Область техники, к которой относится настоящее изобретение
Настоящее изобретение относится к новому соединению, одновременно характеризующемуся эффектами тромболизиса, акцептирования свободных радикалов и направленного действия на тромб, а также к способу его получения и применению. Настоящее изобретение также относится к новому трехкомпонентному конъюгату «пептид, содержащий последовательность PAK/имидазолин/пептид, содержащий последовательность RGD», образованному путем связывания вместе тромболитического олигопептида, содержащего последовательность PAK (Pro-Ala-Lys), 1-(4-оксиацетилфенил)-3,3,4,4-тетраметилимидазолина и пептида с направленным действием на тромб/антитромботического олигопептида, содержащего последовательность RGD (Arg-Gly-Asp), посредством связующего звена, содержащего карбоксильную и аминогруппы. Настоящее изобретение также относится к содержащей вышеупомянутое соединение фармацевтической композиции, применяемой для акцептирования свободных радикалов NO, тромболизиса, направленного действия на тромб/антитромботической терапии и лечения инсульта/инфаркта головного мозга. Настоящее изобретение также относится к способу получения соединения.
Предшествующий уровень техники настоящего изобретения
В глобальном масштабе тромботические заболевания занимают первое место по заболеваемости и смертности. Тромбоз коронарных артерий приводит к инфаркту миокарда. Тромбоз сосуда головного мозга приводит к инфаркту головного мозга, т.е. к клиническому ишемическому инсульту. Пациентам с инфарктом миокарда могут проводиться внутривенные инъекции тромболитических средств или операции коронарного шунтирования. Следует отметить, что положительный эффект внутривенной инъекции тромболитических средств пациентам с инфарктом миокарда представляет собой ишемию/реперфузию. Поскольку в процессе ишемии/реперфузии генерируется огромное количество свободных радикалов NO, процесс тромболизиса ассоциирован с повреждением миокарда и смертью пациента. Это является серьезной проблемой для современного тромболитического лечения инфаркта миокарда. В настоящее время, лечение инфаркта головного мозга сопряжено с еще более сложными проблемами. Например, все современные тромболитические средства не способны проникать через гематоэнцефалический барьер, а потому эффективность внутривенной инъекции тромболитических средств пациентам с инфарктом головного мозга довольно ограничена. Кроме того, например, на сегодняшний день не существует доступной подходящей хирургической процедуры, позволяющей спасти пациентов с инфарктом головного мозга. По аналогии, даже при наличии положительного эффекта от внутривенной инъекции тромболитических средств пациентам с инфарктом головного мозга, в процессе ишемии/реперфузии может генерироваться огромное количество свободных радикалов NO, так что процесс тромболизиса ассоциирован с повреждением тканей головного мозга и смертью пациента. Это представляет собой серьезную проблему в современном тромболитическом лечении инфаркта головного мозга. Более того, существует четыре серьезных проблемы в клиническом лечении пациентов с инсультом: 1) никакое лекарственное средство, за исключением tPA (тканевой активатор плазминогена), не демонстрирует свою эффективность у пациентов с инсультом; 2) лечение tPA эффективно лишь в течение 3 часов от начала инсульта, т.е. для лечения tPA существует лишь 3-часовое окно; 3) лечение tPA часто приводит к системному кровотечению; 4) посредством лечения tPA невозможно избежать повреждения ткани головного мозга пациентов и смерти пациентов, ассоциированных с огромным количеством свободных радикалов NO, продуцируемых в процессе ишемии/реперфузии. Поэтому, для достижения существенного прогресса в клиническом лечении пациентов с инсультом существует необходимость решить указанные четыре проблемы.
В патентных публикациях Китая CN 102807604 и CN 102807605 раскрыты два соединения, Nα-(1,3-диоксо-4,4,5,5-тетраметилимидазолин-2-фенил-4′-оксиацетил)-nω-(ацил жирной кислоты)-Lys-Arg-Gly-Asp-Val и Nα-(1,3-диоксо-4,4,5,5-тетраметилимидазолин-2-фенил-4′-оксиацетил)-nω-(ацил жирной кислоты)-Lys-Arg-Gly-Asp-Phe. Оба соединения получают путем конъюгирования имидазолина, характеризующегося способностью к акцептированию свободных радикалов NO, с антитромботическим олигопептидом, содержащим последовательность RGD (Arg-Gly-Asp), посредством лизина. В отличие от соединения согласно настоящему изобретению, указанные два соединения не содержат тромболитического пептида, присоединенного к ним. Указанные два соединения не участвуют в тромболизисе, а потому не подходят для производства тромболитических лекарственных средств и не подходят для лечения пациентов с ишемическим инсультом.
Для решения вышеупомянутых проблем, существует потребность в новом соединении, одновременно характеризующемся эффектами тромболизиса, акцептирования свободных радикалов и направленного действия на тромб. Кроме того, необходимо, чтобы такое новое соединение было эффективно даже при введении спустя 3 часа от начала инсульта у пациентов, т.е. не было ограничено 3-часовым окном, как в случае лечения с применением tPA; не вызывало системное кровотечение, как в случае лечения tPA; и было способно устранять огромное количество свободных радикалов NO, генерируемых в процессе ишемии/реперфузии.
Краткое раскрытие настоящего изобретения
Настоящее изобретение относится к трехкомпонентному конъюгату, одновременно обладающему способностью к проникновению через гематоэнцефалический барьер, тромболизису, антитромботической активности и акцептированию свободных радикалов NO, в котором три составные части трехкомпонентного конъюгата относятся к имидазолину, характеризующемуся способностью к акцептированию свободных радикалов NO, пептиду, характеризующемуся тромболитической активностью, и пептиду с направленным действием на тромб, причем три составные части связаны вместе посредством подходящего связующего звена.
В частности, трехкомпонентный конъюгат согласно настоящему изобретению может быть представлен соединением формулы I:
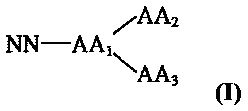
в которой NN представляет собой имидазолин, характеризующийся способностью к акцептированию свободных радикалов NO; AA1 представляет собой связующее звено, содержащее, по меньшей мере, три группы для связывания; АА2 представляет собой пептид, характеризующийся тромболитической активностью; и АА3 представляет собой пептид с направленным действием на тромб.
Используемый в настоящем изобретении имидазолин может содержать имидазолнитроксилнитроксидные (NN) радикалы, которые могут устранять NO и выполнять функции по устранению свободных радикалов кислорода, обеспечивая мощную защиту клеток, поврежденных свободными радикалами кислорода. Имидазолин согласно настоящему изобретению, характеризующийся способностью к акцептированию свободных радикалов NO, предпочтительно представляет собой 1,3-диоксо-2-[(4-оксиацетокси)фенил]-4,4,5,5-тетраметилимидазолин, который характеризуется превосходной химической и физической стабильностью, и не только подходит для любой химической реакции конъюгирования пептида, характеризующегося тромболитической активностью, с пептидом с направленным действием на тромб, но также не склонного к разрушению в процессе хранения, тем самым удовлетворяя требованиям к лекарственным формам.
Используемое в настоящем изобретении связующее звено может содержать, по меньшей мере, три группы для связывания, например, карбоксильные и аминогруппы, которые используются для того, чтобы связать имидазолин, пептид, характеризующийся тромболитической активностью, и пептид с направленным действием на тромб. Связующее звено согласно настоящему изобретению может представлять собой натуральные аминокислоты, например L-Lys, L-Asp и L-Glu. Если связующее звено (AA1), используемое в настоящем изобретении, содержит три или более групп для связывания, то посредством него могут быть связаны один или несколько NN, АА2 или АА3, причем два или несколько NN, АА2 или АА3 могут быть одинаковыми или разными. Например, если AA1 содержит четыре группы для связывания, то посредством него могут быть связаны один NN, два АА2 и один АА3, причем два АА2 могут представлять собой одинаковые или разные пептиды, характеризующиеся тромболитической активностью.
Используемый в настоящем изобретении пептид, характеризующийся тромболитической активностью может представлять собой олигопептид, содержащий последовательность PAK (Pro-Ala-Lys), последовательность AKP (Ala-Lys-Pro) или последовательность KAP (Lys-Ala-Pro), или пептид, содержащий повторяющиеся элементы последовательности PAK, последовательности AKP или последовательности KAP. Олигопептид относится к низкомолекулярному пептиду, имеющему молекулярный вес 1000 Дальтон (D) или менее, который, как правило, состоит из 3-8 аминокислот. Олигопептид согласно настоящему изобретению, характеризующийся тромболитической активностью, может представлять собой трипептид - октопептид, который содержит последовательность PAK, последовательность AKP или последовательность KAP, предпочтительно трипептид - пентапептид, который содержит последовательность PAK, последовательность AKP или последовательность KAP. Например, используемый в настоящем изобретении олигопептид, который содержит последовательность PAK, последовательность AKP или последовательность KAP, может представлять собой PAK, RPAK (Arg-Pro-Ala-Lys), ARPAK (Ala-Arg-Pro-Ala-Lys), GRPAK (Gly-Arg-Pro-Ala-Lys), QRPAK (Gln-Arg-Pro-Ala-Lys), AKP, KAP, KPAK (Lys-Pro-Ala-Lys), PAKP (Pro-Ala-Lys-Pro), AKPAK (Ala-Lys-Pro-Ala-Lys) или PAKPA (Pro-Ala-Lys-Pro-Ala). Например, используемый в настоящем изобретении пептид, содержащий повторяющиеся элементы последовательности PAK, последовательности AKP или последовательности KAP, может представлять собой любой из таких пептидов, описанных в патентной публикации Китая CN 101190941, в качестве пептида, характеризующегося тромболитической активностью, включая пептид, содержащий повторяющиеся элементы последовательности PAK, такой как (PAK)2, (PAK)3, (PAK)4, (PAK)5 и (PAK)6; пептид, содержащий повторяющиеся элементы последовательности AKP, такой как (AKP)2, (AKP)3, (AKP)4, (AKP)5 и (AKP)6; пептид, содержащий повторяющиеся элементы последовательности KPA, такой как (KPA)2, (KPA)3, (KPA)4, (KPA)5 и (KPA)6.
Используемый в настоящем изобретении пептид с направленным действием на тромб/антитромботический пептид может представлять собой олигопептид, содержащий последовательность RGD (Arg-Gly-Asp). Олигопептид, содержащий последовательность RGD, может представлять собой тетрапептид на основе RGD, такой как RGDS (Arg-Gly-Asp-Ser), RGDV (Arg-Gly-Asp-Val) и RGDF (Arg-Gly-Asp-Phe). Специфическое связывание фибриногена (Fg) с активированным рецептором гликопротеина (GP) IIb/IIIa мембраны тромбоцита представляет собой приводящий к агрегации тромбоцитов общеизвестный конечный путь метаболизма, запускаемый различными физиологическими индукторами, и играет важную роль в формировании тромба. Кроме того, последовательности RGD служат в качестве активных сайтов для связывания Fg лигандов и активированных GPIIb/IIIa рецепторов и характеризуются направленным действием на активированные тромбоциты. Структуры, содержащие последовательность RGD, могут конкурентно ингибировать и блокировать связывание Fg и GPIIb/IIIa рецепторов, предотвращая тем самым агрегацию тромбоцитов и образование тромба, что позволяет содержащему RGD олигопептиду стать эффективной молекулой с направленным действием на тромб и антитромботическим средством.
Кроме того, используемый в настоящем изобретении пептид с направленным действием на тромб может представлять собой любой из таких полипептидов, описанных в патентной публикации Китая CN 101190940, в качестве полипептида, характеризующегося направленным действием и антитромботической активностью, включая полипептиды, полученные в результате модификации путем конъюгирования пептида RGD с пептидом YIGS (Tyr-Ile-Gly-Ser). Полученные путем модификации полипептиды включают в себя YIGSRRGDS, YIGSRRGDV, YIGSRRGDF, YIGSRYIGSK, YIGSRYIGSR, YIGSKRGDS, YIGSKRGDF, YIGSICRGDV, YIGSKYIGSK, YIGSKYIGSR, RGDSRGDS, RGDVRGDV, RGDFRGDF, RGDSYIGSR, RGDSYIGSK, RGDVYIGSR, RGDVYIGSK, RGDFYIGSR или RGDFYIGSK.
Согласно предпочтительному варианту осуществления, в соединении согласно настоящему изобретению имидазолин, характеризующийся способностью к акцептированию свободных радикалов NO, представляет собой 1,3-диоксо-2-[(4-оксиацетокси)фенил]-4,4,5,5-тетраметилимидазолин, пептид, характеризующийся тромболитической активностью, представляет собой олигопептид, содержащий последовательность PAK (Pro-Ala-Lys), и пептид с направленным действием на тромб представляет собой олигопептид, содержащий последовательность RGD (Arg-Gly-Asp). Таким образом, настоящее изобретение относится к трехкомпонентному конъюгату «пептид, содержащий последовательность PAK/имидазолин/пептид, содержащий последовательность RGD», одновременно характеризующемуся способностью к проникновению через гематоэнцефалический барьер, тромболизису, антитромботической активности и акцептированию свободных радикалов NO.
Согласно одному варианту осуществления, в соединении согласно настоящему изобретению, имидазолин, характеризующийся способностью к акцептированию свободных радикалов NO, представляет собой 1,3-диоксо-2-[(4-оксиацетокси)фенил]-4,4,5,5-тетраметилимидазолин, связующее звено представляет собой L-Lys, пептид, характеризующийся тромболитической активностью, представляет собой олигопептид, содержащий последовательность PAK (Pro-Ala-Lys), и пептид с направленным действием на тромб представляет собой олигопептид, содержащий последовательность RGD (Arg-Gly-Asp). В этом случае олигопептид, содержащий последовательность PAK, может представлять собой пентапептид ARPAK, пентапептид GRPAK, тетрапептид RPAK или трипептид PAK; олигопептид, содержащий последовательность RGD (Arg-Gly-Asp), может представлять собой тетрапептид на основе RGD, такой как RGDS, RGDV или RGDF. Если в качестве связующего звена используется L-Lys, то соединение согласно настоящему изобретению может характеризоваться следующей общей формулой I-1 или I-2:
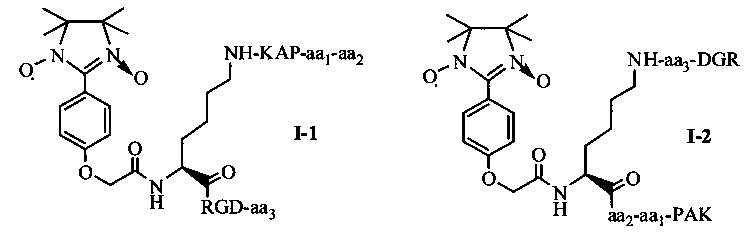
в которой aa1 и аа2 могут оба присутствовать, или оба отсутствовать, или aa1 присутствует, но аа2 отсутствует; если aa1 и аа2 оба присутствуют, то aa1 представляет собой R (Arg), и аа2 представляет собой G (Gly), A (Ala) или Q (Gln); если aa1 присутствует, но аа2 отсутствует, то aa1 представляет собой R (Arg); аа3 может представлять собой S (Ser), V (Val) или F (Phe).
Для примеров, относящихся к соединению общей формулы I-1, согласно предпочтительному примеру, соединение согласно настоящему изобретению может представлять собой трехкомпонентный конъюгат ARPAK/имидазолин/RGD, представленный следующей формулой I-1-1; согласно другому предпочтительному примеру, соединение согласно настоящему изобретению может представлять собой трехкомпонентный конъюгат GRPAK/имидазолин/RGD, представленный следующей формулой I-1-2; согласно еще одному предпочтительному примеру, соединение согласно настоящему изобретению может представлять собой трехкомпонентный конъюгат RPAK/имидазолин/RGD, представленный следующей формулой I-1-3; и согласно еще одному предпочтительному примеру, соединение согласно настоящему изобретению может представлять собой трехкомпонентный конъюгат PAK/имидазолин/RGD, представленный следующей формулой I-1-4:
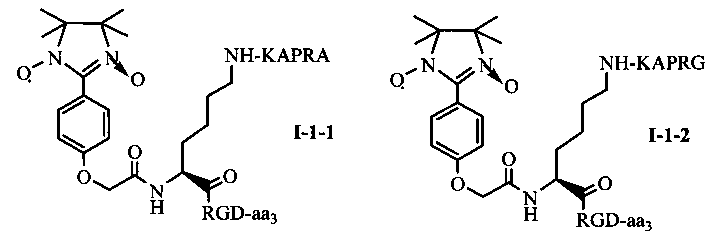
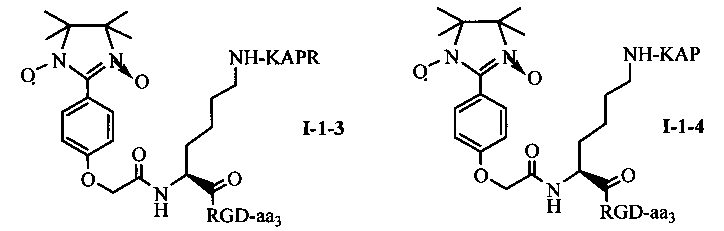
в которых аа3 может представлять собой S (Ser), V (Val) или F (Phe), предпочтительно V (Val).
Для примеров, относящихся к соединению общей формулы I-2, соединение согласно настоящему изобретению может предпочтительно характеризоваться следующей общей формулой I-2-1, I-2-2, I-2-3 или I-2-4:
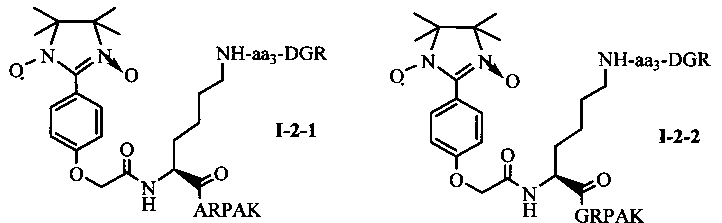
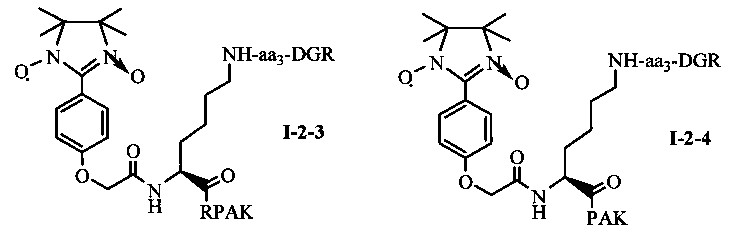
в которых аа3 может представлять собой S (Ser), V (Val) или F (Phe), предпочтительно V (Val).
Согласно другому варианту осуществления, в соединении согласно настоящему изобретению, имидазолин, характеризующийся способностью к акцептированию свободных радикалов NO, представляет собой 1,3-диоксо-2-[(4-оксиацетокси)фенил]-4,4,5,5-тетраметилимидазолин, связующее звено представляет собой L-Asp, пептид, характеризующийся тромболитической активностью, представляет собой олигопептид, содержащий последовательность PAK (Pro-Ala-Lys), и пептид с направленным действием на тромб представляет собой олигопептид, содержащий последовательность RGD (Arg-Gly-Asp). Если в качестве связующего звена используется L-Asp, то соединение согласно настоящему изобретению может характеризоваться следующей общей формулой I-3 или I-4:
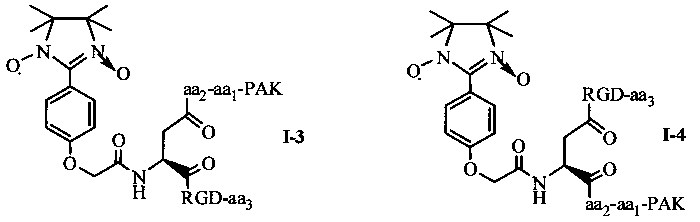
в которых aa1 и аа2 могут оба присутствовать, или оба отсутствовать, или aa1 присутствует, но аа2 отсутствует; если aa1 и аа2 оба присутствуют, то aa1 представляет собой R (Arg), и аа2 представляет собой G (Gly), A (Ala) или Q (Gln); если aa1 присутствует, но аа2 отсутствует, то aa1 представляет собой R (Arg); аа3 может представлять собой S (Ser), V (Val), или F (Phe). aa1 предпочтительно представляет собой R (Arg), аа2 предпочтительно представляет собой G (Gly), и аа3 предпочтительно представляет собой V (Val).
Для примеров, относящихся к соединению общей формулы 1-3, соединение согласно настоящему изобретению может предпочтительно характеризоваться следующей общей формулой I-3-1, I-3-2, I-3-3 или I-3-4:
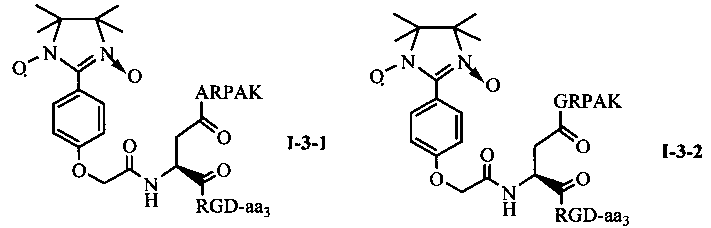
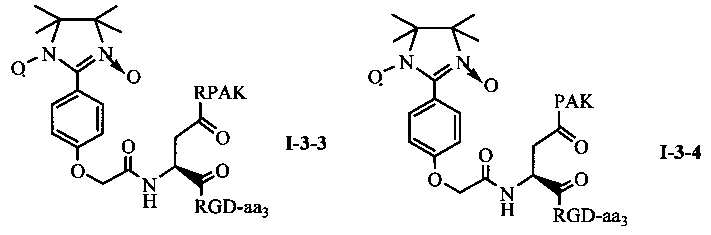
в которых аа3 может представлять собой S (Ser), V (Val) или F (Phe), предпочтительно V (Val).
Для примеров, относящихся к соединению общей формулы 1-4, соединение согласно настоящему изобретению может предпочтительно характеризоваться следующей общей формулой I-4-1, I-4-2, I-4-3 или I-4-4:
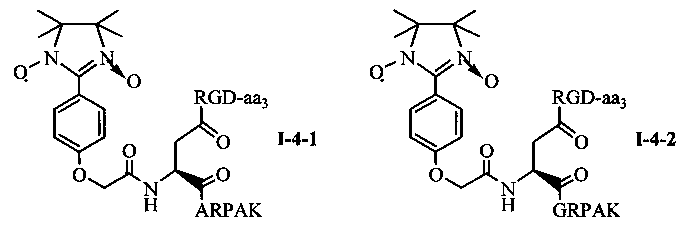
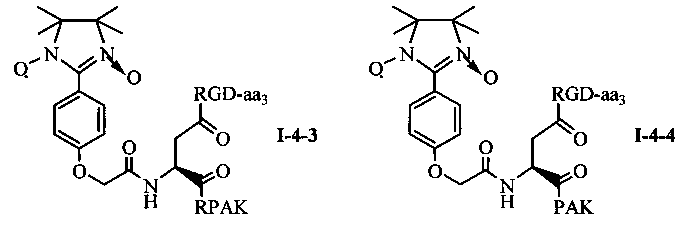
в которых аа3 может представлять собой S (Ser), V (Val) или F (Phe), предпочтительно V (Val).
Согласно еще одному варианту осуществления, в соединении согласно настоящему изобретению, имидазолин, характеризующийся способностью к акцептированию свободных радикалов NO, представляет собой 1,3-диоксо-2-[(4-оксиацетокси)фенил]-4,4,5,5-тетраметилимидазолин, связующее звено представляет собой L-Glu, пептид, характеризующийся тромболитической активностью, представляет собой олигопептид, содержащий последовательность PAK (Pro-Ala-Lys), и пептид с направленным действием на тромб представляет собой олигопептид, содержащий последовательность RGD (Arg-Gly-Asp). Если в качестве связующего звена используется L-Glu, то соединение согласно настоящему изобретению может характеризоваться следующей общей формулой I-5 или I-6:
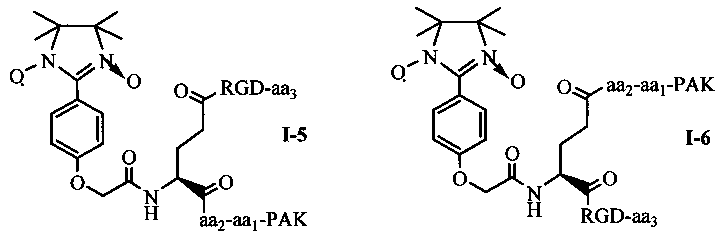
в которых aa1 и аа2 могут оба присутствовать, или оба отсутствовать, или aa1 присутствует, но аа2 отсутствует; если aa1 и аа2 оба присутствуют, то aa1 представляет собой R (Arg), и аа2 представляет собой G (Gly), A (Ala) или Q (Gln); если aa1 присутствует, но аа2 отсутствует, то aa1 представляет собой R (Arg); аа3 может представлять собой S (Ser), V (Val), или F (Phe). aa1 предпочтительно представляет собой R (Arg), аа2 предпочтительно представляет собой G (Gly), и аа3 предпочтительно представляет собой V (Val).
Для примеров, относящихся к соединению общей формулы 1-5, соединение согласно настоящему изобретению может предпочтительно характеризоваться следующей общей формулой I-5-1, I-5-2, I-5-3 или I-5-4:
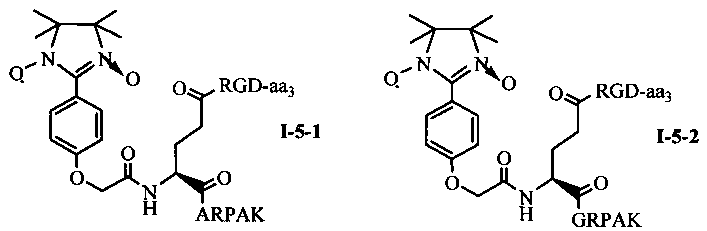
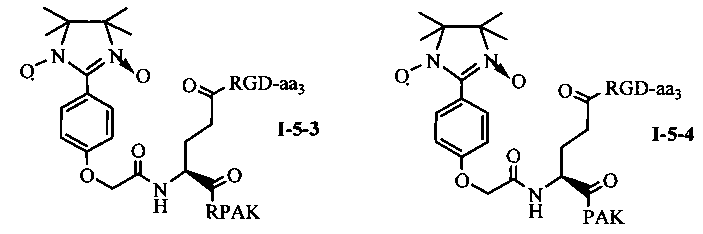
в которых аа3 может представлять собой S (Ser), V (Val) или F (Phe), предпочтительно V (Val).
Для примеров, относящихся к соединению общей формулы I-6, соединение согласно настоящему изобретению может предпочтительно характеризоваться следующей общей формулой I-6-1, I-6-2, I-6-3 или I-6-4:
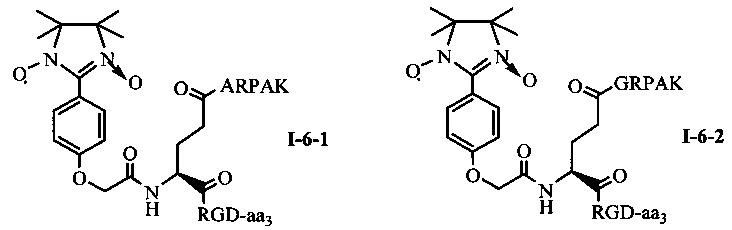
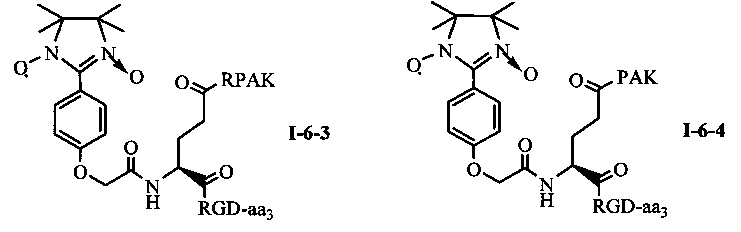
в которых аа3 может представлять собой S (Ser), V (Val) или F (Phe), предпочтительно V (Val).
Согласно другому аспекту, настоящее изобретение также относится к фармацевтической композиции, содержащей вышеупомянутое соединение согласно настоящему изобретению и фармацевтически приемлемый носитель. Предпочтительно, фармацевтическая композиция согласно настоящему изобретению содержит соединение вышеупомянутой общей формулы I-1, I-2, I-3, I-4, I-5 или I-6. Более предпочтительно, фармацевтическая композиция согласно настоящему изобретению содержит соединение вышеупомянутой общей формулы I-1-1, I-1-2, I-1-3 или I-1-4. Если фармацевтическая композиция согласно настоящему изобретению содержит соединение общей формулы I-1-1, I-1-2, I-1-3 или I-1-4, то соединение может находиться в фармацевтической композиции в форме димерной, тримерной или тетрамерной структуры, и может находиться в форме наносферы с диаметром от 2 до 300 нм. В фармацевтической композиции согласно настоящему изобретению, наносферическая структура может предпочтительно иметь диаметр от 2 до 100 нм. В нанофармакологии хорошо известен тот факт, что наносферы, имеющие диаметр менее 100 нм, менее склонны быть поглощенными макрофагами в процессе транспортировки в кровяном русле и могут легко проникать через стенку кровеносных капилляров. Такие свойства позволяют соединению согласно настоящему изобретению проникать через гематоэнцефалический барьер. Фармацевтическая композиция согласно настоящему изобретению может быть использована в качестве тромболитического лекарства при лечении заболеваний, таких как инфаркт миокарда, ишемический инсульт, тромбоз глубоких вен, легочная эмболия, окклюзионная болезнь периферических артерий, окклюдированные центральные магистрали сосудистого доступа, коагулированная артериовенозная фистула и шунты, и каротидный стеноз. Фармацевтическая композиция согласно настоящему изобретению также может быть использована в качестве лекарства для акцептирования свободных радикалов NO при лечении нейродегенеративных заболеваний, таких как болезнь Альцгеймера, болезнь Паркинсона, заболевания моторных нейронов, боковой амиотрофический склероз, потеря слуха от воздействия шума, болезнь Лу Герига или болезнь Гентингтона; при лечении сердечно-сосудистых заболеваний, таких как атеросклероз, коронарная болезнь сердца или инфаркт миокарда; при лечении психических заболеваний, таких как биполярное нарушение, шизофрения или аутизм; и при лечении заболеваний, включающих в себя высотную болезнь, сахарный диабет, ревматоидный артрит, травматическое повреждение головного мозга, злокачественную опухоль, синдром ломкой X хромосомы, серповидно-клеточную анемию, красный плоский лишай, витилиго, синдром хронической усталости и т.д. Фармацевтическая композиция согласно настоящему изобретению также может быть использована в качестве лекарства с направленным действием на тромб/антитромботического лекарства при лечении заболеваний, таких как тромбоцитоз, миелопролиферативное заболевание, истинная полицитемия или синдром Бадда-Киари. Фармацевтическая композиция согласно настоящему изобретению также может быть использована в качестве лекарства при лечении инсульта или инфаркта головного мозга, предпочтительно при лечении инсульта или инфаркта головного мозга спустя 3, 4, 6 и 24 часа от начала симптомов с последовательными введениями. Фармацевтическая композиция/соединение согласно настоящему изобретению одновременно характеризуется функциями акцептирования свободных радикалов NO, тромболизиса и антитромботического/направленного действия на тромб, а потому демонстрирует эффективность даже при введении позже 3 часов от начала инсульта у пациентов; т.е. они не ограничены 3-часовым окном, как при лечении с использованием tPA, не вызывают системное кровотечение, как tPA, и могут устранять огромное количество свободных радикалов NO, генерируемых в процессе ишемии/реперфузии, предотвращая у пациентов повреждение тканей черепных нервов в процессе лечения. В фармацевтической композиции согласно настоящему изобретению наносферические структуры соединений способны максимизировать эффекты проникновения через гематоэнцефалический барьер, тромболизиса, направленного действия на тромб/антитромботического действия, а также эффект устранения свободных радикалов NO, генерируемых в процессе ишемии/реперфузии.
Фармацевтическая композиция согласно настоящему изобретению может представлять собой любую клинически приемлемую лекарственную форму, например, инъекционную лекарственную форму (порошок для инъекции, лиофилизированный порошок для инъекции, жидкость для инъекции, инфузия и т.п.), таблетку, пероральную жидкость, гранулу, капсулу, мягкую капсулу, микропилюлю и т.п., в которой фармацевтически приемлемые носители могут представлять собой один или несколько из ксилита, маннита, лактозы, фруктозы, декстрана, глюкозы, поливинилпирролидона, низкомолекулярного декстрана, хлорида натрия, глюконата кальция или фосфата кальция. Кроме того, фармацевтическая композиция согласно настоящему изобретению может также содержать наполнитель, который может представлять собой антиоксидант, комплексообразователь, начинку, каркасное вещество и т.п.
Согласно другому аспекту, настоящее изобретение также относится к способу получения вышеупомянутого соединения формулы I, включающему в себя следующие стадии:
(1) получения имидазолина, характеризующегося способностью к акцептированию свободных радикалов NO, связующего звена, содержащего, по меньшей мере, три группы для связывания (AA1), пептида, характеризующегося тромболитической активностью (АА2), и пептида с направленным действием на тромб (АА3), где связующее звено содержит первую группу для связывания, вторую группу для связывания и третью группу для связывания;
(2) связывание в соответствующих реакционных условиях имидазолина, характеризующегося способностью к акцептированию свободных радикалов NO (NN), с первой группой для связывания на связующем звене (AA1) с образованием соединения общей формулы IM-1:

(3) связывание в соответствующих реакционных условиях пептида, характеризующегося тромболитической активностью (АА2), с соединением общей формулы IM-1, где один конец пептида, характеризующегося тромболитической активностью, связан со второй группой для связывания на связующем звене, с образованием соединения общей формулы IM-2:

(4) связывание в соответствующих реакционных условиях пептида с направленным действием на тромб (АА3), с соединением общей формулы IM-2, где один конец пептида с направленным действием на тромб связан с третьей группой для связывания на связующем звене, с образованием соединения общей формулы I;
где стадии (3) и (4) взаимозаменяемы по порядку проведения.
В способе получения согласно настоящему изобретению стадия (1) также включает в себя защиту защитными группами второй и третьей группы для связывания на связующем звене (AA1), и защиту защитными группами активных групп пептида, характеризующегося тромболитической активностью (АА2), и пептида с направленным действием на тромб (АА3), отличных от групп, используемых для связывания; стадия (3) также включает в себя сначала снятие защиты с защищенной второй группы для связывания, а затем связывание пептида, характеризующегося тромболитической активностью, с второй группой для связывания со снятой защитой; стадия (4) также включает в себя сначала снятие защиты с защищенной третьей группы для связывания, а затем связывание пептида с направленным действием на тромб с третьей группой для связывания со снятой защитой; и после стадии (4) также дополнительно присутствует стадия снятия защиты с защищенных активных групп пептида, характеризующегося тромболитической активностью (АА2), и пептида с направленным действием на тромб (АА3). Посредством применения методик введения и удаления защитных групп, порядок, в котором NN, АА2 и АА3 связываются со связующим звеном, и их положение связывания являются регулируемыми. Затем, после завершения связывания, удаляют защитные группы на других активных группах. Соответствующие реакционные условия относятся к условиям, традиционно используемым при синтезе пептидов. Определения имидазолина, характеризующегося способностью к акцептированию свободных радикалов NO (NN), связующего звена, содержащего, по меньшей мере, три группы для связывания (AA1), пептида, характеризующегося тромболитической активностью (АА2) и пептида с направленным действием на тромб (АА3) являются теми же самыми, что и определенные выше для соединения формулы I согласно настоящему изобретению.
Способ получения согласно настоящему изобретению станет более понятен исходя из последующего более детального описания.
Согласно одному варианту осуществления, в способе получения согласно настоящему изобретению, первая группа для связывания на связующем звене представляет собой аминогруппу, тогда как вторую и третью группы для связывания выбирают из группы, состоящей из карбоксильной группы и аминогруппы.
Согласно предпочтительному варианту осуществления способа получения согласно настоящему изобретению, имидазолин, характеризующийся способностью к акцептированию свободных радикалов NO, представляет собой 1,3-диоксо-2-[(4-оксиацетокси)фенил]-4,4,5,5-тетраметилимидазолин, связующее звено представляет собой L-Lys, пептид, характеризующийся тромболитической активностью, представляет собой олигопептид, содержащий последовательность PAK (Pro-Ala-Lys), и пептид с направленным действием на тромб представляет собой олигопептид, содержащий последовательность RGD (Arg-Gly-Asp). Если связующее звено представляет собой L-Lys, то возможны два следующих пути конъюгирования:
(1) 1,3-диоксо-2-[(4-оксиацетокси)фенил]-4,4,5,5-тетраметилимидазолин связывают с аминогруппой на связующем звене L-Lys, карбоксильную группу на олигопептиде, содержащем последовательность PAK, связывают с другой аминогруппой на связующем звене L-Lys, и аминогруппу на олигопептиде, содержащем последовательность RGD, связывают с карбоксильной группой на связующем звене L-Lys (как представлено в вышеупомянутом соединении формулы I-1); или
(2) 1,3-диоксо-2-[(4-оксиацетокси)фенил]-4,4,5,5-тетраметилимидазолин связывают с аминогруппой на связующем звене L-Lys, аминогруппу на олигопептиде, содержащем последовательность PAK, связывают с карбоксильной группой на связующем звене L-Lys, и карбоксильную группу на олигопептиде, содержащем последовательность RGD, связывают с другой аминогруппой на связующем звене L-Lys (как представлено в вышеупомянутом соединении формулы I-2).
Для примеров, относящихся к соединению формулы I-1, в том случае, если получают соединение общей формулы I-1-1, I-1-2, I-1-3 или I-1-4, то способ получения согласно настоящему изобретению может проводиться в соответствии со схемами синтеза, представленными на фиг. 1-4. На фиг. 1 представлена схема синтеза соединения общей формулы I-1-1. На фиг. 2 представлена схема синтеза соединения общей формулы I-1-2. На фиг. 3 представлена схема синтеза соединения общей формулы I-1-3. На фиг. 4 представлена схема синтеза соединения общей формулы I-1-4. На фиг. 1-4, аа3 может представлять собой S (Ser), V (Val) или F (Phe), как описано выше. Для примеров, относящихся к соединению формулы I-1-2, способ получения согласно настоящему изобретению описывается следующим образом:
(1) получение 1,3-диоксо-2-[(4-оксиацетокси)фенил]-4,4,5,5-тетраметилимидазолина;
(2) получение 1,3-диоксо-2-[(4′-оксиацетил-Lys-ОМе)фенил]-4,4,5,5-тетраметилимидазолина (карбоксильная группа на связующем звене Lys защищена защитной группой);
(3) получение HCl·Arg(NO2)-Gly-Asp(OBzl)-Ser(Bzl)-Obzl, HCl·Arg(NO2)-Gly-Asp(OBzl)-Val-Obzl или HCl·Arg(NO2)-Gly-Asp(OBzl)-Phe-Obzl;
(4) получение Boc-Gly-Arg(NO2)-Pro-Ala-Lys(Z);
(5) связывание Boc-Gly-Arg(NO2)-Pro-Ala-Lys(Z) с лизином 1,3-диоксо-2-[(4′-оксиацетил-Lys-OMe)фенил]-4,4,5,5-тетраметилимидазолина с получением 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Boc-Gly-Arg(NO2)-Pro-Ala-Lys(Z)]-Lys}фенил}-4,4,5,5-тетраметилимидазолина;
(6) соответствующее конъюгирование HCl·Arg(NO2)-Gly-Asp(OBzl)-Ser(Bzl)-Obzl, HCl·Arg(NO2)-Gly-Asp(OBzl)-Val-Obzl или HCl·Arg(NO2)-Gly-Asp(OBzl)-Phe-Obzl с 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Boc-Gly-Arg(NO2)-Pro-Ala-Lys(Z)]-Lys}фенил}-4,4,5,5-тетраметилимидазолином с получением 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Boc-Gly-Arg(NO2)-Pro-Ala-Lys(Z)]-Lys-Arg(NO2)-Gly-Asp(OBzl)-Ser(Bzl)-OBzl}фенил}-4,4,5,5-тетраметилимидазолина, 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Boc-Gly-Arg(NO2)-Pro-Ala-Lys(Z)]-Lys-Arg-(NO2)-Gly-Asp-(OBzl)-Val-OBzl}фенил}-4,4,5,5-тетраметилимидазолина или 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Boc-Gly-Arg(NO2)-Pro-Ala-Lys(Z)]-Lys-Arg(NO2)-Gly-Asp(OBzl)-Phe-OBzl}фенил}-4,4,5,5-тетраметилимидазолина, соответственно;
(7) снятие защиты с соединений, полученных на стадии (6), с получением 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Gly-Arg-Pro-Ala-Lys]-Lys-Arg-Gly-Asp-Ser}фенил}-4,4,5,5-тетраметилимидазолина, 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Gly-Arg-Pro-Ala-Lys]-Lys-Arg-Gly-Asp-Val}фенил}-4,4,5,5-тетраметилимидазолина или 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Gly-Arg-Pro-Ala-Lys]-Lys-Arg-Gly-Asp-Phe}фенил}-4,4,5,5-тетраметилимидазолина.
В случае получения соединений общей формулы I-1-1, I-1-3 и I-1-4, вышеупомянутый процесс получения повторяют, заменяя «Boc-Gly-Arg(NO2)-Pro-Ala-Lys(Z)» на стадии (4) на «Boc-Ala-Arg(NO2)-Pro-Ala-Lys(Z)», «Boc-Arg(NO2)-Pro-Ala-Lys(Z)» и «Boc-Pro-Ala-Lys(Z)».
В целях планирования конъюгирования, активные группы в соответствующих положениях олигопептида, содержащем последовательность PAK, и олигопептида, содержащем последовательность RGD, могут быть защищены, так что один конец выбранных последовательностей (включая активную группу, подлежащую связыванию со связующим звеном) используется для связывания с активной группой на связующем звене. Стадия присоединения олигопептида, содержащего последовательность PAK, и стадия присоединения олигопептида, содержащего последовательность RGD, взаимозаменяемы по порядку проведения. Например, сначала к связующему звену присоединяют олигопептид, содержащий последовательность RGD, а затем к ним присоединяют олигопептид, содержащий последовательность PAK.
Активные группы включают в себя группы, которые могут быть подвергнуты реакции конденсации, такие как аминогруппа или карбоксильная группа. Амино-защитные группы могут представлять собой карбоксибензил (CBz), трет-бутоксикарбонил (Boc), 9-флуоренилметоксикарбонил (Fmoc), бензил (Bn) или пара-метоксифенил (РМР). Карбоксил-защитные группы могут представлять собой сложный метиловый эфир (ОМе), сложный бензиловый эфир (OBn), сложный бензилметиловый эфир (Obzl), сложный трет-бутиловый эфир (OBUT) или сложный силиловый эфир (OSi(CH3)3).
Для примеров, относящихся к соединению формулы I-2, в том случае, если получают соединение общей формулы I-2-1, I-2-2, I-2-3 или I-2-4, то способ получения согласно настоящему изобретению может проводиться в соответствии со схемами синтеза, представленными на фиг. 5-8. На фиг. 5 представлена схема синтеза соединения общей формулы I-2-1. На фиг. 6 представлена схема синтеза соединения общей формулы I-2-2. На фиг. 7 представлена схема синтеза соединения общей формулы I-2-3. На фиг. 8 представлена схема синтеза соединения общей формулы I-2-4. На фиг. 5-8, аа3 может представлять собой S (Ser), V (Val) или F (Phe), как описано выше. В том случае, если получают соединение общей формулы I-2-1, I-2-2, I-2-3 или I-2-4, то сначала может быть получен 1,3-диоксо-2-[(4′-оксиацетил-Lys-ОМе)фенил]-4,4,5,5-тетраметилимидазолин, затем С-конец олигопептида, содержащего последовательность RGD, связывают с аминогруппой на связующем звене Lys; и, в завершение, N-конец олигопептида, содержащего последовательность PAK, связывают с карбоксильной группой со снятой защитой на связующем звене.
Согласно другому варианту осуществления, в способе получения согласно настоящему изобретению, имидазолин, характеризующийся способностью к акцептированию свободных радикалов NO, представляет собой 1,3-диоксо-2-[(4-оксиацетокси)фенил]-4,4,5,5-тетраметилимидазолин, связующее звено представляет собой L-Asp, пептид, характеризующийся тромболитической активностью, представляет собой олигопептид, содержащий последовательность PAK (Pro-Ala-Lys), и пептид с направленным действием на тромб представляет собой олигопептид, содержащий последовательность RGD (Arg-Gly-Asp), причем 1,3-диоксо-2-[(4-оксиацетокси)фенил]-4,4,5,5-тетраметилимидазолин связан с аминогруппой на связующем звене L-Asp, аминогруппа олигопептида, содержащего PAK последовательность, связана с карбоксильной группой на связующем звене L-Asp, и аминогруппа олигопептида, содержащего последовательность RGD, связана с другой карбоксильной группой на связующем звене L-Asp (как представлено в вышеупомянутом соединении формулы I-3 или I-4).
Для примеров, относящихся к соединению формулы 1-3, в том случае, если получают соединение общей формулы I-3-1, I-3-2, I-3-3 или I-3-4, то способ получения согласно настоящему изобретению может проводиться в соответствии со схемами синтеза, представленными на фиг. 9-12. На фиг. 9 представлена схема синтеза соединения общей формулы I-3-1. На фиг. 10 представлена схема синтеза соединения общей формулы I-3-2. На фиг. 11 представлена схема синтеза соединения общей формулы I-3-3. На фиг. 12 представлена схема синтеза соединения общей формулы I-3-4. На фиг. 9-12, аа3 может представлять собой S (Ser), V (Val) или F (Phe), как описано выше. В том случае, если получают общей формулы I-3-1, I-3-2, I-3-3 или I-3-4, то сначала может быть получен 1,3-диоксо-2-[(4′-оксиацетил-Asp-ОМе)фенил]-4,4,5,5-тетраметилимидазолин, затем N-конец олигопептида, содержащего последовательность PAK, связывают к одной карбоксильной группой на связующем звене Asp; и, в завершении, N-конец олигопептида, содержащего последовательность RGD, связывают с другой карбоксильной группой со снятой защитой на связующем звене Asp.
Для примеров, относящихся к соединению формулы I-4, в том случае, если получают соединение общей формулы I-4-1, I-4-2, I-4-3 или I-4-4, то способ получения согласно настоящему изобретению может проводиться в соответствии со схемами синтеза, представленными на фиг. 13-16. На фиг. 13 представлена схема синтеза соединения общей формулы I-4-1. На фиг. 14 представлена схема синтеза соединения общей формулы I-4-2. На фиг. 15 представлена схема синтеза соединения общей формулы I-4-3. На фиг. 16 представлена схема синтеза соединения общей формулы I-4-4. На фиг. 13-16, аа3 может представлять собой S (Ser), V (Val) или F (Phe), как описано выше. В том случае, если получают соединение общей формулы I-4-1, I-4-2, I-4-3 или I-4-4, то сначала может быть получен 1,3-диоксо-2-[(4′-оксиацетил-Asp-ОМе)фенил]-4,4,5,5-тетраметилимидазолин, затем N-конец олигопептида, содержащего последовательность RGD, связывают с одной карбоксильной группой на связующем звене Asp; и, в завершение, N-конец олигопептида, содержащего последовательность PAK, связывают с другой карбоксильной группой со снятой защитой на связующем звене Asp.
Согласно еще одному варианту осуществления, в способе получения согласно настоящему изобретению, имидазолин, характеризующийся способностью к акцептированию свободных радикалов NO, представляет собой 1,3-диоксо-2-[(4-оксиацетокси)фенил]-4,4,5,5-тетраметилимидазолин, связующее звено представляет собой L-Glu, пептид, характеризующийся тромболитической активностью, представляет собой олигопептид, содержащий последовательность PAK (Pro-Ala-Lys), и пептид с направленным действием на тромб представляет собой олигопептид, содержащий последовательность RGD (Arg-Gly-Asp), причем 1,3-диоксо-2-[(4-оксиацетокси)фенил]-4,4,5,5-тетраметилимидазолин связан с аминогруппой на связующем звене L-Glu, аминогруппа олигопептида, содержащего последовательность PAK, связана с карбоксильной группой на связующем звене L-Glu, и аминогруппа олигопептида, содержащего последовательность RGD, связана с другой карбоксильной группой на связующем звене L-Glu (как представлено в вышеупомянутом соединении формулы I-5 или I-6).
Для примеров, относящихся к соединению формулы I-5, в том случае, если получают соединение общей формулы I-5-1, I-5-2, I-5-3 или I-5-4, то способ получения согласно настоящему изобретению может проводиться в соответствии со схемами синтеза, представленными на фиг. 17-20. На фиг. 17 представлена схема синтеза соединения общей формулы I-5-1. На фиг. 18 представлена схема синтеза соединения общей формулы I-5-2. На фиг. 19 представлена схема синтеза соединения общей формулы I-5-3. На фиг. 20 представлена схема синтеза соединения общей формулы I-5-4. На фиг. 17-20, ааз может представлять собой S (Ser), V (Val) или F (Phe), как описано выше. В том случае, если получают соединение общей формулы I-5-1, I-5-2, I-5-3 или I-5-4, то сначала может быть получен 1,3-диоксо-2-[(4′-оксиацетил-Glu-ОМе)фенил]-4,4,5,5-тетраметилимидазолин, затем N-конец олигопептида, содержащего последовательность RGD, связывают с одной карбоксильной группой на связующем звене Glu; и, в завершении, N-конец олигопептида, содержащего последовательность PAK, связывают с другой карбоксильной группой со снятой защитой на связующем звене Glu.
Для примеров, относящихся к соединению формулы I-6, в том случае, если получают соединение общей формулы I-6-1, I-6-2, I-6-3 или I-6-4, то способ получения согласно настоящему изобретению может проводиться в соответствии со схемами синтеза, представленными на фиг. 21-24. На фиг. 21 представлена схема синтеза соединения общей формулы I-6-1. На фиг. 22 представлена схема синтеза соединения общей формулы I-6-2. На фиг. 23 представлена схема синтеза соединения общей формулы I-6-3. На фиг. 24 представлена схема синтеза соединения общей формулы I-6-4. На фиг. 21-24, aa3 может представлять собой S (Ser), V (Val) или F (Phe), как описано выше. В том случае, если получают соединение общей формулы I-6-1, I-6-2, I-6-3 или I-6-4, то сначала может быть получен 1,3-диоксо-2-[(4′-оксиацетил-Glu-ОМе)фенил]-4,4,5,5-тетраметилимидазолин, затем N-конец олигопептида, содержащего последовательность PAK, связывают с одной карбоксильной группой на связующем звене Glu; и, в завершении, N-конец олигопептида, содержащего последовательность RGD, связывают с другой карбоксильной группой со снятой защитой на связующем звене Glu.
В описанном выше способе получения, олигопептид, содержащий последовательность PAK, может представлять собой ARPAK (Ala-Arg-Pro-Ala-Lys), GRPAK (Gly-Arg-Pro-Ala-Lys), QRPAK (Gln-Arg-Pro-Ala-Lys), RPAK (Arg-Pro-Ala-Lys) или PAK (Pro-Ala-Lys), и олигопептид, содержащий последовательность RGD, может представлять собой RGDS (Arg-Gly-Asp-Ser), RGDV (Arg-Gly-Asp-Val) или RGDF (Arg-Gly-Asp-Phe).
Для соединения или фармацевтической композиции согласно настоящему изобретению высокая способность к акцептированию свободных радикалов NO демонстрируется на крысах в in vivo моделях акцептирования свободных радикалов NO; более высокие показатели тромболизиса и антитромботической активности демонстрируются в экспериментах по тромболизису и антитромботическому действию in vivo и in vitro; нейропротективная эффективность и более высокие показатели активности против инсульта демонстрируются на крысах в in vivo моделях инсульта; и эффективность по снижению объема инфаркта головного мозга демонстрируется на крысах в моделях инсульта.
Описание чертежей
На фиг. 1 представлена схема синтеза варианта осуществления соединения согласно настоящему изобретению (соединение общей формулы I-1-1);
На фиг. 2 представлена схема синтеза варианта осуществления соединения согласно настоящему изобретению (соединение общей формулы I-1-2);
На фиг. 3 представлена схема синтеза варианта осуществления соединения согласно настоящему изобретению (соединение общей формулы I-1-3);
На фиг. 4 представлена схема синтеза варианта осуществления соединения согласно настоящему изобретению (соединение общей формулы I-1-4);
На фиг. 5 представлена схема синтеза варианта осуществления соединения согласно настоящему изобретению (соединение общей формулы I-2-1);
На фиг. 6 представлена схема синтеза варианта осуществления соединения согласно настоящему изобретению (соединение общей формулы I-2-2);
На фиг. 7 представлена схема синтеза варианта осуществления соединения согласно настоящему изобретению (соединение общей формулы I-2-3);
На фиг. 8 представлена схема синтеза варианта осуществления соединения согласно настоящему изобретению (соединение общей формулы I-2-4);
На фиг. 9 представлена схема синтеза варианта осуществления соединения согласно настоящему изобретению (соединение общей формулы I-3-1);
На фиг. 10 представлена схема синтеза варианта осуществления соединения согласно настоящему изобретению (соединение общей формулы I-3-2);
На фиг. 11 представлена схема синтеза варианта осуществления соединения согласно настоящему изобретению (соединение общей формулы I-3-3);
На фиг. 12 представлена схема синтеза варианта осуществления соединения согласно настоящему изобретению (соединение общей формулы I-3-4);
На фиг. 13 представлена схема синтеза варианта осуществления соединения согласно настоящему изобретению (соединение общей формулы I-4-1);
На фиг. 14 представлена схема синтеза варианта осуществления соединения согласно настоящему изобретению (соединение общей формулы I-4-2);
На фиг. 15 представлена схема синтеза варианта осуществления соединения согласно настоящему изобретению (соединение общей формулы I-4-3);
На фиг. 16 представлена схема синтеза варианта осуществления соединения согласно настоящему изобретению (соединение общей формулы I-4-4);
На фиг. 17 представлена схема синтеза варианта осуществления соединения согласно настоящему изобретению (соединение общей формулы I-5-1);
На фиг. 18 представлена схема синтеза варианта осуществления соединения согласно настоящему изобретению (соединение общей формулы I-5-2);
На фиг. 19 представлена схема синтеза варианта осуществления соединения согласно настоящему изобретению (соединение общей формулы I-5-3);
На фиг. 20 представлена схема синтеза варианта осуществления соединения согласно настоящему изобретению (соединение общей формулы I-5-4);
На фиг. 21 представлена схема синтеза варианта осуществления соединения согласно настоящему изобретению (соединение общей формулы I-6-1);
На фиг. 22 представлена схема синтеза варианта осуществления соединения согласно настоящему изобретению (соединение общей формулы I-6-2);
На фиг. 23 представлена схема синтеза варианта осуществления соединения согласно настоящему изобретению (соединение общей формулы I-6-3);
На фиг. 24 представлена схема синтеза варианта осуществления соединения согласно настоящему изобретению (соединение общей формулы I-6-4);
На фиг. 25 представлены наноструктуры соединения Ia согласно настоящему изобретению в водных растворах с концентрацией 1×10-6 М, 1×10-9 М и 1×10-12 М;
На фиг. 26 представлены наноструктуры соединения Ib согласно настоящему изобретению в водных растворах с концентрацией 1×10-6 М, 1×10-9 М и 1×10-12 М;
На фиг. 27 представлены наноструктуры соединения Ic согласно настоящему изобретению в водных растворах с концентрацией 1×10-6 М, 1×10-9 М и 1×10-12 М;
На фиг. 28 представлены наноструктуры соединения Id согласно настоящему изобретению в водных растворах с концентрацией 1×10-6 М, 1×10-9 М и 1×10-12 М;
На фиг. 29 представлены наноструктуры соединения Ie согласно настоящему изобретению в водных растворах с концентрацией 1×10-6 М, 1×10-9 М и 1×10-12 М;
На фиг. 30 представлены наноструктуры соединения If согласно настоящему изобретению в водных растворах с концентрацией 1×10-6 М, 1×10-9 М и 1×10-12 М;
На фиг. 31 представлены наноструктуры соединения Ig согласно настоящему изобретению в водных растворах с концентрацией 1×10-6 М, 1×10-9 М и 1×10-12 М;
На фиг. 32 представлены наноструктуры соединения Ih согласно настоящему изобретению в водных растворах с концентрацией 1×10-6 М, 1×10-9 М и 1×10-12 М;
На фиг. 33 представлены наноструктуры соединения Ii согласно настоящему изобретению в водных растворах с концентрацией 1×10-6 М, 1×10-9 М и 1×10-12 М;
На фиг. 34 представлены наноструктуры соединения Ij согласно настоящему изобретению в водных растворах с концентрацией 1×10-6 М, 1×10-9 М и 1×10-12 М;
На фиг. 35 представлены наноструктуры соединения Ik согласно настоящему изобретению в водных растворах с концентрацией 1×10-6 М, 1×10-9 М и 1×10-12 М;
На фиг. 36 представлены наноструктуры соединения Il согласно настоящему изобретению в водных растворах с концентрацией 1×10-6 М, 1×10-9 М и 1×10-12 М;
На фиг. 37 представлен FT-MS спектр высокого разрешения соединения Ie согласно настоящему изобретению в концентрации 0.01 мкМ;
На фиг. 38 представлен FT-MS спектр высокого разрешения соединения Ie согласно настоящему изобретению в концентрации 0.1 мкМ;
На фиг. 39 представлен FT-MS спектр высокого разрешения соединения Ie согласно настоящему изобретению в концентрации 1 мкМ;
На фиг. 40 представлен FT-MS спектр высокого разрешения соединения Ie согласно настоящему изобретению в концентрации 10 мкМ.
Подробное описание вариантов осуществления
Настоящее изобретение будет далее описано вместе с последующими конкретными примерами, и его преимущества и характерные черты станут очевидны в сете описания. Указанные примеры имеют чисто иллюстративный характер и никоим образом не ограничивают объем настоящего изобретения. Специалист в данной области техники может иметь ввиду, что в детали и установленный порядок технических решений согласно настоящего изобретения могут быть внесены изменение или замена без отступления от духа и объема настоящего изобретения, и указанные изменения и замены предполагаются к включению в объем притязаний по настоящему изобретению.
Получение имидазолинов, характеризующихся способностью к акцептированию свободных радикалов NO: 1,3-диоксо-2-[(4-оксиацетокси)фенил]-4,4,5,5-тетраметилимидазолин
Пример 1. Получение 2,3-диметил-2,3-динитробутана
К 130 мл водного раствора NaOH (6н) добавляли 69 г (0,78 моль) 2-нитропропана. При перемешивании на льдосоляной бане в течение 1 ч по каплям добавляли 20 мл (0,38 моль) Br2. После завершения добавления Br2 добавляли 240 мл этанола, и нагревали с обратным холодильником при 90°С в течение 3 ч. Реакционной раствор, еще будучи горячим, незамедлительно вливали в 800 мл воды со льдом, а затем фильтровали с получением 55 г указанного в заголовке соединения (81%) в виде бесцветного чешуйчатого кристаллического вещества, т.пл. 110-112°С.
Пример 2. Получение 2,3-диметил-2,3-дигидроксиаминобутана
В 80 мл водного раствора этанола (50%) смешивали и суспендировали 7 г (40 ммоль) 2,3-диметил-2,3-динитробутана и 4 г NH4Cl, и перемешивали на бане со льдом, в которую в течение 3 ч добавляли 16 г цинкового порошка. После завершения добавления цинкового порошка баню со льдом удаляли, и продолжали реакцию в течение 3 ч при комнатной температуре (к.т.) при перемешивании, а затем подвергали реакционную смесь вакуумному фильтрованию. Осадок на фильтре неоднократно промывали водным раствором этанола (50%). Фильтрат и смыв объединяли, корректировали до pH=2 добавлением конц. HCl, а затем подвергали перегонке в условиях пониженного давления до состояния суспензии. После добавления соответствующего количества карбоната калия суспензию равномерно смешивали и экстрагировали в течение 6 ч с использованием экстрактора Сокслета и хлороформа в качестве экстрагента. Экстракт концентрировали в условиях пониженного давления до небольшого объема, в который добавляли петролейный эфир, с выпадением в осадок 2,60 г указанного в заголовке соединения (44%) в виде бесцветного кристаллического вещества, т.пл. 157-159°С.
Пример 3. Получение 1,3-Дигидрокси-2-(4′-гидроксифенил)-4,4,5,5-тетраметилимидазолидина
В 10 мл метанола растворяли 1,22 г (10 ммоль) пара-гидроксибензальдегида и 1,48 г (10 ммоль) 2,3-диметил-2,3-дигидроксиаминобутана, и перемешивали при к.т. в течение 8 ч до исчезновения пятна исходного вещества согласно анализу методом TLC. После вакуумного фильтрования получали 1,29 г (51%) указанного в заголовке соединения в виде бесцветного кристаллического вещества. EI-MS (m/z) 252 [М]+. 1Н-ЯМР (DMSO-d6) δ (м.д.) = 1,03 (с, 6Н), 1,05 (с, 6Н), 4,39 (с, 1H), 6,70 (д, J=6,9 Гц, 2Н), 7,23 (д, J=6,9 Гц, 2Н), 7,63 (с, 1Н), 7,85 (с, 2Н).
Пример 4. Получение 1,3-дигидрокси-2-(4′-гидроксифенил)-4,4,5,5-тетраметилимидазолина
В 30 мл метанола растворяли 504 мг (2 ммоль) 1,3-дигидрокси-2-(4′-гидроксифенил)-4,4,5,5-имидазолидина, затем добавляли 3 г PbO2, и перемешивали при к.т. в течение 40 мин до исчезновения пятна исходного вещества согласно анализу методом TLC. После удаления исходных веществ путем вакуумного фильтрования фильтрат перегоняли досуха в условиях пониженного давления при к.т., и очищали остаток методом колоночной хроматографии (с хлороформом в качестве элюента) с получением 260 мг (52%) указанного в заголовке соединения в виде голубого твердого вещества, т.пл. 134-135°С, EI-MS (m/z) 249 [М]+. IR (KBr) 3250, 1610, 1500, 1490, 840.
Пример 5. Получение 1,3-диоксо-2-(4′-(оксиацетатэтиловый эфир)фенил)-4,4,5,5-тетраметилимидазолина
В 5 мл безводного THF растворяли 250 мг (1 ммоль) 1,3-дигидрокси-2-(4′-гидроксифенил)-4,4,5,5-тетраметилимидазолина, 0,32 мл этилбромацетата и 32 мг NaH. Смесь перемешивали при 60°С в течение 5 ч до исчезновения пятна исходного вещества согласно анализу методом TLC. После фильтрования в условиях пониженного давления при к.т.фильтрат концентрировали досуха в условиях пониженного давления, и очищали остаток методом колоночной хроматографии (этилацетат : петролейный эфир = 1:5) с получением 300 мг (90%) целевого соединения, т.пл. 107-109°С.
Пример 6. Получение 1,3-диоксо-2-(4′-оксиацетоксифенил)-4,4,5,5-тетраметилимидазолина (TMMZ)
В раствор 33 мг (0,1 ммоль) 1,3-диоксо-2-(4′-(оксиацетатэтиловый эфир)фенил)-4,4,5,5-тетраметилимидазолина в 3 мл метанола добавляли 7 капель водного раствора NaOH (2н), а затем перемешивали при к.т. в течение 30 мин до исчезновения пятна исходного вещества согласно анализу методом TLC. Реакционную смесь концентрировали в условиях пониженного давления, остаток разбавляли добавлением 2 мл насыщенного солевого раствора, корректировали до pH 6 добавлением 2н HCl, а затем трижды экстрагировали этилацетатом (3×3 мл). Слои этилацетатной фазы объединяли и сушили над безводным сульфатом натрия, а затем фильтровали. Фильтрат концентрировали досуха в условиях пониженного давления при к.т. с получением 30 мг (99%) указанного в заголовке соединения в виде голубого кристаллического вещества, т.пл. 155-157°С. EI-MS (m/z) 307 [М]+.
Сочетание имидазолинов, характеризующихся способностью к акцептированию свободных радикалов NO, со связующим звеном: 1,3-диоксо-2-[(4′-оксиацетил-Lys-ОМе)фенил]-4,4,5,5-тетраметилимидазолин
Пример 7. Получение 1,3-Диоксо-2-[(4′-оксиацетил-Nω-Boc-Lys-ОМе)фенил]-4,4,5,5-тетраметилимидазолина
Раствор 307 мг (1 ммоль) 1,3-диоксо-2-(4′-оксиацетил-фенил)-4,4,5,5-тетраметилимидазолина в 30 мл безводного THF перемешивали на бане со льдом, к нему добавляли 250 мг (1,2 ммоль) DCC и 135 мг (1 ммоль) HOBt, и перемешивали на бане со льдом в течение 10 мин. Затем, к нему добавляли раствор, полученный из 300 мг (1 ммоль) HCl·Lys(Boc)-OMe, 122 мг (1 ммоль) N-метилморфолина и 6 мл безводного THF, и проводили реакцию в реакционной смеси при к.т. в течение 24 ч. Методом TLC (этилацетат : петролейный эфир = 2:1) обнаруживали исчезновение HCl·Lys(Boc)-OMe. Реакционную смесь концентрировали досуха в условиях пониженного давления, остаток растворяли в этилацетате, и удаляли нерастворимое вещество путем фильтрования. Фильтрат последовательно промывали насыщенным водным раствором бикарбоната натрия и насыщенным водным раствором NaCl, разделенную этилацетатную фазу сушили над безводным сульфатом натрия и фильтровали, а затем концентрировали фильтрат в условиях пониженного давления при 37°С (указанные операции здесь и далее в документе обобщенно называют «стандартной процедурой»). Остаток очищали методом колоночной хроматографии (этилацетат : петролейный эфир = 2:1) с получением 433 мг (65%) указанного в заголовке соединения в виде голубого твердого вещества. ESI-MS (m/z) 550 [М+Н]+.
Пример 8. Получение 1,3-диоксо-2-[(4′-оксиацетил-Lys-ОМе)фенил]-4,4,5,5-тетраметилимидазолина
В 15 мл безводного хлороводорода в этилацетате (4н) растворяли 625 мг (1 ммоль) 1,3-диоксо-2-[(4′-оксиацетил-Nω-Boc-Lys-ОМе)фенил]-4,4,5,5-тетраметилимидазолина, и перемешивали при к.т. в течение 3 ч до исчезновения пятна исходного вещества согласно анализу методом TLC (CHCl3:МеОН, 20:1). Затем, реакционную смесь подвергали стандартной процедуре. Остаток кристаллизовали в безводном этиловом эфире с получением указанного в заголовке соединения.
Получение пептида, характеризующегося тромболитической активностью: соответствующим образом защищенный ARPAK
Пример 9. Получение Boc-Ala-Lys(Z)-OBzl
В 10 мл безводного THF растворяли 473 мг (2,5 ммоль) Boc-Ala. На бане со льдом к нему добавляли раствор, полученный из 338 мг (2,5 ммоль) HOBt, 619 мг (3 ммоль) DCC и 10 мл безводного THF. Реакционную смесь перемешивали на бане со льдом в течение 20 мин, после чего к нему добавляли раствор, полученный из 936 мг (2,3 ммоль) HCl·Lys(Z)-Obzl и 232 мг (2,3 ммоль) N-метилморфолина и 6 мл безводного THF. В полученной реакционной смеси проводили реакцию при к.т. в течение 24 ч до исчезновения HCl·Lys(Z)-Obzl согласно анализу методом TLC (CHCl3:МеОН=30:1). Реакционную смесь подвергали стандартной процедуре с получением 1,204 г (97%) указанного в заголовке соединения в виде бесцветного твердого вещества, т.пл. 88-90°С. 
Пример 10. Получение HCl·Ala-Lys(Z)-OBzl
Приблизительно в 10 мл раствора безводного хлороводорода в этилацетате (4н) растворяли 1,354 г (2,5 ммоль) Boc-Ala-Lys(Z)-Obzl, и перемешивали при к.т. в течение 3 ч до исчезновения пятна исходного вещества согласно анализу методом TLC (CHCl3:МеОН, 30:1). Реакционный раствор концентрировали в условиях пониженного давления при к.т., растворяли остаток в этилацетате, а затем концентрировали при к.т.; описанный выше процесс повторяли несколько раз до удаления всего свободного хлороводорода (указанные операции здесь и далее в документе называют «стандартной процедурой»). Остаток кристаллизовали в безводном этиловом эфире с получением указанного в заголовке соединения, которое непосредственно использовали в реакции на следующей стадии.
Пример 11. Получение Boc-Pro-Ala-Lys(Z)-OBzl
В соответствующем количестве безводного THF растворяли 538 мг (2,5 ммоль) Boc-Pro, затем на бане со льдом добавляли 338 мг (2,5 ммоль) HOBt и 619 мг (3 ммоль) DCC в безводном THF, а затем проводили реакцию в течение 20 мин. К этому раствору добавляли раствор, полученный из 1,099 г (2,3 ммоль) HCl·Ala-Lys(Z)-Obzl и 232 мг (2,3 ммоль) N-метилморфолина в 10 мл безводного THF, и проводили реакцию при к.т. в течение 24 ч. Методом TLC (CHCl3:МеОН, 20:1) обнаруживали исчезновение пятна исходного вещества. Соединения в реакционной смеси подвергали стандартной процедуре с получением 2,847 г (98%) указанного в заголовке соединения, т.пл. 82-83°С. 
Пример 12. Получение HCl·Pro-Ala-Lys(Z)-OBzl
В 15 мл раствора безводного хлороводорода в этилацетате (4н) растворяли 1,596 г (2,5 ммоль) Boc-Pro-Ala-Lys(Z)-Obzl, и перемешивали при к.т.в течение 3 ч до исчезновения пятна исходного вещества согласно анализу методом TLC (CHCl3:МеОН, 20:1). Реакционную смесь подвергали стандартной процедуре, и кристаллизовали остаток в безводном этиловом эфире с получением указанного в заголовке соединения, которое непосредственно использовали в реакции на следующей стадии.
Пример 13. Получение Boc-Arg(NO2)-Pro-Ala-Lys(Z)-OBzl
На бане со льдом раствор 798 мг (2,5 ммоль) Boc-Arg(NO2), 338 мг (2,5 ммоль) HOBt, 619 мг (3 ммоль) DCC в 10 мл безводного THF перемешивали в течение 20 мин, к нему добавляли раствор, полученный из 1,322 г (2,3 ммоль) HCl·Pro-Ala-Lys(Z)-Obzl и 232 мг (2,3 ммоль) N-метилморфолина в 5 мл безводного THF, и проводили реакцию при к.т. в течение 24 часов до исчезновения пятна исходного вещества согласно анализу методом TLC (CHCl3:МеОН, 20:1). Проводили стандартную процедуру с получением 1,642 г (85%) указанного в заголовке соединения, т.пл. 84-85°С. 
Пример 14. Получение HCl·Arg(NO2)-Pro-Ala-Lys(Z)-OBzl
В 20 мл раствора безводного хлороводорода в этилацетате (4н) растворяли 2,099 г (2,5 ммоль) Boc-Arg(NO2)-Pro-Ala-Lys(Z)-Obzl, и перемешивали при к.т. в течение 3 ч до исчезновения пятна исходного вещества согласно анализу методом TLC (CHCl3:МеОН, 20:1). Реакционную смесь подвергали стандартной процедуре, и кристаллизовали остаток в безводном этиловом эфире с получением указанного в заголовке соединения, которое непосредственно использовали в реакции на следующей стадии.
Пример 15. Получение Boc-Ala-Arg(NO2)-Pro-Ala-Lys(Z)-OBzl
На бане со льдом раствор 473 мг (2,5 ммоль) Boc-Ala, 338 мг (2,5 ммоль) HOBt, 619 мг (3 ммоль) DCC в 10 мл безводного THF перемешивали в течение 20 мин, к нему добавляли раствор, полученный из 1,785 г (2,3 ммоль) HCl·Arg(NO2)-Pro-Ala-Lys(Z)-Obzl и 232 мг (2,3 ммоль) N-метилморфолина в 5 мл безводного THF, и проводили реакцию в течение 24 часов с получением 1,802 г (86%) указанного в заголовке соединения, т.пл. 87-89°С. 
Пример 16. Получение Boc-Ala-Arg(NO2)-Pro-Ala-Lys(Z)
В 3 мл метанола растворяли 921 мг (1 ммоль) Boc-Ala-Arg(NO2)-Pro-Ala-Lys(Z)-OBzl, на бане со льдом к нему добавляли водный раствор NaOH (2н), и перемешивали при к.т.в течение 30 мин. При поддержании pH 12 реакционную смесь перемешивали в течение 10 мин на бане со льдом до исчезновения пятна исходного вещества согласно анализу методом TLC. Значение pH корректировали до 7 добавлением 2н HCl, и концентрировали реакционный раствор в условиях пониженного давления. Остаток разбавляли 2 мл насыщенного солевого раствора и корректировали до pH 2 добавлением 2н HCl, а затем трижды экстрагировали этилацетатом (3×5 мл). Слои этилацетатой фазы объединяли и сушили над безводным сульфатом натрия, и концентрировали в условиях пониженного давления при к.т. с получением 767 мг (80%) указанного в заголовке соединения в виде бесцветного твердого вещества. EI-MS (m/z) 830 [М-Н]-.
Получение пептида с направленным действием на тромб/антитромботического пептида: соответствующим образом защищенные RGDS, RGDV, RGDF
Пример 17. Получение Boc-Asp(OBzl)-Ser(Bzl)-OBzl
На бане со льдом перемешивали раствор 808 мг (2,5 ммоль) Boc-Asp(OBzl), 338 мг (2,5 ммоль) HOBt, 619 мг (3 ммоль) DCC в 10 мл безводного THF, проводили реакцию в течение 20 мин, затем к нему добавляли раствор, полученный из 740 мг (2,3 ммоль) HCl·Ser(Bzl)-Obzl и 232 мг (2,3 ммоль) N-метилморфолина в 5 мл безводного THF, и проводили реакцию при к.т. в течение 24 ч до исчезновения исходного вещества согласно анализу методом TLC (CHCl3:МеОН, 20:1). Соединения в реакционной смеси подвергали стандартной процедуре с получением 1,29 г (95%) указанного в заголовке соединения в виде бесцветного маслянистого вещества. ESI-MS (m/z) 591 [М+Н]+.
Пример 18. Получение HCl·Asp(OBzl)-Ser(Bzl)-OBzl
В 15 мл раствора безводного хлороводорода в этилацетате (4н) растворяли 1,477 г (2,5 ммоль) Boc-Asp(OBzl)-Ser(Bzl)-Obzl, и перемешивали при к.т. в течение 3 ч до исчезновения исходного вещества согласно анализу методом TLC (CHCl3:МеОН, 20:1). Реакционную смесь подвергали стандартной процедуре, и кристаллизовали остаток в безводном этиловом эфире с получением указанного в заголовке соединения, которое непосредственно использовали в реакции на следующей стадии.
Пример 19. Получение Boc-Gly-Asp(OBzl)-Ser(Bzl)-OBzl
На бане со льдом раствор 438 мг (2,5 ммоль) Boc-Gly, 338 мг (2,5 ммоль) HOBt, 619 мг (3 ммоль) DCC в 10 мл безводного THF перемешивали в течение 20 мин, затем к нему добавляли раствор, полученный из 1,212 г (2,3 ммоль) HCl·Asp(OBzl)-Ser(Bzl)-Obzl и 232 мг (2,3 ммоль) N-метилморфолина в 5 мл безводного THF, и проводили реакцию при к.т. в течение 24 ч до исчезновения исходного вещества согласно анализу методом TLC (CHCl3:МеОН, 20:1). Реакционную смесь подвергали стандартной процедуре с получением 1,461 г (98%) указанного в заголовке соединения в виде бесцветного твердого вещества, т.пл. 53-55°С. 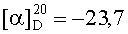
Пример 20. Получение HCl· Gly-Asp(OBzl)-Ser(Bzl)-OBzl
В 15 мл раствора безводного хлороводорода в этилацетате (4н) растворяли 1,619 г (2,5 ммоль) Boc-Gly-Asp(OBzl)-Ser(Bzl)-Obzl, и перемешивали при к.т. в течение 3 ч до исчезновения пятна исходного вещества согласно анализу методом TLC (CHCl3:МеОН, 20:1). Реакционную смесь подвергали стандартной процедуре, и кристаллизовали остаток в безводном этиловом эфире с получением указанного в заголовке соединения, которое непосредственно использовали в реакции на следующей стадии.
Пример 21. Получение Boc-Arg(NO2)-Gly-Asp(OBzl)-Ser(Bzl)-OBzl
На бане со льдом раствор 798 мг (2,5 ммоль) Boc-Arg(NO2), 338 мг (2,5 ммоль) HOBt, 619 мг (3 ммоль) DCC в 10 мл безводного THF перемешивали в течение 20 мин, затем к нему добавляли раствор, полученный из 1,343 г (2,3 ммоль) HCl·Gly-Asp(OBzl)-Ser(Bzl)-Obzl и 232 мг (2,3 ммоль) N-метилморфолина в 5 мл безводного THF, и проводили реакцию при к.т. в течение 24 ч до исчезновения исходного вещества согласно анализу методом TLC (CHCl3:МеОН, 20:1). После проведения стандартной процедуры получали 1,66 г (85%) указанного в заголовке соединения в виде бесцветного твердого вещества, т.пл. 74-75°С. 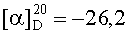
Пример 22. Получение HCl·Arg(NO2)-Gly-Asp(OBzl)-Ser(Bzl)-OBzl
Смесь 2,122 г (2,5 ммоль) Boc-Arg(NO2)-Gly-Asp(OBzl)-Ser(Bzl)-Obzl и 20 мл раствора хлороводорода в этилацетате (4н) перемешивали при к.т. в течение 3 ч до исчезновения пятна исходного вещества согласно анализу методом TLC (CHCl3:МеОН, 20:1). Реакционную смесь подвергали стандартной процедуре, и кристаллизовали остаток в безводном этиловом эфире с получением указанного в заголовке соединения.
Пример 23. Получение Boc-Asp(OBzl)-Val-OBzl
На бане со льдом раствор 808 мг (2,5 ммоль) Boc-Asp(OBzl), 338 мг (2,5 ммоль) HOBt, 619 мг (3 ммоль) DCC в 10 мл безводного THF перемешивали в течение 20 мин, затем к нему добавляли раствор, полученный из 558 мг (2,3 ммоль) HCl·Val-Obzl и 232 мг (2,3 ммоль) N-метилморфолина в 5 мл безводного THF, и проводили реакцию при к.т. в течение 24 ч до исчезновения исходного вещества согласно анализу методом TLC (CHCl3:МеОН, 20:1). Реакционную смесь подвергали стандартной процедуре с получением 1,129 г (96%) указанного в заголовке соединения в виде бесцветной маслянистой жидкости. ESI-MS(m/z) 512 [М+Н]+.
Пример 24. Получение HCl·Asp(OBzl)-Val-OBzl
В 15 мл раствора безводного хлороводорода в этилацетате (4н) растворяли 1,278 г (2,5 ммоль) Boc-Asp(OBzl)-Val-Obzl, и перемешивали при к.т. в течение 3 ч до исчезновения пятна исходного вещества согласно анализу методом TLC (CHCl3:МеОН, 20:1). Реакционную смесь подвергали стандартной процедуре, и кристаллизовали остаток в безводном этиловом эфире с получением указанного в заголовке соединения, которое непосредственно использовали в реакции на следующей стадии.
Пример 25. Получение Boc-Gly-Asp(OBzl)-Val-OBzl
На бане со льдом раствор 438 мг (2,5 ммоль) Boc-Gly, 338 мг (2,5 ммоль) HOBt, 619 мг (3 ммоль) DCC в 10 мл безводного THF перемешивали в течение 20 мин, затем к нему добавляли раствор, полученный из 1,03 г (2,3 ммоль) HCl·Asp(OBzl)-Val-Obzl и 232 мг (2,3 ммоль) N-метилморфолина в 5 мл безводного THF, и проводили реакцию при к.т.в течение 24 ч до исчезновения исходного вещества согласно анализу методом TLC (CHCl3:МеОН, 20:1). Реакционную смесь подвергали стандартной процедуре с получением 1,242 г (95%) указанного в заголовке соединения в виде бесцветного твердого вещества, т.пл. 66-68°С. 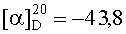
Пример 26. Получение HCl·Gly-Asp(OBzl)-Val-OBzl
В 15 мл раствора безводного хлороводорода в этилацетате (4н) растворяли 1,421 г (2,5 ммоль) Boc-Gly-Asp(OBzl)-Val-Obzl, и перемешивали при к.т.в течение 3 ч до исчезновения пятна исходного вещества согласно анализу методом TLC (CHCl3:МеОН, 20:1). Реакционную смесь подвергали стандартной процедуре, и кристаллизовали остаток в безводном этиловом эфире с получением указанного в заголовке соединения, которое непосредственно использовали в реакции на следующей стадии.
Пример 27. Получение Boc-Arg(NO2)-Gly-Asp(OBzl)-Val-OBzl
На бане со льдом раствор 798 мг (2,5 ммоль) Boc-Arg(NO2), 338 мг (2,5 ммоль) HOBt, 619 мг (3 ммоль) DCC в 10 мл безводного THF перемешивали в течение 20 мин, затем к нему добавляли раствор, полученный из 1,162 г (2,3 ммоль) HCl·Gly-Asp(OBzl)-Val-Obzl и 232 мг (2,3 ммоль) N-метилморфолина в 5 мл безводного THF, и проводили реакцию при к.т. в течение 24 ч до исчезновения исходного вещества согласно анализу методом TLC (CHCl3:МеОН, 20:1). Реакционную смесь подвергали стандартной процедуре с получением 1,523 г (86%) указанного в заголовке соединения в виде бесцветного твердого вещества, т.пл. 107-109°С. 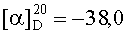
Пример 28. Получение HCl·Gly-Asp(OBzl)-Val-OBzl
В 20 мл раствора безводного хлороводорода в этилацетате (4н) растворяли 1,925 г (2,5 ммоль) Boc-Arg(NO2)-Gly-Asp(OBzl)-Val-Obzl, и перемешивали при к.т. в течение 3 ч до исчезновения пятна исходного вещества согласно анализу методом TLC (CHCl3:МеОН, 20:1). Реакционную смесь подвергали стандартной процедуре, и кристаллизовали остаток в безводном этиловом эфире с получением указанного в заголовке соединения.
Пример 29. Получение Boc-Asp(OBzl)-Phe-OBzl
На бане со льдом раствор 808 мг (2,5 ммоль) Boc-Asp(OBzl), 338 мг (2,5 ммоль) HOBt, 619 мг (3 ммоль) DCC в 10 мл безводного THF перемешивали в течение 20 мин, затем к нему добавляли раствор, полученный из 668 мг (2,3 ммоль) HCl·Phe-Obzl и 232 мг (2,3 ммоль) N-метилморфолина в 5 мл безводного THF, и проводили реакцию при к.т.в течение 24 ч до исчезновения исходного вещества согласно анализу методом TLC (CHCl3:МеОН, 20:1). После проведения стандартной процедуры 1,222 г (95%) указанного в заголовке соединения получали в виде бесцветного твердого вещества, т.пл. 79-80°C. 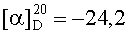
Пример 30. Получение HCl·Asp(OBzl)-Phe-OBzl
В 15 мл раствора безводного хлороводорода в этилацетате (4н) растворяли 1,398 г (2,5 ммоль) Boc-Asp(OBzl)-Phe-Obzl, и перемешивали при к.т.в течение 3 ч до исчезновения пятна исходного вещества согласно анализу методом TLC (CHCl3:МеОН, 20:1). Реакционную смесь подвергали стандартной процедуре, и кристаллизовали остаток в безводном этиловом эфире с получением указанного в заголовке соединения, которое непосредственно использовали в реакции на следующей стадии.
Пример 31. Получение Boc-Gly-Asp(OBzl)-Phe-OBzl
На бане со льдом раствор 438 мг (2,5 ммоль) Boc-Gly, 338 мг (2,5 ммоль) HOBt, 619 мг (3 ммоль) DCC в безводном THF перемешивали в течение 20 мин, затем к нему добавляли раствор, полученный из 1,141 г (2,3 ммоль) HCl·Asp(OBzl)-Phe-Obzl и 232 мг (2,3 ммоль) N-метилморфолина в 5 мл безводного THF, и проводили реакцию при к.т. в течение 24 ч до исчезновения исходного вещества согласно анализу методом TLC (CHCl3:МеОН, 20:1). Реакционную смесь подвергали стандартной процедуре с получением 1,29 г (91%) указанного в заголовке соединения в виде бесцветного твердого вещества, т.пл. 70-71°С. 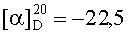
Пример 32. Получение HCl·Gly-Asp(OBzl)-Phe-OBzl
В 15 мл раствора безводного хлороводорода в этилацетате (4н) растворяли 1,541 г (2,5 ммоль) Boc-Gly-Asp(OBzl)-Phe-Obzl, и перемешивали при к.т. в течение 3 ч до исчезновения пятна исходного вещества согласно анализу методом TLC (CHCl3:МеОН, 20:1). Реакционную смесь подвергали стандартной процедуре, и кристаллизовали остаток в безводном этиловом эфире с получением указанного в заголовке соединения, которое непосредственно использовали в реакции на следующей стадии.
Пример 33. Boc-Arg(NO2)-Gly-Asp(OBzl)-Phe-OBzl
На бане со льдом раствор 798 мг (2,5 ммоль) Boc-Arg(NO2), 338 мг (2,5 ммоль) HOBt, 619 мг (3 ммоль) DCC в 10 мл безводного THF перемешивали в течение 20 мин, затем к нему добавляли раствор, полученный из 1,272 г (2,3 ммоль) HCl·Gly-Asp(OBzl)-Phe-Obzl и 232 мг (2,3 ммоль) N-метилморфолина в 5 мл безводного THF, и проводили реакцию при к.т.в течение 24 ч до исчезновения исходного вещества согласно анализу методом TLC (CHCl3:МеОН, 20:1). Реакционную смесь подвергали стандартной процедуре с получением 1,637 г (87%) указанного в заголовке соединения в виде бесцветного твердого вещества, т.пл. 77-79°С. 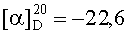
Пример 34. Получение HCl·Arg(NO2)-Gly-Asp(OBzl)-Phe-OBzl
В 15 мл раствора безводного хлороводорода в этилацетате (4н) растворяли 2,045 г (2,5 ммоль) Boc-Arg(NO2)-Gly-Asp(OBzl)-Phe-Obzl, и перемешивали при к.т. в течение 3 ч до исчезновения пятна исходного вещества согласно анализу методом TLC (CHCl3:МеОН, 20:1). Реакционную смесь подвергали стандартной процедуре, и кристаллизовали остаток в безводном этиловом эфире с получением указанного в заголовке соединения.
Получение трехкомпонентных конъюгатов ARPAK/имидазолин/RGD (соединения общей формулы I-1-1): Ia, Ib, Ic
Пример 35. Получение 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Boc-Ala-Arg(NO2)-Pro-Ala-Lys(Z)]-Lys-ОМе}фенил}-4,4,5,5-тетраметилимидазолина
На бане со льдом раствор 821 мг (1 ммоль) Boc-Ala-Arg(NO2)-Pro-Ala-Lys(Z), 135 мг (1 ммоль) HOBt и 250 мг (1 ммоль) DCC в 10 мл безводного THF перемешивали в течение 20 мин, затем к нему добавляли раствор, полученный из 480 мг (1 ммоль) 1,3-диоксо-2-[(4′-оксиацетил-Lys-ОМе)фенил]-4,4,5,5-тетраметилимидазолина и 100 мг (1 ммоль) N-метилморфолина в 5 мл безводного THF, и проводили реакцию при к.т. в течение 24 ч до исчезновения исходного вещества согласно анализу методом TLC (CHCl3:МеОН, 40:1). Реакционную смесь подвергали стандартной процедуре с получением 925 мг (83%) указанного в заголовке соединения в виде голубого твердого вещества, т.пл. 179-182°С. 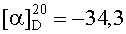
Пример 36. Получение 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Вос-Ala-Arg(NO2)-Pro-Ala-Lys(Z)]-Lys}фенил}-4,4,5,5-тетраметилимидазолина
На бане со льдом в 3 мл метанола растворяли 1260 мг (1 ммоль) 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Вос-Ala-Arg(NO2)-Pro-Ala-Lys(Z)]-Lys-ОМе}фенил}-4,4,5,5-тетраметилимидазолина, затем добавляли водный раствор NaOH (2н), а затем перемешивали при к.т. в течение 30 мин. При поддержании pH 12 реакционную смесь перемешивали в течение 10 мин на бане со льдом до исчезновения исходного вещества согласно анализу методом TLC. Значение pH корректировали до 7 добавлением 2н HCl, реакционный раствор концентрировали в условиях пониженного давления, остаток разбавляли 2 мл насыщенного солевого раствора, корректировали до pH 2 добавлением 2н HCl, а затем трижды экстрагировали этилацетатом (3×5 мл). Объединенную этилацетатную фазу сушили над безводным сульфатом натрия и фильтровали, и концентрировали фильтрат в условиях пониженного давления при к.т. с получением 945 мг (82%) указанного в заголовке соединения в виде голубого твердого вещества. EI-MS (m/z) 1238 [М-Н]-.
Пример 37. Получение 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Boc-Ala-Arg(NO2)-Pro-Ala-Lys(Z)]-Lys-Arg-(NO2)-Gly-Asp(OBzl)-Ser(Bzl)-OBzl}фенил}-4,4,5,5-тетраметилимидазолина
На бане со льдом раствор 618 мг (0,5 ммоль) 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Вос-Ala-Arg(NO2)-Pro-Ala-Lys(Z)]-Lys}фенил}-4,4,5,5-тетраметилимидазолина, 69 мг (0,5 ммоль) HOBt и 126 мг (0,6 ммоль) DCC в 20 мл безводного THF перемешивали в течение 20 мин, затем к нему добавляли раствор, полученный из 442 мг (0,5 ммоль) HCl·Arg(NO2)-Gly-Asp(OBzl)-Ser(Bzl)-Obzl и 50 мг (0,5 ммоль) N-метилморфолина в 5 мл безводного THF, и проводили реакцию при к.т. в течение 24 ч до исчезновения исходного вещества согласно анализу методом TLC (CHCl3:МеОН, 20:1). Реакционную смесь подвергали стандартной процедуре с получением 300 мг (31%) указанного в заголовке соединения в виде голубого твердого вещества, т.пл. 138-140°С. 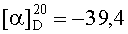
Пример 38. Получение 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Boc-Ala-Arg(NO2)-Pro-Ala-Lys(Z)]-Lys-Arg-(NO2)-Gly-Asp(OBzl)-Val-OBzl}фенил}-4,4,5,5-тетраметилимидазолина
На бане со льдом раствор 618 мг (0,5 ммоль) 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Boc-Ala-Arg(NO2)-Pro-Ala-Lys(Z)]-Lys}фенил}-4,4,5,5-тетраметилимидазолина, 69 мг (0,5 ммоль) HOBt и 126 мг (0,6 ммоль) DCC в 20 мл безводного THF перемешивали в течение 20 мин, затем к нему добавляли раствор, полученный из 421 мг (0,5 ммоль) HCl·Arg(NO2)-Gly-Asp(OBzl)-Val-Obzl и 50 мг (0,5 ммоль) N-метилморфолина в 5 мл безводного THF, и проводили реакцию при к.т. в течение 24 ч до исчезновения исходного вещества согласно анализу методом TLC (CHCl3:МеОН, 20:1). Реакционную смесь подвергали стандартной процедуре с получением 389 мг (36%) указанного в заголовке соединения в виде голубого твердого вещества, т.пл. 117-120°С. 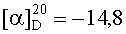
Пример 39. Получение 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Boc-Ala-Arg(NO2)-Pro-Ala-Lys(Z)]-Lys-Arg-(NO2)-Gly-Asp(OBzl)-Phe-OBzl}фенил}-4,4,5,5-тетраметилимидазолина
На бане со льдом раствор 618 мг (0,5 ммоль) 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Вос-Ala-Arg(NO2)-Pro-Ala-Lys(Z)]-Lys}фенил}-4,4,5,5-тетраметилимидазолина, 69 мг (0,5 ммоль) HOBt и 126 мг (0,6 ммоль) DCC в 20 мл безводного THF перемешивали в течение 20 мин, затем к нему добавляли раствор, полученный из 445 мг (0,5 ммоль) HCl·Arg(NO2)-Gly-Asp(OBzl)-Phe-Obzl и 50 мг (0,5 ммоль) N-метилморфолина в 5 мл безводного THF, и проводили реакцию при к.т. в течение 24 ч до исчезновения исходного вещества согласно анализу методом TLC (CHCl3:МеОН, 20:1). Реакционную смесь подвергали стандартной процедуре с получением 320 мг (36%) указанного в заголовке соединения в виде голубого твердого вещества, т.пл. 115-118°С. 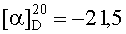
Пример 40. Получение 1,3-диоксо-2-{4′-оксиацетил-[Nω-(Ala-Arg-Pro-Ala-Lys)-Lys-Arg-Gly-Asp-Ser]фенил}-4,4,5,5-тетраметилимидазолина (Ia)
На бане со льдом 199 мг (0,1 ммоль) 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Boc-Ala-Arg(NO2)-Pro-Ala-Lys(Z)]-Lys-Arg-(NO2)-Gly-Asp(OBzl)-Ser(Bzl)-OBzl}фенил}-4,4,5,5-тетраметилимидазолина смешивали с 6 мл трифторуксусной кислоты и 1,5 мл трифторметансульфоновой кислоты, и перемешивали в течение 1 ч до исчезновения исходного вещества согласно анализу методом TLC (CHCl3:МеОН, 1:1). Реакционную смесь концентрировали в условиях пониженного давления, остаток неоднократно промывали безводным этиловым эфиром и концентрировали в условиях пониженного давления. Остаток растворяли в воде, корректировали до pH=8 добавлением 25% водного раствора аммиака, обессоливали с использованием Sephadex G10, а затем очищали на С18 колонке. Собранные фракции лиофилизировали с получением 109 мг (85%) указанного в заголовке соединения в виде голубого твердого вещества, т.пл. 134-135°С. 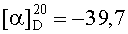
Пример 41. Получение l,3-диоксо-2-{4′-оксиацетил-[Nω-(Ala-Arg-Pro-Ala-Lys)-Lys-Arg-Gly-Asp-Val]фенил}-4,4,5,5-тетраметилимидазолина (Ib)
На бане со льдом 190 мг (0,1 ммоль) 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Вос-Ala-Arg(NO2)-Pro-Ala-Lys(Z)]-Lys-Arg-(NO2)-Gly-Asp(OBzl)-Val-OBzl}фенил}-4,4,5,5-тетраметилимидазолина смешивали с 6 мл трифторуксусной кислоты и 1,5 мл трифторметансульфоновой кислоты, и перемешивали в течение 1 ч до исчезновения исходного вещества согласно анализу методом TLC (CHCl3:МеОН, 1:1). Реакционную смесь концентрировали в условиях пониженного давления, остаток неоднократно промывали безводным этиловым эфиром и концентрировали в условиях пониженного давления. Остаток растворяли в воде, корректировали до pH 8 добавлением 25% водного раствора аммиака, обессоливали с использованием Sephadex G10, а затем очищали на С18 колонке. Собранные фракции лиофилизировали с получением 96 мг (82%) указанного в заголовке соединения в виде голубого твердого вещества, т.пл. 143-144°С. 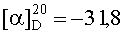
Пример 42. Получение 13-диоксо-2-{4′-оксиацетил-[Nω-(Ala-Arg-Pro-Ala-Lys)-Lys-Arg-Gly-Asp-Phe]фенил}-4,4,5,5-тетраметилимидазолина (Ic)
На бане со льдом 194 мг (0,1 ммоль) 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Вос-Ala-Arg(NO2)-Pro-Ala-Lys(Z)]-Lys-Arg-(NO2)-Gly-Asp(OBzl)-Phe-OBzl}фенил}-4,4,5,5-тетраметилимидазолина смешивали с 6 мл трифторуксусной кислоты и 1,5 мл трифторметансульфоновой кислоты, и перемешивали в течение 1 ч до исчезновения исходного вещества согласно анализу методом TLC (CHCl3:МеОН, 1:1). Реакционную смесь концентрировали в условиях пониженного давления, остаток неоднократно промывали безводным этиловым эфиром и концентрировали в условиях пониженного давления. Остаток растворяли в воде, корректировали до pH 8 добавлением 25% водного раствора аммиака, обессоливали с использованием Sephadex G10, а затем очищали на С18 колонке. Собранные фракции лиофилизировали с получением 106 мг (81%) указанного в заголовке соединения в виде голубого твердого вещества, т.пл. 96-97°С. 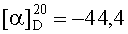
Получение пептида, характеризующегося тромболитической активностью: соответствующим образом защищенный GRPAK
Пример 43. Получение Boc-Gly-Arg(NO2)-Pro-Ala-Lys(Z)-OBzl
На бане со льдом раствор 438 мг (2,5 ммоль) Boc-Gly, 338 мг (2,5 ммоль) HOBt, 619 мг (3 ммоль) DCC в 10 мл безводного THF перемешивали в течение 20 мин, затем к нему добавляли раствор, полученный из 1,785 г (2,3 ммоль) HCl·Arg(NO2)-Pro-Ala-Lys(Z)-Obzl и 232 мг (2,3 ммоль) N-метилморфолина в 5 мл безводного THF, и проводили реакцию при к.т. в течение 24 ч с получением 1,857 г (90%) указанного в заголовке соединения, т.пл. 85-87°С. 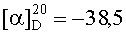
Пример 44. Получение Boc-Gly-Arg(NO2)-Pro-Ala-Lys(Z)
В 3 мл метанола растворяли 907 мг (1 ммоль) Boc-Gly-Arg(NO2)-Pro-Ala-Lys(Z)-Obzl, затем на бане со льдом добавляли водный раствор NaOH (2н), а затем перемешивали при к.т. в течение 30 мин. При поддержании pH 12 реакционную смесь перемешивали в течение 10 мин на бане со льдом до исчезновения исходного вещества согласно анализу методом TLC. Значение pH корректировали до 7 добавлением 2н HCl, реакционный раствор концентрировали в условиях пониженного давления, остаток разбавляли 2 мл насыщенного солевого раствора, корректировали до pH 2 добавлением 2н HCl, а затем трижды экстрагировали этилацетатом (3×5 мл). Объединенную этилацетатную фазу сушили над безводным сульфатом натрия, а затем концентрировали в условиях пониженного давления при к.т. с получением 785 мг (82%) указанного в заголовке соединения в виде бесцветного твердого вещества. EI-MS (m/z) 816 [М-Н]-.
Получение трехкомпонентных конъюгатов GRPAK/имидазолин/RGD (соединения общей формулы I-1-2): Id, Ie, If
Пример 45. Получение 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Вос-Gly-Arg(NO2)-Pro-Ala-Lys(Z)]-Lys-OMe}фенил}-4,4,5,5-тетраметилимидазолина
На бане со льдом раствор 817 мг (1 ммоль) Boc-Gly-Arg(NO2)-Pro-Ala-Lys(Z), 135 мг (1 ммоль) HOBt и 250 мг (1 ммоль) DCC в 10 мл безводного THF перемешивали в течение 20 мин, затем к нему добавляли раствор, полученный из 480 мг (1 ммоль) 1,3-диоксо-2-[(4′-оксиацетил-Lys-ОМе)фенил]-4,4,5,5-тетраметилимидазолина и 100 мг (1 ммоль) N-метилморфолина в 5 мл безводного THF, и проводили реакцию при к.т.в течение 24 ч до исчезновения исходного вещества согласно анализу методом TLC (CHCl3:МеОН, 40:1). Реакционную смесь подвергали стандартной процедуре с получением 680 мг (52%) указанного в заголовке соединения в виде голубого твердого вещества, т.пл. 79-82°С. 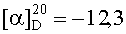
Пример 46. Получение 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Вос-Gly-Arg(NO2)-Pro-Ala-Lys(Z)]-Lys}фенил}-4,4,5,5-тетраметилимидазолина
На бане со льдом в 3 мл метанола растворяли 1260 мг (1 ммоль) 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Boc-Gly-Arg(NO2)-Pro-Ala-Lys(Z)]-Lys-OMe}фенил}-4,4,5,5-тетраметилимидазолина, затем добавляли водный раствор NaOH (2н), а затем перемешивали при к.т. в течение 30 мин. При поддержании pH 12 реакционную смесь перемешивали в течение 10 мин на бане со льдом до исчезновения исходного вещества согласно анализу методом TLC. Значение pH корректировали до 7 добавлением 2н HCl, реакционный раствор концентрировали в условиях пониженного давления, остаток разбавляли 2 мл насыщенного солевого раствора, корректировали до pH 2 добавлением 2н HCl, а затем трижды экстрагировали этилацетатом (3×5 мл). Объединенную этилацетатную фазу сушили над безводным сульфатом натрия и фильтровали, и концентрировали фильтрат в условиях пониженного давления при к.т. с получением 945 мг (82%) указанного в заголовке соединения в виде бесцветного твердого вещества. EI-MS (m/z) 1223 [М-Н]-.
Пример 47. Получение 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Boc-Gly-Arg(NO2)-Pro-Ala-Lys(Z)]-Lys-Arg-(NO2)-Gly-Asp(OBzl)-Ser(Bzl)-OBzl}фенил}-4,4,5,5-тетраметилимидазолина
На бане со льдом раствор 611 мг (0,5 ммоль) 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Boc-Gly-Arg(NO2)-Pro-Ala-Lys(Z)]-Lys}фенил}-4,4,5,5-тетраметилимидазолина, 69 мг (0,5 ммоль) HOBt и 126 мг (0,6 ммоль) DCC в 20 мл безводного THF перемешивали в течение 20 мин, затем к нему добавляли раствор, полученный из 442 мг (0,5 ммоль) HCl·Arg(NO2)-Gly-Asp(OBzl)-Ser(Bzl)-Obzl и 50 мг (0,5 ммоль) N-метилморфолина в 5 мл безводного THF, и проводили реакцию при к.т. в течение 24 ч до исчезновения исходного вещества согласно анализу методом TLC (CHCl3:МеОН, 20:1). Реакционную смесь подвергали стандартной процедуре с получением 500 мг (48%) указанного в заголовке соединения в виде голубого твердого вещества, т.пл. 127-129°С. 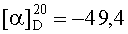
Пример 48. Получение 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Boc-Gly-Arg(NO2)-Pro-Ala-Lys(Z)]-Lys-Arg-(NO2)-Gly-Asp(OBzl)-Val-OBzl}фенил}-4,4,5,5-тетраметилимидазолина
На бане со льдом раствор 611 мг (0,5 ммоль) 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Boc-Gly-Arg(NO2)-Pro-Ala-Lys(Z)]-Lys}фенил}-4,4,5,5-тетраметилимидазолина, 69 мг (0,5 ммоль) HOBt и 126 мг (0,6 ммоль) DCC в 20 мл безводного THF перемешивали в течение 20 мин, затем к нему добавляли раствор, полученный из 421 мг (0,5 ммоль) HCl·Arg(NO2)-Gly-Asp(OBzl)-Val-Obzl, 50 мг (0,5 ммоль) N-метилморфолина в 5 мл безводного THF, и проводили реакцию при к.т. в течение 24 ч до исчезновения исходного вещества согласно анализу методом TLC (CHCl3:МеОН, 20:1). Реакционную смесь подвергали стандартной процедуре с получением 392 мг (35%) указанного в заголовке соединения в виде голубого твердого вещества, т.пл. 147-150°С. 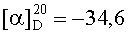
Пример 49. Получение 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Boc-Gly-Arg(NO2)-Pro-Ala-Lys(Z)]-Lys-Arg-(NO2)-Gly-Asp(OBzl)-Phe-OBzl}фенил}-4,4,5,5-тетраметилимидазолина
На бане со льдом раствор 611 мг (0,5 ммоль) 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Boc-Gly-Arg(NO2)-Pro-Ala-Lys(Z)]-Lys}фенил}-4,4,5,5-тетраметилимидазолина, 69 мг (0,5 ммоль) HOBt и 126 мг (0,6 ммоль) DCC в 20 мл безводного THF перемешивали в течение 20 мин, затем к нему добавляли раствор, полученный из 445 мг (0,5 ммоль) HCl·Arg(NO2)-Gly-Asp(OBzl)-Phe-Obzl и 50 мг (0,5 ммоль) N-метилморфолина в 5 мл безводного THF, и проводили реакцию при к.т. в течение 24 ч до исчезновения исходного вещества согласно анализу методом TLC (CHCl3:МеОН, 20:1). Реакционную смесь подвергали стандартной процедуре с получением 336 мг (31%) указанного в заголовке соединения в виде голубого твердого вещества, т.пл. 125-128°С. 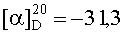
Пример 50. Получение 1,3-диоксо-2-{4′-оксиацетил-[Nω-(Gly-Arg-Pro-Ala-Lys)-Lys-Arg-Gly-Asp-Ser]фенил}-4,4,5,5-тетраметилимидазолина (Id)
На бане со льдом 195 мг (0,1 ммоль) 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Вос-Gly-Arg(NO2)-Pro-Ala-Lys(Z)]-Lys-Arg-(NO2)-Gly-Asp(OBzl)-Ser(Bzl)-OBzl}фенил}-4,4,5,5-тетраметилимидазолина смешивали с 6 мл трифторуксусной кислоты и 1,5 мл трифторметансульфоновой кислоты, и перемешивали в течение 1 ч до исчезновения исходного вещества согласно анализу методом TLC (CHCl3:MeOH, 1:1). Реакционную смесь концентрировали в условиях пониженного давления, остаток неоднократно промывали безводным этиловым эфиром и концентрировали в условиях пониженного давления. Остаток растворяли в воде, корректировали до pH 8 добавлением 25% водного раствора аммиака, обессоливали с использованием Sephadex G10, а затем очищали на С18 колонке. Собранные фракции лиофилизировали с получением 102 мг (82%) указанного в заголовке соединения в виде голубого твердого вещества, т.пл. 142-145°C. 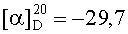
Пример 51. Получение 1,3-диоксо-2-{4′-оксиацетил-[Nω-(Gly-Arg-Pro-Ala-Lys)-Lys-Arg-Gly-Asp-Val]фенил}-4,4,5,5-тетраметилимидазолина (Ie)
На бане со льдом 190 мг (0,1 ммоль) 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Boc-Gly-Arg(NO2)-Pro-Ala-Lys(Z)]-Lys-Arg-(NO2)-Gly-Asp(OBzl)-Val-OBzl}фенил}-4,4,5,5-тетраметилимидазолина смешивали с 6 мл трифторуксусной кислоты и 1,5 мл трифторметансульфоновой кислоты, и перемешивали в течение 1 ч до исчезновения исходного вещества согласно анализу методом TLC (CHCl3:МеОН, 1:1). Реакционную смесь концентрировали в условиях пониженного давления, остаток неоднократно промывали безводным этиловым эфиром и концентрировали в условиях пониженного давления. Остаток растворяли в воде, корректировали до pH 8 добавлением 25% водного раствора аммиака, обессоливали с использованием Sephadex G10, а затем очищали на С18 колонке. Собранные фракции лиофилизировали с получением 99 мг (84%) указанного в заголовке соединения в виде голубого твердого вещества, т.пл. 147-149°С. 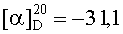
Пример 52. Получение 1,3-диоксо-2-{4′-оксиацетил-[Nω-(Gly-Arg-Pro-Ala-Lys)-Lys-Arg-Gly-Asp-Phe]фенил}-4,4,5,5-тетраметилимидазолина (If)
На бане со льдом 192 мг (0,1 ммоль) 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Вос-Gly-Arg(NO2)-Pro-Ala-Lys(Z)]-Lys-Arg-(NO2)-Gly-Asp(OBzl)-Phe-OBzl}фенил}-4,4,5,5-тетраметилимидазолина смешивали с 6 мл трифторуксусной кислоты и 1,5 мл трифторметансульфоновой кислоты, и перемешивали в течение 1 ч до исчезновения исходного вещества согласно анализу методом TLC (CHCl3:МеОН, 1:1). Реакционную смесь концентрировали в условиях пониженного давления, остаток неоднократно промывали безводным этиловым эфиром и концентрировали в условиях пониженного давления. Остаток растворяли в воде, корректировали до pH 8 добавлением 25% водного раствора аммиака, обессоливали с использованием Sephadex G10, а затем очищали на С18 колонке. Собранные фракции лиофилизировали с получением 106 мг (81%) указанного в заголовке соединения в виде голубого твердого вещества, т.пл. 84-85°С. 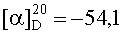
Получение пептида, характеризующегося тромболитической активностью: соответствующим образом защищенный RPAK
Пример 53. Получение Boc-Arg(NO2)-Pro-Ala-Lys(Z)
На бане со льдом в 3 мл метанола растворяли 850 мг (1 ммоль) Boc-Arg(NO2)-Pro-Ala-Lys(Z)-Obzl, затем добавляли водный раствор NaOH (2н), а затем перемешивали при к.т. в течение 30 мин. При поддержании pH 12 реакционную смесь перемешивали в течение 10 мин на бане со льдом до исчезновения исходного вещества согласно анализу методом TLC. Значение pH корректировали до 7 добавлением 2н HCl, реакционный раствор концентрировали в условиях пониженного давления, остаток разбавляли 2 мл насыщенного солевого раствора, корректировали до pH 2 добавлением 2н НСl, а затем трижды экстрагировали этилацетатом (3×5 мл). Объединенную этилацетатную фазу сушили над безводным сульфатом натрия и фильтровали, а затем концентрировали фильтрат в условиях пониженного давления при к.т. с получением 742 мг (92%) указанного в заголовке соединения в виде бесцветного твердого вещества. EI-MS (m/z)849 [M-H]-.
Получение трехкомпонентных конъюгатов RPAK/имидазолин/RGD (соединения общей формулы I-1-3): Ig, In, Ii
Пример 54. Получение 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Вос-Arg(NO2)-Pro-Ala-Lys(Z)]-Lys-ОМе}фенил}-4,4,5,5-тетраметилимидазолина
На бане со льдом раствор 760 мг (1 ммоль) Boc-Arg(NO2)-Pro-Ala-Lys(Z), 135 мг (1 ммоль) HOBt и 250 мг (1 ммоль) DCC в 10 мл безводного THF перемешивали в течение 20 мин, затем к нему добавляли раствор, полученный из 480 мг (1 ммоль) 1,3-диоксо-2-[(4′-оксиацетил-Lys-ОМе)фенил]-4,4,5,5-тетраметилимидазолина и 100 мг (1 ммоль) N-метилморфолина в 5 мл безводного THF, и проводили реакцию при к.т. в течение 24 ч до исчезновения исходного вещества согласно анализу методом TLC (CHCl3:МеОН, 40:1). Реакционную смесь подвергали стандартной процедуре с получением 920 мг (83%) указанного в заголовке соединения в виде голубого твердого вещества, т.пл. 72-76°С. 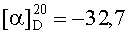
Пример 55. Получение 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Вос-Arg(NO2)-Pro-Ala-Lys(Z)]-Lys}фенил}-4,4,5,5-тетраметилимидазолина
На бане со льдом в 3 мл метанола растворяли 1200 мг (1 ммоль) 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Boc-Arg(NO2)-Pro-Ala-Lys(Z)]-Lys-ОМе}фенил}-4,4,5,5-тетраметилимидазолин, затем добавляли водный раствор NaOH (2н), а затем перемешивали при к.т. в течение 30 мин. При поддержании pH 12 реакционную смесь перемешивали в течение 10 мин на бане со льдом до исчезновения исходного вещества согласно анализу методом TLC. Значение pH корректировали до 7 добавлением 2н HCl, реакционный раствор концентрировали в условиях пониженного давления, остаток разбавляли 2 мл насыщенного солевого раствора, корректировали до pH 2 добавлением 2н HCl, а затем трижды экстрагировали этилацетатом (3×5 мл). Объединенную этилацетатную фазу сушили над безводным сульфатом натрия и фильтровали, и концентрировали фильтрат в условиях пониженного давления при к.т. с получением 899 мг (80%) указанного в заголовке соединения в виде голубого твердого вещества. EI-MS (m/z) 1116 [M-H]-.
Пример 56. Получение 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Boc-Arg(NO2)-Pro-Ala-Lys(Z)]-Lys-Arg-(NO2)-Gly-Asp(OBzl)-Ser(Bzl)-OBzl}фенил}-4,4,5,5-тетраметилимидазолина
На бане со льдом раствор 583 мг (0,5 ммоль) 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Boc-Arg(NO2)-Pro-Ala-Lys(Z)]-Lys}фенил}-4,4,5,5-тетраметилимидазолина, 69 мг (0,5 ммоль) HOBt и 126 мг (0,6 ммоль) DCC в 20 мл безводного THF перемешивали в течение 20 мин, затем к нему добавляли раствор, полученный из 442 мг (0,5 ммоль) HCl·Arg(NO2)-Gly-Asp(OBzl)-Ser(Bzl)-Obzl и 50 мг (0,5 ммоль) N-метилморфолина в 5 мл безводного THF, и проводили реакцию при к.т. в течение 24 ч до исчезновения исходного вещества согласно анализу методом TLC (CHCl3:МеОН, 20:1). Реакционную смесь подвергали стандартной процедуре с получением 421 мг (40%) указанного в заголовке соединения в виде голубого твердого вещества, т.пл. 77-79°С. 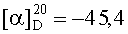
Пример 57. Получение 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Boc-Arg(NO2)-Pro-Ala-Lys(Z)]-Lys-Arg-(NO2)-Gly-Asp(OBzl)-Val-OBzl}фенил}-4,4,5,5-тетраметилимидазолина
На бане со льдом раствор 583 мг (0,5 ммоль) 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Boc-Arg(NO2)-Pro-Ala-Lys(Z)]-Lys}фенил}-4,4,5,5-тетраметилимидазолина, 69 мг (0,5 ммоль) HOBt и 126 мг (0,6 ммоль) DCC в 20 мл безводного THF перемешивали в течение 20 мин, затем к нему добавляли раствор, полученный из 421 мг (0,5 ммоль) HCl·Arg(NO2)-Gly-Asp(OBzl)-Val-Obzl и 50 мг (0,5 ммоль) N-метилморфолина в 5 мл безводного THF, и проводили реакцию при к.т. в течение 24 ч до исчезновения исходного вещества согласно анализу методом TLC (CHCl3:МеОН, 20:1). Реакционную смесь подвергали стандартной процедуре с получением 472 мг (42%) указанного в заголовке соединения в виде голубого твердого вещества, т.пл. 107-109°С. 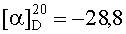
Пример 58. Получение 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Boc-Arg(NO2)-Pro-Ala-Lys(Z)]-Lys-Arg-(NO2)-Gly-Asp(OBzl)-Phe-OBzl}фенил}-4,4,5,5-тетраметилимидазолина
На бане со льдом раствор 583 мг (0,5 ммоль) 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Вос-Arg(NO2)-Pro-Ala-Lys(Z)]-Lys}фенил}-4,4,5,5-тетраметилимидазолина, 69 мг (0,5 ммоль) HOBt и 126 мг (0,6 ммоль) DCC в 20 мл безводного THF перемешивали в течение 20 мин, затем к нему добавляли раствор, полученный из 445 мг (0,5 ммоль) HCl·Arg(NO2)-Gly-Asp(OBzl)-Phe-Obzl и 50 мг (0,5 ммоль) N-метилморфолина в 5 мл безводного THF, и проводили реакцию при к.т. в течение 24 ч до исчезновения исходного вещества согласно анализу методом TLC (CHCl3:МеОН, 20:1). Реакционную смесь подвергали стандартной процедуре с получением 420 мг (47%) указанного в заголовке соединения в виде голубого твердого вещества, т.пл. 141-144°С. 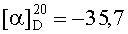
Пример 59. Получение 1,3-диоксо-2-{4′-оксиацетил-[Nω-(Arg-Pro-Ala-Lys)-Lys-Arg-Gly-Asp-Ser]фенил}-4,4,5,5-тетраметилимидазолина (Ig)
На бане со льдом 170 мг (0,1 ммоль) 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Вос-Arg(NO2)-Pro-Ala-Lys(Z)]-Lys-Arg-(NO2)-Gly-Asp(OBzl)-Ser(Bzl)-OBzl}фенил}-4,4,5,5-тетраметилимидазолина смешивали с 6 мл трифторуксусной кислоты и 1,5 мл трифторметансульфоновой кислоты, и перемешивали в течение 1 ч до исчезновения исходного вещества согласно анализу методом TLC (CHCl3:МеОН, 1:1). Реакционную смесь концентрировали в условиях пониженного давления, остаток неоднократно промывали безводным этиловым эфиром и концентрировали в условиях пониженного давления. Остаток растворяли в воде, корректировали до pH 8 добавлением 25% водного раствора аммиака, обессоливали с использованием Sephadex G10, а затем очищали на С18 колонке. Собранные фракции лиофилизировали с получением 102 мг (82%) указанного в заголовке соединения в виде голубого твердого вещества, т.пл. 148-150°С. 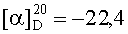
Пример 60. Получение 13-диоксо-2-{4′-оксиацетил-[Nω-(Arg-Pro-Ala-Lys)-Lys-Arg-Gly-Asp-Val]фенил}-4,4,5,5-тетраметилимидазолина (Ih)
На бане со льдом 182 мг (0,1 ммоль) 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Boc-Arg(NO2)-Pro-Ala-Lys(Z)]-Lys-Arg-(NO2)-Gly-Asp(OBzl)-Val-OBzl}фенил}-4,4,5,5-тетраметилимидазолина смешивали с 6 мл трифторуксусной кислоты и 1,5 мл трифторметансульфоновой кислоты, и перемешивали в течение 1 ч до исчезновения исходного вещества согласно анализу методом TLC (CHCl3:МеОН, 1:1). Реакционную смесь концентрировали в условиях пониженного давления, остаток неоднократно промывали безводным этиловым эфиром и концентрировали в условиях пониженного давления. Остаток растворяли в воде, корректировали до pH 8 добавлением 25% водного раствора аммиака, обессоливали с использованием Sephadex G10, а затем очищали на С18 колонке. Собранные фракции лиофилизировали с получением 99 мг (84%) указанного в заголовке соединения в виде голубого твердого вещества, т.пл. 137-139°С. 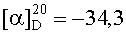
Пример 61. Получение 1,3-диоксо-2-{4′-оксиацетил-[Nω-(Arg-Pro-Ala-Lys)-Lys-Arg-Gly-Asp-Phe]фенил}-4,4,5,5-тетраметилимидазолина (Ii)
На бане со льдом 187 мг (0,1 ммоль) 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Boc-Arg(NO2)-Pro-Ala-Lys(Z)]-Lys-Arg-(NO2)-Gly-Asp(OBzl)-Phe-OBzl}фенил}-4,4,5,5-тетраметилимидазолина смешивали с 6 мл трифторуксусной кислоты и 1,5 мл трифторметансульфоновой кислоты, и перемешивали в течение 1 ч до исчезновения исходного вещества согласно анализу методом TLC (CHCl3:МеОН, 1:1). Реакционную смесь концентрировали в условиях пониженного давления, остаток неоднократно промывали безводным этиловым эфиром и концентрировали в условиях пониженного давления. Остаток растворяли в воде, корректировали до pH 8 добавлением 25% водного раствора аммиака, обессоливали с использованием Sephadex G10, а затем очищали на С18 колонке. Собранные фракции лиофилизировали с получением 96 мг (81%) указанного в заголовке соединения в виде голубого твердого вещества, т.пл. 99-100°С. 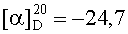
Получение пептида, характеризующегося тромболитической активностью: соответствующим образом защищенный PAK
Пример 62. Получение Boc-Pro-Ala-Lys(Z)
На бане со льдом в 3 мл метанола растворяли 638 мг (1 ммоль) Вос-Pro-Ala-Lys(Z)-Obzl, затем добавляли водный раствор NaOH (2н), а затем перемешивали при к.т. в течение 30 мин. При поддержании pH 12 реакционную смесь перемешивали в течение 10 мин на бане со льдом до исчезновения исходного вещества согласно анализу методом TLC. Значение pH корректировали до 7 добавлением 2н HCl, реакционный раствор концентрировали в условиях пониженного давления, остаток разбавляли 2 мл насыщенного солевого раствора, корректировали до pH 2 добавлением 2н HCl, а затем трижды экстрагировали этилацетатом (3×5 мл). Объединенную этилацетатную фазу сушили над безводным сульфатом натрия и фильтровали, а затем концентрировали фильтрат в условиях пониженного давления при к.т. с получением 509 мг (91,6%) указанного в заголовке соединения в виде бесцветного твердого вещества. EI-MS (m/z) 547 [М-Н]-.
Получение трехкомпонентных конъюгатов PAK/имидазолин/RGD (соединения общей формулы I-1-4): Ij, Ik, Il
Пример 63. Получение 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Boc-Pro-Ala-Lys(Z)]-Lys-ОМе}фенил}-4,4,5,5-тетраметилимидазолина
На бане со льдом раствор 548 мг (1 ммоль) Boc-Pro-Ala-Lys(Z), 135 мг (1 ммоль) HOBt и 250 мг (1 ммоль) DCC в 10 мл безводного THF перемешивали в течение 20 мин, затем к нему добавляли раствор, полученный из 480 мг (1 ммоль) 1,3-диоксо-2-[(4′-оксиацетил-Lys-ОМе)фенил]-4,4,5,5-тетраметилимидазолина и 100 мг (1 ммоль) N-метилморфолина в 5 мл безводного THF, и проводили реакцию при к.т.в течение 24 ч до исчезновения исходного вещества согласно анализу методом TLC (CHCl3:МеОН, 40:1). Реакционную смесь подвергали стандартной процедуре с получением 876 мг (87%) указанного в заголовке соединения в виде голубого твердого вещества, т.пл. 77-80°C. 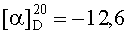
Пример 64. Получение 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Вос-Pro-Ala-Lys(Z)]-Lys}фенил}-4,4,5,5-тетраметилимидазолина
На бане со льдом в 3 мл метанола растворяли 980 мг (1 ммоль) 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Boc-Pro-Ala-Lys(Z)]-Lys-OMe}фенил}-4,4,5,5-тетраметилимидазолина, затем добавляли водный раствор NaOH (2н), а затем перемешивали при к.т.в течение 30 мин. При поддержании pH 12 реакционную смесь перемешивали в течение 10 мин на бане со льдом до исчезновения исходного вещества согласно анализу методом TLC. Значение pH корректировали до 7 добавлением 2н HCl, реакционный раствор концентрировали в условиях пониженного давления, остаток разбавляли 2 мл насыщенного солевого раствора, корректировали до pH 2 добавлением 2н HCl, а затем трижды экстрагировали этилацетатом (3×5 мл). Объединенную этилацетатную фазу сушили над безводным сульфатом натрия и фильтровали, и концентрировали фильтрат в условиях пониженного давления при к.т. с получением 867 мг (80%) указанного в заголовке соединения в виде голубого твердого вещества. EI-MS (m/z) 965 [М-Н]-.
Пример 65. Получение 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Вос-Pro-Ala-Lys(Z)]-Lys-Arg-(NO2)-Gly-Asp(OBzl)-Ser(Bzl)-OBzl}фенил}-4,4,5,5-тетраметилимидазолина
На бане со льдом раствор 483 мг (0,5 ммоль) 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Вос-Pro-Ala-Lys(Z)]-Lys}фенил}-4,4,5,5-тетраметилимидазолина, 69 мг (0,5 ммоль) HOBt и 126 мг (0,6 ммоль) DCC в 20 мл безводного THF перемешивали в течение 20 мин, затем к нему добавляли раствор, полученный из 442 мг (0,5 ммоль) HCl·Arg(NO2)-Gly-Asp(OBzl)-Ser(Bzl)-Obzl и 50 мг (0,5 ммоль) N-метилморфолина в 5 мл безводного THF, и проводили реакцию при к.т.в течение 24 ч до исчезновения исходного вещества согласно анализу методом TLC (CHCl3:МеОН, 20:1). Реакционную смесь подвергали стандартной процедуре с получением 421 мг (42%) указанного в заголовке соединения в виде голубого твердого вещества, т.пл. 97-100°С. 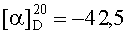
Пример 66. Получение 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Вос-Pro-Ala-Lys(Z)]-Lys-Arg-(NO2)-Gly-Asp(OBzl)-Val-OBzl}фенил}-4,4,5,5-тетраметилимидазолина
На бане со льдом раствор 483 мг (0,5 ммоль) 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Вос-Pro-Ala-Lys(Z)]-Lys}фенил}-4,4,5,5-тетраметилимидазолина, 69 мг (0,5 ммоль) HOBt и 126 мг (0,6 ммоль) DCC в 20 мл безводного THF перемешивали в течение 20 мин, затем к нему добавляли раствор, полученный из 432 мг (0,5 ммоль) HCl·Arg(NO2)-Gly-Asp(OBzl)-Val-Obzl и 50 мг (0,5 ммоль) N-метилморфолина в 5 мл безводного THF, и проводили реакцию при к.т. в течение 24 ч до исчезновения исходного вещества согласно анализу методом TLC (CHCl3:МеОН, 20:1). Реакционную смесь подвергали стандартной процедуре с получением 357 мг (37%) указанного в заголовке соединения в виде голубого твердого вещества, т.пл. 117-120°С. 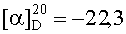
Пример 67. Получение 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Boc-Pro-Ala-Lys(Z)]-Lys-Arg-(NO2)-Gly-Asp(OBzl)-Phe-OBzl}фенил}-4,4,5,5-тетраметилимидазолина
На бане со льдом раствор 483 мг (0,5 ммоль) 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Вос-Pro-Ala-Lys(Z)]-Lys}фенил}-4,4,5,5-тетраметилимидазолина, 69 мг (0,5 ммоль) HOBt и 126 мг (0,6 ммоль) DCC в 20 мл безводного THF перемешивали в течение 20 мин, затем к нему добавляли раствор, полученный из 439 мг (0,5 ммоль) HCl·Arg(NO2)-Gly-Asp(OBzl)-Phe-Obzl и 50 мг (0,5 ммоль) N-метилморфолина в 5 мл безводного THF, и проводили реакцию при к.т.в течение 24 ч до исчезновения исходного вещества согласно анализу методом TLC (CHCl3:МеОН, 20:1). Реакционную смесь подвергали стандартной процедуре с получением 472 мг (48%) указанного в заголовке соединения в виде голубого твердого вещества, т.пл. 111-114°С. 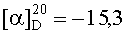
Пример 68. Получение 1,3-диоксо-2-{4′-оксиацетил-[Nω-(Pro-Ala-Lys)-Lys-Arg-Gly-Asp-Ser]фенил}-4,4,5,5-тетраметилимидазолина (Ij)
На бане со льдом 169 мг (0,1 ммоль) 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Вос-Pro-Ala-Lys(Z)]-Lys-Arg-(NO2)-Gly-Asp(OBzl)-Ser(Bzl)-OBzl}фенил}-4,4,5,5-тетраметилимидазолина смешивали с 6 мл трифторуксусной кислоты и 1,5 мл трифторметансульфоновой кислоты, и перемешивали в течение 1 ч до исчезновения исходного вещества согласно анализу методом TLC (CHCl3:МеОН, 1:1). Реакционную смесь концентрировали в условиях пониженного давления, остаток неоднократно промывали безводным этиловым эфиром и концентрировали в условиях пониженного давления. Остаток растворяли в воде, корректировали до pH 8 добавлением 25% водного раствора аммиака, обессоливали с использованием Sephadex G10, а затем очищали на С18 колонке. Собранные фракции лиофилизировали с получением 98 мг (80%) указанного в заголовке соединения в виде голубого твердого вещества, т.пл. 127-128°С. 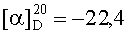
Пример 69. Получение 1,3-диоксо-2-{4′-оксиацетил-[Nω-(Pro-Ala-Lys)-Lys-Arg-Gly-Asp-Val]фенил}-4,4,5,5-тетраметилимидазолина (Ik)
На бане со льдом 162 мг (0,1 ммоль) 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Вос-Pro-Ala-Lys(Z)]-Lys-Arg-(NO2)-Gly-Asp(OBzl)-Val-OBzl}фенил}-4,4,5,5-тетраметилимидазолина смешивали с 6 мл трифторуксусной кислоты и 1,5 мл трифторметансульфоновой кислоты, и перемешивали в течение 1 ч до исчезновения исходного вещества согласно анализу методом TLC (CHCl3:МеОН, 1:1). Реакционную смесь концентрировали в условиях пониженного давления, остаток неоднократно промывали безводным этиловым эфиром и концентрировали в условиях пониженного давления. Остаток растворяли в воде, корректировали до pH 8 добавлением 25% водного раствора аммиака, обессоливали с использованием Sephadex G10, а затем очищали на С18 колонке. Собранные фракции лиофилизировали с получением 96 мг (81%) указанного в заголовке соединения в виде голубого твердого вещества, т.пл. 123-124°С. 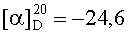
Пример 70. Получение 1,3-диоксо-2-{4′-оксиацетил-[Nω-(Pro-Ala-Lys)-Lys-Arg-Gly-Asp-Phe]фенил}-4,4,5,5-тетраметилимидазолина (Il)
На бане со льдом 169 мг (0,1 ммоль) 1,3-диоксо-2-{4′-оксиацетил-{Nω-[Boc-Pro-Ala-Lys(Z)]-Lys-Arg-(NO2)-Gly-Asp(OBzl)-Phe-OBzl}фенил}-4,4,5,5-тетраметилимидазолина смешивали с 6 мл трифторуксусной кислоты и 1,5 мл трифторметансульфоновой кислоты, и перемешивали в течение 1 ч до исчезновения исходного вещества согласно анализу методом TLC (CHCl3:МеОН, 1:1). Реакционную смесь концентрировали в условиях пониженного давления, остаток неоднократно промывали безводным этиловым эфиром и концентрировали в условиях пониженного давления. Остаток растворяли в воде, корректировали до pH 8 добавлением 25% водного раствора аммиака, обессоливали с использованием Sephadex G10, а затем очищали на С18 колонке. Собранные фракции лиофилизировали с получением 96 мг (81%) указанного в заголовке соединения в виде голубого твердого вещества, т.пл. 153-154°С. 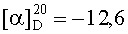
Экспериментальный пример 1. Эксперименты по акцептированию радикалов NO соединениями Ia-Il согласно настоящему изобретению
Самцов крыс Wistar весом от 250 до 300 г подвергали голоданию в течение 12 ч до операции со свободным доступом к питьевой воде и умерщвляли смещением шейных позвонков. Сразу осуществляли торакотомию и вынимали грудную аорту, прикрепленные к ней соединительные ткани рассекали и сосуды разрезали на аортальные кольца длиной от 3 до 5 мм, и помещали в баню для перфузии. Баня содержала 15 мл раствора Кребса-Хенселейта и поддерживалась при постоянной температуре 37°С, в которой поддерживался газовый состав 95% О2-5% CO2. Якорь, к которому были иммобилизованы кольца аорты, подключали к датчику напряжения, а кривые коронарных сосудов записывали на двухлучевом записывающем устройстве при скорости бумаги 1 мм/мин. С помощью статического напряжения, доведенного до 1,0 г и уравновешивания 30 мин, давали дозу норадреналина в конечной концентрации 10-8 М, чтобы обеспечить сокращение аорты для предвозбуждения. Норэпинефрин отмывали с последующим уравновешиванием в течение 30 мин, и в баню добавляли норадреналин до конечной концентрации 10-8 М. Когда напряжение сокращения оставалось стабильным на уровне плато, в баню добавляли 20 мкл физиологического раствора (контроль), 20 мкл раствора любого из соединений от Ia до Il в физиологическом растворе (с конечной концентрацией 5×10-6 М) или 20 мкл акцептора свободных радикалов NO (1,3-диоксо-2-(4′-оксиацетоксилфенил)-4,4,5,5-тетраметилимидазолин, TMMZ) в физиологическом растворе (с конечной концентрацией 5×10-6 М). После стабилизации добавляли 20 мкл ацетилхолина в физиологическом растворе (с конечной концентрацией 10-6 М). Способность лекарственных средств акцептировать NO выражали в виде процента ингибирования индуцированной ацетилхолином вазодилатации. Результаты эксперимента приведены в таблице 1.
Как показано в экспериментальных результатах, соединения Ia-Il были способны ингибировать вазодилатирующий эффект ацетилхолина на части сосудов путем акцептирования NO. Таким образом, с помощью связывания тромболитического пептида ARPAK, GRPAK, RPAK или PAK и пептида с направленным действием RGDS, RGDV или RGDF с акцептором свободных радикалов (1,3-диоксо-2-(4′-оксиацетоксилфенил)-4,4,5,5-тетраметилимидазолин, TMMZ) через Lys, 9 соединений характеризовались значительно более высокой активностью в ингибировании индуцированной ацетилхолином вазодилатации, чем TMMZ, 2 соединения характеризовались той же активностью в ингибировании индуцированной ацетилхолином вазодилатации, что и TMMZ, и одно соединение характеризовалось меньшей активностью в ингибировании индуцированной ацетилхолином вазодилатации, чем TMMZ. Среди 12 подвергнутых оцениванию соединений, 4 соединения характеризовались процентом ингибирования выше, чем 30%, и эти четыре соединения были ранжированы по активности в ингибировании индуцированной ацетилхолином вазодилатации таким образом: Ie>Ih>Ib>If. Это показывает, что активность фрагмента TMMZ в акцептировании свободных радикалов NO в целом улучшалась с помощью связывания тромболитического пептида ARPAK, GRPAK, RPAK или PAK и пептида с направленным действием RGDS, RGDV или RGDF с акцептором свободных радикалов TMMZ через Lys.
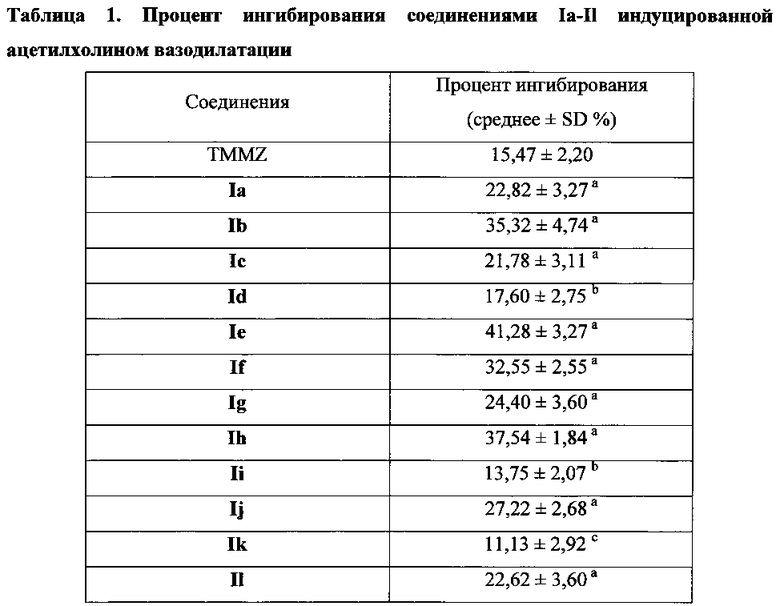
n=6; а) p<0,01 по отношению к TMMZ; b) p>0,05 по отношению к TMMZ; с) p<0,05 по отношению к TMMZ
Экспериментальный пример 2. Эксперименты по лизису эуглобулиновых сгустков соединениями Ia-Il согласно настоящему изобретению
Свиную кровь забирали и смешивали с цитратом натрия в концентрации 3,8% в объемном соотношении 9:1, сразу же центрифугировали при 3000 об/мин в течение 10 мин и отделяли обедненную тромбоцитами плазму. 2 мл обедненной тромбоцитами плазмы свиньи и 36 мл ультрачистой воды добавляли в центрифужную пробирку объемом 50 мл. В каждую пробирку добавляли 0,4 мл уксусной кислоты (1%) и тщательно перемешивали, и пробирку помещали в холодильник с температурой 4°С на 10 мин и затем центрифугировали при 3000 об/мин в течение 10 мин. Центрифужные пробирки переворачивали и затем внутреннюю стенку пробирок высушивали с использованием фильтровальной бумаги после сливания жидкости. Эуглобулиновые осадки, полученные в результате центрифугирования, сушили вымораживанием в течение примерно 40 мин и выскребали. Брали приблизительно 35 мг эуглобулина и растворяли в 7 мл боратного буфера (pH 9,28). Эуглобулин в основном растворялся через 1 ч, в который добавляли 0,7 мл раствора CaCl2 (25 мМ) и немедленно помещали на стеклянную пластину размером 10×10 см с толщиной около 1 мм. После образования сгустка 10 мкл физиологического раствора или 10 мкл раствора одного из соединений Ia-Il в физиологическом растворе (1 мМ), или 10 мкл раствора урокиназы в физиологическом растворе (0,8 мг/мл) пипетировали и наносили на пластину сгустка с интервалом между каждыми двумя каплями более чем 1,5 см, и каждый образец наносили 3 раза. Диаметр окружности лизиса сгустка измеряли через 4 ч, и показания представлены в таблице 2.
Как показано в экспериментальных результатах, с помощью связывания тромболитического пептида ARPAK, GRPAK, RPAK или PAK и пептида с направленным действием RGDS, RGDV или RGDF с акцептором свободных радикалов TMMZ через Lys, все соединения проявляли значительную активность лизиса эуглобулиновых сгустков.

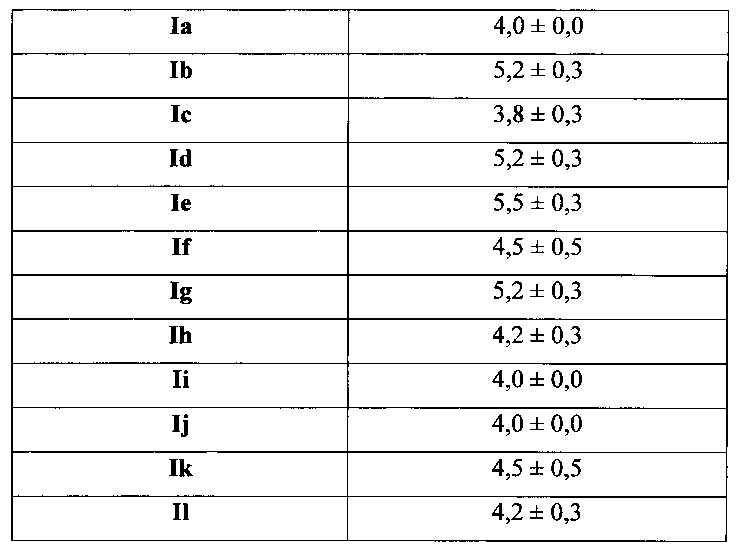
n=3; a) p<0,01 по отношению к Ia-l
Экспериментальный пример 3. Эксперименты тромболиза in vitro для соединений Ia-Il согласно настоящему изобретению
Крыс SD (самцов, от 350 до 400 г) анестезировали путем внутрибрюшинной инъекции раствора уретана в дозе 1200 мг/кг. Анестезированных крыс фиксировали в положении на спине, и правую общую сонную артерию рассекали, зажимали на проксимальном конце артериальной клипсой и пронизывали нитью на проксимальном и дистальном концах, соответственно. Шов на дистальном конце крепко зажимали гемостатическим зажимом на шерсти. Канюли проводили на дистальном конце, артериальный зажим удаляли, и всю артериальную кровь выливали в контейнер объемом 50 мл, ранее обработанной силиконовым маслом. 0,8 мл артериальной крови крыс вводили в вертикально фиксированную стеклянную трубку (20 мм в длину, с внутренним диаметром 4 мм и внешним диаметром 5 мм, закрытую каучуковой пробкой на дне), в которую сразу же помещали винт иммобилизации тромба из нержавеющей стали. Винт иммобилизации тромба, образованный путем намотки проволоки из нержавеющей стали диаметром 0,2 мм, содержал спиральную часть 18 мм в длину, 15 намоток, каждая из которых характеризовалась диаметром 1,8 мм, и ствол 7,0 мм в длину, который подсоединяли к спиральной части, и характеризовался формой подобной вопросительному знаку. Через 40 мин после того, как кровь коагулировала, резиновую пробку в нижней части стеклянной трубки удаляли, ствол винта иммобилизации тромба зажимали щипцами и обернутый тромбом винт иммобилизации тромба аккуратно забирали из стеклянной трубки. Затем винт суспендировали и погружали в тройную дистиллированную воду, чтобы удалить излишнюю кровь, и точно взвешивали через 1 ч. Тромб суспендировали в 8 мл физиологического раствора или раствора соединений Iа-Il в физиологическом растворе (в концентрации 100 нМ), или раствора ARPAK, GRPAK, RPAK или PAK в физиологическом растворе (в концентрации 100 нМ), или раствора урокиназы в физиологическом растворе (100 МЕ/мл), а затем встряхивали при 37°С в термостатическом шейкере (63 г/мин) и удаляли через 2 ч и точно взвешивали, чтобы определить вес тромба. Рассчитывали разницу в массе тромба до и после введения, и результаты приведены в таблице 3.
Как показано в экспериментальных результатах, с помощью связывания тромболитического пептида ARPAK, GRPAK, RPAK или PAK и пептида с направленным действием RGDS, RGDV или RGDF с акцептором свободных радикалов TMMZ через Lys все соединения проявляли значительную тромболитическую активность in vitro. Поскольку активность Ia-Ic была сопоставима с ARPAK, активность Id-If была сопоставима с GRPAK, активность Ig-Ii была сопоставима с RPAK и активность Ij-Ii была сопоставима с таковой PAK, с одной стороны, тромболитическая активность in vitro Ia-Il могла быть отнесена к активности тромболитического пептида, а с другой стороны связывание тромболитического пептида ARPAK, GRPAK, RPAK или PAK и пептида с направленным действием RGDS, RGDV или RGDF с акцептором свободных радикалов TMMZ через Lys не уменьшало активность тромболитического пептида.
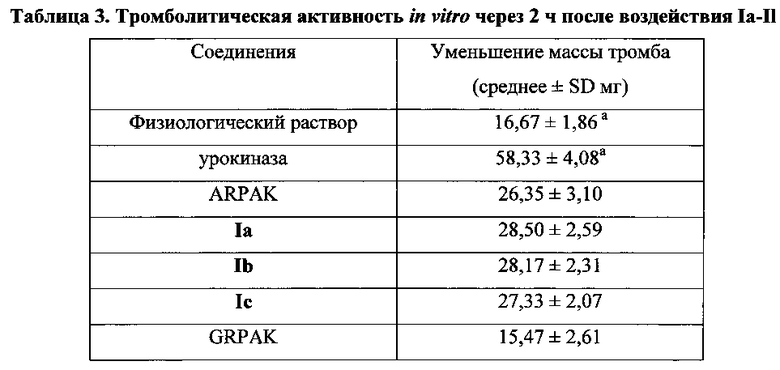
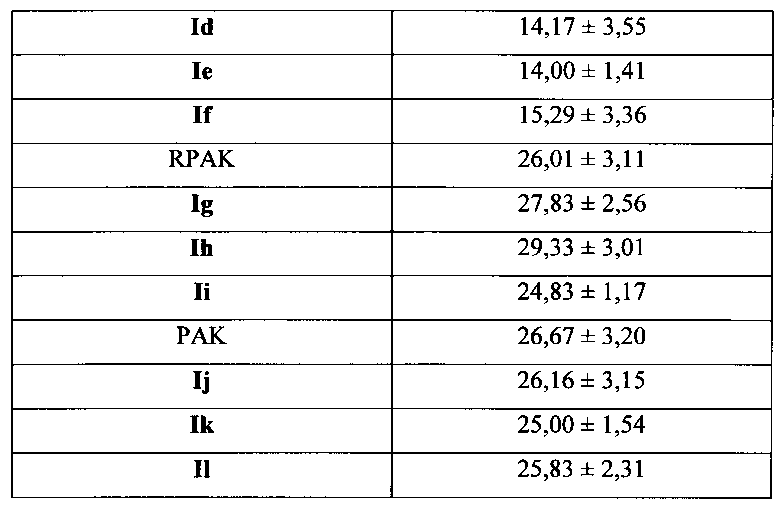
N=6; а) p<0,01 по отношению к Ia-Il
Экспериментальный пример 4. Эксперименты тромболиза in vivo для соединений Ia-Il согласно настоящему изобретению
Крыс SD (самцов, от 220 до 230 г) анестезировали путем внутрибрюшинной инъекции раствора уретана в дозе 1200 мг/кг. Анестезированных крыс фиксировали в положении на спине, и правую общую сонную артерию рассекали, зажимали на проксимальном конце артериальной клипсой и пронизывали нитью на проксимальном и дистальном концах, соответственно. Шов на дистальном конце крепко зажимали гемостатическим зажимом на шерсти. Канюли проводили на дистальном конце, артериальный зажим удаляли и приблизительно 1 мл артериальной крови выливали в пробирку эппендорф объемом 1 мл. 0,1 мл артериальной крови крыс вводили в вертикально фиксированную стеклянную трубку (15 мм в длину, с внутренним диаметром 2,5 мм и внешним диаметром 5 мм, закрытую резиновой пробкой на дне), в которую сразу же помещали винт иммобилизации тромба из нержавеющей стали. Винт иммобилизации тромба, образованный путем намотки проволоки из нержавеющей стали диаметром 0,2 мм, содержал спиральную часть 12 мм в длину, 15 намоток, каждая из которых характеризуется диаметром 1,8 мм, и ствол 1,0 мм в длину, который подсоединяли к спиральной части, и характеризовался формой подобной вопросительному знаку. Через 15 мин после того, как кровь коагулировала, резиновую пробку в нижней части стеклянной трубки удаляли, ствол винта иммобилизации тромба зажимали щипцами и обернутый тромбом винт иммобилизации тромба аккуратно забирали из стеклянной трубки и затем точно взвешивали.
Обходная канюля состояла из 3 сегментов. Средний сегмент представлял собой трубку из полиэтилена длиной 60,0 мм и внутренним диаметром 3,5 мм. Сегменты на обоих концах представляли собой подобные полиэтиленовые трубки длиной 100,0 мм, внутренним диаметром 1,0 мм и внешним диаметром 2,0 мм, один конец которой был разобран, чтобы образовать наконечник, с наружным диаметром 1,0 мм, который может быть вставлен в сонную артерию или вену крысы, а другой конец которой был обшит полиэтиленовой трубкой длиной 7,0 мм и внешним диаметром 3,5 мм (утолщенной, с тем, чтобы быть вставленным в полиэтиленовую трубку в середине сегмент). Внутренняя стенка 3-сегментной канюли была полностью силилирована (1% силиконовым маслом в этиловом эфире). Обернутый тромбом винт иммобилизации тромба помещали в полиэтиленовую трубку среднего сегмента, и оба конца трубки заключали в утолщенные концы двух полиэтиленовых трубок. Канюлю заполняли раствором гепарина в изотоническом растворе (50 МЕ/кг) через конец наконечника с использованием шприца и она была готова к использованию. Трахеи анестезированной крысы затем рассекали и проводили катетеризацию трахеи. Левую наружную сонную вену крысы рассекали и пронизывали швом на проксимальном и дистальном концах, соответственно. Неравномерный открытый разрез осторожно делали на выделенной левой наружной сонной вене, и кончик обходной канюли, полученной, как описано выше, вводили в проксимальный конец открытого разреза в левой наружной сонной вене, в противоположную сторону от ствола винта иммобилизации тромба в среднем сегменте обходной канюли (которая размещала точно взвешенный винт иммобилизации тромба). Точное количество гепарина в солевом растворе (50 МЕ/кг) вводили через наконечник на другом конце с помощью шприца. В этот момент, без удаления шприца из этиленовой трубы, трубку между шприцом и полиэтиленовой трубкой зажимали щипцами. Кровоток останавливали зажимом проксимального конца правой общей сонной артерии с помощью артериального зажима, и неравномерно открытый разрез аккуратно разрезали по общей сонной артерии вблизи зажима. Шприц вытаскивали из кончика полиэтиленовой трубки, и кончик полиэтиленовой трубки затем вставляли в проксимальный конец открытого разреза артерии. Оба конца обходной канюли фиксировали к артерии или вене швами №4.
Физиологический раствор (3 мл/кг) или раствор урокиназы в физиологическом растворе (в дозе 20000 МЕ/кг), или раствор одного из соединений Ia-Il в физиологическом растворе (в дозе 0,1 мкмоль/кг), или раствор ARPAK, GRPAK, RPAK или PAK в физиологическом растворе (в дозе 1 мкмоль/кг) соединяли с положением, близким к вене в стороне, противоположной от винта иммобилизации тромба с использованием скальпельной иглы, чтобы проколоть средний сегмент обходной канюли (в котором размещался точно взвешенный винт иммобилизации тромба). Зажим артерии удаляли, чтобы позволить крови течь из артерии в вену через обходную канюлю. Таким образом, была создана крысиная модель артериовенозного обходного тромболиза. Раствор в шприце медленно вводили в кровь, что позволяло физиологическому раствору (пустой контроль), урокиназе (положительный контроль), ARPAK, GRPAK, RPAK или PAK (контроль компонента), или Ia-Il влиять на тромб путем циркуляции крови в порядке вена-сердце-артерия. Процесс хронометрировали в начале инъекции, и винт иммобилизации тромба удаляли из обходной канюли через 1 ч и точно взвешивали. Определяли разницу в массе винта иммобилизации тромба у крыс с обходной канюлей до и после введения, и полученные результаты представлены в таблице 4.
Как показано в экспериментальных результатах, не только соединения Ia-Ic, полученные путем связывания тромболитического пептида ARPAK, GRPAK, RPAK или PAK и пептида с направленным действием RGDS, RGDV или RGDF с акцептором свободных радикалов TMMZ через Lys, проявляли тромболитическую активностью в дозе 0,1 мкмоль/кг, потенциальная возможность их активности также была сопоставима с соответствующим тромболитическим пептидом ARPAK, GRPAK, RPAK или PAK в дозе 1 мкмоль/кг. Таким образом, с помощью связывания тромболитического пептида ARPAK, GRPAK, RPAK или PAK и пептида с направленным действием RGDS, RGDV или RGDF с акцептором свободных радикалов TMMZ через Lys эффективная доза может быть уменьшена в 10 раз.

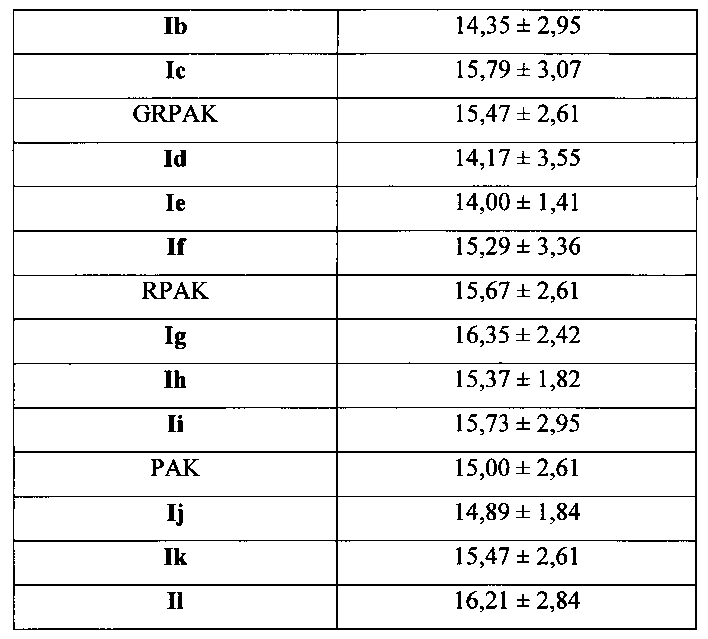
n=10; a) p<0,01 по отношению к Ia-Il
Экспериментальный пример 5. Антитромботические эксперименты in vivo для соединений Ia-Il согласно настоящему изобретению
Крыс SD (самцов, от 220 до 230 г) случайным образом разделяли на группы с 11 крысами в каждой группе. В 1-й день крыс кормили в состоянии покоя и подвергали голоданию в течение ночи. Крысы получали физиологический раствор (в дозе 3 мл/кг), раствор одного из соединений Ia-Il в физиологическом растворе (в дозе 0,1 мкмоль/кг), раствор пептида с направленным действием RGDS, RGDV или RGDF в физиологическом растворе (в дозе 10 мкмоль/кг) или аспирин (в дозе 33 мг/кг) через желудочный зонд. Через 30 мин крыс анестезировали с помощью 20% уретанового раствора, и рассекали правую сонную артерию и левую сонную вену. Канюлю наполняли гепарином натрия в физиологическом растворе, и один ее конец вставляли в левую вену, тогда как через другой конец вводили определенное количество гепарина натрия для антикоагуляции с помощью шприца, а затем вставляли в правую артерию. Кровь перетекала из правой артерии в левую вену через полиэтиленовую трубку, прикрепленную лигатура с тромбом вынимали через 15 мин и ее вес записывали. Массу влажного тромба определяли путем вычитания массы лигатуры от общей массы. Полученные результаты приведены в таблице 5.
Как показано в экспериментальных результатах, не только соединения Ia-Ic, полученные путем связывания тромболитического пептида ARPAK, GRPAK, RPAK или PAK и пептида с направленным действием RGDS, RGDV или RGDF с акцептором свободных радикалов TMMZ через Lys, проявляли тромболитическую активностью в пероральной дозе 0,1 мкмоль/кг, потенциальная возможность их активности также была сопоставима с таковой тромболитического пептида ARPAK, GRPAK, RPAK или PAK в дозе 10 мкмоль/кг. Таким образом, с помощью связывания тромболитического пептида ARPAK, GRPAK, RPAK или PAK и пептида с направленным действием RGDS, RGDV или RGDF с акцептором свободных радикалов TMMZ через Lys, эффективная доза может быть уменьшена в 100 раз.
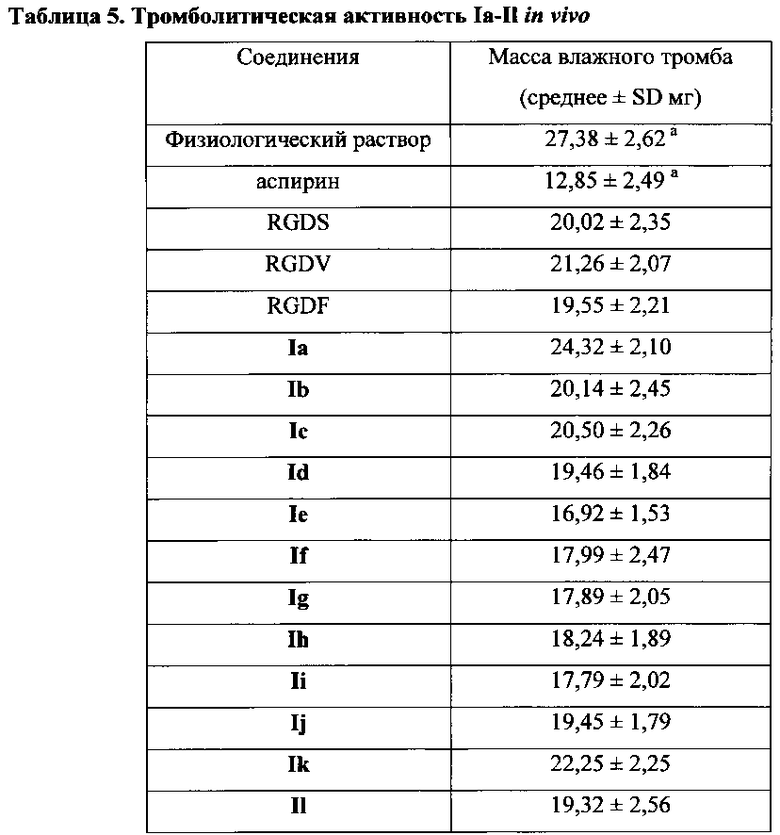
n=11; а) p<0,01 в отношении Ia-Il
Создание животных моделей для оценки эффективности соединений согласно настоящему изобретению в лечении пациентов с инсультом
(1) описанный в настоящем документе экспериментальной протокол крыс соответствовал Женевскому руководству в экспериментах на животных и был утвержден коллегией этического комитета. Чистопородных здоровых самцов крыс SD, весом от 280 до 305 г, приобретали в лабораториях экспериментальных животных Vital River. Эти крысы случайным образом использовали для получения тромбов или создания моделей инсульта.
(2) 10%-ный раствор хлоралгидрата внутрибрюшинно вводили крысам SD в дозе 400 мг/кг массы тела для анестезии. Сонную артерию рассекали, отбирали 15 мл свежей артериальной крови и аликвоты по 10 мкл каждая затем добавляли в 1,5 мл флаконы ЕР. Образованный тромб выдерживали при комнатной температуре в течение 2 ч, а затем при -20°С в холодильнике в течение 22 ч. При использовании добавляли 0,5 мл физиологического раствора к тромбу, который разбивали с использованием стеклянной палочки, чтобы приготовить раствор суспензии гомогената тромба с объемом около 0,1 мм3 для каждой части тромба.
(3) 10%-ный раствор хлоралгидрата внутрибрюшинно вводили крысам SD в дозе 400 мг/кг массы тела для анестезии. Сонную артерию рассекали, отбирали 15 мл свежей артериальной крови и аликвоты по 10 мкл каждая затем добавляли в 1,5 мл флаконы ЕР. Образованный тромб выдерживали при комнатной температуре в течение 24 ч. При использовании добавляли 0,5 мл физиологического раствора к тромбу, который разбивали с использованием стеклянной палочки, чтобы приготовить раствор суспензии гомогената тромба с объемом около 0,1 мм3 для каждой части тромба.
(4) 10%-ный раствор хлоралгидрата внутрибрюшинно вводили самцам крыс SD в дозе 400 мг/кг массы тела для анестезии. Делали продольный открытый надрез в центре шеи и правый общий ствол сонной артерии рассекали (около 3 см в длину). Каждую наружную ветвь сонной артерии рассекали и лигировали на подъязычном уровне, и внутреннюю сонную артерии рассекали в опухшей части шеи. Открытые разрезы во внутренней сонной артерии и проксимальном конце общей сонной артерии пережимали соответственно неинвазивными артериальными зажимами, и дистальный конец наружной сонной артерии лигировали. Катетер, содержащий 0,5 мл суспензии тромба в физиологическом растворе, вставляли в ствол наружной сонной артерии. В то же время, когда освобождали зажим на внутренней сонной артерии, 0,5 мл суспензии тромба в физиологическом растворе в катетере медленно протекало из наружной сонной артерии к ее проксимальному концу, а затем вводилось в артерии в мозге через внутреннюю сонную артерию. Впоследствии, проксимальный конец внутренней сонной артерии лигировали, артериальные зажимы на внутренней сонной артерии и общей сонной артерии высвобождали и поток крови восстанавливали. Основную наружную яремную вену рассекали, и физиологический раствор (пустой контроль) или раствор соединений согласно настоящему изобретению в физиологическом растворе вводили через наружную яремную вену. После зашивания раны вводили 20000 ME пенициллина внутримышечно для профилактики заражения. Таким образом, создавали модель немедленного лечения после начала инсульта.
(5) 10%-ный раствор хлоралгидрата внутрибрюшинно вводили самцам крыс SD в дозе 400 мг/кг массы тела для анестезии. Делали продольный открытый надрез в центре шеи и правый общий ствол сонной артерии рассекали (около 3 см в длину). Каждую наружную ветвь сонной артерии рассекали и лигировали на подъязычном уровне, и внутреннюю сонную артерию рассекали в опухшей части шеи. Открытые разрезы во внутренней сонной артерии и проксимальном конце общей сонной артерии пережимали соответственно неинвазивными артериальными зажимами, и дистальный конец наружной сонной артерии лигировали. Катетер, содержащий 0,5 мл суспензии тромба в физиологическом растворе, вставляли в ствол наружной сонной артерии. В то же время, когда освобождали зажим на внутренней сонной артерии, 0,5 мл суспензии тромба в физиологическом растворе в катетере медленно протекало из наружной сонной артерии к ее проксимальному концу, а затем вводилось в артерии в мозге через внутреннюю сонную артерию. Впоследствии, проксимальный конец внутренней сонной артерии лигировали, артериальные зажимы на внутренней сонной артерии и общей сонной артерии высвобождали и поток крови восстанавливали. После зашивания раны вводили 20000 ME пенициллина внутримышечно для профилактики заражения. Через 4 ч, 6 ч или 24 ч физиологический раствор (пустой контроль) или раствор соединений согласно настоящему изобретению в физиологическом растворе вводили через хвостовую вену. Таким образом, создавали крысиные модели лечения инсульта через 4 ч, 6 ч и 24 ч после начала.
Создание животных моделей для оценки эффективности соединений согласно настоящему изобретению через 4 ч, 6 ч и 24 ч после начала инсульта
(1) Эффективность после немедленного лечения, лечения через 4 ч после начала и через 6 ч после начала инсульта у крыс с помощью соединений согласно настоящему изобретению означает результат оценивания поведения крыс через 24 ч после возвращения крыс в сознание. Поведение включает в себя походку, степень опускания века правого глаза, степень жесткости хвоста, напряжение мышц, степень разгибания головы, поддержание силы конечностей и состояние смерти.
(2) Эффективность лечения через 24 ч после начала инсульта у крыс с помощью соединений согласно настоящему изобретению означает результат наблюдения за поведением крыс через 24 ч после возвращения крыс в сознание. Поведение включает в себя походку, степень опускания века правого глаза, степень жесткости хвоста, напряжение мышц, степень разгибания головы, поддержание силы конечностей и состояние смерти.
(3) Эффективность у крыс с инсультом, получавших только соединение согласно настоящему изобретению, по сравнению с эффективностью у крыс с инсультом, получавших только солевой раствор.
(4) Крысам с инсультом при непрерывном лечении вводили соединения согласно настоящему изобретению в физиологическом растворе каждые 24 ч через хвостовую вену. На следующий день записывали видео и производили сравнение среди записанных результатов.
Результаты исследований соединений Ia-Il согласно настоящему изобретению на вышеуказанных животных моделях следующие:
Экспериментальный пример 6. Эксперименты на крысах, получавших немедленное лечение после начала инсульта в виде соединений Ia-Il согласно настоящему изобретению
Противоинсультная активность in vivo согласно настоящему изобретению представляли с помощью оценки нервной функции с более низкой оценкой, указывающей на более высокую эффективность. 10%-ный раствор хлоралгидрата (400 мг/кг) вводили внутрибрюшинно крысам-самцам SD (250-300 g) для анестезии. Открытый разрез 2 см в длину делали в продольном направлении немного справа от центра шеи, и ствол правой общей сонной артерии, наружной сонной артерии и внутренней сонной артерии рассекали по краю внутренней стороны грудино-ключично-сосцевидной мышцы. Открытые разрезы во внутренней сонной артерии и проксимальном конце общей сонной артерии пережимали соответственно неинвазивными артериальными зажимами. Небольшой открытый надрез делали через наружную сонную артерию и дистальный конец наружной сонной артерии лигировали. Артериальный зажим на проксимальном конце наружной сонной артерии отпускали, и 10 мкл крови отбирали до повторного пережимания проксимального конца общей сонной артерии неинвазивным артериальным зажимом. 10 мкл отобранной крови помещали во флакон ЕР объемом 1 мл и хранили при комнатной температуре в течение 30 мин для коагуляции крови, а затем переносили в холодильник при -20°С в течение 1 ч, чтобы сделать возможной коагуляцию. Через 1 ч сгустки крови отбирали, в которые добавляли 1 мл физиологического раствора, а затем разбивали на однообразные микротромбы с использованием стального шпателя. Суспензию микротромбов затем переносили в шприц объемом 1 мл до использования. В то же время, когда отпускали зажим на внутренней сонной артерии крысы, 1 мл суспензии тромба в шприце медленно вводили в наружную сонную артерию крысы в ее проксимальном конце, и суспензию вводили в мозг крысы через внутреннюю сонную артерию. Впоследствии, проксимальный конец наружной сонной артерии лигировали, артериальные зажимы на внутренней сонной артерии и общей сонной артерии высвобождали и поток крови восстанавливали. Общую яремную вену крыс вскрывали. Вену сразу лигировали, 3 капли пенициллина капали в месте раны, рану зашивали, и животным позволяли прийти в себя в виде имитации операционной группы. Или выполняли инъекцию урокиназы в физиологическом растворе (положительная контрольная группа, в дозе 20000 МЕ/кг), физиологического раствора (нейтральная группа, в дозе 3 мл/кг), TMMZ в физиологическом растворе (контрольная группа компонента, в дозе 1 мкмоль/кг), тромболитического пептида ARPAK, GRPAK, RPAK или PAK в физиологическом растворе (контрольная группа компонента, в дозе 1 мкмоль/кг) или одного из соединений Ia-Il в физиологическом растворе (в дозе 0,1 мкмоль/кг). Через 24 ч после пробуждения крыс степень повреждения нервной функции оценивали по способу Zealonga. Балл 0 указывал на отсутствие признаков потери нервной функции, 1 указывал на то, что передние конечности на неповрежденной стороне не могут вытягиваться, 2 указывал на ходьбу в сторону неповрежденной стороны, 3 указывал на верчение по кругу в сторону неповрежденной стороны, 4 указывал на непроизвольную ходьбу с нарушением сознания и 5 указывал на смерть. Результаты эксперимента приведены в таблице 6.
Как показано в экспериментальных результатах, соединения Ia-Ic, полученные путем связывания тромболитического пептида ARPAK, GRPAK, RPAK или PAK и пептида с направленным действием RGDS, RGDV или RGDF с акцептором свободных радикалов TMMZ через Lys, проявляли противоинсультную активность при дозировке 0,1 мкмоль/кг, в то время как урокиназа не проявляла противоинсультную активность в дозе 20000 МЕ/кг. Аналогичным образом, тромболитический пептид ARPAK, GRPAK, RPAK или PAK не проявлял противоинсультную активность в дозе 1 мкмоль/кг. Таким образом, с помощью связывания тромболитического пептида ARPAK, GRPAK, RPAK или PAK и пептида с направленным действием RGDS, RGDV или RGDF с акцептором свободных радикалов TMMZ через Lys, соединения были снабжены противоинсультной функцией. Специально, в дозе 1 мкмоль/кг, 4 соединения характеризовались противоинсультной активностью, сравнимой с урокиназой в дозе 20000 МЕ/кг, и 8 соединений характеризовались значительно более высокой противоинсультной активностью, чем урокиназа в дозе 20000 МЕ/кг.
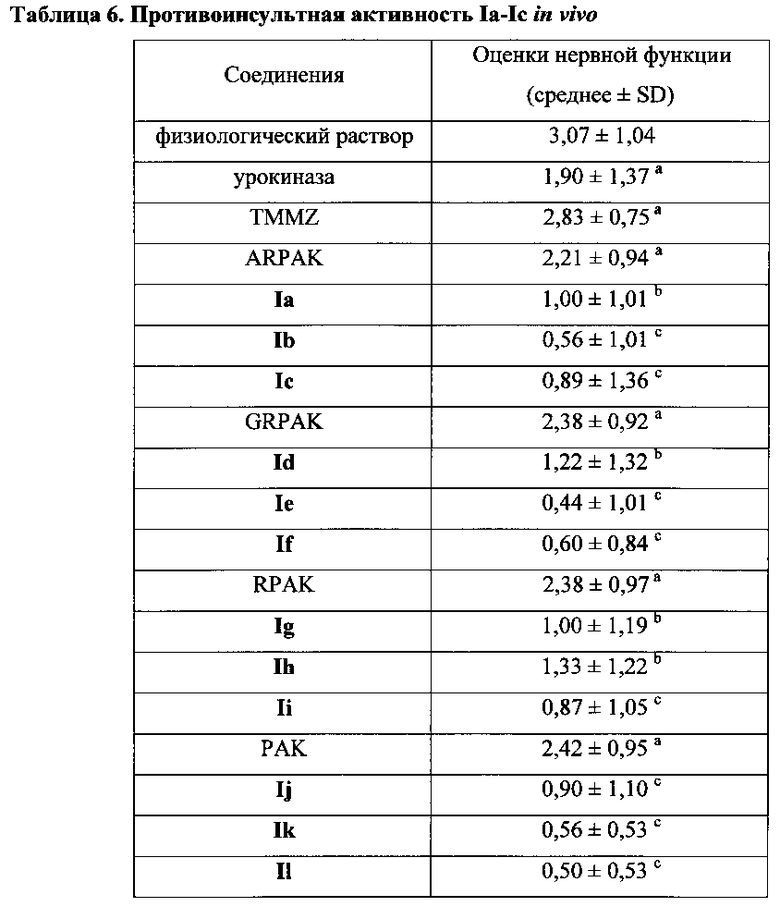
n=10; a) p>0,05 по отношению к физиологическому раствору; b) p>0,05 по отношению к урокиназе; p<0,01 по отношению к физиологическому раствору; с) p<0,01 по отношению к физиологическому раствору или урокиназе
Экспериментальный пример 7. Эксперименты по объему ишемического инсульта у крыс, которые получали немедленное лечение соединениями Ia-Il согласно настоящему изобретению после начала инсульта
После того, как крысы находились в сознании в течение 24 ч и их оценивали по степени повреждения нервной функции в экспериментальном примере 6, их анестезировали уретаном с последующей немедленной декапитацией и удалением мозга. Ткани мозга хранили при -20°С холодильнике в течение 2 ч, и фронтальные срезы около 2 мм последовательно нарезали от префронтального конца, производя в общей сложности шесть срезов, а затем помещали в 2% раствор ТТС, чтобы инкубировать в темноте при 37°С в течение 30 мин. Наблюдалось изменение цвета в срезах мозга: нормальные ткани мозга окрашивались в красный цвет с помощью ТТС, в то время как ишемические ткани мозга, т.е. ткани мозга с инфарктами, проявлялись в белом цвете. Фотографии делали с использованием цифровой камеры и обрабатывали с помощью программного обеспечения SPSS Statistics, и рассчитывали объем инфаркта в тканях головного мозга и объем нормальных тканей мозга во фронтальных срезах. Результаты эксперимента приведены в таблице 7.
Как показано в экспериментальных результатах, не только соединения Ia-Ic, полученные путем связывания тромболитического пептида ARPAK, GRPAK, RPAK или PAK и пептида с направленным действием RGDS, RGDV или RGDF с акцептором свободных радикалов TMMZ через Lys проявляли эффект в уменьшении объема ишемического инсульта у крыс с инсультом в дозе 0,1 мкмоль/кг, такой эффект был значительно более мощным, чем у урокиназы в дозе 20000 МЕ/кг.
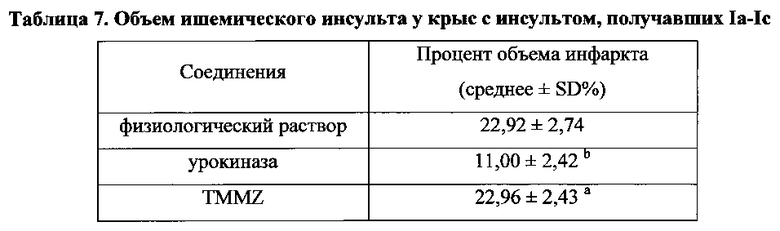
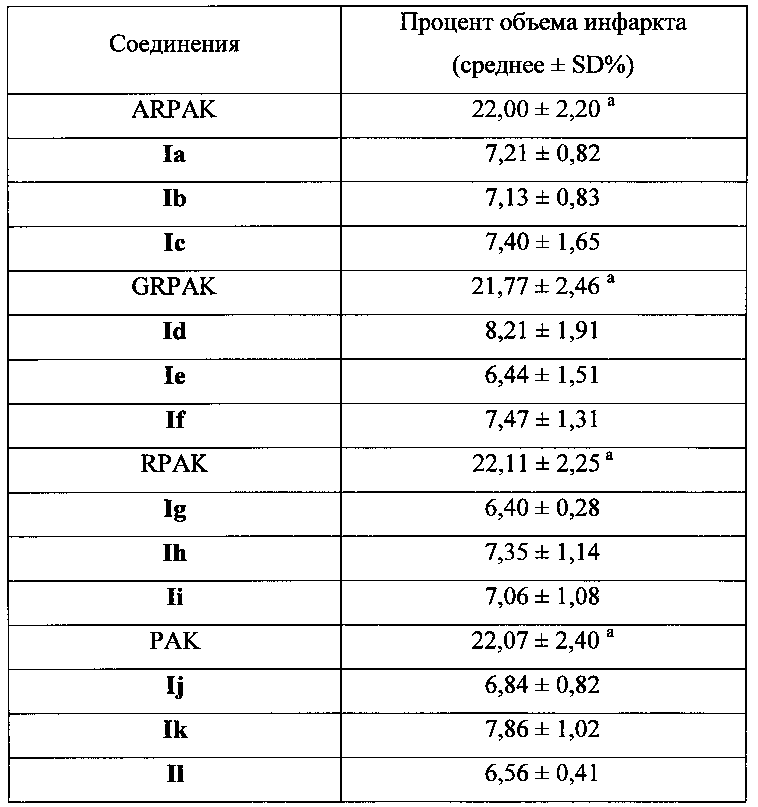
n=10; a) p>0,05 по отношению к физиологическому раствору; b) p<0,01 по отношению к физиологическому раствору и Ia-Ic
Экспериментальный пример 8. Эксперименты на крысах, получавших немедленное лечение различными дозами соединения Ie согласно настоящему изобретению после начала инсульта
После анализа и сравнения всех экспериментальных результатов в настоящем изобретении, соединение Ie использовали в качестве эталонного, с тем чтобы продемонстрировать зависимый от дозы терапевтический эффект, проявляемый соединениями Ia-Il в приведенных выше экспериментах. Следует отметить, что другие соединения Ia-Il могли достигать подобного зависимого от дозы терапевтического эффекта, что и соединение Ie, поскольку другие соединения Ia-Il достигли того же эффекта, что и соединение Ie в акцептировании свободных радикалов NO, лизисе эуглобулиновых сгустков, тромболизисе, антитромботическом действии и лечении инсульта у крыс.
10%-ный раствор хлоралгидрата (400 мг/кг) вводили внутрибрюшинно крысам-самцам SD (250-300 g) для анестезии. Открытый разрез 2 см в длину делали в продольном направлении немного справа от центра шеи, и ствол правой общей сонной артерии, наружной сонной артерии и внутренней сонной артерии рассекали по краю внутренней стороны грудино-ключично-сосцевидной мышцы. Открытые разрезы во внутренней сонной артерии и проксимальном конце общей сонной артерии пережимали, соответственно, неинвазивными артериальными зажимами. Небольшой открытый надрез делали через наружную сонную артерию и дистальный конец наружной сонной артерии лигировали. Артериальный зажим на проксимальном конце наружной сонной артерии отпускали и 10 мкл крови отбирали до повторного пережимания проксимального конца общей сонной артерии неинвазивным артериальным зажимом. 10 мкл отобранной крови помещали во флакон ЕР объемом 1 мл и хранили при комнатной температуре в течение 30 мин до коагуляции крови, а затем переносили в холодильник при -20°С на 1 ч, чтобы сделать возможной твердую коагуляцию. Через 1 ч сгустки крови отбирали, добавляли в 1 мл физиологического раствора, а затем разбивали на однообразные микротромбы с использованием стального шпателя. Суспензию микротромбов затем переносили в шприц объемом 1 мл до использования. В то же время, когда отпускали зажим на внутренней сонной артерии крысы, 1 мл суспензии тромба в шприце медленно вводили в наружную сонную артерию крысы в ее проксимальном конце, и суспензию вводили в мозг крысы через внутреннюю сонную артерию. Впоследствии, проксимальный конец наружной сонной артерии лигировали, артериальные зажимы на внутренней сонной артерии и общей сонной артерии освобождали и поток крови восстанавливали. Проводили введение урокиназы в физиологическом растворе (положительная контрольная группа, в дозе 20000 МЕ/кг), tPA в физиологическом растворе (положительная контрольная группа, в дозе 3 мг/кг), физиологического раствора (нейтральная группа, в дозе 3 мл/кг) или соединения Ie в физиологическом растворе (в дозе 1 мкмоль/кг, 0,1 мкмоль/кг или 0,01 мкмоль/кг). Через 24 ч после пробуждения крыс степень повреждения нервной функции оценивали по способу Zealonga. Оценка 0 указывала на отсутствие признаков потери нервной функции, 1 указывала на то, что передние конечности на неповрежденной стороне не могут вытягиваться, 2 указывала на ходьбу в сторону неповрежденной стороны, 3 указывала на верчение по кругу в сторону неповрежденной стороны, 4 указывала на непроизвольную ходьбу с нарушением сознания и 5 указывала на смерть. Результаты эксперимента приведены в таблице 8. Как показано в результатах, у крыс, получавших немедленное лечение после начала инсульта в дозе 1 мкмоль/кг, 0,1 мкмоль/кг и 0,01 мкмоль/кг соединения Ie, процент крыс с оценкой 0 нервной функции составлял 60%, 30% и 0% соответственно; и процент крыс с оценкой 1 нервной функции составлял 20%, 30% и 10%, соответственно. Таким образом, это показывает, что противоинсультная активность соединения Ie зависит от дозы. Кроме того, у крыс с инсультом, обработанных урокиназой в дозе 20000 МЕ/кг и tPA в дозе 3 мг/кг, процент крыс с оценкой 0 нервной функции составлял 10% и 40%, соответственно, а процент крыс с оценкой 1 нервной функции составлял 50% и 10%, соответственно; в сравнении, эффективность 1 мкмоль/кг и 0,1 мкмоль/кг соединения 1е была, очевидно, лучше.
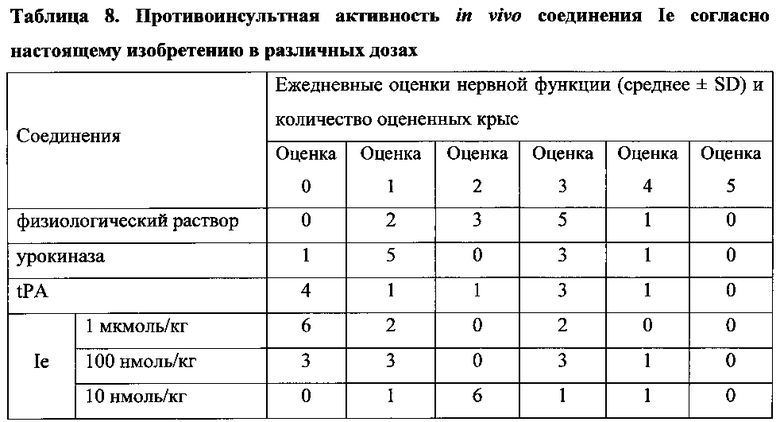
n=10;а) p<0,01 по отношению к физиологическому раствору
Экспериментальный пример 9. Эксперименты на крысах, получающих 6 последовательных воздействий соединением Ie согласно настоящему изобретению в дозе 1 мкмоль/кг через 4 часа после начала инсульта
Эффективность представляли с помощью оценки нервной функции, и более низкая оценка указывает на более высокую эффективность. 10%-ный раствор хлоралгидрата внутрибрюшинно вводили самцам крыс SD в дозе 400 мг/кг массы тела для анестезии. Делали продольный открытый надрез в центре шеи и правый общий ствол сонной артерии рассекали (около 3 см в длину). Каждую наружную ветвь сонной артерии рассекали и лигировали на подъязычном уровне, и внутреннюю сонную артерию рассекали в опухшей части шеи. Открытые разрезы во внутренней сонной артерии и проксимальном конце общей сонной артерии пережимали соответственно неинвазивными артериальными зажимами, и дистальный конец наружной сонной артерии лигировали. Катетер, содержащий 0,5 мл суспензии тромба в физиологическом растворе, вставляли в ствол наружной сонной артерии. В то же время, когда освобождали зажим на внутренней сонной артерии, 0,5 мл суспензии тромба в физиологическом растворе в катетере медленно протекало из наружной сонной артерии к ее проксимальному концу, а затем вводилось в артерии в мозге через внутреннюю сонную артерию. Впоследствии, проксимальный конец внутренней сонной артерии лигировали, артериальные зажимы на внутренней сонной артерии и общей сонной артерии высвобождали и поток крови восстанавливали. После зашивания раны вводили 20000 ME пенициллина внутримышечно для профилактики заражения. Через 4 ч соединение 1е в физиологическом растворе (в дозе 1 мкмоль/кг, n=11), урокиназу в физиологическом растворе (в дозе 20000 МЕ/кг, n=6) или tPA в физиологическом растворе (в дозе 3 мг/кг, n=6) вводили через хвостовую вену. Инфузию соединения 1е в физиологическом растворе через хвостовую вену крысы проводили один раз в день в течение 6 дней подряд, наблюдали в течение 7 дней. Крыс сравнивали с собой каждый день и оценивали на степень повреждения нервной функции способом Zealonga. Альтернативно, инфузию урокиназы в физиологическом растворе через хвостовую вену крысы проводили один раз в день в течение двух последовательных дней, крыс сравнивали с собой каждый день и оценивали на степень повреждения нервной функции способом Zealonga. Альтернативно, инфузию tPA в физиологическом растворе через хвостовую вену крысы проводили один раз в день в течение двух последовательных дней, крыс сравнивали с собой каждый день и оценивали на степень повреждения нервной функции способом Zealonga. Оценка 0 указывала на отсутствие признаков потери нервной функции, 1 указывала на то, что передние конечности на неповрежденной стороне не могут вытягиваться, 2 указывала на ходьбу в сторону неповрежденной стороны, 3 указывала на верчение по кругу в сторону неповрежденной стороны, 4 указывала на непроизвольную ходьбу с нарушением сознания и 5 указывала на смерть. Результаты эксперимента приведены в таблицах 9-1, 9-2 и 9-3.
Приведенные в таблице 9-1 данные показали, что у крыс, получавших лечение через 4 ч после начала инсульта в виде одной дозы соединения 1е в концентрации 1 мкмоль/кг каждый день в течение 6 дней подряд, за исключением одной, которая случайно погибла на 2 день, 8 из оставшихся 10 крыс восстановились до отсутствия любых признаков потери нервной функции, в то время как остальные 2 крысы характеризовались только признаком небольшой потери нервной функции. Таким образом, соединение Ie проявляло терапевтический эффект в дозе 1 мкмоль/кг при инсульте за пределами золотого терапевтического окна.
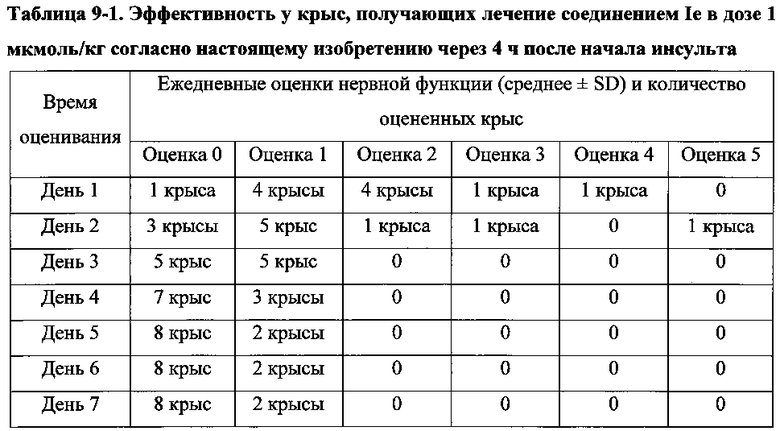
Приведенные в таблице 9-2 данные показали, что у крыс, получавших лечение через 4 ч после начала инсульта в виде одной дозы урокиназы в концентрации 20000 МЕ/кг каждый день, 2 из 6 крыс погибли в течение 48 ч. При вскрытии мертвых крыс было показано кровоизлияние во внутренних органах, в частности тяжелое кровоизлияние в легких. Таким образом, режим дозирования был прекращен после двух доз. Никакие крысы после приема двух доз не восстанавливались до отсутствия признаков потери нервной функции или наличия только признака небольшой потери нервной функции.

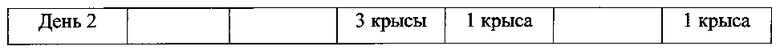
Данные, приведенные в таблице 9-3 показали, что у крыс, которые получали лечение через 4 ч после начала инсульта в виде одной дозы tPA в концентрации 3 мг/кг каждый день, 1 из 6 крыс погибла в течение 24 ч. При вскрытии мертвой крысы было показано кровоизлияние во внутренних органах, в частности тяжелое кровоизлияние в легких. Таким образом, режим дозирования был прекращен после двух доз. Никакие крысы после приема двух доз не восстанавливались до отсутствия признаков потери нервной функции и только 2 крысы восстанавливались до наличия признака небольшой потери нервной функции.
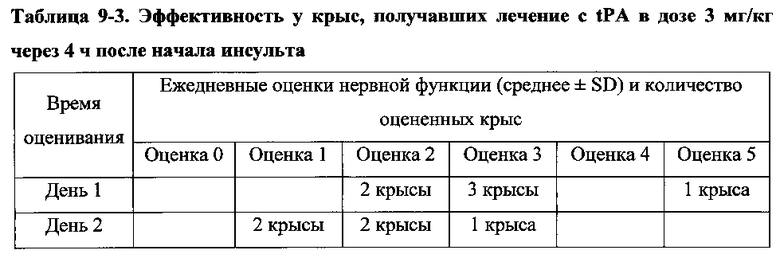
Подводя итог относительно данных в таблице 9-1, 9-2 и 9-3, даже для крыс, получавших лечение через 4 ч после начала инсульта в течение двух дней подряд, соединение Ie в дозе 1 мкмоль/кг показало намного более высокую эффективность, чем урокиназа в дозе 20000 МЕ/кг и tPA в дозе 3 мг/кг.
Экспериментальный пример 10. Эксперименты на крысах, получающих 6 последовательных воздействий соединением Ie согласно настоящему изобретению в дозе 1 мкмоль/кг через 6 ч после начала инсульта
Эффективность представляли с помощью оценки нервной функции, и более низкая оценка указывает на более высокую эффективность. 10%-ный раствор хлоралгидрата вводили внутрибрюшинно самцам крыс SD в дозе 400 мг/кг массы тела для анестезии. Делали продольный открытый надрез в центре шеи и правый общий ствол сонной артерии рассекали (около 3 см в длину). Каждую наружную ветвь сонной артерии рассекали и лигировали на подъязычном уровне, и внутреннюю сонную артерию рассекали в опухшей части шеи. Открытый разрез во внутренней сонной артерии и проксимальный конец общей сонной артерии пережимали соответственно неинвазивными артериальными зажимами, и дистальный конец наружной сонной артерии лигировали. Катетер, содержащий 0,5 мл суспензии тромба в физиологическом растворе, вставляли в ствол наружной сонной артерии. В то же время, когда освобождали зажим на внутренней сонной артерии, 0,5 мл суспензии тромба в физиологическом растворе в катетере медленно протекало из наружной сонной артерии к ее проксимальному концу, а затем вводилось в артерии в мозге через внутреннюю сонную артерию. Впоследствии, проксимальный конец внутренней сонной артерии лигировали, артериальные зажимы на внутренней сонной артерии и общей сонной артерии высвобождали и поток крови восстанавливали. После зашивания раны вводили 20000 ME пенициллина внутримышечно для профилактики заражения. Через 6 ч соединение 1е в физиологическом растворе (в дозе 1 мкмоль/кг, n=11), урокиназу в физиологическом растворе (в дозе 20000 МЕ/кг, n=6) или tPA в физиологическом растворе (в дозе 3 мг/кг, n=6) вводили через хвостовую вену. Инфузию соединения Ie в физиологическом растворе через хвостовую вену крысы проводили один раз в день в течение 6 дней подряд, наблюдали в течение 7 дней. Крыс сравнивали с собой каждый день и оценивали на степень повреждения нервной функции способом Zealonga. Альтернативно, инфузию урокиназы в физиологическом растворе через хвостовую вену крысы проводили один раз в день в течение двух дней подряд, крыс сравнивали с собой каждый день и оценивали на степень повреждения нервной функции способом Zealonga. Альтернативно, инфузию tPA в физиологическом растворе через хвостовую вену крысы проводили один раз в день в течение двух дней подряд, крыс сравнивали с собой каждый день и оценивали на степень повреждения нервной функции способом Zealonga. Оценка 0 указывала на отсутствие признаков потери нервной функции, 1 указывала на то, что передние конечности на неповрежденной стороне не могут вытягиваться, 2 указывала на ходьбу в сторону неповрежденной стороны, 3 указывала на верчение по кругу в сторону неповрежденной стороны, 4 указывала на непроизвольную ходьбу с нарушением сознания и 5 указывала на смерть. Результаты эксперимента приведены в таблицах 10-1,10-2 и 10-3.
Приведенные в таблице 10-1 данные показали, что у крыс, получавших лечение через 6 ч после начала инсульта в виде одной дозы соединения Ie в концентрации 1 мкмоль/кг каждый день в течение 6 дней подряд, за исключением двух, которые случайно погибли на 2 день, 2 из оставшихся 9 крыс восстановились до отсутствия любых признаков потери нервной функции, одна крыса характеризовались только признаком небольшой потери нервной функции и 6 демонстрировали признак верчения по кругу в сторону неповрежденной стороны. Таким образом, соединение Ie проявляло терапевтический эффект в дозе 1 мкмоль/кг при инсульте за пределами золотого терапевтического окна.
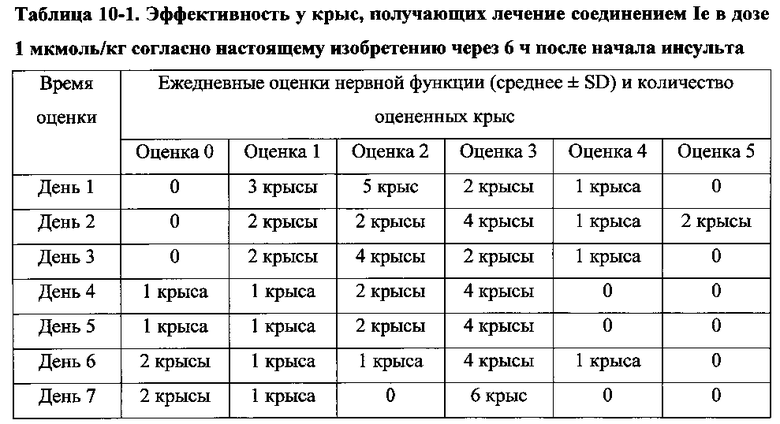
Приведенные в таблице 10-2 данные показали, что у крыс, получавших лечение через 6 ч после начала инсульта с одной дозой урокиназы в концентрации 20000 МЕ/кг каждый день, 4 из 6 крыс погибли в течение 24 ч. При вскрытии мертвых крыс было показано кровоизлияние во внутренних органах, в частности тяжелое кровоизлияние в легких. Таким образом, режим дозирования был прекращен после двух доз. Никакие крысы после приема двух доз не восстанавливались до отсутствия признаков потери нервной функции или наличия только признака небольшой потери нервной функции.
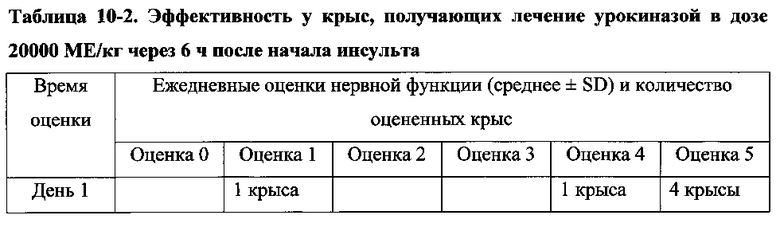

Данные, приведенные в таблице 10-3 показали, что у крыс, которые получали лечение через 6 ч после начала инсульта в виде одной дозы tPA в концентрации 3 мг/кг каждый день, 2 из 6 крыс погибли в течение 24 ч. При вскрытии мертвых крыс было показано кровоизлияние во внутренних органах, в частности тяжелое кровоизлияние в легких. Таким образом, режим дозирования был прекращен после двух доз. Никакие крысы после приема двух доз не восстанавливались до отсутствия признаков потери нервной функции, 2 крысы восстанавливались до наличия только признака небольшой потери нервной функции, одна крыса демонстрировала признак верчения по кругу в сторону неповрежденной стороны и одна крыса демонстрировала непроизвольную ходьбу с нарушением сознания.
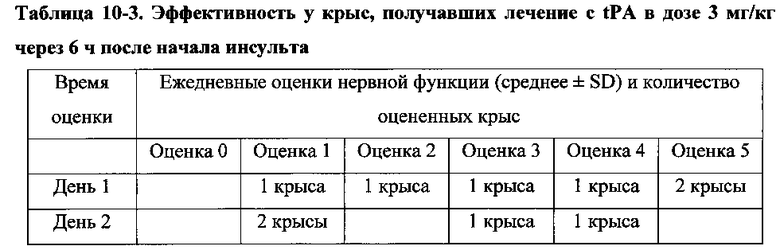
Подводя итог относительно данных в таблице 10-1, 10-2 и 10-3, даже для крыс, получавших лечение через 6 ч после начала инсульта в течение двух дней подряд, соединение 1е в дозе 1 мкмоль/кг показало намного более высокую эффективность, чем урокиназа в дозе 20000 МЕ/кг и tPA в дозе 3 мг/кг.
Экспериментальный пример 11. Эксперименты на крысах, получающих лечение через 6 ч после начала инсульта соединением Ie согласно настоящему изобретению с начальной дозой 5 мкмоль/кг и 5 последующими дозами по 2 мкмоль/кг каждая
Эффективность представляли с помощью оценки нервной функции, и более низкая оценка указывает на более высокую эффективность. 10%-ный раствор хлоралгидрата внутрибрюшинно вводили самцам крыс SD в дозе 400 мг/кг массы тела для анестезии. Делали продольный открытый надрез в центре шеи и правый общий ствол сонной артерии рассекали (около 3 см в длину). Каждую наружную ветвь сонной артерии рассекали и лигировали на подъязычном уровне, и внутреннюю сонную артерию рассекали в опухшей части шеи. Открытый разрез во внутренней сонной артерии и проксимальный конец общей сонной артерии пережимали соответственно неинвазивными артериальными зажимами, и дистальный конец наружной сонной артерии лигировали. Катетер, содержащий 0,5 мл суспензии тромба в физиологическом растворе, вставляли в ствол наружной сонной артерии. В то же время, когда освобождали зажим на внутренней сонной артерии, 0,5 мл суспензии тромба в физиологическом растворе в катетере медленно протекало из наружной сонной артерии к ее проксимальному концу, а затем вводилось в артерии в мозге через внутреннюю сонную артерию. Впоследствии, проксимальный конец внутренней сонной артерии лигировали, артериальные зажимы на внутренней сонной артерии и общей сонной артерии высвобождали и поток крови восстанавливали. После зашивания раны вводили 20000 ME пенициллина внутримышечно для профилактики заражения. Через 6 ч соединение Ie в физиологическом растворе (в начальной дозе 5 мкмоль/кг, n=12) вводили через хвостовую вену. Затем инфузию соединения Iе в физиологическом растворе (в дозе 2 мкмоль/кг, n=12) через хвостовую вену крысы проводили один раз в день в течение 6 дней подряд, наблюдаемых в течение 7 дней. Крыс сравнивали с собой каждый день и оценивали на степень повреждения нервной функции способом Zealonga. Оценка 0 указывала на отсутствие признаков потери нервной функции, 1 указывала на то, что передние конечности на неповрежденной стороне не могут вытягиваться, 2 указывала на ходьбу в сторону неповрежденной стороны, 3 указывала на верчение по кругу в сторону неповрежденной стороны, 4 указывала на непроизвольную ходьбу с нарушением сознания и 5 указывала на смерть. Результаты эксперимента приведены в таблице 11.
Приведенные в таблице 11 данные показали, что эффективность была показана у крыс, получавших лечение через 6 ч после начала инсульта одной дозой соединения Ie в концентрации 5 мкмоль/кг в первый день и одной дозой соединения Ie в концентрации 2 мкмоль/кг в день в течение 5 дней. Среди 12 получавших лечение крыс две погибли, в то время как 6 из оставшихся 10 крыс восстановились до отсутствия любых признаков потери нервной функции, две характеризовались только признаком небольшой потери нервной функции, одна демонстрировала признак ходьбы в сторону неповрежденной стороны и одна демонстрировала признак верчения по кругу в сторону неповрежденной стороны. Таким образом, продолжительное лечение соединением Ie показало терапевтический эффект при инсульте за пределами золотого терапевтического окна.
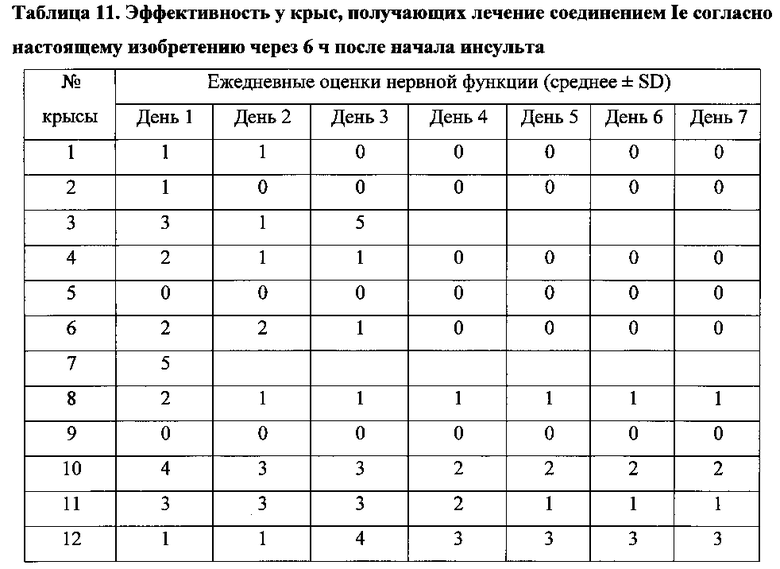
Экспериментальный пример 12. Эксперименты на крысах, получающих лечение через 24 ч после начала инсульта соединением Ie согласно настоящему изобретению с начальной дозой 5 мкмоль/кг и 5 последующими дозами по 2 мкмоль/кг каждая
Эффективность представляли с помощью оценки нервной функции, и более низкая оценка указывает на более высокую эффективность. 10%-ный раствор хлоралгидрата вводили внутрибрюшинно самцам крыс SD в дозе 400 мг/кг массы тела для анестезии. Делали продольный открытый надрез в центре шеи и правый общий ствол сонной артерии рассекали (около 3 см в длину). Каждую наружную ветвь сонной артерии рассекали и лигировали на подъязычном уровне, и внутреннюю сонную артерию рассекали в опухшей части шеи. Открытые разрезы во внутренней сонной артерии и проксимальном конце общей сонной артерии пережимали соответственно неинвазивными артериальными зажимами, и дистальный конец наружной сонной артерии лигировали. Катетер, содержащий 0,5 мл суспензии тромба в физиологическом растворе, вставляли в ствол наружной сонной артерии. В то же время, когда освобождали зажим на внутренней сонной артерии, 0,5 мл суспензии тромба в физиологическом растворе в катетере медленно протекало из наружной сонной артерии к ее проксимальному концу, а затем вводилось в артерии в мозге через внутреннюю сонную артерию. Впоследствии, проксимальный конец внутренней сонной артерии лигировали, артериальные зажимы на внутренней сонной артерии и общей сонной артерии высвобождали и поток крови восстанавливали. После зашивания раны вводили 20000 ME пенициллина внутримышечно для профилактики заражения. Через 24 ч соединение Ie в физиологическом растворе (в начальной дозе 5 мкмоль/кг, n=12) вводили через хвостовую вену. Затем инфузию соединения Ie в физиологическом растворе (в дозе 2 мкмоль/кг, n=12) через хвостовую вену крысы проводили один раз в день в течение 6 дней подряд, наблюдаемых в течение 7 дней. Крыс сравнивали с собой каждый день и оценивали на степень повреждения нервной функции способом Zealonga. Оценка 0 указывала на отсутствие признаков потери нервной функции, 1 указывала на то, что передние конечности на неповрежденной стороне не могут вытягиваться, 2 указывала на ходьбу в сторону неповрежденной стороны, 3 указывала на верчение по кругу в сторону неповрежденной стороны, 4 указывала на непроизвольную ходьбу с нарушением сознания и 5 указывала на смерть. Результаты эксперимента приведены в таблице 12.
Приведенные в таблице 12 данные показали, что эффективность была показана у крыс, получавших лечение через 24 ч после начала инсульта одной дозой соединения Iе в концентрации 5 мкмоль/кг в первый день и одной дозой соединения Ie в концентрации 2 мкмоль/кг в день в течение 5 дней. Среди 12 получавших лечение крыс три погибли, в то время как 8 из оставшихся 9 крыс восстановились до отсутствия любых признаков потери нервной функции и одна характеризовалась только признаком небольшой потери нервной функции. Таким образом, продолжительное лечение соединением Ie показало терапевтический эффект при инсульте за пределами золотого терапевтического окна.
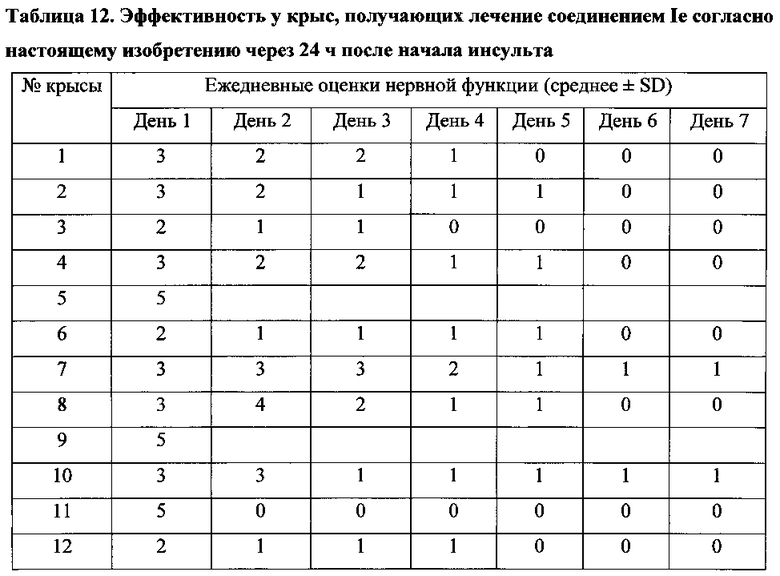
Экспериментальный пример 13. Эксперименты на крысах, получающих лечение в виде 6 последовательных введений соединения Ie согласно настоящему изобретению в дозе 2 мкмоль/кг через 6 ч после начала инсульта
Эффективность представляли с помощью оценки нервной функции, и более низкая оценка указывает на более высокую эффективность. 10%-ный раствор хлоралгидрата вводили внутрибрюшинно самцам крыс SD в дозе 400 мг/кг массы тела для анестезии. Делали продольный открытый надрез в центре шеи и правый общий ствол сонной артерии рассекали (около 3 см в длину). Каждую наружную ветвь сонной артерии рассекали и лигировали на подъязычном уровне, и внутреннюю сонную артерию рассекали в опухшей части шеи. Открытые разрезы во внутренней сонной артерии и проксимальном конце общей сонной артерии пережимали соответственно неинвазивными артериальными зажимами, и дистальный конец наружной сонной артерии лигировали. Катетер, содержащий 0,5 мл суспензии тромба в физиологическом растворе, вставляли в ствол наружной сонной артерии. В то же время, когда освобождали зажим на внутренней сонной артерии, 0,5 мл суспензии тромба в физиологическом растворе в катетере медленно протекало из наружной сонной артерии к ее проксимальному концу, а затем вводилось в артерии в мозге через внутреннюю сонную артерию. В отличие от предыдущих экспериментальных примеров, используемые в этом экспериментальном примере тромбовые сгустки представляли собой суспензию удивительно твердого тромба в физиологическом растворе, полученного при использовании более выдержанного тромба, хранившегося при комнатной температуре в течение 24 ч, вместо суспензии тромба в физиологическом растворе, полученного с использованием тромба, хранившегося при -24°С. Впоследствии, проксимальный конец внутренней сонной артерии лигировали, артериальные зажимы на внутренней сонной артерии и общей сонной артерии высвобождали и поток крови восстанавливали. После зашивания раны вводили 20000 ME пенициллина внутримышечно для профилактики заражения. Через 6 ч соединение Ie в физиологическом растворе (в начальной дозе 5 мкмоль/кг, n=12) вводили через хвостовую вену. Затем инфузию соединения Ie в физиологическом растворе (в дозе 2 мкмоль/кг, n=12) через хвостовую вену крысы проводили один раз в день в течение 6 дней подряд, наблюдаемых в течение 7 дней. Крыс сравнивали с собой каждый день и оценивали на степень повреждения нервной функции способом Zealonga. Оценка 0 указывала на отсутствие признаков потери нервной функции, 1 указывала на то, что передние конечности на неповрежденной стороне не могут вытягиваться, 2 указывала на ходьбу в сторону неповрежденной стороны, 3 указывала на верчение по кругу в сторону неповрежденной стороны, 4 указывала на непроизвольную ходьбу с нарушением сознания и 5 указывала на смерть. Результаты эксперимента приведены в таблице 13.
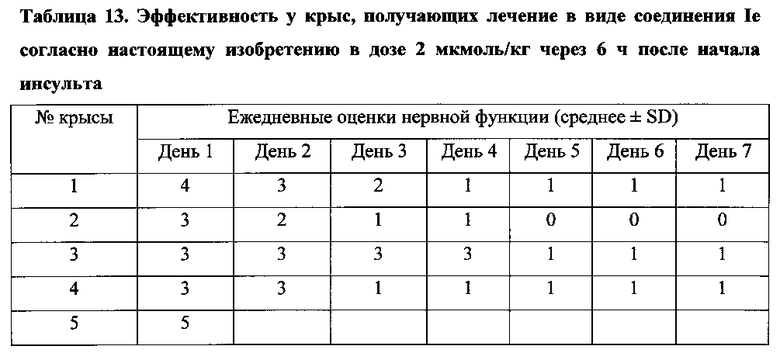
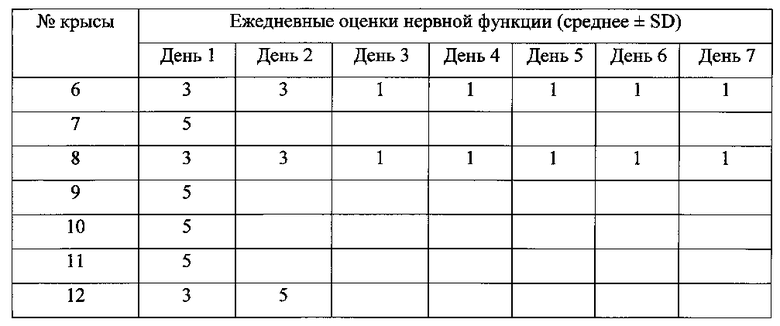
Приведенные в таблице 13 данные показали, что в крысиных моделях через 6 часов после начала индуцированного выдержанным тромбом инсульта эффективность была показана после 6 последовательных воздействий одной дозой соединения Ie в концентрации 2 мкмоль/кг в день в течение 6 дней подряд. Среди 12 получавших лечение крыс шесть погибли, в то время как 1 из оставшихся 6 крыс восстановилась до отсутствия любых признаков потери нервной функции и пять характеризовались только признаком небольшой потери нервной функции. Таким образом, продолжительное лечение соединением Ie показало терапевтический эффект при давнем инсульте.
Следует отметить, что, поскольку соединения Ia-Il, за исключением Ie, в экспериментальных примерах с 1 по 7 достигали эффектов в акцептировании свободных радикалов NO, лизисе эуглобулиновых сгустков, тромболизисе, антитромботическом действии и лечении крыс с инсультом, аналогично таковым соединения Ie, другие соединения Ia-Il могут достигать тех же терапевтических эффектов на давний инсульт, что и соединение Ie.
Экспериментальный пример 14. Эксперименты по наноструктурам соединений Ia-Il согласно настоящему изобретению в концентрации 1×10-6 М, 1×10-9 М и 1×10-12М
Соединения Ia-Il согласно настоящему изобретению получали в растворах с концентрацией 1×10-6 М, 1×10-9 М и 1×10-12 М, соответственно. 10 мкл раствора брали и капали на медную сетку с размещенной внизу фильтровальной бумагой, сушили на воздухе, а затем исследовали под трансмиссионным электронным микроскопом (ТЕМ) (JEOL, JEM-1230). Фотографии были сделаны таким образом, чтобы записать морфологию и размер частиц.
1. Исследуемое соединение: соединения Ia-Il согласно настоящему изобретению
2. Способ исследования: исследуемое соединение (Ia-Il) готовили в растворах с концентрацией 1×10-6 М, 1×10-9 М и 1×10-12 М с тройной дистиллированной водой, соответственно. Небольшое количество (около 10 мкл) брали и капали на медную сетку с размещенной внизу фильтровальной бумагой, сушили на воздухе, а затем исследовали под трансмиссионным электронным микроскопом (ТЕМ) (JEOL, JEM-1230). Фотографии были сделаны таким образом, чтобы записать морфологию и размер частиц.
3. Результаты исследований: результаты показаны на фиг. 25-36. На фиг. 25 представлены наноструктуры соединения Ia согласно настоящему изобретению в водных растворах с концентрацией 1×10-6 М, 1×10-9 М и 1×10-12 М, и наноструктуры Ia в водных растворах представляли собой наносферы с диаметром от 3,1 до 86,1 нм; на фиг. 26 представлены наноструктуры соединения Ib согласно настоящему изобретению в водных растворах с концентрацией 1×10-6 М, 1×10-9 М и 1×10-12 М, и наноструктуры Ib в водных растворах представляли собой наносферы с диаметром от 4,3 до 297,9 нм; на фиг. 27 представлены наноструктуры соединения Ic согласно настоящему изобретению в водных растворах с концентрацией 1×10-6 М, 1×10-9 М и 1×10-12 М, и наноструктуры Ic в водных растворах представляли собой наносферы с диаметром от 2,2 до 165,7 нм; на фиг. 28 представлены наноструктуры соединения Id согласно настоящему изобретению в водных растворах с концентрацией 1×10-6 М, 1×10-9 М и 1×10-12 М, и наноструктуры Id в водных растворах представляли собой наносферы с диаметром от 16,2 до 201,2 нм; на фиг. 29 представлены наноструктуры соединения Ie согласно настоящему изобретению в водных растворах с концентрацией 1×10-6 М, 1×10-9 М и 1×10-12 М, и наноструктуры Ie в водных растворах представляли собой наносферы с диаметром от 3,3 до 138,9 нм; на фиг. 30 представлены наноструктуры соединения If согласно настоящему изобретению в водных растворах с концентрацией 1×10-6 М, 1×10-9 М и 1×10-12 М, и наноструктуры If в водных растворах представляли собой наносферы с диаметром от 3,6 до 82,4 нм; на фиг. 31 представлены наноструктуры соединения Ig согласно настоящему изобретению в водных растворах с концентрацией 1×10-6 М, 1×10-9 М и 1×10-12 М, и наноструктуры Ig в водных растворах представляли собой наносферы с диаметром от 6,3 до 264,5 нм; на фиг. 32 представлены наноструктуры соединения Ih согласно настоящему изобретению в водных растворах с концентрацией 1×10-6 М, 1×10-9 М и 1×10-12 М, и наноструктуры Ih в водных растворах представляли собой наносферы с диаметром от 5,1 до 149,8 нм; на фиг. 33 представлены наноструктуры соединения Ii согласно настоящему изобретению в водных растворах с концентрацией 1×10-6 М, 1×10-9 М и 1×10-12 М, и наноструктуры Ii в водных растворах представляли собой наносферы с диаметром от 4,7 до 107,7 нм; на фиг. 34 представлены наноструктуры соединения Ij согласно настоящему изобретению в водных растворах с концентрацией 1×10-6 М, 1×10-9 М и 1×10-12 М, и наноструктуры Ij в водных растворах представляли собой наносферы с диаметром от 9,1 до 73,7 нм; на фиг. 35 представлены наноструктуры соединения Ik согласно настоящему изобретению в водных растворах с концентрацией 1×10-6 М, 1×10-9 М и 1×10-9 М, и наноструктуры Ik в водных растворах представляли собой наносферы с диаметром от 10,1 до 66,7 нм; на фиг. 36 представлены наноструктуры соединения Il согласно настоящему изобретению в водных растворах с концентрацией 1×10-6 М, 1×10-9 М и 1×10-12 М, и наноструктуры Il в водных растворах представляли собой наносферы с диаметром от 6,1 до 153,3 нм.
Экспериментальный пример 15. Эксперименты по FT-MS высокого разрешения соединений Ia-Il согласно настоящему изобретению в концентрациях 1×10-6 М, 1×10-9 М и 1×10-12 М
Соединения Ia-Il получали в растворе с концентрацией 12,5 мкМ с тройной дистиллированной водой, и образцы по 10 мкл наносили на масс-спектроскопию solariX FT-ICR (Bruker Daltonik). Наблюдали статус межмолекулярной ассоциации и получали данные. Результаты приведены в таблицах 14-16.
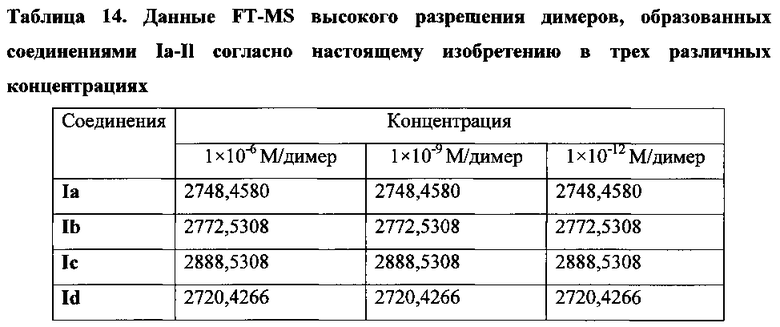
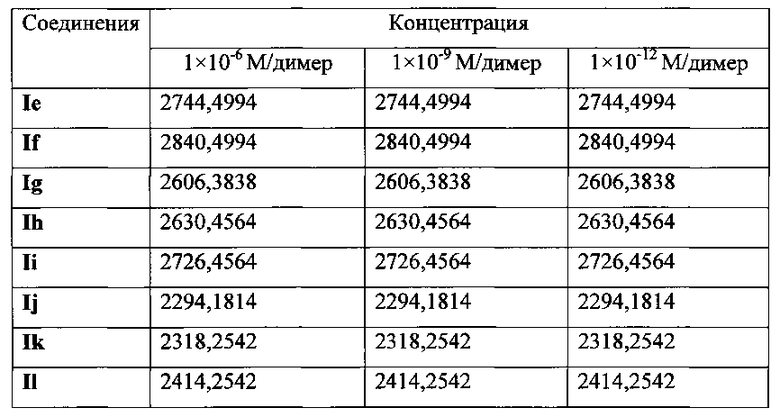
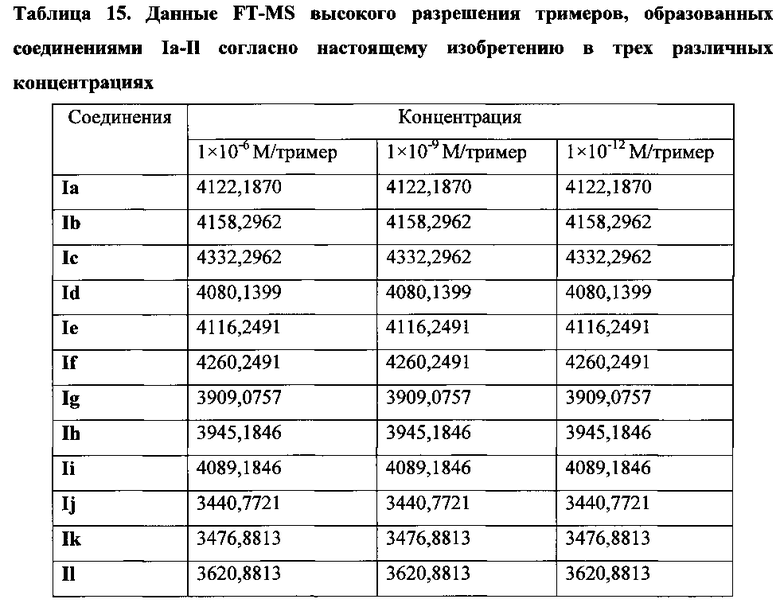

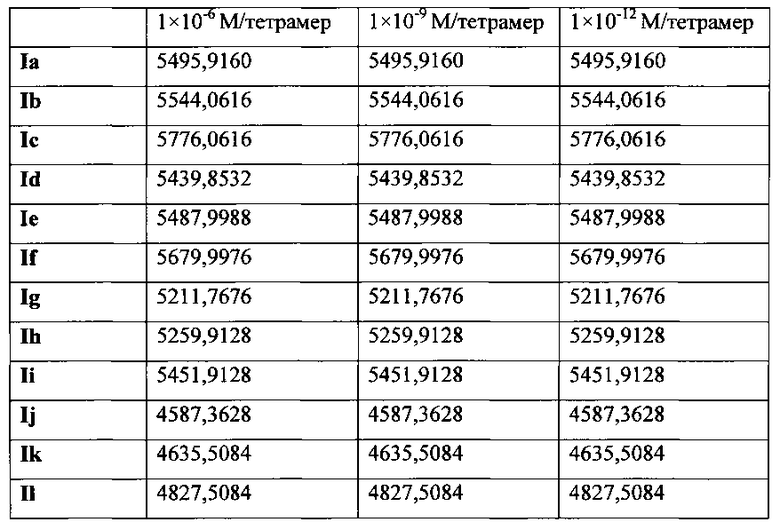
Таблицы 14-16 показывают точные массовые числа, измеренные масс-спектрометрией с Фурье-преобразованием высокого разрешения. Эти массовые числа указывают на то, что димеры, тримеры и тетрамеры были обнаружены в трех различных концентрациях соединений Ia-Il согласно настоящему изобретению. Таким образом, соединения согласно настоящему изобретению могут образовывать димеры, тримеры и тетрамеры в водном растворе в одно и то же время.
Экспериментальный пример 16. Эксперименты по FT-MS высокого разрешения соединения 1е согласно настоящему изобретению в концентрациях 10,0 мкМ, 1,0 мкМ, 0,1 мкМ и 0,01 мкМ
Для МС визуализации соединение 1е получали в растворах с концентрацией 10,0 мкМ, 1,0 мкМ, 0,1 мкМ и 0,01 мкМ с тройной дистиллированной водой, и образец объемом 10 мкл наносили на масс-спектроскопию solariX FT-ICR (Bruker Daltonik). Наблюдали статус межмолекулярной ассоциации и получали данные. Результаты приведены на фиг 37-40. Фиг. 37 представляет собой спектр FT-MS высокого разрешения соединения 1е согласно настоящему изобретению в концентрации 0,01 мкМ: 915,84146 представляет собой трехзарядный ион димера, 1030,32114 представляет собой четырехзарядный ион тримера и 1099,00914 представляет собой пятизарядный ион тетрамера; фиг. 38 представляет собой спектр FT-MS высокого разрешения соединения Ie согласно настоящему изобретению в концентрации 0,1 мкМ: 915,84124 представляет собой трехзарядный ион димера, 1030,32208 представляет собой четырехзарядный ион тримера и 1099,00829 представляет собой пятизарядный ион тетрамера; фиг. 39 представляет собой спектр FT-MS высокого разрешения соединения Ie согласно настоящему изобретению в концентрации 1 мкМ: 915,84095 представляет собой трехзарядный ион димера, 1030,32067 представляет собой четырехзарядный ион тримера и 1099,00914 представляет собой пятизарядный ион тетрамера; фиг. 40 представляет собой спектр FT-MS высокого разрешения соединения Ie согласно настоящему изобретению в концентрации 10 мкМ: 915,84163 представляет собой трехзарядный ион димера, 1030,32067 представляет собой четырехзарядный ион тримера и 1099,00914 представляет собой пятизарядный ион тетрамера.
Димеры, тримеры и тетрамеры, образованные соединениями согласно настоящему изобретению, дополнительно собираются в наносферы с диаметром от 2 до 300 нм. Среди таких наносфер таких размеров наносферы с диаметром менее 100 нм составляли более 99%. В нанофармакологии хорошо известен тот факт, что наносферы, имеющие диаметр менее 100 нм, менее склонны быть поглощенными макрофагами в процессе транспортировки в кровяном русле и могут легко проникать через стенку кровеносных капилляров. Такие свойства позволяют соединению согласно настоящему изобретению проникать через гематоэнцефалический барьер. Это свойство проникновения соединениями согласно настоящему изобретению через гематоэнцефалический барьер позволяет продуктам метаболизма соединений согласно настоящему изобретению быть обнаруженными в тканях мозга у крыс, получавших лечение инсульта.
Экспериментальный пример 17. Эксперименты по контролю с использованием FT-MS высокого разрешения метаболических продуктов в тканях мозга у крыс, получавших соединение Ie согласно настоящему изобретению
Весь мозг крыс вынимали и помещали в центрифужную пробирку объемом 50 мл, в которую добавляли 10 мл 0,9% NaCl и гомогенизировали для получения однородной суспензии, которую затем центрифугировали при 3000 оборотах в минуту в течение 10 мин. 5 мл супернатанта добавляли в 10 мл метанола и равномерно смешивали путем встряхивания, и центрифугировали при 3000 оборотах в минуту в течение 10 мин. Супернатант концентрировали при пониженном давлении до сухого с последующим добавлением 1 мл метанола и снова центрифугировали при 12000 оборотах в минуту в течение 10 мин. Полученный супернатант использовали для контроля содержания продуктов метаболизма в тканях мозга у крыс, получавших соединение 1е.
Экспериментальные результаты по FT-MS высокого разрешения показали два метаболических продукта M1 и М2 в головном мозге. Среди них, M1 характеризовалось [М+1]+ 291,06971 и молекулярной формулой C15H19O5N2; и М2 характеризовалось [М+1]+ 307,04350 и молекулярной формулой C15H19O4N2. (условия МС: загрузка: 10 мкл; режим ионизации: ES+; напряжение на конусе: 30 В; скорость потока подвижной фазы: 0,2 мл/мин). В соответствии с вышеуказанными данными, метаболические продукты M1 и М2 считались следующими соединениями:
Это показывает, что соединение Ie согласно настоящему изобретению действительно пересекает гематоэнцефалический барьер, делая возможным эффект акцептирования свободных радикалов NO, тромболизис и антитромботический эффект в головном мозге.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ СОЕДИНЕНИЯ, ОБЛАДАЮЩИЕ ТРОЙНОЙ АКТИВНОСТЬЮ, ТРОМБОЛИЗИСНОЙ, АНТИТРОМБОТИЧЕСКОЙ И ЗАХВАТА РАДИКАЛОВ, И ИХ СИНТЕЗ, НАНОСТРУКТУРЫ И ПРИМЕНЕНИЕ | 2014 |
|
RU2660901C2 |
| ПЕПТИД И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1994 |
|
RU2067000C1 |
| ПЕПТИДЫ, ОБЛАДАЮЩИЕ АНТИКОАГУЛЯНТНО-ФИБРИНОЛИТИЧЕСКОЙ, АНТИТРОМБОЦИТАРНОЙ И АНТИТРОМБОТИЧЕСКОЙ АКТИВНОСТЬЮ | 2005 |
|
RU2286166C1 |
| ПЕПТИД, ОБЛАДАЮЩИЙ АНТИКОАГУЛЯНТНОЙ, ФИБРИНДЕПОЛИМЕРИЗАЦИОННОЙ, АНТИТРОМБОТИЧЕСКОЙ И ФИБРИНОЛИТИЧЕСКОЙ АКТИВНОСТЯМИ | 2005 |
|
RU2288736C1 |
| ГЕПТАПЕПТИД, ОБЛАДАЮЩИЙ СВОЙСТВАМИ ПСИХОСТИМУЛЯТОРА ПРОЛОНГИРОВАННОГО ДЕЙСТВИЯ С ИММУНОТРОПНОЙ АКТИВНОСТЬ | 1983 |
|
RU1124544C |
| Способ получения полипептидов | 1977 |
|
SU904518A3 |
| RGD-СОДЕРЖАЩИЕ ПЕПТИДЫ | 2001 |
|
RU2214416C2 |
| КОНЪЮГАТЫ АНТИТЕЛА И ЛЕКАРСТВЕННОГО СРЕДСТВА (ADC) И КОНЪЮГАТЫ АНТИТЕЛА И ПРОЛЕКАРСТВА (APDC), СОДЕРЖАЩИЕ ФЕРМЕНТАТИВНО РАСЩЕПЛЯЕМЫЕ ГРУППЫ | 2016 |
|
RU2751512C2 |
| Способ получения пептидов | 1988 |
|
SU1598881A3 |
| ПРОИЗВОДНЫЕ ГЕМИНА И ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ПОЛУЧЕНИЯ, ПРИМЕНЕНИЕ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2002 |
|
RU2238950C2 |
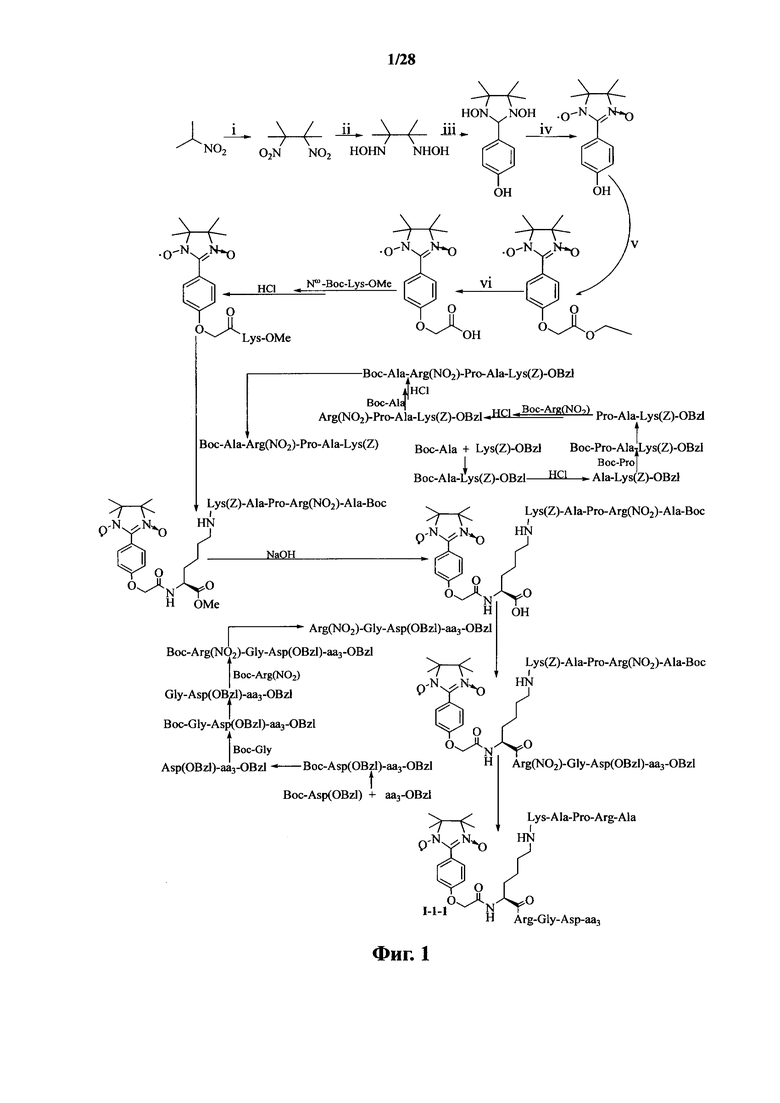
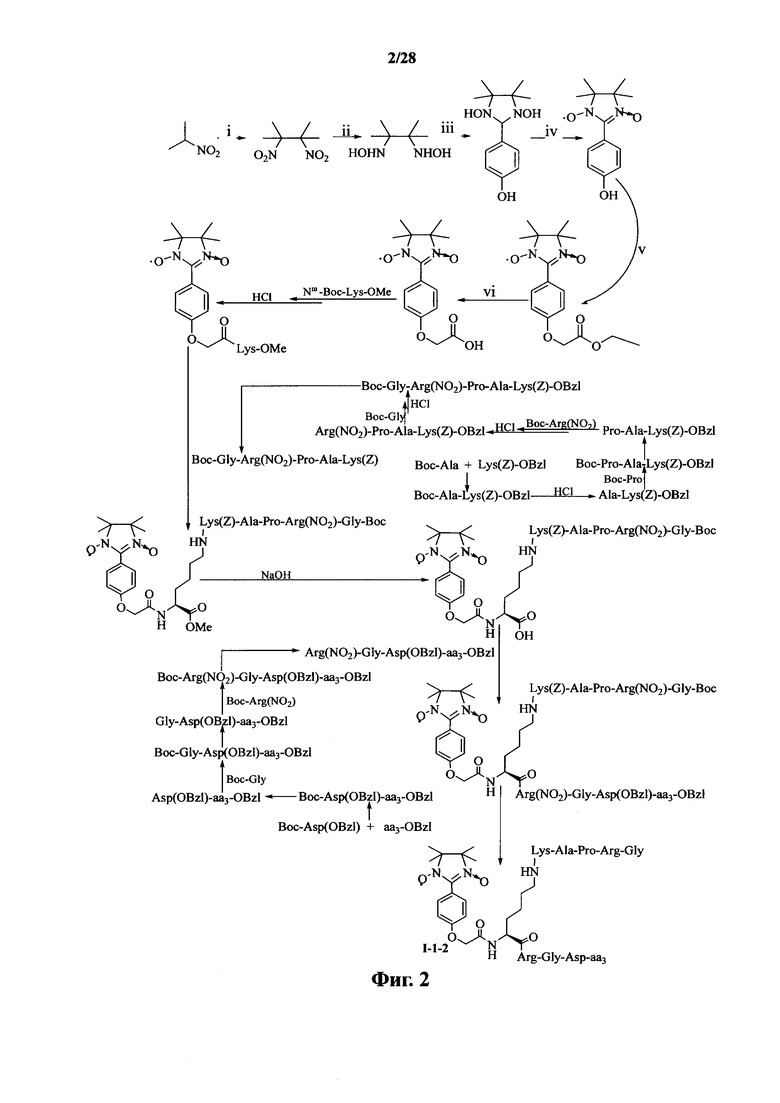
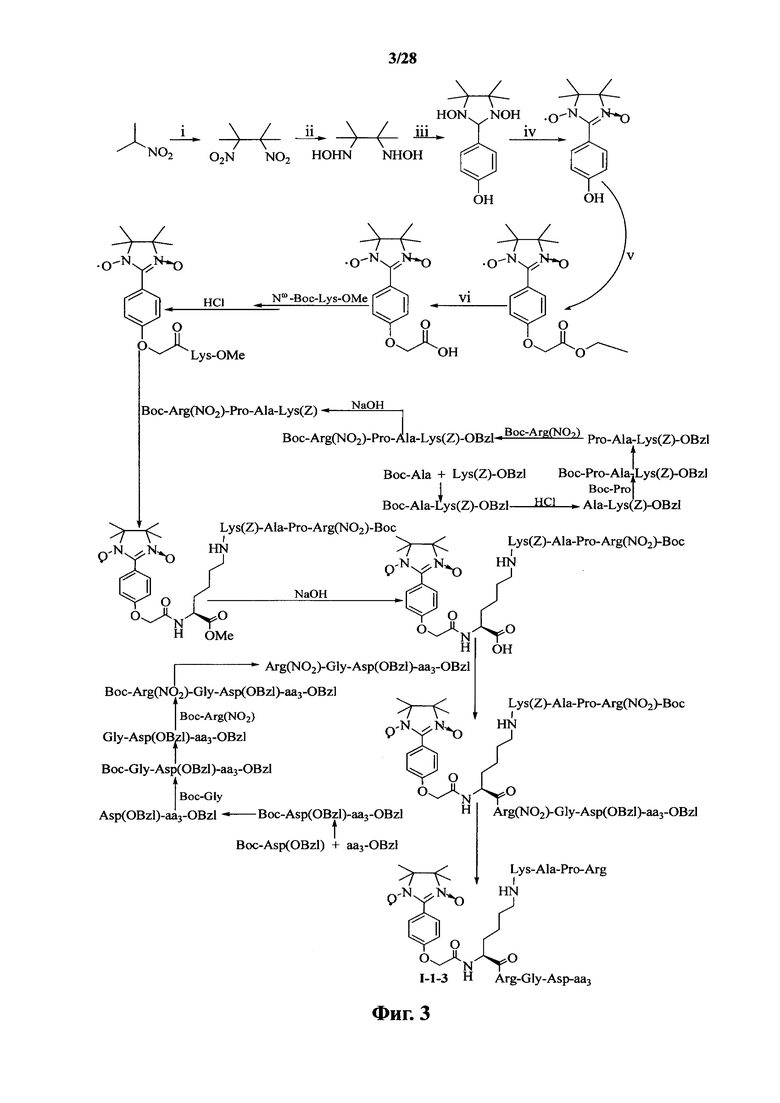
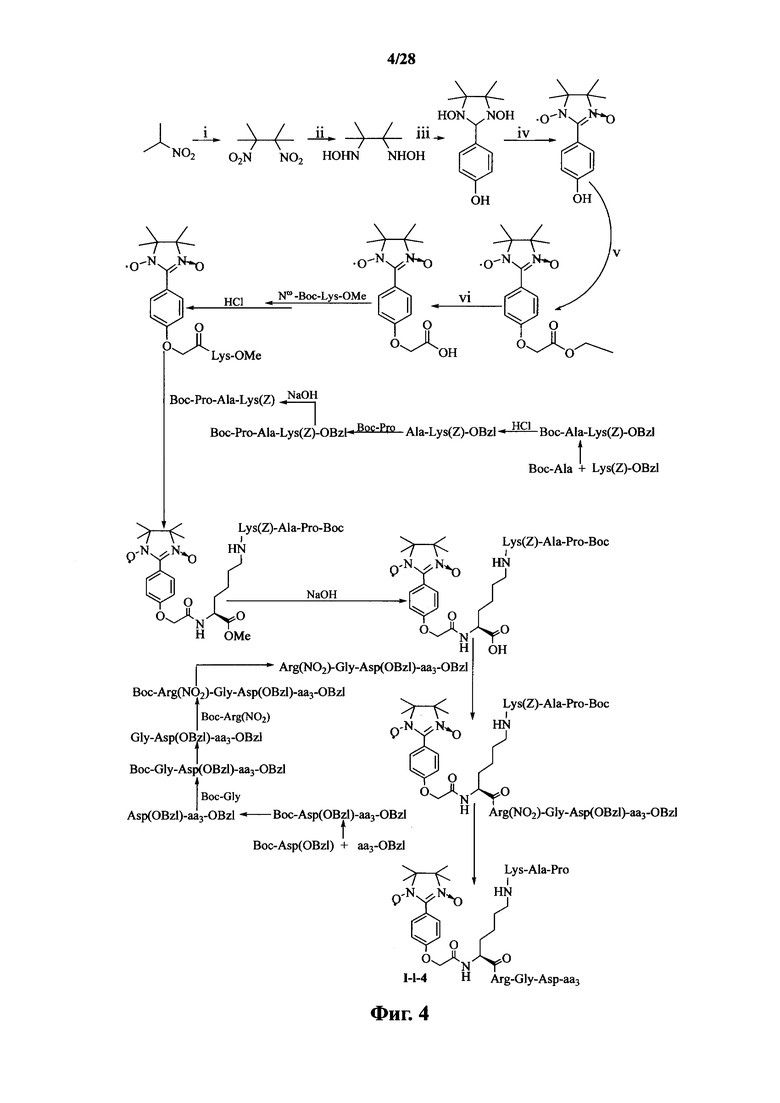
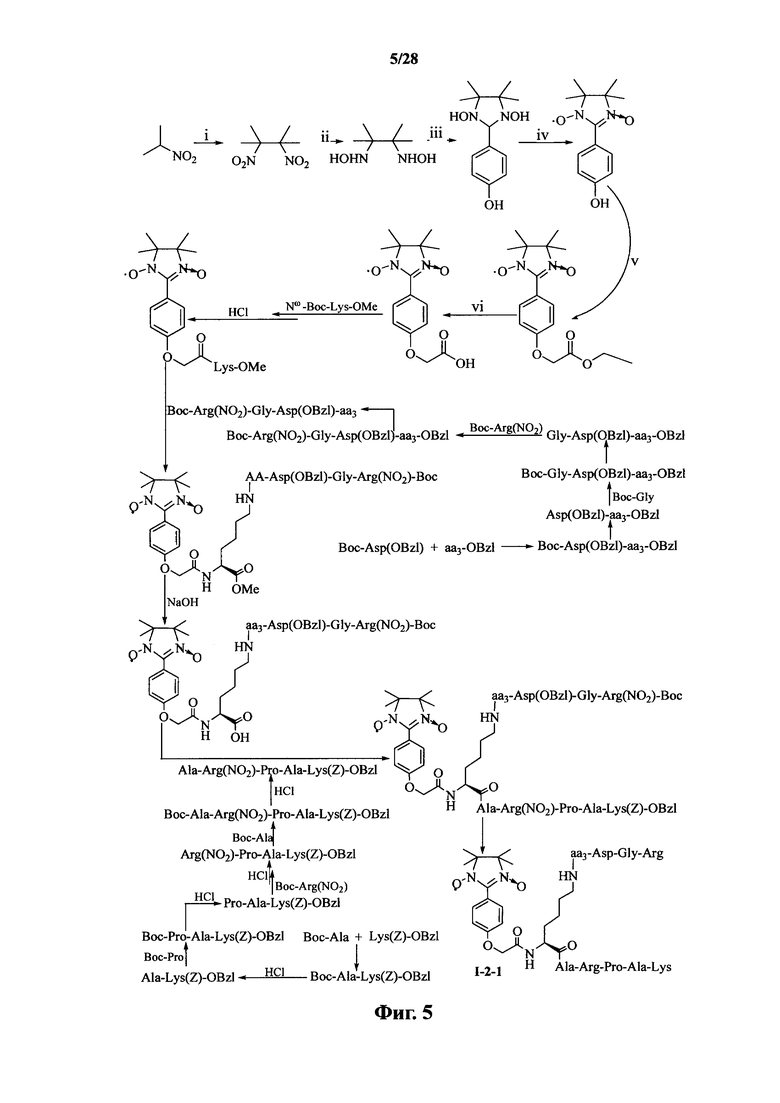
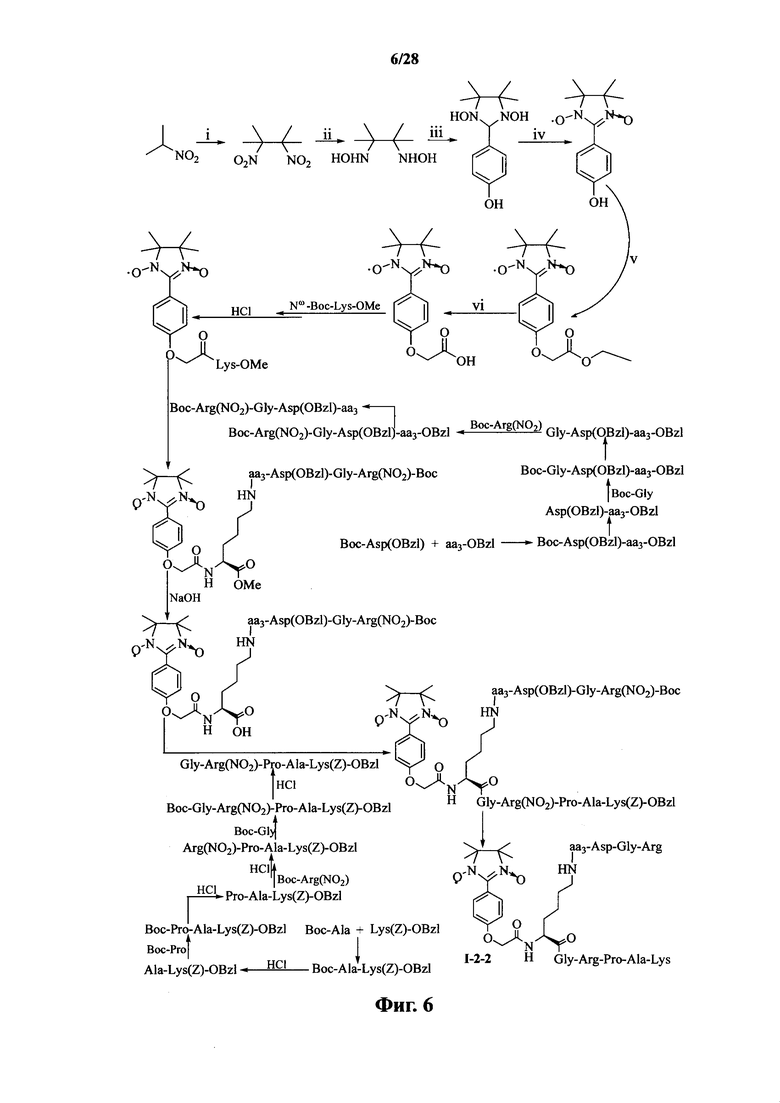
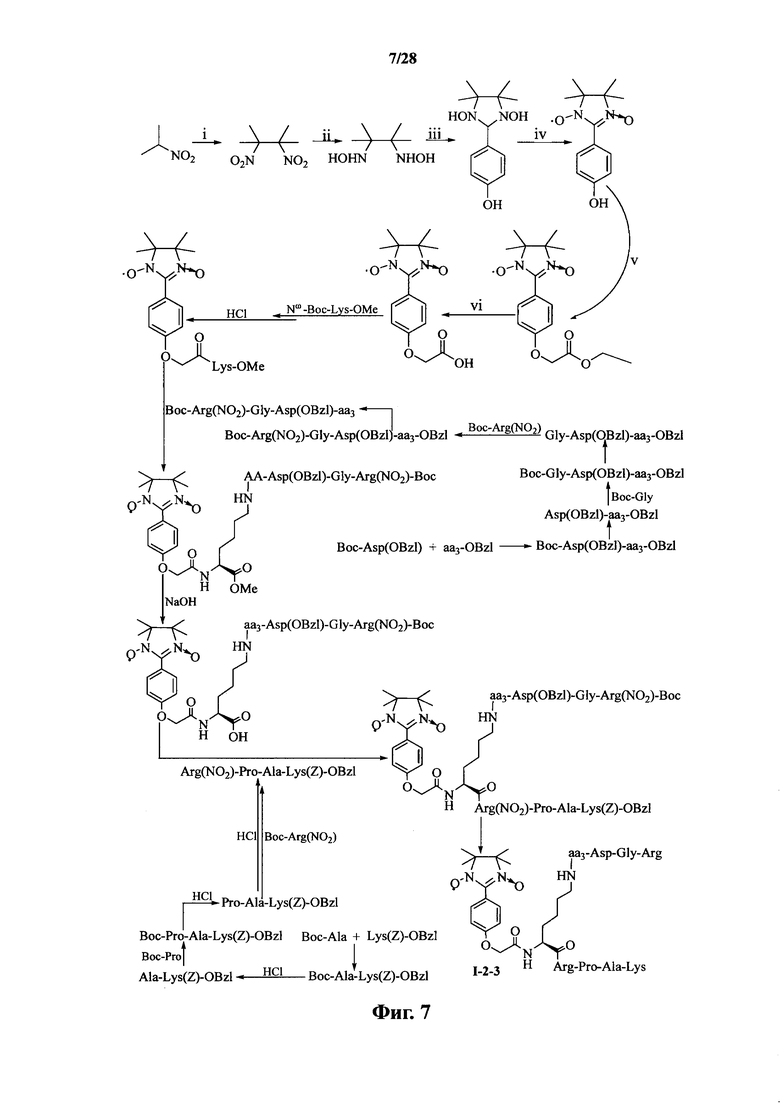
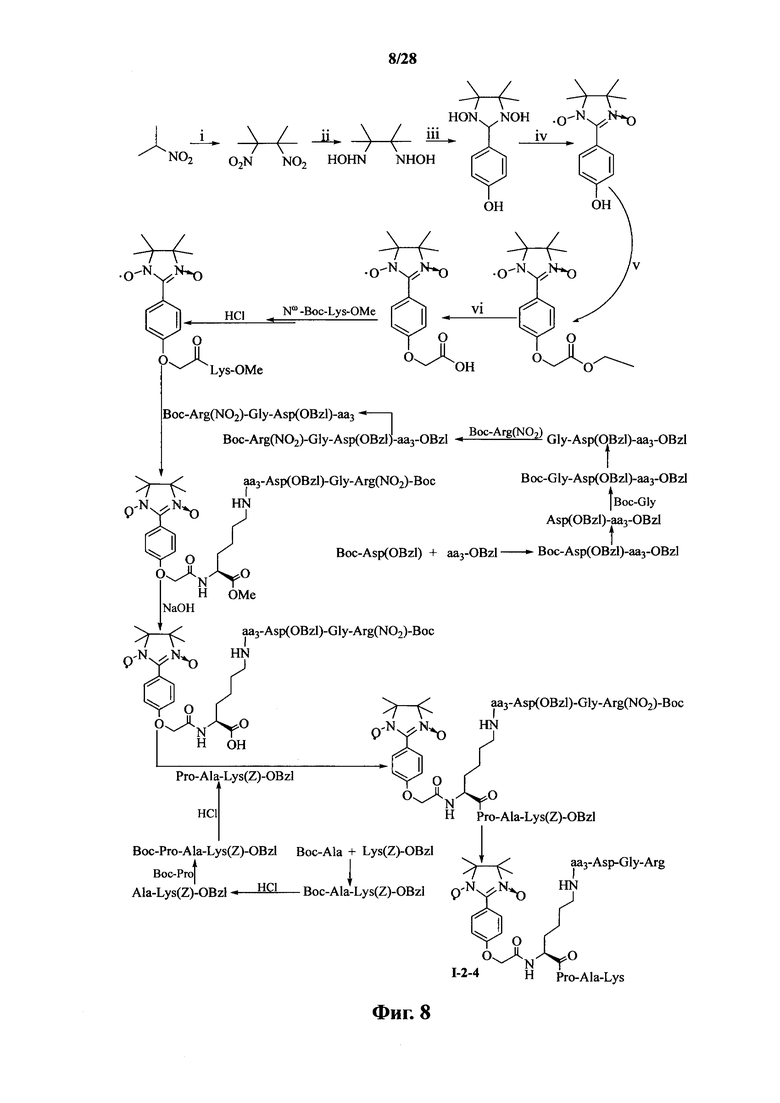
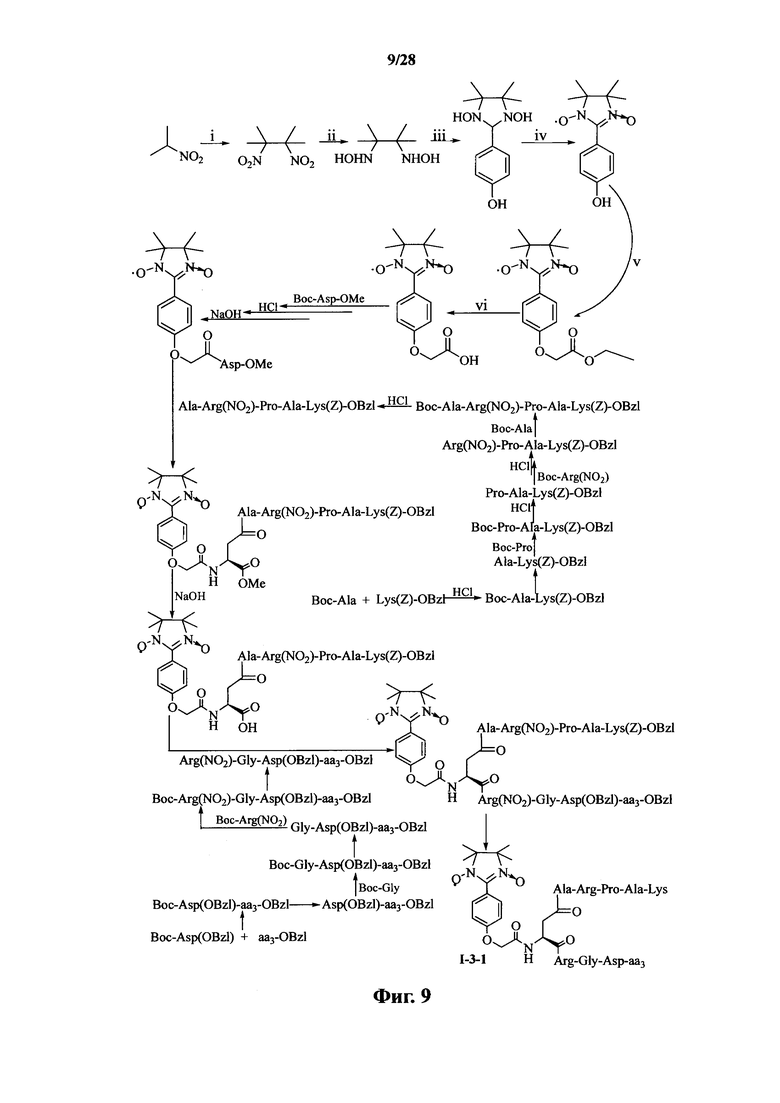
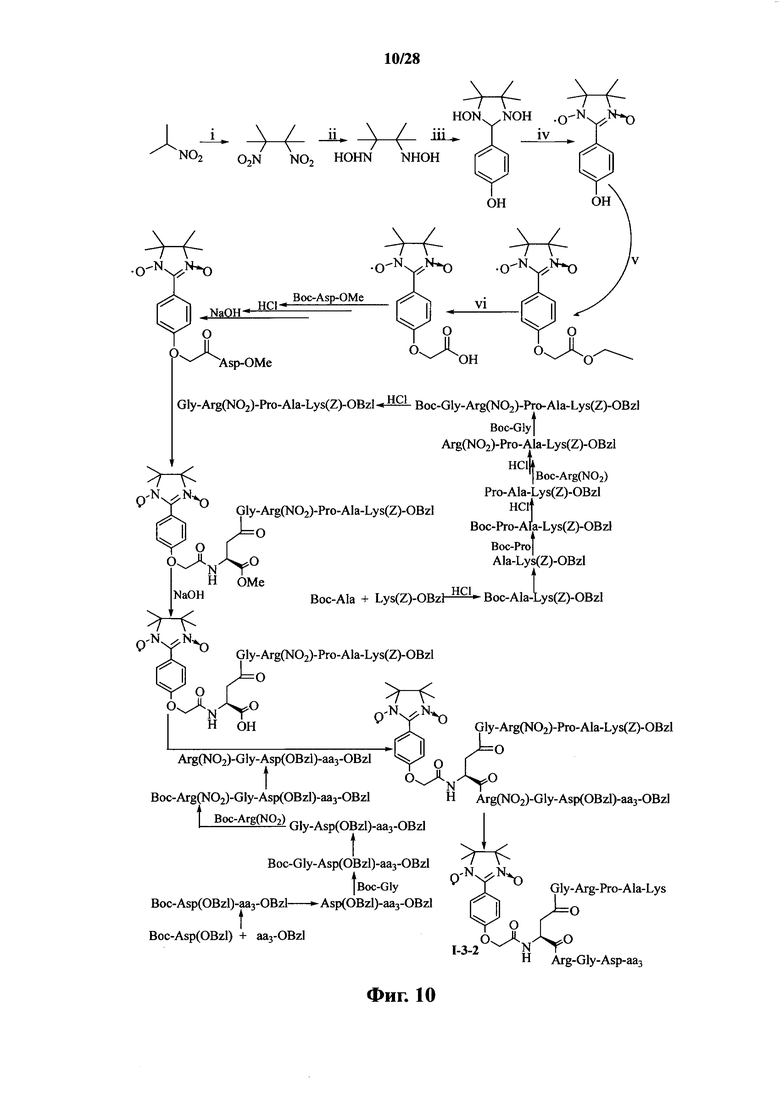
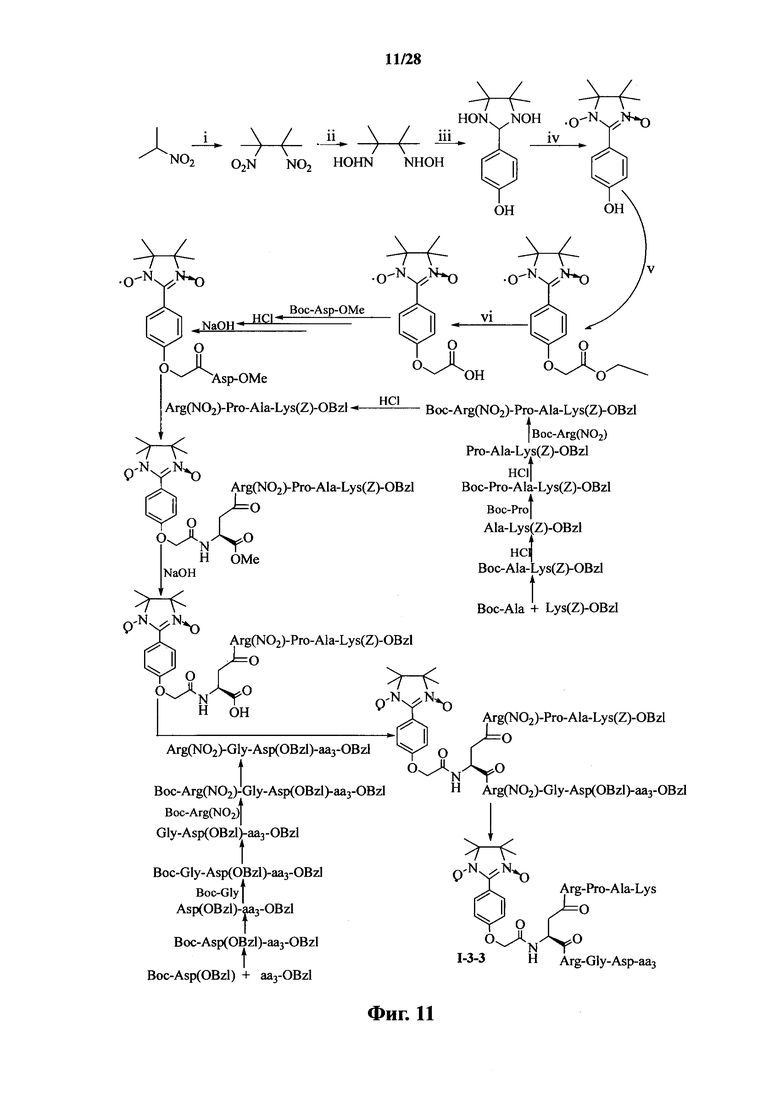
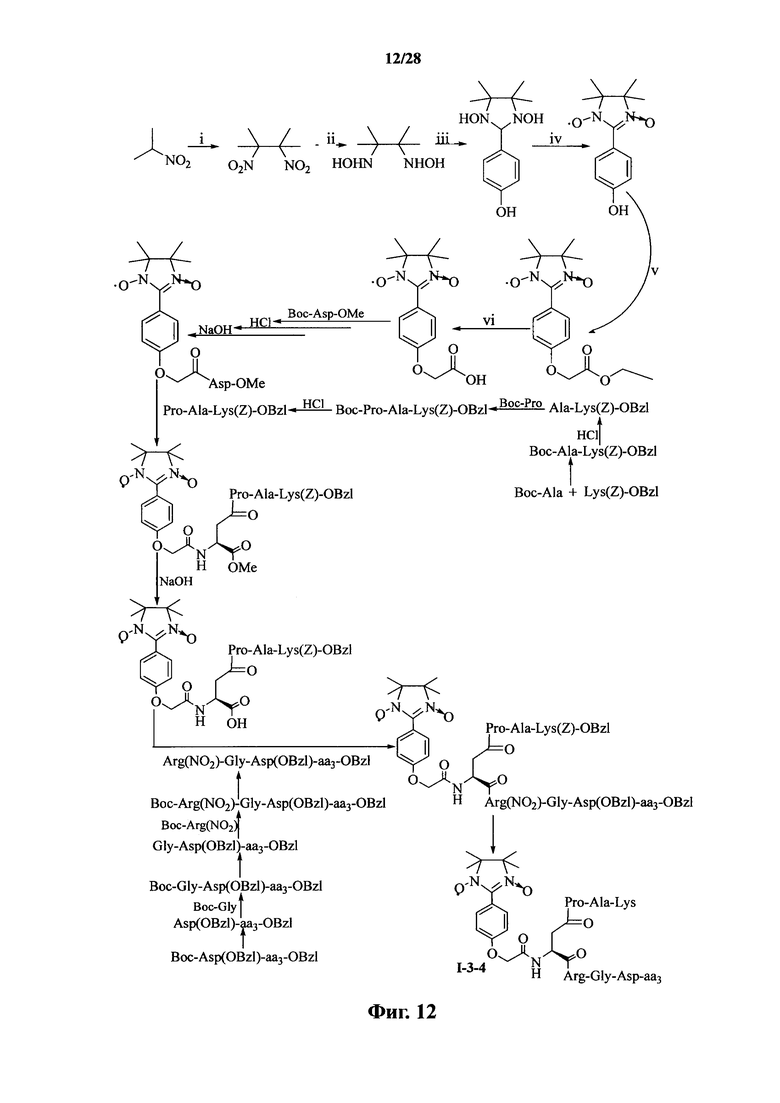
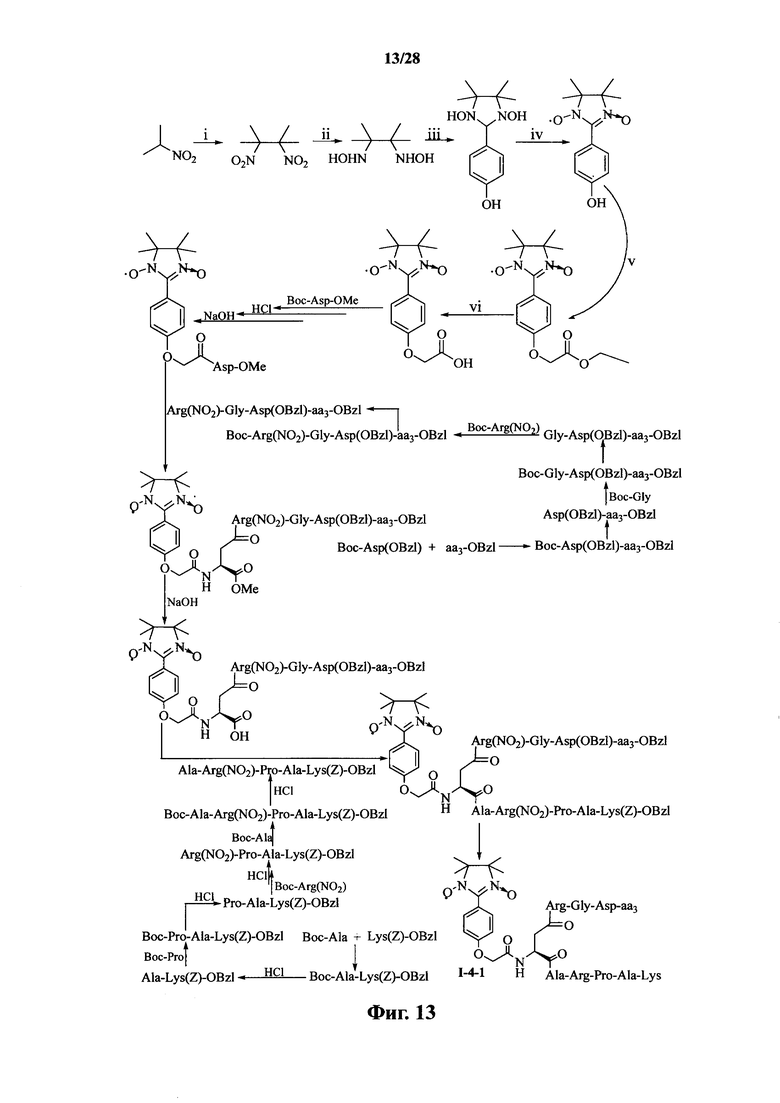
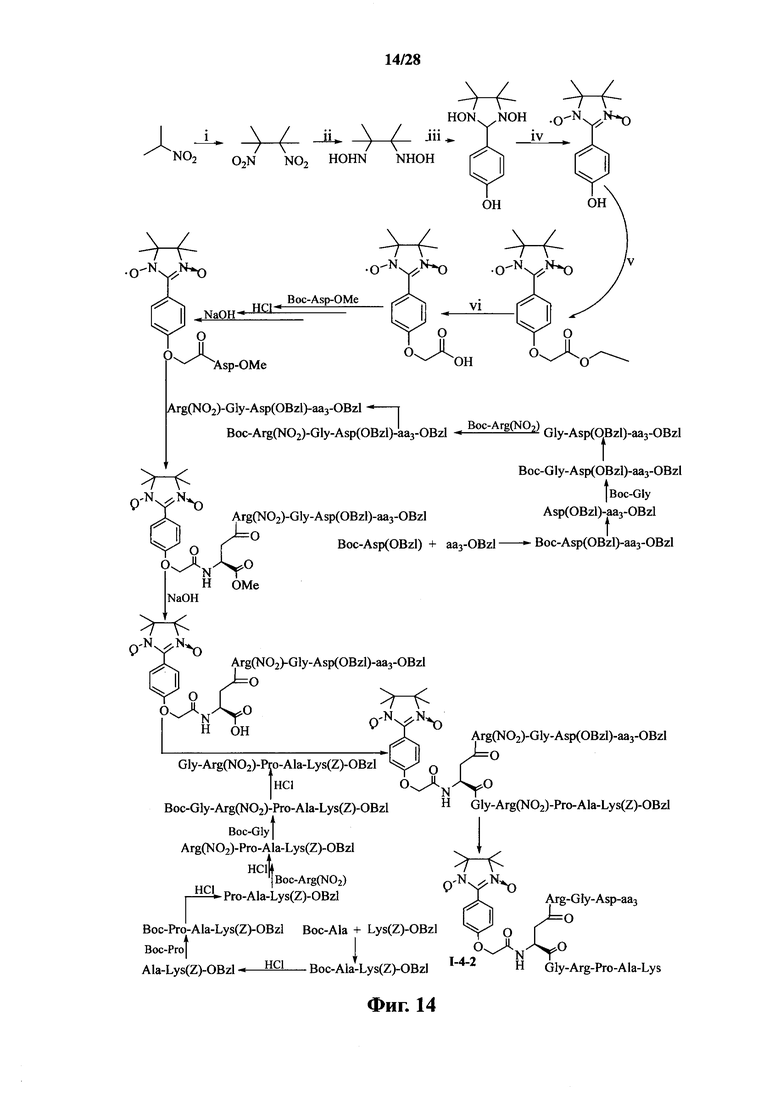
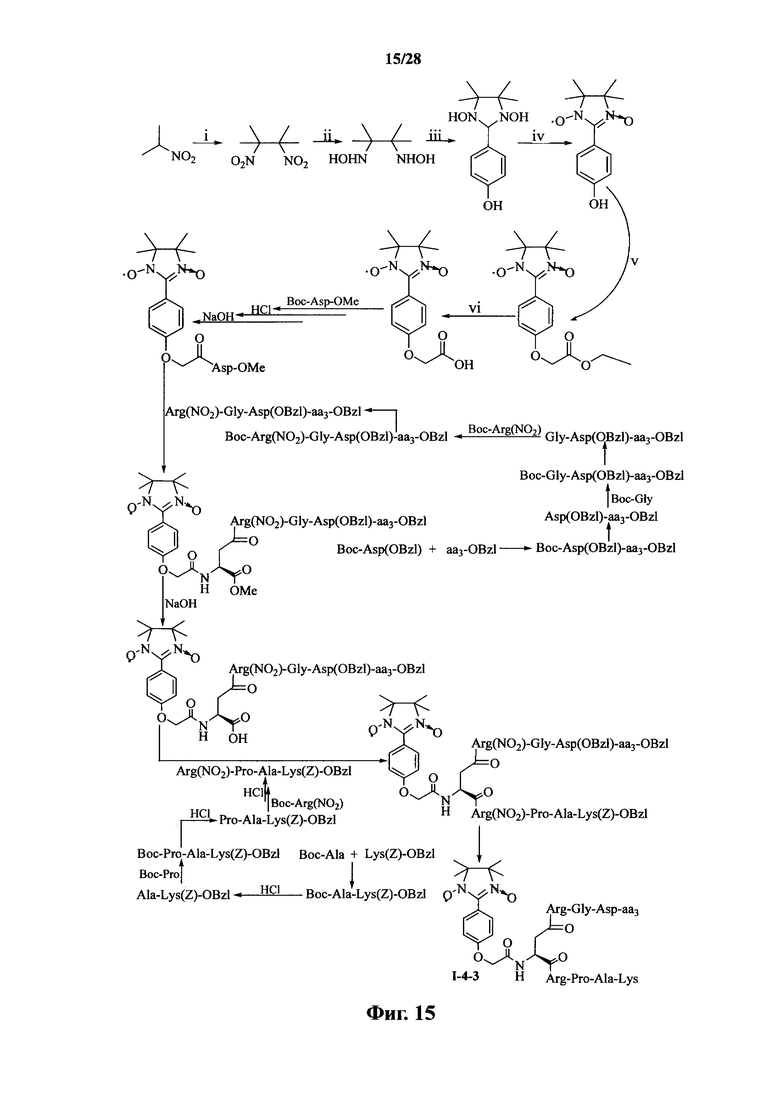
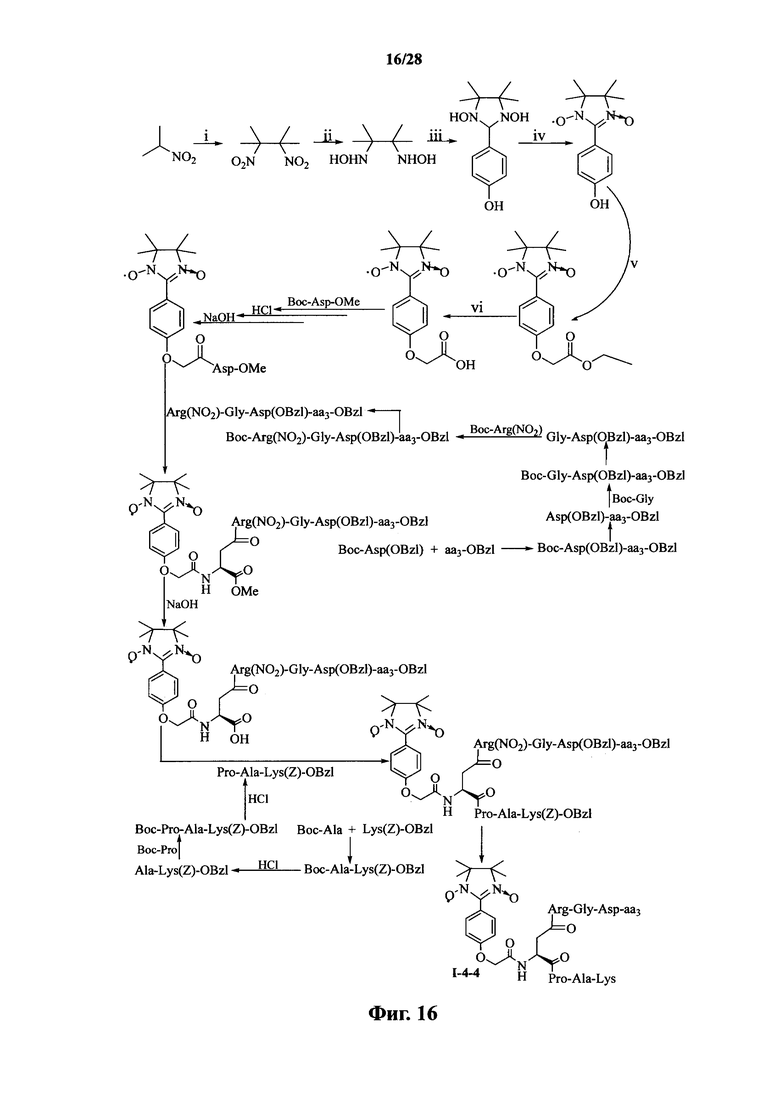
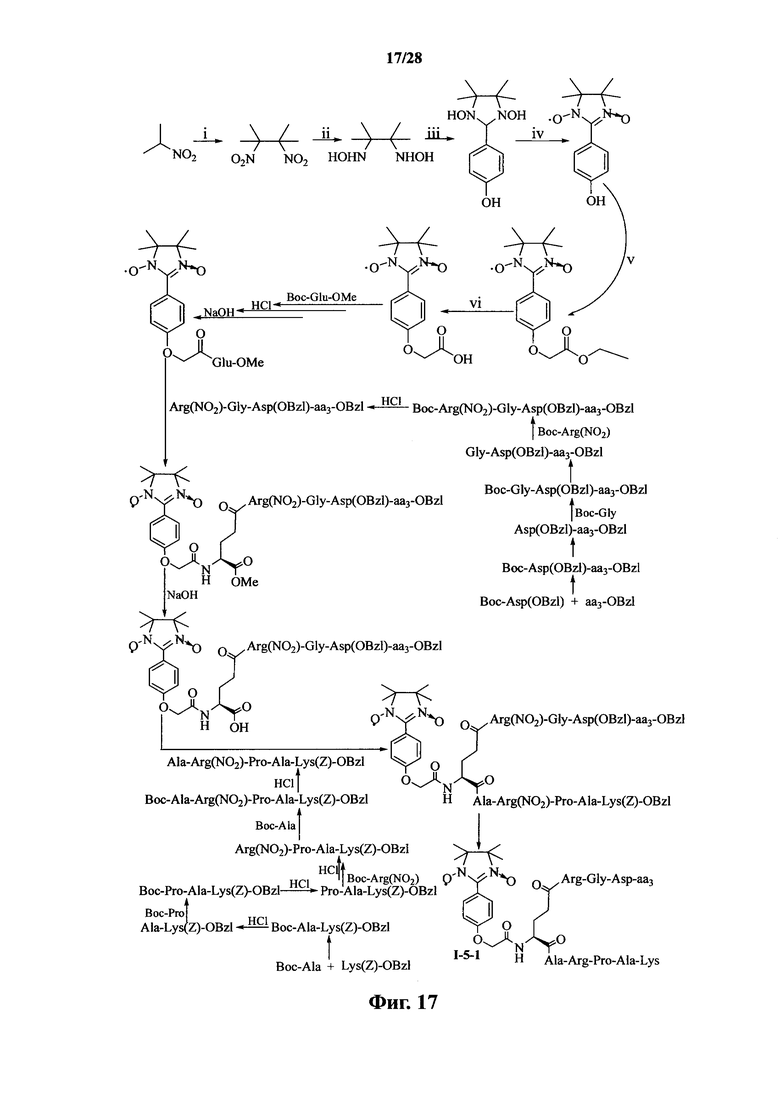
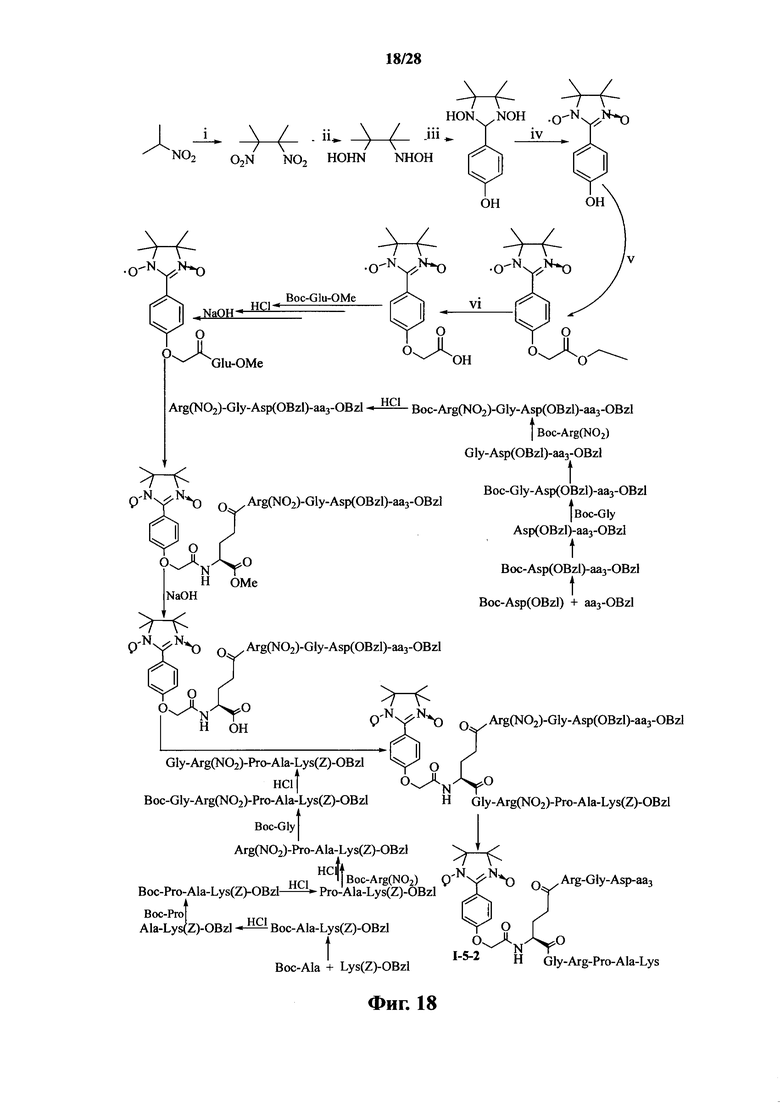
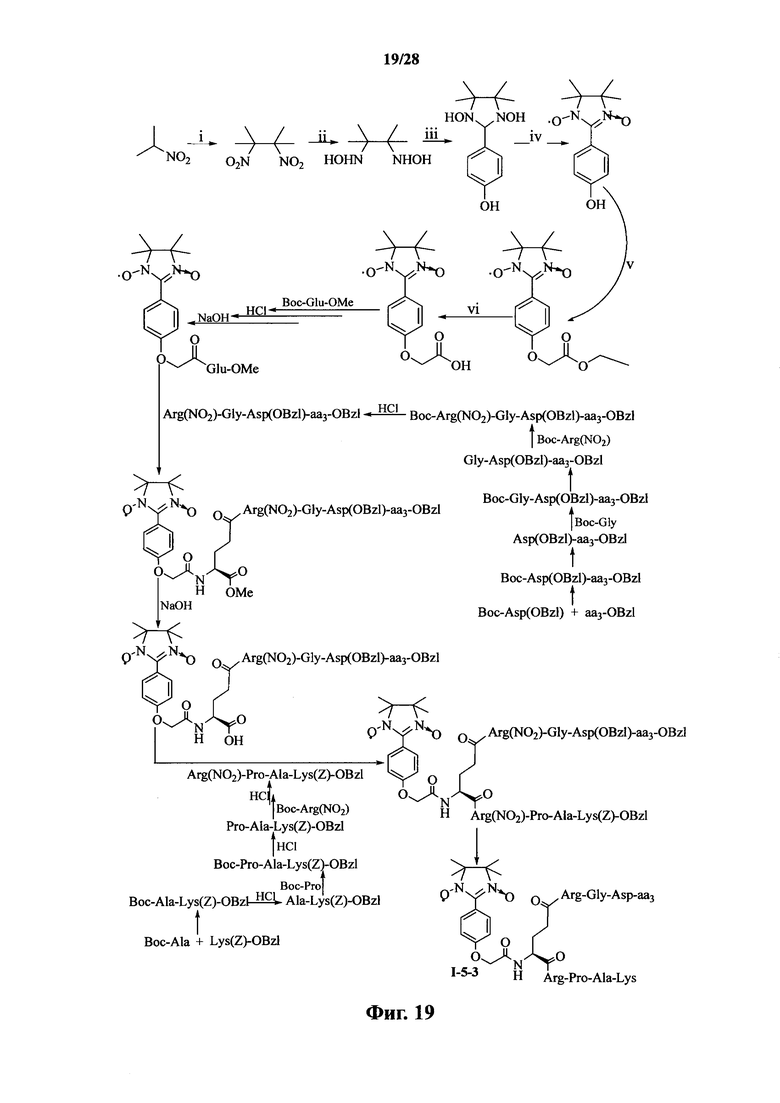
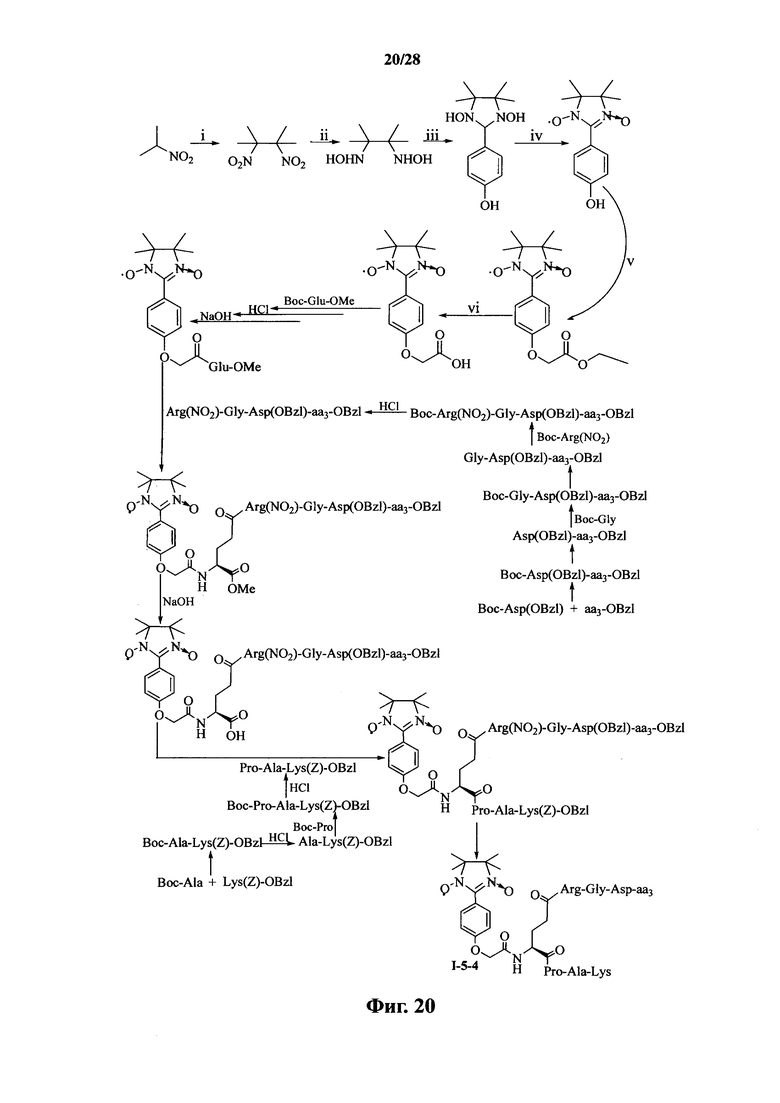
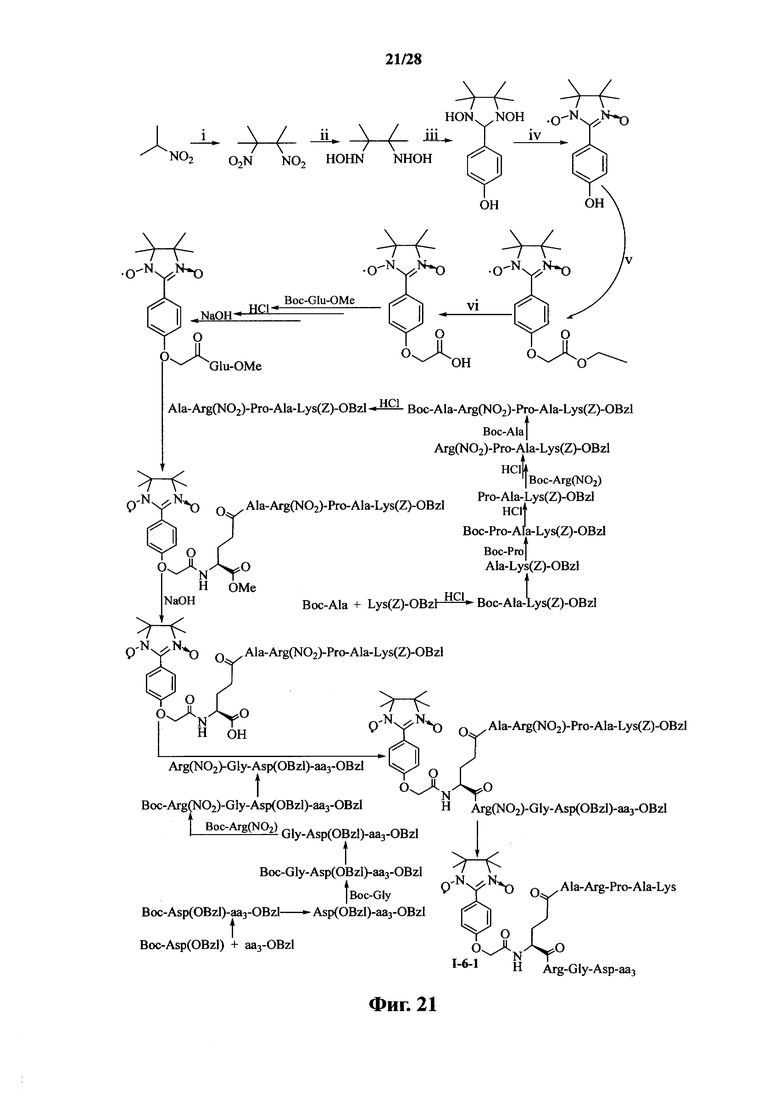
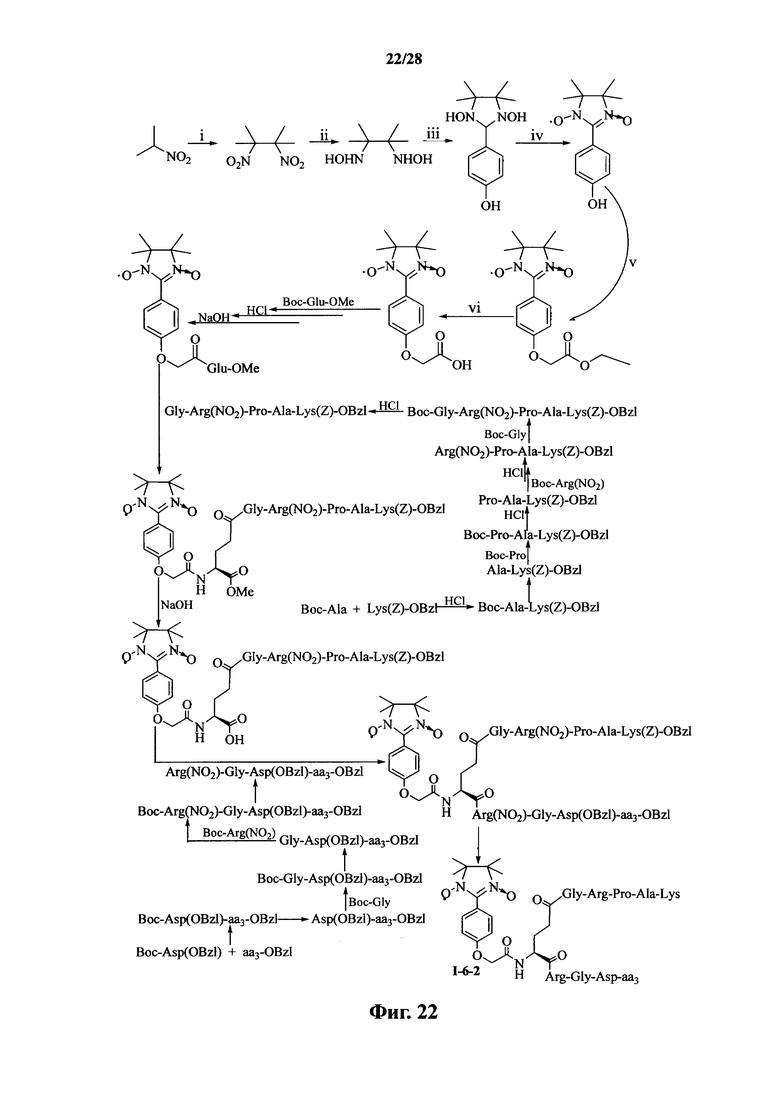
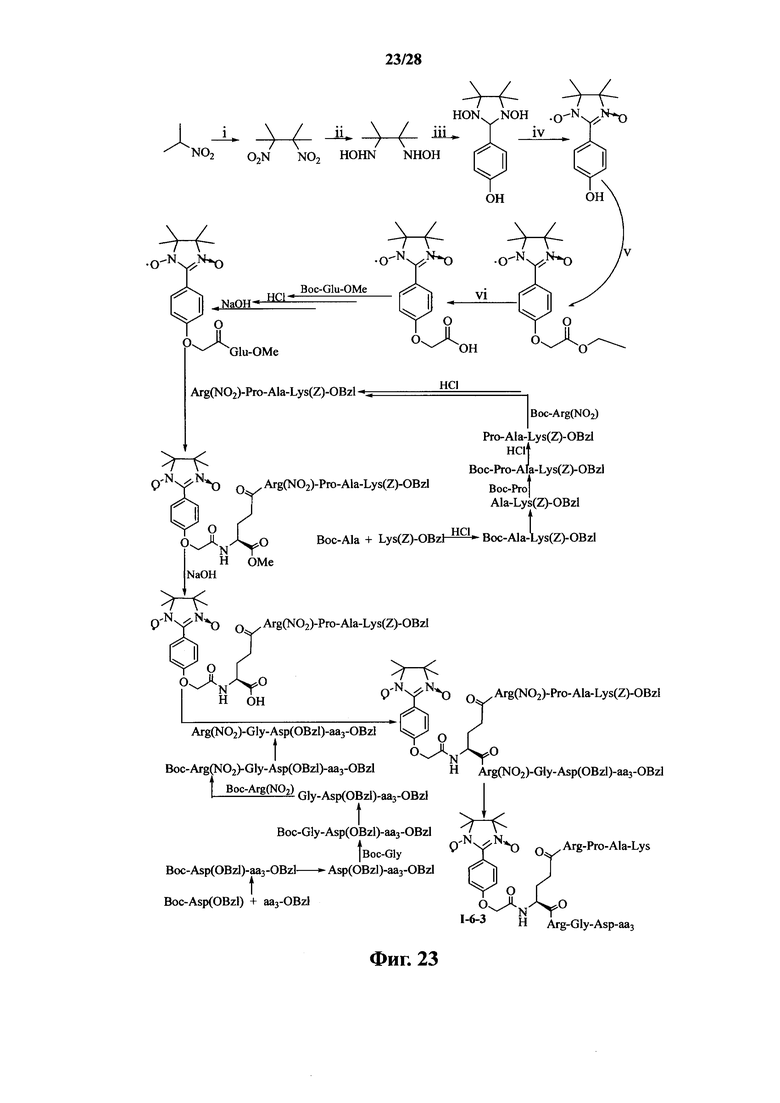
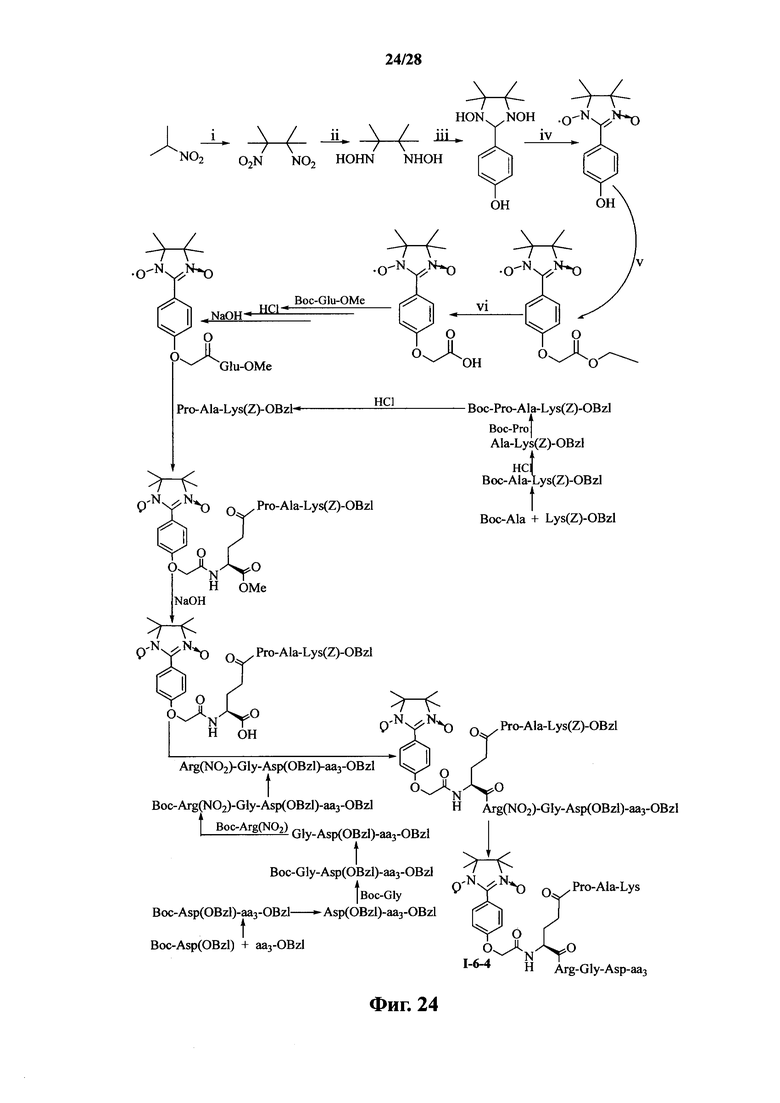


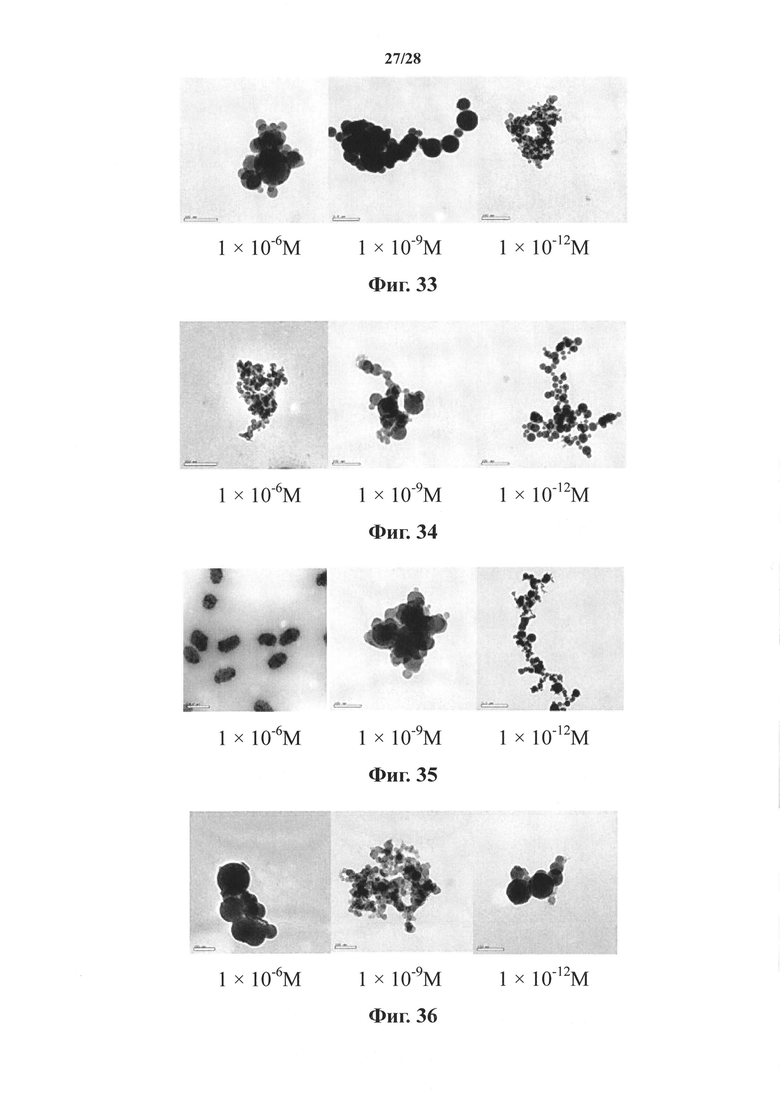
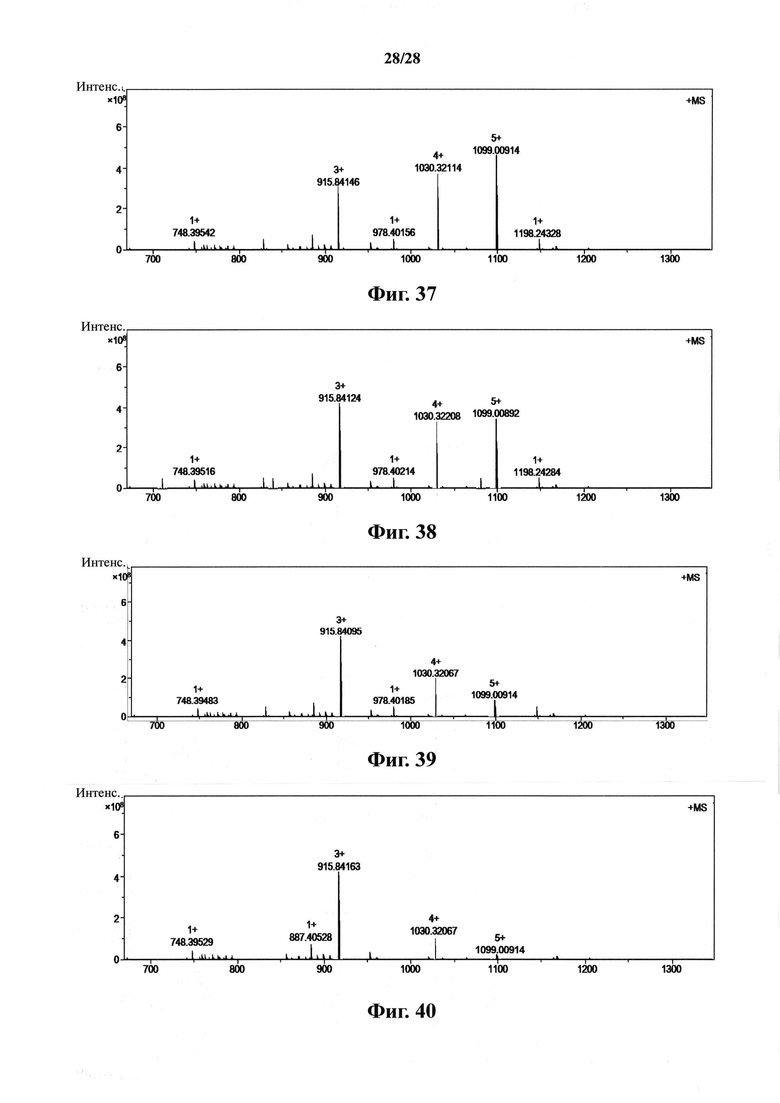
Изобретение относится к новому соединению формулы I-1, характеризующемуся эффектами тромболизиса, акцептирования свободных радикалов и направленного действия на тромб. Соединение представляет собой трехкомпонентный конъюгат, образованный путем конъюгирования тромболитического олигопептида, акцептора свободных радикалов и пептида с направленным действием на тромб/антитромботического олигопептида посредством связующего звена. Настоящее изобретение также относится к фармацевтической композиции, содержащей соединения, причем соединения образуют наносферическую структуру. 2 н. и 11 з.п. ф-лы, 40 ил., 21 табл., 17 пр.
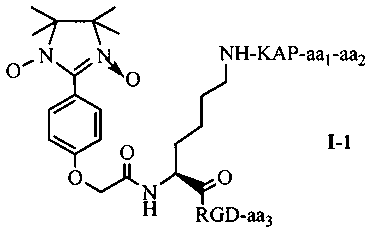
1. Трехкомпонентный конъюгат, состоящий из
1,3-диоксо-2-[(4-оксиацетокси)фенил]-4,4,5,5-тетраметилимидазолина, характеризующегося способностью к акцептированию свободных радикалов NO, пептида, характеризующегося тромболитической активностью, и тетрапептида с направленным действием на тромб, имеющего последовательность RGD (Arg-Gly-Asp),
которые связаны вместе посредством L-Lys связующего звена, при этом пептид, характеризующийся тромболитической активностью, представляет собой ARPAK (Ala-Arg-Pro-Ala-Lys), GRPAK (Gly-Arg-Pro-Ala-Lys), RPAK (Arg-Pro-Ala-Lys) или PAK (Pro-Ala-Lys),
характеризующийся следующей структурной формулой I-1:
в которой aa1 и аа2 могут оба присутствовать, или aa1 присутствует, но аа2 отсутствует, или оба могут отсутствовать; если aa1 и аа2 оба присутствуют, то aa1 представляет собой R(Arg), и аа2 представляет собой G(Gly) или A(Ala); если aa1 присутствует, но аа2 отсутствует, то aa1 представляет собой R(Arg); аа3 может представлять собой S(Ser), V(Val) или F(Phe).
2. Трехкомпонентный конъюгат по п. 1, характеризующийся следующей структурной формулой I-1-1:
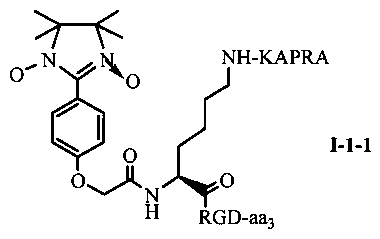
при этом аа3 может представлять собой S(Ser), V(Val) или F(Phe).
3. Трехкомпонентный конъюгат по п. 1, характеризующийся следующей структурной формулой I-1-2:
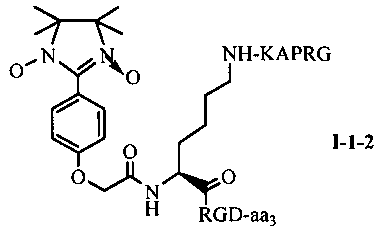
при этом aa3 может представлять собой S (Ser), V(Val) или F(Phe).
4. Трехкомпонентный конъюгат по п. 3, в котором аа3 представляет собой V(Val).
5. Трехкомпонентный конъюгат по п. 1, характеризующийся следующей структурной формулой I-1-3:
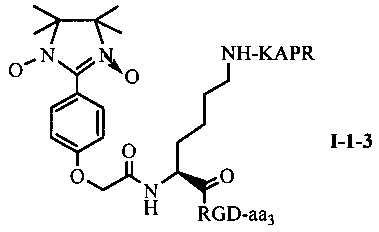
при этом аа3 может представлять собой S(Ser), V(Val) или F(Phe).
6. Трехкомпонентный конъюгат по п. 1, характеризующийся следующей структурной формулой I-1-4:
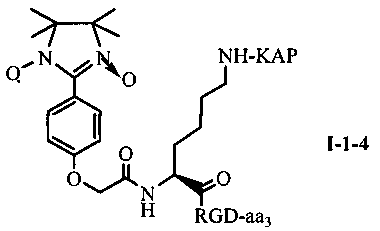
при этом аа3 может представлять собой S(Ser), V(Val) или F(Phe).
7. Фармацевтическая композиция, содержащая эффективную дозу трехкомпонентного конъюгата по любому из пп. 1-6 и фармацевтически приемлемый носитель, предназначенная для применения в качестве тромболитического лекарства, лекарства для акцептирования свободных радикалов NO, антитромботического лекарства или лекарства при лечении инсульта или инфаркта головного мозга.
8. Фармацевтическая композиция по п. 7, где трехкомпонентный конъюгат находится в форме наносферической структуры.
9. Фармацевтическая композиция по п. 7 для применения в качестве тромболитического лекарства, лекарства для акцептирования свободных радикалов NO или антитромботического лекарства.
10. Фармацевтическая композиция по п. 9 для применения в качестве лекарства при лечении инсульта или инфаркта головного мозга.
11. Фармацевтическая композиция по п. 10 для применения в качестве лекарства при лечении инсульта или инфаркта головного мозга, при этом трехкомпонентный конъюгат характеризуются следующей структурной формулой I-1-2:
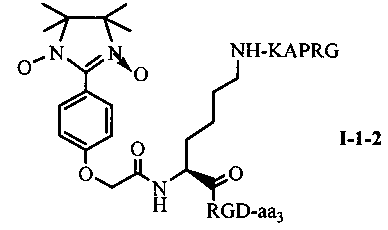
при этом аа3 может представлять собой S(Ser), V(Val) или F(Phe).
12. Фармацевтическая композиция по п. 11 для применения в качестве лекарства при лечении инсульта или инфаркта головного мозга, при этом аа3 представляет собой V(Val).
13. Фармацевтическая композиция по п. 10 для применения при лечении инсульта или инфаркта головного мозга спустя 3 часа после возникновения симптомов.
| CN 1978462 A, 13.06.2007 | |||
| CN 101591376 A, 02.12.2009 | |||
| RGD-СОДЕРЖАЩИЕ ПЕПТИДЫ | 2001 |
|
RU2214416C2 |

