помощи 1M водного раствора гидроксида натрия. Этот раствор очищали на ODS колонке. (ODS гель: YMC GEL ODS-A 12 нм S-50 мкм AA12S50 20 см2, растворитель для элюирования:ацетонитрил:вода=5:95→20:80→30:70→40:60→45:55, детектор: УФ 210 нм, скорость потока 50 мл/мин), фракции, содержащие целевой продукт, объединяли и концентрировали при пониженном давлении и затем остаток подвергали сушке замораживанием, с получением 48,1 мг соединения Примера 45.
Таким же способом, как в описанных выше Примерах, соединения Примеров, представленные в следующих далее Таблицах, получали с использованием соответствующих исходных веществ, соответственно. Структуры соединений Примера 1 по Пример 75 представлены в Таблицах 24-37 и Таблицах 54-64, способы получения и физико-химические данные представлены в Таблице 38 и Таблице 39, и в Таблице 65 и Таблице 66.
Кроме того, следующие аббревиатуры использовали в Примерах получения, Примерах и следующих далее Таблицах. Пр.пол.: номер примера получения, Пр.: номер примера, Стр.: структурная формула, Синт.: способ получения (для Примеров/Примеров получения, описанных выше, указан номер Примера получения, который получают таким же способом, или Способ получения указан как Пример. Например, показано, что соединение Примера получения 37 было получено таким же способом, как соединение Примера получения 20, Пример 4 был получен таким же способом, как Пример 1). Данные: Физико-химические данные (ЯМР1: δ(м.д.) в 1H ЯМР DMSO-d6, ЯМР2: δ(м.д.) в 1H ЯМР DMSO-d6+D2O, ЯМР3: δ(м.д.) в 1H ЯМР CDCl3, ESI: ESI+(катион), ESI-(анион), EI: EI-MS(катион)), Соль: когда соединение представляет собой кислотно-аддитивную соль, представлено указание кислоты и основания (“-” означает свободную форму), Me: метил, Et: этил, Boc: трет-бутоксикарбонил, Bn: бензил, Z: бензилоксикарбонильная группа, Fmoc: (9H-флуорен-9-илметокси)карбонильная группа.



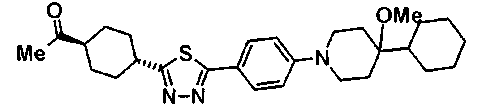


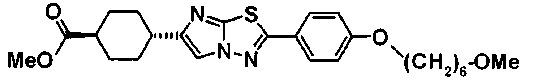


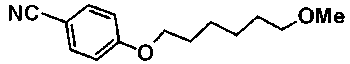



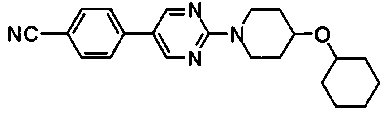


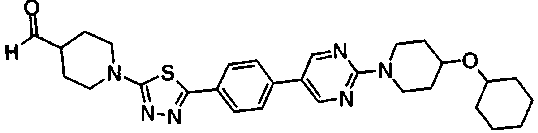

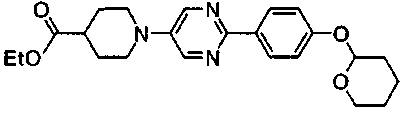
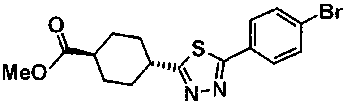








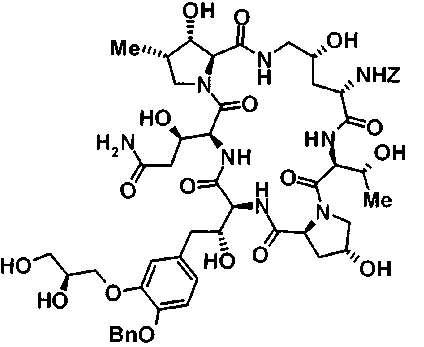



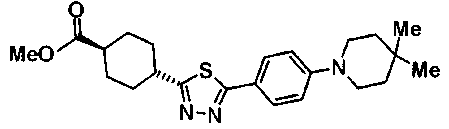





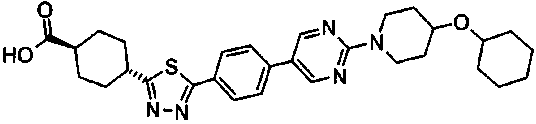
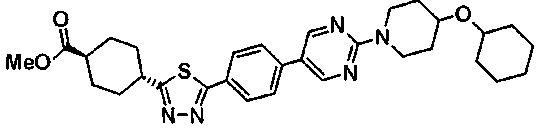
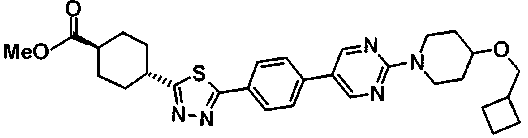

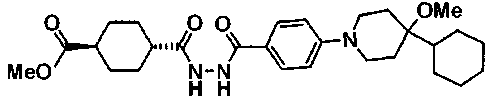
















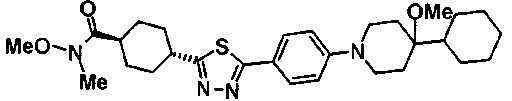

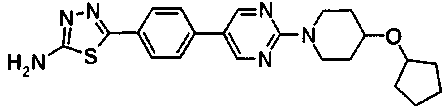






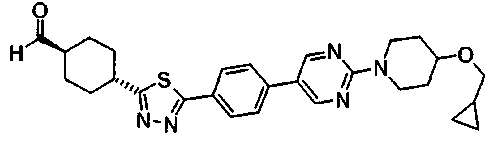




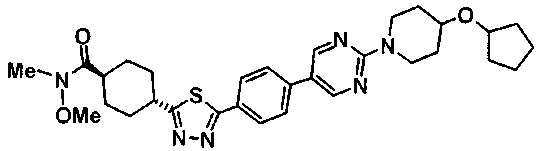
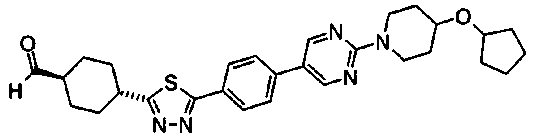







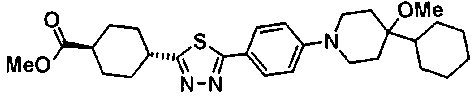
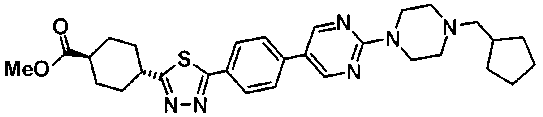


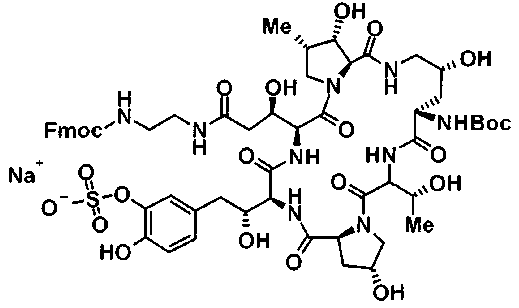

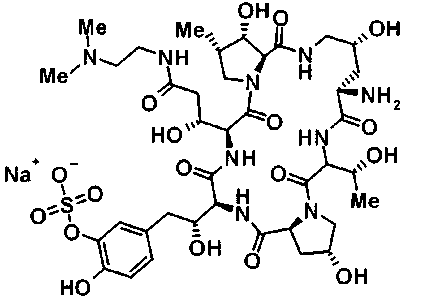

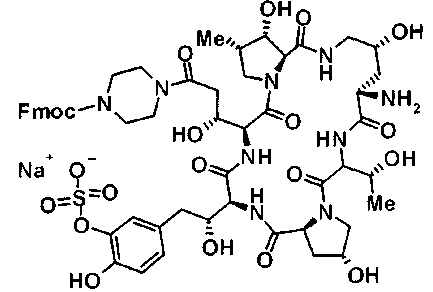
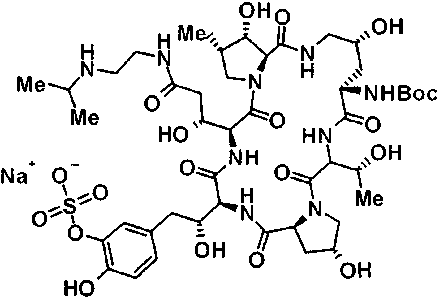



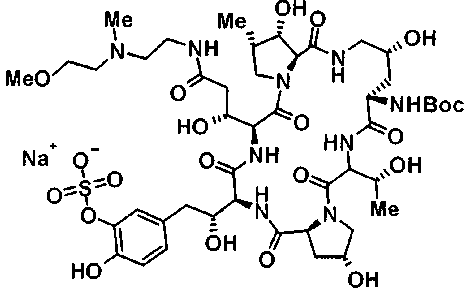


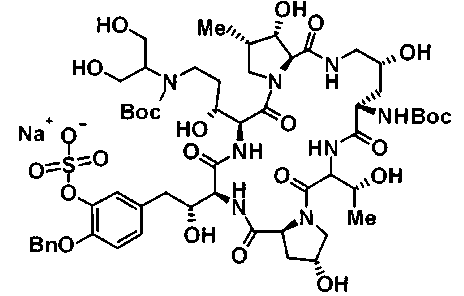
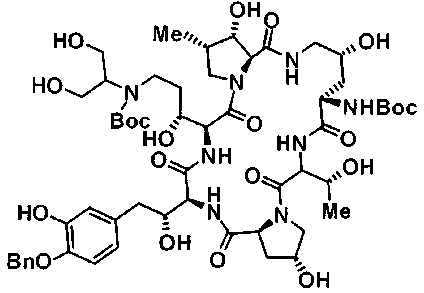

пол.

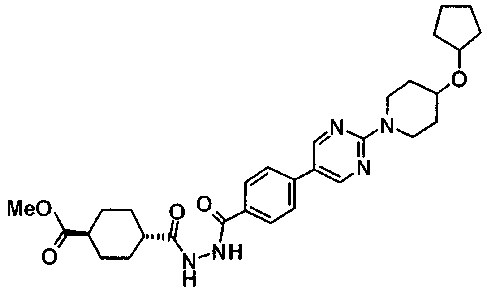
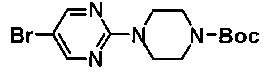



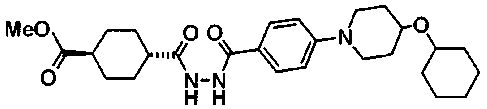


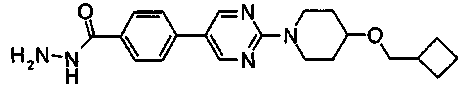




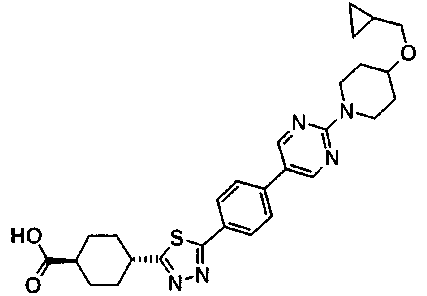
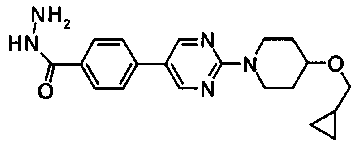

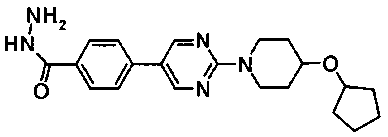



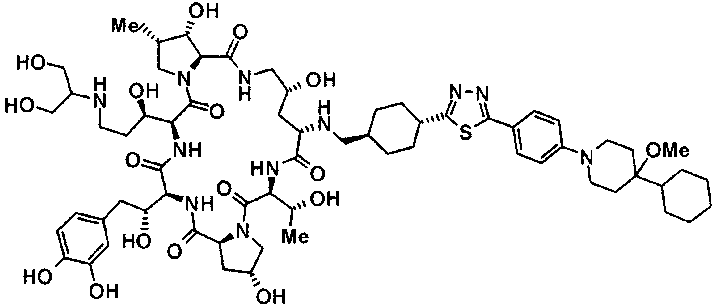


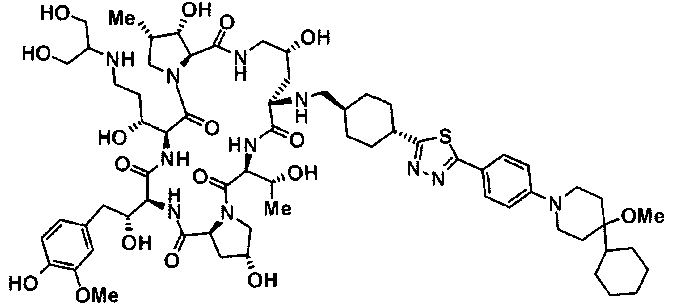
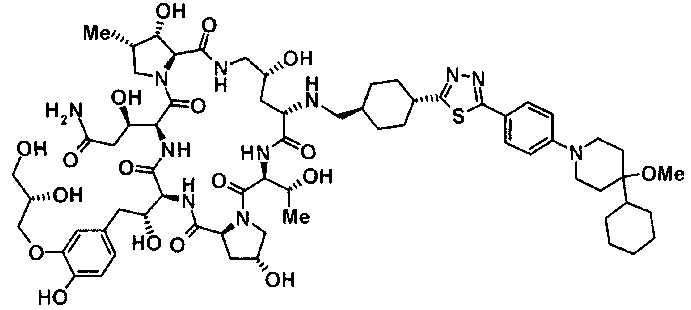






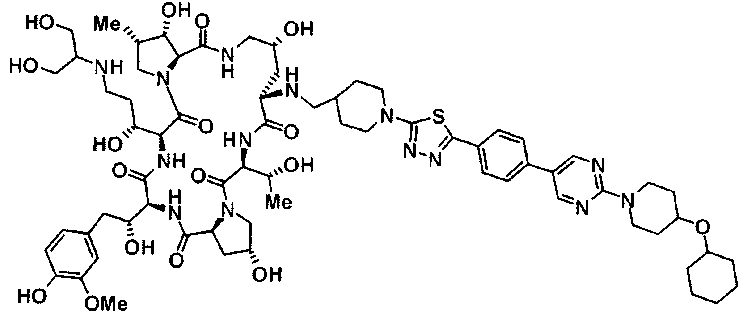


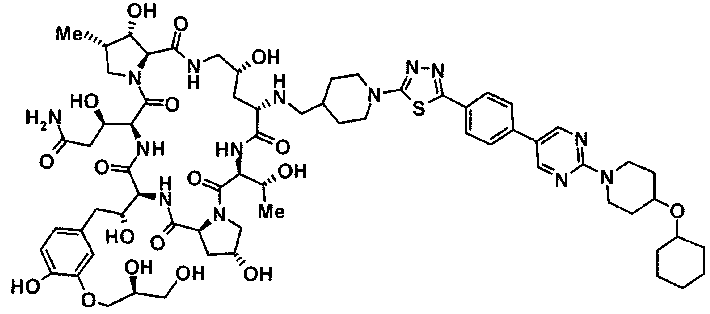


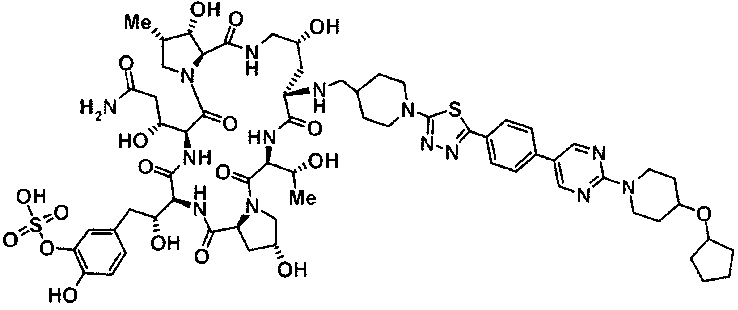


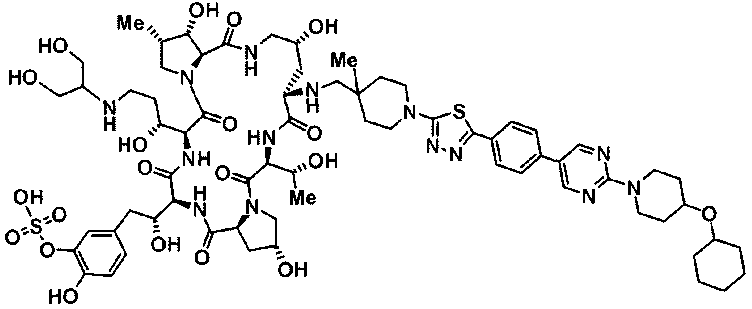



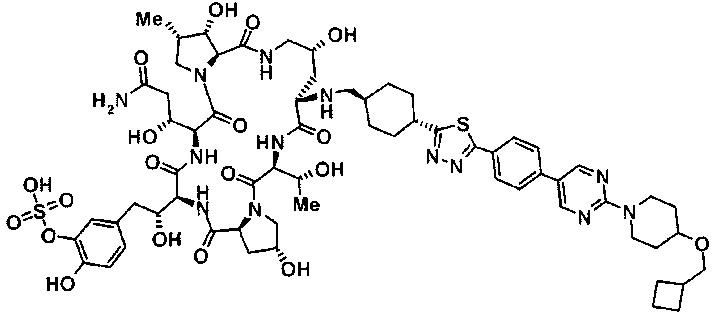
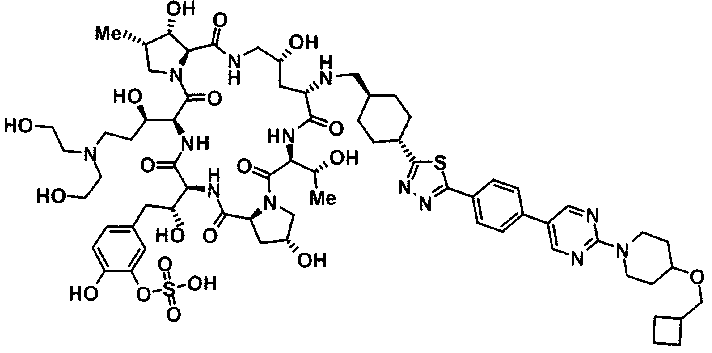
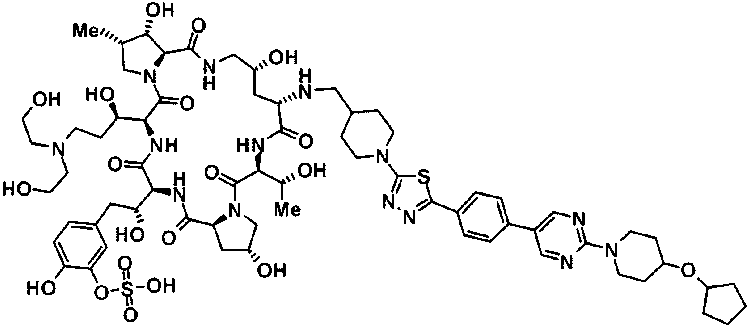



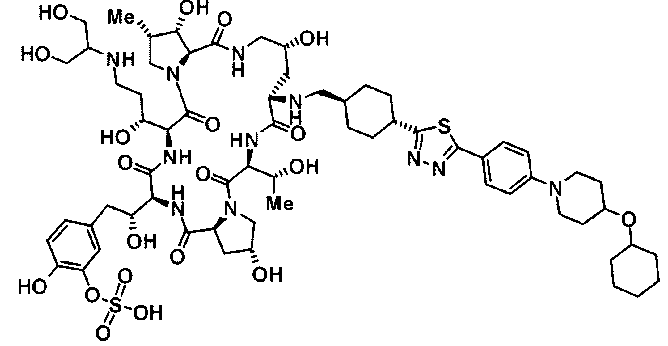



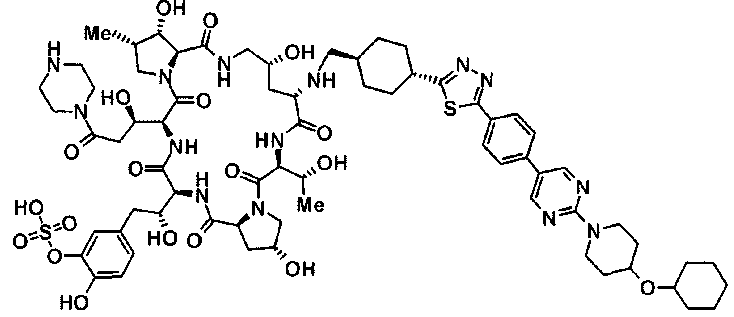
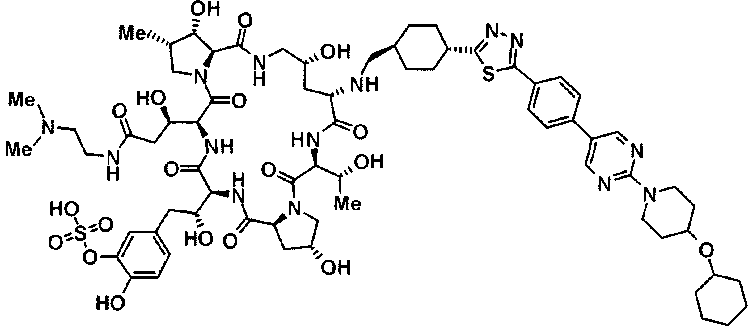


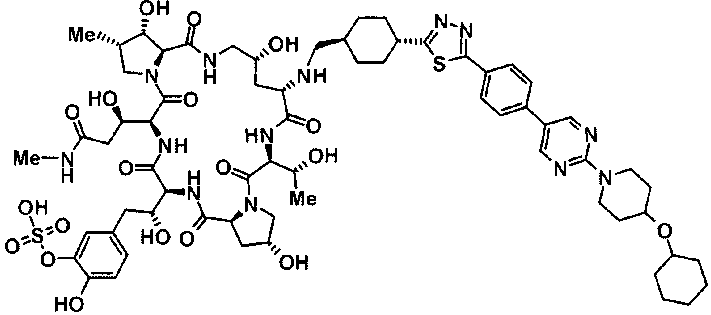



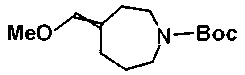






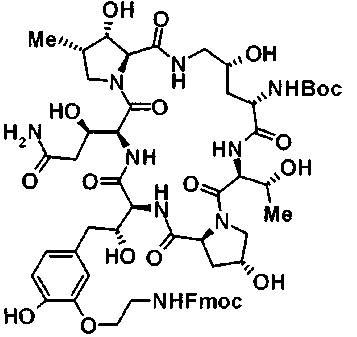














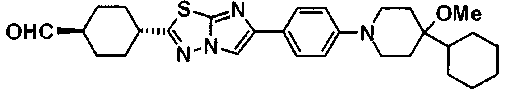













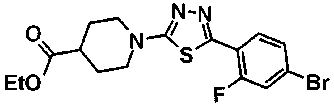
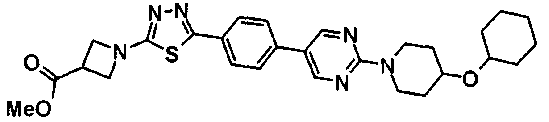




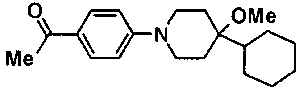






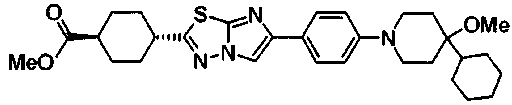







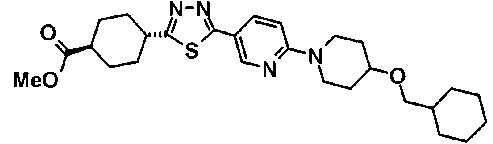









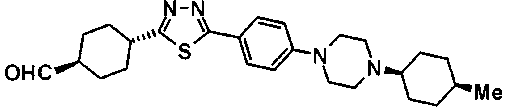


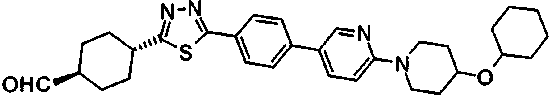
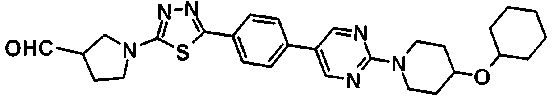
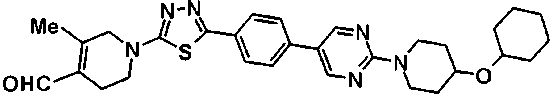
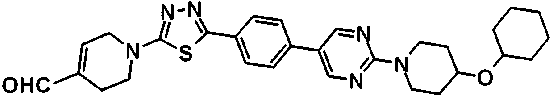


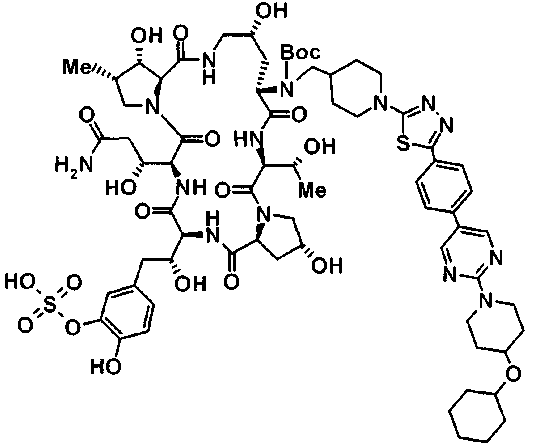






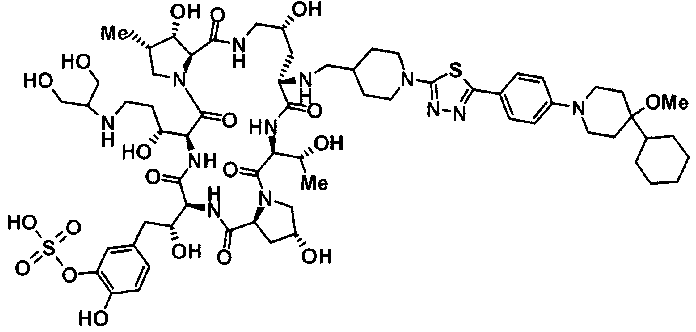

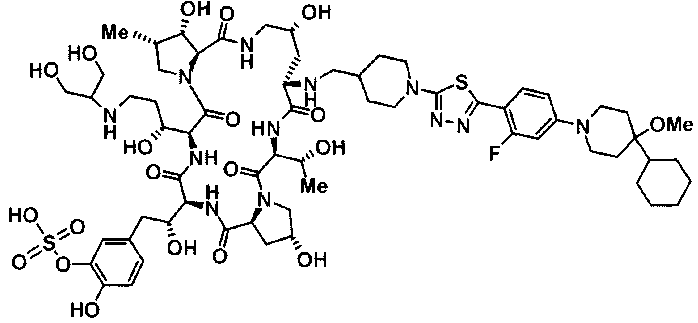





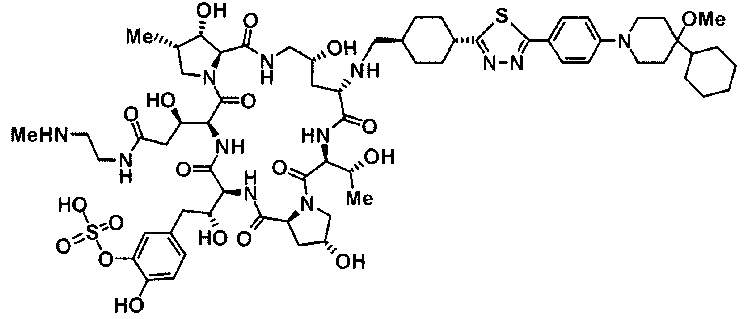
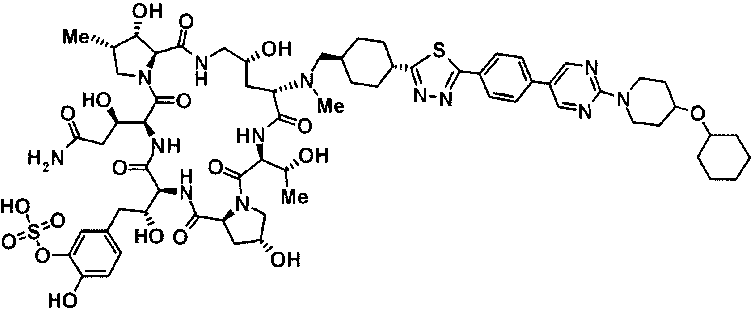



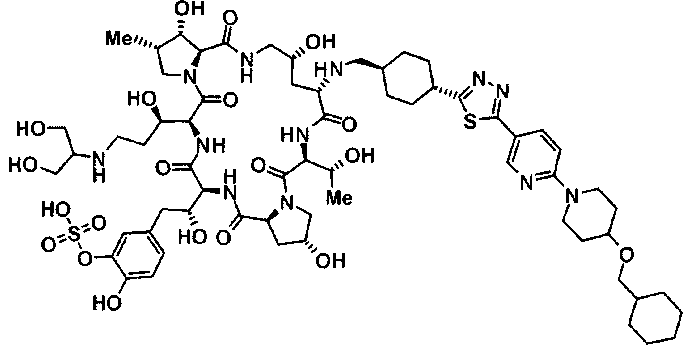

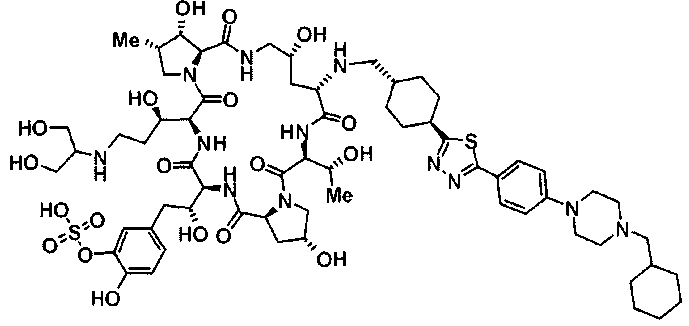




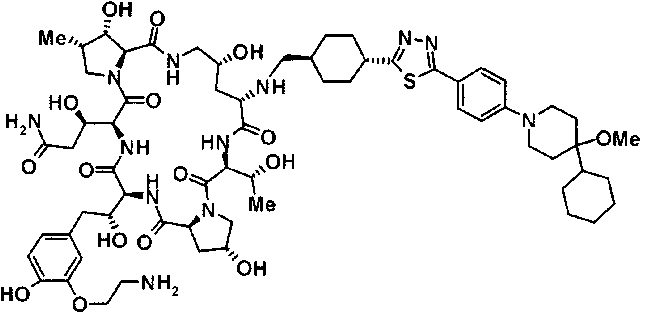







ECI+: 682,8 [(М+2H)/2]
ECI-: 1414,6
ECI+: 660,4 [(М+H)/2]
ECI-: 1418,1
| название | год | авторы | номер документа |
|---|---|---|---|
| ДИАМИНОГЕТЕРОЦИКЛИЧЕСКОЕ КАРБОКСАМИДНОЕ СОЕДИНЕНИЕ | 2010 |
|
RU2526253C2 |
| ПРОИЗВОДНЫЕ ПИПЕРИДИНОВ | 2011 |
|
RU2554353C2 |
| Новое тетерагидропиримидиновое соединение или его соль | 2015 |
|
RU2636310C1 |
| АЗААДАМАНТАНОВЫЕ ПРОИЗВОДНЫЕ И СПОСОБЫ ПРИМЕНЕНИЯ | 2007 |
|
RU2450002C2 |
| ЗАМЕЩЕННЫЕ ГЕТЕРОАРИЛОМ ПИРИДИНЫ И СПОСОБЫ ПРИМЕНЕНИЯ | 2017 |
|
RU2756743C2 |
| ПРОИЗВОДНЫЕ 5-ОКСА-2-АЗАСПИРО[3.4]ОКТАНА В КАЧЕСТВЕ АГОНИСТОВ M4 | 2020 |
|
RU2820477C1 |
| СОЕДИНЕНИЯ, ОБЛАДАЮЩИЕ ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ ПО ОТНОШЕНИЮ К PDE9A, И ИХ ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2019 |
|
RU2788148C2 |
| Соединение, обладающее агонистической активностью в отношении GPR119, способ его получения и фармацевтическая композиция, содержащая его в качестве эффективного компонента | 2015 |
|
RU2670197C1 |
| ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА, СОДЕРЖАЩИЕ ИХ КОМПОЗИЦИИ, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2004 |
|
RU2346938C2 |
| АЗОЛЬНЫЕ СОЕДИНЕНИЯ | 2009 |
|
RU2493154C2 |
Изобретение относится к новому полипептидному соединению, обладающему противогрибковой активностью. Заявленные полипептидные соединения содержат в стуктуре циклогексилметиламиногруппы или пиперидилметиламиногруппы, что обеспечивает отличную активность против глубокой грибковой инфекции. 14 н. и 2 з.п. ф-лы, 66 табл., 45 пр.
1. Соединение формулы (I) или его фармацевтически приемлемая соль

(где символы в формуле имеют следующие значения:
R1 представляет собой -CO-NR5R6, -R00-NR5R6 или -СO-(азотсодержащий гетероцикл), такой как -СО-пиперазинил;
R5 и R6 являются одинаковыми или отличными друг от друга и представляют собой H, R0, RA или RB,
RA представляет собой низший алкил, который может быть замещен одной-тремя гидроксильными группами,
RB представляет собой низший алкил, который может быть замещен одной-тремя группами, выбранными из -NH2, -NHR0 и -N(R0)2,
R2 представляет собой Н, -S(O)2-OH, R0, RA, RB или -R00-NH-R00-OH,
R4 представляет собой Н или -ОН,
R7 в каждом случае имеет одинаковое или отличное друг от друга значение и представляет собой Н или R0;
R9 представляет собой простую связь,
R13 и R14 являются одинаковыми или отличными друг от друга и представляют собой Н или R0,
кольцо Е представляет собой группу, представленную следующими формулами (II), (III), (IV), (V) или (XVIII)

где Х представляет собой СН или N,
R8 в каждом случае имеет одинаковое или отличное друг от друга значение и представляет собой Н или R0,
R10 представляет собой Н;
кольцо А представляет собой азотсодержащий гетероцикл, такой как пиримидинил, оксадиазолил, имидазотиадиазолил или тиадиазолил;
R3 представляет собой группу, представленную следующими формулами (VI), (VII) или (VIII)

где кольцо В и кольцо D являются одинаковыми или отличными друг от друга и представляют собой арил, который может быть замещен галогеном, где арил представляет собой фенил или нафтил, кольцо С представляет собой азотсодержащий гетероцикл, такой как пиридил, пиримидинил, пиперазинил или пиперидинил;
R11 и R12 являются одинаковыми или отличными друг от друга и представляют собой группу, выбранную из Н, R0, С3-С8циклоалкила,
-(С3-С8циклоалкил)-R0, -R00-(С3-С8циклоалкил), -OR0, -O-(С3-С8циклоалкил), -O-(С2-10алкилен)-ОR0, -О-R00-(С3-С8циклоалкил), -(азотсодержащий гетероцикл)-R00-(С3-С8циклоалкил), где азотсодержащий гетероцикл представляет собой пиперазинил, -(азотсодержащий гетероцикл)-O-(С3-С8циклоалкил), где азотсодержащий гетероцикл представляет собой пиперидинил, и -(азотсодержащий гетероцикл)-O-R00-(С3-С8циклоалкил), где азотсодержащий гетероцикл представляет собой пиперидинил;
R0 представляет собой низший алкил, и R00 представляет собой низший алкилен).
2. Соединение по п.1 или его фармацевтически приемлемая соль, где R1 представляет собой -CO-NR5R6 или -R00-NR5R6, R2 представляет собой -S(O)2-OH или R0, R4 представляет собой Н, R7 представляет собой Н или R0, R9 представляет собой простую связь, кольцо Е представляет собой группу, представленную формулой (II), кольцо А представляет собой моноциклический азотсодержащий гетероцикл, такой как пиримидинил, оксадиазолил или тиадиазолил, R8 представляет собой Н или R0, R3 представляет собой группу, представленную формулой (VII) или (VIII), кольцо В и кольцо D, которые представляют собой фенил, кольцо С представляет собой моноциклический азотсодержащий гетероцикл, такой как пиридил, пиримидинил, пиперазинил или пиперидинил, и R11 и R12, которые являются одинаковыми или отличными друг от друга, представляют собой каждый Н, С3-С8циклоалкил, -O-R0, -O-(С3-С8циклоалкил), -(азотсодержащий гетероцикл)-O-(С3-С8циклоалкил), где азотсодержащий гетероцикл представляет собой пиперидинил, или -(азотсодержащий гетероцикл)-O-R00- (С3-С8циклоалкил), где азотсодержащий гетероцикл представляет собой пиперидинил.
3. Соединение по п.1 или его фармацевтически приемлемая соль, где R1 представляет собой -CO-NH2, -CH2-NH2 или -CH2-NH-CH(CH2OH)2, R2 представляет собой -S(O)2-OH или метил, R4 представляет собой Н, R7 представляет собой H, кольцо Е представляет собой циклогександиил, кольцо А представляет собой тиадиазолил или имидазотиадиазолил, R3 представляет собой группу, представленную формулой (VII), кольцо В представляет собой фенил, и кольцо С представляет собой вместе с R11 и R12 группу, представленную следующими формулами (XX) или (XXI)
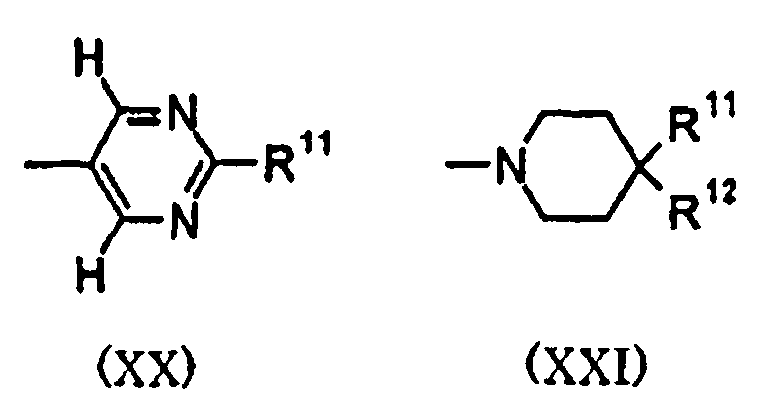
где любой из R11 и R12 представляет собой H или -О-(метил), а другой представляет собой группу, выбранную из циклогексила, -O-(циклогексил), -О-СН2-(циклобутил) и 4-(циклогексилокси) пиперидин-1-ила.
4. Фармацевтическая композиция, обладающая антифунгицидной активностью, включающая эффективное количество соединения или его фармацевтически приемлемой соли по п.1 и фармацевтически приемлемый эксципиент.
5. Фармацевтическая композиция для предупреждения или лечения грибковой инфекции, включающая эффективное количество соединения или его фармацевтически приемлемой соли по п.1 и фармацевтически приемлемый эксципиент.
6. Применение соединения или его фармацевтически приемлемой соли по п.1 для получения средства для предупреждения или лечения грибковой инфекции.
7. Способ предупреждения или лечения грибковой инфекции, включающий введение пациенту эффективного количества соединения или его фармацевтически приемлемой соли по п.1.
8. (3R)-3-{(2R,6S,11R,14aS,15S,16S,20S,23S,25aS)-9-[({тpaнc-4-[5-(4-{2-[4-(циклопентилметил)пиперазин-1-ил]пиримидин-5-ил}фенил)-1,3,4-тиадиазол-2-ил]циклогексил} метил)амино]-2,11,15-тригидрокси-6-[(1R)-1-гидроксиэтил]-23-[(1R)-1-гидрокси-2-(4-гидрокси-3-метоксифенил)этил]-16-метил-5,8,14,19,22,25-гексаоксотетракозагидро-1Н-дипирроло[2,1-с:2',1'-1][1,4,7,10,13,16]гексаазациклогеникозин-20-ил}-3-гидроксипропанамид или его фармацевтически приемлемая соль.
9. (2R,6S,9S,11R,14aS,15S,16S,20S,23S,25aS)-9-{[(тpaнc-4-{5-[4-(4-циклогексил-4-метоксипиперидин-1-ил)фенил]-1,3,4-тиадиазол-2-ил}циклогексил)метил]амино}-20-{(1R)-3-[(1,3-дигидроксипропан-2-ил)амино]-1-гидроксипропил}-2,11,15-тригидрокси-6-[(1R)-1-гидроксиэтил]-23-[(1R)-1-гидрокси-2-(4-гидрокси-3-метоксифенил)этил]-16-метилгексадекагидро-1Н-дипирроло[2,1-с:2',1'-1] [1,4,7,10,13,16]-гексаазациклогеникозин-5,8,14,19,22,25(9Н,25аН)-гексон или его фармацевтически приемлемая соль.
10. 2-гидрокси-5-{(2R)-2-гидрокси-2-[(2R,11R,14aS,15S,16S,20S,23S,25aS)-2,11,15-тригидрокси-6-[(1R)-1-гидроксиэтил]-20-[(1R)-1-гидрокси-3-{[2-гидрокси-1-(гидроксиметил)этил]амино}пропил]-9-({[транс-4-(2-{4-[(6-метоксигексил)окси]фенил}имидазо[2,1-b] [1,3,4]тиадиазол-6-ил)циклогексил]метил}амино)-16-метил-5,8,14,19,22,25-гексаоксотетракозагидро-1Н-дипирроло[2,1-с:2',1'-1][1,4,7,10,13,16]гексаазациклогеникозин-23-ил]этил}фенил гидросульфат или его фармацевтически приемлемая соль.
11. (2R,6S,11R,14aS,15S,16S,20S,23S,25aS)-20-[(1R)-3-aмино-1-гидроксипропил]-9-{[(транс-4-{5-[4-(4-циклогексил-4-метоксипиперидин-1-ил)фенил]-1,3,4-тиадиазол-2-ил}циклогексил)метил]амино}-2,11,15-тригидрокси-6-[(1R)-1-гидроксиэтил]-23-[(1R)-1-гидрокси-2-(4-гидрокси-3-метоксифенил)этил]-16-метилгексадекагидро-1Н-дипирроло[2,1-с:2',1'-1] [1,4,7,10,13,16]гексаазациклогеникозин-5,8,14,19,22,25(9Н, 25аН)-гексон или его фармацевтически приемлемая соль.
12. 5-[(2R)-2-{(2R,6S,9S,11R,14aS,15S,16S,20S,23S,25aS)-20-[(1R)-3-aмино-1-гидрокси-3-оксопропил]-9-[({транс-4-[5-(4-{2-[4-(циклогексилокси)пиперидин-1-ил]пиримидин-5-ил}фенил)-1,3,4-тиадиазол-2-ил]циклогексил}метил)амино]-2,11,15-тригидрокси-6-[(1R)-1-гидроксиэтил]-16-метил-5,8,14,19,22,25-гексаоксотетракозагидро-1Н-дипирроло[2,1-с:2',1'-1] [1,4,7,10,13,16]гексаазациклогеникозин-23-ил}-2-гидроксиэтил]-2-гидроксифенил гидросульфат или его фармацевтически приемлемая соль.
13. 5-[(2R)-2-{(2R,6S,9S,11R,14aS,15S,16S,20S,23S,25aS)-20-{(1R)-3-[(2-аминоэтил)амино]-1-гидрокси-3-оксопропил}-9-[({1-[5-(4-{2-[4-(циклогексилокси)пиперидин-1-ил]пиримидин-5-ил} фенил)-1,3,4-тиадиазол-2-ил]пиперидин-4-ил}метил)амино]-2,11,15-тригидрокси-6-[(1R)-1-гидроксиэтил]-16-метил-5,8,14,19,22,25-гексаоксотетракозагидро-1Н-дипирроло[2,1-с:2',1'-1][1,4,7,10,13,16]гексаазациклогеникозин-23-ил}-2-гидроксиэтил]-2-гидроксифенил гидросульфат или его фармацевтически приемлемая соль.
14. 5-[(2R)-2-{(2R,6S,11R,14aS,15S,16S,20S,23S,25aS)-9-({[tpaнс-4-(5-{4-[4-(циклогексилокси)пиперидин-1-ил]фенил}-1,3,4-тиадиазол-2-ил)циклогексил]метил}амино)-2,11,15-тригидрокси-6-[(1R)-1-гидроксиэтил]-20-[(1R)-1-гидрокси-3-{[2-гидрокси-1-(гидроксиметил)этил]амино}пропил]-16-метил-5,8,14,19,22,25-гексаоксотетракозагидро-1Н-дипирроло[2,1-с:2',1'-1][1,4,7,10,13,16]гексаазациклогеникозин-23-ил-2-гидроксиэтил]-2-гидроксифенил гидросульфат или его фармацевтически приемлемая соль.
15. 5-[(2R)-2-{(2R,6S,11R,14aS,15S,16S,20S,23S,25aS)-20-[(1R)-3-{[карбоки(гидрокси)метил]амино}-1-гидрокси-3-оксопропил]-9-[({транс-4-[5-(4-{2-[4-(циклогексилокси)пиперидин-1-ил]пиримидин-5-ил}фенил)-1,3,4-тиадиазол-2-ил]циклогексил}метил)амино]-2,11,15-тригидрокси-6-[(1R)-1-гидроксиэтил]-16-метил-5,8,14,19,22,25-гексаоксотетракозагидро-1Н-дипирроло[2,1-с:2',1'-1][1,4,7,10,13,16]гексаазациклогеникозин-23-ил-2-гидроксиэтил]-2-гидроксифенил сульфат или его фармацевтически приемлемая соль.
16. 5-[(2R)-2-{(2R,6S,11R,14aS,15S,16S,20S,23S,25aS)-9-[({1-[5-(4-{2-[4-(циклогексилокси)пиперидин-1-ил]пиримидин-5-ил}фенил)-1,3,4-тиадиазол-2-ил]пиперидин-4-ил}метил)амино]-2,11,15-тригидрокси-6-[(1R)-1-гидроксиэтил]-20-[(1R)-1-гидрокси-3-({2-[(2-метоксиэтил)(метил)амино]этил}амино)-3-оксопропил]-16-метил-5,8,14,19,22,25-гексаоксотетракозагидро-1Н-дипирроло[2,1-с:2',1'-1][1,4,7,10,13,16]гексаазациклогеникозин-23-ил-2-гидроксиэтил]-2-гидроксифенил сульфат или его фармацевтически приемлемая соль.
| ПОЛИПЕПТИДНОЕ СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1995 |
|
RU2165423C2 |
| Полипептидное соединение, способы его получения, фармацевтическая композиция и способ профилактики или лечения грибковых заболеваний | 2001 |
|
RU2224765C1 |
| WO 2004113368 A1, 29.12.2004 | |||
| TAWARA S | |||
| ET AL.: "In Vitro Activities of a New Lipopeptide Antifungal Agent, FK463, against a Variety of Clinically Important Fungi" ANTIMICROBIAL AGENTS AND CHEMOTHERAPY, 2000, vol.44, pages 57-62 | |||
| OKI H | |||
| ET AL.: "Koshinkinsei Tennenbutsu о Lead to sum Soyaku | |||

