ОБЛАСТЬ ТЕХНИКИ
Изобретение относится к способу получения соединения формулы (I):
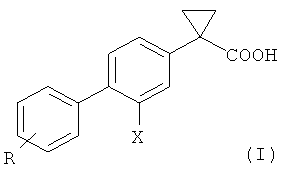
Изобретение также относится к промежуточным соединениям, используемым в этом способе.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Болезнь Альцгеймера является нейродегенеративным расстройством, которое характеризуется с гистопатологической точки зрения распространенным присутствием внеклеточных и периваскулярных нейритических (амилоидных) бляшек и внутриклеточных нейрофибриллярных клубков в церебральной паренхиме пациентов с болезнью Альцгеймера.
Нейритические бляшки состоят в основном из агрегатов белка с 39-43 аминокислотными остатками, известного как β-амилоид (βА) и, в зависимости от числа аминокислот, Аβ39, Аβ40, Аβ42 и Аβ43.
В данной области известны соединения, которые могут снижать продуцирование наиболее нейротоксичной изоформы β-амилоида, а именно формы, содержащей 42 аминокислоты (Аβ42), взаимодействуя с макромолекулярным/мультибелковым ферментативным комплексом с аспартилпротеазной активностью, известным как γ-секретаза.
В частности, в WO 2004/074232 раскрыты производные 1-(2-галогенобифенил-4-ил)-циклопропанкарбоновой кислоты общей формулы (I),
где Х и R такие, как определено ниже,
способные модулировать γ-секретазную активность, не оказывая влияния на другие важные метаболические процессы, такие как активность ферментов-циклооксигеназ.
Ключевой промежуточной стадией получения указанных соединений является реакция Сузуки подходящей фенилбороновой кислоты или ее сложного эфира с 3,4-дигалогено-циклопропанкарбоновой кислотой.
В WO 2004/074232 3,4-дигалогено-циклопропанкарбоновую кислоту получают, начиная с 3,4-дигалогено-толуола, который превращают в соответствующий бензилбромид путем радикального бромирования в четыреххлористом углероде (CCl4), полученный бромид превращают в 3,4-дигалогенофенилацетонитрил, последний подвергают взаимодействию с 1,2-дибромэтаном с получением соответствующего 3,4-дигалогено-фенилциклопропаннитрила, который в конце гидролизуют с получением целевой 3,4-дигалогено-циклопропанкарбоновой кислоты.
Однако способ, описанный в WO 2004/074232, дает низкий суммарный выход (12-14%) и страдает серьезными ограничениями для промышленного использования.
Например, на стадии радикального бромирования образуется значительное количество бис-галогенированного побочного продукта, что отрицательно сказывается на выходе, и используется ССЦ, который является высокотоксичным газом, истощающим озоновый слой и вызывающим парниковый эффект.
Кроме того, конечная реакция сочетания Сузуки дает низкий выход, и полученный продукт трудно поддается очистке кристаллизацией без снижения выхода. Например, для такой очистки используют хроматографию на силикагеле, но увеличение масштабов хроматографии на силикагеле затягивает процесс и требует больших объемов растворителей.
Таким образом, задача настоящего изобретения заключается в создании способа получения производных 1-(2-галогенобифенил-4-ил)-циклопропанкарбоновой кислоты формулы (I), альтернативного способу, раскрытому в WO 2004/074232, и не имеющего всех вышеупомянутых недостатков.
Настоящее изобретение решает эту задачу путем проведения реакции Сузуки в качестве первой стадии.
Кроме того, введены другие условия увеличения выхода на других стадиях, в частности на стадии радикального бромирования.
Оказалось, что способ по изобретению более эффективен, особенно для крупномасштабного производства, поскольку обеспечивает более высокий выход соединений формулы (I) с высокой химической чистотой без стадии хроматографической очистки.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТНИЯ
Объектом настоящего изобретения является способ получения соединения общей формулы (I) и его солей
где
Х представляет собой атом галогена, предпочтительно атом фтора;
R представляет собой одну или более групп, независимо выбранных из:
- атомов галогенов, предпочтительно атома хлора;
- CF3;
- СН=СН2;
- CN;
- CH2OH;
- NO2;
- метилендиокси;
- этилендиокси;
- циклоалкила, предпочтительно С3-С6циклоалкила;
- фенила;
- OR1 или NHCOR1, где R1 выбран из группы, состоящей из CF3, алкенила, алкинила, бензила и фенила;
- SR2, SOR2 или COR2, где R2 представляет собой алкил;
включающий следующие стадии в соответствии со схемой 1:
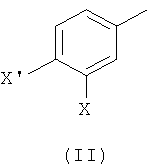
1) взаимодействие соединения формулы (II)
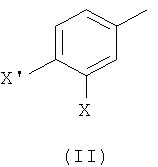
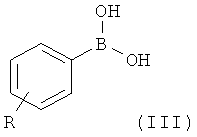
где Х такой, как определено выше, и X' выбран из группы, состоящей из атомов хлора, брома, йода и трифлатной группы (CF3SO3), с соединением формулы (III)
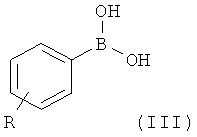
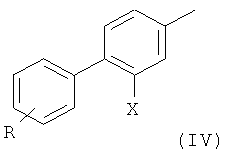
где R такой, как определено выше, с образованием соединения формулы
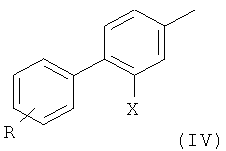
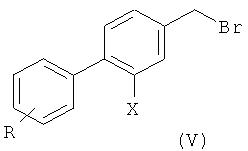
2) радикальное бромирование соединения формулы (IV) с образованием соединения формулы (V);
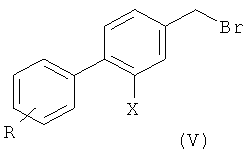
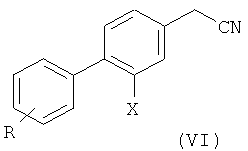
3) превращение соединения формулы (V) в соответствующее нитрильное производное формулы (VI);
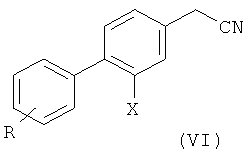
4) взаимодействие соединения формулы (VI) с 1,2-дибромэтаном с образованием соединения формулы (VII); и
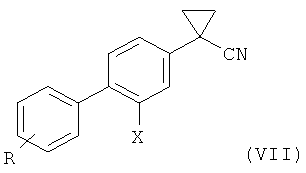
5) гидролиз соединения формулы (VII) с получением соединения формулы (I).
Предпочтительно, радикальное бромирование проводят с N-бромсукцинимидом (NBS) в присутствии каталитического количества бензоилпероксида [PhCOO)2] и ацетонитрила в качестве растворителя.
Изобретение также относится к соединению формулы (VII), которое получено как стабильный промежуточный продукт реакции, описанной выше.
Изобретение также относится к способу приготовления фармацевтической композиции, включающему стадии (1)-(5) и дополнительную стадию (6), включающую смешивание с одним или более фармацевтически приемлемыми эксципиентами.
ОПРЕДЕЛЕНИЯ
Термины, использованные в описании изобретения, имеют следующие значения:
Термин "атомы галогенов" охватывает атомы фтора, хлора, брома и йода.
Термин "алкил" означает прямоцепочечный или разветвленный C1-С4алкил, такой как метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил.
Термин "алкенил" означает прямоцепочечный или разветвленный С2-С6алкенил, такой как винил, 1-пропенил, 2-пропенил, 1-бутенил, изобутенил, или прямоцепочечный или разветвленный пентенил и гексенил. Термин "алкинил" толкуется аналогичным образом.
Термин "циклоалкил" означает циклическую углеводородную группу, содержащую от 3 до 8 атомов углерода. Примеры включают циклопропил, циклобутил, циклопентил, циклогексил и циклогептил.
Термин "насыщенная гетероциклическая группа" означает насыщенную гетероциклическую группу, имеющую по меньшей мере 4 атома углерода и по меньшей мере один гетероатом, предпочтительно от одного до четырех гетероатомов, выбранных из атомов азота, кислорода и серы. Примеры включают пиперидил или тетрагидрофурил.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Согласно настоящему изобретению предложен способ получения соединения общей формулы (I) в соответствии со схемой 1,
где
X и R такие, как определено выше.
В случаях, когда R представляет собой циклоалкил, указанное кольцо возможно замещено одной или более группами, независимо выбранными из групп алкил, CF3, ОН и оксо.
Предпочтительно, циклоалкильная группа представляет собой С3-С6циклоалкил.
В случаях, когда R представляет собой фенил, указанное кольцо возможно замещено одной или более группами, независимо выбранными из атомов галогенов, CF3, OCF3, ОН, алкила и насыщенной гетероциклической группы.
Насыщенная гетероциклическая группа представляет собой предпочтительно моноциклическое кольцо, имеющее 5 или 6 атомов и один или два атома азота или один атом азота и один атом кислорода, например прирролидин, имидазолидин и изоксазолидин.

На первой стадии (стадия 1) соединение, имеющее формулу (II), где Х представляет собой атом галогена, предпочтительно атом фтора, и X' выбран из группы, состоящей из атомов хлора, брома, йода и группы CF3SO3 (трифлат), подвергают взаимодействию с фенилбороновой кислотой формулы (III), где R представляет собой одну или более групп, независимо выбранных из атомов галогенов, предпочтительно атома хлора; CF3; СН=СН2; CN; CH2OH; NO2; метилендиокси; этилендиокси; циклоалкила; фенила; OR1 или NHCOR1, где R1 выбран из группы, состоящей из CF3, алкенила, алкинила, бензила, фенила; SR2, SOR2 и COR2, где R2 представляет собой алкил.
Соединения формулы (II) и (III) коммерчески доступны или могут быть получены способами, известными специалисту.
Предпочтительно, это взаимодействие, известное как реакция Сузуки или реакция Мияура-Сузуки, проводят с использованием 4-бром-3-фтор-толуола в качестве соединения формулы (II) и 3,4-дихлор-фенилбороновой кислоты в качестве соединения формулы (III).
Указанное взаимодействие, которое протекает на палладиевом катализаторе, можно также проводить с использованием алкилбороновых эфиров вместо бороновых кислот.
Предпочтительно, в качестве катализатора может быть использован любой палладиевый катализатор, например тетракис(трифенилфосфин)палладий [Pd(PPh)3], палладий на активированной угле, также известный как палладий на углероде (Pd на С), палладий на оксиде алюминия.
Предпочтительно, используют Pd на С, поскольку он дешевле и легче в обращении.
Обычно стадию (1) проводят в присутствии органического растворителя. Органические растворители, которые могут быть предпочтительно использованы, включают этанол, ацетон, тетрагидрофуран (THF), изопропиловый спирт, N-метилпирролидон (NMP), диоксан и их смеси с водой.
Комбинация органических растворителей также может быть использована.
Предпочтительно, взаимодействие проводят при температуре дефлегмации растворителя.
Когда используют Pd(PPh)3, предпочтительным растворителем является смесь диоксан/вода 2:1 об./об., а когда используют Pd/C, предпочтительным растворителем является этанол.
Предпочтительно, стадию (1) проводят в присутствии основания.
Основания, которые могут быть предпочтительно использованы, включают Na2CO3, К2СО3, K3PO4, Cs2CO3, NaOH и КОН. Предпочтительным основанием является Na2CO3.
Возможно, в реакционную среду могут быть добавлены добавки, такие как трифенилфосфин (Р(Ph3)), полиметилгидросилоксан (PMHS), бромид тетрабутиламмония (ТВАВ), 1,4-диазабицикло[2.2.2]октан (DABCO) или Nal.
Предпочтительно, стадию (1) проводят с использованием небольшого молярного избытка соединения формулы (III) по отношению к соединению формулы (II).
Предпочтительные условия взаимодействия на стадии (1) следующие:
- растворитель: 20 объемов этанола;
- основание: 2 эквивалента Na2CO3;
- катализатор: 13% масс./масс.10% Pd на С.
- температура: температура дефлегмации.
Обычно соединение формулы (IV) получают с выходом выше 70%, предпочтительно выше 80%.
Соединение формулы (IV) предпочтительно представляет собой 3',4'-дихлор-2-фтор-4-метил-бифенил.
На второй стадии (стадия 2) соединение формулы (IV) подвергают радикальному бромированию с образованием соединения формулы (V), где Х и R такие, как определено выше.
Соединение формулы (IV) может представлять собой неочищенный продукт или может быть предварительно подвергнуто кристаллизации стандартными методами.
Предпочтительно, радикальное бромирование проводят с N-бромсукцинимидом (NBS) в присутствии каталитического количества бензоилпероксида [PhCOO)2] и ацетонитрила в качестве растворителя.
Как правило, реакцию проводят при температуре дефлегмации растворителей.
Предпочтительно, для того чтобы минимизировать образование дибромированного продукта, стадию (2) проводят с небольшим избытком NBS, предпочтительно от 1,05 моль эквивалентов до 1 моль эквивалента соединения формулы (IV), и в присутствии 0,04 эквивалента PhCOOO2.
Как правило, соединение формулы (V), которое предпочтительно представляет собой 3',4'-дихлор-2-фтор-4-бром-метил-бифенил, получают с выходом выше 85%, предпочтительно выше 90%.
Возможно, соединение формулы (V) может быть дополнительно очищено кристаллизацией стандартными методами.
На третьей стадии (стадия 3) соединение формулы (V) превращают в соответствующее нитрильное производное формулы (VI), где Х и R такие, как определено выше.
Могут быть использованы цианид натрия или другие подходящие соли.
Предпочтительно, стадию (3) проводят в органическом растворителе, таком как этанол или ацетонитрил, предпочтительно этанол.
Температура на стадии (3) предпочтительно составляет от 20°С до примерно 60°С, более предпочтительно от примерно 40°С до примерно 50°С.
Предпочтительно, стадию (3) проводят с молярным избытком цианида натрия. Используют предпочтительно от 1,2 моль эквивалента до 1,0 моль эквивалента цианида натрия, и предпочтительно от 1,05 моль эквивалента до 1 эквивалента соединения формулы (V).
Как правило, соединение формулы (VI), которое предпочтительно представляет собой 3',4'-дихлор-2-фтор-4-цианометил-бифенил, получают с выходом выше 50%, предпочтительно примерно 55-60%.
Возможно, указанное соединение может быть дополнительно очищено кристаллизацией стандартными методами, предпочтительно путем суспендирования в этаноле.
На четвертой стадии (4) соединение формулы (VI) подвергают взаимодействию с 1,2-дибромэтаном с образованием соединения формулы (VII), где Х и R такие, как определено выше.
Предпочтительно, стадию (4) проводят в органическом растворителе, таком как этанол или ацетонитрил или их смеси с водой.
Предпочтительно, указанную стадию циклопропанирования проводят как каталитическую реакцию фазового перехода в присутствии 30% NaOH и хлорида тетрабутиламмония (ТВАС) или бромида тетрабутиламмония (ТВАВ).
Температуру на стадии (4) предпочтительно поддерживают от примерно 20°С до примерно 50°С.
Как правило, соединение формулы (VII), которое предпочтительно представляет собой 1-(3',4'-дихлор-2-фтор[1,1'-бифенил]-4-ил)-циклопропаннитрил, получают с выходом выше 60%, предпочтительно примерно 65-70%.
Возможно, указанное соединение может быть дополнительно очищено кристаллизацией стандартными методами, предпочтительно с использованием н-гептана в качестве кристаллизационного растворителя.
На пятой стадии (стадия 5) соединение формулы (VII) гидролизуют с получением целевого соединения формулы (I) способами, известными специалисту в данной области.
Предпочтительно, гидролиз проводят в смеси метанола и воды в присутствии сильного основания, предпочтительно КОН, в условиях дефлегмации.
Как правило, соединение формулы (I), которое предпочтительно представляет собой 1-(3',4'-дихлор-2-фтор[1,1'-бифенил]-4-ил)-циклопропанкарбоновую кислоту, получают с выходом выше 65%.
Промывка, фильтрование и выделение соединения формулы (I) могут быть проведены различными методами, известными в данной области.
Указанное соединение может быть дополнительно очищено кристаллизацией стандартными методами и получено с высокой химической чистотой, например с чистотой выше 95%, без использования конечной очистки хроматографией.
Кристаллизация из смеси н-гептана и изопропилового спирта особенно предпочтительна.
Суммарный выход при осуществлении данного способа обычно составляет по меньшей мере 20%, предпочтительно 25% или выше, более предпочтительно выше 30%.
В предпочтительном воплощении изобретения предложен способ получения соединения формулы (I), где Х представляет собой атом фтора, и R представляет собой атом хлора.
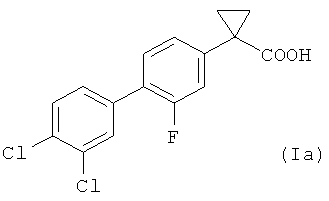
В более предпочтительном воплощении изобретения предложен способ получения 1-(3',4'-дихлор-2-фтор[1,1'-бифенил]-4-ил)-циклопропан-карбоновой кислоты, имеющей формулу (Ia)
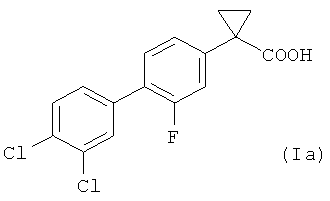 .
.
Полученное соединение формулы (I) может быть дополнительно превращено в соответствующие фармацевтически приемлемые соли различными способами, известными в данной области.
Фармацевтически приемлемые соли включают соли, в которых кислотную функциональную группу подвергают взаимодействию с соответствующим основанием с образованием, например, натриевой, калиевой, кальциевой, магниевой и аммониевой соли.
Соединения формулы (I), полученные способом по изобретению, могут быть использованы в приготовлении фармацевтических композиций для лечения и/или предупреждения нейродегенеративных заболеваний, таких как болезнь Альцгеймера.
Указанные фармацевтические композиции, предпочтительно для перорального применения, содержат по меньшей мере одно соединение формулы (I) в смеси с фармацевтически приемлемыми эксципиентами и/или носителями, описанными, например, в Remington's Pharmaceutical Sciences Handbook, XVII Ed., Mack Pub., N.Y., U.S.A.
Изобретение иллюстрируется приведенными ниже конкретными Примерами.
Пример 1
Получение 3',4'-дихлор-2-фтор-4-метил-бифенила
3-Фтор-4-бромтолуол (50 г, 0,265 моль) и 3,4-дихлорфенилбороновую кислоту (53 г, 0,278 моль) растворяют в этаноле (970 мл) и добавляют карбонат натрия (56,1 г, 0,529 моль). Добавляют 10% палладий на угле (6,6 г), и смесь нагревают с обратным холодильником в течение 4 часов в атмосфере азота. Реакционную смесь охлаждают, фильтруют и концентрируют, добавляют изопропилацетат (250 мл), и затем раствор снова концентрируют. Остаток растворяют в изопропилацетате (250 мл) и 1М растворе гидроксида натрия (250 мл). Органическую фазу отделяют, промывают водой (125 мл), нейтрализуют 3М раствором соляной кислоты, промывают рассолом (250 мл) и концентрируют.
В остаток добавляют смесь ацетонитрил/вода 1/1 об./об. (150 мл), нагревают до 40°С до растворения, а затем охлаждают до 0-5°С и перемешивают в течение 30 минут при этой температуре.
Соединение 3',4'-дихлор-2-фтор-4-метил-бифенил кристаллизуется в виде порошка, который отфильтровывают, промывают смесью ацетонитрил/вода 1/1 об./об. (25 мл) и сушат при 40°С (выход 56 г, 86%).
ВЭЖХ-УФ (высокоэффективная хроматография в сочетании со спектрометрией в ультрафиолетовой области) чистота (210 нм): 95,0%.
1H ЯМР (DMSO-d6, 300 МГц): 7.73 (m, 2Н); 7.49 (m, 2H); 7.14 (m, 2H); 2.36 (s, 3H).
Пример 2
Получение 3',4'-дихлор-2-фтор-4-бром-метил-бифенила
3',4'-Дихлор-2-фтор-4-метил-бифенил (29 г, 0,114 моль), N-бромсукцинимид (21,2 г, 0,119 моль), бензоилпероксид (1,4 г, 0,004 моль) растворяют в ацетонитриле (190 мл).
Смесь нагревают с обратным холодильником в течение 3 часов, затем охлаждают, добавляют раствор сульфита натрия (2,2 г) в воде (54 мл), перемешивают в течение 30 минут и затем оставляют стоять для разделения фаз.
Нижнюю водную фазу отделяют и экстрагируют дихлорметаном (29 мл).
Верхнюю фазу концентрируют под вакуумом, добавляют воду (10 мл) и дихлорметан (58 мл) и перемешивают. Органические фазы отделяют и объединяют, промывают дважды водой (29 мл) и концентрируют под вакуумом.
Соединение 3',4'-дихлор-2-фтор-4-бром-метил-бифенил выделяют в виде оранжевого масла (выход 35,7 г, 94%).
ВЭЖХ-УФ чистота (250 нм); 77,1%
1H ЯМР (DMSO-d6, 300 МГц): 7.87-7.12 (m, 6H); 4.76 (s, 2H).
Пример 3
Получение 3',4'-дихлор-2-фтор-4-цианометил-бифенила
3',4'-Дихлор-2-фтор-4-бром-метил-бифенил (35,0 г, 0,105 моль) и цианид натрия (5,4 г, 0,110 моль) растворяют в смеси этанола (228 мл) и воды (25 мл), затем нагревают при 50°С в течение 3 часов. Раствор концентрируют под вакуумом, и остаток суспендируют в смеси этанол/вода 1/1 об./об. (35 мл) и охлаждают при 0-5°С в течение 30 мин.
Полученное твердое вещество отфильтровывают и сушат при 40°С под вакуумом. Неочищенный продукт суспендируют в этаноле (56 мл) при 20-25°С в течение 30 минут, фильтруют и сушат при 40°С под вакуумом.
Соединение 3',4'-дихлор-2-фтор-4-цианометил-бифенил получают в виде светло-коричневого порошка (выход 16,8 г, 57%).
ВЭЖХ-УФ чистота (250 нм): 92,3%.
1H ЯМР (DMSO-d6, 300 МГц): 7.78 (m, 2Н); 7.60 (m, 2H); 7.34 (m, 2H); 4.14(s, 1H).
Пример 4
Получение 1-(3',4'-дихлор-2-фтор[1,1'-бифенил]-4-ил)-циклопропаннитрила
3',4'-Дихлор-2-фтор-4-цианометил-бифенил (9,0 г, 0,032 моль), 1,2-дибромметан (9,0 г, 0,048 моль), хлорид тетрабутиламмония (1,2 г, 0,043 моль), толуол (60 мл) и воду (9 мл) загружают в реактор.
По каплям добавляют 30% водный раствор гидроксида натрия (60 г, 0,45 моль) в течение 30 минут при 20-25°С, и реакционную смесь перемешивают в течение 6 часов. Органическую фазу отделяют и промывают последовательно водой (12 мл), 3М водным раствором соляной кислоты (36 мл) и в конце водой (12 мл).
Раствор концентрируют, затем добавляют н-гептан (18 мл) при 80°С.
Раствор охлаждают до 0-5°С и перемешивают в течение 30 мин.
Продукт кристаллизуется из раствора, его отфильтровывают, промывают холодным н-гептаном (5 мл) и сушат при 40°С под вакуумом.
Соединение 1-(3',4'-дихлор-2-фтор[1,1'-бифенил]-4-ил)-циклопропаннитрил получают в виде желтого порошка (выход 6,4 г, 65%).
ВЭЖХ-УФ чистота (250 нм): 98,2%.
1H ЯМР (DMSO-d6, 300 МГц): 7.78 (m, 2H); 7.60 (m, 2H); 7.30 (m, 2H); 1.84 (m,2H); 1.63(m, 2H).
Пример 5
Получение 1-(3',4'-дихлор-2-фтор[1,1'-бифенил]-4-ил)-циклопропанкарбоновой кислоты
1-(3',4'-Дихлор-2-фтор[1,1'-бифенил]-4-ил)-циклопропаннитрил (14,3 г, 0,047 моль) растворяют в смеси метанола (143 мл) и воды (71,5 мл), порциями добавляют гидроксид калия (35,1 г, 0,563 моль), и смесь кипятят с обратным холодильником в течение 48 часов.
Реакционную смесь охлаждают и вливают в раствор 36% водной соляной кислоты (57 мл) в воде (57 мл) при 20-25°С. Суспензию перемешивают и фильтруют. Твердое вещество повторно промывают водой и сушат при 40°С под вакуумом. Неочищенный продукт растворяют в 2-пропаноле (178 мл) при температуре дефлегмации, к этому раствору добавляют активированный уголь (0,3 г), перемешивают при температуре дефлегмации, фильтруют, концентрируют и добавляют н-гептан (116 мл). Горячий раствор охлаждают до 0-5°С, и кристаллическое твердое вещество отфильтровывают, промывают 2-пропанолом и сушат при 40°С под вакуумом.
Соединение 1-(3',4'-дихлор-2-фтор[1,1'-бифенил]-4-ил)-циклопропанкарбоновую кислоту получают в виде белого порошка (выход 10,3 г, 68%).
ВЭЖХ-УФ чистота (255 нм): 99,8%.
1H ЯМР (DMSO-d6, 300 МГц): 12.51 (bs, 1Н); 7.78 (m, 2H); 7.54 (m, 2H); 7.30 (m, 2H); 1.48 (m, 2H); 1.22 (m, 2H).
MC (ЭРИ-, 40 В) (масс-спектрометрия с электрораспылительной ионизацией с регистрацией отрицательных ионов при 40 В): 323 (М-); 279.
Температура плавления: 199-200°С.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 1-(2-ГАЛОГЕНОБИФЕНИЛ-4-ИЛ)-ЦИКЛОПРОПАНКАРБОНОВОЙ КИСЛОТЫ | 2010 |
|
RU2540076C2 |
| СПОСОБ ПОЛУЧЕНИЯ ФЕНИЛЗАМЕЩЕННОГО ГЕТЕРОЦИКЛИЧЕСКОГО ПРОИЗВОДНОГО ПОСРЕДСТВОМ СОЧЕТАНИЯ С ИСПОЛЬЗОВАНИЕМ ПЕРЕХОДНОГО МЕТАЛЛА В КАЧЕСТВЕ КАТАЛИЗАТОРА | 2010 |
|
RU2510393C9 |
| КОНДЕНСИРОВАННЫЕ ЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ | 2007 |
|
RU2444521C2 |
| ПОЛУЧЕНИЕ ЗАМЕЩЕННЫХ ИНДЕНОВ | 2003 |
|
RU2323921C2 |
| РЕАКЦИИ СОЧЕТАНИЯ, КОТОРЫЕ МОГУТ БЫТЬ ИСПОЛЬЗОВАНЫ ПРИ ПОЛУЧЕНИИ ПРОИЗВОДНЫХ (1Н-ТЕТРАЗОЛ-5-ИЛ)БИФЕНИЛА | 2005 |
|
RU2426728C2 |
| ГАЛОГЕНЗАМЕЩЕННОЕ ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ | 2013 |
|
RU2756506C2 |
| ГАЛОГЕНЗАМЕЩЕННОЕ ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ | 2013 |
|
RU2649398C2 |
| СОЛЬ ГЕТЕРОЦИКЛИЧЕСКОГО СОЕДИНЕНИЯ, ЗАМЕЩЕННОГО ГАЛОГЕНОМ | 2015 |
|
RU2689315C2 |
| ЗАМЕЩЕННЫЕ КАРБОЦИКЛИЧЕСКИЕ АМИДЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ПРОИЗВОДНЫЕ ИНДАНА, ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ И СПОСОБ ПРОФИЛАКТИКИ ИЛИ ЛЕЧЕНИЯ | 1995 |
|
RU2145954C1 |
| 4-ОКСО-1-(3-ЗАМЕЩЕННЫЙ ФЕНИЛ)-1,4-ДИГИДРО-1,8-НАФТИРИДИН-3-КАРБОКСАМИДЫ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ, СПОСОБ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ И СПОСОБ УЛУЧШЕНИЯ ПОЗНАВАТЕЛЬНОЙ СПОСОБНОСТИ У ЗДОРОВОГО СУБЪЕКТА | 2003 |
|
RU2312865C2 |
Изобретение относится к усовершенствованному способу получения соединения общей формулы (Ia), включающему следующие стадии: 1) взаимодействие соединения формулы (II), где Х представляет собой атом фтора, и X' выбран из группы, состоящей из атомов хлора, брома, йода и трифлатной группы (CF3SO3), с соединением формулы (III), где R представляет собой два атома хлора, в присутствии палладиевого катализатора, с образованием соединения формулы (IV); 2) радикальное бромирование соединения формулы (IV) с использованием N-бромсукцинимида в присутствии каталитического количества бензоилпероксида, с образованием соединения формулы (V); 3) превращение соединения формулы (V) в соответствующее нитрильное производное формулы (VI); 4) взаимодействие соединения формулы (VI) с 1,2-дибромэтаном с образованием соединения формулы (VII); и 5) гидролиз соединения формулы (VII) с получением соединения формулы (Ia). Способ обеспечивает более высокий выход соединения формулы (Ia) с высокой химической чистотой без стадии хроматографической очистки. 2 н. и 6 з.п. ф-лы, 5 пр.
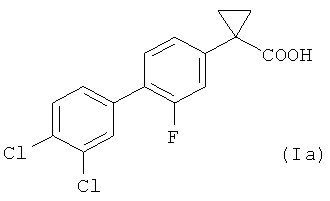

1. Способ получения соединения общей формулы (Ia)
включающий следующие стадии:
1) взаимодействие соединения формулы (II),
где Х представляет собой атом фтора, и X' выбран из группы, состоящей из атомов хлора, брома, йода и трифлатной группы (CF3SO3), с соединением формулы (III),
где R представляет собой два атома хлора, в присутствии палладиевого катализатора, с образованием соединения формулы (IV);
2) радикальное бромирование соединения формулы (IV) с использованием N-бромсукцинимида в присутствии каталитического количества бензоилпероксида, с образованием соединения формулы (V);
3) превращение соединения формулы (V) в соответствующее нитрильное производное формулы (VI);
4) взаимодействие соединения формулы (VI) с 1,2-дибромэтаном с образованием соединения формулы (VII); и
5) гидролиз соединения формулы (VII) с получением соединения формулы (Ia).
2. Способ по п.1, дополнительно включающий стадии выделения и кристаллизации соединения формулы (Ia).
3. Способ по п.2, где кристаллизацию проводят с использованием смеси н-гептана и изопропилового спирта.
4. Способ по п.1, где палладиевый катализатор на стадии (1) выбран из группы, состоящей из тетракис(трифенилфосфин)палладия, палладия на активированном угле и палладия на оксиде алюминия.
5. Способ по п.4, где палладиевый катализатор представляет собой палладий на активированном угле.
6. Способ по п.1, где стадию (2) проводят с использованием ацетонитрила в качестве растворителя.
7. Способ по п.1, включающий следующие стадии:
1) взаимодействие 4-бром-3-фтор-толуола с 3,4-дихлорфенилбороновой кислотой с образованием 3',4'-дихлор-2-фтор-4-метил-бифенила;
2) радикальное бромирование 3',4'-дихлор-2-фтор-4-метил-бифенила с образованием 3',4'-дихлор-2-фтор-4-бромметил-бифенила;
3) превращение 3',4'-дихлор-2-фтор-4-бромметил-бифенила в соответствующий 3',4'-дихлор-2-фтор-4-цианометил-бифенил;
4) взаимодействие 3',4'-дихлор-2-фтор-4-цианометил-бифенила с 1,2-дибромэтаном с образованием 1-(3',4'-дихлор-2-фтор[1,1'-бифенил]-4-ил)-циклопропаннитрила; и
5) гидролиз 1-(3',4'-дихлор-2-фтор[1,1'-бифенил]-4-ил)-циклопропаннитрила с получением 1-(3',4'-дихлор-2-фтор[1,1'-бифенил]-4-ил)-циклопропанкарбоновой кислоты.
8. Способ приготовления фармацевтической композиции, включающий стадии (1)-(5) из п.1 и дополнительную стадию (6), включающую смешивание с одним или более фармацевтически приемлемыми эксципиентами.
| PERETTO, I | |||
| ET | |||
| AL: "Synthesis and Biological Activity of Flurbiprofen Analogues as Selective Inhibitors of beta-Amyloid Secretion" JOURNAL OF MEDICINAL CHEMISTRY, vol | |||
| Приспособление для автоматической односторонней разгрузки железнодорожных платформ | 1921 |
|
SU48A1 |
| Способ использования делительного аппарата ровничных (чесальных) машин, предназначенных для мериносовой шерсти, с целью переработки на них грубых шерстей | 1921 |
|
SU18A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| EA 200700860 A1, 26.10.2007. | |||

