Настоящее изобретение относится к гетероарилзамещенным пиразоло-пиридиновым ингибиторам протеинкиназ, которые ингибируют митоген-активируемую протеинкиназу 4 (MKK4) и в частности селективно ингибируют MKK4 по сравнению с протеинкиназами JNK1 и MKK7.
УРОВЕНЬ ТЕХНИКИ
Заболевания печени могут быть вызваны инфекцией, травмой, воздействием токсичных соединений, таких как алкоголь или лекарственные средства, аутоиммунными процессами, генетическими дефектами и другими факторами. Печень обладает замечательной регенеративной способностью, которая, однако, может быть нарушена при болезненном состоянии и, следовательно, может быть недостаточной для компенсации потери гепатоцитов и функции органа.
WO 2007/002433 описывает соединения, которые являются ингибиторами протеинкиназ, пригодными для лечения заболеваний и состояний, связанных с аберрантной активностью протеинкиназ. Эти соединения являются ингибиторами протеинкиназы Raf, в частности B-Raf и c-Raf и их мутаций и, таким образом, являются пригодными для лечения рака. Кроме того, считается, что они ингибируют большое количество других протеинкиназ, среди них c-Jun N-концевые киназы (JNK) и в частности JNK1. WO 2007/002325 содержит аналогичное раскрытие, и WO 2012/109075 и WO 2014/194127 раскрывают модифицированные соединения, обладающие активностью ингибирования протеинкиназы Raf. H. Vin et al. ссылаются на два соединения WO 2007/002433 в качестве ингибиторов B-Raf, которые подавляют апоптоз посредством нецелевого ингибирования передачи сигналов JNK. WO 2010/111527 описывает пиразоло[3,4-b]пиридиновые соединения, которые являются ингибиторами протеинкиназ, пригодными для лечения заболевания или состояния, опосредованного протеинкиназой Raf, такого как рак. Кроме того, считается, что они ингибируют большое количество других протеинкиназ, среди них c-Jun N-концевые киназы (JNK) и в частности JNK1. WO 2012/136859 раскрывает некоторые соединения, которые описаны как ингибиторы митоген-активируемой протеинкиназы 4 (MKK4) и как пригодные при лечении печеночной недостаточности, для защиты гепатоцитов от апоптоза и для регенерации гепатоцитов. Wuestefeld et al. (Cell 153:389-401, 2013) и Willebring et al. (Cell 153:283-284) описывают функциональный генетический подход к идентификации генов-мишеней, которые можно использовать для увеличения регенеративной способности гепатоцитов. В частности, Wuestefeld et al. идентифицируют протеинкиназу MKK4 как ключевой регулятор регенерации печени и сообщают, что подавление MKK4 увеличивало регенерацию гепатоцитов за счет компенсаторной повышенной регуляции MKK7 и JNK1-зависимой активации ATF2 и ELK1.
На основании результатов предшествующего уровня техники был сделан вывод, что ингибиторы MKK4 и JNK1 могут быть пригодными для лечения заболеваний, опосредованных JNK1. Однако, несмотря на признание того, что ингибирование JNK1 может быть предпочтительным для лечения заболеваний печени, клинических исследований не проводилось. В WO 2018/134254 раскрыты пирроло-пиридиновые соединения, которые являются ингибиторами протеинкиназ для стимуляции регенерации печени или снижения или предотвращения гибели гепатоцитов.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Проблема, лежащая в основе изобретения, заключалась в предоставлении соединений, которые являются пригодными ингибиторами MKK4, в частности ингибиторами MKK4, которые селективно ингибируют MKK4 по сравнению с MKK7 и JNK1. Дополнительная проблема заключалась в предоставлении соединений, которые являются ингибиторами MKK4, которые селективно ингибируют MKK4 по сравнению с MKK7 и JNK1, которые являются пригодными для лечения заболеваний печени и особенно для стимуляции регенерации печени или снижения или предотвращения гибели гепатоцитов.
Эта проблема была решена путем предоставления соединений формулы (I).
Таким образом, изобретение относится к следующим вариантам осуществления:
1. Соединение, имеющее формулу (I)
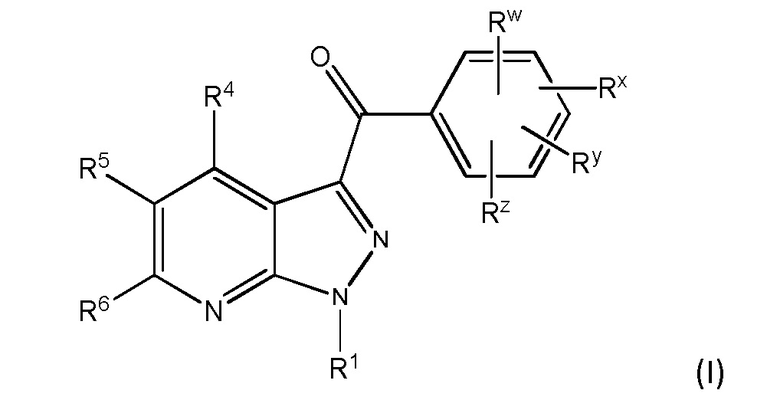
и его фармацевтически приемлемые соли, пролекарства, биологически активные метаболиты, сольваты и стереоизомеры,
в котором переменные в формуле (I) имеют следующие значения:
R1 представляет собой H или алкил;
R4 представляет собой Н или алкил;
R5 выбран из
а) пиримидинила, который замещен 1 или 2 заместителями, независимо выбранными из циклоалкила, алкила, -COOR10, -ОН, алкилсульфанила, алкилсульфинила, алкилсульфонила,
тетразолила, CN, галогена, алкокси, -(NR10=)S(=O)-алкил[S-алкилсульфонимидоила] и
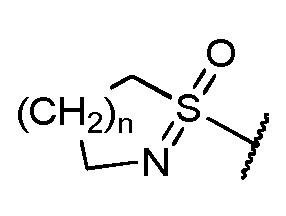 и
и
b1) пиридила, который замещен 1 или 2 заместителями, независимо выбранными из алкила и галогена, и который необязательно дополнительно замещен группой, выбранной из -OH, алкокси, CN, -COOR10, CF3, -(NR10=)S(=O)-алкила и
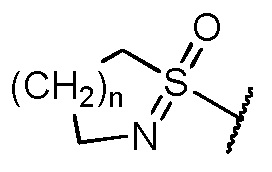 и
и
b2) пиридила, замещенного -COOR10 и дополнительно замещенного -OH, CN или CF3;
R6 представляет собой Н или алкил;
Rw представляет собой -NR10SO2R12;
Rx представляет собой Н, галоген или алкил;
Ry представляет собой Н, галоген или алкил;
Rz представляет собой Н, галоген или алкил;
в которой один, или два, или три из Rx, Ry или Rz представляют собой галоген и другой(ие) из Rx, Ry или Rz представляет собой H или алкил;
R10 в каждом случае представляет собой независимо Н или алкил;
R12 представляет собой Н, алкил или фенилалкил и
n равно 1 или 2.
2. Соединение и его фармацевтически приемлемые соли, пролекарства, сольваты и оптические изомеры по варианту осуществления 1, в котором R1 представляет собой H.
3. Соединение и его фармацевтически приемлемые соли, пролекарства, сольваты и оптические изомеры по варианту осуществления 1 или 2, в котором 2 или 3 из Rx, Ry или Rz представляют собой галоген.
4. Соединение и его фармацевтически приемлемые соли, пролекарства, сольваты и оптические изомеры по любому одному из вариантов осуществления 1-3, в котором атом галогена или атомы галогена Rx, Ry или Rz представляют собой независимо F или Cl, в частности F.
5. Соединение и его фармацевтически приемлемые соли, пролекарства, сольваты и оптические изомеры по любому одному из предшествующих вариантов осуществления, в котором R4 и R6 представляют собой Н.
6. Соединение и его фармацевтически приемлемые соли, пролекарства, сольваты и оптические изомеры по варианту осуществления 6, в котором R5 представляет собой пиримидинил, замещенный 1 или 2 заместителями, независимо выбранными из циклоалкила, алкила, -COOR10, алкокси, ОН, алкилсульфанила, алкилсульфинила, алкилсульфонила, галогена, CN и тетразолила.
7. Соединение и его фармацевтически приемлемые соли, пролекарства, сольваты и оптические изомеры по варианту осуществления 6, в котором R5 представляет собой пиримидинил, замещенный 1 или 2 заместителями, независимо выбранными из циклоалкила, алкила, алкокси, ОН, алкилсульфанила, алкилсульфинила, алкилсульфонила, галогена, CN и тетразолила.
8. Соединение и его фармацевтически приемлемые соли, пролекарства, сольваты и оптические изомеры по варианту осуществления 7, в котором R5 представляет собой пиримидинил, замещенный 1 или 2 заместителями, независимо выбранными из циклоалкила, алкокси, ОН, алкилсульфанила, алкилсульфинила, алкилсульфонила, галогена, CN и тетразолила.
9. Соединение и его фармацевтически приемлемые соли, пролекарства, сольваты и оптические изомеры по варианту осуществления 8, в котором R5 представляет собой пиримидинил, замещенный 1 или 2 заместителями, независимо выбранными из циклоалкила, алкокси, ОН, алкилсульфанила, галогена, CN и тетразолила.
10. Соединение и его фармацевтически приемлемые соли, пролекарства, сольваты и оптические изомеры по варианту осуществления 9, в котором R5 представляет собой пиримидинил, замещенный 1 или 2 заместителями, независимо выбранными из циклоалкила, алкокси, -ОН, галогена и алкилсульфанила.
11. Соединение и его фармацевтически приемлемые соли, пролекарства, сольваты и оптические изомеры по любому одному из вариантов осуществления 1-5, в котором R5 представляет собой пиримидинил, замещенный группой, выбранной из циклоалкила, алкила, -COOR10, алкокси, ОН, алкилсульфанила, алкилсульфинила и алкилсульфонила, и дополнительно замещенный группой, выбранной из галогена, CN, тетразолила, -(NR10=)S(=O)-алкила и 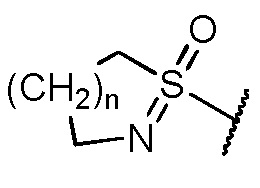 .
.
12. Соединение и его фармацевтически приемлемые соли, пролекарства, сольваты и оптические изомеры по варианту осуществления 7, в котором R5 представляет собой пиримидинил, замещенный группой, выбранной из циклоалкила, алкила, алкокси, -ОН и алкилсульфанила, и дополнительно замещенный группой, выбранной из алкила, алкокси и алкилсульфанила.
13. Соединение и его фармацевтически приемлемые соли, пролекарства, сольваты и оптические изомеры по варианту осуществления 12, в котором R5 представляет собой пиримидинил, замещенный группой, выбранной из циклоалкила, алкокси, -ОН и алкилсульфанила, и дополнительно замещенный группой, выбранной из алкила, алкокси и алкилсульфанила.
14. Соединение и его фармацевтически приемлемые соли, пролекарства, сольваты и оптические изомеры по любому одному из вариантов осуществления 1-5, в котором R5 представляет собой пиримидинил, который замещен циклоалкильной группой и который необязательно дополнительно замещен группой, выбранной из циклоалкила, алкила, -COOR10, -OH, алкилсульфанила, алкилсульфинила, алкилсульфонила, тетразолила, CN, галогена, алкокси, -(NR10=)S(=O)-алкил[S-алкилсульфонимидоила] и 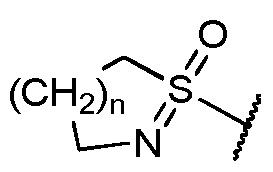 .
.
15. Соединение и его фармацевтически приемлемые соли, пролекарства, сольваты и оптические изомеры по варианту осуществления 14, в котором R5 представляет собой пиримидинил, который замещен циклоалкильной группой и который необязательно дополнительно замещен группой, выбранной из циклоалкила, алкила, -COOR10, -ОН, алкилсульфанила, алкилсульфинила, алкилсульфонила, тетразолила, CN, галогена и алкокси.
16. Соединение и его фармацевтически приемлемые соли, пролекарства, сольваты и оптические изомеры по варианту осуществления 15, в котором R5 представляет собой пиримидинил, который замещен циклоалкильной группой и который необязательно дополнительно замещен группой, выбранной из циклоалкила, алкила, -COOR10, -ОН, алкилсульфанила, тетразолила, CN, галогена и алкокси.
17. Соединение и его фармацевтически приемлемые соли, пролекарства, сольваты и оптические изомеры по варианту осуществления 16, в котором R5 представляет собой пиримидинил, который замещен циклоалкильной группой и который необязательно дополнительно замещен группой, выбранной из алкила, -COOR10, -ОН, алкилсульфанила, CN, галогена и алкокси.
18. Соединение и его фармацевтически приемлемые соли, пролекарства, сольваты и оптические изомеры по любому одному из предшествующих вариантов осуществления, в котором R5 представляет собой пиримидинил, замещенный во 2-м положении.
19. Соединение и его фармацевтически приемлемые соли, пролекарства, сольваты и оптические изомеры по любому одному из вариантов осуществления 1-5, в котором R5 представляет собой пирид-3-ил (пиридильная группа связана в 3-м положении с пиразолопиридиновой группой), замещенный -COOR10 во 2-м положении и дополнительно замещенный -OH, CN или CF3.
20. Соединение и его фармацевтически приемлемые соли, пролекарства, сольваты и оптические изомеры по любому одному из вариантов осуществления 1-5, в котором R5 представляет собой пиридил-4-ил (пиридильная группа связана в 4-м положении с пиразолопиридиновой группой), замещенный алкилом или галогеном в 3-м положении.
21. Соединение и его фармацевтически приемлемые соли, пролекарства, сольваты и оптические изомеры по любому одному из предшествующих вариантов осуществления, в котором R10 представляет собой H или алкил, в частности H.
22. Соединение и его фармацевтически приемлемые соли, пролекарства, сольваты и оптические изомеры по любому одному из предшествующих вариантов осуществления, в котором R12 представляет собой алкил или фенилалкил.
23. Соединение и его фармацевтически приемлемые соли, пролекарства, сольваты и оптические изомеры по варианту осуществления 22, в котором R12 представляет собой C1-C3-алкил или бензил.
24. Соединение и его фармацевтически приемлемые соли, пролекарства, сольваты и оптические изомеры по любому одному из предшествующих вариантов осуществления, имеющее формулу (Ia)
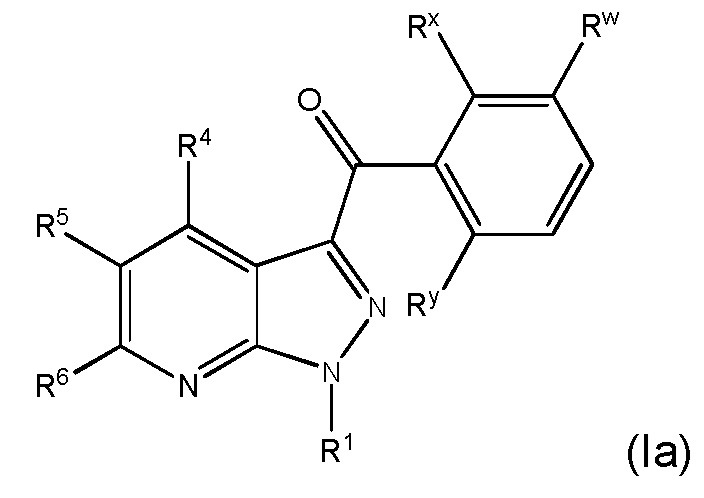
в которой
Rx представляет собой галоген;
Ry представляет собой галоген и
R1, R4, R5, R6 и Rw представляют собой, как определено по любому одному из предшествующих вариантов осуществления.
25. Соединение и его фармацевтически приемлемые соли, пролекарства, сольваты и оптические изомеры по любому одному из вариантов осуществления 1-23, имеющее формулу (Ib)
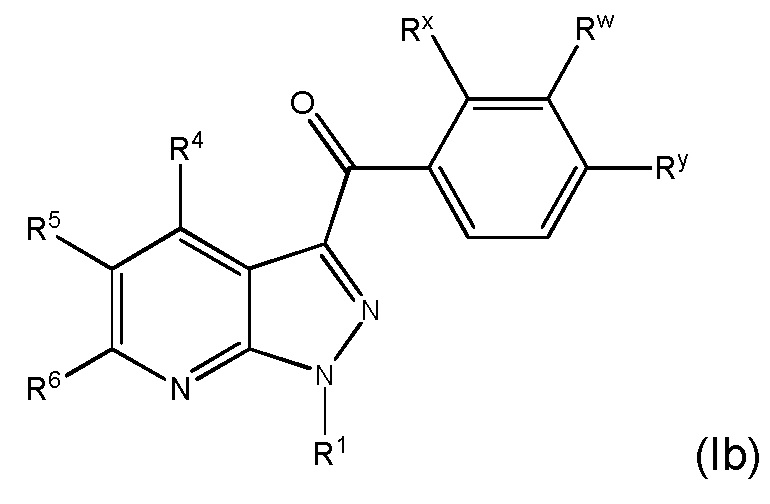
в которой
Rx представляет собой галоген;
Ry представляет собой галоген и
R1, R4, R5, R6 и Rw представляют собой, как определено по любому одному из вариантов осуществления 1-15.
26. Соединение и его фармацевтически приемлемые соли, пролекарства, сольваты и оптические изомеры по любому одному из вариантов осуществления 1-23, имеющее формулу (Ic)
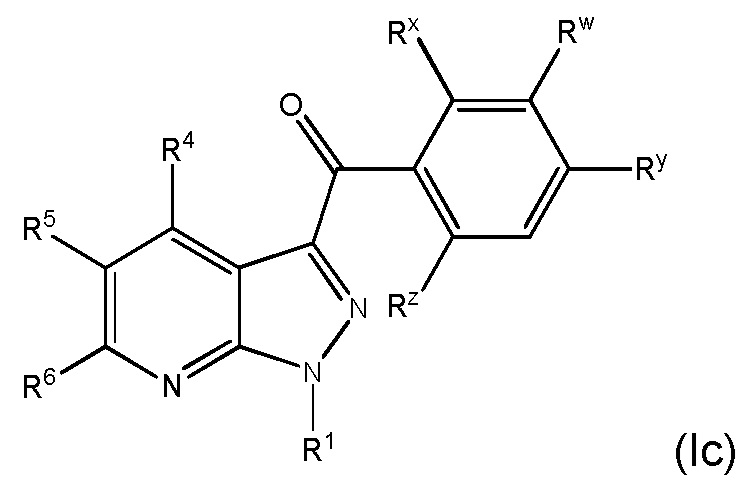
в которой
Rx представляет собой галоген;
Ry представляет собой галоген;
Rz представляет собой галоген и
R1, R4, R5, R6 и Rw представляют собой, как определено по любому одному из вариантов осуществления 1-15.
27. Соединение и его фармацевтически приемлемые соли, пролекарства, сольваты и оптические изомеры по любому одному из вариантов осуществления 24-26, в котором Rx, Ry и Rz (если присутствуют) представляют собой F или Cl, в частности F.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В варианте осуществления изобретение относится к соединению формулы I и его фармацевтически приемлемым солям, пролекарствам, сложным эфирам, сольватам и оптическим изомерам, в котором R1, R4-R6, Rw, Rx, Ry и Rz представляют собой, как определено выше в любой комбинации.
В дополнительном варианте осуществления изобретение относится к соединению формулы (Ia), (Ib) и (Ic) и его фармацевтически приемлемым солям, пролекарствам, сложным эфирам, сольватам и оптическим изомерам, в котором переменные представляют собой, как определено в вариантах осуществления выше.
В дополнительном варианте осуществления, по меньшей мере, два из Rx, Ry или Rz представляют собой галоген и другой из Rx, Ry и Rz представляет собой Н, галоген или алкил, в частности алкил или галоген. Галоген представляет собой предпочтительно F или Cl, и в частности F.
В дополнительном варианте осуществления R1, R4 и R6 представляют собой H.
В дополнительном варианте осуществления R12 представляет собой метил, этил или пропил.
В варианте осуществления изобретение относится к ингибиторам MKK4 формулы (I) и (Ia)-(Ic) и их фармацевтически приемлемым солям, пролекарствам, сольватам и оптическим изомерам, и в частности к ингибиторам MKK4, которые селективно ингибируют протеинкиназу MKK4 по сравнению с протеинкиназами JNK1 и MKK7.
Кроме того, изобретение также относится к соединениям изобретения для применения при ингибировании протеинкиназы MKK4, и в частности для применения при селективном ингибировании протеинкиназы MKK4 по сравнению с протеинкиназами JNK1 и MKK7.
Кроме того, изобретение также относится к указанным соединениям для использования в стимулировании регенерации печени или снижении или предотвращении гибели гепатоцитов и в то же время увеличении пролиферации гепатоцитов.
Изобретение также включает фармацевтически приемлемые соли соединений, упомянутых выше. Фармацевтически приемлемые соли представляют собой, в частности, кислотно- или основно-аддитивные соли с фармацевтически приемлемыми кислотами или основаниями. Примеры подходящих фармацевтически приемлемых органических и неорганических кислот представляют собой хлористоводородную кислоту, бромистоводородную кислоту, фосфорную кислоту, серную кислоту, сульфаминовую кислоту, C1-C4-алкилсульфокислоты, такие как метансульфокислота, циклоалифатические сульфокислоты, такие как S-(+)-10-камфорсульфокислота, ароматические сульфокислоты, такие как бензолсульфокислота и толуолсульфокислота, ди- и трикарбоновые кислоты и гидроксикарбоновые кислоты, содержащие от 2 до 10 атомов углерода, такие как щавелевая кислота, малоновая кислота, малеиновая кислота, фумаровая кислота, молочная кислота, винная кислота, лимонная кислота, гликолевая кислота, адипиновая кислота и бензойная кислота. Другие пригодные для использования кислоты описаны, например, в Fortschritte der Arzneimittelforschung [Advances in drug research], Volume 10, pages 224 ff., Birkhauser Verlag, Basel and Stuttgart, 1966. Примеры подходящих фармацевтически приемлемых органических и неорганических оснований представляют собой гидроксиды щелочных металлов, такие как гидроксид натрия или гидроксид калия, гидроксиды щелочноземельных металлов, такие как гидроксид кальция или магния, гидроксид аммония, органические азотсодержащие основания, такие как диметиламин, триметиламин, этаноламин, диэтаноламин, триэтаноламин, холин, 2-амино-2-гидроксиметилпропан-1,3-диол, меглумин, прокаин и т. д., L-аргинин, L-лизин, этилендиамин или гидроксиэтилпирролидин.
Изобретение также включает любую таутомерную, кристаллическую и полиморфную форму соединений и солей настоящего изобретения и их смеси.
Изобретение также включает сольваты, такие как гидраты.
Соединения изобретения могут содержать один или более хиральных центров и существовать в различных оптически активных формах, таких как энантиомеры и диастереомеры.
Используемый в настоящем описании термин «пролекарство» относится к агенту, который превращается в исходное лекарственное средство in vivo в результате какого-либо физиологического химического процесса. Примером, без ограничения, пролекарства может быть соединение настоящего изобретения в форме сложного эфира.
Пролекарства обладают многими полезными свойствами. Например, пролекарство может быть более растворимым в воде, чем конечное лекарство, что облегчает внутривенное введение лекарственного средства. Пролекарство также может иметь более высокий уровень пероральной биодоступности, чем конечное лекарственное средство. После введения пролекарство подвергается ферментативному или химическому расщеплению для доставки готового лекарственного средства в кровь или ткань. Иллюстративные пролекарства включают, но не ограничиваются ими, соединения с заместителями карбоновой кислоты, в которых свободный водород замещен на (C1-C4)алкил, (C1-C12)алканоилоксиметил, (C4-C9)1-(алканоилокси)этил, 1-метил-1-(алканоилокси)этил, содержащий от 5 до 10 атомов углерода, алкоксикарбонилоксиметил, содержащий от 3 до 6 атомов углерода, 1-(алкоксикарбонилокси)этил, содержащий от 4 до 7 атомов углерода, 1-метил-1-(алкоксикарбонилокси)этил, содержащий от 5 до 8 атомов углерода, N-(алкоксикарбонил)аминометил, содержащий от 3 до 9 атомов углерода, 1-(N-(алкоксикарбонил)амино)этил, содержащий от 4 до 10 атомов углерода, 3-фталидил, 4-кротонолактонил, гамма-бутиролактон-4-ил, ди-N, N-(C1-C2)алкиламино(C2-C3)алкил (такой как β-диметиламиноэтил), карбамоил-(C1-C2)алкил, N, N-ди(C1-C2)-алкилкарбамоил-(C1-C2)алкил и пиперидино-, пирролидино- или морфолино(C2-C3)алкил. Другие иллюстративные пролекарства высвобождают спирт Формулы (I), в которой свободный водород гидроксильного заместителя (например, группа R содержит гидроксил) замещен на (C1-C6)алканоилоксиметил, 1-((C1-C6)алканоилокси)этил, 1-метил-1-((C1-C6)алканоилокси)этил, (C1-C12)алкоксикарбонилоксиметил, N-(C1-C6)-алкоксикарбониламинометил, сукциноил, (C1-C6)алканоил, α-амино(C1-C4)алканоил, арилактил и α-аминоацил или α-аминоацил-α-аминоацил, в котором указанные α-аминоацильные фрагменты независимо представляют собой любую из встречающихся в природе L-аминокислот, обнаруженных в белках, P(O)(OH)2, -P(O)(O(C1-C6)алкил)2 или гликозил (радикал, образующийся в результате отщепления гидроксила полуацеталя углевода).
Выражение «ингибитор MKK4» означает, что киназная активность MKK4 ингибируется при IC50˂10 мкмоль/л, предпочтительно ˂1 мкмоль/л, и в частности ˂0,5 мкмоль/л. Выражение «селективно ингибировать протеинкиназу MKK4 по сравнению с протеинкиназами JNK1 и MKK7», используемое в настоящем описании, означает, что соотношение ингибирующей активности MKK7 и ингибирующей активности MKK4 или соотношение ингибирующей активности JNK1 и ингибирующей активности MKK4, выраженное или в процентах от контроля, или Kd, составляет ≥10, как измерено с помощью KINOMEscan™.
Используемое в настоящем описании выражение «стимулирование регенерации печени или снижение или предотвращение гибели гепатоцитов» означает увеличение относительного количества пролиферирующих гепатоцитов, по меньшей мере, на 30%, предпочтительно, по меньшей мере, на 50% по сравнению с количеством пролиферирующих клеток в начале терапии. В частности, экспрессия означает увеличение на ≥100% по сравнению с количеством пролиферирующих клеток в начале терапии. В этом контексте экспериментальное определение и количественная оценка будут выполняться с использованием стандартных способов, например, количественное определение белка Ki67, который тесно связан с пролиферацией клеток. Для количественного определения пролиферирующих гепатоцитов на предметном стекле доступно несколько стандартных иммуногистохимических методов, в которых используется первичное анти-Ki67 антитело с последующей визуализацией анти-Ki67-связывания с использованием, например, вторичного антитела, конъюгированного с пероксидазой хрена. Величина пероксидазной активности, которая визуализируется ферментативной конверсией хромогенных субстратов, коррелирует с количеством белка Ki67 и количеством пролиферирующих клеток.
В экспериментах, описанных ниже, пролиферацию гепатоцитов количественно определяли с помощью окрашивания Ki67 с использованием первичного поликлонального кроличьего анти-Ki67 антитела от Abcam (статья № ab15580, Abcam, Кембридж, США) и флуорофора тетраметилродамина, содержащего вторичное козье поликлональное антитело от Invitrogen (статья № 16101, Invitrogen/ThermoFisher). На основании данных, полученных на нескольких доклинических мышиных моделях, было обнаружено, что опосредованная мшРНК (малая шпилечная РНК) супрессия MKK4 на мышиной модели хронического CCl4 (тетрахлорид углерода) опосредованного повреждения печени увеличивала пролиферацию гепатоцитов от 13% до 27% (по сравнению с контрольной мшРНК) и была связана с пониженным повреждением печени (трансаминазы) и пониженным фиброзом печени. В соответствии с определением в предшествующей главе относительное увеличение пролиферирующих клеток составляло 108%. На модели алкогольного стеатогепатита (АСГ) опосредованное мшРНК подавление MKK4 приводило к скорости пролиферации гепатоцитов 4% по сравнению с 2% при использовании контрольной мшРНК (относительное увеличение: 100%). Дублирование пролиферации гепатоцитов ассоциировалось с пониженным стеатозом (отложением жира) и пониженным повреждением печени, как измерено с помощью трансаминаз. Аналогично, опосредованное мшРНК подавление MKK4 увеличивало пролиферацию гепатоцитов от 16% (контрольная мшРНК) до 33% (относительное увеличение: 106%) на модели частичной гепатэктомии (48 часов после хирургического удаления двух третей печени). В свою очередь, повышенная пролиферация гепатоцитов была связана с улучшенной регенерацией печени и более быстрым восстановлением массы печени. В заключение, эти исследования подтверждают, что MKK4 является терапевтической мишенью для лечения острых и хронических заболеваний печени. Кроме того, в WO 2018/134254 раскрыты новые соединения, которые ингибируют MKK4 селективно по сравнению с MKK7 и JNK1. На экспериментальных моделях регенерации печени in vitro и in vivo эти соединения оказались эффективными для предотвращения острой печеночной недостаточности, вызванной введением Jo2 антитела, и индуцировали пролиферацию выделенных первичных мышиных гепатоцитов.
Новые соединения, раскрытые в настоящей заявке, представляют собой эффективные ингибиторы MKK4 с селективностью в отношении MKK7 и JNK1, и, следовательно, по аналогии с соединениями, раскрытыми в WO 2018/134254, их можно использовать для лечения заболеваний печени и для стимуляции регенерации печени или снижения или предотвращения гибели гепатоцитов.
Органические фрагменты, упомянутые в вышеприведенных определениях переменных представляют собой, подобно термину «галоген», собирательные термины для индивидуальных списков отдельных членов группы. Префикс Cn-Cm указывает в каждом случае возможное количество атомов углерода в группе.
Термин галоген означает в каждом случае фтор, бром, хлор или иод, в частности фтор или хлор и предпочтительно фтор.
Алкил представляет собой алкильную группу с прямой или разветвленной цепью, которая представляет собой предпочтительно C1-C6-алкильную группу, т. е. алкильную группу, содержащую от 1 до 6 атомов углерода, и более предпочтительно C1-C4-алкильную группу, и в частности C1-C3-алкильную группу. Примеры алкильной группы представляют собой метил, этил, н-пропил, изопропил, н-бутил, 2-бутил, изобутил, трет-бутил, пентил, 1-метилбутил, 2-метилбутил, 3-метилбутил, 2,2-диметилпропил, 1-этилпропил, гексил, 1,1-диметилпропил, 1,2-диметилпропил, 1-метилпентил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 1,1-диметилбутил, 1,2-диметилбутил, 1,3-диметилбутил, 2,2-диметилбутил, 2,3-диметилбутил, 3,3-диметилбутил, 1-этилбутил, 2-этилбутил, 1,1,2-триметилпропил, 1,2,2-триметилпропил, 1-этил-1-метилпропил и 1-этил-2-метилпропил.
Определение алкила подобным образом применимо к любой группе, которая включает алкильную группу, такую как алкокси, алкилсульфинил, фенилалкил и т. д.
Галогеналкил представляет собой галогенированную алкильную группу, как определено выше, в которой, по меньшей мере, один, например, 1, 2, 3, 4 или все атомы водорода замещены 1, 2, 3, 4 или соответствующим количеством одинаковых или различных атомов галогена, такую как трифторметил, хлорметил, бромметил, дифторметил, фторметил, дифторэтил и т. д. Определенные примеры включают фторированные C1-C4 алкильные группы, как определено, такие как трифторметил, дифторметил, фторметил или дифторэтил.
Циклоалкил представляет собой циклоалифатический радикал, который представляет собой предпочтительно C3-C8-циклоалкил, т. е. циклоалкильную группу, содержащую от 3 до 8 атомов углерода. В частности, от 3 до 6 атомов углерода образуют циклическую структуру, такую как циклопропил, циклобутил, циклопентил и циклогексил. Циклическая структура может быть незамещенной или может содержать 1, 2, 3 или 4 C1-C4алкильных радикала, предпочтительно один или более метильных радикалов.
Соединения изобретения могут быть получены, как раскрыто в WO 2010/111527, которая включена в настоящее описание в полном объеме посредством ссылки или в соответствии с аналогичными методиками. Кислотно- или основно-аддитивные соли получают стандартным способом, смешивая свободное основание с соответствующей кислотой или смешивая свободную кислоту с желаемым основанием. Необязательно реакцию проводят в растворе в органическом растворителе, например, низшем спирте, таком как МеОН, этанол или пропанол, эфире, таком как метил-трет-бутиловый эфир или диизопропиловый эфир, кетоне, таком как ацетон или метилэтилкетон или сложном эфире, таком как EtOAc.
Соединения изобретения являются пригодными для стимуляции регенерации печени или снижения или предотвращения гибели гепатоцитов и в то же время для увеличения пролиферации гепатоцитов. Таким образом, соединения являются пригодными для лечения, модулирования, улучшения или предотвращения заболеваний, которые включают острые или хронические повреждения печени, которые могут быть вызваны инфекцией, травмой, воздействием токсических соединений, аномальным накоплением нормальных веществ в крови, аутоиммунным процессом, генетическим нарушением или неизвестными причинами.
Такие заболевания печени включают все заболевания, при которых повышенная регенерация печени и снижение или предотвращение гибели гепатоцитов могут быть полезными для достижения потенциального терапевтического эффекта, т. е. частичного или полного восстановления функций печени. Такие заболевания включают острые и хронические или острые при хронических заболеваниях печени, такие как острые и хронические вирусные гепатиты, такие как гепатиты В, С, Е, гепатиты, вызванные вирусом Эпштейна-Барр, цитомегаловирусом, вирусом простого герпеса и другими вирусами, все виды аутоиммунного гепатита, первичный склерозирующий гепатит, алкогольный гепатит;
метаболические заболевания печени, такие как метаболический синдром, жировая дистрофия печени, такая как неалкогольная жировая болезнь печени (НАЖБП), неалкогольный стеатогепатит (НАСГ), алкогольный стеатогепатит (АСГ), болезнь Вильсона, гемохроматоз, дефицит альфа-1-антитрипсина, болезни накопления гликогена;
все виды цирроза печени, такие как первичный билиарный цирроз, этилтоксический цирроз печени, криптогенный цирроз;
острая (фульминантная) или хроническая печеночная недостаточность, такая как токсическая печеночная недостаточность, такая как печеночная недостаточность, вызванная ацетаминофеном (парацетамолом), печеночная недостаточность, вызванная альфа-аманитином, гепатотоксичность, вызванная лекарственными средствами, печеночная недостаточность, вызванная, например, антибиотиками, нестероидными противовоспалительными средствами и противосудорожными средствами, острая печеночная недостаточность, вызванная растительными добавками (кава, эфедра, шлемник, мята болотная и т. д.), заболевание печени и недостаточность вследствие сосудистых заболеваний, таких как синдром Бадда-Киари, острая печеночная недостаточность неизвестного происхождения, хронические заболевания печени вследствие правожелудочковой недостаточности;
галактоземия, кистозный фиброз, порфирия, нарушение перфузии при ишемии печени, синдром малой доли после трансплантации печени, первичный склерозирующий холангит или печеночная энцефалопатия.
Для стимуляции регенерации печени или уменьшения или предотвращения гибели гепатоцитов соединения изобретения вводят пациенту, нуждающемуся в таком лечении, в терапевтически эффективном количестве. Различные диагностические методы являются доступными для выявления наличия заболевания печени. Известно, что уровни аланинаминотрансферазы (АЛТ) и аспартатаминотрансферазы (АСТ) в крови, превышающие клинически приемлемые нормальные диапазоны, указывают на продолжающееся повреждение печени. Уровни билирубина в крови или другие ферменты печени могут использоваться в качестве критериев обнаружения или диагностики. Повседневный мониторинг уровней АЛТ и АСТ в крови пациентов с заболеваниями печени используется для измерения прогрессирования заболевания печени во время лечения. Снижение повышенных уровней АЛТ и АСТ до допустимого нормального диапазона расценивается как клиническое свидетельство, отражающее уменьшение степени тяжести поражения печени у пациентов. Коммерческие анализы, такие как FibroTest/FibroSURE, HepaScore®, FibroMeter или Cirrhometer, оценивают комбинированные результаты пяти и более биохимических параметров для выявления стеатоза, фиброза и цирроза печени. Кроме того, неинвазивные, инновационные методы физической визуализации, такие как методы магнитно-резонансной томографии, сонографии, и в частности эластографии, доступны для выявления и мониторинга состояния и прогрессирования заболеваний печени.
Кроме того, было обнаружено, что супрессия MKK4, опосредованная мшРНК, ослабляет управляемую ФНО-α деградацию хрящевого матрикса при остеоартрите (Cell Death and Disease (2017) 8, e3140). Таким образом, ингибирование активности MKK4 с использованием соединений изобретения также является пригодным для лечения остеоартрита и ревматоидного артрита.
Кроме того, ингибиторы MKK4 также могут быть пригодными для лечения нейродегенеративных заболеваний, таких как болезнь Альцгеймера и болезнь Паркинсона. Grueninger et al. обнаружили, что в клетках нейробластомы человека MKK4 играет ключевую роль в фосфорилировании тау-белка по серину 422, что способствует агрегации тау (Mol Cell Biochem (2011) 357:199-207). Ингибиторы фосфорилирования тау, которые предотвращают агрегацию тау, считаются пригодными для предотвращения или лечения болезни Альцгеймера.
Недавно был описан ингибитор MKK4 с активными нейропротекторными эффектами in vitro и in vivo. В культурах гиппокампа инкубация с ингибитором MKK4 предотвращала гибель клеток, индуцированную глутаматом, и активацию каспазы-3, а также ингибировала гибель клеток, индуцированную N-метил-4-фенилпиридиния иодидом и амилоидом β1-42, в клетках SH-SY5Y. Это же соединение также облегчало индуцированную 1-метил-4-фенил-1,2,3,6-тетрагидропиридином дегенерацию нигростриарных дофаминергических нейронов у мышей (Biochemical Pharmacology (2018), Vol. 162, April 2019, 109-122; doi: https://doi.org/10.1016/j.bcp.2018.10.008).
Соединения изобретения обычно вводят в форме фармацевтических композиций, которые содержат, по меньшей мере, одно соединение в соответствии с изобретением, необязательно вместе с инертным носителем (например, фармацевтически приемлемым эксципиентом) и в случае необходимости с другими лекарственными средствами. Эти композиции можно, например, вводить перорально, ректально, трансдермально, подкожно, внутрибрюшинно, внутривенно, внутримышечно или интраназально.
Примеры подходящих фармацевтических композиций представляют собой твердые лекарственные формы, такие как порошки, гранулы, таблетки, в частности таблетки с пленкой, пастилки, саше, облатки, таблетки с сахарным покрытием, капсулы, такие как твердые желатиновые капсулы и мягкие желатиновые капсулы, или суппозитории, полутвердые лекарственные формы, такие как мази, кремы, гидрогели, пасты или пластыри, а также жидкие лекарственные формы, такие как растворы, эмульсии, в частности эмульсии масло-в-воде, суспензии, например, лосьоны, препараты для инъекций и препараты для инфузий. Кроме того, также можно использовать липосомы или микросферы.
При получении композиций соединения в соответствии с изобретением необязательно смешивают или разбавляют одним или более носителями (эксципиентами). Носители (эксципиенты) могут быть твердыми, полутвердыми или жидкими материалами, которые служат наполнителями, носителями или средой для активного соединения.
Подходящие носители (эксципиенты) перечислены в специализированных медицинских монографиях. Кроме того, составы могут включать фармацевтически приемлемые вспомогательные вещества, такие как смачивающие агенты; эмульгаторы и суспендирующие агенты; консерванты; антиоксиданты; антираздражающие агенты; хелатирующие агенты; вспомогательные вещества для покрытия; стабилизаторы эмульсии; пленкообразователи; гелеобразователи; средства, маскирующие запах; корректоры вкуса; смолы; гидроколлоиды; растворители; солюбилизаторы; нейтрализующие агенты; ускорители диффузии; пигменты; четвертичные аммониевые соединения; пережиривающие агенты и пережириватели; сырье для мазей, кремов или масел; производные силикона; вспомогательные средства для растекания; стабилизаторы; стерилизующие агенты; суппозиторные основы; вспомогательные вещества для таблеток, такие как связующие вещества, наполнители, глиданты, разрыхлители или покрытия; пропелленты; сушильные агенты; замутнители; загустители; воски; пластификаторы и белые минеральные масла. Состав в этом отношении основан на специальных знаниях, как описано, например, в Fiedler, H.P., Lexikon der Hilfsstoffe für Pharmazie, Kosmetik und angrenzende Gebiete [Encyclopedia of auxiliary substances for pharmacy, cosmetics and related fields], 4th edition, Aulendorf: ECV-Editio-Cantor-Verlag, 1996.
Соединения изобретения также могут подходить для комбинации с другими терапевтическими агентами. Таким образом, изобретение дополнительно относится к комбинации, включающей соединение изобретения с одним или более дополнительными терапевтическими агентами, в частности, для применения для стимуляции регенерации печени или снижения или предотвращения гибели гепатоцитов. Комбинированные терапии изобретения могут вводиться дополнительно. Под дополнительным введением подразумевается граничащее или совмещенное введение каждого из компонентов в виде отдельных фармацевтических композиций или устройств. Этот режим терапевтического введения двух или более терапевтических агентов обычно упоминается специалистом в данной области техники и в настоящем описании как дополнительное терапевтическое введение; он также известен как дополнительное терапевтическое введение. Любые и все схемы лечения, при которых пациент получает раздельное, но граничащее или совмещенное терапевтическое введение соединений изобретения и, по меньшей мере, одного дополнительного терапевтического агента, входят в объем настоящего изобретения. В одном варианте осуществления дополнительного терапевтического введения, как описано в настоящей заявке, пациент обычно стабилизируется при терапевтическом введении одного или более компонентов в течение периода времени и затем получает введение другого компонента.
Комбинированные терапии изобретения также можно вводить одновременно. Под одновременным введением подразумевается схема лечения, при которой отдельные компоненты вводят вместе или в форме одной фармацевтической композиции или устройства, включающего или содержащего оба компонента, или в виде отдельных композиций или устройств, каждое из которых содержит один из компонентов, вводимых одновременно. Такие комбинации отдельных индивидуальных компонентов для одновременной комбинации могут быть предоставлены в форме набора из частей.
Подходящие агенты для применения в комбинации с соединениями изобретения включают, например:
ингибиторы АСС, такие как TOFA (5-(тетрадецилокси)-2-фурановая кислота), фирсокостат (ранее известный как GS 0976), PF-05221304 и ингибиторы АСС, как раскрыто в WO 2016/112305,
антагонисты рецепторов ангиотензина II,
ингибиторы ангиотензинпревращающего фермента (АПФ), такие как эналаприл,
ингибиторы ASK1 (регулирующая сигнал апоптоза киназа 1, MAP3K5), такие как селонсертиб (ранее известный как GS-4997) или SRT-015
ингибиторы каспаз, такие как эмриказан,
ингибиторы катепсина B, такие как смешанный ингибитор катепсина B/протеазы NS3 вируса гепатита C, такой как VBY-376,
антагонисты хемокинов CCR2, такие как смешанный антагонист хемокинов CCR2/CCR5, такой как ценикривирок,
антагонисты хемокинов CCR5,
стимуляторы хлоридных каналов, такие как кобипростон,
солюбилизаторы холестерина,
ингибиторы аминоксидазы меди 3 (AOC3), такие как BI 1467335 (ранее известный как PXS-4728A)
ингибиторы диацилглицерол-О-ацилтрансферазы 1 (DGAT1), такие как LCQ908 или GSK-3008356, ингибиторы диацилглицерол-О-ацилтрансферазы 2 (DGAT2), такие как PF-06865571,
ингибиторы дипептидилпептидазы IV (DPPIV), такие как линаглиптин,
агонисты фарнезоидного X-рецептора (FXR), такие как INT-747 (обетихолевая кислота), клиофексор (ранее известный как GS-9674 или PX-102), тропифексор (ранее известный как LJN452), EDP-305 или LMB-763,
факторы роста фибробластов (FGF) и их аналоги, такие как аналоги длительного действия FGF19 (например, альдафермин, ранее известный как NGM-282) или аналоги длительного действия FGF21 (например, TEV-47948, также называемый Bio89-100, или ARK01, или PF-05231023)
двойные агонисты FXR/TGR5, такие как INT-767,
ингибиторы галектина-3, такие как GR-MD-02,
агонисты глюкагоноподобного пептида 1 (GLP1), такие как лираглутид или эксенатид,
двойные агонисты глюкагоноподобного пептида 1 (GLP1)/глюкагона, такие как котадутид,
агонисты двойного рецептора глюкозозависимого инсулинотропного полипептида (GIP) и глюкагоноподобного пептида-1 (GLP-1), такие как тирзепатид (ранее известный как LY3298176)
предшественники глутатиона,
ингибиторы протеазы NS3 вируса гепатита С, такие как смешанный ингибитор катепсина В/протеазы NS3 вируса гепатита С, такой как VBY-376,
ингибиторы ГМГ-КоА-редуктазы, такие как статин, такой как аторвастатин,
ингибиторы 11β-гидроксистероиддегидрогеназы (11β-HSD1), такие как R05093151,
антагонисты ИЛ-1β,
антагонисты ИЛ-6, такие как смешанный ингибитор лиганда ИЛ-6/ИЛ-1β/ФНОα, такой как BLX-1002,
агонисты ИЛ-10, такие как пегилодекакин,
анти-ИЛ-11 антитела или антагонисты ИЛ-11
антагонисты ИЛ-17, такие как KD-025,
ингибиторы котранспортера натрия/желчных кислот в подвздошной кишке, такие как воликсибат (ранее известный как SHP-626),
ингибиторы интегрина, такие как селективные αvβ1-ингибиторы (например, PLN-1474 или описанные в Wilkinson et al., Eur. J. Pharmacol., 842, 239-247 (2019)),
ингибиторы кетогексокиназы, такие как PF-06835919
аналоги лептина, такие как метрелептин,
ингибиторы 5-липоксигеназы, такие как смешанный ингибитор 5-липоксигеназы/ФДЭ3/ФДЭ4/PLC, такой как типелукаст,
cтимуляторы гена LPL, такие как алипоген типарвовек,
ингибиторы гомолога 2 лизилоксидазы (LOXL2), такие как анти-LOXL2 антитело, такое как симтузумаб (ранее известный как GS-6624), или низкомолекулярные ингибиторы, такие как раскрытые в WO 2017/136870,
домен пирина семейства nod-подобных рецепторов, содержащий 3 низкомолекулярных ингибитора инфламмасомы (NLRP3), такие как MCC950,
омега-3 полиненасыщенные жирные кислоты и их производные, такие как икосабутат и примеры, раскрытые в US 8735436 B2,
оксистеролсульфаты, такие как 25-гидроксихолестерин 3-сульфат и 25-гидроксихолестерин 3,25-дисульфат,
ингибиторы ФДЭ4, такие как ASP-9831
агонисты PPARα, такие как смешанный агонист PPARα/δ элафибранор (ранее известный как GFT-505), смешанный агонист PPARα/γ/δ ланифибранор или смешанный агонист PPARα/γ сароглитазар),
агонисты PPARγ, такие как пиоглитазон,
агонисты PPARδ, такие как селаделпар,
ингибиторы Rho-ассоциированной протеинкиназы 2 (ROCK2), такие как KD-025,
ингибиторы транспортера натрия-глюкозы-2 (SGLT2), такие как ремоглифлозина этабонат,
ингибиторы транспортера натрия-глюкозы-1/2 (SGLT1/2), такие как ликоглифлозин
ингибиторы стеароил-КоА-десатуразы-1, такие как арамхол или CVT-12805,
агонисты β-рецептора гормона щитовидной железы, такие как MGL-3196 или VK2809,
ингибиторы лиганда фактора некроза опухоли α (ФНОα),
ингибиторы трансглутаминазы и предшественники ингибиторов трансглутаминазы, такие как
меркаптамин,
ингибиторы PTPIb, такие как A119505, A220435, A321842, CPT633, ISIS-404173, JTT-551, MX-7014, MX-7091, MX-7102, NNC-521246, OTX-001, OTX-002 или TTP814, и
намацизумаб, антитело, которое стабилизирует каннабиноидный рецептор 1 (CB1) в неактивной конформации.
В некоторых вариантах осуществления один или более дополнительных терапевтических агентов выбраны из ацетилсалициловой кислоты, алипогена типарвовека, арамхола, аторвастатина, BLX-1002, ценикривирока, кобипростона, колесевелама, эмрикасана, эналаприла, GFT-505, GR-MD-02, гидрохлоротиазида, икозапентэтилового эфира (этилэйкозапентаеновой кислоты), IMM-124E, KD-025, линаглиптина, лираглутида, меркаптамина, MGL-3196, обетихолевой кислоты, олезоксима, пегилодекакина, пиоглитазона, GS-9674, ремоглифлозина этабоната, SHP-626, солитромицина, типелукаста, TRX-318, урсодезоксихолевой кислоты и VBY-376.
В некоторых вариантах осуществления один из одного или более дополнительных терапевтических агентов выбран из ацетилсалициловой кислоты, алипогена типарвовека, арамхола, аторвастатина, BLX-1002 и ценикривирока.
В варианте осуществления изобретение относится к способу
ингибирования протеинкиназы MKK4,
селективного ингибирования протеинкиназы MKK4 по сравнению с протеинкиназами JNK1 и MKK7, стимуляции регенерации печени или предотвращения гибели гепатоцитов,
лечения острого, острого при хроническом или хронического заболевания печени,
лечения острых и хронических или острых при хронических заболеваний печени, таких как острые и хронические вирусные гепатиты, такие как гепатиты В, С, Е, гепатиты, вызванные вирусом Эпштейна-Барр, цитомегаловирусом, вирусом простого герпеса и другими вирусами, все виды аутоиммунного гепатита, первичный склерозирующий гепатит, алкогольный гепатит;
лечения метаболических заболеваний печени, таких как метаболический синдром, жировая дистрофия печени, такая как неалкогольная жировая болезнь печени (НАЖБП), неалкогольный стеатогепатит (НАСГ), алкогольный стеатогепатит (АСГ), болезнь Вильсона, гемохроматоз, дефицит альфа-1-антитрипсина, болезни накопления гликогена;
лечения всех типов цирроза печени, таких как первичный билиарный цирроз, этилтоксический цирроз печени, криптогенный цирроз;
лечения острой (фульминантной) или хронической печеночной недостаточности, такой как токсическая печеночная недостаточность, такая как печеночная недостаточность, вызванная ацетаминофеном (парацетамолом), печеночная недостаточность, вызванная альфа-аманитином, гепатотоксичность, вызванная лекарственными средствами, и печеночная недостаточность, вызванная, например, антибиотиками, нестероидными противовоспалительными средствами, противосудорожными средствами, острая печеночная недостаточность, вызванная растительными добавками (кава, эфедра, шлемник, мята болотная и т. д.), заболевание печени и недостаточность вследствие сосудистых заболеваний, таких как синдром Бадда-Киари, острая печеночная недостаточность неизвестного происхождения, хронические заболевания печени вследствие правожелудочковой недостаточности;
лечения галактоземии, кистозного фиброза, порфирии, нарушения перфузии при ишемии печени, синдрома малой доли после трансплантации печени, первичного склерозирующего холангита или печеночной энцефалопатии,
лечения остеоартрита, ревматоидного артрита или заболеваний, связанных с ЦНС, таких как болезнь Альцгеймера и болезнь Паркинсона,
который включает введение эффективного количества соединения или композиции, как определено выше, объекту, нуждающемуся в таком лечении.
В варианте осуществления соединения изобретения вводят в дозе от 0,2 до 15 мг/кг или от 0,5 до 12 мг/кг объекту, который подвергается лечению. Соединения можно вводить один или несколько раз в день. Соединения вводят в течение от 4 до 12 недель.
Следующие примеры иллюстрируют изобретение, не ограничивая его.
ПРИМЕРЫ
Сокращения:
Пример 1: Синтез N-(3-(5-(2-циклопропилпиримидин-5-ил)-1H-пиразоло[3,4-b]пиридин-3-карбонил)-2,4-дифторфенил)-1-фенилметансульфонамида
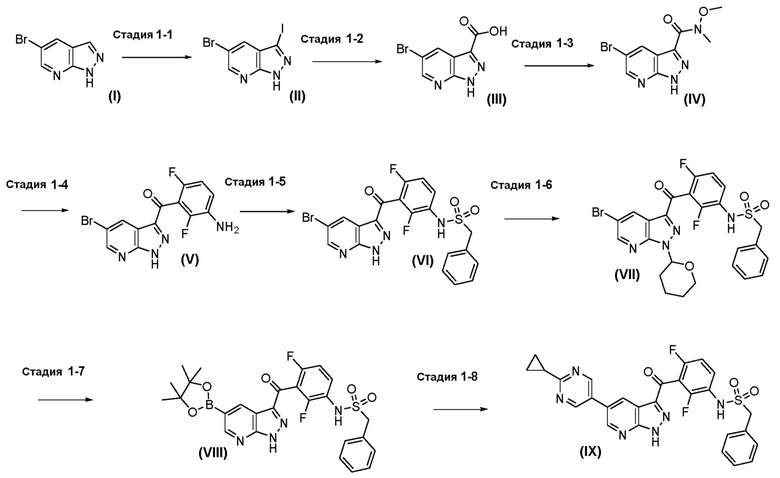
Стадия 1-1: 5-Бром-3-иод-1H-пиразоло[3,4-b]пиридин (II)
К перемешиваемой смеси 5-бром-1H-пиразоло[3,4-b]пиридина ((I), 6,81 г, 34,4 ммоль) и гидроксида калия (КОН, 6,75 г, 120,4 ммоль) в ДМФ (45 мл) добавляли иод (9,60 г, 37,8 ммоль) одной порцией при КТ. После короткого индукционного периода начиналась экзотермическая реакция. Через 1 ч добавляли дополнительную порцию 1 г иода и смесь перемешивали при 45°С в течение 1 ч. Смесь выливали в 300 мл разбавленного раствора Na2SO3 и подкисляли 2 н. HCl. Твердые вещества собирали фильтрованием с отсасыванием, промывали водой и высушивали в cушильном шкафу при 110°С. Выход: 10,92 г,
Аналитические данные:
ВЭЖХ чистота: 95%,
1H ЯМР (200 МГц, ДМСО) δ 14,29 (с, 1H), 8,62 (с, 1H), 8,17 (с, 1H); 13С ЯМР (50 МГц, ДМСО) δ 150,53, 150,17, 131,86, 120,58, 112,43, 91,95;
МС(ESI-): m/z 322,0/324,0 [M-H]-
Стадия 1-2: 5-Бром-1H-пиразоло[3,4-b]пиридин-3-карбоновая кислота (III)
5-Бром-3-иод-1H-пиразоло[3,4-b]пиридин ((II), 10,44 г, 32,2 ммоль) смешивали с ДМФ, МеОН и триэтиламином (ТЭА, по 75 мл каждого). Сосуд вакуумировали и продували аргоном (4×). Добавляли Ксантфос (1,12 г, 1,93 ммоль) и Pd(OAc)2 (217 мг, 0,97 ммоль) и через раствор барботировали монооксид углерода (полученный из муравьиной кислоты и серной кислоты) при нагревании до 60°C. Смесь перемешивали в атмосфере монооксида углерода (баллон) в течение 8 ч. Через каждые 1,5 ч барботировали монооксид углерода через раствор в течение 5 минут. Смесь концентрировали при пониженном давлении и остаток растирали с 2 н. HCl. Твердые вещества нагревали при 95°С приблизительно в 100 мл 1 н. NaOH в течение ночи (O/N). После охлаждения до КТ смесь подкисляли конц. HCl, и осадок собирали фильтрованием с отсасыванием, и промывали водой. Твердые вещества высушивали в сушильном шкафу при 110°С до постоянной массы. Твердые вещества обрабатывали ультразвуком в 100 мл толуола в течение 5 минут и перемешивали в течение 30 минут. Продукт отфильтровывали, промывали дополнительными 20 мл толуола и высушивали при 110°С. Выход: 7,92 г.
Аналитические данные:
ВЭЖХ чистота: ˃99%,
1H ЯМР (200 МГц, ДМСО) δ 8,64 (д, J=7,9 Гц, 2H), 5,69 (уш.с, 1H); 13С ЯМР (50 МГц, ДМСО) δ 163,27, 150,97, 149,67, 136,69, 132,65, 115,73, 113,6;
МС(ESI-): m/z 239,9/241,9 [M-H]-.
Стадия 1-3: 5-бром-N-метокси-N-метил-1H-пиразоло[3,4-b]пиридин-3-карбоксамид (IV)
5-Бром-1H-пиразоло[3,4-b]пиридин-3-карбоновую кислоту ((III), 7,91 г, 32,7 ммоль) и 1,1'-карбонилдиимидазол (5,83 г, 35,9 ммоль) перемешивали в 200 мл ДМФ при 60°С в течение 45 минут. К полученной суспензии добавляли N, O-диметилгидроксиламина гидрохлорид (3,51 г, 35,9 ммоль) и смесь перемешивали в течение 4 ч при 65°C. Большую часть растворителя удаляли под вакуумом и к остатку добавляли половину насыщ. раствора NaHCO3. Твердые вещества собирали фильтрованием с отсасыванием, промывали водой и высушивали при 110°С. Выход: 7,94 г.
Аналитические данные:
ВЭЖХ чистота: 96%,
1H ЯМР (200 МГц, ДМСО) δ 14,46 (с, 1H), 8,62 (д, J=20,4 Гц, 2H), 3,76 (с, 3H), 3,44 (с, 3H),
МС(ESI-): m/z 283,0/285,0 [M-H]-.
Стадия 1-4: Синтез (3-амино-2,6-дифторфенил)(5-бром-1H-пиразоло[3,4-b]пиридин-3-ил)метанона (V)
2,4-Дифторанилин (6,25 г, 48,4 ммоль) растворяли в 50 мл сухого ТГФ и охлаждали до -78°С в атмосфере аргона. Добавляли по каплям 2,5 М н-бутиллития в гексане (19,4 мл, 48,4 ммоль). Через 15 минут по каплям добавляли 1,2-бис(хлордиметилсилил)этан (10,9 г, 49,5 ммоль) в 15 мл сухого ТГФ и смесь перемешивали в течение 30 минут. Добавляли по каплям 2,5 М н-бутиллития в гексане (19,4 мл, 48,4 ммоль) и смесь оставляли достичь КТ в течение 1 ч. После охлаждения до -78°С по каплям добавляли 2,5 М н-бутиллития в гексане (19,4 мл, 48,4 ммоль) и перемешивали в течение 1 ч при -78°С. (= раствор А).
5-бром-N-метокси-N-метил-1H-пиразоло[3,4-b]пиридин-3-карбоксамид ((IV), 6,00 г, 21,1 ммоль) суспендировали в 50 мл сухого ТГФ и охлаждали до 0°С в атмосфере аргона. Порциями добавляли NaH (60% в минеральном масле, 0,88 г, 22,1 ммоль) и раствор перемешивали при КТ в течение 1 ч. (= раствор В).
Раствор В добавляли по каплям к раствору А при -78°С. После завершения добавления смесь нагревали до КТ в течение 30 минут. Осторожно добавляли 12 мл конц. HCl и смесь перемешивали в течение 30 минут. Для нейтрализации раствора добавляли твердый NaHCO3, твердые вещества отфильтровывали и промывали ТГФ. Фильтрат испаряли, и остаток растирали с МеОН и водой и высушивали при 110°С. Выход: 4,03 г;
Аналитические данные:
ВЭЖХ чистота: 97%,
1H ЯМР (200 МГц, ДМСО-d6) δ 14,91 (с, 1H), 8,77 (дд, J=5,4, 2,1 Гц, 2H), 7,18-6,59 (м, 2H), 5,25 (с, 2H); 13С ЯМР (50 МГц, ДМСО) δ 183,95, 151,04, 150,79, 150,27 (дд, J=161,0, 6,8 Гц), 145,50 (дд, J=167,3, 6,8 Гц), 141,34, 133,35 (дд, J=12,8, 2,6 Гц), 132,28, 117,45 (дд, J=8,4, 6,5 Гц), 116,24 (дд, J=22,7, 19,1 Гц), 115,55, 114,81, 111,26 (дд, J=21,7, 3,5 Гц); МС(ESI-): m/z 351,1/353,1 [M-H]-.
Стадия 1-5: N-(3-(5-бром-1H-пиразоло[3,4-b]пиридин-3-карбонил)-2,4-дифторфенил)пропан-1-сульфонамид (VI)
(3-Амино-2,6-дифторфенил)-(5-бром-1Н-пиразоло[3,4-b]пиридин-3-ил)метанон (579 мг, 1,64 ммоль) и 4-диметиламинопиридин (4-ДМАП, 0,401 г, 3,28 ммоль) растворяли в пиридине (3,31 мл, 41,0 ммоль) при нагревании. После охлаждения до -10°C к образовавшейся суспензии добавляли фенилметансульфонилхлорид (406 мг, 2,13 ммоль). Смесь перемешивали в течение 10 минут при -10°С, еще 10 минут при КТ с последующим нагреванием до 50°С и перемешиванием в течение 30 минут. Смесь концентрировали при пониженном давлении, восстанавливали в 2 н. NaOH (2,46 мл, 4,92 ммоль) и перемешивали при КТ в течение 10 минут. Смесь разбавляли водой и медленно добавляли к 25 мл 2 н. HCl при перемешивании. Через 10 минут образовавшееся твердое вещество собирали фильтрованием с отсасыванием, промывали водой и высушивали при 75°С (0,590 г, 1,16 ммоль, выход 71%).
Аналитические данные:
1H ЯМР (200 МГц, CDCl3) δ 14,22 (с, 1H), 8,96 (с, 1H), 8,79 (д, J=2,2 Гц, 1H), 8,59 (д, J=2,1 Гц, 1H), 7,52-7,25 (м, 6Н), 6,87 (тд,=9,1, 1,6 Гц, 1Н), 4,32 (с, 2Н);
МС(ESI-): m/z 504,7 [M-1]-.
Стадия 1-6: N-[3-[5-бром-1-(оксан-2-ил)пиразоло[3,4-b]пиридин-3-карбонил]-2,4-дифторфенил]-1-фенилметансульфонамид (VII)
N-[3-(5-бром-1H-пиразоло[3,4-b]пиридин-3-карбонил)-2,4-дифторфенил]-1-фенилметансульфонамид (0,419 г, 0,826 ммоль), п-толуолсульфокислоты (п-ТСК) моногидрат (15,7 мг, 0,0826 ммоль) и дигидропиран (0,0904 мл, 0,991 ммоль) растворяли в 4,13 мл ДХМ и нагревали с обратным холодильником в течение 1,5 ч. После охлаждения смесь разбавляли ДХМ, промывали насыщенным раствором NaHCО3, высушивали и отфильтровывали. Добавляли н-гептан и удаляли ДХМ. После охлаждения на бане со льдом продукт собирали фильтрованием с отсасыванием. (0,393 г, 0,6650 ммоль, выход 80%).
Аналитические данные:
1H ЯМР (200 МГц, CDCl3) δ 8,88 (д, J=1,8 Гц, 1H), 8,68 (д, J=1,8 Гц, 1H), 7,62 (тд, J=9,0, 5,6 Гц, 1H), 7,34 (с, 5H), 6,96 (т, J=8,1 Гц, 1H), 6,48 (с, 1H), 6,14 (дд, J=9,8, 2,0 Гц, 1H), 4,39 (с, 2H), 4,03 (д, J=11,1 Гц, 1H), 3,90-3,56 (м, 1H), 2,56-2,36 (м, 1H), 2,09-1,46 (м, 5H);
МС(ESI-): m/z 591,3/589,4 [M-H]-.
Стадия 1-7: N-[2,4-дифтор-3-[1-(оксан-2-ил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиразоло[3,4-b]пиридин-3-карбонил]фенил]-1-фенилметансульфонамид (VIII)
В сосуд загружали N-[3-[5-бром-1-(оксан-2-ил)пиразоло[3,4-b]пиридин-3-карбонил]-2,4-дифторфенил]-1-фенилметансульфонамид (0,365 г, 0,617 ммоль), бис(пинаколато)диборон (0,313 г, 1,23 ммоль), ацетат калия (0,182 г, 1,85 ммоль) и ДМФ (3,09 мл). Смесь нагревали до 90°С, и сосуд вакуумировали, и заполняли аргоном (3×). Добавляли бис(трифенилфосфин)палладия(II) дихлорид (PdCl2(PPh3)2, 4,33 мг, 0,00617 ммоль) и реакционную смесь перемешивали в течение 4 ч при 90°С. После охлаждения реакционную смесь концентрировали, растворяли в EtOAc и промывали водой, полунасыщенным солевым раствором и солевым раствором, высушивали и отфильтровывали. К фильтрату добавляли активированный уголь и смесь нагревали с обратным холодильником в течение 15 минут. После охлаждения смесь отфильтровывали через целит и удаляли растворитель. Остаток растворяли в небольшом количестве ДХМ, добавляли н-гептан и удаляли ДХМ при пониженном давлении. Твердые вещества собирали фильтрованием с отсасыванием, промывали н-гексаном и высушивали продукт в виде белого с желтоватым или сероватым оттенком твердого вещества (0,298 г, 0,4670 ммоль, выход 76%), который использовали без дополнительной очистки.
Аналитические данные:
1H ЯМР (200 МГц, CDCl3) δ 9,16 (с, 1H), 8,97 (с, 1H), 7,63 (тд, J=9,1, 5,7 Гц, 1H), 7,34 (с, 5H), 6,96 (т, J=8,6 Гц, 1H), 6,45 (с, 1H), 6,23 (д, J=8,8 Гц, 1H), 4,39 (с, 2H), 4,12-3,94 (м, 1H), 3,80 (т, J=9,9 Гц, 1H), 2,60-2,31 (м, 1H), 2,09-1,55 (м, 5H), 1,39 (с, 12H).
Стадия 1-8: N-(3-(5-(2-циклопропилпиримидин-5-ил)-1H-пиразоло[3,4-b]пиридин-3-карбонил)-2,4-дифторфенил)-1-фенилметансульфонамид (IX)
В сосуд загружали N-[2,4-дифтор-3-[1-(оксан-2-ил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиразоло[3,4-b]пиридин-3-карбонил]фенил]-1-фенилметансульфонамид (0,275 г, 0,431 ммоль), 5-бром-2-циклопропилпиримидин (0,111 г, 0,560 ммоль), PdCl2(PPh3)2 (3,02 мг, 0,00431 ммоль) и 1,4-диоксан (1,44 мл). Сосуд вакуумировали и заполняли аргоном (3×). Добавляли дегазированный 3М водный K2CO3 (0,431 мл, 1,29 ммоль) и смесь перемешивали при 60°C в течение 1 ч. После охлаждения реакционную смесь разбавляли EtOAc и нейтрализовали раствором NH4Cl. Органическую фазу высушивали, испаряли и выделяли основной продукт флэш-хроматографией (ДХМ/EtOAc, от 10 до 50%). Для удаления защитной группы THP выделенный продукт нагревали с обратным холодильником в 2,5 н. HCl (3 мл) в течение 1 ч.
Аналитические данные:
1H ЯМР (200 МГц, ДМСО-d6) δ 15,01 (с, 1H), 9,86 (с, 1H), 9,12 (с, 2H), 9,08 (д, J=2,0 Гц, 1H), 8,90 (д, J=2,0 Гц, 1H), 7,60-7,16 (м, 7H), 4,54 (с, 2H), 2,39-2,19 (м, 1H), 1,27-1,04 (м, 4H).
МС(ESI-): m/z 545,5 [M-H]-.
Пример 2: Синтез N-(3-(5-(4-хлорфенил)-1H-пиразоло[3,4-b]пиридин-3-карбонил)-2,6-дифторфенил)метансульфонамида
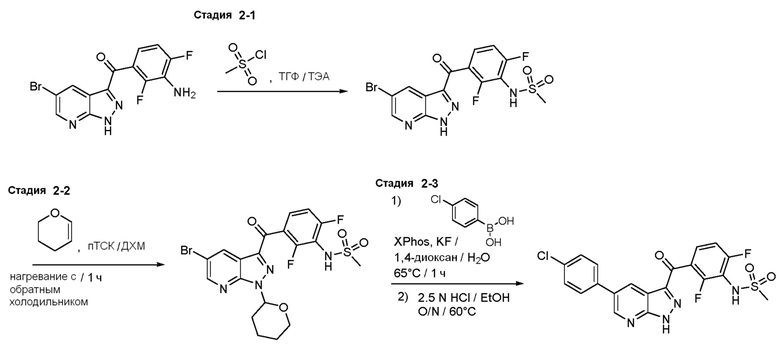
Стадия 2-1: N-[3-(5-бром-1H-пиразоло[3,4-b]пиридин-3-карбонил)-2,6-дифторфенил]метансульфонамид
(3-Амино-2,4-дифторфенил)(5-бром-1H-пиразоло[3,4-b]пиридин-3-ил)метанон (1,29 г, 3,65 ммоль) растворяли в 18 мл ТГФ и 5,09 мл ТЭА (36,5 ммоль, 10 экв.). Смесь нагревали до полного растворения. Затем раствор охлаждали до 0°С с последующим добавлением 0,99 мл мезилхлорида (12,8 ммоль, 3,5 экв.). Через 10 мин смесь оставляли нагреваться до КТ, добавляли 12 мл 2 н. КОН и смесь перемешивали в течение 10 мин. После подкисления 3 н. HCl ТГФ удаляли при пониженном давлении, твердое вещество собирали фильтрованием с отсасыванием, промывали 3 н. HCl/MeOH (1+1) и высушивали (1,46 г, 93%).
Аналитические данные:
1H ЯМР (400 МГц, ДМСО-d6) δ 14,87 (с, 1H), 9,75 (с, 1H), 8,71 (д, J=13,9 Гц, 2H), 7,85 (д, J=6,6 Гц, 1Н), 7,39 (т, J=8,5 Гц, 1Н), 3,12 (с, 3Н).
МС(ESI-): m/z 431,1/429,1 [M-H]-, 411,1/409,1 [M-H-HF]-.
Стадия 2-2: N-[3-[5-бром-1-(оксан-2-ил)пиразоло[3,4-b]пиридин-3-карбонил]-2,6-дифторфенилил]метансульфонамид
К суспензии дигидропирана (0,614 мл, 6,73 ммоль) и N-[3-(5-бром-1Н-пиразоло[3,4-b]пиридин-3-карбонил)-2,6-дифторфенил]метансульфонамида (1,45 г, 3,36 ммоль) в 16,8 мл ДХМ добавляли п-ТСК моногидрат (64,0 мг, 0,336 ммоль) и смесь нагревали с обратным холодильником в течение 1 ч. Смесь разбавляли ДХМ, промывали насыщенным раствором NaHCО3, высушивали над Na2SО4 и концентрировали. Остаток обрабатывали небольшим количеством ацетона и при перемешивании переносили в диэтиловый эфир. Продукт собирали фильтрованием с отсасыванием и промывали диэтиловым эфиром с получением продукта в виде белого порошка (1,07 г, 2,08 ммоль, выход 62%).
Аналитические данные:
1H ЯМР (200 МГц, ДМСО-d6) δ 9,78 (с, 1H), 8,80 (дд, J=13,4, 1,6 Гц, 2H), 7,88 (дд, J=14,6, 7,8 Гц, 1H), 7,43 (т, J=9,0 Гц, 1H), 6,14 (д, J=8,6 Гц, 1H), 3,94 (д, J=11,6 Гц, 1H), 3,72 (дд, J=14,6, 9,1 Гц, 1H), 3,13 (с, 3Н), 2,46-2,19 (м, 1Н), 2,00-1,18 (м, 5Н);
МС(ESI-): m/z 515,2/513,2 [M-H]-.
Стадия 2-3: N-[3-[5-(4-хлорфенил)-1Н-пиразоло[3,4-b]пиридин-3-карбонил]-2,6-дифторфенилил]метансульфонамид
В сосуд загружали N-[3-[5-бром-1-(оксан-2-ил)пиразоло[3,4-b]пиридин-3-карбонил]-2,6-дифторфенил]метансульфонамид (119 мг, 0,231 ммоль), (4-хлорфенил)бороновую кислоту (36,1 мг, 0,231 ммоль), PdCl2(PPh3)2 (3,24 мг, 0,00462 ммоль) и продували аргоном. Добавляли дегазированный 1,4-диоксан (0,770 мл) и дегазированный фторид калия (95,7 мг, 0,693 ммоль) и реакционную смесь перемешивали при 60°С в течение 15 минут. После охлаждения добавляли EtOAc и раствор NH4Cl и водную фазу удаляли. Органическую фазу высушивали над Na2SО4, концентрировали и остаток растворяли в 2,5 н. HCl (3 мл) в EtOH и перемешивали при 60°C O/N. Добавляли 3 мл изопропанола, твердое вещество собирали центрифугированием и растворитель удаляли. Твердое вещество помещали в ТГФ/раствор NaHCО3 и встряхивали. Органическую фазу высушивали, растворитель испаряли и остаток растирали с ДХМ (выход: 44,0 мг, 0,0941 ммоль, 41%).
Аналитические данные:
1H ЯМР (400 МГц, ДМСО-d6) δ 14,82 (с, 1H), 9,75 (с, 1H), 9,00 (д, J=2,2 Гц, 1H), 8,76 (д, J=2,2 Гц, 1Н), 7,90-7,81 (м, 3Н), 7,61-7,57 (м, 2Н), 7,40 (т, J=8,4 Гц, 1Н), 3,11 (с, 3Н);
Рассчитанная точная масса: 462,04, МС(ESI-): m/z: 461,0 [М-1]-.
Пример 3: Синтез N-(3-(5-(4-хлорфенил)-1H-пиразоло[3,4-b]пиридин-3-карбонил)-2,6-дифторфенил)этансульфонамида
По аналогии с Примером 2 получали N-(3-(5-(4-хлорфенил)-1H-пиразоло[3,4-b]пиридин-3-карбонил)-2,6-дифторфенил)этансульфонамид.
Аналитические данные:
1H ЯМР (400 МГц, ДМСО-d6) δ 9,00 (д, J=2,2 Гц, 1H), 8,76 (д, J=2,2 Гц, 1H), 7,84 (дд, J=13,4, 8,4 Гц, 3H), 7,60 (д, J=8,6 Гц, 2H), 7,38 (т, J=8,6 Гц, 1H), 3,17 (к, J=7,3 Гц, 2H), 1,32 (т, J=7,3 Гц, 3H)
Рассчитанная точная масса: 476,05, МС(ESI-): m/z: 474,9 [M-1]-.
Пример 4: Синтез этил-4-(3-(2,4-дифтор-3-(метилсульфонамидо)бензоил)-1H-пиразоло[3,4-b]пиридин-5-ил)бензоата
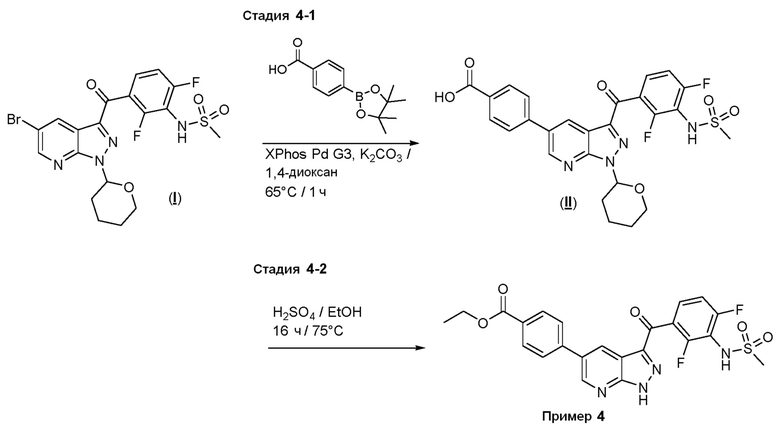
Стадия 4-1: 4-[3-[2,4-дифтор-3-(метансульфонамидо)бензоил-1-(оксан-2-ил)пиразоло[3,4-b]пиридин-5-ил]бензойная кислота
N-[3-[5-бром-1-(оксан-2-ил)пиразоло[3,4-b]пиридин-3-карбонил]-2,6-дифторфенил]метансульфонид ((I), 355 мг, 0,689 ммоль) и 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензойную кислоту (188 мг, 0,758 ммоль) XPhos Pd G3 ((2-дициклогексилфосфино-2',4',6'-триизопропил-1,1'-бифенил)[2-(2'-амино-1,1'-бифенил)]палладия(II) метансульфонат, коммерчески доступный от Aldrich; 17,5 мг, 0,0207 ммоль) объединяли в дегазированном 1,4-диоксане (2,30 мл) и 1,5 М карбонате калия (2,07 мл, 3,10 ммоль). Реакционную смесь вакуумировали и продували аргоном (3×). Добавляли XPhos Pd G3 (17,5 мг, 0,0207 ммоль) и реакционную смесь перемешивали при 60°C (температура масляной бани) в течение 3 ч. После охлаждения смесь подкисляли 2 н. HCl и экстрагировали EtOAc. Экстракт промывали солевым раствором, высушивали над сульфатом натрия (Na2SO4) и концентрировали. Остаток очищали флэш-хроматографией (ДХМ+МеОН, от 3% до 25%) и растирали с н-гексаном (276 мг, 0,4960 ммоль, выход 72%).
Стадия 4-2: Этил-5-(3-(2,4-дифтор-3-(метилсульфонамидо)бензоил)-1 Н -пиразоло[3,4-b]пиридин-5-ил)бензоат
50 мг (II) (0,09 ммоль) растворяли в смеси 300 мкл H2SO4 и 800 мкл EtOH и перемешивали при 75°С в течение 16 ч. После охлаждения смеси до КТ ее выливали в водный NaHCО3, осадок собирали центрифугированием и промывали водой и диэтиловым эфиром (22 мг, 46%, химическая чистота (ВЭЖХ/УФ): 95%).
Аналитические данные:
1H ЯМР (400 МГц, ДМСО) δ 14,86 (с, 1H), 9,76 (с, 1H), 9,06 (с, 1H), 8,83 (с, 1H), 8,10 (д, J=7,8 Гц, 2H), 8,04-7,94 (м, 2H), 7,87 (дд, J=14,5, 7,6 Гц, 1H), 7,40 (т, J=8,8 Гц, 1H), 4,35 (к, J=7,0 Гц, 2H), 3,11 (с, 3H), 1,35 (т, J=7,1 Гц, 3H);
Рассчитанная точная масса: 500,10, МС(ESI-): m/z: 499,4 [M-1]-.
Пример 5: Синтез метил-5-(3-(2,4-дифтор-3-(метилсульфонамидо)бензоил)-1H-пиразоло[3,4-b]пиридин-5-ил)пиколината
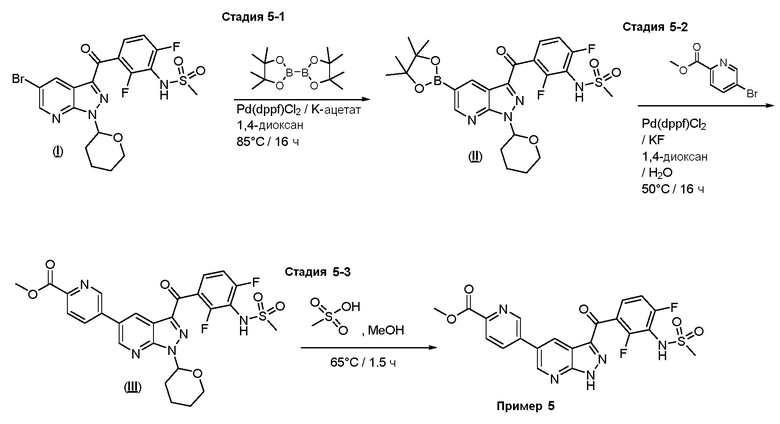
Стадия 5-1: N-[2,6-дифтор-3-[1-(оксан-2-ил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиразоло[3,4-b]пиридин-3-карбонил]фенил]метансульфонамид
В сосуд загружали N-[3-[5-бром-1-(оксан-2-ил)пиразоло[3,4-b]пиридин-3-карбонил]-2,6-дифторфенил]метансульфонамид ((I), 0,630 г, 1,22 ммоль), бис(пинаколато)диборон (341 мг, 1,34 ммоль), безводный ацетат калия (360 мг, 3,67 ммоль) и сухой 1,4-диоксан (4,08 мл). Сосуд вакуумировали и заполняли аргоном (3×). Добавляли 1,1’-Бис(дифенилфосфино)-ферроцен-дихлорпалладий (1:1 комплекс с ДХМ) (Pd(dppf)Cl2, 17,9 мг, 0,0245 ммоль) и реакционную смесь перемешивали при 80°C O/N. После охлаждения добавляли EtOAc, суспензию перемешивали в течение 30 минут и отфильтровывали через целит. Растворитель концентрировали, добавляли н-гептан и твердые вещества собирали фильтрованием с отсасыванием, промывали гексаном и высушивали (0,690 г, 1,23 ммоль, выход 100%).
Стадия 5-2: Метил-5-(3-(2,4-дифтор-3-(метилсульфонамидо)бензоил)-1-(тетрагидро-2H-пиран-2-ил)-1H-пиразоло[3,4-b]пиридин-5-ил)пиколинат
В сосуд загружали N-[2,6-дифтор-3-[1-(оксан-2-ил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиразоло[3,4-b]пиридин-3-карбонил]фенил]метансульфонамид ((II), 116 мг, 0,206 ммоль), метил-5-бромпиридин-2-карбоксилат (49,0 мг, 0,227 ммоль), фторид калия (36,0 мг, 0,619 ммоль), Pd(dppf)Cl2⋅ДХМ (8,42 мг, 0,0103 ммоль) и дегазированный 1,4-диоксан/вода (4+1) (0,6 мл), и сосуд вакуумировали, и заполняли аргоном (3×). Смесь перемешивали O/N при 50°C, затем разбавляли EtOAc, промывали солевым раствором и удаляли растворители. Продукт выделяли флэш-хроматографией (ДХМ+EtOAc, от 0% до 20%); 81,0 мг белого твердого вещества (0,1420 ммоль, выход 69%).
Стадия 5-3: Метил-5-(3-(2,4-дифтор-3-(метилсульфонамидо)бензоил)-1Н-пиразоло[3,4-b]пиридин-5-ил)пиколинат
К раствору метил-5-[3-[2,4-дифтор-3-(метансульфонамидо)бензоил]-1-(оксан-2-ил)пиразоло[3,4-b]пиридин-5-ил]пиридин-2-карбоксилата ((III), 60,0 мг, 0,105 ммоль) в метаноле (0,525 мл) добавляли метансульфоновую кислоту (0,0273 мл, 0,420 ммоль) и смесь перемешивали при 65°С в течение 1,5 ч. Смесь охлаждали до КТ и медленно добавляли к диэтиловому эфиру (15 мл). Твердое вещество собирали фильтрованием с отсасыванием, промывали диэтиловым эфиром и высушивали под вакуумом; 36,0 мг белого твердого вещества (0,0739 ммоль, выход 70%).
Аналитические данные:
1H ЯМР (200 МГц, CDCl3) δ 14,90 (с, 1H), 9,74 (с, 1H), 9,16 (дд, J=12,4, 1,9 Гц, 2H), 8,93 (д, J=2,0 Гц, 1H), 8,47 (дд, J=8,2, 2,3 Гц, 1H), 8,19 (д, J=8,3 Гц, 1H), 7,88 (дд, J=14,8, 7,7 Гц, 1H), 7,41 (т, J=8,7 Гц, 1H), 3,93 (с, 3H), 3,12 (с, 3H);
Рассчитанная точная масса: 487,08; МС(ESI+): m/z: 510,4 [M+Na+]+.
Пример 6: Синтез N-(2,6-дифтор-3-(5-(пиримидин-5-ил)-1H-пиразоло[3,4-b]пиридин-3-карбонил)фенил)пропан-1-сульфонамида
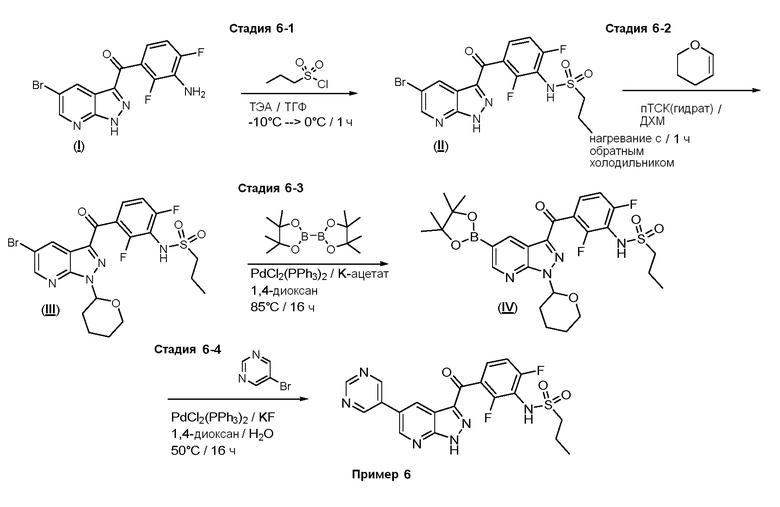
Стадия 6-1: N-[3-(5-бром-1Н-пиразоло[3,4-b]пиридин-3-карбонил)-2,6-дифторфенил]пропан-1-сульфонамид
К суспензии (3-амино-2,4-дифторфенил)-(5-бром-1Н-пиразоло[3,4-b]пиридин-3-ил)метанона ((I), 65,0 г, 184 ммоль) и ТЭА (282 мл, 2020 ммоль) в ТГФ (614 мл) по каплям добавляли смесь 1:1 (об./об.) 1-пропансульфонилхлорида (68,4 мл, 607 ммоль) и ДХМ при -10°С, поддерживая температуру ниже -5°С. После завершения добавления смесь перемешивали при 0°С в течение 1 ч. Добавляли 2 н. NaOH (736 мл, 1470 ммоль) и смесь перемешивали при КТ еще 30 минут. ТГФ и ТЭА испаряли из смеси при 45°С при пониженном давлении. Раствор охлаждали до 20°С и осторожно промывали EtOAc (4×300 мл) при кручении (без встряхивания). Остаточный EtOAc удаляли при пониженном давлении и раствор медленно добавляли к 3 н. HCl (736 мл, 2210 ммоль) при эффективном перемешивании. Твердые вещества собирали фильтрованием с отсасыванием, промывали большим количеством воды и высушивали при 100°C с получением N-[3-(5-бром-1H-пиразоло[3,4-b]пиридин-3-карбонил)-2,6-дифторфенил]пропан-1-сульфонамида (73,9 г, 161 ммоль, выход 87%) в виде белого с желтоватым или сероватым оттенком твердого вещества.
Стадия 6-2: N-[3-[5-бром-1-(оксан-2-ил)пиразоло[3,4-b]пиридин-3-карбонил]-2,6-дифторфенил]пропан-1-сульфонамид
К суспензии (II) (70,7 г, 154 ммоль) в ДХМ (440 мл) добавляли п-ТСК моногидрат (2,93 г, 15,4 ммоль) и дигидропиран (15,5 мл, 169 ммоль) и смесь нагревали с обратным холодильником в течение 1 ч. После охлаждения смесь разбавляли 200 мл ДХМ, промывали насыщ. раствором NaHCО3 и солевым раствором, и высушивали над Na2SО4, и испаряли. Маслянистый остаток растворяли в теплом (45°С) МеОН (150 мл), добавляли затравочный кристалл из предшествующего синтеза и смесь перемешивали при КТ. После наблюдаемого осаждения смесь разбавляли холодным МеОН, суспензию охлаждали до -20°С в течение 2 ч, твердое вещество собирали фильтрованием с отсасыванием и промывали 50 мл МеОН при -20°С. Продукт высушивали при 50°C в вакуумной печи с получением N-[3-[5-бром-1-(оксан-2-ил)пиразоло[3,4-b]пиридин-3-карбонил]-2,6-дифторфенил]пропан-1-сульфонамида (73,4 г, 135 ммоль, выход 88%).
Стадия 6-3: N-[2,6-дифтор-3-[1-(оксан-2-ил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиразоло[3,4-b]пиридин-3-карбонил]фенил]пропан-1-сульфонамид
В сосуд загружали N-[3-[5-бром-1-(оксан-2-ил)пиразоло[3,4-b]пиридин-3-карбонил]-2,6-дифторфенил]пропан-1-сульфонамид ((III), 4,74 г, 8,72 ммоль), бис(пинаколато)диборон (4,65 г, 18,3 ммоль) и безводный ацетат калия (2,57 г, 26,2 ммоль). Сосуд вакуумировали и заполняли аргоном (3×). Добавляли сухой ДМФ (29,1 мл) и смесь нагревали до 90°С. Сосуд вакуумировали и снова заполняли аргоном (3×). Добавляли PdCl2(PPh3)2 (30,6 мг, 0,0436 ммоль) в потоке аргона, сосуд закрывали и реакционную смесь перемешивали при 90°С в течение 4 ч. После охлаждения реакционную смесь концентрировали при пониженном давлении, остаток растворяли в прибл. 50 мл EtOAc и промывали водой, полунасыщенным раствором NaCl и солевым раствором. После высушивания экстракт отфильтровывали, добавляли активированный уголь и смесь нагревали с обратным холодильником в течение 15 минут. После охлаждения смесь отфильтровывали через целит, к фильтрату добавляли н-гептан (50 мл) и удаляли растворители. Твердые вещества перемешивали в н-гексане (100 мл) в течение 30 минут и продукт собирали фильтрованием с отсасыванием. После сушки получали N-[2,6-дифтор-3-[1-(оксан-2-ил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиразоло[3,4-b]пиридин-3-карбонил]фенил]пропан-1-сульфонамид (4,37 г, 7,4 ммоль, выход 85%) в виде бесцветного твердого вещества.
Стадия 6-4: N-[2,6-дифтор-3-(5-пиримидин-5-ил-1H-пиразоло[3,4-b]пиридин-3-карбонил)фенил]пропан-1-сульфонамид
В сосуд загружали N-[2,6-дифтор-3-[1-(оксан-2-ил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиразоло[3,4-b]пиридин-3-карбонил]фенил]пропан-1-сульфонамид ((IV), 120 мг, 0,203 ммоль), 5-бромпиримидин (35,5 мг, 0,224 ммоль), фторид калия (35,4 мг, 0,610 ммоль), PdCl2(PPh3)2 (3,32 мг, 0,00406 ммоль) и дегазированный 1,4-диоксан/вода (0,5 мл, (4+1)). Из сосуда выкачивали воздух, и заполняли аргоном (3×), и нагревали до 60°C в течение 2 ч. Реакцию подкисляли конц. HCl (0,3 мл), разбавляли MeOH (0,2 мл) и нагревали до 60°C O/N. Добавляли дополнительные 0,3 мл конц. HCl и продолжали перемешивание в течение 4 ч. После охлаждения смесь разбавляли водой, нейтрализовали раствором NaHCO3 и экстрагировали ТГФ. Органическую фазу промывали солевым раствором, высушивали над Na2SO4 и испаряли. Остаток очищали флэш-хроматографией (ДХМ/MeOH, от 2% до 8%) и растирали с ацетоном с получением N-[2,6-дифтор-3-(5-пиримидин-5-ил-1H-пиразоло[3,4-b]пиридин-3-карбонил)фенил]пропан-1-сульфонамида (35,0 мг, 0,0741 ммоль, выход 36%).
Аналитические данные:
1H ЯМР (400 МГц, ДМСО-d6) δ 14,87 (с, 1H), 9,71 (с, 1H), 9,28 (д, J=10,4 Гц, 3H), 9,10 (д, J=2,2 Гц, 1H), 8,94 (д, J=2,2 Гц, 1H), 7,87 (дд, J=14,7, 7,6 Гц, 1H), 7,40 (т, J=8,6 Гц, 1H), 3,20-3,07 (м, 2Н), 1,89-1,71 (м, 2Н), 0,99 (т, J=7,4 Гц, 3Н);
Рассчитанная точная масса: 458,10 для C20H16F2N6O3S (молекулярная масса: 458,44);
МС(ESI-): m/z: 456,9 [M-1]-.
Пример 7: Синтез N-(2,6-дифтор-3-(5-(2-метилпиримидин-5-ил)-1H-пиразоло[3,4-b]пиридин-3-карбонил)фенил)пропан-1-сульфонамида
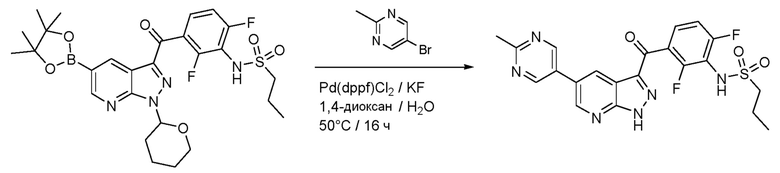
В сосуд загружали N-[2,6-дифтор-3-[1-(оксан-2-ил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиразоло[3,4-b]пиридин-3-карбонил]фенил]пропан-1-сульфонамид (120 мг, 0,203 ммоль), 5-бром-2-метилпиримидин (38,7 мг, 0,224 ммоль), фторид калия (35,4 мг, 0,610 ммоль), Pd(dppf)Cl2⋅ДХМ (3,32 мг, 0,00406 ммоль) и дегазированный 1,4-диоксан/вода (0,5 мл, (4+1)). Из сосуда выкачивали воздух, и заполняли аргоном (3×), и нагревали до 60°C в течение 2 ч. Смесь подкисляли конц. HCl (0,3 мл), разбавляли MeOH (0,2 мл) и нагревали до 60°C O/N. Добавляли дополнительные 0,3 мл конц. HCl и продолжали перемешивание в течение 3 ч. После охлаждения смесь разбавляли EtOAc и водой. Органическую фазу испаряли и продукт очищали флэш-хроматографией.
Аналитические данные:
1H ЯМР (400 МГц, ДМСО-d6) δ 14,82 (с, 1H), 9,66 (с, 1H), 9,16 (с, 2H), 9,06 (д, J=2,1 Гц, 1H), 8,89 (д, J=2,2 Гц, 1H), 7,86 (дд, J=14,6, 7,6 Гц, 1H), 7,40 (т, J=8,7 Гц, 1H), 3,15 (дд, J=8,7, 6,5 Гц, 2H), 2,70 (с, 3H), 1,81 (дкв, J=14,9, 7,4 Гц, 2H), 0,99 (т, J=7,5 Гц, 3Н);
Рассчитанная точная масса: 472,11 для C21H18F2N6O3S (молекулярная масса: 472,47);
МС(ESI-): m/z: 471,0 [M-1]-.
Пример 8: Синтез 5-(3-(2,4-дифтор-3-(пропилсульфонамидо)бензоил)-1H-пиразоло[3,4-b]пиридин-5-ил)пиримидин-2-карбоновой кислоты
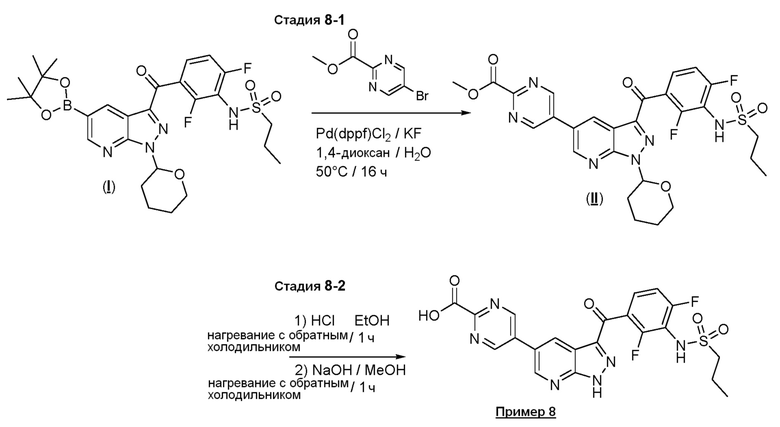
Стадия 8-1: Метил-5-(3-(2,4-дифтор-3-(пропилсульфонамидо)бензоил)-1-(тетрагидро-2H-пиран-2-ил)-1H-пиразоло[3,4-b]пиридин-5-ил)пиримидин-2-карбоксилат
В сосуд загружали N-[2,6-дифтор-3-[1-(оксан-2-ил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиразоло[3,4-b]пиридин-3-карбонил]фенил]пропан-1-сульфонамид ((I), 134 мг, 0,227 ммоль), метил-5-бромпиримидин-2-карбоксилат (54,2 мг, 0,250 ммоль), фторид калия (39,6 мг, 0,681 ммоль), Pd(dppf)Cl2⋅ДХМ (3,71 мг, 0,00454 ммоль) и дегазированный 1,4-диоксан/вода (0,5 мл, (4+1)). Из сосуда выкачивали воздух, и заполняли аргоном (3×), и нагревали до 60°C в течение 2 ч. Добавляли раствор NH4Cl и EtOAc и смесь испаряли над целитом. Продукт выделяли флэш-хроматографией (ДХМ/EtOAc, от 30% до 100%); 121 мг твердого вещества (0,2010 ммоль, выход 89%).
Стадия 8-2: 5-(3-(2,4-дифтор-3-(пропилсульфонамидо)бензоил)-1Н-пиразоло[3,4-b]пиридин-5-ил)пиримидин-2-карбоновая кислота
Метил-5-[3-[2,4-дифтор-3-(пропилсульфониламино)бензоил]-1H-пиразоло[3,4-b]пиридин-5-ил]пиримидин-2-карбоксилат ((II), 87,0 мг 0,1640 ммоль, выход 82%) растворяли в 3 мл 2,5 М HCl в EtOH и нагревали с обратным холодильником в течение 1 ч. Растворитель удаляли под вакуумом и остаток растворяли в 2 мл MeOH и 2 мл 2 н. NaOH и нагревали с обратным холодильником в течение 1 ч. После охлаждения смесь подкисляли до рН 3 с помощью 1 н. HCl, добавляли солевой раствор и смесь экстрагировали ТГФ и ТГФ/EtOAc (1:1). Экстракты промывали солевым раствором (подкисленным некоторым количеством 1 н. HCl), высушивали над Na2SO4 и отфильтровывали. Растворитель удаляли с получением 5-[3-[2,4-дифтор-3-(пропилсульфониламино)бензоил]-1H-пиразоло[3,4-b]пиридин-5-ил]пиримидин-2-карбоновой кислоты (87,0 мг, 0,1640 ммоль, выход 82%).
Аналитические данные:
1H ЯМР (400 МГц, ДМСО-d6) δ 14,94 (с, 1H), 9,68 (д, J=4,6 Гц, 1H), 9,45 (с, 2H), 9,16 (д, J=1,7 Гц, 1H), 9,04 (с, 1H), 7,88 (дд, J=14,5, 7,9 Гц, 1H), 7,41 (т, J=8,9 Гц, 1H), 3,15 (дд, J=8,7, 6,6 Гц, 3Н), 1,87-1,77 (м, 2Н), 1,00 (т, J=7,5 Гц, 3Н);
Рассчитанная точная масса: 502,09 для C21H16F2N6O5S (молекулярная масса: 502,45);
МС(ESI-): m/z: 501,0 [M-1]-.
Пример 9: Синтез N-(3-(5-(2-цианопиримидин-5-ил)-1H-пиразоло[3,4-b]пиридин-3-карбонил)-2,6-дифторфенил)пропан-1-сульфонамида
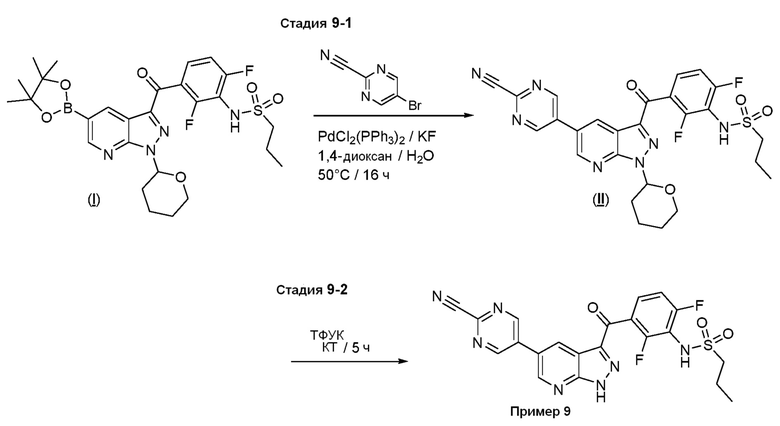
Стадия 9-1: N-[3-[5-(2-цианопиримидин-5-ил)-1-(оксан-2-ил)пиразоло[3,4-b]пиридин-3-карбонил]-2,6-дифторфенил]пропан-1-сульфонамид
В сосуд загружали N-[2,6-дифтор-3-[1-(оксан-2-ил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиразоло[3,4-b]пиридин-3-карбонил]фенил]пропан-1-сульфонамид ((I), 398 мг, 0,674 ммоль), 5-бромпиримидин-2-карбонитрил (136 мг, 0,741 ммоль), фторид калия (117 мг, 2,02 ммоль), PdCl2(PPh3)2 (11,8 мг, 0,0169 ммоль) и дегазированный диоксан/вода (4+1, 2,5 мл). Из сосуда выкачивали воздух, и заполняли аргоном (3×), и нагревали до 60°C в течение 2 ч. После охлаждения реакционную смесь разбавляли раствором NH4Cl и экстрагировали EtOAc. Экстракт промывали солевым раствором, высушивали над Na2SО4 и испаряли. Остаток очищали флэш-хроматографией (ДХМ/EtOAc, от 5% до 30%) с получением N-[3-[5-(2-цианопиримидин-5-ил)-1-(оксан-2-ил)пиразоло[3,4-b]пиридин-3-карбонил]-2,6-дифторфенил]пропан-1-сульфонамида (270 мг, 0,4760 ммоль, выход 71%).
Стадия 9-2: N-[3-[5-(2-цианопиримидин-5-ил)-1Н-пиразоло[3,4-b]пиридин-3-карбонил]-2,6-дифторфенил]пропан-1-сульфонамид
N-[3-[5-(2-цианопиримидин-5-ил)-1-(оксан-2-ил)пиразоло[3,4-b]пиридин-3-карбонил]-2,6-дифторфенил]пропан-1-сульфонамид ((II), 50,0 мг, 0,0881 ммоль) перемешивали в 1 мл ТФУК при КТ в течение 5 ч. Смесь концентрировали, растворяли в воде и pH доводили до 4 с помощью 1 н. NaOH. Водную фазу экстрагировали EtOAc, экстракт промывали солевым раствором, высушивали над Na2SO4 и испаряли. Остаток растирали с диэтиловым эфиром с получением N-[3-[5-(2-цианопиримидин-5-ил)-1H-пиразоло[3,4-b]пиридин-3-карбонил]-2,6-дифторфенил]пропан-1-сульфонамида (27,0 мг, 0,0542 ммоль, выход 61%).
Аналитические данные:
1H ЯМР (400 МГц, ДМСО-d6) δ 14,98 (с, 1H), 9,68 (с, 1H), 9,55 (с, 2H), 9,31 (с, 1H), 9,17 (д, J=2,2 Гц, 1H), 9,08 (д, J=2,1 Гц, 1H), 7,88 (дд, J=14,5, 7,8 Гц, 1H), 7,41 (т, J=8,8 Гц, 1H), 3,19-3,10 (м, 2Н), 1,86-1,74 (м, 2Н), 1,00 (т, J=7,5 Гц, 3Н);
Рассчитанная точная масса: 483,09 для C21H15F2N7O3S (молекулярная масса: 483,45);
МС(ESI-): m/z: 482,0 [M-1]-.
Пример 10: Синтез N-(3-(5-(2-(1H-тетразол-5-ил)пиримидин-5-ил)-1H-пиразоло[3,4-b]пиридин-3-карбонил)-2,6-дифторфенил)пропан-1-сульфонамида
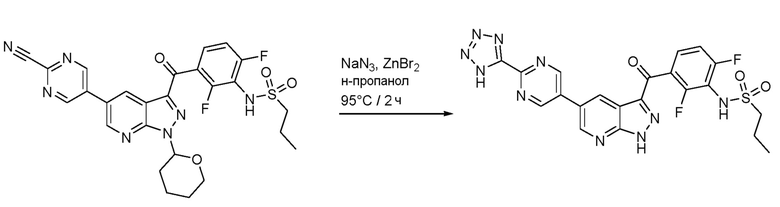
Подготовлено на основе: Vorona, S., et al. (Synthesis-Stuttgart 46(6): 781-786 (2014)), N-[3-[5-(2-цианопиримидин-5-ил)-1-(оксан-2-ил)пиразоло[3,4-b]пиридин-3-карбонил]-2,6-дифторфенил]пропан-1-сульфонамид (75,0 мг, 0,132 ммоль), бромид цинка (29,8 мг, 0,132 ммоль) и азид натрия (9,45 мг, 0,145 ммоль) нагревали до 95°С в 1-пропаноле (0,661 мл) в течение 2 ч. Реакционную смесь охлаждали, добавляли 0,25 н. NaOH (2,64 мл, 0,661 ммоль), удаляли н-пропанол при пониженном давлении и суспензию перемешивали в течение 15 минут при КТ. Смесь отфильтровывали и фильтр промывали 0,25 н. NaOH (2,64 мл, 0,661 ммоль). Фильтрат подкисляли 2 н. HCl и экстрагировали EtOAc. Экстракт промывали солевым раствором, высушивали над Na2SO4 и испаряли. Остаток растворяли в 2,5 н. HCl в EtOH (2 мл) и нагревали до 70°С в течение 2 ч. Реакционную смесь концентрировали, разбавляли диэтиловым эфиром и твердые вещества собирали фильтрованием с отсасыванием с получением N-[2,6-дифтор-3-[5-[2-(1H-тетразол-5-ил)пиримидин-5-ил]-1H-пиразоло[3,4-b]пиридин-3-карбонил]фенил]пропан-1-сульфонамида гидрохлорида (44,0 мг, 0,0782 ммоль, выход 59%).
Аналитические данные:
1H ЯМР (400 МГц, ДМСО-d6) δ 14,96 (с, 1H), 9,68 (с, 1H), 9,57 (с, 2H), 9,22 (д, J=2,1 Гц, 1H), 9,08 (д, J=2,1 Гц, 1H), 7,88 (дд, J=14,8, 7,6 Гц, 1H), 7,41 (т, J=8,5 Гц, 1H), 3,21-3,09 (м, 2H), 1,89-1,72 (м, 2H), 1,00 (т, J=7,4 Гц, 3H);
Рассчитанная точная масса: 526,11 для C21H16F2N10O3S (молекулярная масса: 526,48);
МС(ESI-): m/z: 525,0 [M-1]-.
Пример 11: Синтез N-(3-(5-(2-циклопропилпиримидин-5-ил)-1H-пиразоло[3,4-b]пиридин-3-карбонил)-2,6-дифторфенил)пропан-1-сульфонамида
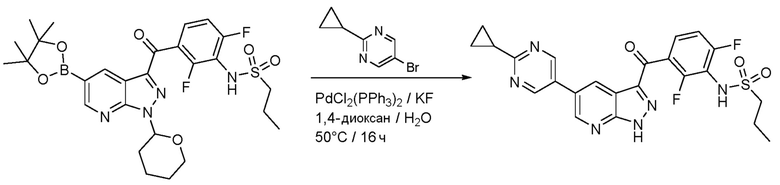
В сосуд загружали N-[2,6-дифтор-3-[1-(оксан-2-ил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиразоло[3,4-b]пиридин-3-карбонил]фенил]пропан-1-сульфонамид (176 мг, 0,298 ммоль), 5-бром-2-циклопропилпиримидин (65,3 мг, 0,328 ммоль), фторид калия (52,0 мг, 0,894 ммоль), PdCl2(PPh3)2 (4,18 мг, 0,00596 ммоль) и дегазированный 1,4-диоксан/вода (1 мл, (4+1)). Из сосуда выкачивали воздух, и заполняли аргоном (3×), и нагревали до 60°C в течение 2 ч. После охлаждения смесь распределяли между насыщ. раствором NH4Cl и EtOAc, органическую фазу высушивали над Na2SО4 и испаряли. Основной продукт выделяли флэш-хроматографией (ДХМ/EtOAc, от 20% до 80%), и помещали в 2 мл 2,5 н. HCl в EtOH, и нагревали с обратным холодильником в течение 2 ч. Реакционную смесь концентрировали и твердые вещества растирали с EtOAc с получением N-[3-[5-(2-циклопропилпиримидин-5-ил)-1H-пиразоло[3,4-b]пиридин-3-карбонил]-2,6-дифторфенил]пропан-1-сульфонамида гидрохлорида (55,0 мг, 0,1030 ммоль, выход 34%).
Аналитические данные:
1H ЯМР (400 МГц, ДМСО) δ 14,90 (с, 1H), 9,68 (с, 1H), 9,11 (с, 2H), 9,04 (д, J=2,2 Гц, 1H), 8,87 (д, J=2,2 Гц, 1H), 7,86 (дд, J=14,9, 7,6 Гц, 1H), 7,40 (т, J=8,5 Гц, 1H), 3,20-3,10 (м, 2H), 2,35-2,26 (м, 1H), 1,86-1,74 (м, 2H), 1,16-1,04 (м, 4H), 0,99 (т, J=7,5 Гц, 3H);
Рассчитанная точная масса: 498,13; для C23H20F2N6O3S (молекулярная масса: 498,51);
МС(ESI-): m/z: 497,0 [M-1]-.
Пример 12: Синтез N-(3-(5-(2-хлорпиримидин-5-ил)-1H-пиразоло[3,4-b]пиридин-3-карбонил)-2,6-дифторфенил)пропан-1-сульфонамида
В сосуд загружали N-[2,6-дифтор-3-[1-(оксан-2-ил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиразоло[3,4-b]пиридин-3-карбонил]фенил]пропан-1-сульфонамид (120 мг, 0,203 ммоль), 5-бром-2-хлорпиримидин (43,2 мг, 0,224 ммоль), фторид калия (35,4 мг, 0,610 ммоль), Pd(dppf)Cl2⋅ДХМ (3,32 мг, 0,00406 ммоль) и дегазированный 1,4-диоксан/вода (0,5 мл, (4+1)). Из сосуда выкачивали воздух, и заполняли аргоном (3×), и нагревали до 60°C в течение 2 ч. После охлаждения смесь распределяли между насыщ. раствором NH4Cl и EtOAc, органическую фазу высушивали над Na2SО4 и испаряли. Остаток растворяли в 1 мл ДХМ и 1 мл ТФУК и перемешивали O/N. Смесь выливали в воду, экстрагировали EtOAc и экстракт промывали раствором NН4Cl, высушивали над Na2SО4 и испаряли. Продукт очищали флэш-хроматографией (растворитель: МеОН; ДХМ/EtOAc, от 20% до 80% в качестве системы растворителей продукт не элюировали с колонки). Фракции, содержащие продукт, испаряли досуха и остаток растирали с толуолом и МеОН. ВЭЖХ показала чистоту только 87%. Твердые вещества промывали ДХМ, MeCN и диэтиловым эфиром и высушивали (33,0 мг, 0,0596 ммоль, выход 29%).
Аналитические данные:
1H ЯМР (400 МГц, ДМСО-d6) δ 14,92 (с, 1H), 9,67 (с, 1H), 9,28 (с, 2H), 9,09 (д, J=2,1 Гц, 1H), 8,98 (д, J=2,2 Гц, 1H), 7,87 (дд, J=14,9, 7,6 Гц, 1H), 7,40 (т, J=8,7 Гц, 1H), 3,22-3,10 (м, 2H), 1,86-1,74 (м, 2H), 1,00 (т, J=7,5 Гц, 3H);
Рассчитанная точная масса: 492,06 для C20H15ClF2N6O3S (молекулярная масса: 492,89);
МС(ESI-): m/z: 490,9 [M-1]-.
Пример 13: Синтез N-(3-(5-(4-хлорфенил)-1H-пиразоло[3,4-b]пиридин-3-карбонил)-2,4,6-трифторфенил)пропан-1-сульфонамида
Стадия 13-1: (3-амино-2,4,6-трифторфенил)-(5-бром-1Н-пиразоло[3,4-b]пиридин-3-ил)метанон
Раствор 2,4,6-трифторанилина (941 мг, 6,40 ммоль) и хлортриметилсилана (1,62 мл, 12,8 ммоль) в ТГФ (5,82 мл) охлаждали до -78°C и добавляли по каплям 2М диизопропиламид лития (6,40 мл, 12,8 ммоль) в смеси ТГФ/гептан/этилбензол. Смесь нагревали до КТ и перемешивали в течение 30 минут. Добавляли 5-бром-N-метокси-N-метил-1Н-пиразоло[3,4-b]пиридин-3-карбоксамид (829 мг, 2,91 ммоль) и смесь охлаждали до -30°С. Добавляли по каплям диизопропиламид лития (4,65 мл, 9,30 ммоль) в смеси ТГФ/гептан/этилбензол и смесь перемешивали при -15°С в течение 20 минут. Добавляли конц. HCl (3 мл) и смесь перемешивали в течение 10 минут при КТ. Реакционную смесь нейтрализовали 2 н. NaOH, водную фазу отделяли и экстрагировали ТГФ. Объединенные органические фазы высушивали над Na2SО4, отфильтровывали и концентрировали. Остаток очищали флэш-хроматографией (ДХМ/EtOAc, 0%-25%) и растирали с небольшим количеством ДХМ с получением (3-амино-2,4,6-трифторфенил)-(5-бром-1H-пиразоло[3,4-b]пиридин-3-ил)метанона (0,423 г, 1,14 ммоль, выход 39%) в виде ярко-желтого твердого вещества.
Аналитические данные:
1H ЯМР (200 МГц, CDCl3) δ 14,96 (с, 1H), 8,77 (д, J=2,8 Гц, 3H), 7,21 (т, J=10 Гц, 2H), 5,33 (с, 4H).
МС(ESI+): m/z= 370,8/368,8 [M-H]-, 350,8/348,8 [M-H-HF]-.
Стадия_13-2: N-[3-(5-бром-1H-пиразоло[3,4-b]пиридин-3-карбонил)-2,4,6-трифторфенил]пропан-1-сульфонамид
К раствору (3-амино-2,4,6-трифторфенил)-(5-бром-1Н-пиразоло[3,4-b]пиридин-3-ил)метанона (0,378 г, 1,02 ммоль) и ТЭА (1,56 мл, 11,2 ммоль) в ТГФ (5,09 мл) медленно добавляли при -10°С 1-пропансульфонилхлорид (0,378 мл, 3,36 ммоль) в 1 мл ТГФ. Реакционную смесь перемешивали при 0°С в течение 30 минут и добавляли 2 н. NaOH (4,07 мл, 8,15 ммоль). После перемешивания при КТ в течение 10 минут ТГФ испаряли из смеси и водный раствор промывали EtOAc. Раствор подкисляли 2 н. HCl и экстрагировали EtOAc. Экстракт высушивали над Na2SО4, испаряли и остаток растирали с н-гексаном с получением N-[3-(5-бром-1H-пиразоло[3,4-b]пиридин-3-карбонил)-2,4,6-трифторфенил]пропан-1-сульфонамида (0,344 г, 0,7210 ммоль, выход 71%).
Аналитические данные:
1H ЯМР (200 МГц, ДМСО-d6) δ 8,78 (дд, J=5,7, 2,2 Гц, 2H), 7,57 (тд, J=9,8, 2,0 Гц, 1H), 3,18-3,05 (м, 3H), 1,90-1,68 (м, 2Н), 0,98 (т, J=7,4 Гц, 3Н);
МС(ESI-): 476,8/474,8 [M-H]-, 456,9/454,9 [M-H-HF]-.
Стадия_13-3: N-[3-[5-(4-хлорфенил)-1H-пиразоло[3,4-b]пиридин-3-карбонил]-2,4,6-трифторфенил]пропан-1-сульфонамид
В сосуд для микроволнового облучения загружали N-[3-(5-бром-1H-пиразоло[3,4-b]пиридин-3-карбонил)-2,4,6-трифторфенил]пропан-1-сульфонамид (78,0 мг, 0,163 ммоль), (4-хлорфенил)бороновую кислоту (26,8 мг, 0,172 ммоль) и продували аргоном. Добавляли дегазированный 1,4-диоксан (0,545 мл) и дегазированный 1,5 М водный карбонат калия (0,327 мл, 0,490 ммоль) и реакционную смесь нагревали до 110°С под микроволновым облучением в течение 1 ч. Реакционную смесь распределяли между раствором NH4Cl и EtOAc и органическую фазу испаряли. Остаток очищали флэш-хроматографией (ДХМ/EtOAc, от 5% до 35%) и растирали со смесью ДХМ и н-гексана с получением N-[3-[5-(4-хлорфенил)-1H-пиразоло[3,4-b]пиридин-3-карбонил]-2,4,6-трифторфенил]пропан-1-сульфонамида (44,0 мг, 0,0865 ммоль, выход 53%).
Аналитические данные:
1H ЯМР (400 МГц, ДМСО-d6) δ 14,97 (с, 1H), 9,66 (с, 1H), 9,04 (д, J=2,2 Гц, 1H), 8,77 (д, J=2,2 Гц, 1H), 8,05-7,69 (м, 2H), 7,71-7,44 (м, 3H), 3,21-3,02 (м, 2H), 1,92-1,62 (м, 2H), 0,99 (т, J=7,4 Гц, 3H);
Рассчитанная точная масса: 508,06 для C22H16ClF3N4O3S (молекулярная масса: 508,90);
МС(ESI-): m/z: 507,0 [M-1]-.
Пример 14: Синтез N-(3-(5-(2-циклопропилпиримидин-5-ил)-1H-пиразоло[3,4-b]пиридин-3-карбонил)-2,6-дифторфенил)-1-фенилметансульфонамида
Пример 14 синтезировали аналогично Примеру 11.
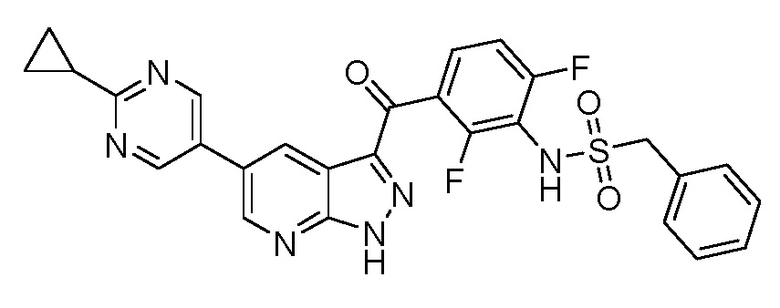
Аналитические данные:
1H ЯМР (400 МГц, ДМСО) δ 15,02-14,46 (м, 1H), 9,77 (с, 1H), 9,10 (с, 2H), 9,05 (д, J=2,1 Гц, 1H), 8,88 (д, J=2,1 Гц, 1H), 7,86 (дд, J=14,3, 7,5 Гц, 1H), 7,46-7,34 (м, 6H), 4,51 (с, 2H), 2,35-2,24 (м, 1H), 1,15-1,06 (м, 4Н).
Рассчитанная точная масса: 546,13 для C27H20F2N6O3S (молекулярная масса: 546,55)
МС(ESI+): m/z 546,0 [M+H]+.
Пример 15: Синтез N-(2,6-дифтор-3-(5-(2-метоксипиримидин-5-ил)-1H-пиразоло[3,4-b]пиридин-3-карбонил)фенил)пропан-1-сульфонамида
Пример 15 синтезировали аналогично Примеру 7.
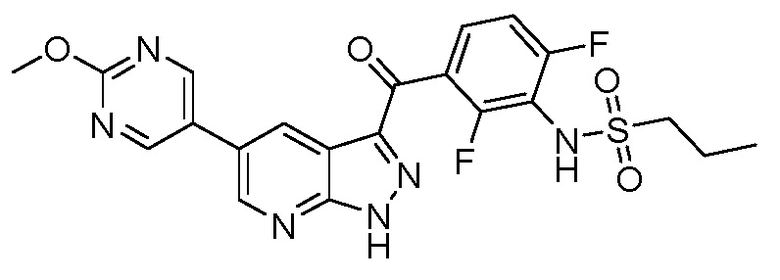
Аналитические данные:
1H ЯМР (400 МГц, ДМСО-d6) δ 14,87 (с, 1H), 9,67 (с, 1H), 9,09 (с, 2H), 9,04 (д, J=1,7 Гц, 1H), 8,86 (д, J=1,6 Гц, 1H), 7,86 (дд, J=14,3, 7,6 Гц, 1H), 7,40 (т, J=8,8 Гц, 1H), 4,00 (с, 3H), 3,19-3,09 (м, 2H), 1,81 (тд, J=14,8, 7,3 Гц, 2H), 0,99 (т, J=7,3 Гц, 3H).
Рассчитанная точная масса: 488,11 для C21H18F2N6O4S (молекулярная масса: 488,47)
МС(ESI+): m/z 489,05 [M+H]+.
Пример 16: Синтез N-(2,6-дифтор-3-(5-(2-гидроксипиримидин-5-ил)-1H-пиразоло[3,4-b]пиридин-3-карбонил)фенил)пропан-1-сульфонамида
Пример 16 синтезировали аналогично Примеру 7.
Аналитические данные:
1H ЯМР (400 МГц, ДМСО-d6) δ 14,78 (с, 1H), 12,38 (с, 1H), 9,66 (с, 1H), 8,95 (с, 1H), 8,77 (с, 1H), 8,49 (с, 2H), 7,85 (дд, J=14,7, 7,7 Гц, 1H), 7,39 (т, J=9,0 Гц, 1H), 3,18-3,11 (м, 2H), 1,81 (тд, J=14,8, 7,3 Гц, 2H), 1,00 (т, J=7,4 Гц, 3H).
Рассчитанная точная масса: 474,09 для C20H16F2N6O4S (молекулярная масса: 474,44)
МС(ESI+): m/z 475,00 [M+H]+.
Пример 17: Синтез N-(3-(5-(3-хлорпиридин-4-ил)-1H-пиразоло[3,4-b]пиридин-3-карбонил)-2,6-дифторфенил)пропан-1-сульфонамида
Пример 17 синтезировали аналогично Примеру 7.
Аналитические данные:
1H ЯМР (400 МГц, ДМСО-d6) δ 15,05-14,58 (м, 1H), 9,66 (с, 1H), 8,83 (с, 2H), 8,76 (д, J=1,6 Гц, 1H), 8,68 (д, J=4,8 Гц, 1H), 7,87 (дд, J=14,3, 8,0 Гц, 1H), 7,70 (д, J=4,9 Гц, 1H), 7,40 (т, J=8,9 Гц, 1H), 3,19-3,10 (м, 2H), 1,81 (дкв, J=14,8, 7,3 Гц, 2H), 1,00 (т, J=7,4 Гц, 3H).
Рассчитанная точная масса: 491,06 для C21H16ClF2N5O3S (молекулярная масса: 491,90)
МС(ESI+): m/z 491,95 [M+H]+.
Пример 18: Синтез N-(2,6-дифтор-3-(5-(3-метилпиридин-4-ил)-1H-пиразоло[3,4-b]пиридин-3-карбонил)фенил)пропан-1-сульфонамида
Пример 18 синтезировали аналогично Примеру 7.
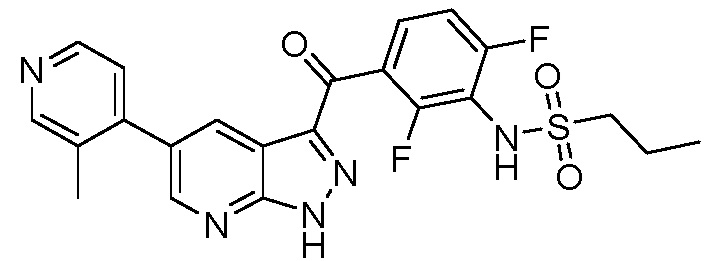
Аналитические данные:
1H ЯМР (400 МГц, ДМСО-d6) δ 14,82 (с, 1H), 9,88-9,37 (м, 1H), 8,75 (д, J=1,9 Гц, 1H), 8,56 (д, J=1,6 Гц, 1H) , 8,55 (д, J=0,9 Гц, 1H), 7,85 (дд, J=14,7, 7,5 Гц, 1H), 7,78 (д, J=7,6 Гц, 1H), 7,39 (дд, J=10,8, 5,9 Гц , 2H), 3,17-3,11 (м, 2H), 2,48 (с, 3H), 1,81 (дкв, J=14,9, 7,4 Гц, 2H), 1,00 (т, J=7,4 Гц, 3H).
Рассчитанная точная масса: 471,12 для C22H19F2N5O3S (молекулярная масса: 471,48)
МС(ESI+): m/z 471,9 [M+H]+.
Пример 19: Синтез N-(3-(5-(4-(трет-бутил)фенил)-1H-пиразоло[3,4-b]пиридин-3-карбонил)-2,6-дифторфенил)пропан-1-сульфонамида
Пример 19 синтезировали аналогично Примеру 7.
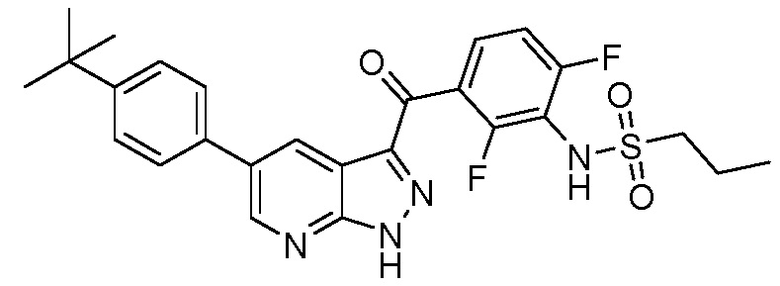
Аналитические данные:
1H ЯМР (400 МГц, ДМСО-d6) δ 14,78 (с, 1H), 9,67 (с, 1H), 9,00 (д, J=2,1 Гц, 1H), 8,73 (д, J=2,1 Гц, 1H), 7,86 (дд, J=14,7, 7,6 Гц, 1H), 7,75 (д, J=8,3 Гц, 2H), 7,57 (д, J=8,4 Гц, 2H), 7,40 (т, J=8,8 Гц, 1H), 3,18-3,12 (м, 2Н), 1,81 (тд, J=15,0, 7,4 Гц, 2Н), 1,34 (с, 9Н), 1,00 (т, J=7,4 Гц, 3Н).
Рассчитанная точная масса: 512,17 для C26H26F2N4O3S (молекулярная масса: 512,56)
МС(ESI+): m/z 513,0 [M+H]+.
Пример 20: Синтез N-(3-(5-(2-хлор-4-метоксифенил)-1H-пиразоло[3,4-b]пиридин-3-карбонил)-2,6-дифторфенил)пропан-1-сульфонамида
Пример 20 синтезировали аналогично Примеру 7.
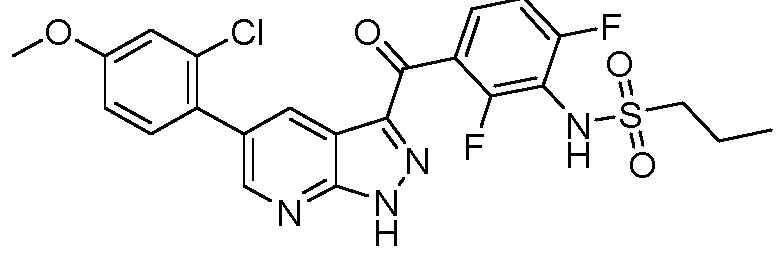
Аналитические данные:
1H ЯМР (400 МГц, ДМСО-d6) δ 14,82 (с, 1H), 9,66 (с, 1H), 8,72 (д, J=1,8 Гц, 1H), 8,57 (д, J=1,8 Гц, 1H), 7,86 (дд, J=15,0, 7,4 Гц, 1H), 7,53 (д, J=8,6 Гц, 1H), 7,39 (т, J=8,7 Гц, 1H), 7,25 (д, J=2,3 Гц, 1H), 7,10 (дд, J=8,6, 2,4 Гц, 1H), 3,86 (с, 3H), 3,15 (т, J=7,6 Гц, 2H), 1,81 (дкв, J=15,0, 7,5 Гц, 2H), 1,00 (т, J=7,4 Гц, 3H).
Рассчитанная точная масса: 520,08 для C23H19ClF2N4O4S (молекулярная масса: 520,94)
МС(ESI+): m/z 521,0 [M+H]+.
Пример 21: Синтез N-(3-(5-(2-(трет-бутил)пиримидин-5-ил)-1H-пиразоло[3,4-b]пиридин-3-карбонил)-2,6-дифторфенил)пропан-1-сульфонамида
Пример 21 синтезировали аналогично Примеру 7.
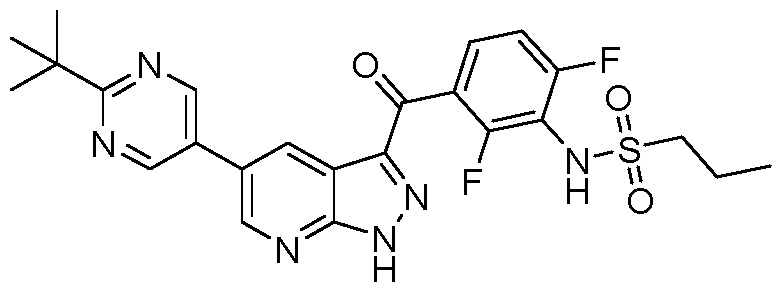
Аналитические данные:
1H ЯМР (400 МГц, ДМСО-d6) δ 14,88 (с, 1H), 9,66 (с, 1H), 9,22 (с, 2H), 9,08 (с, 1H), 8,91 (с, 1H), 7,86 (дд, J=14,7, 7,3 Гц, 1H), 7,40 (т, J=8,7 Гц, 1H), 3,18-3,11 (м, 2H), 1,87-1,74 (м, 2H), 1,43 (с, 9H), 1,00 (т, J=7,2 Гц, 3H).
Рассчитанная точная масса: 514,16 для C24H24F2N6O3S (молекулярная масса: 514,55)
МС(ESI+): m/z 515,15 [M+H]+.
Пример 22: Синтез N-(2,6-дифтор-3-(5-(2-(метилтио)пиримидин-5-ил)-1H-пиразоло[3,4-b]пиридин-3-карбонил)фенил)пропан-1-сульфонамида
Пример 22 синтезировали аналогично Примеру 7.
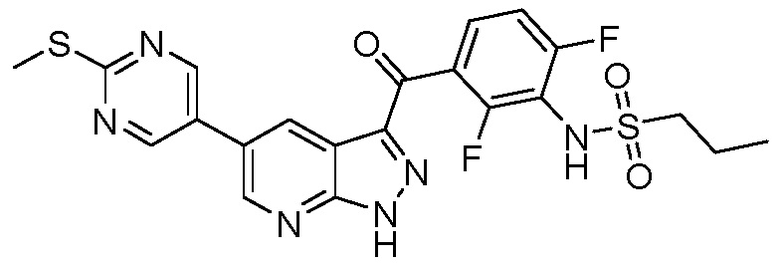
Аналитические данные:
1H ЯМР (400 МГц, ДМСО-d6) δ 14,87 (с, 1H), 9,67 (с, 1H), 9,13 (с, 2H), 9,06 (д, J=1,9 Гц, 1H), 8,90 (д, J=1,9 Гц, 1H), 7,86 (дд, J=14,3, 7,5 Гц, 1H), 7,40 (т, J=8,8 Гц, 1H), 3,19-3,12 (м, 2H), 2,60 (с, 3H), 1,87-1,75 (м, 2H), 1,00 (т, J=7,4 Гц, 3H).
Рассчитанная точная масса: 504,08 для C21H18F2N6O3S2 (молекулярная масса: 504,53)
МС(ESI+): m/z 505,05 [M+H]+.
Пример 23: Синтез N-(2,6-дифтор-3-(5-(2-изопропилпиримидин-5-ил)-1H-пиразоло[3,4-b]пиридин-3-карбонил)фенил)пропан-1-сульфонамида
Пример 23 синтезировали аналогично Примеру 7.
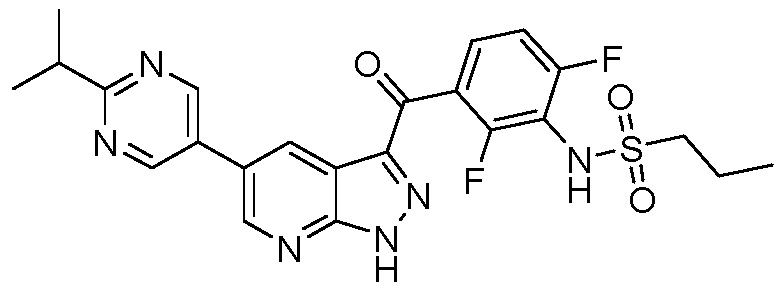
Аналитические данные:
1H ЯМР (400 МГц, ДМСО-d6) δ 14,87 (с, 1H), 9,67 (с, 1H), 9,13 (с, 2H), 9,06 (д, J=1,9 Гц, 1H), 8,90 (д, J=1,9 Гц, 1H), 7,86 (дд, J=14,3, 7,5 Гц, 1H), 7,40 (т, J=8,8 Гц, 1H), 3,19-3,12 (м, 2H), 2,60 (с, 3H), 1,87-1,75 (м, 2H), 1,00 (т, J=7,4 Гц, 3H).
Рассчитанная точная масса: 500,14 для C23H22F2N6O3S (молекулярная масса: 500,52)
МС(ESI+): m/z 501,05 [M+H]+.
Пример 24: Синтез N-(2,6-дифтор-3-(5-(4-фтор-2-метилфенил)-1H-пиразоло[3,4-b]пиридин-3-карбонил)фенил)пропан-1-сульфонамида
Пример 24 синтезировали аналогично Примеру 7.
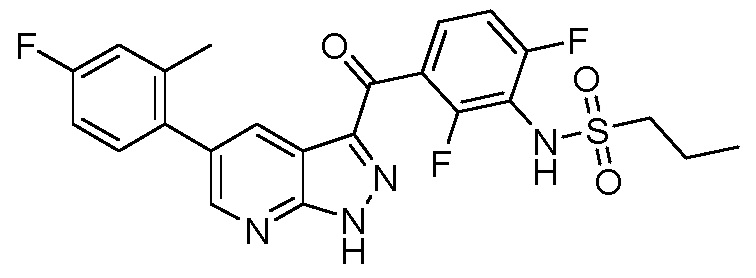
Аналитические данные:
1H ЯМР (400 МГц, ДМСО-d6) δ 14,82 (с, 1H), 9,67 (с, 1H), 8,68 (д, J=2,1 Гц, 1H), 8,48 (д, J=2,1 Гц, 1H), 7,86 (дд, J=14,8, 7,6 Гц, 1H), 7,42-7,36 (м, 2H), 7,26 (дд, J=10,1, 2,5 Гц, 1H), 7,17 (тд, J=8,5, 2,6 Гц, 1H), 3,18-3,11 (м, 2Н), 2,28 (с, 3Н), 1,81 (дкв, J=15,0, 7,4 Гц, 2Н), 1,00 (т, J=7,4 Гц, 3Н).
Рассчитанная точная масса: 488,11 для C23H19F3N4O3S (молекулярная масса: 488,49).
МС(ESI+): m/z 489,05 [M+H]+.
Пример 25 и 26: Синтез N-(2,6-дифтор-3-(5-(4-метокси-2-метилпиримидин-5-ил)-1H-пиразоло[3,4-b]пиридин-3-карбонил)фенил)пропан-1-сульфонамида (Пример 25) и N-(2,6-дифтор-3-(5-(4-гидрокси-2-метилпиримидин-5-ил)-1H-пиразоло[3,4-b]пиридин-3-карбонил)фенил)пропан-1-сульфонамида (Пример 26)
Стадия 1: Превращение N-(2,6-дифтор-3-(1-(тетрагидро-2H-пиран-2-ил)-5-(4,4,5,5-тетраметил-1,3,2)-диоксаборолан-2-ил)-1H-пиразоло[3,4-b]пиридин-3-карбонил)фенил)пропан-1-сульфонамида с 5-бром-4-хлор-2-метилпиримидином проводили аналогично Примеру 9, Стадия 9-1.
Две различные методики снятия защиты приводили или к Примеру 25, или к Примеру 26.
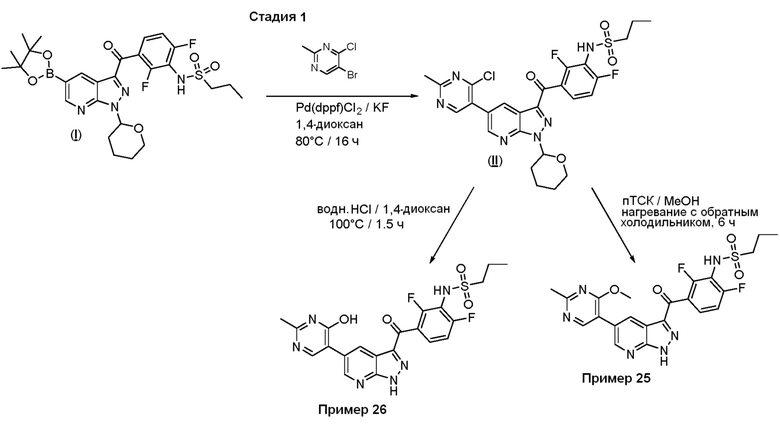
Аналитические данные N-(2,6-дифтор-3-(5-(4-метокси-2-метилпиримидин-5-ил)-1H-пиразоло[3,4-b]пиридин-3-карбонил)фенил)пропан-1-сульфонамида:
1H ЯМР (400 МГц, ДМСО-d6) δ 14,84 (с, 1H), 9,66 (с, 1H), 8,86 (д, J=2,0 Гц, 1H), 8,75 (д, J=2,0 Гц, 1H), 8,64 (с, 1H), 7,85 (дд, J=15,4, 7,4 Гц, 1H), 7,39 (т, J=8,7 Гц, 1H), 3,99 (с, 3H), 3,18-3,10 (м, 2Н), 2,62 (с, 3Н), 1,81 (тд, J=15,3, 7,7 Гц, 2Н), 1,00 (т, J=7,5 Гц, 3Н).
Рассчитанная точная масса: 502,12 для C22H20F2N6O4S (молекулярная масса: 502,50)
МС(ESI+): m/z 502,95 [M+H]+.
Аналитические данные N-(2,6-дифтор-3-(5-(4-гидрокси-2-метилпиримидин-5-ил)-1H-пиразоло[3,4-b]пиридин-3-карбонил)фенил)пропан-1-сульфонамида:
1H ЯМР (400 МГц, ДМСО-d6) δ 14,84 (с, 1H), 9,66 (с, 1H), 8,86 (д, J=2,0 Гц, 1H), 8,75 (д, J=2,0 Гц, 1H), 8,64 (с, 1H), 7,85 (дд, J=15,4, 7,4 Гц, 1H), 7,39 (т, J=8,7 Гц, 1H), 3,99 (с, 3H), 3,18-3,10 (м, 2H), 2,62 (с, 3H), 1,81 (тд, J=15,3, 7,7 Гц, 2H), 1,00 (т, J=7,5 Гц, 3H).
Рассчитанная точная масса: 488,11 для C21H18F2N6O4S (молекулярная масса: 488,47)
МС(ESI+): m/z 489,15 [M+H]+.
Пример 27: Синтез N-(2,6-дифтор-3-(5-(пиридин-3-ил)-1H-пиразоло[3,4-b]пиридин-3-карбонил)фенил)пропан-1-сульфонамида
Пример 27 синтезировали аналогично Примеру 11.
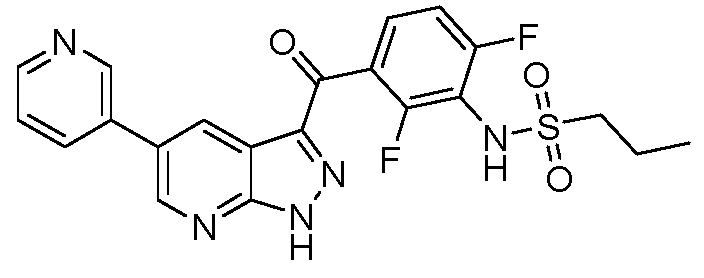
Аналитические данные:
1H ЯМР (400 МГц, ДМСО-d6) δ 14,86 (с, 1H), 9,67 (с, 1H), 9,05 (дд, J=10,7, 2,0 Гц, 2H), 8,83 (д, J=2,2 Гц, 1H), 8,66 (дд, J=4,8, 1,5 Гц, 1H), 8,30-8,23 (м, 1H), 7,87 (дд, J=14,9, 7,5 Гц, 1H), 7,57 (дд, J=7,5, 4,8 Гц, 1H), 7,40 (т, J=8,5 Гц, 1H), 3,17-3,12 (м, 2H), 1,86-1,75 (м, 2H), 1,00 (т, J=7,5 Гц, 3H);
Рассчитанная точная масса: 457,10 для C21H17F2N5O3S (молекулярная масса: 457,46)
МС(ESI-): m/z: 455,9 [M-1]-.
Пример 28: Синтез 5-(3-(2,4-дифтор-3-(пропилсульфонамидо)бензоил)-1H-пиразоло[3,4-b]пиридин-5-ил)пиколиновой кислоты
Пример 28 синтезировали аналогично Примеру 11.
Аналитические данные:
1H ЯМР (400 МГц, ДМСО-d6) δ 14,92 (с, 1H), 9,68 (с, 1H), 9,15 (дд, J=16,8, 2,0 Гц, 2H), 8,93 (д, J=2,0 Гц, 1H), 8,45 (дд, J=8,1, 2,3 Гц, 1H), 8,18 (д, J=8,1 Гц, 1H), 7,88 (дд, J=14,5, 7,7 Гц, 1H), 7,41 (т, J=8,7 Гц, 1H), 3,19-3,10 (м, 2H), 1,86-1,75 (м, 2H), 1,00 (т, J=7,5 Гц, 3H);
Рассчитанная точная масса: 501,09 для C22H17F2N5O5S (молекулярная масса: 501,46);
МС(ESI-): m/z: 500,1 [M-1]-.
Пример 29: Синтез N-[2,6-дифтор-3-[5-(4-метилпиримидин-5-ил)-1H-пиразоло[3,4-b]пиридин-3-карбонил]фенил]пропан-1-сульфонамида
Пример 29 синтезировали аналогично Примеру 7.
Аналитические данные:
1H ЯМР (400 МГц, ДМСО-d6) δ 14,90 (с, 1H), 9,66 (с, 1H), 9,12 (с, 1H), 8,80 (д, J=2,1 Гц, 1H), 8,76 (с, 1H), 8,68 (д, J=2,1 Гц, 1H), 7,90-7,82 (м, 1H), 7,43-7,36 (м, 1H), 3,30 (с, 3H), 3,19-3,11 (м, 3H), 1,88-1,73 (м, 3Н), 1,00 (т, J=7,4 Гц, 3Н);
Рассчитанная точная масса: 472,11 для C21H18F2N6О3S (молекулярная масса: 472,47);
МС(ESI+): m/z: 473,55 [M+H]+.
Пример 30: Синтез N-[3-[5-(2,4-диметилпиримидин-5-ил)-1H-пиразоло[3,4-b]пиридин-3-карбонил]-2,6-дифторфенил]пропан-1-сульфонамида
Пример 30 синтезировали аналогично Примеру 7.
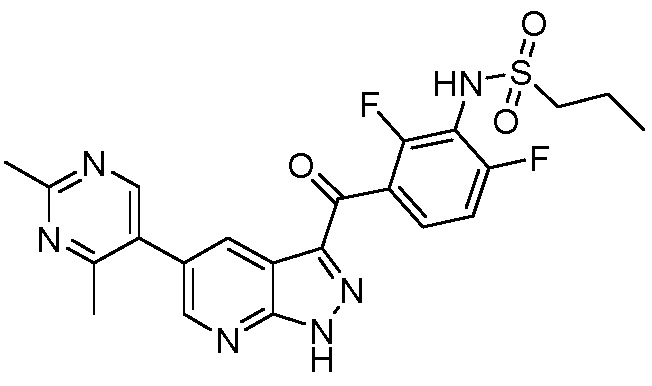
Аналитические данные:
1H ЯМР (400 МГц, ДМСО-d6) δ 14,88 (с, 1H), 9,67 (с, 1H), 8,77 (д, J=1,6 Гц, 1H), 8,67-8,59 (м, 2H), 7,86 (дд, J=14,6, 7,5 Гц, 1H), 7,40 (т, J=8,8 Гц, 1H), 3,20-3,10 (м, 2H), 2,66 (с, 3H), 2,45 (с, 3H), 1,87-1,74 (м, 2H), 0,99 (т, J=7,4 Гц, 3H).
Рассчитанная точная масса: 486,13 для C22H20F2N6O3S (молекулярная масса: 486,49);
МС(ESI+): m/z: 486,9 [M+H]+.
Пример 31: Синтез N-(3-(5-(2-циклопропил-4-метилпиримидин-5-ил)-1H-пиразоло[3,4-b]пиридин-3-карбонил)-2,6-дифторфенил)пропан-1-сульфонамида
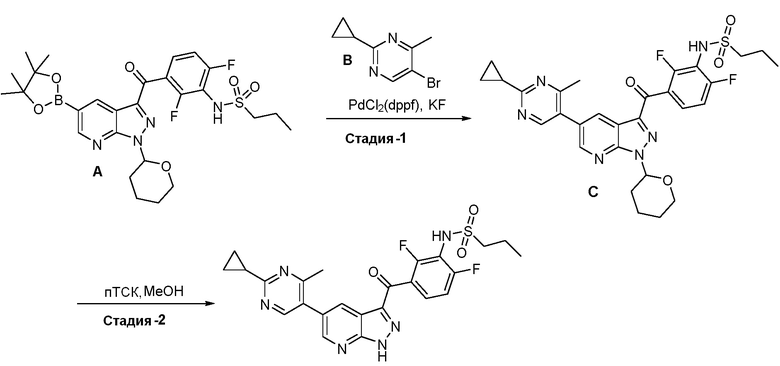
Стадия 1: Синтез N-(3-(5-(2-циклопропил-4-метилпиримидин-5-ил)-1-(тетрагидро-2H-пиран-2-ил)-1H-пиразоло[3,4-b]пиридин-3-карбонил)-2,6-дифторфенил)пропан-1-сульфонамида (С)
N-(2,6-дифтор-3-(1-(тетрагидро-2H-пиран-2-ил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразоло[3,4-b]пиридин-3-карбонил)фенил)пропан-1-сульфонамид (300 мг, 0,51 ммоль), соединение B (119 мг, 0,56 ммоль) и KF (144 мг, 1,53 ммоль) в 1,4-диоксан/H2O (10 мл) перемешивали при комнатной температуре и затем продували аргоном в течение 5 мин. Добавляли PdCl2(dppf).CH2Cl2 (42 мг, 0,051 ммоль) и смесь снова продували аргоном в течение 5 мин. Затем реакционную смесь нагревали с обратным холодильником в течение 8 ч. За ходом реакции следили с помощью ТСХ с использованием (20% EtOAc в гексане, об./об.). После завершения реакционную смесь постепенно охлаждали до комнатной температуры и разбавляли этилацетатом (20 мл), отфильтровывали через слой целита и концентрировали при пониженном давлении с получением неочищенного соединения. Неочищенный продукт окончательно очищали с помощью FCC с получением 135 мг (45%) желаемого соединения C.
1H ЯМР (400 МГц, ДМСО-d6) δ: 9,69 (с, 1H), 8,81 (д, 1H, J=2,0 Гц), 8,64 (д, 1H, J=2,0 Гц), 8,56 (с, 1H), 7,93-7,85 (м, 1H), 7,43 (т, 1H, J=8,8 Гц), 6,21 (д, 1H, J=9,6 Гц), 3,93-3,97 (м, 1H), 3,71-3,80 (м, 1H), 3,14 (т, 2H, J=7,6 Гц), 2,43 (уш.с, 3H), 2,21-2,27 (м, 1H), 1,95-2,01 (м, 2H), 1,67-1,75 (м, 3Н), 1,55-1,62 (м, 3Н), 1,10-1,20 (м, 4Н), 0,99 (т, 3Н, J=7,2 Гц).
МС(ESI+): m/z 597,1 (М+Н+).
Стадия 2: Синтез N-(3-(5-(2-циклопропил-4-метилпиримидин-5-ил)-1H-пиразоло[3,4-b]пиридин-3-карбонил)-2,6-дифторфенил)пропан-1-сульфонамида
К раствору соединения С (135 мг, 0,22 ммоль) в МеОН (10 мл) при комнатной температуре добавляли п-ТСК (28 мг, 0,24 ммоль), реакционную смесь нагревали до 50°С в течение 6 ч. За ходом реакции следили с помощью ТСХ с использованием (5% МеОН в ДХМ об./об.). После завершения реакции растворитель испаряли под вакуумом, добавляли воду (5 мл) и смесь нейтрализовали с использованием водн. насыщ. NaHCО3 и экстрагировали EtOAc (3×20 мл). Объединенный органический слой высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением неочищенного соединения, которое при растирании предоставляло 55 мг (48%) целевого соединения в виде белого с желтоватым или сероватым оттенком твердого вещества.
1H ЯМР (400 МГц, ДМСО-d6) δ: 9,67 (уш.с, 1H), 8,76 (д, 1H, J=2,0 Гц), 8,61 (д, 1H, J=2,0 Гц), 8,55 (с, 1H), 7,82-7,88 (м, 1H), 7,39 (т, 1H, J=8,80 Гц), 3,14 (т, 2H, J=7,60 Гц), 2,42 (с, 3H), 2,20-2,28 (м, 1H), 1,80 (кв, 2H, J=7,60 Гц), 1,05-1,11 (м, 4H), 0,99 (т, 3H, J=7,20 Гц).
Рассчитанная точная масса: 512,14 для C24H22F2N6O3S (молекулярная масса: 512,53);
МС(ESI+): m/z 513,60 [M+H]+.
Пример 32: Синтез N-(3-(5-(2-циклопропил-4-(метилтио)пиримидин-5-ил)-1H-пиразоло[3,4-b]пиридин-3-карбонил)-2,6-дифторфенил)пропан-1-сульфонамида
Часть А. Синтез 5-бром-2-циклопропил-4-(метилтио)пиримидина (D)
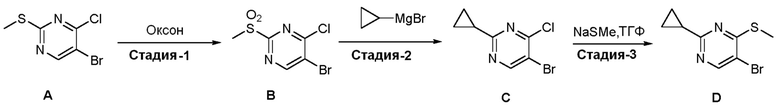
Стадия-1: 5-бром-4-хлор-2-(метилсульфонил)пиримидин (В)
К перемешиваемому раствору 5-бром-4-хлор-2-(метилтио)пиримидина (3 г, 12,6 моль) в ТГФ (25 мл) при комнатной температуре добавляли оксон (1,16 г, 37,8 ммоль), растворенный в воде (15 мл). Затем смесь оставляли перемешиваться в течение 4 ч. После завершения реакции по данным ТСХ с использованием EtOAc/н-гексана (30/70% об./об.) в качестве растворителя реакционную смесь разбавляли водой (50 мл) и экстрагировали EtOAc (3×25 мл). Объединенные разделенные органические слои промывали солевым раствором, высушивали над безводным Na2SО4 и испаряли при пониженном давлении с получением 3,2 г (94%) соединения В в виде белого с желтоватым или сероватым оттенком твердого вещества.
1H ЯМР (400 МГц, ДМСО-d6) δ: 9,38 (с, 1H), 3,41 (с, 3H).
МС(ESI+): m/z 272,80 [М+Н]+.
Стадия 2: 5-бром-4-хлор-2-циклопропилпиримидин (С)
К перемешиваемому раствору соединения В (1,5 г, 5,55 ммоль) в сухом ТГФ (20 мл) при 0°С медленно добавляли 1М циклопропилмагния бромида (6,66 мл, 6,66 ммоль). Затем смесь постепенно нагревали до КТ и перемешивали в течение 2 ч. После завершения реакции по данным ТСХ с использованием EtOAc/н-гексана (20/80% об./об.) в качестве растворителя ее гасили добавлением водн. раствора NН4Cl и экстрагировали с использованием EtOAc (2×25 мл). Разделенные объединенные органические слои промывали солевым раствором (15 мл), высушивали над безводным Na2SО4 и испаряли при пониженном давлении с получением неочищенного соединения. Неочищенный продукт окончательно очищали с помощью FCC с получением 600 мг (52%) желаемого соединения С в виде белого с желтоватым или сероватым оттенком твердого вещества.
1H ЯМР (400 МГц, ДМСО-d6) δ: 8,59 (с, 1H), 2,05-2,25 (м, 1H), 1,05-1,10 (м, 4H).
МС(ESI+): m/z 234,8 [M+H]+.
Стадия 3: 5-бром-2-циклопропил-4-(метилтио)пиримидин (D)
К перемешиваемому раствору соединения С (1,2 г, 5,17 ммоль) в ДМФ (10 мл) при КТ добавляли NaSMe (544 мг, 7,75 моль) и смесь перемешивали в течение 16 ч при КТ. После завершения реакции по данным ТСХ с использованием EtOAc/н-гексана (10/90% об./об.) добавляли воду (30 мл) и экстрагировали EtOAc (2×25 мл), отделенный органический слой промывали солевым раствором, высушивали над безводным Na2SO4 и испаряли при пониженном давлении с получением 800 мг (80%) соединения D в виде желтой жидкости.
1H ЯМР (400 МГц, CDCl3) δ: 8,25 (с, 1H), 2,52 (с, 3H), 4,10-4,23 (м, 1H), 1,10-1,20 (м, 4H).
МС(ESI+): m/z 246,8 [M+H]+.
Часть B: Синтез N-(3-(5-(2-циклопропил-4-(метилтио)пиримидин-5-ил)-1H-пиразоло[3,4-b]пиридин-3-карбонил)-2,6-дифторфенил)пропан-1-сульфонамида
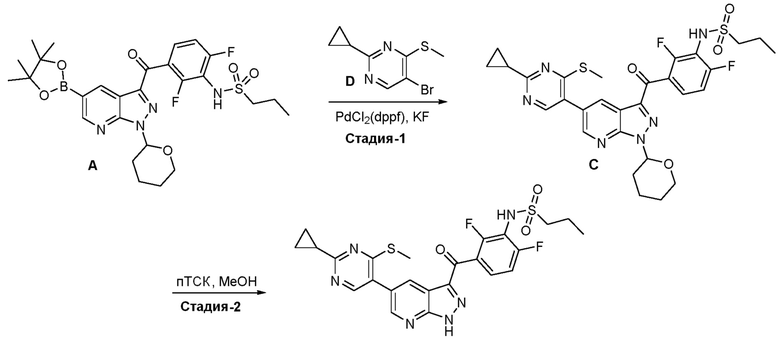
Стадия 1: N-(3-(5-(2-циклопропил-4-(метилтио)пиримидин-5-ил)-1-(тетрагидро-2H-пиран-2-ил)-1H-пиразоло[3,4-b]пиридин-3-карбонил)-2,6-дифторфенил)пропан-1-сульфонамид (С)
К перемешиваемому раствору N-(2,6-дифтор-3-(1-(тетрагидро-2H-пиран-2-ил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразоло[3,4-b]пиридин-3-карбонил)фенил)пропан-1-сульфонамида (400 мг, 0,67 ммоль) и соединения D (182 мг, 0,74 ммоль) в 5 мл 1,4-диоксан/H2O (75/25% об./об.) добавляли 118 мг (2,03 ммоль) KF, растворенного в 1 мл воды. Полученную смесь продували аргоном в течение 5 мин, затем добавляли Pd(dppf)Cl2⋅ДХМ (0,1 экв., 55 мг, 0,067 ммоль) и смесь снова продували аргоном в течение 5 мин. Затем реакционную смесь нагревали при 90°C в течение 6 ч. После завершения реакции добавляли воду (10 мл) и рН доводили до 6 с помощью 1 н. HCl. Смесь экстрагировали EtOAc (3×15 мл), органический слой отделяли, высушивали и концентрировали с получением неочищенного остатка. Неочищенный материал очищали препаративной ВЭЖХ с обращенной фазой с получением 60 мг (выход 14%) соединения C.
МС(ESI+): m/z 629,20 [М+Н]+.
Стадия 2: N-(3-(5-(2-циклопропил-4-(метилтио)пиримидин-5-ил)-1H-пиразоло[3,4-b]пиридин-3-карбонил)-2,6-дифторфенил)пропан-1-сульфонамид
К перемешиваемому раствору соединения C (90 мг, 0,143 ммоль) в MeOH (2 мл) добавляли пТСК (74 мг, 0,42 ммоль) при КТ и реакционную смесь нагревали до 65°C в течение 16 ч. Ход реакции контролировали с помощью ТСХ с использованием EtOAc/н-гексана (70/30% об./об.). После завершения реакции растворитель испаряли под вакуумом, добавляли воду (5 мл), затем нейтрализовали (pH=7) водн. раствором NaHCО3 и экстрагировали EtOAc (3×5 мл). Отделенный органический слой высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением неочищенного продукта, который очищали SFC с получением 25 мг (32%) желаемого соединения.
1H ЯМР (400 МГц, ДМСО-d6) δ: 9,66 (с, 1H), 8,72 (с, 1H), 8,69 (с, 1H), 8,37 (с, 1H), 7,86 (кв, 1H, J=6,8 Гц), 7,39 (т, 1H, J=8,8 Гц), 3,14 (т, 2H, J=8,0 Гц), 2,20-2,30 (м, 1H), 1,70-1,86 (м, 2H), 1,10-1,15 (уш.с, 4H), 0,99 (т, 3H, J=7,2 Гц).
Рассчитанная точная масса: 512,14 для C24H22F2N6О3S (молекулярная масса: 512,53);
МС(ESI+): m/z 545,10 [M+H]+.
Пример 33: Синтез N-[2,6-дифтор-3-[5-(4-метил-2-метилсульфанилпиримидин-5-ил)-1H-пиразоло[3,4-b]пиридин-3-карбонил]фенил]пропан-1-сульфонамида
Пример 33 синтезировали аналогично Примеру 7.
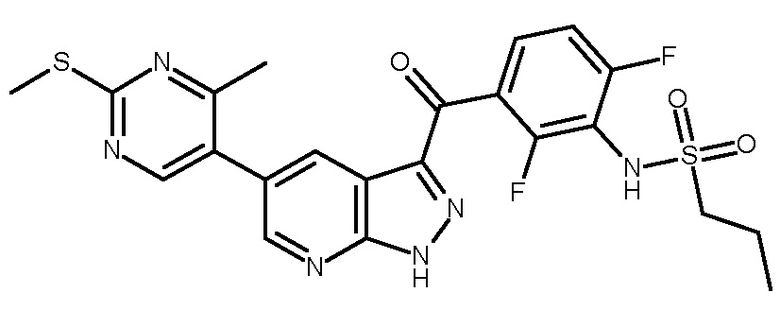
Аналитические данные:
1H ЯМР (400 МГц, ДМСО-d6) δ 14,79 (с, 1H), 9,73 (с, 1H), 8,77 (д, J=2,0 Гц, 1H), 8,65 (д, J=2,0 Гц, 1H), 8,58 (с, 1H), 7,86 (дд, J=14,6, 7,8 Гц, 1H), 7,39 (т, J=9,0 Гц, 1H), 3,21-3,05 (м, 2H), 2,57 (с, 3Н), 2,44 (с, 3Н), 1,91-1,68 (м, 2Н), 0,99 (т, J=7,4 Гц, 3Н).
Рассчитанная точная масса: 518,10 для C22H20F2N6O3S2 (молекулярная масса: 518,56);
МС(ESI-): m/z: 517,5 [M-1]-.
Пример 34: Синтез N-[3-[5-(2,4-диметоксипиримидин-5-ил)-1H-пиразоло[3,4-b]пиридин-3-карбонил]-2,6-дифторфенил]пропан-1-сульфонамида
Пример 34 синтезировали аналогично Примеру 7.
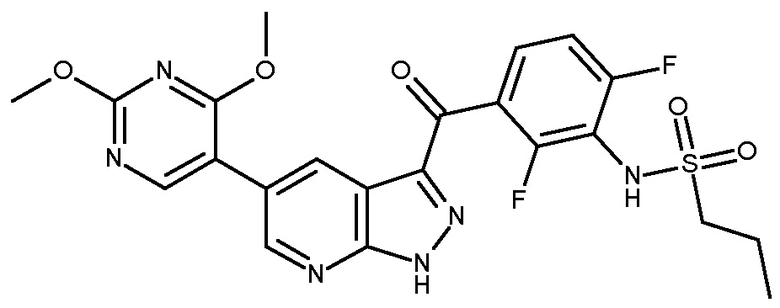
Аналитические данные:
1H ЯМР (400 МГц, ДМСО-d6) δ 14,80 (с, 1H), 9,66 (с, 1H), 8,81 (д, J=2,1 Гц, 1H), 8,69 (д, J=2,1 Гц, 1H), 8,54 (с, 1H), 7,84 (дд, J=14,5, 7,7 Гц, 1H), 7,38 (т, J=8,8 Гц, 1H), 4,05-3,89 (м, 6H), 3,18-3,09 (м, 2Н), 1,86-1,72 (м, 2Н), 0,98 (т, J=7,4 Гц, 3Н).
Рассчитанная точная масса: 518,12 для C22H20F2N6O5S (молекулярная масса: 518,49);
МС(ESI+): m/z 519,15 [M+H]+
Пример 35: Синтез N-(3-(5-(2-циклопропилпиримидин-5-ил)-1H-пиразоло[3,4-b]пиридин-3-карбонил)-2,4,6-трифторфенил)пропан-1-сульфонамида
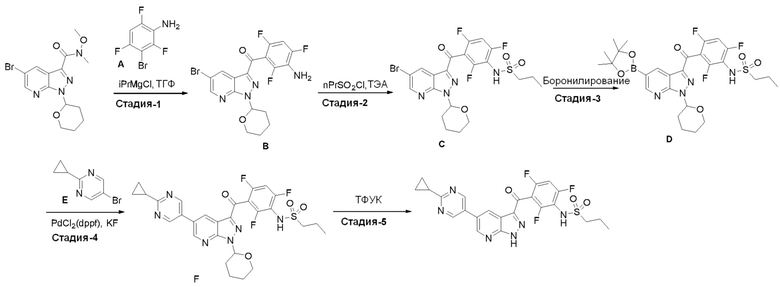
Стадия 1: Синтез (3-амино-2,4,6-трифторфенил)(5-бром-1-(тетрагидро-2H-пиран-2-ил)-1H-пиразоло[3,4-b]пиридин-3-ил)метанона (B)
Раствор A : К соединению A (4,22 г, 18,68 ммоль) в сухом ТГФ (24 мл) по каплям добавляли 2М изопропилмагния хлорида в ТГФ (9,33 мл, 1 экв., 18,68 ммоль) при -10°C, поддерживая температуру ниже 10°С. После перемешивания в течение 20 минут при 25°С реакционную смесь охлаждали до 0°С и медленно добавляли хлортриметилсилан (2,02 г, 1 экв., 18,68 ммоль). Смесь нагревали до 25°С и перемешивали в течение 30 минут. Раствор снова охлаждали до -10°С и добавляли по каплям 2М изопропилмагния хлорида в ТГФ (9,33 мл, 1 экв., 18,68 ммоль), поддерживая температуру ниже 10°С. После перемешивания в течение 20 минут при 25°С реакционную смесь охлаждали до 0°С и медленно добавляли хлортриметилсилан (2,02 г, 1 экв., 18,68 ммоль). Смесь нагревали до 25°С и перемешивали в течение 30 минут. Реакционную смесь охлаждали до -10°С и по каплям добавляли 2М изопропилмагния хлорид в ТГФ (9,33 мл, 1 экв., 18,68 ммоль). После завершения добавления смесь перемешивали при 0°С в течение 20 минут.
Раствор B : Во второй колбе 2М изопропилмагния хлорид в ТГФ (9,33 мл, 1 экв., 18,68 ммоль) добавляли к суспензии 5-бром-N-метокси-N-метил-1-(тетрагидро-2H-пиран-2-ил)-1H-пиразоло[3,4-b]пиридин-3-карбоксамида (3 г, 8,12 ммоль) в ТГФ (30 мл) при -10°С по каплям, поддерживая температуру ниже 0°С. После завершения добавления смесь перемешивали при 0°С в течение 10 минут.
Раствор В добавляли к раствору А и реакционную смесь перемешивали при 25°С в течение 2 ч. Затем осторожно добавляли водн. 4 н. HCl до растворения всех солей магния. Водный раствор доводили до pH 5 с помощью 30% NaOHводн., насыщали NaCl и разделяли слои. Водный слой экстрагировали смесью ТГФ и EtOAc (добавление EtOAc было необходимо для разделения слоев). Экстракт испаряли, повторно растворяли в ТГФ и объединяли с органической фазой. Добавляли конц. HClводн. (5 мл) и смесь перемешивали в течение 10 мин при КТ. Смесь нейтрализовали твердым NaHCО3, нижний водн. слой удаляли и органический слой высушивали над Na2SО4. После испарения растворителя остаток суспендировали в диэтиловом эфире (15 мл), растирали с ДХМ (20 мл) и твердые вещества собирали фильтрованием с отсасыванием. Фильтрат испаряли, остаток растворяли в (20 мл) диэтилового эфира и твердые вещества собирали фильтрованием с отсасыванием. Объединенные твердые вещества очищали с помощью FCC с использованием 0-5% MeOH в ДХМ с получением 900 мг (38%) желаемого белого с желтоватым или сероватым оттенком твердого вещества.
1H ЯМР (400 МГц, ДМСО-d6) δ: 8,86 (д, 1H, J=2,0 Гц), 8,80 (д, 1H, J=2,0 Гц), 7,25-7,35 (м, 1H), 6,14 (д, 1H, J=10 Гц), 5,36 (уш.с, 2H), 3,90-3,95 (м, 1H), 3,70-3,85 (м, 1H), 2,25-2,33 (м, 1H), 1,65-1,85 (м, 1H), 1,50-1,62 (м, 2H), 1,10-1,15 (м, 2H).
МС(ESI+): m/z 455,18 [M+H]+.
Стадия 2: Синтез N-(3-(5-бром-1-(тетрагидро-2H-пиран-2-ил)-1H-пиразоло[3,4-b]пиридин-3-карбонил)-2,4,6-трифторфенил)пропан-1-сульфонамида (С)
К раствору соединения В (900 мг, 1,98 ммоль) и триэтиламина (13,7 мл, 10 ммоль) в ТГФ (20 мл) медленно добавляли 1-пропансульфонилхлорид (0,8 мл, 7 ммоль) в равном объеме ТГФ при - 10°С и реакционную смесь перемешивали при -10°С в течение 30 минут. Добавляли 2 н. NaOH (3,75 мл, 12 ммоль) и испаряли ТГФ при 45°С при пониженном давлении. Водный раствор промывали EtOAc (2×20 мл) и диэтиловым эфиром. Остаточные органические вещества испаряли, раствор подкисляли 2 н. HCl и энергично перемешивали в течение 30 минут для разрушения более крупных комков. Твердое вещество собирали фильтрованием с отсасыванием, промывали водой и высушивали при 100°С с получением соединения С в виде белого с желтоватым или сероватым оттенком твердого вещества (960 г, выход 87%). 1H ЯМР выявил смесь моно- и дисульфонамида, которую использовали как таковую в следующей стадии.
Стадия 3: Синтез N-(2,4,6-трифтор-3-(1-(тетрагидро-2H-пиран-2-ил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразоло[3,4-b]пиридин-3-карбонил)фенил)пропан-1-сульфонамида (D)
К перемешиваемому раствору соединения C (900 мг, 1,60 ммоль) в 1,4-диоксане (15 мл) добавляли бис(пинаколато)диборон (612 мг, 2,41 ммоль) и KF (278 мг, 4,8 ммоль) и полученную смесь дегазировали аргоном в течение 10 мин. Добавляли Pd(dppf)Cl2⋅ДХМ (65 мг, 0,08 ммоль), смесь дополнительно дегазировали аргоном в течение 5 мин и перемешивали при 85°С в течение 8 ч. Ход реакции контролировали с помощью ТСХ, используя EtOAc/н-гексан (60/40% об./об.) в качестве растворителя. После завершения реакционную смесь охлаждали до КТ, разбавляли EtOAc (20 мл), отфильтровывали через слой целита и органический слой концентрировали при пониженном давлении с получением неочищенного соединения, которое очищали с помощью FCC с получением (960 мг , 87%) соединения D.
МС(ESI+): m/z 609,0 [M+H]+.
Стадия 4: Синтез N-(3-(5-(2-циклопропилпиримидин-5-ил)-1-(тетрагидро-2H-пиран-2-ил)-1H-пиразоло[3,4-b]пиридин)-3-карбонил)-2,4,6-трифторфенил)пропан-1-сульфонамида (F)
К перемешиваемому раствору соединения D (950 мг, 1,56 ммоль) и соединения E (342 мг, 1,71 ммоль) в 1,4-диоксан/H2O (10 мл) добавляли KF (271 мг, 4,68 ммоль) и полученную смесь дегазировали аргоном в течение 10 мин. Добавляли Pd(dppf)Cl2⋅ДХМ (64 мг, 0,078 ммоль), реакционную смесь дополнительно дегазировали аргоном в течение 5 мин и перемешивали при 90°С в течение 8 ч. Ход реакции контролировали с помощью ТСХ, используя EtOAc/н-гексан (40/60% об./об.) в качестве растворителя. После завершения реакционную смесь охлаждали до КТ и разбавляли EtOAc (20 мл), отфильтровывали через слой целита и органический слой концентрировали при пониженном давлении с получением неочищенного соединения, которое при растирании с использованием диэтилового эфира предоставляло (650 мг, 61%) соединения F.
1H ЯМР (400 МГц, ДМСО-d6) δ: 9,10-9,15 (м, 3H), 8,89 (д, 1H, J=2,4 Гц), 7,80 (т, 1H, J=8,8 Гц), 6,66 (дд, 1H, J=2,4 и 10,4 Гц), 3,60-3,80 (м, 4H), 2,22-2,38 (м, 3H), 1,95-2,00 (м, 2H), 1,72-1,93 (м, 3H), 1,55-1,60 (м, 2Н), 1,06-1,09 (м, 2Н), 0,95-1,05 (м, 5Н).
Стадия 5: Синтез N-(3-(5-(2-циклопропилпиримидин-5-ил)-1H-пиразоло[3,4-b]пиридин-3-карбонил)-2,4,6-трифторфенил)пропан-1-сульфонамида
К соединению F (650 г, 1,08 ммоль) добавляли ТФУК (10 мл) при КТ и смесь перемешивали в течение ночи. После завершения реакции по данным ТСХ с использованием (MeOH/ДХМ 5/95% об./об.) реакционную смесь испаряли с использованием потока N2 при КТ, получая неочищенное соединение. Этот неочищенный материал нейтрализовали до pH 8, используя водн. насыщ. NaHCО3 с последующей экстракцией ДХМ (2×20 мл), органический слой отделяли и высушивали над безводным Na2SО4, испарение растворителя предоставляло неочищенный материал. Неочищенный продукт окончательно очищали с помощью FCC с использованием 0-5% МеОН в ДХМ (об./об.) с получением 175 мг (32%) желаемого соединения в виде белого с желтоватым или сероватым оттенком твердого вещества.
1H ЯМР (400 МГц, ДМСО-d6) δ: 13,05 (уш.с, 1H), 9,66 (уш.с, 1H), 9,11 (с, 2H), 9,07 (с, 1H), 8,88 (с, 1H), 7,59 (т, 1H, J=8,0 Гц), 3,13 (т, 1H, J=8,0 Гц), 2,25-3,35 (м, 1H), 1,70-1,85 (м, 2H), 1,05-1,15 (м, 4H), 0,98 (т, 3H, J=7,60 Гц)
МС(ESI+): m/z 517,06 [M+H]+.
Пример 36: Синтез N-[3-[5-(3-этил-4-пиридил)-1H-пиразоло[3,4-b]пиридин-3-карбонил]-2,6-дифторфенил]пропан-1-сульфонамида
Пример 36 синтезировали аналогично Примеру 7.
Аналитические данные:
1H ЯМР (400 МГц, ДМСО-d6) δ: 14,90 (с, 1H), 9,67 (с, 1H), 8,72 (д, J=1,9 Гц, 1H), 8,64 (с, 1H), 8,59-8,50 (м, 2H), 7,87 (дд, J=14,7, 7,6 Гц, 1H), 7,39 (дд, J=14,1, 6,6 Гц, 2H), 3,21-3,06 (м, 2H), 2,66 (кв, J=7,5 Гц, 2H), 1,88-1,73 (м, 2H), 1,06 (т, J=7,5 Гц, 3H), 1,00 (т, J=7,4 Гц, 3Н).
Рассчитанная точная масса: 485,13 для C23H21F2N5О3S (молекулярная масса: 485,51);
МС(ESI-): m/z: 484,2 [M+H]-.
Пример 37: Синтез N-[3-[5-(3-циано-4-пиридил)-1H-пиразоло[3,4-b]пиридин-3-карбонил]-2,6-дифторфенил]пропан-1-сульфонамида
Пример 37 синтезировали аналогично Примеру 7.
Аналитические данные:
1H ЯМР (400 МГц, ДМСО-d6) δ: 9,19 (с·1H), 9,04-8,91 (м, 3H), 7,95-7,84 (м, 2H), 7,41 (т, J=8,8 Гц, 1H), 3,19-3,10 (м, 2H), 1,81 (дкв, J=15,0, 7,5 Гц, 2H), 1,00 (т, J=7,4 Гц, 3H).
Рассчитанная точная масса: 482,10 для C22H16F2N6О3S (молекулярная масса: 482,47);
МС(ESI-): m/z: 481,3 [M+H]-.
Пример 38: Биологическая активность - эффективность ингибирования и селективность в функциональных ферментных анализах
Киназные активности соединений изобретения измеряли с использованием сервиса для анализа 33PanQinase®, предоставленного ProQinase GmbH, Фрайбург, Германия. Подробная информация об условиях анализа раскрыта на веб-сайте ProQinase (https://www.proqinase.com/products-services-biochemical-assay-services/kinase-assays). Вкратце, все анализы киназы проводили в 96-луночных планшетах FlashPlate™ от PerkinElmer (Бостон, Массачусетс, США) в 50 мкл реакционном объеме. Реакционный коктейль пипетировали в четыре стадии в следующем порядке:
• 20 мкл буфера для анализа (стандартный буфер)
• 5 мкл раствора АТФ (в H2O)
• 5 мкл исследуемого соединения (в 10% ДМСО)
• 20 мкл смеси фермент/субтрат
Анализ для всех протеинкиназ содержал 70 мМ HEPES-NaOH pH 7,5, 3 мМ MgCl2, 3 мМ MnCl2, 3 мкМ Na-ортованадата, 1,2 мМ DTT, 50 мкг/мл PEG20000, АТФ (переменные концентрации, соответствующие кажущейся АТФ-Km соответствующей киназы), [γ-33Р]-АТФ (приблизительно 8×1005 импульсов в минуту на лунку), протеинкиназу и субстрат.
Реакционные коктейли инкубировали при 30°С в течение 60 минут. Реакцию останавливали 50 мкл 2% (об./об.) H3PO4, планшеты аспирировали и дважды промывали 200 мкл 0,9% (масс./об.) NaCl. Включение 33Pi определяли с помощью сцинтилляционного счетчика для микропланшетов (Microbeta, Wallac).
Все анализы проводили с использованием базовой системы BeckmanCoulter/SAGIANTM.
В случае анализов с одной концентрацией эффективность исследуемого соединения выражается в виде % остаточной активности. Для определения значений IC50 тестировали серийные разведения в диапазоне конечных концентраций от 100 мкМ до 3 нМ (10 концентраций). Подходящей моделью для определений IC50 была «Сигмоидальная характеристика (переменный наклон)» с параметрами «вверху», зафиксированными при 100%, и «внизу» при 0%. Используемый метод подбора представлял собой подгонку методом наименьших квадратов.
Эффективность MKK4:
Эффективность ингибирования в отношении MKK4 классифицируется следующим образом:
Анализы с определением значений IC50:
Селективность исследуемых соединений в отношении BRaf, JNK1 и MKK7, в целом обозначенных как нецелевые, рассчитывали с помощью соотношения IC50 (нецелевое)/IC50 (MKK4) и классифицировали следующим образом:
Таблица 1: Биохимическая эффективность характерных Примеров в отношении MKK4 и селективность в отношении BRaf, MKK7 и JNK1 на основании значений IC50.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ ПРОТЕИНКИНАЗЫ MKK4 ДЛЯ СТИМУЛЯЦИИ РЕГЕНЕРАЦИИ ПЕЧЕНИ ИЛИ УМЕНЬШЕНИЯ ИЛИ ПРЕДОТВРАЩЕНИЯ ГИБЕЛИ ГЕПАТОЦИТОВ | 2019 |
|
RU2788000C2 |
| ИНГИБИТОРЫ ПРОТЕИНКИНАЗ ДЛЯ УСИЛЕНИЯ РЕГЕНЕРАЦИИ ПЕЧЕНИ ИЛИ СНИЖЕНИЯ ИЛИ ПРЕДОТВРАЩЕНИЯ ГИБЕЛИ ГЕПАТОЦИТОВ | 2019 |
|
RU2822216C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2014 |
|
RU2681849C2 |
| ПРОИЗВОДНЫЕ ХИНАЗОЛИН-4-ОНА, ПОЛЕЗНЫЕ ДЛЯ ЛЕЧЕНИЯ BRAF-АССОЦИИРОВАННЫХ ЗАБОЛЕВАНИЙ И НАРУШЕНИЙ | 2020 |
|
RU2797606C1 |
| ПОЛИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБЫ ЦЕЛЕНАПРАВЛЕННОЙ ДЕГРАДАЦИИ ПОЛИПЕПТИДОВ БЫСТРО УСКОРЕННОЙ ФИБРОСАРКОМЫ | 2019 |
|
RU2830173C2 |
| ИНГИБИТОРНЫЕ СОЕДИНЕНИЯ | 2013 |
|
RU2673079C2 |
| ПРОИЗВОДНЫЕ ПИРИДАЗИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ RORc | 2017 |
|
RU2757571C2 |
| БЕНЗОКСЕПИНОВЫЕ ИНГИБИТОРЫ PI3 И СПОСОБЫ ПРИМЕНЕНИЯ | 2010 |
|
RU2557658C2 |
| ПРОИЗВОДНЫЕ БЕНЗИЗОКСАЗОЛСУЛЬФОНАМИДА | 2020 |
|
RU2813356C2 |
| ПИРАЗОЛ-4-ИЛ-ГЕТЕРОЦИКЛИЛ-КАРБОКСАМИДНЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ПРИМЕНЕНИЯ | 2012 |
|
RU2638552C2 |
Изобретение относится к соединению, имеющему формулу (Iа), или его фармацевтически приемлемой соли, которые селективно ингибируют митоген-активируемую протеинкиназу 4 (MKK4) по сравнению с протеинкиназами JNK1 и MKK7. В формуле (Iа) R1 представляет собой H; R4 представляет собой Н; R5 выбран из а) пиримидинила, который замещен 1 или 2 заместителями, независимо выбранными из C3-C6-циклоалкила, C1-C4-алкила, -COOR10, -ОН, C1-C4-алкилсульфанила, тетразолила, CN, галогена и C1-C4-алкокси, и b) пиридила, который замещен 1 или 2 заместителями, независимо выбранными из C1-C4-алкила и галогена; R6 представляет собой Н; Rw представляет собой -NR10SO2R12; Rx представляет собой F; Ry представляет собой F; Rz представляет собой Н или F; R10 в каждом случае представляет собой независимо Н или C1-C4-алкил; R12 представляет собой C1-C4-алкил или фенил-C1-C4-алкил. Изобретение относится также к конкретным соединениям, фармацевтическим композициям для селективного ингибирования протеинкиназы MKK4 по сравнению с протеинкиназами JNK1 и MKK7, для стимулирования регенерации печени и для снижения или предотвращения гибели гепатоцитов и к применению указанных соединений для получения таких фармацевтических композиций. 8 н. и 6 з.п. ф-лы, 1 табл., 38 пр.
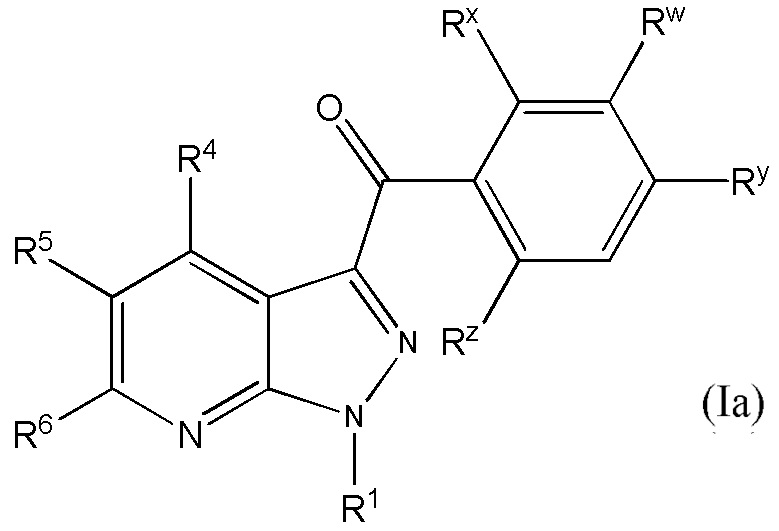
1. Соединение, имеющее формулу (Iа)
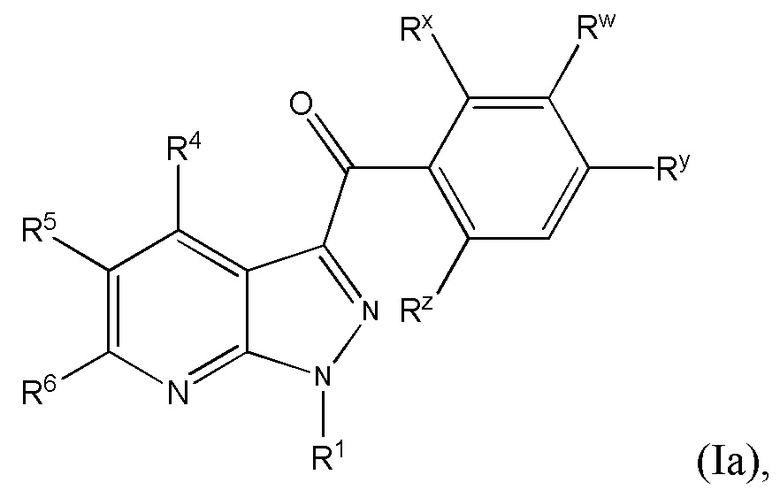
или его фармацевтически приемлемая соль,
в котором переменные в формуле (Iа) имеют следующие значения:
R1 представляет собой H;
R4 представляет собой Н;
R5 выбран из
а) пиримидинила, который замещен 1 или 2 заместителями, независимо выбранными из C3-C6-циклоалкила, C1-C4-алкила, -COOR10, -ОН, C1-C4-алкилсульфанила, тетразолила, CN, галогена и C1-C4-алкокси, и
b) пиридила, который замещен 1 или 2 заместителями, независимо выбранными из C1-C4-алкила и галогена;
R6 представляет собой Н;
Rw представляет собой -NR10SO2R12;
Rx представляет собой F;
Ry представляет собой F;
Rz представляет собой Н или F;
R10 в каждом случае представляет собой независимо Н или C1-C4-алкил;
R12 представляет собой C1-C4-алкил или фенил-C1-C4-алкил.
2. Соединение или его фармацевтически приемлемая соль по п. 1, в котором R5 представляет собой пиримидинил, замещенный 1 или 2 заместителями, независимо выбранными из C3-C6-циклоалкила, C1-C4-алкила, C1-C4-алкокси, -ОН, C1-C4-алкилсульфанила, галогена, CN и тетразолила.
3. Соединение или его фармацевтически приемлемая соль по п. 2, в котором R5 представляет собой пиримидинил, замещенный 1 или 2 заместителями, независимо выбранными из C3-C6-циклоалкила, C1-C4-алкила, C1-C4-алкокси, -ОН, C1-C4-алкилсульфанила, галогена и CN.
4. Соединение или его фармацевтически приемлемая соль по любому из предшествующих пунктов, в котором R10 представляет собой H.
5. Соединение или его фармацевтически приемлемая соль по п. 1, в котором R12 представляет собой C1-C3-алкил или бензил.
6. Соединение или его фармацевтически приемлемая соль по любому из пп. 1-5, имеющее формулу (Ib)
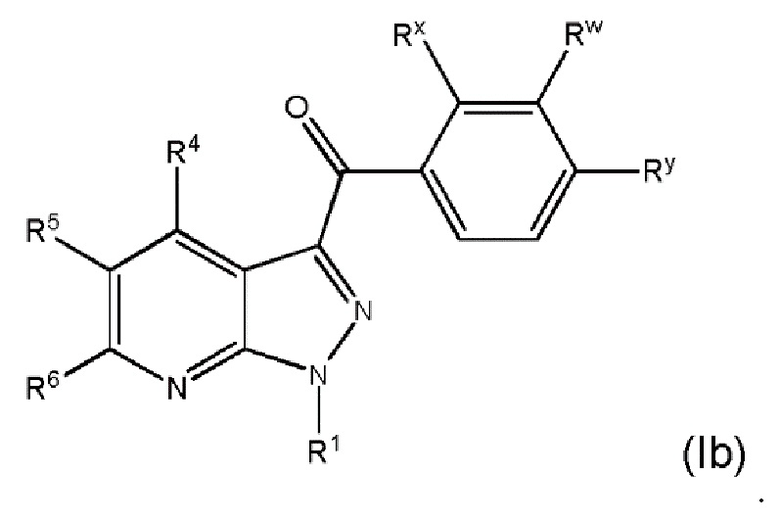
7. Соединение по п. 1, выбранное из:
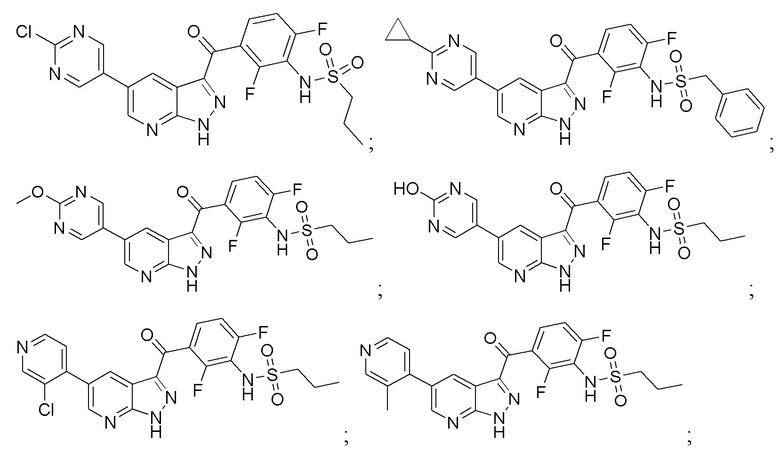
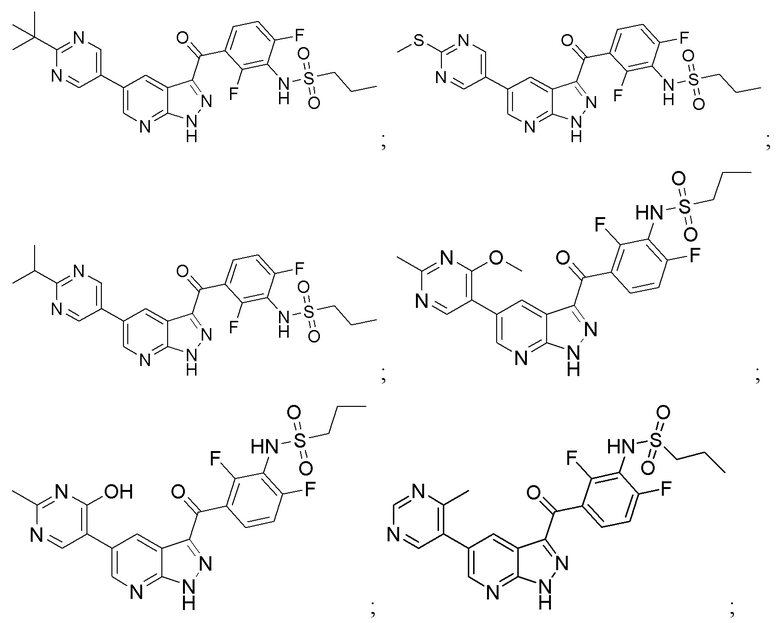
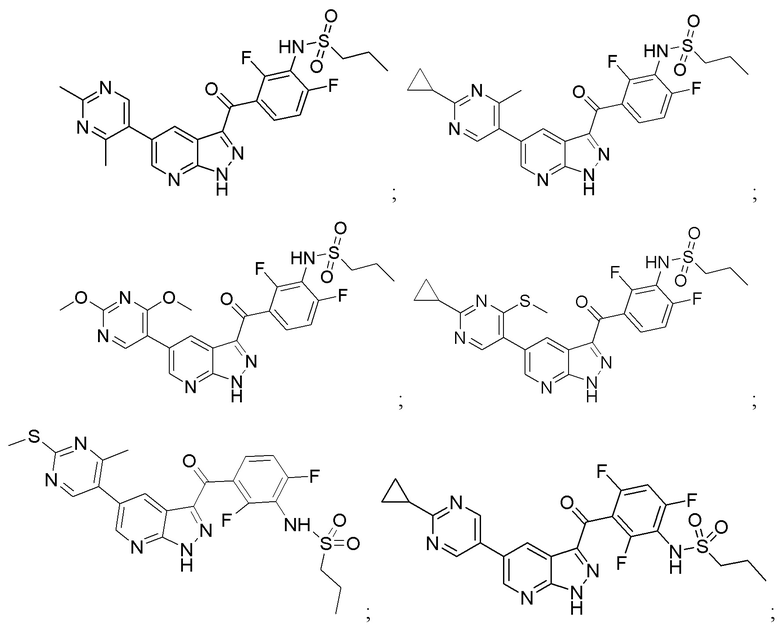
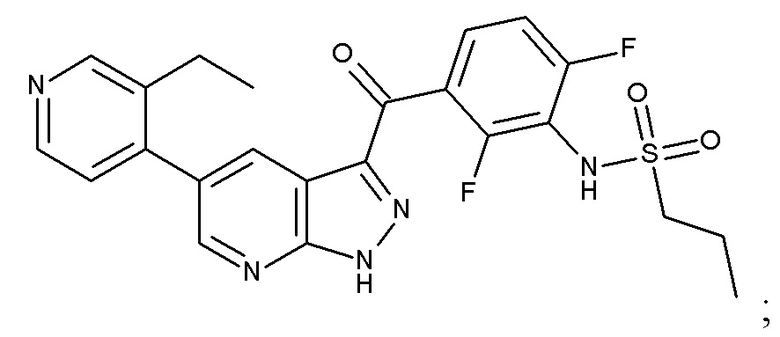
или его фармацевтически приемлемая соль.
8. Соединение, выбранное из:
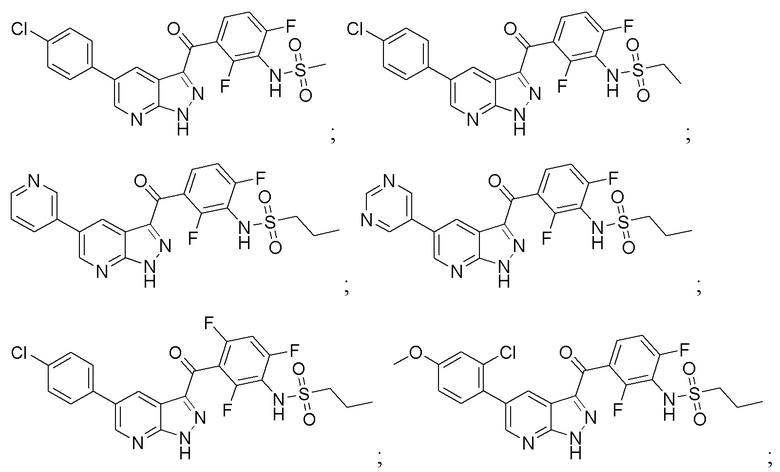
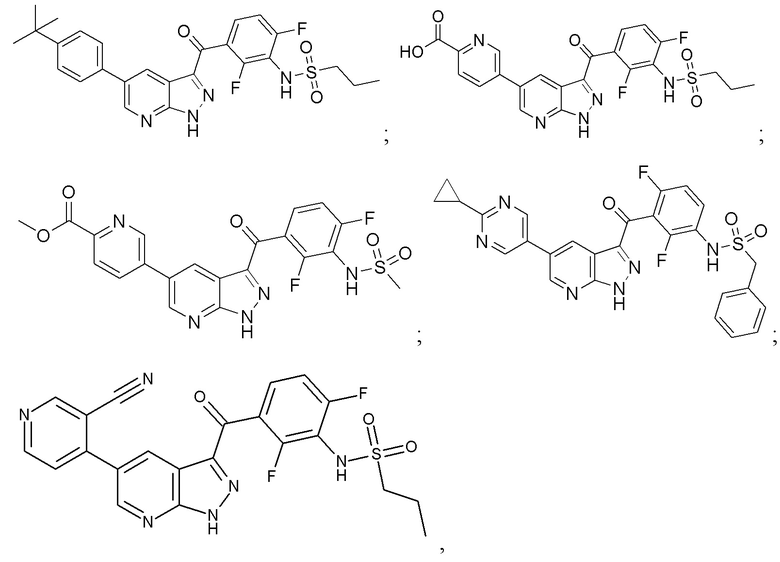
или его фармацевтически приемлемая соль.
9. Фармацевтическая композиция для селективного ингибирования протеинкиназы MKK4 по сравнению с протеинкиназами JNK1 и MKK7, содержащая соединение по любому из пп. 1-8 или его фармацевтически приемлемую соль.
10. Фармацевтическая композиция для стимулирования регенерации печени, содержащая соединение по любому из пп. 1-8 или его фармацевтически приемлемую соль.
11. Фармацевтическая композиция для снижения или предотвращения гибели гепатоцитов, содержащая соединение по любому из пп. 1-8 или его фармацевтически приемлемую соль.
12. Применение соединения или его фармацевтически приемлемой соли по любому из пп. 1-8 для получения фармацевтической композиции для селективного ингибирования протеинкиназы MKK4 по сравнению с протеинкиназами JNK1 и MKK7.
13. Применение соединения или его фармацевтически приемлемой соли по любому из пп. 1-8 для получения фармацевтической композиции для стимулирования регенерации печени.
14. Применение соединения или его фармацевтически приемлемой соли по любому из пп. 1-8 для получения фармацевтической композиции для снижения или предотвращения гибели гепатоцитов.

