Область техники
Настоящее описание относится к новым кристаллическим кислотно-аддитивным солям трициклического производного или ее гидратам и способу их получения.
Известный уровень техники
Лекарства, вводимые перорально, показывают медицинские эффекты благодаря абсорбции, распределению, метаболизму или элиминации, а при разработке лекарств также существенными являются присущие свойства твердого состояния, состояния соли, специфического состояния кандидатов в лекарства.
В отличие от термодинамически стабильного кристаллического состояния, аморфное твердое вещество имеет термодинамически очень нестабильное состояние. Так, аморфное твёрдое вещество имеет быструю скорость элюирования и высокую растворимость по сравнению с кристаллическим твердым веществом. Соответственно, даже хотя они являются тем же самым химическим соединением, может получаться иная биодоступность.
В частности, поскольку на скорость попадания перорально вводимых активных ингредиентов в кровь пациента оказывает влияние скорость элюирования, а скорость элюирования активных ингредиентов из желудочно-кишечной жидкости пациента играет важную роль в достижении лечебных эффектов, важной является скорость элюирования в водном растворе. Среди лекарственных состояний аморфная форма растворяется быстро и работает быстро при коротком времени длительности действия, а кристаллическая форма растворяется медленно и работает медленно с длительным временем продолжительности действия.
Каждое из твердых состояний (кристаллического или аморфного) лекарственных кандидатов имеет разные физические и химические свойства, такие как растворимость, стабильность или способность воспроизводства. Данные свойства могут оказывать влияние на конечный тип введения лекарства, оптимизированный процесс производства и абсорбцию в теле человека, и обнаружение наиболее подходящего типа для разработки лекарства теперь может сокращать время и расходы, необходимые для разработки.
Существенно, получение чистого кристаллического состояния и аморфного состояния или даже иного некристаллического состояния является очень благоприятным при разработке лекарств. Данные состояния могут придавать кандидатам в лекарства даже лучшие химические и физические свойства. Таким образом, может стать возможным образование или идентификация состояний для сочетания желаемых лечебных эффектов, а получение лекарств может стать относительно легким. Кристаллическое состояние твердой фазы имеет более благоприятные фармакологические свойства, легко получается и имеет лучшую стабильность при хранении.
Что касается кристаллических соединений, имеющих любые состояния, такие как растворенное состояние, количество остаточных растворителей в конечном лекарстве может быть небольшим. В дополнение к сказанному, благодаря кристаллизации может получаться дополнительный эффект очистки. В дополнение, поскольку данное состояние является очень стабильным во время производства лекарства, обращение с ним во время производства является удобным.
Изобретатели настоящей заявки подали патентную заявку с названием ″дихлорид 10-этокси-8-(морфолинометил)-1,2,3,4-тетрагидробензо[h][1,6]нафтидин-5(6Н)-она и способ его получения″, представленный следующей химической формулой, которая зарегистрирована Корейской патентной регистрацией под №10-0968175.
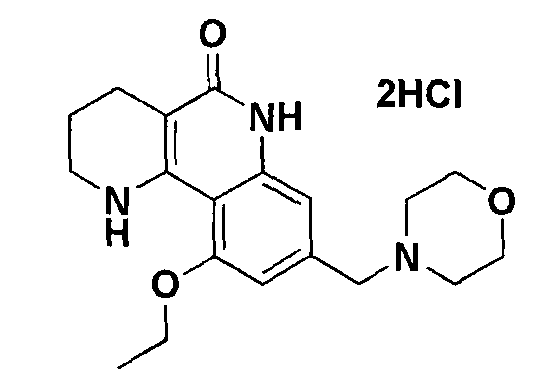
Дихлорид 10-этокси-8-(морфолинометил)-1,2,3,4-тетрагидробензо[h][1,6]нафтидин-5(6Н)-она является трициклическим производным, имеющим активность ингибирования полимеразы поли(ADP-рибозы), которое может успешно использоваться в качестве эффективного ингредиента в фармацевтической композиции для профилактики или лечения невропатической боли, эпилепсии, удара, болезни Альцгеймера, болезни Паркинсона, амиотрофического бокового склероза (ALS), болезни Хантингтона, шизофрении, хронической и острой боли, ишемического повреждения головного мозга, нейронной потери после гипоксии, травм и нервных повреждений, которые являются медицинскими состояниями, вызываемыми повышенной активностью PARP.
Однако для получения соединения в случае массового производства с использованием колоночной хроматографии используется избыточное количество растворителей. Таким образом, массовое производство является трудным вследствие проблем окружающей среды и больших расходов и времени.
Кроме того, поскольку ангидрид дихлорида 10-этокси-8-(морфолинометил)-1,2,3,4-тетрагидробензо[h][1,6]нафтидин-5(6Н)-она абсорбирует влагу в воздухе, и вес его может увеличиваться, требуется осторожное обращение с ним. Аморфный дихлорид 10-этокси-8-(морфолинометил)-1,2,3,4-тетрагидробензо[h][1,6]нафтидин-5(6Н)-она быстро или сильно абсорбирует влагу в воздухе и изменяется нестабильно в кристаллическое состояние.
Поэтому изобретатели настоящей заявки исследовали стабильное кристаллическое состояние дихлорида 10-этокси-8-(морфолинометил)-1,2,3,4-тетрагидробензо[h][1,6]нафтидин-5(6Н)-она для решения описанных выше дефектов и получения стабильного кристаллического состояния соединения 10-этокси-8-(морфолинометил)-1,2,3,4-тетрагидробензо[h][1,6]нафтидин-5(6Н)-она с высоким выходом и предложили способ разделения, для завершения настоящего изобретения.
Раскрытие изобретения
Техническая проблема
Одним объектом настоящего изобретения является предоставление новых кристаллических кислотно-аддитивных солей трициклического производного или их гидратов.
Еще одним объектом настоящего изобретения является предоставление способа получения кристаллических кислотно-аддитивных солей трициклических производных или их гидратов.
Еще одним объектом настоящего изобретения является предоставление фармацевтической композиции, включающей в себя кристаллическую кислотно-аддитивную соль трициклического производного или ее гидрат и фармацевтически приемлемый носитель, для профилактики или лечения заболеваний, вызываемых сверхактивностью PARP (полимеразы поли(ADP-рибозы)).
Техническое решение
Для того чтобы получить данные объекты, настоящее изобретение предоставляет новые кристаллические кислотно-аддитивные соли трициклических производных или их гидраты, представленные следующей химической формулой 1:
[Химическая формула 1]
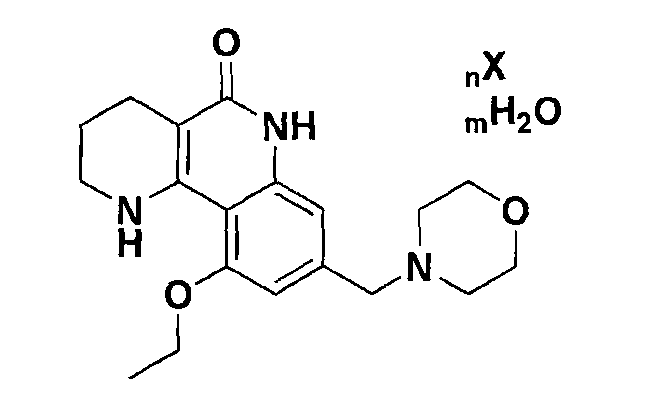 ,
,
где n, m и X имеют значения, определенные в настоящем описании.
Настоящее изобретение предоставляет также способ получения кристаллической кислотно-аддитивной соли трициклического производного или ее гидрата, включающий в себя добавление кислоты в трициклическом производном следующей химической формулы 1А в воде или органическом растворителе, проведение реакции, завершение реакции и сначала перекристаллизацию с использованием органического растворителя (стадия 1); и еще одну перекристаллизацию твердого вещества, полученного на стадии 1 с использованием воды и органического растворителя (стадия 2), как показано в следующей реакционной формуле 1:
[Реакционная формула 1]
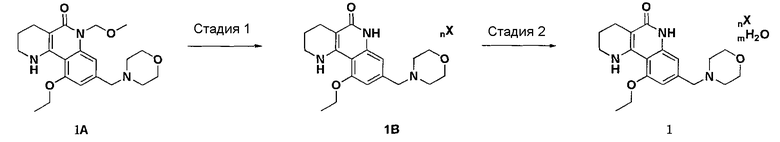 ,
,
где n, m и X имеют значения, определенные в настоящем описании.
Кроме того, настоящее изобретение предоставляет фармацевтическую композицию, включающую кристаллическую кислотно-аддитивную соль трициклического производного или ее гидрат и фармацевтически приемлемый носитель, для профилактики или лечения заболеваний, вызываемых сверхактивностью PARP.
Преимущественные эффекты
Кристаллическая кислотно-аддитивная соль или ее гидрат, согласно настоящему изобретению, является стабильной в отношении влажности и стабильной в отношении гигроскопичности, и контроль качества во время производства лекарств является благоприятным. В дополнение, кристаллическая кислотно-аддитивная соль или ее гидрат могут использоваться в фармацевтической композиции для профилактики или лечения невропатической боли, эпилепсии, удара, болезни Альцгеймера, болезни Паркинсона, амиотрофического бокового склероза (ALS), болезни Хантингтона, шизофрении, хронической и острой боли, ишемического повреждения головного мозга, нейронной потери после гипоксии, травм и нервных повреждений, которые являются медицинскими состояниями, вызываемыми сверхактивностью PARP.
Краткое описание рисунков
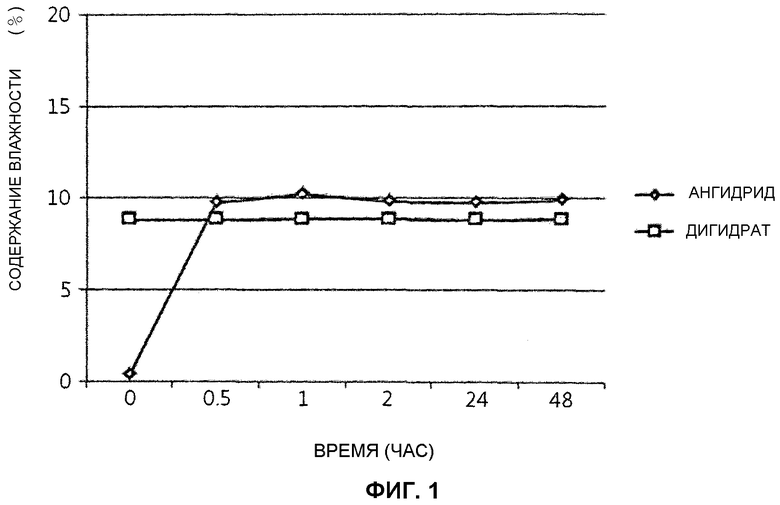
Фиг. 1 иллюстрирует содержание влаги дигидрата дихлорида согласно примеру 1 настоящего изобретения и ангидрида согласно сравнительному примеру 1;
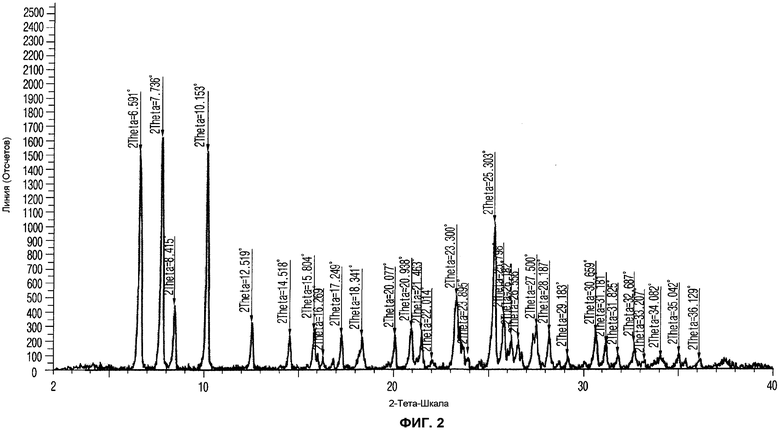
Фиг. 2 иллюстрирует XRD данные дигидрата дихлорида согласно примеру 1 настоящего изобретения; и
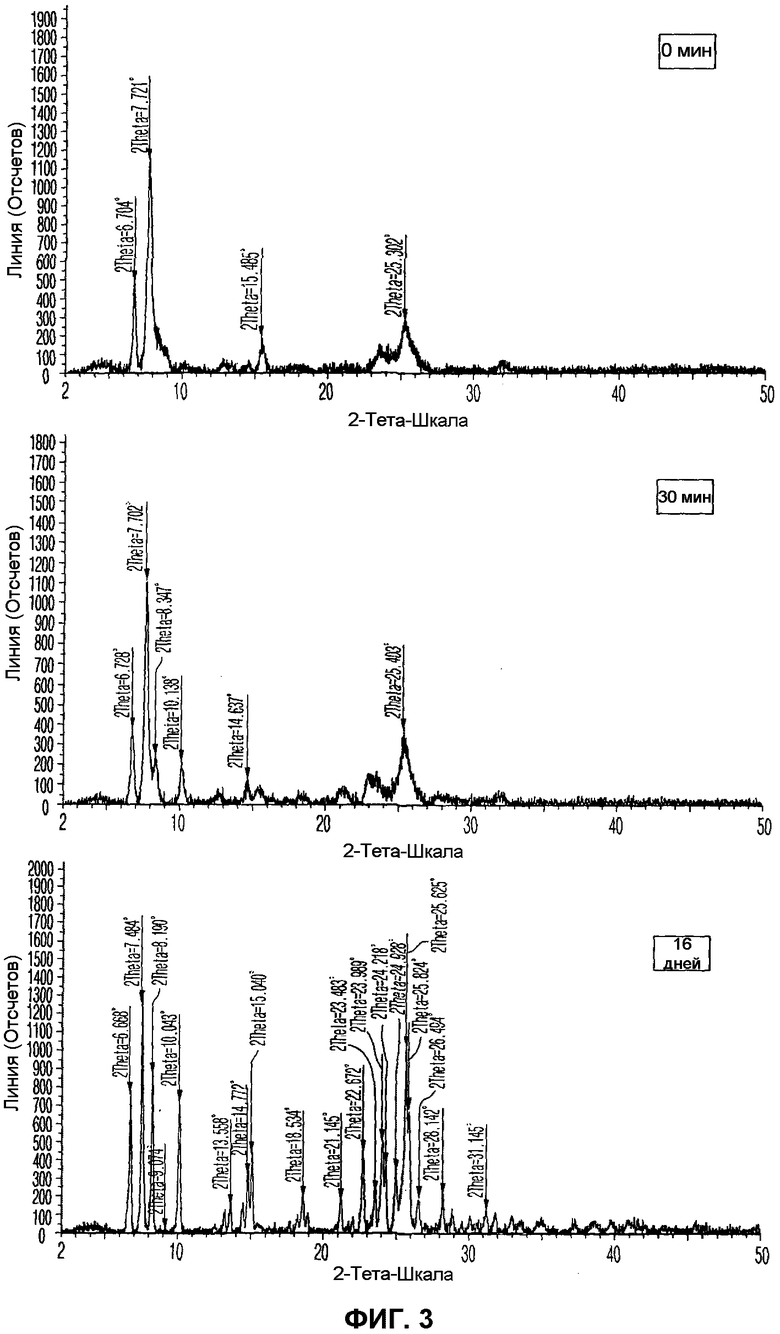
Фиг. 3 иллюстрирует изменения XRD пика аморфного соединения согласно сравнительному примеру 2 в отношении времени.
Наилучший способ осуществления изобретения
Далее ниже настоящее изобретение будет описано подробно.
Настоящее изобретение предоставляет новые кристаллические кислотно-аддитивные соли трициклического производного или их гидраты, представленные следующей химической формулой 1.
[Химическая формула 1]
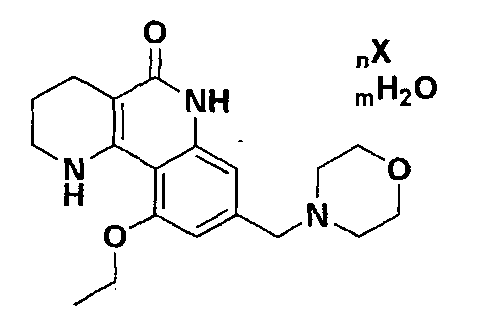 ,
,
где n или m представляет собой целое число от 0 до 3; и
Х представляет фармацевтически приемлемую неорганическую кислоту или органическую кислоту.
Предпочтительно, в приведенной выше Химической Формуле 1
n или m представляет собой целое число от 0 до 3; и
Х имеет значение, выбранное из группы, состоящей из соляной кислоты, бензолсульфоновой кислоты, малеиновой кислоты, диметансульфоновой кислоты, бис[(7,7-диметил-2-оксобицикло[2,2,1]гептан-1-ил)метансульфоновой кислоты], винной кислоты, 2,6-диоксо-1,2,3,6-тетрагидропиримидин-4-карбоновой кислоты, адипиновой кислоты, диазотной кислоты, фумаровой кислоты, (S)-2-аминоянтарной кислоты, 2-гидроксипропан-1,2,3-трикарбоновой кислоты, циклогексилсульфаминовой кислоты, серной кислоты, янтарной кислоты, муравьиной кислоты, глютаминовой кислоты и дифосфорной кислоты.
Более предпочтительно, новая кристаллическая кислотно-аддитивная соль трициклического производного или ее гидрат, представленные приведенной выше химической формулой 1, является соединением, выбранным из группы, состоящей из:
(1) дигидрата дихлорида 10-этокси-8-(морфолинометил)-1,2,3,4-тетрагидробензо[h][1,6]-нафтиридин-5(6H)-она;
(2) бензолсульфоната 10-этокси-8-(морфолинометил)-1,2,3,4-тетрагидробензо[h][1,6]нафтиридин-5(6Н)-она;
(3) малеата 10-этокси-8-(морфолинометил)-1,2,3,4-тетрагидробензо[h][1,6]нафтиридин-5(6Н)-она;
(4) диметансульфоната 10-этокси-8-(морфолинометил)-1,2,3,4-тетрагидробензо[h][1,6]нафтиридин-5(6Н)-она;
(5) бис[(7,7-диметил-2-оксобицикло[2,2,1]гептан-1-ил)метансульфоната] 10-этокси-8-(морфолинометил)-1,2,3,4-тетрагидробензо[h][1,6]нафтиридин-5(6Н)-она;
(6) тартрата 10-этокси-8-(морфолинометил)-1,2,3,4-тетрагидробензо[h][1,6]нафтиридин-5(6Н)-она;
(7) 2,6-диоксо-1,2,3,6-тетрагидропиримидин-4-карбоксилата 10-этокси-8-(морфолинометил)-1,2,3,4-тетрагидробензо[h][1,6]нафтиридин-5(6Н)-она;
(8) адипата 10-этокси-8-(морфолинометил)-1,2,3,4- тетрагидробензо[h][1,6]нафтиридин-5(6Н)-она;
(9) нитрита 10-этокси-8-(морфолинометил)-1,2,3,4- тетрагидробензо[h][1,6]нафтиридин-5(6Н)-она;
(10) фумарата 10-этокси-8-(морфолинометил)-1,2,3,4-тетрагидробензо[h][1,6]нафтиридин-5(6Н)-она;
(11) (S)-2-аминосукцината 10-этокси-8-(морфолинометил)-1,2,3,4-тетрагидробензо[h][1,6]нафтиридин-5(6Н)-она;
(12) 2-гидроксипропан-1,2,3-трикарбоксилата 10-этокси-8-(морфолинометил)-1,2,3,4-тетрагидробензо[h][1,6]нафтиридин-5(6Н)-она;
(13) циклогексилсульфамата 10-этокси-8-(морфолинометил)-1,2,3,4-тетрагидробензо[h][1,6]нафтиридин-5(6Н)-она;
(14) сульфата 10-этокси-8-(морфолинометил)-1,2,3,4-тетрагидробензо[h][1,6]нафтиридин-5(6Н)-она;
(15) сукцината 10-этокси-8-(морфолинометил)-1,2,3,4-тетрагидробензо[h][1,6]нафтиридин-5(6Н)-она;
(16) формиата 10-этокси-8-(морфолинометил)-1,2,3,4-тетрагидробензо[h][1,6]нафтиридин-5(6Н)-она;
(17) глютамата 10-этокси-8-(морфолинометил)-1,2,3,4-тетрагидробензо[h][1,6]нафтиридин-5(6Н)-она; и
(18) дифосфата 10-этокси-8-(морфолинометил)-1,2,3,4-тетрагидробензо[h][1,6]нафтиридин-5(6Н)-она.
Более предпочтительно, новой кристаллической кислотно- аддитивной солью трициклического производного или его гидрата выше указанной химической формулы 1 является дигидрат дихлорида 10-этокси-8-(морфолинометил)-1,2,3,4-тетрагидробензо[h][1,6]-нафтиридин-5(6H)-она, представленный следующей химической формулой 2.
[Химическая формула 2]
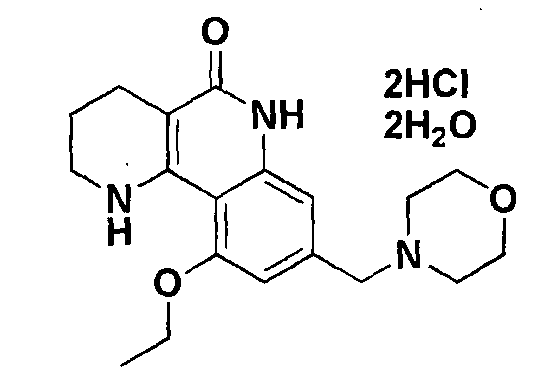
Кристаллический дигидрат дихлорида химической формулы 2 имеет 2θ значения пиков дифракции рентгеновских лучей 6,59°, 7,74°, 8,42°, 10,15°, 12,52°, 23,30° и 25,30° по характеру дифракции рентгеновских лучей Cu целевого облучения.
В дополнение, настоящее изобретение предоставляет способ получения кристаллической кислотно-аддитивной соли трициклического производного или ее гидрата, представленных приведенной выше химической формулой 1.
В частности, способ получения включает в себя:
добавление кислоты в трициклическом производном следующей химической формулы 1А в воде или органическом растворителе, проведение реакции, завершение реакции и сначала перекристаллизацию с использованием органического растворителя (стадия 1); и
еще одну перекристаллизацию твердого вещества, полученного на стадии 1 с использованием воды и органического растворителя (стадия 2), как показано на следующей Реакционной Формуле 1:
[Реакционная формула 1]
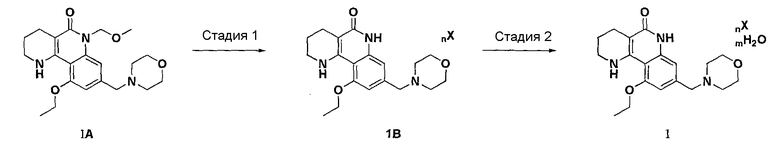 ,
,
где n, m и X имеют значения, определенные в приведенной выше Химической Формуле 1.
Показанная выше Стадия 1 является стадией получения целевого соединения растворением трициклического производного приведенной выше химической формулы 1А в органическом растворителе и добавлением к нему кислоты.
В данном случае органическим растворителем может быть С1-С4 спирт, и может быть метанол или этанол.
Кроме того, кислотой может быть фармацевтически приемлемая неорганическая кислота или органическая кислота, и может быть кислотой, выбранной из группы, состоящей из соляной кислоты, бензолсульфоновой кислоты, малеиновой кислоты, диметансульфоновой кислоты, бис[(7,7-диметил-2-оксобицикло[2,2,1]гептан-1-ил)метаносульфоновой] кислоты, винной кислоты, 2,6-диоксо-1,2,3,6-тетрагидропиримидин-4-карбоновой кислоты, адипиновой кислоты, диазотной кислоты, фумаровой кислоты, (S)-2-аминоянтарной кислоты, 2-гидроксипропан-1,2,3-трикарбоновой кислоты, циклогексилсульфаминовой кислоты, серной кислоты, янтарной кислоты, муравьиной кислоты, глютаминовой кислоты и дифосфорной кислоты.
Далее, органический растворитель, используемый при первой перекристаллизации в приведенной выше стадии 1, обозначает растворитель, который не растворяет соединение настоящего изобретения, и может быть, по крайней мере, растворителем, выбранным из группы, состоящей из ацетона, гексана, гептана, толуола, этилацетата, дихлорметана, тетрагидрофурана и хлороформа. Предпочтительно может использоваться смесь этанола и этилацетата.
В данном случае соотношение добавления этанола и этилацетата может составлять 1:8-10. В случае, когда соотношение добавления отклоняется от указанного интервала, образование целевого соединения в твердом состоянии может быть затруднено.
В дополнение, приведенная выше стадия 2 является еще одной стадией перекристаллизации путем добавления воды и органического растворителя в твердое вещество, получаемое на приведенной выше стадии 1. После растворения твердого вещества, получаемого на показанной выше стадии 1, добавляется анти-растворитель с последующим перемешиванием или оставлением смеси для получения осадка.
В данном случае используемый органический растворитель обозначает растворитель, в котором соединение настоящего изобретения является нерастворимым, т.е. анти-растворитель, и может быть растворителем, выбранным из группы, состоящей из ацетона, гексана, гептана, толуола, этилацетата, дихлорметана, тетрагидрофурана и хлороформа. Предпочтительно может использоваться ацетон.
Соотношение добавления воды и ацетона может составлять 1:8-10. В случае, когда соотношение добавления отклоняется от указанного интервала, образование целевого соединения в твердом состоянии может быть затруднено.
Далее, настоящее изобретение предоставляет фармацевтическую композицию, включающую в свой состав кристаллическую кислотно-аддитивную соль трициклического производного или ее гидрат, представленные приведенной выше химической формулой 1, и фармацевтически приемлемый носитель, для профилактики или лечения заболеваний, вызываемых сверхактивностью PARP.
Кристаллическая кислотно-аддитивная соль или ее гидрат согласно настоящему изобретению является стабильной в отношении влажности и стабильной в отношении гигроскопичности, и контроль качества во время производства лекарств является благоприятным (См. экспериментальные примеры 1 и 2). В дополнение, поскольку трициклическое производное оказывает хорошие лечебные действия на невропатическую боль, эпилепсию, удар, болезнь Альцгеймера, болезнь Паркинсона, амиотрофический боковой склероз (ALS), болезнь Хантингтона, шизофрению, хроническую и острую боль, ишемическое повреждение головного мозга, нейронную потерю после гипоксии, травмы и нервные повреждения, которые являются медицинскими состояниями, вызываемыми сверхактивностью PARP (Корейская патентная публикация № 2010-0053468), трицилическое производное может использоваться в виде фармацевтической композиции для профилактики или лечения заболеваний, вызываемых сверхактивностью PARP.
В случае, когда композиция настоящего изобретения используется в качестве медицинских средств, фармацевтическая композиция, содержащая трициклическую кристаллическую кислотно-аддитивную соль или ее гидрат в качестве эффективного ингредиента, может формулироваться в виде разнообразных типов перорального или парентерального введения для клинического введения без ограничений.
Рецептурная форма для перорального введения может включать в себя таблетки, пилюли, твердые/мягкие капсулы, жидкие рецептурные формы, суспензии, эмульсии, сиропы, гранулы, эликсиры, троши и проч. Рецептурная форма включает в себя разбавитель (например, лактозу, декстрозу, сахарозу, манит, сорбит, целлюлозу и/или глицерин) и модификатор скольжения (например, двуокись кремния, тальк, стеариновую кислоту и ее магниевую или кальциевую соль, и/или полиэтиленгликоль), отличный от эффективных ингредиентов. Рецептурная форма может содержать связующий агент, такой как магниевый силикат алюминия, крахмальная паста, желатин, метилцеллюлоза, натриевая карбоксиметилцеллюлоза и/или поливинилпирролидин, и может содержать крахмал, агар-агар, дезинтегрирующий агент, такой как альгиновая кислота и ее натриевая соль, или кипящую смесь и/или абсорбент, окрашивающий агент, вкусовой агент и подслащивающий агент, в зависимости от потребностей.
Фармацевтическая композиция, включающая в свой состав трициклическую кристаллическую кислотно-аддитивную соль или ее гидрат в качестве эффективного ингредиента, могут вводиться парентерально, и парентеральное введение может включать в себя гиподермальную инъекцию, внутривенную инъекцию или внутриторакальную (внутригрудную) инъекцию.
Для рецептурной формы для парентерального введения трициклическая кристаллическая кислотно-аддитивная соль или ее гидрат смешивается со стабилизатором или буферным агентом в воде с получением жидкости или суспензии, и может производиться для типа введения в виде ампул или пузырьков. Композиция может стерилизоваться и/или может содержать антисептик, стабилизатор, диспергируемый в воде порошок или эмульсионный промотор, вспомогательный агент, такой как соль для регулирования осмотического давления и/или буферный агент, и другие полезные материалы для лечения. Рецептурная форма может получаться обычным способом, предусматривающим смешение, гранулирование или нанесение покрытия.
Дозировка фармацевтической композиции, содержащей в качестве эффективного ингредиента трициклическую кристаллическую кислотно-аддитивную соль или ее гидрат, в организме человека зависит от возраста, веса, пола, типа введения, физических состояний и степени заболеваний пациента. Предпочтительно, композиция может вводиться перорально или парентерально несколько раз в день, предпочтительно 1-3 раза в день, с постоянным интервалом времени в количестве 0,01-200 мг/кг/день согласно решению врача или фармацевта.
Способ осуществления изобретения
Далее, данное изобретение будет объяснено более подробно со ссылкой на примеры и сравнительные примеры.
Однако примеры и сравнительные примеры являются иллюстративными, и данное изобретение не должно расцениваться как ограниченное примерами и сравнительными примерами.
<Пример 1> Получение дигидрата дихлорида 10-этокси-8-(морфолинометил)-1,2,3,4-тетрагидробензо[h][1,6]нафтиридин-5(6Н)-она
Стадия 1: Получение метил 3-гидрокси-5-нитробензоата
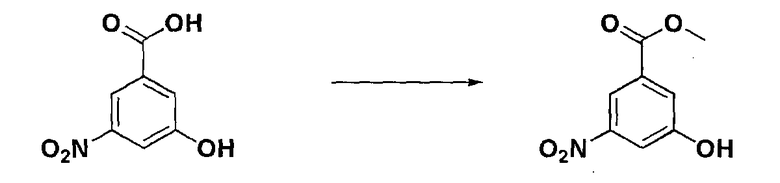
К 3-гидрокси-5-нитробензойной кислоте (5,9 кг, 32,2 моль) добавляли метанол (60 л), и к ним медленно добавляли серную кислоту (375 мл, каталитическое количество) с последующим нагреванием с обратным холодильником и перемешиванием при 80°С в течение 18 часов. После завершения реакции, реагирующее вещество охлаждали до комнатной температуры. К нему добавляли очищенную воду (50 л), и метанол отгоняли при пониженном давлении. Полученное таким образом твердое вещество перемешивали при 10°С в течение 1 часа и фильтровали. Фильтрат промывали очищенной водой и сушили при 60°С, получая указанное в заголовке целевое соединение (5,52 кг, выход:87%, желтое твердое вещество).
1Н-ЯМР (400 МГц, ДМСО-d6) δ 10,91 (с, 1H), 8,04 (с, 1H), 7,74 (с, 1H), 7,66 (с, 1H), 3,88 (с, 3H).
Стадия 2: Получение метил 3-этокси-5-нитробензоата
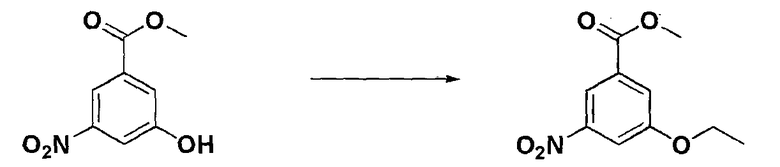
К соединению, полученному в стадии 1 (5,52 кг, 28,0 ммоль), добавляли ацетонитрил (36 л) и к ним добавляли этилиодид (6,1 кг, 39,2 моль) и карбонат калия (5,8 кг, 42,0 моль) с последующим нагреванием с обратным холодильником и перемешиванием при 95°С в течение 18 часов. После завершения реакции, реагирующее вещество охлаждали до комнатной температуры. К нему добавляли очищенную воду (36 л), и ацетонитрил отгоняли при пониженном давлении. Полученное таким образом твердое вещество фильтровали, промывали очищенной водой и сушили при 60°С, получая целевое соединение (6,1 кг, выход: 96,9%, желтое твердое вещество).
1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,18 (с, 1H), 7,93 (с, 1H), 7,78 (с, 1H), 4,21 (кв, J=6,6 Гц, 2H), 3,91 (с, 3H), 1,37 (т, J=6,6 Гц, 3H).
Стадия 3: Получение метил 3-амино-5-этоксибензоата
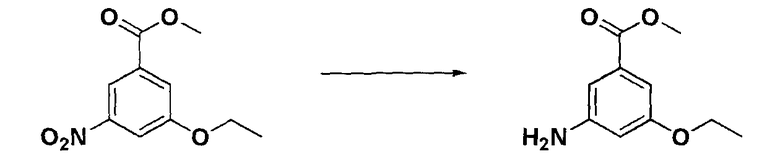
К соединению, полученному в стадии 2 (6,1 кг, 27,1 моль), добавляли метанол (54 л) и очищенную воду (54 л), и к ним добавляли 10%-палладий на угле (6,1 кг, 10% вес/вес) с последующим перемешиванием в условиях газообразного водорода при 4 атм. в течение 2,5 часов при комнатной температуре. После подтверждения завершения реакции, добавляли ацетон (54 л) и проводили фильтрование для удаления 10%-палладия на угле. Метанол и ацетон отгоняли при пониженном давлении. Полученное таким образом твердое вещество фильтровали и промывали очищенной водой, получая целевое соединение (4,88 кг, выход: 92,2%, желтое твердое вещество).
1Н-ЯМР (400 МГц, CDCl3) δ 6,95 (с, 2H), 6,39 (с, 1H), 4,01 (кв, J=6,8 Гц, 2H), 3,87 (с, 3H), 3,76 (с, 2H), 1,38 (т, J=6,8 Гц, 3H).
Стадия 4: Получение метил 3-(2-хлорникотинамидо)-5-этоксибензоата
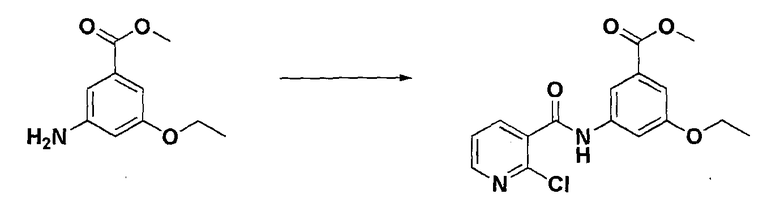
К соединению, полученному в стадии 3 (4,88 кг, 25,0 моль), добавляли дихлорметан (60 л), и к ним добавляли 2-хлорникотиновую кислоту (5,5 кг, 35,0 моль), EDC·HCl (6,7 кг, 35 моль) и гидроксибензотриазол (1,0 кг, 7,5 моль) с последующим перемешиванием при комнатной температуре в течение 2 часов. После подтверждения завершения реакции добавляли карбонат калия (1,0 кг) и очищенную воду (30 л), и органический слой отгоняли при пониженном давлении. К полученной таким образом суспензии добавляли этилацетат (5 л) и н-гексан (50 л) и перемешивали. Полученное твердое вещество фильтровали и сушили, получая целевое соединение (8,98 кг, выход: четвертичный выход, не совсем белое твердое вещество).
1H-ЯМР (400 МГц, CDC13) δ 8,53 (с, 1H), 8,49 (д, J=4,8 Гц, 1H), 8,14 (д, J=7,6 Гц, 1H), 7,78 (с, 1H), 7,66 (с, 1H), 7,40-7,37 (м, 2H), 4,33 (кв т, J=7,6 Гц, 2H), 3,88 (с, 3H), 1,38 (т, J=7,6 Гц, 3H).
Стадия 5: Получение метил 3-(2-хлор-N-(метоксиметил)никотинамидо)-5-этоксибензоата
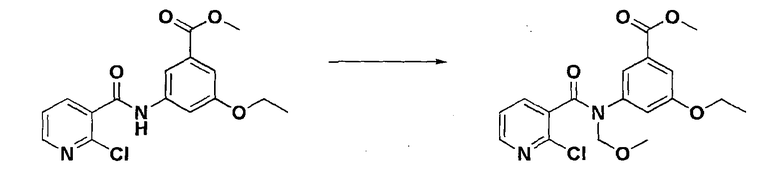
К соединению, полученному в стадии 4 (8,98 кг, 25,0 моль), добавляли дихлорметан (90 л) и растворяли, и к ним добавляли метоксиметилхлорид (5,03 кг, 50,0 моль) и тетрабутиламмонийбромид (3,2 кг, 10,0 моль). Реакционную смесь охлаждали до 10°С, и по каплям добавляли гидроксид натрия (4,0 кг, 10,0 моль), растворенного в очищенной воде (9 л) с последующим энергичным перемешиванием при комнатной температуре в течение 4 часов. После подтверждения завершения реакции добавляли очищенную воду (30 л), и органический слой отгоняли при пониженном давлении. К полученной таким образом суспензии добавляли этилацетат (50 л) и растворяли, и полученное в результате вещество промывали очищенной водой (40 л) 6 раз, отгоняли при пониженном давлении и сушили в вакууме, получая целевое соединение (9,0 кг, выход: 95%, желтое твердое вещество).
1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,24 (д, 1H, J=2,4 Гц), 7,93 (д, 1H, J=4,4 Гц), 7,41 (с, 1H), 7,30-7,29 (м, 1H), 7,21 (с, 1H), 7,15 (с, 1H), 5,17 (с, 2H), 3,94 (кв, 2H, J=6,8 Гц), 3,77 (с, 3H), 3,41 (с, 3H), 1,23 (т, 3H, J=6,8 Гц).
Стадия 6: Получение метил 10-этокси-6-(метоксиметил)-5-оксо-5,6-дигидробензо[h][1,6]нафтиридин-8-карбоксилата
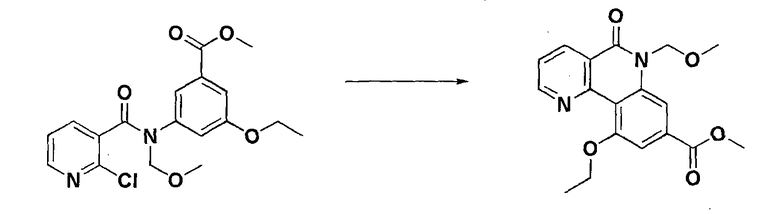
К соединению, полученному в стадии 5 (11,4 кг, 30,0 моль), добавляли N,N-диметилформамид (54 л), и к ним добавляли ацетат палладия (1,84 кг, 7,52 моль), трибутилфосфин (6,1 кг, 30 моль) и карбонат натрия (8,3 кг, 60 моль) с последующим перемешиванием при 130°С в течение 2 часов. После подтверждения завершения реакции реагент охлаждали и добавляли очищенную воду (108 л). Полученное таким образом твердое вещество фильтровали, промывали очищенной водой и сушили при 60°С, получая целевое соединение (6,88 кг, выход: 67,0%, черное твердое вещество).
1H-ЯМР (400 МГц, CDCl3) δ 9,11-9,10 (м, 1H), 8,84 (тд, J=2,0 Гц, 8,0 Гц, 1H), 7,97 (с, 1H), 7,60 (с, 1H), 7,53-7,50 (м, 1H), 5,85 (с, 2H), 4,34 (кв, J=6,8 Гц, 2H), 3,98 (с, 3H), 3,49 (с, 3H), 1,60 (т, J=6,8 Гц, 3H).
Стадия 7: Получение метил 10-этокси-6-(метоксиметил)-5-оксо-1,2,3,4,5,6-гексагидробензо[h][1,6]нафтиридин-8-карбоксилата

К соединению, полученному в стадии 6 (6,88 кг, 27,1 моль), добавляли тетрагидрофуран (54 л) и очищенную воду (54 л), и к ним добавляли 10%-палладий на угле (13,8 кг, 20% вес/вес) с последующим перемешиванием в условиях газообразного водорода при 4 атм. в течение 4,5 часов при комнатной температуре. После подтверждения завершения реакции, добавляли дихлорметан (54 л). Проводили фильтрование для удаления 10%-палладия на угле, и органический слой отгоняли при пониженном давлении. К полученной таким образом суспензии добавляли н-гексан (50 л) и перемешивали в течение 1 часа. Полученное твердое вещество фильтровали и сушили, получая целевое соединение (5,01 кг, выход: 72,2%, не совсем белое твердое вещество).
1Н-ЯМР (400 МГц, CDCl3) δ 7,85 (с, 1H), 7,63 (с, 1H), 7,29 (с, 1H), 5,73 (с, 2H), 4,26 (кв, J=7,2 Гц, 2H), 3,94 (с, 3H), 3,42 (с, 3H), 3,42-3,37 (м, 2H), 2,69 (т, J=6,0 Гц, 2H), 1,93-1,90 (м, 2H), 1,53 (т, J=7,2 Гц, 3H).
Стадия 8: Получение 10-этокси-8-(гидроксиметил)-6-(метоксиметил)-1,2,3,4-тетрагидробензо[h][1,6]нафтиридин-5-(6Н)-она

К соединению, полученному в стадии 7 (500 г, 1,44 моль), добавляли тетрагидрофуран (4 л) и при 10°С медленно добавляли литийалюминий гидрид (50,0 г, 1,34 моль) с последующим перемешиванием при комнатной температуре в течение 1 часа. После подтверждения завершения реакции, одно за другим медленно добавляли очищенную воду (50 мл), 15% водный раствор гидроксида натрия (50 мл) и очищенную воду (150 мл) с последующим перемешиванием при комнатной температуре в течение 2 часов. Полученную таким образом суспензию фильтровали, и фильтрат концентрировали при пониженном давлении. К полученному твердому веществу добавляли этилацетат (2,0 л) и перемешивали в течение 1 часа. Проводили фильтрование и сушку, получая целевое соединение (361 г, выход: 78,7%, не совсем белое твердое вещество).
1H-ЯМР (400 МГц, CDCl3) δ 7,63 (с, 1H), 7,00 (с, 1H), 6,73 (с, 1H), 5,55 (с, 2H), 4,69 (с, 2H), 4,19 (кв, J=7,2 Гц, 2H), 3,36-3,35 (м, 2H), 3,36 (с, 3H), 2,65 (т, J=6,0 Гц, 2H), 1,91-1,88 (м, 2H), 1,51 (т, J=7,2 Гц, 3H).
Стадия 9: Получение 8-(хлорметил)-10-этокси-6-(метоксиметил)-1,2,3,4-тетрагидробензо[h][1,6]нафтиридин-5-(6Н)-она
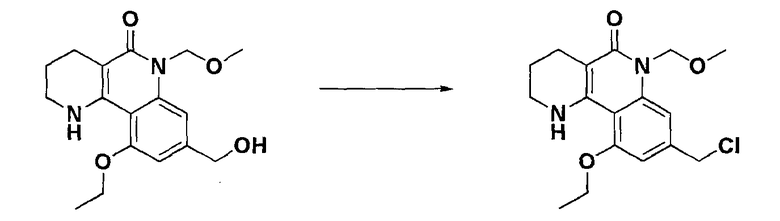
К соединению, полученному в стадии 8 (310,1 г, 0,97 моль), добавляли дихлорметан (1,5 л) и медленно, при 0°С добавляли тионилхлорид (106,5 мл, 1,46 моль) с последующим нагреванием с обратным холодильником и перемешиванием при 50°С в течение 2 часов. После подтверждения завершения реакции, добавляли очищенную воду (500 мл) и проводили нейтрализацию с применением водного раствора бикарбоната натрия. Органический слой концентрировали при пониженном давлении. Полученное таким образом твердое вещество перекристаллизовывали с использованием этилацетата (0,5 л) и н-гексана (1,0 л), получая целевое соединение (305,4 г, выход: 93,1%, не совсем белое твердое вещество).
1H-ЯМР (400 МГц, CDCl3) δ 7,59 (с, 1H), 7,14 (с, 1H), 6,69 (с, 1H), 5,67 (с, 2H), 4,59 (с, 2H), 4,20 (кв, 2H, J=6,8 Гц), 3,40 (с, 3H), 3,40-3,35 (м, 2H), 2,66 (т, 2H, J=6,0 Гц), 1,92- 1,86 (м, 2H), 1,53 (т, 3H, J=6,8 Гц).
Стадия 10: Получение 10-этокси-6-(метоксиметил)-8-(морфолинометил)-1,2,3,4-тетрагидробензо[h][1,6]нафтиридин-5-(6Н)-она
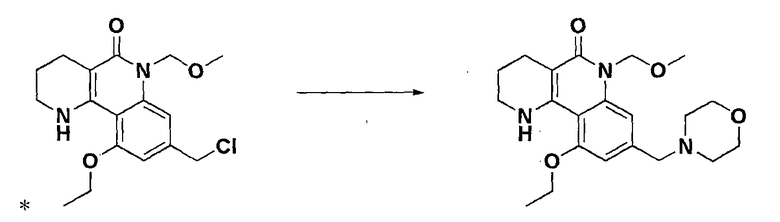
К соединению, полученному в стадии 9 (305,4 г, 0,907 моль), один за другим добавляли метанол (3,0 л) и морфолин (395 мл, 4,53 моль) с последующим нагреванием с обратным холодильником и перемешиванием при 80°С в течение 18 часов. После подтверждения завершения реакции, добавляли очищенную воду (3,0 л), и органический слой концентрировали при пониженном давлении. Полученное таким образом твердое вещество перекристаллизовывали с использованием этилацетата (1,0 л) и н-гексана (2,0 л), получая целевое соединение (326,9 г, выход: 96,5%, не совсем белое твердое вещество).
1Н-ЯМР (400 МГЦ, ДМСО) δ 7,61 (С, 1H), 7,08 (С, 1H), 6,72 (с, 1H), 5,68 (с, 2H), 4,19 (кв, 2H, J=6,8 Гц), 3,72-3,69 (м, 4H), 3,51 (с, 2H), 3,40 (с, 3H), 3,40-3,35 (м, 2H), 2,66 (т, 2H, J=6,0 Гц), 2,45 (м, 4H), 1,90-1,87 (м, 2H), 1,52 (т, 3H, J=6,8 Гц).
Стадия 11: Получение дигидрата дихлорида 10-этокси-8-(морфолинометил)-1,2,3,4-тетрагидробензо[h][1,6]нафтиридин-5-(6Н)-она

К соединению, полученному в стадии 10 (326,9 г, 0,875 моль) добавляли этанол (2,3 л) и с-соляную кислоту (230 мл) с последующим нагреванием с обратным холодильником и перемешиванием при 90°С в течение 3 часов. После подтверждения завершения реакции, реакционную смесь концентрировали при пониженном давлении и перекристаллизовывали в этаноле (100 мл) и этилацетате (900 мл). Полученное таким образом твердое вещество перекристаллизовывали с использованием воды (1,0 л) и ацетона (8,0 л), получая целевое соединение (351 г, выход: 88,7%, влагосодержание: 8,5%, белое твердое вещество).
1Н-ЯМР (400 МГЦ, ДМСО-d6) δ 12,21 (с, 1H), 12,13 (с, 1H), 7,55 (с, 1H), 7,11 (с, 1H), 4,39-4,34 (м, 4H), 3,91-3,90 (м, 4H), 3,44-3,41 (м, 2H), 3,19-3,13 (м, 4H), 2,50 (т, 2H, J=6,0 Гц), 1,80-1,77 (м, 2H), 1,44 (т, 3H, J=6,8 Гц).
<Пример 2> Получение бензолсульфоната 10-этокси-8-(морфолинометил)-1,2,3,4-тетрагидробензо[h][1,6]нафтиридин-5-(6Н)-она
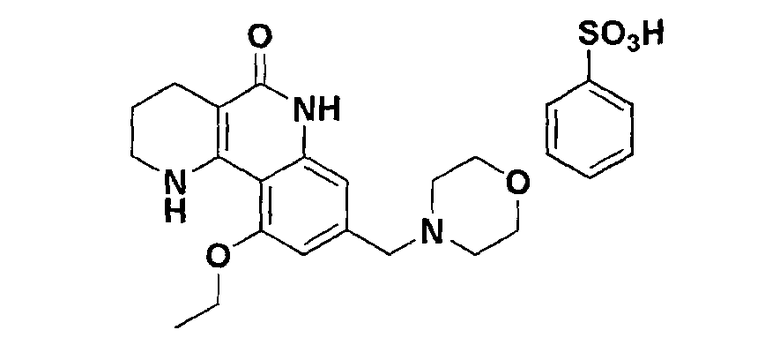
Осуществляли ту же самую процедуру, как описано в примере 1, за исключением того, что в стадии 10 использовали бензолсульфоновую кислоту вместо с-соляной кислоты, получая целевое соединение (716 мг, 98%).
1H-ЯМР (400 МГц, ДМСО-d6) δ 11,34 (с, 1H), 9,87 (ушир., 1H), 7,61-7,59 (м, 2H), 7,35-7,30 (м, 3H), 6,92 (с, 1H), 6,87 (с, 1H), 4,34 (ушир., 2H), 4,23 (кв, J=3,4 Гц, 2H), 3,98-3,95 (м, 2H), 3,63 (т, J=12,0 Гц, 2H), 3,36 (ушир., 2H), 3,28 (д, J=6,2 Гц, 2H), 3,15-3,13 (м, 2H), 2,48-2,45 (м, 2H), 1,78-1,75 (м, 2H), 1,44 (т, J=6,8 Гц, 3H).
<Пример 3> Получение малеата 10-этокси-8-(морфолинометил)-1,2,3,4-тетрагидробензо[h][1,6]нафтиридин-5-(6Н)-она
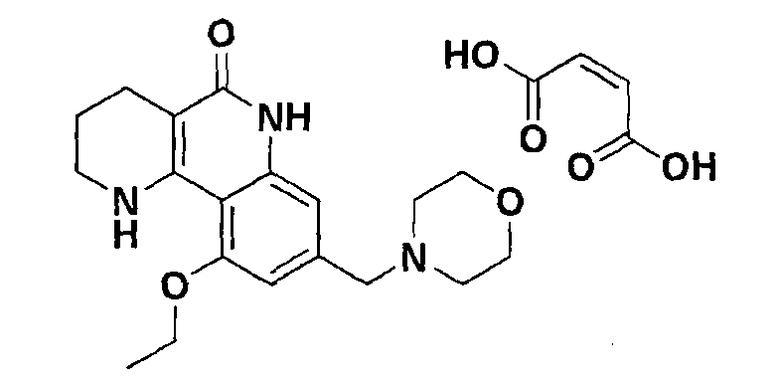
Осуществляли ту же самую процедуру, как описано в примере 1, за исключением того, что в стадии 10 использовали малеиновую кислоту вместо с-соляной кислоты, получая целевое соединение (569 мг, 85%).
1H-ЯМР (400 МГц, ДМСО-d6) δ 10,92 (ушир., 1H), 7,38 (с, 1H), 6,83 (с, 1H), 6,72 (с, 1H), 6,10 (с, 1H), 4,21 (кв, J=3,6 Гц, 2H), 3,70 (ушир., 4H), 3,32 (ушир., 6H), 2,94 (ушир., 2H), 2,43 (т, J=6,4 Гц, 2H), 1,75 (т, J=5,6 Гц, 2H), 1,05 (т, J=7,2 Гц, 3H).
<Пример 4> Получение диметансульфоната 10-этокси-8-(морфолинометил)-1,2,3,4-тетрагидробензо[h][1,6]нафтиридин-5-(6Н)-она
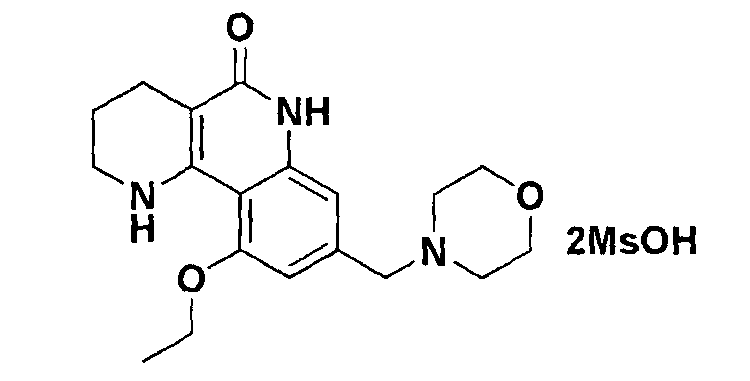
Осуществляли ту же самую процедуру, как описано в примере 1, за исключением того, что в стадии 10 использовали диметансульфоновую кислоту вместо с-соляной кислоты, получая целевое соединение (760 мг, 97%).
1Н-ЯМР (400 МГц, ДМСО-d6) δ 11,69 (с, 1H), l0,01 (с, 1H), 7,88-7,85 (ушир., 2H), 6,98 (с, 1H), 6,97 (с, 1H), 4,36 (с, 2H), 4,29 (кв, J=6,8 Гц, 2H), 3,96 (м, 2H), 3,65 (м, 2H), 3,38 (м, 2H), 3,27 (м, 2H), 3,15 (м, 2H), 2,48 (м, 2H), 2,38 (с, 6H), 1,77 (м, 2H), 1,45 (т, J=6,8 Гц, 3H).
<Пример 5> Получение бис[(7,7-диметил-2-оксобицикло[2,2,1]гептан-1-ил)метансульфоната] 10-этокси-8-(морфолинометил)-1,2,3,4-тетрагидробензо[h][1,6]нафтиридин-5-(6Н)-она
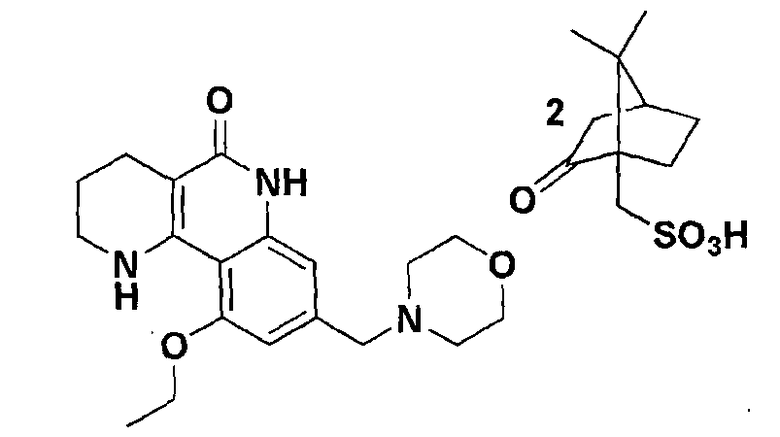
Осуществляли ту же самую процедуру, как описано в примере 1, за исключением того, что в стадии 10 использовали бис[(7,7-диметил-2-оксобицикло[2,2,1]гептан-1-ил)метансульфоновую кислоту вместо с-соляной кислоты, получая целевое соединение (900 мг, 77%).
1H-ЯМР (400 МГц, ДМСО-d6) δ 11,60 (с, 1H), 9,98 (с, 1H), 7,82-7,23 (ушир.,2H), 6,95 (м, 2H), 4,33 (с, 2H), 4,27 (кв, J=6,8 Гц, 2H), 3,94 (м, 2H), 3,65 (м, 2H), 3,36 (м, 2H), 3,25 (м, 2H), 3,14 (м, 2H), 2,90 (с, 1H), 2,87 (с, 1H), 2,62 (м, 2H), 2,45 (м, 2H), 2,42 (с, 1H), 2,38 (с, 1H), 2,24 (м, 1H), 2,19 (м, 1H), 1,92 (т, J=4,4 Гц, 2H), 1,85-1,74 (м, 6H), 1,43 (т, J=7,2 Гц, 3H), 1,31-1,22 (м, 4H), 1,01 (с, 6H), 0,71 (с, 6H).
<Пример 6> Получение тартрата 10-этокси-8-(морфолинометил)-1,2,3,4-тетрагидробензо[h][1,6]нафтиридин-5-(6Н)-она
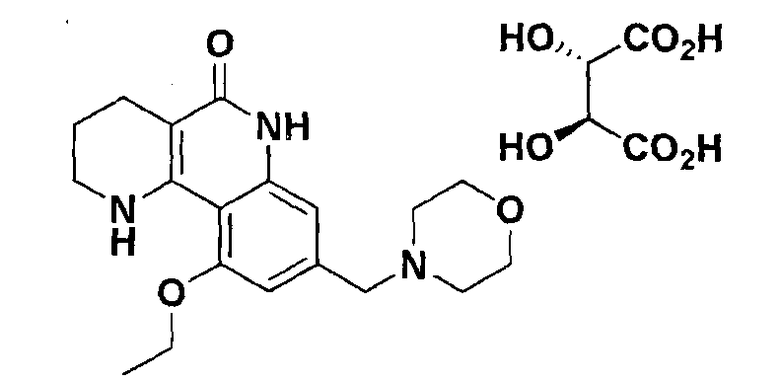
Осуществляли ту же самую процедуру, как описано в примере 1, за исключением того, что в стадии 10 использовали виннокаменную кислоту вместо с-соляной кислоты, получая целевое соединение (640 мг, 89%).
1Н-ЯМР (400 МГц, ДМСО-d6) δ 10,69 (с, 1H), 7,36 (с, 1H), 6,76 (с, 1H), 6,60 (с, 1H), 4,29 (с, 2H), 4,16 (кв, J=6,8 Гц, 2H), 3,58 (м, 4H), 3,44 (с, 2H), 3,31 (м, 2H), 2,41-2,38 (м, 6H), 1,74 (м, 2H), 1,40 (т, J=6,8 Гц, 3H).
<Пример 7> Получение 2,6-диоксо-1,2,3,6-тетрагидропиримидин-4-карбоксилата 10-этокси-8-(морфолинометил)-1,2,3,4-тетрагидробензо[h][1,6]нафтиридин-5-(6Н)-она
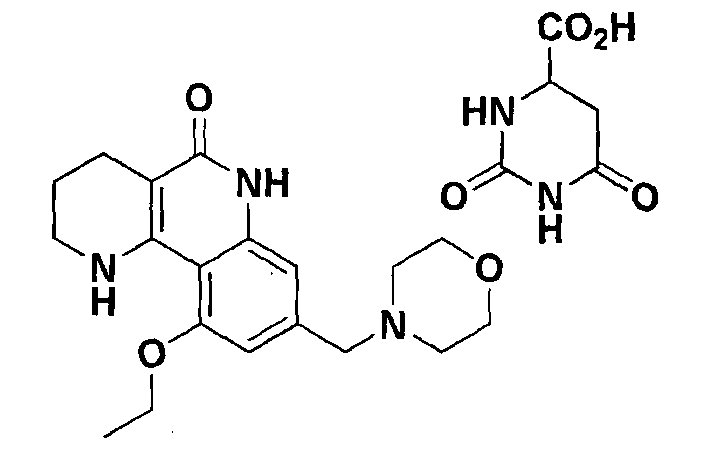
Осуществляли ту же самую процедуру, как описано в примере 1, за исключением того, что в стадии 10 использовали 2,6-диоксо-1,2,3,6-тетрагидропиримидин-4-карбоновую кислоту вместо с-соляной кислоты, получая целевое соединение (640 мг, 88%).
1Н-ЯМР (400 МГЦ, ДМСО-d6) δ 11,19 (с, 1H), 10,85 (с, 1H), 10,42 (с, 1H), 7,36 (с, 1H), 6,81 (с, 1H), 6,71 (с, 1H), 5,89 (с, 1H), 4,16 (кв, J=6,8 Гц, 2H), 3,81 (с, 2H), 3,68 (м, 4H), 3,31 (м, 2H), 2,72 (м, 4H), 2,42 (т, J=6,0 Гц, 2H), 1,74 (м, 2H), 1,40 (т, J=6,8 Гц, 3H).
<Пример 8> Получение адипата 10-этокси-8-(морфолинометил)-1,2,3,4-тетрагидробензо[h][1,6]нафтиридин-5-(6Н)-она
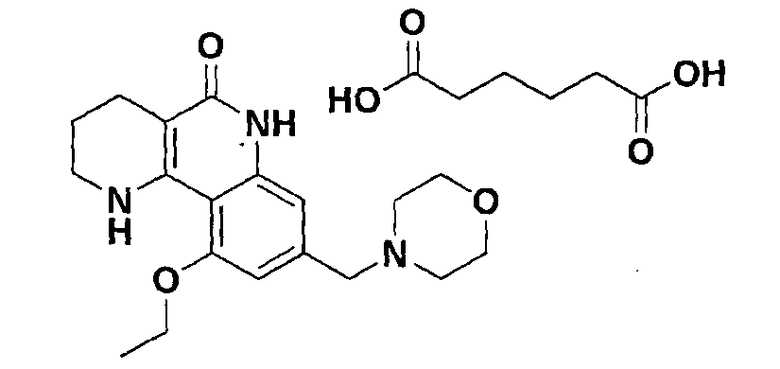
Осуществляли ту же самую процедуру, как описано в примере 1, за исключением того, что в стадии 10 использовали адипиновую кислоту вместо с-соляной кислоты, получая целевое соединение (400 мг, 56%).
1H-ЯМР (400 МГц, ДМСО-d6) δ 12,04 (с, 1H), 10,69 (с, 1H), 7,36 (с, 1H), 6,76 (с, 1H), 6,59 (с, 1H), 4,17 (кв, J=3,4 Гц, 2H), 3,58 (ушир., 4H), 3,41 (ушир., 2H), 3,31 (ушир., 2H), 2,42 (т, J=6,0 Гц, 2H), 2,35 (ушир., 4H), 1,74 (ушир., 2H), 1,49 (ушир., 4H), 1,41 (т, J=7,2 Гц, 3H).
<Пример 9> Получение нитрита 10-этокси-8-(морфолинометил)-1,2,3,4-тетрагидробензо[h][1,6]нафтиридин-5-(6Н)-она
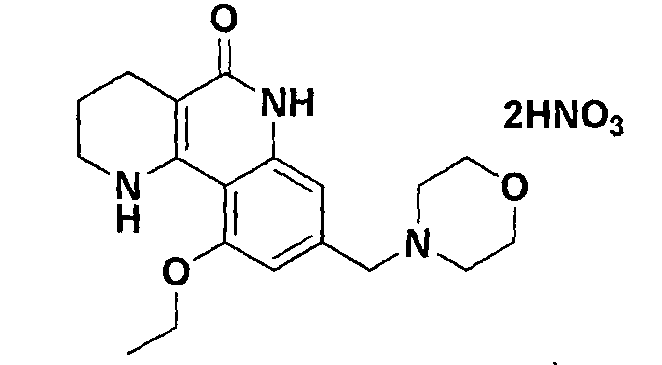
Осуществляли ту же самую процедуру, как описано в примере 1, за исключением того, что в стадии 10 использовали динитро кислоту вместо с-соляной кислоты, получая целевое соединение (638 мг, 96%).
1H-ЯМР (400 МГц, ДМСО-d6) δ 11,61 (с, 1H), 9,91 (ушир., 1H), 7,85 (ушир., 1H), 6,96 (с, 1H), 6,89 (с, 1H), 4,36-4,35 (м, 2H), 4,27 (кв, J=3,4 Гц, 2H), 3,97 (д, J=5,6 Гц, 2H), 3,61 (т, J=11,6 Гц, 2H), 3,41-3,37 (м,, 2H), 3,28 (д, J=6,0 Гц, 2H), 3,16-3,13 (м, 2H), 2,50-2,48 (м, 2H), 1,79-1,76 (м, 2H), 1,45 (т, J=7,2 Гц, 3H).
<Пример 10> Получение фумарата 10-этокси-8-(морфолинометил)-1,2,3,4-тетрагидробензо[h][1,6]нафтиридин-5-(6Н)-она
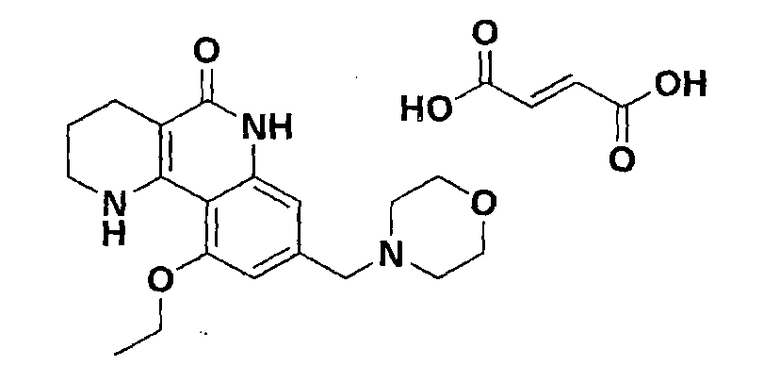
Осуществляли ту же самую процедуру, как описано в примере 1, за исключением того, что в стадии 10 использовали фумаровую кислоту вместо с-соляной кислоты, получая целевое соединение (426 мг, 64%).
1H-ЯМР (400 МГц, ДМСО-d6) δ 13,15 (ушир., 1H), 10,68 (с, 1H), 7,34 (с, 1H), 6,74 (с, 1H), 6,60-6,58 (м, 3H), 4,14 (кв, J=3,8 Гц, 2H), 3,56 (ушир., 4H), 3,41 (с, 2H), 3,29 (ушир., 2H), 2,41-2,35 (м, 6H), 1,73-1,71 (м, 2H), 1,38 (т, J=6,8 Гц, 3H).
<Пример 11> Получение (S)-2-аминосукцината 10-этокси-8-(морфолинометил)-1,2,3,4-тетрагидробензо[h][1,6]нафтиридин-5-(6Н)-она
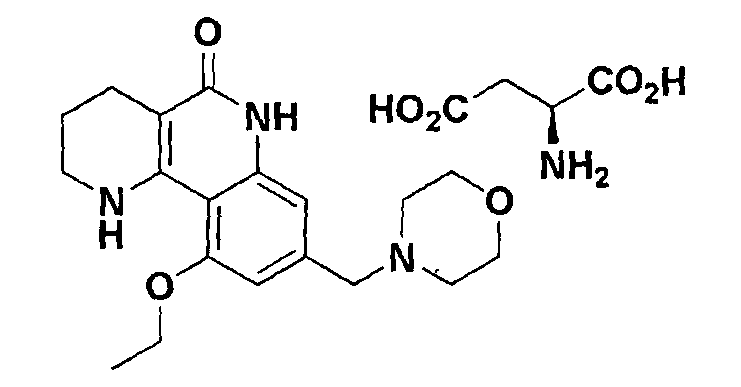
Осуществляли ту же самую процедуру, как описано в примере 1, за исключением того, что в стадии 10 использовали (S)-2-амино янтарную кислоту вместо с-соляной кислоты, получая целевое соединение (620 мг, 89%).
1Н-ЯМР (400 МГц, ДМСО-d6) δ 10,68 (с, 1H), 7,35 (с, 1H), 6,75 (с, 1H), 6,58 (с, 1H), 4,16 (кв, J=6,8 Гц, 2H), 3,77 (м, 1H), 3,57 (м, 4H), 3,40 (с, 2H), 3,30 (м, 2H), 2,72 (м, 1H), 2,43-2,39 (м, 3H), 2,34 (м, 4H), 1,74 (м, 2H), 1,40 (т, J=6,8 Гц, 3H).
<Пример 12> Получение 2-гидроксипропан-1,2,3-трикарбоксилата 10-этокси-8-(морфолинометил)-1,2,3,4-тетрагидробензо[h][1,6]нафтиридин-5-(6Н)-она
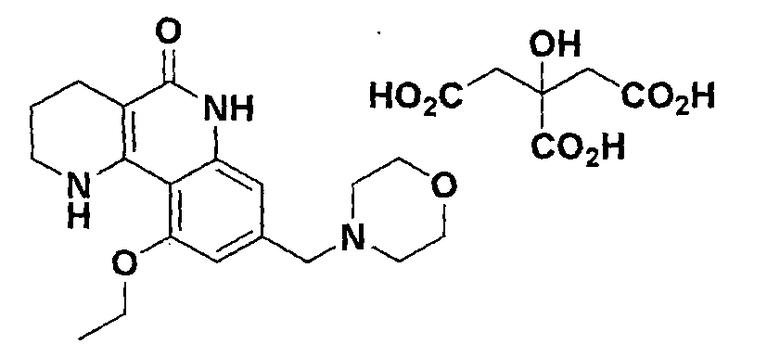
Осуществляли ту же самую процедуру, как описано в примере 1, за исключением того, что в стадии 10 использовали 2-гидроксипропан-1,2,3-трикарбоновую кислоту вместо с-соляной кислоты, получая целевое соединение (665 мг, 85%).
1Н-ЯМР (400 МГц, ДМСО-d6) δ 10,75 (с, 1H), 7,37 (с, 1H), 6,78 (с, 1H), 6,62 (с, 1H), 4,17 (кв, J=3,8 Гц, 2H), 3,61 (ушир., 4H), 3,53 (с, 2H), 3,31 (ушир., 2H), 2,68 (дд, J=12,4 Гц, 7,6 Гц, 4H), 2,50-2,40 (м, 6H), 1,76-1,73 (м, 2H), 1,41 (т, J=6,0 Гц, 3H).
<Пример 13> Получение циклогексилсульфамата 10-этокси-8-(морфолинометил)-1,2,3,4-тетрагидробензо[h][1,6]нафтиридин-5-(6Н)-она
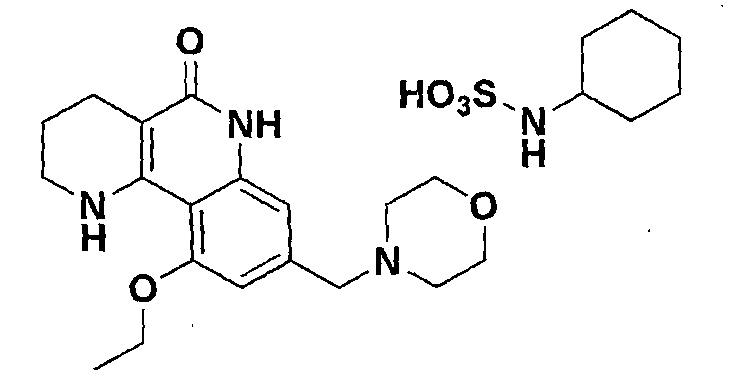
Осуществляли ту же самую процедуру, как описано в примере 1, за исключением того, что в стадии 10 использовали циклогексилсульфаминовую кислоту вместо с-соляной кислоты, получая целевое соединение (453 мг, 75%).
1H-ЯМР (400 МГц, ДМСО-d6) δ 10,76 (ушир.с, 1H), 7,37 (с, 1H), 6,78 (с, 1H), 6,64 (с, 1H), 4,21-4,16 (м, 2H), 3,34-3,31 (м, 5H), 3,09 (ушир.с, 1H), 2,43-2,40 (м, 4H), 2,08-2,06 (м, 2H), 1,76-1,69 (м, 4H), 1,55-1,52 (м, 2H), 1,42 (т, J=6,8 Гц, 3H), 1,27-1,07 (м, 6H).
<Пример 14> Получение сульфата 10-этокси-8-(морфолинометил)-1,2,3,4-тетрагидробензо[h][1,6]нафтиридин-5-(6Н)-она
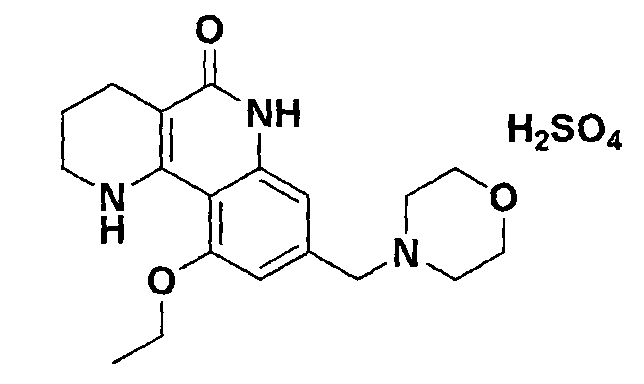
Осуществляли ту же самую процедуру, как описано в примере 1, за исключением того, что в стадии 10 использовали серную кислоту вместо с-соляной кислоты, получая целевое соединение (511 мг, 99%).
1Н-ЯМР (400 МГЦ, ДМСО-d6) δ 11,07 (с, 1H), 9,83 (ушир. с, 1H), 7,40 (ушир. с, 1H), 6,86 (с, H), 6,79 (с, 1H), 4,30-4,26 (м, 2H), 4,24-4,20 (м, 2H), 3,98-3,94 (м, 4H), 3,61 (т, J=11,9 Гц, 2H), 3,32-3,25 (м, 4H), 3,19-3,13 (м, 2H), 1,75-1,62 (м, 2H), 1,45 (т, J=6,9 Гц, 3H).
<Пример 15> Получение сукцината 10-этокси-8-(морфолинометил)-1,2,3,4-тетрагидробензо[h][1,6]нафтиридин-5-(6Н)-она
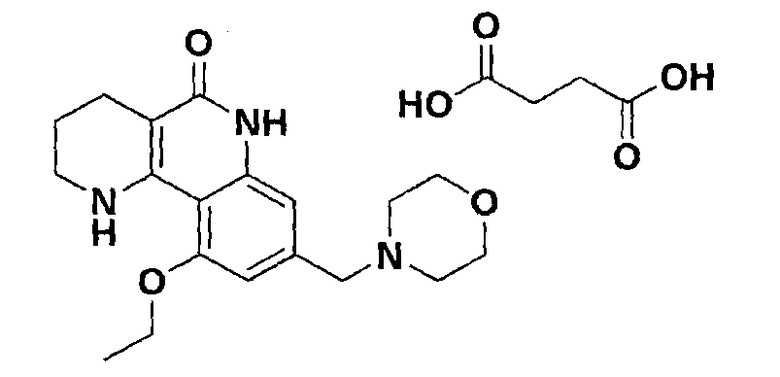
Осуществляли ту же самую процедуру, как описано в примере 1, за исключением того, что в стадии 10 использовали янтарную кислоту вместо с-соляной кислоты, получая целевое соединение (453 мг, 86%).
1H-ЯМР (400 МГц, ДМСО-d6) δ 10,68 (ушир. с, 1H), 7,36 (ушир. с, 1H), 6,76 (с, 1H), 6,59 (2, 1H), 4,17 (кв, J=6,9, 7,3 Гц, 2H), 3,59-3,56 (м, 4H), 3,42 (с, 2H), 3,34-3,31 (м, 4H), 2,41 (с, 8H), 2,35 (ушир. с, 4H), 1,75-1,72 (м, 2H), 1,40 (т, J=6,8 Гц, 3H).
<Пример 16> Получение формиата 10-этокси-8-(морфолинометил)-1,2,3,4-тетрагидробензо[h][1,6]нафтиридин-5-(6Н)-она
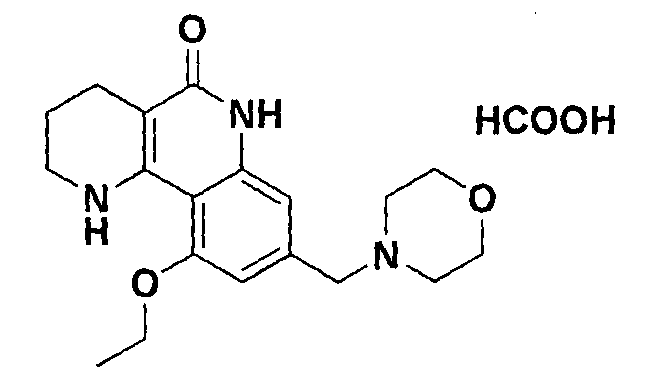
Осуществляли ту же самую процедуру, как описано в примере 1, за исключением того, что в стадии 10 использовали муравьиную кислоту вместо с-соляной кислоты, получая целевое соединение (427 мг, 94%).
1Н-ЯМР (400 МГц, ДМСО-d6) δ 10,69 (ушир. с, 1H), 8,13 (с, 1H), 7,36 (ушир. с, 1H), 6,76 (с, 1H), 6,59 (с, 1H), 4,17 (кв, J=6,8, 7,2 Гц, 2H), 3,59-3,56 (м, 4H), 3,42 (с, 2H), 3,32-3,30 (м, 2H), 2,49-2,35 (м, 6H), 1,75-1,73 (м, 2H), 1,40 (т, J=6,9 Гц, 3H).
<Пример 17> Получение глютамата 10-этокси-8-(морфолинометил)-1,2,3,4-тетрагидробензо[h][1,6]нафтиридин-5-(6Н)-она
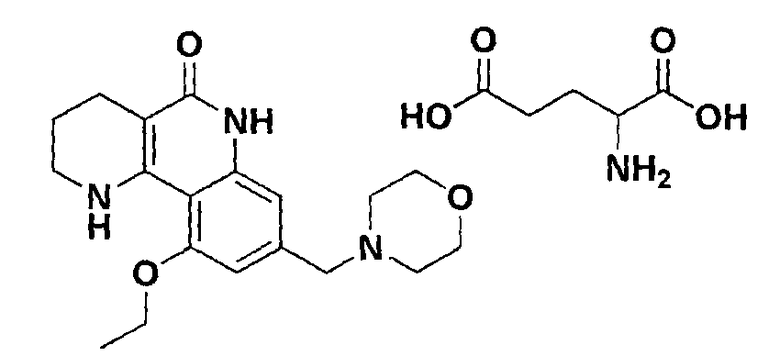
Осуществляли ту же самую процедуру, как описано в примере 1, за исключением того, что в стадии 10 использовали глютаминовую кислоту вместо с-соляной кислоты, получая целевое соединение (545 мг, 96%).
1H-ЯМР (400 МГц, ДМСО-d6) δ 10,68 (ушир. с, 1H), 7,36 (ушир. с, 1H), 6,76 (с, 1H), 6,59 (с, 1H), 4,17 (кв, J=6,9, 7,2 Гц, 2H), 3,59-3,52 (м, 4H), 3,41-3,27 (м, 8H), 2,42-2,34 (м, 6H), 1,87-1,81 (м, 1H), 1,75-1,72 (м, 2H), 1,40 (т, J=6,9 Гц, 3H).
<Пример 18> Получение дифосфата 10-этокси-8-(морфолинометил)-1,2,3,4-тетрагидробензо[h][1,6]нафтиридин-5-(6Н)-она
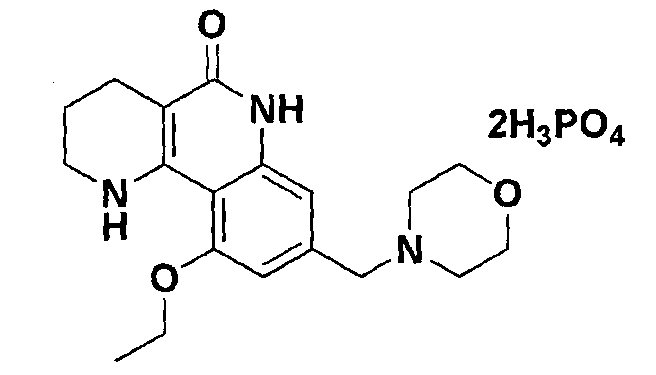
Осуществляли ту же самую процедуру, как описано в примере 1, за исключением того, что в стадии 10 использовали пирофосфорную кислоту вместо с-соляной кислоты, получая целевое соединение (785 мг, 99%).
1Н-ЯМР (400 МГц, ДМСО-d6) δ 10,80 (с, 1H), 7,38 (с, 1H), 6,77 (с, 1H), 6,62 (с, 1H), 4,17 (кв, J=6,8 Гц, 2H), 3,59 (м, 4H), 3,48 (с, 2H), 3,31 (м, 2H), 2,42 (м, 6H), 1,75 (м, 2H), 1,41 (т, J=6,8 Гц, 3H).
<Сравнительный пример 1> Получение ангидрида дихлорида 10-этокси-8-(морфолинометил)-1,2,3,4-тетрагидробензо[h][1,6]нафтиридин-5-(6Н)-она
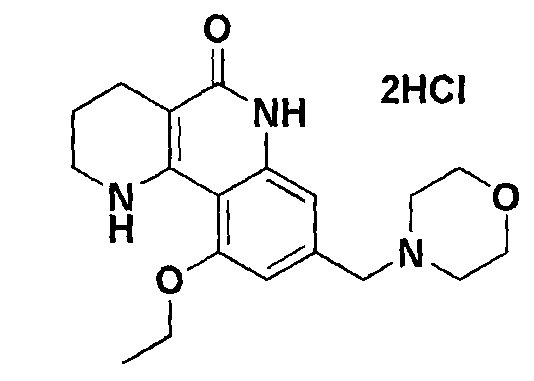
Дигидрат дихлорида 10-этокси-8-(морфолинометил)-1,2,3,4-тетрагидробензо[h][1,6]нафтиридин-5(6Н)-она, полученный в примере 1 (3,0 г, 6,63 ммоль), и 15,0 г пентоксида фосфора (Р2О5) помещали в вакуумную печь (Daihan Labtech, LVO-2060) и сушили при 100°С, при 10 ммHg в течение 5 часов, получая целевое соединение.
<Сравнительный пример 2> Получение аморфного дихлорида 10-этокси-8-(морфолинометил)-1,2,3,4-тетрагидробензо[h][1,6]нафтиридин-5-(6Н)-она
Дигидрат дихлорида 10-этокси-8-(морфолинометил)-1,2,3,4-тетрагидробензо[h][1,6]нафтиридин-5(6Н)-она, полученный в примере 1 (3,0 г, 6,63 ммоль), растворяли в 30 мл дистиллированной воды и сушили вымораживанием. Затем, полученный таким образом продукт и 15,0 г пентоксида фосфора (Р2О5) помещали в вакуумную печь (Daihan Labtech, LVO-2060) и повторно сушили при 100°С, при 10 ммHg в течение 5 часов, получая целевое соединение.
<Экспериментальный пример 1> Сравнение стабильности дигидрата дихлорида 10-этокси-8-(морфолинометил)-1,2,3,4-тетрагидробензо[h][1,6]нафтиридин-5(6Н)-она и ангидрида дихлорида 10-этокси-8-(морфолинометил)-1,2,3,4-тетрагидробензо[h][1,6]нафтиридин-5-(6Н)-она
Для сравнения стабильности дигидрата дихлорида 10-этокси-8-(морфолинометил)-1,2,3,4-тетрагидробензо[h][1,6]-нафтиридин-5(6H)-она, полученного в примере 1, и ангидрида дихлорида 10-этокси-8-(морфолинометил)-1,2,3,4-тетрагидробензо[h][1,6]-нафтиридин-5(6H)-она, полученного в сравнительном примере 1, был проведен следующий эксперимент.
3,0 г каждого из соединений помещали в чашку Петри, и чашку Петри оставляли в термо-гидростате, поддерживающем 25°С и 60% RH в открытом состоянии. Содержание влаги относительно времени измеряли с использованием аппарата Карла Фишера и регистрировали. С помощью измерения изменения содержания влаги относительно времени в описанных выше условиях сравнивали стабильность дигидрата и стабильность ангидрида. Результаты иллюстрируются в следующей таблице 1 и на Фиг. 1.
Как показано в таблице 1, по результатам измерения стабильности дигидрата примера 1 согласно настоящему изобретению и ангидрида сравнительного примера 1, содержание влаги соединения сравнительного примера 1 увеличилось в пределах 30 минут по сравнению с начальным состоянием и на воздухе абсорбируется влага. Таким образом, соединение сравнительного примера 1 быстро изменяется в кристаллический тип и является нестабильным. Однако содержание влаги соединения согласно настоящему изобретению является постоянным, и соединение является стабильным в отношении влаги и в результате стабильным в отношении гигроскопичности и поэтому является очень полезным в поддержании качества при производстве лекарства. В дополнение, соединение согласно изобретению может быть использовано в фармацевтической композиции для лечения невропатической боли, эпилепсии, удара, болезни Альцгеймера, болезни Паркинсона, амиотрофического бокового склероза (ALS), болезни Хантингтона, шизофрении, хронической и острой боли, ишемического повреждения головного мозга, нейронной потери после гипоксии, травм и нервных повреждений, которые являются медицинскими состояниями, вызываемыми сверхактивностью PARP.
<Экспериментальный пример 2> сравнение стабильности аморфного дихлорида 10-этокси-8-(морфолинометил)-1,2,3,4-тетрагидробензо[h][1,6]-нафтиридин-5(6H)-она
Для сравнения стабильности кристаллического соединения примера 1 согласно настоящему изобретению и аморфного соединения сравнительного примера 2 проводили следующий эксперимент.
Стабильность каждого кристаллического соединения примера 1 согласно настоящему изобретению и аморфного соединения сравнительного примера 2 измеряли путем измерения изменения характера дифракции с помощью дифракционного анализа рентгеновских лучей (XRD).
В результате, как показано на Фиг. 2, для кристаллического соединения примера 1 согласно настоящему изобретению получены были постоянные данные без изменения характера XRD дифракции относительно времени. Для аморфного соединения сравнительного примера 2 точный и конкретный XRD характер аморфного типа не был показан, однако аморфный дихлорид 10-этокси-8-(морфолинометил)-1,2,3,4-тетрагидробензо[h][1,6]-нафтиридин-5(6H)-она, полученный после сушки вымораживанием, абсорбирует окружающую влагу во время проведения XRD, тем самым увеличивая кристалличность и показания XRD пиков. Таким образом, сочли, что соединение сравнительного примера 2 абсорбирует окружающую влагу, и кристалличность его увеличивается (см. Фиг. 3).
Для ссылки, 2θ величины, полученные путем измерения XRD дигидрата дихлорида соединения химической формулы 2 согласно примеру 1, суммированы и проиллюстрированы в следующей таблице 2.
Между тем, содержание влаги соединения сравнительного примера 2 составляло 0,4% сразу после сушки и увеличивалось до 5,2% после измерения XRD.
Следовательно, соединение согласно настоящему изобретению является стабильным в отношении гигроскопичности и поэтому очень полезным в поддержании качества при получении лекарства. Кроме того, соединение согласно изобретению может быть использовано в фармацевтической композиции для лечения невропатической боли, эпилепсии, удара, болезни Альцгеймера, болезни Паркинсона, амиотрофического бокового склероза (ALS), болезни Хантингтона, шизофрении, хронической и острой боли, ишемического повреждения головного мозга, нейронной потери после гипоксии, травм и нервных повреждений, которые являются медицинскими состояниями, вызываемыми сверхактивностью PARP.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВОЕ ТРИЦИКЛИЧЕСКОЕ ПРОИЗВОДНОЕ ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2009 |
|
RU2470934C1 |
| ТРИЦИКЛИЧЕСКОЕ ПРОИЗВОДНОЕ СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ТАКОЕ СОЕДИНЕНИЕ | 2016 |
|
RU2715413C2 |
| ТРИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2004 |
|
RU2326864C2 |
| АНТАГОНИСТЫ РЕЦЕПТОРА СОМАТОСТАТИНА ПОДТИПА 5 (SSTR5) | 2014 |
|
RU2671958C2 |
| ДИАМИНОВЫЕ ПРОИЗВОДНЫЕ | 2003 |
|
RU2333203C2 |
| ИНГИБИТОРЫ ИНТЕГРИНА AVB6 | 2018 |
|
RU2769702C2 |
| ПРОИЗВОДНОЕ ТИОФЕНА И ЕГО ПРИМЕНЕНИЕ | 2019 |
|
RU2781643C2 |
| ФЕНИЛТЕТРАГИДРОИЗОХИНОЛИНОВОЕ СОЕДИНЕНИЕ, ЗАМЕЩЕННОЕ ГЕТЕРОАРИЛОМ | 2015 |
|
RU2703456C2 |
| ИНГИБИТОР FGFR, МЕТОД ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2019 |
|
RU2771526C1 |
| ЗАМЕЩЕННЫЕ 4-(АРИЛАМИНО) СЕЛЕНОФЕНОПИРИМИДИНОВЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2011 |
|
RU2566293C2 |
Настоящее изобретение относится к новой кристаллической кислотно-аддитивной соли трициклического производного в форме ее гидрата, представленной следующей химической формулой 2:
[Химическая формула 2]
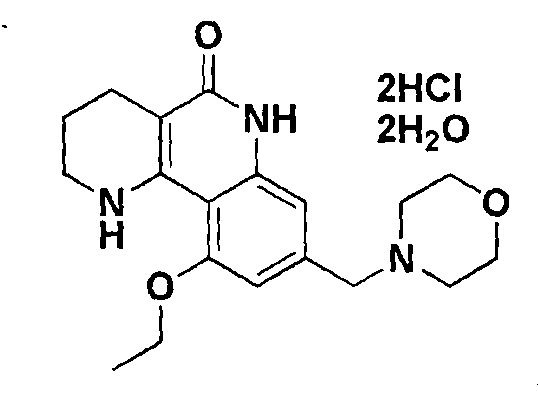 , способу ее получения, а также фармацевтической композиции на ее основе для профилактики или лечения заболеваний, вызываемых сверхактивностью PARP. Технический результат: получена новая кристаллическая кислотно-аддитивная соль трициклического производного в форме ее гидрата, которая является стабильной в отношении гигроскопичности и поэтому полезной в поддержании качества при получении лекарства. 3 н. и 1 з.п. ф-лы, 3 ил., 2 табл., 22 пр.
, способу ее получения, а также фармацевтической композиции на ее основе для профилактики или лечения заболеваний, вызываемых сверхактивностью PARP. Технический результат: получена новая кристаллическая кислотно-аддитивная соль трициклического производного в форме ее гидрата, которая является стабильной в отношении гигроскопичности и поэтому полезной в поддержании качества при получении лекарства. 3 н. и 1 з.п. ф-лы, 3 ил., 2 табл., 22 пр.
1. Новая кристаллическая кислотно-аддитивная соль трициклического производного в форме ее гидрата, представленного следующей химической формулой 2:
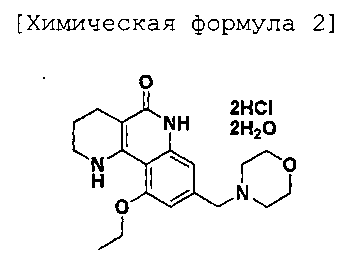
2. Новая кристаллическая кислотно-аддитивная соль трициклического производного в форме ее гидрата по п. 1, в которой кристаллический дигидрат дихлорида химической формулы 2 показывает конкретные пики, представленные 2θ значениями пиков в характере дифракции рентгеновских лучей Cu целевого облучения при 6,59°, 7,74°, 8,42°, 10,15°, 12,52°, 23,30° и 25,30°.
3. Способ получения кристаллической кислотно-аддитивной соли трициклического производного в форме ее гидрата по п. 1, включающий в себя:
добавление соляной кислоты к трициклическому производному следующей химической формулы 1А в воде или органическом растворителе, проведение реакции, завершение реакции и сначала перекристаллизацию с использованием органического растворителя (стадия 1); и
еще одну перекристаллизацию твердого вещества, полученного на стадии 1, с использованием воды и органического растворителя (стадия 2), как показано на следующей реакционной формуле 1:
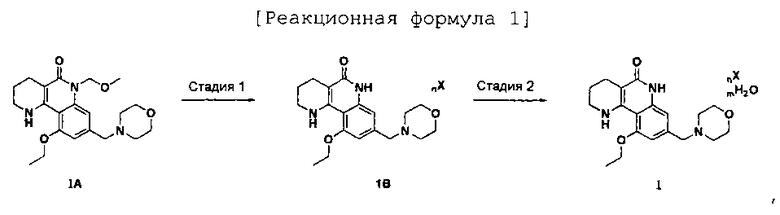
где n представляет собой целое число, равное 2;
m представляет собой целое число, равное 2; и
X представляет собой HCl.
4. Фармацевтическая композиция, включающая терапевтически эффективное количество кристаллической кислотно-аддитивной соли трициклического производного в форме ее гидрата по п. 1, и фармацевтически приемлемый носитель, для профилактики или лечения заболеваний, вызываемых сверхактивностью PARP (полимеразы поли(ADP-рибозы)).
| KR 20100053468 A, 20.05.2010 | |||
| US 2005074470 A1, 07.04.2005 | |||
| US 2004067949 A1, 08.04.2004 | |||
| US 6696437 B1, 24.02.2004 | |||
| RU 99128081 A, 20.10.2001. |

