Изобретение относится к технологии получения солей олова (II) и может быть использовано в различных областях химической и иных видов практики, в аналитическом контроле и в научных исследованиях.
Известно получение карбоксилатов ряда металлов, включая и олово (II), на основе водных растворов солей металлов, C8-C22 карбоновых кислот и щелочного агента при нагревании, когда процесс проводят путем ввода соответствующих кислот в нагретый водный раствор солей металлов, поставляющих катион карбоксилата, и нагревание осуществляют до температуры плавления или предплавления кислоты, после чего в полученную суспензию или эмульсию расплава жирных кислот в водном растворе солей вводят при указанной температуре щелочной агент и процесс проводят при молярном соотношении соль:кислота:щелочной агент 1:n:n или (n+0,4), где n – заряд соответствующего катиона металла. При этом в качестве щелочного агента используется едкий натр или едкое кали, процесс проводят при 40-100°C или при температуре ниже температуры плавления соответствующего металлического мыла на 40-45°C (Пат. РФ 2088570, опубл. 27.08.1997).
Недостатками данного способа получения солей методом обменного разложения являются:
1. Использование в качестве реагентов кислоты, щелочи и водорастворимой соли металла, карбоксилат которого предполагается получить. При этом, если кислота и соль поливалентного металла поставляет свои фрагменты в целевой карбоксилат, то NaOH или KOH в последнем никак не представлен и превращается в сопутствующую соль с анионом взятой водорастворимой соли поливалентного металла, являющуюся в ряде случаев просто компонентом сточных вод.
2. Водорастворимая соль поливалентного металла является продуктом более глубокой переработки природного сырья в сравнении с оксидом и (или) металлом. Следовательно, она более дефицитна.
3. Рассматриваемый способ сориентирован на C8-C22 жирные кислоты. Совсем не очевидно, что их можно просто так заменить на ароматические.
4. Данный способ требует подогрева до температур вплоть до 100˚C, причем в несколько этапов, с довольно жестким поддержанием температуры в период ввода щелочного агента и плавления и предплавления кислоты. И хотя данные температуры невелики (до 100°C), их поддержание в требуемом диапазоне - задача непростая.
5. Процесс требует больших расходов воды, что приводит к большим объемам требующих очистки сточных вод.
Известен способ получения карбоксилатов олова (II), в соответствии с которым металлическое олово в виде порошка и других форм вводят в контакт с карбоновой кислотой и промотором при нагревании до 60°C, с подачей воздуха или другого кислородсодержащего газа, дальнейшим нагреванием до 140-180°C и выдержкой при такой температуре до накопления определенного количества олова-продукта. На определенном этапе кислородсодержащий газ меняют на инертный и в такой атмосфере проводят завершение и прекращение процесса (Patent USA №6303808, опубл. 10/16/2001).
Недостатками данного способа являются:
1. Использование довольно высоких температур в основной стадии процесса, обеспечивающих поддержание кислоты-реагента в расплавленном состоянии или же вблизи него.
2. Замена среды свободного контакта с воздухом на инертную в определенный момент времени по ходу процесса, что требует соответствующих мер текущего контроля и соответствующих возможностей для проведения такой операции.
3. В продуктах реакции всегда присутствуют карбоксилаты олова (II) и олова (IV), которые нужно разделять, что значительно усложняет процесс. К тому же нет никаких оснований полагать, что это будут только средние соли, а не смесь средних и основных, которых в 2 раза больше (одна для олова (II) и 3 для олова (IV), не считая различных аддуктов основных и средних солей. Поскольку металлическое олово бралось в избытке, просчитать благоприятную загрузку кислоты на получение доминирования карбоксилатов олова (II) просто не представляется возможным.
4. Процесс проводят в присутствии промотора в количестве до 20% от металлического олова (предпочтительно 1-2%). В качестве последних рекомендованы 4-трет-бутилкатехол; 2,5-ди-трет-бутилгидрохинон, либо стерически затрудненные фенолы, пероксиды, гидропероксиды и способные легко окисляться с образованием пероксидов и гидроксидов углеводороды. Среди них есть довольно сильные окислители соединений олова (II) в соединения олова (IV), что дополнительно осложняет управление составом получаемых продуктов.
5. При разделении реакционных смесей рекомендуется отгонка непрореагировавшей кислоты под вакуумом, что гораздо более сложно, время- и материальнозатратно в сравнении с отделением твердой фазы путем фильтрования.
Наиболее близким к заявляемому является способ трибохимического получения металлсодержащих мыл – компонентов жирующих смесей (патент РФ №2092533, опубл. 10.10.1997; БИ №28), в соответствии с которым соли переходных и иных металлов получают путем прямого взаимодействия оксидов, гидроксидов и карбонатов металлов I-VIII групп Периодической системы с C7 и выше карбоновыми кислотами как индивидуально, так и в виде содержащих эти кислоты композиций и отходов, в присутствии масляной составляющей и растворителя в условиях полного измельчения твердой фазы системы по ходу процесса в бисерной мельнице в присутствии 1-2% по массе воды и 0,08-0,10% по массе трибохимического катализатора, в качестве которого используют азотсодержащие соединения, лучшими из которых являются нитраты аммония и щелочных металлов, вторичные и третичные амины и амиды кислот. При этом в качестве растворителя используют парафиновые и ароматические углеводороды, их технические смеси и масло ПОД. В него вводят масляную составляющую (рыбий жир и подсолнечное масло, сложноэфирные и неомыляемые составляющие талового и кислых масел, смолистые компоненты отходов СЖК, индустриальные масла), жидкие и твердые компоненты кислотсодержащих композиций, воду и трибохимический катализатор и при интенсивном перемешивании и нагревании все перечисленное превращают в раствор, в который дозируют металлсодержащий реагент и ведут процесс получения целевого продукта.
Недостатками данного способа являются:
1. Предложенный способ направлен на получение металлсодержащих мыл для мыловаренного и кожевенного производства, для чего в реакционную смесь вводят масляную композицию, составляющую значительную по массе часть загрузки. Способ не предполагает выделения полученных карбоксилатов в чистом виде, т.е. предполагает использование полученной реакционной смеси как единого целого. Уже сам этот факт исключает прямое применение данного способа для получения индивидуального карбоксилата металла с выделением, соответствующей очисткой и подтверждающей идентификацией.
2. В цитируемом способе используется много многокомпонентного технического сырья, компоненты и примеси которого могут оказывать на ход процесса значимое, но непредсказуемое, а на данный момент и практически неизученное влияние. Отказ от специального ввода таких компонентов также непредсказуем, вплоть до того, что процесс станет неработоспособным.
3. В рассматриваемом способе вводится 1-2% по массе воды, которая в соизмеримых количествах как реакционная образуется по ходу процесса. Такой ввод воды с точки зрения целесообразности не очевиден и требует экспериментального подтверждения или же надежного опровержения.
4. Наличие масляной составляющей, да еще в больших количествах, может существенно нивелировать влияния других составляющих объемных фаз рассматриваемых процессов, в частности, парафиновых и ароматических углеводородов в качестве компонентов комбинированных растворителей. Нет оснований считать, что все эти компоненты, будучи самостоятельными растворителями, т.е. без масляной составляющей, окажутся благоприятными для проведения процесса и не потребуют изменений в природе и количествах других составляющих загрузок на процесс. Все это может определиться лишь при подтверждении или опровержении целенаправленным экспериментом.
5. То же самое можно сказать и о природе и количестве трибохимического катализатора или стимулирующей добавки. Тем более, когда не ясен детальный механизм данного стимулирования.
Задачей предлагаемого решения является подобрать также условия проведения процесса взаимодействия оксида олова (II) с бензойной кислотой, которые бы обеспечили при комнатной и близких к ней температурах высокие выход и избирательность получения бензоата олова (II), а также приемлемые для практических целей степени превращения исходных реагентов и скорости получения средней соли олова (II).
Поставленная задача достигается тем, что оксид олова (II) дозируют в количестве 0,15÷1,0 моль/кг исходной загрузки в мольном соотношении с бензойной кислотой 1:2,10, в качестве растворителя объемной фазы берут уайт-спирит, а в качестве перетирающего агента - фехраль в массовом соотношении с загрузкой 1:1, загрузку ведут в последовательности уайт-спирит, перетирающий агент, кислота, после чего включают механическое перемешивание и подогрев и в течение 10-24 мин ведут приготовление раствора кислоты с температурой в момент завершения 40-45°C, после чего подогрев удаляют и, не прекращая перемешивания, вводят трибохимический катализатор в количестве (1-25)·10-3 моль/кг и оксид олова (II), далее проводят процесс получения карбоксилата при текущем контроле за ходом протекания вплоть до полного превращения оксида в целевую соль, затем перемешивание прекращают, суспензию твердой соли отделяют от перетирающего агента и фильтруют, осадок на фильтре промывают промывным растворителем после отмывки перетирающего агента и элементов реактора от остатков реакционной смеси уайт-спиритом, хорошо отжимают, снимают с фильтра и сушат на воздухе до постоянного веса. При этом в качестве трибохимического катализатора берут п-аминоазобензол, бензидин, нафтиламин, п-аминофенол, 2-аминопиридин и 4-амино-1,2,4-триазол. А фильтрат и промывной растворитель возвращают в повторный процесс.
Характеристика используемого сырья:
Оксид олова (II) ТУ 6-09-1503-87
Бензойная кислота ГОСТ 6413-77
п-Аминоазобензол ГОСТ 4681-70
Бензидин ТУ 6-09-4221-76
Нафтиламин ГОСТ 8827-74
п-Аминофенол ГОСТ 5209-77
2-Аминопиридин ТУ 6-09-672-77
4-амино-1,2,4-триазол ТУ 6-09-08-949-75
Уайт-спирит ГОСТ 3134-78
Фехраль ГОСТ 8803-89.
Проведение процесса заявляемым способом следующее. В бисерную мельницу вертикального типа с высокооборотной механической мешалкой лопастного типа и пластмассовым корпусом объемом 285 мл и диаметром входного отверстия 49 мм вводят расчетные количества растворителя, фехраля в виде колец диаметром 8-10 мм, выполненных из провода диаметром 1 мм, и бензойной кислоты. Корпус с загруженными компонентами помещают на свое место в каркасной раме, соединяют с крышкой, снабженной сальниковой коробкой, механической мешалкой с лопастью из нержавеющей стали, отверстиями для помещения датчика температуры и пробоотборника, и должным образом крепят. Включают механическое перемешивание, подводят снизу жидкостную обогревательную баню и ведут обогрев, перемешивание и приготовление раствора бензойной кислоты таким образом, чтобы температура к концу операции достигала 40-45°C. По завершении стадии растворения обогревающую баню удаляют, в реактор загружают расчетные количества трибохимического катализатора и оксида олова (II) и этот момент принимают за начало процесса получения карбоксилата. Контроль за ходом процесса осуществляют методом отбора проб и определения в них содержания соединений олова (II) и текущих концентраций кислоты. Как только последние приближаются к расчетным значениям, отвечающим практически 100%-ному превращению оксида в средний карбоксилат, перемешивание прекращают, отсоединяют корпус от его крышки и опускают таким образом, чтобы была возможность остаткам реакционной смеси стечь с вала и лопасти механической мешалки в корпус. Далее корпус реактора вынимают из гнезда каркасной рамы, а его содержимое выливают в воронку с сеткой с ячейками 0,3×0,3 мм. Оставшийся на сетке перетирающий агент аккуратно снимают и возвращают в корпус реактора. Проводят снова сборку бисерной мельницы и ее крепление в каркасной раме. Далее вводят некоторое количество растворителя жидкой фазы, включают механическое перемешивание и проводят отмывку корпуса, мешалки и перетирающего агента от остатков реакционной смеси (РС). После завершения этой операции перемешивание прекращают и проводят повторное отделение перетирающего агента от промывного растворителя, собирая последний в специально предусмотренную емкость. Параллельно отделенную от перетирающего агента суспензию реакционной смеси фильтруют, осадок на фильтре промывают промывным растворителем, тщательно отжимают и отправляют на сушку или очистку путем перекристаллизации. А фильтрат и промывной растворитель анализируют и используют в загрузке повторного процесса. Последний может быть использован и в обозначенных выше промывках осадка продукта на фильтре.
Пример №1
В бисерную мельницу вертикального типа с высокооборотной (1560 об/мин) мешалкой с размерами лопасти 48×36×1,8 мм из нержавеющей стали вводят 90,12 г уайт-спирита, 100 г фехраля как перетирающего агента и 6,41 г бензойной кислоты. Корпус реактора помещают в свое гнездо в каркасной раме, соединяют с крышкой, проверяют прокручивание вала мешалки, подводят снизу предварительно нагретую до 50-70°C нагревательную баню, включают механическое перемешивание и начинают приготовление раствора кислоты. Начальная температура 23°C. Через 15 мин она достигла 41°C и практически вся твердая фаза кислоты перешла в раствор. Не прекращая перемешивания, убирают, путем опускания вниз, нагревательную баню, через предназначенное для отбора проб отверстие вводят 0,1 г п-аминоазобензола и 3,37 г оксида олова (II) и начинают процесс получения целевого продукта. Процесс ведут при естественно складывающейся температуре и текущем контроле методом отбора проб и определения в них содержаний накопившихся соединений олова (II) и неизрасходованной кислоты. На основе этих данных строят зависимость выхода продукта от времени протекания процесса. Для рассматриваемого примера она приводится в табличной форме записи:
По достижении по результатам анализа превышающего 98% выхода продукта процесс его получения прекращают, останавливают механическое перемешивание реактора, отсоединяют корпус от крышки с механической мешалкой и опускают его вниз таким образом, чтобы нижняя кромка лопасти мешалки оказалась над реакционной смесью в корпусе. В таком положении выдерживают 5 мин, давая возможность остаткам РС стечь. После этого корпус с перетирающим агентом и РС вынимают из гнезда каркасной рамы и его содержимое выливают в воронку с сеткой в качестве фильтровальной перегородки для отделения фехраля. Перетирающий агент тщательно снимают с сетки и возвращают в корпус реактора. Установку собирают заново, вводят 30 г уайт-спирита, включают механическое перемешивание и в течение 10 мин проводят отмывку стенок и содержимого реактора от остатков РС. Далее перемешивание прекращают и проводят повторное отделение перетирающего агента. Промывной растворитель собирают в специально предусмотренную емкость.
Отделенную ранее от перетирающего агента суспензию РС подвергают фильтрованию, осадок промывают на фильтре частью собранного промывного растворителя, тщательно отжимают, аккуратно снимают и отправляют на воздушную сушку до постоянного веса. После завершения сушки полученную массу взвешивают и определяют выход продукта с учетом потерь при выделении. Он оказался равным 96%, т.е. потери при выделении составили ~ 3%. Твердую фазу тщательно измельчают, определяют эквивалентную массу и помещают в емкость для хранения. При необходимости ее направляют на очистку путем перекристаллизации.
Фильтрат и промывной растворитель взвешивают, определяют в них содержания соли олова (II) и остаточной кислоты и отправляют на загрузку повторного процесса. В данном случае с ними возвращается ~ 1% полученного бензоата олова (II) как целевого продукта.
Примеры №2-5
Реактор, исходные реагенты, трибохимический катализатор, растворитель жидкой фазы, мольное соотношение оксид:кислота, дозировка п-аминоазобензола, масса загрузки и ее соотношение с массой перетирающего агента, природа последнего, последовательности операций при загрузке, приготовлении раствора бензойной кислоты, проведении процесса, его завершении, отделении перетирающего агента, отмывке его и внутренних поверхностей корпуса и элементов реактора от остатков реакционной смеси, выделении из последней твердого целевого продукта и возврате фильтрата и промывного растворителя на загрузку повторного процесса аналогичны описанным в примере 1. Отличается величинами загрузок оксида олова (II) и бензойной кислоты. Указанные различия и другие характеристики процесса сведены в табл. (РС – реакционная смесь).
Температура РС, °C
Примеры №6-9
Реактор, исходные реагенты и их загрузки, растворитель жидкой фазы, масса загрузки и ее соотношение с массой перетирающего агента, природа последнего, последовательность операций при загрузке, приготовлении раствора бензойной кислоты, проведении процесса, его завершении, отделении перетирающего агента, отмывке его и внутренних поверхностей корпуса и элементов реактора от остатков реакционной смеси, выделении из последней твердого целевого продукта и возврате фильтрата и промывного растворителя на загрузку повторного процесса аналогичны описанным в примере 1. Отличается отсутствием п-аминоазобензола в загрузке и дозировками этого трибохимического катализатора, что вместе с другими характеристиками процесса сведено в таблицу.
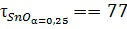
* - максимально достигнутая степень превращения реагента в недостатке (т.е. SnO) равна 0,4, а продукты представлены основной и средней солью в мольном соотношении 1:1,8.
Примеры №10-14
Реактор, исходные реагенты и их загрузки, растворитель жидкой фазы, масса загрузки и ее соотношение с массой перетирающего агента, природа последнего, последовательности операций при загрузке, приготовлении раствора кислоты, проведении процесса, разделении реакционной смеси, работе с жидкой и твердой фазами, а также загрузка трибохимического катализатора аналогичны описанным в примере 8. Отличаются природой трибохимического катализатора, которая вместе с другими характеристиками процесса приведена в таблице.
Температура РС, °C
32
Положительный эффект предлагаемого решения состоит в том:
1. В предлагаемом решении не требуется большой избыток кислоты и достигается высокий процент перехода масс исходных реагентов в массу целевого продукта. Сопутствующий продукт вода имеет небольшую в сравнении с целевым карбоксилатом молекулярную массу и накапливается в небольших по массе количествах.
2. Основная масса продукта из-за плохой растворимости в жидкой составляющей объемной фазы накапливается в суспендированном виде и легко отделяется путем простого фильтрования. Такой продукт при практически количественном превращении в него реагента в недостатке и отмывке остатков кислоты промывным растворителем на фильтре имеет довольно высокую степень чистоты и во многих случаях своего использования в дополнительной очистке путем перекристаллизации просто не нуждается.
3. Процесс не осложнен переработкой фильтратов и промывного растворителя. Они как единое целое могут быть переработаны в самом процессе.
4. Процесс проводится при низких температурах и не требует котлонадзорного оборудования.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения ацетата или оксалата свинца из его оксида (II) | 2023 |
|
RU2807759C1 |
| Способ получения основного бензоата олова (II) | 2017 |
|
RU2650893C1 |
| Двухстадийный способ получения карбоксилатов олова (II) из металла | 2017 |
|
RU2678092C1 |
| Способ получения фторида олова (II) из металла и его диоксида | 2018 |
|
RU2713840C1 |
| Способ получения карбоксилатов олова (II) | 2017 |
|
RU2671197C1 |
| Способ получения бензоата и замещенных бензоатов олова (IV) из вторичного сырья | 2017 |
|
RU2673470C1 |
| Способ получения бензоата и замещенных бензоатов олова (IV) | 2017 |
|
RU2660905C1 |
| Способ получения нитрата олова (IV) | 2017 |
|
RU2655142C1 |
| Способ получения соли олова (IV) с анионами азотной и бензойной кислот | 2020 |
|
RU2735433C1 |
| Способ получения нитрата олова (IV) путем окисления нитрата олова (II) | 2019 |
|
RU2717810C1 |
Изобретение относится к способу получения бензоата олова (II) путем прямого взаимодействия оксида олова (II) с карбоновой кислотой в условиях интенсивного механического перемешивания и использования перетирающего агента, объемной фазы на основе органического растворителя и трибохимического катализатора при соизмеримых с комнатной температурах, где оксид олова (II) дозируют в количестве 0,15-1,00 моль/кг исходной загрузки в мольном соотношении с бензойной кислотой 1:2,10, растворителем объемной фазы берут уайт-спирит, а в качестве перетирающего агента фехраль в массовом соотношении с загрузкой 1:1, загрузку ведут в последовательности уайт-спирит, перетирающий агент, кислота, после чего включают механическое перемешивание и подогрев и в течение 10-24 мин ведут приготовление раствора кислоты с температурой в момент завершения 40-45˚C, после чего подогрев удаляют и, не прекращая перемешивания, вводят трибохимический катализатор в количестве (1÷25)·10-3 моль/кг и оксид олова (II), далее проводят процесс получения карбоксилата при текущем контроле за ходом протекания вплоть до полного превращения оксида в целевую соль, затем перемешивание прекращают, суспензию твердой соли отделяют от перетирающего агента и фильтруют, осадок на фильтре промывают промывным растворителем после отмывки перетирающего агента и элементов реактора от остатков реакционной смеси уайт-спиритом, хорошо отжимают, снимают с фильтра и сушат на воздухе до постоянного веса. Способ позволяет увеличить выход целевого продукта и достигается высокий процент перехода масс исходных реагентов в массу целевого продукта. 2 з.п. ф-лы, 14 пр.
1. Способ получения бензоата олова (II) путем прямого взаимодействия оксида олова (II) с карбоновой кислотой в условиях интенсивного механического перемешивания и использования перетирающего агента, объемной фазы на основе органического растворителя и трибохимического катализатора при соизмеримых с комнатной температурах, отличающийся тем, что оксид олова (II) дозируют в количестве 0,15-1,00 моль/кг исходной загрузки в мольном соотношении с бензойной кислотой 1:2,10, растворителем объемной фазы берут уайт-спирит, а в качестве перетирающего агента фехраль в массовом соотношении с загрузкой 1:1, загрузку ведут в последовательности уайт-спирит, перетирающий агент, кислота, после чего включают механическое перемешивание и подогрев и в течение 10-24 мин ведут приготовление раствора кислоты с температурой в момент завершения 40-45°C, после чего подогрев удаляют и, не прекращая перемешивания, вводят трибохимический катализатор в количестве (1÷25)·10-3 моль/кг и оксид олова (II), далее проводят процесс получения карбоксилата при текущем контроле за ходом протекания вплоть до полного превращения оксида в целевую соль, затем перемешивание прекращают, суспензию твердой соли отделяют от перетирающего агента и фильтруют, осадок на фильтре промывают промывным растворителем после отмывки перетирающего агента и элементов реактора от остатков реакционной смеси уайт-спиритом, хорошо отжимают, снимают с фильтра и сушат на воздухе до постоянного веса.
2. Способ по п.1, отличающийся тем, что в качестве трибохимического катализатора берут п-аминоазобензол, бензидин, нафтиламин, п-аминофенол, 2-аминопиридин или 4-амино-1,2,4-триазол.
3. Способ по п.1, отличающийся тем, что фильтрат и промывной растворитель возвращают в повторный процесс.
| EP 644223 A2, 22.03.1995 | |||
| CN 104892564 A, 09.09.2015 | |||
| DE 3428404 A1, 21.02.1985 | |||
| Газоанализатор | 1973 |
|
SU636545A1 |
| СПОСОБ ТРИБОХИМИЧЕСКОГО ПОЛУЧЕНИЯ МЕТАЛЛСОДЕРЖАЩИХ МЫЛ - КОМПОНЕНТОВ ЖИРУЮЩИХ СМЕСЕЙ | 1995 |
|
RU2092533C1 |

