Настоящее изобретение относится в общем к способу получения новых йодированных контрастных агентов и в частности к способу получения йопамидола с высоким выходом и высокой степенью чистоты. Йопамидол, а также другие неионогенные йодированные контрастные агенты используют в методах рентгенографической диагностики.
Уровень техники
Соединение N,N'-бис[2-гидрокси-1-(гидроксиметил)этил]-5-[[(2S)-2-гидрокси-1-оксопропил]-амино]-2,4,6-трийодо-1,3-бензолдикарбоксамид (см. формулу ниже), общеизвестное как йопамидол (указатель «Мерк», издание XIII, 2001 г., № 5073), широко используется в методах диагностики:
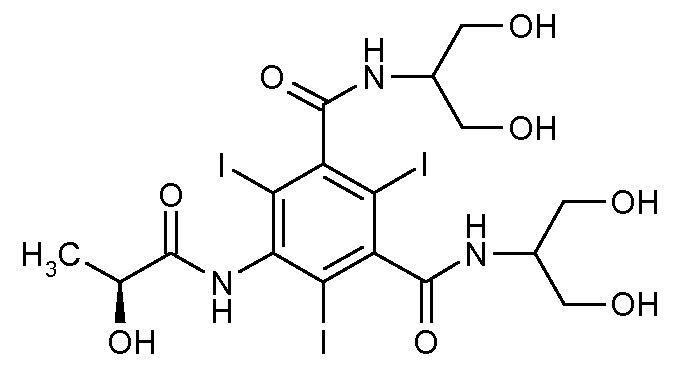
Из литературы известно несколько способов получения йопамидола, например из 5-нитроизофталевой кислоты, что предполагает использование многочисленных реагентов и систем растворителей, необязательное выделение промежуточных продуктов синтеза и очистку конечного продукта. Как возможное исходное вещество, 5-нитроизофталевую кислоту удобно восстанавливать в соответствующее аминопроизводное, например, каталитическим гидрированием, а затем йодировать в фенильное кольцо для получения соответствующего 2,4,6-трийодопроизводного.
Затем последнее превращают, например, в присутствии тионилхлорида, в соответствующий дихлорангидрид 5-амино-2,4,6-трийодоизофталевой кислоты (см., например, международные заявки WO 96/037458, WO 96/037459, WO 96/016927 и WO 96/036590).
Способ получения йопамидола на основе дихлорангидрида 5-амино-2,4,6-трийодоизофталевой кислоты, с учетом возможных вариантов, можно описать следующей схемой синтеза (см., например, международные патенты WO 96/037460, WO 97/047590, WO 98/24757, WO 98/028259 и WO 99/058494, а также патент США US 5362905):
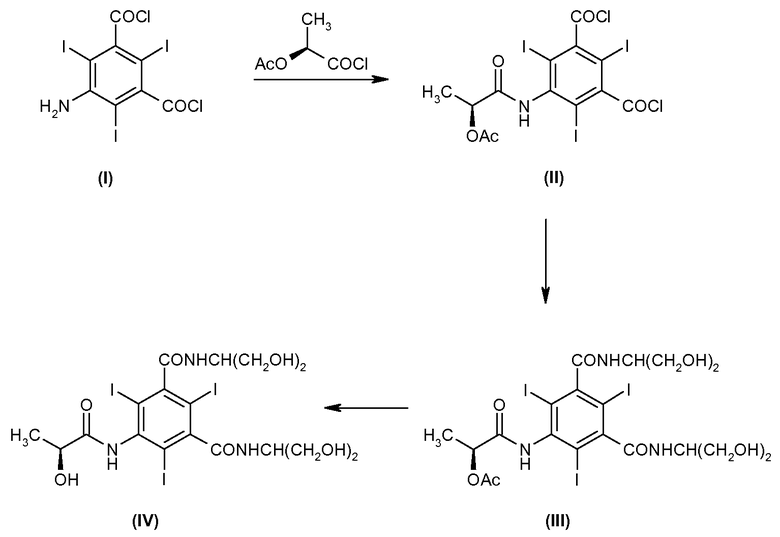
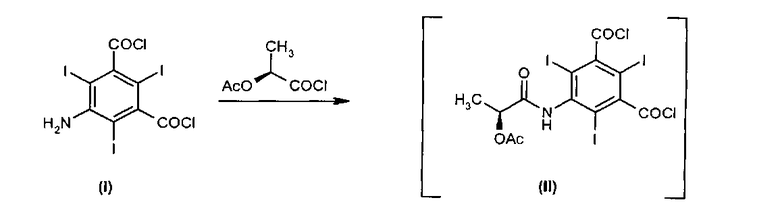
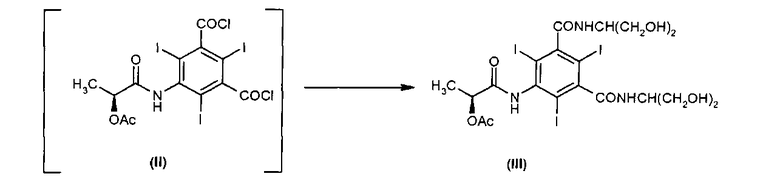
Дихлорангидрид 5-амино-2,4,6-трийодоизофталевой кислоты (I) превращают в присутствии хлорангидрида (S)-[2-(ацетилокси)]пропионовой кислоты в соответствующее соединение (II). Полученный промежуточный продукт (II) затем превращают в ацетилйопамидол (III) в присутствии 2-амино-1,3-пропандиола.
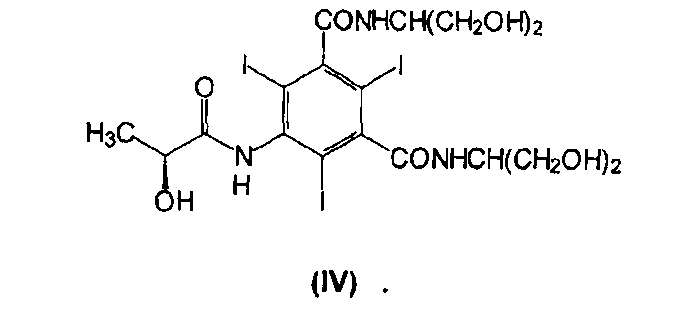
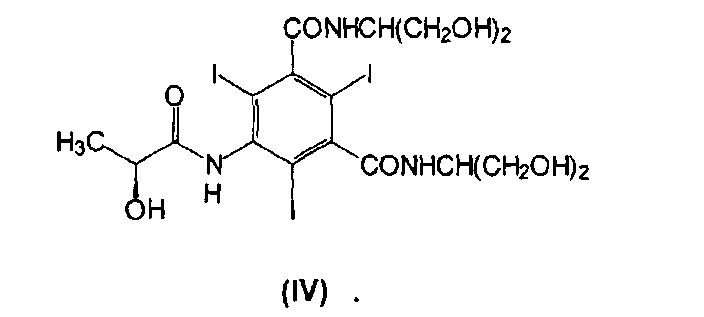
Наконец, после гидролиза соединения (III) и последующей стадии очистки выделяют йопамидол (IV).
Из литературы известно применение соответствующих оснований в реакции конденсации 2-амино-1,3-пропандиола и соединения (II), в частности третичный амин добавляют в реакционную систему перед введением 2-амино-1,3-пропандиола (см., например, международный патент WO 98/24757).
Такая добавка очень полезна, потому что она обеспечивает нейтрализацию кислоты, образующейся в реакции соединения (II) и 2-амино-1,3-пропандиола, и предотвращает возможное и нежелательное образование соли указанной кислоты и 2-амино-1,3-пропандиола.
Из числа пригодных для использования третичных аминов, согласно сообщениям, выбирали, в частности, алифатические третичные амины, например триэтиламин, трипропиламин, трибутиламин и диизопропилэтиламин.
Согласно вышеуказанным патентным заявкам применение неорганических оснований вместо третичных аминов не дает таких высоких результатов, как применение третичных аминов, потому что существенно увеличивается время реакции вследствие нерастворимости неорганических оснований в органических растворителях, например, в диметилацетамиде (ДМА).
Кроме того, соли, образующиеся в реакции при избытке хлорангидрида (S)-[2-(ацетилокси)]пропионовой кислоты в реакционной системе, вероятно, будут отфильтровываться вместе с продуктом, что привело бы к снижению общего выхода и загрязнению конечного продукта.
Насколько известно авторам, в литературе описано применение отдельных оснований в реакции конденсации аминоспирта и соответствующего дихлорангидрида изофталевой кислоты для получения и других неионогенных йодированных контрастных агентов. Обратимся к примеру способа получения йоверсола (указатель «Мерк», издание XIII, 2001 г., №5085) в реакции 3-амино-1,2-пропандиола с дихлорангидридом 5-ацетоксиацетамидо-2,4,6-трийодоизофталевой кислоты в присутствии оснований и апротонных полярных органических растворителей, например диметилацетамида (ДМА), диметилформамида (ДМФА) или диметилсульфоксида (ДМСО).
Однако единственное найденное экспериментальное свидетельство данной реакции конденсации, проводимой в присутствии неорганических оснований, как описано, например, в индийском патенте IN 187816, предполагает применение гидроксида натрия в присутствии изопропанола как возможного растворителя или, в качестве альтернативы, применение карбоната калия в ДМФА.
К своему удивлению, авторы обнаружили, что отдельные неорганические основания, например оксиды или гидроксиды щелочных или щелочноземельных металлов, в указанных условиях реакции удобно использовать в способе получения йопамидола.
Такой способ позволяет одновременно нейтрализовать кислоту, образующуюся в ходе вышеуказанной реакции конденсации, а также использовать мягкие условия в выбранном растворителе. Данный способ, кроме того, обеспечивает получение йопамидола с высоким выходом и высокой степенью чистоты. В данном отношении, как и для других контрастных агентов, которые используются для диагностики, в случае йопамидола очень важно, чтобы исходный материал можно было получать с высокой степенью чистоты, что позволяет оптимизировать, даже с замечательными результатами, стадии очистки, которые требуются для получения конечного продукта, предназначаемого для введения в организм человека, что, соответственно, требует соблюдения предусмотренных фармакопейных ограничений и условий.
Среди известных примесей в отношении йопамидола следует отметить соединение N'-[2-гидрокси-1-(гидроксиметил)этил]-5-[[(2S)-2-гидроксипропаноил]амино]-2,4,6-трийодо-N,N-диметилбензол-1,3-дикарбоксамид, структурная формула которого приведена ниже:
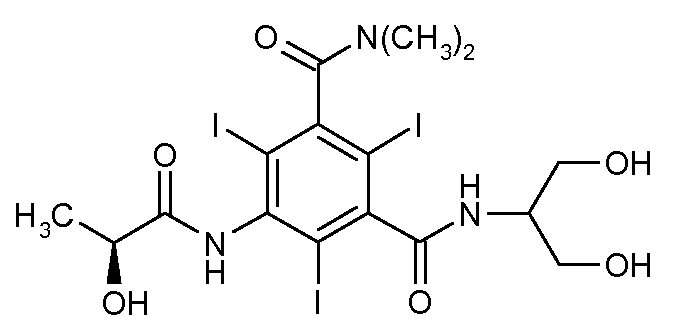
Вышеуказанное соединение, далее именуемое «примесь F», входит в список возможных побочных продуктов, образующихся в синтезе йопамидола (см., например, Европейскую Фармакопею, издание 6.0/01, 2008 г., 1115).
Преимущество способа по настоящему изобретению заключается в том, что он позволяет получать йопамидол с весьма низким содержанием примеси F, а также других побочных продуктов при использовании неорганических оснований. Все это позволяет удалять данные побочные продукты на стадиях обычной очистки и получать конечный продукт в соответствии с установленными ограничениями, как указано выше.
Описание изобретения
Таким образом, целью настоящего изобретения является способ получения соединения (III) по реакции конденсации дихлорангидрида кислоты (II) и 2-амино-1,3-пропандиола, которая проводится в апротонном полярном растворителе в присутствии оксида или гидроксида щелочного или щелочноземельного металла:
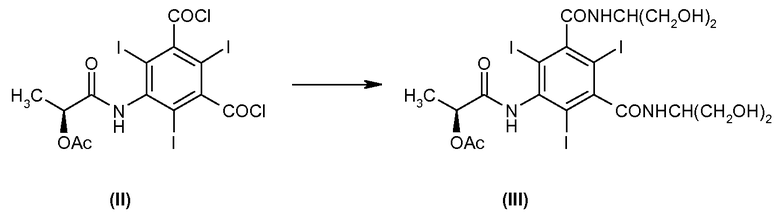
Как подробно описано в экспериментальной части, применение указанных неорганических оснований позволяет получать продукт (III) без недостатков, известных в литературе.
Если не указано иное, термин «оксид или гидроксид щелочного или щелочноземельного металла» означает оксид или гидроксид лития, калия, магния или кальция.
В отношении оксидов предпочтение отдается оксидам кальция и магния.
Предпочтительно проводить реакцию в присутствии гидроксида натрия или кальция, но предпочтительнее в присутствии гидроксида кальция.
Гидроксид добавляют согласно методике, обычно используемой в промышленных условиях, т.е. введением соответствующего количества выбранного основания в твердой форме или в водном растворе.
Необходимое количество гидроксида добавляют единовременно или, в качестве альтернативы, порциями.
Как правило, выбранное основание добавляют в эквивалентном отношении не менее 2:1 к соединению (II) [т. е. в эквивалентном отношении 1:1 к 2-амино-1,3-пропандиолу], но предпочтительнее в избытке.
Как указано в экспериментальной части, основание можно добавлять к раствору соединения (II) до, во время или после добавления 2-амино-1,3-пропандиола.
При введении основания и 2-амино-1,3-пропандиола в реакционную систему смесь удобным образом перемешивают согласно общим методам, принятым в промышленной практики.
По предпочтительному варианту данного изобретения, 2-амино-1,3-пропандиол и основание добавляют постепенно, следя за температурой реакционной смеси, которую поддерживают не выше 30°C.
Как указано выше, реакцию проводят в присутствии апротонного полярного органического растворителя, предпочтительно диметилацетамида (ДМА).
Как реагенты, так и растворители, используемые в данной реакции, известны и широко применяются в промышленных условиях. Исходное вещество, т. е. соединение (II) также хорошо известно, и в данном случае его можно получить по приведенной выше схеме из соответствующего дихлорангидрида 5-амино-2,4,6-трийодоизофталевой кислоты (I).
В связи с этим следует отметить, что соединение (III) можно получить по способу, составляющему предмет настоящего изобретения, на основе соединения (II) или, в качестве варианта, из соответствующего соединения (I), без необходимости выделения образующегося промежуточного продукта (II). Следовательно, другой целью настоящего изобретения является способ получения соединения (III), включающий стадии:
а) реакция дихлорангидрида 5-амино-2,4,6-трийодоизофталевой кислоты (I) с хлорангидридом (S)- [2-(ацетилокси)]пропионовой кислоты в апротонном полярном органическом растворителе с получением раствора соединения (II), и
b) введение оксида или гидроксида щелочного или щелочноземельного металла и 2-амино-1,3-пропандиола в раствор соединения (II) с получением соединения (III).
Реакцию конденсации дихлорангидрида 5-амино-2,4,6-трийодоизофталевой кислоты (I) с хлорангидридом (S)-[2-(ацетилокси)]пропионовой кислоты на стадии (а) проводят известными в литературе способами, предпочтительно в ДМА как возможном апротонном полярном органическом растворителе.
Затем прямо в полученную реакционную смесь добавляют выбранный оксид или гидроксид щелочного или щелочноземельного металла, а также соответствующее количество 2-амино-1,3-пропандиола, как описано ранее.
Однако, в отличие от проведения реакции с выделением производного (II), в реакции, проводимой «в одном флаконе», используют значительно большее количество основания.
В таком случае основание используют не только для нейтрализации кислоты, образующейся в реакции амидирования дихлорангидрида 5-амино-2,4,6-трийодоизофталевой кислоты (I) и хлорангидрида (S)-[2-(ацетилокси)]пропионовой кислоты, но и для нейтрализации кислоты, которая может образоваться из возможного избытка хлорангидрида (S)-[2-(ацетилокси)]пропионовой кислоты, а также для нейтрализации кислоты, образующейся в реакции амидирования соединения (II) и 2-амино-1,3-пропандиола.
Следовательно, по предпочтительному варианту данного изобретения, когда процесс проводят без выделения промежуточного продукта (II), выбранное основание используют в эквивалентном отношении не менее 3:1 к дихлорангидриду 5-амино-2,4,6-трийодоизофталевой кислоты (I), а предпочтительнее в избытке.
Как правило, вследствие присутствия кислоты в исходном реагенте, содержащем соединение (II), основание вводят в реакционную систему перед добавлением 2-амино-1,3-пропандиола.
Кроме того, в этом случае реагенты, например гидроксид или оксид, в твердой форме или в водном растворе, а также 2-амино-1,3-пропандиол предпочтительно добавлять медленно или порциями при перемешивании и поддержании температуры приблизительно ниже 30°С.
Еще предпочтительнее добавлять основание к исходному реагенту, который содержит промежуточный продукт (II), при температуре от 5°С до 20°С.
Соединение (III), полученное из выделенного соединения (II) или соответствующего исходного соединения (I), затем превращают в йопамидол (IV) способами, известными из литературы.
Как правило, например, по описанию в экспериментальной части соединение (III) сначала отделяют от исходного реакционного материала, очищают, возможно, пропусканием через ионообменные смолы, а затем гидролизуют в щелочной среде с разрушением ацетильной группы.
В связи с этим следующая цель настоящего изобретения способ получения йопамидола (IV) гидролизом в щелочной среде соответствующего ацетилйопамидола (III), полученного, как указано выше, в реакции конденсации соединения (II) в чистом виде или в смеси реагентов, полученной в реакции дихлорангидрида 5-амино-2,4,6-трийодоизофталевой кислоты (I) и хлорангидрида (S)-[2-(ацетилокси)]пропионовой кислоты с 2-амино-1,3-пропандиолом в апротонном полярном растворителе в присутствии оксида или гидроксида щелочного или щелочноземельного металла.
Определенные условия и возможные варианты используемых условий, например, в отношении сроков добавления реагентов, возможного разведения реакционной смеси, избытка основания, концентрации выбранного гидроксида, если его добавляют в виде водного раствора, или другие возможные изменения в отношении температуры и/или времени реакции должны, насколько возможно, оптимизировать способ, который является целью данного изобретения.
Данный способ обеспечивает высокий выход и высокую степень чистоты целевого продукта. Содержание существующих примесей, в том числе примеси F, как указано выше, настолько низкое, что данные примеси можно удалить традиционными способами очистки.
Кроме того, данный способ может широко применяться в промышленности и иметь преимущества при получении других йодированных неионогенных контрастных агентов, синтез которых предусматривает реакцию конденсации соответствующего производного дихлорангидрида 5-амино-2,4,6-трийодоизофталевой кислоты и аминоспирта.
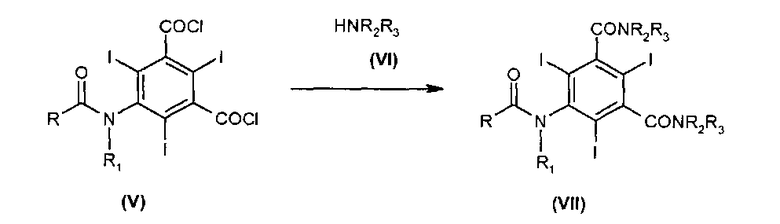
Поэтому еще одной целью настоящего изобретения является способ получения соединения (VII) в реакции конденсации дихлорангидрида кислоты (V) и аминоспирта (VI), которую проводят в апротонном полярном растворителе в присутствии оксида или гидроксида щелочного или щелочноземельного металла
где:
R - линейная или разветвленная алкильная группа, содержащая от 1 до 6 атомов углерода, необязательно замещенная одной или несколькими гидроксильными группами, возможно в защищенной форме, причем данная алкильная группа может прерываться одним или несколькими гетероатомами, выбранными из -О- и -NH-;
R1 - атом водорода или группа R;
R2 - линейная или разветвленная алкильная группа, содержащая от 1 до 4 атомов углерода, замещенная по меньшей мере одной гидроксильной группой;
R3 - атом водорода или группа R2.
В данном описании, если не указано иное, термин «линейная или разветвленная алкильная группа, содержащая от 1 до 6 атомов углерода» означает такие группы, как, например, метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, н-гексил и т.д.
Такая алкильная группа может содержать один или несколькими гидроксильных заместителей, возможно в защищенной форме.
Термин «защищенная форма» означает, что в гидроксильной группе атом водорода замещен соответствующим образом, что обеспечивает защиту данной группы в ходе химической реакции, а затем заместитель удаляется известными способами с восстановлением исходной формы гидроксильной группы. Гидроксильную группу предпочтительно защищать ацильной группой, например, ацетильной (-СОСН3), в результате чего образуется соответствующая ацетоксигруппа (-ОСОСН3). Общую информацию об использовании защитных групп в органическом синтезе см. например, в книге Т.W. Green «Защитные группы в органическом синтезе» (Нью-Йорк, издательство Wiley, 1981 г.).
Если не указано иное, в качестве аминоспирта (VI) предпочтительно выбирать 2-амино-1,3-пропандиол или 3-амино-1,2-пропандиол, поэтому группа R2 означает -СН(СН2ОН)2 или СН2СНОНСН2ОН соответственно.
В отношении апротонного полярного органического растворителя, соответствующего оксида или гидроксида, а также основных параметров данного способа см. описание выше.
По примеру практического осуществления, к раствору дихлорангидрида 5-амино-2,4,6-трийодоизофталевой кислоты (I) в апротонном полярном растворителе, предпочтительно ДМА, добавляют необходимое количество хлорангидрида (S)-[2-(ацетилокси)]пропионовой кислоты при перемешивании и поддержании температуры предпочтительно ниже 30°С.
После окончания реакции реакционную смесь разбавляют достаточным количеством растворителя и по выбору охлаждают, до температуры около 15°С.
Затем при перемешивании и сохранении температуры под контролем добавляют соответствующее количество выбранного основания, например, порошка гидроксида кальция вместе с необходимым количеством 2-амино-1,3-пропандиола.
Смесь оставляют на ночь и перемешивают при выбранной температуре, например, около 30°С до окончания реакции. Полученный неочищенный материал упаривают в вакууме для удаления большей части растворителя, а сырой остаток разбавляют водой и очищают с помощью катионообменной смолы обычными способами.
Элюат, содержащий продукт (III), затем гидролизуют в щелочной среде, а после окончания реакции доводят среду до практически нейтральной (т.е. рН около 7).
Полученную смесь, которая содержит йопамидол (IV), окончательно очищают известными способами, в том числе, например, с помощью смол, нанофильтрации и кристаллизации в присутствии соответствующего спирта, например 2-бутанола.
На последующих стадиях фильтрации и сушки получают йопамидол (IV), который соответствует указанным выше условиям чистоты.
В целях лучшей иллюстрации настоящего изобретения, без установления каких-либо ограничений, далее приведены соответствующие примеры.
Пример 1
Получение соединения (III) из выделенного соединения (II) в присутствии гидроксида кальция
Гидроксид кальция (12,8 г, 0,17 3 моль) медленно добавляли при перемешивании и поддержании температуры ниже 25°С к раствору соединения (II) (120 г, 0,169 моль) в 305 г ДМА.
Затем к реакционной смеси по каплям добавляли раствор 2-амино-1,3-пропандиола в ДМА (133 г, 28 мас.%, 0,406 моль) в течение приблизительно 4 5 минут. Смесь выдерживали около 30°С в течение 10 часов до окончания реакции. Неочищенный продукт реакции, содержащий производное (III), можно очищать и гидролизовать по методике в приведенном ниже примере 3.
Профиль ВЭЖХ смеси после обработки образца NaOH:
йопамидол (IV): 97,9%;
примесь F: 0,2%.
Пример 2
Получение соединения (III) из выделенного соединения (II) в присутствии гидроксида натрия
При перемешивании и поддержании температуры ниже 25°С гидроксид натрия (13,9 г, 0,346 моль) медленно добавляли к раствору соединения (II) (120 г, 0,169 моль) в 305 г ДМА.
Затем раствор 2-амино-1,3-пропандиола в ДМА (133 г, 28 мас.%, 0,406 моль) по каплям добавляли к реакционной смеси в течение около 4 5 минут. Смесь выдерживали при температуре около 30°С в течение 10 часов до окончания реакции.
Неочищенный продукт реакции, содержащий производное (III), можно очистить и гидролизовать по методике следующего примера 3.
Профиль ВЭЖХ смеси после обработки образца NaOH:
йопамидол (IV): 97,4%;
примесь F: 0,3%.
Пример 3
Получение йопамидола (IV) из выделенного соединения (II), в присутствии гидроксида кальция.
Раствор 2-амино-1,3-пропандиола в ДМА (610 г, 28 мас.%, 1,87 моль) добавляли к раствору соединения (II) (600 г, 0,845 моль) в ДМА (1510 г) при перемешивании в течение около 4 5 минут.
Затем гидроксид кальция (70,0 г, 0,945 моль) медленно добавляли к реакционной смеси при поддержании температуры ниже 30°С. После этого смесь выдерживали при температуре около 30°С в течение 10 часов до окончания реакции.
Затем неочищенный продукт реакции упаривали в вакууме (95°С, 10 мбар или 7,5 мм рт. ст.) для удаления большей части растворителя с получением вязкого остатка. Полученный горячий остаток далее обрабатывали деионизированной водой (1455 г) и доводили рН до 1,7 добавлением соляной кислоты (33 г, 34 мас.%).
Полученный раствор элюировали через 1500 мл сильной катионообменной смолы (Dowex С350™ от фирмы Dow) в форме Na+ для удаления ионов Са2+ вместе с избытком 2-амино-1,3-пропандиола. Затем элюат обрабатывали раствором гидроксида натрия (250 г, 30%) и выдерживали при температуре около 35°С в течение 7 часов для гидролиза эфира уксусной кислоты. После этого рН доводили приблизительно до 7 добавлением соляной кислоты, а полученный раствор обрабатывали сульфитом натрия (0,25 г), очищали на смоле ПС-ДВБ (1300 мл, Amberlite XAD100™ от фирмы Rohm and Haas), а затем удаляли соли нанофильтрацией.
Стадию заключительного обессоливания проводили на двух последовательных пластинах из смолы, содержащих 425 мл сильной катионообменной смолы в форме Н+ (Dowex С350™) и 500 мл слабой анионообменной смолы (Relite MG1® от фирмы Mitsubishi).
Полученный раствор затем концентрировали в вакууме до содержания воды 30 мас.%, а остаток перекристаллизовали из 2-бутанола (1250 г). Кристаллизацию завершали добавлением достаточного количества 2-бутанола с одновременной отгонкой азеотропной смеси при атмосферном давлении до содержания воды в суспензии около 3 мас.%. После охлаждения системы при температуры около 25°С продукт отфильтровывали и высушивали в вакууме при температуре около 50°С, получая йопамидол (591 г, выход 90,0% в расчете на соединение (II). Полученный йопамидол соответствовал требуемым параметрам чистоты.
Пример 4
Получение соединения (II)
Дихлорангидрид 5-амино-2,4,6-трийодоизофталевой кислоты (I) (506 г, 0,85 моль) и гидрохлорид диметилацетамида (37 г) растворяли в ДМА (690 г) при 25°С. Затем к полученному раствору добавляли хлорангидрид (S)-[2-(ацетокси)]пропионовой кислоты в течение 4 часов, поддерживая температуру от 10° до 15°С. После этого температуру раствора доводили до 20°С и перемешивали в течение 30 часов. Продукт использовали в неочищенном виде, как указано в примерах 5-9.
Профиль ВЭЖХ:
соединение (II): 94%;
дихлорангидрид 5-амино-2,4,6-трийодоизофталевой кислоты (I): <0,1%.
Пример 5
Получение соединения (III) из неочищенного продукта реакции, содержащего соединение (II)
Раствор 423 г неочищенного продукта реакции в ДМА по примеру 4, содержащий теоретически 0,25 моль соединения (II), разбавляли ДМА (199 г), и реакционную смесь охлаждали при температуре около -5°С. Затем гидроксид кальция (42 г, 0,567 моль) медленно добавляли к реакционной смеси при перемешивании и поддержании температуры ниже 5°С.
После этого в течение 1 часа добавляли раствор 2-амино-1,3-пропандиола в ДМА (231 г, 24 мас.%, 0,608 моль). Затем смесь нагревали до 70°С и выдерживали в таких условиях 3 часа до окончания реакции.
Профиль ВЭЖХ смеси после обработки образца NaOH:
йопамидол (IV): 89,6%;
примесь F: 0,4%.
Пример 6
Получение соединения (III) из неочищенного продукта реакции, содержащего соединение (II)
Методику по примеру 5 изменяли так, чтобы работать при более низкой температуре, как описано ниже.
Раствор 423 г неочищенного продукта реакции в ДМА по примеру 4, содержащий теоретически 0,25 моль соединения (II), разбавляли ДМА (200 г) и смесь охлаждали при температуре около -5°С. Затем гидроксид кальция (46,3 г, 0,625 моль) медленно добавляли к реакционной смеси при перемешивании и поддержании температуры ниже 5°С.
После этого в течение 1 часа добавляли раствор 2-амино-1,3-пропандиола в ДМА (231 г, 24 мас.%, 0,61 моль) . Затем смесь нагревали примерно до 30°С и выдерживали в таких условиях 20 часов до окончания реакции.
Профиль ВЭЖХ смеси после обработки образца NaOH:
йопамидол (IV): 93,9%;
примесь F: 0,3%.
Пример 7
Получение йопамидола (IV)
Раствор 1440 г неочищенного продукта реакции в ДМА по примеру 4, содержащий теоретически 0,8 5 моль соединения (II), разбавляли ДМА (677 г) и охлаждали реакционную смесь до температуры около 15°С. Затем гидроксид кальция (157 г, 2,12 моль) добавляли к реакционной смеси пятью порциями при перемешивании и поддержании температуры ниже 20°С. После этого в течение 1 часа добавляли раствор 2-амино-1,3-пропандиола в ДМА (785 г, 24 мас.%, 2,06 моль). Затем смесь слегка нагревали до 30°С и выдерживали в таких условиях 20 часов до окончания реакции. Неочищенный продукт реакции упаривали в вакууме (95°С, 10 мбар или 7,5 мм рт.ст.) для удаления большей части растворителя с получением вязкого остатка. Полученный горячий остаток обрабатывали деионизованной водой (14 60 г) и доводили рН до 2 добавлением соляной кислоты (108 г, 34 мас.%).
Полученный раствор затем элюировали через 2500 мл сильной катионообменной смолы (Dowex С350™ от фирмы Dow) в форме Na+ для удаления ионов Са++ вместе с избытком 2-амино-1,3-пропандиола.
После этого элюат обрабатывали раствором гидроксида натрия (260 г, 30%) и выдерживали при 35°С в течение 7 часов для гидролиза эфира уксусной кислоты. Уровень рН доводили до 7 добавлением соляной кислоты, а полученный раствор обрабатывали сульфитом натрия (0,25 г), очищали на смоле ПС-ДВБ (1300 мл, Amberlite XAD1600™ от фирмы Rohm and Haas), а затем обессоливали нанофильтрацией. Стадию заключительного обессоливания проводили на двух последовательных пластинах из смолы, содержащих 425 мл сильной катионообменной смолы в форме Н+ (Dowex С350™) и 500 мл слабой анионообменной смолы (Relite MG1S от фирмы Mitsubishi). Полученный раствор затем концентрировали в вакууме до содержания воды 30 мас.%, а остаток перекристаллизовали из 2-бутанола (1250 г) . Кристаллизацию завершали добавлением достаточного количества 2-бутанола с одновременной отгонкой азеотропной смеси при атмосферном давлении до содержания воды в суспензии около 3 мас.%. После охлаждения системы до 25°С продукт отфильтровывали и высушивали в вакууме при температуре около 50°С, получая йопамидол (57 6 г, выход 87,3% в расчете на соединение (II) . Полученный йопамидол соответствовал требуемым параметрам чистоты.
Сравнительные примеры
Пример 3а
Получение йопамидола (IV) из выделенного соединение (II), в присутствии оксида кальция или магния
Выполняли такие же операции, как в предыдущем примере 3, за исключением того, что гидроксид кальция заменяли таким же молярным количеством оксида кальция или оксида магния. Результаты приведены ниже:
Пример 7а
Получение йопамидола (IV) в присутствии оксида кальция Выполняли такие же операции, как в предыдущем примере 7 за исключением того, что гидроксид кальция заменяли таким же молярным количеством оксида кальция. Йопамидол, полученный с выходом 83,0% в расчете на исходное соединение II, соответствовал требуемым параметрам чистоты. Пример 8
Получение соединения (III) в присутствии карбоната натрия Методику, приведенную в предыдущем примере 5, изменили, используя карбонат натрия вместо гидроксида кальция следующим образом.
Раствор 423 г неочищенного продукта реакции по примеру 4, содержащий теоретически 0,25 моль соединения II) разбавляли ДМА (200 г) и охлаждали смесь до -5°С. Затем карбонат натрия (119 г, 1,13 моль) медленно добавляли к реакционной смеси при перемешивании и поддержании температуры ниже 5°С. После этого раствор 2-амино-1,3-пропандиола в ДМА (231 г, 24 мас.%, 0,61 моль) добавляли в течение 1 часа. Полученную смесь нагревали до 70°С и выдерживали в таких условиях в течение 3 часов до окончания реакции.
Профиль ВЭЖХ:
йопамидол (IV): 92,0%;
примесь F: 0,2%;
всего примесей: 8%.
После отфильтровывания солей, согласно примеру 3, получали йопамидол (IV) (выход 78,0%) путем очистки и гидролиза соединения (III). Продукт не соответствовал параметрам чистоты.
Пример 9
Сравнительный пример получения соединения (III) в присутствии триэтиламина
Методику, приведенную в предыдущем примере 5, изменили, используя триэтиламин вместо гидроксида кальция следующим образом.
Раствор 423 г неочищенного продукта реакции в ДМА по примеру 4, содержащий теоретически 0,25 моль соединения II, разбавляли ДМА (199 г) и смесь охлаждали при температуре около -5°С. Затем триэтиламин (114 г, 1,13 моль) медленно добавляли к реакционной смеси при перемешивании и поддержании температуры ниже 5°С. После этого раствор 2-амино-1,3-пропандиола в ДМА (231 г, 24 мас.%, 0,61 моль) добавляли в течение 1 часа. Полученную смесь нагревали до 70°С и выдерживали в таких условиях в течение 3 часов до окончания реакции.
При выполнении операций, аналогичных примеру 3, йопамидол (IV) получали путем очистки и гидролиза соединения (III).
Профиль ВЭЖХ:
йопамидол (IV): 89,1%;
примесь F: не обнаружена;
всего примесей: 10,9%.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ЙОПАМИДОЛА | 2014 |
|
RU2657238C2 |
| СПОСОБ ПОЛУЧЕНИЯ ТРИЙОДИРОВАННЫХ КАРБОКСИЛЬНЫХ АРОМАТИЧЕСКИХ ПРОИЗВОДНЫХ | 2010 |
|
RU2512358C2 |
| СПОСОБ ИОДИРОВАНИЯ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ | 2010 |
|
RU2506254C2 |
| Способ получения амидов 5-оксипропиониламино-2,4,6-трийодизофталевой кислоты | 1975 |
|
SU628813A3 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 5-АМИНО-2,4,6-ТРИИОДО- 1,3-БЕНЗОЛКАРБОНОВОЙ КИСЛОТЫ | 1990 |
|
RU2046795C1 |
| ГИДРОКСИЗАМЕЩЕННЫЕ СТЕРИЧЕСКИ ЗАТРУДНЕННЫЕ N-АЛКОКСИАМИНЫ | 2000 |
|
RU2243216C2 |
| N-(5-АЦЕТИЛ-2-ФТОРФЕНИЛ)-N-МЕТИЛАЦЕТАМИД, СПОСОБ ПОЛУЧЕНИЯ N-[5-(3-ДИМЕТИЛАМИНО-АКРИЛОИЛ)-2-ФТОРФЕНИЛ]-N-МЕТИЛАЦЕТАМИДА И СПОСОБ ПОЛУЧЕНИЯ N-{2-ФТОР-5-[3-ТИОФЕН-2-КАРБОНИЛ-ПИРАЗОЛО [1,5-а]ПИРИМИДИН-7-ИЛ]ФЕНИЛ}-N-МЕТИЛАЦЕТАМИДА | 2010 |
|
RU2503655C2 |
| СПОСОБ ПОЛУЧЕНИЯ КЛОПИДОГРЕЛА И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, ИСПОЛЬЗУЕМЫЕ В СПОСОБЕ | 2005 |
|
RU2357970C1 |
| Рентгеноконтрастное средство | 1980 |
|
SU1087052A3 |
| СМЕСЬ БЛОК-ОЛИГОМЕРОВ, СПОСОБ ЕЕ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ И КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ЭТУ СМЕСЬ | 1996 |
|
RU2175660C2 |
Изобретение относится к улучшенному способу получения соединения формулы (VII) по реакции конденсации соединения формулы (V) и аминоспирта формулы (VI) при проведении реакции в диметилацетамиде (ДМА) в присутствии оксида или гидроксида щелочного или щелочноземельного металла:
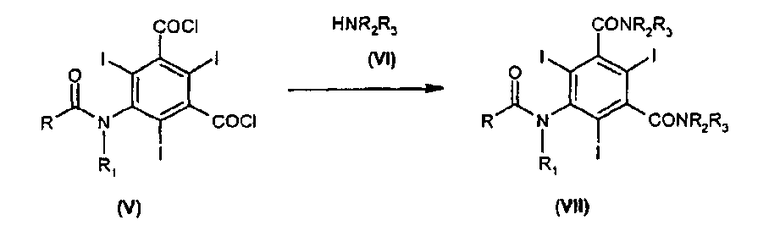
где R - линейная или разветвленная алкильная группа, содержащая от 1 до 6 атомов углерода, необязательно замещенная одним или несколькими гидроксильными заместителями, необязательно в защищенной форме, причем данная алкильная группа может прерываться одним или несколькими гетероатомами, выбранными из -О- и -NH-; R1 - атом водорода или группа R; R2 - линейная или разветвленная алкильная группа, содержащая от 1 до 4 атомов углерода, замещенная по меньшей мере одной гидроксильной группой; R3 - атом водорода или группа R2. Способ позволяет получать продукт с высоким выходом и высокой степенью чистоты. 8 з.п. ф-лы, 9 пр., 1 табл.
1. Способ получения соединения формулы (VII) по реакции конденсации соединения формулы (V) и аминоспирта формулы (VI), причем данную реакцию проводят в диметилацетамиде (ДМА) в присутствии оксида или гидроксида щелочного или щелочноземельного металла
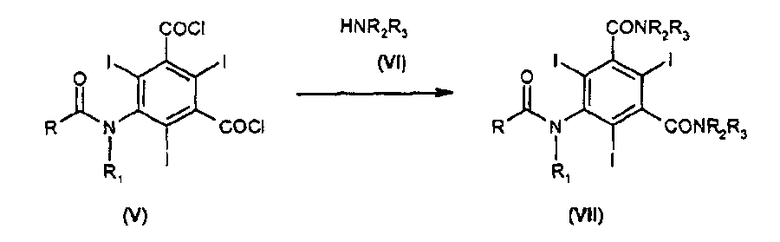
где R - линейная или разветвленная алкильная группа, содержащая от 1 до 6 атомов углерода, необязательно замещенная одним или несколькими гидроксильными заместителями, необязательно в защищенной форме, причем данная алкильная группа может прерываться одним или несколькими гетероатомами, выбранными из -О- и -NH-;
R1 - атом водорода или группа R;
R2 - линейная или разветвленная алкильная группа, содержащая от 1 до 4 атомов углерода, замещенная по меньшей мере одной гидроксильной группой;
R3 - атом водорода или группа R2.
2. Способ по п.1, в котором гидроксид щелочного или щелочноземельного металла выбирают из гидроксида натрия или кальция.
3. Способ по п.2, в котором гидроксидом является гидроксид кальция.
4. Способ по п.1, в котором оксид щелочного или щелочноземельного металла выбирают из оксида магния или кальция.
5. Способ по п.1, в котором:
R - -СН3;
R1 - -ОСОСН3; и
R2, R3 - -СН(СН2ОН)2.
6. Способ по п.1, отличающийся тем, что соединение формулы (V) получают взаимодействием дихлорангидрида 5-амино-2,4,6-трийодоизофталевой кислоты (I) с хлорангидридом (S)-[2-(ацетилокси)]пропионовой кислоты.
7. Способ по п.1, в котором:
R - -СН3;
R1 - -ОСОСН3;
R2 - -СН(СН2ОН)2; и
R3 - Н,
который дополнительно включает стадию гидролиза соединения (VII) в щелочной среде с получением йопамидола (IV)
8. Способ по п.7, в котором соединение формулы (V) получают взаимодействием дихлорангидрида 5-амино-2,4,6-трийодоизофталевой кислоты (I) с хлорангидридом (S)-[2-(ацетилокси)]пропионовой кислоты.
9. Способ по п.8, который дополнительно включает стадию гидролиза соединения (VII) в щелочной среде с получением йопамидола (IV)
| Способ производства сиропов из спиртованных соков | 1987 |
|
SU1472050A1 |
| US 4364921 А, 21.12.1982 | |||
| WO 9824757 А1, 11.06.1998 | |||
| WO 9854124 А1, 03.12.1998 | |||
| WO 9958494 A2, 18.11.1999 | |||
| Дальномер | 1926 |
|
SU5922A1 |
| J | |||
| HAAVALDSEN et al | |||
| Кипятильник для воды | 1921 |
|
SU5A1 |

