Изобретение относится к области аналитической химии и может быть использовано для контроля содержания токсичных примесей в пищевых продуктах.
В течение длительного времени для борьбы с паразитарными и грибковыми заболеваниями в рыбоводстве широко применялись трифенилметановые красители, в частности: малахитовый зеленый (МЗ), кристаллический фиолетовый (КФ) и бриллиантовый зеленый (БЗ). Попадая в организм рыбы, эти красители превращаются в соответствующие им метаболиты: лейкоформы (Л-МЗ) и (Л-КФ) [К. Bauer et al., "Aufnahme und ausscheidung von malachitgrun bei regenbogenforellen" Arch. Lebensmittelhyg. 39, 97-102. (1988); S.M. Plakas, et al., "Uptake, tissue distribution, and metabolism of malachite green in the channel catfish (Ictalurus punctatus)" Can J. Fish. Aquat. Sci. 53, 1427-1433 (1996)].
В настоящее время установлено, что трифенилметановые красители и их метаболиты проявляют канцерогенное и генотоксическое действия, в связи с чем, использование этих веществ запрещено в РФ, США, Канаде и странах ЕС (Commission Decision 2004/25/ЕС as regards the setting of minimum required performance limits (MRPLs) for certain residues in food of animal origin, (2004) Off. J. Euro. Union, L6, 38-39. Однако в ряде стран Азии и Латинской Америки, откуда возможно поступление рыбной продукции в РФ, эти красители широко применяются
Описаны различные методы анализа содержания красителей в тканях рыб (International Food Research Journal 20(4):1511-1519 (2013)). Так, известен способ биологического мониторинга МЗ (TW 201115147 (A) ZHANG WEN-XING, LIU QI-RU; NAT CHIAYI UNIVERSITY, 01.05.2011), в котором остаточное содержание МЗ определяют по спектру флуоресценции с использованием генно-инженерных методов.
Известен способ иммунологического анализа с подтверждением результатов жидкостной хроматографией и масс-спектрометрией, который позволяет определить указанные красители: МЗ, Л-МЗ, а также карбинол в пробах тканей рыб и воды (US 2007254323 (A1) WANG JUN, Et al., 01.11.2007). Соединения и процедуры, использованные для подготовки реагентов, представляют собой конъюгированные по нескольким позициям хроматические производные МЗ, присоединенные к полипептидным антигенам для выработки антител, используемых в иммуноферментном анализе. Результаты определения хроматического производного МЗ в образцах воды и тканей рыб могут быть подтверждены методами ВЭЖХ и масс-спектрометрии.
В заявке (CN 103304662 (A), SHANGHAI OCEAN UNIVERSITY, 18.09.2013) описан метод получения реагентов на основе моноклональных антител, обладающих повышенной специфичностью к Л-МЗ: раствор антител и ИФА-набор. Моноклональные антитела имеют высокую чувствительность и специфичность, с перекрестной чувствительностью только к МЗ в отсутствие таковой к парафуксину, метиленовому синему, КФ, ципрофлоксацину и хлорамфениколу, таким образом разработанная технология получения и использования антител позволяет проводить быстрое определение остаточного содержания лейкомалахитового зеленого в продукции аквакультуры и предоставляет технологию и материал для создания ИФА-набора для определения лейкомалахитового зеленого.
Однако описанные выше методы не касаются КФ и его метаболита Л-КФ, не используют процедуру преобразования метаболитов, обладают перекрестной специфичностью, что препятствует однозначной идентификации соединений, а также являются полуколичественными методами, положительные результаты которых требуют дополнительного подтверждения.
Описан способ определения остаточных количеств трифенилметановых красителей в мышечной ткани рыб (Wendy С. Andersen, et al., Determination of Malachite Green and Leucomalachite Green in Salmon with In-Situ Oxidation and Liquid Chromatography with Visible Detection. Laboratory Information Bulletin No. 4334 p.p. 1-13, 2005; Journal of AOAC International 88 (5):1292-1298, 2006). Красители МЗ и Л-МЗ извлекали из тканей рыбы ацетонитрилом в присутствии гидроксиламина (0.25 г/мл) и 100 мкл паратолуола сульфоновой кислоты (1 М), с дальнейшим переводом органической части в дихлорметан с помощью делительной воронки и концентрировании до 3 мл, и переводом метаболитов в исходные соединения 3 миллилитрами 0.001 М 2,3-дихлор-5,6-дициано-p-бензохиноном (ДДБ) при комнатной температуре. При детектировании используют жидкостную хроматографию с флюориметрическим детектором, однако исследование касается только определения МЗ.
Наиболее близким по назначению является способ определения остаточных количеств трифенилметановых красителей в мышечной ткани рыб (Jonathan A. Tarbin, et al., Multiresidue determination of triarylmethane and phenothiazine dyes in fish tissues by LC-MS/MS. Analytica chimica acta 625 (2008) 188-194 - прототип). Экстракцию Л-МЗ, МЗ и КФ из тканей рыб проводят смесью раствора ацетата аммония и ацетонитрила с дальнейшим переводом органической составляющей в слой дихлорметана. Затем следует 15 минутное окисление при комнатной температуре 3 мл 0,005 M раствора 2,3-дихлор-5,6-дициано-p-бензохиноном (ДЦБ), при котором метаболиты исходных соединений (Л-КФ, Л-МЗ), переходят в сами соединения, что позволяет, после дополнительной очистки на катионообменном сорбенте, определять суммарное содержание КФ и Л-КФ, МЗ и Л-МЗ, по сигналу от исходных соединений. При этом используются установки, основанные на высокоэффективной жидкостной хроматографии с масс-спектрометрическим детектором.
Недостаток способа-прототипа состоит в том, что используется неоптимизированная процедура преобразования метаболитов в исходные соединения, что приводит к снижению полноты окисления, и как следствие, повышению порога количественного определения. Это влечет невоспроизводимость результатов анализа при использовании технологии очистки на колонках с обращеннофазным сорбентом, так как доля органической составляющей смеси при ее нанесении на сорбент не должна превышать 15-20%, во избежание потерь определяемых веществ. Кроме того, проверка рекомендованной в прототипе процедуры окисления лейкоформ Л-МЗ и Л-КФ показала, что полнота их окисления недостаточна и варьируется в диапазоне от 57% для МЗ и от 35,3% для КФ.
Настоящее изобретение направлено на устранение недостатков прототипа, а именно повышение полноты перевода лейкоформ указанных красителей непосредственно в красители.
Патентуемый способ определения остаточных количеств трифенилметановых красителей в мышечной ткани рыб включает: извлечение аналитов из ткани смесью ацетонитрила и буфера с получением экстракта в результате центрифугирования; введение дихлорметана в полученный экстракт и перевод органической части экстракта в слой дихлорметана при перемешивании и центрифугировании с отделением надосадочного раствора; перевод метаболитов в начальные формы красителей путем введения в надосадочный раствор окислителя на основе 2,3-дихлоро-5,6-дициано-парабензохинона (ДДБ) с последующей очисткой смеси методом твердофазной экстракции, концентрирование аналитов и их анализ методом жидкостной хроматографии с масс-спектрометрическим детектированием.
Способ отличается от прототипа следующими признаками. В качестве буфера при извлечении аналитов используют смесь раствора лимонной кислоты и натрия фосфорнокислого двузамещенного. Перед введением дихлорметана в полученный экстракт добавляют расслаивающий агент. В качестве окислителя используют смесь ДДБ и 2,3,5,6-тетрахлор-п-бензохинона (далее хлоранил) в молярном соотношении 1:3, процесс перевода метаболитов проводят в потоке азота, а при твердофазной экстракции используют гидрофильно-липофильный сбалансированный обращеннофазный сорбент. Процесс концентрирования аналитов проводят в потоке азота при температуре 40-60°C в течение 30-50 мин.
Способ может характеризоваться тем, что трифенилметановые красители включают малахитовый зеленый и/или кристаллический фиолетовый и/или бриллиантовый зеленый.
Способ может характеризоваться и тем, что используют смесь растворов лимонной кислоты с концентрацией 0,1 моль/дм3 и натрия фосфорнокислого двузамещенного с концентрацией 0,2 моль/дм3, а также тем, что используют гидрофильно-липофильный сбалансированный обращеннофазный сорбент марки Oasis HLB фирмы Waters.
Способ может характеризоваться, кроме того, тем, что в качестве расслаивающего агента используют сульфат аммония в количестве 0,4-0,6 на 1 объемную часть экстракта, а также тем, что давление азота составляет 610-630 мм. рт.ст.
Технический результат изобретения - повышение чувствительности и понижение порога количественного определения за счет повышения полноты перевода лейкоформ красителей непосредственно в исходные красители.
Патентуемые режимы позволяют значительно, до 80-90%, повысить полноту перевода метаболитов указанных красителей непосредственно в сами одноименные красители, что дает возможность соответственно увеличить в среднем на 30-40% чувствительность определения трифенилметановых красителей и их метаболитов.
Рекомендуемые режимы выполнения способа и реагенты подобраны в результате экспериментов, проведенных заявителем: исследованы зависимости изменения полноты перевода метаболитов в исходные красители от температуры, времени воздействия, концентрации и объемного соотношения компонентов окисляющей смеси.
Было установлено, что в результате взаимодействия метаболитов с компонентами смеси окислителей, состоящей из 2,3-дихлор-5,6-дициано-p-бензохинона (ДДБ) и 2,3,5,6- тетрахлор-п-бензохинона (хлоранил) с мольными концентрациями 0,001 и 0,003, соответственно, при объемном соотношении 100/400 мкл и нагреве 50°C в токе азота, происходит их перевод в сами красители. При этом полнота перевода Л-МЗ приближается к 91,3%, а полнота перевода Л-КФ - к 85,1%, что превышает эффективность метода перевода метаболитов в красители, описанного в прототипе, на 34,3% и 50,1%, соответственно.
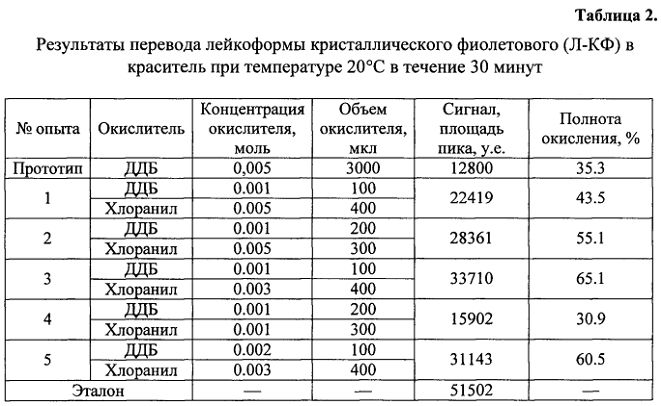
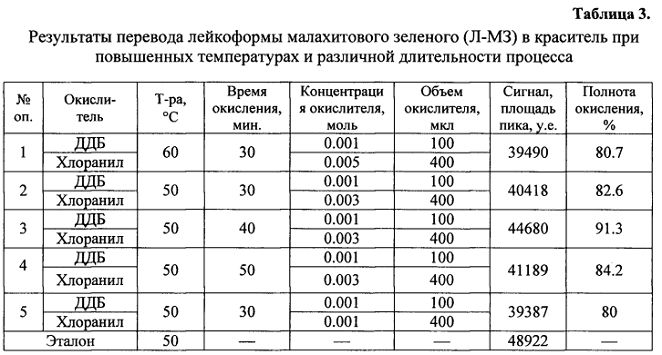
Дальнейшее описание экспериментов по выбору оптимальных параметров способа поясняется графическими материалами, где:
на фиг. 1 показана зависимость логарифма суммы хроматографических пиков от условий перевода метаболитов в исходные красители при комнатной температуре, приведенных в табл. 1, 2;
фиг. 2 - зависимость логарифма суммы хроматографических пиков от условий перевода метаболитов в исходные красители с нагреванием, приведенных в табл. 3, 4;
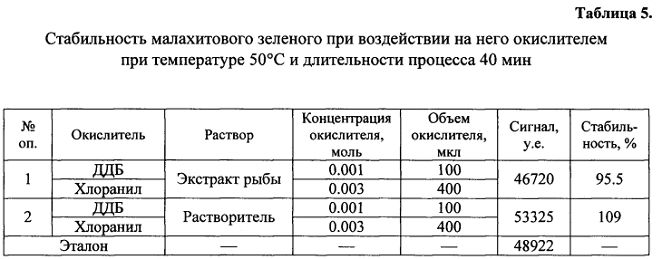
фиг. 3 - обобщенная зависимость полноты перевода метаболитов в исходные красители от условий перевода.
Установление оптимальных условий перевода метаболитов непосредственно в исходные красители проводилось в несколько этапов.
На первом этапе определено влияние концентрации и объемного соотношения компонентов окисляющей смеси на полноту перевода. Для этого в экстракты мышечной ткани рыб вводились добавки метаболитов Л-МЗ и Л-КФ, с концентрацией, эквивалентной эталону, и добавлялись смеси окислителей с параметрами, указанными в табл. 1, 2.
Данные растворы выдерживали при комнатной температуре (20°C) одинаковое время (30 мин). После процедуры перевода, экстракты обрабатывались методом твердофазной экстракции на гидрофильно-липофильном сбалансированном обращеннофазном сорбенте, а анализируемые компоненты определялись методом жидкостной хроматографии с масс-спектрометрическим детектированием. За эталон принимался аналитический стандарт исходной формы красителей, прошедший все стадии, за исключением окисления. Концентрация аналитического стандарта выбиралась равноценной экспериментальной. На фиг. 1 показана зависимость логарифма суммы хроматографических пиков от условий перевода лейкоформы МЗ. Сигнал (площадь пика, у.е) является средней величиной нескольких опытов, при этом полнота окисления в % рассчитана относительно аналитического стандарта.
На втором этапе исследовано влияние температуры и времени воздействия на полноту перевода. Для этого в экстракты мышечной ткани рыб вводились добавки метаболитов Л-МЗ и Л-КФ с концентрацией, эквивалентной эталону и добавлялись смеси окислителей в соответствии с табл. 3, 4. Аналогично первому этапу, растворы подвергались концентрированию в токе азота, но при различных температурах и времени. После процедуры перевода, экстракты обрабатывались методом твердофазной экстракции, а анализируемые компоненты определялись методом жидкостной хроматографии с масс-спектрометрическим детектированием. За эталон принимался аналитический стандарт исходной формы красителей, прошедший все стадии, за исключением окисления. Концентрация аналитического стандарта являлась равноценной экспериментальной. Результаты представлены в таблицах 3 и 4, на фиг. 2.
Исходя из полученных данных была получена обобщенная зависимость полноты перевода метаболитов в исходные соединения от изменяемых условий, которая представлена на фиг. 3. Согласно этой зависимости можно заключить, что оптимальными условиями являются: соотношение мольных концентраций 0,001 и 0,003 смеси окислителей ДДБ и хлоранила, соответственно, при их вносимом объеме 100 мкл и 400 мкл, соответственно, при нагреве смеси при 50°C в токе азота, то есть характеризуют режимы опыта №3, приведенного в табл. 3 и 4.
Также была проверена стабильность исходных веществ при воздействии на них окислителем по одной из оптимальных схем. В качестве тестируемого компонента был выбран МЗ. Результаты приведены в таблице 5. Из полученных данных видно, что МЗ, а следовательно и другие красители, стабильны и не разрушаются в процессе перевода метаболитов.
Способ определения остаточных количеств трифенилметановых красителей в мышечной ткани рыб осуществляют следующим образом.
Анализируемые компоненты (Л-МЗ, МЗ, Л-КФ, КФ и др.) извлекают из гомогенизированных тканей рыбы смесью ацетонитрила и лимоннокислого буфера (смесь растворов лимонной кислоты с концентрацией 0,1 моль/дм3 и натрия фосфорнокислого двузамещенного с концентрацией 0,2 моль/дм3).
Полученный экстракт разделяют центрифугированием и надосадочную жидкость переносят в слой дихлорметана, эквивалентный 50% объему экстракта в присутствии водорастворимой соли в виде сульфата аммония в количестве 0,4-0,6 на 1 объемную часть экстракта, для перевода целевых компонентов.
После перемешивания и разделения образовавшихся слоев центрифугированием, полученный органический слой, расположенный сверху, переносят в новую пробирку и добавляют 0,5 мл смеси окислителей, состоящей из 0,1 мл 0,001 M ДДБ и 0,4 мл 0,003 М хлоранила в ацетонитриле. Полученный раствор перемешивают и концентрируют в токе азота при температуре 40-60° и давлении азота 620 мм. рт.ст. в течение 30-50 минут.
К сухому остатку добавляют 1 см3 ацетонитрила, 3 см3 деионизованной воды, 0,2 см3 муравьиной кислоты и 2 см3 гексана. Содержимое пробирки перемешивают, и слои разделяют центрифугированием. После этого отбрасывают гексан, а остаток очищают методом твердофазной экстракции на гидрофильно-липофильном сбалансированном обращеннофазном сорбенте марки Oasis HLB фирмы Waters.
После процедуры очистки анализируемые компоненты концентрируют в токе азота и определяют их содержание методом жидкостной хроматографии с масс-спектрометрическим детектированием.
Таким образом, представленные материалы подтверждают возможность достижения технического результата - повышения полноты до 80-90% перевода метаболитов красителей непосредственно в сами красители, что дает возможность соответственно увеличить в среднем на 30-40% чувствительность определения трифенилметановых красителей и их метаболитов.


| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ ГЛИФОСАТА, ЕГО МЕТАБОЛИТА И ГЛЮФОСИНАТА В ПРОДУКЦИИ ЖИВОТНОВОДСТВА | 2021 |
|
RU2783283C1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ КЛАВУЛАНОВОЙ КИСЛОТЫ В МЫШЕЧНЫХ ТКАНЯХ ЖИВОТНОГО ПРОИСХОЖДЕНИЯ | 2021 |
|
RU2781486C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ОСТАТОЧНЫХ КОЛИЧЕСТВ АВИЛАМИЦИНА В БИОЛОГИЧЕСКИХ ТКАНЯХ ЖИВОТНОГО ПРОИСХОЖДЕНИЯ | 2021 |
|
RU2783284C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ОСТАТОЧНЫХ КОЛИЧЕСТВ ПИПЕРАЗИНА В БИОЛОГИЧЕСКИХ ТКАНЯХ И ОБЪЕКТАХ ЖИВОТНОГО ПРОИСХОЖДЕНИЯ | 2024 |
|
RU2837303C1 |
| Способ определения массовых концентраций фенола и пирокатехина в крови методом высокоэффективной жидкостной хроматографии | 2022 |
|
RU2786509C1 |
| Способ подготовки пробы мочи для определения монометилфталата, моноэтилфталата, монобутилфталата, монобензилфталата, моноэтилгексилфталата методом высокоэффективной жидкостной хроматографии/масс-спектрометрии | 2019 |
|
RU2687738C1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ БЕНЗ(А)ПИРЕНА В МОЧЕ МЕТОДОМ ЖИДКОСТНОЙ ХРОМАТОГРАФИИ | 2011 |
|
RU2466406C1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ БЕНЗ(А)ПИРЕНА В КРОВИ МЕТОДОМ ЖИДКОСТНОЙ ХРОМАТОГРАФИИ | 2014 |
|
RU2546530C1 |
| Способ идентификации наркотических и психоактивных веществ в биосубстрате человека | 2019 |
|
RU2723907C1 |
| СПОСОБ ОБНАРУЖЕНИЯ 1,4-ДИОКСАНА И 2-МЕТИЛ-1,3-ДИОКСОЛАНА В МОЛОКЕ | 2021 |
|
RU2776197C1 |
Изобретение относится к области аналитической химии и касается способа определения остаточных количеств трифенилметановых красителей в мышечной ткани рыб. Сущность способа заключается в том, что производят извлечение аналитов из ткани смесью ацетонитрила и буфера с получением экстракта в результате центрифугирования, введение дихлорметана в полученный экстракт и перевод органической части экстракта в слой дихлорметана при перемешивании и центрифугировании с отделением надосадочного раствора. Перевод метаболитов в начальные формы красителей осуществляют путем введения в надосадочный раствор окислителя на основе 2,3-дихлоро-5,6-дициано-пара-бензохинона (ДДБ) с последующей очисткой смеси методом твердофазной экстракции. В качестве буфера при извлечении аналитов используют смесь раствора лимонной кислоты и натрия фосфорнокислого двузамещенного; перед введением дихлорметана в полученный экстракт добавляют расслаивающий агент; в качестве окислителя используют смесь ДДБ и 2,3,5,6-тетрахлор-п-бензохинона в молярном соотношении 1:3, процесс перевода метаболитов проводят в потоке азота, а при твердофазной экстракции используют гидрофильно-липофильный сбалансированный обращеннофазный сорбент. Использование способа позволяет с высокой точностью определить содержание остаточных количеств трифенилметановых красителей в мышечной ткани рыб. 1 з.п. ф-лы, 3 ил., 5 табл.
1. Способ определения остаточных количеств трифенилметановых красителей малахитового зеленого, кристаллического фиолетового и бриллиантового зеленого в мышечной ткани рыб, включающий извлечение аналитов из ткани смесью ацетонитрила и буфера с получением экстракта в результате центрифугирования, введение дихлорметана в полученный экстракт и перевод органической части экстракта в слой дихлорметана при перемешивании и центрифугировании с отделением надосадочного раствора, перевод метаболитов в начальные формы красителей путем введения в надосадочный раствор окислителя на основе 2,3-дихлоро-5,6-дициано-пара-бензохинона (ДДБ) с последующей очисткой смеси методом твердофазной экстракции, концентрирование аналитов и их анализ методом жидкостной хроматографии с масс-спектрометрическим детектированием, отличающийся тем, что в качестве буфера при извлечении аналитов используют смесь растворов лимонной кислоты с концентрацией 0,1 моль/дм3 и натрия фосфорнокислого двузамещенного с концентрацией 0,2 моль/дм3, перед введением дихлорметана в полученный экстракт добавляют расслаивающий агент сульфат аммония в количестве 0,4-0,6 на 1 объемную часть экстракта, в качестве окислителя используют смесь ДДБ и 2,3,5,6-тетрахлор-п-бензохинона в молярном соотношении 1:3, процесс перевода метаболитов проводят концентрированием в потоке азота, а при твердофазной экстракции используют гидрофильно-липофильный сбалансированный обращеннофазный сорбент, при этом процесс концентрирования аналитов проводят в потоке азота под давлением 610-630 мм рт. ст. при температуре 40-60°C в течение 30-50 минут.
2. Способ по п. 1, отличающийся тем, что используют гидрофильно-липофильный сбалансированный обращеннофазный сорбент марки Oasis HLB фирмы Waters.
| TABRIN J.A., et al., Multiresidue determination of triaylmethane and phenothiazine dyes in fish tissues by LC-MC/MS//Anal.Chim.Acta, 2008, 625, P.188-194 | |||
| ANDERSEN W | |||
| C | |||
| et al., Malachite Green and Leucomalachite Green in Salmon // LIB No | |||
| ПРИСПОСОБЛЕНИЕ ДЛЯ РЕГУЛИРОВАНИЯ СКОРОСТИ СЪЕМКИ КИНОАППАРАТА | 1925 |
|
SU4334A1 |
| Походная разборная печь для варки пищи и печения хлеба | 1920 |
|
SU11A1 |
| US 2007254323 A1, 01.11.2007. | |||

