Изобретение относится к способу получения хиральных гетероциклических лигандов на основе 1,2-диаминоциклогексана, содержащих гетероциклические фрагменты: тиенил-2-, тиенил-3-, фурил-2-, 5-метилфурил-2-, (2,2'-битиофен)-5-ил-, 5-(4'-метилциклогекс-1'-ен-1'-ил)тиофен-2-, которые могут входить в структуру комплексов для проведения энантиоселективных реакций и асимметрического катализа, а также обладать люминесцентными свойствами [1, 2].
Данным изобретением решена задача получения лигандов на основе оптически чистого 1,2-диаминоциклогексана, содержащих гетероциклические фрагменты.
Известные методы синтеза хиральных структур на основе 1,2-диаминоциклогексана [2-4], имеют ряд существенных недостатков.
1. Введение в реакцию оптически чистого (1R,2R)-диминоциклогексана или (1S,2S)-диминоциклогексана.
2. Использование дополнительных реагентов для адсорбции выделяющейся воды.
3. Длительное время протекания реакции (до 48 часов).
4. Использование органических растворителей.
5. Сложность технологического исполнения в ряде случаев.
Одним из методов конденсации цис-/транс-1,2-диаминоциклогексана с альдегидами является использование в качестве растворителя хлористого метилена и сульфата магния для абсорбции выделяющейся в реакции воды [1, 2]. В случае бензальдегида реакция проходит при температуре 40°С в течение 15 минут [1]. Показано, что уменьшение растворимости бифенил-4-карбальдегида в органическом растворителе приводит к резкому увеличению времени реакции конденсации с (1R,2R)-диаминоциклогексаном с 15 мин до 32 ч [2]. Конденсацию салицилового альдегида с (1R,2R)-диаминоциклогексаном проводят при комнатной температуре в среде этанола [3]. В патентах [4, 5] описан метод синтеза 1,2-циклогександииминов на основе транс-1,2-диаминоциклогексана из бензальдегида, тиофен-2-карбальдегида или пиридин-2-карбальдегида, где в качестве растворителя применяют этанол и используют сульфат магния, при этом во всех случаях реакционную массу кипятят, и в зависимости от альдегидной компоненты время реакции возрастает от 1 часа (для бензальдегида) до 18 часов (для 2-тиофенкарбальдегилда). Использование дихлорметана в качестве растворителя для сочетания 2-формилтиофена и свободного амина - (1R,2R)-диаминоциклогексана при комнатной температуре приводит к увеличению времени реакции до 32 часов [2, 3, 6]. В аналогичных условиях получены диимины на основе 5'-замещенного 2,2'-битиофен-5-карбальдегида и свободного основания - (1R,2R)-диаминоциклогексана, стоит отметить что в некоторых случаях реакция проходила в течение 48 часов и в зависимости от заместителей выход варьировался в диапазоне от 31 до 70% [7]. Вышеописанные методы содержат ряд недостатков, которые в основном заключаются в длительном проведении реакции, использовании только одного изомера хирального 1,2'-диаминоциклогексана, при этом выходы целевых продуктов не всегда приемлемы. Стоит отметить, что в зависимости от структуры исходного альдегида условия реакции изменяются для достижения больших выходов.
Конденсацию ароматических альдегидов можно проводить не только с 1,2-диаминоциклогексаном, но и с (R,R)-(-)-1,2-диаминоциклогексан моно-(+)-тартратом [8]. Однако в данном случае необходимо использование основания для получения свободного амина и осуществления реакции [8]. Наиболее близким по техническому исполнению методом является конденсация соли (R,R)-(-)-1,2-диаминоциклогексан моно-(+)-тартрата с ароматическими альдегидами в водно-этанольной смеси (1:1 по объему) в присутствии карбоната калия при температуре 80°C [9]. Стоит отметить, что растворимость многих карбальдегидов резко снижается в водно-этанольной смеси и, соответственно, уменьшает выход целевого соединения. Использование данной методики конденсации в случае 2-(4'-метилциклогекс-1'-ен-1'-ил)тиофен-5-карбальдегида приводит к значительному осмолению реакционной массы и использованию большого модуля растворителя.
Известно, что воздействие на некоторые реакции микроволнового или ультразвукового излучения может способствовать прохождению реакции и ускорять процесс, что отмечено в случае построения трициклических макроструктур [10].
Полученный нами результат является более простым в техническом отношении, экономически эффективным и безопасным методом синтеза дииминов на основе смеси транс-(1R,2R)-диаминоциклогексанов. Методика синтеза азометинов, описанная в статье [3], была отработана на реакции конденсации 5'-метил-2,2'-битиофен-5-карбальдегида со смесью транс-(1R,2R)-диаминоциклогексана. В результате замены растворителя - хлористого метилена на ацетонитрил удалось резко сократить время проведения реакции с 48 ч до 6 ч, что, возможно, связано с понижением растворимости азометина и возможностью выделения из реакционной массы хорошим выходом.
Преимуществом предлагаемой методики является возможность использования коммерчески более доступной смеси транс-(1R,2R)-диаминоциклогексанов. Для получения оптически чистых продуктов использовали описанную методику разделения смеси транс-изомеров 1,2-диаминоциклогексана - метод раскристаллизации с помощью оптически чистой природной L-(+)-винной кислоты для получения соли (R,R)-(-)-l,2-диаминоциклогексан моно-(+)-тартрата в виде кристаллического осадка и соли (S,S)-(-)-1,2-диаминоциклогексан моно-(+)-тартрата, находящейся в маточном растворе [11].
Преимуществом метода является то, что синтез целевых азометинов проводили следующим образом: в одногорлую круглодонную колбу, помещенную в ультразвуковую ванну, вносят соль (S,S)-(-)-1,2-диаминоциклогексан моно-(+)-тартрата (1 моль), насыщенный раствор карбоната калия (2 моль) и диспергируют в течение 5 минут, затем порционно добавляют карбальдегид (2 моль) в минимальном объеме этилового спирта и на полученную смесь воздействуют ультразвуком в течение 5 часов, температура реакции 40-45°C. Образующийся осадок отфильтровывают, промывают водой, сушат и при необходимости азометин перекристаллизовывают из этилового спирта. Стоит отметить, что проведение реакции в аналогичных условиях при перемешивании и без использования ультразвука не позволяет достичь хороших выходов соединений и резко увеличивает время реакции.
Основные отличительные признаки предлагаемого метода получения промежуточных азометинов можно сформулировать следующим образом.
1. В предлагаемом методе в качестве исходного соединения используются оптически чистая соль (R,R)-(-)-1,2-диаминоциклогексан моно-(+)-тартрата, что более удобно в техническом исполнении, при этом полученная соль более стабильна и может храниться в течение длительного времени, в отличие от самих 1,2-диаминоциклогексанов.
2. Наиболее приемлемым мольным соотношением соль (R,R)-(-)-1,2-диаминоциклогексан моно-(+)-тартрата/гетероароматический карбальдегид/карбонат калия (безводный) является 1:2:2. При этом карбальдегид растворяют в минимальном объеме этанола.
3. Значительно сокращается время проведения реакции (с 48 ч [8] или 32 ч [2, 3, 7] до 3 ч). Сокращение времени реакции приводит к упрощению технологической схемы получения и увеличению производительности.
4. Предлагаемый метод не предполагает использование сложных лабораторных установок, что существенно упрощает схему реактора, и не требует применения сложного и дорогостоящего оборудования.
5. Предлагаемый метод может использоваться на различных количествах реагентов, существует возможность увеличения масштабов производства.
Получение 2,2'-битиофен-5-карбальдегида и 5'-метил-2,2'-битиофен-5-карбальдегида проводили по ранее описанным методам [12a,b].
Интерес представляет создание не только азометинов, но и дальнейшее восстановление азометиновой связи и образованию структур, которые могут быть использованы в качестве лигандов в комплексах с металлами для энантиоселективных реакций [13].
В литературе описан ряд методов восстановления азометинов, содержащих как ароматический фрагмент, так и гетероциклический фрагмент, в методиках используется боргидрид натрия в качестве восстановителя, однако в качестве среды применяют метиловый спирт или смеси метиловый спирт/эфир, время реакции составляет от 1 до 4 часов при комнатной температуре [1-3, 8]. Стоит отметить, токсичные свойства метилового спирта в данном методе. Описан метод восстановления иминной связи с помощью боргидрида натрия в среде этанола, однако реакцию проводят при 0°С с подъемом температуры до комнатной в течение 16 часов [14].
Преимуществом предлагаемой методики восстановления азометинов и получения целевых хиральных гетероциклических лигандов является использование коммерчески доступного и более безопасного этилового спирта и сокращение реакции до 3 часов с возможностью использования технического азометина. Преимуществом предлагаемого метода является:
1. Использование коммерчески доступного и более безопасного этилового спирта;
2. Наиболее приемлемыми мольным соотношением является: азометин/боргидрид натрия - 1:10 и использование минимального объема этилового спирта;
3. Сокращено время реакции с 16 до 3 часов с сохранением выхода продуктов;
4. Метод не предполагает использование сложных лабораторных установок, что существенно упрощает схему реактора, и не требует применения сложного и дорогостоящего оборудования.
Технический результат достигается тем, что способ получения хиральных гетероциклических лигандов на основе 1,2-диаминоциклогексана основан на взаимодействии гетероароматических альдегидов и (R,R)-(-)-1,2-диаминоциклогексан моно-(+)-тартрата в присутствии карбоната калия при мольном соотношении 1:2:2, соответственно, в водно-этанольной среде под действием ультразвука при комнатной температуре в течение 3 часов, с последующим восстановлением под действием боргидрида натрия при мольном соотношении азометин/боргидрид натрия 1:10 в этиловом спирте при температуре 60-70°С в течение 3 часов, затем отгоняют 1/2 этилового спирта, остаток выливают в лед, продукт экстрагируют хлористым метиленом (3 раза), сушат, растворитель удаляют, в остатке получают продукт с выходом 90-95% и чистотой 97-98%.
Выполнение метода
Строение синтезированных соединений подтверждено данными ИК-спектров, контроль над ходом реакции и индивидуальность соединений определялись с помощью TCX на пластинках Marchery Nagel. Проявление осуществлялось парами йода и облучением УФ лампой 254/365. ИК спектр записан на спектрометре Shimadzu IRAffinity-1 в таблетках бромида калия либо в тонком слое в призмах. Элементный анализ выполнен на автоматическом CHNS-анализаторе "Euro Vector ЕА-3000". Спектры ЯМР 1Н записаны на приборе JEOL JNM ECX 400 (400 МГц), в CDCl3, ДМСО-d6, внутренний стандарт - тетраметилсилан. Температура плавления измерена на приборе ПТП.
Способ получения азометинов. Общая методика.
В одногорлую круглодонную колбу, помещенную в ультразвуковую ванну, вносят соль (R,R)-(-)-1,2-диаминоциклогексан моно-(+)-тартрата (1 моль), насыщенный раствор карбоната калия (2 моль) и диспергируют в течение 5 минут, затем порционно добавляют карбальдегид (2 моль) в минимальном объеме этилового спирта и на полученную смесь воздействуют ультразвуком в течение 5 часов, температура реакции 40-45°С. Образующийся осадок отфильтровывают, промывают водой, сушат и при необходимости азометин перекристаллизовывают из этилового спирта.
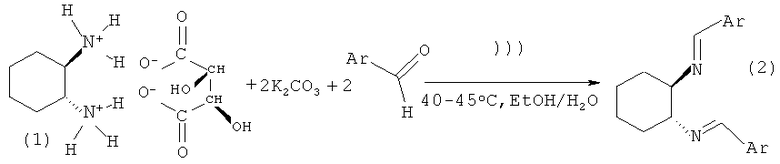






Пример 1. Синтез (1R,2R)-N,N-{ди-(тиофен-2-ил)метилиден}-циклогексан-1,2-диимина (2a)

4.1 г (15.5 ммоль) (R,R)-(-)-1,2-диаминоциклогексан моно-(+)-тартрата, 3.5 г (31 ммоль) тиофен-2-карбальдегида, 35 мл этилового спирта, 4.4 г (31 ммоль) карбоната калия в 10 мл воды. Выход 4.21 г (90%), т.пл. 128-133°C. ИК спектр (KBr), ν, см-1: 2926, 2873, 2843; 1635 (C=N); 796, 759 (Cap-Cap). Спектр ЯМР 1H (400 MHz, CDCl3), δ, м. д. (J, Гц): 8.25 (2H, c, N=CH) 7.27 (2H, дд, 3J=5.04, 4J=1.4, H-5), 7.12 (2H, дд, 3J=3.65, 4J=1.4, H-3), 6.94 (2H, дд, 3J=3.65, 3J=5.04, H-4), 3.3 (2H, м, CH циклогекс); 1.82 (6H, м, CH2 циклогекс); 1.43 (2H, м, CH2 циклогекс). Найдено, %: C 63.52, Н 6.01, N 9.28, S 21.19. C16H18N2S2. Вычислено, %: С 63.54, H 6.00, N 9.26, S 21.20.
Синтез (1R,2R)-N,N-{ди-(тиофен-3-ил)метилиден}циклогексан-1,2-диимина (2b).
Выход 4.21 г (94%), т.пл. 128-132°C. ИК спектр (KBr), ν, см-1: 2927, 2854, 2843; 1639 (C=N), 800, 783 (Cap-Cap). Спектр ЯМР 1H (400 MHz, CDCl3), δ, м. д. (J, Гц): 8.18 (2H, с, 
Синтез (1R,2R)-N,N-{ди-(фуран-2-ил)метилиден}-циклогексан-1,2-диимина (2c).
Выход 5.4 г (85%). ИК спектр (KBr), ν, см-1: 2931, 2858; 1647 (C=N), 1014 (Cap-Cap). Спектр ЯМР 1H (400 MHz, CDCl3), δ, м.д. (J, Гц): 7.21 (2H, с, C=N), 7.41 (2H, дд, 3J=5.2, 4J=1.8 H-5), 6.55 (2H, д, 3J=3.4, H-3), 6.37 (2H, д, 3J=5.2, 3J=3.4, H-4), 3.34-3.32 (2H, м, CH циклогекс), 1.82-1.79 (6H, м, CH2 циклогекс), 1.42-1.41 (2H, м, CH2 циклогекс). Найдено, %: C 71.10, H 6.73, N 10.34, O 11.83. C16H18N2O2. Вычислено, %: С 71.09, Н 6.71, N 10.36, О 11.84.
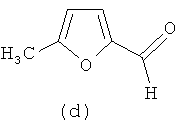
Синтез (1R,2R)-N,N-{ди-(5-метилфуран-2-ил)метилиден}цикло-гексан-1,2-диимина (2d). Выход 5.25 г (97%). ИК спектр (KBr), ν, см-1: 2931, 2858; 1643 (C=N), 1022 (Cap-Cap). Спектр ЯМР 1H (400 MHz, CDCl3), δ, м. д. (J, Гц): 7.86 (2H, c, N=CH), 6.37 (2H, д, 3J=3.2, H-3), 5.90 (2H, д, 3J=3.2, H-4), 3.3-3.23 (2H, м, CH циклогекс), 2.22 (6H, с, CH3), 1.72 (6H, м, CH2 циклогекс), 1.34 (2H, м, CH2 циклогекс). Найдено, %: C 72.45, H 7.41, N 9.41, O 10.73. C18H22N2O2. Вычислено, %: C 72.46, H 7.43, N 9.39, O 10.72.
Синтез (1R,2R)-N,N-{ди-(2,2'-битиофен-5-ил)метилиден}-циклогексан-1,2-диимина (2e). Выход 1.82 г (91%), т.пл. 110-113°C. ИК спектр спектр (KBr), ν, см-1: 2955, 2878, 1624 (C=N), 855, 815, 699 (Cap-Cap). Спектр ЯМР 1H (400 MHz, CDCl3), δ, м. д. (J, Гц): 8.19 (2H, c, N=CH), 7.22 (2H, дд, 3J=5.2, 4J=1.16, H-5), 7.18 (2H, дд, 3J=3.67, 4J=1.16, H-4'), 7.0 (4H, м, H-3, H-3'), 6.99 (2H, дд, 3J=3.67,3J=5.2, H-4); 3.3 (2H, м, CH циклогексен.); 1.83 (6H, м, CH2 циклогекс); 1.46 (2H, м, CH2 циклогекс). Найдено, %: C 61.78, H 4.74, N 6.02, S 27.46. C24H22N2S4. Вычислено, %: С 61.76, H 4.76, N 6.00, S 27.48.
Синтез (1R,2R)-N,N-{ди-(5-метил-2,2'-битиофен-5'-ил)метилиден}-циклогексан-1,2-диимина (2f). Выход 1.8 г (90%), т.пл. 90-93°C. ИК спектр (KBr), ν, см-1: 2927, 2856, 1622 (C=N), 1481, 1226, 812, 790. Спектр ЯМР 1H (400 MHz, CDCl3), δ, м.д. (J, Гц): 8.17 (2H, c, N=CH), 7.00 (2H, д, 3J=3.68, H-4), 6.97 (2H, д, 3J=3.68, H-3'), 6.93 (2H, д, 3J=3.68, H-3); 6.64 (2H, д, 3J=3.68, H-4'); 3.3 (2H, м, CH циклогексен.); 1.83 (6H, м, CH2 циклогекс); 1.46 (2H, м, CH2 циклогекс). Найдено, %: C 63.11, H 5.33, N 5.65, S 25.91. C26H26N2S4. Вычислено, %: C 63.12, H 5.30, N 5.66, S 25.92.
Синтез N,N-{ди(5-(4-метилциклогекс-1-ен-1-ил)тиофен-2-ил)метилиден}-циклогексан-1,2-диимина (2g). Выход 0.34 г (39%), т.пл. 147-149°C. ИК спектр (KBr), ν, см-1: 2914, 2850; 1627 (C=N), 1490 (C=C), 792 (Cap-Cap). Спектр ЯМР 1H (400 MHz, CDCl3), δ, м. д. (J, Гц): 8.14 (2H, c, N=CH), 6.97 (2H, д, 3J=3.68, H-3), 6.78 (2H, д, 3J=3.68, H-4), 6.18 (2H, уш. с, C=CH циклогексен), 3.26 (2H, м, CH циклогекс); 2.35 (6H, м, CH2 циклогекс); 1.74 (10H, м, CH2 циклогекс); 1.33 (4H, м, CH2 циклогекс); 0.98 (6H, д, 3J=6.43, CH3). Найдено, %: C 73.40, H 7.81, N 5.72, S 13.07. C30H38N2S2. Вычислено, %: C 73.42, H 7.80, N5.71, S 13.07.
Восстановление азометинов. Общая методика.
В колбе, снабженной магнитной мешалкой, растворяют азометин (1 моль) в минимальном объеме этилового спирта. При интенсивном перемешивании при комнатной температуре добавляют боргидрид натрия (10 моль) и кипятят с обратным холодильником в течение 3 часов. Удаляют ½ этилового спирта, остаток выливают в лед, продукт экстрагируют хлористым метиленом (3 раза), сушат, растворитель удаляют, в остатке получают продукт с выходом 90-95% и чистотой 97-98%.

Пример 2. Синтез (1R,2R)-N,N-ди-(2-тиенилметил)циклогексан-1,2-диамина (3a)

2.7 г (8.94 ммоль) (1R,2R)-N,N-{Ди-(тиофен-2-ил)метилиден}-циклогексан-1,2-диимина, 7.05 г (178.8 ммоль) боргидрида натрия, 140 мл этанола. Выход 1.5 г (98.7%). Т.пл. 70-72°C. ПК спектр (KBr), ν, см-1: 2924, 2850; 3298 (N-H). Спектр ЯМР 1H, δ, м.д. (J, Гц): 7.39 (2H, дд, 3J=4.82, 4J=1.40, H-5), 6.94-6.91 (4H, м, H-3, H-4), 4.11 (2H, д, 3J=14.19, CH2-NH), 3.88 (2H, д, 3J=14.19, CH2-NH), 2.27-2.30 (2H, м, циклогекс.), 2.07-2.15 (2H, м, циклогекс), 1.71-1.73 (2H, м, циклогекс), 1.20-1.25 (2Н, м, циклогекс), 1.02-1.04 (2H, м, циклогекс). Найдено, %: C 62.73, H 7.22, N 9.15, S 20.90. C16H22N2S2. Вычислено, %: C 62.70, H 7.24, N9.14, S 20.92.
Синтез (1R,2R)-N,N-ди-(3-тиенилметил)циклогексан-1,2-диамина (3b). Выход 1.3 г (85.5%). ИК спектр (KBr), ν, см-1: 2924; 3294 (N-H). Спектр ЯМР 1H, δ, м.д. (J, Гц): 7.24 (2H, дд, 3J=5.04, 4J=3.29, H-5), 7.09-7.11 (2H, м, H-4), 7.03 (2H, д, 4J=1.36, H-2), 3.91 (2H, д, 3J=13.85, CH2-NH), 3.68 (2H, д, 3J=13.85, CH2-NH), 2.22-2.29 (2H, м, циклогекс), 2.04-2.15 (2H, м, циклогекс); 1.71-1.73 (2H, м, циклогекс), 1.20-1.27 (2H, м, циклогекс), 1.01-1.04 (2H, м, циклогекс). Найдено, %: C 62.73, H 7.22, N 9.15, S 20.90. C16H22N2S2. Вычислено, %: C 62.70, H 7.24, N 9.14, S 20.92.
Синтез (1R,2R)-N,N-{ди-(фуран-2-ил)метил}-циклогексан-1,2-диамина (3c). Выход 3.5 г (83%). ИК спектр (KBr), ν, см-1: 3302 (N-H), 2931, 2858; 1010 (Сар-Сар). Спектр ЯМР 1H, δ, м.д. (J, Гц): 7.19 (2H, дд, 3J=4.82, 4J=1.40, H-5), 6.07 (2H, д, 3J=4.82, 3J=3.24, H-4), 6.16 (2H, дд, 3J=3.24, 4J=1.40, H-3), 3.75 (2H, д, 3J=14.43, CH2-NH), 3.62 (2H, д, 3J=14.43, CH2-NH), 2.11-2.16 (2H, м, CH циклогекс), 1.89-1.93 (2H, м, циклогекс), 1.57-1.64 (2H, м, циклогекс), 1.06-1.1 (2H, м, циклогекс), 0.93-0.98 (2H, м, циклогекс). Найдено, %: C 70.01, H 8.07, N 10.23, О 11.69. C16H22N2O2. Вычислено, %: C 70.04, H 8.08, N 10.21, O 11.67.
Синтез (1R,2R)-N,N-{ди-(5-метилфуран-2-ил)метил}-циклогексан-1,2-диамина (3d). Выход 3.3 г (94%). ИК спектр (KBr), ν, см-1: 3302 (N-H), 2931, 2858; 1010 (Cap-Cap). Спектр ЯМР 1H, δ, м. д. (J, Гц): 5.77 (2H, д, 3J=3.2, H-4), 5.95 (2H, д, 3J=3.2, H-3), 3.7 (2H, д, 3J=14.19, CH2-NH), 3.54 (2Н, д, 3J=14.19, CH2-NH), 2.16 (2H, с, CH3), 1.61-1.63 (2H, м, CH циклогекс), 1.95-1.98 (4H, м, циклогекс), 1.11-1.2 (2H, м, циклогекс), 0.93-0.96 (2H, м, циклогекс). Найдено, %: C 71.50, Н 8.66, N 9.25, О 10.59. C18H26N2O2. Вычислено, %: C 71.49, H 8.67, N 9.26, O 10.58.
Синтез (1R,2R)-N,N-ди-(2,2'-битиенил-5-метил)циклогексан-1,2-диамина (3e). Выход 0.82 г (88%). ИК спектр (KBr), ν, см-1: 2920, 2850, 2819; 3290 (N-H), 821, 798, 698 (Cap-Cap). Спектр ЯМР 1H, δ, м.д. (J, Гц): 7.15 (2H, дд, 3J=5.04, 4J=1.36, H-5), 7.09 (2H, д, 3J=3.64, Н-4'), 6.99 (2Н, д, 3J=3.52, H-3), 6.94-6.96 (2H, м, H-3'), 6.82 (2H, д, 3J=3.52, 3J=5.04, H-4), 4.07 (2H, д, 3J=14.05, CH2-NH), 3.86 (2H, д, 3J=14.05, CH2-NH), 2.32-2.34 (2H, м, CH циклогекс). 2.12-2.16 (2H, м, циклогекс). 1.72-1.74 (2H, м, циклогекс), 1.21-1.26 (2h, м, циклогекс), 1.05-1.08 (2H, м, циклогекс). Найдено, %: C 61.22, H 5.58, N 5.91, S 27.26. C24H26N2S4. Вычислено, %: C 61.24, H 5.57, N 5.94, S 27.25.
Синтез (1R,2R)-N,N-ди-((5-метил-2,2'-битиенил)-5-метил)циклогексан-1,2-диамина (3f). Выход 1.6 г (88%). ИК спектр (KBr), ν, см-1: 3437 (N-H), 3290, 2916, 2852, 1448, 1197, 895, 790. Спектр ЯМР 1H (400 MHz, CDCl3), δ, м.д. (J, Гц): 6.89 (4Н, м, Н-4, H-3), 6.79 (2H, м, H-3'), 6.60 (2H, м, H-3); 4.08 (2H, д. 3J=14.19, CH2-NH), 3.85 (2H, д, 3J=14.19, CH2-NH), 2.32-2.34 (2H, м, циклогекс), 2.14-2.11 (2H, м, циклогекс), 1.72-1.74 (2H, м, циклогекс), 1.30-1.19 (4H, м, циклогекс), 1.05-1.07 (2Н, м, циклогекс.). Найдено, %: C 62.62, H 6.04, N 5.61, S 25.73. C26H30N2S4. Вычислено, %: C 62.61, H 6.06, N 5.62, S 25.71.
Синтез 1R,2R)-N,N-{ди-[5-(4'-метилциклогекс-1'-ен-1'-ил)тиофен-2-ил)метил}-циклогексан-1,2-диамина (3g). Выход 1.1 г (91%). ИК спектр (KBr), ν, см-1: 3290 (N-H), 2924, 2862, 2819; 1454 (C=C), 786 (Cap-Cap). Спектр ЯМР 1H, δ, м. д. (J, Гц): 6.74 (4H, м, H-3, Н-4), 6.06 (2H, уш. с, C=CH), 4.02 (2H, д, 3J=14.19, CH2-NH), 3.88 (2H, д, 3J=14.19, CH2-NH), 2.35-2.28 (10Н, м, циклогекс), 1.79-1.78 (14H, м, циклогекс), 1.24-1.23 (2H, м, CH3-CH циклогекс), 0.97 (2H, д, 3J=4.3, CH3). Найдено, %: C 72.80, H 8.57, N 5.63, S 12.97. C30H42N2S2. Вычислено, %: C 72.82, H 8.56, N 5.66, S 12.96.
Литература:
[1] Albano V.G. et all. Controlling Stereochemical Outcomes of Asymmetric Processes by Catalyst Remote Molecular Functionalizations: Chiral Diamino-oligothiophenes (DATs) as Ligands in Asymmetric Catalysis // Chem. Eur. J. - 2006. №12. - P.667-675.
[2] Albano V., Bandini M., Melucci M. Novel Chiral Diamino-Oligothiophenes as Valuable Ligands in Pd-Catalyzed Allylic Alkylations. On the "Primary" Role of "Secondary" Interactions in Asymmetric Catalysis. // Adv. Synth. Catal. - 2005. №347. - P.1507-1512.
[3] Ambroziak K., Rozwadowski Z., Dziembowska T. Synthesis and spectroscopic study of Schiff bases derived from trans-1,2-diaminocyclohexane. Deuterium isotope effect on 13C chemical shift. // Journal of Molecular Structure. - 2002. №615. - P.109-120.
[4] Патент 0234239 A1 США. Method Of Forming Carbon-Heteroatom Linkage. / Taillefer M., Cristau H., Cellier P. - Заявлено 2.06.2003. - Опубл. 20.10.2005.
[5] Патент 0236413Al США. Process for arylating of vinylating or alkynating nucleophilic compound. / Cellier P., Cristau H., Spindler J. - Заявлено 31.05.2002. - Опубл. 25.12.2003.
[6] Lere-Porte J., Moreau J., Serein-Spirau F. A chiral polymer with alternating conjugated segments and (1R,2R)-1,2-diaminocyclohexane as a unit with C2 symmetry. // Tetrahedron. - 2001. №42. - P.3073-3076.
[7] Bandini M., Benaglia M., Quinto T. New Recoverable Poly (ethylene glycol)-Supported C1-Diaminooligothiophene Ligands for Palladium-Promoted Asymmetric Allylic Alkylation (AAA) Reactions. // Adv. Synth. Catal. - 2006. №348. - P.1521-1527.
[8] Zhang X., Li Y., Dong Z. Asymmetric transfer hydrogenation of aromatic ketones with chiral diamino-thiophene/iridium catalyst systems. // Journal of Molecular Catalysis A: Chemical. - 2009. №307. - P.149-153.
[9] Duguet N., Donaldson A., Leckie S. Chiral relay in NHC-mediated asymmetric β-lactam synthesis I; substituent effects in NHCs derived from (1R,2R)-cyclohexane-1,2-diamine. // Tetrahedron: Asymmetry. - 2010. №21. - P.582-600.
[10] Srimurugan S., Viswanathan B. Microwave assisted cyclocondensation of dialdehydes with chiral diamines forming calixsalen type macrocycles. // Tetrahedron Letters. - 2005. №46. - P.3151-3155.
[11] Larrow F., Jacobsen E. A Practical Method for the Large-Scale Preparation of E-N,N-Bis(3,5-di-tert-butylsalicylidene)-1,2-cyclohexanediaminato-2)]manganese Chloride, a Highly Enantioselective Epoxidation Catalyst. // J. Org. Chem. - 1994. №59. - P.1939-1942.
[12] a) Nakayama J., Murabayashi S., Hoshino M. Preparation of (E)-1,2-bis(2,2'-bithiophene-5-yl)ethylene and (E)-1,2-bis-(2,2':5',2”-terthiophene-5-yl)ethylene // Heterocycles. - 1986. V.24. №9. - P.2639-2643. b) Lescot E., Buu-Hoi Ng. Ph., Xuong N.D., Thiophene Derivatives. Part XIV. Some problems of substitution in the 2,2'-bithienyl series. // J. Chem. Soc. - 1959. V.656. - P.3234-3237.
[13] Cooper Ch., Jones M., Brayshaw S. When is an imine not an imine? Unusual reactivity of a series of Cu(II) imine-pyridine complexes and their exploitation for the Henry reaction. // Dalton Trans. - 2011. №40. - P.3677-3682.
[14] Kowalczyk R., Skarzewski J. Asymmetric nitroaldol reaction catalyzed by copper-diamine complexes: selective construction of two contiguous stereogenic centers. // Tetrahedron: Asymmetry. - 2009. №20. - P.2467-2473.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ 3-АЛКИЛ(АРИЛ)-2,2'-БИТИОФЕН-5-КАРБОНОВЫХ КИСЛОТ И ИХ ЭФИРОВ | 2011 |
|
RU2470930C1 |
| СОЕДИНЕНИЯ ТРИАЗОЛА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ИХ СОДЕРЖАЩАЯ, ПРИМЕНЕНИЕ СОЕДИНЕНИЙ ТРИАЗОЛА И СПОСОБ ЛЕЧЕНИЯ ГРИБКОВОЙ ИНФЕКЦИИ | 2002 |
|
RU2276670C2 |
| СПОСОБ ПОЛУЧЕНИЯ 4-АЛКОКСИ-4Н-ТИЕНО[3,2-с]ХРОМЕН-2-КАРБАЛЬДЕГИДОВ, ОБЛАДАЮЩИХ ПРОТИВОЯЗВЕННОЙ АКТИВНОСТЬЮ | 2015 |
|
RU2571094C1 |
| СПОСОБ ПОЛУЧЕНИЯ ТРЕТИЧНЫХ ЦИКЛИЧЕСКИХ СПИРТОВ РЯДА 2,2'-БИТИОФЕНА | 2011 |
|
RU2495018C2 |
| ПРОИЗВОДНЫЕ 1-(4-ИЗОКСАЗОЛ-5-ИЛ)-1Н-ПИРАЗОЛ-1-ИЛ)-2-МЕТИЛПРОПАН-2-ОЛА И РОДСТВЕННЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИРОРОВ ИЛ-17 И ИФН-ГАММА ДЛЯ ЛЕЧЕНИЯ АУТОИММУННЫХ ЗАБОЛЕВАНИЙ И ХРОНИЧЕСКОГО ВОСПАЛЕНИЯ | 2018 |
|
RU2785342C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 7-(ГЕТЕРО)АРИЛ-4,5,6,7-ТЕТРАГИДРО[1,2,3]ТРИАЗОЛО[1,5-A]ПИРИДИНА | 2013 |
|
RU2563254C2 |
| Хиральные монотерпеновые сульфинамиды | 2017 |
|
RU2646959C1 |
| ПРОИЗВОДНОЕ ДИ- И ТРИАЗОЛИЛПРОПАНА, СПОСОБЫ ЕГО ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ | 1996 |
|
RU2145605C1 |
| 2,6-ДИИЗОБОРНИЛФЕНОЛЫ | 2011 |
|
RU2502719C2 |
| Хиральные трифторметилированные монотерпеновые тиоацетаты и тиолы на основе миртеналя | 2020 |
|
RU2743302C1 |
Изобретение относится к способу получения хиральных гетероциклических лигандов на основе 1,2-диаминоциклогексана, содержащих гетероциклические фрагменты: тиенил-2-, тиенил-3-, фурил-2-, 5-метилфурил-2-, (2,2'-битиофен)-5-ил-, 5-(4'-метилциклогекс-1'-ен-1'-ил)тиофен-2-, которые могут входить в структуру комплексов для проведения энантиоселективных реакций и асимметрического катализа, а также обладать люминесцентными свойствами. Упрощение технологического процесса достигается за счет использования доступных реагентов, использование приемлемого мольного соотношения реагентов, значительного сокращения общего времени проведения реакции, уменьшение стоимости производства достигается за счет упрощения схемы реактора и времени проведения реакции. 2 пр.
Способ получения хиральных гетероциклических лигандов на основе 1,2-диаминоциклогексана основан на взаимодействии гетероароматических альдегидов и (R,R)-(-)-1,2-диаминоциклогексан моно-(+)-тартрата в присутствии карбоната калия при мольном соотношении 1:2:2, соответственно, в водно-этанольной среде под действием ультразвука при комнатной температуре в течение 3 часов, с последующим восстановлением под действием боргидрида натрия при мольном соотношении азометин/боргидрид натрия 1:10 в этиловом спирте при температуре 60-70°C в течение 3 часов, затем отгоняют ½ этилового спирта, остаток выливают в лед, продукт экстрагируют хлористым метиленом (3 раза), сушат, растворитель удаляют, в остатке получают продукт с выходом 90-95% и чистотой 97-98%.
| RU 2011114665, A, 13.04.2011 | |||
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| Способ и приспособление для нагревания хлебопекарных камер | 1923 |
|
SU2003A1 |

