ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к новому замещенному дейтерием производному пиримидина и к содержащей его фармацевтической композиции.
УРОВЕНЬ ТЕХНИКИ
Немелкоклеточный рак легкого (NSCLC) представляет собой заболевание, которое среди онкологических заболеваний в последние годы характеризуется во всем мире очень высокой распространенностью и смертностью. Немелкоклеточный рак легкого вызывают несколько факторов, но основной причиной его развития является мутация, сверхэкспрессия и другими подобные процессы в гене тирозинкиназы или гене киназы анапластической лимфомы, и для лечения немелкоклеточного рака легкого в настоящее время разрабатываются противораковые препараты, нацеленные на ингибирование активности этих ферментов.
Известно, что немелкоклеточный рак легкого, который в основном встречается в Восточной Азии, в том числе и в Южной Корее, часто характеризуется мутациями гена рецептора эпидермального фактора роста (EGFR), и было обнаружено, что активирующие мутации в области киназы рецептора эпидермального фактора роста (EGFR) являются онкогенными у некоторых пациентов с немелкоклеточным раком легкого, и гефитиниб, эрлотиниб и другие подобные лекарственные средства используют в качестве терапевтических средств, то есть низкомолекулярных ингибиторов киназы рецептора эпидермального фактора роста (EGFR), для лечения таких пациентов (Science 2004, 304: 1497-500; and New England Journal of Medicine 2004, 350: 2129-39). У пациентов с немелкоклеточным раком легкого, у которых подтверждена активность мутации EGFR, применение гефитиниба и эрлотиниба в качестве терапевтических средств приводит к лекарственной резистентности у большинства пациентов в течение одного года (Clinical Cancer Research 2013, 19: 2240-7). Среди этих механизмов резистентности, частота мутаций рецептора T790M эпидермального фактора роста наблюдалась на уровне до 60%. Поэтому был разработан ингибитор EGFR третьего поколения, который нацелен на мутантный рецептор T790M эпидермального фактора роста (EGFR) при раке легкого. Типичные лекарственные средства включают осимертиниб, лазертиниб и другие подобные средства, которые нацелены на мутацию T790M и проявляют относительно низкую токсичность, и, в силу этого, используются в клинике для лечения немелкоклеточного рака легкого (J Thorac Dis. 2018 Jul 2018);10(7): 3909-3921). Однако постоянно появлялись сообщения о лекарственной резистентности ингибиторов EGFR 3-го поколения, и сообщалось о мутации C797S, амплификации МЕТ и других подобных процессах в качестве основных механизмов резистентности (J Hematol Oncol. 2016, Jul 22, 9(1): 59; Nature Medicine 2015, 21: 560-562; Lung Cancer 2018, 118: 105-110; and ASCO 2017, abstract 2572, 9020). Сообщалось, что мутация C797S и амплификация MET обнаруживаются по отдельности, но иногда и одновременно.
У ряда пациентов с немелкоклеточным раком легкого наблюдается нарушение в гене киназы анапластической лимфомы (ALK) (слияние EML4-ALK), и для лечения этих видов рака в клинической практике используют различные ингибиторы тирозинкиназы (TKI) и другие подобные лекарственные средства. ALK-положительный немелкоклеточный рак легкого возникает в результате слияния генов ALK и EML4, и, поскольку латентный в обычных условиях ген ALK в результате слияния двух генов начинает увеличивать скорость роста клеток, получающие этот сигнал клетки быстро метастазируют в раковые клетки. В 2011 году в качестве многоцелевого противоопухолевого средства Управлением по контролю качества продовольствия и медикаментов США был одобрен типичный терапевтический препарат кризотиниб. Этот препарат применяют для лечения метастатического ALK-положительного немелкоклеточного рака легкого и других подобных типов рака путем ингибирования активности MET, ALK, ROS1 и других подобных ферментов. Анализ результатов клинического исследования кризотиниба показывает, что в этом исследовании в основном принимали участие пациенты, страдающие раком легкого с аденокарциномой тканевого типа, и 46% из них относились к уроженцам Азии. Этот препарат показал очень высокую эффективность, характеризующуюся частотой ответа опухоли приблизительно 65% и периодом выживаемости без прогрессирования заболевания 7,7 месяца (3 месяца в группе химиотерапии), а наиболее частыми нежелательными явлениями были нарушения поля зрения, диарея, рвота, отек, тошнота и другие подобные нежелательные явления (J Thorac Oncol 2012;7(7):1086-90). При применении кризотиниба неизбежно возникает резистентность, и сообщалось в основном о вторичных мутациях в домене киназы ALK (около 30%), амплификации мутаций слитого гена ALK, активации обходного сигнального пути и других подобных процессах. Наряду с существованием большого разнообразий мутаций, существуют вторичные мутации, включающие L1196M и G1269A, а также L1196M, которые наиболее часто встречаются в остатке привратника, препятствующие связыванию кризотиниба с ALK (J Clin Oncol 2013;31(8):1105-11).
Сообщается, что главный механизм резистентности основан на том, что все виды немелкоклеточного рака легкого, вызванные мутацией ALK или мутацией EGFR (или и той, и другой), характеризуются наличием вторичной мутации, которая ингибирует силу связывания киназы с лекарственным средством, и эти мутации влияют на внутриклеточную передачу сигналов в нисходящем направлении (Eur Med Chem. 2017 Aug 18;136:497-510). Несмотря на постоянно ведущиеся разработки различных ингибиторов ALK и EGFR, разработка ингибиторов, которые одновременно ингибируют две киназы, пока не дает ощутимых результатов. В связи с этим, существует необходимость в разработке лекарственного средства, которое эффективно ингибирует рост ALK-мутированных или EGFR-мутированных раковых клеток, вызывающих лекарственную резистентность по основным механизмам, описанные выше.
Кроме того, немелкоклеточный рак легкого возникает в результате экспрессии, реаранжировки или другого подобного процесса различных онкогенов, и их примеры включают KRAS, ROS1, RET и другие подобные онкогены (Lancet Oncol 2011;12(2):175-80).
Ссылки на известный уровень техники
Патентные документы
PCT International Publication No. WO 2009/143389 A1
ОПИСАНИЕ ИЗОБРЕТЕНИЯ
ТЕХНИЧЕСКАЯ ЗАДАЧА
Авторы настоящего изобретения ставили перед собой задачу разработать новое соединение, которое эффективно ингибирует ALK-мутированные и EGFR-мутированные типы рака. В результате проведенных исследований было подтверждено, что новое замещенное дейтерием производное пиримидина обладает очень высокой эффективностью при лечении рака легкого.
Поэтому, задачей настоящего изобретения является разработка нового замещенного дейтерием производного пиримидина, обладающего очень высокой эффективностью при лечении рака легкого, и фармацевтической композиции, включающей это производное.
Еще одной задачей настоящего изобретения является разработка нового замещенного дейтерием производного пиримидина, обладающего очень высокой эффективностью при лечении рака легкого, экспрессирующего мутацию ALK или мутацию EGFR, наряду с другими типами рака легкого, и фармацевтической композиции, включающей это производное.
РЕШЕНИЕ ТЕХНИЧЕСКОЙ ЗАДАЧИ
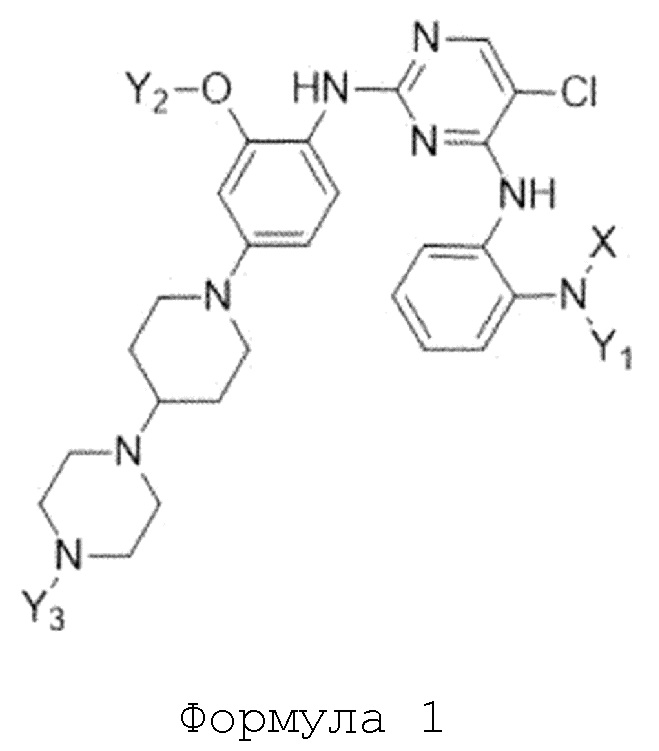
Для решения указанных выше задач, в настоящем изобретении предлагается соединение, представленное следующей формулой 1:
Формула 1
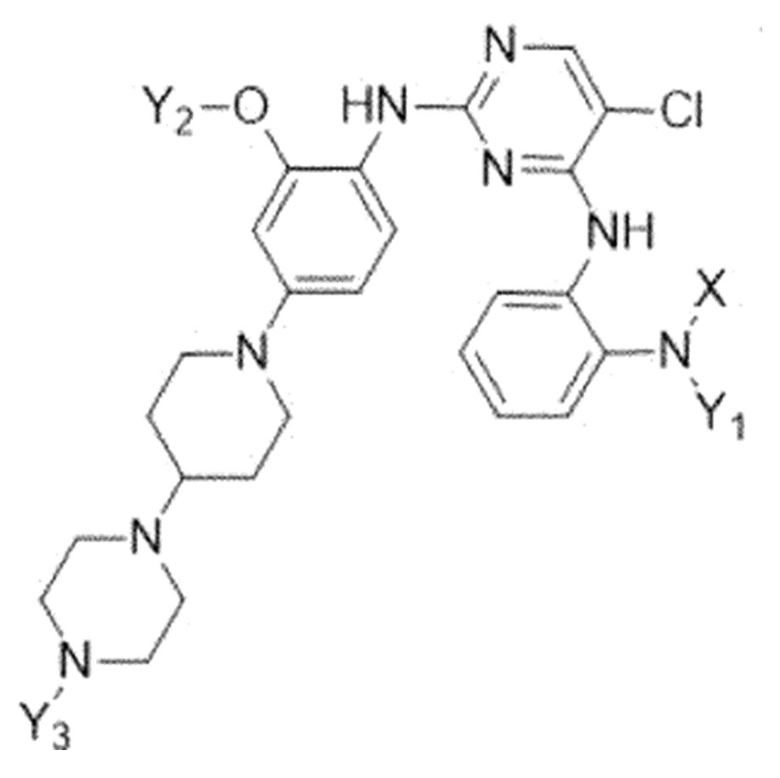
где
X представляет собой C1-C4 алкилсульфонильную группу, незамещенную или замещенную дейтерием, или C1-C4 диалкилфосфорильную группу, незамещенную или замещенную дейтерием; и
Y1, Y2 и Y3 независимо представляют собой C1-C4 алкильную группу, незамещенную или замещенную дейтерием,
где соединение формулы 1 содержит один или более атомов дейтерия,
или его фармацевтически приемлемая соль.
Кроме того, в настоящем изобретении предлагается фармацевтическая композиция для лечения рака легкого, включающая соединение, представленное формулой 1 выше, или его фармацевтически приемлемую соль в качестве активного ингредиента, и фармацевтически приемлемый носитель.
Кроме того, в настоящем изобретении предлагается соединение, представленное формулой 1 выше, или его фармацевтически приемлемая соль, которое применяют для лечения рака легкого.
Кроме того, в настоящем изобретении предлагается способ лечения животного, страдающего от рака легкого, включающий введение эффективного количества соединения, представленного формулой 1 выше, или его фармацевтически приемлемой соли животному.
ПОЛЕЗНЫЕ ЭФФЕКТЫ ИЗОБРЕТЕНИЯ
Новое замещенное дейтерием производное пиримидина по настоящему изобретению и фармацевтическая композиция, включающая это производное, обеспечивает высокую эффективность лечения рака легкого.
Кроме того, замещенное дейтерием производное пиримидина и фармацевтическая композиция, включающая это производное, в частности, эффективно ингибирует рост ALK-мутированных или EGFR-мутированных раковых клеток.
Описание чертежей
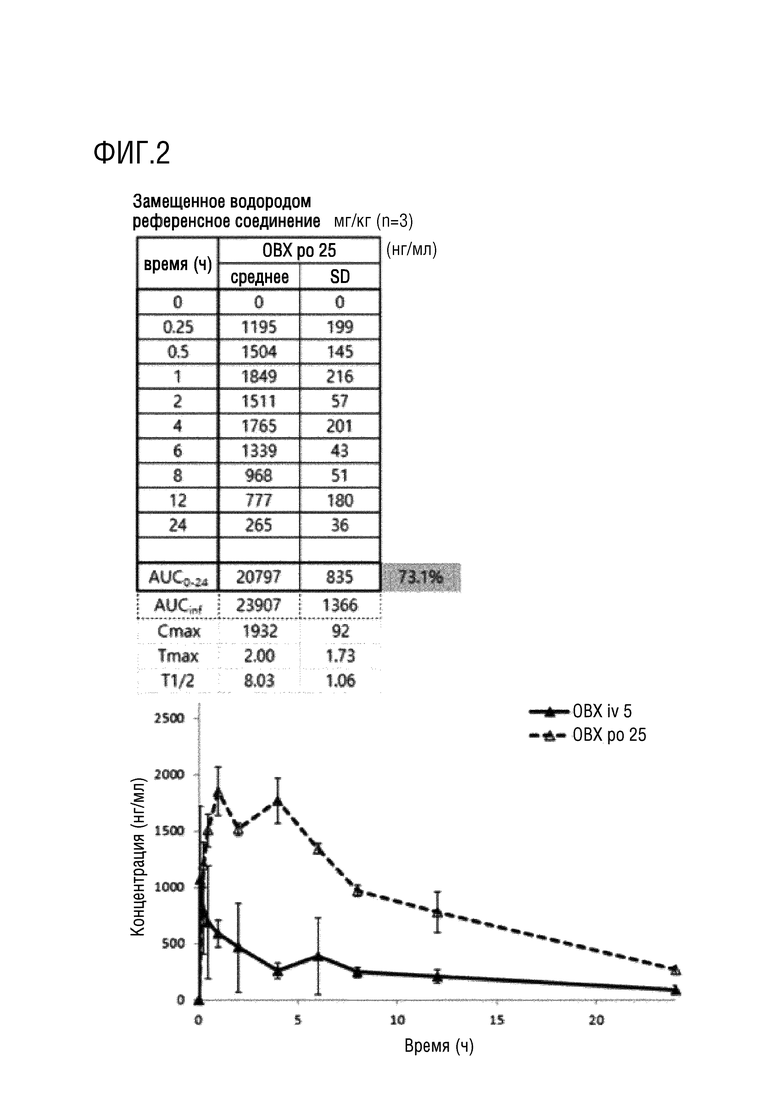
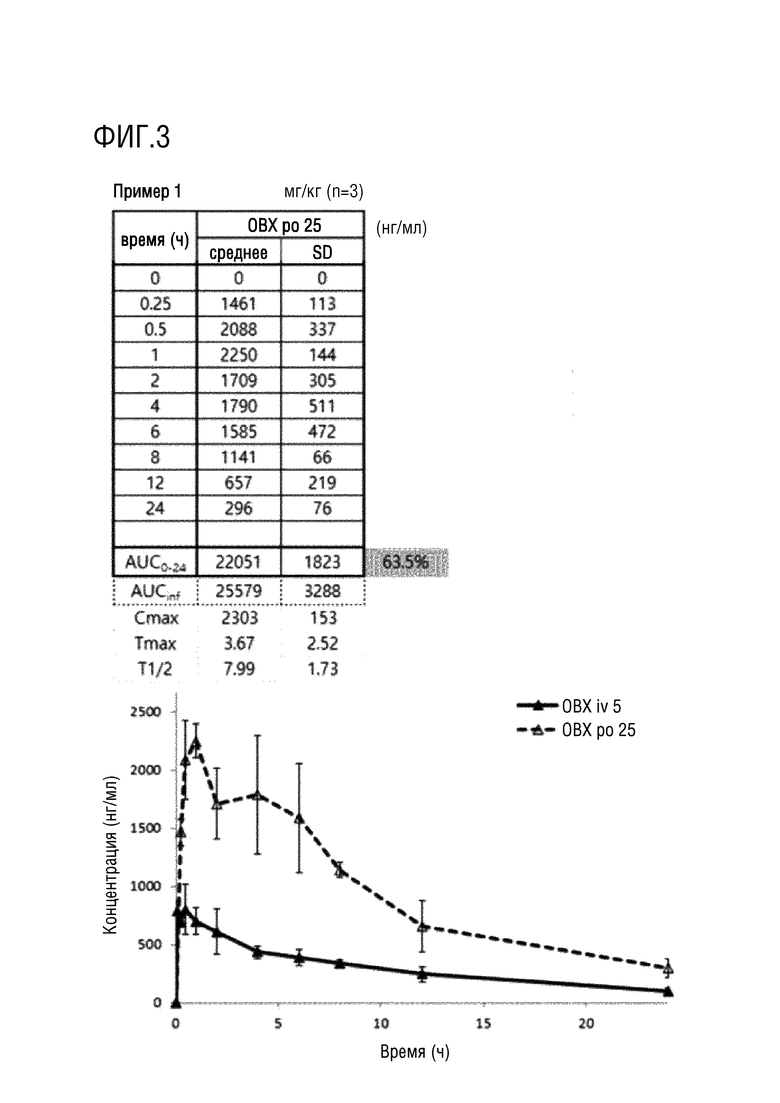
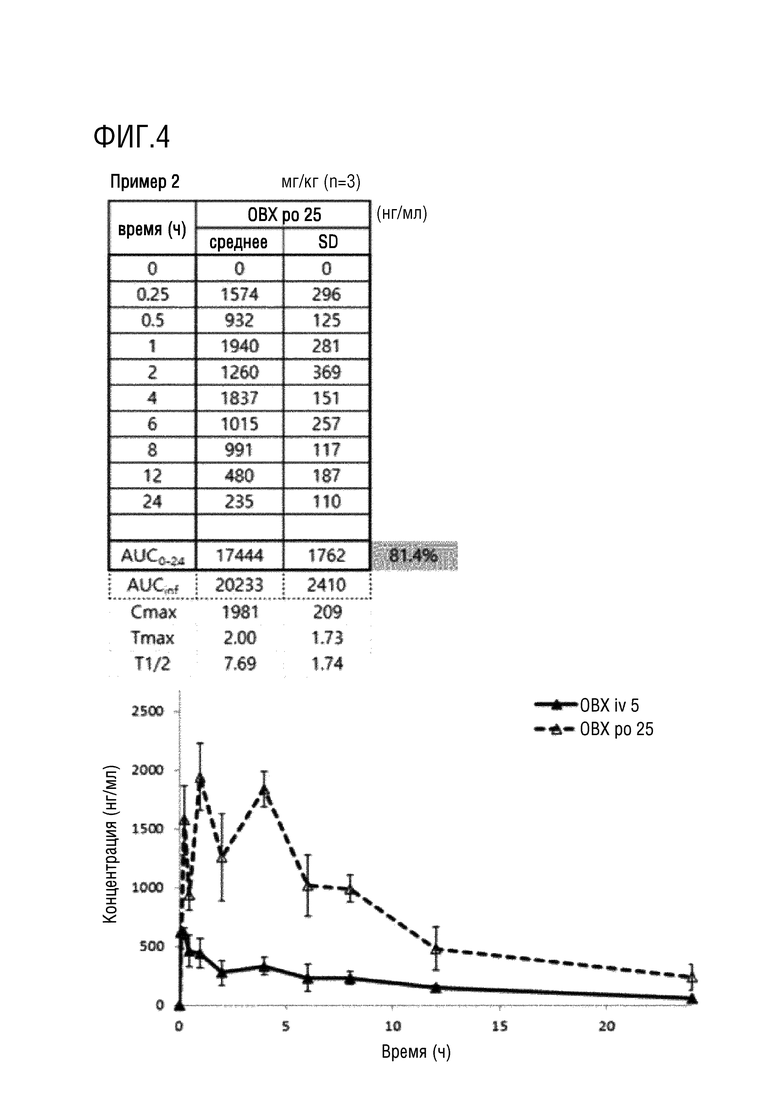
На фигурах 1-4 представлены результаты исследования на крысах фармакокинетических параметров соединений по настоящему изобретению, полученных в примерах 1 и 2.
ВАРИАНТ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Далее настоящее изобретение будет описано более подробно на примерах его вариантов осуществления. Однако настоящее изобретение не ограничивается вариантами осуществления, которые были представлены в качестве примеров, и объем настоящего изобретения определяется только объемом прилагаемых пунктов формулы изобретения. Кроме того, если даже вариант осуществления является достаточно важным для применения настоящего изобретения на практике, но этот вариант является очевидным для специалиста в данной области и может быть легко применен им на практике, то, в данном случае, подробное описание этого варианта осуществления не будет приводиться в изобретении.
Если не казано далее иное, то термин "соединение по настоящему изобретению" или "соединение формулы 1" применяют в качестве концептуального представления, включающего как само по себе соединение, так и его фармацевтически приемлемую соль.
Используемый в изобретении термин "алкильная группа" относится к линейным и разветвленным углеводородным группам, имеющим указанное число углеродных атомов. Алкильная группа может представлять собой, например, метил, этил, n-пропил, изопропил, н-бутил, вторбутил, изобутил, третбутил, и другие подобные алкильные группы.
Используемый в изобретении термин "алкилсульфонил" относится к алкил-S(O2)-. В этом случае, алкил определен выше.
Настоящее изобретение относится к соединению, представленному следующей формулой 1:
Формула 1
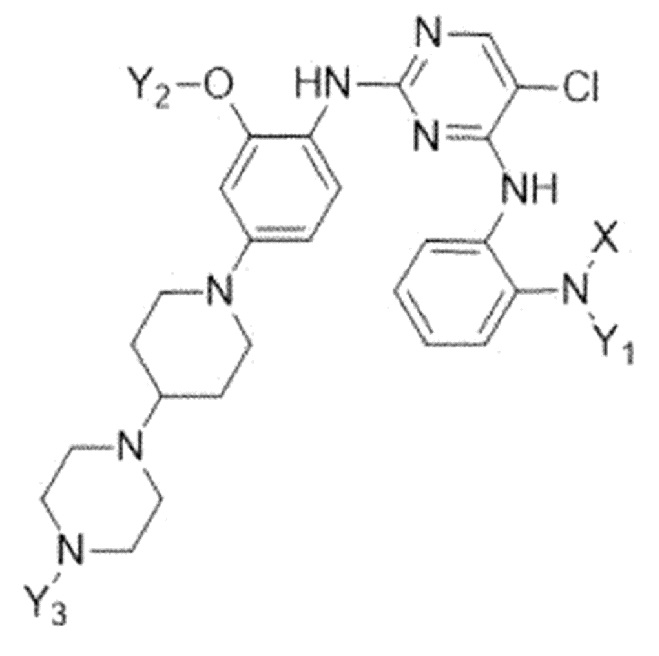
где
X представляет собой C1-C4 алкилсульфонильную группу, незамещенную или замещенную дейтерием, или C1-C4 диалкилфосфорильную группу, незамещенную или замещенную дейтерием; и
Y1, Y2 и Y3 независимо представляют собой C1-C4 алкильную группу, незамещенную или замещенную дейтерием,
где соединение формулы 1 содержит один или более атомов дейтерия,
или к его фармацевтически приемлемой соли.
В соединение формулы 1, любой один или более из Y1, Y2 и Y3 могут представлять собой C1-C4 алкильную группу, замещенную дейтерием.
В соединении формулы 1, алкильная группа в C1-C4 алкильной группе, незамещенной или замещенной дейтерием, предпочтительно может представлять собой метильную группу.
В соединении формулы 1, X может представлять собой C1-C4 алкилсульфонильную группу, незамещенную или замещенную дейтерием.
Описанное выше соединение формулы 1 представляет собой предпочтительно любое одно из следующих соединений:
N-(2-((5-хлор-2-((2-метокси-4-(4-(4-(метил-d3)пиперазин-1-ил)пиперидин-1-ил)фенил)амино)пиримидин-4-ил)амино)фенил)-N-метилметансульфонамид (соединение 1);
N-(2-((5-хлор-2-((2-(метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)амино)пиримидин-4-ил)амино)фенил)-N-(метил-d3)метансульфонамид (соединение 2);
N-(2-((5-хлор-2-((2-(метокси-d3)-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)амино)пиримидин-4-ил)амино)фенил)-N-метилметансульфонамид (соединение 3);
N-(2-((5-хлор-2-((2-метокси-4-(4-(4-(метил-d3)пиперазин-1-ил)пиперидин-1-ил)фенил)амино)пиримидин-4-ил)амино)фенил)-N-(метил-d3)метансульфонамид (соединение 4);
N-(2-((5-хлор-2-((2-(метокси-d3)-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)амино)пиримидин-4-ил)амино)фенил)-N-(метил-d3)метансульфонамид (соединение 5);
N-(2-((5-хлор-2-((2-метокси-d3)-4-(4-(4-(метил-d3)-пиперазин-1-ил)пиперидин-1-ил)фенил)амино)пиримидин-4-ил)амино)-фенил)-N-метилметансульфонамид (соединение 6) и
N-(2-((5-хлор-2-((2-(метокси-d3)-4-(4-(4-(метил-d3)-пиперазин-1-ил)пиперидин-1-ил)фенил)амино)пиримидин-4-ил)амино)-фенил)-N-(метил-d3)метансульфонамид (соединение 7).
Дейтерий, входящий в соединение формулы 1 по настоящему изобретению, является одним из изотопов водорода и содержит в атоме водорода один протон и один нейтрон и, поэтому, имеет атомное ядро, имеющее в два раза большую массу, чем масса обычного водорода. Дейтерий, условно обозначаемый буквой D, является стабильным элементом, не распадающимся и существующим в природе в очень малых количествах. Дейтерий в основном производят путем электролиза и концентрирования воды, и он образуется за счет разницы в скорости реакции, при которой легкая жесткая вода сначала реагирует с электродом с разложением, а затем тяжелая вода реагирует с электродом с относительно низкой скоростью с электролитическим разложением. В случае тяжелого элемента, на его массу не оказывает заметного влияния количество нейтронов, и, в силу этого, разница в химических свойствах между изотопами невелика. Однако в случае водорода с малой массой, изменение массы за счет изменения числа нейтронов очень существенно влияет на химические и физические свойства, такие как реакционная способность и скорость диффузии. Это явление называют изотопным эффектом, и известно, что этот эффект особенно ярко проявляется в случае водорода и дейтерия.
Настоящее изобретение характеризуется тем, что в результате использования таких свойств дейтерия повышается противораковая активность соединения формулы 1 его фармацевтически приемлемой соли.
Соль соединения, представленного формулой 1, по настоящему изобретению может представлять собой соль, образованную неорганической кислотой или органической кислотой, и в этом случае, предпочтительные соли включают соли хлористоводородной кислоты, бромистоводородной кислоты, серной кислоты, фосфорной кислоты, азотной кислоты, уксусной кислоты, гликолевой кислоты, молочной кислоты, пировиноградной кислоты, малоновой кислоты, янтарной кислоты, глутаровой кислоты, фумаровой кислоты, яблочной кислоты, миндальной кислоты, винной кислоты, лимонной кислоты, аскорбиновой кислоты, пальмитиновой кислоты, малеиновой кислоты, бензойной кислоты, гидроксибензойной кислоты, фенилуксусной кислоты, коричной кислоты, салициловой кислоты, метансульфоновой кислоты, этансульфоновой кислоты, бензолсульфоновой кислоты, толуолсульфоновой кислоты или другой подобной кислоты.
Кроме того, настоящее изобретение относится к фармацевтической композиции для лечения рака легкого, включающей соединение, представленное формулой 1, или его фармацевтически приемлемую соль в качестве активного ингредиента и фармацевтически приемлемый носитель.
В частности, фармацевтическая композиция может быть эффективно применена для лечения ALK-мутированного и рецептор эпидермальный фактор роста (EGFR)-мутированного рака легкого.
Фармацевтическая композиция по настоящему изобретению может быть применена для лечения как немелкоклеточного рака легкого, так и мелкоклеточного рака легкого.
Соединение, представленное формулой 1, по настоящему изобретению может быть применено в форме фармацевтически приемлемой соли, образованной неорганической кислотой или органической кислотой, и в этом случае, предпочтительные соли включают хлористоводородной кислоты, бромистоводородной кислоты, серной кислоты, фосфорной кислоты, азотной кислоты, уксусной кислоты, гликолевой кислоты, молочной кислоты, пировиноградной кислоты, малоновой кислоты, янтарной кислоты, глутаровой кислоты, фумаровой кислоты, яблочной кислоты, миндальной кислоты, винной кислоты, лимонной кислоты, аскорбиновой кислоты, пальмитиновой кислоты, малеиновой кислоты, бензойной кислоты, гидроксибензойной кислоты, фенилуксусной кислоты, коричной кислоты, салициловой кислоты, метансульфоновой кислоты, этансульфоновой кислоты, бензолсульфоновой кислоты, толуолсульфоновой кислоты или другой подобной кислоты.
Фармацевтическая композиция по настоящему изобретению может быть приготовлена традиционными методами, и может быть приготовлена в различных пероральных формах, таких как таблетки, пилюли, порошки, капсулы, сиропы, эмульсии, микроэмульсии и другие подобные пероральные формы, или в парентеральных формах, таких как препараты для внутривенной инфузии, подкожной инфузии, внутримышечной инфузии, интраперитонеальной инфузии, трансдермальной инфузии и инфузии непосредственно в ткань.
В случае приготовления фармацевтической композиции по настоящему изобретению в форме перорального препарата, в качестве фармацевтически приемлемого носителя могут быть использованы известные ингредиенты, на которые не накладывают конкретных ограничений, при условии, что они не оказывают отрицательного влияния на действие активного ингредиента.
Носитель может включать, но этим не ограничивая, например, вспомогательные вещества, разбавители, разрыхлители, связующие вещества, скользящие вещества, поверхностно-активные вещества, эмульгаторы, суспендирующие средства и другие подобные средства.
В случае приготовления фармацевтической композиции по настоящему изобретению в форме инъекции, в качестве фармацевтически приемлемого носителя могут быть использованы известные ингредиенты, на которые не накладывают конкретных ограничений, при условии, что они не оказывают отрицательного влияния на действие активного ингредиента.
В частности, носитель может включать, но этим не ограничивая, например, воду, физиологический раствор, водный раствор глюкозы, водный раствор псевдосахара, спирт, гликоль, простой эфир (например, полиэтиленгликоль 400), масло, жирную кислоту, сложный эфир жирной кислоты, глицерид, поверхностно-активное вещество, суспендирующее средство, эмульгатор и другие подобные вещества.
Предпочтительно, когда дозирование фармацевтической композиции по настоящему изобретению определяют с учетом возраста, пола и состояния пациента, степени абсорбции активного ингредиента в организме, степени инактивации и использования в комбинации лекарственного средства, и доза может составлять от 0,0001 мг/кг (массы тела) до 100 мг/кг (массы тела) в единицу времени в расчете на соединение, представленное формулой 1. Целесообразно, если число введений составляет приблизительно от 1 до 3 раз в сутки.
Кроме того, настоящее изобретение относится к соединению, представленному формулой 1, или его фармацевтически приемлемой соли, которое применяют для лечения рака легкого.
Кроме того, настоящее изобретение относится к способу лечения животного, страдающего от рака легкого, включающему введение эффективного количества соединения, представленного формулой 1 выше, или его фармацевтически приемлемой соли животному.
Животное может представлять собой человека, а рак легкого может представлять собой рак легкого, имеющий ALK-мутированные или EGFR-мутированные раковые клетки.
Вариант осуществления изобретения
Далее настоящее изобретение будет описано более подробно с помощью примеров. Для специалиста в данной области является очевидным, что эти примеры приведены только для более подробной иллюстрации настоящего изобретения, и они никоим образом не ограничивают объем настоящего изобретения в соответствии с сущностью настоящего изобретения.
Соединение, представленное формулой 1, по настоящему изобретению может быть получено методом, проиллюстрированным на следующей схеме, но который не является ограничением.
Схема
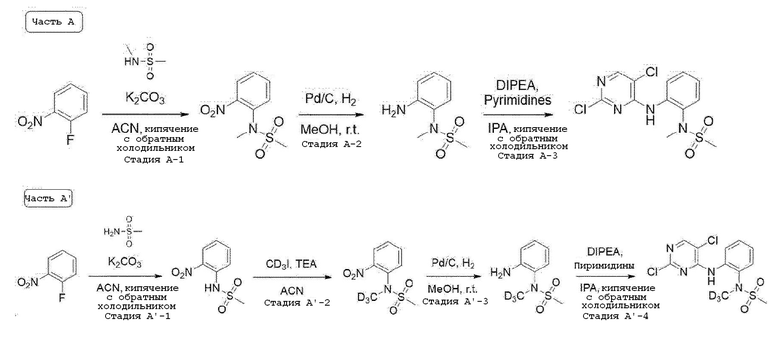
Стадия A-1. Синтез N-метил-N-(2-нитрофенил)метансульфон-амида
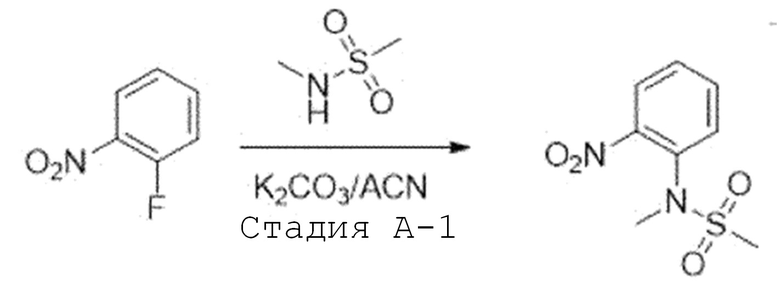
1-Фтор-2-нитробензол (1,0 экв) растворяют в ацетонитриле и добавляют при комнатной температуре карбонат калия (2,0 экв) и N-метилметансульфонамид (1,4 экв). Затем перемешивают при 80°C в течение ночи. После завершения реакции, температуру понижают до комнатной температуры и проводят фильтрацию. Фильтрат испаряют при пониженном давлении с получением соединения, которое используют в следующей реакции без проведения какого-либо процесса разделения.
Стадия A-2. Синтез N-(2-аминофенил)-N-метилметансульфонамида
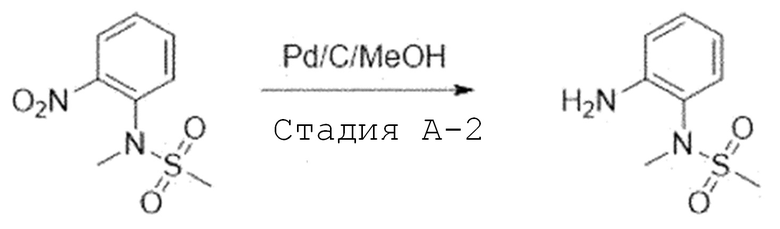
N-метил-N-(2-нитрофенил)метансульфонамид (1,0 экв) растворяют в смешанном растворе метаноле и этилацетата (1:1) и добавляют 10% палладия на угле (0,2 экв). Затем перемешивают в течение 2 часов в атмосфере водорода. После завершения реакции, реакционную смесь фильтруют через целит. Фильтрат испаряют при пониженном давлении и отверждают, используя этиловый эфир и пентан. Затем фильтруют с получением целевого соединения, которое используют в следующей реакции без проведения какого-либо процесса разделения.
Стадия A-3. Синтез N-(2-((2,5-дихлорпиримидин-4-ил)амино)-фенил)-N-метилметансульфонамида
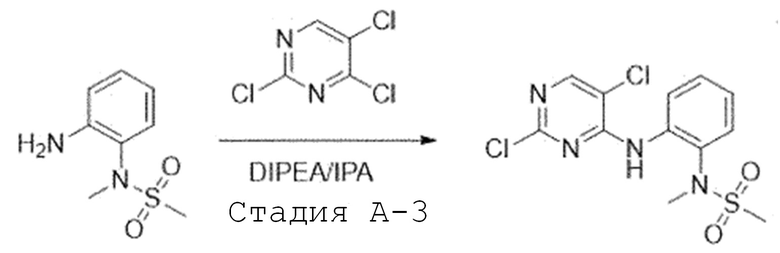
N-(2-аминофенил)-N-метилметансульфонамид (1,0 экв) растворяют в изопропиловом спирте и добавляют при комнатной температуре 2,4,5-трихлорпиримидин (1,1 экв) и N,N-диизопропил-этиламин (2,5 экв). Перемешивают при 80°C в течение ночи. После завершения реакции, испаряют при пониженном давлении и экстрагируют, используя воду и дихлорметан. Органический слой промывают 2N раствором хлористоводородной кислоты. Органический слой испаряют при пониженном давлении с получением целевого соединения, которое используют в следующей реакции без проведения какого-либо процесса разделения.
Стадия A'-1. Синтез N-(2-нитрофенил)метансульфонамида
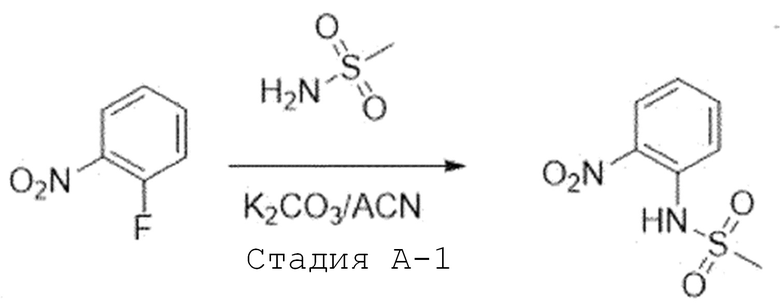
1-Фтор-2-нитробензол (1,0 экв) растворяют в ацетонитриле и добавляют при комнатной температуре карбонат калия (2,0 экв) и метансульфонамид (1,4 экв). Затем перемешивают при 80°C в течение ночи. После завершения реакции, температуру понижают до комнатной температуры и проводят фильтрацию. Фильтрат испаряют при пониженном давлении с получением целевого соединения, которое используют в следующей реакции без проведения какого-либо процесса разделения.
Стадия A'-2. Синтез N-(метил-d3)-N-(2-нитрофенил)метан-сульфонамида
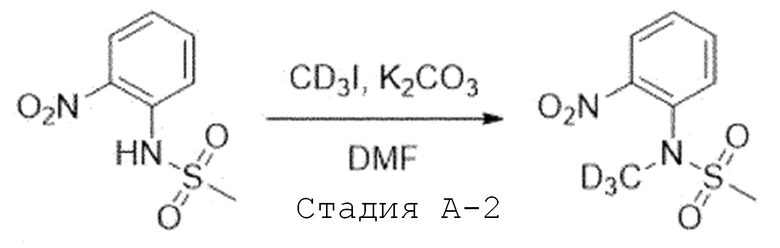
N-(2-нитрофенил)метансульфонамид (1,0 экв) растворяют в N,N-диметилформамиде и добавляют при комнатной температуре карбонат калия (1,2 экв) и йодметан-d3 (1,1 экв). Затем перемешивают при 70°C в течение 2 часов, реакцию завершают, и температуру понижают до комнатной температуры, и добавляют воду. Полученное твердое вещество фильтруют, промывают достаточным количеством воды и используют в следующей реакции без проведения какого-либо процесса разделения.
Стадия A'-3. Синтез N-(2-аминофенил)-N-(метил-d3)метан-сульфонамида
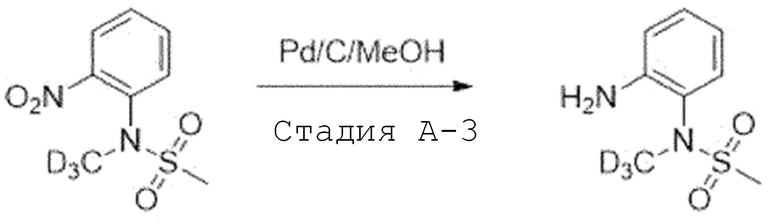
N-метил-N-(2-нитрофенил)метансульфонамид (1,0 экв) растворяют в метаноле и этилацетате (1:1) и добавляют 10% палладий на угле (0,2 экв). Смесь перемешивают в течение 2 часов в атмосфере водорода. После завершения реакции, реакционную смесь фильтруют через целит. Фильтрат испаряют при пониженном давлении. Затем отверждают, используя этиловый эфир и пентан, фильтруют с получением целевого соединения, которое используют в следующей реакции без проведения какого-либо процесса разделения.
Стадия A'-4. Синтез N-(2-((2,5-дихлорпиримидин-4-ил)амино)-фенил)-N-(метил-d3)метансульфонамида
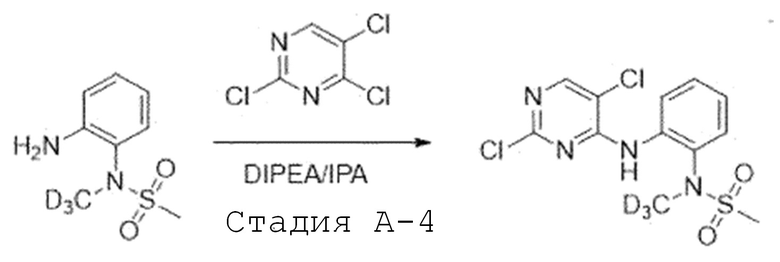
N-(2-аминофенил)-N-метилметансульфонамид (1,0 экв) растворяют в изопропиловом спирте и добавляют при комнатной температуре 2,4,5-трихлорпиримидин (1,1 экв) и N,N-диизопропилэтиламин (2,5 экв). Затем перемешивают при 80°C в течение ночи. После завершения реакции, реакционную смесь испаряют при пониженном давлении и экстрагируют, используя воду и дихлорметан. Органический слой промывают 2 N раствором хлористоводородной кислоты, и органический слой испаряют при пониженном давлении с получением целевого соединения. Его используют в следующей реакции без проведения какого-либо процесса разделения.
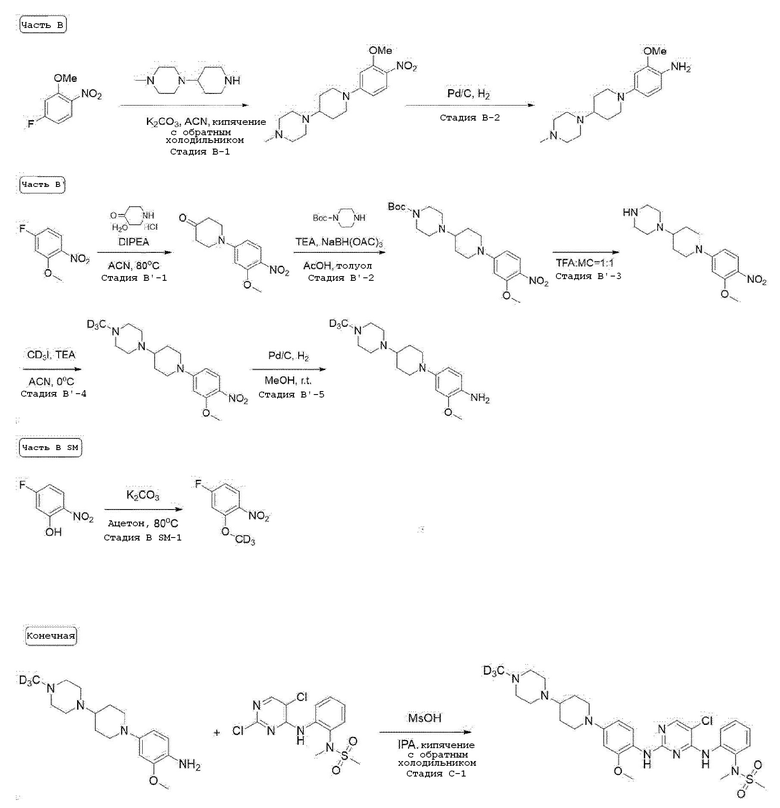
Стадия B-1. Синтез 1-(1-(3-метокси-4-нитрофенил)пиперидин-4-ил)-4-метилпиперазина
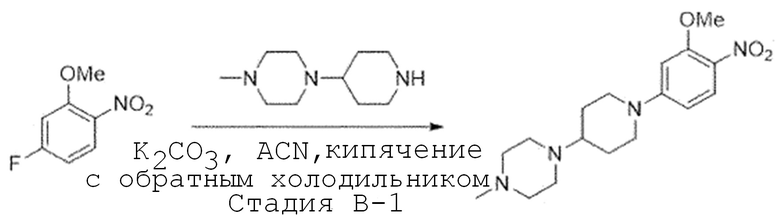
4-Фтор-2-метокси-1-нитробензол (1,0 экв) растворяют в ацетонитриле и добавляют при комнатной температуре карбонат калия (2,5 экв) и производное пиперазина (1,1 экв). Затем перемешивают в течение ночи при кипячении с обратным холодильником. После завершения реакции, температуру понижают до комнатной температуры и проводят фильтрацию. Фильтрат испаряют при пониженном давлении с получением целевого соединения, которое используют в следующей реакции без проведения какого-либо процесса разделения.
Стадия B-2. Синтез 2-метокси-4-(4-(4-метилпиперазин-1-ил)-пиперидин-1-ил)анилина
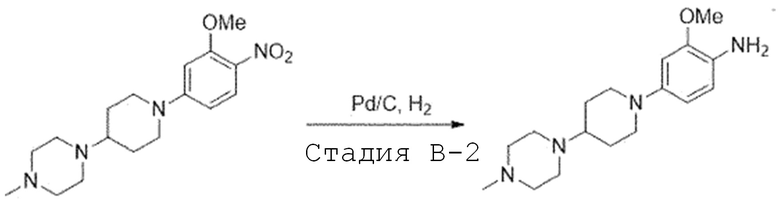
1-(1-(3-Метокси-4-нитрофенил)пиперидин-4-ил)-4-метил-пиперазин (1,0 экв) растворяют в смешанном растворителе метилового спирта и дихлорметана (1:1) и добавляют 10% палладий на угле (0,2 экв). Затем перемешивают в течение 2 часов в атмосфере водорода. После завершения реакции, реакционную смесь фильтруют через целит, и фильтрат испаряют при пониженном давлении. Отверждают, используя гексан, и полученное твердое вещество используют в следующей реакции без проведения какого-либо процесса разделения.
Стадия B'-1. Синтез 1-(3-метокси-4-нитрофенил)пиперидин-4-она
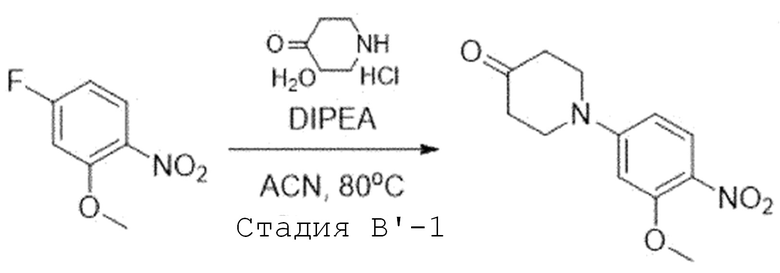
4-Фтор-2-метокси-1-нитробензол (1,0 экв) растворяют в ацетонитриле. Добавляют при комнатной температуре N,N-диизопропилэтиламин (3,0 экв) и гидрохлорид моногидрата 4-пиперидона (1,2 экв). Затем перемешивают при 80°C в течение ночи. После завершения реакции, растворитель испаряют при пониженном давлении и затем проводят экстракцию водой и дихлорметаном. Органический слой собирают и испаряют при пониженном давлении с получением целевого соединения. Его используют в следующей реакции без проведения какого-либо процесса разделения.
Стадия B'-2. Синтез третбутил 4-(1-(3-метокси-4-нитрофенил)пиперидин-4-ил)пиперазин-1-карбоксилата
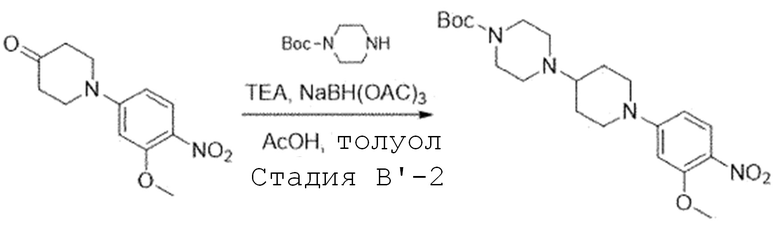
1-(3-Метокси-4-нитрофенил)пиперидин-4-он (1,0 экв) растворяют в толуоле и добавляют при комнатной температуре третбутил пиперазин-1-карбоксилат (1,97 экв), триэтиламин (2,58 экв) и уксусную кислоту (1,53 экв). Затем перемешивают при комнатной температуре в течение 30 минут и добавляют триацетоксиборгидрид натрия (0,83 экв). Этот процесс повторяют еще два раза. После завершения добавления, смесь перемешивают при комнатной температуре в течение ночи. После завершения реакции, реакционную смесь экстрагируют водой и этилацетатом, и органический слой собирают. Органический слой испаряют при пониженном давлении с получением названного соединения. Его используют в следующей реакции без проведения какого-либо процесса разделения.
Стадия B'-3. Синтез 1-(1-(3-метокси-4-нитрофенил)пиперидин-4-ил)пиперазина
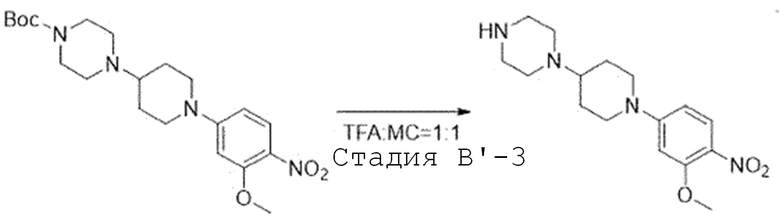
Третбутил 4-(1-(3-метокси-4-нитрофенил)пиперидин-4-ил)-пиперазин-1-карбоксилат (1,0 экв) растворяют в смешанном растворителе трифторуксусной кислоты и дихлорметана (1:1) и перемешивают при комнатной температуре. После завершения реакции, реакционную смесь испаряют при пониженном давлении. Затем проводят экстракцию 2 N раствором гидроксида калия и дихлорметаном, и органический слой собирают. Органический слой испаряют при пониженном давлении с получением названного соединения. Его используют в следующей реакции без проведения какого-либо процесса разделения.
Стадия B'-4. Синтез 1-(1-(3-метокси-4-нитрофенил)пиперидин-4-ил)-4-(метил-d3)пиперазина
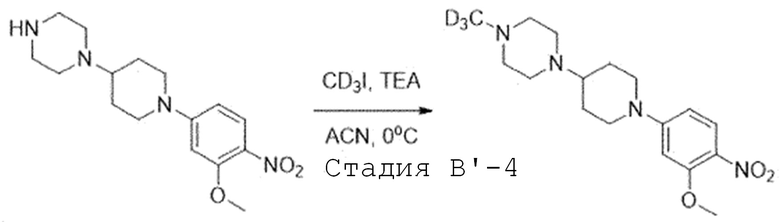
1-(1-(3-Метокси-4-нитрофенил)пиперидин-4-ил)пиперазин (1,0 экв) растворяют в ацетонитриле и добавляют при 0°C триэтиламин (1,2 экв) и йодметан-d3 (1,1 экв). Затем перемешивают при этой же температуре. После завершения реакции, проводят экстракцию водой и этилацетатом, и органический слой собирают. Органический слой испаряют при пониженном давлении, и получают целевое соединение, используя колоночную хроматографию (10% метиловый спирт/дихлорметан).
Стадия B'-5. Синтез 2-метокси-4-(4-(4-(метил-d3)пиперазин-1-ил)пиперидин-1-ил)анилина
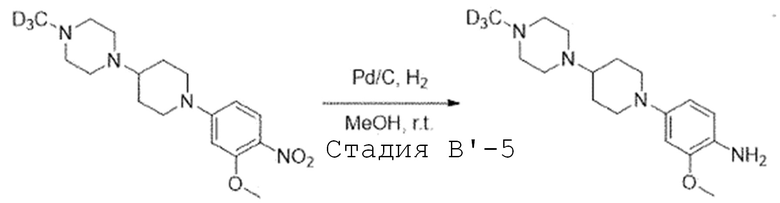
1-(1-(3-Метокси-4-нитрофенил)пиперидин-4-ил)-4-(метил-d3)пиперазин (1,0 экв) растворяют в смешанном растворителе метилового спирта и этилацетата (1:1) и добавляют 10% палладий на угле (0,2 экв). Затем перемешивают в течение 2 часов в атмосфере водорода. После завершения реакции, реакционную смесь фильтруют через целит. Фильтрат испаряют при пониженном давлении. Затем отверждают, используя гексан, и полученное твердое вещество используют в следующей реакции без проведения какого-либо процесса разделения.
Стадия B SM-1. 4-Фтор-2-(метокси-d3)-1-нитробензол
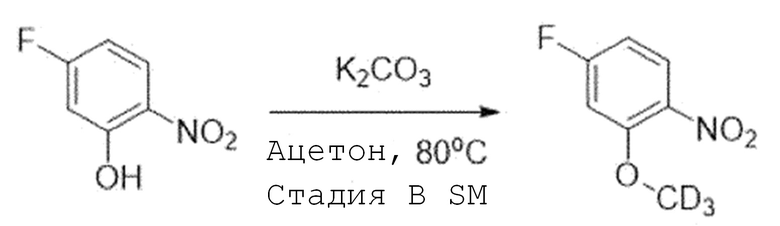
1-(1-(3-Метокси-4-нитрофенил)пиперидин-4-ил)пиперазин (1,0 экв) растворяют в ацетонитриле и добавляют при комнатной температуре карбонат калия (2,0 экв) и йодметан-d3 (1,3 экв). Затем перемешивают при 60°C в течение 2 часов. После завершения реакции, реакционную смесь испаряют при пониженном давлении и экстрагируют водой и этилацетатом, и органический слой собирают. Органический слой испаряют при пониженном давлении, и целевое соединение получают, используя колоночную хроматографию (25% этилацетат/н-гексан).
Стадия C-1. Синтез конечного соединения
Производное пиримидина (1,0 экв) растворяют в изопропиловом спирте и добавляют при комнатной температуре производное анилина (1,0 экв) и метансульфониловую кислоту (1,3 экв). Затем перемешивают при 80°C в течение ночи. После завершения реакции, реакционную смесь испаряют при пониженном давлении с удалением растворителя растворитель и экстрагируют водой и смешанным растворителем 10% метанол/дихлорметан. Органический слой испаряют при пониженном давлении, и целевое соединение получают, используя колоночную хроматографию (10% метиловый спирт/дихлорметан).
Пример 1. N-(2-((5-Хлор-2-((2-метокси-4-(4-(4-(метил-d3)пиперазин-1-ил)пиперидин-1-ил)фенил)амино)пиримидин-4-ил)-амино)фенил)-N-метилметансульфонамид (соединение 1)
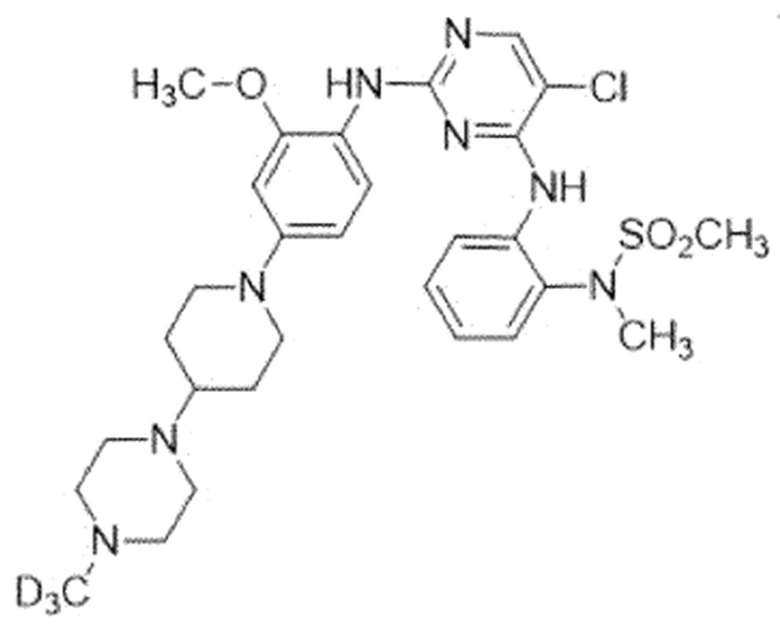
Для синтеза соединения использовали стадию A, стадию B' и стадию C.
Выход: 22,5%, белое твердое вещество,
1H ЯМР (400 МГц, DMSO-d6) δ 8,23(м, 2H), 8,07-8,06(м, 2H), 7,54(дд, J=7,9, 1,6 Гц, 1H), 7,32(д, J=8,6 Гц, 1H), 7,21(т, J=7,8 Гц, 1H), 7,12(тд, J=7,6, 1,5 Гц, 1H), 6,57(д, J=2,5 Гц, 1H), 6,41(дд, J=8,7, 2,6 Гц, 1H), 3,71- 3,66(м, 5H), 3,14(с, 3H), 3,06(с, 3H), 2,62(т, J=11,7 Гц, 3H), 2,52-2,43(м, 4H), 2,29-2,23(м, 5H), 1,85-1,79(м, 2H), 1,54-1,41(м, 2H). MS: ESI m/z 618,20 [M+H]+
Пример 2. N-(2-((5-Хлор-2-((2-(метокси-4-(4-(4-метил-пиперазин-1-ил)пиперидин-1-ил)фенил)амино)пиримидин-4-ил)амино)-фенил)-N-(метил-d3)метансульфонамид (соединение 2)
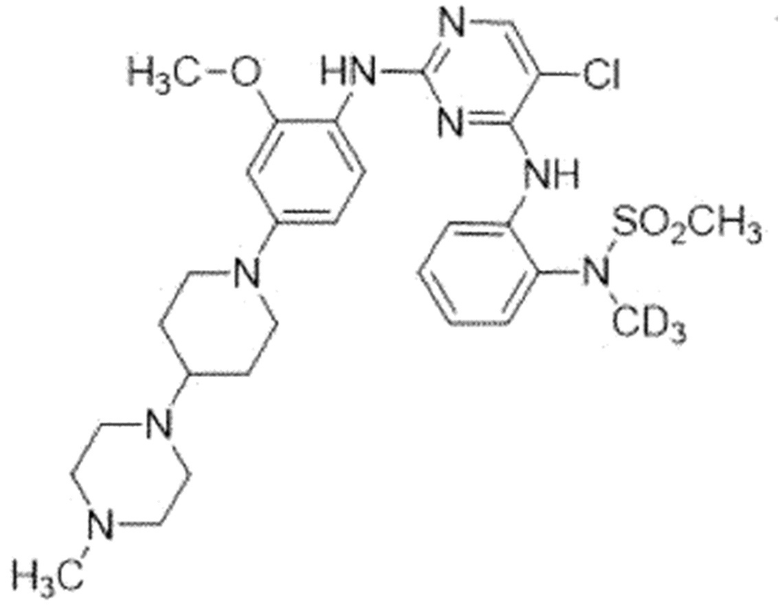
Для синтеза соединения использовали стадию A', стадию B и стадию C.
Выход: 25,5%, белое твердое вещество,
1H ЯМР(400 МГц, DMSO-d6) δ 8,24-8,22(м, 2H), 8,06(с, 2H), 7,54(дд, J=7,9, 1,6 Гц, 1H), 7,32(д, J=8,6 Гц, 1H), 7,21(т, J=7,7 Гц, 1H), 7,12(тд, J=7,6, 1,5 Гц, 1H), 6,58(д, J=2,5 Гц, 1H), 6,41(дд, J=8,8, 2,6 Гц, 1H), 3,71-3,66(м, 5H), 3,06(с, 3H), 2,65-2,60(м, 2H), 2,53-2,43(м, 4H), 2,29-2,24(м, 5H), 2,10(с, 3H), 1,86-1,79(м, 2H), 1,53-1,43(м, 2H). MS: ESI m/z 618,20 [M+H]+
Пример 3. N-(2-((5-Хлор-2-((2-(метокси-d3)-4-(4-(4-метил-пиперазин-1-ил)пиперидин-1-ил)фенил)амино)пиримидин-4-ил)амино)-фенил)-N-метилметансульфонамид (Соединение 3)
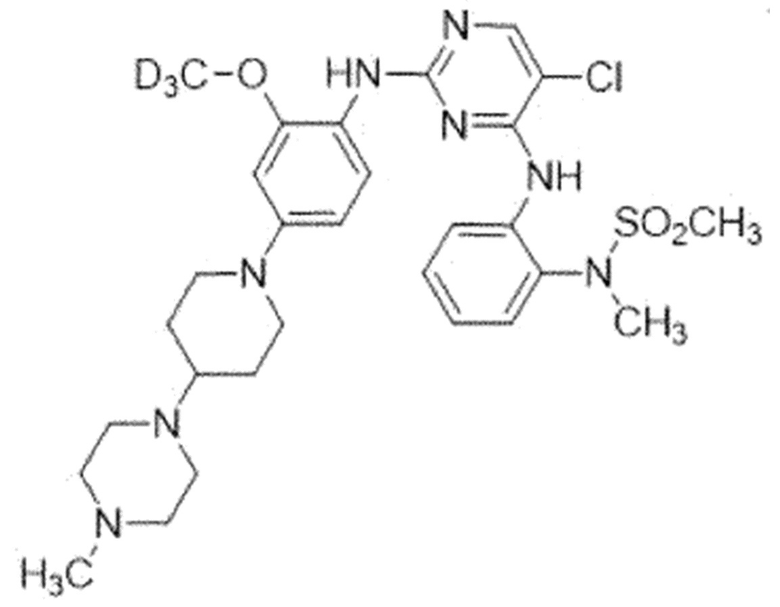
Для синтеза соединения использовали стадию A', стадию B SM, стадию B и стадию C.
Выход: 40,3%, белое твердое вещество,
1H ЯМР(400 МГц, DMSO-d6) δ 8,23(м, 2H), 8,07-8,06(м, 2H), 7,54(дд, J=7,9, 1,6 Гц, 1H), 7,32(д, J=8,6 Гц, 1H), 7,21(т, J=7,8 Гц, 1H), 7,12(тд, J=7,6, 1,5 Гц, 1H), 6,57(д, J=2,5 Гц, 1H), 6,41(дд, J=8,7, 2,6 Гц, 1H), 3,70- 3,66(м, 2H), 3,14(с, 3H), 3,06(с, 3H), 2,62(т, J=11,7 Гц, 3H), 2,52-2,43(м, 4H), 2,29-2,23(м, 5H), 2,10(с, 3H), 1,85-1,79(м, 2H), 1,54-1,41(м, 2H). MS: ESI m/z 618,20 [M+H]+
Пример 4. N-(2-((5-Хлор-2-((2-метокси-4-(4-(4-(метил-d3)-пиперазин-1-ил)пиперидин-1-ил)фенил)амино)пиримидин-4-ил)амино)-фенил)-N-(метил-d3)метансульфонамид
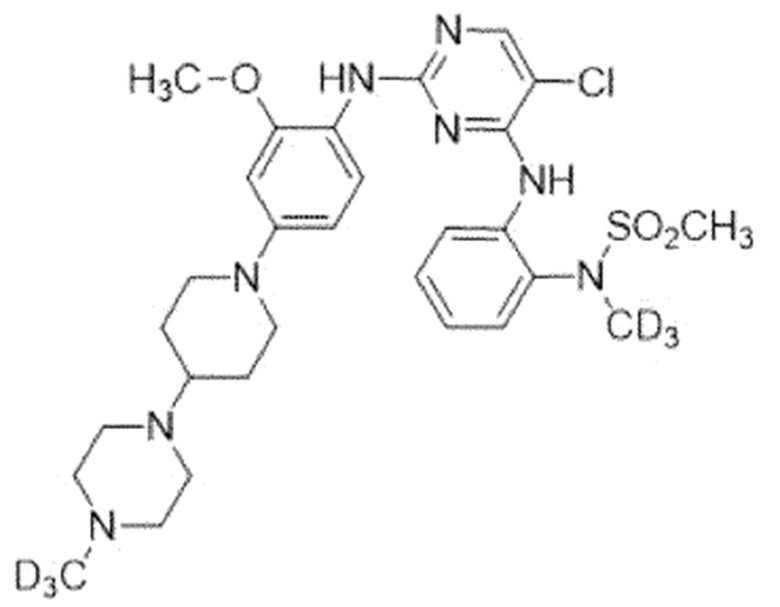
Для синтеза соединения использовали стадию A', стадию B' и стадию C.
Выход: 20,2%, белое твердое вещество,
1H ЯМР(400 МГц, DMSO-d6) δ 8,26-8,23(м, 2H), 8,08-8,06(м, 2H), 7,54(дд, J=7,9, 1,6 Гц, 1H), 7,32(д, J=8,6 Гц, 1H), 7,21(т, J=7,7 Гц, 1H), 7,12(тд, J=7,6, 1,5 Гц, 1H), 6,58(д, J=2,5 Гц, 1H), 6,41(дд, J=8,8, 2,6 Гц, 1H), 3,71-3,66(м, 5H), 3,06(с, 3H), 2,66-2,59(м, 2H), 2,53-2,43(м, 4H), 2,37-2,25(м, 5H), 1,87-1,80(м, 2H), 1,52-1,44(м, 2H). MS: ESI m/z 621,20 [M+H]+
Пример 5. N-(2-((5-Хлор-2-((2-(метокси-d3)-4-(4-(4-метил-пиперазин-1-ил)пиперидин-1-ил)фенил)амино)пиримидин-4-ил)амино)-фенил)-N-(метил-d3)метансульфонамид
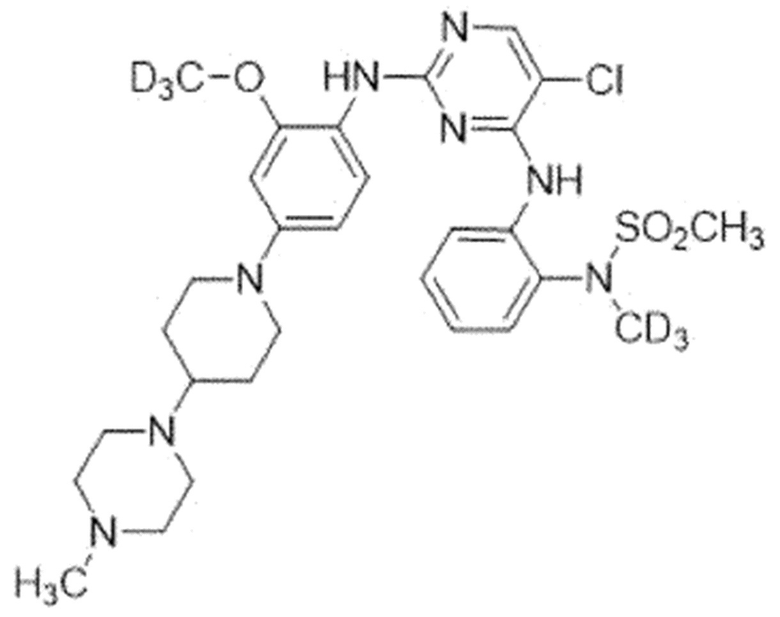
Для синтеза соединения использовали стадию A', стадию B SM, стадию B и стадию C.
Выход: 38,9%, белое твердое вещество,
1H ЯМР(400 МГц, DMSO-d6) δ 8,26-8,23(м, 2H), 8,08-8,06(м, 2H), 7,54(дд, J=7,9, 1,6 Гц, 1H), 7,32(д, J=8,6 Гц, 1H), 7,21(т, J=7,7 Гц, 1H), 7,12(тд, J=7,6, 1,5 Гц, 1H), 6,58(д, J=2,5 Гц, 1H), 6,41(дд, J=8,8, 2,6 Гц, 1H), 3,70-3,66(м, 2H), 3,06(с, 3H), 2,66-2,59(м, 2H), 2,53-2,43(м, 4H), 2,37-2,25(м, 5H), 2,10(с, 3H), 1,87-1,80(м, 2H), 1,52-1,44(м, 2H). MS: ESI m/z 621,20 [M+H]+
Пример 6. N-(2-((5-Хлор-2-((2-метокси-4-(4-(4-(метил-d3)-пиперазин-1-ил)пиперидин-1-ил)фенил)амино)пиримидин-4-ил)амино)-фенил)-N-(метил-d3)метансульфонамид
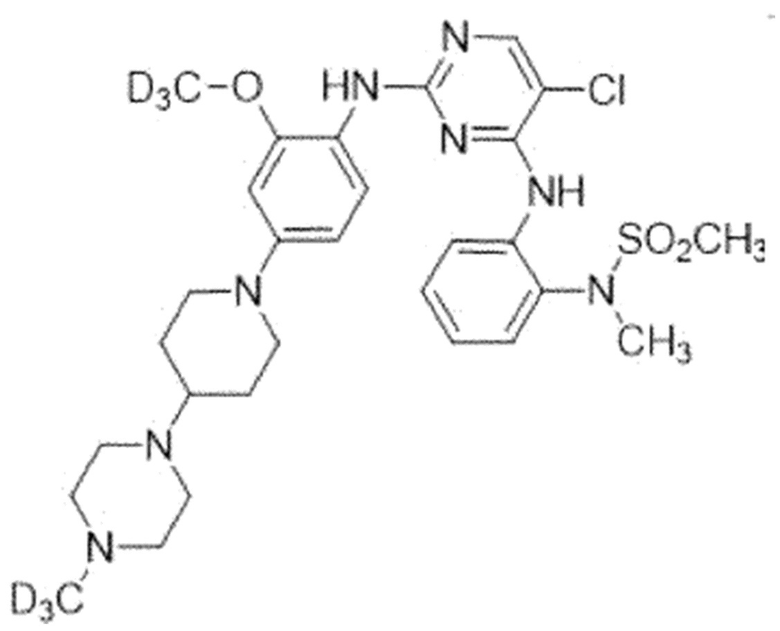
Для синтеза соединения использовали стадию A, стадию B SM, стадию B' и стадию C.
Выход: 41,3%, белое твердое вещество,
1H ЯМР(400 МГц, DMSO-d6) δ 8,26-8,23(м, 2H), 8,08-8,06(м, 2H), 7,54(дд, J=7,9, 1,6 Гц, 1H), 7,32(д, J=8,6 Гц, 1H), 7,21(т, J=7,7 Гц, 1H), 7,12(тд, J=7,6, 1,5 Гц, 1H), 6,58(д, J=2,5 Гц, 1H), 6,41(дд, J=8,8, 2,6 Гц, 1H), 3,70-3,66(м, 2H), 3,14(с, 3H), 3,06(с, 3H), 2,66-2,59(м, 2H), 2,53-2,43(м, 4H), 2,37-2,25(м, 5H), 1,87-1,80(м, 2H), 1,52-1,44(м, 2H). MS: ESI m/z 621,20 [M+H]+
Пример 7. N-(2-((5-Хлор-2-((2-(метокси-d3)-4-(4-(4-(метил-d3)пиперазин-1-ил)пиперидин-1-ил)фенил)амино)пиримидин-4-ил)-амино)фенил)-N-(метил-d3)метансульфонамид (соединение 7)
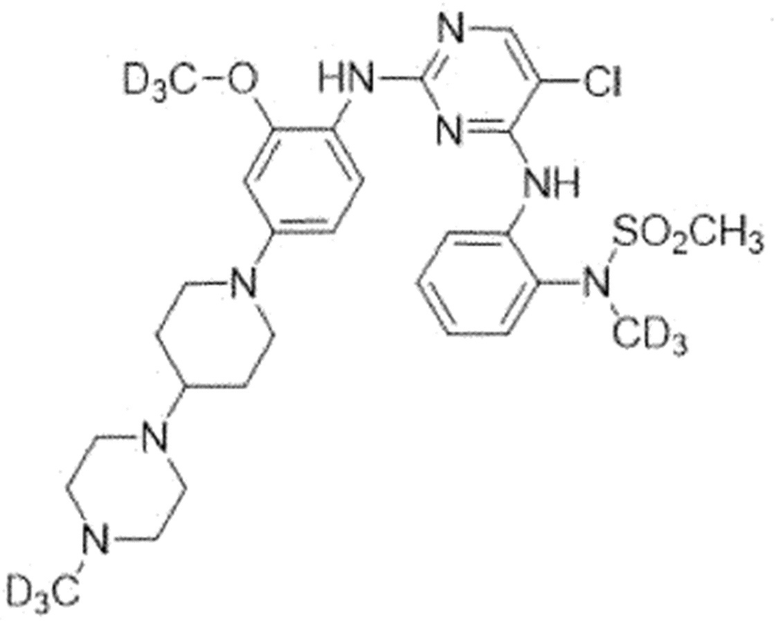
Для синтеза соединения использовали стадию A', стадию B SM, стадию B' и стадию C.
Выход: 38,1%, белое твердое вещество,
1H ЯМР(400 МГц, DMSO-d6) δ 8,26-8,23(м, 2H), 8,08-8,06(м, 2H), 7,54(дд, J=7,9, 1,6 Гц, 1H), 7,32(д, J=8,6 Гц, 1H), 7,21(т, J=7,7 Гц, 1H), 7,12(тд, J=7,6, 1,5 Гц, 1H), 6,58(д, J=2,5 Гц, 1H), 6,41(дд, J=8,8, 2,6 Гц, 1H), 3,70-3,66(м, 2H), 3,06(с, 3H), 2,66-2,59(м, 2H), 2,53-2,43(м, 4H), 2,37-2,25(м, 5H), 1,87-1,80(м, 2H), 1,52-1,44(м, 2H). MS: ESI m/z 624,30 [M+H]+
Пример 8. Синтез N-(2-((5-хлор-2-((2-метокси-4-(4-(4-метил-пиперазин-1-ил)пиперидин-1-ил)фенил)амино)пиримидин-4-ил)амино)-фенил)-N-(метил-d3)метансульфонамида метансульфоната
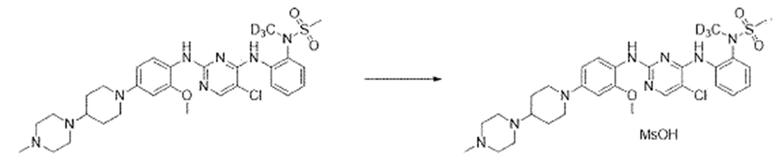
N-(2-((5-Хлор-2-((2-метокси-4-(4-(4-метилпиперазин-1-ил)-пиперидин-1-ил)фенил)амино)пиримидин-4-ил)амино)фенил)-N-(метил-d3)метансульфонамид (соединение, полученное в примере 2, 24,0 мг, 0,039 ммоль) растворяли в этиловом спирте (1 мл) и добавляли при комнатной температуре метансульфоновую кислоту (3,8 мг, 0,039 ммоль). Затем перемешивали до образования твердого вещества. После образования твердого вещества добавляли гептан (1 мл) и перемешивали в течение 2 часов. После завершения перемешивания, проводили фильтрацию, и осадок на фильтре промывали гептаном. Отфильтрованное твердое вещество сушили при 70°C с получением целевого соединения.
Выход: 97,4%; светло-серый порошок;
1H ЯМР (400 МГц, DMSO-d6) δ 8,35 (с, 1H), 8,20 (м, 2H), 8,08 (с, 1H), 7,56 (дд, J=7,9, 1,5 Гц, 1H), 7,36 (с, 1H), 7,24 (м, 1H), 7,15 (т, J=7,6 Гц, 1H), 6,63 (с, 1H), 6,47 (с, 1H), 3,83-3,65 (м, 5H), 3,10-2,89 (с, 7H), 2,84-2,63 (с, 6H), 2,31-2,27 (м, 1H), 2,27 (с, 3H), 1,91 (д, J=32,1 Гц, 2H), 1,58 (с, 2H), 1,31-1,07 (м, 2H). MS: ESI m/z 714,05 [M+H]+
Пример испытания 1. Измерение ингибирующего действия на рост раковых клеток
Измеряли ингибирующее действие на рецептор эпидермального фактора роста (EGFR) киназы, содержащей C797S, в случае использования соединений, полученных в примерах 1 и 2 выше, соединения, в котором дейтерий замещен на водород в соединении примера 2 (замещенного водородом референсного соединения), и контрольного лекарственного средства третьего поколения осимертиниба, и результаты представлены в таблице 1 ниже. Метод измерения ингибирующего действия в отношении киназы заключался в следующем.
1. Каждую киназу инкубировали в среде 8 мМ MOPS (pH 7,0), 0,2 мМ EDTA, 250 мкМ KKKGQEEEYVFIE, 1 мМ ортованадата натрия, 5 мМ натрий-6-глицерофосфата, 10 мМ ацетата магния и [η-33P]-ATP.
2. Для проведения реакции добавляли исследуемое соединение (раствор в DMSO) и Mg/ATP.
3. Приблизительно через 40 минут добавляли при комнатной температуре 10 мкл 0,5% фосфорной кислоты для завершения реакции.
4. 0,5% реакционный раствор разделяли на порции по 10 мкл и наносили в виде капли на фильтр P30 Filtermat.
5. Промывали 4 раза с помощью 0,425% раствора фосфорной кислоты в течение приблизительно 4 минут.
6. Промывали один раз метанолом и затем сушили и проводили измерение активности сцинтилляционным методом для определения величины IC50.
7. Для каждого соединения рассчитывали величину GI50, которая представляет собой концентрацию, при которой происходит 50% ингибирование клеточного роста, и результаты представлены в форме условных обозначений A, B, C и D в таблице 1 ниже.
Условные обозначения оценочных критериев
A: GI50 ≤ 500 нМ, B: 500 нМ<GI50 ≤ 1,000 нМ, C: GI50>1,000 нМ
Как показано в таблице 1 выше, соединения 1 и 2, полученные в примерах 1 и 2, по настоящему изобретению проявляли высокую активность в отношении линий раковых клеток, экспрессирующих C797S мутантный рецептор эпидермального фактора роста. С другой стороны, осимертиниб, который представляет собой противораковое средство третьего поколения, характеризовался низкой активностью. Таким образом, эти экспериментальные данные подтверждают, что соединения по настоящему изобретению, которые являются новыми производными пиримидина, могут эффективно применяться для лечения C797S-мутированного рака легкого, который проявляет резистентность к терапии с использованием существующих противораковых лекарственных средств третьего поколения.
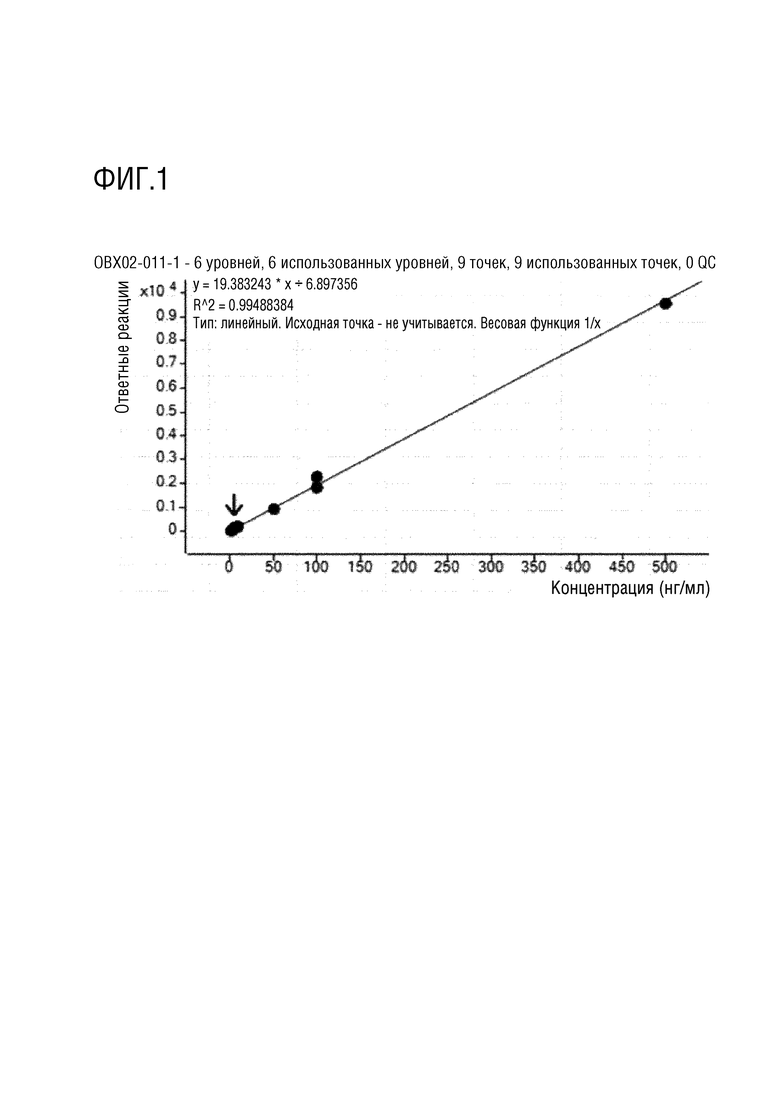
Пример испытания 2. Фармакокинетическое исследование
Фармакокинетические исследования соединений, полученных в примерах 1 и 2 выше, и соединения, в котором дейтерий замещен на водород в соединении примера 2 (замещенного водородом референсного соединения) проводили на крысах следующим образом.
После приготовления испытуемых соединений в форме растворов с концентраций 2,5 или 5 мг/мл в WFI (воде для инъекций), их вводили в форме заданной разовой дозы (10 мл/кг) и собирали кровь в заданный момент времени (0, 0,25, 0,5, 1, 2, 4, 6, 8, 10, 24 часа), и затем отделяли плазму. Анализ лекарственного средства проводили методом высокоэффективной жидкостной хроматографии (HPLC) (колонка XBridge C18, Waters, подвижная фаза 0,1% муравьиная кислота:ацетонитрил (30:70, %/%)) и тандемной масс-спектрометрии (МС/МС) (ESI с положительными ионами, MRM), готовили растворы с концентрациями 5, 50, 100, 500, 1000 и 5000 нг/мл и проводили измерения путем смешивания контрольного раствора плазмы крысы и каждого выпускаемого промышленностью стандартного раствора в соотношении 9:1. Кроме того, готовили препараты образцов для контроля качества в концентрациях 100, 750 и 2500 нг/мл путем смешивания контрольного раствора плазмы крысы и стандартного раствора для контроля качества в соотношении 9:1. Что касается метода предварительной подготовки образцов для анализа, то 100 мкл образца плазмы переносили в центрифужную пробирку, добавляли в нее 10 мкл раствора внутреннего стандарта и 300 мкл метанола, а затем перемешивали в течение примерно 30 секунд. Пробирку центрифугировали в течение приблизительно 5 минут при скорости вращения 12 000 об/мин (4°C) в центрифуге, отбирали надосадочную жидкость, переносили в виалу для жидкостной хроматографии и затем вводили в хроматограф. После чего количественно определяли концентрацию лекарственного средства в плазме крысы ранее верифицированным методом анализа. Для определения фармакокинетических параметров использовали программу WinNonlin 5,2 (Pharsight, США), и рассчитывали величины AUC0-t, AUC0-∞, Cmax, Tmax и t1/2 методом некомпартментного моделирования (методом наилучшего приближения). Полученные значения фармакокинетических параметров выражали в виде среднего значения и стандартного отклонения (SD) и статистически обрабатывали с использованием программы SPSS (Statistical Package for the Social Sciences, 10,0K, USA).
Экспериментальные результаты представлены на фигурах 1-4. Как показано на фигурах 1-4 выше, соединения примеров 1 и 2 характеризовались степенью абсолютной абсорбции, эквивалентной или большей, чем для контрольного лекарственного средства, соединения примера 2, в котором дейтерий замещен водородом (замещенное водородом референсное соединение). В частности, соединение примера 2 характеризовалось улучшенной относительной степенью абсорбции 111,4% по сравнению с замещенным водородом референсным соединением.
Изобретение относится к соединению формулы 1, где X представляет собой C1-C4 алкилсульфонильную группу, каждый Y2 и Y3 независимо представляет собой C1-C4 алкильную группу, не замещенную или замещенную дейтерием, и Y1 представляет собой метильную группу, замещенную одним или более атомами дейтерия, или его фармацевтически приемлемой соли. Также предложена фармацевтическая композиция для лечения рака легкого, включающая указанное соединение. Соединение, представленное формулой 1, эффективно ингибирует рост ALK-мутированных и EGFR-мутированных раковых клеток. 2 н. и 5 з.п. ф-лы, 4 ил., 1 табл., 8 пр.
1. Соединение, представленное следующей формулой 1:
Формула 1
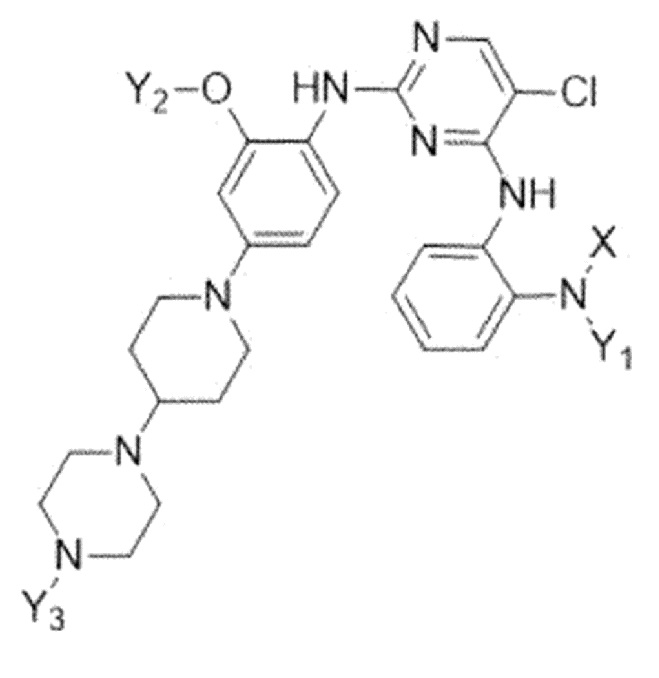
где
X представляет собой C1-C4 алкилсульфонильную группу,
каждый Y2 и Y3 независимо представляет собой C1-C4 алкильную группу, незамещенную или замещенную дейтерием, и
Y1 представляет собой метильную группу, замещенную одним или более атомами дейтерия,
или его фармацевтически приемлемая соль.
2. Соединение, представленное формулой 1 по п. 1, отличающееся тем, что соединение формулы 1 представляет собой любое одно из следующих соединений:
N-(2-((5-хлор-2-((2-(метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)амино)пиримидин-4-ил)амино)фенил)-N-(метил-d3)метансульфонамид (соединение 2);
N-(2-((5-хлор-2-((2-метокси-4-(4-(4-(метил-d3)пиперазин-1-ил)пиперидин-1-ил)фенил)амино)пиримидин-4-ил)амино)фенил)-N-(метил-d3)метансульфонамид (соединение 4);
N-(2-((5-хлор-2-((2-(метокси-d3)-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)амино)пиримидин-4-ил)амино)фенил)-N-(метил-d3)метансульфонамид (соединение 5); и
N-(2-((5-хлор-2-((2-(метокси-d3)-4-(4-(4-(метил-d3)пиперазин-1-ил)пиперидин-1-ил)фенил)амино)пиримидин-4-ил)амино)-фенил)-N-(метил-d3)метансульфонамид (соединение 7),
или его фармацевтически приемлемая соль.
3. Соединение, представленное формулой 1, или его фармацевтически приемлемая соль по п. 1, отличающиеся тем, что соль представляет собой соль, образованную одной или более кислотами, выбранными из группы, состоящей из хлористоводородной кислоты, бромистоводородной кислоты, серной кислоты, фосфорной кислоты, азотной кислоты, уксусной кислоты, гликолевой кислоты, молочной кислоты, пировиноградной кислоты, малоновой кислоты, янтарной кислоты, глутаровой кислоты, фумаровой кислоты, яблочной кислоты, миндальной кислоты, винной кислоты, лимонной кислоты, аскорбиновой кислоты, пальмитиновой кислоты, малеиновой кислоты, бензойной кислоты, гидроксибензойной кислоты, фенилуксусной кислоты, коричной кислоты, салициловой кислоты, метансульфоновой кислоты, этансульфоновой кислоты, бензолсульфоновой кислоты и толуолсульфоновой кислоты.
4. Фармацевтическая композиция для лечения рака легкого, включающая соединение по любому одному из пп. 1-3 или его фармацевтически приемлемую соль в качестве активного ингредиента и фармацевтически приемлемый носитель.
5. Фармацевтическая композиция для лечения рака легкого по п. 4, отличающаяся тем, что фармацевтически приемлемый носитель представляет собой один или более фармацевтически приемлемых носителей, выбранных из группы, состоящей из вспомогательных веществ, разбавителей, разрыхлителей, связующих веществ, смазывающих веществ, поверхностно-активных веществ, эмульгаторов и суспендирующих веществ.
6. Фармацевтическая композиция для лечения рака легкого по п. 4, отличающаяся тем, что фармацевтически приемлемый носитель представляет собой один или более фармацевтически приемлемых носителей, выбранных из группы, состоящей из воды, физиологического раствора, водного раствора глюкозы, водного раствора псевдосахара, спирта, гликоля, эфира, масла, жирной кислоты, эфира жирной кислоты, глицерида, поверхностно-активного вещества, суспендирующего вещества и эмульгатора.
7. Фармацевтическая композиция для лечения рака легкого по п. 4, отличающаяся тем, что рак легкого представляет собой рак легкого, экспрессирующий мутацию ALK и мутацию рецептора эпидермального фактора роста (мутацию EGFR).
| WO 2019190259 A1, 03.10.2019 | |||
| KR 20190067699 A, 17.06.2019 | |||
| WO 2018230934 A1, 20.12.2018 | |||
| US 20170226065 A1, 10.08.2017 | |||
| WO 2009112490 A1, 17.09.2009 | |||
| WO 2016022460 A1, 11.02.2016 | |||
| WO 2009032703 A1, 12.03.2009. |

