Настоящее изобретение относится к новым фармацевтическим композициям, предназначенным для лечения заболеваний, включающих пролиферацию клеток, миграцию или апоптоз раковых клеток, или ангиогенез, и к их получению. Настоящее изобретение также относится к способу лечения заболеваний, включающих пролиферацию клеток, миграцию или апоптоз раковых клеток, или ангиогенез, и этот способ включает совместное введение лицу, нуждающемуся в таком лечении, и/или совместное лечение лица, нуждающегося в таком лечении, эффективным количеством:
соединения 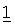
в которой группы L, R1, R2, R3, R4 и R5 обладают значениями, приведенными в формуле изобретения и описании, необязательно в форме его таутомеров, рацематов, энантиомеров, диастереоизомеров и их смесей и необязательно в форме его фармакологически приемлемых солей присоединения с кислотами, сольватов, гидратов, полиморфных форм, физиологически функциональных производных или пролекарств; и
по меньшей мере дополнительного химиотерапевтического, иммунотерапевтического или иммуномодулирующего, антиангиогенного, гормонального или природного, полусинтетического или синтетического терапевтического средства 
лучевой терапии или радиоиммунотерапии.
УРОВЕНЬ ТЕХНИКИ
Polo-подобные киназы (ППК) представляют собой серин/треонинкиназы, которые играют важную роль в процессах регуляции клеточного цикла. В настоящее время известны четыре ППК, т.е. ППК-1, ППК-2, ППК-3 и ППК-4. ППК играют роль при возникновении и заканчивании митоза для клеток млекопитающих. В особенности для ППК-1 показано, что она играет главную роль в регуляции митоза (Glover et al. 1998, Genes Dev. 12:3777-87; Qian et al. 2001, Mol Biol Cell. 12:1791-9). Сверхэкспрессирование ППК-1, видимо, сильно связано с опухолевыми клетками, включая раковые (WO 2004/014899). Сверхэкспрессирование ППК-1 обнаружено для разных типов опухолей, таких как немелкоклеточный рак легких, плоскоклеточные карциномы, карциномы молочной железы, яичников и папиллярные карциномы, а также колоректальные типы рака (Wolf et al. 1997, Oncogene 14, pages 543-549; Knecht et al. 1999, Cancer Res. 59, pages 2794-2797; Wolf et al. 2000, Pathol Res Pract. 196, pages 753-759; Weichert et al. 2004, Br. J. Cancer 90, pages 815-821; Ito et al. 2004, Br. J. Cancer 90, pages 414-418; Takahashi et al. 2003, Cancer Sci. 94, pages 148-152).
Для лечения заболеваний онкологической природы уже предложено большое количество химиотерапевтических, иммунотерапевтических или иммуномодулирующих, антиангиогенных или гормональных средств, которые можно применять в виде монотеорапии (лечение одним средством) или в виде комбинированной терапии (одновременное, раздельное или последовательное лечение более, чем одним средством) и/или которые можно объединить с лучевой терапией или радиоиммунотерапией. В этом контексте химиотерапевтическое средство означает природное, полусинтетическое или синтетическое химическое соединение, которое, само по себе или с помощью последующей активации, например, облучением в случае радиоиммунотерапии, подавляет или уничтожает растущие клетки и которое можно применять или которое утверждено к применению для лечения заболеваний онкологической природы, которые обычно также называют раками. В литературе эти средства обычно классифицируют в соответствии с механизмом их действия. В связи с этим можно указать, например, классификацию, выполненную в публикации "Cancer Chemotherapeutic Agents", American Chemical Society, 1995, W.O. Foye Ed.
Эффективность химиотерапевтических средств можно улучшить путем использования комбинационной терапии с другими химиотерапевтическими, иммунотерапевтическими или иммуномодулирующими, антиангиогенными или гормональными соединениями. Комбинированные терапии являются "золотым стандартом" во многих медицинских учреждениях, в которых лечат рак.
Несмотря на то, что уже предложена концепция комбинирования различных терапевтических средств или методик лечения и различные методики комбинированного лечения уже изучаются и проводятся клинические исследования, все же необходимы новые и эффективные терапевтические композиции, предназначенные для лечения раковых заболеваний, которые обладали бы преимуществами по сравнению со стандартными методиками лечения.
Настоящее изобретение относится к комбинированной терапии с использованием ингибиторов ППК формулы (I), предназначенной для лечения различных раковых заболеваний.
ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Таким образом, в контексте настоящего изобретения особый интерес представляют следующие классы химиотерапевтических средств, хотя перечень не является ограничивающим:
- синтетические небольшие молекулы-антагонисты рецептора СЭФР (сосудистого эндотелиального фактора роста)
- небольшие молекулы-антагонисты рецептора фактора роста (ФР)
- ингибиторы рецептора ЭФР (эндотелиального фактора роста) и/или рецептора СЭФР и/или рецепторов интегрина или рецепторов любой другой протеинкиназы, которые не отнесены к классу небольших синтетических молекул
- небольшие молекулы-ингибиторы путей Ras/Raf/MAPK или PI3K/AKT или любых других серин/треонинкиназ
- ингибиторы путей Ras/Raf/MAPK или PI3K/AKT или любых других серин/треонинкиназ, которые не отнесены к классу небольших синтетических молекул
- ингибиторы рецептора ЭФР и/или рецептора СЭФР и/или рецепторов интегрина или рецепторов любой другой протеинкиназы, которые являются полученными синтетически антителами, фрагментами антител или белками слияния
- соединения, которые взаимодействуют с нуклеиновыми кислотами и которые отнесены к классу алкилирующих средств, или соединения платины
- соединения, которые взаимодействуют с нуклеиновыми кислотами и которые отнесены к классам антрациклинов, интеркаляторов ДНК или агентов, сшивающих ДНК
- антиметаболиты
- природные, полусинтетические или синтетические антибиотики типа блеомицина (антибиотики группы БЛМ)
- ингибиторы транскрибирующих ДНК ферментов, в особенности ингибиторы топоизомеразы I или топоизомеразы II
- модифицирующие хроматин агенты
- ингибиторы митоза, антимитотические средства или ингибиторы клеточного цикла
- соединения, взаимодействующие с тубулином или связывающие его
- соединения, ингибирующие митотические кинезины или другие двигательные белки, включая, но не ограничиваясь только ими, Eg5, CENP-E, MCAK, Kid, MKLP-1
- ингибиторы протеосом
- ингибиторы белка теплового шока
- соединения, влияющие на антиапоптозную функцию Bcl-2, Bcl-x1 и аналогичные молекулы
- ферменты, гормоны, антагонисты гормонов или ингибиторы гормонов, или ингибиторы биосинтеза стероидов
- стероиды
- цитокины, селективные для гипоксии цитокины, ингибиторы цитокинов, лимфокины, антитела против цитокинов или средства, обеспечивающие переносимость при пероральном и парентеральном введении
- поддерживающие средства
- противовоспалительные соединения, такие как, но не ограничиваясь только ими, ингибиторы СОХ-2
- химические сенсибилизаторы радиации и средства защиты
- фотохимически активированные лекарственные средства
- синтетические поли- или олигонуклеотиды
- другие химиотерапевтические или природные, полусинтетические или синтетические терапевтические средства, такие как цитотоксические антибиотики, антитела, воздействующие на поверхностные молекулы раковых клеток, антитела, воздействующие на факторы роста или их рецепторы, ингибиторы металлопротеиназ, ингибиторы онкогенеза, ингибиторы транскрипции генов или трансляции РНК или экспрессирования белков, или комплексы редкоземельных элементов.
Преимущества настоящего изобретения в основном базируются на аддитивных и синергетических эффектах комбинированного лечения или на улучшенной переносимости лечения пациентом вследствие, например, введения меньших доз использующихся терапевтических средств.
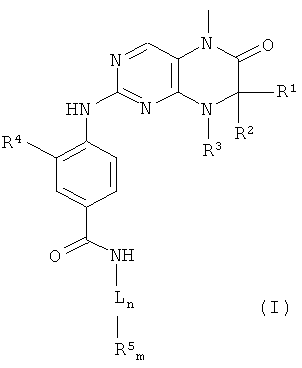
В контексте настоящего изобретения соединение 
в которой
R1, R2, которые могут быть одинаковыми или разными, обозначают водород или необязательно замещенный С1-С6-алкил, или
R1 и R2 совместно обозначают 2- - 5-членный алкильный мостик, который может содержать 1-2 гетероатома,
R3 обозначает водород или группу, выбранную из группы, включающей необязательно замещенный С1-С12-алкил, С2-С12-алкенил, С2-С12-алкинил и С6-С14-арил, или
группу, выбранную из группы, включающей необязательно замещенный и/или мостиковый С3-С12-циклоалкил, С3-С12-циклоалкенил, С7-С12-полициклоалкил, С7-С12-полициклоалкенил, С5-С12-спироциклоалкил, С3-С12-гетероциклоалкил, который содержит 1-2 гетероатома, и С3-С12-гетероциклоалкенил, который содержит 1-2 гетероатома, или
R1 и R3 или R2 и R3 совместно обозначают насыщенный или ненасыщенный С3-С4-алкильный мостик, который может содержать 1 гетероатом,
R4 обозначает группу, выбранную из группы, включающей водород, -CN, гидроксигруппу, -NR6R7 и галоген, или
группу, выбранную из группы, включающей необязательно замещенный С1-С6-алкил, С2-С6-алкенил, С2-С6-алкинил, С1-С5-алкилоксигруппу, С2-С5-алкенилоксигруппу, С2-С5-алкинилоксигруппу, C1-С6-алкилтиогруппу, C1-С6-алкилосульфоксогруппу и С1-С6-алкилсульфонил,
L обозначает соединительный фрагмент, выбранный из группы, включающей необязательно замещенный С2-С10-алкил, С2-С10-алкенил, С6-С14-арил, -С2-С4-алкил-С6-С14-арил, -С6-С14-арил-С1-С4-алкил, необязательно мостиковый С3-С12-циклоалкил и гетероарил, который содержит 1 или 2 атома азота,
n равно 0 или 1
m равно 1 или 2
R5 обозначает группу, выбранную из группы, включающей необязательно замещенный морфолинил, пиперидинил, пиперазинил, пиперазинилкарбонил, пирролидинил, тропенил, R8-дикетометилпиперазинил, сульфоксоморфолинил, сульфонилморфолинил, тиоморфолинил, -NR8R9 и азациклогептил,
R6, R7, которые могут быть одинаковыми или разными, обозначают водород или С1-С4-алкил,
и
R8, R9 обозначают незамещенные азотсодержащие заместители у R5, которые могут быть одинаковыми или разными, обозначают водород или группу, выбранную из группы, включающей C1-С6-алкил, -С1-С4-алкил-С3-С10-циклоалкил, С3-С10-циклоалкил, С6-С14-арил, -С1-С4-алкил-С6-С14-арил, пиранил, пиридинил, пиримидинил, С1-С4-алкилоксикарбонил, С6-С14-арилкарбонил, С1-С4-алкилкарбонил, С6-С14-арилметилоксикарбонил, С6-С14-арилсульфонил, С1-С4-алкилсульфонил- и С6-С14-арил-С1-С4-алкилсульфонил-, необязательно в форме его таутомеров, рацематов, энантиомеров, диастереоизомеров и их смесей и необязательно в форме его фармакологически приемлемых солей присоединения с кислотами, сольватов, гидратов, полиморфных форм, физиологически функциональных производных или пролекарств.
Предпочтительными соединениями формулы (I) являются такие, в которых R1-R4, R6 и R7 являются такими, как определено выше, и
L обозначает соединительный фрагмент, выбранный из группы, включающей необязательно замещенный С2-С10-алкил, С2-С10-алкенил, С6-С14-арил, -С2-С4-алкил-С6-С14-арил, -С6-С14-арил-С1-С4-алкил, необязательно мостиковый С3-С12-циклоалкил и гетероарил, который содержит 1 или 2 атома азота
n равно 1
m равно 1 или 2
R5 обозначает группу, которая связана с L через атом азота, выбранную из группы, включающей необязательно замещенный морфолинил, пиперидинил, R8-пиперазинил, пирролидинил, тропенил, R8-дикетометилпиперазинил, сульфоксоморфолинил, сульфонилморфолинил, тиоморфолинил, -NR8R9 и азациклогептил,
R8, R9 обозначают незамещенные азотсодержащие заместители у R5, которые могут быть одинаковыми или разными, обозначают водород или группу, выбранную из группы, включающей С1-С6-алкил, -С1-С4-алкил-С3-С10-циклоалкил, С3-С10-циклоалкил, С6-С14-арил, -С1-С4-алкил-С6-С14-арил, пиранил, пиридинил, пиримидинил, С1-С4-алкилоксикарбонил, С6-С14-арилкарбонил, С1-С4-алкилкарбонил, С6-С14-арилметилоксикарбонил, C6-C14-арилсульфонил, С1-С4-алкилсульфонил и С6-С14-арил-С1-С4-алкилсульфонил.
Также предпочтительными являются соединения формулы (I), в которой R1-R4, R6 и R7 являются такими, как определено выше,
L обозначает соединительный фрагмент, выбранный из группы, включающей необязательно замещенный С2-С10-алкил, С2-С10-алкенил, С6-С14-арил, -С2-С4-алкил-С6-С14-арил, -С6-С14-арил-С1-С4-алкил, необязательно мостиковый С3-С12-циклоалкил и гетероарил, который содержит 1 или 2 атома азота
n равно 0 или 1
m равно 1 или 2
R5 обозначает группу, которая связана с L через атом углерода, выбранную из группы, включающей R8-пиперидинил, R8R9-пиперазинил, R8-пирролидинил, R8-пиперазинилкарбонил, R8-тропенил, R8-морфолинил и R8-азациклогептил,
и
R8, R9 обозначают незамещенные азотсодержащие заместители у R5, которые могут быть одинаковыми или разными, обозначают водород или группу, выбранную из группы, включающей C1-С6-алкил, -С1-С4-алкил-С3-С10-циклоалкил, С3-С10-циклоалкил, С6-С14-арил, -С1-С4-алкил-С6-С14-арил, пиранил, пиридинил, пиримидинил, С1-С4-алкилоксикарбонил, С6-С14-арилкарбонил, С1-С4-алкилкарбонил, С6-С14-арилметилоксикарбонил, C6-C14-арилсульфонил, С1-С4-алкилсульфонил и С6-С14-арил-С1-С4-алкилсульфонил, необязательно в форме их таутомеров, рацематов, энантиомеров, диастереоизомеров и их смесей и необязательно в форме их фармакологически приемлемых солей присоединения с кислотами.
Также предпочтительными являются соединения формулы (I), в которой
L, m, n и R3-R9 являются такими, как определено выше, и
R1, R2, которые могут быть одинаковыми или разными, обозначают группу, выбранную из группы, включающей водород, Me, Et, Pr, или
R1 и R2 совместно образуют С2-С4-алкильный мостик,
необязательно в форме их таутомеров, рацематов, энантиомеров, диастереоизомеров и их смесей и необязательно в форме их фармакологически приемлемых солей присоединения с кислотами.
Особенно предпочтительными являются соединения формулы (I), в которой
R1, R2, m, n и R5-R8 являются такими, как определено выше, и
R3 обозначает группу, выбранную из группы, включающей необязательно замещенный C1-С10-алкил, С3-С7-циклоалкил, С3-С6-гетероциклоалкил и С6-С14-арил или
R1 и R3 или R2 и R3 совместно обозначают насыщенный или ненасыщенный С3-С4-алкильный мостик, который может содержать 1-2 гетероатома,
R4 обозначает группу, выбранную из группы, включающей водород, ОМе, ОН, Me, Et, Pr, OEt, NHMe, NH2, F, CL, Br, O-пропаргил, O-бутинил, CN, SMe, NMe2, CONH2, этинил, пропинил, бутинил и аллил,
и
L обозначает соединительный фрагмент, выбранный из группы, включающей необязательно замещенный фенил, фенилметил, циклогексил и разветвленный С1-С6-алкил,
необязательно в форме их таутомеров, рацематов, энантиомеров, диастереоизомеров и их смесей и необязательно в форме их фармакологически приемлемых солей присоединения с кислотами.
В другом варианте осуществления соединение 
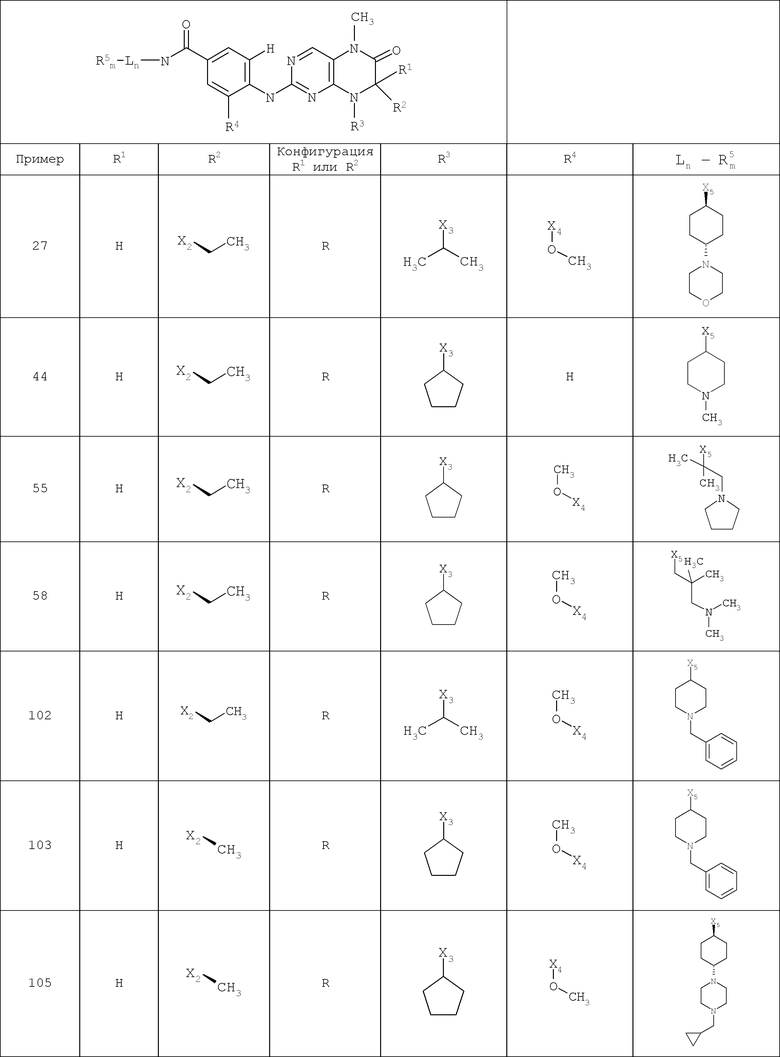
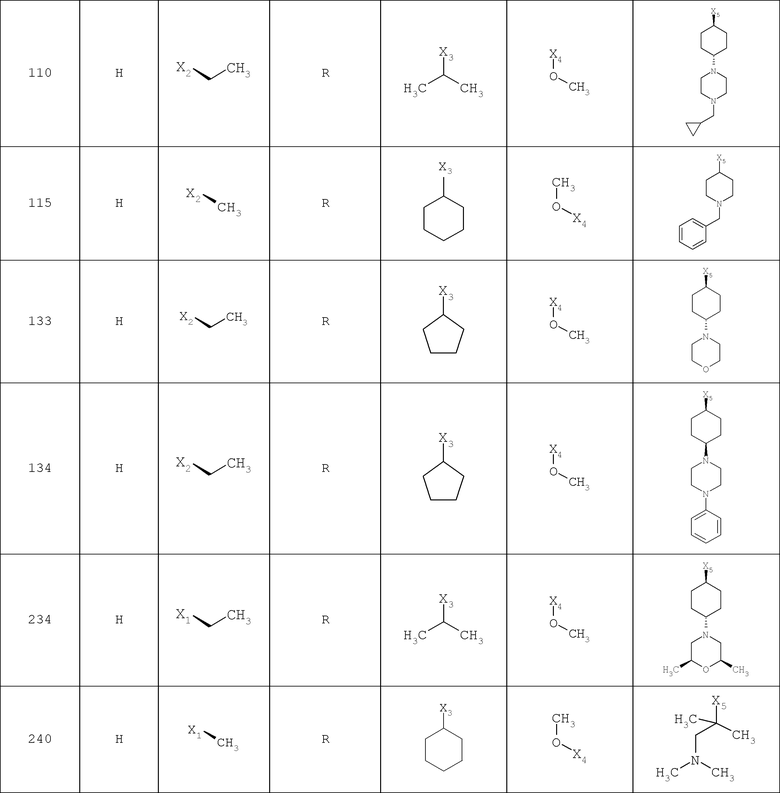
где обозначения X1, Х2, Х3, Х4 и Х5, использованные в таблице, в каждом случае обозначают связывание в положении в общей формуле, приведенной в таблице, а не соответствующие группы R1, R2, R3, R4 и L-R5.
Таким образом, настоящее изобретение относится к фармацевтической композиции, включающей эффективные количества:
(i) соединения 
(ii) по меньшей мере одного дополнительного химиотерапевтического или природного, полусинтетического или синтетического терапевтического средства 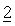
необязательно в комбинации с одним или большим количеством фармацевтически приемлемых инертных наполнителей, и необязательно приспособленной для совместного лечения с лучевой терапией или радиоиммунотерапией, в форме комбинированного препарата, предназначенного для одновременного, раздельного или последовательного применения для лечения заболеваний, включающих пролиферацию клеток, миграцию или апоптоз раковых клеток, или ангиогенез, предпочтительно - включающих пролиферацию клеток или апоптоз раковых клеток.
В предпочтительном варианте осуществления настоящее изобретение относится к фармацевтической композиции, в которой дополнительное химиотерапевтическое или природное, полусинтетическое или синтетическое терапевтическое средство 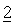
Предпочтительные соединения включают небольшие молекулы-ингибиторы тирозинкиназы или серин/треонинкиназы, соединения, взаимодействующие с нуклеиновыми кислотами и отнесенные к классам алкилирующих средств или антрациклинов, антиметаболиты, ингибиторы транскрибирующих ДНК ферментов, таких как топоизомераза I или II, лекарственные средства, связывающиеся с тубулином, антимитотические средства, антитела, воздействующие на факторы роста или их рецепторы, и антитела, связывающиеся с поверхностными молекулами раковых клеток или лигандами этих поверхностных молекул, в форме гидратов и/или сольватов и необязательно в форме их индивидуальных оптических изомеров, смесей индивидуальных энантиомеров или рацематов.
В другом предпочтительном варианте осуществления настоящее изобретение относится к фармацевтической комбинации, в которой дополнительное химиотерапевтическое или природное, полусинтетическое или синтетическое терапевтическое средство 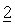
Предпочтительные соединения включают небольшие молекулы-антагонисты рецептора СЭФР, такие как ваталаниб (PTK-787/ZK222584), SU-5416, SU-6668, SU-11248, SU-14813, AZD-6474, антагонисты EGFR/HER2, такие как CI-1033 или GW-2016, антагонист EGFR, такой как иресса (гефитиниб, ZD-1839), тарцева (эрлотиниб, OSI-774), PKI-166, EKB-569, HKI-272 или герцептин, антагонист активированной митогеном протеинкиназы, такой как BAY-43-9006 или BAY-57-9006, атрасентан, ритуксимаб, цетуксимаб, авастин™ (бевацизумаб), IMC-1C11, эрбитукс (С-225), DC-101, EMD-72000, витаксин, иматиниб, алкилирующее средство или соединение платины, такое как мелфалан, циклофосфамид, цисплатин, карбоплатин, оксалиплатин, сатраплатин, даунорубицин, доксорубицин (адриамицин), липосомный доксорубицин (доксил), эпирубицин, идарубицин, аналог пиримидина или пурина или антагонист или ингибитор нуклеозиддифосфатредуктазы, такой как цитарабин, 5-фторурацил (5-FU), пеметрексед, тегафур/урацил, гемцитабин, капецитабин, меркаптопурин, метотрексат, противораковое лекарственное средство, такое как паклитаксел (таксол) или доцетаксел, алкалоид барвинка, такой как навелбин, винбластин, винкристин, виндезин или винорелбин, антимитотический пептид, такой как доластатин, эпиподофиллотоксин или производное подофиллотоксина, такое как этопозид или тенипозид, нестероидное противовоспалительное лекарственное средство, такое как мелоксикам, целекоксиб, рофекоксиб, антитело, воздействующее на поверхностные молекулы раковых клеток, такое как аполизумаб или 1D09C3 или модулятор белка теплового шока HSP90 гелданамицин и его производное 17-аллиламиногелданамицин или 17-AAG.
В другом предпочтительном варианте осуществления настоящее изобретение относится к фармацевтической композиции, в которой дополнительное химиотерапевтическое или природное, полусинтетическое или синтетическое терапевтическое средство 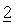
Предпочтительные соединения включают небольшие молекулы-антагонисты рецептора, такие как ваталаниб, SU 11248 или AZD-6474, антагонисты EGFR или HER2, такие как гефитиниб, эрлотиниб, CI-1033 или герцептин, антитела, такие как бевацизумаб, цетуксимаб, ритуксимаб, алкилирующие ДНК лекарственные средства, такие как цисплатин, оксалиплатин или карбоплатин, антрациклины, такие как доксорубицин или эпирубицин, антиметаболит, такой как 5-FU, пеметрексед, гемцитабин или капецитабин, камптотецин, такой как иринотекан или топотекан, противораковое лекарственное средство, такое как паклитаксел или доцетаксел, эпиподофиллотоксин, такой как этопозид или тенипозид, ингибитор протеосом, такой как бортезомиб или противовоспалительные лекарственные средства, такие как целекоксиб или рофекоксиб, необязательно в форме фармацевтически приемлемых солей, в форме гидратов и/или сольватов и необязательно в форме их индивидуальных оптических изомеров, смесей индивидуальных энантиомеров или рацематов.
В другом предпочтительном варианте осуществления настоящее изобретение относится к фармацевтической композиции, определенной выше в настоящем изобретении, в которой дополнительное химиотерапевтическое или природное, полусинтетическое или синтетическое терапевтическое средство 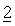
В другом предпочтительном варианте осуществления настоящее изобретение относится к фармацевтической композиции, определенной выше в настоящем изобретении, в которой дополнительное химиотерапевтическое или природное, полусинтетическое или синтетическое терапевтическое средство 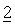
В другом предпочтительном варианте осуществления настоящее изобретение относится к фармацевтической композиции, определенной выше в настоящем изобретении, в которой дополнительное химиотерапевтическое или природное, полусинтетическое или синтетическое терапевтическое средство 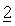
В другом предпочтительном варианте осуществления настоящее изобретение относится к фармацевтической композиции, определенной выше в настоящем изобретении, в которой дополнительное химиотерапевтическое или природное, полусинтетическое или синтетическое терапевтическое средство 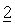
В другом предпочтительном варианте осуществления настоящее изобретение относится к фармацевтической композиции, определенной выше в настоящем изобретении, в которой дополнительное химиотерапевтическое или природное, полусинтетическое или синтетическое терапевтическое средство 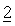
В другом предпочтительном варианте осуществления настоящее изобретение относится к фармацевтической композиции, определенной выше в настоящем изобретении, в которой дополнительное химиотерапевтическое или природное, полусинтетическое или синтетическое терапевтическое средство 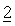
В другом предпочтительном варианте осуществления настоящее изобретение относится к фармацевтической композиции, в которой дополнительное химиотерапевтическое или природное, полусинтетическое или синтетическое терапевтическое средство 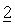
В другом предпочтительном варианте осуществления настоящее изобретение относится к фармацевтической композиции, определенной выше в настоящем изобретении, в которой дополнительное природное, полусинтетическое или синтетическое терапевтическое средство 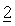
Кроме того, когда соединения 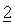
Соединения 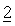
Настоящее изобретение включает в свой объем пролекарства соединения 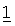
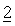
В другом варианте осуществления настоящее изобретение относится к композиции, определенной выше в настоящем изобретении, которая ингибирует пролиферацию различных линий опухолевых клеток человека, включая, но не ограничиваясь только ими, Saos-2, H4, MDA-MB-435S, MDA-MB453, MCF7, HeLa S3, НСТ116, Colo 205, HT29, FaDu, HL-60, K-562, THP-1, HepG2, A549, NCI-H460, GRANTA-519, Raji, Ramos, BRO, SKOV-3, BxPC-3, Mia CaPa-2, DU145, PC-3, NCI-N87, MES-SA, SK-UT-1B и А431.
В другом варианте осуществления настоящее изобретение относится к применению фармацевтической композиции, определенной выше в настоящем изобретении, для приготовления лекарственного средства, предназначенного для лечения онкологических заболеваний, таких как злокачественные неоплазии человека.
В предпочтительном варианте осуществления настоящее изобретение относится к применению фармацевтической композиции определенной выше в настоящем изобретении, в которой онкологическое заболевание выбрано из группы, включающей солидные опухоли.
В другом предпочтительном варианте осуществления настоящее изобретение относится к применению фармацевтической композиции определенной выше в настоящем изобретении, в которой онкологическое заболевание выбрано из группы, включающей раковые заболевания мочеполовой системы (такие как рак предстательной железы, раковые заболевания клеток почечного эпителия, раковые заболевания мочевого пузыря), гинекологические раковые заболевания (такие как раковые заболевания яичников, раковые заболевания шейки матки, раковые заболевания эндометрия), рак легких, раковые заболевания желудочно-кишечного тракта (такие как колоректальные раковые заболевания, рак поджелудочной железы, рак желудка, раковые заболевания пищевода, гепатоцеллюлярные раковые заболевания, холангиоцеллюлярные раковые заболевания), рак головы и шеи, злокачественную мезотелиому, рак молочной железы, злокачественную меланому или саркомы костей и мягких тканей.
В другом предпочтительном варианте осуществления настоящее изобретение относится к применению фармацевтической композиции, определенной выше в настоящем изобретении, в котором онкологическое заболевание выбрано из группы, включающей стойкую или рецидивирующую множественную миелому, острый или хронический миелогенный лейкоз, миелодиспластический синдром, острый лимфобластный лейкоз, ходжкинскую или неходжкинскую лимфому.
В другом предпочтительном варианте осуществления заболевание представляет собой чувствительный к гомонам или стойкий к гормонам рак предстательной железы, карциному яичников, или мелкоклеточный рак легких.
В другом предпочтительном варианте осуществления настоящее изобретение относится к применению композиции, определенной выше в настоящем изобретении, в котором онкологическое заболевание характеризуется неприемлемой пролиферацией, миграцией, апоптозом или ангиогенезом клеток, предпочтительно - неприемлемой пролиферацией клеток. Неприемлемая пролиферация клеток означает пролиферацию клеток, обусловленную неприемлемым ростом клеток, обусловленную избыточным делением клеток, делением клеток, происходящим с повышенной скоростью, и/или неприемлемой продолжительностью существования клеток.
В другом предпочтительном варианте осуществления настоящее изобретение относится к применению, предлагаемому в настоящем изобретении, в котором заболеванием является рак, выбранный из группы, включающей карциномы, саркомы, меланомы, миеломы, опухоли кроветворной системы, лимфомы и раковые заболевания детей.
Примеры карцином в объеме настоящего изобретения включают, но не ограничиваются только ими, аденокарциному (АК), плоскоклеточную карциному (ПКК) и смешанные или недифференцированные карциномы. Карциномы в объеме настоящего изобретения включают, но не ограничиваются только ими, следующие гистологически обнаруживаемые опухоли:
- опухоли головы и шеи: ПКК, АК, раковые заболевания переходных клеток, слизеобразующие плоскоклеточные раковые заболевания, недифференцированные карциномы;
- опухоли центральной нервной системы: астроцитому, глиобластому, менингиому, невриному, шванному, эпендимому, гипофизому, олигодендроглиому, медуллобластому;
- опухоли бронхов и медиастинальные опухоли:
- опухоли бронхов:
- мелкоклеточные раковые заболевания легких (МКРЛ): овсяно-клеточный рак легких, промежуточный рак легких, комбинированный овсяно-клеточный рак легких;
- немелкоклеточные раковые заболевания легких (НМКРЛ): ПКК, веретеноклеточный рак, АК, бронхоальвеолярную карциному, крупноклеточный НМКРЛ, светлоклеточный НМКРЛ;
- мезотелиому;
- тимому;
- карциномы щитовидной железы: папиллярную, фолликулярную, анапластическую, медуллярную;
- опухоли желудочно-кишечного тракта:
- раковые заболевания пищевода: ПКК, АК, анапластический, карциноидный, саркому;
- раковые заболевания желудка: АК, аденосквамозный, анапластический;
- колоректальные раковые заболевания: АК, включая наследственные формы АК, карциноидный, саркому;
- раковые заболевания ануса: ПКК, переходный эпителиальный рак, АК, базально-клеточную карциному;
- раковые заболевания поджелудочной железы: АК, включая раковые заболевания протоков и ацинозные раковые заболевания, папиллярные, аденосквамозные, недифференцированные опухоли поджелудочной железы;
- гепатоцеллюлярную карциному, холангиоцеллюлярную карциному, ангиосаркому, гепатобластому;
- билиарные карциномы: АК, ПКК, мелкоклеточные, недифференцированные;
- желудочно-кишечные стромальные опухоли (ЖКСО);
- гинекологические раковые заболевания:
- раковые заболевания молочной железы: АК, включая инвазивные раковые заболевания протоков, дольковые и медуллярные раковые заболевания, трубчатые, слизеобразующие раковые заболевания, карциному Педжетта, воспалительную карциному, раковые заболевания протоков и дольковую карциному in situ;
- раковые заболевания яичников: эпителиальные опухоли, опухоли половых клеток, недифференцированные опухоли;
- раковые заболевания шейки матки: ПКК, АК, смешанные и недифференцированные опухоли;
- раковые заболевания эндометрия: АК, ПКК, смешанные, недифференцированные опухоли;
- раковые заболевания вульвы: ПКК, АК;
- раковые заболевания влагалища: ПКК, АК;
- раковые заболевания мочевых путей и тестикулярные раковые заболевания:
- тестикулярные раковые заболевания: семиному;
- несеминомные опухоли половых клеток: тератому, карциному эмбриональных клеток, хориокарциному, опухоль желточного мешка, смешанные опухоли клеток Сертоли и лейдиговских клеток;
- опухоли внегонадных половых клеток;
- раковые заболевания предстательной железы: АК, мелкоклеточные, ПКК;
- раковые заболевания клеток почечного эпителия: АК, включая светлоклеточные, папиллярные и хромофобные карциномы, наследственные формы (например, синдром Гиппеля-Линдау), нефробластому;
- раковые заболевания мочевого пузыря: раковые заболевания переходных клеток (уротелиальных), ПКК, АК;
- раковые заболевания уретры: ПКК, раковые заболевания переходных клеток, АК;
- раковые заболевания пениса: ПКК;
- опухоли эндокринных тканей:
- раковые заболевания щитовидной железы: папиллярную, фолликулярную, анапластическую, медуллярную карциномы, включая множественный эндокринный аденоматоз;
- опухоли поджелудочной железы;
- карциноиды;
- опухоли надпочечников, например, феохромоцитому.
Примеры сарком в объеме настоящего изобретения включают, но не ограничиваются только ими, саркому Эвинга, остеосаркому или остеогенную саркому, хондросаркому, синовиальную саркому, лейомиосаркому, рабдомиосаркому, мезотелиальную саркому или мезотелиому, фибросаркому, ангиосаркому или гемангиоэндотелиому, липосаркому, глиому или астроцитому, миксосаркому, злокачественную фиброзную гистиоцитому, мезенхимальную или смешанную мезодермальную опухоль, нейробластому и светлоклеточную саркому.
Примеры опухолей кожи в объеме настоящего изобретения включают, но не ограничиваются только ими, базально-клеточную карциному, карциному клеток Меркеля, сальную карциному, фиброксантому, злокачественную фиброзную гистиоцитому и саркому кожи.
Примеры меланом в объеме настоящего изобретения включают, но не ограничиваются только ими, поверхностную меланому, узелковую и лентиго-злокачественную меланому.
Примеры миелом в объеме настоящего изобретения включают, но не ограничиваются только ими, иммуноцитому, плазмоцитому и множественную миелому.
Примеры раковых заболеваний детей в объеме настоящего изобретения включают, но не ограничиваются только ими, опухоль Вилмса, нейробластому, ретинобластому, рабдомиосаркому, саркому Эвинга и периферические примитивные нейроэктодермальные опухоли, опухоли половых клеток и лимфомы и лейкозы у детей.
В другом предпочтительном варианте осуществления настоящее изобретение относится к применению композиции, определенной выше в настоящем изобретении, в котором раком кроветворной системы является лейкоз.
Другие примеры опухолей кроветворной системы в объеме настоящего изобретения включают, но не ограничиваются только ими, острые и хронические лейкозы миелоидного, эритроидного или лимфатического происхождения, миелодиспластические синдромы (МДС) и миелопролиферативные синдромы (МПС, такие как хронический миелогенный лейкоз, остеомиелофиброз, истинная полицитемия и идиопатическая тромбоцитемия).
Примеры лимфом в объеме настоящего изобретения включают, но не ограничиваются только ими:
- ходжкинскую лимфому;
- неходжкинские лимфомы: Т- и В-клеточные лимфомы
- В-клеточные лимфомы:
- Низкой и средней степени злокачественности: хронический лимфоцитарный лейкоз (ХЛЛ), пролимфоцитарный лейкоз (ПЛЛ), В-клеточную лимфому, волосатоклеточный лейкоз, плазмацитарную лимфому, лимфому клеток ткани, одевающей спорангий, фолликулярную лимфому, лимфому краевого пояса включая лимфому лимфоидной ткани слизистых оболочек;
- Высокой степени злокачественности: диффузную большую В-клеточную лимфому (ДБВКЛ, включая иммунобластные и центробластные варианты), лимфобластную, лимфому Беркитта;
- Т-клеточные лимфомы:
- Низкой степени злокачественности: Т-ХЛЛ, Т-ПЛЛ, грибовидный микоз, синдром Сезари;
- Высокой степени злокачественности: анапластическую крупноклеточную, Т-иммунобластную и лимфобластную.
В другом предпочтительном варианте осуществления настоящее изобретение относится к применению, предлагаемому в настоящем изобретении, в котором заболеванием является рак, выбранный из группы, включающей смешанные опухоли, недифференцированные опухоли и их метастазы.
Примеры смешанных опухолей в объеме настоящего изобретения включают, но не ограничиваются только ими, аденосквамозные карциномы, смешанные мезодермальные опухоли, карциносаркомы и тератокарциномы.
Примеры недифферинцированных, других опухолей или их метастазов в объеме настоящего изобретения включают, но не ограничиваются только ими, недифференцированные опухоли, карциномы неизвестной первичной локализации (КНЛ), метастазы неизвестной первичной локализации (МНЛ) и феохромоцитома, карциноиды.
В другом варианте осуществления настоящее изобретение относится к применению композиции, определенной выше в настоящем изобретении, для приготовления лекарственного средства, предназначенного для лечения аутоиммунных нарушений, выбранных из группы, включающей амилоидоз, системную красную волчанку, ревматоидный артрит, болезнь Крона, рассеянный склероз, системный склероз (склеродерму), смешанное заболевание соединительной ткани, синдром Шегрена, анкилозирующий спондилит, аутоиммунный васкулит, синдром Бехчета, псориаз, аутоиммунный артрит, саркоидоз и сахарный диабет.
В другом варианте осуществления настоящее изобретение относится к применению фармацевтической композиции, определенной выше в настоящем изобретении, для приготовления лекарственного средства, предназначенного для лечения других неонкологических заболеваний, таких как диабетическая ретинопатия и ревматоидный артрит.
В другом варианте осуществления настоящее изобретение относится к применению композиции, определенной выше в настоящем изобретении, в котором композицию, предлагаемую в настоящем изобретении, вводят перорально, энтерально, чрескожно, внутривенно, внутрибрюшинно или путем инъекции, предпочтительно - внутривенно.
В другом варианте осуществления настоящее изобретение относится к набору препаратов фармацевтической комбинации, предназначенному для лечения заболеваний, включающих пролиферацию клеток, миграцию или апоптоз миеломных клеток, или ангиогенез, включающему терапевтически эффективное количество соединения 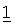
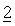
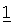
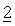
В предпочтительном варианте осуществления настоящее изобретение относится к набору препаратов фармацевтической комбинации, в котором препарат соединения 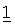
В другом варианте осуществления настоящее изобретение относится к применению фармацевтической комбинации или набора препаратов фармацевтической комбинации для приготовления лекарственного средства, необязательно приспособленного для совместного лечения с лучевой терапией или радиоиммунотерапией, для лечения заболеваний, включающих пролиферацию клеток, миграцию или апоптоз раковых клеток, или ангиогенез, в организме человека или млекопитающего, не являющегося человеком.
В другом варианте осуществления настоящее изобретение относится к применению эффективного количества соединения 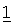
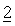
В другом варианте осуществления настоящее изобретение относится к способу лечения заболеваний, включающих пролиферацию клеток, миграцию или апоптоз раковых клеток, или ангиогенез, и этот способ включает одновременное, раздельное или последовательное совместное введение эффективного количества:
(i) соединения 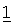
(ii) по меньшей мере дополнительного химиотерапевтического или природного, полусинтетического или синтетического терапевтического средства 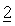
В другом варианте осуществления настоящее изобретение относится к применениям, описанным выше, характеризующимся тем, что соединение 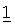
Термин "терапевтически эффективное количество" означает количество лекарственного средства или фармацевтического агента, которое обеспечивает биологический или лекарственный ответ ткани, системы, животного или человека, необходимый для исследователя или клинициста.
При использовании в настоящем изобретении термин "композиция" означает продукт, содержащий указанные ингредиенты в указанных количествах, а также любой продукт, который прямо или косвенно образуется из комбинации указанных ингредиентов в указанных количествах.
Как уже отмечено выше, в контексте настоящего изобретения компоненты 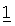
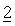
В контексте настоящего изобретения элементы комбинации 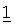
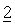
В предпочтительном варианте осуществления элемент 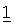
Фармацевтические композиции, предназначенные для введения компонентов 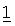
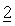
Фармацевтические композиции, содержащие активные ингредиенты 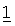
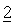
Дозированные формы, предназначенные для перорального применения, можно приготовить с помощью любой из методик, известных в области приготовления фармацевтических препаратов и таких композиций.
Инертными наполнителями могут быть, например: (а) инертные разбавители, такие как маннит, сорбит, карбонат кальция, предварительно желатинизированный крахмал, лактоза, фосфат кальция или фосфат натрия; (b) гранулированные и измельченные агенты, такие как повидон, коповидон, гидроксипропилметилцеллюлоза, кукурузный крахмал, альгиновая кислота, кросповидон, натриевая соль гликолята крахмала, кроскармелоза или полакрилин калия; (с) связующие агенты, такие как микрокристаллическая целлюлоза или камедь акации; и (d) смазывающие агенты, такие как стеарат магния, стеариновая кислота, фумаровая кислота или тальк.
В некоторых случаях препараты, предназначенные для перорального применения, могут находиться в форме капсул из твердого желатина или ГПМЦ (гидроксипропилметилцеллюлоза), в которых активные ингредиенты 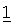
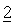
Таблетки, капсулы или пеллеты могут не содержать покрытия или на них по известным методикам может быть нанесено покрытие, предназначенное для задержки распада и всасывания в желудочно-кишечном тракте и тем самым обеспечения задержанного действия или пролонгированного действия в течение более длительного периода времени. Например, можно использовать вещество, обеспечивающее задержку по времени, такое как ацетат-фталат целлюлозы или ацетат-сукцинат гидроксипропилцеллюлозы, или вещество, обеспечивающее пролонгированное высвобождение, такое как этилцеллюлоза или аммонийметакрилатный сополимер (типа В).
Жидкие дозированные формы, предназначенные для перорального введения, предлагаемые в настоящем изобретении, включают фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы и эликсиры, содержащие инертные разбавители, обычно применяющиеся в данной области техники, такие как воду. Кроме таких инертных разбавителей композиции также могут включать вспомогательные вещества, такие как смачивающие агенты, эмульгирующие и суспендирующие агенты и подсластители, вкусовые добавки, отдушки и консерванты.
Водные суспензии, предлагаемые в настоящем изобретении, обычно содержат активные соединения 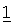
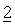
Водные суспензии также могут содержать: один или большее количество консервантов, например, этил- или н-пропил-п-гидроксибензоат; один или большее количество красителей; одну или большее количество вкусовых добавок; и один или большее количество подсластителей, таких как сахароза или сахарин.
Масляные суспензии, предлагаемые в настоящем изобретении, можно приготовить путем суспендирования активных ингредиентов 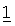
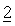
Диспергирующиеся порошки и гранулы являются композициями, пригодными для приготовления водных суспензий, предлагаемых в настоящем изобретении. В этих композициях активные ингредиенты 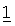
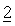
Фармацевтические композиции, предлагаемые в настоящем изобретении, также могут находиться в форме эмульсий типа масло-в-воде. Масляной фазой может быть растительное масло, такое как оливковое масло или арахисовое масло, или минеральное масло, такое как жидкий парафин, или их смеси.
Подходящими эмульгирующими агентами могут быть (а) природные камеди, такие как камедь акации и трагакантовая камедь, (b) природные фосфатиды, такие как соя и лецитин, (с) полные или частичные сложные эфиры, полученные из жирных кислот и ангидридов гексита, например, сорбитмоноолеат, (d) продукты конденсации указанных частичных сложных эфиров с этиленоксидом, например, полиоксиэтиленсорбитмоноолеат. Эмульсии также могут содержать подсластители и вкусовые добавки.
Сиропы и эликсиры, предлагаемые в настоящем изобретении, можно приготовить с подсластителями, например, глицерином, пропиленгликолем, сорбитом или сахарозой. Такие композиции также могут содержать консерванты и вкусовые добавки и красители.
Фармацевтические композиции, содержащие 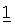
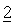
Препараты для парентерального введения, предлагаемые в настоящем изобретении, содержащие 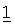
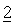
Примерами неводных растворителей или разбавителей, подходящих для препаратов, предлагаемых в настоящем изобретении, являются пропиленгликоль, полиэтиленгликоль, растительные масла, такие как оливковое масло и кукурузное масло, желатин, и пригодные для инъекций органические сложные эфиры, такие как этилолеат. Такие дозированные формы также могут содержать вспомогательные вещества, такие как консерванты, смачивающие, эмульгирующие и диспергирующие агенты. Их можно стерилизовать, например, путем фильтрования через удерживающий бактерии фильтр, путем включения в композиции стерилизующих агентов, путем облучения композиций или путем нагревания композиций. Их также можно приготовить в виде стерильных твердых композиций, которые можно восстановить в стерильной воде или какой-либо другой стерильной среде непосредственно перед применением.
Элементы 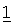
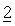
Композиции для буккального, назального или сублингвального введения, предлагаемые в настоящем изобретении, можно получить с использованием стандартных инертных наполнителей, хорошо известных в данной области техники.
Для местного введения элементы 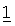
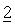
Дозировка активных ингредиентов в композициях, предлагаемых в настоящем изобретении, может меняться, хотя количество активных ингредиентов 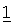
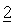
Ниже настоящее изобретение иллюстрируется с помощью примеров фармацевтических композиций, включающих соединение 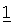
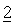
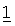
Комбинация приведенного в качестве примера соединения №46, представленного в таблице 1, и иринотекана (исследование модели рака ободочной кишки НСТ 116 с использованием комбинации)
Задача исследования
Приведенное в качестве примера соединение №46, представленное в таблице 1, является активным и селективным ингибитором серин/треонинкиназы ППК-1. Иринотекан (продается под торговым названием Campto®) является стандартным химиотерапевтическим средством для лечения колоректальной карциномы. Предшествующие исследования показали, что приведенное в качестве примера соединение №46, представленное в таблице 1, и иринотекан активны по отношению к полученным из НСТ 116 опухолям голых мышей. Задачей настоящего исследования является оценка противораковой эффективности субоптимальных доз приведенного в качестве примера соединения №46, представленного в таблице 1, иринотекана и комбинации приведенного в качестве примера соединения №46, представленного в таблице 1, и иринотекана, при карциноме ободочной кишки человека, модель НСТ 116, выращенной в виде подкожных ксенотрансплантатов у голых мышей. Субоптимальные дозы обоих соединений использовали, чтобы облегчить обнаружение аддитивных, синергетических или антагонистических эффектов.
Схема исследования
Модель: Карцинома ободочной кишки человека, модель НСТ 116, выращенная в виде подкожных ксенотрансплантатов у голых мышей.
Группы подопытных животных (10 животных в группе):
Контроли - растворитель, ВВ (внутривенно), один раз в неделю в течение 6 недель ((q7d)×6)
Приведенное в качестве примера соединение №46, представленное в таблице 1 30 мг/кг, ВВ, один раз в неделю в течение 10 недель ((q7d)×10)
Иринотекан 12,5 мг/кг, ВБ, один раз в неделю в течение 10 недель ((q7d)×10)
Комбинация 30 мг/кг приведенного в качестве примера соединения №46, представленного в таблице 1, ВВ, один раз в неделю в течение 10 недель ((q7d)×10) и 12,5 мг/кг иринотекана, ВБ, один раз в неделю (~1 ч после приведенного в качестве примера соединения №46, представленного в таблице 1) в течение 10 недель ((q7d)×10)
Объемы опухолей и массы животных регистрировали 3 раза в неделю. Оценка результатов лечения основывалась на абсолютных объемах отдельных опухолей.
Материалы и методики:
Использовали самок мышей BomTac:NMRI-nu/nu. Приведенное в качестве примера соединение №46, представленное в таблице 1, растворяли в хлористоводородной кислоте (0,1 н.) разбавляли с помощью 0,9% NaCl и внутривенно вводили в хвостовую вену. Концентрат для вливания иринотекана разбавляли с помощью 0,9% NaCl и вводили путем внутрибрюшинной инъекции. Вводимый объем составлял 10 мл/(кг массы тела) для обоих соединений. Опухоли НСТ 116 образовывали из клеток культур НСТ 116. Объемы опухолей определяли три раза в неделю с помощью штангенциркуля. В те же дни определяли массы мышей, являющиеся показателями переносимости. Пробы плазмы брали в последний день лечения.
Основные результаты (см. фиг.1.1-1.3)
Краткое описание чертежей
Фиг.1.1
Ответы опухоли НСТ 116 на лечение с помощью 30 мг/кг приведенного в качестве примера соединения №46, представленного в таблице 1, 12,5 мг/кг иринотекана или обоих.
Мышь, у которой имелась опухоль НСТ 116, лечили внутривенно с помощью 30 мг/кг приведенного в качестве примера соединения №46, представленного в таблице 1, один раз в неделю ((q7d)×10), с помощью 12,5 мг/кг иринотекана один раз в неделю ((q7d)×10), с помощью обоих средств параллельно ((q7d)×10) или один раз в неделю с помощью только растворителя, и строили зависимости средних объемов опухолей от времени. День 1 был первым днем, день 64 был последним днем лечения и день 121 был заключительным днем исследования. Треугольники указывают дни лечения.
Фиг.1.2
Количество дней, через которое объемы опухолей НСТ 116 достигают 1000 мм3.
Мышь, у которой имелась опухоль НСТ 116, лечили с помощью 30 мг/кг приведенного в качестве примера соединения №46, представленного в таблице 1, ВВ один раз в неделю ((q7d)×10), с помощью 12,5 мг/кг иринотекана ВБ (внутрибрюшинно) один раз в неделю ((q7d)×10), или комбинации обоих соединений ((q7d)×10) в соответствующих дозах. Мышей, которых лечили растворителем (один раз в неделю), использовали в качестве контрольных. На график наносили количества дней, через которое объемы индивидуальных опухолей НСТ 116 достигают 1000 мм3. Каждый символ относится к одной индивидуальной опухоли. Горизонтальные отрезки указывают среднее количество дней.
Фиг.1.3
Изменение массы тела в ответ на лечение с помощью 30 мг/кг приведенного в качестве примера соединения №46, представленного в таблице 1, 12,5 мг/кг иринотекана или обоих.
Мышь, у которой имелась опухоль НСТ 116, лечили внутривенно с помощью 30 мг/кг приведенного в качестве примера соединения №46, представленного в таблице 1, один раз в неделю ((q7d)×10), с помощью 12,5 мг/кг иринотекана один раз в неделю ((q7d)×10), с помощью обоих средств параллельно ((q7d)×10) или один раз в неделю с помощью только растворителя и построены зависимости средних изменений массы тела от времени. День 1 был первым днем, день 64 был последним днем лечения и день 121 был заключительным днем исследования. Треугольники указывают дни лечения.
Результаты на день 39 (завершение для контрольных животных):
30 мг/кг Приведенного в качестве примера соединения №46, представленного в таблице 1, ВВ значительно задерживает рост опухоли НСТ 116 (Т/С=20%, p<0,001)
12,5 мг/кг Иринотекана ВБ значительно задерживает рост опухоли (Т/С=25%, p<0,001)
Совместное введение 30 мг/кг приведенного в качестве примера соединения №46, представленного в таблице 1, и 12,5 мг/кг иринотекана значительно задерживает рост опухоли (Т/С=8%, p<0,001).
30 мг/кг Приведенного в качестве примера соединения №46, представленного в таблице 1, 12,5 мг/кг иринотекан и их комбинация хорошо переносятся. Увеличение массы тела контрольных мышей составило 10,3%. У мышей, которых лечили с помощью 30 мг/кг приведенного в качестве примера соединения №46, представленного в таблице 1, увеличение массы тела составило 8,6%, у мышей, которых лечили с помощью 12,5 мг/кг иринотекана, увеличение массы тела в среднем составило 5,9% и у мышей, которых лечили комбинацией увеличение массы тела составило 5,5%.
Результаты на день 121 (конец исследования):
Проводимое один раз в неделю лечение (до дня 64) с помощью приведенного в качестве примера соединения №46, представленного в таблице 1, иринотекана или их комбинации задерживало среднее время достижения опухолью объема, равного 1000 мм3, до 33,7 дней, 35,1 дней или 56,0 дней соответственно.
Выводы
Лечение с помощью субоптимальных доз приведенного в качестве примера соединения №46, представленного в таблице 1, или иринотекана значительно задерживает рост опухоли и хорошо переносится.
Лечение комбинацией субоптимальных доз приведенного в качестве примера соединения №46, представленного в таблице 1, и иринотекана приводит к значительной задержке роста и большей эффективности, чем при использовании любого из соединений по отдельности, без ухудшения переносимости.
Сопоставление задержек роста (время, через которое объемы опухолей достигают 1000 мм3) свидетельствует об аддитивном/синергетическом эффекте.
Комбинация приведенного в качестве примера соединения №46, представленного в таблице 1, и доцетаксела (модель NCI-H460 легких)
Задача исследования
С помощью приведенного в качестве примера соединения №46, представленное в таблице 1, является активным и селективным ингибитором серин/треонинкиназы ППК-1. Доцетаксел (продается под торговым названием Таксотер®) является стандартным химиотерапевтическим средством для лечения рака легких. Предшествующие исследования показали, что приведенное в качестве примера соединение №46, представленное в таблице 1, для голых мышей активно по отношению к ксеротрансплантатам, полученным из линии клеток NCI-H460 рака легких человека. Задачей настоящего исследования является оценка противоракового воздействия субоптимальных доз приведенного в качестве примера соединения №46, представленного в таблице 1, и доцетаксела на рост опухоли NCI-H460 при введении по отдельности или в комбинации.
Субоптимальные дозы обоих соединений использовали, чтобы облегчить обнаружение аддитивных, синергетических или антагонистических эффектов.
Схема исследования
Модель: Немелкоклеточная карцинома легких человека, модель NCI-H460, выращенная в виде подкожных ксенотрансплантатов у голых мышей.
Группы подопытных животных (внутривенное введение, 10 животных в группе):
Контроли - растворитель, один раз в неделю в течение 4 недель ((q7d)×4)
Приведенное в качестве примера соединение №46, представленное в таблице 1 50 мг/кг, один раз в неделю в течение 4 недель ((q7d)×4)
Доцетаксел 15 мг/кг, один раз в неделю в течение 4 недель ((q7d)×4)
Комбинация 50 мг/кг приведенного в качестве примера соединения №46, представленного в таблице 1, один раз в неделю в течение 4 недель ((q7d)×4) и 15 мг/кг доцетаксела, один раз в неделю (через 3 дня после приведенного в качестве примера соединения №46, представленного в таблице 1) в течение 4 недель ((q7d)×4)
Объемы опухолей и массы животных регистрировали 3 раза в неделю. Оценка результатов лечения основывалась на абсолютных объемах отдельных опухолей.
Материалы и методики:
Использовали самок мышей BomTac:NMRI-nu/nu. Приведенное в качестве примера соединение №46, представленное в таблице 1, растворяли в хлористоводородной кислоте (0,1 н.) разбавляли с помощью 0,9% NaCl и внутривенно вводили в хвостовую вену. Концентрат для вливания доцетаксела разбавляли с помощью 0,9% NaCl и вводили внутривенно. Вводимый объем составлял 10 мл/(кг массы тела). Опухоли NCI-H460 образовывали из клеток культур NCI-H460. Объемы опухолей определяли три раза в неделю с помощью штангенциркуля. В те же дни определяли массы мышей, являющиеся показателями переносимости. Пробы плазмы брали в последний день лечения.
Основные результаты (см. фиг.2.1-2.3)
Краткое описание чертежей
Фиг.2.1
Ответы опухоли NCI-H460 на лечение с помощью 50 мг/кг приведенного в качестве примера соединения №46, представленного в таблице 1, 15 мг/кг доцетаксела или обоих.
Мышь, у которой имелась опухоль NCI-H460, лечили внутривенно с помощью 50 мг/кг приведенного в качестве примера соединения №46, представленного в таблице 1, один раз в неделю ((q7d)×4), с помощью 15 мг/кг доцетаксела один раз в неделю ((q7d)×4), с помощью обоих средств параллельно или один раз в неделю с помощью только растворителя, и строили зависимости средних объемов опухолей от времени. День 1 был первым днем, день 25 был последним днем лечения и день 43 был последним днем расчета среднего объема опухоли. Треугольники указывают дни лечения.
Фиг.2.2
Количество дней, через которое объемы опухолей NCI-H4606 достигают 1000 мм3.
Мышь, у которой имелась опухоль NCI-H460, лечили с помощью 50 мг/кг приведенного в качестве примера соединения №46, представленного в таблице 1, ВВ один раз в неделю ((q7d)×4), с помощью 15 мг/кг доцетаксела ВВ один раз в неделю ((q7d)×4), или комбинации обоих соединений в таких же дозах. Мышей, которых лечили растворителем (один раз в неделю), использовали в качестве контрольных. На график наносили количества дней, через которое объемы индивидуальных опухолей NCI-H460 достигают 1000 мм3. Каждый символ относится к одной индивидуальной опухоли. Горизонтальные отрезки указывают среднее количество дней.
Фиг.2.3
Изменение массы тела в ответ на лечение с помощью 50 мг/кг приведенного в качестве примера соединения №46, представленного в таблице 1, 15 мг/кг доцетаксела или обоих.
NCI-H460 Мышь, у которой имелась опухоль, лечили внутривенно с помощью 50 мг/кг приведенного в качестве примера соединения №46, представленного в таблице 1, один раз в неделю ((q7d)×4), с помощью 15 мг/кг доцетаксела один раз в неделю ((q7d)×4), с помощью обоих средств параллельно или один раз в неделю с помощью только растворителя и построены зависимости средних изменений массы тела от времени. День 1 был первым днем, день 25 был последним днем лечения и день 43 был последним днем расчета средних масс тела. Треугольники указывают дни лечения.
Результаты на день 17 (завершение для контрольных животных)
50 мг/кг приведенного в качестве примера соединения №46, представленного в таблице 1, ВВ один раз в неделю не приводило к значительной задержке роста опухоли NCI-H460 (Т/С=65%, p>0,05).
15 мг/кг Доцетаксела ВВ один раз в неделю значительно задерживает рост опухоли (Т/С=42%, p<0,05).
Совместное введение 50 мг/кг приведенного в качестве примера соединения №46, представленного в таблице 1, и 15 мг/кг доцетаксела значительно задерживает рост опухоли (Т/С=26%, p<0,001).
50 мг/кг Приведенного в качестве примера соединения №46, представленного в таблице 1, хорошо переносится.
15 мг/кг Доцетаксела, введенного отдельно или в комбинации, не характеризуется хорошей переносимостью. У мышей, которых лечили с помощью 15 мг/кг доцетаксела, ко дню 17 уменьшение массы тела в среднем составляло 4,8%. Комбинация приведенного в качестве примера соединения №46, представленного в таблице 1, и доцетаксела приводила к уменьшению массы тела на 8,3%.
Результаты к дню 71 (конец исследования)
Проводимое один раз в неделю лечение (до дня 25) с помощью приведенного в качестве примера соединения №46, представленного в таблице 1, доцетаксела или их комбинации задерживало среднее время достижения опухолью объема, равного 1000 мм3, до 4,0 дней, 10,5 дней или 28,0 дней по сравнению с контрольными животными соответственно.
У мышей, которых лечили с помощью 15 мг/кг доцетаксела, происходило дополнительное уменьшение массы тела (до 9,4% к дню 24) и одну мышь пришлось умертвить вследствие сильного уменьшения массы тела. У мышей, которых лечили одновременно с помощью приведенного в качестве примера соединения №46, представленного в таблице 1, и доцетаксела, происходило дополнительное уменьшение массы тела (до 10,4% к дню и двух мышей пришлось умертвить вследствие сильного уменьшения массы тела.
Выводы
Лечение с помощью субоптимальных доз приведенного в качестве примера соединения №46, представленного в таблице 1, не приводило к значительной задержке роста опухоли (Т/С=65%, p>0,05).
Доцетаксел значительно задерживает рост опухоли (Т/С=42%, p<0,05).
Комбинация приведенного в качестве примера соединения №46, представленного в таблице 1, и доцетаксела приводит к значительной задержке роста по сравнению с контрольными животными (Т/С=26%, p<0,001). Отличие от лечения с помощью только приведенного в качестве примера соединения №46, представленного в таблице 1, также является статистически значимым (p<0,01) и указывает на то, что эти два средства могут действовать по меньшей мере аддитивно.
Комбинация приведенного в качестве примера соединения №46, представленного в таблице 1, и гемцитабина (модель для ВхРС-3 поджелудочной железы)
Задача исследования
Приведенное в качестве примера соединение №46, представленное в таблице 1, является активным и селективным ингибитором серин/треонинкиназы ППК-1. Гемцитабин (продается под торговым названием Gemzar®) является стандартным химиотерапевтическим средством для лечения аденокарциномы поджелудочной железы. Задачей настоящего исследования является оценка противораковой эффективности субоптимальных доз приведенного в качестве примера соединения №46, представленного в таблице 1, гемцитабина и их комбинации при аденокарциноме поджелудочной железы человека, модель ВхРС-3, выращенной в виде подкожных ксенотрансплантатов у голых мышей. Субоптимальные дозы обоих соединений использовали, чтобы облегчить обнаружение аддитивных, синергетических или антагонистических эффектов.
Схема исследования
Модель: Аденокарцинома человека, модель ВхРС-3, выращенная в виде подкожных ксенотрансплантатов у голых мышей.
Группы подопытных животных (10 животных в группе):
Контроли - растворитель, ВВ, один раз в неделю в течение 4 недель ((q7d)×4)
С помощью приведенного в качестве примера соединения №46, представленное в таблице 1
50 мг/кг, ВВ, один раз в неделю в течение 6 недель ((q7d)×6)
Гемцитабин 100 мг/кг, ВБ, один раз в неделю в течение 6 недель ((q7d)×6)
Комбинация 50 мг/кг приведенного в качестве примера соединения №46, представленного в таблице 1, ВВ, один раз в неделю в течение 6 недель ((q7d)×6) и 100 мг/кг гемцитабина, ВБ, один раз в неделю (~1 ч после приведенного в качестве примера соединения №46, представленного в таблице 1) в течение 6 недель ((q7d)×6)
Объемы опухолей и массы животных регистрировали 3 раза в неделю. Оценка результатов лечения основывалась на абсолютных объемах отдельных опухолей.
Материалы и методики:
Использовали самок мышей BomTac:NMRI-nu/nu. Приведенное в качестве примера соединение №46, представленное в таблице 1, растворяли в хлористоводородной кислоте (0,1 н.) разбавляли с помощью 0,9% NaCl и внутривенно вводили в хвостовую вену. Концентрат для вливания гемцитабина разбавляли с помощью 0,9% NaCl и вводили путем внутрибрюшинной инъекции. Вводимый объем составлял 10 мл/(кг массы тела) для обоих соединений. Опухоли ВхРС-3 образовывали из клеток культур ВхРС-3. Объемы опухолей определяли три раза в неделю с помощью штангенциркуля. В те же дни определяли массы мышей, являющиеся показателями переносимости. Пробы плазмы брали в последний день лечения.
Основные результаты (см. фиг.3.1-3.2)
Краткое описание чертежей
Фиг.3.1
Ответы опухоли ВхРС-3 на лечение с помощью 50 мг/кг приведенного в качестве примера соединения №46, представленного в таблице 1, 100 мг/кг гемцитабина или обоих.
Мышь, у которой имелась опухоль ВхВС-3, лечили внутривенно с помощью 50 мг/кг приведенного в качестве примера соединения №46, представленного в таблице 1, один раз в неделю ((q7d)×6), с помощью 100 мг/кг гемцитабин один раз в неделю ((q7d)×6), с помощью обоих средств параллельно или один раз в неделю с помощью только растворителя, и строили зависимости средних объемов опухолей от времени. День 1 был первым днем, день 36 был последним днем лечения и день 26 был последним днем расчета среднего объема опухоли.
Фиг.3.2
Изменение массы тела в ответ на лечение с помощью 50 мг/кг приведенного в качестве примера соединения №46, представленного в таблице 1, 100 мг/кг гемцитабина или обоих.
Мышь, у которой имелась опухоль ВхРС-3, лечили внутривенно с помощью 50 мг/кг приведенного в качестве примера соединения №46, представленного в таблице 1, один раз в неделю ((q7d)×6), с помощью 100 мг/кг гемцитабина один раз в неделю ((q7d)×6), с помощью обоих средств параллельно или один раз в неделю с помощью только растворителя и построены зависимости средних изменений массы тела от времени. День 1 был первым днем, день 36 был последним днем лечения и день 43 был последним днем расчета средних масс тела. Треугольники указывают дни лечения.
Результаты на день 26 (завершение для контрольных животных):
50 мг/кг Приведенного в качестве примера соединения №46, представленного в таблице 1, ВВ значительно задерживает рост опухоли НСТ 116 (Т/С=28%).
100 мг/кг Гемцитабина ВБ лишь незначительно задерживают рост опухоли (Т/С=65%). Более значительные дозы гемцитабина не являлись переносимыми.
Совместное введение 50 мг/кг приведенного в качестве примера соединения №46, представленного в таблице 1, и 100 мг/кг гемцитабина задерживает рост опухоли в такой степени, как и введение только одного приведенного в качестве примера соединения №46, представленного в таблице 1 (Т/С=24%).
50 мг/кг Приведенного в качестве примера соединения №46, представленного в таблице 1, 100 мг/кг гемцитабина и их комбинация хорошо переносятся. Увеличение массы тела контрольных мышей составило 6,2%. У мышей, которых лечили с помощью 50 мг/кг приведенного в качестве примера соединения №46, представленного в таблице 1, увеличение массы тела составило 8,2%, у мышей, которых лечили с помощью 100 мг/кг гемцитабин увеличение массы тела в среднем составило 8,8% и у мышей, которых лечили комбинацией увеличение массы тела составило 8,5%.
Результаты на день 43 (конец исследования):
Проводимое один раз в неделю лечение (до дня 36) с помощью приведенного в качестве примера соединения №46, представленного в таблице 1, или комбинации приведенного в качестве примера соединения №46, представленного в таблице 1, и гемцитабина задерживало среднее время достижения опухолью объема, равного 1000 мм3, в такой же степени. Не обнаружено статистически значимых различий между двумя группами подопытных животных.
Выводы
Лечение с помощью субоптимальных доз приведенного в качестве примера соединения №46, представленного в таблице 1, значительно задерживает рост опухоли и хорошо переносится. В отличие от этого, максимальная переносимая доза гемцитабина не характеризуется значительной противоопухолевой активностью.
Лечение комбинацией субоптимальных доз приведенного в качестве примера соединения №46, представленного в таблице 1, и гемцитабина, приводит к задержке роста, сравнимой с наблюдающейся для лечения с помощью только одного приведенного в качестве примера соединения №46, представленного в таблице 1, что указывает на то, что в данной модели эти два средства не воздействуют антагонистически.
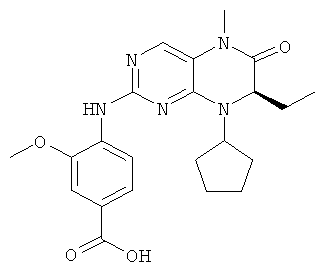
Методика получения приведенного в качестве примера соединения №46, представленного в таблице 1, т.е. соединения 4-[[(7R)-8-циклопентил-7-этил-5,6,7,8-тетрагидро-5-метил-6-оксо-2-птеридинил]амино]-3-метокси-N-(1-метил-4-пиперидинил)-бензамида, описана в WO 03/20722, а также в WO 04/76454, которые включены в настоящее изобретение в качестве ссылки.
Однако для полноты ниже также описана методика получения соединения 4-[[(7R)-8-циклопентил-7-этил-5,6,7,8-тетрагидро-5-метил-6-оксо-2-птеридинил]амино]-3-метокси-N-(1-метил-4-пиперидинил)-бензамида. Следует понимать, что эта методика является иллюстрацией настоящего изобретения и не налагает ограничения на его объект.
Синтез 4-[[(7R)-8-циклопентил-7-этил-5,6,7,8-тетрагидро-5-метил-6-оксо-2-птеридинил]амино]-3-метокси-N-(1-метил-4-пиперидинил)-бензамида
Для синтеза сначала получают промежуточное соединение Z3, как описано ниже.
54,0 г (0,52 моля) D-2-Аминомасляной кислоты суспендируют в 540 мл метанола и медленно объединяют с 132 г (1,1 моля) тионилхлорида при охлаждении льдом. Смесь кипятят с обратным холодильником в течение 1,5 ч и затем выпаривают. Оставшееся масло объединяют с 540 мл трет-бутилметилового эфира и образовавшиеся бесцветные кристаллы отфильтровывают с отсасыванием.
Выход: 78,8 г соединения Z3a (бесцветные кристаллы)
74,2 г Соединения Z3a и 43,5 мл (0,49 моля) циклопентанона растворяют в 800 мл дихлорметана. После прибавления 40,0 г (0,49 моля) ацетата натрия и 150,0 г (0,71 моля) триацетоксиборогидрида натрия при 0°С смесь перемешивают в течение 12 ч при температуре окружающей среды и затем прибавляют 500 мл 20% раствора гидрокарбоната натрия. Водную фазу экстрагируют дихлорметаном. Объединенные органические фазы промывают водой, сушат над MgSO4 и выпаривают.
Выход: 85,8 г соединения Z3b (светло-желтое масло)
40,0 г Соединения Z3b и 30,0 г (0,22 моля) карбоната калия суспендируют в 600 мл ацетона и объединяют с 45,0 г (0,23 моля) 2,4-дихлор-5-нитропиримидина в 200 мл ацетона при охлаждении льдом. Через 12 ч прибавляют еще 5,0 г 2,4-дихлор-5-нитропиримидина и перемешивают в течение 3 ч. Реакционную смесь выпаривают, растворяют в 800 мл этилацетата и 600 мл воды и водную фазу экстрагируют этилацетатом. Объединенные органические фазы промывают водой, сушат над MgSO4 и выпаривают.
Выход: 75,0 г соединения Z3c (коричневое масло)
100 г Соединения Z3c растворяют в 650 мл ледяной уксусной кислоты и при 70°С порциями прибавляют 20 г порошкообразного железа. Смесь перемешивают в течение 1 ч при 70°С, затем в течение 1,5 ч при 100°С и затем в горячем виде фильтруют через диатомит (кизельгур). Реакционную смесь выпаривают, растворяют в смеси метанол/дихлорметан, вносят в силикагель и очищают этилацетатом путем экстракции в аппарате Сокслетта. Растворитель удаляют и остаток перемешивают с метанолом.
Выход: 30,0 г соединения Z3d (светло-коричневые кристаллы)
25,0 г Соединения Z3d и 6,5 мл (0,1 моля) метилйодида помещают в 250 мл диметилацетамида и при -10°С прибавляют 3,8 г (0,95 моля) гидрида натрия в виде 60% дисперсии в минеральном масле. Смесь перемешивают в течение 20 мин при 0°С, затем в течение 30 мин при температуре окружающей среды и в заключение прибавляют лед. Реакционную смесь выпаривают и объединяют с 300 мл воды. Образовавшийся осадок отфильтровывают с отсасыванием и промывают петролейным эфиром.
Выход: 23,0 г соединения Z3e (бесцветное твердое вещество)
6,0 г Соединения Z3e и 5,1 г (31 ммоля) 4-амино-3-метоксибензойной кислоты суспендируют в 90 мл этанола и 350 мл воды, объединяют с 3,5 мл концентрированной хлористоводородной кислоты и кипятят с обратным холодильником в течение 48 ч. Реакционную смесь выпаривают, остаток перемешивают со смесью метанол/диэтиловый эфир и образовавшийся осадок отфильтровывают с отсасыванием.
Выход: 6,3 г соединения Z3 (светло-бежевые кристаллы)
4-[[(7R)-8-Циклопентил-7-этил-5,6,7,8-тетрагидро-5-метил-6-оксо-2-птеридинил]амино]-3-метокси-N-(1-метил-4-пиперидинил)-бензамид получают так, как описано ниже.
0,15 г Соединения Z3, 0,12 г ТМУТ, 0,12 мл ДИПЭА растворяют в 5 мл дихлорметана и перемешивают в течение 30 мин при 25°С. Затем прибавляют 50 мг 1-метил-4-аминопиперидина и смесь перемешивают в течение еще 2,5 ч при 25°С. Затем раствор экстрагируют водой и затем выпаривают. Остаток растворяют в теплом этилацетате и кристаллизуют из эфира и петролейного эфира.
Выход: 0,025 г белых кристаллов. Т.пл. (температура плавления): 203°С в виде основания.
Все соединения формулы (I), предлагаемые в настоящем изобретении, можно получить по методикам синтеза А, описанным ниже в настоящем изобретении, где заместители общих формул (А1)-(А9) обладают приведенными ниже значениями. Следует понимать, что эта методика является иллюстрацией настоящего изобретения и не налагает ограничения на его объект.
Методика А
Стадия 1А
Соединение формулы (А1) вводят в реакцию с соединением формулы (А2) и получают соединение формулы (A3) (диаграмма 1А). Эту реакцию можно проводить в соответствии с WO 00/43369 или WO 00/43372. Соединение (А1) продает, например, фирма City Chemical LLC, расположенная по адресу 139 Allings Crossing Road, West Haven, CT, 06516, USA. Соединение (А2) можно получить по методикам, известным из литературы: (a) F. Effenberger, U. Burkhart, J. Willfahrt Liebigs Ann. Chem. 1986, 314-333; (b) Т. Fukuyama, С.-K. Jow, M. Cheung, Tetrahedron Lett. 1995, 36, 6373-6374; (с) R.K. Olsen, J. Org. Chem. 1970, 35, 1912-1915; (d) F.E. Dutton, B.H. Byung Tetrahedron Lett. 1998, 30, 5313-5316; (e) J.M. Ranajuhi, M.M. Joullie Synth. Commun. 1996, 26, 1379-1384.).
Диаграмма 1А
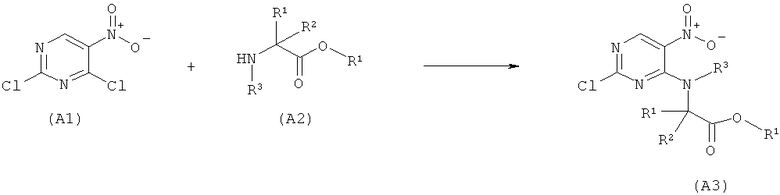
На стадии 1А 1 экв. соединения (А1) и 1-1,5 экв., предпочтительно - 1,1 экв. основания, предпочтительно - карбоната калия, гидрокарбоната калия, карбоната натрия или гидрокарбоната натрия, карбоната кальция, наиболее предпочтительно - карбоната калия, перемешивают в разбавителе, необязательно смешанном с водой, например, ацетоне, тетрагидрофуране, диэтиловом эфире, циклогексане, петролейном эфире или диоксане, предпочтительно - циклогексане или диэтиловом эфире.
При температуре, равной от 0 до 15°С, предпочтительно - от 5 до 10°С, по каплям прибавляют 1 экв. аминокислоты формулы (А2), растворенной в органическом растворителе, например, ацетоне, тетрагидрофуране, диэтиловом эфире, циклогексане или диоксане. Реакционную смесь при перемешивании нагревают при температуре, равной от 18°С до 30°С, предпочтительно - примерно 22°С, и затем перемешивают в течение еще от 10 до 24 ч, предпочтительно - примерно 12 ч. Затем разбавитель отгоняют, остаток объединяют с водой и смесь 2-3 раза экстрагируют органическим растворителем, таким как диэтиловый эфир или этилацетат, предпочтительно - этилацетат. Объединенные органические экстракты сушат и растворитель отгоняют. Остаток (соединение A3) можно использовать на стадии 2 без какой-либо предварительной очистки.
Стадия 2А
Соединение, полученное на стадии 1А (A3), восстанавливают по нитрогруппе и циклизуют с образованием соединения формулы (А4) (диаграмма 2А).
Диаграмма 2А

На стадии 2А 1 экв. нитросоединения (A3) растворяют в кислоте, предпочтительно - ледяной уксусной кислоте, муравьиной кислоте или хлористоводородной кислоте, предпочтительно - ледяной уксусной кислоте, и нагревают при температуре от 50 до 70°С, предпочтительно - примерно 60°С. Затем восстановительный реагент, например, цинк, олово или железо, предпочтительно - железные опилки, прибавляют для завершения экзотермической реакции и смесь перемешивают в течение от 0,2 до 2 ч, предпочтительно - 0,5 ч, при от 100 до 125°С, предпочтительно - примерно при 117°С. После охлаждения до температуры окружающей среды соль железа отфильтровывают и растворитель отгоняют. Остаток растворяют в растворителе или смеси растворителей, например, этилацетате или смеси дихлорметан/метанол 9/1 и разбавленном вдвое насыщенном растворе NaCl, и фильтруют, например, через кизельгур. Органическую фазу сушат и выпаривают. Остаток (соединение (А4)) можно очистить с помощью хроматографии или с помощью кристаллизации или использовать в виде неочищенного продукта на стадии 3А синтеза.
Стадия 3А
Соединение, полученное на стадии 2А (А4), можно ввести в реакцию электрофильного замещения, как это показано на диаграмме 3А, с получением соединения формулы (А5).
Диаграмма 3А

На стадии 3А 1 экв. амида формулы (А4) растворяют в органическом растворителе, например, диметилформамиде или диметилацетамиде, предпочтительно - диметилацетамиде, и охлаждают до температуры, равной примерно от -5 до 5°С, предпочтительно - 0°С.
Затем прибавляют от 0,9 до 1,3 экв. гидрида натрия и от 0,9 до 1,3 экв. метилирующего реагента, например, метилйодида. Реакционную смесь перемешивают в течение 0,1-3 ч, предпочтительно - примерно 1 ч, при температуре примерно при от 0 до 10°С, предпочтительно - примерно при 5°С, и ее необязательно можно выдержать в течение еще 12 ч при этой температуре. Реакционную смесь выливают в воду со льдом и осадок отделяют. Остаток (соединение (А5)) можно очистить с помощью хроматографии, предпочтительно - на силикагеле, или с помощью кристаллизации, или использовать в виде неочищенного продукта на стадии 4А синтеза.
Стадия 4А
Аминирование соединения (А5), полученного на стадии 3А, с образованием соединения формулы (А9) (диаграмма 4А) можно проводить по методикам, известным из литературы, для вариантов 4.1А, например, из (a) M.P.V. Boarland, J.F.W. McOmie J. Chem. Soc. 1951, 1218-1221 или (b) F.H.S. Curd, F.C. Rose J. Chem. Soc. 1946, 343-348, для вариантов 4.2А, например, из (a) Banks J. Am. Chem. Soc. 1944, 66, 1131, (b) Ghosh and Dolly J. Indian Chem. Soc. 1981, 58, 512-513 или (с) N.P. Reddy and M. Tanaka Tetrahedron Lett. 1997, 38, 4807-4810.
Диаграмма 4А
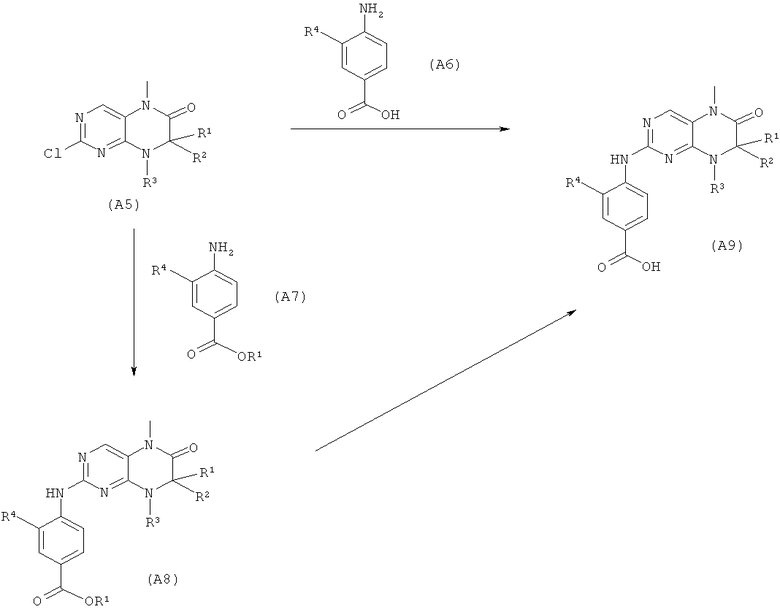
Например, в варианте 4.1 А 1 экв. соединения (А5) и от 1 до 3 экв., предпочтительно - примерно 2 экв. соединения (А6) нагревают без растворителя или в органическом растворителе, таком как, например, сульфолан, диметилформамид, диметилацетамид, толуол, N-метилпирролидон, диметилсульфоксид или диоксан, предпочтительно - сульфолан, в течение от 0,1 до 4 ч, предпочтительно - 1 ч, при температуре от 100 до 220°С, предпочтительно - примерно при 160°С. После охлаждения продукт (А9) кристаллизуют путем прибавления органических растворителей или смесей растворителей, например, смеси диэтиловый эфир/метанол, этилацетата, метиленхлорида или диэтилового эфира, предпочтительно - смеси диэтиловый эфир/метанол 9/1, или очищают с помощью хроматографии.
Например, в варианте 4.2 А 1 экв. соединения (А5) и от 1 до 3 экв. соединения (А6) перемешивают с кислотой, например, 1-10 экв. 10-38% хлористоводородной кислоты и/или спиртом, например, этанолом, пропанолом, бутанолом, предпочтительно - этанолом, при кипячении с обратным холодильником в течение от 1 до 48 ч, предпочтительно - примерно 5 ч.
Осадившийся продукт (А9) отфильтровывают и необязательно промывают водой, сушат и кристаллизуют из подходящего органического растворителя.
Например, в варианте 4.3 А 1 экв. соединения (А5) и от 1 до 3 экв. соединения (А7) растворяют в растворителе, например, толуоле или диоксане, и объединяют с фосфиновым лигандом, например, 2,2′-бис-(дифенилфосфино)-1,1′-бинафтилом и палладиевым катализатором, например, трис(дибензилиденацетон)-дипалладием(0), и основанием, например, карбонатом цезия, и кипятят с обратным холодильником в течение 1-24 ч, предпочтительно - 17 ч. Реакционную смесь очищают, например, на силикагеле и продукт (А8) выделяют из раствора или получают с помощью подходящей кристаллизации. Продукт (А8) растворяют в подходящем растворителе, например, диоксане, и смешивают с кислотой, например, вдвое разбавленной концентрированной хлористоводородной кислотой, например, при отношении количества растворителя к количеству кислоты, составляющем 3:1. Затем смесь кипятят с обратным холодильником в течение 1-48 ч, например, 12 ч, и образовавшийся осадок отделяют. При необходимости продукт (А9) очищают с помощью кристаллизации.
Стадия 5А
Диаграмма 5А
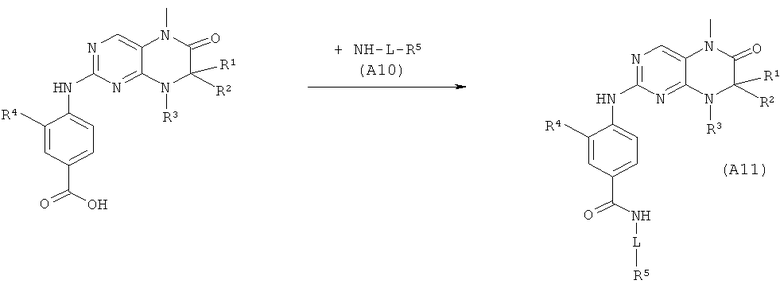
Например, 1 экв. соединения (А9) растворяют совместно с 1 экв. активирующего реагента, например, O-бензотриазолил-N,N,N′,N′-тетраметилуронийтетрафторбората (ТМУТ) и основания, например, 1,5 экв. диизопропилэтиламина (ДИПЭА) в органическом разбавителе, например, дихлорметане, тетрагидрофуране, диметилформамиде, N-метилпирролидоне, диметилацетамиде, предпочтительно - дихлорметане или диметилформамиде. После прибавления 1 экв. амина (А10) реакционную смесь перемешивают в течение от 0,1 до 24 ч, предпочтительно - примерно 2 ч при температуре от 20°С до 100°С. Продукт формулы (A11) получают, например, с помощью кристаллизации или хроматографической очистки.
Соединения общей формулы (I) можно синтезировать аналогично приведенным ниже примерам синтеза. Нумерация примеров соответствует нумерации, использованной в таблице 1. Однако эти примеры приведены только в качестве примеров методик для дополнительного иллюстрирования настоящего изобретения без наложения ограничений на его объект.
Ниже также описано получение некоторых промежуточных соединений, применяющихся для синтеза соединений.
Получение кислот
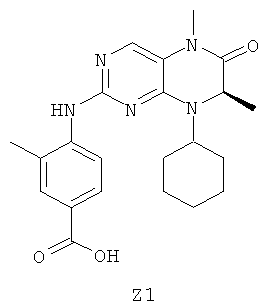
Для синтеза соединений примеров 94 и 95, приведенных в таблице 1, сначала получают промежуточное соединение Z1, как это описано ниже.
50,0 г (0,48 моля) Гидрохлорида метилового эфира D-аланина, 49,1 г (0,50 моля) циклогексанона помещают в 300 мл дихлорметана и затем объединяют с 41,0 г (0,50 моля) ацетата натрия и 159,0 г (0,75 моля) триацетоксиборогидрида натрия. Смесь перемешивают в течение ночи и затем прибавляют 300 мл 10% раствора гидрокарбоната натрия. Водную фазу экстрагируют дихлорметаном. Объединенные органические фазы промывают 10% раствором бикарбоната натрия, сушат над Na2SO4 и выпаривают.
Выход: 72,5 г соединения Z1a (прозрачная жидкость)
72,5 г Соединения Z1a помещают в 500 мл воды и прибавляют 76,6 г (0,39 моля) 2,4-дихлор-5-нитропиримидина в 500 мл диэтилового эфира. При температуре, равной от -5°С, по каплям прибавляют 100 мл 10% раствора гидрокарбоната калия. Смесь перемешивают в течение 3 ч при -5°С и в течение еще 12 ч при температуре окружающей среды. Органическую фазу отделяют и сушат над Na2SO4. После выпаривания продукт кристаллизуется.
Выход: 48,0 г соединения Z1b (желтые кристаллы)
48,0 г Соединения Z1b растворяют в 350 мл ледяной уксусной кислоты и нагревают при 60°С. Прибавляют 47,5 г порошкообразного железа, и температура повышается до 105°С. Реакционную смесь перемешивают в течение 3 ч при 80°С, затем в горячем виде фильтруют через целлюлозу и выпаривают. Остаток перемешивают в воде и этилацетате, отфильтровывают с отсасыванием и светло-серый осадок промывают этилацетатом. Фильтрат промывают разбавленным раствором аммиака и водой, органическую фазу сушат над Na2SO4, фильтруют через активированный древесный уголь и выпаривают. Получают дополнительное количество светло-серого твердого вещества.
Выход: 29,5 г соединения Z1c (светло-серые кристаллы)
32,1 г Соединения Z1c помещают в 300 мл диметилацетамида и объединяют с 13 мл (0,2 моля) метилйодида. При -5°С порциями прибавляют 6,4 г (0,16 моля) гидрида натрия в виде 60% дисперсии в минеральном масле. Через 2 ч реакционную смесь выливают в 800 мл воды со льдом. Образовавшийся осадок отфильтровывают с отсасыванием и промывают петролейным эфиром.
Выход: 33,0 г соединения Z1d (бежевые кристаллы)
4,0 г Соединения Z1d и 2,3 г (15 ммолей) 4-амино-3-метилбензойной кислоты суспендируют в 50 мл этанола и 120 мл воды, объединяют с 2 мл концентрированной хлористоводородной кислоты и кипятят с обратным холодильником в течение 48 ч. Образовавшийся осадок при охлаждении отфильтровывают с отсасыванием и промывают водой, этанолом и диэтиловым эфиром.
Выход: 2,9 г соединения Z1 (бесцветные кристаллы)
Для синтеза соединений примера 188 и примера 203, приведенных в таблице 1, сначала получают промежуточное соединение Z2, как это описано ниже.
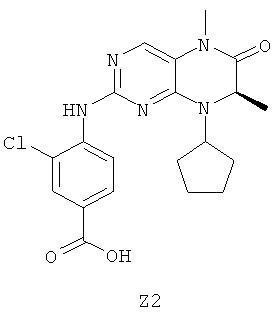
Раствор 128,2 г (0,83 моля) гидрохлорида этилового эфира D-аланина и 71,5 г (0,85 моля) циклопентанон в 1500 мл дихлорметана объединяют с 70,1 (0,85 моля) ацетата натрия и 265,6 г (1,25 моля) триацетоксиборогидрида натрия. Реакционную смесь перемешивают в течение 12 ч и затем выливают в 1,5 л 10% раствора гидрокарбоната натрия. Водную фазу экстрагируют дихлорметаном. Объединенные органические фазы сушат над Na2SO4 и выпаривают.
Выход: 143,4 г соединения Z2a (бесцветное масло)
66,0 г Соединения Z2a помещают в 500 мл воды и объединяют с 85,0 г (0,44 моля) 2,4-дихлор-5-нитропиримидина в 500 мл диэтилового эфира. При -5°С по каплям прибавляют 100 мл 10% раствора гидрокарбоната калия и реакционную смесь перемешивают в течение 48 ч при температуре окружающей среды. Водную фазу экстрагируют диэтиловым эфиром, объединенные органические фазы сушат над Na2SO4 и выпаривают. Темно-красное твердое вещество перемешивают с петролейным эфиром и отфильтровывают с отсасыванием.
Выход: 88,0 г соединения Z2b (желтые кристаллы)
88,0 г Соединения Z2b растворяют в 1000 мл ледяной уксусной кислоты и при 60°С порциями объединяют с 85 г порошкообразного железа и температура повышается до 110°С. Смесь перемешивают в течение 1 ч при 60°С, затем отфильтровывают с отсасыванием в горячем виде через целлюлозу и выпаривают. Коричневое твердое вещество перемешивают с 700 мл воды и отфильтровывают с отсасыванием.
Выход: 53,3 г соединения Z2c (светло-коричневые кристаллы)
53,3 г Соединения Z2c растворяют в 300 мл диметилацетамида и объединяют с 13 мл (0,21 моля) метилйодида. При -5°С порциями прибавляют 5,0 г (0,21 моля) гидрида натрия в виде 60% дисперсии в минеральном масле. Через 12 ч реакционную смесь выливают в 1000 мл воды со льдом и образовавшийся осадок отфильтровывают с отсасыванием.
Выход: 40,0 г соединения Z2d (бесцветные кристаллы)
4,0 г Соединения Z2d и 2,8 г (16 ммолей) 4-амино-3-хлорбензойной кислоты суспендируют в 25 мл этанола и 60 мл воды, объединяют с 3 мл концентрированной хлористоводородной кислоты и кипятят с обратным холодильником в течение 43 ч. Образовавшийся осадок при охлаждении отфильтровывают с отсасыванием и промывают водой, этанолом и диэтиловым эфиром.
Выход: 0,9 г соединения Z2 (бесцветные кристаллы)
Для синтеза соединений примеров 19, 21, 22, 23, 45, 46, 55, 58, 116, 128, 131, 133, 134, 136, 138, 177, 217, 231, 239, 46, 184, 166 и 187, приведенных в таблице 1, сначала получают промежуточное соединение Z3, как описано ниже.
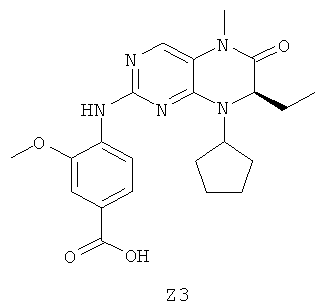
54,0 г (0,52 моля) D-2-Аминомасляной кислоты суспендируют в 540 мл метанола и медленно объединяют с 132 г (1,1 моля) тионилхлорида при охлаждении льдом. Смесь кипятят с обратным холодильником в течение 1,5 ч и затем выпаривают. Оставшееся масло объединяют с 540 мл трет-бутилметилового эфира и образовавшиеся бесцветные кристаллы отфильтровывают с отсасыванием.
Выход: 78,8 г соединения Z3a (бесцветные кристаллы)
74,2 г Соединения Z3a и 43,5 мл (0,49 моля) циклопентанон растворяют в 800 мл дихлорметана. После прибавления 40,0 г (0,49 моля) ацетата натрия и 150,0 г (0,71 моля) триацетоксиборогидрида натрия при 0°С смесь перемешивают в течение 12 ч при температуре окружающей среды и затем прибавляют 500 мл 20% раствора гидрокарбоната натрия. Водную фазу экстрагируют дихлорметаном. Объединенные органические фазы промывают водой, сушат над MgSO4 и выпаривают.
Выход: 85,8 г соединения Z3b (светло-желтое масло)
40,0 г Соединения Z3b и 30,0 г (0,22 моля) карбоната калия суспендируют в 600 мл ацетона и объединяют с 45,0 г (0,23 моля) 2,4-дихлор-5-нитропиримидина в 200 мл ацетона при охлаждении льдом. Через 12 ч прибавляют еще 5,0 г 2,4-дихлор-5-нитропиримидина и перемешивают в течение 3 ч. Реакционную смесь выпаривают, растворяют в 800 мл этилацетата и 600 мл воды и водную фазу экстрагируют этилацетатом. Объединенные органические фазы промывают водой, сушат над MgSO4 и выпаривают.
Выход: 75,0 г соединения Z3c (коричневое масло)
100 г Соединения Z3c растворяют в 650 мл ледяной уксусной кислоты и при 70°С порциями прибавляют 20 г порошкообразного железа. Смесь перемешивают в течение 1 ч при 70°С, затем в течение 1,5 ч при 100°С и затем в горячем виде фильтруют через кизельгур. Реакционную смесь выпаривают, растворяют в метанол/дихлорметан, вносят в силикагель и очищают этилацетатом путем экстракции в аппарате Сокслетта. Растворитель удаляют и остаток перемешивают с метанолом.
Выход: 30,0 г соединения Z3d (светло-коричневые кристаллы)
25,0 г Соединения Z3d и 6,5 мл (0,1 моля) метилйодида помещают в 250 мл диметилацетамида и при -10°С прибавляют 3,8 г (0,95 моля) гидрида натрия в виде 60% дисперсии в минеральном масле. Смесь перемешивают в течение 20 мин при 0°С, затем в течение 30 мин при температуре окружающей среды и в заключение прибавляют лед. Реакционную смесь выпаривают и объединяют с 300 мл воды. Образовавшийся осадок отфильтровывают с отсасыванием и промывают петролейным эфиром.
Выход: 23,0 г соединения Z3e (бесцветное твердое вещество)
6,0 г Соединения Z3e и 5,1 г (31 ммоль) 4-амино-3-метоксибензойной кислоты суспендируют в 90 мл этанола и 350 мл воды, объединяют с 3,5 мл концентрированной хлористоводородной кислоты и кипятят с обратным холодильником в течение 48 ч. Реакционную смесь выпаривают, остаток перемешивают со смесью метанол/диэтиловый эфир и образовавшийся осадок отфильтровывают с отсасыванием.
Выход: 6,3 г соединения Z3 (светло-бежевые кристаллы)
Для синтеза соединений примеров 81, 82, 93 и 137, приведенных в таблице 1, сначала получают промежуточное соединение Z4, как это описано ниже.
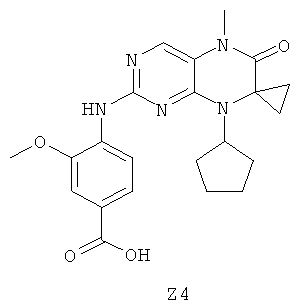
25,0 г (0,19 моля) Этил-1-аминоциклопропан-1-карбоксилата × HCl и 16,8 г (0,20 моля) циклопентанона растворяют в 300 мл дихлорметана и объединяют с 16,4 г (0,20 моля) ацетата натрия и 61,7 г (0,29 моля) триацетоксиборогидрида натрия. Смесь перемешивают в течение ночи и затем реакционную смесь выливают в 400 мл 10% раствора гидрокарбоната натрия. Водную фазу экстрагируют дихлорметаном. Объединенные органические фазы сушат над Na2SO4 и выпаривают.
Выход: 34,5 г соединения Z4a (бесцветное масло)
42,5 г (0,22 моля) 2,4-Дихлор-5-нитропиримидина в 350 мл диэтилового эфира прибавляют к смеси 34,5 г соединения Z4a в 350 мл воды. При -5°С смесь объединяют с 80 мл 10% раствора гидрокарбоната калия и перемешивают в течение ночи при температуре окружающей среды. Водную фазу экстрагируют диэтиловым эфиром. Объединенные органические фазы сушат над Na2SO4 и выпаривают.
Выход: 53,8 г соединения Z4b (коричневое масло)
20,1 г Соединения Z4b растворяют в 200 мл ледяной уксусной кислоты и порциями объединяют при 60°С с 19,1 г порошкообразного железа, и за это время температура повышается до 100°С. Смесь перемешивают в течение 3 ч при 60°С, затем отфильтровывают с отсасыванием через целлюлозу и выпаривают. Остаток перемешивают в воде и этилацетате и желтый осадок отфильтровывают с отсасыванием. Фильтрат промывают разбавленным раствором аммиака и водой, органическую фазу сушат над Na2SO4 и выпаривают. После прибавления диэтилового эфира кристаллизуется дополнительное количество продукта.
Выход: 4,0 г соединения Z4c (желтые кристаллы)
7,8 г Соединения Z4c и 2,6 мл (0,04 моля) метилйодида растворяют в 100 мл диметилацетамида и при -5°С порциями прибавляют 1,5 г (0,04 моля) гидрида натрия в виде 60% дисперсии в минеральном масле. Через 2 ч реакционную смесь выливают в воду со льдом и образовавшийся осадок отфильтровывают с отсасыванием.
Выход: 7,5 г соединения Z4d (светло-коричневые кристаллы)
3,0 г Соединения Z4d и 1,9 г (11 ммолей) 4-амино-3-метоксибензойной кислоты суспендируют в 40 мл этанола и 80 мл воды, объединяют с 2 мл концентрированной хлористоводородной кислоты и кипятят с обратным холодильником в течение 20 ч. Прибавляют еще 0,5 г 4-амино-3-метоксибензойной кислоты и кипятят с обратным холодильником в течение 48 ч. Образовавшийся осадок при охлаждении отфильтровывают с отсасыванием и промывают водой, этанолом и диэтиловым эфиром.
Выход: 2,1 г соединения Z4 (бесцветные кристаллы) т.пл.: 222-223°С
Для синтеза соединений примеров 162, 43, 53, 161, 202, 211, 215 и 212, приведенных в таблице 1, сначала получают промежуточное соединение Z5, как это описано ниже.
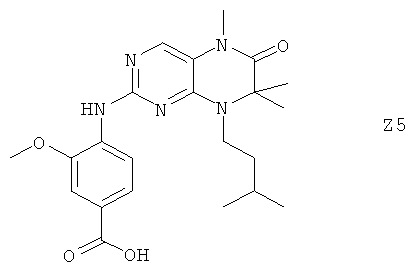
Смесь 73,4 мл (0,5 моля) Этил-2-бромизобутирата, 87,1 мл (0,75 моля) 3-метил-1-бутиламина, 82,5 г (0,6 моля) йодида натрия и 76,0 г (0,6 моля) карбоната калия в 1000 мл этилацетата кипятят с обратным холодильником в течение 3 дней. Содержащиеся соли отфильтровывают и фильтрат выпаривают.
Выход: 97,0 г соединения Z5a (красное масло)
49,0 г (0,25 моля) 2,4-Дихлор-5-нитропиримидина и 38,3 г (0,28 моля) карбоната калия суспендируют в 500 мл ацетона и при 0°С объединяют с 93,0 г соединения Z5a в 375 мл ацетона. Реакционную смесь перемешивают в течение ночи при температуре окружающей среды, фильтруют и выпаривают. Остаток, растворенный в этилацетате, промывают водой и органическую фазу сушат над MgSO4 и выпаривают.
Выход: 102,7 г соединения Z5b (коричневое масло)
22,7 г Соединения Z5b растворяют в 350 мл ледяной уксусной кислоты и при 60°С порциями объединяют с 17,4 г порошкообразного железа. После завершения прибавления смесь кипятят с обратным холодильником в течение 0,5 ч, фильтруют в горячем виде и выпаривают. Остаток растворяют в 200 мл дихлорметана/метанол (9:1) и промывают раствором хлорида натрия. Органическую фазу отфильтровывают с отсасыванием через кизельгур, сушат над MgSO4, выпаривают и очищают с помощью хроматографии на колонке (элюент: этилацетат/циклогексан 1:1).
Выход: 1,9 г соединения Z5c (бесцветные кристаллы)
1,9 г Соединения Z5c растворяют в 32 мл диметилацетамида и при охлаждении льдом объединяют с 0,3 г (7 ммолей) гидрида натрия в виде 60% дисперсии в минеральном масле. Через 10 мин прибавляют 0,5 мл (7 ммолей) метилйодида и перемешивают в течение 3 ч при температуре окружающей среды. Реакционную смесь выпаривают и объединяют с водой. Образовавшийся осадок отфильтровывают с отсасыванием и промывают петролейным эфиром.
Выход: 1,6 г соединения Z5d (бесцветные кристаллы)
14,0 г Соединения Z5d и 10,0 г (0,06 моля) 4-амино-3-метоксибензойной кислоты суспендируют в 200 мл диоксан и 80 мл воды, объединяют с 10 мл концентрированной хлористоводородной кислоты и кипятят с обратным холодильником в течение 40 ч. Образовавшийся осадок при охлаждении отфильтровывают с отсасыванием и промывают водой, диоксаном и диэтиловым эфиром.
Выход: 13,9 г соединения Z5 (бесцветные кристаллы)
Для синтеза соединений примеров 88, 194, 229 и 89, приведенных в таблице 1, сначала получают промежуточное соединение Z6, как это описано ниже.
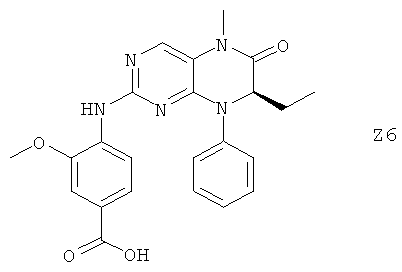
6,0 г (0,06 моля) L-2-Аминомасляной кислоты помещают в 80 мл 0,5 М серной кислоты и при 0°С объединяют с 5,5 г (0,08 моля) нитрита натрия в 15 мл воды. Реакционную смесь перемешивают в течение 22 ч при 0°С, объединяют с сульфатом аммония и фильтруют. Фильтрат экстрагируют диэтиловым эфиром и объединенные органические фазы сушат над MgSO4 и выпаривают.
Выход: 6,0 г соединения Z6a (желтое масло)
200 мл Метанола последовательно объединяют с 65,0 мл (0,89 моля) тионилхлорида и 76,0 г соединения Z6a в 50 мл метанола при охлаждении льдом. Полученную смесь перемешивают в течение 1 ч при 0°С и 2 ч при температуре окружающей среды и затем метанол и оставшийся тионилхлорид удаляют в вакууме при 0°С.
Выход: 40,0 г соединения Z6b (желтое масло)
30,0 мл (0,17 моля) ангидрида трифторметансульфоновой кислоты помещают в 150 мл дихлорметана и при охлаждении льдом в течение 1 ч прибавляют раствор 20,0 г соединения Z6b и 14,0 мл (0,17 моля) пиридина в 50 мл дихлорметана. Смесь перемешивают в течение 2 ч при температуре окружающей среды, все образовавшиеся соли отфильтровывают с отсасыванием и затем промывают с помощью 100 мл воды. Органическую фазу сушат над MgSO4 и выпаривают.
Выход: 42,0 г соединения Z6c (светло-желтое масло)
42,0 г Соединения Z6c в 200 мл дихлорметана по каплям прибавляют в течение 1 ч к раствору 15,5 мл (0,17 моля) анилина и 24,0 мл (0,17 моля) триэтиламина в 400 мл дихлорметана при охлаждении льдом. Смесь перемешивают в течение 1 ч при температуре окружающей среды и еще 2 ч при 35°С. Реакционную смесь промывают водой, сушат над MgSO4 и выпаривают. Полученный остаток очищают с помощью перегонки (95-100°С, 1*10-3 мбар).
Выход: 14,0 соединения Z6d (бесцветное масло)
14,0 г Соединения Z6d и 16,0 г (0,1 моля) карбоната калия суспендируют в 100 мл ацетона и при 10°С объединяют с 16,0 г (0,08 моля) 2,4-дихлор-5-нитропиримидина. Смесь перемешивают в течение 4 ч при 40°С, все образовавшиеся соли отфильтровывают с отсасыванием и фильтрат выпаривают. Остаток растворяют в 300 мл этилацетата и промывают водой. Органическую фазу сушат над MgSO4 и выпаривают.
Выход: 31,0 г соединения Z6e (коричневое масло)
31,0 г Соединения Z6e растворяют в 200 мл ледяной уксусной кислоты и при 60°С порциями объединяют с 10 г порошкообразного железа и за это время температура повышается до 85°С. Смесь перемешивают в течение еще 1 ч при 60°С, фильтруют через кизельгур и выпаривают. Остаток перемешивают с метанолом.
Выход: 4,5 г соединения Z6f (коричневые кристаллы)
При -20°С 0,6 г (16 ммолей) гидрида натрия в виде 60% дисперсии в минеральном масле порциями прибавляют к смеси 4,5 г соединения Z6f и 1,0 мл (16 ммолей) метилйодида в 100 мл диметилацетамида. Через 1 ч реакционную смесь объединяют с 50 мл воды и выпаривают. Остаток перемешивают с 200 мл воды, осадок отфильтровывают с отсасыванием и промывают петролейным эфиром.
Выход: 4,5 г соединения Z6g (бесцветные кристаллы)
Суспензию 1,5 г соединения Z6g и 1,4 г (8 ммолей) метил 4-амино-3-метоксибензоата в 30 мл толуола объединяют с 0,4 г (0,6 ммоля) 2,2′-бис-(дифенилфосфино)-1,1′-бинафтила, 0,23 г (0,3 ммоля) трис(дибензилиденацетон)-дипалладия(0) и 7,0 г (21 ммоль) карбоната цезия и кипятят с обратным холодильником в течение 17 ч. Реакционную смесь вносят в силикагель и очищают с помощью хроматографии на колонке (элюент: дихлорметан/метанол 9:1).
Выход: 1,7 г соединения Z6h (желтые кристаллы)
1,7 г Соединения Z6h растворяют в 50 мл диоксана, объединяют с 15 мл вдвое разбавленной концентрированной хлористоводородной кислоты и кипятят с обратным холодильником в течение 12 ч. После охлаждения образовавшийся осадок отфильтровывают с отсасыванием.
Выход: 1,1 г соединения Z6 (бесцветное твердое вещество)
Для синтеза соединений примеров 26, 20, 32, 56, 101, 112 и 209, приведенных в таблице 1, сначала получают промежуточное соединение Z7, как это описано ниже.
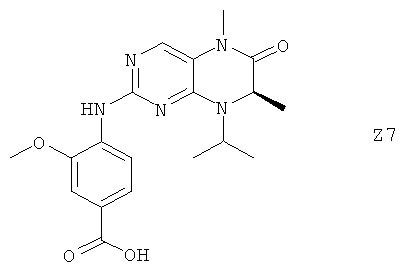
50,0 г (0,36 моля) Гидрохлорида метилового эфира D-аланина суспендируют в 500 мл дихлорметана и 35 мл ацетона и объединяют с 30,0 г (0,37 моля) ацетата натрия и 80,0 г (0,38 моля) триацетоксиборогидрида натрия. Смесь перемешивают в течение 12 ч и затем выливают в 400 мл 10% раствора гидрокарбоната натрия. Органическую фазу сушат над Na2SO4 и выпаривают.
Выход: 51,0 г соединения Z7a (желтое масло)
Суспензию 51,0 г соединения Z7a в 450 мл воды объединяют с 80,0 г (0,41 моля) 2,4-дихлор-5-нитропиридина в 450 мл диэтилового эфира. При -5°С по каплям прибавляют 100 мл 10% раствора гидрокарбоната калия. Реакционную смесь перемешивают в течение 3 ч, органическую фазу сушат над Na2SO4 и выпаривают.
Выход: 74 г соединения Z7b (желтое масло)
18,6 г Соединения Z7b растворяют в 200 мл ледяной уксусной кислоты и при 60°С порциями объединяют с 20,0 г порошкообразного железа. Смесь перемешивают в течение 2 ч при 60°С и затем отфильтровывают с отсасыванием через целлюлозу. Остаток растворяют в этилацетат и промывают водой и концентрированным раствором аммиака. Органическую фазу сушат над Na2SO4 и выпаривают. Остаток кристаллизуют из диэтилового эфира.
Выход: 9,8 г соединения Z7c (бесцветные кристаллы)
17,0 г Соединения Z7c и 7 мл (0,1 моля) метилйодида растворяют в 200 мл диметилацетамида и при -5°С объединяют с 4,0 г (0,1 моля) гидрида натрия в виде 60% дисперсии в минеральном масле. Реакционную смесь перемешивают в течение 30 мин и затем выливают в 300 мл воды со льдом. Образовавшийся осадок отфильтровывают с отсасыванием и перемешивают с петролейным эфиром.
Выход: 14,8 г соединения Z7d (бежевые кристаллы)
0,9 г Соединения Z7d и 1,5 г (9 ммолей) 4-амино-3-метоксибензойной кислоты нагревают при 210°С в течение 30 мин. После охлаждения остаток перемешивают с этилацетатом и полученный осадок отфильтровывают с отсасыванием.
Выход: 1,2 г соединения Z7 (серые кристаллы)
Указанные ниже кислоты можно, например, получить по методикам синтеза, аналогичным описанным выше в настоящем изобретении.
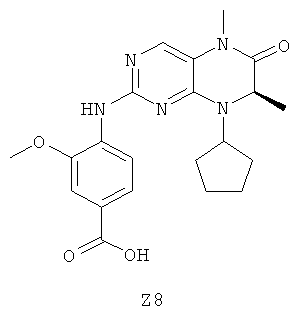
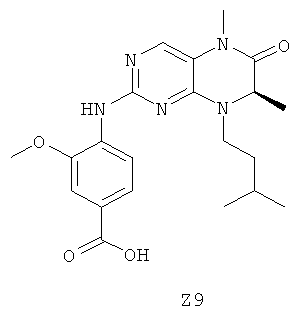
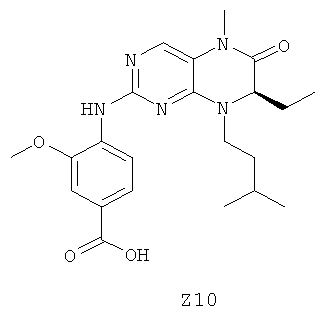
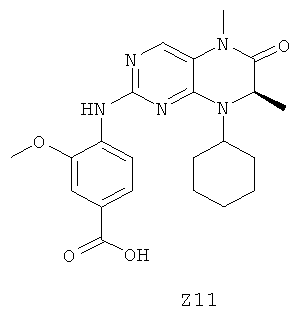
Синтез аминных компонентов L-R5
Указанные ниже амины,
1,1-диметил-2-диметиламино-1-илэтиламин и 1,1-диметил-2-пиперидин-1-илэтиламин, можно получить следующим образом.
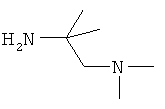
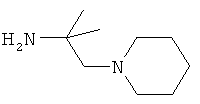
Эти соединения можно получить в соответствии со следующими публикациями: (a) S. Schuetz et al. Arzneimittel-Forschung 1971, 21, 739-763, (b) V.M. Belikov et al. Tetrahedron 1970, 26, 1199-1216 и (с) Е.В. Butler and McMillan J. Amer. Chem. Soc. 1950, 72, 2978.
Другие амины можно получить, как это описано ниже, по методикам, измененным по сравнению с приведенными в указанных выше публикациях.
1,1-Диметил-2-морфолин-1-илэтиламин
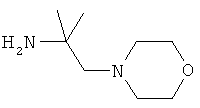
8,7 мл Морфолина и 9,3 мл 2-нитропропан вводят в реакцию при охлаждении реакционной смеси льдом, по каплям медленно прибавляют (<10°С) 7,5 мл формальдегида (37%) и 4 мл 0,5 моль/л раствора NaOH. Затем смесь перемешивают в течение 1 ч при 25°С и 1 ч при 50°С. Раствор обрабатывают водой и эфиром и водную фазу трижды экстрагируют эфиром. Объединенные органические фазы сушат над NaSO4 и объединяют с HCl в диоксане (4 моль/л), образовавшийся осадок отфильтровывают с отсасыванием. Выход: 21,7 белого порошкообразного вещества. 5 г Белого порошкообразного вещества растворяют в 80 мл метанола и с прибавлением 2 г RaNi (Ni Ренея) обрабатывают водородом при 35°С и давлении 50 фунт-сила/дюйм2 в течение 40 мин. Это дает 3,6 г 1,1-диметил-2-морфолин-1-илэтиламина.
Указанные ниже амины можно получить аналогичным образом.
1,1-диметил-N-метилпиперазин-1-илэтиламин
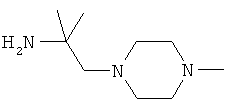
1,1-диметил-2-пирролидин-1-илэтиламин
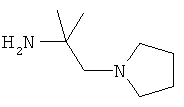
1,3-Диморфолин-2-аминопропан
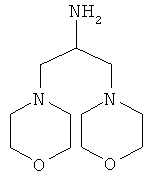
5 г 1,3 Диморфолин-2-нитропропан, полученный у фирмы Messrs. Aldrich, растворяют в 80 мл метанола и обрабатывают водородом в течение 5,5 ч при 30°С и давлении 50 фунт-сила/дюйм2 с прибавлением 2 г RaNi. Это дает 4,2 г 1,3 диморфолин-2-аминопропана.
4-Аминобензилморфолин
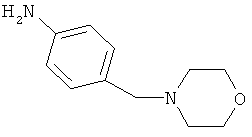
Получение этого амина описано в следующей публикации:
S. Mitsuru et al. J. Med. Chem. 2000, 43, 2049-2063.
4-Амино-1-тетрагидро-4Н-пиран-4-илпиперидин
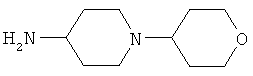
20 г (100 ммолей) 4-Трет-бутоксикарбониламинопиперидина растворяют в 250 мл CH2Cl2 и перемешивают в течение 12 ч при комнатной температуре с 10 г (100 ммолей) тетрагидро-4Н-пиран-4-она и 42 г (200 ммолей) NaBH(ОАс)3. Затем прибавляют воду и карбонат калия, органическую фазу отделяют, сушат и растворитель удаляют в вакууме. Остаток растворяют в 200 мл CH2Cl2 и перемешивают в течение 12 ч при комнатной температуре с 100 мл трифторуксусной кислоты. Растворитель удаляют в вакууме, остаток растворяют в CHCl3 и повторно выпаривают, затем растворяют в ацетоне и гидрохлорид осаждают эфирным раствором HCl. Выход: 14,3 г (56%).
Цис- и транс-4-морфолиноциклогексиламин
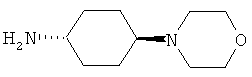
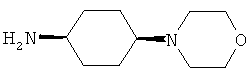
Дибензил-4-морфолиноциклогексиламин
3,9 г (30 ммолей) 4-Дибензилциклогексанона растворяют в 100 мл CH2Cl2 и перемешивают в течение 12 ч при комнатной температуре с 3,9 г (45 ммолей) морфолина и 9,5 г (45 ммолей) NaBH(ОАс)3. Затем прибавляют воду и карбонат калия, органическую фазу отделяют, сушат и растворитель удаляют в вакууме. Остаток очищают на колонке с силикагелем (примерно 20 мл силикагеля; примерно 500 мл смеси этилацетат 90/метанол 10 + 1% концентрированного раствора аммиака). Соответствующие фракции выпаривают в вакууме. Выход: 6,6 г (60%) цис-изомера и 2 г (18%) транс-изомера.
Альтернативно, транс-дибензил-4-морфолиноциклогексиламин можно получить по следующей методике:
33 г (112 ммолей) 4-Дибензилциклогексанона растворяют в 300 мл МеОН, объединяют с 17,4 г (250 ммолей) гидроксиламингидрохлорида и перемешивают в течение 4 ч при 60°С. Растворитель выпаривают в вакууме, объединяют с 500 мл воды и 50 г карбоната калия и дважды экстрагируют с помощью 300 мл дихлорметана. Органическую фазу сушат, выпаривают в вакууме, остаток кристаллизуют из петролейного эфира, растворяют в 1,5 л EtOH и нагревают при 70°С. Порциями прибавляют 166 г натрия и смесь кипятят с обратным холодильником до растворения натрия. Растворитель удаляют в вакууме, остаток объединяют с 100 мл воды и дважды экстрагируют с помощью 400 мл эфира. Органическую фазу промывают водой, сушат, выпаривают в вакууме и транс-изомер отделяют с помощью колонки (примерно 1,5 л силикагеля; примерно 2 л смеси этилацетат 80/метанол 20 + 2% концентрированного раствора аммиака). Выход: 12,6 г (41,2%).
6,8 г (23 ммолей) Транс-1-амино-4-дибензиламиноциклогексанона растворяют в 90 мл ДМФ (диметилформамид) и перемешивают в течение 8 ч при 100°С с 5 мл (42 ммолей) 2,2'-дихлорэтилового эфира и 5 г карбоната калия. После охлаждения прибавляют 30 мл воды, осадившиеся кристаллы отфильтровывают с отсасыванием и очищают на короткой колонке (примерно 20 мл силикагеля, примерно 100 мл этилацетата). Остаток кристаллизуют из метанола и концентрированной HCl в виде гидрохлорида. Выход: 7,3 г (72,4%).
Транс-4-морфолиноциклогексиламин
7,2 г (16,4 ммоля) Транс-дибензил-4-морфолиноциклогексиламина растворяют в 100 мл МеОН и гидрируют над 1,4 г Pd/C (10%) при 30-50°С. Растворитель удаляют в вакууме и остаток кристаллизуют из этанола и концентрированной HCl. Выход: 3,9 г (93%).
Цис-изомер можно получить аналогичным образом.
Цис- и транс-4-пиперидиноциклогексиламин
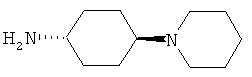
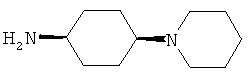
Транс-дибензил-4-пиперидиноциклогексиламин
2,0 г (6,8 ммоля) Транс-1-амино-4-дибензиламиноциклогексанона (см. пример 2) растворяют в 50 мл ДМФ и в течение 48 ч при комнатной температуре перемешивают с 1,6 г (7 ммолей) 1,5-дибромпентана и 2 г карбоната калия. Смесь охлаждают, объединяют с водой, дважды экстрагируют с помощью 100 мл дихлорметана, сушат и растворитель удаляют в вакууме. Остаток очищают на колонке (примерно 100 мл силикагеля, примерно 500 мл смеси этилацетат 80/метанол 20 + 1% концентрированного раствора аммиака). Необходимые фракции выпаривают в вакууме и кристаллизуют из петролейного эфира. Выход: 1,2 г (49%).
Транс-4-пиперидиноциклогексиламин
1,7 г (4,8 ммоля) Транс-дибензил-4-пиперидиноциклогексиламина растворяют в 35 мл МеОН и гидрируют над 350 мг Pd/C (10%) при 20°С. Растворитель удаляют в вакууме и остаток кристаллизуют из этанола и концентрированной HCl. Выход: 1,1 г (78%).
Цис-изомер можно получить аналогичным образом.
Цис- и транс-4-(4-фенилпиперазин-1-ил)-циклогексиламин
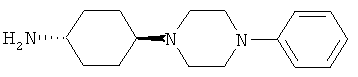
4,1 г (25,3 ммоля) 4-Дибензилциклогексанона растворяют в 50 мл дихлорметана и перемешивают в течение 12 ч при комнатной температуре с 7,4 г (25,3 ммоля) N-фенилпиперазина и 7,4 г (35 ммолей) NaBH(OAc)3. Затем прибавляют воду и карбонат калия, органическую фазу отделяют, сушат и растворитель удаляют в вакууме. Остаток очищают на колонке с силикагелем (этилацетат 80/метанол 20 + 0,5% концентрированного раствора аммиака). Выход: 1,7 г (15,8%) цис-изомера и 0,27 (2,5%) транс-изомера.
Транс-4-(4-фенилпиперазин-1-ил)-циклогексиламин
270 мг (0,61 ммоля) Транс-дибензил-[4-(4-фенилпиперазин-1-ил)-циклогексил]-амина растворяют в 5 мл МеОН и гидрируют над 40 мг Pd/C (10%) при 20-30°С. Растворитель удаляют в вакууме и остаток кристаллизуют из этанола и концентрированной HCl. Выход: 110 мг (69%).
Цис-изомер можно получить аналогичным образом.
Цис- и транс-4-(4-циклопропилметилпиперазин-1-ил)-циклогексиламин
9,8 г (33,4 ммоля) 4-Дибензилциклогексанона растворяют в 100 мл дихлорметана и перемешивают в течение 12 ч при комнатной температуре с 5,6 г (40 ммолей) N-циклопропилметилпиперазина и 8,5 г (40 ммолей) NaBH(OAc)3. Затем прибавляют воду и карбонат калия, органическую фазу отделяют, сушат и растворитель удаляют в вакууме. Остаток очищают на колонке с силикагелем (примерно 50 мл силикагеля, примерно 3 л смеси этилацетат 95/метанол 5 + 0,25% концентрированного раствора аммиака. Соответствующие фракции выпаривают в вакууме. Быстрее элюирующееся цис-соединение кристаллизуют из этилацетата. Транс-соединение кристаллизуют из этанола с прибавлением концентрированной HCl. Выход: 8,5 г (61%) цис-изомера и 2,2 (13%) транс-изомера.
Цис-4-(4-циклопропилметилпиперазин-1-ил)-циклогексиламин
8,5 г (20 ммолей) Цис-дибензил-[4-(4-циклопропилметилпиперазин-1-ил)-циклогексил]-амина растворяют в 170 мл МеОН и гидрируют над 1,7 г Pd/C (10%) при 30-50°С. Растворитель удаляют в вакууме и остаток кристаллизуют из этанола и концентрированной HCl. Выход: 4,4 г (91%).
Транс-изомера можно получить аналогичным образом.
Примеры синтеза
Пример 152
0,15 г Соединения Z10, 0,14 г ТМУТ, 0,13 мл ДИПЭА растворяют в дихлорметане и перемешивают в течение 20 мин при 25°С. Затем прибавляют 90 мкл 1-(3-аминопропил)-4-метилпиперазина и перемешивают в течение еще 2 ч при 25°С. Затем раствор разбавляют дихлорметаном и экстрагируют водой. Продукт осаждают путем прибавления петролейного эфира, эфира и этилацетата к органической фазе. Выход: 0,16 г бежевого твердого вещества.
Пример 164
0,10 г Соединения Z10, 0,1 г ТМУТ, 0,08 мл ДИПЭА растворяют в 4 мл дихлорметана и перемешивают в течение 20 мин при 25°С. Затем прибавляют 44 мкл диметиламинопропиламина и перемешивают в течение еще 2 ч при 25°С. Затем раствор разбавляют дихлорметаном и экстрагируют водой. Продукт осаждают путем прибавления петролейного эфира, эфира и ацетона к органической фазе. Выход: 0,08 г желтого твердого вещества.
Пример 242
0,15 г Соединения Z10, 0,14 г ТМУТ, 0,13 мл ДИПЭА растворяют в 5 мл дихлорметана и перемешивают в течение 20 мин при 25°С. Затем прибавляют 75 мкл 1-(2-аминоэтил)-пиперидина и перемешивают в течение еще 2 ч при 25°С. Затем раствор разбавляют дихлорметаном и экстрагируют водой. Продукт осаждают путем прибавления петролейного эфира, эфира и этилацетата к органической фазе. Выход: 0,14 г желтого твердого вещества.
Пример 188
0,1 г Соединения Z2, 0,09 г ТМУТ, 0,05 мл ДИПЭА растворяют в 15 мл дихлорметана и перемешивают в течение 20 мин при 25°С. Затем прибавляют 33 мг 1-метил-4-аминопиперидина и смесь перемешивают в течение еще 3 ч при 25°С. Раствор экстрагируют с помощью 20 мл воды, затем выпаривают в вакууме. Продукт кристаллизуют из эфира. Выход: 0,047 г белых кристаллов.
Пример 203
0,1 г Соединения Z2, 0,09 г ТМУТ, 0,5 мл ДИПЭА растворяют в 15 мл дихлорметана и перемешивают в течение 30 мин при 25°С. Затем прибавляют 50 мг 4-амино-1-бензилпиперидина и смесь перемешивают в течение еще 3 ч при 25°С. Раствор экстрагируют с помощью 20 мл воды, затем выпаривают в вакууме. Затем остаток хроматографируют на силикагеле и выделенный продукт кристаллизуют из эфира. Выход: 0,015 г белых кристаллов.
Пример 94
0,17 г Соединения Z1, 0,19 г ТМУТ, 0,11 мл ДИПЭА растворяют в 50 мл дихлорметана и перемешивают в течение 30 мин при 25°С. Затем прибавляют 63 мг 1-метил-4-аминопиперидина и смесь перемешивают в течение еще 17 ч при 25°С. К раствору прибавляют 50 мл воды и 1 г карбоната калия и органическую фазу отделяют с помощью патрона для разделения фаз, затем выпаривают в вакууме. Затем продукт очищают с помощью хроматографии на силикагеле и очищенный продукт кристаллизуют из эфира. Выход: 0,1 г белых кристаллов.
Пример 95
0,17 г Соединения Z1, 0,19 г ТМУТ, 0,11 мл ДИПЭА растворяют в 50 мл дихлорметана и перемешивают в течение 30 мин при 25°С. Затем прибавляют 77 мг экзо-3-β-аминотропана и смесь перемешивают в течение еще 17 ч при 25°С. К раствору прибавляют 50 мл воды и 1 г карбоната калия и органическую фазу отделяют с помощью патрона для разделения фаз, затем выпаривают в вакууме. Затем продукт очищают с помощью хроматографии на силикагеле и очищенный продукт кристаллизуют из эфира. Выход: 0,03 г белых кристаллов.
Пример 46
0,15 г Соединения Z3, 0,12 г ТМУТ, 0,12 мл ДИПЭА растворяют в 5 мл дихлорметана и перемешивают в течение 30 мин при 25°С. Затем прибавляют 50 мг 1-метил-4-аминопиперидина и смесь перемешивают в течение еще 2,5 ч при 25°С. Затем раствор экстрагируют водой и затем выпаривают. Остаток растворяют в теплом этилацетате и кристаллизуют из эфира и петролейного эфира. Выход: 0,025 г белых кристаллов.
Пример 80
0,2 г Соединения Z8, 0,2 г ТМУТ, 0,1 мл ДИПЭА растворяют в 10 мл дихлорметана и перемешивают в течение 30 мин при 25°С. Затем прибавляют 100 мг 1-метил-4-аминопиперидина и смесь перемешивают в течение еще 17 ч при 25°С. Затем раствор экстрагируют разбавленным раствором карбоната калия и выпаривают. Остаток кристаллизуют из эфира. Выход: 0,12 г белых кристаллов.
Пример 190
0,2 г Соединения Z8, 0,2 г ТМУТ, 0,3 мл ДИПЭА растворяют в 5 мл дихлорметана и перемешивают в течение 1 ч при 25°С. Затем прибавляют 0,13 г 4-амино-1-бензилпиперидина и смесь перемешивают в течение еще 1 ч при 25°С. Затем раствор разбавляют с помощью 10 мл метиленхлорида и экстрагируют с помощью 20 мл воды. Затем продукт очищают на силикагеле и кристаллизуют из этилацетата и эфира. Выход: 0,23 г соединения Z8.
0,23 г Бензиламина Z8 растворяют в 10 мл метанола, объединяют с 50 мг Pd/C и гидрируют при давлении 3 бар в течение 3 ч при 25°С. Путем прибавления петролейного эфира и этилацетата получают белые кристаллы. Их хроматографируют на силикагеле и кристаллизуют из этилацетата и эфира. Выход: 0,075 г белых кристаллов.
Пример 196
0,1 г Соединения Z10, 0,09 г ТМУТ, 0,3 мл ДИПЭА растворяют в 4 мл дихлорметана и перемешивают в течение 20 мин при 25°С. Затем прибавляют 67 мг амина и перемешивают в течение еще 2 ч при 25°С. Затем раствор разбавляют дихлорметаном и экстрагируют водой. Затем его хроматографируют на силикагеле и остаток растворяют в ацетон, объединяют с эфирным раствором HCl и образовавшийся осадок отделяют. Выход: 0,09 г желтого твердого вещества.
Пример 166
0,1 г Соединения Z10, 0,11 г ТМУТ, 0,14 мл ДИПЭА растворяют в 2 мл диметилформамида и перемешивают в течение 3 ч при 50°С. Затем прибавляют 55 мг 4-морфолинометилфениламина. Затем реакционную смесь охлаждают до температуры окружающей среды в течение 17 ч. Затем диметилформамид удаляют в вакууме, остаток растворяют в дихлорметане и экстрагируют водой. Затем его хроматографируют на силикагеле и продукт кристаллизуют из этилацетата и эфира. Выход: 0,06 г желтоватых кристаллов.
Пример 81
0,2 г Соединения Z4, 0,2 г ТМУТ, 0,1 мл ДИПЭА растворяют в 10 мл дихлорметана и перемешивают в течение 30 мин при 25°С. Затем прибавляют 0,1 г 1-метил-4-аминопиперидина и смесь перемешивают в течение еще 17 ч при 25°С. Затем раствор экстрагируют водным раствором карбоната калия и затем выпаривают. Продукт кристаллизуют из эфира. Выход: 0,16 г белых кристаллов.
Пример 162
0,1 г Соединения Z5, 0,07 г ТМУТ, 0,15 мл ДИПЭА растворяют в 5 мл дихлорметана и перемешивают в течение 20 мин при 25°С. Затем прибавляют 0,04 г 1-метил-4-аминопиперидина и смесь перемешивают в течение еще 2 ч при 25°С. Затем раствор разбавляют с помощью 15 мл дихлорметана и экстрагируют с помощью 20 мл воды. Остаток растворяют в МеОН и ацетоне, объединяют с 1 мл эфирного раствора HCl и выпаривают. Кристаллический продукт получают с использованием эфира, этилацетата и небольшого количества МеОН. Выход: 0,1 г белых кристаллов.
Пример 88
0,1 г Соединения Z6, 0,12 г ТМУТ, 0,12 мл ДИПЭА в 10 мл дихлорметана растворяют и перемешивают в течение 30 мин при 25°С. Затем прибавляют 0,04 г 1-метил-4-аминопиперидина и смесь перемешивают в течение еще 2 ч при 25°С. Затем раствор разбавляют с помощью 10 мл дихлорметана и экстрагируют с помощью 10 мл воды. Кристаллический продукт получают с использованием эфира, этилацетата и петролейного эфира. Выход: 0,6 г белых кристаллов.
Пример 89
0,1 г Соединения Z6, 0,08 г ТМУТ, 0,08 мл ДИПЭА растворяют в 10 мл дихлорметана и перемешивают в течение 30 мин при 25°С. Затем прибавляют 37 мкл г 7 N,N-диметилнеопентандиамина и смесь перемешивают в течение еще 2 ч при 25°С. Затем раствор разбавляют с помощью 10 мл дихлорметана и экстрагируют с помощью 10 мл воды. Затем продукт хроматографируют на силикагеле и кристаллизуют из этилацетата, эфира и петролейного эфира. Выход: 0,005 г белых кристаллов.
Пример 26
0,15 г Соединения Z7, 0,16 г ТМУТ, 1 мл ДИПЭА растворяют в 5 мл дихлорметана и перемешивают в течение 30 мин при 25°С. Затем прибавляют 0,1 г 4-морфолиноциклогексиламина и смесь перемешивают в течение еще 17 ч при 25°С. Остаток затем объединяют с 10 мл 10% раствора карбоната калия, осадок отделяют и промывают водой. Затем его растворяют в дихлорметане и повторно выпаривают. Продукт кристаллизуют из этилацетата. Выход: 0,1 г белых кристаллов.
Пример 9
150 мг Соединения Z9 и 93 мг амина растворяют в 5 мл дихлорметана и перемешивают с 160 мг ТМУТ и 1 мл ДИПЭА в течение 12 ч при комнатной температуре. Растворитель удаляют в вакууме, остаток объединяют с 10 мл 10% раствора карбоната калия. Осадок отфильтровывают с отсасыванием, промывают водой, растворяют в дихлорметане, сушат и растворитель удаляют в вакууме. Остаток кристаллизуют из этилацетата. Выход: 82,0 мг.
Пример 16
150 мг Соединения Z8 и 73 мг транс-4-пиперидиноциклогексиламина растворяют в 5 мл дихлорметана и перемешивают с 160 мг (0,50 ммоля) ТМУТ и 1 мл ДИПЭА в течение 12 ч при комнатной температуре. Растворитель удаляют в вакууме, остаток объединяют с 10 мл 10% раствора карбоната калия. Осадок отфильтровывают с отсасыванием, промывают водой, растворяют в дихлорметане, сушат и растворитель удаляют в вакууме. Остаток кристаллизуют из этилацетата. Выход: 87,0 мг.
Пример 37
100 мг Соединения Z9 и 42 мг 3-амино-1-этилпирролидина растворяют в 10 мл дихлорметана и перемешивают с 90 мг ТМУТ и 0,5 мл ДИПЭА в течение 12 ч при комнатной температуре. Растворитель удаляют в вакууме, остаток объединяют с 10 мл 10% раствора карбоната калия. Осадок отфильтровывают с отсасыванием, промывают водой, растворяют в дихлорметане, сушат и растворитель удаляют в вакууме. Остаток кристаллизуют из смеси этилацетат/петролейный эфир. Выход: 24,0 мг.
Пример 120
100 мг Соединения Z11 и 73 мг 4-аминотетрагидро-4Н-пиран-4-илпиперидина растворяют в 10 мл дихлорметана и перемешивают с 90 мг ТМУТ и 0,5 мл ДИПЭА в течение 1 ч при комнатной температуре. Растворитель удаляют в вакууме, остаток объединяют с 10 мл 10% раствора карбоната калия. Осадок отфильтровывают с отсасыванием, промывают водой, растворяют в дихлорметане, сушат и растворитель удаляют в вакууме. Остаток кристаллизуют из смеси этилацетат/петролейный эфир. Выход: 89 мг.
Пример 212
150 мг Соединения Z5 и 150 мг транс-4-(4-циклопропилметилпиперазин-1-ил)-циклогексиламина (в виде гидрохлорида) растворяют в 5 мл дихлорметана и перемешивают с 160 мг ТМУТ и 2 мл ДИПЭА в течение 2 ч при комнатной температуре. Растворитель удаляют в вакууме, остаток объединяют с 10 мл 10% раствора карбоната калия. Осадок отфильтровывают с отсасыванием, промывают водой, растворяют в дихлорметане, сушат и растворитель удаляют в вакууме. Остаток очищают на колонке (20 мл силикагеля, 300 мл смеси этилацетат 90/метанол 10 + 2% концентрированного раствора аммиака). Соответствующие фракции выпаривают в вакууме и кристаллизуют из этилацетата. Выход: 140 мг.
Пример 232
390 мг Соединения Z11 и 240 мг транс-4-(4-трет-бутоксикарбонилпиперазин-1-ил)-циклогексиламина растворяют в 2,5 мл N-метилпирролидона и перемешивают с 482 мг ТМУТ и 1 мл триэтиламина в течение 2 ч при комнатной температуре. Затем прибавляют 100 мл воды и 200 мг карбоната калия, осадок отфильтровывают с отсасыванием, промывают водой и очищают на колонке с силикагелем. Соответствующие фракции выпаривают в вакууме, растворяют в 2 мл дихлорметана, объединяют с 2 мл трифторуксусной кислоты и перемешивают в течение 2 ч при комнатной температуре, объединяют с еще 100 мл воды и 200 мг карбоната калия и осадок отфильтровывают с отсасыванием и промывают водой. Затем осадок очищают на колонке с силикагелем. Соответствующие фракции выпаривают в вакууме и остаток кристаллизуют из этанола и концентрированной хлористоводородной кислоты. Выход: 95 мг.
Пример 213
60 мг Соединения примера 232 растворяют в 10 мл этилацетата и перемешивают с 1 мл уксусного ангидрида и 1 мл триэтиламина в течение 30 мин при комнатной температуре. Растворитель удаляют в вакууме, остаток объединяют с водой и аммиаком, осадившиеся кристаллы отфильтровывают с отсасыванием и промывают водой и небольшим количеством холодного ацетона. Выход: 40 мг.
Пример 218
1,2 г Соединения Z9 и 0,5 г 1,4-диоксаспиро[4.5]дец-8-иламина растворяют в 20 мл дихлорметана и перемешивают с 1,28 г ТМУТ и 4 мл триэтиламина в течение 12 ч при комнатной температуре. Затем прибавляют 50 мл воды и 0,5 г карбоната калия, органическую фазу отделяют, сушат и выпаривают в вакууме. Остаток кристаллизуют из этилацетата, объединяют с 25 мл 1 н. хлористоводородной кислоты и 20 мл метанола и перемешивают в течение 30 мин при 50°С. Метанол удаляют в вакууме, осадок отфильтровывают с отсасыванием, промывают водой и сушат. Остаток растворяют в 20 мл дихлорметана, перемешивают с 0,5 г тиоморфолина и 0,5 г NaBH(OAc)3 в течение 12 ч при комнатной температуре. Затем прибавляют воду и карбоната калия, органическую фазу отделяют, сушат и растворитель удаляют в вакууме. Остаток очищают на колонке с силикагелем. Соответствующие фракции выпаривают в вакууме и гидрохлорид осаждают эфирным раствором HCl. Выход: 86 мг транс-изомера; аморфное порошкообразное вещество.
Пример 187
200 мг Соединения Z3 в 5 мл дихлорметана объединяют с 0,1 мл диизопропилэтиламина и 180 мг ТМУТ и перемешивают в течение 30 мин. Затем прибавляют 191 мг 4-(4-метилпиперазин-1-ил)-фениламина и смесь перемешивают в течение ночи. Реакционную смесь объединяют с водой и водную фазу экстрагируют дихлорметаном. Объединенные органические фазы сушат над Na2SO4 и выпаривают. Остаток очищают с помощью хроматографии на колонке (элюент: дихлорметан/метанол 100:7). Выход: 128 мг (светло-желтые кристаллы).
Соединения формулы (I), приведенные в таблице 1, в частности, получают по методикам, аналогичным описанным выше в настоящем изобретении. Обозначения X1, Х2, Х3, Х4 и Х5, использованные в таблице 1, в каждом случае обозначают связывание в положении в общей формуле, приведенной в таблице 1, использованные в таблице, а не соответствующие группы R1, R2, R3, R4 и L-R5.
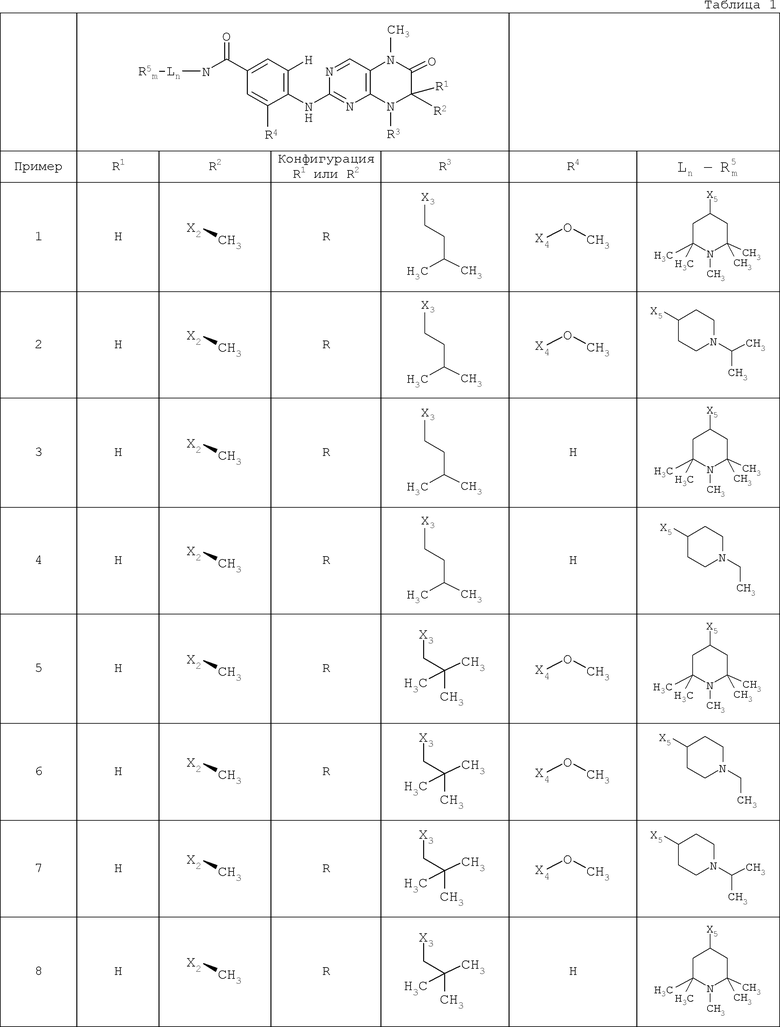
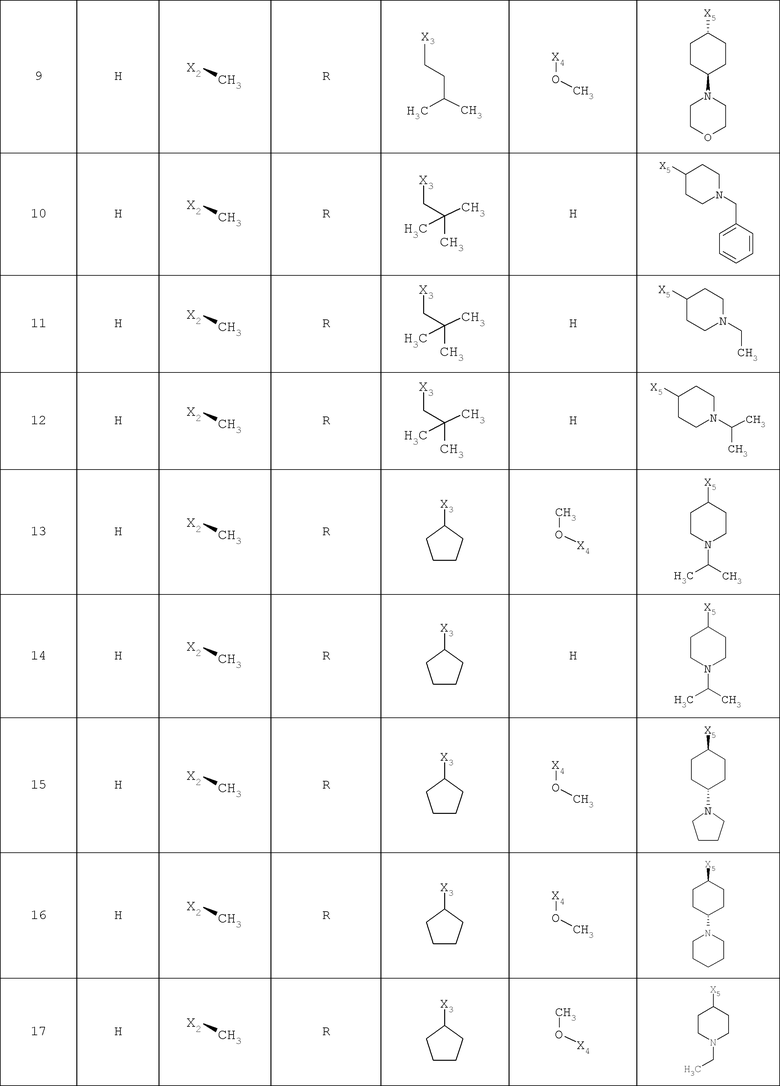
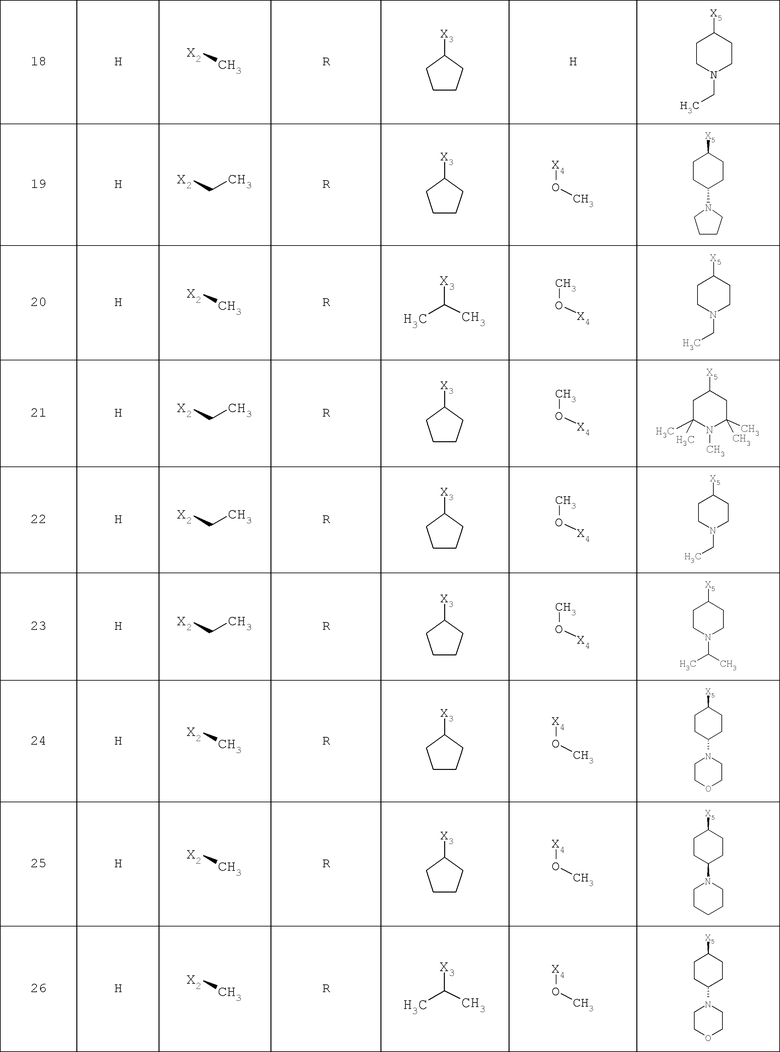
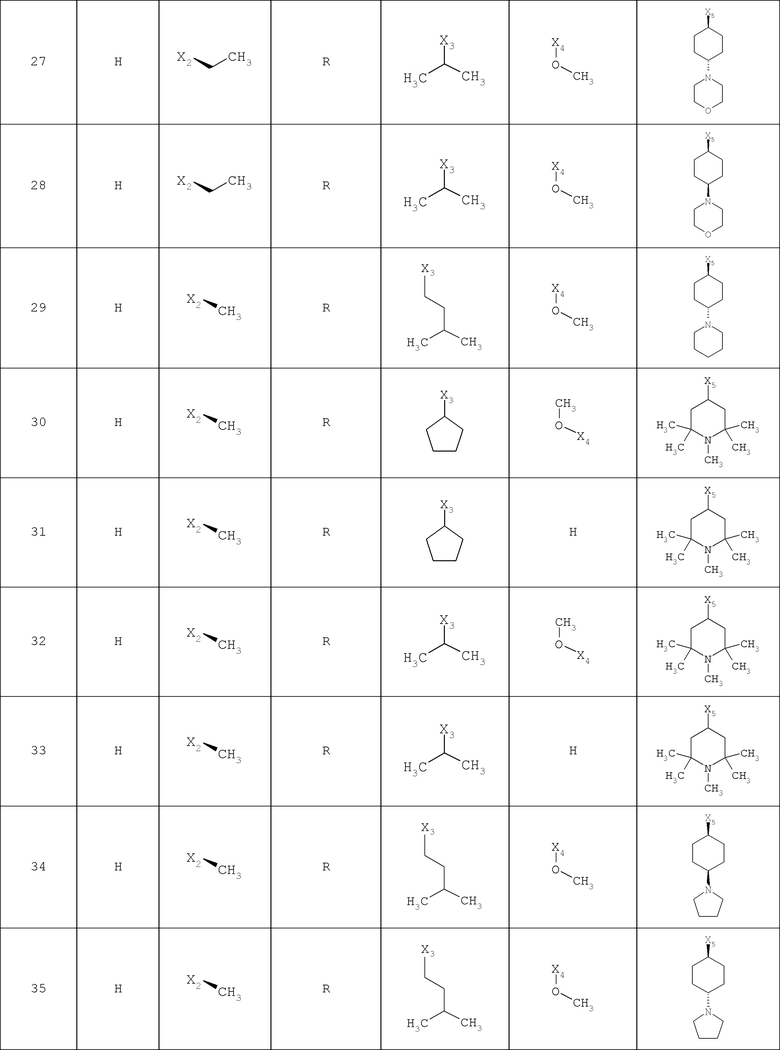
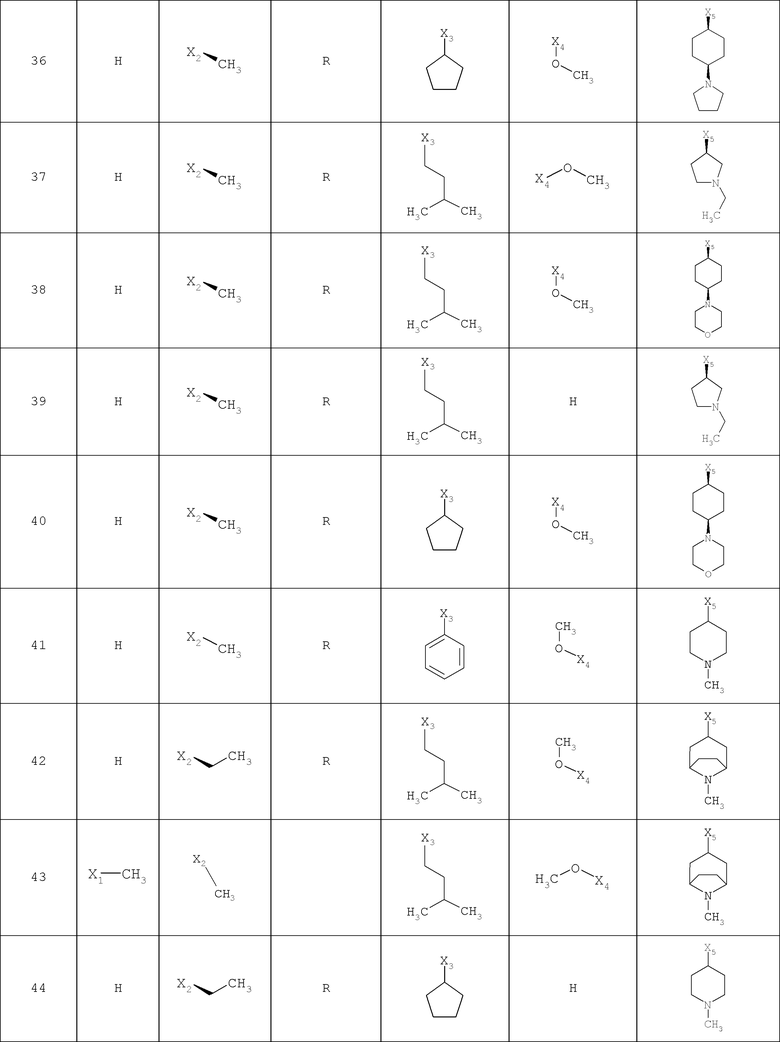
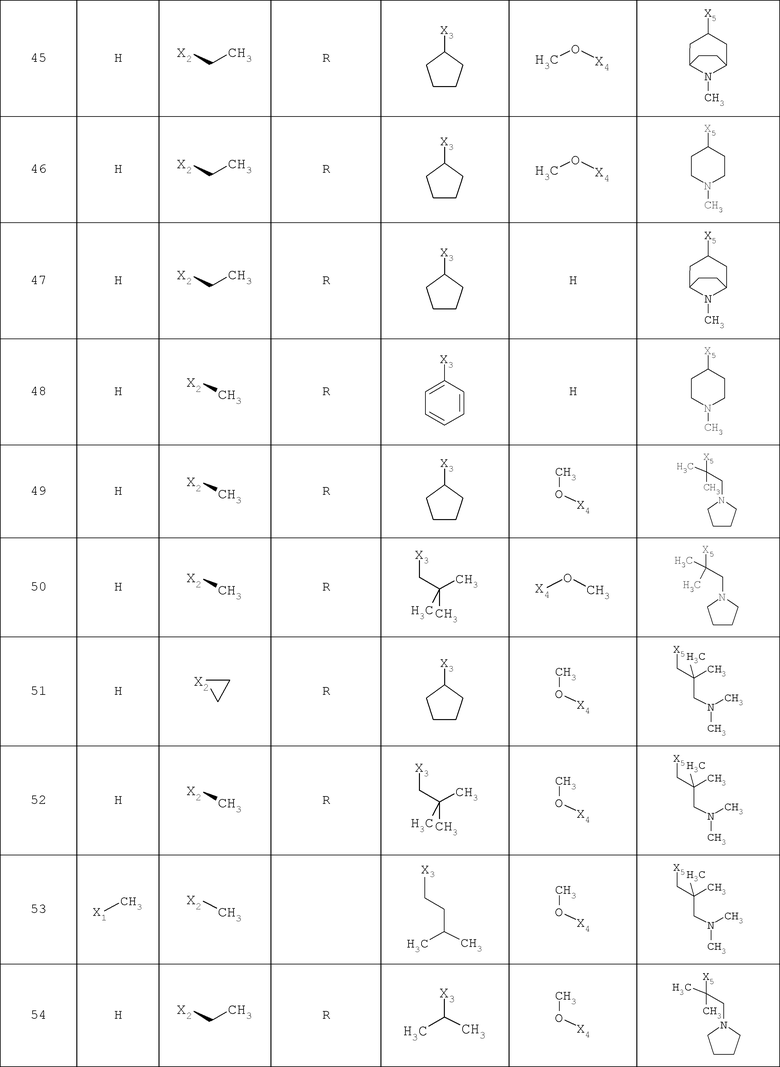
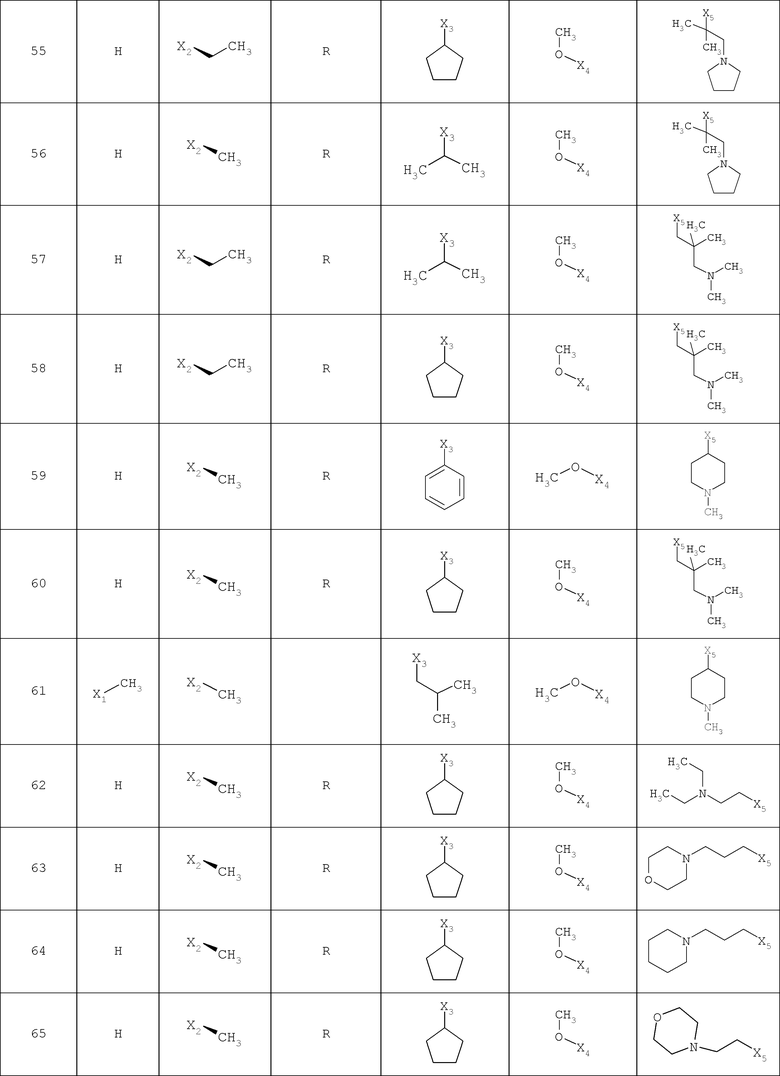
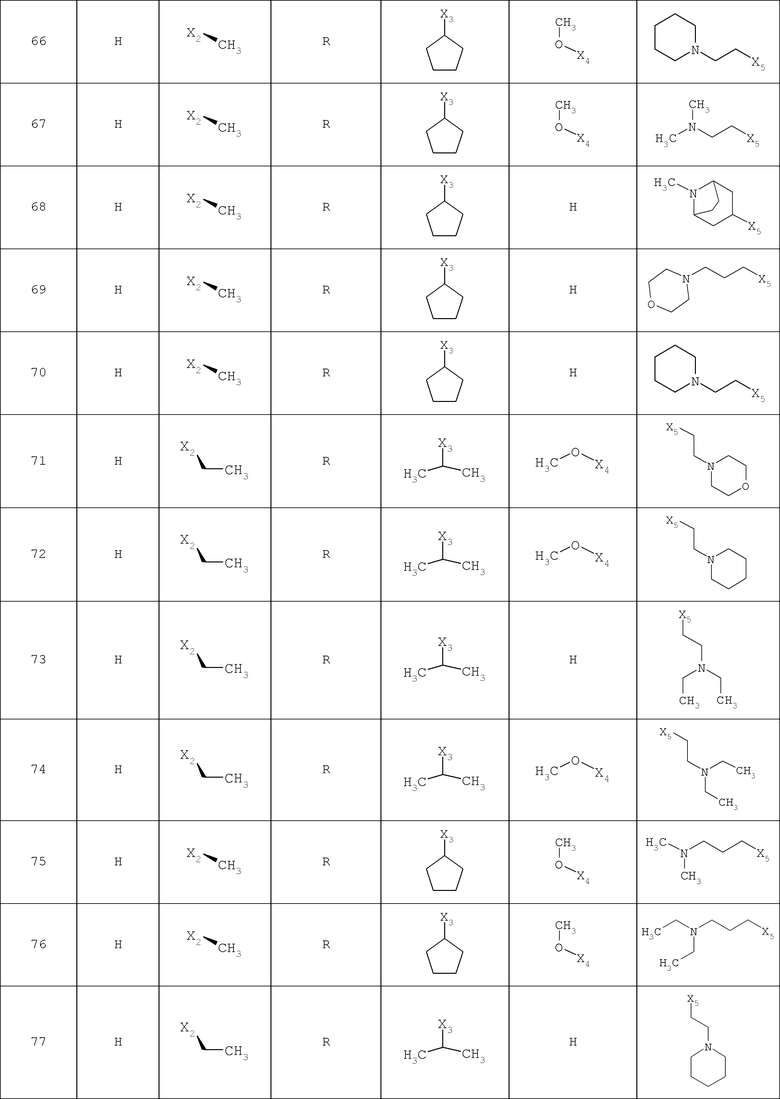
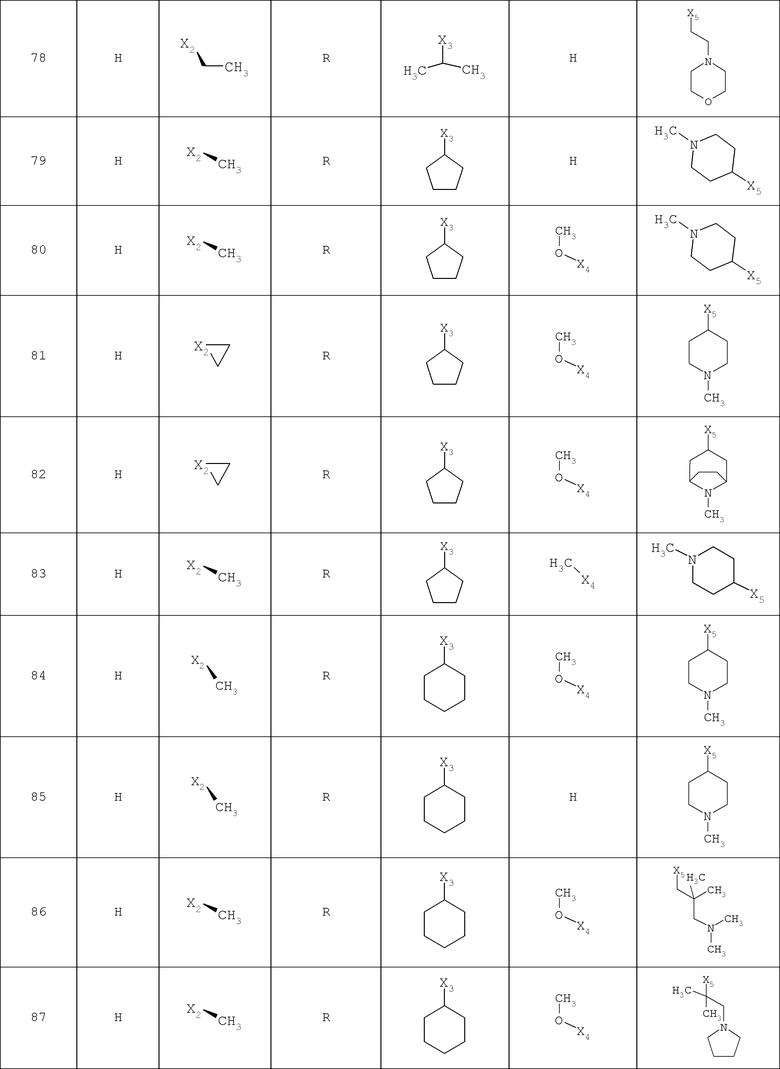
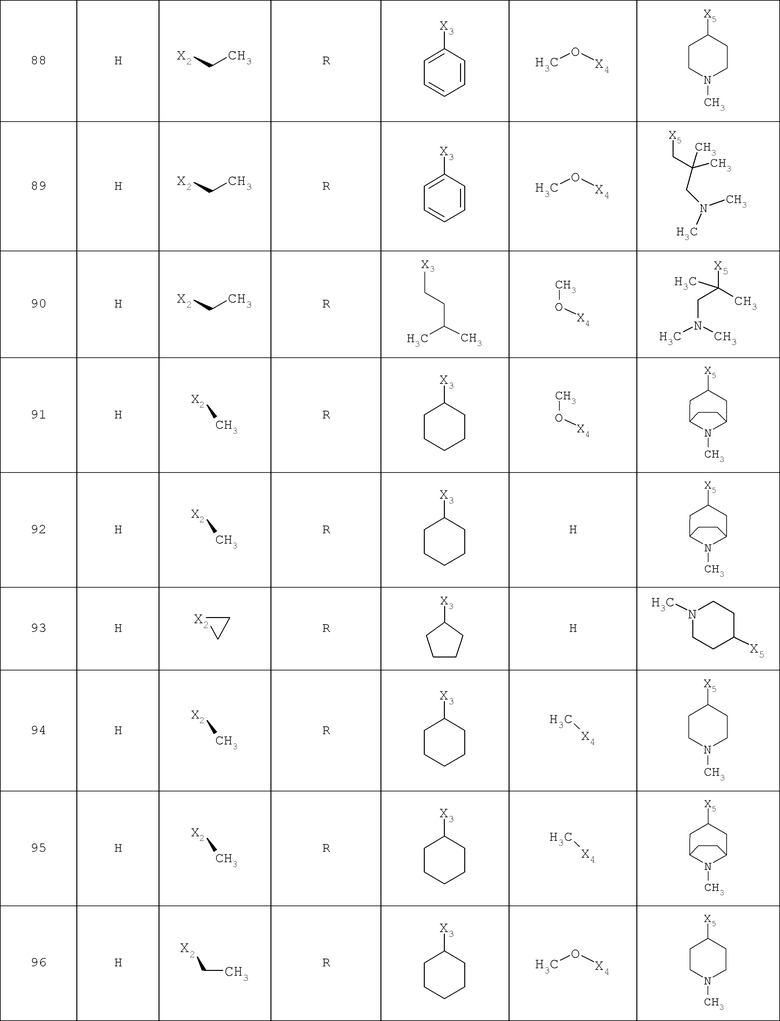
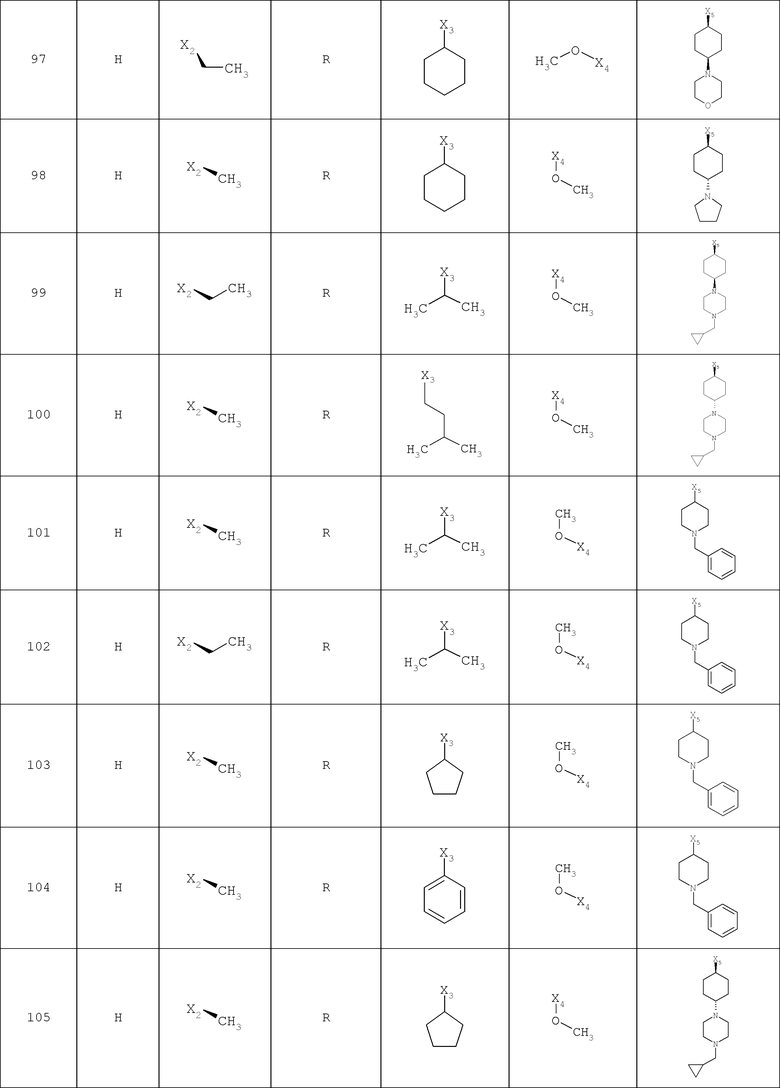
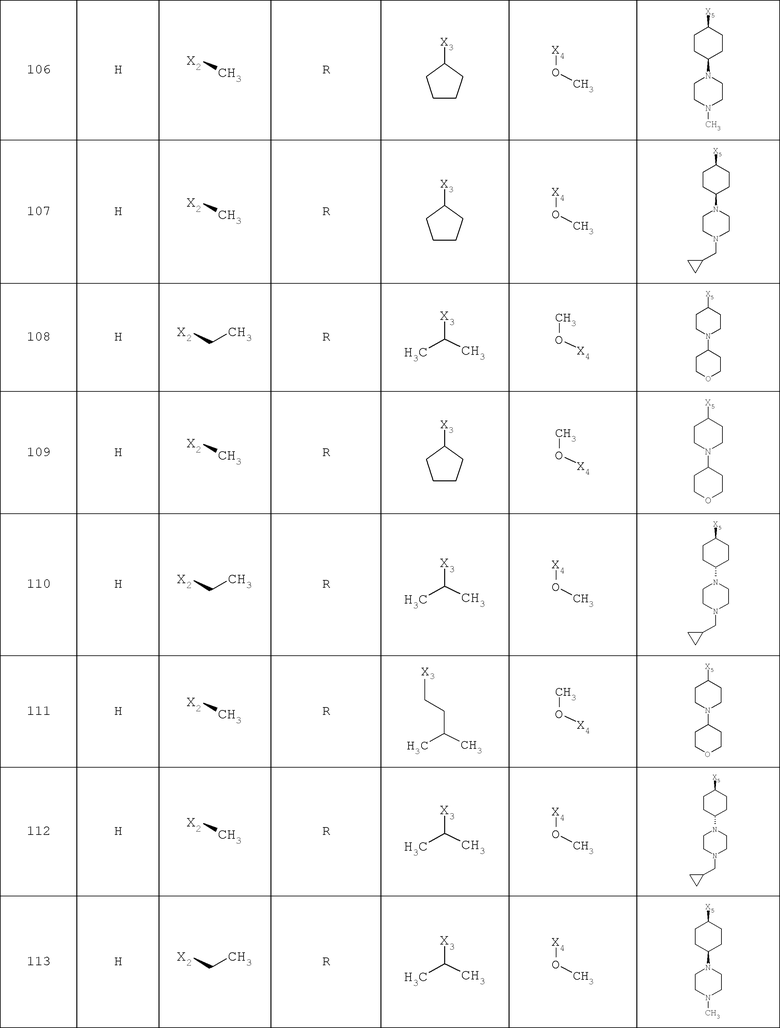
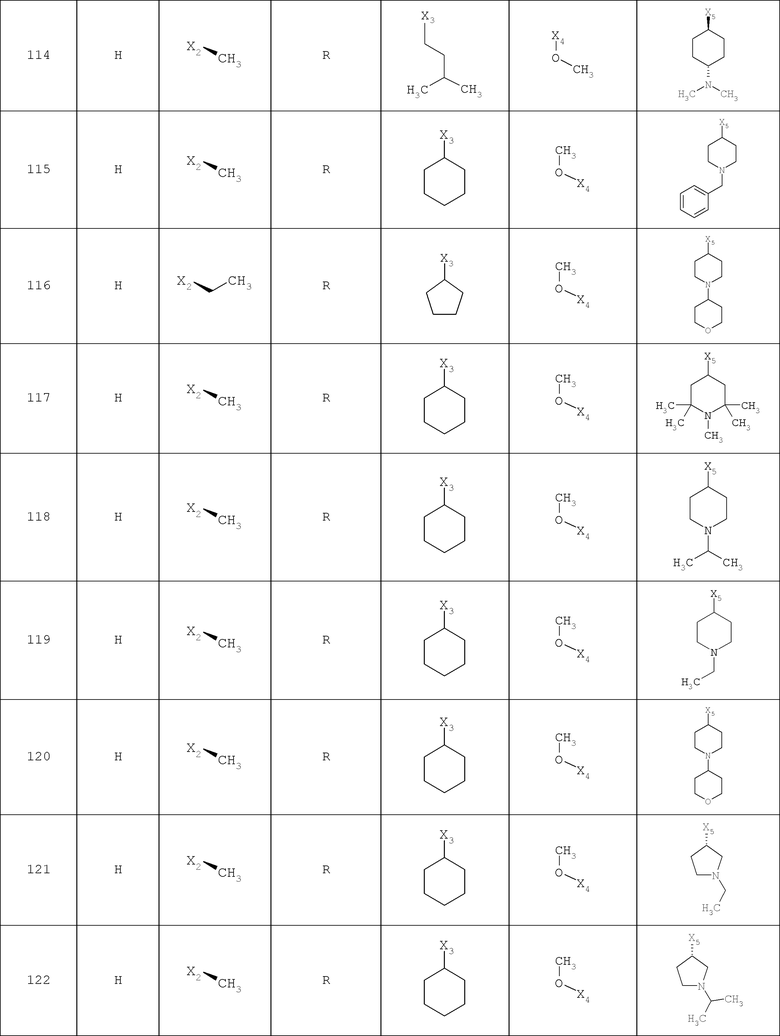
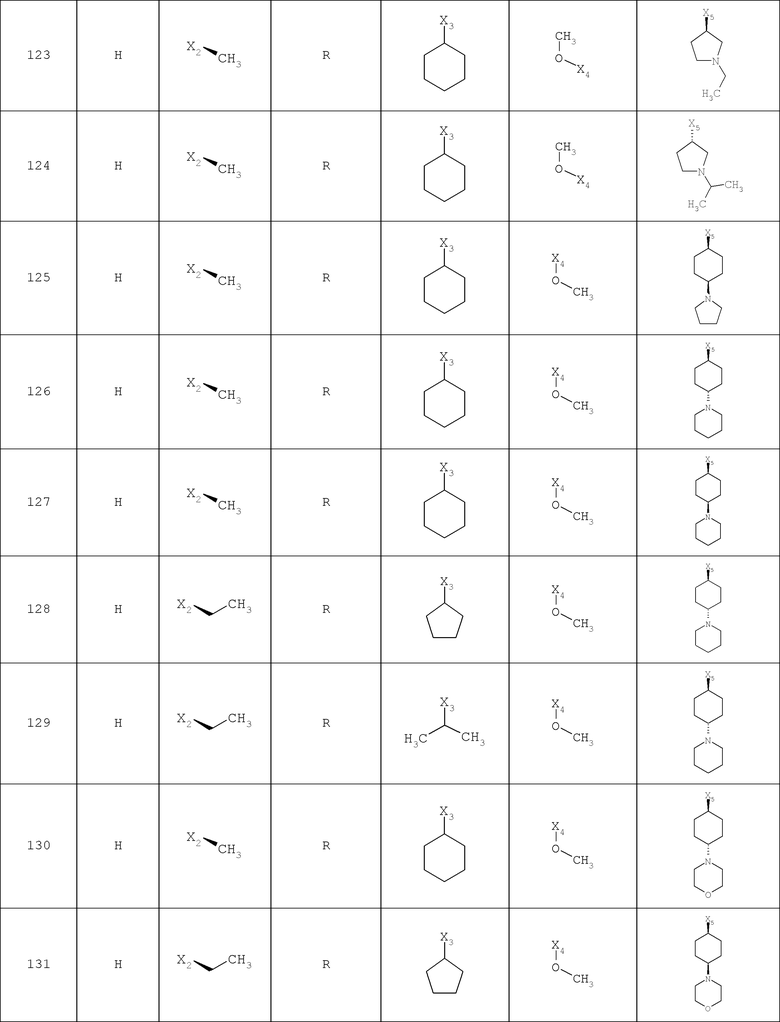
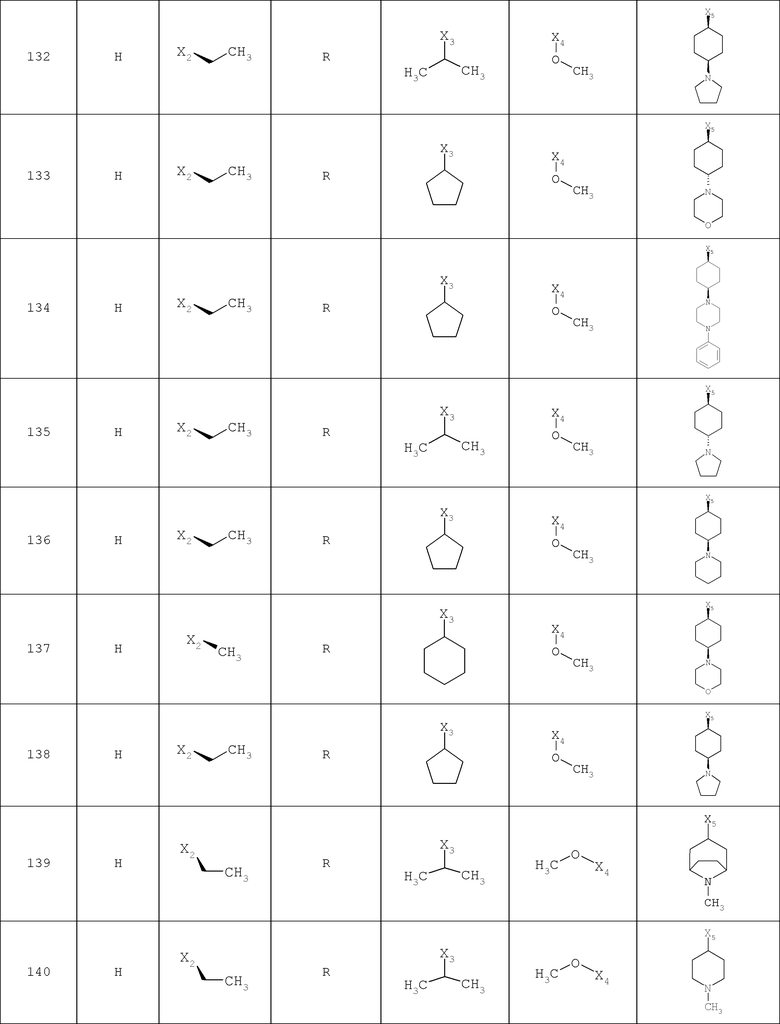
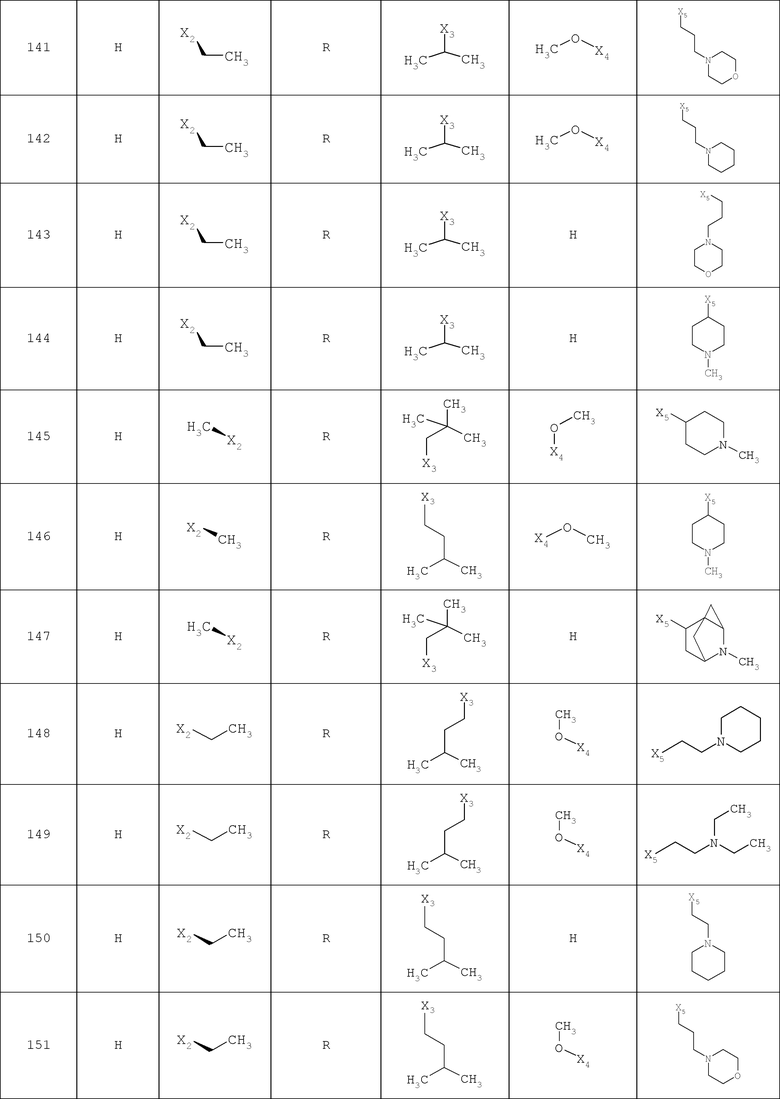
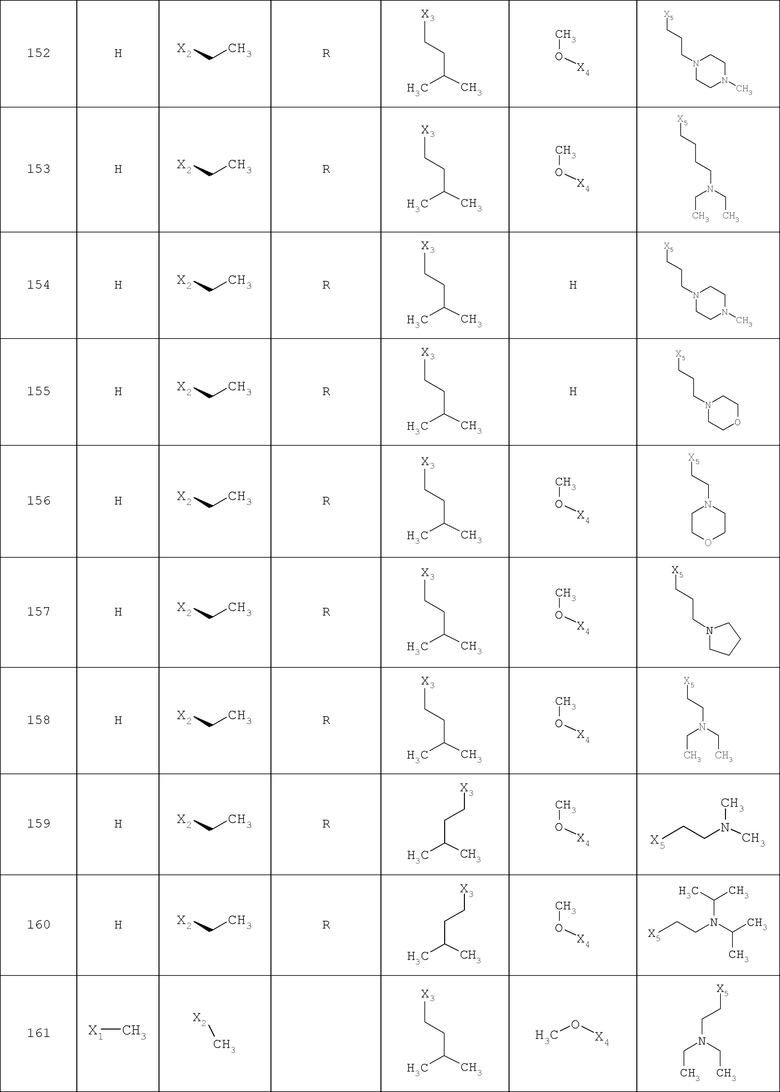
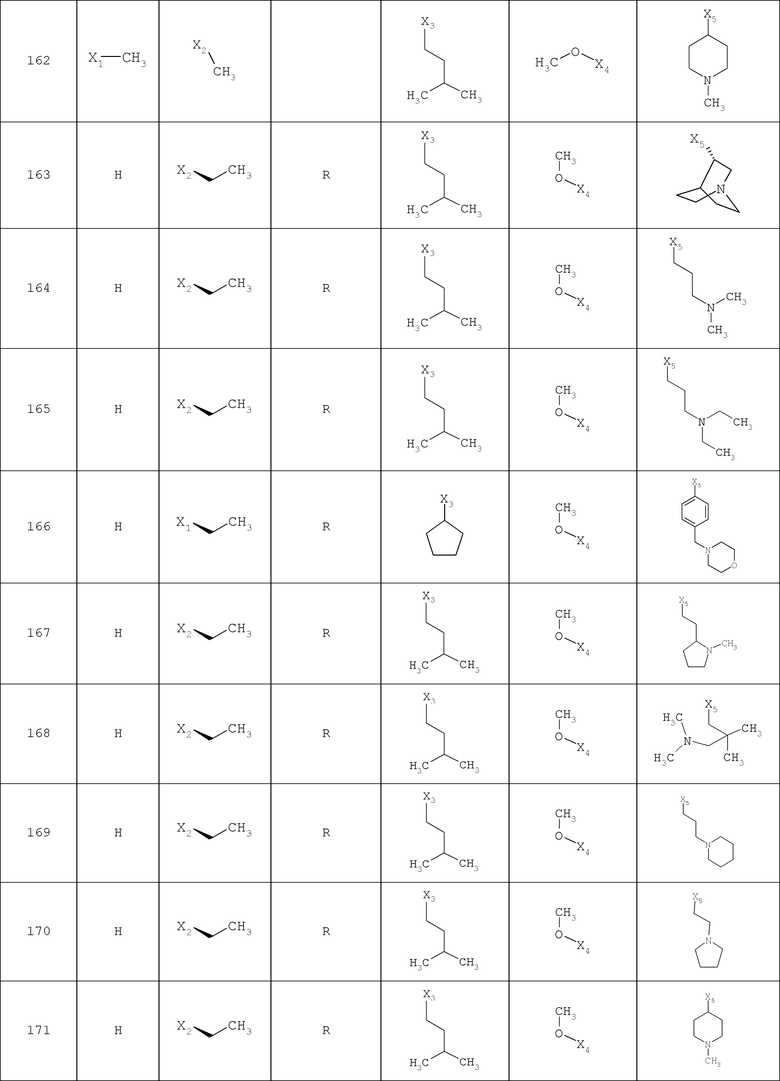
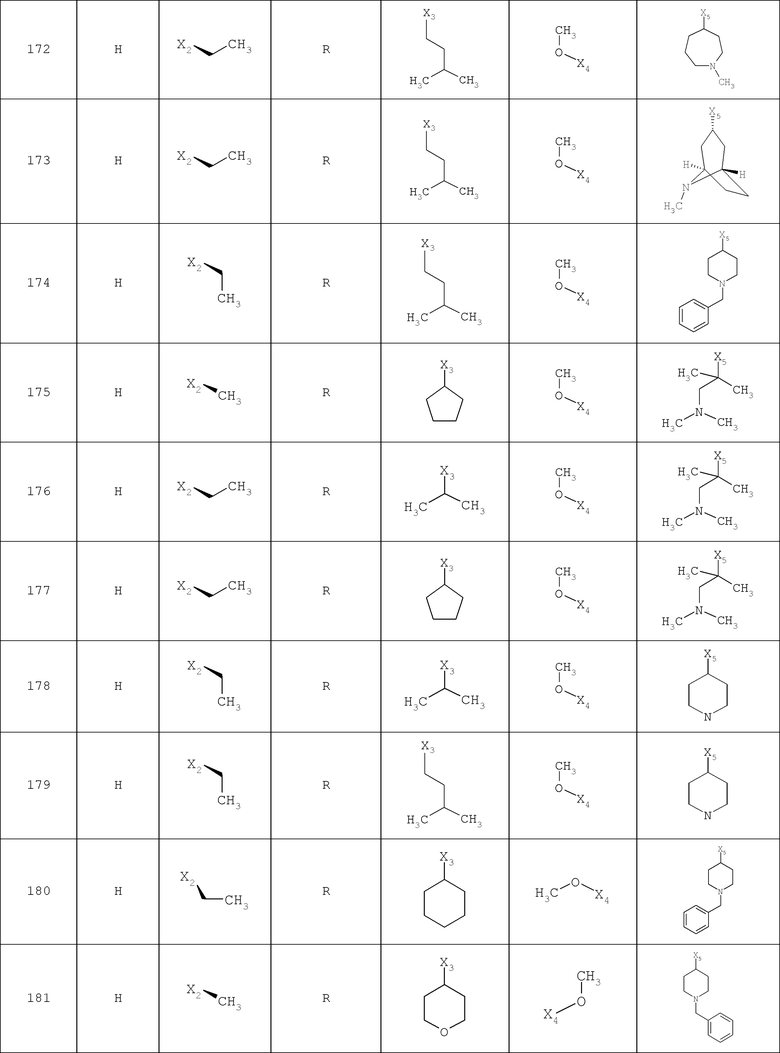
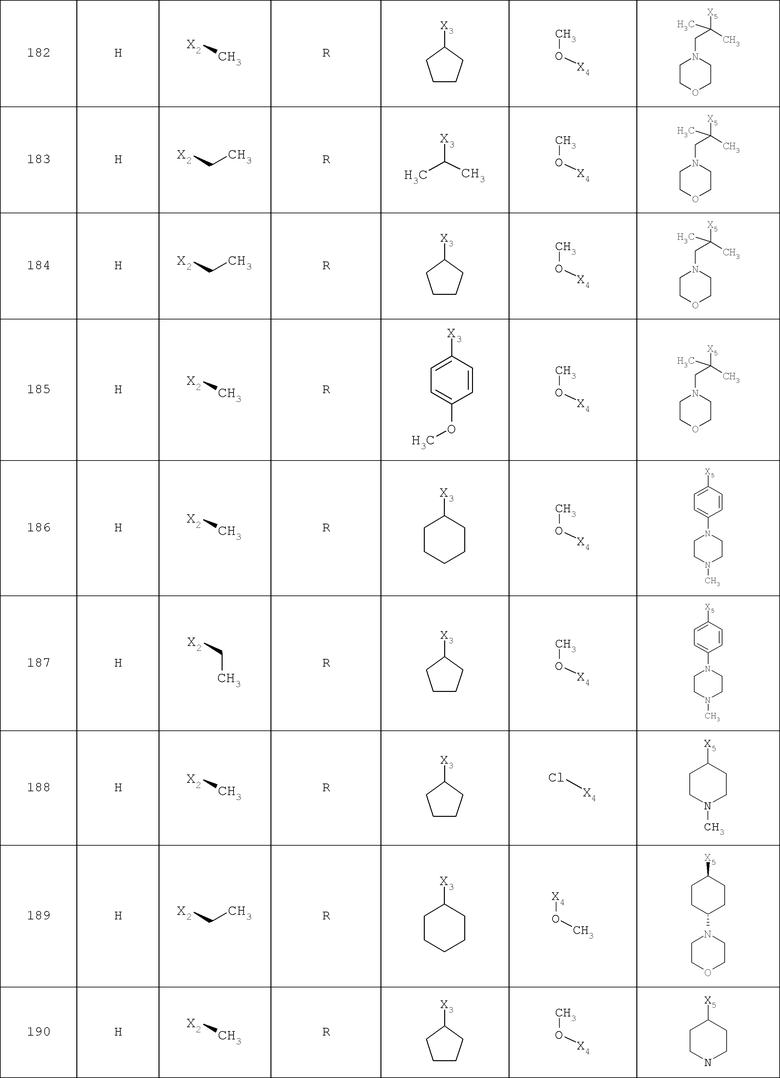
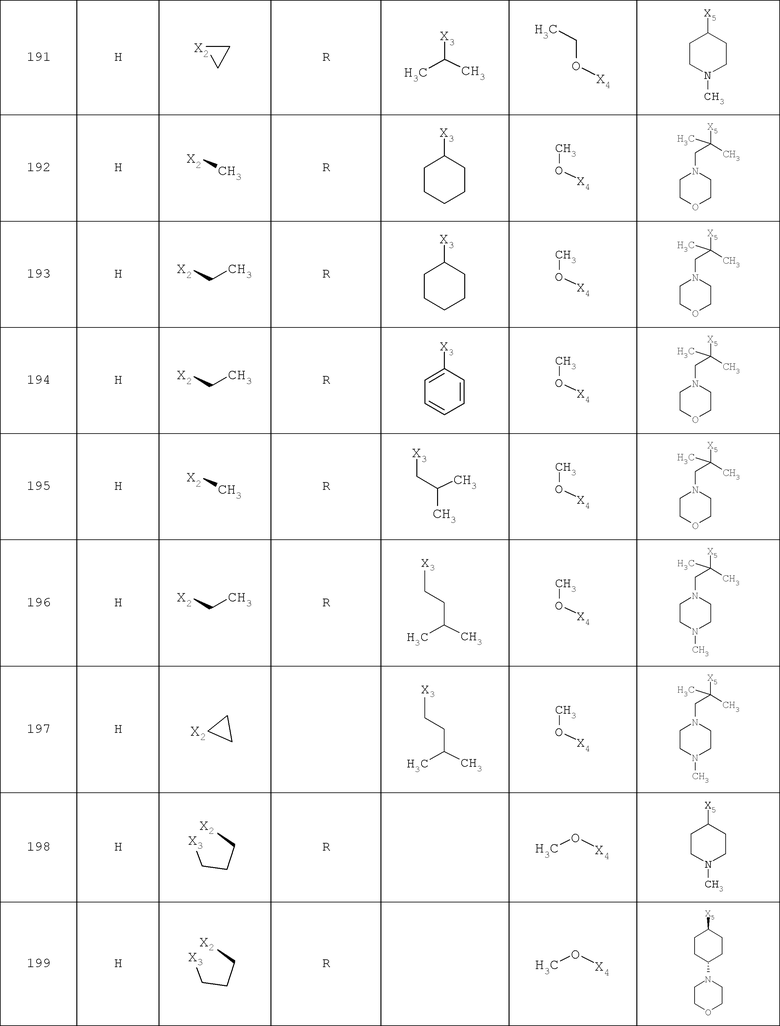
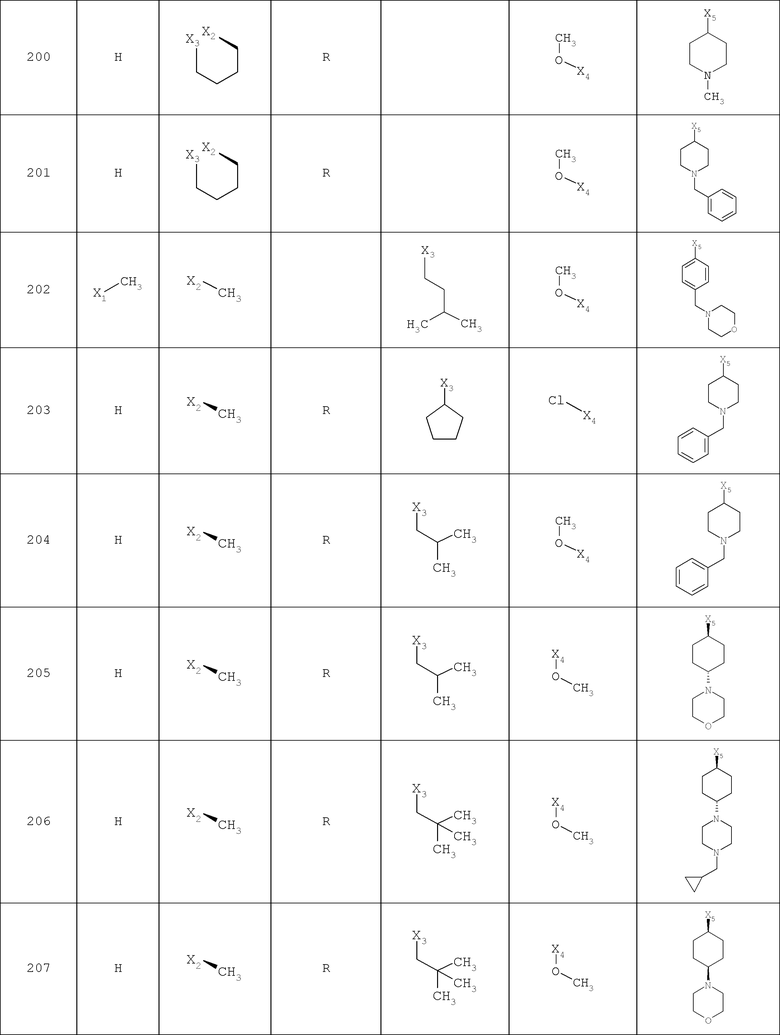
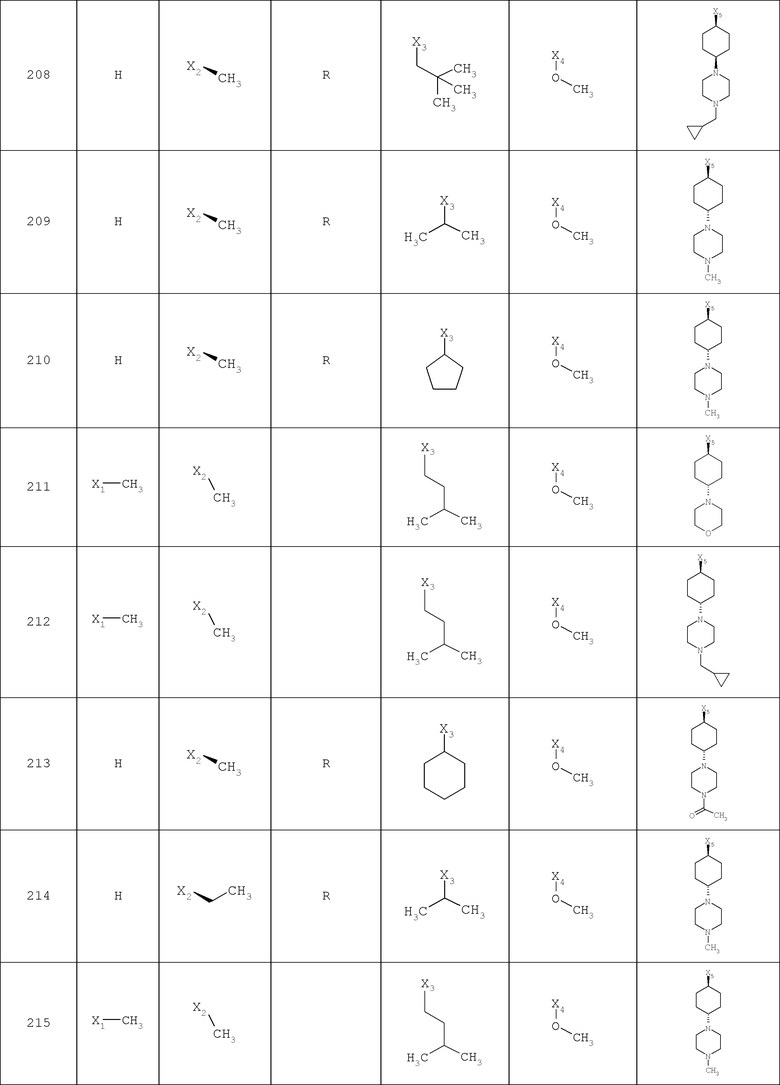
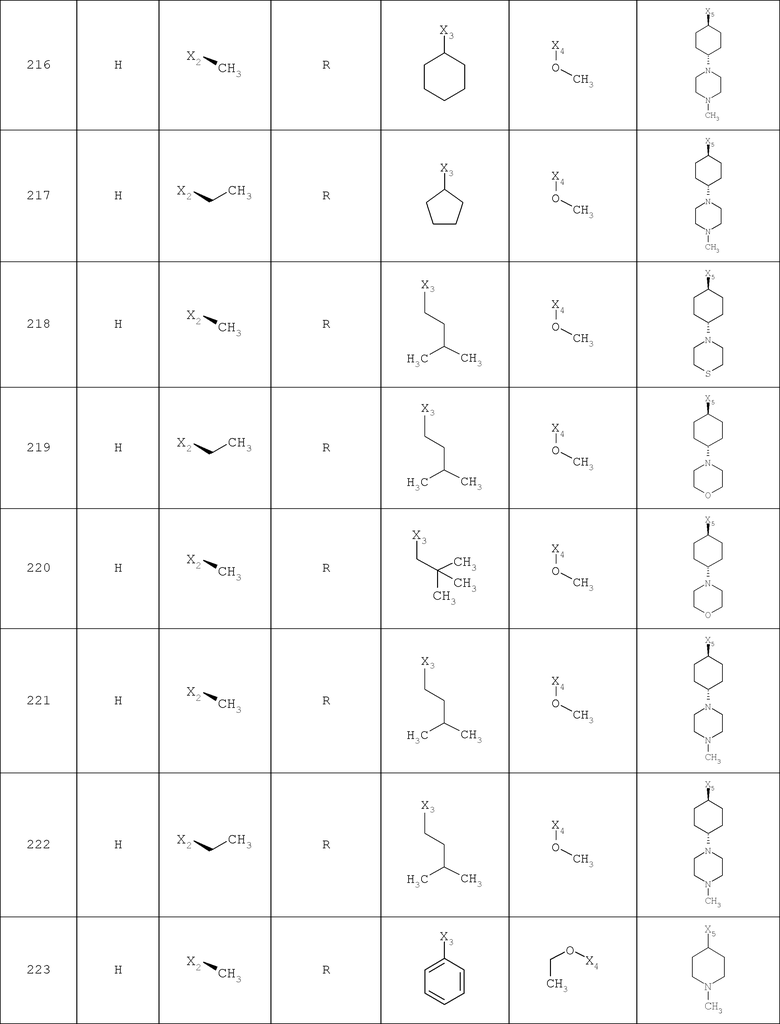
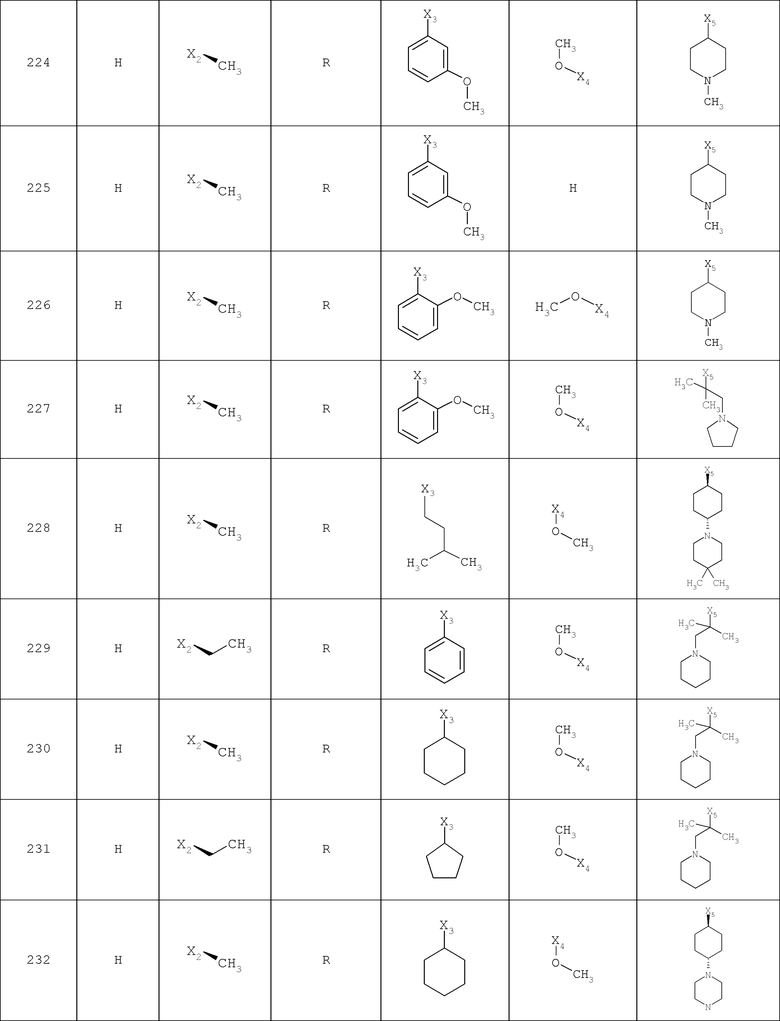
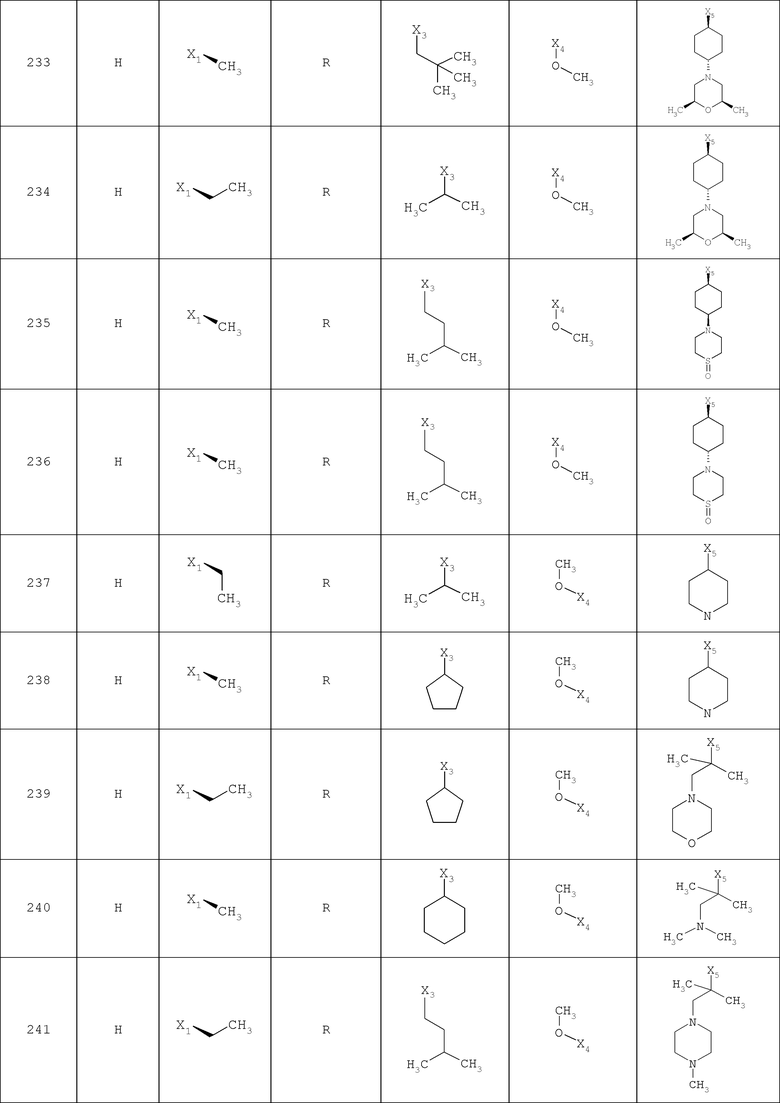
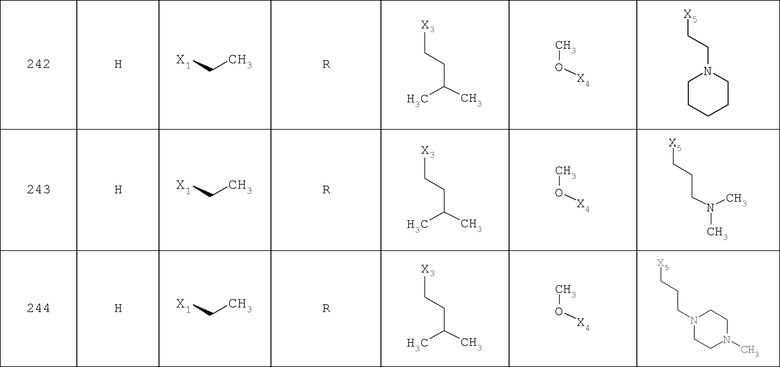
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ ХИМИЧЕСКИЕ СОЕДИНЕНИЯ (ВАРИАНТЫ) И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ ОНКОЛОГИЧЕСКИХ ЗАБОЛЕВАНИЙ | 2013 |
|
RU2550346C2 |
| НОВЫЕ N-СУЛЬФАМОИЛПИПЕРИДИНАМИДЫ, ПРЕДНАЗНАЧЕННЫЕ ДЛЯ ПРОФИЛАКТИКИ ИЛИ ЛЕЧЕНИЯ ОЖИРЕНИЯ И РОДСТВЕННЫХ ПАТОЛОГИЧЕСКИХ СОСТОЯНИЙ | 2006 |
|
RU2442773C2 |
| ТРИЦИКЛИЧЕСКИЕ ПРОТИВООПУХОЛЕВЫЕ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ НА ИХ ОСНОВЕ | 2001 |
|
RU2293734C9 |
| ПИРИМИДИНОВЫЕ СОЕДИНЕНИЯ, ОБЛАДАЮЩИЕ АНТИПРОЛИФЕРАТИВНОЙ АКТИВНОСТЬЮ (II) | 2003 |
|
RU2326882C2 |
| Соединение-ингибитор мультикиназ и его кристаллическая форма и применение | 2017 |
|
RU2723985C1 |
| СОЕДИНЕНИЯ ПИРИМИДИНИЛИНДОЛА | 2010 |
|
RU2552999C2 |
| СОЕДИНЕНИЯ, КОМПОЗИЦИИ И СПОСОБЫ, ПОДХОДЯЩИЕ ДЛЯ МОБИЛИЗАЦИИ ХОЛЕСТЕРИНА | 2011 |
|
RU2576402C2 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА ИНГИБИТОРА ФОСФОДИЭСТЕРАЗЫ, СПОСОБ ЕЕ ПОЛУЧЕНИЯ И ЕЕ ПРИМЕНЕНИЕ | 2020 |
|
RU2814498C2 |
| НОВЫЕ ХИМИЧЕСКИЕ СОЕДИНЕНИЯ ПРОИЗВОДНЫЕ 2,4-ДИАМИНО-1,3,5-ТРИАЗИНА ДЛЯ ПРОФИЛАКТИКИ И ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ ЧЕЛОВЕКА И ЖИВОТНЫХ | 2012 |
|
RU2509770C2 |
| ФУМАРАТ ПИРИДИЛАМИНА И ЕГО КРИСТАЛЛЫ | 2015 |
|
RU2684278C1 |
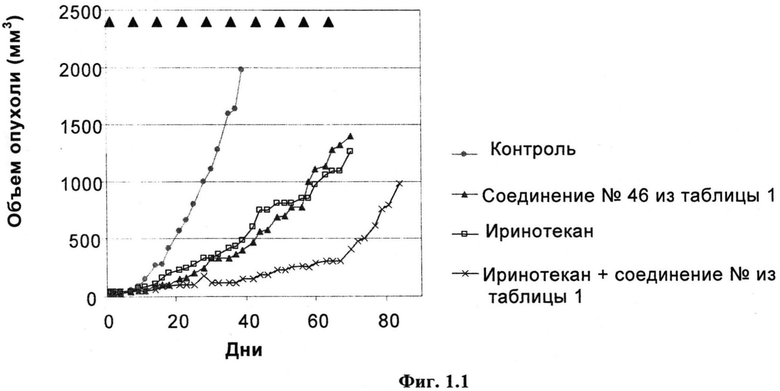
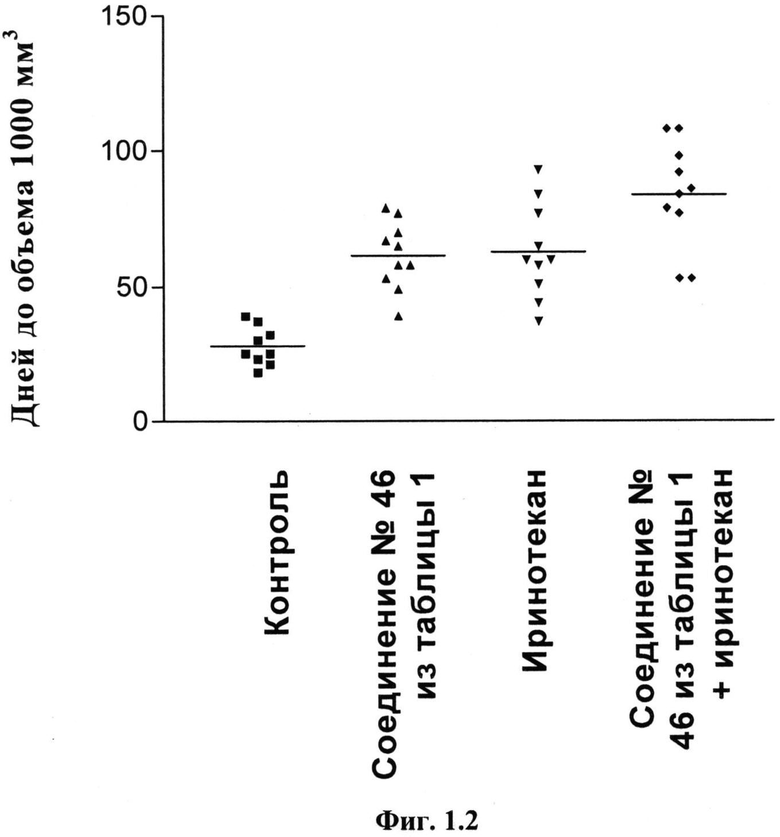
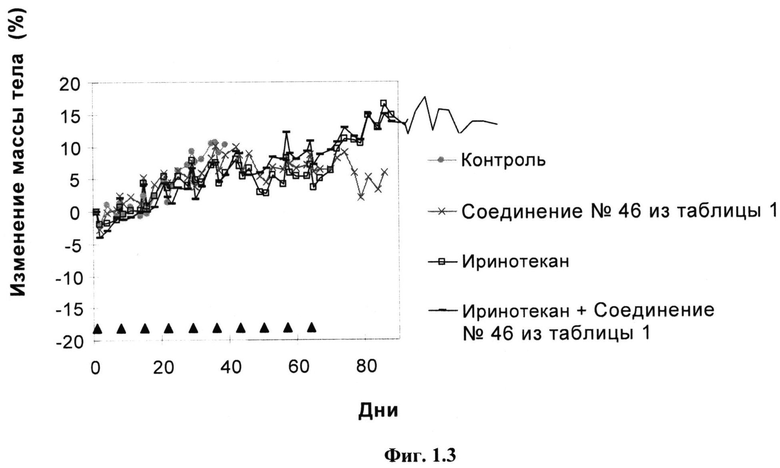
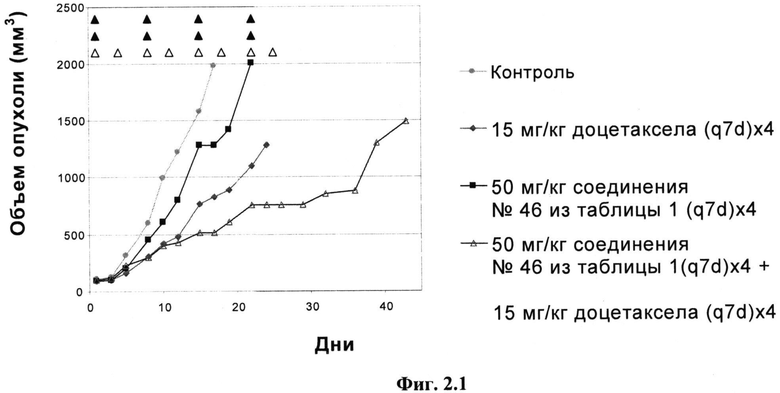
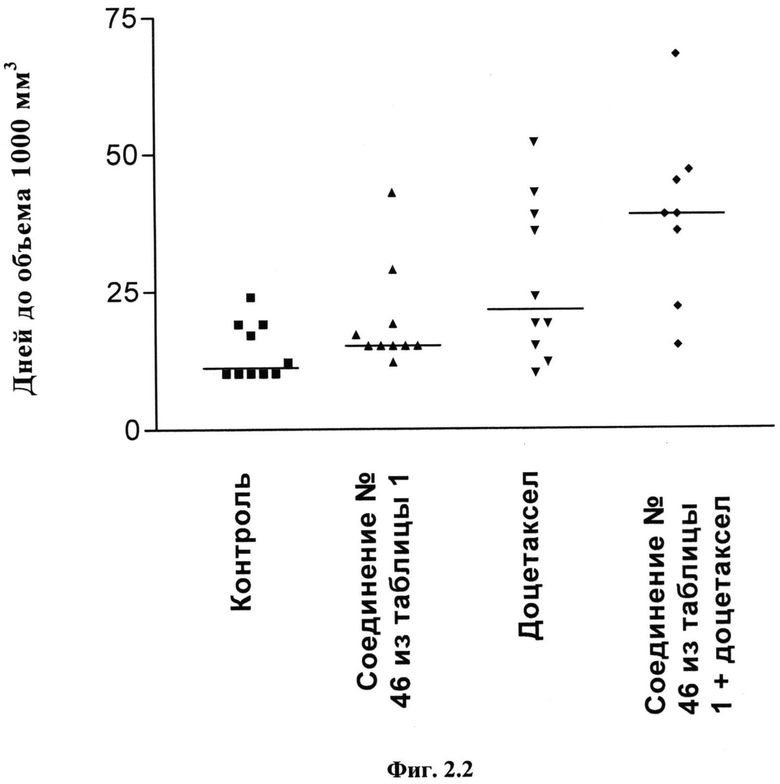
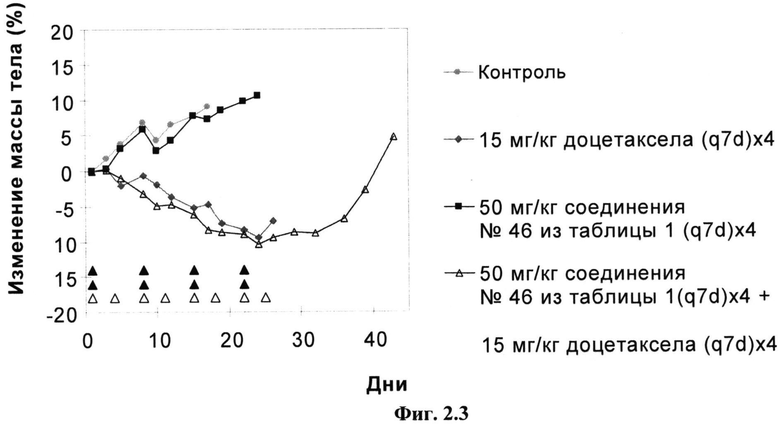
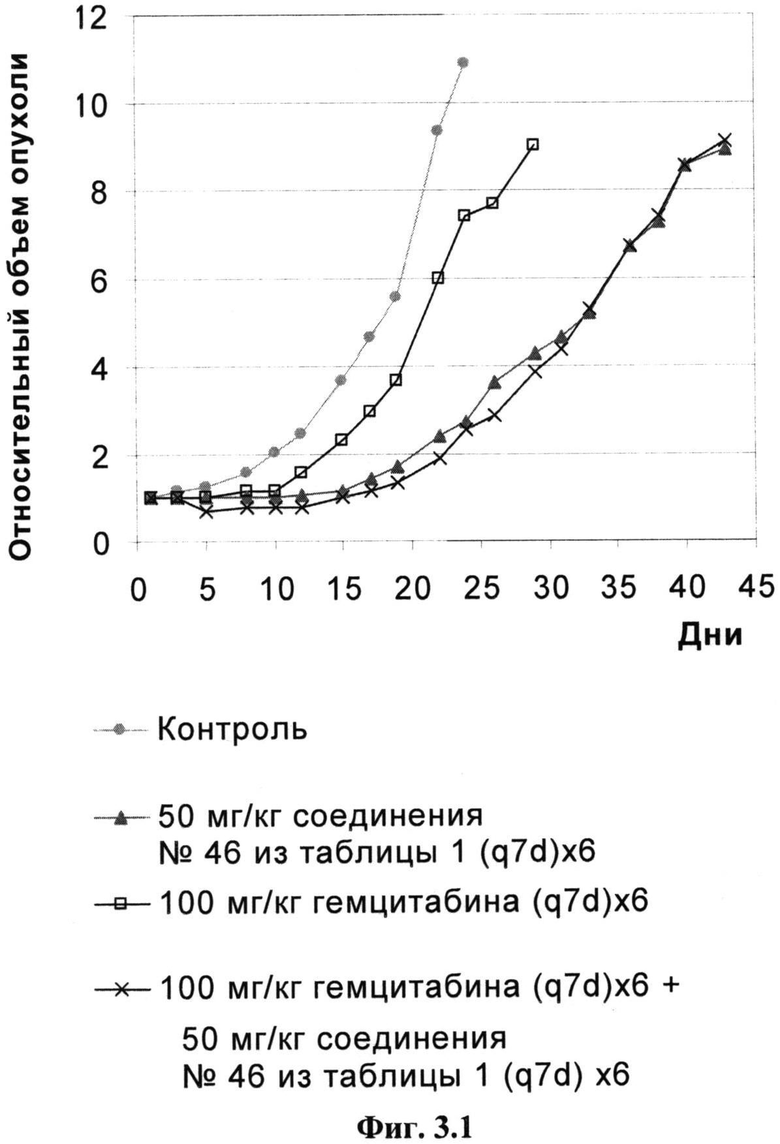
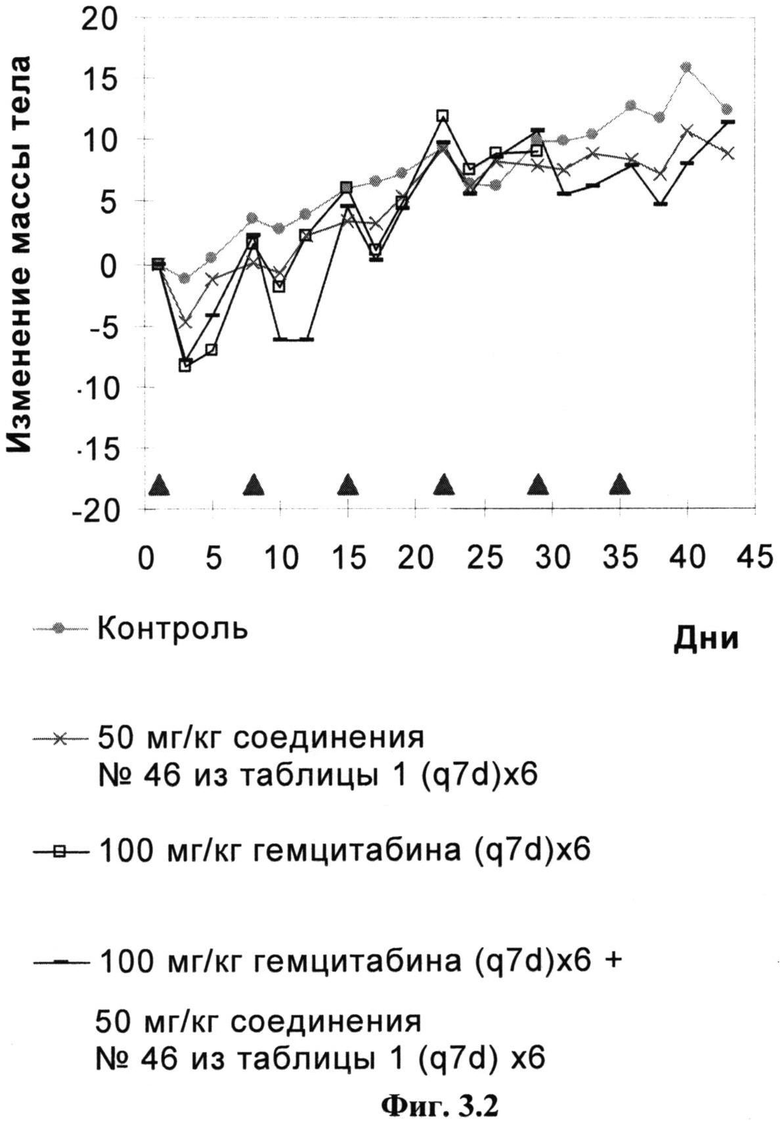
Группа изобретений относится к фармацевтической композиции, предназначенной для лечения заболеваний, которые включают пролиферацию клеток, ее применению и набору, включающему совместное введение соединения  формулы (I)
формулы (I) 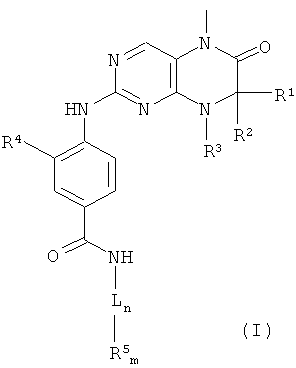 в которой группы L, R1, R2, R3, R4 и R5 обладают значениями, приведенными в формуле изобретения и описании, необязательно в форме его таутомеров, рацематов, энантиомеров, диастереоизомеров и их смесей и необязательно в форме его фармакологически приемлемых солей присоединения с кислотами, сольватов, гидратов, полиморфных форм, физиологически функциональных производных или пролекарств, и эффективного количества активного соединения
в которой группы L, R1, R2, R3, R4 и R5 обладают значениями, приведенными в формуле изобретения и описании, необязательно в форме его таутомеров, рацематов, энантиомеров, диастереоизомеров и их смесей и необязательно в форме его фармакологически приемлемых солей присоединения с кислотами, сольватов, гидратов, полиморфных форм, физиологически функциональных производных или пролекарств, и эффективного количества активного соединения  и/или совместное лечение с лучевой терапией, в соотношении, которое обеспечивает аддитивный и синергетический эффект. 3 н. и 2 з.п. ф-лы, 8 ил., 2 табл., 24 пр.
и/или совместное лечение с лучевой терапией, в соотношении, которое обеспечивает аддитивный и синергетический эффект. 3 н. и 2 з.п. ф-лы, 8 ил., 2 табл., 24 пр.
1. Фармацевтическая комбинация, включающая эффективные количества: (i) соединения 1 формулы (I)
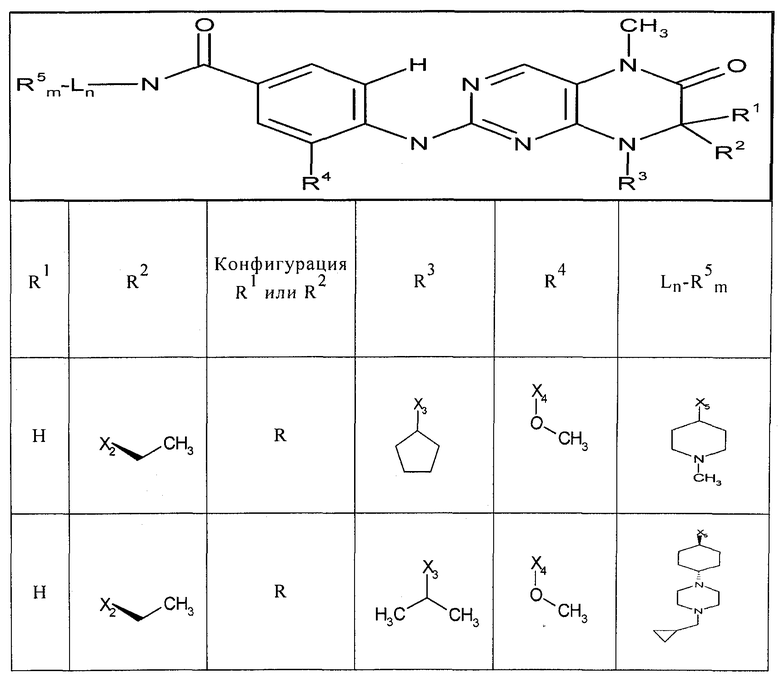
где обозначения X1, Х2, Х3, Х4 и Х5, использованные в таблице, в каждом случае обозначают связывание в положении в общей формуле, приведенной в таблице, а не соответствующие группы R1, R2, R3, R4 и Ln-R5 m,
необязательно в форме его таутомеров, рацематов, энантиомеров, диастереоизомеров и их смесей и необязательно в форме его фармакологически приемлемых солей присоединения с кислотами, сольватов, гидратов или полиморфных форм;
и
(ii) по меньшей мере одного дополнительного терапевтического средства 2, выбранного из группы, состоящей из доцетаксела, гемцитабина и иринотекана,
необязательно в комбинации с одним или более фармацевтически приемлемыми инертными наполнителями, и необязательно приспособленная для совместного лечения с лучевой терапией или радиоиммунотерапией, в форме комбинированного препарата, предназначенного для одновременного, раздельного или последовательного применения для лечения онкологических заболеваний.
2. Применение фармацевтической комбинации по п.1 для приготовления лекарственного средства, предназначенного для лечения онкологического заболевания, выбранного из группы, состоящей из рака ободочной кишки, рака легких и рака поджелудочной железы.
3. Набор, предназначенный для лечения онкологических заболеваний, выбранных из группы, состоящей из рака ободочной кишки, рака легких и рака поджелудочной железы, включающий терапевтически эффективное количество:
(i) соединения 1 формулы (I) по п.1;
и
(ii) по меньшей мере дополнительного терапевтического средства 2 по любому из пп.1 и 2;
и необязательно, приспособленный для совместного лечения с лучевой терапией или радиоиммунотерапией;
характеризующийся тем, что соединение 1 формулы (I) содержится в первом отделении и дополнительное терапевтическое средство 2 содержится во втором отделении, так что введение нуждающемуся в нем пациенту может быть одновременным, раздельным или последовательным.
4. Набор по п.3, в котором препарат соединения 1 формулы (I) предназначен для перорального введения или инъекции.
5. Применение по п.2, характеризующееся тем, что соединение 1, формулы (I) или его полиморфную форму, гидрат или фармацевтически приемлемую соль вводят периодически или в суточной дозе так, что содержание активного вещества в плазме составляет от 10 до 5000 нМ в течение не менее 12 ч после введения.
| WO 2003020722 A1, 13.03.2003 | |||
| БЕЛИКОВ В.Г | |||
| Фармацевтическая химия, М., Высшая школа, 1993, с.43-47 | |||
| МАШКОВСКИЙ М.Д | |||
| "Лекарственные средства", Москва, "Медицина", 1993, ч.1, стр.11 | |||
| ДАЙСОН Г | |||
| "Химия синтетических лекарственных веществ, пер | |||
| с англ | |||
| М.: "Мир", 1964, стр.12-19 |

