УРОВЕНЬ ТЕХНИКИ, ПРЕДШЕСТВУЮЩИЙ ДАННОМУ ИЗОБРЕТЕНИЮ
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Данное изобретение относится к смешанному катализатору и способу получения ненасыщенной кислоты или ненасыщенного нитрила с применением такого смешанного катализатора.
ОПИСАНИЕ ПРЕДШЕСТВУЮЩЕГО УРОВНЯ ТЕХНИКИ
Обычный способ подвергания пропилена или бутилена реакции каталитического окисления в паровой фазе или реакции каталитического аммоксидирования в паровой фазе, чтобы получить соответствующую ненасыщенную карбоновую кислоту или ненасыщенный нитрил, хорошо известен. В последние годы внимание было направлено на способ подвергания пропана или изобутана вместо пропилена или бутилена каталитическому окислению в паровой фазе или каталитическому аммоксидированию в паровой фазе, чтобы получить соответствующую ненасыщенную карбоновую кислоту или ненасыщенный нитрил.
До настоящего времени в качестве катализатора, используемого для каталитического аммоксидирования в паровой фазе, были предложены различные оксидные катализаторы. Хотя оксид, полученный смешиванием и обжигом молибдена и ванадия или т.п., в случае необходимости, обычно используется в качестве катализатора в состоянии «как есть», был также исследован метод дополнительного подвергания обожженного катализатора последующей обработке при производстве ненасыщенной карбоновой кислоты или ненасыщенного нитрила.
Например, выложенная заявка на патент Японии № 10-028862 раскрывает метод импрегнирования катализатора на базе Mo-V-Sb/Te раствором, содержащим один или несколько элементов, выбранных из группы, состоящей из вольфрама, молибдена, хрома, циркония, титана, ниобия, тантала, ванадия, бора, висмута, теллура, палладия, кобальта, никеля, железа, фосфора, кремния, редкоземельных элементов, щелочных металлов и щелочноземельных металлов.
Выложенная заявка на патент Японии № 2008-062231 раскрывает метод приведения смешанного металлооксидного катализатора в соприкосновение с водой и, необязательно, водным предшественником оксида металла, чтобы получить модифицированный смешанный оксид металлов, и обжига полученного модифицированного смешанного оксида металлов.
Выложенная заявка на патент Японии № 2006-077557 раскрывает метод введения вольфрама и марганца в катализатор на базе Mo-V-Sb-Nb методом погружения.
Международная заявка WO 2009-048553 раскрывает метод смешивания катализатора с модификатором, таким как соединение сурьмы, соединение молибдена, соединение теллура и соединение вольфрама, и подвергания полученного катализатора реакционному взаимодействию или смешивания катализатора или предшественника катализатора с модификатором, обжига полученной смеси и подвергания обожженного продукта реакционному взаимодействию.
Однако, когда авторы данного изобретения применяли оксидные катализаторы, раскрытые в выложенной заявке на патент Японии № 10-028862, выложенной заявке на патент Японии № 2008-062231 и выложенной заявке на патент Японии № 2006-077557, для каталитического окисления в паровой фазе или реакции каталитического аммоксидирования в паровой фазе пропана или изобутана, все катализаторы давали недостаточный выход целевого продукта.
Способы получения, описанные в выложенной заявке на патент Японии № 10-028862, выложенной заявке на патент Японии № 2008-062231, выложенной заявке на патент Японии № 2006-077557 и международной заявке WO 2009-048553, описывают улучшение эксплуатационных качеств сложного оксида на базе Mo-V-Te/Sb, уже обладающего активностью в состоянии «как есть», посредством импрегнирования сложного оксида вольфрама или введения вольфрама в сложный оксид методом погружения. Однако сложный оксид перед импрегнированием или погружением не содержит вольфрама, и катализатор, обеспечивающий высокий выход целевого продукта, еще не был получен.
Ввиду вышеуказанной ситуации, целью данного изобретения является предоставление смешанного катализатора для каталитического окисления в паровой фазе или реакции каталитического аммоксидирования в паровой фазе пропана или изобутана, обеспечивающего возможность получения соответствующей ненасыщенной кислоты или соответствующего ненасыщенного нитрила при высоком выходе из пропана или изобутана. Другой целью данного изобретения является предоставление способа производства ненасыщенной кислоты или ненасыщенного нитрила с применением такого смешанного катализатора.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Авторы данного изобретения провели интенсивные исследования для того, чтобы решить вышеуказанные проблемы. В результате, авторы данного изобретения обнаружили, что, когда сложный оксид на базе Mo-V-Nb-W и соединение вольфрама смешиваются, функции вольфрама, содержащегося в сложном оксиде перед смешиванием, и соединения вольфрама в смешанном катализаторе отличаются одна от другой. В результате авторы данного изобретения нашли, что смешанный катализатор, содержащий сложный оксид на базе Mo-V-Nb-W и соединение вольфрама при определенном соотношении, может решить указанные проблемы. Данное изобретение было выполнено на основании этих полученных данных.
А именно, данное изобретение состоит в следующем:
[1] Смешанный катализатор для реакции каталитического окисления в паровой фазе или реакции каталитического аммоксидирования в паровой фазе пропана или изобутана,
где данный смешанный катализатор содержит:
(a) сложный оксид, состав которого представлен приведенной ниже формулой (1):
Mo1VaNbbSbcWdZeOn (1),
где Z представляет собой по меньшей мере один элемент, выбранный из группы, состоящей из La, Ce, Pr, Yb, Y, Sc, Sr и Ba; каждый индекс из a, b, c, d, e и n представляет собой атомную долю каждого элемента в расчете на атом Mo; a находится в интервале 0,01≤a≤1; b находится в интервале 0,01≤b≤1; c находится в интервале 0,01≤c≤1; d находится в интервале 0,001≤d≤1; e находится в интервале 0≤e≤1; и n представляет собой число, определяемое валентностью металлических компонентов; и
(b) соединение вольфрама при соотношении, соответствующем указанной ниже формуле (2):
0,001<w<0,3 (2),
где w представляет собой атомную долю вольфрама в соединении вольфрама как атомную долю в расчете на атом Mo в сложном оксиде.
[2] Смешанный катализатор в соответствии с вышеуказанным пунктом [1], в котором соединение вольфрама содержит оксид вольфрама.
[3] Смешанный катализатор в соответствии с вышеуказанным пунктом [1] или [2], где при этом смешанный катализатор используется для реакции в псевдоожиженном слое.
[4] Способ получения ненасыщенной кислоты или ненасыщенного нитрила, включающий стадию приведения пропана или изобутана и кислорода в соприкосновение со смешанным катализатором по любому из вышеуказанных пунктов с [1] по [3], или приведения пропана или изобутана, а также кислорода и аммиака в соприкосновение со смешанным катализатором по любому из вышеуказанных пунктов с [1] по [3].
[5] Способ в соответствии с вышеуказанным пунктом [4], в котором температура для стадии приведения в соприкосновение устанавливается при 400°C или более.
Соответствующая ненасыщенная кислота или соответствующий ненасыщенный нитрил могут быть получены при высоком выходе из пропана или изобутана посредством применения смешанного катализатора по данному изобретению.
ПОДРОБНОЕ ОПИСАНИЕ ПРЕДПОЧТИТЕЛЬНОГО ВАРИАНТА ОСУЩЕСТВЛЕНИЯ
Далее в данном документе будет подробно описан вариант осуществления данного изобретения (далее в данном документе называемый «представляемый вариант осуществления»). Данное изобретение не ограничивается приведенным ниже вариантом осуществления, и множество изменений может быть сделано в пределах объема данного изобретения.
Смешанный катализатор представляемого варианта осуществления является смешанным катализатором для реакции каталитического окисления в паровой фазе или реакции каталитического аммоксидирования в паровой фазе пропана или изобутана.
Данный смешанный катализатор содержит:
(a) сложный оксид, состав которого представлен приведенной ниже формулой (1):
Mo1VaNbbSbcWdZeOn (1),
где Z представляет собой по меньшей мере один элемент, выбранный из группы, состоящей из La, Ce, Pr, Yb, Y, Sc, Sr и Ba; каждый индекс из a, b, c, d, e и n представляет собой атомную долю каждого элемента в расчете на атом Mo; a находится в интервале 0,01≤a≤1; b находится в интервале 0,01≤b≤1; c находится в интервале 0,01≤c≤1; d находится в интервале 0,001≤d≤1; e находится в интервале 0≤e≤1; и n представляет собой число, определяемое валентностью металлических компонентов; и
(b) соединение вольфрама при соотношении, соответствующем указанной ниже формуле (2):
0,001<w<0,3 (2),
где w представляет собой атомную долю вольфрама в соединении вольфрама как атомную долю в расчете на атом Mo в сложном оксиде.
Хотя способ получения смешанного катализатора представляемого варианта осуществления не ограничивается особым образом, его пример будет описан ниже.
[1] Способ изготовления смешанного катализатора
Сложный оксид, содержащийся в смешанном катализаторе представляемого варианта осуществления, может быть изготовлен, например, следующим способом.
(a) Изготовление сложного оксида
Сложный оксид получают посредством следующих трех стадий.
(1) стадия приготовления исходных материалов, чтобы получить раствор смешанных исходных материалов;
(2) стадия сушки раствора смешанных исходных материалов, полученных на стадии (1), чтобы получить сухой порошок;
(3) стадия обжига сухого порошка, полученного на стадии (2), чтобы получить сложный оксид.
Термин «приготовление» на вышеуказанной стадии (1) означает растворение или диспергирование исходных материалов сложного оксида в водном растворе. Термин «исходный материал» означает соединение, содержащее элемент, образующий сложный оксид.
Исходный материал не ограничивается особым образом и могут быть использованы, например, такие соединения, которые описаны ниже.
В качестве исходных материалов для Mo и V могут подходить для применения гептамолибдат аммония [(NH4)6Mo7O24·4H2O] и метаванадат аммония [NH4VO3], соответственно, хотя исходные материалы не ограничиваются особым образом.
В качестве исходных материалов для Nb могут подходить для применения ниобиевая кислота, неорганический ниобат и органический ниобат. Из них особенно предпочтительной является ниобиевая кислота. Ниобиевая кислота представляет собой соединение формулы Nb2O5·nH2O и ее также называют гидроксидом ниобия или гидратом оксида ниобия. Кроме того, в качестве исходных материалов для Nb также предпочтительно используется раствор исходного материала для Nb, в котором молярное соотношение дикарбоновая кислота/ниобий составляет от 1 до 4. В качестве дикарбоновой кислоты предпочтительной является щавелевая кислота.
В качестве исходных материалов для Sb предпочтительным является оксид сурьмы (III) [Sb2O3], хотя отсутствуют особые ограничения в отношении этих материалов.
Исходные материалы для W не ограничиваются особым образом. Могут быть использованы соединение, содержащее W, и раствор, в котором металлический W растворен в подходящем растворителе. Примеры соединения, содержащего W, включают аммониевую соль, нитрат, карбоксилат, аммониевую соль карбоновой кислоты, пероксокарбоксилат, аммониевую соль пероксокарбоновой кислоты, галогенированную аммониевую соль, галогенид, ацетилацетат и алкоксид W. Из них подходит для применения водный исходный материал, такой как нитрат и карбоксилат W.
Исходные материалы для компонента Z не ограничиваются особым образом при условии, что исходные материалы содержат по меньшей мере один элемент, выбранный из группы, состоящей из La, Ce, Pr, Yb, Y, Sc, Sr и Ba. Могут быть использованы соединения, содержащее вышеуказанный элемент, и раствор, в котором металл вышеуказанного элемента растворен в подходящем растворителе. Примеры соединения, содержащего элемент, включают нитрат, карбоксилат, аммониевую соль карбоновой кислоты, пероксокарбоксилат, аммониевую соль пероксокарбоновой кислоты, галогенированную аммониевую соль, галогенид, ацетилацетат и алкоксид металлического элемента. Из них подходит для применения водный исходный материал, такой как нитрат и карбоксилат.
Когда сложный оксид представляет собой кремнеземистый носитель, в качестве исходного материала для кремнезема может быть использован золь кремниевой кислоты. Однако в качестве кремнеземистого исходного материала порошок кремнезема может быть использован частично или полностью. Порошок кремнезема предпочтительно изготавливается высокотемпературным способом. Порошок кремнезема используется вместе с порошком кремнезема, первоначально диспергированным в воде, чтобы облегчить добавление и смешивание порошка кремнезема с суспензией. Способ диспергирования не ограничивается особым образом. Порошок кремнезема может быть диспергирован посредством применения обычного гомогенизатора, смесителя-гомогенизатора и сверхзвукового вибратора или т.п., по отдельности или в комбинации.
Далее в данном документе будут описаны подходящие примеры изготовления сложного оксида, включающие стадии с (1) по (3).
Стадия (1): Стадия приготовления исходных материалов
На стадии (1) соединение Mo, соединение V, соединение Sb, соединение W, соединение компонента Z и необязательно компонент, который становится любыми другими исходными материалами, добавляют к воде и затем нагревают, посредством чего приготавливают водный смешанный раствор (I). При этом внутреннее пространство контейнера для приготовления смешанного раствора (I) может находиться в атмосфере азота. Соединение Nb и дикарбоновую кислоту затем нагревают в воде при перемешивании, посредством чего приготавливают смешанный раствор (B0). После этого пероксид водорода добавляют к смешанному раствору (B0), посредством чего приготавливают водный смешанный раствор (II). При этом H2O2/Nb (молярное соотношение) составляет предпочтительно от 0,5 до 20 и более предпочтительно от 1 до 10.
Затем, в зависимости от требуемого состава, водный смешанный раствор (I) и водный смешанный раствор (II) смешивают надлежащим образом, посредством чего получают водный смешанный раствор (III). Полученный водный смешанный раствор (III) выдерживают в воздушной атмосфере, посредством чего получают суспензию смешанных исходных материалов (далее в данном документе также называемую просто «суспензией»).
Здесь, выдерживание водного смешанного раствора (III) означает, что водный смешанный раствор (III) оставляется в неподвижном состоянии или при перемешивании в течение заданного периода времени. Когда сложный оксид производится в промышленном масштабе, распылительная сушилка обычно имеет скорость обработки, лимитирующую расход. После того как часть водного смешанного раствора (III) высушена распылением, требуется время, чтобы завершить сушку распылением всего смешанного раствора. Тем временем продолжается перемешивание смешанного раствора, который не высушен распылением. Поэтому время выдерживания включает не только время выдерживания перед сушкой распылением, но также время от начала до завершения сушки распылением.
Время выдерживания составляет предпочтительно 90 минут или более и в пределах 50 часов и более предпочтительно 90 минут или более и в пределах 6 часов с точки зрения выхода целевого продукта.
Температура выдерживания составляет предпочтительно 25°C или более с точки зрения предотвращения конденсации компонента Mo и осаждения V. Температура выдерживания составляет предпочтительно 65°C или менее с точки зрения предотвращения избыточного протекания гидролиза комплекса, содержащего Nb и пероксид водорода, и образования суспензии в предпочтительной форме. Поэтому, температура выдерживания составляет предпочтительно 25°C или более и 65°C или менее и более предпочтительно 30°C или более и 60°C или менее.
Атмосфера в контейнере при выдерживании предпочтительно содержит кислород в достаточной концентрации. Недостаточное содержание кислорода может затруднять существенное изменение водного смешанного раствора (III). Концентрация кислорода (на которую далее в данном документе также делается ссылка как на «концентрацию кислорода в паровой фазе») в парофазной части контейнера составляет предпочтительно 1 об.% или более.
Концентрация кислорода в паровой фазе в контейнере может быть измерена обычными способами. Например, концентрация кислорода в паровой фазе может быть измерена с помощью прибора для измерения содержания кислорода с использованием диоксида циркония. Место, в котором измеряется концентрация кислорода в паровой фазе, предпочтительно находится вблизи поверхности раздела между водным смешанным раствором (III) и паровой фазой. Например, предпочтительно, концентрация кислорода в паровой фазе измеряется три раза в одной и той же точке в течение 1 минуты, и средняя величина результатов трех измерений используется в качестве концентрации кислорода в паровой фазе.
Разбавляющий газ для уменьшения концентрации кислорода в паровой фазе не ограничивается особым образом. Примеры разбавляющего газа включают азот, гелий, аргон, диоксид углерода и пар. В промышленном масштабе предпочтительным является азот. В качестве газа для увеличения концентрации кислорода в паровой фазе предпочтительным является чистый кислород или воздух с высокой концентрацией кислорода.
Как считается, в окислительно-восстановительном состоянии компонента, содержащегося в водном смешанном растворе (III), за счет вышеуказанного выдерживания происходит некоторое изменение. Протекание некоторого изменения предполагается на основании возникновения изменения в цвете и изменения в окислительно-восстановительном потенциале или т.п. водного смешанного раствора (III) во время выдерживания. В результате возникает разница между сложными оксидами, которые образуются при применении или отсутствии выдерживания в течение 90 минут или более и в пределах 50 часов в атмосфере с концентрацией кислорода от 1 до 25 об.%. Например, чрезвычайно трудно корректным образом идентифицировать изменение в форме компонента в жидкости во время выдерживания. Тем не менее, образуются сложные оксиды, имеющие разное время выдерживания, и выход целевого продукта оценивается при использовании сложных оксидов в качестве катализатора, посредством чего выясняется, что предпочтительным является время выдерживания, придающее катализатору способность к высокому выходу. При этом суспензия, имеющая некоторую предпочтительную форму, может быть определена как сформированная.
Считается, что окислительно-восстановительный потенциал водного смешанного раствора (III) регулируется посредством потенциала (600 мВ/AgCl) водного раствора (II) исходного материала и что содержащийся пероксид оксалата Nb и другие компоненты металлов в водном растворе (II) исходного материала вызывают некоторую окислительно-восстановительную реакцию, обусловливая временное уменьшение потенциала. Окислительно-восстановительный потенциал водного смешанного раствора (III) составляет предпочтительно от 450 до 530 мВ/AgCl и более предпочтительно от 470 до 510 мВ/AgCl.
Концентрация кислорода во время выдерживания составляет предпочтительно 1 об.% или более с точки зрения предотвращения чрезмерной задержки в протекании окислительно-восстановительной реакции, оказывающей влияние на изменение в окислительно-восстановительном состоянии компонентов, содержащихся в водном смешанном растворе (III), и предотвращении какого-либо чрезмерного окисления в окислительно-восстановительном состоянии суспензии. С другой стороны, концентрация кислорода во время выдерживания составляет предпочтительно 25 об.% или менее с целью предотвращения некоторого избыточного восстановления суспензии, вызываемого избыточным протеканием окислительно-восстановительной реакции. Так или иначе, необходимо поддерживать концентрацию кислорода в подходящем интервале, поскольку кислород в паровой фазе оказывает влияние на окислительно-восстановительное состояние суспензии. Интервал составляет более предпочтительно от 5 до 23 об.% и еще более предпочтительно от 10 до 20 об.%.
Когда сложный оксид представляет собой кремнеземистый носитель, готовят раствор смешанных исходных материалов, содержащий золь кремниевой кислоты. Золь кремниевой кислоты может быть добавлен к нему подходящим образом. Водная дисперсия порошка кремнезема может быть использована в качестве части золя кремниевой кислоты. Водная дисперсия порошка кремнезема может быть также добавлена подходящим образом.
Пероксид водорода предпочтительно добавляется к водному смешанному раствору (I) или жидкости, содержащей компоненты водного смешанного раствора (I), во время приготовления. При этом H2O2/Nb (молярное соотношение) составляет предпочтительно от 0,01 до 5 и более предпочтительно от 0,05 до 4. При этом перемешивание предпочтительно продолжается при температуре от 30 до 70°C в течение от 30 минут до 2 часов.
Стадия (2): Стадия сушки
На стадии (2) суспензию, полученную на стадии приготовления исходного материала, сушат, посредством чего получают сухой порошок. Сушка может быть выполнена известными способами, такими как сушка распылением или выпаривание досуха. Из них получают сухой порошок в виде мелких сферических частиц, предпочтительно сушкой распылением. Распыление в способе сушки распылением может быть выполнено с помощью центробежного устройства, сопловым устройством с двумя текучими средами или сопловым устройством высокого давления. Воздух, нагретый паром, и электрический нагреватель или т.п. может быть использован в качестве источника тепла для сушки. Температура на входе сушилки устройства для сушки распылением составляет предпочтительно от 150 до 300°C. Температура на выходе сушилки составляет предпочтительно от 100 до 160°C.
Стадия (3): Стадия обжига
На стадии (3) сложный оксид получают обжигом сухого порошка, полученного на стадии сушки. Например, в качестве оборудования для обжига может быть использована барабанная печь. Форма устройства для обжига не ограничивается особым образом. Однако форма устройства для обжига предпочтительно является трубчатой, поскольку может быть выполнен непрерывный обжиг. Форма трубы для обжига не ограничивается особым образом. Однако форма трубы для обжига предпочтительно является цилиндрической.
Система нагревания предпочтительно является внешней системой нагревания. Например, может быть использована соответствующим образом электрическая печь. Размер и материал или т.п. трубы для обжига могут быть выбраны подходящим образом в зависимости от условий обжига и объема производства. Внутренний диаметр трубы для обжига составляет предпочтительно от 70 до 2000 мм и более предпочтительно от 100 до 1200 мм. Длина трубы для обжига составляет предпочтительно от 200 до 10000 мм и более предпочтительно от 800 до 8000 мм. Когда ударное воздействие прикладывается к устройству для обжига, толщина устройства для обжига составляет предпочтительно 2 мм или более и более предпочтительно 4 мм или более, с учетом того, что устройство для обжига должно иметь достаточную толщину, чтобы не ломаться при ударном воздействии. Толщина устройства для обжига составляет предпочтительно 100 мм или менее и более предпочтительно 50 мм или менее, с учетом того, что ударное воздействие в достаточной мере передается в трубу для обжига. Материал трубы для обжига не ограничивается особым образом, при условии, что труба для обжига обладает термостойкостью и такой прочностью, что не ломается при ударном воздействии. Нержавеющая сталь SUS может быть использована подходящим образом в качестве материала трубы для обжига.
Перегородка, имеющая центральную часть с отверстием, через которое проходит порошок, размещается перпендикулярно потоку порошка в трубе для обжига, и тем самым труба для обжига может быть также разделена на две зоны или более. Время выдерживания в трубе для обжига легко обеспечивается размещением такой перегородки. Перегородка может быть одна или их может быть несколько. Материалом перегородки является предпочтительно металл и, соответственно, может быть использована перегородка, изготовленная из того же самого материала, что и труба для обжига. Высота перегородки может быть отрегулирована в соответствии со временем выдерживания, которое должно быть обеспечено. Например, когда порошок подается при 250 г/ч при применении барабанной печи с трубой для обжига, имеющей внутренний диаметр 150 мм и длину 1150 мм и изготовленной из нержавеющей стали SUS, высота перегородки составляет предпочтительно от 5 до 50 мм, более предпочтительно от 10 до 40 мм и еще более предпочтительно от 13 до 35 мм. Толщина перегородки не ограничивается особым образом и предпочтительно устанавливается в соответствии с размером трубы для обжига. Например, в случае барабанной печи с трубой для обжига, имеющей внутренний диаметр 150 мм и длину 1150 мм и изготовленной из нержавеющей стали SUS, толщина перегородки составляет предпочтительно 0,3 мм или более и 30 мм или менее и более предпочтительно 0,5 мм или более и 15 мм или менее.
Для того, чтобы предотвратить растрескивание частиц сухого порошка и образование в них волосных трещин или т.п. и чтобы равномерно обжечь сухой порошок, труба для обжига предпочтительно вращается во время обжига. Скорость вращения трубы для обжига составляет предпочтительно от 0,1 до 30 об/мин, более предпочтительно от 0,5 до 20 об/мин и еще более предпочтительно от 1 до 10 об/мин.
При прокаливании сухого порошка, предпочтительно, температуру нагревания сухого порошка повышают непрерывным или прерывистым образом до температуры в интервале от 550 до 800°C от температуры ниже чем 400°C.
Обжиг может проводиться в воздушной атмосфере или в потоке воздуха. Однако по меньшей мере часть обжига предпочтительно выполняется при протекании инертного газа, который по существу не содержит кислорода, как, например, азот. Количество подаваемого инертного газа составляет предпочтительно 50 норм. литров или более в расчете на 1 кг сухого порошка, более предпочтительно от 50 до 5000 норм. литров и еще более предпочтительно от 50 до 3000 норм. литров (норм. литр означает литр, измеренный при нормальной температуре и нормальном давлении, а именно, при 20°C и 1 атм). При этом потоки инертного газа и сухого порошка могут быть противоточными или прямоточными. Однако противоточное контактирование является предпочтительным, принимая во внимание газообразные компоненты, образующиеся из сухого порошка, и следовое количество воздуха, вовлеченного в сухой порошок.
Стадия обжига может быть выполнена в виде единственного этапа. Однако обжиг предпочтительно включает предварительный обжиг, выполняемый в интервале температур от 250 до 400°C, и основной обжиг, выполняемый в интервале температур от 550 до 800°C. Предварительный обжиг и основной обжиг могут выполняться непрерывным образом. Основной обжиг может быть выполнен сразу же после завершения предварительного обжига. Как предварительный обжиг, так и основной обжиг может быть разделен на несколько этапов.
Предварительный обжиг выполняется предпочтительно в потоке инертного газа при температуре нагревания от 250°C до 400°C и предпочтительно от 300°C до 400°C. Предварительный обжиг предпочтительно выполняется при поддержании постоянной температуры в температурном интервале от 250°C до 400°C. Однако температура может колебаться в температурном интервале от 250°C до 400°C или же постепенно повышаться или понижаться. Время поддержания температуры нагревания составляет предпочтительно 30 минут или более и более предпочтительно от 3 до 12 часов.
Профиль увеличения температуры до достижения температуры предварительного обжига может быть линейно возрастающим, или же температура может возрастать таким образом, что образуется дугообразный профиль с выпуклостью в верхнем или в нижнем направлении.
Средняя скорость увеличения температуры во время подъема температуры до достижения температуры предварительного обжига не ограничивается особым образом. Однако средняя скорость увеличения температуры составляет обычно от примерно 0,1 до 15°C/мин, предпочтительно от 0,5 до 5°C/мин и более предпочтительно от 1 до 2°C/мин.
Основной обжиг может быть выполнен, предпочтительно в потоке инертного газа, предпочтительно при температуре от 550 до 800°C, более предпочтительно от 580 до 750°C, еще более предпочтительно от 600 до 720°C и особенно предпочтительно от 620 до 700°C. Основной обжиг предпочтительно выполняется при поддержании постоянной температуры в температурном интервале от 620°C до 700°C. Однако температура может колебаться в температурном интервале от 620°C до 700°C или же постепенно повышаться или понижаться. Время основного обжига составляет предпочтительно от 0,5 до 20 часов и более предпочтительно от 1 до 15 часов.
Когда труба для обжига разделена перегородкой, сухой порошок и/или сложный оксид непрерывно проходит по меньшей мере через 2 зоны, предпочтительно от 2 до 20 зон, и более предпочтительно от 4 до 15 зон. Температура может регулироваться с помощью одного или нескольких контроллеров. Однако для того, чтобы получить желательный профиль температуры обжига, нагреватель и контроллер предпочтительно размещаются в каждой из зон, разделенных этими перегородками, чтобы регулировать температуру. Например, когда семь перегородок расположены таким образом, что длина части трубы для обжига, введенной в нагревательную печь, разделена равным образом на восемь частей, и используется труба для обжига, разделенная на восемь зон, заданная температура каждой из восьми зон предпочтительно регулируется нагревателем и контроллером, установленными в каждой из зон, так что температура сухого порошка и/или сложного оксида имеет желательный профиль температуры обжига. Окислительный компонент (например, кислород) или восстановительный компонент (например, аммиак) может быть добавлен в атмосферу для обжига в потоке инертного газа, при необходимости.
Профиль увеличения температуры до достижения температуры основного обжига может быть линейно возрастающим, или же температура может возрастать таким образом, что образуется дугообразный профиль с выпуклостью в верхнем или в нижнем направлении.
Средняя скорость увеличения температуры во время подъема температуры до достижения температуры основного обжига не ограничивается особым образом. Однако средняя скорость увеличения температуры составляет обычно от примерно 0,1 до 15°C/мин, предпочтительно от 0,5 до 10°C/мин и более предпочтительно от 1 до 8°C/мин.
Средняя скорость снижения температуры после того, как основной обжиг завершен, составляет от 0,01 до 1000°C/мин, предпочтительно от 0,05 до 100°C/мин, более предпочтительно от 0,1 до 50°C/мин и особенно предпочтительно от 0,5 до 10°C/мин. После того как основной обжиг завершен, также предпочтительно в течение некоторого времени поддерживается температура, которая ниже температуры основного обжига. Поддерживаемая температура ниже, чем температура основного обжига, на 10°C или более, предпочтительно на 50°C или более и более предпочтительно на 100°C или более. Время поддержания составляет 0,5 часа или более, предпочтительно 1 час или более, более предпочтительно 3 часа или более и особенно предпочтительно 10 часов или более.
Когда основной обжиг осуществляется сразу же после завершения предварительного обжига, низкотемпературная обработка предпочтительно выполняется в процессе основного обжига.
Время, требующееся для низкотемпературной обработки, а именно, время, требующееся для снижения температуры сухого порошка и/или сложного оксида и повышения температуры до температуры обжига, может надлежащим образом регулироваться посредством размера, толщины и материалы устройства для обжига, количества производимого катализатора, последовательности периодов для непрерывного обжига сухого порошка и/или сложного оксида и постоянной скорости и постоянного количества или т.п. Например, когда используется труба для обжига, имеющая внутренний диаметр 500 мм, длину 4500 мм и толщину 20 мм, которая изготовлена из нержавеющей стали SUS, время, требующееся для низкотемпературной обработки, находится предпочтительно в пределах 30 дней на протяжении последовательности периодов для непрерывного обжига катализатора, более предпочтительно в пределах 15 дней, еще более предпочтительно в пределах 3 дней и особенно предпочтительно в пределах 2 дней.
Например, когда сухой порошок подается при расходе 35 кг/ч, при том, что барабанная печь, имеющую трубу для обжига с внутренним диаметром 500 мм, длиной 4500 мм и толщиной 20 мм, изготовленную из нержавеющей стали SUS, вращается при 6 об/мин, и основной обжиг выполняется при температуре основного обжига 645°C, стадия снижения температуры до 400°C и повышения температуры до 645°C может быть выполнена в течение примерно 1 дня. Когда обжиг выполняется непрерывно в течение 1 года, обжиг может быть выполнен посредством проведения такой низкотемпературной обработки раз в месяц, наряду с тем, что температура слоя оксида поддерживается стабильным образом.
На стадии обжига, часть соединения, имеющего низкую температуру плавления, в кристалле оксида может быть закристаллизована в выступающем состоянии на поверхности частицы сложного оксида. Поскольку кристаллы соединения с низкой температурой плавления, выделяются в выступающем состоянии на поверхности катализатора, данные кристаллы могут препятствовать текучести, когда катализатор используется в реакции в псевдоожиженном слое. Поэтому в случае катализатора для реакции в псевдоожиженном слое кристаллы соединения с низкой температурой плавления, образованные в выступающем состоянии, предпочтительно физически удаляются перед применением катализатора с целью предотвращения снижения текучести. Когда кристаллы соединения с низкой температурой плавления удаляются с поверхности катализатора, кристаллы могут быть удалены перед стадией смешивания источника вольфрама, как описано ниже, или могут быть удалены после стадии смешивания.
(b) Стадия смешивания источника вольфрама
Сложный оксид представляемого варианта осуществления обладает каталитической активностью в состоянии «как есть». Когда смешанный катализатор содержит соединение вольфрама и сложный оксид при определенном соотношении, выход целевого продукта увеличивается.
В качестве способа получения смешанного катализатора, содержащего соединение вольфрама, будет описан пример стадии смешивания подаваемого источника соединения вольфрама (на который далее в данном документе также делается ссылка как на «источник вольфрама») и сложного оксида.
(b-1) Источник вольфрама
Примеры источника вольфрама включают аммониевую соль, нитрат, карбоксилат, аммониевую соль карбоновой кислоты, пероксокарбоксилат, аммониевую соль пероксокарбоновой кислоты, галогенированную аммониевую соль, галогенид, ацетилацетат, алкоксид, трифенильное соединение, полиоксометалат и аммониевую соль полиоксометалата вольфрама; порошковый исходный материал, такой как оксид вольфрама, вольфрамат, паравольфрамат аммония, вольфрамокремниевая кислота, кремневольфрамомолибденовая кислота и ванадовольфрамокремниевая кислота; и жидкий исходный материал, такой как водный раствор метавольфрамата аммония и золь оксида вольфрама.
Вид источника вольфрама и смеси источника вольфрама в твердотельной форме или жидкой форме может быть выбран в зависимости от стадии смешивания или состава смешанного катализатора, который должен быть приготовлен, или т.п. Как описано ниже, стадия смешивания включает способ подачи источника вольфрама в первоначальном твердотельном состоянии и способ подачи источника вольфрама в жидком состоянии. В случае способа подачи источника вольфрама в жидком состоянии может быть использован коммерчески доступный жидкий исходный материал, такой как водный раствор метавольфрамата аммония. Однако, разумеется, вышеупомянутый порошковый исходный материал может быть использован как порошковый исходный материал, растворенный и/или диспергированный в растворителе. В этом случае подходящее количество порошкового исходного материала может быть растворено и/или диспергировано в воде, ацетоне, метаноле, этаноле и другом полярном/неполярном растворителе. Вода предпочтительно используется в качестве растворителя и/или дисперсионной среды, с учетом простоты обращения, зависимости от растворимости или т.п. Когда порошок смешивается непосредственным образом без применения растворителя, оксид вольфрама является предпочтительным с точки зрения влияния соединения вольфрама на целевой продукт, более предпочтительным является оксид вольфрама, имеющий структуру вольфрамовой бронзы, и еще более предпочтительным является триоксид вольфрама.
Смешанный катализатор представляемого варианта осуществления может быть получен посредством физического или химического смешивания сложного оксида и источника вольфрама при заданном соотношении.
(b-2) Способ физического смешивания
Способ физического смешивания сложного оксида и источника вольфрама не ограничивается особым образом. Примеры способа физического смешивания включают способ добавления сложного оксида в бункерный питатель для подачи катализатора в реактор и добавления в него соответствующего количества источника вольфрама. Сложный оксид и источник вольфрама могут быть первоначально смешаны перед тем как сложный оксид и источник вольфрама помещаются в бункерный питатель. Однако даже если сложный оксид и источник вольфрама не смешиваются первоначально, сложный оксид и источник вольфрама смешиваются естественным образом на этапе подачи катализатора в реактор из бункерного питателя, и, тем самым, первоначальное смешивание не является обязательным. Порядок помещения сложного оксида и источника вольфрама в бункерный питатель также не ограничивается особым образом. Порядок может быть определен надлежащим образом так, что сложный оксид и источник вольфрама в достаточной мере приводятся в соприкосновение один с другим в реакторе с учетом диаметров частиц или т.п. как сложного оксида, так и источника вольфрама. Сложный оксид и источник вольфрама могут быть также, при необходимости, смешаны с помощью потока воздуха и азота. В случае реакционного взаимодействия в псевдоожиженном слое сложный оксид и источник вольфрама не подаются одновременно из бункерного питателя, а сложный оксид и источник вольфрама могут быть поданы последовательно в реактор. В этом случае, сложный оксид и источник вольфрама смешиваются в то время, как сложный оксид и источник вольфрама находятся в подвижном состоянии в реакторе во время развития реакции.
Поскольку в случае способа физического смешивания в источнике вольфрама перед реакцией получения ненасыщенной кислоты или ненасыщенного нитрила химического изменения не происходит, соединение вольфрама, содержащееся в смешанном катализаторе, имеет такую же структуру, что и структура источника вольфрама. С другой стороны, когда реакция получения ненасыщенной кислоты или ненасыщенного нитрила прогрессирует при применении этого смешанного катализатора, тепло или т.п. прикладывается к соединению вольфрама при приведении соединения вольфрама в соприкосновение со сложным оксидом и реакционноспособной основой, и, соответственно, происходит химическое изменение. Смешанный катализатор представляемого варианта осуществления предназначен для воздействия на сложный оксид в соответствии со структурным изменением во время развития реакции, чтобы оказать влияние на эксплуатационные качества катализатора. Поэтому, когда смешанный катализатор присутствует в качестве смеси перед реакцией, то даже если структура части или всего соединения вольфрама изменяется во время развития реакции, например, до структуры части сложного оксида, смешанный катализатор естественным образом относится к категории смешанного катализатора представляемого варианта осуществления.
Даже если источник вольфрама подается в реактор с псевдоожиженным слоем во время развития реакции, имеет место такой же эффект, что и при его подаче перед протеканием реакции. В этом случае, можно сказать, что смешанный катализатор производится сразу же после подачи источника вольфрама, хотя смешанный катализатор не присутствует перед реакцией.
(b-3) Способ химического смешивания
(b-3-i) Жидкофазная стадия
В представляемом варианте осуществления способ просачивания раствора, содержащего растворенный в нем источник вольфрама, в сложный оксид называют «импрегнирование». С другой стороны, способ добавления сложного оксида к раствору, содержащему растворенный в нем источник вольфрама, и приведения сложного оксида в соприкосновение с раствором в течение определенного времени посредством перемешивания или т.п. называют «погружение». В каждом случае, ненужный раствор может быть удален фильтрованием или испарением. Испарение выполняется при температуре примерно от 30 до 300°C и предпочтительно от 40 до 250°C. Затем может быть выполнена обработка обжигом, если это необходимо, и тем самым часть или все количество источника вольфрама может также быть преобразовано в оксид. Тонкие поры сложного оксида заполнены атмосферным газом, присутствующим перед проведением операции контактирования со сложным оксидом, таким как воздух и инертный газ. Атмосферный газ может блокировать диффузию вольфрама в тонкие поры. В этом случае, газ в тонких порах может также быть удален в атмосфере при пониженном давлении перед импрегнированием и погружением или во время импрегнирования и погружения.
Когда выполняются импрегнирование и погружение, может быть осуществлен ионный обмен элемента-металла и вольфрама в сложном оксиде, пока сложный оксид и источник вольфрама присутствуют в жидкой фазе. Когда происходит ионный обмен, ожидается прогрессирование модификации поверхности, обусловленной ионным обменом, во время обработки в жидкой фазе. Однако вольфрам введен в сложный оксид. В этом случае вольфрам не присутствует в качестве простого вещества в виде соединения вольфрама в смешанном катализаторе, а является частью сложного оксида, и едва ли можно сказать, что катализатор представляет собой смесь. Соответственно, данный катализатор не включается в смешанный катализатор представляемого варианта осуществления. При сравнении катализатора, в котором вольфрам введен в сложный оксид, и смешанного катализатора в отношении эксплуатационных качеств катализатора имеет место следующее. Когда катализатор, в котором вольфрам введен в сложный оксид, используется для реакции получения ненасыщенной кислоты или ненасыщенного нитрила, выход целевого продукта представляет собой максимальную величину на первоначальной стадии реакции. Выход может затем уменьшаться, однако он не увеличивается. С другой стороны, в случае смешанного катализатора выход целевого продукта имеет тенденцию к увеличению с течением времени до определенного времени реакции, как описано ниже.
Ионный обмен в жидкой фазе зависит от pH, температуры жидкости, концентрации вольфрама в растворе и времени контактирования раствора и сложного оксида или т.п. Однако, например, ионный обмен может быть подавлен посредством регулирования pH до величины от 1,0 до 7,0 и предпочтительно от 1,0 до 4,0. Предпочтительно, температура жидкости является обычно низкой. Ионный обмен может быть подавлен посредством регулирования температуры жидкости до, например, от 0 до 50°C и более предпочтительно от 0 до 30°C. Когда концентрация вольфрама низкая, ионный обмен может почти не прогрессировать. Его предпочтительная концентрация составляет менее чем 1,0 моль/кг в расчете на концентрацию металлического вольфрама. Время контактирования раствора и сложного оксида является предпочтительно коротким. Время контактирования находится в пределах 1 часа и более предпочтительно в пределах 15 минут.
Хотя смесь, полученная посредством стадии импрегнирования и/или стадии погружения, может быть использована в качестве смешанного катализатора в состоянии «как есть», данная смесь может быть использована после обжига.
Для соединения вольфрама, содержащегося в смешанном катализаторе после обработки жидкой фазы, может также иметь место вариант, в котором источник вольфрама изменен (например, окисление, кристалличность, некристалличность), в дополнение к варианту, в котором соединение вольфрама имеет ту же самую структуру, что и источника вольфрама. Однако смешанный катализатор каждого из вариантов относится к категории смешанного катализатора представляемого варианта осуществления, если только смешанный катализатор данного варианта содержит соединение вольфрама, которое не является частью сложного оксида.
(b-3-ii) Стадия обжига
Когда ионный обмен не происходит в жидкой фазе, реакция обмена может происходить на стадии обжига с преобразованием источника вольфрама в часть сложного оксида. Ожидается, что модификация поверхности прогрессирует в ходе реакции обмена, приводя к состоянию, в котором смешанный катализатор не является смесью, также как при обработке в жидкой фазе. Возможность протекания реакции обмена в основном зависит от температуры обжига. Когда температура обжига является чрезмерно высокой, то реакция обмена имеет тенденцию к прогрессированию. Температура обжига, при которой реакция обмена почти не протекает, составляет предпочтительно от 200 до 400°C и более предпочтительно от 250 до 350°C. Соединение вольфрама, содержащееся в смешанном катализаторе после обработки обжигом, обычно имеет структуру, отличающуюся от структуры источника вольфрама. Однако данный смешанный катализатор относится к категории смешанного катализатора представляемого варианта осуществления, если только смешанный катализатор содержит соединение вольфрама, которое не является частью сложного оксида как после обработки в жидкой фазе.
(c) Смешанный катализатор
Смешанный катализатор представляемого варианта осуществления является смешанным катализатором для реакции каталитического окисления в паровой фазе или реакции каталитического аммоксидирования в паровой фазе пропана или изобутана, содержащим:
(a) сложный оксид, состав которого представлен приведенной ниже формулой (1):
Mo1VaNbbSbcWdZeOn (1),
где Z представляет собой по меньшей мере один элемент, выбранный из группы, состоящей из La, Ce, Pr, Yb, Y, Sc, Sr и Ba; каждый индекс из a, b, c, d, e и n представляет собой атомную долю каждого элемента в расчете на атом Mo; a находится в интервале 0,01≤a≤1; b находится в интервале 0,01≤b≤1; c находится в интервале 0,01≤c≤1; d находится в интервале 0,001≤d≤1; e находится в интервале 0≤e≤1; и n представляет собой число, определяемое валентностью металлических компонентов; и
(b) соединение вольфрама при соотношении, соответствующем указанной ниже формуле (2):
0,001<w<0,3 (2),
где w представляет собой атомную долю вольфрама в соединении вольфрама как атомную долю в расчете на атом Mo в сложном оксиде.
Смешанный катализатор представляемого варианта осуществления является смесью соединения вольфрама и сложного оксида. Смешанный катализатор содержит соединение вольфрама в качестве обязательного компонента, и тем самым выход целевого продукта может быть значительно увеличен на основании указанного ниже принципа. В соответствии с данным принципом, вольфрам как элемент в соединении вольфрама диффундирует на поверхность сложного оксида и фиксируется во время реакции каталитического окисления в паровой фазе пропана или изобутана кислородом и реакции каталитического аммоксидирования в паровой фазе пропана или изобутана кислородом и аммиаком.
Состав сложного оксида представляемого варианта осуществления представлен приведенной ниже формулой (1):
Mo1VaNbbSbcWdZeOn (1),
где Z представляет собой по меньшей мере один элемент, выбранный из группы, состоящей из La, Ce, Pr, Yb, Y, Sc, Sr и Ba; каждый индекс из a, b, c, d, e и n представляет собой атомную долю элемента в расчете на атом Mo; a находится в интервале 0,01≤a≤1; b находится в интервале 0,01≤b≤1; c находится в интервале 0,01≤c≤1; d находится в интервале 0,001≤d≤1; e находится в интервале 0≤e≤1; и n представляет собой число, определяемое валентностью металлических компонентов.
Предпочтительно, a и b, представляющие собой атомные доли V и Nb в расчете на атом Mo, составляют соответственно от 0,1 до 0,4 и от 0,02 до 0,2.
c, представляющий собой атомную долю Sb в расчете на атом Mo, составляет предпочтительно от 0,01 до 0,6 и более предпочтительно от 0,1 до 0,4. Соотношение a/c, которое представляло собой атомное соотношение V и Sb, было тщательно исследовано. В результате было найдено, что a/c предпочтительно находится в интервале от 0,1 до 1 с точки зрения повышения выхода, хотя детальная причина была неясна.
d, представляющий собой атомную долю W в расчете на атом Mo, находится в интервале 0,001≤d≤1 и предпочтительно в интервале 0,001≤d≤0,3. Вольфрам в сложном оксиде (далее в данном документе вольфрам, который присутствует в сложном оксиде, может называться «объемный вольфрам») рассматривается как замещающий места молибдена или ванадия в сложном оксиде. Если сравнить температуры плавления оксидов, то, например, температуры плавления триоксида молибдена и триоксида вольфрама равны соответственно 795°C и 1473°C, и температура плавления оксида вольфрама выше. Соответственно, когда молибден в сложном оксиде замещен вольфрамом, то предполагается, что температура плавления сложного оксида повышается. Поэтому считается, что объемный вольфрам, диспергированный в сложном оксиде, оказывает влияние на кристаллическую структуру сложного оксида, внося вклад в термостойкость и устойчивость к окислению-восстановлению. В результате, сложный оксид, содержащий объемный вольфрам, имеет тенденцию увеличивать срок службы катализатора и быть выгодным для долговременного промышленного применения. С другой стороны, вольфрам в соединении вольфрама оценивается как оказывающий положительное влияние на увеличение выхода ненасыщенной кислоты или ненасыщенного нитрила. Кроме того, считается, что объемный вольфрам оказывает сдерживающее влияние на снижение выхода целевого продукта, обусловленное чрезмерной диффузией вольфрама в соединении вольфрама в сложный оксид.
e, представляющий собой атомную долю компонента Z в расчете на атом Mo, находится в интервале 0≤e≤1, предпочтительно в интервале 0,001≤e≤1, более предпочтительно в интервале 0,001≤e≤0,1 и еще более предпочтительно в интервале 0,002≤e≤0,01. Поскольку компонент Z вызывает неблагоприятную реакцию в суспензии, как указано в выложенной заявке на патент Японии № 11-244702, то предпочтительно содержится следовое количество компонента Z. С другой стороны, поскольку компонент Z оказывает значительное влияние на увеличение выхода целевого продукта, то предпочтительно компонент Z равномерно диспергирован в частицах катализатора. Компонент Z представляет собой по меньшей мере один элемент, выбранный из группы, состоящей из La, Ce, Pr, Yb, Y, Sc, Sr и Ba, предпочтительно один или несколько элементов, выбранных из группы, состоящей из La, Ce, Pr, и Yb, и особенно предпочтительно Ce, поскольку Ce склонен предоставлять наибольший выход целевого продукта.
Сложный оксид предпочтительно поддерживается носителем, содержащим кремнезем в качестве основного компонента. Когда сложный оксид поддерживается носителем, содержащим кремнезем в качестве основного компонента, сложный оксид имеет высокую механическую прочность, и, тем самым, сложный оксид подходит для реакции каталитического окисления в паровой фазе или реакции каталитического аммоксидирования в паровой фазе с применением реактора с псевдоожиженным слоем. Содержание кремнезема в носителе, содержащем кремнезем в качестве основного компонента составляет, в расчете на SiO2 предпочтительно от 20 до 70 масс.% и более предпочтительно от 30 до 60 масс.% по отношению к общей массе поддерживаемого оксида, содержащего оксид элемента, составляющего сложный оксид, и носителя.
Содержание кремнезема в сложном оксиде составляет предпочтительно 20 масс.% или более с точки зрения прочности и предотвращения измельчения в порошок. Когда содержание кремнезема меньше чем 20 масс.%, затрудняется стабильное функционирование сложного оксида при промышленном применении сложного оксида, и необходимо восполнять потерянный сложный оксид. Соответственно, содержание кремнезема меньше чем 20 масс.% является экономически невыгодным. В противоположность этому, когда содержание кремнезема больше чем 70 масс.%, не достигается достаточная активность, вследствие чего увеличивается количество необходимого катализатора. В частности, когда содержание кремнезема больше чем 70 масс.% в случае псевдоожиженного слоя, удельная масса частиц носителя из кремнезема чрезмерно мала, и, соответственно, трудно достигается хорошее текучее состояние.
Смешанный катализатор представляемого варианта осуществления содержит сложный оксид, состав которого представлен вышеуказанной формулой (1), и соединение вольфрама при соотношении, соответствующем указанной ниже формуле (2)
0,001<w<0,3 (2)
В формуле (2) w представляет собой атомную долю вольфрама в соединении вольфрама как атомную долю в расчете на атом Mo в сложном оксиде.
w, который представляет собой атомную долю вольфрама в соединении вольфрама в расчете на атом Mo сложного оксида, находится в интервале 0,001<w<0,3, предпочтительно в интервале 0,01<w<0,2 и более предпочтительно в интервале 0,015<w<0,18. Когда w составляет 0,001 или менее, количество вольфрама является слишком малым, и, таким образом, повышение выхода целевого продукта не может быть поддержано. Когда w составляет 0,3 или более, количество вольфрама является чрезмерным, и, соответственно, поверхность катализатора не преобразуется до предпочтительной формы. Кроме того, не может быть достигнут эффект повышения выхода целевого продукта.
Состав смешанного катализатора и состав сложного оксида определяются с помощью рентгенофлуоресцентного анализа, и величина w определяется из следующей формулы:
w = (доля W в составе смешанного катализатора) - (доля W в составе сложного оксида)
Соединение, имеющее низкую температуру плавления, может быть закристаллизовано в выступающем состоянии на поверхности частиц сложного оксида после основного обжига. Даже когда кристаллы удаляются с поверхности катализатора вследствие взаимного контактирования частиц катализатора или т.п., это почти не оказывает влияние на величину w. Поэтому величина w может быть определена посредством компонентного анализа на любой стадии перед и после удаления кристаллов оксида.
Сложный оксид и соединение вольфрама предпочтительно приводятся в соприкосновение один с другим в смешанном катализаторе. Частицы порядка микрометра или миллиметра могут быть приведены в соприкосновение одни с другими, или же соединение вольфрама и сложный оксид, диспергированные до размера порядка нанометров в тонких порах в частицах сложного оксида, могут быть приведены в соприкосновение одни с другими. Например, первые получают физическим смешиванием частиц сложного оксида и источника вольфрама, а вторые получают обработкой в жидкой фазе при использовании частиц сложного оксида и жидкого источника вольфрама.
Даже когда рентгеноструктурный анализ или т.п. выполняется на смешанном катализаторе представляемого варианта осуществления, трудно подтвердить изменение в структуре смешанного катализатора перед смешиванием и после него. Когда целевой продукт производится с применением смешанного катализатора, в основном не наблюдается разница на стадии непосредственно после инициирования реакции по сравнению с эксплуатационными качествами сложного оксида, который не содержит соединения вольфрама. Например, когда реакция получения ненасыщенного нитрила выполняется при 445°C, по существу не наблюдается разница между выходами целевых продуктов в смешанном катализаторе и сложном оксиде после 5 часов от инициирования реакции. С другой стороны, разница между выходами в обеих реакциях проявляется со временем при протекании реакции. Например, повышение выхода на 1% или более наблюдается по сравнению с первой стадией в реакции с применением смешанного катализатора после 240 ч. Однако в случае сложного оксида первоначальный выход почти не изменяется.
Причина, почему выход целевого продукта повышается во время реакции получения ненасыщенной кислоты или ненасыщенного нитрила с применением смешанного катализатора представляемого варианта осуществления, будет описана ниже. Авторы данного изобретения полагают, что вольфрам в соединении вольфрама диффундирует на поверхность сложного оксида в твердофазной реакции и происходит реакция обмена между вольфрамом и элементом-металлом, таким как Mo, посредством чего поверхность катализатора преобразуется в предпочтительную форму. Предпочтительная форма поверхности катализатора и механизм улучшения эксплуатационных качеств являются неясными. Предполагается, что вольфрам должен быть расположен в определенных местах вблизи поверхности сложного оксида, чтобы подавить последующее разложение целевого продукта или промежуточного продукта. Как описано выше, структура и состояние распределения соединения вольфрама рассматриваются как различающиеся в зависимости от способа приготовления или т.п. смешанного катализатора. Однако по мнению авторов данного изобретения разница в эксплуатационных качествах, зависящая от структуры и состояния распределения соединения вольфрама, почти не наблюдалась в смешанном катализаторе перед твердофазной реакцией. Важно, что смешанный катализатор содержит лишь подходящее количество соединения вольфрама независимо от сложного оксида. Предполагается, что твердофазная реакция происходит во время реакции получения на протяжении длительного периода времени до тех пор, пока содержание соединения вольфрама находится в подходящем интервале независимо от состояния соединения вольфрама. Когда реакция выполняется в течение длительного периода времени при применении обычного смешанного катализатора, содержание Mo в катализаторе уменьшается с течением времени. В этом случае, известен метод введения соединения Mo в реактор и предотвращения снижения активности, обусловленного уменьшением содержания Mo (метод подпитки), в качестве общепринятого метода. Метод подпитки для соединения Mo может быть использован одновременно с проведением реакции получения ненасыщенной кислоты или ненасыщенного нитрила с применением смешанного катализатора представляемого варианта осуществления.
[2] Ненасыщенная кислота или ненасыщенный нитрил
В присутствии смешанного катализатора представляемого варианта осуществления пропан или изобутан подвергается реакции каталитического окисления в паровой фазе или реакции каталитического аммоксидирования в паровой фазе, посредством чего производится соответствующая ненасыщенная кислота или соответствующий ненасыщенный нитрил.
А именно, способ получения ненасыщенной кислоты или ненасыщенного нитрила представляемого варианта осуществления включает стадию приведения пропана или изобутана и кислорода в соприкосновение с вышеуказанным смешанным катализатором или приведение пропана или изобутана, а также кислорода и аммиака в соприкосновение со смешанным катализатором.
Когда используется смешанный катализатор представляемого варианта осуществления, выход целевого продукта приближается к максимальной величине в ходе протекания реакции получения ненасыщенной кислоты или ненасыщенного нитрила. Хотя детальная причина является неясной, предполагается, что твердофазная реакция вольфрама в соединении вольфрама прогрессирует надлежащим образом в ходе реакции каталитического окисления в паровой фазе с применением пропана или изобутана и воздуха или в ходе реакции каталитического аммоксидирования в паровой фазе с применением пропана или изобутана, а также воздуха и аммиака, и посредством этого смешанный катализатор активируется. Это связано с тем, что выход целевого продукта не повышается совсем при обжиге в воздушной атмосфере и при обжиге в атмосфере азота при температуре от 300 до 550°C в основной концепции представляемого варианта осуществления. Предпочтительное условие для активации не ограничивается особым образом. Однако пропан или изобутан и кислород предпочтительно нагреваются до температуры от 350 до 550°C и более предпочтительно от 400 до 500°C при приведении пропана или изобутана и кислорода в соприкосновение со смешанным катализатором. В качестве альтернативы, пропан или изобутан, а также кислород и аммиак предпочтительно нагреваются до температуры от 350 до 550°C и более предпочтительно от 400 до 500°C при приведении пропана или изобутана, а также кислорода и аммиака в соприкосновение со смешанным катализатором. С другой стороны, время активирующей обработки составляет предпочтительно от 1 до 1500 часов, более предпочтительно от 5 до 750 часов и еще более предпочтительно от 50 до 500 часов.
Подаваемые в качестве исходных материалов пропан или изобутан и аммиак не обязательно являются высокочистыми и могут быть использованы газы технического сорта.
Воздух, обогащенный кислородом, воздух или чистый кислород могут быть использованы в качестве подаваемого источника кислорода. Кроме того, в качестве разбавляющего газа могут подаваться гелий, аргон, диоксид углерода, пар и азот или т.п.
Реакция каталитического окисления в паровой фазе пропана или изобутана может быть выполнена при следующих условиях.
Молярное отношение кислорода, подаваемого для реакции, к пропану или изобутану составляет от 0,1 до 6 и предпочтительно от 0,5 до 4.
Температура реакции составляет от 300 до 500°C и предпочтительно от 350 до 450°C.
Давление реакции составляет от 5×104 до 5×105 Па и предпочтительно от 1×105 до 3×105 Па.
Время контактирования составляет от 0,1 до 10 (с·г/см3) и предпочтительно от 0,5 до 5 (с·г/см3). В представляемом варианте осуществления время контактирования определяется посредством следующей формулы.
Время контактирования (с·г/см3) = (W/F)×273/(273+T)
При этом W, F и T определяются следующим образом:
W = Количество (г) размещенного катализатора
F = Расход (норм. см3/с) исходного материала, смешанного с газом при нормальных условиях (0°C, 1,013×105 Па)
T = Температура реакции (°C).
Реакция каталитического аммоксидирования в паровой фазе пропана или изобутана может быть выполнена при следующих условиях.
Молярное отношение кислорода, подаваемого для реакции, к пропану или изобутану составляет от 0,1 до 6 и предпочтительно от 0,5 до 4.
Молярное отношение аммиака, подаваемого для реакции, к пропану или изобутану составляет от 0,3 до 1,5 и предпочтительно от 0,7 до 1,2.
Температура реакции составляет от 350 до 500°C и предпочтительно от 380 до 470°C.
Давление реакции составляет от 5×104 до 5×105 Па и предпочтительно от 1×105 до 3×105 Па.
Время контактирования составляет от 0,1 до 10 (с·г/см3) и предпочтительно 0,5 до 5 (с·г/см3).
В качестве метода реакции может быть приспособлен любой из обычных методов, такой как метод с неподвижным слоем, метод с псевдоожиженным слоем и метод с подвижным слоем. Однако реактор с псевдоожиженным слоем, предоставляющий упрощенное удаление теплоты реакции, является предпочтительным. Реакция каталитического окисления или аммоксидирования в паровой фазе может представлять собой либо однопоточную систему, либо систему с рециркуляцией.
ПРИМЕРЫ
Далее в данном документе представляемый вариант осуществления будет дополнительно описан подробно со ссылками на примеры и сравнительные примеры. Однако диапазон представляемого варианта осуществления не ограничивается данными примерами.
В примерах и сравнительных примерах степень превращения пропана и выход акрилонитрила, соответственно, определяются указанным ниже образом.
Степень превращения пропана (%) = (Число молей прореагировавшего пропана)/(Число молей поданного пропана)×100
Выход акрилонитрила (AN)(%) = (Число молей произведенного акрилонитрила)/(Число молей поданного пропана)×100
(Приготовление смешанного раствора ниобия)
Смешанный раствор ниобия готовили описанным ниже способом.
К 10 кг воды добавляли 0,765 кг ниобиевой кислоты, содержащей 80,0 масс.% ниобия в расчете на Nb2O5 и 2,633 кг дигидрата щавелевой кислоты [H2C2O4·2H2O]. Молярное соотношение щавелевая кислота/ниобий в качестве исходных материалов составляло 5,0, и концентрация исходного ниобия составляла 0,50 (молей Nb/кг раствора). Полученный раствор нагревали в течение двух часов при 95°C при перемешивании, посредством чего получают смешанный раствор, в котором был растворен ниобий. Этот смешанный раствор оставляли для выдерживания, охлаждали льдом и подвергали вакуумной фильтрации для удаления твердотельного вещества, посредством чего получают равномерный смешанный раствор ниобия. Молярное соотношение щавелевая кислота/ниобий в этом смешанном растворе ниобия составляло 2,71 в соответствии с анализом, описанным ниже.
10 г этого смешанного раствора ниобия точно отвешивали и помещали в тигель, сушили в течение ночи при 95°C и подвергали термообработке в течение одного часа при 600°C, посредством чего получали 0,771 г Nb2O5. В результате этого концентрация ниобия составляла 0,580 (молей Nb/кг раствора).
3 г этого смешанного раствора ниобия точно отвешивали и помещали в стеклянный лабораторный стакан емкостью 300 мл, добавляли 200 мл горячей воды, имеющей температуру примерно 80°C, и затем добавляли 10 мл серной кислоты (1:1). Полученный смешанный раствор титровали при использовании 1/4 н. раствора KMnO4 при перемешивании и поддержании при температуре 70°C на обогреваемой мешалке. Момент времени, при котором бледнеет ярко-розовый цвет, обусловленный KMnO4, при продолжении в течение примерно 30 секунд или более, определялся как конечный момент. Концентрацию щавелевой кислоты определяли на основании полученного титра в соответствии с приведенной ниже формулой и, в качестве результата, она составляла 1,570 (молей щавелевой кислоты/кг).
2KMnO4+3H2SO4+5H2C2O4→K2SO4+2MnSO4+10CO2+8H2O
Полученный смешанный раствор ниобия использовали в качестве смешанного раствора ниобия (B0) для применения при изготовлении катализатора, как будет описано ниже.
[Пример 1]
(Приготовление сложного оксида)
Сложный оксид, имеющий состав исходных материалов, представленный формулой Mo1V0,21Nb0,10Sb0,24W0,03Ce0,005On/47,0 масс.% SiO2, изготавливали как указано ниже.
К 1,902 кг воды добавляли 424,3 г гептамолибдата аммония [(NH4)6Mo7O24·4H2O], 59,0 г метаванадата аммония [NH4VO3], 5,22 г гексагидрата нитрата церия и 87,6 г оксида сурьмы (III) [Sb2O3] и нагревали в течение 1 часа при 95°C при перемешивании, посредством чего получали водный раствор исходного материала (I).
К 414,3 г смешанного раствора ниобия (B0) добавляли 54,5 г раствора пероксида водорода, содержащего 30 масс.% H2O2, и смешивали в течение 10 минут при комнатной температуре при перемешивании, посредством чего приготавливали водный раствор исходного материала (II).
После охлаждения полученного водного раствора исходного материала (I) до 70°C к нему добавляли 760,3 г золя кремниевой кислоты, содержащего 34,0 масс.% SiO2, и затем добавляли 102,2 г раствора пероксида водорода, содержащего 30 масс.% H2O2, после чего полученную смесь непрерывно перемешивали в течение 30 минут при 55°C. Затем последовательно добавляли водный раствор исходного материала (II), 33,4 г водного раствора метавольфрамата аммония, содержащего 50 масс.% WO3, и жидкостную дисперсию, в которой 211,5 г порошка кремнезема было диспергировано в 2,750 кг воды, посредством чего получали водный смешанный раствор (III). Водный смешанный раствор (III) выдерживали при 50°C в течение 2 часов и 30 минут после добавления водного раствора исходного материала (II), посредством чего получали суспензию.
Полученную суспензию подавали в центробежную распылительную сушилку и сушили, посредством чего получали микросферический сухой порошок. Температуры на входе и выходе сушилки составляли соответственно 210°C и 120°C.
800 г полученного сухого порошка помещали в трубу для обжига диаметром 3 дюйма (7,62 см), изготовленную из нержавеющей стали SUS, и затем обжигали в течение 2 часов при 680°C в потоке газообразного азота с расходом 8,0 норм. л/мин при вращении трубы, посредством чего получали сложный оксид.
(Приготовление смешанного катализатора)
100 г полученного сложного оксида добавляли к водному раствору (концентрация W: 0,7 моль/кг) при перемешивании и смешивали. Водный раствор получали разбавлением 46,2 г водного раствора метавольфрамата аммония водой в количестве 453,8 г. Полученный смешанный раствор перемещали в аспирационный контейнер и затем подвергали обработке при пониженном давлении при 100 кПа в течение 10 минут. Смешанный раствор в аспираторе затем фильтровали и полученное твердое вещество подвергали сушке при 50°C в течение 12 часов в сушилке, посредством чего получали смесь сложного оксида и соединения вольфрама. Составы полученного сложного оксида и смешанного катализатора подвергали анализу. Для компонентного анализа был использован рентгенофлуоресцентный анализатор (RIX1000, производства Rigaku Corporation).
Состав сложного оксида представлял собой Mo1V0,21Nb0,10Sb0,24W0,03Ce0,005On, и состав смешанного катализатора представлял собой Mo1V0,21Nb0,10Sb0,24W0,10Ce0,005On. Из разницы между долями содержания W в составе сложного оксида и смешанного катализатора было подтверждено, что w составляло 0,07.
(Реакция аммоксидирования пропана)
35 г полученного смешанного катализатора помещали в реакционную трубу с псевдоожиженным слоем из викора (кварцоидного стекла), имеющую внутренний диаметр 25 мм. Смешанный газ, имеющий молярное соотношение пропан:аммиак:кислород:гелий, составляющее 1:1:3:18, подавали в реакционную трубу при величине времени контактирования 3,4 (с·г/см3) при температуре реакции 445°C и при давлении реакции 0,05 МПа. По прошествии 5 часов после начала подачи смешанного газа степень превращения пропана составляла 88,3% и выход акрилонитрила (AN) составлял 52,4%.
Реакция затем продолжалась при тех же самых условиях. По прошествии 240 часов после начала подачи смешанного газа степень превращения пропана составляла 88,8%, и выход акрилонитрила (AN) составлял 54,4%.
[Пример 2]
(Изготовление смешанного катализатора)
Смешанный катализатор был изготовлен при использовании сложного оксида, полученного в примере 1. Смешанный катализатор изготавливали таким же образом, что и в примере 1, за исключением того, что концентрация W в разбавленном водном растворе вольфрамата аммония была изменена до 1,5 моль/кг.
В результате определения состава полученного смешанного катализатора таким же образом, что и в примере 1, было подтверждено, что состав был следующий: Mo1V0,21Nb0,10Sb0,24W0,18Ce0,005On, и w составляло 0,15.
(Реакция аммоксидирования пропана)
Реакцию аммоксидирования пропана выполняли таким же образом, что и в примере 1, при использовании полученного смешанного катализатора. По прошествии 5 часов после начала подачи смешанного газа степень превращения пропана составляла 88,2% и выход акрилонитрила (AN) составлял 52,3%.
Реакция затем продолжалась при тех же самых условиях. По прошествии 240 часов после начала подачи смешанного газа степень превращения пропана составляла 88,6% и выход акрилонитрила (AN) составлял 53,8%.
[Сравнительный пример 1]
(Приготовление сложного оксида)
Сложный оксид, имеющий состав исходных материалов, представленный формулой Mo1V0,21Nb0,10Sb0,24Ce0,005On/47,0 масс.% SiO2, изготавливали как указано ниже.
К 1,964 кг воды добавляли 441,1 г гептамолибдата аммония [(NH4)6Mo7O24·4H2O], 61,4 г метаванадата аммония [NH4VO3], 5,42 г гексагидрата нитрата церия и 87,4 г оксида сурьмы (III) [Sb2O3] и нагревали в течение 1 часа при 95°C при перемешивании, посредством чего получали водный раствор исходного материала (I).
К 430,8 г смешанного раствора ниобия (B0) добавляли 56,7 г раствора пероксида водорода, содержащего 30 масс.% H2O2, и смешивали в течение 10 минут при комнатной температуре при перемешивании, посредством чего приготавливали водный раствор исходного материала (II).
После охлаждения полученного водного раствора исходного материала (I) до 70°C к нему добавляли 760,3 г золя кремниевой кислоты, содержащего 34,0 масс.% SiO2, и затем добавляли 102,2 г раствора пероксида водорода, содержащего 30 масс.% H2O2, после чего полученную смесь непрерывно перемешивали в течение 30 минут при 55°C. Затем последовательно добавляли водный раствор исходного материала (II) и жидкостную дисперсию, в которой 211,5 г порошка кремнезема было диспергировано в 2,750 кг воды, посредством чего получали водный смешанный раствор (III). Водный смешанный раствор (III) выдерживали при 50°C в течение 2 часов и 30 минут после добавления водного раствора исходного материала (II), посредством чего получали суспензию.
Полученную суспензию подавали в центробежную распылительную сушилку и сушили, посредством чего получали микросферический сухой порошок. Температуры на входе и выходе сушилки составляли соответственно 210°C и 120°C.
800 г полученного сухого порошка помещали в трубу для обжига диаметром 3 дюйма (7,62 см), изготовленную из нержавеющей стали SUS, и затем обжигали в течение 2 часов при 680°C в потоке газообразного азота с расходом 8,0 норм. л/мин при вращении трубы, посредством чего получали сложный оксид.
(Изготовление смешанного катализатора)
100 г полученного сложного оксида добавляли к водному раствору (концентрация W: 1,0 моль/кг) при перемешивании и смешивали. Водный раствор получали разбавлением 231,0 г водного раствора метавольфрамата аммония, содержащего 50,0 масс.% WO3, водой в количестве 453,8 г. Полученный смешанный раствор перемещали в аспирационный контейнер и затем подвергали обработке при пониженном давлении при 100 кПа в течение 10 минут. Смешанный раствор в аспираторе затем фильтровали и полученное твердое вещество подвергали сушке при 50°C в течение 12 часов в сушилке, посредством чего получали смесь сложного оксида и соединения вольфрама. Составы полученного сложного оксида и смешанного катализатора подвергали анализу. Для компонентного анализа был использован рентгенофлуоресцентный анализатор (RIX1000, производства Rigaku Corporation).
Состав сложного оксида представлял собой Mo1V0,21Nb0,10Sb0,24Ce0,005On, и состав смешанного катализатора представлял собой Mo1V0,21Nb0,10Sb0,24W0,10Ce0,005On. Из разницы между долями содержания W в составе сложного оксида и смешанного катализатора было подтверждено, что w составляло 0,10.
(Реакция аммоксидирования пропана)
Реакцию аммоксидирования пропана выполняли таким же образом, что и в примере 1, при использовании полученного смешанного катализатора. По прошествии 5 часов после начала подачи смешанного газа степень превращения пропана составляла 87,3% и выход акрилонитрила (AN) составлял 52,2%.
Реакция затем продолжалась при тех же самых условиях. По прошествии 240 часов после начала подачи смешанного газа степень превращения пропана составляла 86,3% и выход акрилонитрила (AN) составлял 51,8%.
[Сравнительный пример 2]
(Изготовление смешанного катализатора)
Смешанный катализатор изготавливали таким же образом, что и в сравнительном примере 1, за исключением того, что концентрация W в разбавленном водном растворе вольфрамата аммония была изменена до 1,5 моль/кг.
В результате определения состава полученного смешанного катализатора таким же образом, что и в сравнительном примере 1, было подтверждено, что состав был следующий: Mo1V0,21Nb0,10Sb0,24W0,15Ce0,005On, и w составляло 0,15.
(Реакция аммоксидирования пропана)
Реакцию аммоксидирования пропана выполняли таким же образом, что и в примере 1, при использовании полученного смешанного катализатора. По прошествии 5 часов после начала подачи смешанного газа степень превращения пропана составляла 86,9% и выход акрилонитрила (AN) составлял 51,8%.
Реакция затем продолжалась при тех же самых условиях. По прошествии 240 часов после начала подачи смешанного газа степень превращения пропана составляла 85,2% и выход акрилонитрила (AN) составлял 50,3%.
[Пример 3]
(Изготовление смешанного катализатора)
Приготавливали водный раствор, в котором 500 г водного раствора метавольфрамата аммония был разбавлен водой в количестве 500 г. Полученный водный раствор подавали в центробежную распылительную сушилку и сушили, посредством чего получали микросферический сухой порошок. Температуры на входе и выходе сушилки составляли соответственно 210°C и 120°C. 100 г полученного продукта, высушенного распылением и содержащего вольфрам, обжигали при 500°C в воздушной атмосфере в течение 2 часов, посредством чего получали порошкообразное соединение вольфрама. Измерение с применением рентгеновской дифракции использовали, чтобы подтвердить то, что соединение вольфрама являлось триоксидом вольфрама.
100 г сложного оксида, полученного в примере 1, и 3,37 г полученного соединения вольфрама смешивали в виде порошка, посредством чего получали смешанный катализатор. Состав полученного смешанного катализатора подвергали анализу. Для компонентного анализа был использован рентгенофлуоресцентный анализатор (RIX1000, производства Rigaku Corporation).
Было подтверждено, что состав полученного смешанного катализатора представлял собой Mo1V0,21Nb0,10Sb0,24W0,09Ce0,005On, и w составляло 0,06.
(Реакция аммоксидирования пропана)
Реакцию аммоксидирования пропана выполняли таким же образом, что и в примере 1, при использовании полученного смешанного катализатора. По прошествии 5 часов после начала подачи смешанного газа степень превращения пропана составляла 88,7% и выход акрилонитрила (AN) составлял 52,6%.
Реакция затем продолжалась при тех же самых условиях. По прошествии 240 часов после начала подачи смешанного газа степень превращения пропана составляла 89,2% и выход акрилонитрила (AN) составлял 54,0%.
[Пример 4]
(Изготовление смешанного катализатора)
Смешанный катализатор получали таким же образом, что и в примере 3, за исключением того, что массу смешиваемого соединения вольфрама изменяли до 10,1 г. Состав полученного смешанного катализатора анализировали таким же образом, что и в примере 1. Для компонентного анализа был использован рентгенофлуоресцентный анализатор (RIX1000, производства Rigaku Corporation).
Было подтверждено, что состав полученного смешанного катализатора представлял собой Mo1V0,21Nb0,10Sb0,24W0,18Ce0,005On, и w составляло 0,15.
(Реакция аммоксидирования пропана)
Реакцию аммоксидирования пропана выполняли таким же образом, что и в примере 1, при использовании полученного смешанного катализатора. По прошествии 5 часов после начала подачи смешанного газа степень превращения пропана составляла 88,6% и выход акрилонитрила (AN) составлял 52,5%.
Реакция затем продолжалась при тех же самых условиях. По прошествии 240 часов после начала подачи смешанного газа степень превращения пропана составляла 89,0% и выход акрилонитрила (AN) составлял 53,6%.
[Сравнительный пример 3]
(Изготовление смешанного катализатора)
Смешанный катализатор изготавливали таким же образом, что и в примере 3, за исключением того, что использовали сложный оксид, полученный в сравнительном примере 1, и массу смешиваемого соединения вольфрама изменяли до 6,74 г.
В результате определения состава полученного смешанного катализатора таким же образом, что и в примере 1, было подтверждено, что состав был следующий: Mo1V0,21Nb0,10Sb0,24W0,10Ce0,005On, и w составляло 0,10.
(Реакция аммоксидирования пропана)
Реакцию аммоксидирования пропана выполняли таким же образом, что и в примере 1, при использовании полученного смешанного катализатора. По прошествии 5 часов после начала подачи смешанного газа степень превращения пропана составляла 88,6% и выход акрилонитрила (AN) составлял 52,5%.
Реакция затем продолжалась при тех же самых условиях. По прошествии 240 часов после начала подачи смешанного газа степень превращения пропана составляла 87,6% и выход акрилонитрила (AN) составлял 52,1%.
[Сравнительный пример 4]
(Изготовление смешанного катализатора)
Смешанный катализатор приготавливали таким же образом, что и в примере 3, за исключением того, что использовали сложный оксид, полученный в сравнительном примере 1, и массу смешиваемого соединения вольфрама изменяли до 10,1 г.
В результате определения состава полученного смешанного катализатора таким же образом, что и в примере 1, было подтверждено, что состав был следующий: Mo1V0,21Nb0,10Sb0,24W0,15Ce0,005On, и w составляло 0,15.
(Реакция аммоксидирования пропана)
Реакцию аммоксидирования пропана выполняли таким же образом, что и в примере 1, при использовании полученного смешанного катализатора. По прошествии 5 часов после начала подачи смешанного газа степень превращения пропана составляла 88,5% и выход акрилонитрила (AN) составлял 52,4%.
Реакция затем продолжалась при тех же самых условиях. По прошествии 240 часов после начала подачи смешанного газа степень превращения пропана составляла 86,3% и выход акрилонитрила (AN) составлял 50,9%.
[Сравнительный пример 5]
(Реакция аммоксидирования пропана)
Реакцию аммоксидирования пропана выполняли таким же образом, что и в примере 1, при использовании сложного оксида, полученного в примере 1, в состоянии «как есть», без изготовления смешанного катализатора. По прошествии 5 часов после начала подачи смешанного газа степень превращения пропана составляла 88,4% и выход акрилонитрила (AN) составлял 52,2%.
Реакция затем продолжалась при тех же самых условиях. По прошествии 240 часов после начала подачи смешанного газа степень превращения пропана составляла 88,3% и выход акрилонитрила (AN) составлял 52,1%.
[Пример 5]
(Приготовление сложного оксида)
Сложный оксид, имеющий состав исходных материалов, представленный формулой Mo1V0,21Nb0,10Sb0,24W0,12Ce0,005On/47,0 масс.% SiO2, изготавливали таким же способом, что и в примере 1, за исключением того, что добавленное количество водного раствора метавольфрамата аммония было изменено до 133,6 г.
(Изготовление смешанного катализатора)
Смешанный катализатор изготавливали таким же образом, что и в примере 1, при использовании полученного сложного оксида. В результате определения составов сложного оксида и смешанного катализатора таким же методом, что и в примере 1, состав сложного оксида составлял Mo1V0,21Nb0,10Sb0,24W0,12Ce0,005On, и состав смешанного катализатора составлял Mo1V0,21Nb0,10Sb0,24W0,19Ce0,005On. Из долей содержания W в составе сложного оксида и смешанного катализатора было подтверждено, что w составляло 0,07.
(Реакция аммоксидирования пропана)
Реакцию аммоксидирования пропана выполняли таким же образом, что и в примере 1, при использовании полученного смешанного катализатора. По прошествии 5 часов после начала подачи смешанного газа степень превращения пропана составляла 87,6% и выход акрилонитрила (AN) составлял 52,1%.
Реакция затем продолжалась при тех же самых условиях. По прошествии 240 часов после начала подачи смешанного газа степень превращения пропана составляла 87,7% и выход акрилонитрила (AN) составлял 54,1%.
[Пример 6]
Смешанный катализатор изготавливали таким же образом, что и в примере 2, при использовании сложного оксида, полученного в примере 5.
В результате определения состава полученного смешанного катализатора таким же образом, что и в примере 1, было подтверждено, что состав был следующий: Mo1V0,21Nb0,10Sb0,24W0,27Ce0,005On, и w составляло 0,15.
(Реакция аммоксидирования пропана)
Реакцию аммоксидирования пропана выполняли таким же образом, что и в примере 1, при использовании полученного смешанного катализатора. По прошествии 5 часов после начала подачи смешанного газа степень превращения пропана составляла 87,5% и выход акрилонитрила (AN) составлял 52,0%.
Реакция затем продолжалась при тех же самых условиях. По прошествии 240 часов после начала подачи смешанного газа степень превращения пропана составляла 87,6% и выход акрилонитрила (AN) составлял 53,6%.
[Пример 7]
(Изготовление смешанного катализатора)
Смешанный катализатор был изготовлен при использовании сложного оксида, полученного в примере 1. Смешанный катализатор изготавливали таким же образом, что и в примере 1, за исключением того, что концентрация W в разбавленном водном растворе вольфрамата аммония была изменена до 0,2 моль/кг.
В результате определения состава полученного смешанного катализатора таким же образом, что и в примере 1, было подтверждено, что состав был следующий: Mo1V0,21Nb0,10Sb0,24W0,05Ce0,005On, и w составляло 0,02.
(Реакция аммоксидирования пропана)
Реакцию аммоксидирования пропана выполняли таким же образом, что и в примере 1, при использовании полученного смешанного катализатора. По прошествии 5 часов после начала подачи смешанного газа степень превращения пропана составляла 88,3% и выход акрилонитрила (AN) составлял 52,3%.
Реакция затем продолжалась при тех же самых условиях. По прошествии 240 часов после начала подачи смешанного газа степень превращения пропана составляла 88,9% и выход акрилонитрила (AN) составлял 55,1%.
[Пример 8]
(Изготовление смешанного катализатора)
Смешанный катализатор был изготовлен при использовании сложного оксида, полученного в примере 1. Смешанный катализатор изготавливали таким же образом, что и в примере 1, за исключением того, что концентрация W в разбавленном водном растворе вольфрамата аммония была изменена до 0,3 моль/кг.
В результате определения состава полученного смешанного катализатора таким же образом, что и в примере 1, было подтверждено, что состав был следующий: Mo1V0,21Nb0,10Sb0,24W0,06Ce0,005On, и w составляло 0,03.
(Реакция аммоксидирования пропана)
Реакцию аммоксидирования пропана выполняли таким же образом, что и в примере 1, при использовании полученного смешанного катализатора. По прошествии 5 часов после начала подачи смешанного газа степень превращения пропана составляла 88,4% и выход акрилонитрила (AN) составлял 52,4%.
Реакция затем продолжалась при тех же самых условиях. По прошествии 240 часов после начала подачи смешанного газа степень превращения пропана составляла 89,2% и выход акрилонитрила (AN) составлял 55,2%.
[Сравнительный пример 6]
(Изготовление смешанного катализатора)
Смешанный катализатор был изготовлен при использовании сложного оксида, полученного в сравнительном примере 1. Смешанный катализатор изготавливали таким же образом, что и в сравнительном примере 1, за исключением того, что концентрация W в разбавленном водном растворе вольфрамата аммония была изменена до 0,05 моль/кг.
В результате определения состава полученного смешанного катализатора таким же образом, что и в сравнительном примере 1, было подтверждено, что состав был следующий: Mo1V0,21Nb0,10Sb0,24W0,005Ce0,005On, и w составляло 0,005.
(Реакция аммоксидирования пропана)
Реакцию аммоксидирования пропана выполняли таким же образом, что и в примере 1, при использовании полученного смешанного катализатора. По прошествии 5 часов после начала подачи смешанного газа степень превращения пропана составляла 88,3% и выход акрилонитрила (AN) составлял 52,1%.
Реакция затем продолжалась при тех же самых условиях. По прошествии 240 часов после начала подачи смешанного газа степень превращения пропана составляла 88,2% и выход акрилонитрила (AN) составлял 52,0%.
[Сравнительный пример 7]
(Изготовление смешанного катализатора)
Смешанный катализатор был изготовлен при использовании сложного оксида, полученного в примере 1. Смешанный катализатор получали таким же образом, что и в примере 3, за исключением того, что массу смешиваемого соединения вольфрама изменяли до 44,9 г.
В результате определения состава полученного смешанного катализатора таким же образом, что и в примере 1, было подтверждено, что состав был следующий: Mo1V0,21Nb0,10Sb0,24W0,43Ce0,005On, и w составляло 0,40.
(Реакция аммоксидирования пропана)
Реакцию аммоксидирования пропана выполняли таким же образом, что и в примере 1, при использовании полученного смешанного катализатора. По прошествии 5 часов после начала подачи смешанного газа степень превращения пропана составляла 86,2% и выход акрилонитрила (AN) составлял 51,9%.
Реакция затем продолжалась при тех же самых условиях. По прошествии 240 часов после начала подачи смешанного газа степень превращения пропана составляла 83,5% и выход акрилонитрила (AN) составлял 49,3%.
[Пример 9]
(Приготовление сложного оксида)
Сложный оксид, имеющий состав исходных материалов, представленный формулой Mo1V0,21Nb0,10Sb0,24W0,03On/47,0 масс.% SiO2, изготавливали таким же способом, что и в примере 1, за исключением того, что не добавляли 5,22 г гексагидрата нитрата церия.
(Изготовление смешанного катализатора)
Смешанный катализатор изготавливали таким же образом, что и в примере 7, при использовании полученного сложного оксида. В результате определения составов сложного оксида и смешанного катализатора таким же методом, что и в примере 1, состав сложного оксида составлял Mo1V0,21Nb0,10Sb0,24W0,03On, и состав смешанного катализатора составлял Mo1V0,21Nb0,10Sb0,24W0,05On. Из долей содержания W в составе сложного оксида и смешанного катализатора было подтверждено, что w составляло 0,02.
(Реакция аммоксидирования пропана)
Реакцию аммоксидирования пропана выполняли таким же образом, что и в примере 1, при использовании полученного смешанного катализатора. По прошествии 5 часов после начала подачи смешанного газа степень превращения пропана составляла 85,8% и выход акрилонитрила (AN) составлял 51,0%.
Реакция затем продолжалась при тех же самых условиях. По прошествии 240 часов после начала подачи смешанного газа степень превращения пропана составляла 86,3% и выход акрилонитрила (AN) составлял 53,1%.
[Пример 10]
(Приготовление сложного оксида)
Сложный оксид, имеющий состав исходных материалов, представленный формулой Mo1V0,21Nb0,10Sb0,24W0,03La0,005On/47,0 масс.% SiO2, изготавливали таким же способом, что и в примере 1, за исключением того, что 5,21 г гексагидрата нитрата лантана добавляли вместо 5,22 г гексагидрата нитрата церия.
(Изготовление смешанного катализатора)
Смешанный катализатор изготавливали таким же образом, что и в примере 7, при использовании полученного сложного оксида. В результате определения составов сложного оксида и смешанного катализатора таким же методом, что и в примере 1, состав сложного оксида составлял Mo1V0,21Nb0,10Sb0,24W0,03La0,005On, и состав смешанного катализатора составлял Mo1V0,21Nb0,10Sb0,24W0,05La0,005On. Из долей содержания W в составе сложного оксида и смешанного катализатора было подтверждено, что w составляло 0,02.
(Реакция аммоксидирования пропана)
Реакцию аммоксидирования пропана выполняли таким же образом, что и в примере 1, при использовании полученного смешанного катализатора. По прошествии 5 часов после начала подачи смешанного газа степень превращения пропана составляла 87,6% и выход акрилонитрила (AN) составлял 51,8%.
Реакция затем продолжалась при тех же самых условиях. По прошествии 240 часов после начала подачи смешанного газа степень превращения пропана составляла 87,9% и выход акрилонитрила (AN) составлял 53,6%.
[Пример 11]
(Приготовление сложного оксида)
Сложный оксид, имеющий состав исходных материалов, представленный формулой Mo1V0,21Nb0,10Sb0,24W0,03Pr0,005On/47,0 масс.% SiO2, изготавливали таким же способом, что и в примере 1, за исключением того, что 5,23 г гексагидрата нитрата празеодима добавляли вместо 5,22 г гексагидрата нитрата церия.
(Изготовление смешанного катализатора)
Смешанный катализатор изготавливали таким же образом, что и в примере 7, при использовании полученного сложного оксида. В результате определения составов сложного оксида и смешанного катализатора таким же методом, что и в примере 1, состав сложного оксида составлял Mo1V0,21Nb0,10Sb0,24W0,03Pr0,005On, и состав смешанного катализатора составлял Mo1V0,21Nb0,10Sb0,24W0,05Pr0,005On. Из долей содержания W в составе сложного оксида и смешанного катализатора было подтверждено, что w составляло 0,02.
(Реакция аммоксидирования пропана)
Реакцию аммоксидирования пропана выполняли таким же образом, что и в примере 1, при использовании полученного смешанного катализатора. По прошествии 5 часов после начала подачи смешанного газа степень превращения пропана составляла 87,9% и выход акрилонитрила (AN) составлял 52,4%.
Реакция затем продолжалась при тех же самых условиях. По прошествии 240 часов после начала подачи смешанного газа степень превращения пропана составляла 88,5% и выход акрилонитрила (AN) составлял 54,4%.
[Пример 12]
(Приготовление сложного оксида)
Сложный оксид, имеющий состав исходных материалов, представленный формулой Mo1V0,21Nb0,10Sb0,24W0,03Yb0,005On/47,0 масс.% SiO2, изготавливали таким же способом, что и в примере 1, за исключением того, что 4,97 г тригидрата нитрата иттербия добавляли вместо 5,22 г гексагидрата нитрата церия.
(Изготовление смешанного катализатора)
Смешанный катализатор изготавливали таким же образом, что и в примере 7, при использовании полученного сложного оксида. В результате определения составов сложного оксида и смешанного катализатора таким же методом, что и в примере 1, состав сложного оксида составлял Mo1V0,21Nb0,10Sb0,24W0,03Yb0,005On и состав смешанного катализатора составлял Mo1V0,21Nb0,10Sb0,24W0,05Yb0,005On. Из долей содержания W в составе сложного оксида и смешанного катализатора было подтверждено, что w составляло 0,02.
(Реакция аммоксидирования пропана)
Реакцию аммоксидирования пропана выполняли таким же образом, что и в примере 1, при использовании полученного смешанного катализатора. По прошествии 5 часов после начала подачи смешанного газа степень превращения пропана составляла 87,7% и выход акрилонитрила (AN) составлял 52,4%.
Реакция затем продолжалась при тех же самых условиях. По прошествии 240 часов после начала подачи смешанного газа степень превращения пропана составляла 88,2% и выход акрилонитрила (AN) составлял 54,3%.
Данная заявка основана на заявках на патент Японии (№ 2010-111422 и 2010-111444), поданных в Патентное ведомство Японии 13 мая 2010 г., содержание которых включено в данный документ посредством ссылки.
Смешанный катализатор по данному изобретению может быть выгодным с практической точки зрения, применен для промышленного производственного процесса подвергания пропана или изобутана реакции каталитического окисления в паровой фазе или реакции каталитического аммоксидирования в паровой фазе, чтобы получить соответствующую ненасыщенную кислоту или соответствующий ненасыщенный нитрил.
Изобретение относится к смешанному катализатору для реакции каталитического окисления в паровой фазе или реакции каталитического аммоксидирования в паровой фазе пропана или изобутана. Данный смешанный катализатор содержит: (a) сложный оксид, состав которого представлен формулой Mo1VaNbbSbcWdZeOn (1), где Z представляет собой по меньшей мере один элемент, выбранный из группы, состоящей из La, Ce, Pr, Yb, Y, Sc, Sr и Ba; каждый индекс из a, b, c, d, e и n представляет собой атомную долю каждого элемента в расчете на атом Mo; a находится в интервале 0,01≤a≤1; b находится в интервале 0,01≤b≤1; c находится в интервале 0,01≤c≤1; d находится в интервале 0,001≤d≤1; e находится в интервале 0≤e≤1; и n представляет собой число, определяемое валентностью металлических компонентов; и (b) соединение вольфрама при соотношении, соответствующем формуле 0,001<w<0,3 (2), где w представляет собой атомную долю вольфрама в соединении вольфрама как атомную долю в расчете на атом Mo в сложном оксиде. При этом элементарный вольфрам из указанного соединения вольфрама (b) диффундирует на поверхность сложного оксида (а) и фиксируется на указанной поверхности сложного оксида (а) во время реакции каталитического окисления в паровой фазе пропана или изобутана кислородом или во время реакции каталитического аммоксидирования в паровой фазе пропана или изобутана кислородом и аммиаком. Предлагаемый смешанный катализатор обладает повышенной термостойкостью и устойчивостью к окислению/восстановлению. Изобретение также относится к способу получения ненасыщенной кислоты или ненасыщенного нитрила с участием такого смешанного катализатора. 2 н. и 3 з.п. ф-лы, 12 пр.
1. Смешанный катализатор для реакции каталитического окисления в паровой фазе или реакции каталитического аммоксидирования в паровой фазе пропана или изобутана,
где данный смешанный катализатор содержит:
(a) сложный оксид, состав которого представлен приведенной ниже формулой (1):
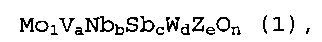
где Z представляет собой по меньшей мере один элемент, выбранный из группы, состоящей из La, Ce, Pr, Yb, Y, Sc, Sr и Ba; каждый индекс из a, b, c, d, e и n представляет собой атомную долю каждого элемента в расчете на атом Mo; а находится в интервале 0,01≤a≤1; b находится в интервале 0,01≤b≤1; с находится в интервале 0,01≤c≤1; d находится в интервале 0,001≤d≤1; е находится в интервале 0≤e≤1; и n представляет собой число, определяемое валентностью металлических компонентов; и
(b) соединение вольфрама при соотношении, соответствующем указанной ниже формуле (2):

где w представляет собой атомную долю вольфрама в соединении вольфрама как атомную долю в расчете на атом Mo в сложном оксиде,
причем элементарный вольфрам из указанного соединения вольфрама (b) диффундирует на поверхность сложного оксида (a) и фиксируется на указанной поверхности сложного оксида (a) во время реакции каталитического окисления в паровой фазе пропана или изобутана кислородом или во время реакции каталитического аммоксидирования в паровой фазе пропана или изобутана кислородом и аммиаком.
2. Смешанный катализатор по п.1, в котором соединение вольфрама содержит оксид вольфрама.
3. Смешанный катализатор по п.1 или 2, где данный смешанный катализатор используется для реакции в псевдоожиженном слое.
4. Способ получения ненасыщенной кислоты или ненасыщенного нитрила, включающий стадию приведения пропана или изобутана и кислорода в контакт со смешанным катализатором по п.1 или 2, или приведения пропана или изобутана, а также кислорода и аммиака в соприкосновение со смешанным катализатором по п.1 или 2.
5. Способ по п.4, в котором температура для стадии приведения в контакт устанавливается при 400°C или более.
| Колосоуборка | 1923 |
|
SU2009A1 |
| EP 1997554 A1, 03.12.2008 | |||
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| СПОСОБ ПРИГОТОВЛЕНИЯ КАТАЛИЗАТОРА ДЛЯ ОКИСЛЕНИЯ И АММОКСИДАЦИИ ОЛЕФИНОВ | 2003 |
|
RU2341327C2 |

