ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к кристаллическим формам (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ола и способам его производства.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
(3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ол (далее в настоящем документе называемый Соединением (1)) Формулы (1):
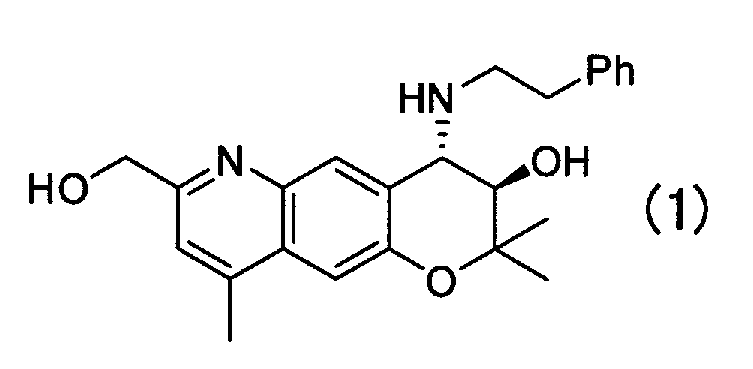
проявляет антиаритмическое действие, и, как известно, его можно применять в качестве лекарственного средства.
В качестве лекарственных средств требуются соединения, обладающие постоянными свойствами, благодаря которым всегда можно ожидать, что они будут проявлять постоянную эффективность, и поэтому их, как правило, кристаллизуют. Кроме того, кристаллизация соединения дает возможность получать такое преимущество, как повышение его химической стабильности (см., например, Непатентный документ 1). Между тем известно, что твердый материал обладает способностью принимать две или более разных кристаллических структур. Эти две или более кристаллические структуры, как описано выше, называют кристаллическим полиморфизмом. Кроме того, известно, что в тех случаях, когда соединение кристаллизуют в органическом растворителе или в воде, органический растворитель или вода, используемые для кристаллизации, иногда включаются в соединение, так что соединение становится кристаллическим сольватом (кристаллосольватом) (сольватом) или кристаллогидратом (гидратом). В настоящем описании для кристаллического полиморфизма, кристаллических сольватов (кристаллосольватов) и кристаллогидратов принято наименование «кристаллические формы». Эти кристаллические формы обычно имеют разную растворимость, скорость растворения, стабильность, гигроскопичность, точки плавления, способность подвергаться обработке и т.п., поэтому при разработке лекарственного средства в виде кристаллической формы некоторого соединения необходимо всесторонне оценивать эти характеристики и отбирать такую кристаллическую форму, которая подходит для создания лекарственного средства (см., например, Непатентный документ 2).
Однако в отношении Соединения (1) до настоящего времени имеется только сообщение о том, что это соединение получено посредством очистки с применением колоночной хроматографии, а кристаллическая форма Соединения (1) неизвестна (см., например, Патентный документ 1). Необходимо было выяснить, способно ли данное соединение кристаллизоваться и иметь кристаллические формы. Затем, если кристаллические формы существуют, необходимо было найти воспроизводимые способы получения химически стабильных кристаллических форм и выявить новые кристаллические формы данного соединения.
ДОКУМЕНТЫ ПО РОДСТВЕННЫМ ОБЛАСТЯМ ТЕХНИКИ
ПАТЕНТНЫЕ ДОКУМЕНТЫ
[Патентный документ 1]
Публикация международной заявки № WO 2005/090357.
НЕПАТЕНТНЫЕ ДОКУМЕНТЫ
[Непатентный документ 1]
«Iyakuhin no kesshoutakei to shouseki no kagaku (Science of crystal polymorphism and crystallization of drugs)» edited and written by Kazuhide Ashizawa, p. 392, published by Maruzen Planet Co., Ltd.
[Непатентный документ 2]
«Farumashia» vol. 45, No. 4, p. 327 (2009).
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
ЗАДАЧИ, РЕШАЕМЫЕ ИЗОБРЕТЕНИЕМ
Для разработки (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ола в качестве лекарственного средства необходимо подтвердить возможность кристаллизации данного соединения и существование его кристаллических форм; при наличии кристаллических форм требуется найти воспроизводимые способы получения химически стабильных кристаллических форм и выявить новые кристаллические формы данного соединения.
СРЕДСТВА РЕШЕНИЯ ПОСТАВЛЕННЫХ ЗАДАЧ
В результате тщательного исследования, предпринятого для решения этих задач, авторы настоящего изобретения обнаружили, что (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ол можно кристаллизовать, и, кроме того, было установлено, что существует не менее шести типов кристаллических форм этого соединения.
Конкретно, настоящее изобретение заключается в следующем.
(I)
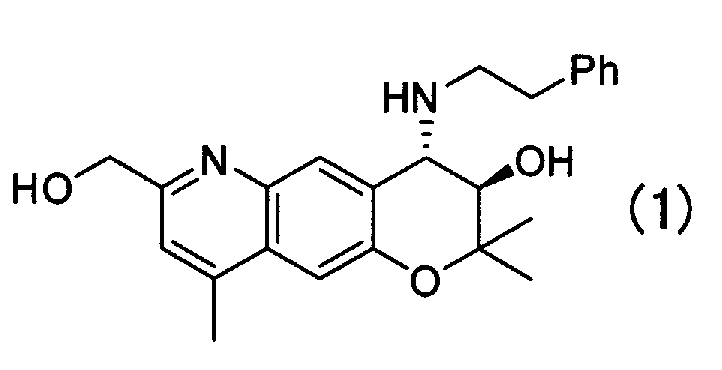
Кристаллическая форма А (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ола Формулы (I):
 ,
,
имеющая характеристические пики при углах дифракции (2θ) 5,6, 8,2, 12,0, 14,7, 16,6, 16,9, 17,9, 18,4, 22,5, 24,5, 27,6 на порошковой рентгеновской дифрактограмме кристаллов.
(II)
Способ получения кристаллической формы А (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ола, характеризующийся тем, что включает кристаллизацию соединения Формулы (1) в сложном эфире в качестве растворителя.
(III)
Способ получения кристаллической формы А (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ола, характеризующийся тем, что включает кристаллизацию соединения Формулы (1) в алифатическом углеводороде в качестве растворителя.
(IV)
Способ получения кристаллической формы А (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ола, характеризующийся тем, что включает кристаллизацию соединения Формулы (1) в нитриле в качестве растворителя.
(V)
Способ получения кристаллической формы А (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ола, характеризующийся тем, что включает кристаллизацию соединения Формулы (1) в ароматическом углеводороде в качестве растворителя.
(VI)
Способ получения кристаллической формы А (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ола, характеризующийся тем, что включает кристаллизацию соединения Формулы (1) в кетоне в качестве растворителя.
(VII)
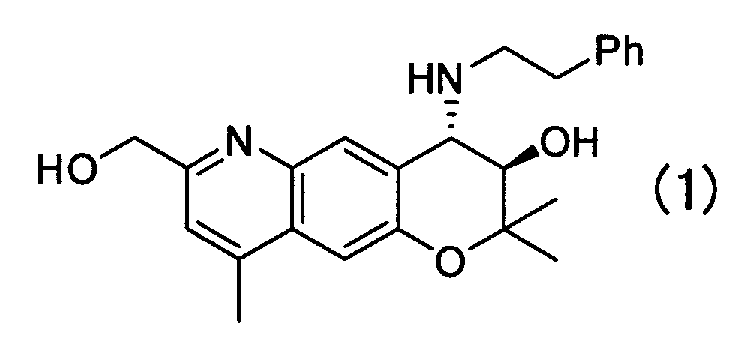
Кристаллическая форма В (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ола Формулы (1):
 ,
,
имеющая характеристические пики при углах дифракции (2θ) 6,4, 8,7, 12,8, 17,5, 19,1, 20,7, 22,0, 24,8, 26,6 на порошковой рентгеновской дифрактограмме кристаллов.
(VIII)
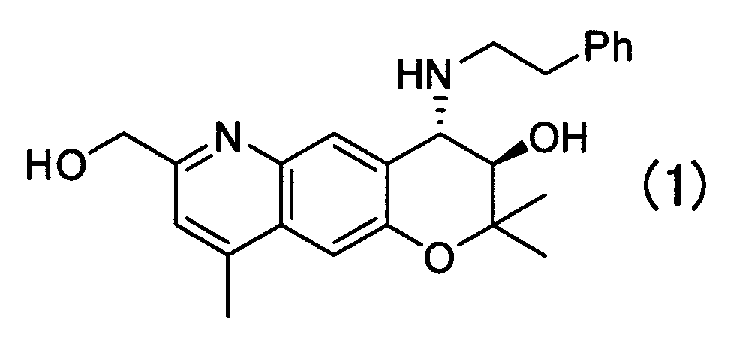
Кристаллическая форма Е (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ола Формулы (1):
 ,
,
имеющая характеристические пики при углах дифракции (2θ) 9,1, 12,8, 13,1, 14,6, 15,2, 16,4, 22,1, 23,6, 24,8 на порошковой рентгеновской дифрактограмме кристаллов.
(IX)
Кристаллическая форма F (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ола Формулы (1):
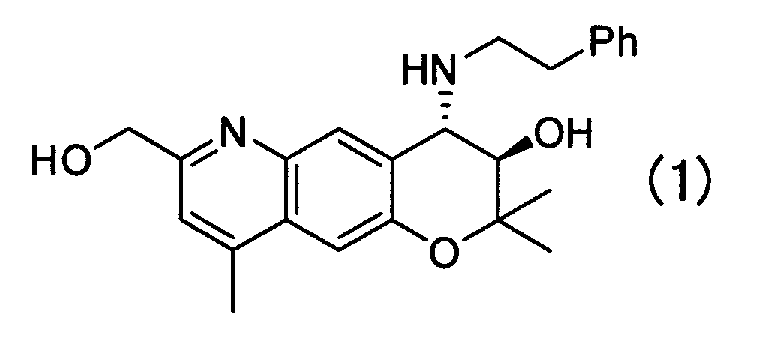 ,
,
имеющая характеристические пики при углах дифракции (2θ) 6,8, 11,7, 13,7, 16,8, 18,0, 19,3, 20,4, 20,8, 24,6, 25,6 на порошковой рентгеновской дифрактограмме кристаллов.
(X)
Кристаллическая форма G (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ола Формулы (1):
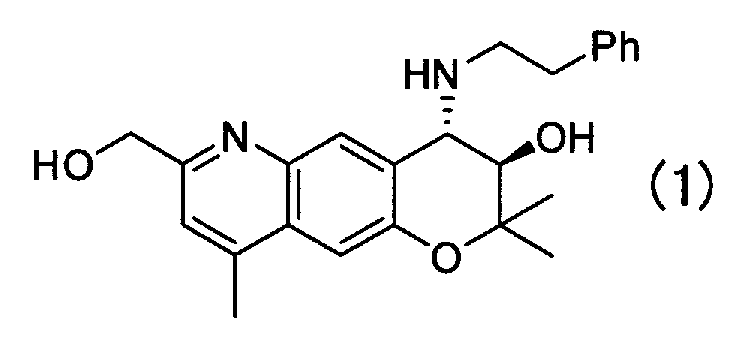 ,
,
имеющая характеристические пики при углах дифракции (2θ) 6,7, 11,6, 11,9, 13,6, 16,6, 17,7, 18,6, 19,1, 19,8, 20,1, 20,8 на порошковой рентгеновской дифрактограмме кристаллов.
(XI)
Кристаллическая форма H (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ола Формулы (I):
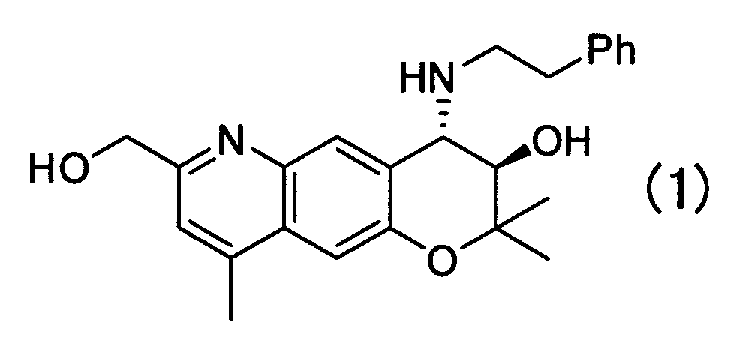 ,
,
имеющая характеристические пики при углах дифракции (2θ) 6,0, 16,4, 17,0, 19,2, 19,8, 20,3, 21,0, 22,8 на порошковой рентгеновской дифрактограмме кристаллов.
(XII)
Способ получения кристаллической формы В (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ола, характеризующийся тем, что включает кристаллизацию соединения Формулы (1) из органического растворителя, содержащего воду.
(XIII)
Способ получения кристаллической формы Е (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ола, характеризующийся тем, что включает нагревание и растворение соединения Формулы (1) в сложном эфире уксусной кислоты в качестве растворителя или в кетоне в качестве растворителя и одно быстрое добавление алифатического углеводорода в качестве растворителя и мгновенное охлаждение.
(XIV)
Способ получения кристаллической формы F (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ола, характеризующийся тем, что включает кристаллизацию соединения Формулы (1) из этанола.
(XV)
Способ получения кристаллической формы G (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ола, характеризующийся тем, что включает кристаллизацию соединения Формулы (1) из 2-пропанола.
(XVI)
Способ получения кристаллической формы H (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ола, характеризующийся тем, что включает нагревание и растворение соединения Формулы (1) в простом эфире в качестве растворителя и быстрое добавление алифатического углеводорода в качестве растворителя и мгновенное охлаждение.
НАИЛУЧШИЙ СПОСОБ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Первым описан способ получения кристаллической формы А.
Примеры сложных эфиров, применяемых в качестве растворителей, включают в себя: сложные эфиры муравьиной кислоты, имеющие C1-3-алкоксильную группу, такие как метилформиат, этилформиат и н-пропилформиат; и сложные эфиры уксусной кислоты, имеющие C1-4-алкоксильную группу, такие как метилацетат, этилацетат, н-пропилацетат, изопропилацетат, н-бутилацетат, изобутилацетат и трет-бутилацетат; среди них предпочтительны этилацетат, н-пропилацетат и изопропилацетат.
Примеры алифатических углеводородов, применяемых в качестве растворителя, включают в себя C5-7 -алифатические углеводороды, такие как пентан, н-гексан, циклогексан, н-гептан и метилциклогексан; среди них предпочтителен н-гептан.
Примеры нитрилов, применяемых в качестве растворителя, включают в себя C2-4-нитрилы, такие как ацетонитрил, пропионитрил и бутиронитрил; среди них предпочтителен ацетонитрил.
Примеры ароматических углеводородов, применяемых в качестве растворителя, включают в себя C6-8-ароматические углеводороды, такие как бензол, толуол и ксилол; среди них предпочтительны толуол и ксилол.
Примеры кетонов, применяемых в качестве растворителя, включают в себя C3-6-кетоны, такие как ацетон, метилэтилкетон, метилизопропилкетон и метилизобутилкетон; среди них предпочтительны ацетон и метилизобутилкетон.
Кроме того, можно применять только один из этих растворителей или смесь этих растворителей и можно добавлять другие растворители.
Количество применяемого растворителя составляет от 1- до 50-кратного количества по массе, предпочтительно от 2- до 20-кратного количества по массе, более предпочтительно от 5- до 10-кратного количества по массе, относительно единицы массы Соединения (1).
Если кристаллизацию проводят, охлаждая раствор Соединения (1), то температурой для этого охлаждения может быть определенная температура от 0°С до температуры, при которой растворитель кипит с обратным холодильником; однако предпочтительно кристаллизацию проводить, охлаждая раствор до 0-5°С.
Если кристаллизацию проводят, концентрируя раствор Соединения (1), то кристаллизацию можно проводить, оставляя любое количество растворителя или удаляя растворитель полностью.
Кроме того, кристаллизацию можно проводить, комбинируя обе операции охлаждения и концентрирования.
Для кристаллизации можно применять кристаллы-затравки. Кристаллы для затравки можно получать хорошо известными способами, например трением шпателем внутренней стенки колбы, содержащей кристаллизуемый раствор.
Следующим описан способ получения кристаллической формы В, которая представляет собой гидрат Соединения (1).
Применяемый органический растворитель никак не ограничивают, при условии что этот растворитель может смешиваться с водой; примеры применимых органических растворителей включают в себя спирт, применяемый в качестве растворителя, нитрил, применяемый в качестве растворителя, простой эфир, применяемый в качестве растворителя, кетон, применяемый в качестве растворителя, амид, применяемый в качестве растворителя, и сульфоксид, применяемый в качестве растворителя. Предпочтительные примеры таких растворителей включают в себя: метанол, этанол, 1-пропанол и 2-пропанол, применяемые в качестве спиртовых растворителей; ацетонитрил и пропионитрил, применяемые в качестве нитрильных растворителей; тетрагидрофуран, 1,2-диметоксиэтан и 1,4-диоксан, применяемые в качестве эфирных растворителей; ацетон и метилэтилкетон, применяемые в качестве кетоновых растворителей; N,N-диметилформамид и N,N-диметилацетамид, применяемые в качестве амидных растворителей; и диметилсульфоксид, применяемый в качестве сульфоксидного растворителя. Кроме того, предпочтительные примеры органических растворителей включают в себя метанол, этанол, 2-пропанол, ацетонитрил и ацетон.
Если в составе водосодержащего органического растворителя используют некоторое количество воды, то содержание такой воды можно выбирать на любом уровне, способном временно обеспечивать растворение Соединения (1). Учитывая баланс между эффективностью кристаллизации и эффективностью очистки, соотношение органического растворителя и воды, предпочтительно, находятся в диапазоне от 1:4 до 10:1, более предпочтительно от 1:2 до 3:1.
Кристаллизацию можно проводить либо способом, включающим в себя первоначальное приготовление водосодержащего органического растворителя и последующее нагревание и растворение Соединения (1) в подготовленном органическом растворителе для охлаждения полученного раствора, либо способом, включающим в себя растворение Соединения (1) в органическом растворителе и последующее добавление воды в полученный раствор. В обоих способах кристаллизацию можно проводить, применяя охлаждение, концентрирование или сочетание охлаждения и концентрирования.
Относительно количества применяемого органического растворителя, допустимо любое количество, достаточное для растворения Соединения (1). Однако предпочтительно количество, составляющее от 1- до 100-кратного количества по массе, более предпочтительно от 2- до 50-кратного количества по массе, еще более предпочтительно от 5- до 20-кратного количества по массе, относительно единицы массы Соединения (1).
Следующим описан способ получения кристаллической формы Е.
Примеры сложных эфиров уксусной кислоты, применяемых в качестве растворителя, включают в себя сложные эфиры уксусной кислоты, имеющие C1-4-алкоксильную группу, такие как метилацетат, этилацетат, н-пропилацетат, изопропилацетат, н-бутилацетат, изобутилацетат и трет-бутилацетат. Среди них предпочтительны этилацетат, н-пропилацетат и изопропилацетат; особо предпочтителен этилацетат.
Относительно количества сложного эфира уксусной кислоты, применяемого в качестве растворителя, допустимо любое количество, достаточное для растворения Соединения (1); однако предпочтительно количество, составляющее от 4- до 20-кратного количества по объему, особо предпочтительно от 5- до 10-кратного количества по объему относительно единицы массы Соединения (1).
Примеры кетонов, применяемых в качестве растворителя, включают в себя C3-6-кетоны, такие как ацетон, метилэтилкетон, метилизопропилкетон и метилизобутилкетон. Среди них предпочтительны ацетон и метилизобутилкетон и особо предпочтителен метилизобутилкетон.
Относительно количества кетона, применяемого в качестве растворителя, допустимо любое количество, достаточное для растворения Соединения (1). Однако предпочтительно количество, составляющее от 5- до 50-кратного количества по объему, наиболее предпочтительно от 10- до 30-кратного количества по объему, относительно единицы массы Соединения (1).
Примеры алифатических углеводородов, применяемых в качестве растворителей, включают в себя C5-7-алифатические углеводороды, такие как пентан, н-гексан, циклогексан, н-гептан и метилциклогексан. Среди них предпочтительны н-гексан и н-гептан и особо предпочтителен н-гептан.
Количество алифатического углеводорода, применяемого в качестве растворителя, составляет от 1- до 100-кратного количества по объему, более предпочтительно от 5- до 50 кратного количества по объему, наиболее предпочтительно от 10- до 30-кратного количества по объему, относительно единицы массы Соединения (1).
Кристаллизацию Соединения (1) можно проводить при определенной температуре в диапазоне от 0°С до температуры, при которой растворитель кипит с обратным холодильником. Однако предпочтителен способ, включающий в себя растворение Соединения (1) в сложном эфире уксусной кислоты, применяемом в качестве растворителя, или в кетоне, применяемом в качестве растворителя, при 60-70°С и быстрое добавление алифатического углеводорода, применяемого в качестве растворителя, к полученному раствору и мгновенное охлаждение кристаллизуемого раствора до комнатной температуры. В данном случае «быстро» означает «в пределах 30 секунд».
Далее описан способ получения кристаллической формы F, которая представляет собой сольват (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ола с этанолом.
В качестве растворителя применяют этанол.
Количество применяемого этанола составляет от 1- до 50-кратного количества по массе, предпочтительно от 2- до 20-кратного количества по массе, более предпочтительно от 3- до 10-кратного количества по массе, относительно единицы массы Соединения (1).
Кристаллизацию Соединения (1) можно проводить при определенной температуре в диапазоне от 0°С до температуры, при которой растворитель кипит с обратным холодильником. Однако предпочтительно кристаллизацию проводить, применяя способ, включающий в себя нагревание и растворение Соединения (1) в этаноле, взятом в как можно меньшем количестве, и охлаждение полученного раствора; применяя способ, включающий в себя растворение Соединения (1) в этаноле и концентрирование полученного раствора; или применяя способ, в котором сочетают два вышеуказанных способа.
Следующим описан способ получения кристаллической формы G, которая представляет сольват (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ола с 2-пропанолом.
В качестве растворителя применяют 2-пропанол.
Количество применяемого 2-пропанола составляет, предпочтительно, от 2- до 20-кратного количества по массе, более предпочтительно от 3- до 10-кратного количества по массе, относительно единицы массы Соединения (1).
Кристаллизацию Соединения (1) можно проводить при определенной температуре в диапазоне от 0°С до температуры, при которой растворитель кипит с обратным холодильником. Однако предпочтительно кристаллизацию проводить, применяя способ, включающий в себя нагревание и растворение Соединения (1) в 2-пропаноле, взятом в как можно меньшем количестве, и охлаждение полученного раствора; применяя способ, включающий в себя растворение Соединения (1) в 2-пропаноле и концентрирование полученного раствора; или применяя способ, в котором сочетают два вышеуказанных способа.
Следующим описан способ получения кристаллической формы Н, которая представляет собой сольват (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ола с циклогексаном.
В качестве растворителя применяют растворитель на основе простого эфира, предпочтительно диоксан.
Количество применяемого диоксана составляет от 1- до 5-кратного количества по массе, предпочтительно от 2- до 20-кратного количества по массе, более предпочтительно от 3- до 10-кратного количества по массе, относительно единицы массы Соединения (1).
В качестве растворителя применяют циклогексан.
Количество применяемого циклогексана составляет от 1- до 50-кратного количества по массе, предпочтительно от 2- до 20-кратного количества по массе, относительно единицы массы Соединения (1).
Кристаллизацию Соединения (1), предпочтительно, проводят, применяя способ, включающий в себя растворение Соединения (1) в растворителе на основе простого эфира, взятом в как можно меньшем количестве, при 60-70°С, быстрое добавление циклогексана к полученному раствору и мгновенное охлаждение кристаллизуемого раствора до комнатной температуры. В данном случае «быстро» означает «в пределах 30 секунд».
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
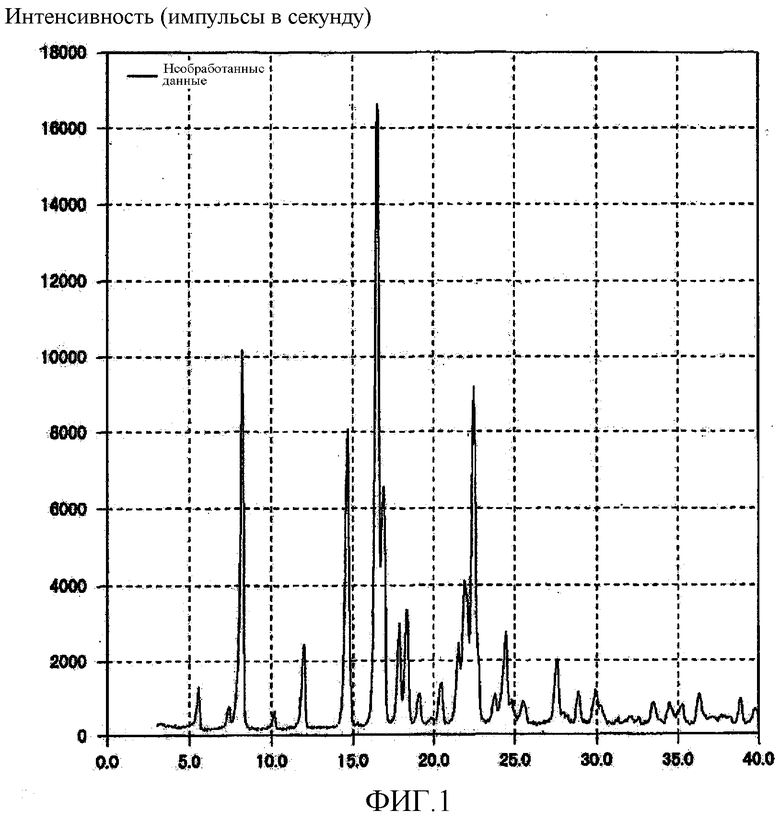
[Фиг. 1] Фиг. 1 представляет собой порошковую рентгеновскую дифрактограмму кристаллической формы А (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ола, полученной согласно настоящему изобретению.
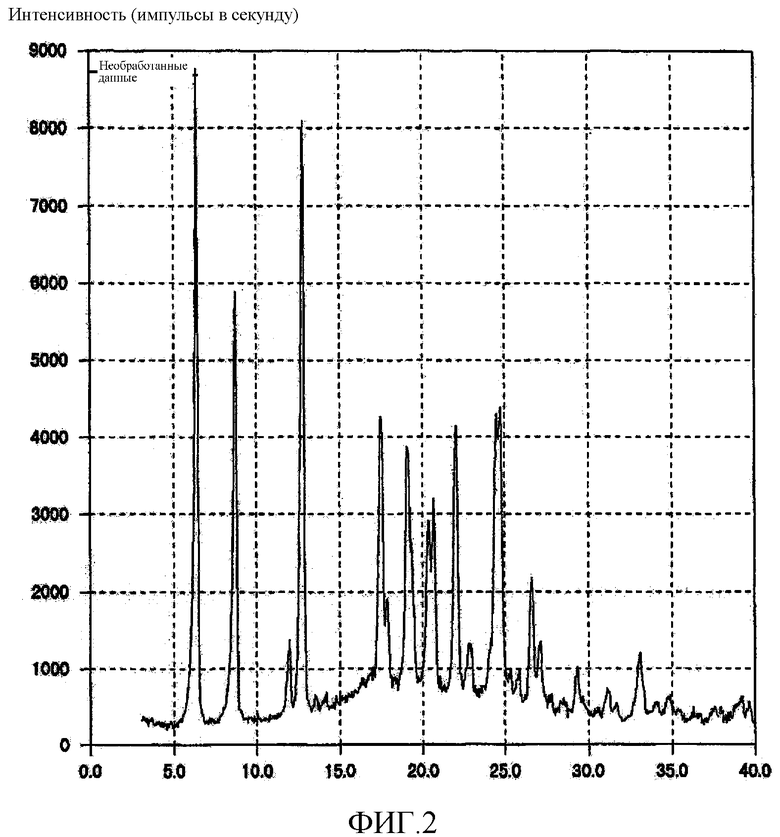
[Фиг. 2] Фиг. 2 представляет собой фигуру, показывающую порошковую рентгеновскую дифрактограмму кристаллической формы B (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ола, полученной согласно настоящему изобретению.
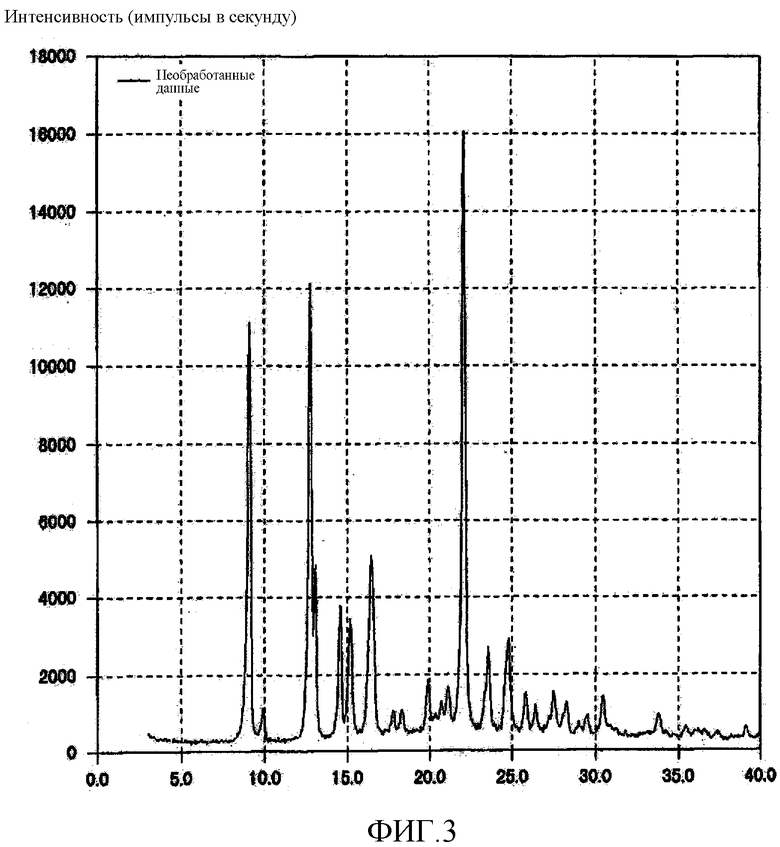
[Фиг. 3] Фиг. 3 представляет собой фигуру, показывающую порошковую рентгеновскую дифрактограмму кристаллической формы E (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ола, полученной согласно настоящему изобретению.
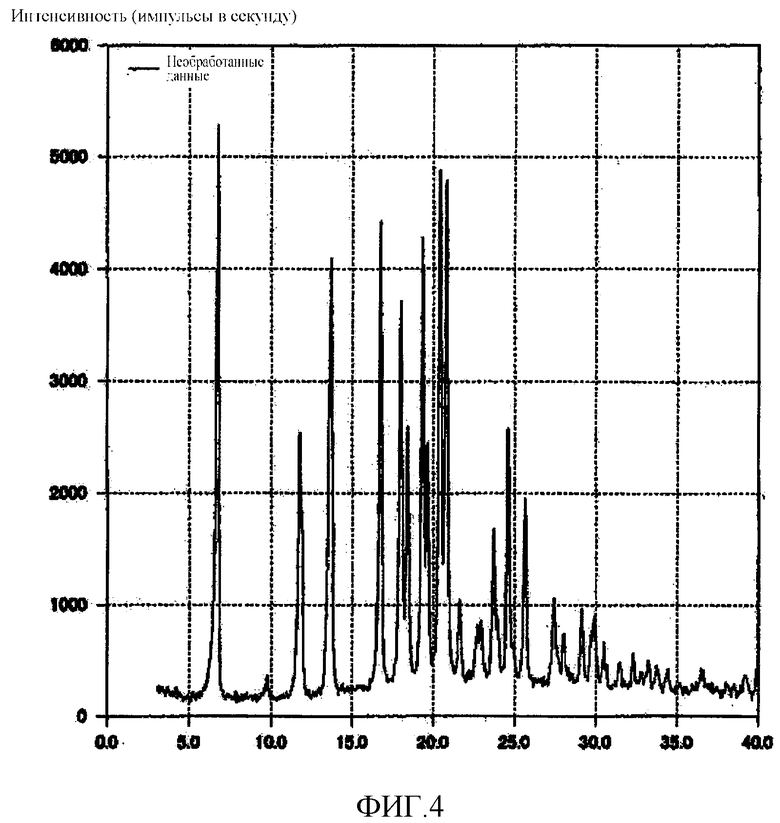
[Фиг. 4] Фиг. 4 представляет собой фигуру, показывающую порошковую рентгеновскую дифрактограмму кристаллической формы F (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ола, полученной согласно настоящему изобретению.
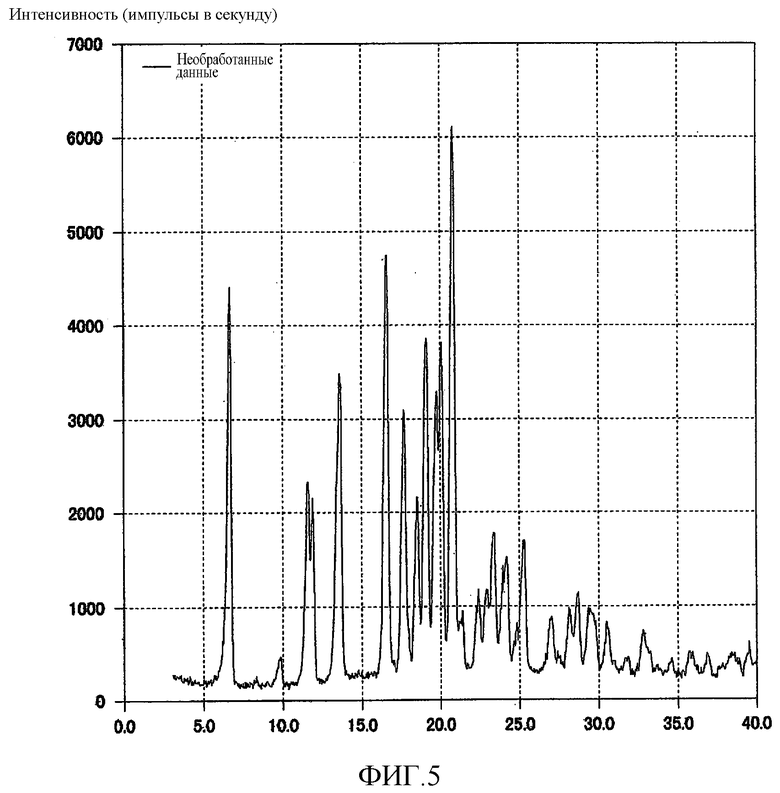
[Фиг. 5] Фиг. 5 представляет собой фигуру, показывающую порошковую рентгеновскую дифрактограмму кристаллической формы G (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ола, полученной согласно настоящему изобретению.
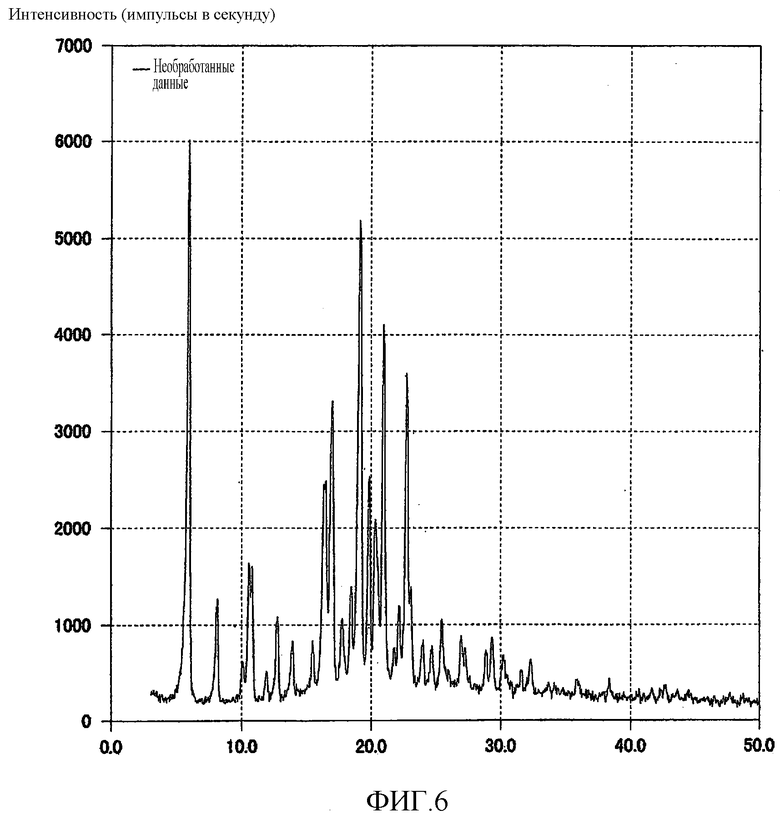
[Фиг. 6] Фиг. 6 представляет собой фигуру, показывающую порошковую рентгеновскую дифрактограмму кристаллической формы H (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ола, полученной согласно настоящему изобретению.
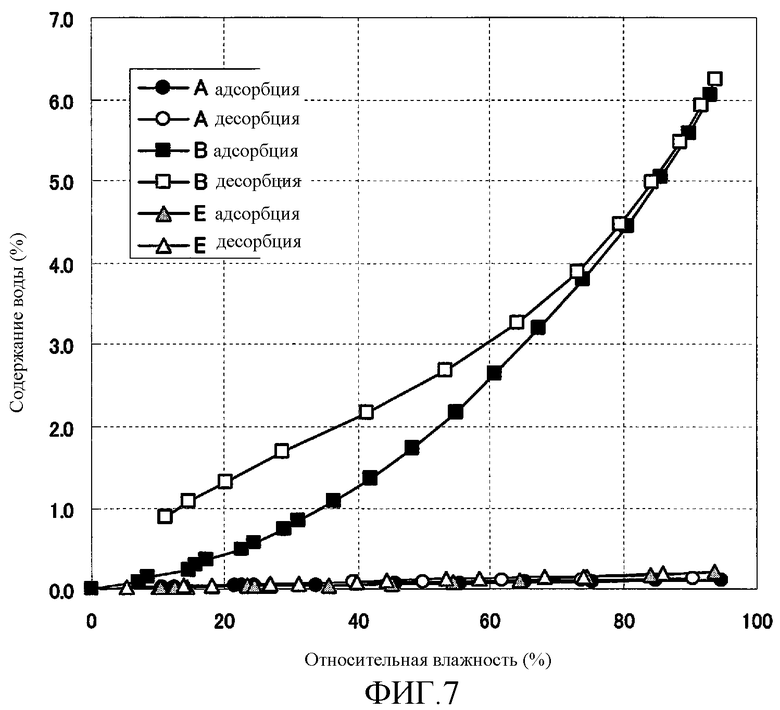
[Фиг. 7] Фиг. 7 представляет собой фигуру, показывающую изотермы адсорбции воды на кристаллах форм А, В и Е (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ола, полученных согласно настоящему изобретению.
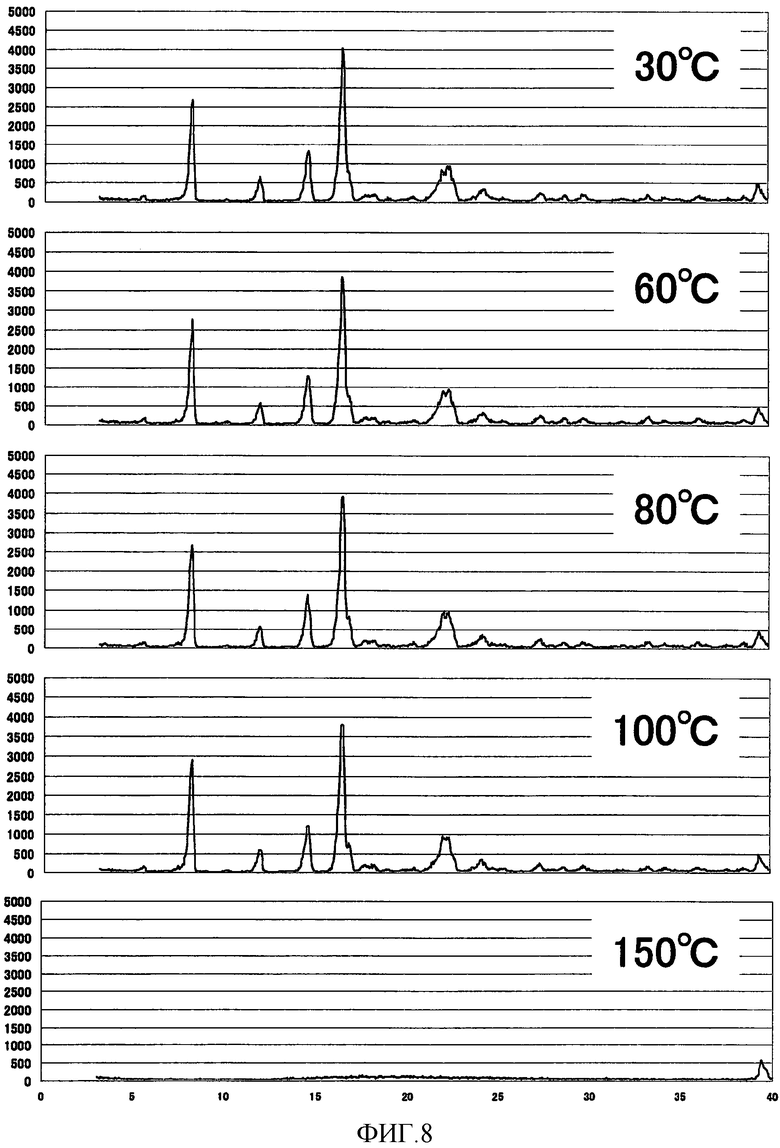
[Фиг. 8] Фиг. 8 представляет собой фигуру, показывающую высокотемпературную рентгеновскую дифрактограмму кристаллической формы A (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ола, полученной согласно настоящему изобретению.
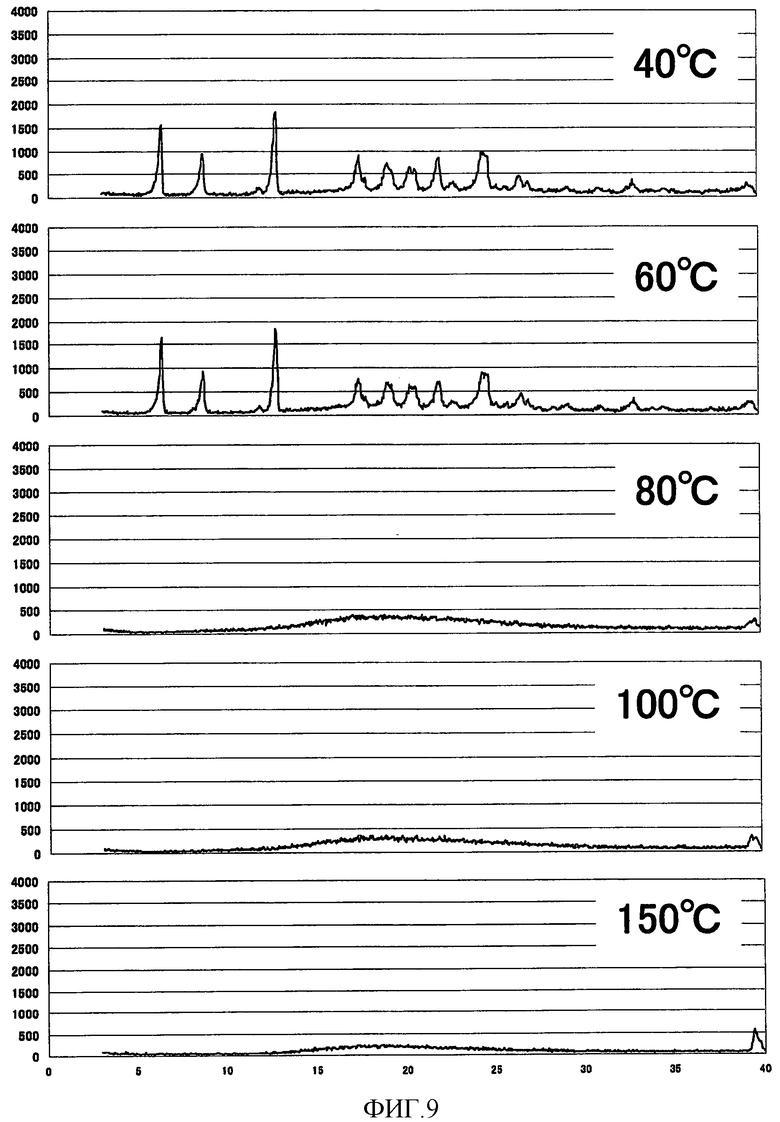
[Фиг. 9] Фиг. 9 представляет собой фигуру, показывающую высокотемпературную рентгеновскую дифрактограмму кристаллической формы B (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ола, полученной согласно настоящему изобретению.
ПРИМЕРЫ
Ниже в настоящем документе настоящее изобретение будет описано более подробно со ссылками на Примеры, которые не следует истолковывать как ограничивающие объем настоящего изобретения. В Примерах измерения точки плавления проводили капиллярным способом, применяя прибор B-545 (изготовленный Shibata Scientific Technology Ltd.) (скорость повышения температуры: 1°C/мин); дифференциальную сканирующую калориметрию (DSC) проводили на приборе DSC 8230 (изготовленном Rigaku Corporation) (скорость повышения температуры: 5°C/мин); а измерения порошковой рентгеновской дифракции проводили на приборе MXLabo (изготовленном Mac Science Co., Ltd.; источник излучения: Cu·Kα, длина волны: 1,54056 (10-10 м)).
Неочищенный продукт (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ола (Соединение (1)) можно получать способом (Способом синтеза А), описанным в публикации международной заявки № WO 2005/090357.
Кроме того, Соединение (1) можно также синтезировать нижеследующим способом (Способом синтеза В).
Ссылочный пример синтеза 1
Синтез (Способ синтеза В) (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ола
10,05 г (32,1 ммоль) (3R,4S)-(3,4-эпокси-2,2,9-триметил-3,4-дигидро-2H-пирано[2,3-g]хинолин-7-ил)метилацетата (синтезированного согласно способу, описанному в публикации международной заявки № WO 2005/090357) растворяли в 99,79 г метанола и в раствор, полученный в результате этого, добавляли по каплям 40 мл (40 ммоль) 1 М водного раствора гидроксида натрия, после чего полученную смесь перемешивали при комнатной температуре в течение 30 минут. К реакционной смеси, полученной таким образом, добавляли 60,06 г хлороформа и 60,30 г H2O и разделяли слои, после чего реакционную смесь два раза экстрагировали хлороформом. Органический слой концентрировали, получая 9,53 г светло-коричневого твердого вещества. К этому твердому веществу добавляли 49,85 г толуола и полученную суспензию перемешивали при 60°C в течение 10 минут и охлаждали до 5°C или ниже, отфильтровывая кристаллическое вещество. Кристаллы промывали 10,0 г толуола и сушили при 50°C при пониженном давлении, получая 7,98 г (выход: 91,7%) (3R,4S)-3,4-эпокси-7-гидроксиметил-2,2,9-триметил-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ола в виде белого твердого вещества.
Внешний вид: белое твердое вещество
11H-ЯМР (CDCl3, TMS)
δ м.д.: 1,31 (3H, с, Me), 1,65 (3H, с, Me), 2,59 (3H, с, Me), 3,60 (1Н, д, J=4,3 Гц, C3), 4,15 (1Н, д, J=4,3 Гц, C4), 4,42 (1Н, т, J=4,0 Гц, CH2OH), 4,83 (2H, д, J=4,0 Гц, CH2OH), 7,07 (1Н, с, Ar), 7,31 (1Н, с, Ar), 8,08 (1Н, с, Ar).
Точка плавления: 143-144°C
К жидкой смеси 7,98 г (29,4 ммоль) полученного (3R,4S)-3,4-эпокси-7-гидроксиметил-2,2,9-триметил-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ола и 16,07 г 1-пропанола добавляли 4,30 г (35,5 ммоль, 1,2 эквивалента) 2-фенилэтиламина и смесь, полученную таким образом, нагревали и кипятили с обратным холодильником в течение 5 часов. Отогнав растворитель, получали 14,52 г (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ола в виде коричневого маслянистого вещества.
Пример 1
Производство кристаллической формы A (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ола
К 14,52 г неочищенного продукта (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ола, полученного в ссылочном примере синтеза 1, добавляли 12,02 г этилацетата и смесь, полученную таким образом, для растворения нагревали до 50°C, после чего в полученный раствор по каплям добавляли 59,94 г н-гептана при 49-58°C для кристаллизации. Смесь, полученную таким образом, непрерывно охлаждали до 3°C и затем отфильтровывали кристаллы, после чего кристаллы промывали смесью 1,5 г этилацетата и 7,5 г н-гептана, а затем 8,0 г н-гептана, получая 10,02 г кристаллической формы A Соединения (1) в виде белого кристаллического вещества. Точка плавления полученного кристаллического вещества находилась при 124-125°C. Полученное кристаллическое вещество подвергали анализу DSC, результат которого подтвердил наличие эндотермического пика при 130°C. На порошковой рентгеновской дифрактограмме кристаллического вещества наблюдали характеристические пики при углах дифракции (2θ) 5,6, 8,2, 12,0, 14,7, 16,6, 16,9, 17,9, 18,4, 22,5, 24,5, 27,6. Результат показан на Фиг. 1.
Пример 2
Производство кристаллической формы B (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ола
62,23 г кристаллической формы A (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ола растворяли в 247,73 г этанола, нагревая смесь до 62°C. К раствору, полученному в результате этого, по каплям добавляли 275,19 г воды при температуре от 55 до 67°С в течение 20 минут и полученную смесь охлаждали до 5°C в течение 3 часов, после чего смесь перемешивали при той же температуре в течение 30 минут и отфильтровывали кристаллическое вещество. Кристаллы сушили при 50°C при пониженном давлении до тех пор, пока масса кристаллов не переставала уменьшаться при дальнейшем высушивании, в результате чего было произведено 59,22 г белого твердого вещества. Полученное кристаллическое вещество подвергали анализу DSC, результат которого подтвердил наличие эндотермического пика при 91°C. На порошковой рентгеновской дифрактограмме кристаллического вещества наблюдали характеристические пики при углах дифракции (2θ) 6,4, 8,7, 12,8, 17,5, 19,1, 20,7, 22,0, 24,8, 26,6. Результат показан на Фиг. 2. Кроме того, в кристаллическом веществе измеряли содержание воды с применением влагомера Карла Фишера (объемный анализ), в результате чего было обнаружено, что содержание воды составляло 3,2% по массе.
Пример 3
Производство кристаллической формы E (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ола
14,99 г кристаллической формы A (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ола растворяли в 75 мл этилацетата, нагревая смесь до 63°C. После этого нагревание прекращали и при комнатной температуре к полученному раствору одной порцией добавляли 300 мл н-гептана, после чего полученную смесь охлаждали до 26°C. В таком состоянии полученную смесь перемешивали в течение 1 часа и отфильтровывали кристаллы. Эти кристаллы сушили при 40°С при пониженном давлении, получая 12,67 г зернистого твердого вещества желтого цвета. Полученное кристаллическое вещество подвергали анализу DSC, результат которого подтвердил наличие эндотермического пика при 119°C. На порошковой рентгеновской дифрактограмме кристаллического вещества наблюдали характеристические пики при углах дифракции (2θ) 9,1, 12,8, 13,1, 14,6, 15,2, 16,4, 22,1, 23,6, 24,8. Результат показан на Фиг. 3.
Пример 4
Производство кристаллической формы F (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ола
10,01 г кристаллической формы A (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ола растворяли в 60,20 г этанола, нагревая смесь до 76°C. Раствор, полученный в результате этого, охлаждали до 5°C в течение 1 часа и 45 минут и перемешивали при 5°C в течение 1 часа, отфильтровывая кристаллы. Эти кристаллы сушили при 50°C при пониженном давлении до тех пор, пока масса кристаллов не переставала уменьшаться при дальнейшем высушивании, в результате чего было произведено 10,96 г белого твердого вещества. Полученное кристаллическое вещество подвергали анализу DSC, результат которого подтвердил наличие эндотермического пика при 99°C. Кроме того, на порошковой рентгеновской дифрактограмме кристаллического вещества наблюдали характеристические пики при углах дифракции (2θ) 6,8, 11,7, 13,7, 16,8, 18,0, 19,3, 20,4, 20,8, 24,6, 25,6. Результат показан на Фиг. 4.
В дополнение к этому раствор кристаллов в CDCl3 с тетраметилсиланом в качестве внутреннего стандарта исследовали методом 1H-ЯМР-спектроскопии. Отношение интегральной величины двух протонов этанола, детектированной при δ 3,7 м.д., к соответствующей величине одного протона в положении 4 Соединения (1) составляло 2,4 к 1, что свидетельствовало о существовании кристаллического вещества в виде сольвата с 1,2 экв. этанола.
Пример 5
Производство кристаллической формы G (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ола
1,00 г кристаллической формы A (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ола суспендировали в 5 мл 2-пропанола и перемешивали при 21°C в течение 19,7 часов. Кристаллы отфильтровывали и сушили при 40°C в течение 6 часов при пониженном давлении до тех пор, пока масса кристаллов не переставала уменьшаться при дальнейшем высушивании, в результате чего было произведено 0,97 г белого кристаллического вещества. Полученное кристаллическое вещество подвергали анализу DSC, результат которого подтвердил наличие эндотермического пика при 75,9°C. На порошковой рентгеновской дифрактограмме кристаллического вещества наблюдали характеристические пики при углах дифракции (2θ) 6,7, 11,6, 11,9, 13,6, 16,6, 17,7, 18,6, 19,1, 19,8, 20,1, 20,8. Результат показан на Фиг. 5.
В дополнение к этому раствор кристаллов в CDCl3 с тетраметилсиланом в качестве внутреннего стандарта исследовали методом 1H-ЯМР-спектроскопии. Отношение интегральной величины одного протона 2-пропанола, детектированной при δ 4,0 м.д., к соответствующей величине одного протона в положении 4 Соединения (1) составляло 0,83 к 1, что свидетельствовало о существовании кристаллического вещества в виде сольвата с 0,83 экв. 2-пропанола.
Пример 6
Производство кристаллической формы H (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ола
19,95 г кристаллической формы A (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ола растворяли в 140 мл диоксана. Нерастворимый материал отфильтровывали и промывали 60 мл диоксана. Растворитель отгоняли при пониженном давлении, получая 50 г остаточного раствора. Остаточный раствор нагревали до 60°С, затем одной порцией добавляли 600 мл циклогексана и раствор подвергали мгновенному охлаждению от 60°C до 28°C. Раствор перемешивали при 25-28°C в течение 5,1 часа и отфильтровывали кристаллы. Кристаллы сушили при 50°C в течение 2 часов при пониженном давлении до тех пор, пока масса кристаллов не переставала уменьшаться при дальнейшем высушивании, в результате чего было произведено 21,96 г белого кристаллического вещества. Полученное кристаллическое вещество подвергали анализу DSC, результат которого подтвердил наличие эндотермического пика при 80,1°C и 86,1°C.
Кроме того, на порошковой рентгеновской дифрактограмме кристаллического вещества наблюдали характеристические пики при углах дифракции (2θ) 6,0, 16,4, 17,0, 19,2, 19,8, 20,3, 21,0, 22,8. Результат показан на Фиг. 6.
В дополнение к этому раствор кристаллов в CDCl3 с тетраметилсиланом в качестве внутреннего стандарта исследовали методом 1H-ЯМР-спектроскопии. Отношение интегральной величины 12 протонов циклогексана, детектированной при δ 51,4 м.д., к соответствующей величине трех протонов метила в Соединении (1) составляло 2,69 к 1, что свидетельствовало о существовании кристаллического вещества в виде сольвата с 0,67 экв. циклогексана.
Примеры испытаний
Ниже в настоящем документе описаны примеры испытаний, проведенных с кристаллическими формами A, B, E и F, полученными способами, указанными выше в Примерах 1-4.
Пример испытаний 1
Испытывали термостойкость, влагостойкость и светостойкость кристаллических форм A, B и F Соединения (1).
Условия проведения каждого испытания были следующими.
Испытание на термостойкость: 60°C, влажность не регулировали, 2 недели, в воздухонепроницаемом контейнере.
Испытание на влагостойкость: 25°C, относительная влажность 90%, 2 недели, в открытом контейнере.
Испытание на светостойкость: 200 Вт/м2·час, 25°C, относительная влажность 60%, 57 часов, в открытом контейнере.
Кроме того, стабильность кристаллов оценивали по степени увеличения общего количества загрязнений посредством ВЭЖХ-анализа. Результат показан в Таблице 1.
В условиях проведения испытаний увеличение содержания загрязнений в каждой кристаллической форме составляло не более 1,5%. Подтверждено, что среди испытанных кристаллических форм кристаллы формы А являются наиболее стабильными, поскольку в условиях испытаний эта форма не показала никакого увеличения содержания загрязнений.
Пример испытаний 2
Кристаллические формы А, В и Е Соединения (1) подвергнуты испытанию в условиях ускоренного старения. Условия испытания были следующими.
Испытание в условиях ускоренного старения: 40°C, относительная влажность 75%, 6 месяцев, в воздухонепроницаемом контейнере.
[Условия измерений]
Кроме того, стабильность кристаллов оценивали по степени увеличения общего количества загрязнений посредством ВЭЖХ-анализа. Результат показан в Таблице 2.
При проведении испытаний в условиях ускоренного старения увеличение содержания загрязнений в каждой кристаллической форме составляло не более 1,5%. Подтверждено, что наиболее стабильными являются кристаллические формы А и Е, поскольку в них не наблюдали никакого увеличения содержания загрязнений.
Пример испытаний 3
Кристаллические формы A, B и E Соединения (1) оценивали посредством анализа адсорбции воды.
[Условия измерений] количество образца: 0,2 г; предварительная обработка: 60°C, 20 часов; температура 25°C, применяли волюмометрический аппарат исследования адсорбции (торговое наименование: BELSORP 18, изготовлен BEL Japan, Inc.).
В кристаллах формы В наблюдали обратимую адсорбцию воды. В кристаллах форм А и Е не наблюдали никакой адсорбции воды. Наблюдали, что адсорбция воды на кристаллах формы А была меньшей, чем адсорбция воды на кристаллах формы Е. Результат показан на Фиг. 7.
Пример испытаний 4
Определяли растворимость и удельную поверхность кристаллов формы А, формы В, формы Е и формы F Соединения (1).
Растворимость
[Условия измерений] JP2, pH 6,8 (3,40 г однозамещенного фосфата калия и 3,55 г безводного двузамещенного фосфата натрия растворяли в воде, получая 1 литр раствора. Один объем раствора смешивали с одним объемом воды, получая раствор согласно JP2, рН 6,8), 60 минут.
Согласно пятнадцатому изданию Фармакопеи Японии испытание на растворимость (Протокол испытания на растворимость; количество образца: 10 мг, испытательный раствор: согласно JP2, pH 6,8, 500 мл, время отбора образцов: 5, 15, 30, 60 мин, скорость вращения лопастной мешалки: 100 об/мин, температура бани: 37°C).
Удельная поверхность
[Условия измерений] температура адсорбции: 77K, время установления адсорбционного равновесия: 300 секунд.
[Единица измерения] квадратные метры на грамм.
Измерения проводили методом БЭТ, используя адсорбцию азота в жидком азоте после предварительной обработки в условиях рефлюкса жидкого азота. Результат показан в Таблице 3.
Каждая кристаллическая форма проявляла способность растворяться в растворе, приготовленном согласно JP2. Конкретно, высокой растворимостью обладала кристаллическая форма А. Кроме того, кристаллическая форма А демонстрировала также и высокое значение удельной поверхности.
Оказалось неожиданным, что кристаллическая форма А обладала превосходной стабильностью и высокой растворимостью. Кроме того, можно указать, что кристаллическая форма А обладала превосходными характеристиками в качестве лекарственного средства.
Пример испытаний 5
С кристаллическими формами А и В Соединения (I) были проведены измерения методом высокотемпературной XRD.
Как показано на Фиг. 8 и Фиг. 9, ниже 100°С никаких изменений на порошковой рентгеновской дифрактограмме кристаллической формы А не наблюдали, так же как и на порошковой рентгеновской дифрактограмме кристаллической формы В при температуре ниже 60°С. В частности, кристаллическая форма А демонстрировала удовлетворительную стабильность в качестве лекарственного средства.
ПРИМЕНЯЕМОСТЬ В ПРОИЗВОДСТВЕННЫХ УСЛОВИЯХ
Согласно настоящему изобретению можно предоставить способ, посредством которого можно производить химически стабильное соединение, применимое в качестве лекарственного средства, обладающее таким же качеством и такой же кристаллической формой, как и (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ол, и его новые кристаллические формы.
| название | год | авторы | номер документа |
|---|---|---|---|
| ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ БЕНЗОПИРАНА В КАЧЕСТВЕ ПРОТИВОАРИТМИЧЕСКИХ АГЕНТОВ | 2005 |
|
RU2380370C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО ДИФЕНИЛМЕТАНА | 2017 |
|
RU2797392C2 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА БЕНЗИЛ-БЕНЗОЛЬНОГО ИНГИБИТОРА SGLT | 2011 |
|
RU2569491C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО ДИФЕНИЛМЕТАНА | 2023 |
|
RU2814846C1 |
| ПОЛИМОРФНАЯ ФОРМА 4-{ [4-({ [4-(2,2,2-ТРИФТОРЭТОКСИ)-1,2-БЕНЗИЗОКСАЗОЛ-3-ИЛ]ОКСИ} МЕТИЛ)ПИПЕРИДИН-1-ИЛ]МЕТИЛ} -ТЕТРАГИДРО-2Н-ПИРАН-4-КАРБОНОВОЙ КИСЛОТЫ | 2012 |
|
RU2616978C2 |
| КРИСТАЛЛЫ ПРОИЗВОДНЫХ ДИСПИРОПИРРОЛИДИНА | 2013 |
|
RU2647840C2 |
| КРИСТАЛЛИЧЕСКАЯ 7-{ (3S,4S)-3-[(ЦИКЛОПРОПИЛАМИНО)МЕТИЛ]-4-ФТОРПИРРОЛИДИН-1-ИЛ} -6-ФТОР-1-(2-ФТОРЭТИЛ)-8-МЕТОКСИ-4-ОКСО-1,4-ДИГИДРОХИНОЛИН-3-КАРБОНОВАЯ КИСЛОТА | 2012 |
|
RU2615509C2 |
| НОВЫЕ ТВЕРДЫЕ ФОРМЫ (2S,3S,4S,5R,6S)-3,4,5-ТРИГИДРОКСИ-6-(((4AR,10AR)-7-ГИДРОКСИ-1-ПРОПИЛ-1,2,3,4,4A,5,10,10A-ОКТАГИДРОБЕНЗО[G]ХИНОЛИН-6-ИЛ)ОКСИ)ТЕТРАГИДРО-2H-ПИРАН-2-КАРБОНОВОЙ КИСЛОТЫ | 2020 |
|
RU2820501C2 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА И СПОСОБ ЕЕ ОЧИСТКИ | 2011 |
|
RU2604734C2 |
| СОДЕРЖАЩАЯ ПРОИЗВОДНОЕ С ТРЕМЯ КОНДЕНСИРОВАННЫМИ КОЛЬЦАМИ СОЛЬ ИЛИ КРИСТАЛЛИЧЕСКАЯ ФОРМА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2020 |
|
RU2835105C1 |
Данное изобретение относится к кристаллическим формам A, B, E и F (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ола формулы I:
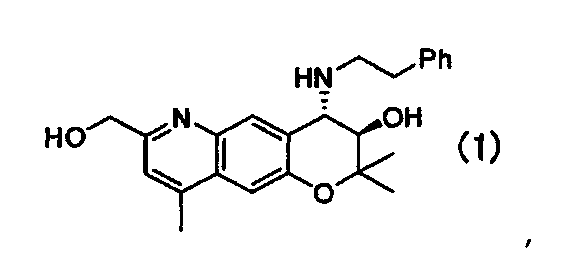
представляющего собой превосходное лекарственное средство антиаритмического действия, и способам их получения, включающим в себя кристаллизацию (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ола из сложного эфира уксусной кислоты, алифатического углеводорода, или этанола, применяемых в качестве растворителя. Эти формы обладают хорошей стабильностью и высокой растворимостью. 8 н.п. ф-лы, 9 ил., 3 табл., 6 пр.
1. Кристаллическая форма A (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2Н-пирано[2,3-g]хинолин-3-ола Формулы (I):
имеющая характеристические пики при углах дифракции (2θ) 5,6, 8,2, 12,0, 14,7, 16,6, 16,9, 17,9, 18,4, 22,5, 24,5, 27,6 на порошковой рентгеновской дифрактограмме кристаллов.
2. Способ получения кристаллической формы A (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ола, характеризующийся тем, что способ включает кристаллизацию соединения Формулы (1) в сложном эфире и в алифатическом углеводороде в качестве растворителя.
3. Кристаллическая форма В (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2Н-пирано[2,3-g]хинолин-3-ола Формулы (1):
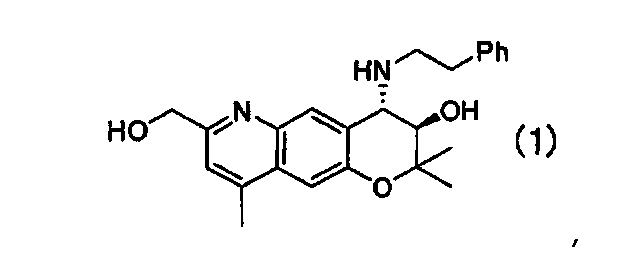
имеющая характеристические пики при углах дифракции (2θ) 6,4, 8,7, 12,8, 17,5, 19,1, 20,7, 22,0, 24,8, 26,6 на порошковой рентгеновской дифрактограмме кристаллов.
4. Кристаллическая форма Е (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ола
Формулы (1):
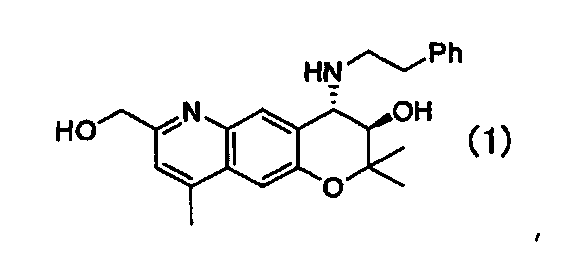
имеющая характеристические пики при углах дифракции (2θ) 9,1, 12,8, 13,1, 14,6, 15,2, 16,4, 22,1, 23,6, 24,8 на порошковой рентгеновской дифрактограмме кристаллов.
5. Кристаллическая форма F (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ола Формулы (1):
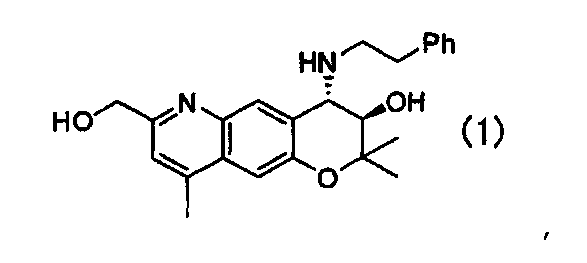
имеющая характеристические пики при углах дифракции (2θ) 6,8, 11,7, 13,7, 16,8, 18,0, 19,3, 20,4, 20,8, 24,6, 25,6 на порошковой рентгеновской дифрактограмме кристаллов.
6. Способ получения кристаллической формы В (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ола, характеризующийся тем, что способ включает кристаллизацию соединения Формулы (1) из содержащего воду этанола.
7. Способ получения кристаллической формы Е (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ола, характеризующийся тем, что способ включает нагревание и растворение соединения Формулы (1) в сложном эфире уксусной кислоты в качестве растворителя или в кетоне в качестве растворителя, быстрое добавление алифатического углеводорода в качестве растворителя и мгновенное охлаждение.
8. Способ получения кристаллической формы F (3R,4S)-7-гидроксиметил-2,2,9-триметил-4-(фенэтиламино)-3,4-дигидро-2H-пирано[2,3-g]хинолин-3-ола, характеризующийся тем, что способ включает кристаллизацию соединения Формулы (1) из этанола.
| WO 2005090357 A1 29.09.2005 | |||
| ПРОИЗВОДНЫЕ ПИРАНО-ХИНОЛИНОВ, СПОСОБ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2125573C1 |
| ПРОИЗВОДНЫЕ 8-ПИРИДОНО [5,6g] ХИНОЛИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБЫ С ИХ ИСПОЛЬЗОВАНИЕМ | 1997 |
|
RU2214412C2 |
| Способ получения производных 4-оксо4н-пирано-(3,2-с) хинолин-2-карбоновых кислот или их солей | 1975 |
|
SU545262A3 |

