Область техники
Настоящее изобретение относится в общем к способу получения производных 5-[(2-гидроксиацил)амино]-2,4,6-трийодо-1,3-бензолдикарбоксамида, используемых в качестве контрастных агентов в диагностических методах.
Предшествующий уровень техники
Контрастными агентами или контрастными средами являются вещества, которые могут изменить способ, по которому анализируется область тела при медицинской визуализации. В частности, они способны изменить контраст органа, раны или любой другой окружающей структуры, чтобы сделать видимыми такие детали, которые иным образом трудно обнаружить или оценить.
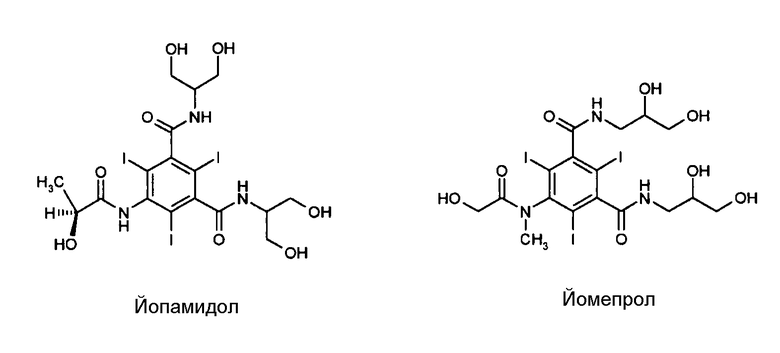
Контрастные агенты используются главным образом в области радиологической или ядерно-магнитно-резонансной диагностики. В зависимости от области применения эти производные представляют свойства, такие как в случае молекул, используемых в качестве контрастных агентов для анализа рентгеновскими лучами, присутствие одного или нескольких атомов с высоким атомным числом (например, йода или бария). Йопамидол (N,N'-бис[2-гидрокси-1-(гидроксиметил)этил]-5-[(2S)(2-гидрокси-1-оксопропил)амино]-2,4,6-трийодо-1,3-бензолдикарбоксамид) и йомепрол (N,N'-бис(2,3-дигидроксипропил)-5-[(гидроксиацетил)метиламино]-2,4,6-трийодо-1,3-бензолдикарбоксамид), структурные формулы которых показаны ниже, являются двумя из многочисленных трийодированных диагностических агентов, доступных в продаже и широко используемых для этой цели.
Среди различных синтетических методик, известных в практике для получения трийодированных ароматических производных, используемых в радиологических применениях, некоторые из указанных методик привлекают перегруппировку подходящего трийодофенилэфирного прекурсора для получения желаемого продукта, при которой новую амидную функциональную группу получают через структурную перестройку соответствующей простой эфирной группы (такая перегруппировка известна как “перегруппировка Смайлса (Smiles)”, см. в качестве основной ссылки S.Smiles et al., J.Chem.Soc. 1931, 3264).
В частности, в этом отношении WO 97/05097 описывает йопамидола путем перегруппировки Смайлса исходя из определенного простоэфирного промежуточного соединения (это последнее получено SN2 реакцией прекурсора в форме его соли) с производным (R)-2-пропанамида в различных органических растворителях согласно следующей схеме:
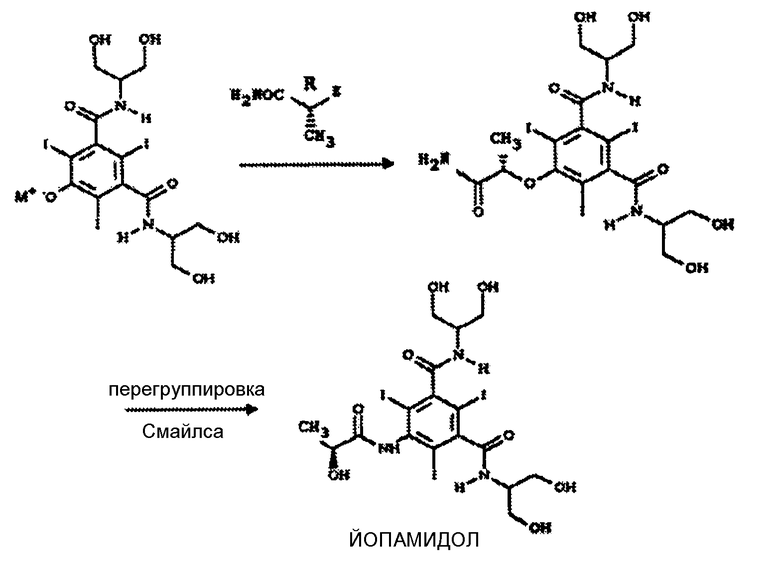
Указанную перегруппировку проводят в щелочно-спиртовой смеси, обычно состоящей из метанола в присутствии КОН, при кипячении с обратным холодильником в течение 2 часов, что дает йопамидол с общим выходом с двух стадий 56%. Кроме того, в примерах WO 97/05097 указаны производные (R)-2-пропанамида, в которых замещаемая группа Z, вовлеченная в первую стадию конденсации, является замещаемой группой, обычно выбираемой из тозилата (TsO), мезилата (MsO) и хлора.
Anelli et al. (Tetrahedron, Vol. 53, № 34, 1997, pp. 11919-11928) описывает получение производных 5-[(2-гидроксиацил)амино]-2,3,6-трийодо-1,3-бензолдикарбоксамида, также включающих йомепрол и йопамидол, перегруппировкой Смайлса подходящего простоэфирного прекурсора. В частности, описаны два способа (способы А и В), включающие использование основания в присутствии соответственно воды или органического растворителя, такого как ДМФ. Оба способа, однако, приводят к одновременному образованию побочных продуктов в различных количествах вследствие конкурирующих реакций циклизации и/или гидролиза исходного материала. Кроме того, использование водного растворителя при получении йопамидола приводит в результате к резкому снижению выхода по сравнению с той же реакцией, проводимой в ДМФ (17,9% против 99%, способ А против способа В).
Должно быть наконец отмечено, что в случае веществ, предназначенных для парентерального использования в качестве контрастных агентов, конечная химическая и оптическая чистота являются фундаментально важными, как особо указывается органами здравоохранения.
Авторы теперь открыли новый способ получения производных 5-[(2-гидроксиацил)амино]-2,4,6-трийодо-1,3-бензолдикарбоксамида, таких как йопамидол и йомепрол, перегруппировкой Смайлса подходящего трийодированного простоэфирного прекурсора при контактировании водного раствора указанного прекурсора с твердой фазой анионообменника. Способ по настоящему изобретению, включающий систему водного растворителя, позволяет выгодно провести реакцию в более благоприятных условиях даже с точки зрения охраны окружающей среды, чем в предшествующем уровне техники, и еще более выгодно позволяет получить конечные продукты, используемые в качестве контрастных агентов, например в радиологии, с высоким выходом, высокой степенью оптической чистоты и практически не содержащими побочных продуктов.
Сущность изобретения
Настоящее изобретение относится к способу получения 5-[(2-гидроксиацил)амино]-2,4,6-трийодо производного общей формулы (5) или его фармацевтически приемлемой соли

где:
R независимо в каждом случае представляет группу, выбранную из -COOR' и -CON(R')2;
R' независимо в каждом случае представляет водород или линейную или разветвленную (С1-С4) алкильную группу, необязательно замещенную одной или несколькими гидроксильными группами как таковыми или в их защищенной форме; и
X представляет водород или линейную или разветвленную (С1-С4) алкильную группу;
путем перегруппировки Смайлса (Smiles) соединения общей формулы (4) или его соли
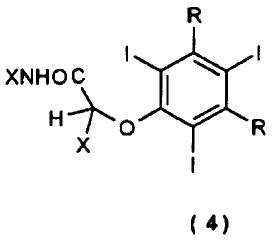
где:
R, R' и X являются такими, как определено выше;
и указанная перегруппировка достигается путем контактирования соединения (4) с твердой фазой анионообменника в присутствии водного растворителя.
Более подробно, твердой фазой может быть анионообменная смола, должным образом выбранная из смол, известных в практике, таких как смола с каркасом из стирола или полиаминоакрила, функционализированная различным образом, например группами четвертичного аммония, такими как Amberlite® или эквиваленты, или Dowex® или эквивалентные типы, или смолы типа Purolite® или эквиваленты, доступные в разных ситовых характеристиках и пористости. Предпочтительно смолу выбирают из Amberlite® IRA 400 (номер Chemical Abstracts (CAS No.) 9002-24-8) и Purolite® A830 (номер Chemical Abstracts (CAS No.) 457070-04-1).
Предпочтительно выбранную смолу соответствующим образом упаковывают в колонку, чтобы сделать возможным контакт с водным раствором или суспензией, содержащими прекурсор (4), обычно при элюировании через колонку.
В связи с этим водным растворителем предпочтительно является вода.
В предпочтительном аспекте изобретения в формулах (4) и (5), приведенных выше,
R представляет группу, выбранную из

и Х независимо представляет метил или водород.
Согласно еще более предпочтительному осуществлению настоящий способ относится к получению соединения формулы (5), где:
R представляет
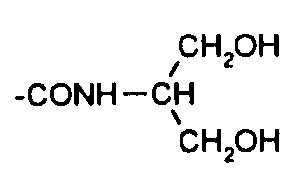
и Х представляет водород;
или где:
R представляет 
и Х представляет метил.
Согласно следующему аспекту настоящее изобретение относится к способу получения соединения (5) в основном так же, как описано выше, в котором соединение формулы (4) получают нуклеофильным замещением соединения формулы (2) или его соли амидным нитрофенилсульфонильным производным формулы (3) в присутствии растворителя, выбранного из воды и смеси воды с одним или несколькими органическими полярными растворителями:
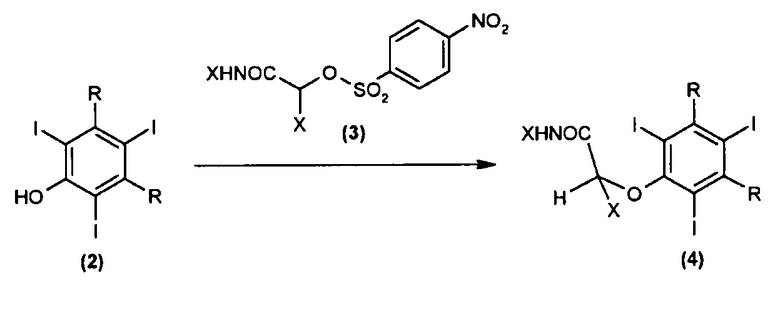
где:
R независимо в каждом случае представляет группу, выбранную из -COOR' и -CON(R')2;
R' независимо в каждом случае представляет водород или линейную или разветвленную (С1-С4) алкильную группу, необязательно замещенную одной или несколькими гидроксильными группами как таковыми или в их защищенной форме; и
X представляет водород или линейную или разветвленную (С1-С4) алкильную группу.
Предпочтительно соединение формулы (2) находится в форме соли, обычно соли щелочного металла, предпочтительно соли натрия.
Согласно следующему предпочтительному осуществлению соединение формулы (3) выбирают из:
(R)-2-[[(4-нитрофенил)сульфонил)]окси]пропиламида или
2-[[(4-нитрофенил)сульфонил)]окси]этиламида,
в то время как соединение формулы (5) предпочтительно является йопамидолом или йомепролом.
Выгодным является то, что способ по настоящему изобретению позволяет выделить конечный продукт (5), йопамидол, с выходами и оптической чистотой выше, чем в предшествующем уровне техники (как описано, например, в WO 97/05097).
Подробное описание изобретения
Настоящее изобретение относится в целом к способу получения 5-[(2-гидроксиацил)амино]-2,4,6-трийодо производных, реакцией перегруппировки Смайлса при контактировании прекурсора формулы (4) или его соли с подходящей твердой фазой анионообменника.
Если не указано иное, термин “линейная или разветвленная (С1-С4) алкильная группа” означает линейную или разветвленную алкильную группу, имеющую от 1 до 4 атомов углерода, такую как, например, метил, этил, пропил, изопропил, бутил, изобутил и т.п., предпочтительно метил.
Термины “твердая фаза анионообменника”, или “анионообменник”, или “твердая фаза” означают твердый носитель, способный осуществлять обмен анионов с находящимися с ним в контакте раствором или суспензией.
Согласно общему осуществлению и как описано более подробно в экспериментальной части, соединение формулы (4), необязательно полученное реакцией соединения формулы (2) с соответствующим производным амида формулы (3), контактирует с подходящей твердой фазой, что приводит к селективному образованию конечного соединения (5) с высокими выходами, практически не содержащего побочных продуктов.
Указанный контакт может быть обеспечен элюированием через колонку, заполненную надлежащей твердой фазой, или, альтернативно, так называемым периодическим способом. Под последним имеется в виду любой способ, который включает подходящий реактор, в котором вовлеченные в процесс реагенты находятся в контакте и реагируют друг с другом, обычно при перемешивании.
Согласно предпочтительному осуществлению контакт между соединением (4) и твердой фазой достигается при проходе через колонну, и в этом отношении твердая фаза может быть использована как таковая и загружена в колонку при необходимости или, альтернативно, твердая фаза может быть представлена в уже предварительно заполненной колонке, без труда доступной на рынке. Как упомянуто выше, прекурсор (4), присутствующий в водной среде, контактирует с твердой фазой при элюировании через колонку или при интенсивном перемешивании в случае периодического процесса при заданной скорости элюирования или перемешивания соответственно и в течение предназначенного периода времени, обычно варьирующегося от нескольких часов до нескольких суток в зависимости главным образом от количества исходного материала. В случае колонного процесса соединение (4) в водной среде обычно несколько раз элюируют через колонку согласно известным способам, таким как, например, гравитационный способ или способ высокого давления при постоянном расходе, например, около 600-800 мл/сек.
Согласно предпочтительному аспекту соединение (4) растворяют или суспендируют в водной среде, выбранной из воды и водных смесей полярного органического растворителя, например спирта, такого как метанол, этанол или подобное, или полярного простого эфира, такого как диоксан, тетрагидрофуран или подобное. Предпочтительно соединение (4) растворяют или суспендируют в воде.
Обычно и в соответствии с предпочтительным осуществлением твердой фазой является сильная анионообменная смола или слабая анионообменная смола, обе доступные в продаже и предпочтительно имеющие стирол-дивинилбензольный каркас. Примерами таких подходящих смол являются смолы Amberlite® (доступные от Rohm and Haas Company, Philadelphia, USA), Dowex® или Purolite® (доступные от The Purolite Company, Bala Cynwyd, PA, USA). Более точно, предпочтительными смолами являются Purolite® A-830 (CAS No. 457070-04-1) и Amberlite® IRA 400 (CAS No. 9002-24-8), последняя является более предпочтительной.
Выбранная смола может быть различным образом функционализирована, или использована как таковая, или даже, сверх того, предварительно активирована методами, известными специалистам в данной области, такими как активация промывкой кислотой, например хлористоводородной кислотой.
Нужно отметить, что сила анионообменника, время контакта и температура должны быть выбраны для того, чтобы максимизировать выход желаемого конечного продукта, с особым вниманием к применению настоящего процесса в промышленном масштабе. Замечено в действительности, что при условиях реакции этого процесса использование слабой анионообменной смолы может привести к конечному продукту с особой склонностью к достижимой высокой степени чистоты. С другой стороны, использование сильной анионообменной смолы может быть удобным в тех случаях, когда предпочтительна эффективность перегруппировки Смайлса в показателях выхода.
В соответствии с настоящим изобретением перегруппировку Смайлса для получения производных формулы (5) проводят, выбрав подходящую анионообменную смолу, при рН реакционной среды, составляющем от примерно 6 до примерно 9, предпочтительно от примерно 6 до примерно 7, в течение времени реакции в интервале от 24 до 40 часов, работая обычно при комнатной температуре, например при температуре около 15-30°С. Еще более предпочтительно настоящий способ описывает получение соединения (5) при контактировании раствора соответствующего прекурсора (4) в воде с молярной концентрацией от примерно 0,05 до 0,07 М путем элюирования через колонку, заполненную смолой Amberlite® IRA 400, работая при рН от примерно 6 до примерно 7 (молярные концентрации данного вещества здесь означают количество молей такого вещества, деленное на общий объем смеси).
Во время колонного процесса для того, чтобы поддерживать рН настолько постоянным, насколько возможно, рекомендовано выпаривать потенциальные аминные остатки (которые, как правило, высвобождаются смолой) из элюированного раствора. Этот прием позволяет легко получить конечный продукт с очень высокими выходами, даже до примерно 90%. В этом случае остающийся таким образом раствор после такого частичного выпаривания на тот случай, если содержит часть еще не прореагировавшего исходного вещества (4), разбавляют водой или водным растворителем, используемым для элюирования через колонну, и повторно элюируют через колонку. Эти последние стадии (т.е. выпаривание и повторное элюирование в колонку) предпочтительно повторяют один или несколько раз, например, через постоянные интервалы в 6 и 24 часа в течение всего процесса для того, чтобы получить конечный продукт с высоким выходом, как описано в прилагаемой экспериментальной части.
Обнаружение конечного соединения (5) в элюированном растворе может быть выполнено любым аналитическим методом, известным в практике, таким как, например, УФ-детектирование или подобное.
Смола в конце процесса может быть легко регенерирована согласно известным способам, таким как, например, промывка низшим спиртом, например метанолом, легко делая таким образом возможным повторное использование той же самой смолы при последующих применениях.
Как подробно изложено здесь, способ по настоящему изобретению делает возможным выгодное получение и выделение соединений формулы (5), таких как йопамидол и йомерпол, с высокими выходами реакции (более 90%) и конечной оптической чистотой (энантиомерный избыток, “ЭИ“ 99%), работая в присутствии водного растворителя.
Следует осознавать, что, поскольку перегруппировка Смайлса в соответствии с настоящим изобретением происходит с сохранением конфигурации, когда Х отличается от водорода, конфигурация стереоцентра в соединении (4) будет сохранена в конечном продукте (5). Таким образом, в подтверждение этого и в качестве примера, настоящим способом можно получить йопамидол, т.е. (N,N'-бис[2-гидрокси-1-(гидроксиметил)этил]-5-[(2S)-(2-гидрокси-1-оксопропил)амино]-2,4,6-трийодо-1,3-бензолдикарбоксамид), а также соответствующий (2R) изомер, исходя из соответствующего прекурсора (4) в (S) или (R) конфигурации соответственно. Согласно предпочтительному осуществлению изобретение относится к перегруппировке Смайлса, приводящей к образованию йопамидола.
В следующем аспекте настоящее изобретение относится к способу получения соединения (5), который был описан перед этим, отличающемуся тем, что соединение формулы (4) получают реакцией соединения формулы (2) или его соли с нитрофенилсульфониламидным производным формулы (3):

где:
R независимо в каждом случае выбирают из группы, состоящей из -COOR' и -CON(R')2;
R' независимо в каждом случае представляет водород или линейную или разветвленную (С1-С4) алкильную группу, необязательно замещенную одной или несколькими гидроксильными группами как таковыми или в их защищенной форме; и
X представляет водород или линейную или разветвленную (С1-С4) алкильную группу;
в присутствии растворителя, выбранного из воды и смеси воды с одним или несколькими органическими полярными растворителями.
Предпочтительно исходное соединение формулы (2) находится в форме его соли, обычно соли щелочного металла, предпочтительно соли натрия.
Согласно предпочтительным осуществлениям, которые описаны выше, соединение формулы (3) предпочтительно выбирают из:
(R)-2-[[(4-нитрофенил)сульфонил)]окси]пропиламида или
2-[[(4-нитрофенил)сульфонил)]окси]этиламида.
Согласно способу получения соединения (4) из соединения (2), который описан выше, конечный продукт (5) выгодно получают с выходами, более высокими, чем в прототипе (82% против 56%, что описано, например, в WО 97/05097) и оптической чистотой (ЭИ) более 99%.
Нитрофенилсульфониламидное производное формулы (3) может быть легко получено известными в практике способами (см., например, Markert et al., Chem. Ber., 1927, 60, 2456) или, альтернативно, куплено как таковое. Обычно исходное соединение формулы (2) присутствует в виде его соли, предпочтительно в виде соли натрия, поскольку рН реакционной смеси поддерживают на уровне от 6 до 9. Предпочтительно указанная величина рН составляет от примерно 7 до примерно 8.
Для измерения рН обычно используют стеклянный электрод, в то время как величину рН можно изменять, используя основание, такое как неорганическое основание, например NaOH.
Согласно иллюстративному осуществлению изобретения и в соответствии с последующей экспериментальной частью соединение (3) добавляют к натриевой соли соединения (2) в водном растворителе, выбранном из воды или воды, смешанной с полярным органическим растворителем, например диоксаном, тетрагидрофураном или подобным в соотношении 1:1, предпочтительно в присутствии избытка воды. В этом отношении выбранным полярным органическим растворителем является диоксан и массовым отношением вода/органический растворитель является 2:1 или более предпочтительно 3:1. Согласно следующему предпочтительному осуществлению водным растворителем является смесь вода/диоксан 3:1 по массе.
Реакционную смесь перемешивают при комнатной температуре или более предпочтительно нагревают до температуры, например, от примерно 50°С до примерно 90°С, более предпочтительно между 70°С и 80°С. Величину рН реакционной среды контролируют и, возможно, корректируют, чтобы иметь значение от примерно 7 до примерно 8, как описано ранее. При мониторинге развития реакции, например, путем анализа методом ТСХ, когда процентное содержание остаточного соединения (2) начинает становиться пренебрежимо малым (обычно меньшим чем примерно 10%), раствор перерабатывают путем очистки и выпаривания растворителя для того, чтобы получить желаемое производное (4).
Полученное таким образом соединение формулы (4) подвергают затем перегруппировке Смайлса путем контактирования с твердой фазой анионообменника в присутствии водного растворителя, как подробно пояснено выше.
Конечный продукт (5), следовательно, получают из соединения (2) с выходами выше чем 70% (общий выход с двух стадий 82%) и с высокой оптической чистотой (ЭИ 99,5%), используя надежный способ, который позволяет работать в присутствии водных реакционных растворителей.
В настоящем изобретении следует осознавать, что, когда Х отличается от водорода, соединение (3) и, следовательно, соединения (4) и (5) имеют хиральный центр и поэтому могут присутствовать в двух конфигурациях (R) или (S), иначе называемых (D) или (L), а также в рацемической форме. Авторы представляют процесс, который включает в себя перегруппировку Смайлса соединения формулы (4), полученного реакцией соответствующего прекурсора с соединением формулы (3), причем это последнее предназначено быть в конфигурации или (R), или (S). Приводя к образованию конечного соединения (5) стереоспецифичным способом. Так, например, настоящий способ может привести к образованию соединения (N,N'-бис-[2-гидрокси-1-(гидроксиметил)этил]-5-[(2-гидрокси-1-изопропил)амино]-2,4,6-трийодо-1,3-бензолдикарбоксамид), имеющего конфигурацию (2S) (а именно, йопамидола), или соответствующего (2R) изомера, исходя из прекурсора (2-[[(4-нитрофенил)сульфонил)]окси]пропиламида (3) с конфигурацией (R) или (S) соответственно. В действительности, после реакции типа SN2 между соединениями (3) и (2) соединение (4) получается инверсией конфигурации. Последующая реакция Смайлса, происходящая с сохранением конфигурации, легко делает весь процесс высокостереоспецифичным.
Поэтому, как подробно изложено выше, настоящее изобретение позволяет получать 5-[(2-гидроксиацил)амино]-2,4,6-трийодо производные общей формулы (5), используемые в качестве контрастных агентов в методах диагностической визуализации, с высокими выходами и оптической чистотой, используя надежный и воспроизводимый процесс, применимый также в промышленном масштабе, который включает перегруппировку Смайлса на твердой фазе анионообменника в присутствии водной среды.
Это изобретение может быть без затруднений применено для получения йопамидола или йомерпола, практически не содержащих побочных продуктов и соответствующих спецификациям, требуемым для их применения в качестве контрастных агентов, обычно в радиологии.
Настоящее изобретение будет теперь проиллюстрировано примерами, которые не предназначены для того, чтобы сформулировать какие-либо ограничения его объема.
Экспериментальная часть
Пример 1: Получение соединения (5) путем перегруппировки Смайлса в присутствии твердой фазы анионообменника в водной среде (общая методика)
Раствор, имеющий рН 6-7, соединения (4) в подходящем водном растворителе загружали в колонку, заполненную твердой фазой анионнообменника, и элюировали через колонку при постоянном расходе. С промежутками в 6 и 24 ч элюирование прекращали и полученный таким образом элюированный раствор, который показывал рН от примерно 9 до примерно 11, выпаривали и разбавляли водным реакционным растворителем, приводя рН к его первоначальному значению путем добавления основания. Реакцию непрерывно контролировали, и анализ методом ЖХВР показывал прогрессирующее убывание соединения (4) в пользу соединения (5).
Пример 1а: Получение йопамидола путем перегруппировки Смайлса в присутствии Amberlite® IRA400
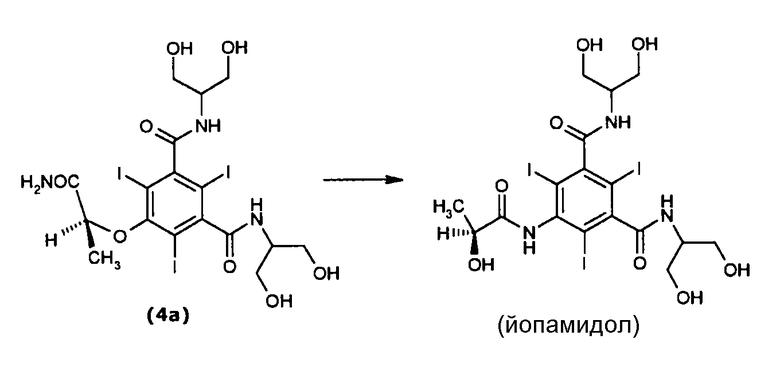
Повторяли общую процедуру примера 1, используя соединение формулы (4а) (7,5 г, 9,6 ммоль) в Н2О (150 мл) в присутствии Amberlite® IRA400, причем последний был загружен в колонку.
Время элюирования составляло 32 часа при постоянном расходе 600 мл/ч. Анализ реакционной смеси методом ЖХВР показал, что концентрация соединения (4а) убывала со временем в пользу йопамидола (выход 93%, энантиомерный избыток > 99%) практически без сопутствующего образования нежелательных продуктов.
Пример 2: Получение соединения (4а) реакцией соединения (2а) с (R)-2-[[(4-нитрофенил)сульфонил)]окси]пропанамидом

(R)-2-[[(4-нитрофенил)сульфонил)]окси]пропанамид (26,7 г, 9,97 ммоль) добавляли порциями за примерно 1,5 ч к раствору 5-гидрокси-N,N'-бис[2-гидрокси-1-(гидроксиметил)этил]-2,4,6-трийод-1,3-бензолдикарбоксамида (2а) в форме натриевой соли (44,2 г, 60,8 ммоль) в растворе Н2О/1,4-диоксан 75:25 (примерно 300 мл) и реакционную смесь перемешивали при 70-80°С в присутствии стеклянного электрода для измерения рН.
Значение рН реакционной среды поддерживали при примерно 7-8 добавлением 1 М NaOH до тех пор, пока остаточное процентное содержание соединения (2) не становилось меньше примерно 10% по измерению анализом ТСХ.
Раствор концентрировали и элюировали через колонку с Amberlite® IRA-120, элюат нейтрализовали 2 М NaOH (30 мл), концентрировали под вакуумом и выдерживали при низкой температуре (около 5°С) в течение 15 часов. После фильтрации и сушки получали первую порцию соединения (4а) (33,3 г, 42,8 ммоль, выход 71%) в виде белого твердого вещества.
Фильтрат дополнительно упаривали, полученный таким образом твердый остаток нагревали в этаноле и полученную суспензию отфильтровывали, чтобы удалить нерастворимый 4-нитробензосульфонат натрия. После 15 часов при примерно 5°С полученную таким образом вторую порцию соединения (4а) в виде сырого продукта растворяли в воде и очищали элюированием через колонку Amberlite® IRA-120, элюируя с водой. Нейтрализованный элюат выпаривали и твердый остаток кристаллизовали из этанола, получая вторую порцию соединения (4а) в виде чистого твердого вещества (6,9 г, 8,8 ммоль, выход 15%), которую соединяли с первой порцией, получая соединение (4а) с суммарным выходом 86%.
Пример 3: Получение йопамидола из соединения (4а), полученного согласно методике примера 2
Соединение формулы (4а), полученное по методике примера 2, подвергали перегруппировке Смайлса согласно общей методике примера 1.
Конечный продукт кристаллизовали из этанола, получая указанное в заголовке соединение с выходом 95%.
Общий выход из соединения (2) (пример 2+пример 3)=82%, ЖХВР 99,9%, [□]20 436 =+144,3 (c 2,5 Cu(II) L2, H2O)=99,5%, выше теоретического значения 145.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ЙОПАМИДОЛА | 2014 |
|
RU2657238C2 |
| СПОСОБ ЙОДИРОВАНИЯ ПРОИЗВОДНЫХ ФЕНОЛА | 2011 |
|
RU2563645C2 |
| СПОСОБ ЙОДИРОВАНИЯ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ | 2009 |
|
RU2469997C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЙОДИРОВАННОГО КОНТРАСТНОГО АГЕНТА | 2009 |
|
RU2493146C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 5-АМИНО-2,4,6-ТРИИОДО- 1,3-БЕНЗОЛКАРБОНОВОЙ КИСЛОТЫ | 1990 |
|
RU2046795C1 |
| КОНТРАСТНЫЕ АГЕНТЫ | 2008 |
|
RU2469021C2 |
| СПОСОБ ПОЛУЧЕНИЯ ТРИЙОДИРОВАННЫХ КАРБОКСИЛЬНЫХ АРОМАТИЧЕСКИХ ПРОИЗВОДНЫХ | 2010 |
|
RU2512358C2 |
| СПОСОБ ИОДИРОВАНИЯ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ | 2010 |
|
RU2506254C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЙОДИРУЮЩЕГО АГЕНТА | 2010 |
|
RU2528402C2 |
| КОМПЛЕКСЫ МЕТАЛЛОВ С БИЦИКЛИЧЕСКИМИ ПОЛИАМИНОКИСЛОТАМИ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ В МЕДИЦИНЕ ДЛЯ ПОЛУЧЕНИЯ ИЗОБРАЖЕНИЯ | 2000 |
|
RU2232763C2 |
Настоящее изобретение относится к способу получения производных 5-[(2-гидроксиацил)амино]-2,4,6-трийодо производного общей формулы (5) или его фармацевтически приемлемой соли, где R независимо в каждом случае представляет группу, выбранную из -COOR' и -CON(R')2; R' независимо в каждом случае представляет водород или линейную или разветвленную (С1-С4) алкильную группу, необязательно замещенную одной или несколькими гидроксильными группами как таковыми или в их защищенной форме; и X представляет водород или линейную или разветвленную (С1-С4) алкильную группу; путем перегруппировки Смайлса (Smiles) соединения общей формулы (4) или его соли, где R, R' и X являются такими, как определено выше, и указанная перегруппировка достигается путем контактирования соединения (4) с твердой фазой анионообменника в присутствии водного растворителя. Технический результат - получение производных 5-[(2-гидроксиацил)амино]-2,4,6-трийодо-1,3-бензолкарбоксамида с высоким выходом, высокой степенью оптической чистоты. 9 з.п. ф-лы, 4 пр.

1. Способ получения 5-[(2-гидроксиацил)амино]-2,4,6-трийодо производного общей формулы (5) или его фармацевтически приемлемой соли
где:
R независимо в каждом случае представляет группу, выбранную из -COOR' и -CON(R')2;
R' независимо в каждом случае представляет водород или линейную или разветвленную (С1-С4) алкильную группу, необязательно замещенную одной или несколькими гидроксильными группами как таковыми или в их защищенной форме; и
X представляет водород или линейную или разветвленную (С1-С4) алкильную группу;
путем перегруппировки Смайлса (Smiles) соединения общей формулы (4) или его соли:
где:
R, R' и X являются такими, как определено выше;
и указанная перегруппировка достигается путем контактирования соединения (4) с твердой фазой анионообменника в присутствии водного растворителя.
2. Способ согласно п. 1, в котором:
R представляет группу, выбранную из 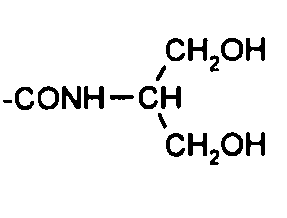 ; и
; и
Х представляет водород.
3. Способ согласно п. 1, в котором:
R представляет группу, выбранную из и
Х представляет метильную группу.
4. Способ согласно п.1, в котором твердой фазой анионообменника является слабая анионообменная смола или сильная анионообменная смола.
5. Способ согласно п.4, в котором твердую фазу анионообменника выбирают из Amberlite® IRA 400 и Purolite® A-830.
6. Способ согласно п.1, в котором водным растворителем является вода.
7. Способ согласно п.1, в котором реакцию проводят при pH от 6 до 9.
8. Способ согласно п.1, в котором получение соединения формулы (4) проводят нуклеофильным замещением соединения формулы (2) или его соли амидным нитрофенилсульфонильным производным формулы (3):

где:
R независимо представляет группу, выбранную из -COOR' и -CON(R')2;
R' независимо представляет водород или линейную или разветвленную (С1-С4) алкильную группу, необязательно замещенную одной или несколькими гидроксильными группами как таковыми или в их защищенной форме; и
X представляет водород или линейную или разветвленную (С1-С4) алкильную группу;
в присутствии растворителя, выбранного из воды и смеси воды с одним или несколькими органическими полярными растворителями.
9. Способ согласно п.8, в котором соединением формулы (3) является (R)-2-[[(4-нитрофенил)сульфонил)]окси]пропанамидом или 2-[[(4-нитрофенил)сульфонил)]окси]этанамидом.
10. Способ согласно любому одному из пп.8 или 9, в котором растворителем является смесь вода/диоксан в массовом соотношении 3:1.
| Бесколесный шариковый ход для железнодорожных вагонов | 1917 |
|
SU97A1 |
| Кипятильник для воды | 1921 |
|
SU5A1 |
| Тепловоз | 1924 |
|
SU3253A1 |

