УРОВЕНЬ ТЕХНИКИ
Область Техники
Настоящее изобретение относится к новому соединению одновременно обладающему тромболитической активностью, способностью захвата свободных радикалов и тромб-таргетной/антитромботической функциями, а также к способу его получения и применению. Настоящее изобретение дополнительно относится к новому бинарному конъюгату, образованному путем связывания тромболитического олигопептида и тетрагидроизохинолинового соединения, имеющего две C1-4 алкильные группы, через линкер. Настоящее изобретение также относится к фармацевтической композиции, содержащей соединение, способу получения и наноструктуре соединения.
Описание Предшествующего Уровня Техники
Частота заболеваемостью тромботическими заболеваниями, такими как инсульт/инфаркт, занимает первое место среди различных заболеваний. Существует тенденция к увеличению частоты заболеваемости этими заболеваниями в последние годы, и эти заболевания становятся серьезной угрозой для здоровья человека. Медикаментозное лечение тромботических заболеваний находится в центре повышенного внимания при лечении тромбоза. В настоящее время существует множество ограничений на клинически применяемые тромботические препараты, и поиск безопасного и эффективного нового тромботического препарата является одним из ведущих научно-исследовательских направлений.
Согласно настоящему исследованию в дополнение к антиагрегантной и антитромботической активностям 3 S-1,1-диметил-6,7-дигидрокси-1,2,3,4-тетрагидроизохинолин-3-карбоновая кислота также обладает активностью в отношении захвата свободных радикалов. Кроме того, в Китайской Патентной Публикации CN 101497651 B, поданной 30 января 2008, раскрыты 10 тетрагидроизохинолиновых соединений, обладающих тромболитической активностью. Эти тетрагидроизохинолиновые соединения включают 3S-6,7-1,2,3,4-тетрагидро-6,7-дигидрокси-изохинолин-3-ацил-Pro-Ala-Lys, 3S-6,7-1,2,3,4-тетрагидро-6,7-дигидроксиизохинолин-3-ацил-Arg-Pro-Ala-Lys, 3S-6,7-1,2,3,4-тетрагидро-6,7-дигидроксиизохинолин-3-ацил-Ala-Arg-Pro-Ala-Lys, 3S-6,7-1,2,3,4-тетрагидро-6,7-дигидроксиизохинолин-3-ацил-Gly-Arg-Pro-Ala-Lys, 3S-6,7-1,2,3,4-тетрагидро-6,7-дигидроксиизохинолин-3-ацил-Gln-Arg-Pro-Ala-Lys, 3S-2-[Pro-Ala-Lys]-1,2,3,4-тетрагидро-6,7-дигидроксиизохинолин-3-карбоновую кислоту, 3S-2-[Arg-Pro-Ala-Lys]-1,2,3,4-тетрагидро-6,7-дигидроксиизохинолин-3-карбоновую кислоту, 3S-2-[Ala-Arg-Pro-Ala-Lys]-1,2,3,4-тетрагидро-6,7-дигидроксиизохинолин-3-карбоновую кислоту, 3S-2-[Gly-Arg-Pro-Ala-Lys]-1,2,3,4-тетрагидро-6,7-дигидроксиизохинолин-3-карбоновую кислоту и 3S-2-[Gln-Arg-Pro-Ala-Lys]-1,2,3,4-тетрагидро-6,7-дигидроксиизохинолин-3-карбоновую кислоту. Соединения, указанные выше, сокращенно называют “6,7-дигидроксиизохинолинами, обладающими тромболитической активностью”. Тем не менее, эффективные дозы этих 6,7-дигидроксиизохинолинов, имеющих тромболитическую активность, выше, и антитромботическая активность и активность в отношении захвата свободных радикалов не были раскрыты или подтверждены. Более того, эффективность в лечении инсульта была продемонстрирована только как эффективность в момент начала инсульта. Что касается лечения инсульта через 30 минут после начала синдрома, то эффективность не была раскрыта или подтверждена.
Таким образом, для эффективного и безопасного лечения тромботических заболеваний в клинической практике существует потребность в новом соединении, которое одновременно обладает тромболитической, антитромботической активностью и активностью в отношении захвата свободных радикалов, может эффективно пересекать гематоэнцефалический барьер (ГЭБ) и может обеспечивать достижение описанных эффектов при низкой дозе.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В первом аспекте настоящее изобретение относится к соединению, имеющему формулу I:
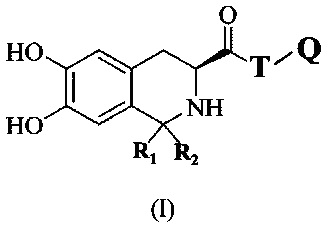
где T представляет собой линкер, имеющий по меньшей мере две группы для связывания, Q представляет собой пептид, имеющий тромболитическую активность, R1 и R2 представляют собой C1-4 алкильные группы, где R1 и R2 могут быть одинаковыми или различными.
В одном варианте выполнения настоящего изобретения по меньшей мере одна из групп для связывания линкера Т представляет собой аминогруппу, а остальные группы для связывания представляют собой карбоксильную группу или аминогруппу.
В предпочтительном варианте выполнения настоящего изобретения линкер Т может быть природной аминокислотой, такой как L-Lys, L-Asp или L-Glu.
В более предпочтительном варианте выполнения настоящего изобретения линкер может быть L-Lys.
В предпочтительном варианте выполнения настоящего изобретения пептид, обладающий тромботической активностью, используемый в настоящем изобретении, выбирается из группы, состоящей из олигопептида, имеющего последовательность РА (Pro-Ala), последовательность РАК (Pro-Ala-Lys), последовательность AKP (Ala-Lys-Pro) или последовательность KAP (Lys-Ala-Pro), или пептида, содержащего повторяющиеся звенья последовательности PAK, последовательности AKP или последовательности КАР.
В одном варианте выполнения настоящего изобретения олигопептид, обладающий тромболитической активностью, может быть от трипептида до октапептида, содержащих последовательность PA (Pro-Ala), последовательность PAK, последовательность AKP или последовательность KAP, предпочтительно трипептидом, содержащим последовательность РА. В более предпочтительном варианте выполнения изобретения трипептид имеет химическую формулу Q1 или Q2, показанную ниже:

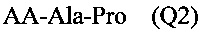
где АА выбирается из группы, состоящей из L-Ala, L-Val, L-Trp, L-Tyr, L-Pro, L-Phe, Gly, L-Ser, L-Ile, L-Thr, L-Lys, L-Leu, L-Gln, L-Asn, L-Asp и L-Glu.
В предпочтительном варианте выполнения изобретения R1 и R2 оба представляют собой метальную группу.
В предпочтительном варианте выполнения изобретения R1 и R2 в формуле I представляют собой метальные группы, линкер представляет собой L-Lys, L-Asp или L-Glu и пептид, обладающий тромболитической активностью, представляет собой трипептид, содержащий последовательность PA (Pro-Ala). Например, соединение может иметь формулу Ia (такую как соединение 5Аа-р на Фиг. 1), Ib (такую как соединение 5 Ва-p р на Фиг. 2), 1с (такую как соединение 5Са-р на Фиг. 3), Id (такую как соединение 5Da-p на Фиг. 4), 1е (такую как соединение 5Еа-р на Фиг. 5), If (такую как соединение 5Fa-p на рис. 6), Ig (такую как соединение 5Ga-p на Фиг. 7) или Ih (такую как соединение 5На-р на Фиг. 8):
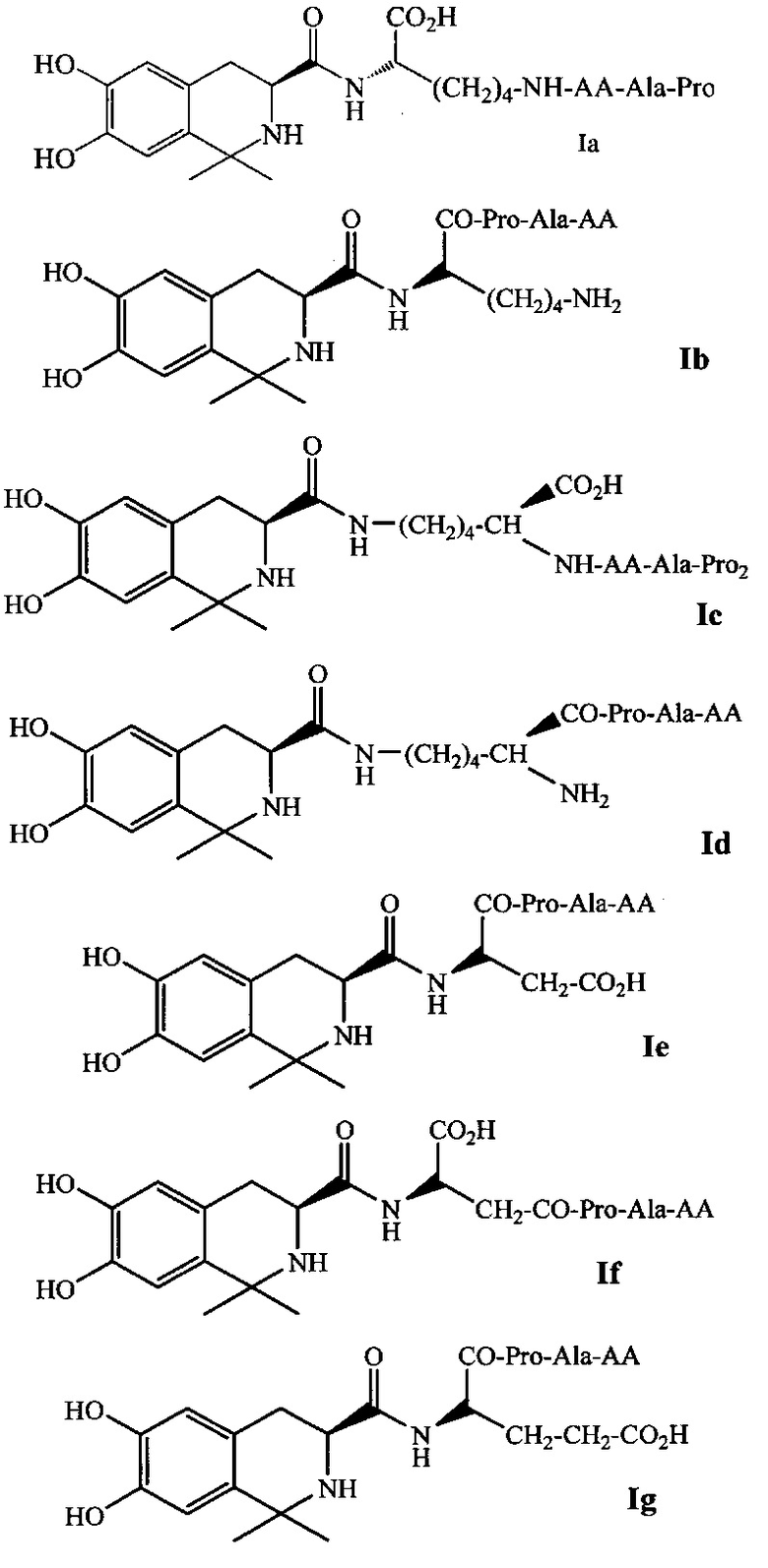
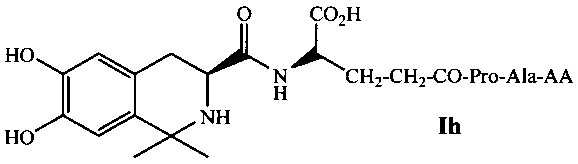
где AA выбирается из группы, состоящей из L-Ala, L-Val, L-Trp, L-Tyr, L-Pro, L-Phe, Gly, L-Ser, L-Ile, L-Thr, L-Lys, L-Leu, L-Gln, L-Asn, L-Asp и L-Glu.
Во втором аспекте настоящее изобретение относится к фармацевтической композиции, содержащей соединение по настоящему изобретению, описанное выше, и фармацевтически приемлемый носитель.
В предпочтительном варианте выполнения настоящего изобретения соединения могут быть в форме наносферической структуры.
В предпочтительном варианте выполнения настоящего изобретения фармацевтическая композиция может быть использована в качестве тромболитического препарата, препарата для захвата свободных радикалов NO или тромб-таргетного/антитромботического препарата.
В другом предпочтительном варианте выполнения настоящего изобретения фармацевтическая композиция может быть использована в качестве препарата для лечения инсульта или церебрального инфаркта, более предпочтительно для лечения инсульта или церебрального инфаркта через 4 часа, 6 часов и 24 часа после начала синдрома, и лечения с помощью последующего введения.
В третьем аспекте настоящее изобретение относится к способу получения соединений, имеющих формулу I. Способ включает в себя следующие стадии:
(1) предоставление соединения, имеющего формулу II:
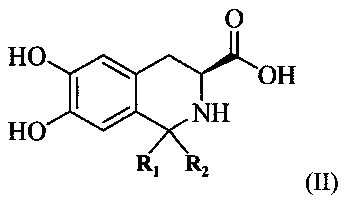
где R1 и R2 представляют собой С1-4 алкильные группы, и R1 и R2 могут быть одинаковыми или разными.
(2) предоставление линкера Т, имеющего по меньшей мере две группы для связывания, и пептида Q, обладающего тромболитической активностью, где линкер имеет первую группу для связывания и вторую группу для связывания.
(3) связывание карбоксильной группы соединения, имеющего формулу II, с первой группой для связывания линкера Т с образованием соединения, имеющего формулу IM-1:
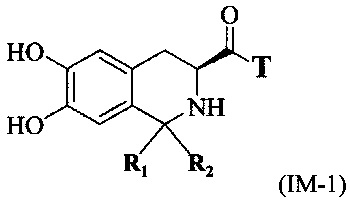
при соответствующих условиях реакции; и
(4) связывание пептида Q, обладающего тромболитической активностью, с соединением, имеющим формулу IM-1, при соответствующих условиях реакции, где один конец пептида Q, обладающего тромболитической активностью, связывается со второй группой для связывания линкера Т с образованием соединения, имеющего формулу I.
В одном варианте выполнения настоящего изобретения первая группа для связывания линкера Т представляет собой аминогруппу, используемую для связывания с карбоксильной группой соединения формулы II по реакции конденсации. Кроме того, вторая группа для связывания является карбоксильной группой или аминогруппой, используемой для связывания с N-концом или С-концом пептида Q, обладающего тромболитической активностью. Определения линкера Т и пептида Q, обладающего тромболитической активностью, используемые в способе получения по настоящему изобретению, являются такими же, как определения соединения, имеющего формулу I выше.
В предпочтительном варианте выполнения настоящего изобретения линкер в настоящем способе получения может быть L-Lys, L-Asp или L-Glu и более предпочтительно L-Lys. Пептид, обладающий тромболитической активностью, может быть трипептидом, содержащим последовательность PA (Pro-Ala), олигопептидом, содержащим последовательность PAK (Pro-Ala-Lys), последовательность AKP (Ala-Lys-Pro) или последовательность КАР (Lys-Ala-Pro), или пептидом, имеющим повторяющуюся последовательность, содержащую последовательность PAK, последовательность AKP или последовательность KAP, и более предпочтительно трипептидом, имеющим формулу Q1 или Q2, показанные ниже:
где АА выбирается из группы, состоящей из L-Ala, L-Val, L-Trp, L-Tyr, L-Pro, L-Phe, Gly, L-Ser, L-Ile, L-Thr, L-Lys, L-Leu, L-Gln, L-Asn, L-Asp и L-Glu.
В другом варианте выполнения настоящего изобретения R1 и R2 соединения, имеющего формулу II, оба представляют собой метальные группы, линкер представляет собой L-Lys, L-Asp или L-Glu и пептид, обладающий тромболитической активностью, представляет собой трипептид, содержащий последовательность PA (Pro-Ala). В более предпочтительном варианте выполнения настоящего изобретения способ получения настоящего изобретения может использоваться для получения соединений, имеющих приведенные выше формулы Ia-h.
Испытания in vivo на крысах показали, что соединения или фармацевтические композиции по настоящему изобретению имеют превосходные тромболитические и антитромботические активности при низкой дозе, и могут эффективно защитить неврологические функции крыс с инсультом, таким образом могут, эффективно и безопасно лечить тромботические заболевания в клинической практике.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Фиг. 1 представляет собой схему синтеза соединения Ia согласно одному из вариантов выполнения настоящего изобретения;
Фиг. 2 представляет собой схему синтеза соединения Ib согласно одному из вариантов выполнения настоящего изобретения;
Фиг. 3 представляет собой схему синтеза соединения Ic согласно одному из вариантов выполнения настоящего изобретения;
Фиг. 4 представляет собой схему синтеза соединения Id согласно одному из вариантов выполнения настоящего изобретения;
Фиг. 5 представляет собой схему синтеза соединения Ie согласно одному из вариантов выполнения настоящего изобретения;
Фиг. 6 представляет собой схему синтеза соединения If согласно одному из вариантов выполнения настоящего изобретения;
Фиг. 7 представляет собой схему синтеза соединения Ig согласно одному из вариантов выполнения настоящего изобретения;
Фиг. 8 представляет собой схему синтеза соединения Ih согласно одному из вариантов выполнения настоящего изобретения; и
Фиг. 9 показывает ТЕМ-снимки наноструктур соединений 5Аа-р согласно одному из вариантов выполнения настоящего изобретения.
ПОДРОБНОЕ ОПИСАНИЕ
Подробное описание, приведенное ниже вместе с прилагаемыми чертежами и вариантами выполнения изобретения, используется для иллюстрации технического решения по настоящему изобретению. Тем не менее, объем настоящего изобретения следует рассматривать как включающий их, но не ограничивающийся ими.
В настоящем изобретении тетрагидроизохинолиновые соединения, имеющие две С1-4 алкильные группы (т.е., соединения формулы II), и пептид, обладающий тромболитической активностью, связаны через линкер с образованием нового бинарного конъюгата, одновременно обладающего тройными функциями тромболитической активности, активностью в захвате свободных радикалов и тромб-таргетинга/антитромботической активностью. Конъюгат, указанный выше, ниже сокращенно называют “новым бинарным конъюгатом по настоящему изобретению”.
Поскольку две С1-4 алкильные группы были введены в положение 1 и линкер был введен в положение 3 соединения, имеющего формулу II, “новый бинарный конъюгат по настоящему изобретению” имеет следующие четыре преимущества по сравнению с “6,7-дигидроксиизохинолином, обладающим тромболитической активностью”. 1) Эффект пространственного затруднения двух С1-4 алкильных групп, введенных в положение 1 соединения, имеющего формулу II, может блокировать приближение карбоксипептидазы и аминопептидазы, таким образом тромболитический олигопептид “нового бинарного конъюгата по настоящему изобретению” не будет легко гидролизоваться; 2) гидрофобный вклад двух С1-4 алкильных групп, введенных в положении 1 соединения, имеющего формулу II, может позволить “новому бинарному конъюгату по настоящему изобретению” пересекать гематоэнцефалический барьер с большей эффективностью; 3) эффект донорства электронов двух С1-4 алкильных групп, введенных в положение 1 соединения, имеющего формулу II, может обеспечить восстанавливающую способность “новому бинарному конъюгату по настоящему изобретению” для удовлетворения потребности в захвате свободных радикалов; 4) линкер, введенный в положение 3 соединения, имеющего формулу II, может позволить “новому бинарному конъюгату по настоящему изобретению” эффективно агрегироваться с образованием наносферических структур, имеющих диаметр 20-210 нм, предпочтительно в диапазоне 20-100 нм. Эта устойчивая наноструктура может помочь “новому бинарному конъюгату по настоящему изобретению” избежать поглощения макрофагами в кровообращении, таким образом оно может быть безопасно транспортировано к области формирования тромба и, в конечном итоге, пересечь гематоэнцефалический барьер. Таким образом, “новый бинарный конъюгат по настоящему изобретению” может образовывать наноструктуру для достижения функции пересечения гематоэнцефалического барьера. В дополнение к тромболитической и антитромботической функциям “новый бинарный конъюгат по настоящему изобретению” может также эффективно захватывать свободные радикалы OH, NO, и супероксиданионов и может достигать эффективного тромболизиса при низкой дозе, тем самым предоставляя хорошие перспективы для клинического применения.
Как используется в настоящем документе, “группа для связывания” означает функциональную группу, такую как карбоксильная группа или аминогруппа, способную вступать в реакцию конденсации.
Как используется в настоящем документе, “линкер” означает молекулу, имеющую группы для связывания, способные связывать соединение, имеющее формулу II, с олигопептидом Q, обладающим тромболитической активностью. По меньшей мере одна группа линкера для связывания представляет собой аминогруппу, а остальные группы для связывания являются карбоксильными группами или аминогруппами. В соответствии с изобретением линкер может быть природной аминокислотой, такой как L-Lys, L-Asp, or L-Glu.
Введение линкера позволяет “новому бинарному конъюгату по настоящему изобретению” формировать стабильную наносферическую структуру, позволяющую избежать поглощения макрофагами. Наносферическая структура может быть безопасно транспортирована к области формироввания тромба и, в конечном итоге, пересечь гематоэнцефалический барьер. В особенности, если линкер представляет собой L-Lys, “новый бинарный конъюгат по настоящему изобретению” может эффективно агрегироваться с формированием наносферических структур, имеющих диаметр 20-210 нм, предпочтительно в диапазоне 20-100 нм. Устойчивая наноструктура может помочь “новому бинарному конъюгату по настоящему изобретению” избежать поглощения макрофагами в кровообращении, таким образом позволяя ему безопасно транспортироваться к области формирования тромба и, в конечном итоге, пересечь гематоэнцефалический барьер.
Как используется в настоящем документе, “олигопептид” означает небольшую пептидную молекулу, имеющую молекулярную массу ниже 1000 Дальтон (D) и, обычно, состоящую из 3-8 аминокислот.
Как используется в настоящем документе, “пептид, обладающий тромболитической активностью” означает олигопептидный тромболитический агент, имеющий функции повышения проницаемости сосудов и тромболизиса, включая Р6А (ARPAK), метаболиты Р6А и родственные производные. В предыдущем исследовании было раскрыто, что Pro-Ala-Lys был самой короткой последовательностью с хорошей активностью, а также самой стабильной последовательностью среди нескольких тромболитических олигопептидов, включая Ala-Arg-Pro-Ala-Lys, Gly-Arg-Pro-Ala-Lys, Gln-Arg-Arg-Pro-Ala-Lys и Pro-Ala-Lys. Введение трипептида, имеющего последовательность Pro-Ala-AA в положении 3 соединения, имеющего формулу II, через линкер может позволить “новому бинарному конъюгату по настоящему изобретению” получить улучшенную стабильность и более сильную тромболитическую активность.
Например, олигопептид, содержащий последовательность PAK, AKP или КАР, используемый в настоящем изобретении, может быть PAK, RPAK (Arg-Pro-Ala-Lys), ARPAK (Ala-Arg-Pro-Ala-Lys), GRPAK (Gly-Arg-Pro-Ala-Lys), QRPAK (Gln-Arg-Pro-Ala-Lys), АКР, КАР, KPAK (Lys-Pro-Ala-Lys), PAKP (Pro-Ala-Lys-Pro), AKPAK (Ala-Lys-Pro-Ala-Lys) или PAKPA (Pro-Ala-Lys-Pro-Ala).
Например, пептид, имеющий повторяющиеся звенья последовательности PAK, последовательности AKP или последовательности KAP, используемый в настоящем изобретении, может быть любым из пептидов, описанных в Китайской Патентной Публикации CN 101190941, как пептидом, обладающим тромболитической активностью, включая пептид, имеющий повторяющиеся звенья последовательности PAK, такие как (PAK)2, (PAK)3, (PAK)4, (PAK)5 и (PAK)6; и пептид, имеющий повторяющиеся звенья последовательности AKP, такие как (AKP)2, (AKP)3, (AKP)4, (AKP)5 и (AKP)6; и пептид, имеющий повторяющиеся звенья последовательности КРА, такие как (KPA)2, (KPA)3, (KPA)4, (KPA)5 и (KPA)6.
Как используется в настоящем документе, “С1-4 алкильная группа” означает алкильную группу, имеющую 1-4 атомов углерода, такую как метил, этил, н-пропил, изопропил, бутил, изобутил или втор-бутил, трет-бутил. Когда R1 и R2 в соединении, имеющем формулу I, оба являются метальными группами, соединение, имеющее формулу II, используемое в качестве исходного вещества, может быть получено конденсацией Пикте-Шпенглера 3,4-дигидрокси-L-фенилаланина и ацетона в присутствии трифторацетата (TFA) и безводного сульфата магния. Преимущество заключается в более легком получении.
В настоящем изобретении фармацевтическая композиция может быть любыми клинически приемлемыми и подходящими составами. Предпочтительно, состав является инъекционными формами (порошок для инъекций, лиофилизированный порошок для инъекций, жидкость для инъекций, инфузионный раствор и т.д.). Фармацевтически приемлемый носитель может быть маннитом, водой, раствором Рингера или изотоническим раствором хлорида натрия и т.д.
Наносферическая структура соединений в соответствии с настоящим изобретением имеет диаметр 20-210 нм и более предпочтительно 20-100 нм, с помощью которой эти соединения могут более эффективно пересекать гематоэнцефалический барьер.
Стабильная наноструктура может помочь соединениям по настоящему изобретению не быть поглощенными макрофагами, в связи с чем соединения могут безопасно транспортироваться к области формирования тромбоза и в конечном итоге пересечь гематоэнцефалический барьер. Фармацевтическая композиция по настоящему изобретению может быть использована в качестве тромболитических препаратов для лечения инфаркта миокарда, ишемического инсульта, тромбоза глубоких вен, легочной эмболии, окклюзионной болезни периферических артерий, закупоренных центральных магистралей сосудистого доступа, сгустков в артериовенозных фистулах и шунтах, стеноза сонной артерии и т.д. Фармацевтическая композиция этого изобретения может быть также использована в качестве препарата для захвата свободных радикалов NO в лечении нейродегенеративных заболеваний, сердечно-сосудистых заболеваний, психических заболеваний, болезни высоты, сахарного диабета, ревматоидногго артрита, травматического повреждения головного мозга, рака, синдрома фрагильной X-хромосомы, серповидно-клеточной анемии, красного плоского лишая, витилиго, синдрома хронической усталости и т.д. Фармацевтическая композиция по настоящему изобретению также может быть использована в качестве тромб-таргетингового/антитромботического препарата для лечения тромбоцитоза, миелопролиферативного заболевания, истинной полицитемии, синдрома Бадда-Киари и т.д.
Фармацевтические композиции/соединения по настоящему изобретению одновременно обладают способностью захвата ОН, NO и супероксиданионных свободных радикалов, тромболитической активностью, а также тромб-таргетной/антитромботической функциями. Соответственно, они могут оставаться терапевтически эффективными у пациентов более 4 часов от начала симптомов инсульта, т.е. их использование не ограничивается 3-часовым окном лечения tPA. Кроме того, применение фармацевтических композиций/соединений по настоящему изобретению не вызывает системных реакций в виде кровотечения, которые вызывает tPA, и может способствовать захвату огромного количества свободных радикалов ОН, NO и супероксиданионов в процессе ишемии-реперфузии, таким образом предотвращая повреждения тканей головного мозга у пациентов, которых лечат. Поскольку две С1-4 алкильные группы и линкер соответственно введены в положения 1 и 3 соединений, имеющих формулу II, по сравнению с “6,7-дигидроксиизохинолином, имеющим тромболитическую активность”, “новый бинарный конъюгат по настоящему изобретению “демонстрирует лучшую тромболитическую активностью, уникальную способность к захвату свободных радикалов и антитромботическую активность в низкой дозе, а также превосходный терапевтический эффект при лечении инсульта через 4 часа от начала инсульта в более высокой дозе.
В выданном Китайском Патенте No. CN 101497651 B было показано, что “6,7-дигидроксиизохинолин, обладающий тромболитической активностью” имеет тромболитическую активность в дозе 10 нмоль/кг. Однако соединения по настоящему изобретению имеют хорошую тромболитическую и антитромботическую активность в дозе 0,1 нмоль/кг. Кроме того, соединения по настоящему изобретению имеют значительный терапевтический эффект при лечении инсульта через 4, 6 и 24 часа после начала инсульта в дозе 1, 2,5 и 5 мкмоль/кг, соответственно.
В способе получения соединений по настоящему изобретению пептид Q, обладающего тромболитической активностью, может быть получен первым, а затем связан со второй группой для связывания линкера Т, в качестве альтернативы, одна или более аминокислот пептида Q, обладающих тромболитической активностью, может быть последовательно соединена с линкером Т в заданном порядке. Например, первая аминокислота на одном конце тромболитического пептида Q связывается со второй группой для связывания линкера Т, и одна или более из остальных аминокислот затем последовательно соединяются с ней.
Способ получения по настоящему изобретению описан более подробно ниже для дальнейшего понимания.
Соединение формулы II может быть получено следующим путем синтеза:
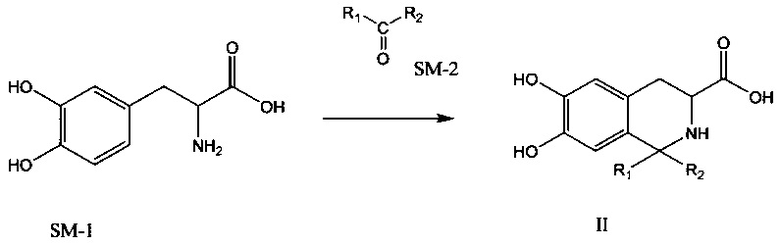
где R1 и R2 оба представляют собой С1-4 алкильную группу и могут быть одинаковыми или различными. Например, 3,4-дигидрокси-L-фенилаланин (SM-1) и соединение SM-2 растворяют в TFA и 3,4-дигидрокси-L-фенилаланин и SM-2 подвергают конденсации Пикте-Шпенглера для получения соединения, имеющего формулу II, в присутствии безводного сульфата магния.
В предпочтительном варианте выполнения изобретения линкером в способе получения по настоящему изобретению является L-Lys, и пептид, обладающий тромболитической активностью, является трипептидом, имеющим последовательность РА (Pro-Ala). Например, карбоксильная группа соединения II соединяется с N-концом L-Lys, а затем трипептид, содержащий последовательность РА, соединяется с остальным N-концом или C-концом линкера L-Lys. В некоторых вариантах выполнения изобретения, когда R1 и R2 оба представляют собой метальную группу (т.е. 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-карбоновая кислота), линкером является L-Lys, и пептид, обладающий тромболитической активностью, является трипептидом, содержащим последовательность PA (Pro-Ala), соединение Ia, Ib, Ic или Id может быть получено в соответствии со способом получения по настоящему изобретению.
Когда соединение Ia получают в соответствии со способом получения по настоящему изобретению, может быть выполнена схема синтеза, раскрытая на Фиг. 1.
В варианте выполнения изобретения, показанном на Фиг. 1, сначала синтезируют трипептид, содержащий последовательность РА, а затем связывают с линкером L-Lys, в котором АА выбирается из остатка L-Ala, L-Val, L-Trp, L-Tyr, L-Pro, L-Phe, Gly, L-Ser, L-Ile, L-Thr, L-Lys, L-Leu, L-Gln, L-Asn, L-Asp и L-Glu (соответственно подходящим соединениям 5Aa-p). Условия реакции перечислены ниже: i) Ацетон, TFA, MgSO4; ii) HCl, Lys(Boc)-OBzl, DCC, HOBt, NMM; iii) 4 M HCl/EA, ледяная баня; iv) DCC, HOBt, NMM, v) EtOH, Pd/C; 4 M HCl/EA, ледяная баня; vi) EtOH, Pd/C; vii) 2 M NaOH. В другом варианте выполнения изобретения одна аминокислота трипептида, содержащего последовательность РА (такую как АА), связывается с линкером L-Lys, а остальные две аминокислоты (такие как Pro-Ala) трипептида, содержащего последовательность РА, затем присоединяются к АА.
Например, в одном варианте формирования соединения Ia способ получения по настоящему изобретению может включать следующие стадии:
1) В присутствии TFA и безводного сульфата магния 3,4-дигидрокси-L-фенилаланин и ацетон подвергаются конденсации Пикте-Шпенглера с получением 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-карбоновой кислоты;
2) В присутствии дициклогексилкарбодиимида (DCC) и N-гидроксибензотриазолтриазола (HOBt), 3s-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-карбоновая кислота конденсировалась с HCl⋅Lys(Boc)-OBzl в безводном N,N-диметилформамиде (DMF) с образованием 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys(Вос)-OBzl. В реакции конденсации N-метилморфолин (NMM) использовался для постоянной регулировки смеси до pH=9;
3) Удаление Boc из 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys(Boc)-OBzl в этилацетатном растворе HCl с получением 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys-OBzl;
4) В присутствии DCC и HOBt Boc-Pro конденсировался с Tos⋅Ala-OBzl в безводном THF с образованием Boc-Pro-Ala-OBzl;
5) В EtOH Boc-Pro-Ala-OBzl гидрогенолизуется с образованием Boc-Pro-Ala;
6) В присутствии DCC и HOBt Boc-Pro-Ala конденсировался с AA-Obzl в безводном THF с образованием Boc-Pro-Ala-AA-OBzl (АА выбирается из остатков L-Ala, Gly, L-Phe, L-Val, L-Leu, L-Ile, L-Trp, L-Ser, L-Thr, L-Tyr, L-Lys(Z), L-Pro, L-Asn и L-Gln);
7) В EtOH Boc-Pro-Ala-AA-OBzl со стадии 6 гидрогенолизуется с образованием Boc-Pro-Ala-АА;
8) В присутствии DCC и HOBt 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys-OBzl конденсировался с Boc-Pro-Ala-AA со стадии 6 в безводном DMF с образованием 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys(Boc-Pro-Ala-AA)-OBzl (определение АА является таким же, как на стадии 6);
9) В присутствии DCC и HOBt 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys-OBzl конденсировался с Boc-AA(OBzl) в безводном DMF с образованием 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys[Boc-AA(OBzl)]-OBzl (АА выбирается из остатка L-Asp, L-Glu);
10) В этилацетатном растворе HCl Boc удаляли из 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys[Boc-AA(OBzl)]-OBzl с получением 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys[AA(OBzl)]-OBzl (определение АА является таким же, как на стадии 9);
11) В присутствии DCC и HOBt 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys[AA(OBzl)]-OBzl конденсировали с Boc-Pro-Ala в безводном DMF с образованием 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys[Boc-Pro-Ala-AA(OBzl)]-OBzl (определение AA является таким же, как на стадии 9);
12) После гидрогенолиза и удаления Boc как с 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys(Boc-Pro-Ala-AA)-OBzl (определение АА является таким же, как на стадии 6), так и с 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys[Boc-Pro-Ala-AA(OBzl)]-OBzl (определение АА является таким же, как на стадии 9) снимали защиту с получением 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys(Pro-Ala-АА).
Когда соединение Ib по настоящему изобретению получают в соответствии со способом получения по настоящему изобретению, может быть использована схема синтеза, показанная на Фиг. 2, где АА выбирается из остатков L-Ala, L-Val, L-Trp, L-Tyr, L-Pro, L-Phe, Gly, L-Ser, L-Ile, L-Thr, L-Lys, L-Leu, L-Gln, L-Asn, L-Asp и L-Glu (соответственно, соответствующих соединениям 5Ba-p). Условия реакции перечислены ниже: i) Ацетон, TFA, MgSO4; ii) HCl, Lys(Boc)-OBzl, DCC, HOBt, NMM; iii) EtOH, Pd/C; iv) DCC, HOBt, NMM, v) EtOH, Pd/C; 4 M HCl/EA, ледяная баня; vi) EtOH, Pd/C; vii) 4 M HCl/EA, ледяная баня.
Когда соединение Ic по настоящему изобретению получают в соответствии со способом получения по настоящему изобретению, может быть использована схема синтеза, показанная на Фиг. 3, где АА выбирается из остатков L-Ala, L-Val, L-Trp, L-Tyr, L-Pro, L-Phe, Gly, L-Ser, L-Ile, L-Thr, L-Lys, L-Leu, L-Gln, L-Asn, L-Asp и L-Glu (соответственно, соответствующих соединениям 5Ca-p). Условия реакции перечислены ниже: i) Ацетон, TFA, MgSO4; ii) HCl, Boc-Lys-OBzl, DCC, HOBt, NMM; iii) 4 M HCl/EA, ледяная баня; iv) DCC, HOBt, NMM, v) EtOH, Pd/C; 4 M HCl/EA, ледяная баня; vi) EtOH, Pd/C; vii) EtOH, Pd/C.
Когда соединение Id по настоящему изобретению получают в соответствии со способом получения по настоящему изобретению, может быть использована схема синтеза, показанная на Фиг. 4, где АА выбирается из остатков L-Ala, L-Val, L-Trp, L-Tyr, L-Pro, L-Phe, Gly, L-Ser, L-Ile, L-Thr, L-Lys, L-Leu, L-Gln, L-Asn, L-Asp и L-Glu (соответственно, соответствующих соединениям 5Da-p). Условия реакции перечислены ниже: i) Ацетон, TFA, MgSO4; ii) HCl, Boc-Lys-OBzl, DCC, HOBt, NMM; iii) EtOH, Pd/C; iv) DCC, HOBt, NMM, v) EtOH, Pd/C; 4 M HCl/EA, ледяная баня; vi) EtOH, Pd/C; vii) EtOH, Pd/C.
В другом предпочтительном варианте выполнения изобретения линкер представляет собой L-Asp и пептид, обладающий тромболитической активностью, является трипептидом, содержащим последовательность PA (Pro-Ala). Например, карбоксильная группа соединения, имеющего формулу II, связывается с N-концом L-Asp, а затем трипептид, содержащий последовательность РА, связывается с одним из остальных С-концов линкера L-Asp. В некоторых вариантах выполнения изобретения когда R1 и R2 соединения, имеющего формулу II, оба представляют собой метальную группу (т.е. 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацилкарбоновую кислоту), линкером является L-Asp, и пептид, обладающий тромболитической активностью, является трипептидом, содержащим последовательность PA (Pro-Ala), способ получения по настоящему изобретению может приводить к соединениям Ie или If выше.
Когда соединение Ie по настоящему изобретению получают в соответствии со способом получения по настоящему изобретению, может быть использована схема синтеза, показанная на Фиг. 5, где АА выбирается из остатков L-Ala, L-Val, L-Trp, L-Tyr, L-Pro, L-Phe, Gly, L-Ser, L-Ile, L-Thr, L-Lys, L-Leu, L-Gln, L-Asn, L-Asp и L-Glu (соответственно, соответствующих соединениям 5Ea-p). Условия реакции перечислены ниже: i) Ацетон, TFA, MgSO4; ii) HCl, Asp(OCH3)-OBzl, DCC, HOBt, NMM; iii) EtOH, Pd/C; iv) DCC, HOBt, NMM, v) EtOH, Pd/C; 2 M NaOH, ледяная баня; vi) EtOH, Pd/C; vii) 4 M HCl/EA, ледяная баня.
Когда соединение If по настоящему изобретению получают в соответствии со способом получения по настоящему изобретению, может быть использована схема синтеза, показанная на Фиг. 6, где АА выбирается из остатков L-Ala, L-Val, L-Trp, L-Tyr, L-Pro, L-Phe, Gly, L-Ser, L-Ile, L-Thr, L-Lys, L-Leu, L-Gln, L-Asn, L-Asp и L-Glu (соответственно, соответствующих соединениям 5Fa-p). Условия реакции перечислены ниже: i) Ацетон, TFA, MgSO4; ii) HCl, Asp(OBzl)-OCH3, DCC, HOBt, NMM; iii) 2 M NaOH; iv) DCC, HOBt, NMM, v) EtOH, Pd/C; 2 M NaOH, ледяная баня; vi) EtOH, Pd/C; vii) 4 M HCl/EA, ледяная баня.
В другом предпочтительном варианте выполнения изобретения линкер представляет собой L-Glu, и пептид, обладающий тромболитической активностью, является трипептидом, содержащим последовательность PA (Pro-Ala). Например, карбоксильная группа соединения, имеющего формулу II, связывается с N-концом L-Glu, а затем трипептид, содержащий последовательность РА, связывается с одним из остальных С-концов линкера L-Glu. В некоторых вариантах выполнения изобретения когда R1 и R2 соединения, имеющего формулу II, оба представляют собой метальную группу (т.е. 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацилкарбоновую кислоту), линкером является L-Glu, и пептид, обладающий тромболитической активностью, является трипептидом, содержащим последовательность PA (Pro-Ala), способ получения по настоящему изобретению может приводить к соединениям Ig или Ih выше.
Когда соединение Ig по настоящему изобретению получают в соответствии со способом получения по настоящему изобретению, может быть использована схема синтеза, показанная на Фиг. 7, где АА выбирается из остатков L-Ala, L-Val, L-Trp, L-Tyr, L-Pro, L-Phe, Gly, L-Ser, L-Ile, L-Thr, L-Lys, L-Leu, L-Gln, L-Asn, L-Asp и L-Glu (соответственно, соответствующих соединениям 5Ga-p). Условия реакции перечислены ниже: i) Ацетон, TFA, MgSO4; ii) HCl, Glu(OCH3)-OBzl, DCC, HOBt, NMM; iii) EtOH, Pd/C; iv) DCC, HOBt, NMM, v) EtOH, Pd/C; 2 M NaOH, ледяная баня; vi) EtOH, Pd/C; vii) 4 M HCl/EA, ледяная баня.
Когда соединение Ih по настоящему изобретению получают в соответствии со способом получения по настоящему изобретению, может быть использована схема синтеза, показанная на Фиг. 8, где АА выбирается из остатков L-Ala, L-Val, L-Trp, L-Tyr, L-Pro, L-Phe, Gly, L-Ser, L-Ile, L-Thr, L-Lys, L-Leu, L-Gln, L-Asn, L-Asp и L-Glu (соответственно, соответствующих соединениям 5Ha-p). Условия реакции перечислены ниже: i) Ацетон, TFA, MgSO4; ii) HCl, Glu(OBzl)-OCH3, DCC, HOBt, NMM; iii) EtOH, Pd/C; iv) DCC, HOBt, NMM, v) EtOH, Pd/C; 2 M NaOH, ледяная баня; vi) EtOH, Pd/C; vii) 4 M HCl/EA, ледяная баня.
Исследования in vivo на крысах соединений и фармацевтических композиций по настоящему изобретению показывают, что соединения и фармацевтические композиции по настоящему изобретению обладают превосходной тромболитической и антитромботической активностью при низкой дозе, и могут эффективно защитить неврологические функции у крыс с инсультом. Таким образом, соединения и фармацевтические композиции по настоящему изобретению могут эффективно и безопасно лечить тромботические заболевания в клинической практике.
Настоящее изобретение теперь будет описано следующими конкретными примерами, и преимущества и особенности станут понятными в свете описания. Эти примеры являются только иллюстративными и никоим образом не ограничивают объем настоящего изобретения. Специалисту в данной области техники может быть понятно, что модификация или замена могут быть сделаны в деталях и формально для технических решений по настоящему изобретению, не отклоняясь от сущности и объема настоящего изобретения, и эти модификации или замены предназначены находиться в пределах объема охраны настоящего изобретения.
Примеры 1-68 иллюстрируют способы получения с Фиг. 1 для получения соединений 5Аа-р по настоящему изобретению.
Пример 1: Общая методика синтеза пептида в жидкой фазе
Аминокислоту, имеющую защищенный N-конец, растворяли в безводном тетрагидрофуране (THF) и в полученный раствор добавляли N-гидроксибензотриазолтриазол (HOBt). Охлаждают раствор на ледяной бане. N,N-дициклогексилкарбодиимид (DCC) растворяли в безводном THF, медленным добавлением к нему, а затем перемешивали при 0°C в течение 15 минут с получением реакционного раствора (I). Аминокислоту, имеющую защищенный С-конец, также растворяли в безводном THF, доводили до pH 9 при помощи N-метилморфолина (NMM), а затем смешивали с реакционным раствором (I) при поддержании pH 9 при помощи N-метилморфолина (NMM). Реакционную смесь перемешивали при комнатной температуре в течение 10 часов и контролировали протекание реакции с помощью TLC. После того как исчезало пятно исходного вещества согласно TLC, реакционную смесь фильтровали и фильтрат концентрировали при пониженном давлении. Полученный вязкий концентрат растворяли в этилацетате (ЕА) или дихлорметане, полученный раствор последовательно промывали 5%-ным водным раствором NaHCO3, 5%-ным водным раствором KHSO4 и насыщенным водным раствором NaCl. Фазу ЕА или дихлорметана сушили безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении с получением целевого соединения.
Пример 2: Общая методика удаления Boc
Вос-защищенный пептид растворяли в небольшом количестве безводного этилацетата, к нему добавляли этилацетатный раствор HCl (4 М) и перемешивали на ледяной бане, пока пятно исходного вещества не исчезало согласно TLC. Реакционный раствор многократно сушили с помощью водяного насоса и также полностью удаляли газ HCl. Остаток многократно растирали с петролейным эфиром или безводным эфиром с получением целевых соединений.
Пример 3: Общая методика удаления бензильной сложноэфирной группы
Полипептид, защищенный бензильной сложноэфирной группой, растворяли в CH3OH, по каплям добавляли к нему водный раствор NaOH (2 М) и перемешивали на ледяной бане, реакционный раствор выдерживали при 0°C до тех пор, пока пятно исходного вещества не исчезало согласно TLC. Реакционный раствор доводили до нейтрального с помощью 1 М HCl и удаляли МеОН при пониженном давлении. Реакционный раствор подкисляли до pH 2 с помощью 1 М HCl и затем экстрагировали EA. Объединенную фазу ЕА промывали насыщенным водным раствором NaCl до нейтральной, сушили безводным Na2SO4 и фильтровали. Фильтрат концентрировали при пониженном давлении с получением целевых соединений.
Пример 4: Общая методика удаления карбобензокси-группы или бензильной сложноэфирной группы
Пептид, защищенный карбобензокси-группой или бензиловой сложноэфирной группой, растворяли в подходящем количестве EtOH, добавляемом к нему Pd/C (10% реагентов) и вводили водород для обеспечения реакции гидрогенолиза при комнатной температуре. После реакции реакционную смесь фильтровали и затем концентрировали при пониженном давлении с получением целевого соединения.
Пример 5: Получение Boc-Pro
5,75 г (50 ммоль) L-Pro растворяли в 1 мл воды. 25 мл водного раствора NaOH (2 M) добавляли к нему по каплям и медленно перемешивали на ледяной бане с получением раствора (I). 13,08 г (60 ммоль) (Boc)2O растворяли в 25 мл диоксана с получением раствора (II). Раствор (II) по каплям добавляли к раствору (I) и перемешивали на ледяной бане, а затем водный раствор NaOH (2 М) добавляли по каплям для доведения до pH 9 и смесь перемешивали на ледяной бане. Через 30 минут измеряли значение pH смеси и использовали водный раствор NaOH (2 М) для поддержания pH 9. Водяной насос использовали для экстрагирования генерируемого газа. После перемешивания в течение 48 часов контроль с помощью TLC (CH2Cl2:MeOH 20:1) показал, что реакция завершилась. Смесь концентрировали при пониженном давлении для удаления диоксана. Остаток растворяли в 5 мл воды, а затем устанавливали pH 2 насыщенным водным раствором KHSO4. Водный раствор экстрагировали ЕА 3 раза. Объединенные фазы ЕА промывали 5% KHSO4 3 раза, а затем промывали насыщенным водным раствором NaCl до нейтрального. Отделенный слой ЕА сушили добавлением безводного сульфата натрия и затем фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток перекристаллизовывали ЕА-петролейным эфиром с получением 10,2 г (95%) указанного в заголовке соединения в виде бесцветных кристаллов. ESI-MS (m/е): 214 [М-Н].
Пример 6: Получение Boc-Pro-Ala-OBzl
Следуя способу Примера 1, 5,19 г (68%) указанного в заголовке соединения в виде бесцветного порошка получали из 4,3 г (20,0 ммоль) Вое-Pro и 8,45 г (24,0 ммоль) Tos⋅Ala-OBzl. ESI-MS (m/e): 377 [М+Н]+.
Пример 7: Получение Boc-Pro-Ala
Следуя способу Примера 4, 3,59 г (91%) указанного в заголовке соединения в виде бесцветного порошка получали из 5,19 г (13,8 ммоль) Boc-Pro-Ala-OBzl. ESI-MS (m/e): 285 [М-Н]-.
Пример 8: Получение Boc-Pro-Ala-Ala-OBzl
Следуя способу Примера 1, 3,04 г (68%) указанного в заголовке соединения в виде бесцветного порошка получали из 3,00 г (10,49 ммоль) Boc-Pro-Ala-OBzl и 3,26 г (12,03 ммоль) Tos⋅Ala-OBzl. ESI-MS (m/e): 448 [M+H]+.
Пример 9: Получение Boc-Pro-Ala-Ala
Следуя способу Примера 3, 3,21 г (90%) указанного в заголовке соединения получали из 4,47 г (10 ммоль) Boc-Pro-Ala-Ala-OBzl. ESI-MS (m/e): 356 [М-Н]-.
Пример 10: Получение Boc-Pro-Ala-Val-OBzl
Следуя способу Примера 1, 3,28 г (69%) указанного в заголовке соединения в виде бесцветного порошка получали из 3,00 г (10,49 ммоль) Boc-Pro-Ala и 3,58 г (12,01 ммоль) Tos⋅Val-OBzl. ESI-MS (m/e): 476 [М+Н]+.
Пример 11: Получение Boc-Pro-Ala-Val
Следуя способу Примера 3, 3,54 г (92%) указанного в заголовке соединения получали из 4,75 г (10 ммоль) Boc-Pro-Ala-Val-OBzl. ESI-MS (m/e): 384 [М-Н]-.
Пример 12: Получение Boc-Pro-Ala-Trp-OBzl
Следуя способу Примера 1, 3,65 г (65%) указанного в заголовке соединения в виде бесцветного порошка получали из 3,00 г (10,49 ммоль) Boc-Pro-Ala и 3,97 г (12,01 ммоль) HCl⋅Trp-OBzl. ESI-MS (m/e): 563 [М+Н]+.
Пример 13: Получение Boc-Pro-Ala-Trp
Следуя способу Примера 3, 4,21 г (89%) указанного в заголовке соединения получали из 5,62 г (10 ммоль) Boc-Pro-Ala-Trp-OBzl. ESI-MS (m/e): 471 [М-Н]-.
Пример 14: Получение Boc-Pro-Ala-Tyr-OBzl
Следуя способу Примера 1, 3,73 г (69%) указанного в заголовке соединения в виде бесцветного порошка получали из 3,15 г (11 ммоль) Boc-Pro-Ala и 4,43 г (10 ммоль) Tos⋅Tyr-OBzl. ESI-MS (m/e): 540 [М+Н]+.
Пример 15: Получение Boc-Pro-Ala-Tyr
Следуя способу Примера 3, 4,13 г (92%) указанного в заголовке соединения получали из 5,39 г (10 ммоль) Boc-Pro-Ala-Tyr-OBzl. ESI-MS (m/e): 448 [М-Н]-.
Пример 16: Получение Boc-Pro-Ala-Phe-OBzl
Следуя способу Примера 1, 3,82 г (66%) указанного в заголовке соединения в виде бесцветного порошка получали из 3,00 г (10,0 ммоль) Boc-Pro-Ala и 2,92 г (11,0 ммоль) Tos⋅Phe-OBzl. ESI-MS (m/e): 524 [М+Н]+.
Пример 17: Получение Boc-Pro-Ala-Phe
Следуя способу Примера 3, 3,94 г (91%) указанного в заголовке соединения получали из 5,23 г (10 ммоль) Boc-Pro-Ala-Phe-OBzl. ESI-MS (m/e): 432 [М-Н]-.
Пример 18: Получение Boc-Pro-Ala-Gly-OBzl
Следуя способу Примера 1, 2,90 г (67%) указанного в заголовке соединения в виде бесцветного порошка получали из 3,15 г (11 ммоль) Boc-Pro-Ala и 3,37 г (10 ммоль) Tos⋅Gly-OBzl. ESI-MS (m/e): 434 [М+Н]+.
Пример 19: Получение Boc-Pro-Ala-Gly
Следуя способу Примера 3, 2,98 г (87%) указанного в заголовке соединения получали из 4,33 г (10 ммоль) Boc-Pro-Ala-Gly-OBzl. ESI-MS (m/e): 342 [М-Н]-.
Пример 20: Получение Boc-Pro-Ala-Ser-OBzl
Следуя способу Примера 1, 2,92 г (63%) указанного в заголовке соединения в виде бесцветного порошка получали из 3,15 г (11 ммоль) Boc-Pro-Ala и 3,67 г (10 ммоль) Tos⋅Ser-OBzl. ESI-MS (m/e): 464 [М+Н]+.
Пример 21: Получение Boc-Pro-Ala-Ser
Следуя способу Примера 3, 3,28 г (88%) указанного в заголовке соединения получали из 4,63 г (10 ммоль) Boc-Pro-Ala-Ser-OBzl. ESI-MS (m/e): 372 [М-Н]-.
Пример 22: Получение Boc-Pro-Ala-Ile-OBzl
Следуя способу Примера 1, 3,33 г (65%) указанного в заголовке соединения в виде бесцветного порошка получали из 3,15 г (11 ммоль) Boc-Pro-Ala и 3,93 г (10 ммоль) Tos⋅Ile-OBzl. ESI-MS (m/e): 490 [М+Н]+.
Пример 23: Получение Boc-Pro-Ala-Ile
Следуя способу Примера 3, 3,67 г (91%) указанного в заголовке соединения получали из 4,89 г (10 ммоль) Boc-Pro-Ala-Ile-OBzl. ESI-MS (m/e): 398 [М-Н]-.
Пример 24: Получение Boc-Pro-Ala-Thr-OBzl
Следуя способу Примера 1, 3,39 г (71%) указанного в заголовке соединения в виде бесцветного порошка получали из 3,15 г (11 ммоль) Boc-Pro-Ala и 3,81 г (10 ммоль) Tos⋅Thr-OBzl. ESI-MS (m/e): 478 [М+Н]+.
Пример 25: Получение Boc-Pro-Ala-Thr
Следуя способу Примера 3, 3,56 г (91%) указанного в заголовке соединения получали из 4,77 г (10 ммоль) Boc-Pro-Ala-Thr-OBzl. ESI-MS (m/e): 386 [М-Н]-.
Пример 26: Получение Boc-Pro-Ala-Lys(Z)-OBzl
Следуя способу Примера 1, 4,15 г (65%) указанного в заголовке соединения в виде бесцветного порошка получали из 3,15 г (11 ммоль) Boc-Pro-Ala и 4,06 г (10 ммоль) HCl⋅Lys(Z)-OBzl. ESI-MS (m/e): 639 [М+Н]+.
Пример 27: Получение Boc-Pro-Ala-Lys(Z)
Следуя способу Примера 3, 4,71 г (86%) указанного в заголовке соединения получали из 6,39 г (10 ммоль) Boc-Pro-Ala-Lys(Z)-OBzl. ESI-MS (m/e): 547[М-Н]-.
Пример 28: Получение Boc-Pro-Ala-Leu-OBzl
Следуя способу Примера 1, 3,37 г (69%) указанного в заголовке соединения в виде бесцветного порошка получали из 3,15 г (11 ммоль) Boc-Pro-Ala и 3,93 г (10 ммоль) Tos⋅Leu-OBzl. ESI-MS (m/e): 490 [М+Н]+.
Пример 29: Получение Boc-Pro-Ala-Leu
Следуя способу Примера 3, 3,67 г (91%) указанного в заголовке соединения получали из 4,89 г (10 ммоль) Boc-Pro-Ala-Leu-OBzl. ESI-MS (m/e): 398 [М-Н]-.
Пример 30: Получение Boc-Pro-Ala-Gln-OBzl
Следуя способу Примера 1, 3,23 г (63%) указанного в заголовке соединения в виде бесцветного порошка получали из 3,15 г (11 ммоль) Boc-Pro-Ala и 2,73 г (10 ммоль) HCl⋅Gln-OBzl. ESI-MS (m/e): 505 [М+Н]+.
Пример 31: Получение Boc-Pro-Ala-Gln
Следуя способу Примера 3, 3,73 г (90%) указанного в заголовке соединения получали из 5,04 г (10 ммоль) Boc-Pro-Ala-Gln-OBzl. ESI-MS (m/e): 413 [М-Н]-.
Пример 32: Получение Boc-Pro-Ala-Asn-OBzl
Следуя способу Примера 1, 3,04 г (61%) указанного в заголовке соединения в виде бесцветного порошка получали из 3,15 г (11 ммоль) Boc-Pro-Ala и 2,59 г (10 ммоль) HCl⋅Asn-OBzl. ESI-MS (m/e): 491 [М+Н]+.
Пример 33: Получение Boc-Pro-Ala-Asn
Следуя способу Примера 3, 3,67 г (92%) указанного в заголовке соединения получали из 4,90 г (10 ммоль) Boc-Pro-Ala-Asn-OBzl. ESI-MS (m/e): 399 [М-Н]-.
Пример 34: Получение 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацилкарбоновой кислоты (1)
5,0 г (25 ммоль) 3,4-дигидрокси-L-фенилаланина растворяли в 250 мл ацетона, а затем в полученный раствор добавляли 6,0 г (30 ммоль) безводного сульфата магния. Через 30 минут в него добавляли 25 мл TFA на ледяной бане. Смесь перемешивали при комнатной температуре в течение 96 часов до тех пор, пока пятно исходного вещества не исчезало согласно TLC (CH2Cl2:МеОН 1:1). Реакционный раствор фильтровали. Фильтрат концентрировали при пониженном давлении и остаток растворяли в ацетоне и продолжали концентрировать при пониженном давлении 3 раза. 200 мл безводного простого эфира добавляли к остатку, чтобы осадить значительное количество бесцветного твердого вещества. После фильтрования с отсасыванием 5,8 г (95%) указанного в заголовке соединения получали в виде бесцветного твердого вещества. ESI-MS (m/e): 238 [М+Н]+; 1Н ЯМР (300 МГц, ДМСО-d6) δ/ppm=6.61 (s, 1Н), 6.45 (s, 1H), 3.70 (dd, J=3.9, 11.4 Гц, 1Н), 2.76 (dd, J=11.7, 15.3 Гц, 1H), 2.62 (m, 1H), 1.41 (s, 3H), 1.32 (s, 3H).
Пример 35: Получение 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys(Boc)-OBzl (2)
Следуя способу Примера 1, 1,19 г (5,01 ммоль) 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацилкарбоновой кислоты растворяли в 10 мл безводного DMF. К полученному раствору добавляли 675 мг (5,00 ммоль) N-гидроксибензотриазолтриазола (HOBt). Через 10 минут туда добавляли 1,20 г (5,83 ммоль) раствора дициклогексилкарбодиимида (DCC) и 5 мл безводного DMF на ледяной бане, с получением реакционного раствора (I). Растворяли 2,83 г (5,53 ммоль) HCl⋅Lys(Boc)-OBzl в 15 мл безводного DMF и затем перемешивали в течение 30 минут с получением реакционного раствора (II). Реакционный раствор (II) добавляли в реакционный раствор (I) при перемешивании на ледяной бане. Реакционную смесь перемешивали при комнатной температуре в течение 12 часов и доводили до рН=9 с помощью МММ, при необходимости, до тех пор, пока 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацилкарбоновая кислота не исчезала согласно TLC (CH2Cl2:МеОН 10:1). Реакционную смесь фильтровали для удаления Дициклогексилмочевины (DCU). Фильтрат концентрировали при пониженном давлении для удаления DMF. Остаток растворяли в 150 мл ЕА. Полученный раствор последовательно промывали насыщенным водным раствором NaHCO3 3 раза, а затем насыщенным водным раствором NaCl 3 раза. Раствор ЕА сушили безводным Na2SO4 и фильтровали. Фильтрат концентрировали при пониженном давлении досуха. Остаток очищали с помощью колоночной хроматографии (CH2Cl2:МеОН, 50:1) с получением 327 мг (59%) указанного в заголовке соединения в виде светло-розового порошка. ESI-MS (m/e): 556 [М+Н]+; 1Н ЯМР (300 МГц, ДМСО-d6) δ/ppm=8.64 (s, 1Н), 8.52 (s, 1H), 8.14 (d, J=7.5 Гц, 1H), 7.36 (m, 5Н), 6.74 (m, 1Н), 6.57 (s, 1H), 6.37 (s, 1Н), 5.14 (m, 2Н), 4.30 (m, 1Н), 3.55 (m, 2Н), 3.32 (m, 2Н), 2.88 (m, 2Н), 2.57 (d, J=3.9 Гц, 1Н), 2.27 (m, 1H), 2.15 (s, 1H), 1.69 (m, 4Н), 1.36 (s, 9Н), 1.33 (s, 3Н), 1.25 (s, 3Н).
Пример 36: Получение 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys-OBzl (3A)
Следуя способу Примера 2, 1,01 г (82%) указанного в заголовке соединения в виде светло-розового твердого вещества получали из 1,50 г (2,73 ммоль) 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys(Boc)-OBzl, который непосредственно использовали в следующей реакции. ESI-MS (m/e): 456 [М+Н]+.
Пример 37: Получение 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys(Boc-Pro-Ala-Ala)-OBzl (4Аа)
Следуя способу Примера 1, 302 мг (38%) указанного в заголовке соединения в виде светло-желтого порошка получали из 482 мг (1,01 ммоль) 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys-OBzl и 424 мг (1,20 ммоль) Boc-Pro-Ala-Ala. ESI-MS (m/e): 795 [М+Н]+; 1Н ЯМР (300 МГц, ДМСО-d6) δ/ppm=9.15 (s, 1Н), 8.89 (s, 2Н), 8.11 (m, 1H), 7.94 (m, 1H), 7.85 (m, 1H), 7.76 (m, 1Н), 7.44-7.31 (m, 5Н), 6.66 (s, 1Н), 6.47 (s, 1Н), 5.14 (m, 2Н), 4.25 (m, 1H), 4.23 (m, 3H), 4.12 (m, 1Н), 3.66 (m, 1H), 3.12 (m, 2Н), 2.96 (m, 2Н), 2.89-2.91 (m, 2H), 2.11-2.08 (m, 1Н), 1.77 (m, 5Н), 1.59 (s, 3H), 1.47-1.27 (m, 17Н), 1.27-1.08 (m, 7Н).
Пример 38: Получение 3S-6,7-дигид рокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys(Boc-Pro-Ala-Val)-OBzl (4Ab)
Следуя способу Примера 1, 604 мг (37%) указанного в заголовке соединения в виде светло-желтого порошка получали из 964 мг (2,01 ммоль) 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys-OBzl и 924 мг (2,40 ммоль) Boc-Pro-Ala-Val. ESI-MS (m/e): 823 [M+H]+; 1H ЯМР (300 МГц, ДМСО-d6) δ/ppm=9.18 (s, 1H), 8.93 (m, 2H), 8.05 (m, 2H), 7.42 (m, 5H), 6.66 (s, 1H), 6.47 (s, 1H), 5.16 (s, 1H), 4.37-4.28 (m, 3H), 4.11 (m, 3H), 3.06-2.89 (m, 5H), 2.75 (m, 6H), 2.04 (s, IH), 1.91 (m, IH), 1.79 (m, 5H), 1.62 (s, 3H), 1.49 (s, 3H), 1.39 (m, 7H), 1.32 (m, 3H), 1.21 (m, 4H), 0.82 (m, 6H).
Пример 39: Получение 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys(Boc-Pro-Ala-Trp)-OBzl (4Ас)
Следуя способу Примера 1, 318 мг (35%) указанного в заголовке соединения в виде светло-желтого порошка получали из 482 мг (1,01 ммоль) 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys-OBzl и 567 мг (1,20 ммоль) Boc-Pro-Ala-Trp. ESI-MS (m/e): 910 [М+Н]+; 1Н ЯМР (300 МГц, ДМСО-d6) δ/ppm=9.51 (s, 1Н), 9.26 (m, 2Н), 8.98 (m, 2Н), 8.05 (d, 1H, J=7.2 Гц), 7.94 (m, 1H), 7.78 (d, 1Н, J=7.2 Гц), 7.54 (m, 1H), 7.38 (m, 5Н), 7.31 (d, 1H, J=4.5 Гц), 7.24 (m, 1H), 7.11 (t, 1H, J=4.5 Гц), 7.05 (t, 1H, J=4.5 Гц), 6.67 (s, 1H), 6.52 (s, 1Н), 5.15 (s, 2Н), 4.44 (m, 1Н), 4.35 (m, 2Н), 4.07 (m, 2Н), 3.23 (m, Ш), 3.17 (m, 5Н), 2.86 (m, 1Н), 1.97 (m, 2Н), 1.76-1.70 (m, 8Н), 1.31 (s, 3Н), 1.24 (s, 3Н), 1.93-1.79 (m, 9Н), 1.16 (m, 4Н).
Пример 40: Получение 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys(Boc-Pro-Ala-Tyr)-OBzl (4Ad)
Следуя способу Примера 1, 549 мг (31%) указанного в заголовке соединения в виде светло-желтого порошка получали из 964 мг (2,01 ммоль) 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys-OBzl и 1077 мг (2,40 ммоль) Boc-Pro-Ala-Tyr. ESI-MS (m/e): 887 [М+Н]+; 1Н ЯМР (300 МГц, ДМСО-d6) δ/ppm=9.17 (m, 1H), 9.02 (m, 1Н), 8.73 (m, 1Н), 8.51 (m, 1H), 8.09 (m, 1Н), 7.97 (m, 1H), 7.73 (m, 1Н), 7.40 (m, 5Н), 6.96 (m, 2Н), 6.61 (m, 3Н), 6.48 (s, 1H), 5.16 (s, 2Н), 4.23 (m, 2Н), 4.07 (m, 2Н), 3.88 (m, 1Н), 3.01 (m, 3Н), 2.74 (m, 3Н), 2.02 (m, 2Н), 1.74 (m, 5Н), 1.46 (s, 3Н), 1.36-1.24 (m, 16Н), 1.16 (m, 3Н), 0.87 (m, 1H).
Пример 41: Получение 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys(Boc-Pro-Ala-Pro)-OBzl (4Ае)
Следуя способу Примера 1, 607 мг (37%) указанного в заголовке соединения в виде светло-желтого порошка получали из 964 мг (2,01 ммоль) 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys-OBzl и 957 мг (2,40 ммоль) Boc-Pro-Ala-Pro. ESI-MS (m/e): 821 [М+Н]+; 1Н ЯМР (300 МГц, ДМСО-d6) δ/ppm=9.54 (m, 1Н), 9.23 (s, 1H), 9.11 (m, 1H), 8.96 (s, 1H), 8.05 (m, 1H), 7.85 (m, 1H), 7.40 (m, 5H), 6.67 (s, 1H), 6.48 (s, 1H), 5.16 (m, 2H), 4.54 (m, 1H), 4.38 (m, 2H), 4.24 (m, 1H), 4.10 (m, 2H), 3.55 (m, 1H), 3.17 (m, 3H), 3.05 (m, 3H), 2.94 (m, 1H), 1.96 (m, 2H), 1.79 (m, 7H), 1.64 (s, 3H), 1.51 (s, 3H), 1.38-1.32 (m, 13H), 1.20 (m, 4H).
Пример 42: Получение 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys(Boc-Pro-Ala-Phe)-OBzl (4Af)
Следуя способу Примера 1, 626 мг (36%) указанного в заголовке соединения в виде светло-желтого порошка получали из 964 мг (2,01 ммоль) 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys-OBzl и 1039 мг (2,40 ммоль) Boc-Pro-Ala-Phe. ESI-MS (m/e): 871 [М+Н]+; 1Н ЯМР (300 МГц, ДМСО-d6) δ/ppm=9.24 (s, 1H), 9.09 (m, 2Н), 8.96 (s, 1Н), 8.01-7.93 (m, 3H), 7.38 (m, 5Н), 7.21 (m, 5Н), 6.67 (s, 1Н), 6.49 (s, 1H), 5.17 (s, 2Н), 4.38 (m, 3Н), 4.09 (m, 2Н), 3.28 (m, 4Н), 2.88 (m, 2Н), 2.79 (m, 5Н), 1.99 (m, 2Н), 1.74 (m, 8Н), 1.50 (m, 3H), 1.32 (m, 14Н), 1.16 (m, 4Н).
Пример 43: Получение 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys(Boc-Pro-Ala-Gly)-OBzl (4Ag)
Следуя способу Примера 1, 304 мг (39%) указанного в заголовке соединения в виде светло-желтого порошка получали из 482 мг (1,01 ммоль) 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys-OBzl и 412 мг (1,20 ммоль) Boc-Pro-Ala-Gly. ESI-MS (m/e): 781 [М+Н]+; 1Н ЯМР (300 МГц, ДМСО-d6) δ/ppm=9.39 (s, 1H), 8.97 (m, 2Н), 8.24-8.14 (m, 2Н), 7.79 (m, 1Н), 7.41 (m, 5Н), 6.66 (s, 1H), 6.47 (s, 1Н), 5.16 (s, 2H), 4.35 (m, 2Н), 4.21 (m, 2Н), 3.71-3.66 (m, 2Н), 3.03 (m, 3Н), 2.89-2.76 (m, 1Н), 2.07 (m, 1Н), 1.81-1.77 (m, 6Н), 1.62 (s, 3H), 1.50 (s, 3Н), 1.38 (m, 7Н), 1.32 (m, 8Н), 1.23 (m, 4Н).
Пример 44: Получение 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys(Boc-Pro-Ala-Ser)-OBzl (4Ah)
Следуя способу Примера 1, 454 мг (29%) указанного в заголовке соединения в виде светло-желтого порошка получали из 965 мг (2,01 ммоль) 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys-OBz1 и 896 мг (2,40 ммоль) Boc-Pro-Ala-Ser. ESI-MS (m/e): 811 [М+Н]+; 1Н ЯМР (300 МГц, ДМСО-d6) δ/ppm=9.48 (m, 1H), 9.23 (s, 2Н), 8.96 (m, 1H), 8.19 (m, 1H), 7.40 (m, 5Н), 6.67 (s, 1H), 6.48 (s, 1Н), 5.16 (m, 2Н), 4.92 (m, 1H), 4.31 (m, 3Н), 4.13 (m, 3Н), 3.53 (m, 2Н), 3.16 (m, 3Н), 3.04-2.81 (m, 4Н), 2.08 (m, 1Н), 1.78 (m, 5H), 1.63 (s, 3Н), 1.51 (s, 3Н), 1.39-1.32 (m, 14Н), 1.22 (m, 3Н).
Пример 45: Получение 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys(Boc-Pro-Ala-Ile)-OBzl (4Ai)
Следуя способу Примера 1, 310 мг (37%) указанного в заголовке соединения в виде светло-желтого порошка получали из 482 мг (1,01 ммоль) 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys-OBzl и 479 мг (1,20 ммоль) Boc-Pro-Ala-Ile. ESI-MS (m/e): 837 [М+Н]+; 1Н ЯМР (300 МГц, ДМСО-d6) δ/ppm=8.84-8.68 (m, 2Н), 8.37 (s, 1Н), 7.99-7.87 (m, 3Н), 7.39 (m, 4Н), 6.60 (s, 1Н), 6.39 (s, 1Н), 5.14 (m, 2Н), 4.32 (m, 2Н), 4.17 (m, 2Н), 3.65 (m, 1H), 3.54 (m, 1H), 3.17 (m, 1Н), 2.96 (m, 2Н), 2.68 (m, 3Н), 2.11 (m, 1H), 1.74-1.68 (m, 7Н), 1.31-1.21 (m, 11Н), 1.17 (m, 5Н), 1.08 (m, 1Н), 0.82 (m, 8Н).
Пример 46: Получение 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys(Boc-Pro-Ala-Thr)-OBzl (4Aj)
Следуя способу Примера 1, 495 мг (30%) указанного в заголовке соединения в виде светло-желтого порошка получали из 964 мг (2,01 ммоль) 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys-OBzl и 929 мг (2,40 ммоль) Boc-Pro-Ala-Thr. ESI-MS (m/e): 825 [М+Н]+; 1Н ЯМР (300 МГц, ДМСО-d6) δ/ppm=9.18 (s, 1H), 8.91 (s, 2Н), 8.27 (m, 1Н),7.64 (m, 1H), 7.41 (m, 6Н), 6.65 (s, 1H), 6.48 (s, 1H), 5.16 (s, 2Н), 4.35 (m, 3Н), 4.23 (m, 3Н), 3.07 (m, 3Н), 2.72 (m, 1H), 1.83-1.76 (m, 5H), 1.61 (s, 3Н), 1.48 (s, 3Н), 1.38-1.32 (m, 14Н), 1.23 (m, 3Н), 1.11 (m, 3Н).
Пример 47: Получение 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys[Boc-Pro-Ala-Lys(Z)]-OBzl (4Ak)
Следуя способу Примера 1, 750 мг (30%) указанного в заголовке соединения в виде светло-желтого порошка получали из 964 мг (2,01 ммоль) 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys-OBzl и 1209 мг (2,40 ммоль) Boc-Pro-Ala-Lys(Z). ESI-MS (m/e): 987 [М+Н]+; 1Н ЯМР (300 МГц, ДМСО-d6) δ/ppm=8.68 (s, 1Н), 8.56 (s, 1H), 8.16 (m, 1H), 8.04 (m, 1Н), 7.78 (m, 1H), 7.68 (m, 1Н), 7.34 (m, 10H), 6.58 (s, 1H), 6.37 (s, 1H), 5.14 (s, 2Н), 4.98 (s, 2Н), 4.29 (m, 2Н), 4.09 (m, 3H), 3.56 (m, 3Н), 3.01 (т, 4Н), 2.62 (m, 1H), 2.40 (m, 1H), 2.27 (m, 1Н), 2.15 (s, 1Н), 2.06 (m, 1Н), 2.01 (s, 1Н), 1.78 (m, 6Н), 1.38-1.31 (m, 18Н), 1.17 (m, 10H).
Пример 48: Получение 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys(Boc-Pro-Ala-Leu)-OBzl (4Al)
Следуя способу Примера 1, 301 мг (36%) указанного в заголовке соединения в виде светло-желтого порошка получали из 482 мг (1,01 ммоль) 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys-OBzl и 479 мг (1,20 ммоль) Boc-Pro-Ala-Leu. ESI-MS (m/e): 837 [М+Н]+; 1Н ЯМР (300 МГц, ДМСО-d6) δ/ppm=8.69-8.57 (m, 2Н), 8.18-8.06 (m, 2Н), 7.85 (m, 1H), 7.69 (m, 5Н), 6.58 (s, 1H), 6.37 (s, 1Н), 5.14 (m, 2Н), 4.25-4.11 (m, 5Н), 3.63-3.55 (m, 1Н), 3.17 (s, 2Н), 3.04-2.98 (m, 2Н), 2.64-2.59 (m, 1Н), 2.15-2.07 (m, 1H), 1.78-1.67 (m, 5Н), 1.36 (m, 14Н), 1.28 (m, 4Н), 1.19 (m, 6Н).
Пример 49: Получение 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys(Boc-Pro-Ala-Gln)-OBzl (4Am)
Следуя способу Примера 1, 665 мг (39%) указанного в заголовке соединения в виде светло-желтого порошка получали из 965 мг (2,01 ммоль) 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys-OBzl и 994 мг (2,40 ммоль) Boc-Pro-Ala-Gln. ESI-MS (m/e): 852 [М+Н]+; 1Н ЯМР (300 МГц, ДМСО-d6) δ/ppm=9.3-9.1 (m, 4Н), 8.16 (m, 1Н), 7.96 (m, 2Н), 7.83 (m, 1H), 7.44-7.38 (m, 7H), 6.89 (s, 1Н), 6.86 (s, 1H), 6.56 (s, 1Н), 5.17 (s, 2Н), 4.4-4.1 (m, 6Н), 3.17 (m, 4Н), 2.98-2.83 (m, 2Н), 2.08 (m, 4Н), 1.91 (m, 1H), 1.64 (s, 3Н), 1.51 (s, 3Н), 1.38-1.31 (m, 17Н), 1.22-1.20 (m, 4Н).
Пример 50: Получение 38-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys(Boc-Pro-Ala-Asn)-OBzl (4An)
Следуя способу Примера 1, 636 мг (38%) указанного в заголовке соединения в виде светло-желтого порошка получали из 965 мг (2,01 ммоль) 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys-OBzl и 960 мг (2,40 ммоль) Boc-Pro-Ala-Asn. ESI-MS (m/e): 838 [M+H]+; 1H ЯМР (300 МГц, ДМСО-d6) δ/ppm=9.26 (s, 1H), 9.04 (m, 2Н), 8.31 (m, 1Н), 8.03 (m, 1Н), 7.57 (m, 1H), 7.44 (m, 6Н), 6.90 (m, 1Н), 6.68 (s, 1H), 6.49 (s, 1Н), 5.16 (s, 2Н), 4.45-4.40 (m, 3Н), 4.14 (m, 2Н), 3.42 (m, 5Н), 3.17 (m, 3Н), 2.89-2.82 (m, 1H), 2.09 (m, 1Н), 1.90 (m, 1Н), 1.79-1.74 (m, 6Н), 1.64 (s, 3Н), 1.52 (s, 3Н), 1.39-1.32 (m, 15Н), 1.22 (m, 3Н).
Пример 51: Получение 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys[Boc-Pro-Ala-Asp(OBzl)]-OBzl (4Ao)
Следуя способу Примера 1, 403 мг (35%) указанного в заголовке соединения в виде светло-желтого порошка получали из 870 мг (1,24 ммоль) 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys[Asp(OBzl)]-Obzl и 429 мг (2,48 ммоль) Boc-Pro-Ala. ESI-MS (m/e): 929 [M+H]+; 1H ЯМР (300 МГц, ДМСО-d6) δ/ppm=8.32 (m, 1H), 8.10 (m, 1H), 7.96 (m, 1H), 7.36 (m, 10H), 6.60 (s, 1H), 6.39 (s, 1H), 5.14 (s, 2H), 5.06 (s, 2H), 4.57 (m, 1H), 4.32 (m, 1H), 4.23 (m, 1H), 4.11 (m, 2H), 3.74 (m, 1H), 3.55 (m, 1H), 3.34-3.28 (m, 2H), 3.00 (m, 2H), 2.89 (s, 1H), 2.81 (m, 1H), 2.73 (m, 2H), 2.58 (m, 1H), 2.16-2.06 (m, 1H), 1.91 (s, 2H), 1.75 (m, 5H), 1.40-1.32 (m, 22H), 1.17 (m, 3H).
Пример 52: Получение 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys[Boc-Pro-Ala-Glu(OBzl)]-OBzl (4 Ap)
Следуя способу Примера 1, 269 мг (38%) указанного в заголовке соединения в виде светло-желтого порошка получали из 530 мг (0,75 ммоль) 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys[Glu(OBzl)]-OBzl и 256 мг (0,90 ммоль) Boc-Pro-Ala. ESI-MS (m/e): 943 [М+Н]+; 1Н ЯМР (300 МГц, ДМСО-d6) δ/ppm=9.26 (s, 1H), 9.10 (m, 1H), 8.97 (s, 1H), 8.79-7.88 (m, 3Н), 7.36 (m, 10H), 6.67 (s, 1Н), 6.48 (s, 1H), 5.14 (s, 2Н), 5.06 (s, 2Н), 4.35 (m, 1H), 4.23 (m, 2Н), 4.10 (m, 1Н), 3.83 (s, 1Н), 3.34 (m, 5Н), 2.83 (m, 1H), 2.72 (m, 1Н), 2.34 (m, 2Н), 1.78 (m, 10Н), 1.51 (s, 3Н), 1.36-1.31 (m, 14Н), 1.21 (m, 3Н).
Пример 53: Получение 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys(Pro-Ala-Ala) (5Аа)
Во-первых, из 300 мг (0,38 ммоль) 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys(Boc-Pro-Ala-Ala)-OBzl удаляли бензиловую сложноэфирую группу по способу Примера 4. Далее, Вое удаляли следя способу Примера 2 с получением 266 мг (78%) указанного в заголовке соединения в виде бесцветного порошка, р 207.2-209.4°C; [α]D25=-4.5 (с=0.25, СН3ОН); ESI-MS (m/e): 630 [М-Н]-; IR(KBr): 3221.1, 3057.2, 2983.8, 2943.3, 2362.8, 1660.7, 1546.9, 1533.4, 1448.5, 1379.1, 1240.2, 1049.3, 867.9, 659.7; 1Н ЯМР (300 МГц, D2O) δ/ppm=6.79 (s, 1H), 6.68 (s, 1H), 4.36-4.29 (m, 5H), 3.32-3.12 (m, 6H), 1.89 (m, 4H), 1.70 (s, 3H), 1.57 (s, 3H), 1.48 (m, 2H), 1.33 (m, 9H).
Пример 54: Получение 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys(Pro-Ala-Ala) (5Ab)
Следуя способу Примера 53, 187 мг (81%) указанного в заголовке соединения в виде бесцветного порошка получали из 300 мг (0,36 ммоль) 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys(Boc-Pro-Ala-Val)-OBzl. Мр 195.8-197.9°C; [α]D25=-3.5 (с=0.25, СН3ОН); ESI-MS (m/e): 630 [М-Н]-; IR(KBr): 3221.1, 3062.9, 2970.4, 1658.8, 1546.9, 1533.4, 1450.5, 1384.9, 1240.2, 1047.4, 991.41, 866.04, 671.2; 1H ЯМР (300 МГц, D2O) δ/ppm=8.13 (m, 1H), 6.79 (s, 1H), 6.68 (s, 1H), 4.36-4.29 (m, 4H), 3.89 (m, 1H), 3.36 (m, 2H), 3.23 (m, 2H), 3.08 (m, 2H), 1.89 (m, 4H), 1.70 (s, 3H), 1.57 (s, 3H), 1.48 (m, 1H), 1.37 (m, 6H), 1.32 (m, 2H), 0.89 (m, 6H).
Пример 55: Получение 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys(Pro-Ala-Trp) (5Ас)
Следуя способу Примера 53, 180 мг (76%) указанного в заголовке соединения в виде светло-розового порошка получали из 300 мг (0,33 ммоль) 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys(Boc-Pro-Ala-Trp)-OBzl. Мр 205.1-207.9°C; [α]D25=-1.3 (c=0.25, СН3ОН); ESI-MS (m/e): 718 [М-Н]-; IR(KBr): 3221.2, 3059.1, 2981.9, 2941.4, 1660.7, 1533.4, 1448.5, 1388.7, 1240.2, 867.9, 748.4, 630.7; 1H ЯМР (300 МГц, D2O) δ/ppm=7.50 (d, J=7.8 Гц, 1H), 7.39 (d, J=7.8 Гц, 1H), 7.12 (t, J=7.2 Гц, 1H), 7.05 (t, J=7.2 Гц, 1H), 6.74 (s, 1H), 6.62 (s, 1H), 4.38 (m, 1H), 4.35 (m, 1H), 4.27 (m, 1H), 4.18 (m, 2H), 3.24 (m, 2H), 3.16 (m, 3H), 3.06 (m, 3H),1.83 (m, 1H), 1.68 (m, 4H), 1.47 (m, 3H), 1.37 (m, 2H), 1.25 (m, 3H), 1.21 (m, 3H).
Пример 56: Получение 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys(Pro-Ala-Tyr) (5Ad)
Следуя способу Примера 53, 174 мг (74%) указанного в заголовке соединения в виде светло-желтого порошка получали из 303 мг (0,34 ммоль) 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys(Boc-Pro-Ala-Tyr)-OBzl. Мр 200.5-202.8°C; [α]D25=-2.3 (c=0.25, СН3ОН); ESI-MS (m/e): 695 [М-Н]-; IR(KBr): 3215.3, 3055.2, 2983.8, 2947.2, 1658.8, 1548.8, 1523.8, 1448.5, 1384.9, 1238.3, 1163.1, 657.7; 1Н ЯМР (300 МГц, D2O) δ/ppm=6.98 (m, 2Н), 6.92 (m, 3Н), 6.65(s, 1H), 4.34-4.26 (m, 6H), 3.27 (m, 2H), 3.15 (m, 1H), 3.08 (m, 1H), 2.96 (m, 2H), 2.85 (m, 12H), 1.89 (m, 5H), 1.70 (s, 3H), 1.51 (m, 3H), 1.33 (m, 3H), 1.25 (m, 4H).
Пример 57: Получение 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys(Pro-Ala-Pro) (5Ае)
Следуя способу Примера 53, 179 мг (78%) указанного в заголовке соединения в виде светло-желтого порошка получали из 301 мг (0,36 ммоль) 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys(Boc-Pro-Ala-Pro)-OBzl. Мр 204.8-205.4°C; [α]D25=-5.0 (c=0.25, СН3ОН); ESI-MS (m/e): 629 [М-Н]-; IR(KBr): 3244.3, 3070.7, 2980.1, 2947.2, 1662.6, 1639.5, 1554.6, 1450.5, 1379.1, 1246.1, 1201.6, 1047.3, 995.3, 871.8, 657.7; 1Н ЯМР (300 МГц, D2O) δ/ppm=6.78 (s, 1H), 6.68 (s, 1H), 4.57 (m, 1H), 4.39-4.33 (m, 5H), 3.74 (m, 1H), 3.60 (m, 1H), 3.26 (m, 2H), 3.17 (m, 1H), 3.04 (m, 2H), 2.19 (m, 3H), 1.85 (m, 5H), 1.69 (m, 4H), 1.54 (m, 6H), 1.36 (m, 6H).
Пример 58: Получение 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys(Pro-Ala-Phe) (5Af)
Следуя способу Примера 53, 183 мг (78%) указанного в заголовке соединения в виде светло-желтого порошка получали из 300 мг (0,34 ммоль) 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys(Boc-Pro-Ala-Phe)-OBzl. Мр 175.8-178.2°C; [α]D25=-3.3 (c=0.25, СН3ОН); ESI-MS (m/e): 679 [М-Н]-; IR(KBr): 3215.3, 3049.5, 2949.2, 1662.6, 1539.2, 1454.3, 1394.5; 1Н ЯМР (300 МГц, D2O) δ/ppm=7.26 (m, 3Н), 7.14 (m, 2Н), 6.77 (s, 1H), 6.65 (s, 1H), 4.39-4.33 (m, 3H), 4.03 (m, 3H), 3.42 (m, 1H), 3.37 (m, 1H), 3..29 (m, 2H), 3.17 (m, IH), 3.09 (m, 4H), 2.95 (m, 3H), 1.91 (m, 5H), 1.74 (s, 3H), 1.53 (s, 3H), 1.33 (m, 6H).
Пример 59: Получение 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys(Pro-Ala-Gly) (5Ag)
Следуя способу Примера 53, 193 мг (85%) указанного в заголовке соединения в виде бесцветного порошка получали из 300 мг (0,38 ммоль) S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys(Boc-Pro-Ala-Gly)-OBzl. Мр 196.4-199.5°C; [α]D25=-3.0 (c=0.25, СН3ОН); ESI-MS (m/e): 588 [М-Н]-; IR(KBr): 3223.1, 3062.9, 2983.8, 1630.2, 1552.7, 1537.3, 1448.5, 1382.9, 1244.1, 1049.3, 1006.8, 869.9, 661.6; 1Н ЯМР (300 МГц, D2O) δ/ppm=6.77 (s, 1H), 6.67 (s, 1H), 4.34-4.28 (m, 4H), 3.78 (s, 2H), 3.30 (m, 2H), 3.15 (m, 3H), 1.92 (m, 3H), 1.70 (s, 3H), 1.55 (s, 3H), 1.33 (m, 6H).
Пример 60: Получение 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys(Pro-Ala-Ser) (5Ah)
Следуя способу Примера 53, 181 мг (79%) указанного в заголовке соединения в виде светло-желтого порошка получали из 300 мг (0,37 ммоль) 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys(Boc-Pro-Ala-Ser)-OBzl. Мр 195.6-197.4°C; [α]D25=-3.8 (с=0.25, СН3ОН); ESI-MS (m/e): 619 [М-Н]-; IR(KBr): 3228.8, 3061.0, 2981.9, 2943.4, 1732.1, 1662.6, 1550.7 1533.3, 1448.5, 1384.9, 1244.1, 1159.2, 1049.3, 869.9, 657.7, 518.8; 1Н ЯМР (300 МГц, D2O) δ/ppm=6.78 (s, 1H), 6.67 (s, 1H), 4.41-4.26 (m, 5H), 3.74 (m, 2H), 3.25-3.13 (m, 6H), 1.91 (m, 3Н), 1.69 (s, 3Н), 1.55 (m, 3Н), 1.46 (m, 2H), 1.37 (m, 6H).
Пример 61: Получение 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys(Pro-Ala-Ile) (5Ai)
Следуя способу Примера 53, 181 мг (78%) указанного в заголовке соединения в виде бесцветного порошка получали из 302 мг (0,36 ммоль) 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys(Boc-Pro-Ala-Ile)-OBzl. Мр 175.6-178.4°C; [α]D25=-3.3 (с=0.25, СН3ОН); ESI-MS (m/e): 645 [М-Н]-; IR(KBr): 3224.9, 3057.2, 2970.4, 2758.2, 2497.8, 2360.87, 1656.9, 1535.3, 1454.3, 1388.7, 1244.1, 1159.1, 1037.7, 871.8, 659.6; 1Н ЯМР (300 МГц, D2O) δ/ppm=6.79 (s, 1H), 6.68 (s, 1H), 4.42 (m, 1H), 4.35 (m, 1H), 4.29 (m, 1H), 4.18 (m, 1H), 3.92 (d, J=1.2 Гц, IH), 3.31 (m, 2H), 3.19 (m, 2H), 3.10 (m, 3H), 2.34 (m, 1H), 2.12 (m, 1H), 1.74 (m, 4H), 1.57 (m, 3H), 1.36 (m, 6H), 1.21 (m, 2H), 0.84 (m, 9H).
Пример 62: Получение 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys(Pro-Ala-Thr) (5Aj)
Следуя способу Примера 53, 175 мг (76%) указанного в заголовке соединения в виде светло-желтого порошка получали из 300 мг (0,36 ммоль) 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys(Boc-Pro-Ala-Thr)-OBzl. Мр 194.8-197.5°C; [α]D25=-2.8 (с=0.25, СН3ОН); ESI-MS (m/e): 633 [М-Н]-; IR(KBr): 3236.5, 3066.8, 2933.7, 1660.7, 1548.8, 1533.4, 1450.8, 1381.0, 1240.2, 1157.3, 1049.3, 869.9, 669.3; 1Н ЯМР (300 МГц, D2O) δ/ppm=6.81 (s, 1H), 6.71 (s, 1H), 4.43-4.31 (m, 4H), 4.11 (m, 2H), 3.33 (m, 2H), 3.24 (m, 1H), 3.24 (m, 2H), 3.21 (m, 1H), 3.18 (m, 1H), 3.09 (m, 1H), 1.94 (m, 3H), 1.74 (s, 3H), 1.58 (s, 3H), 1.51 (m, 2H), 1.23 (m, 1H), 1.19 (m, 1H), 1.14 (m, 3H).
Пример 63: Получение 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys(Pro-Ala-Lys) (5Ak)
Следуя способу Примера 53, 152 мг (75%) указанного в заголовке соединения в виде темно-желтого порошка получали из 305 мг (0,31 ммоль) 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys[Boc-Pro-Ala-LysI(Z)]-OBzl. Мр 185.6-188.1°C; [α]D25=-1.8 (с=0.25, СН3ОН); ESI-MS (m/e): 660 [М-Н]-; IR(KBr): 3853.7, 3738.1, 2956.8, 2349.3, 2017.5, 1668.4, 1537.3, 1400.3, 1257.6, 1043.5, 871.8, 671.2.; 1Н ЯМР (300 МГц, D2O) δ/ppm=6.80 (s, 1H), 6.69 (s, 1H), 4.45 (m, 1H), 4.35 (m, 1H), 4.29 (m, 1H), 4.18 (m, 2H), 3.73 (m, 2H), 3.22-3.13 (m, 8H), 3.09 (m, 4H), 2.95 (m, 1H), 1.93 (m, 6H), 1.74 (m, 9H), 1.58 (m, 6H), 1.47 (m, 2H), 1.32-1.27 (m, 16H), 1.18 (m, 2H).
Пример 64: Получение 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys(Pro-Ala-Leu) (5Al)
Следуя способу Примера 53, 178 мг (77%) указанного в заголовке соединения в виде бесцветного порошка, получали из 300 мг (0,36 ммоль) 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys(Boc-Pro-Ala-Leu)-OBzl. Мр 189.7-192.7°C; [α]D25=-3.5 (с=0.25, СН3ОН); ESI-MS (m/e): 645 [М-Н]-; IR(KBr): 3224.9, 3064.9, 2956.8, 1660.7, 1548.8, 1535.3, 1448.5, 1381.0, 1242.2, 1049.3, 997.2, 869.9, 669.3; 1Н ЯМР (300 МГц, D2O) δ/ppm=6.78 (s, 1H), 6.68 (s, 1H), 4.38-4.29 (m, 3H), 4.13 (m, 2H), 3.31 (m, 3H), 3.21 (m, 1H), 3.08 (m, 2H), 1.89 (m, 4H), 1.70 (s, 3H), 1.57 (m, 4H), 1.46 (m, 5H), 1.32 (m, 6H), 1.32 (m, 1H), 1.16 (m, 6H).
Пример 65: Получение 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys(Pro-Ala-Gln) (5Am)
Следуя способу Примера 53, 184 мг (79%) указанного в заголовке соединения в виде бесцветного порошка получали из 300 мг (0,35 ммоль) 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys(Boc-Pro-Ala-Gln)-OBzl. Мр 181.3-183.3°C; [α]D25=-3.0 (с=0.25, СН3ОН); ESI-MS (m/e): 660 [М-Н]-; IR(KBr): 3213.4, 3059.1, 2981.9, 2943.4, 2362.8, 1739.8, 1662.6, 1548.8, 1537.3, 1450.5, 1375.3, 1244.1, 1047.2, 871.8, 659.7; 1Н ЯМР (300 МГц, D2O) δ/ppm=6.80 (s, IH), 6.69 (s, 1H), 4.40-4.29 (m, 5H), 3.33 (m, 3H), 3.17 (m, 3H), 1.90 (m, 2H), 1.69 (s, 3H), 1.55 (s, 3H), 1.43 (m, 2H), 1.31 (m, 8H).
Пример 66: Получение 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys(Pro-Ala-Asn) (5An)
Следуя способу Примера 53, 174 мг (78%) указанного в заголовке соединения в виде бесцветного порошка получали из 302 мг (0,36 ммоль) 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys(Boc-Pro-Ala-Asn)-OBzl. Мр 202.5-204.7°C; [α]D25=-2.3 (с=0.25, СН3ОН); ESI-MS (m/e): 646 [М-Н]-; IR(KBr): 3207.6, 3059.1, 2943.3, 1674.2, 1537.3, 1448.5, 1381.0, 1247.9, 642.3; 1Н ЯМР (300 МГц, D2O) δ/ppm=6.78 (s, 1H), 6.67 (s, 1H), 4.48 (m, 1H), 4.42-4.29 (m, 4H), 3.29 (m, 2H), 3.16 (m, 4H), 2.65 (m, 2H), 1.90 (m, 2H), 1.69 (s, 3H), 1.55 (s, 3H), 1.31 (m, 6H).
Пример 67: Получение 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys(Pro-Ala-Asp) (5Ao)
Следуя способу Примера 53, 174 мг (83%) указанного в заголовке соединения в виде бесцветного порошка получали из 300 мг (0,32 ммоль) 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys-[Boc-Pro-Ala-Asp(OBzl)]-OBzl. Мр 207.1-209.7°C; [α]D25=-3.5 (с=0.25, СН3ОН); ESI-MS (m/e): 647 [М-Н]-; IR(KBr): 3383.3, 3061.0, 2360.9, 1670.3, 1550.8, 1448.5, 1400.3, 1246.3, 873.8, 611.4; 1Н ЯМР (300 МГц, D2O) δ/ppm=6.76 (s, 1H), 6.66 (s, 1H), 4.52 (m, 1H), 4.37-4.281 (m, 4H), 3.27 (m, 4H), 3.13 (m, 1H), 3.05 (m, 1H), 2.76 (m, 2H), 1.97 (m, 1H), 1.90 (m, 1H), 1.71 (s, 3H), 1.55 (s, 3H), 1.43 (m, 2H), 1.31 (m, 6H).
Пример 68: Получение 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys(Pro-Ala-Glu) (5Ар)
Следуя способу Примера 53, 173 мг (82%) указанного в заголовке соединения в виде бесцветного порошка получали из 301 мг (0,32 ммоль) 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys[Boc-Pro-Ala-Glu(OBzl)]-OBzl. Мр 194.7-196.1°C; [α]D25=-2.3 (с=0.25, СН3ОН); ESI-MS (m/e): 661 [М-Н]-; IR(KBr): 3053.3, 2945.3, 2370.5, 2320.4, 1651.1, 1546.9, 1533.4, 1448.5, 1388.8, 1240.2, 1165.0, 1045.4, 644.2; 1Н ЯМР (300 МГц, D2O) δ/ppm=6.78 (s, 1H), 6.67 (s, 1H), 4.42-4.28 (m, 5H), 3.29 (m, 2H), 3.13 (m, 3H), 2.99 (m, 1H), 2.38 (m, 2H), 2.29 (m, 1H), 1.90 (m, 5H), 1.69 (s, 3H), 1.55 (s, 3H), 1.31 (m, 6H).
Сравнительный Пример 1: Получение 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys (6)
Следуя способу Примера 4, 620 мг (85%) указанного в заголовке соединения в виде светло-розового твердого вещества получали из 1,00 г (2,03 ммоль) 3S-6,7-дигидрокси-1,1-диметил-1,2,3,4-тетрагидроизохинолин-3-ацил-Lys-OBz1. Это соединение 6 использовали в качестве контрольной группы в следующих экспериментальных примерах. ESI-MS(m/e): 363 [М-Н]-.
Экспериментальный Пример 1: Оценка тромболитической активности соединений 5Аа-р по настоящему изобретению с помощью внутривенного введения
1) Способ оценки
200-220 г самцов крыс SD анестезировали 20%-ным уретановым раствором (6 мл/кг, внутрибрюшинно). Анестезированных крыс фиксировали в положении лежа на спине и отделяли правую общую сонную артерию, фиксировали на проксимальном конце артериальным зажимом и пронизывали нитью на проксимальном и дистальном концах, соответственно. Нить на дистальном конце крепко зажимали гемостатическим зажимом на коже/мехе. Выполняли катетеризацию на дистальном конце, ослабляли артериальный зажим и около 1 мл артериальной крови отбирали в пробирку ЕР 1 мл. Вводили 0,1 мл артериальной крови крысы в вертикально зафиксированную стеклянную трубку (длиной 15 мм, с внутренним диаметром 2,5 мм и внешним диаметром 5,0 мм, закрытую резиновой пробкой на дне), в которую незамедлительно вставляли иммобилизирующий тромб винт из нержавеющей стали. Иммобилизирующий тромб винт, образованный намоткой проволоки из нержавеющей стали, имеющей диаметр 0,2 мм, имел спиральную часть 12 мм в длину, включающую 15 витков, каждый из которых имел диаметр 1,0 мм, и стержень 7,0 мм в длину, который был связан со спиральной частью и имел форму знака вопроса. Спустя 40 мин после коагулирования крови, резиновую пробку на дне стеклянной трубки удаляли, стержень иммобилизирующего тромб винта фиксировали щипцами и обернутый тромбом иммобилизирующий тромб винт аккуратно вынимали из стеклянной трубки и точно взвешивали.
Перепускной катетер состоял из 3 сегментов. Средний сегмент представлял собой полиэтиленовую трубку, имеющую длину 60 мм и внутренний диаметр 3,5 мм. Сегменты на обоих концах были подобными полиэтиленовыми трубками, имеющими длину 100 мм, внутренний диаметр 1 мм и наружный диаметр 2 мм, один конец которых был вытянут для формирования наконечника с наружным диаметром 1,0 мм (используются для вставки в сонную артерию или вену крысы), другой конец которых был заключен в полиэтиленовую трубку, имеющую длину 7 мм и наружный диаметр 3,5 мм (утолщенную, используемую для установки в полиэтиленовую трубку среднего сегмента). Внутренняя стенка 3-х сегментного катетера была полностью си лидировала. Обернутый тромбом иммобилизирующий тромб винт помещали в полиэтиленовую трубку среднего сегмента и оба конца трубки заключали в утолщенные концы двух полиэтиленовых труб. Катетер наполняли раствором гепарина в изотоническом растворе (50 МЕ/кг) через верхний конец с помощью инжектора и готовили к использованию.
Отделяли левую наружную яремную вену крысы и пронизывали нитью на проксимальном и дистальном концах, соответственно, и дистальный конец лигировали. Аккуратно выполняли разрез на открытой левой наружной яремной вене и наконечник перепускного катетера, подготовленного как описано выше, вставляли в проксимальный конец разреза в левой наружной яремной вене вдали от иммобилизирующего тромб винта в среднем сегменте перепускного катетера (в котором размещался точно взвешенный иммобилизирующий тромб винт). Точное количество гепарина в солевом растворе (50 МЕ/кг) вводили через наконечник на другом конце с помощью инжектора. В этот момент, не снимая инжектор с полиэтиленовой трубки, трубку между инжектором и полиэтиленовой трубкой зажимали гемостатическим зажимом. Кровоток останавливали зажиманием проксимального конца правой общей сонной артерии артериальным зажимом, и аккуратно производили разрез поперек общей сонной артерии вблизи зажима. Инжектор вытаскивали из полиэтиленовой трубки через наконечник и наконечник полиэтиленовой трубки затем вставляли в проксимальный конец разреза артерии. Оба конца перепускного катетера фиксировали в артерии или вене четырьмя швами.
Иглу-“бабочку” использовали для подачи солевого раствора, урокиназы в солевом растворе или различных концентраций соединений в солевом растворе через среднюю секцию перепускного катетера (в которой размещался точно взвешенный иммобилизирующий тромб винт) в точку на проксимальном конце вены и вдали от иммобилизирующего тромб винта. Артериальный зажим затем удаляли, чтобы позволить крови течь из артерии в вену через перепускной катетер. Таким образом, была создана артериовенозная перепускная модель тромболизиса у крысы. Раствор в инжекторе медленно вводили в кровь (около 6 мин), что позволяло физиологическому раствору, урокиназе (положительный контроль) или соединениям по настоящему изобретению действовать на тромб через кровообращение в порядке вена-сердце-артерия. Засекали время процесса в начале инъецирования и удаляли иммобилизирующий тромб винт из перепускного катетера через 1 час и точно взвешивали. Определяли разницу в массе иммобилизирующего тромб винта в перепускном катетере крысы до и после введения для in vivo оценки тромболитической активности соединений. Снижение массы тромба выражали через средние значения и стандартное отклонение 
2) Способ введения и доза
Способом введения является внутривенная инъекция. Бланковым контролем был физиологический раствор, и доза составляла 3 мл/кг. Положительным контролем была урокиназа, и доза составляла 20000 ЕД/кг, которая была равна 1,68 мг/кг. Доза соединений 5Аа-р по настоящему изобретению составляла 0,1 нмоль/кг.
3) Результаты оценки
Тромболитическую активность выражали через снижение массы тромба  , и результаты сводили в Таблицу 1. Данные показали, что соединения 5Аа-р в дозе 0,1 нмоль/кг через внутривенное введение могут эффективно лизировать тромб (p<0,01 по сравнению с физиологическим раствором). Тромболитические активности соединений 5Аа, 5Af, 5Ag, 5Ak и 5Ао были сопоставимы с активностью урокиназы в дозе 20000 ЕД/кг. Среди соединений с более высокой тромболитической активностью, 5Аа, 5Ad, 5Af, 5Ag и 5Ak, соединение 5Ak показало самую высокую тромболитическую активность, таким образом, далее была изучена дозозависимая связь для него.
, и результаты сводили в Таблицу 1. Данные показали, что соединения 5Аа-р в дозе 0,1 нмоль/кг через внутривенное введение могут эффективно лизировать тромб (p<0,01 по сравнению с физиологическим раствором). Тромболитические активности соединений 5Аа, 5Af, 5Ag, 5Ak и 5Ао были сопоставимы с активностью урокиназы в дозе 20000 ЕД/кг. Среди соединений с более высокой тромболитической активностью, 5Аа, 5Ad, 5Af, 5Ag и 5Ak, соединение 5Ak показало самую высокую тромболитическую активность, таким образом, далее была изучена дозозависимая связь для него.
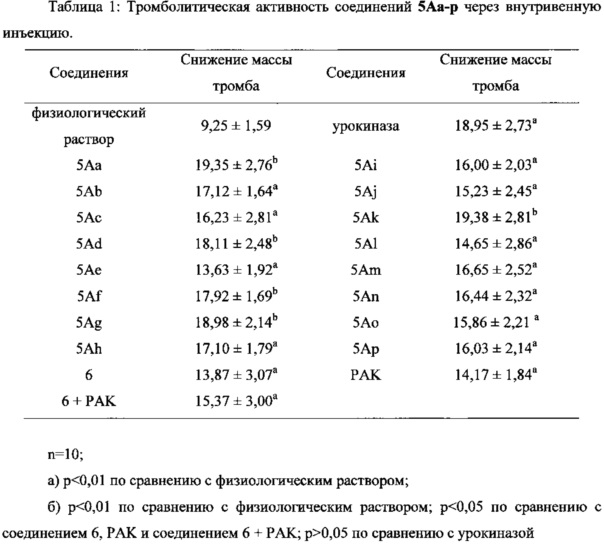
Экспериментальный Пример 2: Оценка взаимосвязи доза-эффект для тромболитической активности соединения 5Ak по настоящему изобретению через внутривенное введение
Введение внутривенной инъекцией проводили в соответствии со способом по примеру 1. Бланковым контролем был физиологический раствор, и доза составляла 3 мл/кг. Положительным контролем была урокиназа, и доза составляла 20000 ЕД/кг, которая была равна 1,68 мг/кг. Высокие, средние и низкие дозы соединения 5Ak были 0,1 нмоль/кг, 0,01 нмоль/кг и 0,001 нмоль/кг, соответственно. Результаты приведены в Таблице 2. Данные показали очевидную взаимосвязь доза-эффект для соединения 5Ak.
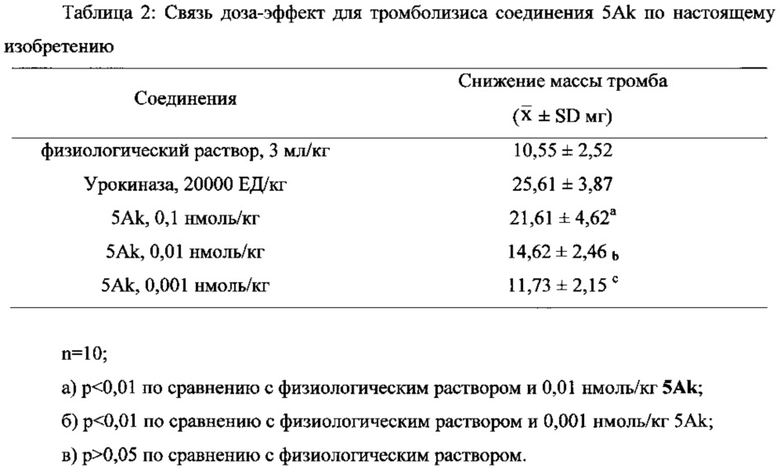
Экспериментальный Пример 3: Оценка антитромботической активности соединений 5Аа-р по настоящему изобретению через внутривенное введение
1) Способ оценки
Катетер состоял из 3 сегментов. Средний сегмент имел длину 80 мм и внутренний диаметр 3,5 мм. Сегменты на обоих концах были подобными полиэтиленовыми трубками, имеющими длину 100 мм, внутренний диаметр 1 мм и наружный диаметр 2 мм, один конец которых был вытянут для формирования наконечника (используется для вставки в сонную артерию или вену крысы), Внутренняя стенка 3-сегментного катетера была полностью силилирована. Предварительно взвешенную нить длиной 60 мм помещали в средний сегмент полиэтиленовой трубки. Оба конца более широкой трубки заключали несуженные концы двух полиэтиленовых трубок, соответственно (0,5 мм нити удерживались одним сегментом, чтобы зафиксировать нить). Катетер наполняли раствором гепарина в изотоническом растворе (50 МЕ/кг) через верхний конец с помощью инжектора и готовили к использованию.
200-220 г самцов крыс SD анестезировали 20%-ным уретановым раствором (6 мл/кг, внутрибрюшинно). Анестезированных крыс фиксировали в положении лежа на спине и отделяли левую наружную яремную вену и пронизывали нитью на проксимальном и дистальном концах, соответственно, и дистальный конец лигировали. Аккуратно выполняли разрез на открытой левой наружной яремной вене, и наконечник без удерживаемой нити перепускного катетера, подготовленного как описано выше, вставляли в проксимальный конец открытого разреза в левой наружной яремной вене. Точное количество гепарина в изотоническом растворе (50 МЕ/кг) вводили через наконечник на другом конце с помощью инжектора. Затем инжектор заменяли, и точное количество препарата вводили таким же образом. В этот момент, не снимая инжектор с этиленовой трубки, отделяли правую общую сонную артерию и зажимали проксимальный конец с помощью артериального зажима. Правую общую сонную артерию пронизывали нитью на проксимальном и дистальном концах, соответственно, и дистальный конец лигировали. Надрез осторожно разрезали через правую общую сонную артерию у артериального зажима. Инжектор вытаскивали из полиэтиленовой трубки через наконечник и наконечник полиэтиленовой трубки затем вставляли в проксимальный конец разреза артерии. Оба конца перепускного катетера фиксировали в артерии или вене четырьмя швами. Затем удаляли артериальный зажим, чтобы позволить крови течь из артерии в вену через перепускной катетер. Таким образом, была создана артериовенозная перепускная модель тромболизиса у крысы. Время процесса было засечено в начале циркуляции, нить с прикрепленным тромбом была удалена из перепускного катетера через 15 минут и точно взвешена. Разница в массе нити до и после была определена как мокрая масса тромба, которая была статистически проанализирована и использована для in vivo оценки антитромботической активности соединений. Мокрую массу тромба выражали через среднее значение и стандартное отклонение 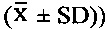 .
.
2) Способ введения препарата и доза
Способом введения препарата является внутривенная инъекция. Бланковым контролем был физиологический раствор, и доза составляла 1 мл/кг. Положительным контролем был аспирин, и доза составляла 9 мг/кг. Доза соединений 5Аа-р по настоящему изобретению составляла 0,1 нмоль/кг.
3) Результаты
Антитромботическую активность выражали через мокрую массу тромба 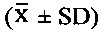 , и результаты сводили в Таблицу 3. Данные показали, что доза 0,1 нмоль/кг соединения 5Ак через внутривенное введение может эффективно ингибировать образование тромба (р<0,001, по сравнению с физиологическим раствором), и таким образом далее оценивали дозозависимую связь 5Ak с более высокой активностью.
, и результаты сводили в Таблицу 3. Данные показали, что доза 0,1 нмоль/кг соединения 5Ак через внутривенное введение может эффективно ингибировать образование тромба (р<0,001, по сравнению с физиологическим раствором), и таким образом далее оценивали дозозависимую связь 5Ak с более высокой активностью.

Экспериментальный Пример 4: Оценка взаимосвязи доза-эффект для антитромботической активности соединения 5Ak по настоящему изобретению через внутривенное введение
1) Способ оценки
Способ был таким же, как в примере 3.
2) Способ введения препарата и доза
Способом введения препарата является внутривенная инъекция. Бланковым контролем был физиологический раствор, и доза составляла 3 мл/кг. Положительным контролем был аспирин, и доза составляла 9 мг/кг. Высокая, средняя и низкая дозы соединения 5Ak составляли 0,1 нмоль/кг, 0,01 нмоль/кг и 0,001 нмоль/кг, соответственно.
3) Результаты
Результаты приведены в Таблице 4, и соединение 5Ak показало очевидную взаимосвязь доза-эффект.

Экспериментальный Пример 5: Оценка терапевтического эффекта соединения 5Ak по настоящему изобретению у крыс с инсультом
Для оценки терапевтического эффекта соединений 5Аа-р у крыс с инсультом, соединение 5Ak было выбрано в качестве представителя и его терапевтический эффект у крыс с инсультом оценивали описанным ниже способом.
1) Способ оценки
Самцов крыс SD (280-300 г) случайным образом разделяли на положительную контрольную группу UK с дозой 20000 МЕ/кг, бланковую контрольную группу с физиологическим раствором, контрольную группу соединения с РАК с дозой 5 мкмоль/кг и группу соединения 5Ak с дозой 1 мкмоль/кг (высокая), 0,1 мкмоль/кг (средняя) и 0,01 мкмоль/кг (низкая). 10% раствор хлоральгидрата (400 мг/кг) вводили интраперитонеально крысам для анестезии. Выполняли вертикальный разрез около 2 см в длину на правой стороне ближе к центру шеи и правую общую сонную артерию (ССА), наружную сонную артерию (ЕСА) и внутреннюю сонную артерию (ВСА) разделяли по краю внутренней стороны грудино-сосцевидных мышц. Разрез на внутренней сонной артерии и проксимальный конец общей сонной артерии зажимали, соответственно, неинвазивными артериальными зажимами. Выполняли небольшой разрез на наружной сонной артерии, а дистальный конец наружной сонной артерии лигировали. Артериальный зажим на проксимальном конце общей сонной артерии разжимали и отбирали 10 мкл крови. После отбора крови проксимальный конец общей сонной артерии вновь зажимали неинвазивным артериальным зажимом. Отобранные 10 мкл крови помещали в 1 мл ампулу ЕР и выдерживали при комнатной температуре в течение 30 мин, чтобы позволить крови свернуться, а затем переносили в холодильник при -20°C на 1 час, чтобы позволить образование твердого коагулята. Через 1 час сгустки крови вынимали, к ним добавляли 1 мл физиологического раствора, а затем сгустки крови разбивали на относительно равномерные микротромбы с помощью стального шпателя. Суспензию микротромбов затем переносили в 1 мл инжектор для использования. Когда зажим на внутренней сонной артерии крысы открывали, медленно инъецировали 1 мл суспензии тромбов инжектором из наружной сонной артерии крысы в ее проксимальный конец, а затем вводили в мозг крысы по внутренней сонной артерии. Впоследствии, проксимальный конец наружной сонной артерии лигировали, снимали артериальные зажимы на внутренней сонной артерии и общей сонной артерии и восстанавливали кровоток. Общую яремную вену отделяли, а затем вводили UK в дозе 20000 МЕ/кг, физиологический раствор или соединение 5Ak в дозе 1 мкмоль/кг. Вену лигировали. Орошали рану 3 каплями пенициллина. Рану зашивали и ждали пробуждения животных.
Через 24 часа после того, как пробуждали крыс, оценивали степень повреждения нервной функции по методу Зиалонга (Zealonga). Оценка 0 указывала на отсутствие признаков потери в нервной функции, 1 указывала на невозможность протянуть передние конечности на неповрежденной стороне, 2 указывала на способность ходить на неповрежденной стороне, 3 указывала на способность гоняться кругами за хвостом на неповрежденной стороне, 4 указывала на непроизвольную ходьбу с нарушением сознания и 5 указывала на смерть. Результаты оценки были статистически проанализированы и подвергнуты t-тесту.
После того как крысы бодрствовали в течение 24 часов и оценивали у них степень повреждения нервной функции методом Зиалонга (Zealonga), их анестезировали уретаном с последующей немедленной декапитацией и удалением мозга. Ткани мозга хранили в холодильнике при -20°C 2 часа, и последовательно нарезали коронарные срезы около 2 мм из префронтального конца, в общей сложности 5 срезов, а затем помещали в 2%-ный раствор ТТС на инкубацию без света при 37°C в течение 30 мин. Наблюдали изменение цвета в срезах мозга: нормальные ткани мозга окрашивались в красный цвет ТТС, а ишемические ткани мозга проявлялись белым. Фотографии сделали с помощью цифровой камеры и обработали с помощью программного обеспечения для статистической обработки изображений, в коронарных срезах рассчитывали объем инфаркта в тканях головного мозга и площадь нормальных тканей мозга. Было статистически рассчитано отношение объема церебрального инфаркта в каждой группе и подвергнуто t-тесту.
2) Результат оценки
Оцененные данные в соответствии с методом Зиалонга (Zealong) после пробуждения крыс перечислены в Таблице 5. Проценты объема церебрального инфаркта перечислены в Таблице 6.



Данные, приведенные в Таблицах 5 и 6, предполагают, что соединение 5Ak может эффективно предотвращать ишемию головного мозга у крыс от возникновения дискинезии и церебрального инфаркта. Эта функция соединения 5Ak зависела от дозы.
Экспериментальный Пример 6: Оценка активности по захвату свободных радикалов для соединений 5Аа-р по настоящему изобретению
1) активность по захвату гидроксильных свободных радикалов 11,316 мг DMPO (диметилпиридин-N-оксид, Sigma) растворяли в 1 мл чистой воды с получением 0,1 М раствора DMPO. 2,78 г FeSO⋅7H2O растворяли в 1 мл чистой воды с получением 10 мМ раствора. 30% медицинской Н2О2 разбавляли до 0,2%.
Измеряли высоту первого пика сигнала ОН и повторяли 6 раз для 2,5 мкл раствора FeSO4⋅7H2O+2,5 мкл раствора DMPO + 5 мкл раствора H2O2 + 5 мкл воды. Эту высоту пика определяли, как известную высоту пика сигнала ОН. Измеряли высоту первого пика сигнала ОН и повторяли 6 раз для 2,5 мкл раствора FeSO4⋅7H2O + 2,5 мкл раствора DMPO + 5 мкл раствора H2O2 + 5 мкл раствора одного из соединений 5Аа-р. Эту высоту пика определяли, как остаточную высоту пика сигнала ОН, захваченного одним из соединений 5Аа-р.
Отношение захвата = (известная высота пика сигнала ОН - остаточная высота пика сигнала ОН для соединений 5Аа-р) / известная высота пика сигнала ОН
1) активность по захвату свободных радикалов NO
7,325 мг MGD (N-метилглюкаминдитиокарбамат, Sigma) растворяли в 1 мл чистой воды с получением 25 мМ раствора MGD. 3,475 г FeSO4⋅7H2O растворяли в 1 мл чистой воды с получением 12,5 мМ раствора. 25 мг SNAP (S-нитрозоацетилпеницилламин) растворяли в 1 мл чистой воды с получением 110 мкм зеленой маточной жидкости, которую разводили в 100 раз с получением 1 мкМ раствора SNAP.
Измеряли высоту первого пика сигнала NO и повторяли 6 раз для 5 мкл раствора MGD + 5 мкл раствора FeSO47H2O + 5 мкл раствора SNAP + 5 мкл воды. Эту высоту пика определяли, как известную высоту пика сигнала NO. Измеряли высоту первого пика сигнала NO и повторяли 6 раз для 5 мкл раствора MGD + 5 мкл раствора FeSO4⋅7H2O + 5 мкл раствора SNAP + 5 мкл раствора одного из соединений 5Аа-р. Эту высоту пика определяли как остаточную высоту пика сигнала NO, захваченного одним из соединений 5Аа-р.
Отношение захвата = (известная высота пика сигнала NO - остаточная высота пика сигнала NO для соединений 5Аа-р) / известная высота пика сигнала NO
1) активность по захвату супероксиданионных радикалов
0,3 г ксантина растворяли в 1 мл чистой воды с получением 0,5 М раствора ксантина (молочно-белый цвет, в основном нерастворим). Коммерчески доступный сырой раствор ксантиноксидазы разбавляли в 10 раз с получением раствора ксантиноксидазы. Насыщенный раствор DETAPAC разбавляли в 20 раз с получением 0,9 мМ раствора. 11,316 мг DMPO растворяли в 1 мл чистой воды с получением 0,1 М раствора DMPO.
5 мкл раствора DMPO + 5 мкл раствора DETAPAC + 5 мкл раствора ксантина + 5 мкл раствора ксантиноксидазы + 5 мкл раствора соединений 5Аа-р
Измеряли высоту первого пика сигнала супероксиданиона и повторяли 6 раз для 5 мкл раствора DMPO+5 мкл раствора DETAPAC + 5 мкл раствора ксантина + 5 мкл раствора ксантиноксидазы + 5 мкл воды. Эту высоту пика определяли, как известную высоту пика сигнала супероксиданиона. Измеряли высоту первого пика сигнала супероксиданиона и повторяли 6 раз для 5 мкл раствора DMPO + 5 мкл раствора DETAPAC + 5 мкл раствора ксантина + 5 мкл раствора ксантиноксидазы + 5 мкл раствора одного из соединений 5Аа-р. Эту высоту пика определяли, как остаточную высоту пика сигнала супероксиданиона, захваченного одним из соединений 5Аа-р.
Отношение захвата = (известная высота пика сигнала супероксиданиона - остаточная высота пика сигнала супероксиданиона для соединений 5Аа-р) / известная высота пика сигнала супероксиданиона
Результаты представлены в Таблице 7. Данные показали, что ЕС50 соединений 5Аа-р для активности по захвату свободных радикалов составила 0,4-0,7 мМ. Соединения 5Аа-р обладают очевидной активностью по захвату свободных радикалов.

Экспериментальный Пример 7: Наблюдения ТЕМ соединений 5Аа-р по настоящему изобретению
Образцы готовили в виде 1×10-7 М, 1×10-9 М, 1×10-11 М растворов в дистиллированной воде, соответственно. Отбирали небольшое количество (около 10 мкл) раствора и капали на поверхность медной сетки с помещенной под ней фильтровальной бумагой для высушивания на воздухе. Затем изучали морфологию и размер частиц под просвечивающим электронным микроскопом (JEOL, JEM- 1230) и записывали в виде фотоснимков.
В водном растворе, соединения 5Аа-р могут быть самоорганизующимися в наносферические структуры, имеющие диаметр 20-210 нм, в основном, диаметр 20-100 нм. Эти наносферы были соединены в различные формы наносетей или наноожерелий и т.д. Например, для концентраций in vivo были 1×10-9 М (теоретическая концентрация препарата в крови) показаны соответствующие фотоснимки ТЕМ (Фиг. 9). На Фиг. 9 соединения 5Аа-р соответственно относятся к номерам 5а, 5b, 5c, 5d, 5е, 5f, 5g, 5h, 5i, 5J, 5k, 51, 5m, 5n, 5o и 5p на фигуре.
Экспериментальный Пример 8: Эксперимент по последовательному 6-ти кратному лечению крыс с инсультом спустя 4 часа от начала инсульта 1 мкмоль/кг 5Ak
Терапевтический эффект оценивали по неврологическим функциональным баллам. Чем ниже был балл, тем лучше был терапевтический эффект. 10% раствора хлоральгидрата внутрибрюшинно вводили самцам крыс SD в дозе 400 мг/кг массы тела для анестезии. Выполняли продольный разрез в центре шеи и отделяли правый ствол общей сонной артерии (около 3 см в длину). Каждую ветвь наружной сонной артерии отделяли и лигировали на подъязычном уровне, и отделяли внутреннюю сонную артерию от набухшей части шеи. Разрез на внутренней сонной артерии и проксимальный конец общей сонной артерии зажимали, соответственно, неинвазивными артериальными зажимами и дистальный конец наружной сонной артерии лигировали. Во внешний ствол сонной артерии вводили катетер, содержащий 0,5 мл суспензии тромбов в физиологическом растворе. Когда открывали зажим на внутренней сонной артерии крысы, медленно инъецировали 0,5 мл суспензии тромбов в физиологическом растворе катетером из наружной сонной артерии крысы в ее проксимальный конец, а затем вводили в артерии в мозгу по внутренней сонной артерии. Впоследствии, проксимальный конец внутренней сонной артерии лигировали, снимали артериальные зажимы на внутренней сонной артерии и общей сонной артерии и восстанавливали кровоток. После того как рану зашивали, внутримышечно вводили 20000 ME пенициллина для профилактики от заражения. Через 4 часа соединение 5Ak в физиологическом растворе (в дозе 1 мкмоль/кг, n=10) вводили последовательно 6 дней и наблюдали в течение 7 дней. Само-сравнение проводили каждый день, и степень неврологического дефицита оценивали по методу Зиалонга (Zealonga). Оценка 0 указывала на отсутствие признаков потери в нервной функции, 1 указывала на невозможность протянуть передние конечности на неповрежденной стороне, 2 указывала на способность ходить на неповрежденной стороне, 3 указывала на способность гоняться кругами за хвостом на неповрежденной стороне, 4 указывала на непроизвольную ходьбу с нарушением сознания и 5 указывала на смерть. Результаты оценки представлены в Таблице 8. Данные показали, что крысы, получавшие последовательно 6-дневное лечение 1 мкмоль/кг 5Ak один раз в день спустя 4 часа от начала инсульта, выживали. Восемь из десяти крыс не демонстрировали неврологического дефицита. Только 2 крысы демонстрировали небольшие неврологические дефициты. Таким образом, доза 1 мкмоль/кг 5Ak имела определенный терапевтический эффект спустя 4 часа от начала инсульта.
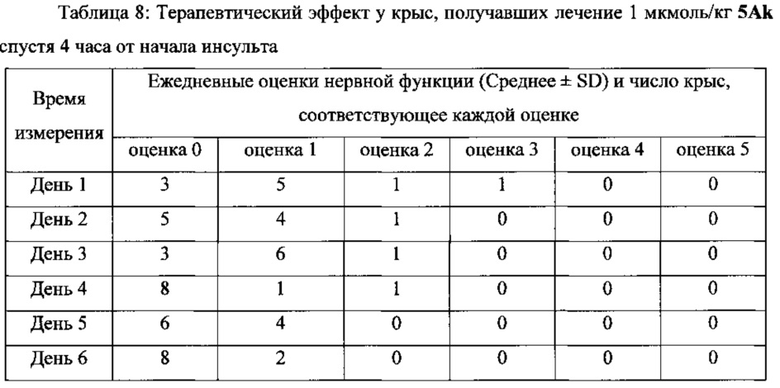
Экспериментальный Пример 9: Эксперимент по последовательному 6-ти кратному лечению крыс с инсультом спустя 6 часов от начала инсульта 1 мкмоль/кг 5Ak
Терапевтический эффект оценивали по неврологическим функциональным показателям. Чем ниже был балл, тем лучше был терапевтический эффект.10% раствор хлоральгидрата внутрибрюшинно вводили самцам крыс SD в дозе 400 мг/кг массы тела для анестезии. Выполняли открытый продольный разрез в центре шеи и отделяли правый ствол общей сонной артерии (около 3 см в длину). Каждую ветвь наружной сонной артерии отделяли и лигировали на подъязычном уровне, и отделяли внутреннюю сонную артерию на набухшей части шеи. Разрез во внутренней сонной артерии и проксимальный конец общей сонной артерии зажимали, соответственно, неинвазивными артериальными зажимами и дистальный конец наружной сонной артерии лигировали. Во внешний ствол сонной артерии вводили катетер, содержащий 0,5 мл суспензии тромбов в физиологическом растворе. Открывали зажим на внутренней сонной артерии, медленно инъецировали 0,5 мл суспензии тромбов в физиологическом растворе катетером из наружной сонной артерии в ее проксимальный конец, а затем вводили в артерии в мозгу по внутренней сонной артерии. Впоследствии, проксимальный конец внутренней сонной артерии лигировали, снимали артериальные зажимы на внутренней сонной артерии и общей сонной артерии и восстанавливали кровоток. После того как рану зашивали, внутримышечно вводили 20000 ME пенициллина для профилактики от заражения. Через 6 часов соединение 5Ak в физиологическом растворе (в дозе 1 мкмоль/кг, n=10) вводили последовательно 6 дней и наблюдали в течение 7 дней. Само-сравнение проводили каждый день, и степень неврологического дефицита оценивали по методу Зиалонга (Zealonga). Оценка 0 указывала на отсутствие признаков потери в нервной функции, 1 указывала на невозможность протянуть передние конечности на неповрежденной стороне, 2 указывала на способность ходить на неповрежденной стороне, 3 указывала на способность гоняться кругами за хвостом на неповрежденной стороне, 4 указывала на непроизвольную ходьбу с нарушением сознания и 5 указывала на смерть. Результаты оценки представлены в Таблице 9. Данные показали, что из крыс, получавших последовательно 6-дневное лечение 1 мкмоль/кг 5Ak один раз в день спустя 6 часа от начала инсульта, только одна крыса умерла случайно. Шесть из девяти крыс не демонстрировали неврологического дефицита. Только 3 крысы демонстрировали небольшие неврологические дефициты. Таким образом, доза 1 мкмоль/кг 5Ak имела определенный терапевтический эффект спустя 6 часов от начала инсульта.
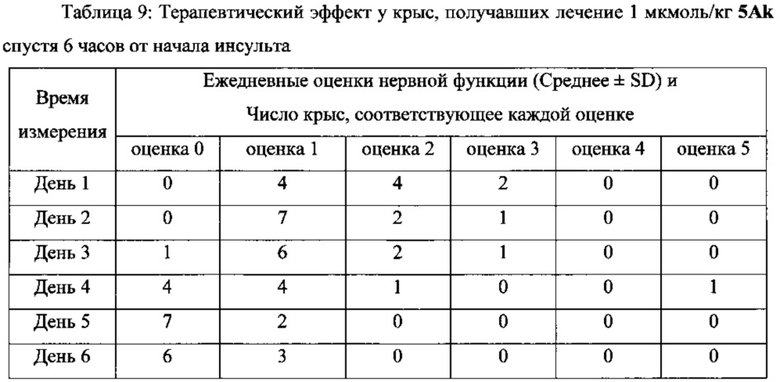
Экспериментальный Пример 10: Эксперимент по последовательному 6-ти кратному лечению крыс с инсультом спустя 24 часа от начала инсульта 1 мкмоль/кг 5Ak
Терапевтический эффект оценивали по неврологическим функциональным показателям. Чем ниже был балл, тем лучше был терапевтический эффект.10% раствор хлоральгидрата внутрибрюшинно вводили самцам крыс SD в дозе 400 мг/кг массы тела для анестезии. Выполняли продольный разрез в центре шеи и отделяли правый ствол общей сонной артерии (около 3 см в длину). Каждую ветвь наружной сонной артерии отделяли и лигировали на подъязычном уровне, и рассекали внутреннюю сонную артерию на набухшей части шеи. Разрезы во внутренней сонной артерии и проксимальный конец общей сонной артерии зажимали, соответственно, неинвазивными артериальными зажимами и дистальный конец наружной сонной артерии лигировали. Во внешний ствол сонной артерии вводили катетер, содержащий 0,5 мл суспензии тромбов в физиологическом растворе. Открывали зажим на внутренней сонной артерии, медленно инъецировали 0,5 мл суспензии тромбов в физиологическом растворе катетером из наружной сонной артерии в ее проксимальный конец, а затем вводили в артерии в мозгу по внутренней сонной артерии. Впоследствии, проксимальный конец внутренней сонной артерии лигировали, снимали артериальные зажимы на внутренней сонной артерии и общей сонной артерии и восстанавливали кровоток. После того как рану зашивали, внутримышечно вводили 20000 ME пенициллина для профилактики от заражения. Через 24 часа соединение 5Ak в физиологическом растворе (в дозе 1 мкмоль/кг, n=10) вводили последовательно 6 дней и наблюдали в течение 7 дней. Само-сравнение проводили каждый день, и степень неврологического дефицита оценивали по методу Зиалонга (Zealonga). Оценка 0 указывала на отсутствие признаков потери в нервной функции, 1 указывала на невозможность протянуть передние конечности на неповрежденной стороне, 2 указывала на способность ходить на неповрежденной стороне, 3 указывала на способность гоняться кругами за хвостом на неповрежденной стороне, 4 указывала на непроизвольную ходьбу с нарушением сознания и 5 указывала на смерть. Результаты оценки представлены в Таблице 10. Данные показали, что 2 крысы умерли через 24 часа от начала инсульта. Остальные 8 крыс, получавших последовательно 6-дневное лечение 1 мкмоль/кг 5Ak один раз в день, выживали. Три из 8 крыс не демонстрировали неврологических дефицитов. Четыре крысы демонстрировали небольшие неврологические дефициты, и одна крыса демонстрировала очевидные неврологические дефициты. Таким образом, доза 1 мкмоль/кг 5Ak все еще имела определенный терапевтический эффект спустя 24 часа от начала инсульта.
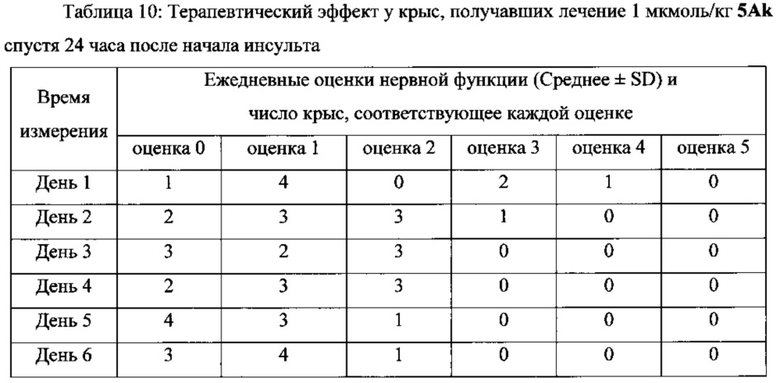
Экспериментальный Пример 11: Эксперимент по последовательному 6-ти кратному лечению крыс с инсультом спустя 6 часов от начала инсульта 2,5 мкмоль/кг 5Ak
Терапевтический эффект оценивали по неврологическим функциональным показателям. Чем ниже был балл, тем лучше был терапевтический эффект. 10% раствора хлоральгидрата внутрибрюшинно вводили самцам крыс SD в дозе 400 мг/кг массы тела для анестезии. Выполняли продольный разрез в центре шеи и отделяли правый ствол общей сонной артерии (около 3 см в длину). Каждую ветвь наружной сонной артерии отделяли и лигировали на подъязычном уровне, и отделяли внутреннюю сонную артерию на набухшей части шеи. Разрез на внутренней сонной артерии и проксимальный конец общей сонной артерии зажимали, соответственно, неинвазивными артериальными зажимами и дистальный конец наружной сонной артерии лигировали. Во внешний ствол сонной артерии вводили катетер, содержащий 0,5 мл суспензии тромбов в физиологическом растворе. В то же время когда открывали зажим на внутренней сонной артерии, медленно инъецировали 0,5 мл суспензии тромбов в физиологическом растворе катетером из наружной сонной артерии в ее проксимальный конец, а затем вводили в артерии в мозгу по внутренней сонной артерии. Впоследствии, проксимальный конец внутренней сонной артерии лигировали, снимали артериальные зажимы на внутренней сонной артерии и общей сонной артерии и восстанавливали кровоток. После того как рану зашивали, внутримышечно вводили 20000 ME пенициллина для профилактики от заражения. Через 6 часов соединение 5Ak в физиологическом растворе (в дозе 2,5 мкмоль/кг, n=10) вводили последовательно 6 дней и наблюдали в течение 7 дней. Самосравнение проводили каждый день, и степень неврологического дефицита оценивали по методу Зиалонга (Zealonga). Оценка 0 указывала на отсутствие признаков потери в нервной функции, 1 указывала на невозможность протянуть передние конечности на неповрежденной стороне, 2 указывала на способность ходить на неповрежденной стороне, 3 указывала на способность гоняться кругами за хвостом на неповрежденной стороне, 4 указывала на непроизвольную ходьбу с нарушением сознания и 5 указывала на смерть. Результаты оценки представлены в Таблице 11. Данные показали, что девять крыс, получавших последовательно 6-дневное лечение 2,5 мкмоль/кг 5Ak один раз в день спустя 6 часов от начала инсульта, выживали. Семь из девяти крыс не демонстрировали неврологического дефицита. Одна крыса демонстрировала небольшие неврологические дефициты. Таким образом, доза 2,5 мкмоль/кг 5Ak имела очевидный улучшенный терапевтический эффект, чем 1 мкмоль/кг 5Ak спустя 6 часов от начала инсульта.
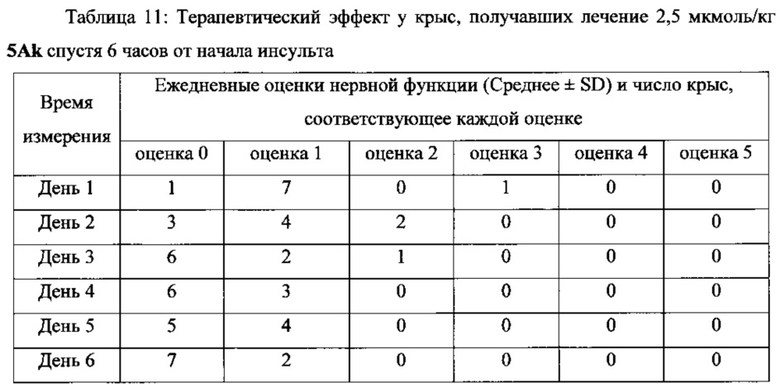
Экспериментальный Пример 12: Эксперимент по последовательному 6-ти кратному лечению крыс с инсультом спустя 24 часа от начала инсульта 2,5 мкмоль/кг 5Ak
Терапевтический эффект оценивали по неврологическим функциональным показателям. Чем ниже был балл, тем лучше был терапевтический эффект. 10% раствор хлоральгидрата внутрибрюшинно вводили самцам крыс SD в дозе 400 мг/кг массы тела для анестезии. Выполняли продольный разрез в центре шеи и отделяли правый ствол общей сонной артерии (около 3 см в длину). Каждую ветвь наружной сонной артерии отделяли и лигировали на подъязычном уровне, и отделяли внутреннюю сонную артерию на набухшей части шеи. Разрез во внутренней сонной артерии и проксимальный конец общей сонной артерии зажимали, соответственно, неинвазивными артериальными зажимами и дистальный конец наружной сонной артерии лигировали. Во внешний ствол сонной артерии вводили катетер, содержащий 0,5 мл суспензии тромбов в физиологическом растворе. Открывали зажим на внутренней сонной артерии, медленно инъецировали 0,5 мл суспензии тромбов в физиологическом растворе катетером из наружной сонной артерии в ее проксимальный конец, а затем вводили в артерии в мозгу по внутренней сонной артерии. Впоследствии, проксимальный конец внутренней сонной артерии лигировали, снимали артериальные зажимы на внутренней сонной артерии и общей сонной артерии и восстанавливали кровоток. После того как рану зашивали, внутримышечно вводили 20000 ME пенициллина для профилактики от заражения. Через 24 часа соединение 5Ak в физиологическом растворе (в дозе 2,5 мкмоль/кг, n=10) вводили последовательно 6 дней и наблюдали в течение 7 дней. Само-сравнение проводили каждый день, и степень неврологического дефицита оценивали по методу Зиалонга (Zealonga). Оценка 0 указывала на отсутствие признаков потери в нервной функции, 1 указывала на невозможность протянуть передние конечности на неповрежденной стороне, 2 указывала на способность ходить на неповрежденной стороне, 3 указывала на способность гоняться кругами за хвостом на неповрежденной стороне, 4 указывала на непроизвольную ходьбу с нарушением сознания и 5 указывала на смерть. Результаты оценки представлены в Таблице 12. Данные показывают, что 3 крысы погибли через 24 часа после начала инсульта. Из остальных семи крыс, получавших последовательно 6-дневное лечение 2,5 мкмоль/кг 5Ak один раз в день, две крысы умерли. Три крысы не демонстрировали неврологических дефицитов. Одна крыса демонстрировала небольшие неврологические дефициты, и одна крыса демонстрировала очевидные неврологические дефициты. Таким образом, доза 2,5 мкмоль/кг 5Ak все еще имела определенный терапевтический эффект спустя 24 часа от начала инсульта.

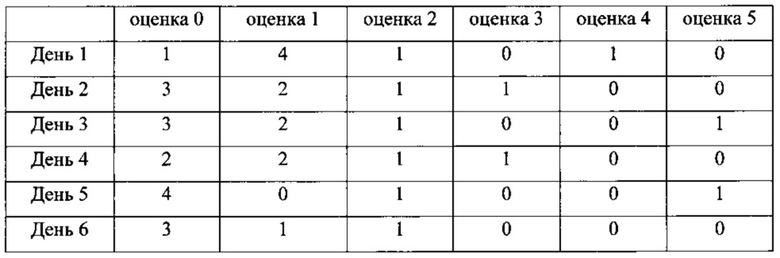
Экспериментальный Пример 13: Эксперимент по последовательному 6-ти кратному лечению крыс с инсультом спустя 24 часа от начала инсульта 5 мкмоль/кг 5Ak
Терапевтический эффект оценивали по неврологическим функциональным показателям. Чем ниже был балл, тем лучше был терапевтический эффект. 10% раствор хлоральгидрата внутрибрюшинно вводили самцам крыс SD в дозе 400 мг/кг массы тела для анестезии. Выполняли продольный разрез в центре шеи и отделяли правый ствол общей сонной артерии (около 3 см в длину). Каждую ветвь наружной сонной артерии отделяли и лигировали на подъязычном уровне, и отделяли внутреннюю сонную артерию на набухшей части шеи. Разрез во внутренней сонной артерии и проксимальный конец общей сонной артерии зажимали, соответственно, неинвазивными артериальными зажимами и дистальный конец наружной сонной артерии лигировали. Во внешний ствол сонной артерии вводили катетер, содержащий 0,5 мл суспензии тромбов в физиологическом растворе. Открывали зажим на внутренней сонной артерии, медленно инъецировали 0,5 мл суспензии тромбов в физиологическом растворе катетером из наружной сонной артерии в ее проксимальный конец, а затем вводили в артерии в мозгу по внутренней сонной артерии. Впоследствии, проксимальный конец внутренней сонной артерии лигировали, снимали артериальные зажимы на внутренней сонной артерии и общей сонной артерии и восстанавливали кровоток. После того как рану зашивали, внутримышечно вводили 20000 ME пенициллина для профилактики от заражения. Через 24 часа соединение 5Ak в физиологическом растворе (в дозе 5 мкмоль/кг, n=10) вводили последовательно 6 дней и наблюдали в течение 7 дней. Само-сравнение проводили каждый день, и степень неврологического дефицита оценивали по методу Зиалонга (Zealonga). Оценка 0 указывала на отсутствие признаков потери в нервной функции, 1 указывала на невозможность протянуть передние конечности на неповрежденной стороне, 2 указывала на способность ходить на неповрежденной стороне, 3 указывала на способность гоняться кругами за хвостом на неповрежденной стороне, 4 указывала на непроизвольную ходьбу с нарушением сознания и 5 указывала на смерть. Результаты оценки представлены в Таблице 13. Данные показывают, что одна крыса погибла через 24 часа после начала инсульта. Остальные десять крыс, получавших последовательно 6-дневное лечение 5 мкмоль/кг 5Ak один раз в день, выживали. Семь крыс не демонстрировали неврологических дефицитов. Три крысы демонстрировали небольшие неврологические дефициты. Таким образом, доза 5 мкмоль/кг 5Ak имела определенный терапевтический эффект спустя 24 часа от начала инсульта и очевидно была лучше, чем доза 2,5 мкмоль/кг.
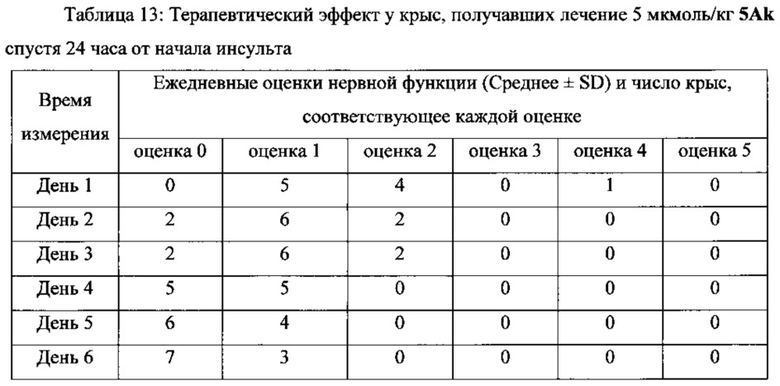
Согласно экспериментальным результатам соединения по настоящему изобретению могут образовывать наноструктуры для достижения функций пересечения гематоэнцефалического барьера. В дополнение к тромболитической и антитромботической функциям эти соединения могут также эффективно захватывать свободные радикалы ОН, NO, супероксиданионные и другие. Кроме того, только низкая доза была необходима для эффективного тромболизиса. Более высокая доза может продемонстрировать отличный эффект в лечении инсульта через 4 часа от начала инсульта. Таким образом, эти соединения имеют хорошие перспективы для клинического применения.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВОЕ СОЕДИНЕНИЕ С ЭФФЕКТАМИ ТРОМБОЛИЗИСА, АКЦЕПТИРОВАНИЯ СВОБОДНЫХ РАДИКАЛОВ И НАПРАВЛЕННОГО ДЕЙСТВИЯ НА ТРОМБ, А ТАКЖЕ СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2013 |
|
RU2604193C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ КОНЪЮГАТ ТРОМБОЛИТИЧЕСКИЙ ПЕПТИД-ТЕТРАГИДРОИЗОХИНОЛИН | 2020 |
|
RU2825937C1 |
| НОВЫЕ ИНДОЛЬНЫЕ И ПИРРОЛЬНЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2014 |
|
RU2693404C2 |
| КОНЪЮГАТЫ АНТИТЕЛА И ЛЕКАРСТВЕННОГО СРЕДСТВА (ADC) И КОНЪЮГАТЫ АНТИТЕЛА И ПРОЛЕКАРСТВА (APDC), СОДЕРЖАЩИЕ ФЕРМЕНТАТИВНО РАСЩЕПЛЯЕМЫЕ ГРУППЫ | 2016 |
|
RU2751512C2 |
| НОВЫЕ ИЗОИНДОЛИНОВЫЕ ИЛИ ИЗОХИНОЛИНОВЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2014 |
|
RU2689305C2 |
| АНТИТЕЛА К В7-Н3 И КОНЪЮГАТЫ АНТИТЕЛА И ЛЕКАРСТВЕННОГО СРЕДСТВА | 2017 |
|
RU2764651C2 |
| СОЕДИНЕНИЯ, ИНГИБИРУЮЩИЕ MASP, И ИХ ПРИМЕНЕНИЯ | 2020 |
|
RU2840441C2 |
| НОВЫЕ ПИРРОЛЬНЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2014 |
|
RU2607788C2 |
| ОПРЕДЕЛЕННЫЕ СОЕДИНЕНИЯ ПЛАДИЕНОЛИДА И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2019 |
|
RU2813597C2 |
| N1/N2-ЛАКТАМНЫЕ ИНГИБИТОРЫ АЦЕТИЛ-КоА-КАРБОКСИЛАЗ | 2011 |
|
RU2540337C2 |
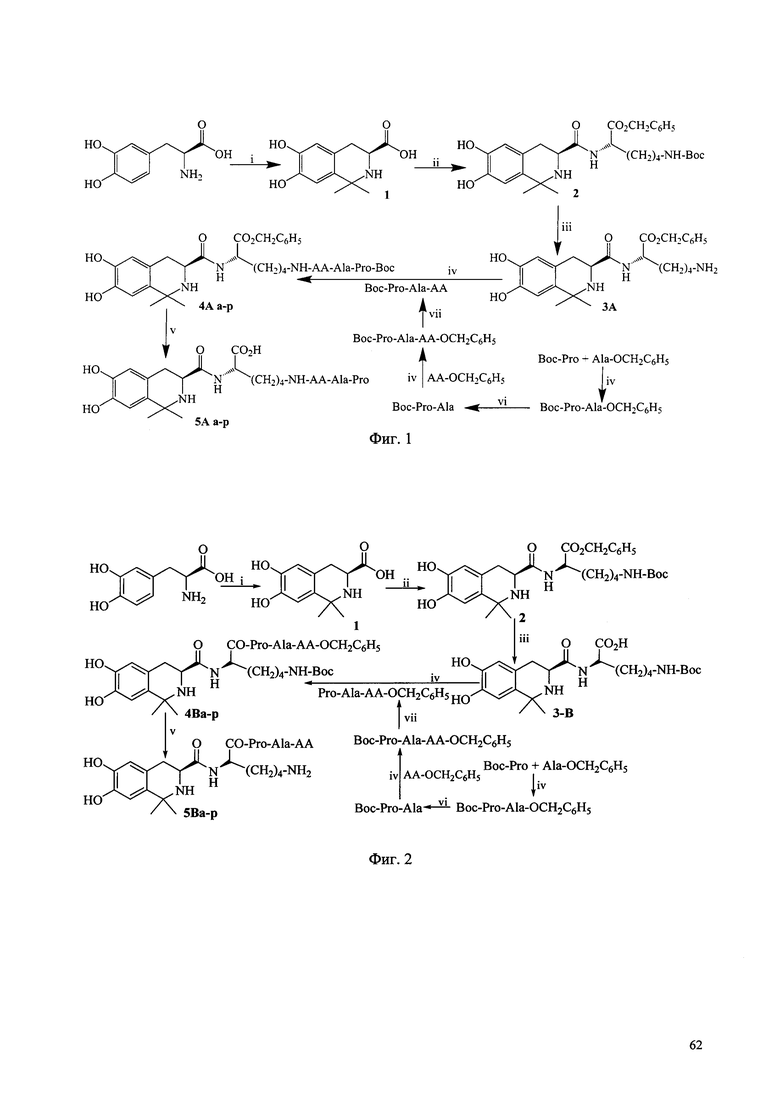
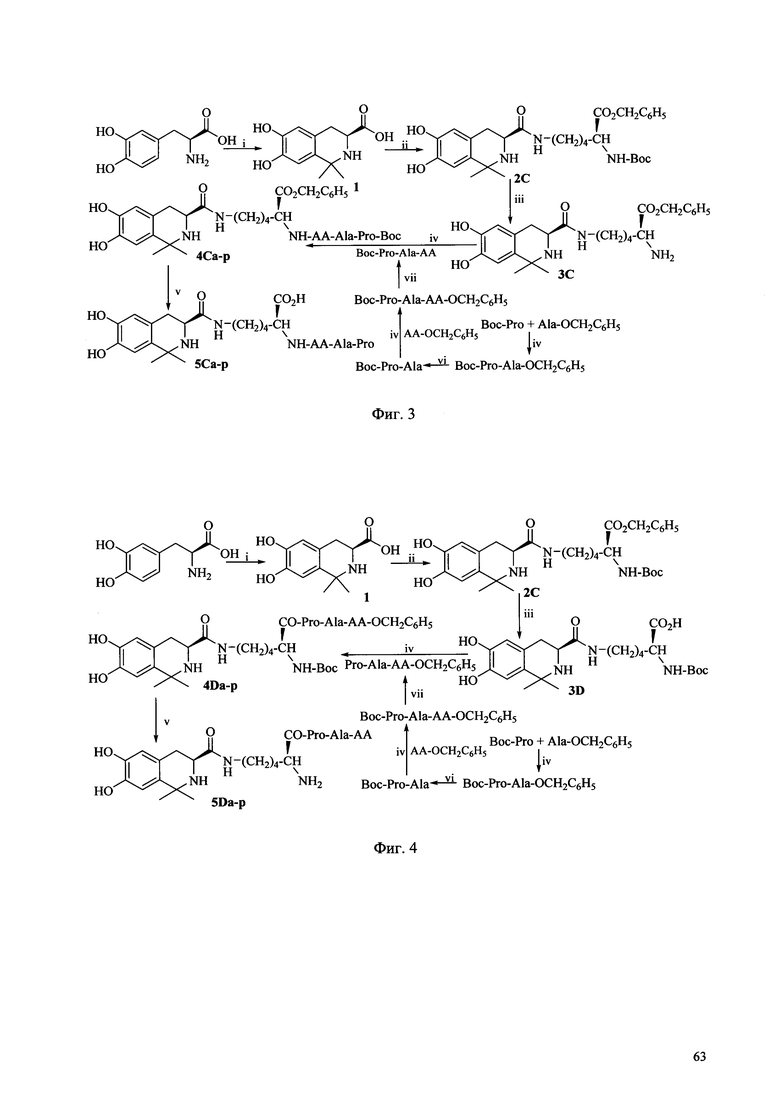
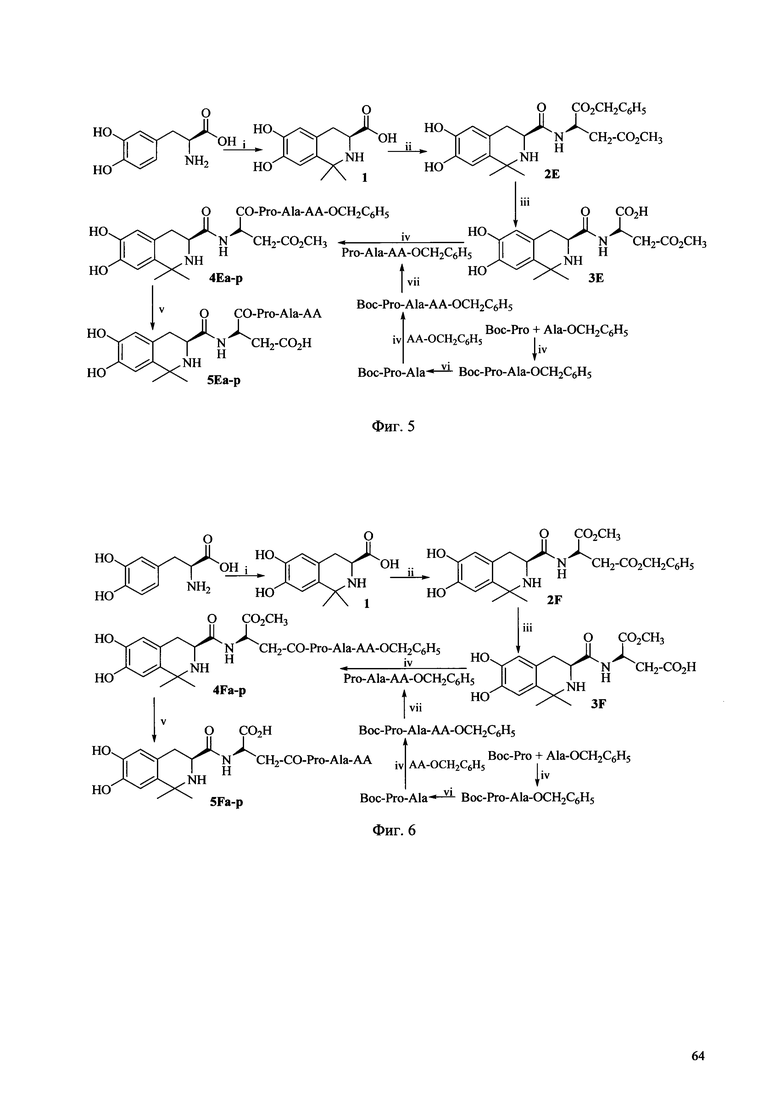
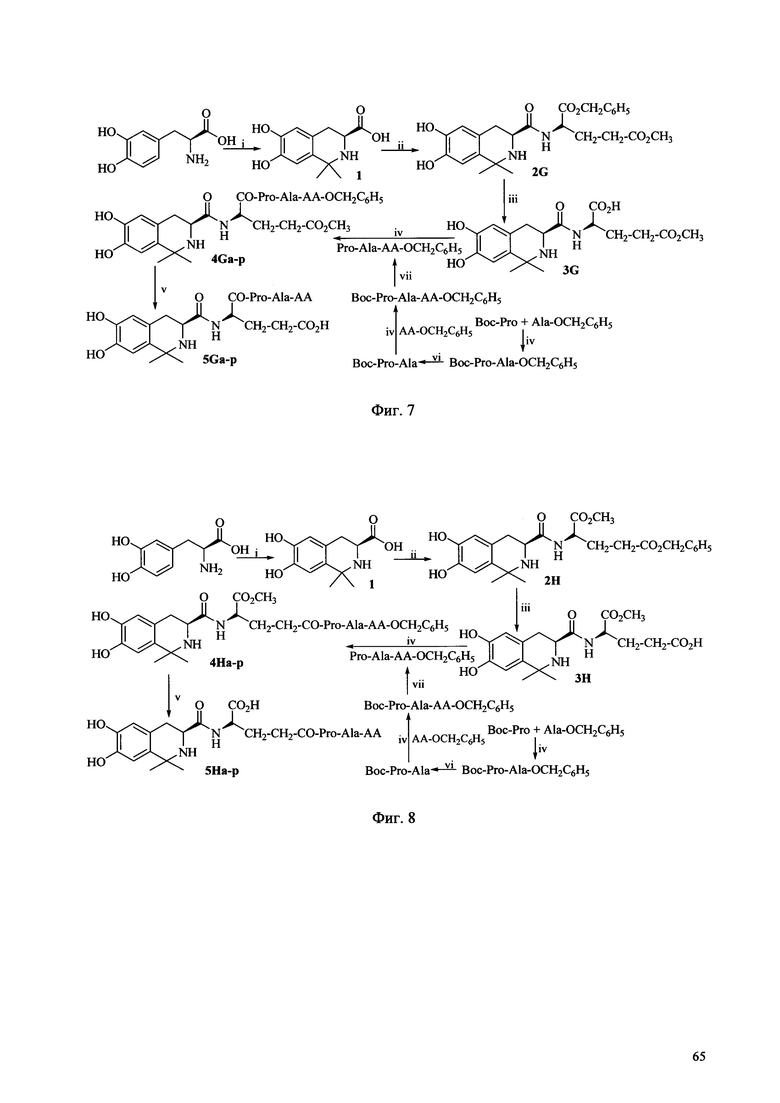

Изобретение относится к соединению формулы I, где АА выбирается из группы, состоящей из L-Ala, L-Val, L-Trp, L-Tyr, L-Pro, L-Phe, Gly, L-Ser, L-Ile, L-Thr, L-Lys, L-Leu, L-Gln, L-Asn, L-Asp и L-Glu, одновременно обладающему тройной активностью в виде тромболизиса, антитромбоза и захвата свободных радикалов, а также к способу получения, композиции. Соединения предназначены для лечения инсульта или церебрального инфаркта. 4 н. и 3 з.п. ф-лы, 9 ил., 13 табл., 68 пр.
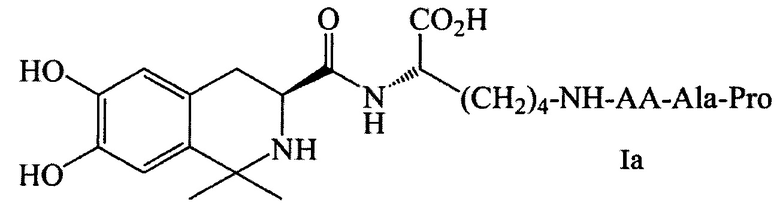
1. Соединение, имеющее следующую формулу 1а:
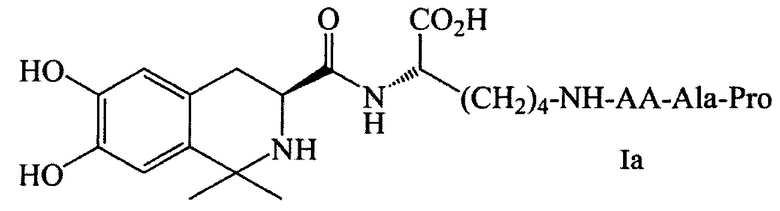
где АА выбирается из группы, состоящей из L-Ala, L-Val, L-Trp, L-Tyr, L-Pro, L-Phe, Gly, L-Ser, L-Ile, L-Thr, L-Lys, L-Leu, L-Gln, L-Asn, L-Asp и L-Glu.
2. Соединение по п. 1, отличающееся тем, что АА представляет собой L-Lys.
3. Фармацевтическая композиция для применения в качестве тромболитического препарата, препарата для захвата свободных радикалов NO или в качестве антитромботического препарата, содержащая терапевтически эффективное количество соединения по любому одному из пп. 1, 2 и фармацевтически приемлемый носитель.
4. Фармацевтическая композиция по п. 3, в которой соединение находится в форме наносферической структуры.
5. Фармацевтическая композиция по п. 3 для применения в качестве препарата при лечении инсульта или церебрального инфаркта.
6. Способ получения соединения, имеющего формулу 1а по п. 1, включающий следующие стадии:
(1) предоставление соединения, имеющего формулу II:
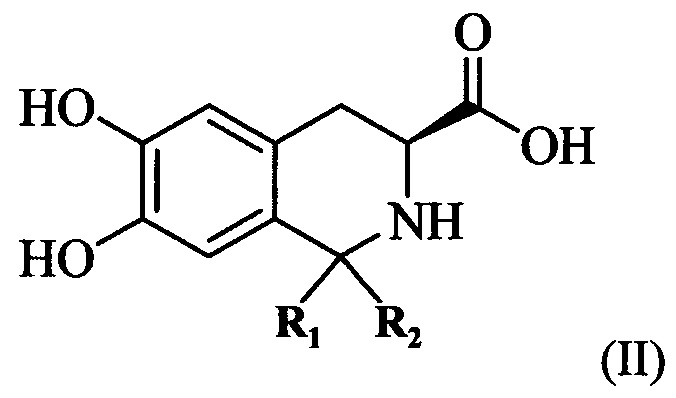
где R1 и R2 представляют собой С14- алкильные группы, и R1 и d R2 являются одинаковыми или различными;
(2) предоставление линкера Т, имеющего по меньшей мере две группы для связывания, и пептида Q, имеющего тромболитическую активность, где линкер имеет первую группу для связывания и вторую группу для связывания;
(3) связывание карбоксильной группы соединения, имеющего формулу II, с первой группой для связывания линкера Т с образованием соединения, имеющего формулу IM-1:
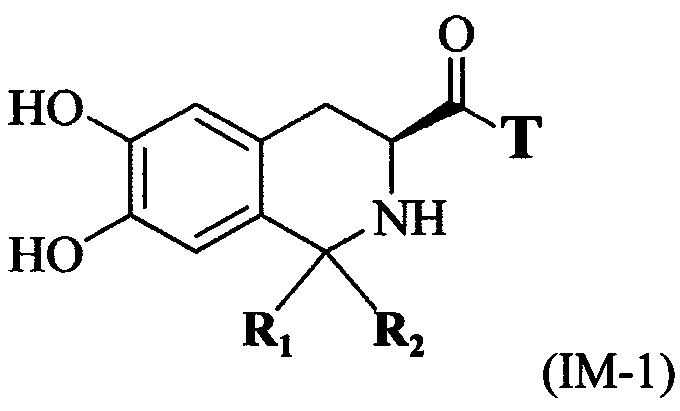
в подходящих реакционных условиях; и
(4) связывание пептида Q, имеющего тромболитическую активность, с соединением, имеющим формулу IM-1, в подходящих реакционных условиях, где один конец пептида Q, имеющего тромболитическую активность, связывается со второй группой для связывания линкера Т с образованием соединения, имеющего формулу Ia,
при этом линкер представляет собой L-Lys, пептид, имеющий тромболитическую активность, выбран из группы, состоящей из олигопептида, имеющего последовательность PA (Pro-Ala), a R1 и R2, оба, представляют собой метальные группы.
7. Способ лечения инсульта или церебрального инфаркта, включающий введение нуждающемуся в этом субъекту терапевтически эффективного количества фармацевтической композиции по п. 3.
| CN 102241740 A, 16.11.2011 | |||
| Прибор для определения силы ветра | 1929 |
|
SU23649A1 |
| CN 101497651 B, 27.06.2012. | |||

