Изобретение относится к области микробиологии и молекулярной генетики и может быть использовано в медицинской промышленности, в частности, в тест-системах по определению микроальбуминурии - основного лабораторного критерия доклинической стадии диабетической нефропатии. Изобретение также может применяться в биотехнологии в качестве доступного реагента для выделения человеческого сывороточного альбумина (HSA) аффинной хроматографией, а также для освобождения сыворотки крови от HSA. Это позволяет определять другие белки, присутствующие в сыворотке в более низких концентрациях.
Одной из важнейших медико-социальных проблем современности является сахарный диабет (СД). Это заболевание играет значительную роль в структуре хронической патологии во всех возрастных группах, приводит к ранней инвалидизации больных из-за развития осложнений, а также значительному увеличению смертности.
Лидирующей причиной смертности больных СД I типа во всем мире является хроническая почечная недостаточность (ХПН) вследствие прогрессирования диабетической нефропатии (ДН) - специфического поражения почек при СД. В США и Японии ДН занимает первое место по распространенности среди всех заболеваний почек (35-40%), оттеснив на вторую-третью позицию такие почечные заболевания, как гломерулонефрит, пиелонефрит, поликистоз и др. В странах Европы ДН носит менее угрожающий характер, но удерживается на уровне 20-25% по потребности в экстракорпоральном лечении. В России смертность от почечной недостаточности при СД I типа по данным Государственного регистра не превышает 18%, что в три раза ниже уровня, регистрируемого в мире на протяжении последних 30 лет. При СД II типа смертность от ХПН в России составляет 1,5%, что в 2 раза ниже мировой. Столь значительные расхождения в показателях распространенности ДН и смертности больных диабетом от ХПН не отражают истинную эпидемиологическую ситуацию в России. Наиболее вероятными причинами подобных расхождений с данными мировой статистики являются: отсутствие повсеместного внедрения программы скрининга ДН в диабетологических учреждениях России; отсутствие единой методологии скрининга ДН; недоступность диализных методов лечения ХПН для больных СД, что приводит к их смерти во внебольничных условиях; регистрация смерти не от почечной недостаточности, а от сердечнососудистых осложнений или других причин.
В 1999-2000 гг. под руководством Федерального диабетологического центра Минздрава РФ были организованы экспедиции в различные регионы России, оснащенные наиболее необходимыми и доступными методами скрининга ДН. При этом было показано, что фактическая распространенность ДН в городах России в 2-8 раз превышает регистрируемую.
Своевременная диагностика ДН представляет важную задачу, стоящую перед диабетологами, поскольку выявление даже самых ранних стадий ДН требует безотлагательного лечения.
Минздрав России утвердил новую классификацию ДН, включающую 3 стадии развития:
1) стадию микроальбуминурии,
2) стадию протеинурии с сохраненной фильтрационной функцией почек,
3) стадию хронической почечной недостаточности.
Наиболее ранним и достоверным методом диагностики ДН является тест на микроальбуминурию.
Под термином «микроальбуминурия» понимают экскрецию альбумина с мочой в низких концентрациях (от 30 до 300 мг/сут). Такое количество белка очень трудно определить, практически оно не определяется при традиционном рутинном исследовании мочи, в связи с чем ранняя стадия ДН может быть не диагностирована. Но это стадия является единственно обратимой при своевременном назначении патогенетической терапии.
Именно выявление микроальбуминурии может предотвратить развитие ДН.
Назначение ингибиторов ангиотензинпревращающего фермента (АПФ) на стадии микроальбуминурии даже при нормальных значениях системного АД позволяет предупредить появление протеинурии у 55% больных СД. Назначение ингибиторов АПФ на стадии протеинурии предупреждает развитие ХПН у 50-55% больных СД. Назначение ингибиторов АПФ на стадии ХПН позволяет продлить додиализный период на 4-5 лет.
По данным опроса краевых, областных и городских эндокринологов различных регионов России, на стадии микроальбуминурии терапия ингибиторами АПФ назначается только в 20% регионов, где исследование микроальбуминурии проводится на постоянной основе. В остальных областях и регионах России отсутствие скрининга больных на наличие микроальбуминурии не позволяет своевременно назначить терапию, вследствие чего патология почек продолжает быстро прогрессировать, переходя в стадию протеинурии и ХПН.
Наиболее перспективным и экономичным для национального здравоохранения направлением в развитии современной диабетологической помощи является профилактика ДН. Такая профилактика возможна лишь при безукоризненном метаболическом контроле СД, начиная с начала возникновения заболевания; при своевременной диагностике диабетического поражения почек, основанной на обязательном регламентированном скрининге больных СД на наличие микроальбуминурии; при своевременном назначении патогенетической терапии, основанной на применении ингибиторов АПФ.
Двукратное определение микроальбуминурии в течение года является обязательным исследованием для каждого больного СД, документально закрепленным в федеральной программе «Диабет».
Все применяемые в настоящее время диагностические тест-системы для выявления микроальбуминурии являются зарубежными и предназначены для иммунологического полуколичественного и количественного определения концентрации альбумина в моче. Полуколичественные - это индикаторные полоски или латекс-системы, в которых применяются различные реагенты с альбумином. Общим принципом является регистрация "положительного" результата только при выявлении в образце мочи концентрации альбумина, соответствующей диапазону микроальбуминурии (20-200 мг/мл).
Известен диагностический набор "Micro-Bumin test" (Mile's Ins Ekkhart, FIN). Он основан на применении с альбумином голубого бромфенола, фиксированного на щелочном матриксе. Окрашивание происходит, если концентрация альбумина в образце превышает 40 мг/л. Низкая специфичность данного теста (<80%) обусловлена способностью реагента связываться с другими белками плазмы.
Известны диагностические наборы "Albu Screen" и "Albusure" ("Cambridge Life Siences", Cambridge, UK), которые представляют собой латекс-агглютинирующий ингибиционный тест на основе использования антисыворотки к альбумину человека и латекс-частиц с альбумином. Если концентрация альбумина в образце мочи менее 30 мг/л, наблюдается латекс-агглютинация. Специфичность и чувствительность данного теста 90%.
Известен диагностический набор "Micral-test" (Boehringer Mannheim, Indiana-polis, Germany), который основан на использовании моноклональных антиальбуминовых антител, меченных галактозидазой, и фиксированных на тест-полоске. Оценка реакции производится спустя 5 минут путем визуального сравнения окрашивания зоны реакции тест-полоски с прилагаемой цветной шкалой, деления которой соответствуют концентрации альбумина 0, 10, 20, 50, 100 мкг/л.
Использование тест-полосок вполне допустимо при скрининге для полуколичественного выявления микроальбуминурии. Однако тест нуждается в количественной оценке экскреции альбумина с мочой.
Для количественного определения уровня экскреции альбумина используются радиоиммунные, иммуноферментные и иммунотурбодиметрические методы. Все они являются иммунохимическими с применением антител к альбумину.
Известен метод радиальной иммунодиффузии (РИД). Он основан на инкорпорации антиальбуминовых антител в агаровый гель с последующей иммунодиффузией альбумина исследуемого образца (Терро F.V. Clin.Chem. 28: 1359-1361? (1982)). Будучи надежным и недорогим, он, тем не менее, не находит широкого применения на практике из-за того, что для реакции требуется весьма длительный инкубационный период. Кроме того, РИД не может быть автоматизирован.
Известен радиоиммунный метод (РИА), который основан на применении изотопа йода и антиальбуминовой антисыворотки, выполняется в жидкой фазе при избытке антигена. Метод чувствительный и точный. Недостатки РИА заключаются в использовании радиоактивных реагентов, имеющих короткий срок годности, метод требует дорогостоящего оборудования и системы утилизации отходов.
Известны методы иммунотурбидиметрии и нефелометрии, которые основаны на свойстве альбумина мочи образовывать иммунные комплексы при взаимодействии с антителами (Watts G. Clin.Chem. 32: 1544-1548, (1986)). Помутнение исследуемого образца, вызванное их образованием, измеряется спектрофотометрически при длине волны 340 нм (при турбидиметрии). Метод прост, точен, легко автоматизируется. Он позволяет определять альбумин в концентрации 5-40 мг/л, а при больших концентрациях альбумина требуется дополнительное разведение исследуемого образца. Чувствительность метода может быть повышена за счет добавления в тест-систему латекс-частиц. Метод требует использования дорогостоящей аппаратуры. Обязательным условием проведения исследования является предварительное центрифугирование образцов, так как любое загрязнение мочи, т.е. любой уровень ее помутнения приводит к ложноположительным результатам. Комплексы альбумина мочи с антителами - основа этих диагностических способов определения микроальбуминурии.
Таким образом, на сегодняшний день отсутствует унифицированная методика для определения микроальбуминурии. Имеющиеся на внутреннем рынке диагностикумы различных западных производителей ("Albu Screen", "Albusure" - "Cambrige Life Science" UK; "Micral-test" Boehringer Mannheim, Germany и др.) представляют собой тест-системы, преимущественно для полуколичественного определения альбумина в моче и являются дорогими для выполнения масштабных программ скрининга и мониторинга ДН в полном объеме. Отечественных аналогов, внедренных в практику, не имеется.
Создание чувствительного и экономически выгодного диагностического способа для определения микроальбуминурии - актуальная проблема.
Сущность заявляемого изобретения состоит в том, что для определения микроальбуминурии предлагается использовать рекомбинантный рецепторный альбумин-связывающий полипептид, полученный из стрептококков.
Альбумин - наиболее распространенный белок плазмы крови человека и млекопитающих. В последние десятилетия выявлены и изучены белки бактерий, обладающие рецепторной активностью в отношении HSA.
Стрептококки (Streptococcus) групп А, С и G экспрессируют поверхностные белки, которые связывают HSA. (Myhre E. and Kronvall G. Infect.Immun. 27: 6-14 (1980)), Wideback К. и др. Acta Pathol. Microbiol. Immunol. Scand. Sect. B. 91: 373-382 (1983)).
Среди таких поверхностных белков стрептококков группы G интерес представляет белок G, полноразмерная молекула которого обладает способностью связывать иммуноглобулин G (IgG) человека и различных млекопитающих, а также HSA человека и животных (Bjorck и др. Mol.Immunol. 24: 1113-1122 (1987)).
Из одного из штаммов Peptostreptococcus magnus выделен РАВ белок, связывающий HSA (de Chateau M., Bjorck L.J. of Biol. Chem. 269: 12147-12151 (1994)). Анализ его аминокислотной последовательности выявил участок из 45 аминокислотных остатков, отвечающий за связывание с HSA. Эта последовательность, названная GA модулем, имеет гомологию с HSA-связывающей областью белка G (Akerstrom В и др. J of Biol. Chem. 262: 13388-13391 (1987), Sjobring U и др. J Immun. 140: 1595-1599. (1988), Nygren Р. И др. J.Mol. Recognit. 1: 69-74 (1988)).
В отличие от РАВ белка, который не имеет гомологичных повторов, белок G, выделенный из штамма G 148, содержит три гомологичные повторяющиеся последовательности, т.е. три GA модуля.
Появление новых рецепторных белков у бактерий часто связывают с переносом генов, их частей или частей генома. Считается, что эти фрагменты кодируют продукты, являющиеся модульными элементами белков. Так, допускается, что HSA-связывающий GA модуль белка РАВ возник при переносе соответствующего элемента из белка G. Структура GA модулей в G белке и GA модуля в белке РАВ одинакова и состоит из трех спиралей (Johansson M. и др. J.Mol.Biol. 316: 1083-1099 (2002)).
Из штамма Streptococcus canis DG12, выделенного из коровьего молока, получен HSA-связывающий белок, который содержит два GA модуля. Он не связывает IgG, но по способности связывать HSA превосходит белок G (Sjobring U. Infection and Immun. 60: 3601-3608 (1992)). Это свойство делает его привлекательным для практического применения.
Изучение структуры HSA-связывающих белков имеет большое значение для технологии создания белковых реагентов, актуальных в иммунохимии, протеомике, биотехнологии и клинической диагностике.
Замена в иммуноферментном анализе определения микроальбуминурии обычно используемых антител на рецепторный рекомбинантный HSA-связывающий полипептид создает определенные преимущества, поскольку позволяет:
1) исключить трудоемкий процесс приготовления специфических антител (для которого используются лабораторные животные),
2) стандартизовать используемый альбуминовый рецептор, и тем самым стабилизировать всю систему анализа.
Созданный авторами метод рецепторноферментного анализа (РФА) является недорогим, простым в употреблении, чувствительным и специфичным способом определения микроальбуминурии у больных СД.
Созданная тест-система выгодно отличается от зарубежных аналогов тем, что не требует иммунных сывороток, высокоспецифична, достаточно чувствительна, не дает фоновых ложных реакций, доступна в использовании и экономична.
Использование микрочиповой технологии в сочетании с HSA-связывающим полипептидом для определения микроальбуминурии может дать быструю количественную оценку содержания альбумина в моче одновременно у большого количества пациентов и тем самым облегчить проведение скрининга и мониторинга диабетической нефропатии.
Практически все штаммы CTG, как человеческого, так и животного происхождения, продуцируют на своей поверхности клеток белок G, который связывает HSA.
Впервые рекомбинантный HSA рецептор, был получен шведскими авторами путем клонирования фрагмента гена HSA рецептора в непатогенном хозяине. (Nygren Р.-А и др. J. Mol. Recognition. 1: 69-74. (1988)). В данной работе для клонирования АВ области (области, ответственной за связывание HSA) рестрицировали плазмиду pSPG2 ферментом EcoRI с последующей обработкой фрагментом Кленова, добавлением синтетического линкера Sail и лигированием. При рестрикции SalI и PstI был получен фрагмент в 640 н.п., который изолировали из агарозы и вставляли между теми же сайтами в вектор pEML8. Фрагмент гена вырезали из полученной плазмиды, используя EcoRI и HindIII сайты и вставляли в плазмиду pEG, конструкция которой описана в работе Eliasson (Eliasson М. И др. J. Biol. Chem. 263: 4323-4327 (1988)).
В результате такой модификации была получена плазмида pB1B2, ответственная за HSA-связывающую активность. Из субклона, продуцирующего HSA-связывающий белок, был выделен белок. Он был очищен аффинной хроматографией.
Описанный авторами белок оказался гетерогенным с деградацией очищенного продукта. Причиной гетерогенности, очевидно, являлся довольно сложный путь его генетического конструирования. Практического применения из-за своих недостатков этот белок не находит.
В 1996 году был проклонирован фрагмент гена, кодирующий третий GA модуль белка G штамма G148. Полученный белок G148-GA3 был использован для изучения структуры HSA-связывающего модуля. (Kraulis P. и др. FEBS Lett. 378: 190-194 (1996)), т.е. только в исследовательских целях.
В 1992 году из CTG животного происхождения, штамма DG 12, был проклонирован фрагмент гена, кодирующий два HSA-связывающих GA модуля, с использованием плазмидного вектора рКК233-2 и определена его нуклеотидная последовательность. (Sjobring U. Infect, and Immun. 60: 3601-3608. (1992)).
Прототипом изобретения является рекомбинантный HSA-связывающий рецепторный полипептид А1. Он был получен из CTG, штамма G148, выделенного от человека в Отделе молекулярной микробиологии в 1996 году (Орлова С.Н. и др. Биотехнология. 8: 13-21.(1996)). Штамм, продуцирующий полипептид А1, задепонирован в коллекции НИИЭМ СЗО РАМН, №357.
Особенностью этого полипептида является то, что он содержит HSA-связывающую область, охватывающую три GA модуля. Был изучен фрагмент гена, который кодирует полипептид А1.
Изучение этого рецепторного полипептида доказало его способность связываться исключительно с основным белком плазмы человека - HSA. Данное свойство полипептида пытались использовать в диагностических тест-системах, позволяющих определять концентрацию HSA в различных биологических жидкостях, в частности, в моче.
Однако при выделении и очистке рекомбинантного полипептида А1 выход активного реагента в достаточном количестве был затруднен, что привело к необходимости создания нового HSA-связывающего препарата, обладающего большей активностью, чем полипептид А1 белка G.
Задачей данного изобретения стало получение рекомбинантного полипептида А2, обладающего высокой способностью связывать HSA и его использование в диагностике при создании чувствительной и экономически выгодной тест-системы для определения микроальбуминурии у больных СД I и II типов.
Задачей изобретения стало также создание штамма-продуцента рекомбинантного полипептида А2.
Поставленная задача решена конструированием праймеров, направленных к участкам гена, кодирующего HSA-связывающий полипептид А2, в состав которого входят два GA модуля. Их использование в полимеразной цепной реакции (ПЦР) позволило получить искомый фрагмент ДНК, лигированный впоследствии с плазмидным вектором pQE 32 (The QIAexpress System, Qiagen, США). Такой путь сделал возможным облегчить выделение и очистку, созданного авторами рекомбинантного полипептида.
Создание штамма-продуцента, обозначенного как Escherichia coli (Е. coli) M 15-A2, осуществляли путем генетической трансформации на основе штаммов E.coli M 15.
Известно, что штамм E.coli M 15 использовали раньше для получения рекомбинантных полипептидов путем трансформации бактерий плазмидной ДНК (Устинович И.А. Биотехнология. 1: 3-10. (2002)), (Дуплик Н.В. Медицинская иммунология, 11: 7-14. (2009)), (Mandel M. J.Mol.Biol. 53: 154-157. (1970)).
Полученный авторами изобретения штамм-продуцент E.coli M 15-A2 включает плазмиду, содержащую участок последовательности гена, кодирующий HSA-связывающую область (последовательность опубликована Sjobring (Sjobring U. Infect. Immun. 60: 3601-3608. (1992)), ассоциированной с ДНК вектора pQE 32. Ранее такой вариант модификации штамма-продуцента не осуществляли.
Именно такая трансформация бактерий E.coli M 15 привела к появлению способности при определенных условиях индуцировать синтез рекомбинантного полипептида А2. Штамм задепонирован в коллекции НИИЭМ СЗО РАМН, №593.
Изобретением является созданная рекомбинантная ДНК (обозначенная как ра2), полученная в результате ПНР, с использованием хромосомной ДНК штамма DG 13 CTG, выделенного из коровьего молока, праймеров РА1 и РА2 и последующего клонирования с использованием экспрессионной плазмиды pQE 32.
Также изобретением является созданная рекомбинантная ДНК (обозначенной как pQE 32-ра2), несущая рекомбинантную ДНКра2.
Фрагмент гена, кодирующего полипептид А2 размером в 999 н.п., получен в ходе ПНР на основе хромосомной ДНК штамма DG 13 CTG с использованием праймеров РА1 и РА2. Авторами осуществлено клонирование этого фрагмента с использованием экспрессионной плазмиды pQE 32 и последующей трансформацией рекомбинантной плазмидной ДНК в гетерологичной системе E.coli М 15.
Рекомбинантный полипептид А2, полученный в результате индуцибельной экспрессии штаммом-продуцентом E.coli М 15-А2, очищали одноступенчатой аффинной хроматографией на Ni-NTA-агарозе (Qiagen, США). Выход рекомбинантного полипептида А2, полученного из одного литра культуральной среды при выращивании штамма E.coli М 15-А2, составил 40 мг, в то время как выход рекомбинантного полипептида А1 составлял 8 мг, т.е выход А2 в 5 раз выше.
В отличие от известных полипептидов для связывания HSA полипептид А2 стабилен, обладает высокой HSA-связывающей активностью, которая не уменьшается при длительном хранении.
Авторами проведена характеристика взаимодействия HSA с полипептидом А2 в сравнении с взаимодействием HSA с ранее полученным (известным) полипептидом А1.
Установлено, что способность связывать HSA у рекомбинантного полипептида А2 более выражена, чем у полипептида А1. Полипептид А2 способен связать количество HSA в два раза больше, чем полипептид А1.
Технический результат предлагаемого изобретения очевиден: полученный и охарактеризованный рекомбинантный полипептид А2 пригоден для использования в тест-системе для определения уровня альбумина в моче, т.е. для диагностики микроальбуминурии, при диабетической нефропатии, осложнении сахарного диабета.
Свойства нового рекомбинантного полипептида А2 позволяют использовать его также как с целью аффинного выделения HSA, так и для освобождения сыворотки крови от HSA.
Получение рекомбинантного полипептида А2.
Источником хромосомной ДНК послужил штамм DG 13 СГО (выделенный из коровьего молока). ДНК, выделенная фенольно-хлороформной экстракцией, была использована в качестве матрицы в полимеразной цепной реакции (ПНР) с целью получения фрагмента генара2 (999 п.н.) Олигонуклеотидные праймеры представлены в таблице 1.

Подчеркнутые звенья нуклеотидной последовательности указывают на сайты рестрикции.
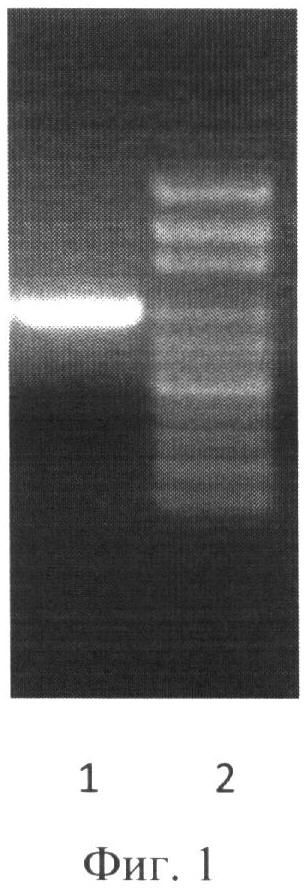
Анализ результатов ПНР осуществлен путем разделения фрагментов ДНК в 1% агарозном геле при помощи горизонтального электрофореза (фиг.1).
Выделение искомого амплифицированного участка ДНК проведено с использованием набора «QIAquick Gel Extraction Kit» (Qiagen, США).
Полученный фрагмент клонировали с использованием экспрессионной плазмиды pQE 32. При подготовке к клонированию была проведена рестрикция выделенного из агарозы фрагмента (999 н.п.) и плазмиды pQE 32 ферментами BamHI и KpnI с образованием липких концов. Продукты рестрикции разделяли с помощью горизонтального электрофореза в 1% агарозном геле, а затем лигировали.
Продуктами лигирования проводили кальциевую трансформацию штамма E.coli M 15. Процесс трансформации осуществляли по методике трансформации кишечной палочки, используя плотные селективные среды, содержащие 50 мкг/мл ампициллина, 25 мкг/мл канамицина. (Maniatis Т, Fritsch E.F., Sambrook J. Molecular cloning: a laboratory manual. - Cold Spring Harbor: Cold Spring Harbor Laboratory Press, 867. (1982)).
Антибиотикоустойчивые трансформанты откалывали на две параллельные чашки Петри с антибиотиками. Колонии с одной чашки переносили на нитроцеллюлозную мембрану и вскрывали раствором, содержащим 0,2 N NaOH, 0,1% SDS и 0,5% β-меркаптоэтанола. Мембрану инкубировали 1 час при комнатной температуре в блокирующем растворе (2 части 3% молока и 1 часть фосфатно-солевого буфера (ФСБ)) для удаления неспецифического связывания и затем в блокирующем растворе, содержащем HSA, меченный пероксидазой хрена (Sigma, USA).
Конъюгирование пероксидазы хрена с HSA проводили периодатным методом (Фримель Г. Иммунологические методы: M. Медицина: 438-439. (1987)).
Затем мембрану последовательно отмывали блокирующим раствором и ФСБ. Пероксидазную активность проявляли раствором парафенилендиамина в концентрации 1 мг/мл с 0,15% перекисью водорода. Клоны-трансформанты, обладающие экспрессией HSA-связывающей активности, представлены на фиг.2.
Из трансформантов с наиболее выраженной экспрессией HSA-связывающей активности выделяли рекомбинантные плазмиды с помощью Mini-prep, plasmid DNA purification kit (Qiagen, USA). Наличие в них рекомбинантной ДНК подтверждали в ПЦР с исходными праймерами РА1 и РА2. Плазмидную ДНК pQE 32-ра2 использовали как матрицу в ПНР с праймерами pQE1 и pQE2. Амплификат выделяли после электрофореза в 1% агарозном геле и секвенировали.
Итоговые данные совпали с известной нуклеотидной последовательностью, кодирующей HSA-связывающий полипептид с двумя GA модулями. (Sjobring U. Infect, and Immun. 60: 3601-3608. (1992)).
На фиг.3 представлена нуклеотидная последовательность, состоящая из 999 п.н. рекомбинантной ДНКра2 и 39 п.н. плазмиды pQE 32
На фиг.4 представлена аминокислотная последовательность, состоящая из 346 аминокислотных остатков, в которой 333 аминокислоты полипептида А2 кодируются рекомбинантной ДНК ра2 и первые 13 аминокислот - плазмидой pQE 32.
Культуру штамма Е. coli М 15, содержащую рекомбинантную плазмиду pQE 32-ра2, обозначенную как Е. coli М 15-А2, культивировали в жидкой среде BHI (Brain Heart Infusion Broth, Gibco, США) с добавлением антибиотиков (ампициллин (100 мкг/мл) и канамицин (25 мкг/мл)) до поздней логарифмической фазы роста (OD600=0,7-0,9). Затем экспрессию рекомбинантного белка индуцировали добавлением изопропил-бета-D-тиогалактопиранозида (IPTG), и клетки культивировали еще 4 часа. После этого клетки осаждали центрифугированием, отмывали лизирующим буфером А (20 мМ Na2HPO4, 20 мМ NaH2PO4, 500 мМ NaCl, 20 мМ имидазола, рН 8,0) и суспендировали в том же буфере, добавляя ингибитор протеаз, фенилметилсульфонилфторид (ФМСФ), до концентрации 1 мМ. Суспензию клеток вскрывали ультразвуком и центрифугировали. Надосадочную жидкость наносили на колонку, заполненную Ni-NTA-агарозой (Qiagen, USA). После того, как белок связывался с Ni-NTA-агарозой, колонку промывали буфером А для удаления неспецифически связавшихся белков. Рекомбинантный белок элюировали буфером Б (20 мМNa2HPO4, 20 мМ NaH2PO4 500 мМ NaCl, 250 мМ имидазола, рН 8,0). После аффинной хроматографии белок диализовали 18 часов против дистиллированной воды.
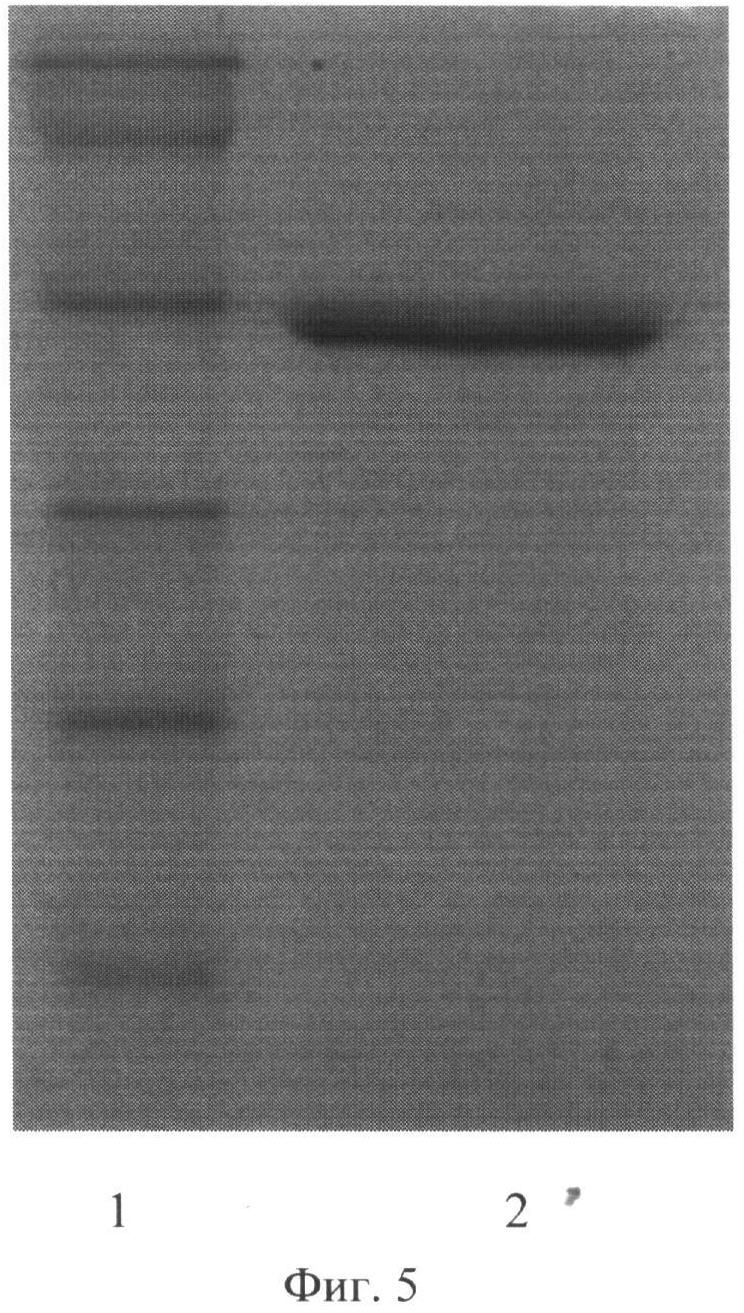
Выделенный рекомбинантный полипептид анализировали методом SDS электрофореза в полиакриламидном геле, который позволил сделать заключение об удовлетворительном качестве очистки полипептида и также об его молекулярной массе, сравнивая пробег полипептида А2 с пробегом белков известной молекулярной массы (Precision Plus Protein standards (161-0373), Bio-Rad, США). Молекулярная масса полипептида А2 оказалась равной (48,0±0,5) кДа (фиг.5).
Таким образом, рекомбинантный полипептид А2 содержит аминокислотную последовательность HSA-связывающего полипептида CTG штамма DG 13, содержащую 333 аминокислоты, ковалентно связанную с 13 аминокислотными остатками, кодируемыми pQE 32.
Изучение особенностей взаимодействия рекомбинантного полипептида А2 с HSA при сравнении с аналогичной реакцией с полипептидом A1.
Авторами получена иммунохимическая характеристика рекомбинантной молекулы А2, представляющей собой HSA-связывающий полипептид. Результаты работы позволяют расширить область применения полипептида А2, дополнив ее использованием при создании диагностикумов, для аффинного выделения HSA и для освобождения сыворотки крови от HSA.
Структурные различия рекомбинантных полипептидов A1 и А2 делают рекомбинантный полипептид А2 более предпочтительным во всех случаях использования.
Проведено сопоставление рецепторной активности рекомбинантных полипептидов A1 и А2 в отношении HSA, представленного следующими образцами: 1) HSA - поликлональный сывороточный альбумин человека меченный пероксидазой хрена. 2) Поликлональный немеченый HSA.
Установлено, что полипептид А2, также как и полипептид А1, обладает способностью к селективному связыванию с HSA в любом из использованных вариантов постановки ИФА (прямой ИФА, непрямой ИФА, ингибиторный ИФА, конкурентное ингибирование). Структурные различия между двумя полипептидами, реализованные методами молекулярной генетики, способствуют существенной дифференцировке рекомбинантных полипептидов А1 и А2 по HSA-связывающим свойствам.
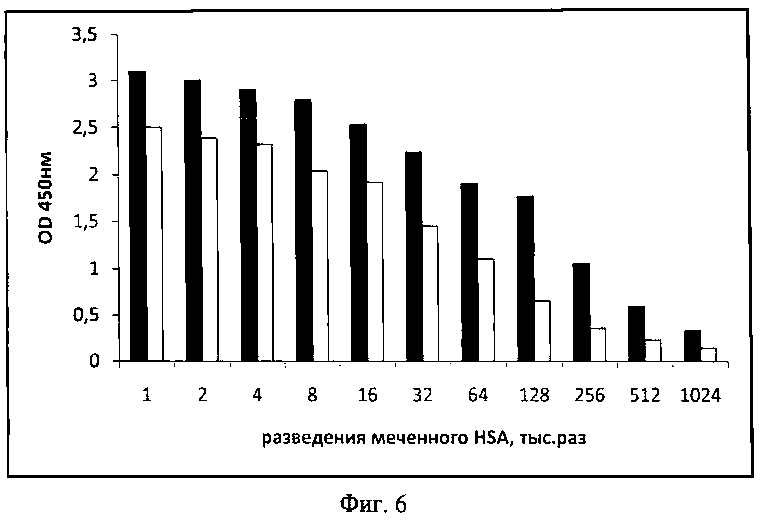
В случае прямого ИФА полипептиды А1 и А2 были иммобилизованы на твердой фазе. При этом оценивали количество связавшегося с ними меченного HSA. На фиг.6 показана гистограмма, отражающая сопоставление HSA-связывающей активности полипептидов А1 и А2. Исследуемые полипептиды А1 и А2 обладают HSA-связывающей активностью в отношении меченного HSA, причем количество HSA, связанного полипептидом А2 выше, чем полипептидом А1 в два раза. Для анализа, проведенного в одинаковых условиях, увеличение связывающей активности в 2 раза, весьма значимо.
Полипептиды А1 и А2, созданные на основе разных видов стрептококков, несомненно имеют, структурные различия, определяющие HSA-рецепторную активность каждого из них. Можно допустить, что на реализацию этой способности влияют и условия постановки ИФА, а именно - нахождение полипептида на твердой фазе или в растворе. Влияние этого фактора объясняется наличием у пептидов пространственной конфигурации, которая неодинакова для молекул, свободно циркулирующих в растворе или фиксированных на твердой фазе.
Изменение конформации гипотетически может привести к увеличению или уменьшению способности молекул связывать HSA. Для проверки этого предположения использовали непрямой и ингибиторный ИФА.
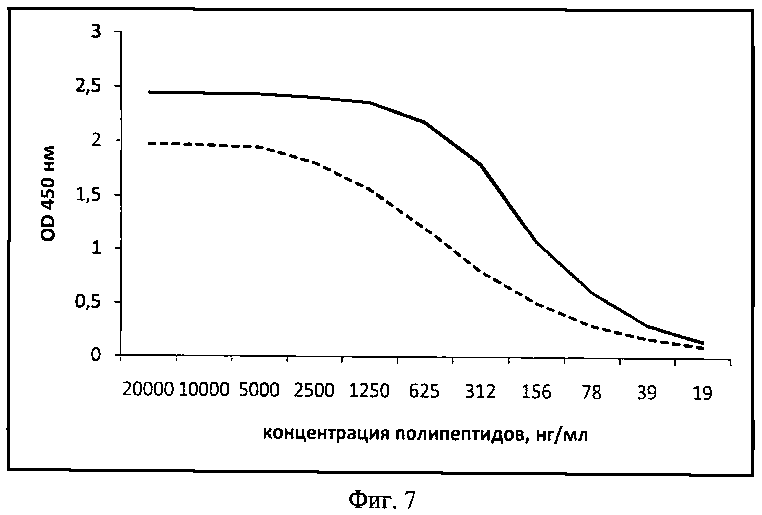
При постановке непрямого ИФА результаты показали, что полипептиды в растворе связывают HSA. Однако А2 связывает HSA с активностью в два раза больше чем А1. (фиг.7).
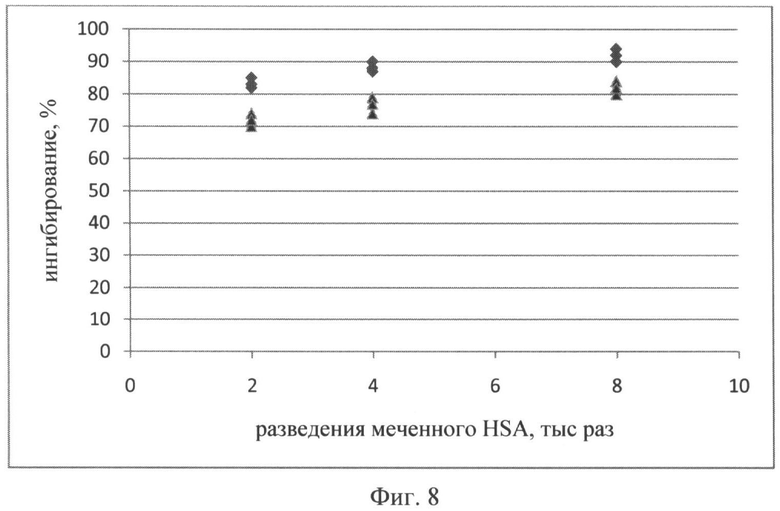
Ингибиторный ИФА показал, что полипептиды А1 и А2, находясь в растворе, препятствовали взаимодействию меченного HSA с адсорбированными молекулами А1 и А2 в пределах 70-94% (фиг.8).
Отмечены особенности в поведении белков в условиях ИФА. Полипептиды А1 и А2 способны вступать во взаимодействие с HSA как в растворе (ингибиторный и непрямой ИФА), так и в адсорбированном состоянии (прямой ИФА). Это свойство позволяет использовать их в качестве иммунохимических реагентов в ИФА, причем, как в иммобилизованном виде, так и в роли выявляющих молекул при наличии ферментной метки.
При этом наибольшая степень связывания отмечена у белка А2 в условиях прямого и непрямого ИФА, несмотря на то, что полипептид А1 имеет три, а полипептид А2 - только два HSA-связывающих GA модуля. Это можно объяснить тем, что все три GA модуля полипептида А1 имеют практически одинаковую последовательность, а GA модули полипептида А2 содержат последовательности с меньшей гомологией между собой и за счет этого отличия более активно связывают HSA.
Положение сайтов связывания антител к HSA и к рекомбинантному белку на молекуле HSA изучали в ингибиторном варианте ИФА. Антитела к HSA адсорбировали на планшет, а затем к ним добавляли заранее приготовленную смесь из меченного HSA и полипептидов А1 или А2 (в соотношении 1:1, инкубация при комнатной температуре в течение двух часов). Инкубацию иммобилизованных антител со смесью полипептидов и меченного HSA проводили в течение 1 часа при 37°С.
О влиянии рекомбинантных полипептидов на взаимодействие меченного HSA с антителами судили по пероксидазной активности ферментной метки HSA, связавшегося с адсорбированными антителами.
Предварительное взаимодействие HSA с полипептидами не препятствовало последующему его связыванию с антителами, из чего следует, что сайты связывания антител и рекомбинантного полипептида А1 или А2 на молекуле HSA полностью независимы.
Одновременное связывание с молекулой HSA двух реагентов открывает возможность создания многоступенчатых тест-систем.
Способ определения экскреции HSA с мочой, т.е. создание тест-системы для определения микроальбуминурии.
Проведенные исследования по взаимодействию полипептидов А1 и А2 с HSA показали, что рекомбинантные молекулы обладают HSA-связывающей способностью. Это свойство послужило основой для создания способа для определения концентрации HSA в моче.
Учитывая больший выход рекомбинантного полипептида А2 в процессе экспрессии белка в Е. coli и его более высокую способность по сравнения с полипептидом А1 связывать HSA, он был выбран для создания диагностических тест-систем для качественного выявления и количественного определения микроальбуминурии.
Для создания количественной тест-системы предпочтителен конкурентный метод ИФА, так как этот метод содержит минимальное число операций, требует незначительного расхода реагентов и легко может быть автоматизирован.
Обычно используемые в ИФА антитела к альбумину были заменены рекомбинантным рецепторным полипептидом А2, а сам иммуноферментный принцип был трансформирован в РФА. Такая замена создала определенные преимущества, поскольку позволила отстроиться от трудоемкого процесса приготовления специфических иммунных сывороток, стандартизовать используемый рекомбинантный полипептид и, тем самым, унифицировать всю систему анализа. Кроме того, применение рекомбинантного рецепторного полипептида позволило избежать фоновых реакций, возможных для иммунологических реакций.
Ранее такой подход для анализа альбумина не применялся.
Принцип РФА метода заключается в следующем: на твердую поверхность планшета иммобилизовывается полипептид А2, на который наносятся анализируемые пробы мочи одновременно с меченным HSA. При такой постановке альбумин пробы и меченный HSA конкурируют за HSA-связывающий полипептид, иммобилизованный на твердой фазе. Концентрация альбумина в анализируемой пробе обратно пропорциональна ферментативной активности твердой фазы.
При разработке тест-системы были подобраны условия анализа, обеспечивающие необходимую чувствительность и специфичность. В результате проведенных экспериментов было выбрано рабочее разведение меченного HSA 1:8000 при концентрации иммобилизованного полипептида А2 от 0,1 до 0,25 мкг/мл. По данным последовательных разведении немеченого HSA строится калибровочная кривая.
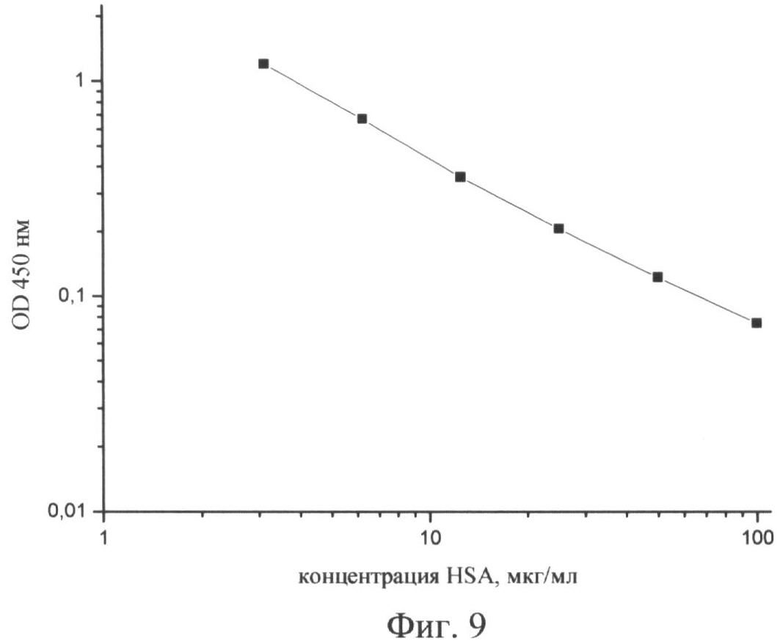
После подбора оптимальных условий строили калибровочную кривую зависимости оптической плотности от концентрации HSA в стандартных пробах (3,125; 6,25; 12,5; 25; 50 и 100 мкг/мл) (фиг.9). Концентрацию HSA в моче находили по значениям оптической плотности на калибровочной кривой.
Таким образом, удалось показать, что рекомбинантный полипептид А2 может быть использован не только для качественного анализа, но и в качестве высоко специфичного реагента для количественного определения альбумина в моче. Минимальная концентрация HSA, которую можно определить в моче, составила 1-2 мкг/мл, а максимальная - 100 мкг/мл. Это очень чувствительный метод детекции. Мочу с большей концентрацией альбумина следует разводить раствором ФСБ. В результате чувствительность такого способа определения позволяет выявлять количество альбумина в моче в пределах, включающих экскрецию альбумина с мочой в норме, т.е. до 20 мкг/мл и в пределах микроальбуминурии от 20 до 200 мкг/мл. Метод является высокоточным.
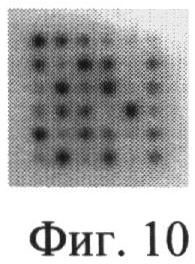
При скрининге и мониторинге больных сахарным диабетом требуется одновременный анализ нескольких десятков образцов. Для решения этой задачи показана возможность создания способа, в котором сочетается использование стабильного высокоспецифичного рекомбинантного HSA-связывающего полипептида и технология микрочипов. В качестве матрицы для нанесения проб мочи пациентов с диабетической нефропатией была выбрана нитроцеллюлозная мембрана. На нитроцеллюлозную мембрану, размером 1 см2 были нанесены 30 проб мочи, был использован автоматический калибратор. Первый ряд на мембране соответствовал разведениям стандартного препарата HSA для построения калибровочной кривой (100; 50; 25; 12,5; 6,25; 3,125 мкг/мл). Мембрана была проинкубирована в блокирующем растворе (2% молоко, разведенное в ФСБ) с меченным пероксидазой HSA-связывающим полипептидом А2. Затем мембрану промывали и окрашивали раствором парафенилдиамина с перекисью водорода. На мембране четко проявлялись пятна, разная степень окрашивания которых соответствовала различным концентрациям альбумина в стандартных пробах и пробах мочи (фиг. 10). Имея считывающее устройство и соответствующую компьютерную программу, можно получать количественную оценку альбумина в моче, причем одновременно в большом количестве проб.
Получение HSA из сыворотки крови аффинной хроматографией и освобождение сыворотки крови от HSA.
Сорбентом служил Affi-Gel 10 (Bio-Rad, США). Была приготовлена колонка, в которой сорбент содержал присоединенный рекомбинантный полипептид А2. Колонку промывали сначала бикарбонатным буфером с 0,5 % NaCl, рН 8,0, затем 0,1 М глициновым буфером, рН 2,2 и снова бикарбонатным буфером с 0.5 % NaCl. Колонку уравновешивали ФСБ, рН 7,4 и хранили при 4°.
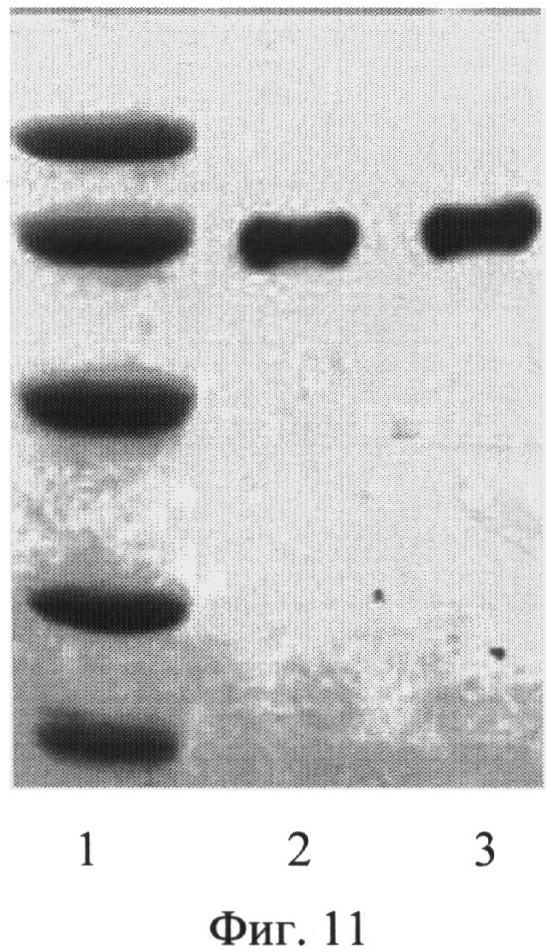
Пробу сыворотки крови человека наносили на колонку Affi-Gel 10-рекомбинантный полипептид А2. Несвязавшиеся белки собирали при промывании колонки раствором ФСБ. Элюцию связавшегося с сорбентом HSA проводили ОД М глициновым буфером, рН 2,2. Элюат собирали фракциями по 500 мкл, концентрацию белка определяли спектрофотометрически по значениям оптической плотности (OD) при длине волны 280 нм. Фракции с наибольшим значением OD объединяли и рН доводили до 7,5 2 N раствором щелочи. Пробы исходной сыворотки, пробы белков сыворотки, не связавшихся с сорбентом на колонке, и пробу, элюированную с колонки, проверяли на наличие HSA с помощью метода электрофореза в полиакриламидном геле (SDS-PAGE).
SDS-PAGE показал, что сыворотка после пропускания через колонку освобождалась от HSA. Освобождение сыворотки от HSA позволяет обнаружить белки, ранее экранированные HSA. В то же время в элюате находился HSA, который был получен в высокоочищенном состоянии (фиг. 11).
Таким образом, полученный нами сорбент с рекомбинантным полипептидом А2 позволяет выявлять белки, находящиеся в низких концентрациях, после освобождения сыворотки от HSA и получать в одну стадию очищенный препарат HSA.
Таким образом, результатом изобретения является:
1. Рекомбинантная ДНК ра2, нуклеотидная последовательность которой составляет 999 п.н.
2. Рекомбинантная ДНК pQE 32-ра2, представляющая собой плазмиду pQE 32, несущая рекомбинантную ДНК ра2 и обеспечивающая получение полипептида А2.
3. Штамм Е. coli М 15-А2, трансформированный рекомбинантной плазмидной ДНК pQE 32-ра2 и экспрессирующий рекомбинантный полипептид А2.
4. Рекомбинантный полипептид А2, имеющий аминокислотную последовательность, представленную на фиг.4 и обладающий способностьюселективно связывать HSA, причем полипептид А2 связывает HSA как в растворе, так и в адсорбированном состоянии.
5. Способность полипептида А2 селективно связывать HSA позволила использовать его в тест-системах для качественного выявления и количественного определения микроальбуминурии.
6. Способность полипептида А2 селективно связывать HSA позволила получить высокоочищенный препарат HSA из сыворотки крови аффинной хроматографией и освободить сыворотку крови от HSA, тем самым позволив определять другие белки крови, присутствующие в более низких концентрациях.
Ниже приводятся конкретные примеры, иллюстрирующие некоторые варианты изобретения, но не ограничивающие его.
Пример 1. Получение фрагмента ДНК/ра2 методом ПЦР.
К 0,25 мкг геномной ДНК, выделенной фенольно-хромосомной экстракцией из штамма DG 13 CTG добавляли по 10 микромолей каждого из специфических праймеров, фланкирующих исследуемую последовательность, буфер с магнием для полимеразы, по 0,2 мМ четырех дезоксирибонуклеотидтрифосфатов, объем доводили водой до 25 мкл и добавляли 0,5 мкл термостабильной ДНК полимеразы. На поверхность жидкости наслаивали 40 мкл минерального масла. Пробирки помещали в амплификатор и инкубировали при 94°C 2 мин. Программа ПЦР состояла из: денатурации при 94°C - 30 сек, отжига праймеров - 60°C - 1 мин и синтеза - 72°С - 1 мин. Этот цикл повторялся 30 раз, после чего смесь инкубировалась при 72°С 10 мин. В работе использовали олигонуклеотидные праймеры, приведенные в Таблице 1. ПНР продукты разделяли в 1% агарозном геле в горизонтальном электрофорезе (фиг.1). Выделение амплифицированного участка ДНК проводили с использованием набора «QIAquick Gel Extraction Kit» (Qiagen, США). Анализ размера полученного фрагмента ДНК проводили, исходя из сравнения его электрофоретической подвижности с электрофоретической подвижностью маркера молекулярных весов (100 п.н. ДНК-маркер, Хеликон).
На фиг.1 (Электрофореграмма амплифицированного ДНК- фрагмента ра2):
1 - продукт ПЦР
2 - 100 п.н. ДНК - маркер, (сверху вниз: 3000, 2000, 1000, 900, 800, 700, 600, 500, 400, 300, 200 и 100 нуклеотидных пар).
В результате ПНР с праймерами РА1 и РА2 был получен фрагмент 999 п.н., что продемонстрировано на фиг.1.
Пример 2. Клонирование ДНК-фрагмента ра2 с использованием экспрессионной плазмиды pQE 32.
Плазмида pQE 32 и фрагмент, полученный в результате ПНР были обработаны двумя рестрикционными ферментами BamHI и KpnI, что привело к образованию липких концов. Продукты рестрикции разделяли с помощью горизонтального электрофореза в 1% агарозном геле, а затем выделяли из агарозы с помощью набора «QIAquick Gel Extraction Kit» (Qiagen, США). В ходе клонирования к 30 мкл фрагмента ДНК ра2, добавляли 2 мкл фрагмента ДНК pQE 32, полученных после рестрикции и вырезанных из агарозы в соотношении концентраций 5:1, 5 мкл десятикратного лигазного буфера и 1 мкл лигазы фага Т4. Объем доводили дистиллированной водой до 50 мкл. Смесь инкубировали при 10° в течение 12 часов. В результате проведенного клонирования была получена рекомбинантная плазмида, обозначенная как pQE 32-ра2, несущая рекомбинантную ДНК (обозначенную как ра2), состоящую из 999 н.п. и 39 н.п. плазмиды pQE 32.
Пример 3. Трансформация культуры Е. coli M 15 плазмидной ДНК pQE 32-ра2.
Рекомбинантную плазмиду трансформировали в гетерологичную систему Е. coli M 15. В качестве положительного контроля параллельно проводили трансформацию Е. coli M 15 исходной плазмидной ДНК pQE 32. Генетическим маркером плазмидной ДНК pQE 32 является маркер amp, кодирующий бета-лактомазу, что обеспечивает устойчивость к ампициллину клеток, несущих эту плазмиду. Культуру клеток Е. coli M 15 культивировали в бульоне BHI (Brain Heart Infusion Broth, Gibco, США) с канамицином (25 мкг/мл) в течение 12 часов при 37° и интенсивном перемешивании. Затем инокулят пересевали на новый объем той же среды (на 10 мл среды - 0,1 мл инокулята) и инкубировали при 37° в течение 2-3 часов при перемешивании до ОД600=0,3. Выращенные клетки в объеме 1,5 мл центрифугировали в течение 1 мин. при 12000 об/мин., и полученный осадок суспендировали в 200 мкл раствора 0,1 М CaCl2. Далее смесь выдерживали во льду 1 час. После центрифугирования осадок ресуспендировали в 187, 5 мкл 0,1 М CaCl2 и 64,5 мкл дистиллированной воды. Затем в пробирку вносили 0,2 мкг плазмидной ДНК и инкубировали смесь во льду 1 час. Далее смесь выдерживали 2 мин. при 42°C и снова переносили смесь в лед на 10 мин. После этого к смеси добавляли 1 мл бульона BHI с канамицином (25 мкг/мл) и инкубировали 1 час при 37°C. После осаждения центрифугированием (8000 об/мин, в течение 1 мин) клетки высевали на чашки с 1% L-агаром (Difco, США), содержащим ампициллин (100 мкг/мл) и канамицин (25 мкг/мл). Через 18 часов роста клеток при 37°C проводили отбор клонов-трансформантов. Антибиотикоустойчивые трансформанты переносили на две параллельные чашки Петри с антибиотиками. Колонии с одной чашки переносили на нитроцеллюлозную мембрану и вскрывали раствором, содержащим 0,2 N NaOH, 0,1% SDS и 0,5% β-меркаптоэтанола. Мембрану инкубировали 1 час в блокирующем растворе, содержащем HSA, меченный пероксидазой хрена (Sigma, США). Затем ее последовательно отмывали блокирующим раствором и раствором ФСБ. Пероксидазную активность проявляли раствором парафенилендиамина в концентрации 1 мкг/мл с 0, 15 М перекисью водорода (фиг.2).
На фиг.2 (Экспрессия HSA-связывающей активности колоний, содержащих рекомбинантный полипептид А2) показана ярко выраженная экспрессия HSA-связывающей активности отдельных клонов.
Таким образом, полученные данные позволяют сделать вывод о том, что в результате проведенного клонирования были получены рекомбинантные клоны, несущие плазмиды pQE 32-pa2 с рекомбинантной ДИК ра2.
Пример 4. Очистка рекомбинантного белка А2.
Культуру штамма Е.coli M 15-A2 выращивали в бульоне BHI с добавлением ампициллина в концентрации 100 мкг/мл и канамицина в концентрации 25 мкг/мл в течение ночи при интенсивном перемешивании. Затем клетки пересевали на 800 мл той же среды и инкубировали при 37°C в течение 2-3 часов при интенсивном перемешивании до ОД600=0,7 - 0,9. Экспрессию рекомбинантного белка индуцировали добавлением раствора изопропил-бета-D-тиогалактопиранозида до конечной концентрации 2 мМ, после чего клетки инкубировали при тех же условиях еще 4 часа. Полученную суспензию клеток центрифугировали при 4000 об/мин. 20 мин. Надосадочную жидкость сливали, а клетки суспендировали в буфере А ((20 мМ Na2HPO4, 20 мМ NaH2PO4, 500 мМ NaCl, 20 мМ имидазола, рН 8,0), добавляя ингибитор протеаз фенилметилсульфонилфторид (ФМСФ) до конечной концентрации 1 мМ. Для лизирования клеток была использована трехкратная ультразвуковая обработка при 4°C в течение 20 сек. с перерывом в 40 сек. в ультразвуковом дезинтеграторе (УЗДН-1У4.2, Англия). Лизат клеток центрифугировали при 20000 об/мин, и 4°C в течение 20 мин. Надосадочную жидкость пропускали через 0,45 мкм фильтр (Millipore, США) и затем ее наносили на колонку с Ni-сефарозой (Qiagen, США), предварительно уравновешенную буфером А. Далее колонку промывали тем же буфером до тех пор, пока значения ОД280 выходящего раствора не были больше, чем 0,01. Белок элюировали раствором буфера Б (20 мМ Na2HPO4, 20 мМ NaH2PO4, 500 мМ NaCl, 250 мМ имидазола, рН 8,0). Элюат собирали по фракциям по 500 мкл и измеряли в них значения ОД280. Фракции с наибольшими значениями ОД280, объединяли и диализовали против дистиллированной воды в течение 12 часов. На фиг.5 представлена электрофореграмма рекомбинантного полипептида А2.
На фиг.5. (Электрофореграмма рекомбинантного полипептида А2):
1 - маркер молекулярной массы, (сверху вниз: 113, 92, 52, 35, 28 и 21 кДа).
2 - препарат очищенного белка А2
Молекулярную массу полипептида А2 определяли, сравнивая пробег белка А2 с пробегом белков известной молекулярной массы (Precision Plus Protein standarts (161-0373), Bio-Rad, США). Молекулярная масса белка А2 оказалась равной (48±0,5) кДа.
Пример 5. Связывание сывороточного меченного HSA адсорбированными полипептидами А1 и А2.
Анализ HSA-связывающей активности проведен методом прямого ИФА. Для проведения ИФА использовали планшеты NUNC MaxiSorb (Danemark).
Приготовление разведенных препаратов меченного HSA осуществляли с применением раствора ФСБ. Удаление несвязавшихся реагентов проводили трехкратным промыванием планшетов раствором ФСБ с 0,05% твин 20 (ФСБТ).
Полипептиды А1 и А2, разведенные в ФСБ, рН 8,0 до 2 мкг/мл, адсорбировали на планшет в течение 18 часов при 4°C. После трехкратного отмывания в планшеты вносили разведенный меченный HSA. Инкубацию проводили при 37°C в течение 1 часа. Далее планшеты 3 раза отмывали от несвязавшихся реагентов. Активность фермента-метки пероксидазы хрена определяли с использованием хромогена - тетраметилбензидина (ТМБ), растворенного в 0,1 М цитрат-фосфатном буфере, рН 5,0, с добавлением перекиси водорода. Реакцию останавливали добавлением 2N серной кислоты. Оптическую плотность определяли при длине волны 450 нм на мультискане.
Установлено, что полипептид А2 имеет более высокую HSA-связывающую активность, чем полипептид А1, что продемонстрировано на фиг.6. Разница в активности составляет: у А2 активность выше, чем у А1 в 2 раза.
На фиг.6. (Сравнение эффективности связывания полипептидов А1 и А2 с HSA):
белый столбик - полипептид А1
черный столбик - полипептид А2
по оси абсцисс - разведения меченного HSA
по оси ординат - оптическая плотность (OD450).
Пример 6. Связывание сывороточного меченного HSA полипептидами А1 и А2 в растворе.
В случае непрямого ИФА на твердой фазе адсорбировали немеченый HSA, к которому из раствора присоединялись молекулы А1 или А2, в свою очередь связывающие из раствора меченный пероксидазой HSA.
Для проведения ИФА использовали планшеты NUNC MaxiSorb (Danemark).
Приготовление разведенных препаратов меченного HSA осуществляли с применением раствора ФСБ. В этом случае сенсибилизировали HSA, разведенный в ФСБ, в течение 18 ч при 4°C. Несвязавшийся HSA отмывали три раза добавлением по 150 мкл ФБСТ в каждую лунку и добавляли двукратные разведения рекомбинантного полипептида, начиная с 20 мкг/мл, инкубировали 1 час при 37°C и вновь три раза отмывали ФБСТ. Полипептид, связавшийся с HSA, определяли с помощью конъюгата HSA с пероксидазой. Результаты регистрировали, как при прямом методе.
В опытах параллельно использовали не менее четырех рядов титрования. Результаты показали, что полипептиды А1 и А2 в растворе связывают HSA. Однако А2 связывает HSA с большей активностью, чем А1 (фиг.7). Активность у А2 выше, чем у А1 в 2 раза.
На фиг.7. (Сравнение эффективности связывания в растворе полипептидов А1 и А2 с HSA):
пунктирная линия - полипептид А1
сплошная линия - полипептид А2
по оси абсцисс - концентрация полипептидов, нг/мл
по оси ординат - оптическая плотность (OD450).
Для постановки ингибиторного ИФА полипептиды А1 и А2 в концентрации 1 мкг/мл были иммобилизованы на планшете. К ним добавляли заранее проинкубированную смесь соответствующего полипептида и разведенного меченного HSA (в соотношении 1:1, инкубация при комнатной температуре в течение двух часов) Инкубацию иммобилизованных полипептидов со смесью полипептидов и меченного HSA проводили в течение 1 часа при 37°C. По значениям оптической плотности рассчитывали процент ингибирования взаимодействия меченного HSA с адсорбированными молекулами А1 и А2 каждым из полипептидов в растворе (фиг.8). Полипептиды А1 и А2, находясь в растворе, препятствовали взаимодействию меченного HSA с адсорбированными молекулами А1 и А2 в пределах 70-94%.
На фиг.8. (Сравнение эффективности связывания HSA адсорбированными полипептидами А1 и А2 и полипептидами А1 и А2 в растворе):
треугольники - полипептид А1
ромбы - полипептид А2
по оси абсцисс - разведения меченного HSA
по оси ординат - ингибирования, %
Пример 7. Способ определения микроальбуминурии.
Количественное определение микроальбуминурии методом РФА проводили следующим образом: на твердую поверхность полистиролового планшета иммобилизовывали полипептид А2, на который наносили анализируемые пробы мочи одновременно с меченным HSA. При такой постановке альбумин пробы и меченный HSA конкурируют за HSA-связывающий полипептид, иммобилизованный на твердой фазе. Концентрация альбумина в анализируемой пробе обратно пропорциональна ферментативной активности твердой фазы. Условия анализа, обеспечивающие необходимую чувствительность и специфичность, следующие: в результате проведенных экспериментов было выбрано рабочее разведение меченного HSA 1:8000 при концентрации иммобилизованного полипептида А2 от 0,1 до 0,25 мкг/мл. После подбора оптимальных условий строили калибровочную кривую зависимости оптической плотности от концентрации HSA в стандартных пробах (3,125; 6,25; 12,5; 25; 50 и 100 мкг/мл) (фиг.9). Концентрацию альбумина в моче находили по значениям оптической плотности на калибровочной кривой.
На фиг.9. (Стандартна кривая зависимости оптической плотности от концентрации HSA):
по оси абсцисс - концентрация HSA, мкг/мл
по оси ординат - оптическая плотность (OD450).
Пример 8. Создание микрочипа для определения микроальбуминурии
В качестве матрицы микрочипа для нанесения проб мочи пациентов с диабетической нефропатией была выбрана нитроцеллюлозная мембрана. На нитроцеллюлозную мембрану, размером 1 см2, были нанесены 30 проб мочи, используя автоматический калибратор. Первый ряд на мембране соответствовал разведениям стандартного препарата HSA (6 двукратных разведении HSA, начиная со 100 мкг/мл) для построения калибровочной кривой. Мембрана была проинкубирована в блокирующем растворе (2% молоко, разведенное в ФСБ) с меченным пероксидазой HSA-связывающим полипептидом А2. Затем мембрана промывалась и окрашивалась раствором парафенилдиамина с перекисью водорода. На мембране четко проявлялись пятна, разная степень окрашивания которых соответствовала различным концентрациям альбумина в стандартных пробах и пробах мочи (фиг.10).
На фиг.10. Нитроцеллюлозная мембрана с проявленными пятнами, которые соответствуют концентрации HSA в пробах мочи пациентов:
первый ряд - разведения стандартного препарата HSA
остальные ряды - пробы мочи пациентов
Пример 9. Получение высокоочищенного препарата HSA из сыворотки крови аффинной хроматографией и освобождение сыворотки крови от HSA. Одновременное освобождение сыворотки крови от HSA и получение высокоочищенного препарата HSA проводили следующим образом: на колонку, заполненную сорбентом Affi-Gel 10 с присоединенным к нему рекомбинантным полипептидом А2, наносили пробу сыворотки крови. Несвязавшиеся белки собирали при промывании колонки раствором ФСБ. Элюцию связавшегося с сорбентом HSA проводили 0,1 М глициновым буфером, рН 2,2. Элюат собирали фракциями по 500 мкл, концентрацию белка определяли спектрофотометрически по значениям оптической плотности (OD) при длине волны 280 нм. Фракции с наибольшим значением OD объединяли и рН доводили до 7,5 2 N раствором щелочи. Пробы исходной сыворотки, пробы белков сыворотки, не связавшихся с сорбентом на колонке и пробу, элюированную с колонки, проверяли на наличие HSA с помощью метода электрофореза SDS-PAGE. SDS-PAGE показал, что сыворотка после пропускания ее через колонку, освобождалась от HSA. В то же время в элюате находился HSA, который был получен в высокоочищенном состоянии (фиг. 11). Освобождение сыворотки от HSA позволяет обнаружить белки, ранее экранированные HSA. На фиг. 11. (SDS-PAGE очищенного препарата HSA):
1 - маркер молекулярной массы, (сверху вниз 97, 66, 36, 21 и 14
кДа)
2 - очищенный препарат HSA
3 - коммерческий препарат HSA (Sigma)


Изобретение относится к области микробиологии и молекулярной генетики и касается рекомбинантного полипептида А2, ДНК, его колирующей, штамма продуцирующего полипептид А2 и способов использования такого рекомбинантного полипептида. Представленный рекомбинантный полипептид А2, характеризуется аминоксилотной последовательностью из 346 аминокислот, в которой первые 13 аминокислот кодируются последовательностью плазмиды pQE 32 и ковалентно связаны с последующими 333 аминокислотами, кодируемыми последовательностью HAS-связывающего фрагмента хромосомной ДНК штамма DG 13 стрептококков группы G-CTG. Представленная группа изобретений может быть использована в диагностике, например, при создании тест-системы по определению микроальбуминурии - основного лабораторного критерия доклинической стадии диабетической нефропатии, а также в качестве реагента для выделения человеческого сывороточного альбумина аффинной хроматографией и для освобождения сыворотки крови от HAS, что позволит определять другие белки, присутствующие в сыворотке в более низких концентрациях. 7 н. и 2 з.п. ф-лы, 11 ил., 1 табл., 9 пр.
1. Рекомбинантный полипептид А2, обладающий способностью селективно связывать человеческий сывороточный альбумин - HSA и характеризующийся аминокислотной последовательностью из 346 аминокислот, представленной на фиг.4, в которой первые 13 аминокислот, кодируются последовательностью плазмиды pQE 32, ковалентно связанные с последующими 333 аминокислотами, кодируемыми последовательностью HSA - связывающего фрагмента хромосомной ДНК штамма DG 13 стрептококков группы G-СГG.
2. Рекомбинантная ДНК ра2, нуклеотидная последовательность которой представлена на фиг.3, кодирует 333 аминокислоты полипептида А2 по п.1, начиная с 14 аминокислоты его последовательности, причем первые 39 п.н. на фиг.3 принадлежат последовательности плазмиды pQE 32, ковалентно связанной с ДНК ра2.
3. Рекомбинантная плазмидная ДНК pQE 32-ра2, представляющая собой плазмидную ДНК pQE 32, несущую рекомбинантную ДНК по п.2 и обеспечивающая получение полипептида А2.
4. Штамм E.coli M15, трансформированный рекомбинантной плазмидной ДНК pQE 32-ра2 по п.3 и экспрессирующий полипептид А2 по п.1.
5. Способ количественного определения HSA в моче, включающий построение калибровочной кривой зависимости оптической плотности от концентрации HSA в стандартных пробах 100; 50; 25; 12,5; 6,25; 3,125 мкг/мл, иммобилизацию полипептида А2 по п.1 на твердую фазу планшета, инкубирование, нанесение на него анализируемых проб мочи одновременно с меченным пероксидазой хрена HSA в разведении 1:8000, проявку тетраметилбензидином (ТМБ), измерение оптической плотности и установление количества HSA в пробах по значениям калибровочной кривой, причем для определения в моче HSA при концентрации более 100 мкг/мл пробы разводят раствором фосфатно-солевого буфера.
6. Способ определения HSA в моче по п.5, отличающийся тем, что твердой фазой является поверхность полистиролового планшета.
7. Способ качественного определения HSA в моче, характеризующийся тем, что определение осуществляют путем нанесения 30 проб мочи на нитроцеллюлозную мембрану размером 1 см2 с первым рядом разведений стандартного HSA 100; 50; 25; 12,5; 6,25; 3,125 мкг/мл, инкубации проб в блокирующем растворе 2% молока в фосфатно-солевом буфере с меченным пероксидазой полипептидом А2 по п.1, промывания указанным буферным раствором, окрашивания HSA раствором парафенилендиамина с перекисью водорода и визуальным сравнением степени окрашивания проб мочи с окрашиванием разведений стандартного HSA.
8. Способ определения HSA в моче по п.7, отличающийся тем, что твердой фазой является нитроцеллюлозная мембрана.
9. Применение рекомбинантного полипептида А2 по п.1 в качестве реагента для удаления HSA из сыворотки крови на колонке с аффинным сорбентом.
| ULF SJOBRING, Isolation and Molecular Characterization of a Novel Albumin-Binding Protein from Group G Streptococci, Infection and Immuni, Sept | |||
| Пуговица для прикрепления ее к материи без пришивки | 1921 |
|
SU1992A1 |
| Способ получения молочной кислоты | 1922 |
|
SU60A1 |
| Разборный с внутренней печью кипятильник | 1922 |
|
SU9A1 |
| УСТРОЙСТВО ДЛЯ ПОГРУЗКИ НА ПОЕЗДНЫЕ ЖЕЛЕЗНОДОРОЖНЫЕ ПЛАТФОРМЫ ШТУЧНЫХ ГРУЗОВ, НАПР., РЕЛЬСОВ И Т. П. МАТЕРИАЛОВ | 1928 |
|
SU23225A1 |
| СЛИТЫЙ ПОЛИПЕПТИД ИЛИ ЕГО СОЛЬ, ЭКСПРЕССИРУЮЩИЙ ВЕКТОР И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПРЕДНАЗНАЧЕННАЯ ДЛЯ ЛЕЧЕНИЯ IGE-ОПОСРЕДОВАННЫХ БОЛЕЗНЕЙ | 1997 |
|
RU2209211C2 |

