Изобретение относится к технологии получения галогенидов олова (II) и может быть использовано в разных областях промышленной и лабораторной практики, в научных исследованиях и в аналитическом контроле.
Известен способ получения фторида олова (II) путем непосредственного взаимодействия оксида олова (II) с 40%-ной плавиковой кислотой в платиновой чашке с последующим выпариванием досуха без доступа воздуха / (Руководство по неорганическому синтезу: в 6-и томах. Т. 1. Пер. с нем. / под ред. Г. Брауэра. - М.: Мир, 1985, С.257).
Недостатками этого способа являются:
1. Рассматриваемый процесс никак не определен ни в количественных соотношениях реагентов, ни в величинах их загрузок, ни в температурных режимах, ни в отношении перемешивания и прочих, включая и временные, характеристиках.
2. Используемый реактор пригоден для лабораторного получения. Но вряд ли его можно рекомендовать для пилотной установки и более крупного масштаба.
3. Выпаривание без доступа воздуха не такая уж простая операция в плане реализации.
В цитируемом выше источнике приводится и вариант получения SnF2 в виде хорошо образованных кристаллов в более мягких условиях. В частности, в полиэтиленовый стакан на 200 мл наливают 15-20 мл не содержащей газов воды и всыпают 0,5 моль SnO. Затем стакан нагревают на паровой бане до 60°C в атмосфере азота, свободного от O2, и медленно по каплям добавляют 46 г 48%-ной плавиковой кислоты (1,1 моль). При этом содержимое стакана все время перемешивают путем покачивания стакана. С началом реакции происходит разогревание реакционной смеси. Когда все растворится, стакан ставят в эксикатор и охлаждают. Через 2 часа маточный раствор декантируют во второй стакан. Оба стакана ставят в эксикатор над CaCl2 и KOH (1:1). Через 2 суток их ставят в эксикатор над Mg(ClO4)2. Еще через 4 суток маточный раствор декантируют и получают порцию кристаллов, которая высушена как первая. Выход 86%.
Недостатками данного способа являются:
1. Он довольно длителен и не обеспечивает приближающийся к количественному выход выделенного продукта.
2. Для проведения химического превращения и выделения целевого продукта требуется много дополнительных операций, реализуемых в специфических условиях. В частности, нужно предварительно получить, сохранить и продозировать лишенную растворенных газов воду; далее нагревание загрузки до 60°C на паровой бане в атмосфере азота с высокой степенью очистки от следов кислорода; для сушки кристаллов используют эксикаторы с разными средствами для поглощения воды из газовой фазы и т.д.
3. Не определена и интенсивность перемешивания реакционной смеси в стакане.
4. В целом предложенный вариант как способ выглядит неопределенным.
Предложен новый двухстадийный метод синтеза фторида олова, включающий стадии получения трифторстанната олова (II) и термического разложения трифторстанната с оловом (Горячева Т.В. Разработка методов синтеза фторидов металлов IV группы с помощью гидродифторида аммония. Дисс. канд. хим. наук. РХТУ им. Д.И. Менделеева. М. 2002. 112 с.). Отмечается, что метод позволяет достигать высоких выходов по олову и фтору.
Недостатками данного варианта являются:
1. Процесс получения обозначенного продукта является двухстадийным, одна из стадий которого (термического разложения) протекает при температурах 180-200°C, что требует соответствующей организации подвода внешнего тепла.
2. Каждая из стадий предложенного метода требует своего аппаратурного оформления и индивидуальных путей управления.
3. Предложенный метод не проработан как способ получения целевого продукта.
Наиболее близким к заявляемому является способ получения фторида олова (II) (заявка CN 103332731 С01G, 19/04, 2013-10-02) в соответствии с которым в реактор вводят фтороводородную кислоту, включают перемешивание, добавляют 8-12% от расчетного количества оксида олова гранулированного олова, после чего дозируют сам оксид со скоростью 15-25 г/мин. При этом загрузку оксида олова (II) и все дальнейшие операции проводят в среде азота. Обращается внимание на возможное разбрызгивание реакционной смесив этот период. По завершении ввода расчетного количества оксида олова (II) перемешивание продолжают в течение 15-30 мин, после чего удаляют гранулированное олово и приступают к концентрированию при 75-95°С и перемешивании и вакуумной сушке полученного продукта. Отмечается высокий выход и чистота, а также низкая стоимость получаемого продукта.
Недостатками данного способа являются:
1. В доступном описании цифровые характеристики представлены таким образом, что не дают представление о величине загрузки в целом, как и о длительности процесса от начала до завершения, либо до момента получения конечного продукта в товарном виде, а также используется ли избыток кислоты и если да, то какой, удаляется ли последний при концентрировании, вакуумной сушке и т.д.
2. Нет ясности и в том, в каком виде продукт накапливается по ходу процесса: в виде раствора или же суспензии, либо в том и другом в разных соотношениях, есть ли отложения продукта на металле и сколько их, участвует ли вводимый металл в получении целевого продукта или же только способствует ускоренному протеканию взаимодействия оксида олова с кислотой, как это имеет место для многих аналогичных взаимодействий с участием не только минеральных, но и карбоновых кислот.
3. Процесс в основной своей фазе проводят в среде азота, что значительно усложняет как технологию проведения, так и аппаратурное оформление.
4. В описании упоминается о необходимости контроля за возможным разбрызгиванием реакционной смеси. Причиной последних могут быть локальные разогревы в зонах с повышенными содержаниями вводимого оксида олова (II), т.е. экзотермичность брутто-процесса в целом, либо гидравлические удары при тех или иных сбоях перемешивания, либо и первое, и второе в комбинированных вариантах. При этом не ясно, в чем состоит контроль за обозначенным явлением и как он отражается на пооперационной схеме процесса и ее аппаратурном оформлении.
5. Нельзя назвать простыми и операции концентрирования при температуре 75-95°С и перемешивании, а также вакуум-сушки при получении конечного продукта. Во-первых, это энергозатратные стадии, что плохо согласуется с постулированием низкой стоимости продукта. Во-вторых, нет состава, удаляемого при концентрировании: одно дело, если удаляется только вода, другое - когда приходится удалять и фтороводородную кислоту, и воду. Что-то можно было бы понять, если бы была информация о количестве и концентрации фтороводородной кислоты в исходной загрузке, а также четкое указание с каким типом процесса (периодическим, промежуточного типа и т.д.) приходится иметь дело.
6. По описанию после удаления гранулированного, а вернее отработанного олова реакционную смесь нужно нагреть до 75-95°С, что также требует определенного аппаратурного оформления, способного работать под вакуумом.
Задачей предлагаемого решения является подобрать такие условия окисления олова его диоксидом в присутствии фтороводородной кислоты и стимулирующей добавки йода с образованием фторида олова (II) в качестве целевого продукта, которые при комнатной и близких к ней температурах позволили в технологически приемлемое время с практически количественным выходом и приближающейся к 100% избирательностью превращать диоксид металла как загружаемый в стехиометрическом недостатке реагент в целевой продукт.
Поставленная задача достигается тем, что SnO берут в количестве 0,25-0,55 моль/(кг загрузки без учета олова) в мольном соотношении с кислотой (1:4,09)÷(1-4,30), в качестве перетирающего агента используют фехраль, загружаемый в массовом соотношении с остальной загрузкой без учета олова, равном 1:1, йод дозируют в количестве 0,035-0,050 моль/(кг загрузки без учета олова), а олово - 10÷22% от остальной загрузки, в качестве растворителя используют этилцеллозольв или уайт-спирит, загрузку ведут в последовательности: растворитель объемной фазы, перетирающий агент, диоксид олова, молекулярный йод, а также в случае этилцеллозольва фтороводородную кислоту и олово, после чего включают механическое перемешивание, либо при использовании в качестве растворителя объемной фазы уайт-спирита сначала включают механическое перемешивание и далее вводят фтороводородную кислоту и олово и в обеих случаях ведут процесс при естественно складывающейся температуре до достижения расчетного содержания продукта в реакционной смеси, после чего перемешивание прекращают, отделяют перетирающий агент и непрореагировавшее олово от реакционной смеси, последнюю фильтруют, осадок на фильтре промывают растворителем жидкой фазы, отжимают и либо сушат, либо подвергают дополнительной перекристаллизации и сушат; при этом непрореагировавшее олово и содержащий избыточную кислоту и основную массу введенного вначале йода и полученных из него соединений фильтрат и промывной растворитель возвращают без всяких очисток и концентрирований на загрузку повторных процессов.
Характеристика используемого сырья:
Олово белое (гранулированное, полоса) ГОСТ 860-75
Оксид олова (IV) ГОСТ 22516-77
Фтороводородная (плавиковая) кислота ГОСТ 10484-78
Молекулярный йод ГОСТ 4159-79
Этилцеллозольв ГОСТ 8313-88
Уайт-спиритГОСТ 3134-78
п-Аминоазобензол ГОСТ 4681-70
Проведение процесса заявляемым способом следующее. В бисерную мельницу вертикального типа с пластмассовым корпусом с входным отверстием не менее 45-50 мм и высокооборотной механической мешалкой лопастного типа вводят расчетные количества растворителя, перетирающего агента, диоксида олова и молекулярного йода. Далее загрузка зависит от растворимости фтороводородной кислоты в выбранном растворителе объемной фазы. При высокой и умеренной растворимости HF продолжают без каких-либо перерывов и других действий загрузку водного раствора кислоты и олова, после чего включают механическое перемешивание, индексируемое как начало процесса. При плохой же и очень плохой растворимости кислоты (в данном случае в уайт-спирите) сначала включают механическое перемешивание, препятствующее возможному попаданию водного раствора кислоты в мертвую зону реактора(реально появляющуюся при превышении зазора между днищем корпуса и нижней кромкой мешалки 0,5 мм), после чего добавляют расчетное количество фтороводородной кислоты и олова, фиксируя уже этот момент как начало проводимого процесса. В обеих случаях процесс ведут до достижения расчетного содержания продукта в реакционной смеси. После этого механическое перемешивание прекращают, отделяют корпус реактора от крышки с мешалкой и опускают вниз, чтобы нижняя кромка мешалки оказалась выше уровня реакционной смеси в корпусе, давая возможность стечь остаткам реакционной смеси с мешалки и прочих элементов. Далее корпус с реакционной смесью (РС) переносят в узел отделения непрореагировавшего металла и перетирающего агента от РС путем пропускания через сетку в качестве фильтровальной перегородки. Отделенный перетирающий агент и непрореагировавшее олововозвращают в корпус бисерной мельницы, собирают установку вновь, добавляют определенное количество растворителя жидкой фазы и проводят отмывку перетирающего агента и элементов реактора от остатков РС. По завершении этой операции проводят повторное отделение перетирающего агента и непрореагировавшего олова,
их разделение, определение массы непрореагировавшего олова и потерь перетирающего агента и далее их возврат на загрузку повторных процессов.
Отделенную реакционную смесь подвергают фильтрованию, осадок на фильтре промывают полученным выше промывным растворителем, отжимают,вместе с фильтровальной перегородкой снимают с фильтра и сушат на воздухе до постоянной массы, периодически дробя для получения порошкообразного состояния. Определяют массу выделенного продукта и его эквивалент.
Полученные совместно (или раздельно) фильтрат и промывной растворитель анализируют на содержание остаточной кислоты, растворенного продукта, молекулярного йода и йодидов, после чего направляют на загрузку повторного процесса.
При необходимости значимо ускорить процесс в начальную реакционную смесь вводят п-аминоазобензол в количестве 10-2 моль/кг в качестве трибохимического катализатора. Место этой операции в последовательности загрузки до момента ввода металла принципиального значения не имеет, а вот выдержка нахождения реакционной смеси с п-аминоазобензолом и всеми другими компонентами до ввода металла или йода, если его вводить последним, может сказываться весьма существенно.
Пример №1.
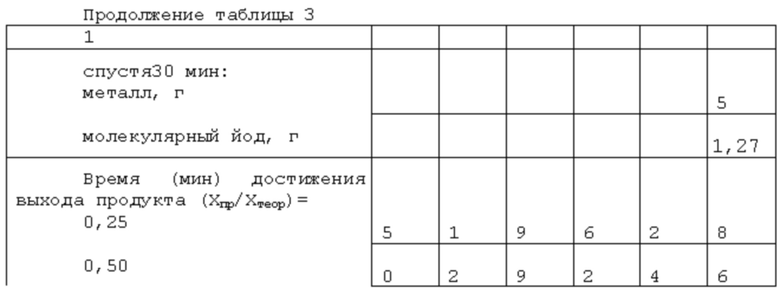
В бисерную мельницу вертикального типа с пластмассовым корпусом с плоским дном, толщиной стенок 2,8 мм и внутренним диаметром 52,3 мм и высотой 137 мм, снабженную высокооборотной (1560 об/мин) лопастной мешалкой с размерами лопасти 50,9×27×2,5 мм из текстолита, сальниковой коробкой и карманами для пробоотборника, дозагрузок после ввода перемешивания и измерения температуры (выполнены в пластмассовой крышке, толщиной 8,4 мм) последовательно загружают 89,80 г этилцеллозольва 100 г перетирающего агента в виде колец диаметром 7-10 мм из провода диаметром 1 мм из фехраля, 3,77 г диоксида олова, 1,27 г молекулярного йода и 5,16 г фтороводородной (плавиковой; 19,8 моль/кг) кислоты и 10 г металла. Корпус с загрузкой соединяют с крышкой, помещают в соответствующее гнездо каркасной рамы, надежно крепят, вручную проворачивают мешалку и включают механическое перемешивание, принимая этот момент за начало проводимого процесса. Далее процесс ведут при естественно складывающейся температуре (без подвода внешнего тепла и принудительного отвода) при текущем контроле за накоплением соли олова (II) и расходованием кислоты. Полученные данные текущего контроля сведены в таблице 1.

По истечении 230 мин, когда значение достигнутой степени расходования диоксида олова (IV) как реагента в недостатке превысила 98,5%, перемешивание прекращают, корпус мельницы отсоединяют от крышки и опускают в гнезде каркасной рамы настолько, чтобы нижняя кромка лопасти мешалки оказалась выше уровня реакционной смеси (РС) в корпусе. В таком положении корпус выдерживают в течение 7 мин, давая возможность остаткам РС с вала и лопасти мешалки и других элементов реактора стечь. После этого корпус с РС вынимают из гнезда каркасной рамы и его содержимое выливают в приемлемую воронку узла фильтрования с сеткой с размерами отверстий 0,3×0,3 мм. Суспензия РС легко проваливается в приемную емкость, а на сетке остается перетирающий агент и непрореагировавший металл. Их возвращают в корпус, собирают бисерную мельницу вновь, вводят 30 мл растворителя жидкой фазы, включают механическое перемешивание и в течение 10 мин проводят отмывку элементов реактора, перетирающего агента и непрореагировавшего металла от остатков РС. Далее проводят повторное отделение твердой фазы на сетке в качестве фильтровальной перегородки, собирая промывной растворитель в специально подготовленную емкость. Перетирающий агент и олово сушат, разделяют и взвешивают; потери перетирающего агента не зафиксированы. А потери металла хорошо коррелируют с количеством полученного продукта, что свидетельствует о хорошем соответствии стехиометрическому уравнению
Sn+4HF+SnO2 →2SnF2+2H2O
Полученную при первом отделении перетирающего агента и остаточного металла РС фильтруют, осадок на фильтре отжимают, далее обрабатывают промывным растворителем, снова отжимают и направляют на воздушную сушку до постоянной массы. После этого его хорошо измельчают до порошкообразного состояния, взвешивают и определяют эквивалентную массу. Фильтрат совместно с промывным растворителем взвешивают, подвергают анализу на содержание йода и его соединений, соли-продукта и остаточной кислоты и как отделенные ранее перетирающий агент и непрореагировавший металл направляют на загрузку повторного(ых) процесса(ов).
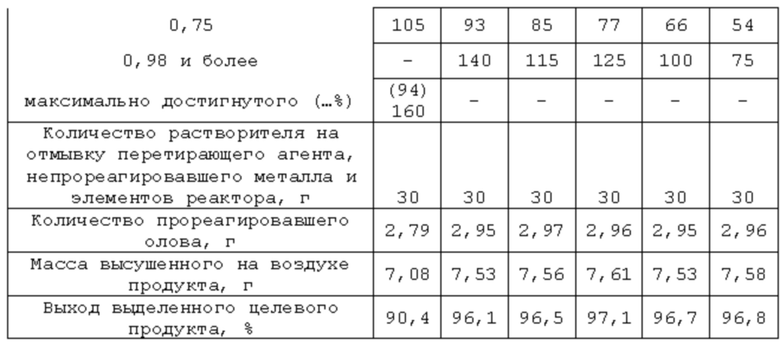
На основе результатов выходного контроля определены масса высушенного на воздухе продукта (7,53 г), массы прореагировавшего Sn (2,96 г) и выход выделенного продукта, который в данном случае составил 96,7%. А эквивалентная масса 157±1, что отвечает SnF2 как целевому продукту.
Примеры №2-7
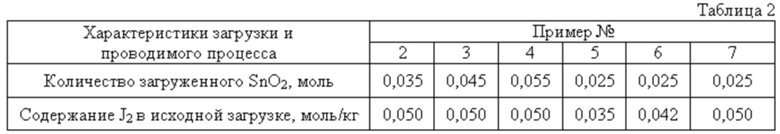
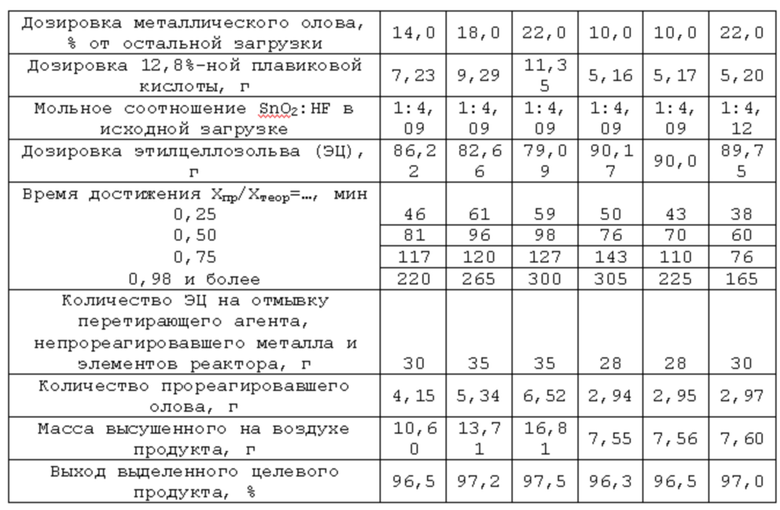
Реактор, исходные реагенты, растворитель жидкой фазы, стимулирующая добавка йода, соотношение масс перетирающего агента и остальной загрузки (металл не входит), мольное соотношение диоксид олова:кислота, последовательность операций при загрузке компонентов реакционной смеси, при запуске процесса, его проведении и прекращении, а также текущем контроле за ходом, отделении перетирающего агента и непрореагировавшего металла, их очистке от остатков РС, разделении РС на фазы, при работе с продуктом и жидкой фазой аналогичны описанным в примере 1. Отличаются начальной дозировкой диоксида олова как реагента в недостатке, количеством стимулирующей добавки йода и массой загруженного металла. Эти различия и другие характеристики процесса сведены в таблицу 2.
Примеры №8-13
Реактор, исходные реагенты, стимулирующая добавка йода, последовательность операций при проведении процесса, текущем контроле за ходом протекания, при определении момента прекращения процесса, отделении перетирающего агента и непрореагировавшего металла, отмывке их и элементов реактора от остатков реакционной смеси, выделения из последней твердого продукта и работе с ним, фильтратом и непрореагировавшим металлом аналогичны описанным в примере 1. Отличаются природой используемого растворителя с плохой растворимостью воды и водных растворов кислоты в нем и связанными с этим изменениями в последовательности дозировок некоторых компонентов реакционной смеси, некоторыми количественными характеристиками их, использованием п-аминоазобензола в качестве трибохимического катализатора взаимодействия диоксида олова с плавиковой кислотой с образованием фторида олова (IV) (более сильного окислителя в сравнении с SnO2), а также сдвигом дозировки металла в сторону большего времени для взаимодействия SnO2 с HF и оценкой последствий такого сдвига. Указанные отличия и характеристики процесса сведены в таблицу 3.
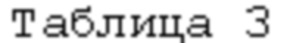
Положительный эффект предлагаемого решения состоит в том, что:
1. Целевой продукт накапливается в основном в суспендированной твердой фазе, что существенно облегчает его выделение и снижает потери в этой операции. А поскольку фильтрат и промывной растворитель возвращаются на загрузку повторных процессов, непроизводительные потери продукта становятся еще меньше.
2. Процесс удается проводить как в хорошо, так и в плохо растворяющих воду органических растворителях. Последние требуют небольших изменений в последовательности загрузок компонентов, но зато вода не накапливается в возвращаемых в повторный процесс фильтратах и промывном растворителе, что позволяет увеличить кратность возврата таких композиций.
3. Избыточная кислота легко вымывается из твердой фазы продукта и не приводит к его существенному загрязнению. Что же касается выхода продукта и селективности по нему, то они не слишком чувствительны к избытку этого реагента в обозначенном диапазоне.
4. Процесс одностадийный, довольно прост в исполнении, не требует ни подвода внешнего тепла, ни отвода реакционного, реализуется в мягких условиях, довольно надежно управляемый и не требует сложного, в том числе и котлонадзорного оборудования.
5. Используемые реагенты доступны: диоксид может быть природного происхождения, а металл как вторичное сырье.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения карбоксилатов олова (II) | 2017 |
|
RU2671197C1 |
| Способ получения ацетата или оксалата свинца из его оксида (II) | 2023 |
|
RU2807759C1 |
| Способ получения хлорида олова (II) путем окисления металла | 2019 |
|
RU2717528C1 |
| Способ получения нитрата олова (IV) | 2017 |
|
RU2655142C1 |
| Способ получения бензоата олова (II) | 2016 |
|
RU2630310C1 |
| Способ получения нитрата олова (IV) путем окисления нитрата олова (II) | 2019 |
|
RU2717810C1 |
| Двухстадийный способ получения карбоксилатов олова (II) из металла | 2017 |
|
RU2678092C1 |
| Способ получения карбоксилатов олова (II) | 2017 |
|
RU2670199C1 |
| Способ получения нитрата олова (II) при окислении металла | 2020 |
|
RU2744006C1 |
| Способ получения основного бензоата олова (II) | 2017 |
|
RU2650893C1 |
Изобретение может быть использовано в химической промышленности. Фторид олова (II) получают из диоксида олова в присутствии фтороводородной кислоты с добавлением металлического олова, а также молекулярного йода в качестве стимулирующей добавки в органической среде в бисерной мельнице вертикального типа. В качестве перетирающего агента используют фехраль, загружаемый в массовом соотношении с остальной загрузкой без учета олова, равном 1:1. В качестве растворителя используют этилцеллозольв или уайт-спирит. Загрузку ведут в следующей последовательности: растворитель объемной фазы, перетирающий агент, диоксид олова, молекулярный йод. Затем при использовании этилцеллозольва вводят фтороводородную кислоту и олово и включают механическое перемешивание. В случае использования в качестве растворителя уайт-спирита сначала включают механическое перемешивание и затем вводят фтороводородную кислоту и олово. Процесс ведут до достижения расчетного содержания продукта в реакционной смеси. Перемешивание прекращают, отделяют перетирающий агент и непрореагировавшее олово от реакционной смеси. Реакционную смесь фильтруют, осадок на фильтре промывают растворителем жидкой фазы, отжимают и сушат. Изобретение позволяет упростить получение фторида олова (II). 1 з.п. ф-лы, 3 табл., 13 пр.
1. Способ получения фторида олова (II) из диоксида олова в присутствии фтороводородной кислоты, отличающийся тем, процесс проводят с добавлением металлического олова, а также молекулярного йода в качестве стимулирующей добавки в органической среде в бисерной мельнице вертикального типа, при этом диоксид олова берут в количестве 0,25-0,55 моль/кг в мольном соотношении с кислотой (1:4,09)÷(1:4,30), в качестве перетирающего агента используют фехраль, загружаемый в массовом соотношении с остальной загрузкой без учета олова, равном 1:1, йод дозируют в количестве 0,035-0,050 моль/кг, а олово - 10÷22% от остальной загрузки, в качестве растворителя используют этилцеллозольв или уайт-спирит, загрузку ведут в следующей последовательности: растворитель объемной фазы, перетирающий агент, диоксид олова, молекулярный йод, затем при использовании этилцеллозольва вводят фтороводородную кислоту и олово и включают механическое перемешивание, а при использовании в качестве растворителя объемной фазы уайт-спирита сначала включают механическое перемешивание и затем вводят фтороводородную кислоту и олово, в обоих случаях ведут процесс при естественно складывающейся температуре до достижения расчетного содержания продукта в реакционной смеси, после чего перемешивание прекращают, отделяют перетирающий агент и непрореагировавшее олово от реакционной смеси, последнюю фильтруют, осадок на фильтре промывают растворителем жидкой фазы, отжимают и либо сушат, либо подвергают дополнительной перекристаллизации и сушат.
2. Способ по п. 1, отличающийся тем, что непрореагировавшее олово и содержащий избыточную кислоту и основную массу введенного вначале йода и полученных из него соединений фильтрат и промывной растворитель возвращают без всяких очисток и концентрирований на загрузку повторных процессов.
| CN 103332731 A, 02.10.2013 | |||
| Способ получения раствора борфторида олова (II) | 1983 |
|
SU1229178A1 |
| DE 1300915 B, 14.08.1969 | |||
| US 2924508 A1, 09.02.1960 | |||
| US 2955914 A, 11.10.1960 | |||
| WO 2018199441 A1, 01.11.2018 | |||
| Устройство для измерения инвариантов эллиптически поляризованного магнитного поля | 1983 |
|
SU1141884A1 |

