Область техники
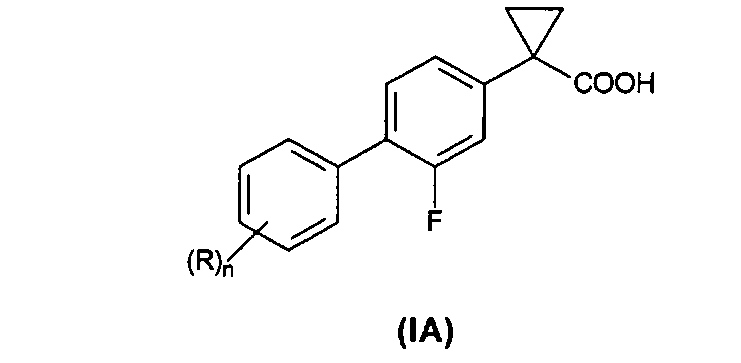
Изобретение относится к способу получения соединений формулы (IA):
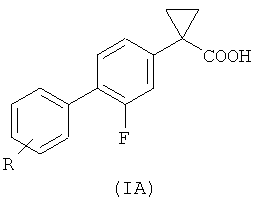
Указанные соединения полезны для предупреждения и/или лечения нейродегенеративных заболеваний, таких как болезнь Альцгеймера.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ИЗОБРЕТЕНИЯ
Болезнь Альцгеймера представляет собой нейродегенеративное расстройство, характеризующееся с гистопатологической точки зрения распространенным наличием внеклеточных и периваскулярных невритных бляшек и внутриклеточных нейрофибриллярных узлов в церебральной паренхиме пациентов с болезнью Альцгеймера.
Невритные бляшки в основном состоят из агрегатов белка с 39-43 аминокислотными остатками, известного как β-амилоид (βА) и, в зависимости от количества аминокислот, Аβ39, Аβ40, Аβ42 и Аβ43.
Известны соединения, которые могут снижать продуцирование наиболее нейротоксичной изоформы β-амилоида, а именно формы, содержащей 42 аминокислоты (Аβ42), посредством их взаимодействия с макромолекулярным/многобелковым ферментативным комплексом с аспартил-протеазной активностью, известным как γ-секретаза.
В WO 2004/074232 раскрыты производные 1-(2-галогенобифенил-4-ил)-циклопропанкарбоновой кислоты формулы (I), способные модулировать активность γ-секретазы, не оказывая воздействия на другие важные метаболические процессы, такие как циклооксигеназная ферментативная активность.
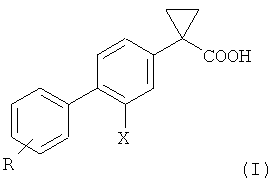
где R такой, как определено ниже, и X предпочтительно представляет собой фтор.
Ключевой промежуточной стадией получения этих соединений является реакция Сузуки соответствующей фенилбороновой кислоты или ее эфира с 3,4-дигалогеноциклопропанкарбоновой кислотой, предпочтительно 3-фтор-4-галогеноциклопропанкарбоновой кислотой.
Из WO 2004/074232 известно, что 3-фтор-4-галогеноциклопропанкарбоновая кислота может быть получена из 3-фтор-4-галогенотолуола, который превращают в соответствующий бензилбромид путем радикального бромирования в тетрахлориде углерода (ССЦ), затем полученный бромид превращают в 3-фтор-4-галогенофенилацетонитрил и последний подвергают взаимодействию с 1,2-дибромэтаном с получением соответствующего 3-фтор-4-галогено-фенилциклопропанонитрила, который в конце подвергают гидролизу до целевой 3-фтор-4-галогеноциклопропанкарбоновой кислоты.
Однако способ, описанный в WO 2004/074232, дает низкий общий выход (12-14%) и имеет жесткие ограничения для промышленного применения.
В частности, конечная реакция сочетания Сузуки дает низкий выход, и полученный продукт трудно очищать кристаллизацией без потери выхода. Для такой очистки применяли хроматографию на силикагеле, однако в промышленных масштабах хроматография на силикагеле является трудоемкой и требует больших объемов растворителей.
Кроме того, радикальное бромирование, используемое для получения производного бензилбромида, дает значительное количество бис-галогенированного побочного продукта, значительно снижая его выход, и включает использование ССl4, который является высокотоксичным веществом, а также озоноразрушающим и парниковым газом.
Настоящее изобретение относится к способу получения производных 1-(2-галогенобифенил-4-ил)-циклопропанкарбоновой кислоты формулы (IA), где атом галогена представляет собой фтор, не имеющему всех упомянутых выше недостатков.
Задача настоящего изобретения решается главным образом путем проведения реакции Сузуки на нитрильном производном с последующим гидролизом до соответствующего производного карбоновой кислоты.
Кроме того, предложены другие условия для повышения выхода на других стадиях, в частности на стадии радикального бромирования.
Способ по данному изобретению оказался более эффективным, особенно для крупномасштабного производства, обеспечивая более высокий выход соединений формулы (IA) с высокой степенью химической чистоты без необходимости проведения стадии хроматографической очистки.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к способу получения соединения формулы (IA):
,
где R представляет собой одну или более групп, независимо выбранных из следующих:
- атомы галогенов, предпочтительно хлора;
- CF3;
- СН=СН2;
- CN;
- СН2ОН;
- NO2;
- метилендиокси;
- этилендиокси;
- циклоалкил, предпочтительно С3-С6циклоалкил;
- фенил;
- OR1 или NHCOR1, где R1 выбран из CF3, алкенила, алкинила; бензила и фенила;
- SR2, SOR2 или COR2, где R2 представляет собой алкил; и его фармацевтически приемлемых солей,
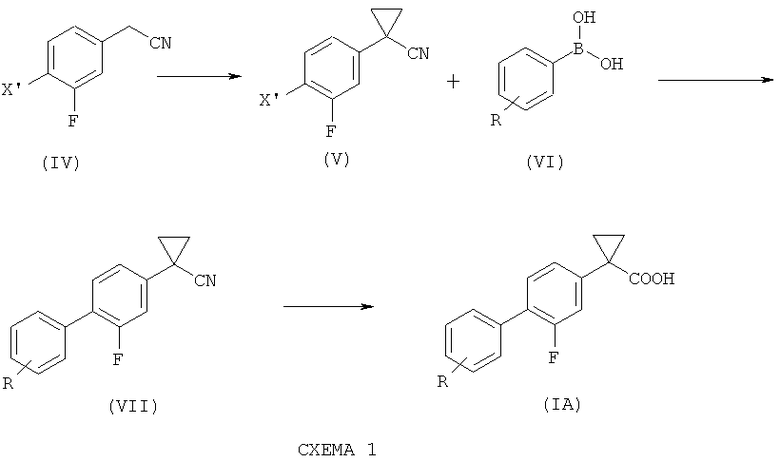
включающему следующие стадии согласно Схеме 1:
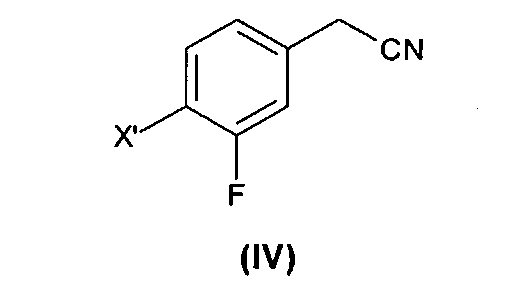
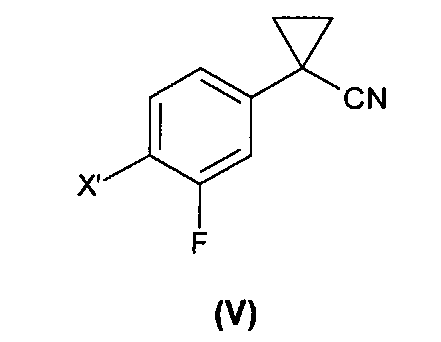
1) взаимодействие соединения формулы (IV), где X' представляет собой хлор, бром, йод или трифлатную группу (CF3SO3), предпочтительно бром, с 1,2-дибромэтаном с образованием соединения формулы (V);
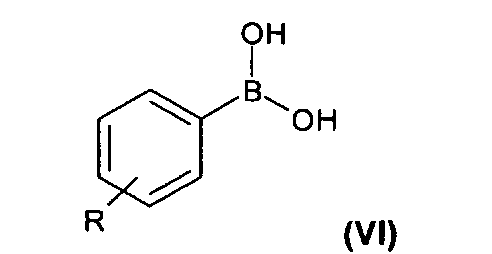
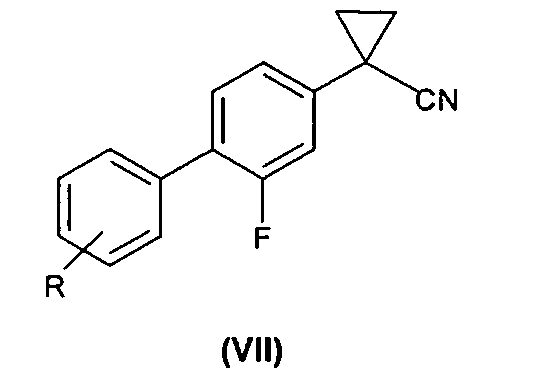
2) связывание соединения формулы (V) с соединением формулы (VI), где R такой, как определено выше, с образованием соединения формулы (VII); и
3) гидролиз соединения формулы (VII) с получением соединения формулы (I).
Предпочтительно, согласно данному изобретению предложен способ получения соединения формулы (IA), где R представляет собой хлор.
Более предпочтительно, согласно данному изобретению предложен способ получения 1-(3',4'-дихлор-2-фтор[1,1'-бифенил]-4-ил)-циклопропанкарбоновой кислоты формулы
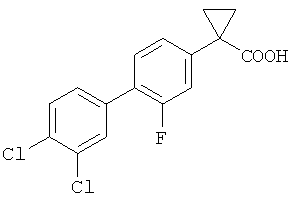
Указанное соединение также упоминается под кодом CHF 5074.
Данное изобретение также относится к способу получения фармацевтической композиции, включающему стадии (1)-(3) и дополнительную стадию, включающую смешивание с одним или более фармацевтически приемлемыми эксципиентами.
ОПРЕДЕЛЕНИЯ
Термин "атомы галогенов" охватывает атомы фтора, хлора, брома и йода.
"Алкил" означает С1-С4алкил с прямой или разветвленной цепью, такой как метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил,
"Алкенил" означает С2-С6алкенил с прямой или разветвленной цепью, такой как винил, 1-пропенил, 2-пропенил, 1-бутенил, изобутенил или прямоцепочечный или разветвленный пентенил и гексенил. Термин "алкинил" следует толковать аналогичным образом.
"Циклоалкил" означает циклическую неароматическую углеводородную группу, содержащую от 3 до 8 атомов углерода. Примеры включают циклопропил, циклобутил, циклопентил, циклогексил и циклогептил.
"Насыщенный гетероциклический" означает насыщенную гетероциклическую группу, имеющую по меньшей мере 4 атома углерода и по меньшей мере один гетероатом, предпочтительно от одного до четырех гетероатомов, выбранных из атомов азота, кислорода и серы. Примеры включают пиперидил или тетрагидрофурил.
Термин "фармацевтически приемлемые соли" относится к солям, получаемым путем взаимодействия главного соединения в форме кислоты с неорганическим или органическим основанием с образованием соли, одобренной для применения у людей, например соли натрия, калия, кальция, магния и аммония.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Согласно настоящему изобретению предложен способ получения соединения формулы (IA), где R такой, как определено выше, включающий стадии согласно Схеме 1.
Когда R представляет собой циклоалкил, тогда он возможно замещен одной или более группами, независимо выбранными из алкила, CF3, ОН и оксогруппы.
Предпочтительно, циклоалкильная группа представляет собой С3-С6циклоалкил.
Когда R представляет собой фенил, тогда он возможно замещен одной или более группами, независимо выбранными из атомов галогенов, CF3, OCF3, ОН, алкила и насыщенной гетероциклической группы.
Насыщенная гетероциклическая группа предпочтительно представляет собой моноциклическое кольцо, имеющее 5 или 6 атомов и один или два атома азота, или один атом азота и один атом кислорода, такое как пирролидиновое, имидазолиновое и изоксазолидиновое кольцо.
В качестве исходного вещества может быть использовано любое соединение формулы (IV) с X', выбранным из группы, состоящей из атомов хлора, брома, йода и группы CF3SO3 (трифлат), которое является коммерчески доступным.
Предпочтительно, в качестве исходного вещества используют соединение, где X' представляет собой бромид.
На первой стадии (стадия 1) соединение формулы (IV) подвергают взаимодействию с 1,2-дибромэтаном с образованием соединения формулы (V), где X' такой, как определено выше.
Преимущественно, стадия (1) может быть проведена в органическом растворителе, таком как этанол или ацетонитрил или их смеси с водой.
Предпочтительно, указанную стадию циклопропанилирования проводят как каталитическую реакцию фазового переноса в присутствии концентрированного NaOH и тетрабутиламмонийхлорида (ТВАС) или тетрабутиламмонийбромида (ТВАВ). Концентрация NaOH преимущественно может составлять от 30 до 50% масс./об.
На стадии (1) можно поддерживать температуру предпочтительно от примерно 20°С до примерно 50°С.
Как правило, соединение формулы (V), которое предпочтительно представляет собой 4-бром-3-фторфенилциклопропаннитрил, получают с выходом выше 80%, предпочтительно с выходом, равным или выше 90%.
Возможно, полученное соединение перед его использованием на следующей стадии способа может быть дополнительно очищено кристаллизацией стандартным способами.
На второй стадии (стадия 2) соединение формулы (V) подвергают взаимодействию с фенилбороновой кислотой формулы (VI), где R представляет собой одну или более групп, независимо выбранных из атомов галогенов, предпочтительно хлора; CF3; СН=СН2; CN; СН2ОН; NO2; метилендиокси; этилендиокси; циклоалкила; фенила; OR1 или NHCOR1, где R1 выбран из группы, состоящей из CF3, алкенила, алкинила, бензила, фенила, SR2, SOR2 и COR2, где R2 представляет собой алкил.
Предпочтительно, реакцию, известную как реакция Сузуки или реакция Мияура-Сузуки, проводят с использованием 4-бром-3-фтор-фенилциклопропаннитрил в качестве соединения формулы (V) и 3,4-дихлор-фенилбороновой кислоты в качестве соединения формулы (VI).
Указанную реакцию, которая протекает в присутствии палладиевого катализатора, можно также проводить с использованием алкилбороновых эфиров вместо бороновых кислот.
Преимущественно, в качестве катализатора может быть использован любой палладиевый катализатор, такой как, например, тетракис(трифенилфосин)палладий [Pd(PPh)3], палладий на активированном угле, также известный как палладий на угле (Pd/C), палладий на оксиде алюминия или смесь Pd(ОСОСН3)2 и PPh3.
Преимущественно, стадия (2) может быть проведена в присутствии органического растворителя, такого как этанол, ацетон, тетрагидрофуран (THF), изопропиловый спирт, N-метилпирролидон (NMP), диоксан и их смеси с водой. Может быть использована также комбинация органических растворителей.
Предпочтительно, при использовании Pd(PPh)3 или смеси Pd(OCOCH3)2 и РРn3 реакцию проводят в присутствии N-метилпирролидона (NMP) или смеси диоксан/вода 2:1.
В случае использования Pd/C предпочтительным растворителем является этанол.
Стадию (2) преимущественно проводят в присутствии от 1 до 4 эквивалентов основания.
Основания, которые преимущественно могут быть использованы, включают Na2CO3, К2СO3, K3PO4, Cs2CO3, NaOH и КОН. Предпочтительными основаниями являются Na2CO3, К2СО3 или К3РO4.
Возможно, в реакционную среду могут быть добавлены добавки, такие как трифенилфосфин (P(Ph3)), полиметилгидросилоксан (PMHS), тетрабутиламмонийбромид (ТВАВ), 1,4-диазабицикло[2.2.2]октан (DABCO) или Nal.
Как правило, реакцию проводят при температуре от 80 до 140°С, предпочтительно при температуре 110°С.
Предпочтительно, стадия (2) может быть проведена с использованием эквимолярного количества соединения (VI) относительно соединения (V) или с небольшим молярным избытком.
Как правило, соединение формулы (VI) получают с выходом выше 60%, предпочтительно выше 70%, еще более предпочтительно выше 80%.
Соединение формулы (VI) предпочтительно представляет собой 3',4'-дихлор-2-фтор-4-цианометил-бифенил.
Предпочтительными условиями на стадии (2) являются:
- растворитель: NMP;
- основание: 4 эквивалента К3РO4 в форме порошка;
- катализатор: смесь 1:2 масс./масс. Pd(OCOCH3)2 и PPh3;
- температура: 110°С.
В этих условиях 3',4'-дихлор-2-фтор-4-цианометил-бифенил получают с выходом выше 90%.
Возможно, полученное соединение перед его использованием на следующей стадии способа может быть дополнительно очищено кристаллизацией стандартными способами.
На третьей стадии (стадия 3) соединение формулы (VII) подвергают гидролизу общеизвестным способом с получением целевого соединения формулы (IA). Предпочтительно, гидролиз проводят в смеси метанола и воды в присутствии сильного основания, предпочтительно КОН, в условиях дефлегмации.
Как правило, соединение формулы (IA), которое предпочтительно представляет собой 1-(3',4'-дихлор-2-фтор[1,1'-бифенил]-4-ил)-циклопропанкарбоновую кислоту, получают с выходом выше 65%.
Соединение формулы (IA) может быть промыто, отфильтровано и выделено различными известными способами.
Указанное соединение может быть дополнительно очищено кристаллизацией стандартными способами и получено с высокой химической чистотой, например с чистотой выше 95%, без использования конечной стадии очистки хроматографией.
Особенно предпочтительной является кристаллизация из смеси н-гептана и изопропилового спирта.
Полученное соединение (IA) затем может быть превращено в соответствующие фармацевтически приемлемые соли различными известными способами.
В альтернативном воплощении, когда для реакции на стадии (2) в качестве растворителя используют NMP, тогда соединение формулы (IA) в форме щелочной соли может быть получено путем прямого осаждения в основном водном растворе без выделения промежуточного соединения (VII).
Это возможно, как было установлено, если все примеси на стадии (2) не содержат группы, которые могут образовывать соли водной фазе.
Это дает возможность увеличить общий выход при осуществлении данного способа.
Щелочная соль может быть превращена в форму свободной кислоты известными способами.
Общий выход, как правило, составляет по меньшей мере 30%, предпочтительно равен или выше 40%, еще более предпочтительно выше 50%.
В частном аспекте, способ по изобретению может дополнительно включать стадии получения соединения (IV) из коммерчески доступного соединения формулы (II) согласно Схеме 2.
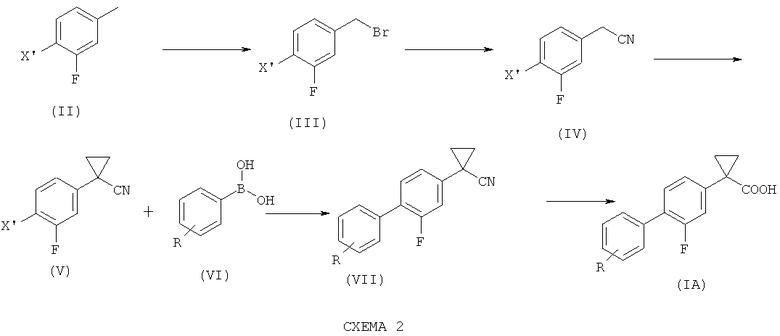
Для того чтобы получить соединение (IV), соединение формулы (II) с X', выбранным из группы, состоящей из хлора, брома йода и группы CF3SO3 (трифлат), подвергают радикальному бромированию с образованием соединения формулы (III).
Радикальное бромирование преимущественно проводят N-бромсукцинимидом (NBS) в присутствии каталитического количества бензоилпероксида [(PhCOO)2] и ацетонитрила в качестве растворителя.
Обычно эту реакцию проводят в растворителе при температуре дефлегмации.
Предпочтительно, для минимизации образования дибромированного продукта указанную стадию проводят с небольшим избытком NBS, предпочтительно 1,05 моль-эквивалента на 1 моль-эквивалент соединения формулы (II), и в присутствии 0,04 эквивалента PhCOOO2.
Как правило, соединение формулы (III), которое предпочтительно представляет собой 3-фтор-4-бромбензилбромид, получают с выходом выше 85%, предпочтительно выше 90%.
Соединение формулы (III) возможно дополнительно подвергают очистке кристаллизацией стандартными способами, а затем превращают в соответствующее нитрильное производное формулы (IV) с использованием цианида натрия или другой подходящей соли.
Преимущественно, указанное превращение проводят в органическом растворителе, таком как этанол или ацетонитрил, предпочтительно этанол, поддерживая температуру от примерно 20°С до примерно 60°С, предпочтительно от примерно 40°С до примерно 50°С.
Предпочтительно, указанное взаимодействие проводят с молярным избытком цианида натрия, преимущественно от 1,2 моль-эквивалента до 1,0 моль-эквивалента цианида натрия, предпочтительно 1,05 моль-эквивалента, на 1 моль-эквивалент соединения формулы (III).
Как правило, соединение формулы (IV), которое предпочтительно представляет собой 4-бром-3-фтор-бензилнитрил, получают с выходом выше 50%.
Возможно, полученное соединение (IV), перед тем, как его подвергают взаимодействию на стадиях, описанных выше, может быть дополнительно очищено кристаллизацией стандартными способами.
Соответственно, настоящее изобретение относится также к способу получения соединения формулы (IA), где R такой, как определено выше, и его фармацевтически приемлемых солей, включающему следующие стадии согласно Схеме 2:
1) подвергание соединения формулы (II), где X' представляет собой атом хлора, брома, йода или трифлатную группу (CF3SO3), предпочтительно атом брома, радикальному бромированию с образованием соединения формулы (III);
2) превращение соединения формулы (III) в соответствующее нитрильное производное формулы (IV);
3) взаимодействие соединения формулы (IV) с 1,2-дибромэтаном с образованием соединения формулы (V);
4) связывание соединения формулы (V) с соединением формулы (VI), где R такой, как определено выше, с образованием соединения формулы (VII); и
5) гидролиз соединения формулы (VII) с получением соединения формулы (IA).
Соединения формулы (IA), полученные способами по изобретению, могут быть использованы в получении фармацевтических композиций для лечения и/или предупреждения нейродегенеративных заболеваний, таких как болезнь Альцгеймера.
Указанные фармацевтические композиции, предпочтительно для перорального применения, содержат по меньшей мере одно соединение формулы (IA) в смеси с фармацевтически приемлемыми эксципиентами и/или носителями, например эксципиентами и/или носителями, которые описаны в Remington Pharmaceutical Sciences Handbook, XVII Ed., Mack Pub., N.Y., U.S.A.
Изобретение иллюстрируется более подробно приведенными ниже Примерами.
Пример 1
Получение 4-бром-3-фторбензилбромида Формулы (III)
В раствор 4-бром-3-фтортолуола (21,5 г, 0,114 моль) в ацетонитриле (200 мл) добавляют N-бромсукцинимид (NBS, 21,2 г, 0,119 моль). Эту смесь кипятят с обратным холодильником, добавляют дибензоилпероксид (1,4 г, 0,004 моль), кипятят с обратным холодильником в течение 3 часов, затем охлаждают при комнатной температуре и экстрагируют водой. Водную фазу отбрасывают, органическую фазу промывают рассолом, сушат над сульфатом натрия и концентрируют в вакууме с получением масла (27,1 г, выход 90%).
Пример 2
Получение 4-бром-3-фторфенилацетонитрила Формулы (IV)
В раствор 4-бром-3-фторбензилбромида (27 г, 0,1 моль) в этаноле (200 мл) добавляют NaCN (5,4 г, 0,11 моль), и эту смесь кипятят с обратным холодильником в течение 3 часов. Смесь концентрируют в вакууме, полученный осадок переносят в воду, затем экстрагируют этилацетатом. Органическую фазу промывают рассолом, сушат над сульфатом натрия и концентрируют в вакууме с получением темного масла (12,1 г, выход 56%).
Пример 3
Получение 4-бром-3-фторфенилциклопропаннитрила формулы (V)
К раствору 4-бром-3-фторфенилацетонитрила (1 г, 4,6 ммоль) в толуоле (4 мл) добавляют 0,6 мл (7 ммоль) 1,2-дибромэтана, 50%-ный водный раствор NaOH (4 мл) и тетрабутиламмонийбромид (0,32 г, 1 ммоль). Эту смесь выдерживают при перемешивании при комнатной температуре в течение 4 часов, затем разбавляют водой и экстрагируют этилацетатом. Органическую фазу выделяют, и растворитель удаляют в вакууме с получением коричневого твердого вещества, которое подвергают очистке хроматографией на силикагеле с получением продукта в твердой форме, имеющего цвет от оранжевого до желтого (1 г, выход 90%).
Пример 4
Получение 1-(3',4'-дихлор-2-фтор[1,1'-бифенил]-4-ил)-циклопропаннитрила формулы (VI)
В инертной атмосфере в колбу с 4-бром-3-фторфенилциклопропаннитрилом (470 мг, 1 экв.) добавляют 3,4-дихлорфенилбороновую кислоту (374 мг, 1 экв.), Pd(OAc)2 (44 мг, 0,1 экв.), PPh3(105 мг, 0,2 экв.) и тонкоизмельченный К3РО4 (1,6 г, 4 экв.).
При комнатной температуре добавляют 5 мл предварительно дегазированного N-метилпирролидона (NMP).
Реакционную смесь нагревают при 110°С в течение 2 часов до завершения взаимодействия (что отслеживают по результатам 19F ЯМР-анализа), затем разбавляют этилацетатом и промывают водой.
Органическую фазу выделяют, и растворитель выпаривают с получением розово-фиолетового порошка (700 мг). К этому твердому веществу добавляют смесь вода:ацетон 1:1 об./об. (40 мл), затем суспензию нагревают до температуры дефлегмации при перемешивании с получением раствора. После выпаривания ацетона получают твердый светло-фиолетовый продукт (560 мг, выход 95%).
Пример 5
Получение 1-(3',4'-дихлор-2-фтор[1,1'-бифенил]-4-ил)-циклопропанкарбоновой кислоты формулы (IA)
1-(3',4'-Дихлор-2-фтор[1,1'-бифенил]-4-ил)-циклопропаннитрил (14,3 г, 0,047 моль) растворяют в смеси метанола (143 мл) и воды (71,5 мл), порциями добавляют гидроксид калия (35,1 г, 0,563 моль), и эту смесь нагревают с обратным холодильником в течение 48 часов.
Реакционную смесь охлаждают и вливают в раствор водного хлористого водорода 36% (57 мл) в воде (57 мл) при 20-25°С. Суспензию перемешивают и фильтруют, твердое вещество неоднократно промывают водой и сушат при 40°С в вакууме. Неочищенный продукт растворяют в 2-пропаноле при температуре дефлегмации (178 мл), в этот раствор добавляют активированный уголь (0,3 г), смесь перемешивают при температуре дефлегмации и фильтруют, концентрируют и добавляют н-гептан (116 мл). Горячий раствор охлаждают до 0-5°С, и кристаллическое твердое вещество отфильтровывают, промывают 2-пропанолом и сушат при 40°С в вакууме.
Соединение 1-(3',4'-дихлор-2-фтор[1,1'-бифенил]-4-ил)-циклопропанкарбоновую кислоту получают в виде белого порошка (10,3 г, выход 68%).
ВЭЖХ-УФ (высокоэффективная жидкостная хроматография с УФ-детектированием): чистота (255 нм): 99,8%
1Н ЯМР (DMSO (диметилсульфоксид)-d6, 300 МГц): 12.51 (bs, 1Н); 7.78 (m, 2Н); 7.54 (m, 2Н); 7.30 (m, 2Н); 1.48 (m, 2Н); 1.22 (m, 2Н)
МС (масс-спектрометрия) (ЭРИ" (электрораспылительная ионизация с регистрацией отрицательных ионов), 40 В): 323 (М-); 279.
Диапазон температуры плавления: 199-200°С.
Изобретение относится к способу получения соединения формулы (IA), в которой R представляет собой одну или более групп, независимо выбранных из атомов галогенов, и n составляет 1 или 2; или его фармацевтически приемлемых солей. Способ включает следующие стадии: (1) взаимодействие соединения формулы (IV), в которой X′ выбран из хлора, брома, йода и трифлатной группы (CF3SO3), с 1,2-дибромэтаном с образованием соединения формулы (V), (2) связывание в присутствии палладиевого катализатора, представляющего собой смесь Pd(OAc)2 и трифенилфосфина, соединения формулы (V) с соединением формулы (VI), в которой R такой, как определено выше, с образованием соединения формулы (VII); (3) гидролиз соединения формулы (VII) с получением соединения формулы (IA). Изобретение также относится к способу получения фармацевтической композиции, включающему вышеуказанные стадии (1)-(3) и дополнительную стадию, включающую смешивание с одним или более фармацевтически приемлемыми эксципиентами. Способ позволяет получать продукты с высоким выходом.
2 н. и 7 з.п. ф-лы, 5 пр.
1. Способ получения соединения формулы (IA)
где R представляет собой одну или более групп, независимо выбранных из атомов галогенов, и n составляет 1 или 2;
и его фармацевтически приемлемых солей, включающий следующие стадии:
(1) взаимодействие соединения формулы (IV)
где X′ выбран из хлора, брома, йода и трифлатной группы (CF3SO3), с 1,2-дибромэтаном с образованием соединения формулы (V)
(2) связывание в присутствии палладиевого катализатора, представляющего собой смесь Pd(OAc)2 и трифенилфосфина, соединения формулы (V) с соединением формулы (VI)
где R такой, как определено выше, с образованием соединения формулы (VII);
(3) гидролиз соединения формулы (VII) с получением соединения формулы (IA).
2. Способ по п.1, где X′ представляет собой бромид.
3. Способ по п.1, дополнительно включающий стадии выделения и кристаллизации соединения формулы (IA).
4. Способ по п.1, где палладиевый катализатор представляет собой смесь 1:2 масс./масс. Pd(OCOCH3)2 и PPh3.
5. Способ по п.1, дополнительно включающий следующие стадии:
(1) подвергание соединения формулы (II)
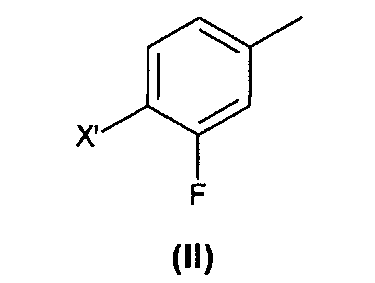
где X′ такой, как определено выше, радикальному бромированию с образованием соединения формулы (III);
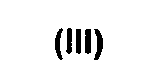
(2) превращение соединения формулы (III) в соответствующее нитрильное производное формулы (IV).
6. Способ по п.5, где стадию бромирования проводят N-бромсукцинимидом в присутствии каталитического количества бензоилпероксида с использованием ацетонитрила в качестве растворителя.
7. Способ по п.1, где атом галогена представляет собой атом хлора.
8. Способ по п.6, где соединение формулы (1А) представляет собой 1-(3′,4′-дихлор-2-фтор[1,1′-бифенил]-4-ил)-циклопропанкарбоновую кислоту.
9. Способ получения фармацевтической композиции, включающий стадии (1)-(3) из п.1 и дополнительную стадию, включающую смешивание с одним или более фармацевтически приемлемыми эксципиентами.
| PERETTO, I | |||
| ET | |||
| AL: "Synthesis and Biological Activity of Flurbiprofen Analogues as Selective Inhibitors of beta-Amyloid Secretion" JOURNAL OF MEDICINAL CHEMISTRY, vol | |||
| Приспособление для автоматической односторонней разгрузки железнодорожных платформ | 1921 |
|
SU48A1 |
| Способ использования делительного аппарата ровничных (чесальных) машин, предназначенных для мериносовой шерсти, с целью переработки на них грубых шерстей | 1921 |
|
SU18A1 |
| Щелевая паровая форсунка | 1926 |
|
SU5705A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| EA 200700860 A1, 26.10.2007 | |||

