Изобретение относится к области аналитической химии и может быть использовано для контроля содержания остаточных количеств авиламицина в продукции животноводства. Сущность способа заключается в извлечении аналита из объекта исследования ацетоном с последующим концентрированием экстракта и применением щелочного гидролиза едким натром, при нагревании. Очистку полученного деривата осуществляют экстракцией этилацетатом с удалением органической составляющей, нейтрализацией остатка ортофосфорной кислотой, с последующим обезжириванием гексаном и удалением органического слоя. Финальную экстракцию проводят этилацетатом с последующим концентрированием досуха и перерастворением в необходимом количестве деионизованной воды, подкисленной муравьиной кислотой. Использование данного способа позволяет с высокой точностью определить содержание остаточного количества авиламицина в мышечной ткани животного происхождения.
Авиламицин является антибиотиком семейства ортомицинов, представители которого, ингибируют рост грамположительных бактерий. Авиламицин по своей химической природе представляет смесь олигосахаридов, которые продуцируются Streptomyces viridochromogenes. На сегодняшний момент известно, что авиламицин имеет один главный изомер (авиламицин А его доля ≥60%) и еще 15 второстепенных (авиламицин А', В, С, D1, D2, Е, F, G, Н, I, J, К, L, М и N) [1].
Молекула авиламицина содержит в себе часть полкетид-дихлор-изо-иверниновой кислоты, связанную с полисахаридной цепочкой.
Несмотря на то, что авиламицин является относительно безопасным лекарственным соединением многие страны устанавливают максимально допустимые уровни (МДУ) на содержание маркерного соединения, по которому оценивается остаточное содержание авиламицина. В Европейском Союзе [2] и Российской Федерации [3] МДУ на маркер авиламицина - дихлоризоэверниновую кислоту составляет 50 мкг/кг для мяса свиней, кроликов и домашней птицы. В Российской Федерации зарегистрирован лекарственный препарат для ветеринарного применения Максус G100 (Maxus G100), содержащий в качестве действующего вещества авиламицин (организация-разработчик: Elanco Animal Health Incorporated, USA; номер регистрационного удостоверения: 840-3-6,16-3548 №ПВИ-3-1.5/01690).
Обращение на рынке препаратов содержащих авиламицин предопределяет необходимость разработки аналитической методики для целей контроля его остаточного содержания.
Описаны способы определения авиламицина А и авиламицина В как целевых аналитов. Так, были опубликованы статьи в которых авиламицин А определяли с помощью жидкостной хроматографии [4, 5]. Однако применяемый метод не является достаточно селективным и имеет высокий предел количественного определения. Предел количественного определения, в данном случае, составляет мг/кг (выше установленных МДУ) [5].
Примечательной была статья в которой в одном методе высокоэффективной жидкостной хроматографии (ВЭЖХ) с УФ-детекцией определяли авиламицин А и авиламицин В [6]. Следует заметить, что по виду, представленных авторами статьи хроматограмм, содержание авиламицина В заметно меньше его основного изомера (что согласуется с литературными данными), но кроме этого хроматографический пик авиламицина В имеет очень низкий аналитический отклик интенсивности, что в перспективе, закрывает возможность применения указанного метода для целей количественного анализа в области низких концентраций. Кроме того, метод ВЭЖХ, является недостаточно селективным: в случае обнаружения остаточных количеств авиламицина в образце, необходимо проводить подтверждение методом высокоэффективной жидкостной хроматографии в сочетании с тандемной масс-спектрометрией (ВЭЖХ-МС/МС), основываясь на данных по наличию фрагментов молекулярного иона аналита.
Начиная с 2010 года возрастает число публикаций определения авиламицина методом высокоэффективной жидкостной хроматографии с масс-спектрометрическим детектированием. Наши соотечественники, в рамках разаработанного ими многокомпонентного подхода к определению остаточного количества соединений, различных по химической природе (492 соединения), опубликовали статью, содержащую в том числе и количественное определение авиламицина [7]. В которой главным недостактом, являлась недоступность определения суммарного количества всех изомеров авиламицина, т.е количественная оценка акцентировалась только на авиламицине А.
Из-за высокой скорости метаболизма авиламицина, акценты были смещены в сторону определения его метаболитов. Это открывало возможности решения проблемы суммарного определения всех изомеров авиламицина т.к. определился главный метаболит авиламицина -дихлоризоэверниновая кислота [8].
По содержанию дихлороизоэверниновой кислоты в объекте исследования можно рассчитать содержание авиламицина. Расчет массовой доли, если требуется, проводится через коэффициент эквивалентности (фактор эквивалентности), который является отношением молярной массы авиламицина к молярной массе дихлороизоэверниновой кислоты (Мав/Мд) и равен 5, 6.
Наиболее близким по назначению, является способ определения авиламицина как дихлороизоэверниновой кислоты в свинине, жире и печени [1] - прототип. Экстракцию дихлороизоэверниновой кислоты производят из 10 г навески тканей 50 см3 ацетона в процессе гомогенизации, с дальнейшим центрифугированием и повторением экстракции новой порцией ацетона (25 см3), с дальнейшим объединением экстрактов в один, и его разбавлением до 100 см3 ацетоном, в колбе. Аликвоту - 10 см3, полученного экстракта, концентрируют досуха в стеклянном флаконе, к остатку приливают 4 см3 1М гидрооксида натрия, перемешивают и помещают для дериватизации в сушильный шкаф на 2 часа при 70°С. Полученный гидролизат переносят в новый флакон, ополаскивают 10 см3 деионизованной воды и 10 см3 этилацетата, последовательно, старый флакон, и объединяют растворы с гидролизатом. Перемешивают флакон с объединенным раствором и центрифугируют при 3000 об/мин в течение 5 минут. Слой этилацетата отбрасывают и вводят в остаток гидролизата 2 см3 ортофосфорной кислоты и 15 см3 этилацетата. Перемешивают флакон и разделяют фракции центрифугированием, при 3000 об/мин в течение 5 минут. Органический слой отбирают в новый флакон и повторяют экстракцию новой порцией этилацетата. Объединенный объем (30 см3) этилацетата концентрируют досуха в токе азота и перерастворяют на ультразвуковой бане в 2 см3 смеси этилацетат/гексан/муравьиная кислота (в соотношении 100/900/1). полученный раствор подвергают твердофазной очистке на колонке с силикагелем, который предварительно промывают 10 см3 смеси этилацетат/гексан/муравьиная кислота (в соотношении 100/900/1). После нанесения гидролизата на колонку, ее промывают 8 см3 смеси этилацетат/гексан/муравьиная кислота (в соотношении 100/900/1). Целевые компоненты смывают с колонки 15 см3 раствора этилацетат/гексан/муравьиная кислота (в соотношении 300/700/1). Раствор экстракта собирают в новый флакон и концентрируют в токе азота досуха, с последующим перерастворением в 1 см3 метанола и анализом методом ВЭЖХ-МС/МС.
1. Недостаток способа-прототипа состоит в том, что в процессе подготовки объекта испытания применяется избыточное количество ацетона, относящегося к прекурсорам, оборот которых в РФ ограничен и в отношении которых допускается исключение некоторых мер контроля.
2. Недостатком способа-прототипа, является применение времязатратного протокола жидкостно-жидкостной экстракции на стадиях, предшествующих твердофазной очистке (далее ТФО) на силикагеле.
3. Недостатком способа-прототипа, является применение стадии ТФО, являющейся времязатратной и требующей наличия специфического сорбента «Inertsep SI» производства GL Sciences (Tokyo, Japan).
Настоящее изобретение направлено на устранение недостатков прототипа, а именно на снижение количества применяемого ацетона при экстракции, на упрощение протокола стадии жидкостно-жидкостной экстракции, на устранение стадии ТФО при сохранении качества получаемых в ходе исследований данных (не более 3-4%).
Патентуемый способ определения остаточных количеств авиламицина в биологических тканях животного происхождения и кормах включает: извлечение анализируемого соединения из образца ацетоном; концентрирование полученного экстракта досуха; гидролиз остатка гидрооксидом натрия с последующей очисткой этилацетататом; восстановление рН ортофосфорной кислотой и обезжиривание гексаном; перевод определяемого соединения в слой этилацетата и его финальное концентрирование досуха, с последующим перерастворением в деионизованной воде с муравьиной кислотой и анализ методом ВЭЖХ-мс/мс.
Способ отличается от прототипа следующими признаками. В качестве экстрагента применяют ацетон, в его соотношении с навеской образца 5:2. Жидкостно-жидкостную экстракцию дериватов проводят однократно, с помощью введения 3-5 см3 этилацетата после стадии гидролиза. Взамен ТФО проводят жидкостно-жидкостную экстракцию мешающих примесей, с помощью 3-4 см3 гексана и его последующим удалением, и конечным концентрированием очищенного деривата в 1 см3 0,5% муравьиной кислоты.
Технический результат изобретения - снижение количества применяемого ацетона и повышение относительной интенсивности сигнала средства измерения за счет упрощения этапа жидкостно-жидкостной экстракции перед стадией ТФО, и замены стадии ТФО на жидкостно-жидкостную экстракцию гексаном.
Патентуемый способ позволяет значительно, на 30%, увеличить аналитический отклик сигнала детектора по метаболиту авиламицина, что позволяет уверенно работать со сложными видами образцов в результате снижения влияния шумов базовой линии; существенно уменьшить расход прекурсора - ацетона; упростить процедуру жидкостно-жидкостной экстракции этилацетатом; избежать процедуры ТФО.
Рекомендуемые режимы выполнения Способа подобраны в результате экспериментов, проведенных заявителем: исследована зависимость изменения полноты извлечения определяемого соединения от количества экстрагента, зависимости относительной интенсивности сигнала на детекторе масс-спектрометра от изменений в выполнении стадии жидкостно-жидкостной экстракции и применения гексана, взамен ТФО.
Перед проведением экспериментов, в гомогенаты мышечной ткани вводили эквивалентное количество дихлороизоэверниновой кислоты. Процедура подготовки образцов к анализу соответствовала прототипу с вариацией количества экстрагента. За эталон принимали образец, прошедший процедуру пробоподготовки согласно прототипу с введением в него определяемого компонента непосредственно перед анализом.
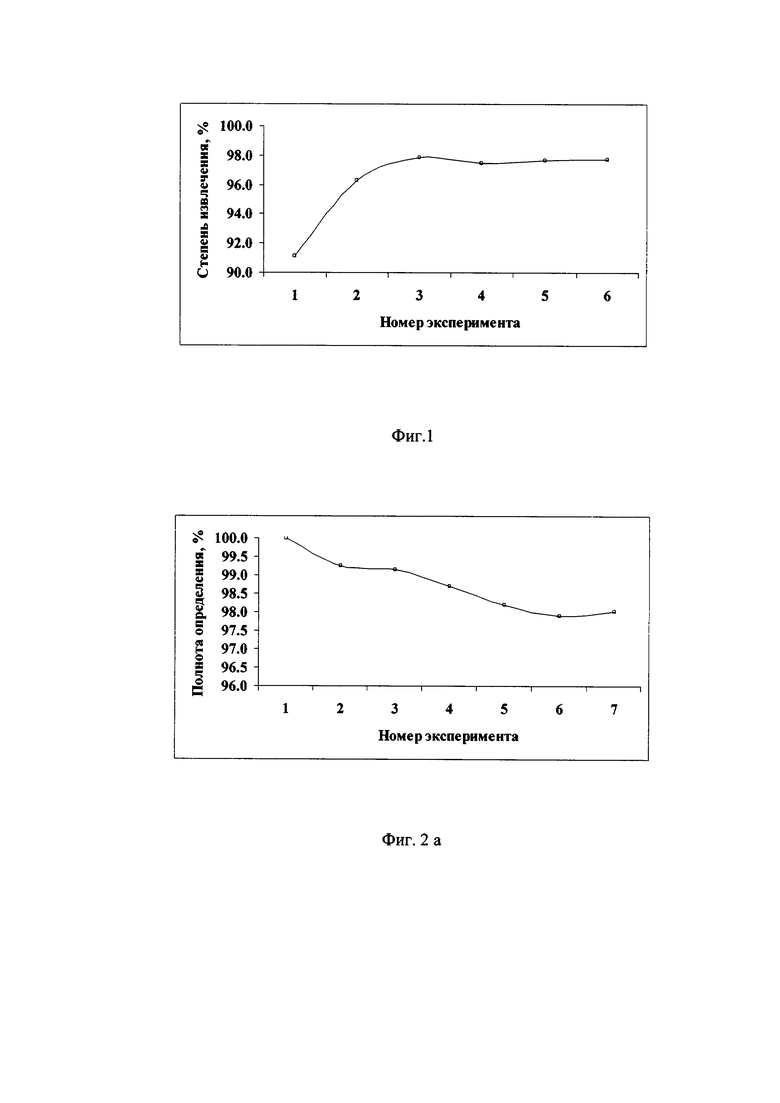
Было установлено, что в результате применения соотношения 5:2 (ацетон, см3/ навеска, г) на стадии экстракции, извлечение определяемого компонента происходит в полной мере, на уровне 97,8% от введенного значения, при сравнении с эталонным значением. Зависимость степени извлечения от соотношения навески и экстрагента отображена графически на фиг. 1 и в табл.1.


Согласно полученным данным можно заключить, что оптимально минимальным объемом ацетона, необходимом для извлечения дихлороизоэверниновой кислоты с минимальными потерями из 2 г навески мышечной ткани, является - 5 см3. Что соответствует опыту №3 (Табл. 1).
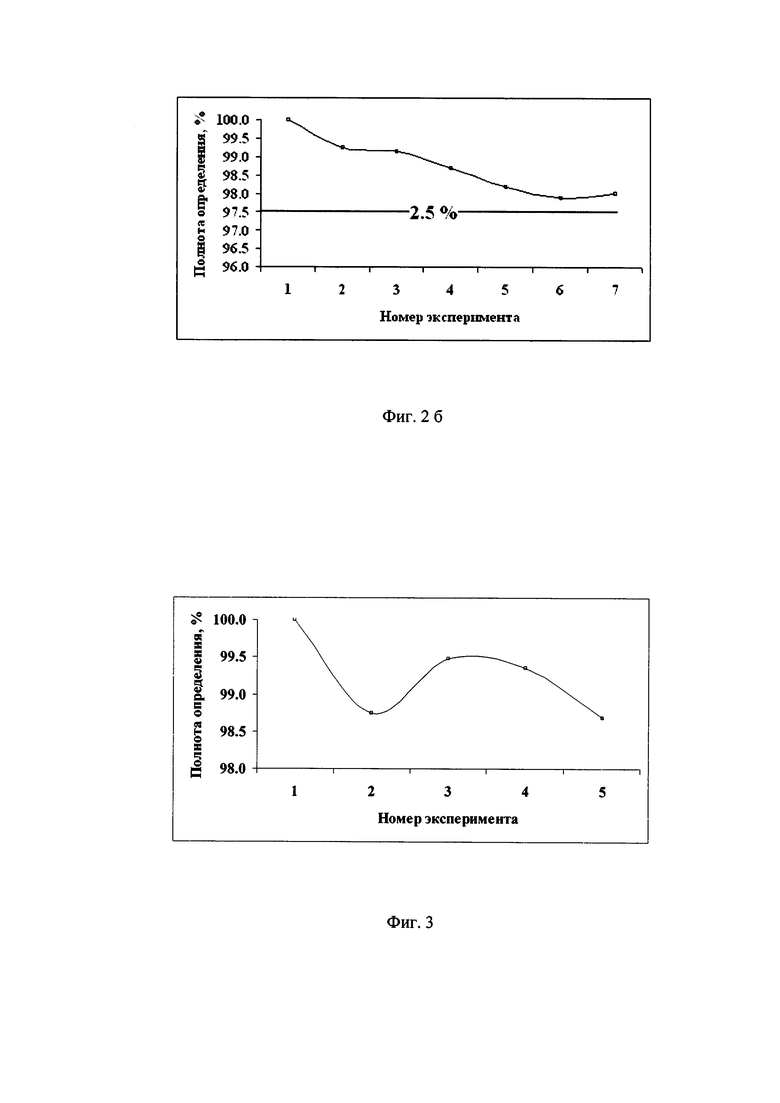
Было установлено, что при использовании измененного протокола применения этилацетата на стадиях, предшествующих ТФО, потери дихлороизоэверниновой кислоты несущественны, и составляют менее 3%. Зависимость потерь определяемого компонента от изменения протокола применения этилацетата отображена графически в табл. 2 и на фиг. 2 а, 2 б.
При проведении экспериментов, варьировали, относительно прототипа, объемы введения в гидролизат деионизованной воды и этилацетата, перед закислением рН среды ортофосфорной кислотой, и после этого. Перед проведением экспериментов, в гомогенаты мышечной ткани вводили эквивалентное количество дихлороизоэверниновой кислоты. Схема эксперимента была следующей:
Гидролизаты, полученные от навесок гомогената мышечной ткани и содержащие эквивалентное количество дихлороизоэверниновой кислоты, переносили в новые флаконы, ополаскивали старые флаконы варьируемым объемом деионизованной воды и варьируемым объемом этилацетата, объединяя эти объемы с гидролизатами в новых флаконах. Перемешивали флаконы с объединенными растворами и центрифугировали. Слои этилацетата отбрасывали, вводили в остаток гидролизатов ортофосфорную кислоту и варьируемый объем этилацетата. Перемешивали флаконы и разделяли фракции центрифугированием. Органический слой отбирали в новые флаконы и повторяли экстракцию новой порцией этилацетата. Объединенные объемы этилацетата концентрировали досуха в токе азота для дальнейшей стадии ТФО.
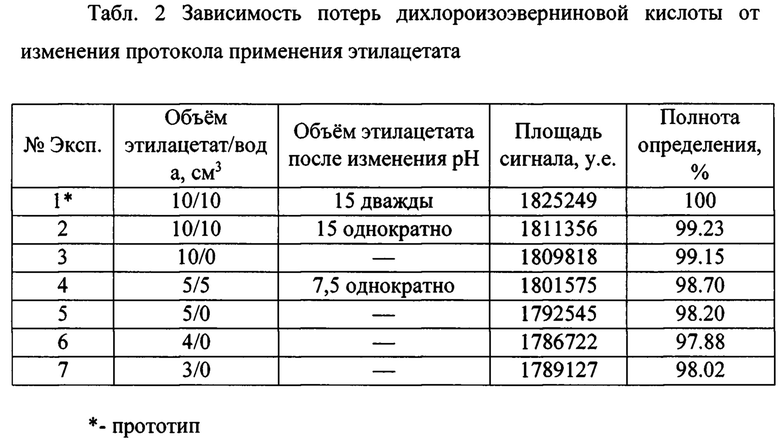
Согласно полученным данным, можно заключить, что наиболее экономичным протоколом применения этилацетата, необходимым для определения дихлороизоэверниновой кислоты с минимальными потерями, в навеске мышечной ткани, является его однократное применение до стадии закисления раствора, в количестве 3-5 см3. Что соответствует опытам №5-7 (Табл. 2).
Было установлено, что при замене процедуры ТФО на жидкостно-жидкостную экстракцию гексаном, достигается необходимая степень чистоты экстракта, с потерями дихлороизоэверниновой кислоты не более 2%, относительно прототипа. Это так же подтверждается отношением сигнала к шуму, равному 46.1 на уровне предела количественного определения (10 мкг/кг), что позволяет проводить количественный анализ данного соединения. Зависимость полноты определения дихлороизоэверниновой кислоты от изменения протокола применения ТФО отображена графически на фиг. 3 и в табл.3. Отношение сигнала к шуму отражено графически на фиг. 4.
Перед проведением экспериментов, в гомогенаты мышечной ткани вводили эквивалентное количество дихлороизоэверниновой кислоты. При проведении эксперимента, в качестве альтернативного протокола очистки, применялся следующий подход:
В гидролизаты, доведенные до необходимого рН раствором ортофосфорной кислоты, вводили варьируемый объем гексана, перемешивали на шейкере и центрифугировали. Гексан отбрасывали и экстрагировали дихлороизоэверниновую кислоту, добавляя к растворам 3 см3 этилацетата, перемешивая на шейкере и центрифугируя. Слой этилацетата переносили в новую полипропиленовую пробирку и упаривали в токе азота досуха, с последующим перерастворением в 1 см3 0,5% муравьиной кислоты и проводили анализ методом ВЭЖХ-МС/МС.
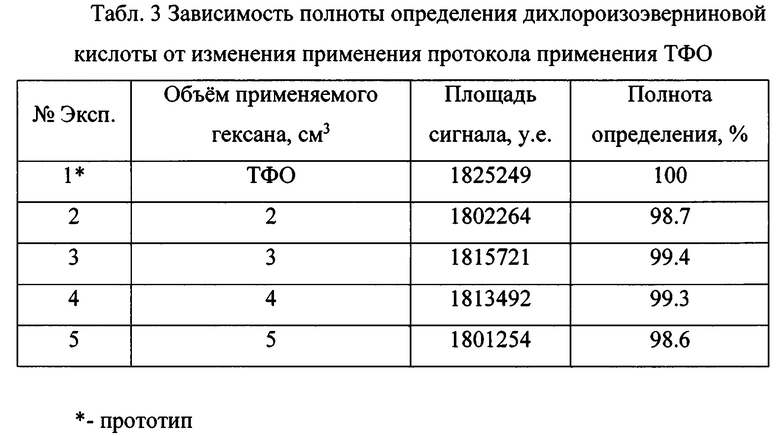
Согласно полученным данным, можно заключить, что наиболее экономичным протоколом дополнительной очистки, является замена протокола Прототипа на применение гексана в объеме 3-4 см3, что соответствует опытам №3-4 (Табл. 3).
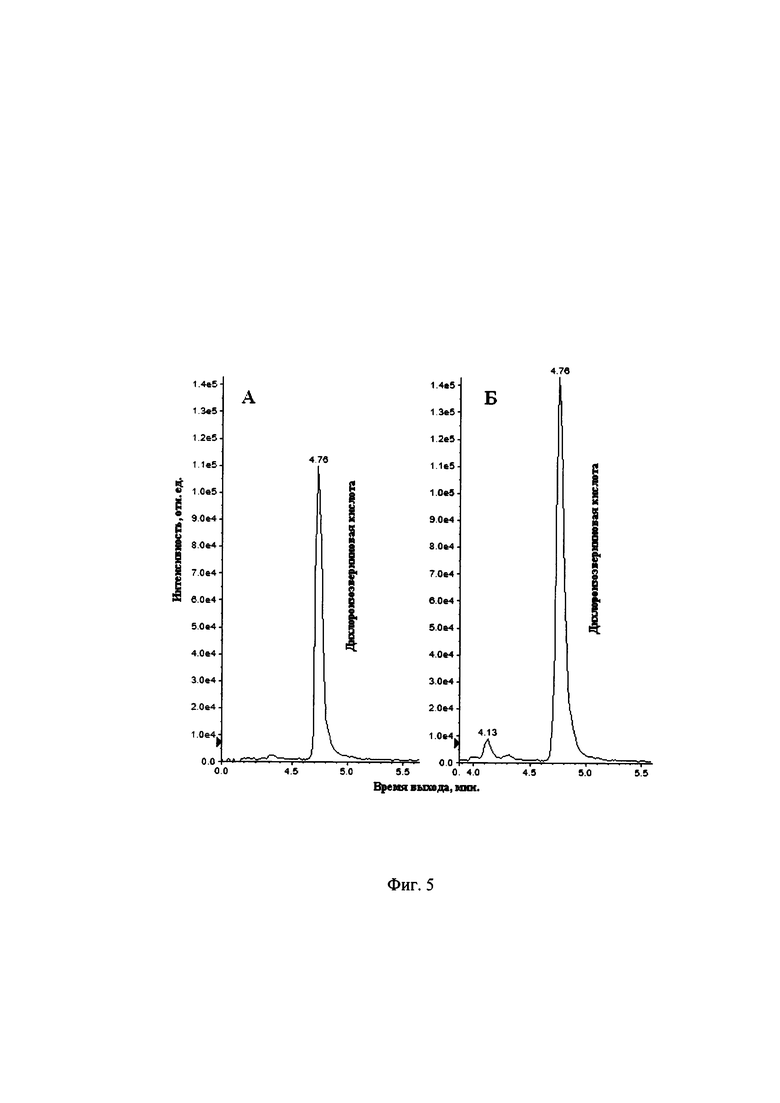
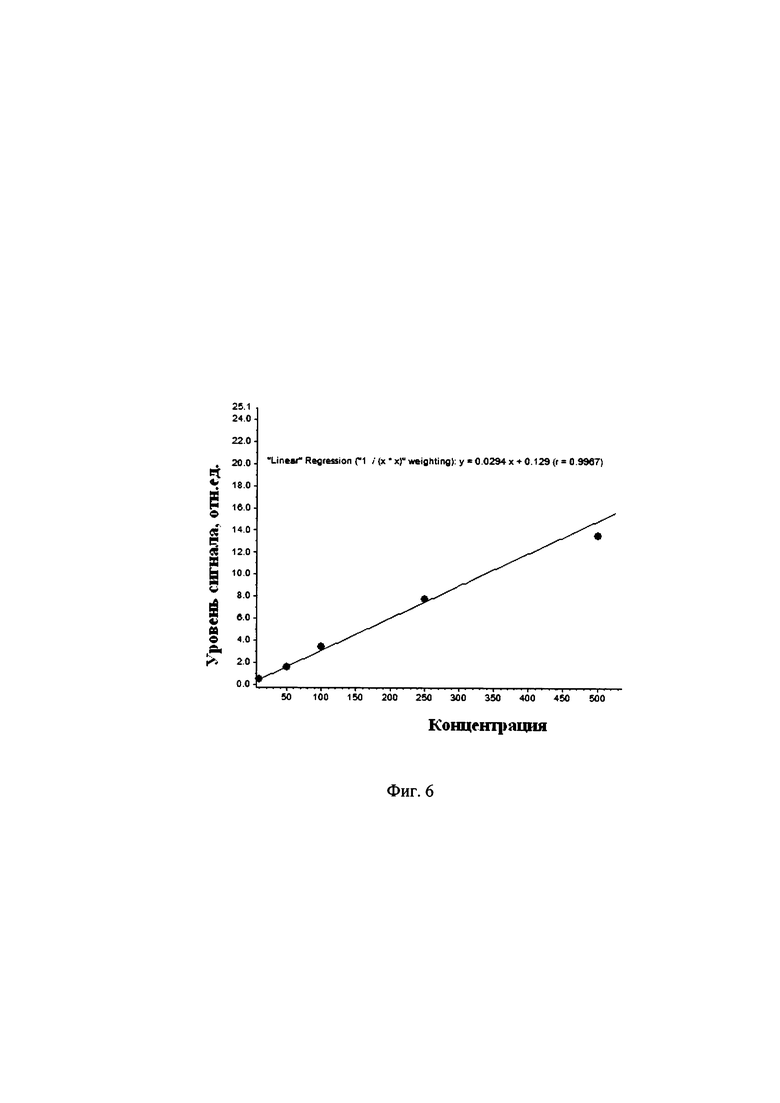
Было установлено, что при использовании новых протоколов экстракции ацетоном, отмывки гидролизата этилацетатом, а так же при замене стадии ТФО на очистку гексаном, с последующим концентрированием в токе азота, значительно повышается аналитический отклик при определении метаболита авиламицина - дихлороизоэверниновой кислоты в мышечных тканях животного происхождения. Сравнение относительной интенсивности сигнала и площади сигнала прототипа с окончательным протоколом способа, при эквивалентной добавке дихлороизоэверниновой кислоты перед процедурой экстракции отражено на фиг. 5. и в табл.4. Градуировочная характеристика с указанным коэффициентом корреляции, построенная по 5 уровням добавок дихлороизоэверниновой кислоты, прошедших процедуру экстракции и очистки в соответствии с патентуемым способом, представлена на фиг. 6.
Литературные источники:
[1] Shizuka Saito-Shida, Tomoko Hayashi, Satoru Nemoto, Hiroshi Akiyama, "S Determination of total avilamycin residues as dichloroisoeverninic acid in porcine muscle, fat, and liver by LC-MS/MS" Food Chemistry, vol. 249, pp.84-90, 2018.
[2] РЕГЛАМЕНТ КОМИССИИ (EU) №37/2010 от 22 декабря 2009 г. по фармакологически активным веществам и их классификации относительно максимальных пределов остатков в пищевых продуктах животного происхождения.
[3] TP ТС 034/2013 Технический регламент Таможенного союза «О безопасности мяса и мясной продукции».
[4] Neely, F.L., & Emerson, С.S." Rapid determination of avilamycin in fermentation broths. Journal of Liquid Chromatography" Journal of Liquid Chromatography, vol. 14, pp.2699-2705, 1991.
[5] Scott, C.A., Yordy, D.W., & Coleman, M.R., «Determination of avilamycin inpoultry feeds by liquid chromatography» Journal of AOAC International, т.82, pp.579-585, 1999.
[6] Sunderland, J., Lovering, A.M., Tobin, С.M., MacGowan, A.P., Roe, J.M., & Delsol, A.A., «Determination of avilamycin A and В in pig faeces by solid phase extraction and reverse-phase HPLC assay. International Journal of Antimicrobial Agents, 24» International Journal of Antimicrobial Agents, т.24, pp. 511-514, 2004.
[7] Amelin, V., Korotkov, A., & Andoralov, A., "Identification and determination of 492 contaminants of different classes in food and feed by high-resolution mass spectrometry using the standard addition method.," Journal of AOAC International, vol. 99, pp.1600-1618, 2016.
[8] Clare Ho & Yiu-Tung Wong, «Determination of avilamycin as dichloroisoeverninic acid in poultry and porcine muscles by isotope dilution liquid chromatography-tandem mass spectrometry.,» Anal Bioanal Chem, т.405, pp. 8633-8643, 2013.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ ГЛИФОСАТА, ЕГО МЕТАБОЛИТА И ГЛЮФОСИНАТА В ПРОДУКЦИИ ЖИВОТНОВОДСТВА | 2021 |
|
RU2783283C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ОСТАТОЧНЫХ КОЛИЧЕСТВ ПИПЕРАЗИНА В БИОЛОГИЧЕСКИХ ТКАНЯХ И ОБЪЕКТАХ ЖИВОТНОГО ПРОИСХОЖДЕНИЯ | 2024 |
|
RU2837303C1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ КЛАВУЛАНОВОЙ КИСЛОТЫ В МЫШЕЧНЫХ ТКАНЯХ ЖИВОТНОГО ПРОИСХОЖДЕНИЯ | 2021 |
|
RU2781486C1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ ОСТАТОЧНОГО СОДЕРЖАНИЯ СТЕРОИДНЫХ ГОРМОНОВ В РЫБЕ | 2021 |
|
RU2776013C1 |
| Способ определения остаточных количеств феноксикарба в почве методом высокоэффективной жидкостной хроматографии | 2020 |
|
RU2760530C1 |
| Способ количественного определения дексаметазона в биологических средах с помощью ВЭЖХ с ультрафиолетовым детектированием | 2022 |
|
RU2792274C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ СОДЕРЖАНИЯ ВИТАМИНА К В ПРОДУКТАХ РАСТИТЕЛЬНОГО ПРОИСХОЖДЕНИЯ | 2017 |
|
RU2647451C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ МИКОТОКСИНОВ В ПРОДУКТАХ ЖИВОТНОГО И РАСТИТЕЛЬНОГО ПРОИСХОЖДЕНИЯ | 2012 |
|
RU2514828C2 |
| СПОСОБ ОПРЕДЕЛЕНИЯ СОДЕРЖАНИЯ ВЫСОКОМОЛЕКУЛЯРНЫХ НАФТЕНОВЫХ КИСЛОТ В НЕФТИ (ВАРИАНТЫ) | 2022 |
|
RU2800377C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ОСТАТОЧНЫХ КОЛИЧЕСТВ ТРИФЕНИЛМЕТАНОВЫХ КРАСИТЕЛЕЙ В МЫШЕЧНОЙ ТКАНИ РЫБ | 2015 |
|
RU2578974C1 |

Изобретение относится к области аналитической химии и касается способа определения остаточного содержания авиламицина в биологических тканях животного происхождения. Способ включает извлечение дихлороизоэверниновой кислоты из тканей ацетоном, концентрирование полученного экстракта досуха с последующим гидролизом остатка едким натром, очистку гидролизата этилацетатом и его закисление ортофосфорной кислотой, последующую дополнительную очистку полученного раствора и перевод определяемого соединения в слой этилацетата для дальнейшего концентрирования и перерастворения перед анализом методом высокоэффективной жидкостной хроматографии в сочетании с тандемной масс-спектрометрией (ВЭЖХ-МС/МС). При этом при экстракции соблюдается соотношение ацетон : навеска образца, равное 5 см3 ацетона на 2 г навески образца, очистку гидролизата перед закислением проводят однократно с использованием 3-5 см3 этилацетата, а подкисленный гидролизат дополнительно очищают 3-4 см3 гексана с дальнейшим удалением органического слоя и добавлением этилацетата, перемешиванием на шейкере и центрифугированием. Затем слой этилацетата упаривают в токе азота досуха, перерастворяют в 1 см3 0,5% муравьиной кислоты и проводят анализ методом ВЭЖХ-МС/МС. Способ позволяет на 30% увеличить аналитический отклик сигнала детектора по метаболиту авиламицина, что позволяет уверенно работать со сложными видами образцов в результате снижения влияния шумов базовой линии; существенно уменьшить расход ацетона при экстракции; упростить процедуру жидкостно-жидкостной экстракции этилацетатом; избежать процедуры твердофазной очистки (ТФО). 4 табл., 6 ил.
Способ определения остаточных количеств авиламицина в биологических тканях животного происхождения, включающий извлечение дихлороизоэверниновой кислоты из тканей ацетоном, концентрирование полученного экстракта досуха с последующим гидролизом остатка едким натром, очистку гидролизата этилацетатом и его закисление ортофосфорной кислотой, последующую дополнительную очистку полученного раствора и перевод определяемого соединения в слой этилацетата для дальнейшего концентрирования и перерастворения перед анализом методом высокоэффективной жидкостной хроматографии в сочетании с тандемной масс-спектрометрией (ВЭЖХ-МС/МС), отличающийся тем, что при экстракции соблюдается соотношение ацетон : навеска образца, равное 5 см3 ацетона на 2 г навески образца, очистку гидролизата перед закислением проводят однократно с использованием 3-5 см3 этилацетата, а подкисленный гидролизат дополнительно очищают 3-4 см3 гексана с дальнейшим удалением органического слоя и добавлением этилацетата, перемешиванием на шейкере и центрифугированием, затем слой этилацетата упаривают в токе азота досуха, перерастворяют в 1 см3 0,5% муравьиной кислоты и проводят анализ методом ВЭЖХ-МС/МС.
| SHIZUKA SAITO-SHIDA ET AL | |||
| Determination of total avilamycin residues as dichloroisoeverninic acid in porcine muscle, fat, and liver by LC-MS/MS | |||
| Food Chemistry, 2018, vol.249, pp.84-90 | |||
| СПОСОБ ОПРЕДЕЛЕНИЯ ОКСИБЕНЗОЛА И ЕГО МОНОМЕТИЛЬНЫХ ПРОИЗВОДНЫХ В БИОЛОГИЧЕСКОМ МАТЕРИАЛЕ | 2004 |
|
RU2269137C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ 2,4-ДИХЛОРФЕНОКСИУКСУСНОЙ КИСЛОТЫ В БИОЛОГИЧЕСКОМ МАТЕРИАЛЕ | 2011 |
|
RU2453848C1 |
| ПРИСПОСОБЛЕНИЕ ДЛЯ УСТАНОВКИ ТАРЫ ПОД МАШИНЫ ДЛЯ ОТМЕРИВАНИЯ ЖИДКИХ И СЫПУЧИХ ТЕЛ | 1929 |
|
SU20001A1 |
| Способ получения изолирующих тел из обжигаемой до спекания окиси алюминия | 1929 |
|
SU21022A1 |
| CN 109932459 A, 25.06.2019. | |||

