Изобретение относится к аналитической химии и может быть использовано для количественного определения биоцидного азотсодержащего органического соединения гидразида изоникотиновой кислоты (изониазида) в водных растворах этого соединения при токсикологическом и техническом анализе субстанции и лекарственных форм этого препарата в фармацевтике, а также в медицине для определения концентрации в биосистемах (сыворотке крови, слюне и др.) с целью регулирования введения оптимальных их доз при лечении активных форм туберкулеза, при исследовании фармакокинетики и др. Изобретение может быть также применено для определения присутствия указанного соединения в пищевых продуктах, сточных водах фармацевтических и сельскохозяйственных производств.
 - лекарственное средство, противотуберкулезный препарат (ПТП),
- лекарственное средство, противотуберкулезный препарат (ПТП),  (ГИНК). Показан для лечения туберкулеза всех форм локализации. Представляет опасность для собак, которые обладают повышенной чувствительностью к препарату. Изониазид - самый эффективный из них для лечения активного туберкулеза, его активность сравнительно ниже в отношении атипичных микобактерий.
(ГИНК). Показан для лечения туберкулеза всех форм локализации. Представляет опасность для собак, которые обладают повышенной чувствительностью к препарату. Изониазид - самый эффективный из них для лечения активного туберкулеза, его активность сравнительно ниже в отношении атипичных микобактерий.
В настоящее время изониазид входит в классификацию Международного союза по борьбе с туберкулезом и наряду с рифампицином относится к препаратам первого ряда (то есть наиболее эффективным). Применяется в составе комбинированных схем лечения совместно со стрептомицином, рифампицином, пиразинамидом и этамбутолом.
Препарат включен в перечень жизненно необходимых и важнейших лекарственных средств.
В связи с этим требуется контроль его содержания в лекарственных формах, а также количественное определение содержания в биологических жидкостях организма человека и животных, продуктов питания, сточных водах фармацевтических предприятий и других объектах.
Известны различные способы количественного определения этого соединения. Согласно общей фармакопейной статье ФС 42-0236-07 (ГФ 12) изониазид качественно обнаруживают по инфракрасному спектру, снятому в пасте с вазелиновым маслом, в области от 4000 до 400 см, который по положению полос поглощения должен соответствовать рисунку спектра изониазида. Для количественного определения около 0,1 г субстанции (точная навеска) растворяют в 20 мл ледяной уксусной кислоты, прибавляют 5 мл уксусного ангидрида и титруют 0,1 М раствором хлорной кислоты до появления зеленого окрашивания (индикатор - 0,1 мл 0,1% раствора кристаллического фиолетового). Параллельно проводят контрольный опыт. 1 мл 0,1 М раствора хлорной кислоты соответствует 13,71 мг C6H7N3O.
Для анализа таблетированных лекарственных форм согласно ФС 42-0236-07 (ГФ 12) около 0,1 г (точная навеска) порошка растертых таблеток помещают в коническую колбу вместимостью 100 мл, встряхивают с 20 мл кислоты уксусной ледяной в течение 3 мин, прибавляют 5 мл уксусного ангидрида, перемешивают и титруют 0,1 М раствором кислоты хлорной до появления зеленого окрашивания (индикатор 0,3 мл раствора кристаллического фиолетового). Параллельно проводят контрольный опыт. 1 мл 0,1 М раствора кислоты хлорной соответствует 0,01371 г изониазида, которого в препарате должно быть соответственно от 0,095 до 0,105 г, от 0,190 до 0,210 г или от 0,285 до 0,315 г, считая на среднюю массу одной таблетки.
Описана аналитическая процедура для экспрессного определения изониазида в трупной крови или плазме с использованием высокоэффективной жидкостной хроматографии с диодно-матричным детектором (Мелентьев А.Б., Лаврентьева А.В. Определение изоцианида в трупной крови и плазие методом высокоэффективной жидкостной хроматографии с диодно-матричным детектором // Судебно-медицинская экспертиза, 2011, №4, с. 27-30). Методика предназначена для токсикологических и судебно-химических анализов. Пробоподготовка включает осаждение белков трихлоруксусной кислотой, образование производного изониазида с коричным альдегидом и хроматографический анализ на колонке Eclipse XDB-C18 с регистрацией при 340 нм. Элюент - метанол - 0,05 М буфер ацетата аммония с рН 3,8 (50:50 по объему). Предел обнаружения в плазме 0,5 мкг/мл, в гемолизированной крови 1 мкг/мл. Диапазон количественного определения от 2 до 200 мкг/мл. Относительное среднеквадратичное отклонение не превышает 12% для концентраций 2 и 50 мкг/мл.
Известен способ количественного определения изониазида в плазме крови с использованием хроматографического метода (Ким М.Е., Мурзагулова К.Б., Степанова Э.Ф. ИССЛЕДОВАНИЕ БИОДОСТУПНОСТИ ЛЕКАРСТВЕННОГО ПРЕПАРАТА ИЗОНИАЗИД-Д ТАБЛЕТКИ ДИСПЕРГИРУЕМЫЕ // Фундаментальные исследования. 2014, №3-4, с. 766-769). Анализ проводили на жидкостном хроматографе «Agilent 1100» с УФ-детектором и компьютером с соответствующим пакетом программ для обсчета результатов. Условия хроматографирования: аналитическая колонка - «Zorbax Bonus-RP», Agilent (150'4,6 мм; 5 мкм); подвижная фаза: градиент растворителей А и В, профильтрованная через мембранный фильтр с размером пор 0,45 мкм и дегазированная на ультразвуковой бане; скорость потока элюента - 1,0 мл/мин; детектирование проводили при длине волны -280 нм; температура колонки +35°С. Для экстракции изониазида из плазмы крови 0,5 мл плазмы помещали в пробирку Centrisart I и центрифугировали в течение 15 мин при 4500 об/мин. По 50 мкл надосадочной жидкости вводили в петлю инжектора. Процент извлечения изониазида составил 84,6%±6,7 (среднее из 3 определений). В диапазоне концентраций 10; 25; 50 и 100 мкг/мл калибровочная зависимость была линейной. Количественное определение изониазида проводили методом абсолютной калибровки. В описанных условиях время удерживания изониазида составило 3,25-4,33 мин. В диапазоне концентраций 10-100 мкг/мл калибровочная кривая была линейной. Стандартная кривая изониазида описывалась уравнением у=46,756x+178,42 (R2=0,9636), где у - площадь хроматографического пика изониазида; х - концентрация, мкг/мл. Минимальная обнаруживаемая концентрация составила 0,01 мкг/мл; относительная ошибка для концентрации 100 мкг/мл не более 32,8%.
Известен спектрофотометрический способ количественного определения пенициллиновых антибиотиков в лекарственных средах, основанный на использовании гидроксамовой реакции при λ=475 нм (Красникова А.В., Иозеп А.А. Спектрофотометрическое определение пенициллиновых антибиотиков // Хим. фарм. журн., 2003, Т.37, №9, с. 49-51). Ацильные соединения, реагируя с гидроксиламином, превращаются в гидроксамовые кислоты, которые с солями железа(III) образуют окрашенные комплексы. Способ заключается в приготовлении растворов антибиотиков, добавлении гидроксиламина для разрушения лактамного кольца в молекулах пенициллинов с образованием гидроксамовых кислот, добавление хлорида железа для образования окрашенного комплекса пенициллинов с ионами железа и измерением оптической плотности окрашенных соединений антибиотиков с ионами железа(III). Количественное содержание антибиотиков определяют по градуировочному графику. Для построения градуировочного графика используют следующую методику. Получают окрашенные комплексы к точной навеске (от 2 до 6⋅10-3 г) антибиотика добавлением 0,4 мл щелочного раствора гидроксиламина, который получают смешением 2 мл 2 М раствора гидроксиламина гидрохлорида с 1,2 мл 4 н. раствора гидроксида натрия и 0,8 мл 2 н. раствора карбоната калия. Смесь оставляют на 20 мин при 0°С, после чего к ней добавляют 0,5 мл 4 н. раствора соляной кислоты и 0,5 мл 10% раствора хлорида железа(III) в 0,1 н. соляной кислоте и доводят объем раствора дистиллированной водой до 20 мл. Оптическую плотность окрашенных растворов измеряют при 485 и 487 нм на фотоколориметре КФК-3 в кювете с толщиной рабочего слоя 10 мм. Раствор сравнения - те же компоненты без антибиотика.
Однако способ характеризуется длительностью процесса, многостадийностью, требует охлаждения раствора. Кроме того, данный способ предназначен только для определения ампициллина тригидрата и ампициллина натриевой соли, амоксициллина тригидрата и амоксициллина натриевой соли и не может быть распространен на другие группы антибиотиков, противомикробные препараты и биологические среды.
Недостатками известных способов является необходимость применения для выполнения анализа разнообразных органических растворителей, уксусного ангидрида, хлорной кислоты, необходимость мембранной фильтрации раствора перед вводом пробы в хроматограф, низкая мобильность (использование стационарного оборудования), необходимость дополнительной подготовки персонала для выполнения анализа.
Наиболее близким к предложенному является способ определения содержания биоцидного азотсодержащего органического соединения - изониазида в водном растворе путем пропускания анализируемой пробы через индикаторную трубку с размещенным в ней сорбентом силикагелем, модифицированным обработкой водным раствором соли переходного металла, предпочтительно меди, при температуре 50-70°С и величине рН от 3 до 5 в течение 1-1,5 ч и высушенным, измеряют длину окрашенной зоны сорбента и определяют по нему концентрацию указанного соединения (RU 2013154042 А, опубл. 10.06.2015).
Недостатками способа является низкая чувствительность и низкая цветовая контрастность изменения окраски (с синей на зеленую), субьективность визуального детектирования аналитического сигнала (длины окрашенной зоны).
Задачей изобретения является создание экспрессного и чувствительного способа количественного определения изониазида в водных и биологических средах (в том числе в жидкости ротовой полости, сыворотке крови и др.) и в лекарственных формах.
Техническим результатом является повышение контрастности количественного определения, упрощение аналитической процедуры, появление возможности количественного внелабораторного контроля, в связи с использованием объективного средства измерений появление возможности выхода метрологических характеристик способа на качественно новый уровень, а именно переход к уровню метрологических характеристик (увеличение точности определения измерений, снижение погрешности определения результата), характерных для количественного анализа.
Технический результат достигается способом количественного определения гидразида изоникотиновой кислоты (изониазида) в водных растворах этого соединения, лекарственных формах, содержащих изониазид, и биологических жидкостях, воздействуют анализируемой пробой водного раствора на сорбент силикагель или оксид алюминия, модифицированный обработкой водным раствором соли меди и высушенный, после развития окраски на сорбенте регистрируют параметр окраски, в котором согласно изобретению в качестве параметра окраски сорбента используют оптический сигнал, регистрацию которого осуществляют путем сканирования поверхности сорбента с помощью оптического сканера, обеспечивающего сканирование по каналам цветности RGB и обработки цветового сигнала образовавшегося окрашенного соединения изониазида с солью меди в графическом редакторе с получением графического изображения, и по интенсивности отклика сигнала по цветовому каналу В графического изображения определяют концентрацию изониазида.
Предложенный способ основан на свойстве азотсодержащих органических соединений образовывать окрашенные соединения с солями переходных металлов в водных растворах, но оно осуществляется в растворах в течение некоторого времени (10-15 мин) и при нагревании в течение не менее 5 мин при температуре свыше 60°С или требует добавления реактивов для осуществления предварительных реакций.
Образование комплексного окрашенного соединения с изониазидом на силикагеле, модифицированном солью меди практически моментально и при комнатной температуре, позволяет предположить, что в этом случае поверхность силикагеля принимает участие в процессе комплексообразования, облегчая образование комплекса.
Комплексообразование в растворе идет по следующей схеме:
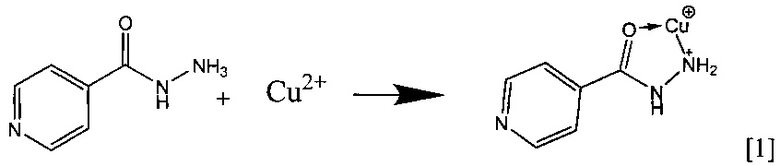
Схема вероятного процесса участия в комплексообразовании поверхностных силанольных групп силикагеля следующая:
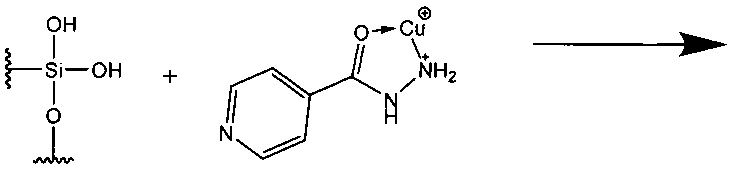
Образование окраски на обработанном солями переходных металлов силикагеле для азотсодержащих органических соединений наблюдалось для различных металлов (медь, железо, кобальт, ртуть, свинец, никель и т.д.), но наиболее интенсивная окраска при более низкой концентрации в растворе определяемого вещества характерна для меди, при этом анионы в составе модифицирующего раствора не влияют на появление окраски, а определяют рН раствора, при котором данное соединение существует в растворенном состоянии при комнатной температуре, поэтому анализ осуществляют при определенном значении рН, сопровождающимся образованием окрашенной зоны сорбции анализируемого соединения.
При изменении характера поверхности сорбента на основную (оксид алюминия) меняется характер взаимодействия изониазида с ионами переходных металлов. При этом при взаимодействии с высшими степенями окисления переходных металлов изониазид окисляется, превращаясь в изоникотиновую кислоту. Например, при реакции с медью (И) происходит выделение оксида меди (I).
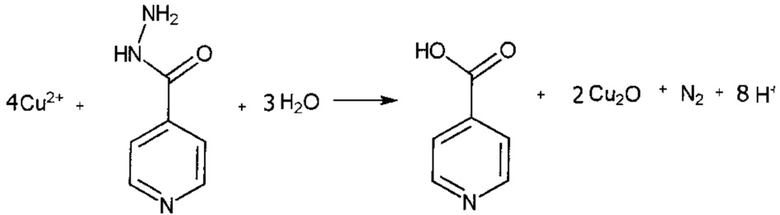
Образование интенсивно-окрашенного оксида меди (I) приводит к контрастному изменению окраски, удобному для измерения с использованием оптического сканера.
Предложенный способ осуществляли следующим образом.
Получали индикаторный сорбент, для чего порошок нейтрального оксида алюминия модифицировали водным раствором (СН3СОО)2Сu. После чего сорбент отфильтровывали и сушили. Высушенный индикаторный сорбент прессовали под давлением в таблетки или на нейтральную подложку.
Раствор изониазида наносили на поверхность индикаторного сорбента в виде таблетки. Через 10 мин проводили сканирование поверхности сканером. По значению интенсивности в цветовом канале В (синий) с использованием градуировочного графика определяли содержание изониазида в воде или в исследуемом растворе лекарственного препарата (в том числе получаемого растворением таблетированных форм). Сканирование и определение интенсивности аналитического сигнала на поверхности осуществляли с использованием сканера CanoScanLIDE500F ("Canon") и графического редактора AdobePhotoshop или другого, обеспечивающего работу с цветовым разрешением не менее 12 бит на цветовой канал, преимущественно 48 бит или 16 бит на каждый из цветовых каналов. Изображения окрашенных поверхностей, полученные сканированием, анализировали по интенсивности (I) в цветовой координате В.
В основе способа лежит измерение интенсивности аналитического сигнала на поверхности индикаторной таблетки, возникающего в результате реакции изониазида и соли меди с образованием хелатного соединения по схеме [1].
Примеры осуществления изобретения
Пример 1. Получение индикаторного сорбента на основе оксида алюминия.
Получали индикаторный сорбент, для чего порошок нейтрального оксида алюминия с удельной поверхностью 130 м2/г, размером частиц 50-200 мкм модифицировали водным раствором (СН3СОО)2Сu следующим образом: 5 г порошка оксида алюминия помещали в коническую колбу, и смешивали с 100 мл 0,3%-ного раствора (СН3СОО)2Сu и выдерживали в течение 30 мин при комнатной температуре и периодическом перемешивании. После чего сорбент отфильтровывали и сушили при температуре 90С° в течение 140 мин.
Пример 2. Получение индикаторного сорбента на основе силикагеля. Так же, как в примере 1, но вместо оксида алюминия модифицировали силикагель.
Пример 3. Получение прессованного индикаторного слоя и индикаторных таблеток.
Так же, как в примере 1, но далее высушенный индикаторный сорбент прессовали под давлением 5⋅107 кг/м2 в таблетки массой 0,260-0,280 г или на нейтральную подложку.
Пример 4. Равномерное нанесение раствора на прессованные поверхности.
Так же, как в примере 1, но индикаторную поверхность получали равномерным нанесением модифицирующего раствора на уплотненную поверхность закрепленного на подложке сорбента.
Пример 5. Процедура получения аналитического сигнала при проведении количественного определения содержания изониазида в жидкости или в растворе лекарственного препарата (в том числе получаемого растворением таблетированных форм) при помощи сканирования оптическим сканером.
Для определения изониазида анализируемый раствор исследуемой жидкости или раствор препарата, содержащего изониазид в объеме 10-200 мкл, наносили на поверхность индикаторного сорбента. Через 10 мин проводили сканирование поверхности сканером. По значению интенсивности в цветовом канале В (синий) (от 236,01 отнимали значение интенсивности окрашивания в цветовом канале В - поправка, учитывающая значение контрольного образца) с использованием градуировочного графика определяли содержание изониазида в жидкости или в исследуемой таблетке лекарственного препарата.
Сканирование и определение интенсивности аналитического сигнала на поверхности осуществляли с использованием сканера CanoScanLIDE500F ("Canon") и графического редактора AdobePhotoshopCS5 Extended. Изображения окрашенных поверхностей, полученные сканированием, анализировали по интенсивности (I) в цветовой координате В. Для этого в изображениях в графическом редакторе AdobePhotoshop Extended выделяли овальный равномерно окрашенный участок поверхности таблетки и при помощи инструмента "Гистограмма" получали усредненное значение интенсивности для каждого из каналов.
Пример 6. Методика построения градуировочного графика для количественного определения содержания изониазида.
Для построения градуировочных графиков точную навеску РСО изониазида растворяли в 10 мл дистиллированной воды. Путем разбавления получали растворы с меньшим содержанием вещества. 10-100 мкл полученных растворов наносили на индикаторную поверхность. Через 10 мин проводили сканирование ее поверхности. Строили градуировочный график зависимости интенсивности окрашивания тест-форм по цветовому каналу В (синий) от концентрации изониазида в растворе.
Уравнение градуировочного графика у=2,025х+12,18
Пример 7. Количественное определение изониазида в модельных растворах.
Получали модельные растворы изониазида в воде: точные навески 0,3 г и 0,2 г РСО изониазида (содержание действующего вещества не менее 99,0%) растворяли в 10 мл дистиллированной воды. Путем разбавления получают растворы с меньшим содержанием вещества. 70 мкл полученных растворов наносят на индикаторную поверхность. Через 10 мин проводят сканирование ее поверхности.
Достоверность результатов, получаемых разработанным методом, оценивали путем определения концентрации изониазида в модельных водных растворах. В таблице 1 приведены расчетные и измеренные значения концентрации изониазида в модельных растворах.
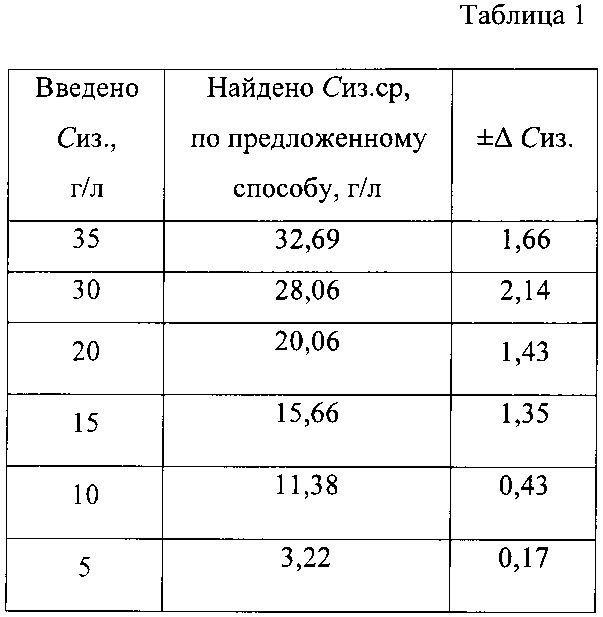
Пример 8. Количественное определение изониазида в лекарственной форме таблетки «Изониазид». ОАО «Татхимпрепараты», серия 20714
Так же, как в примере 3, но определение концентрации изониазида проводят в таблетках препарата «Изониазид».
Для иллюстрации примера взяли таблетки «Изониазид», образец №1, состав таблетки: изониазид 0,3 г, крахмал картофельный - 0,0102 г, магния стеарат 0,00165 г, стеариновая кислота 0,00165 мг, кремния диоксид коллоидный (аэросил) 0,0165 г. Для получения представительной однородной пробы при количественном определении брали 10 таблеток, предварительно взвешенных с точностью до 0,0001 г, затем растирали в фарфоровой ступке, пробу усредняли квартованием и отбирали для анализа массу порошка, равную усредненной массе таблетки. Порошок растворяли в 10 мл дистиллированной воды и 70 мкл полученного раствора наносили на индикаторную поверхность. Через 10 мин проводили сканирование ее поверхности.
В таблице 2 приведены результаты количественного определения содержания изониазида в лекарственном препарате индикаторными таблетками в сочетании с цветометрическим методом (n=3, Р=0,95).
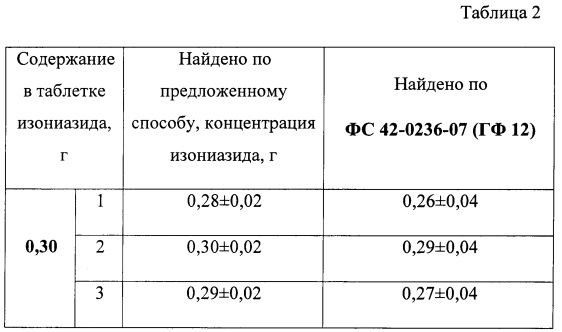
Полученные результаты определения содержания изониазида по предложенному способу хорошо согласуются с общеизвестным способом по ФС 42-0236-07 (ГФ 12) и не уступают ему по сходимости результатов определения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ОПРЕДЕЛЕНИЯ СОДЕРЖАНИЯ БИОЦИДНОГО АЗОТСОДЕРЖАЩЕГО ОРГАНИЧЕСКОГО СОЕДИНЕНИЯ В ВОДНОМ РАСТВОРЕ ЭТОГО СОЕДИНЕНИЯ | 2013 |
|
RU2567335C2 |
| Способ обнаружения биоцидного азотсодержащего органического соединения в водном растворе этого соединения | 2015 |
|
RU2611047C2 |
| Индикатор на носителе для определения содержания серосодержащих соединений в автомобильном топливе, способ определения содержания серосодержащих соединений в автомобильном топливе и способ получения индикатора на носителе | 2017 |
|
RU2649978C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ СОДЕРЖАНИЯ МОНОМЕТИЛАНИЛИНА В УГЛЕВОДОРОДНЫХ ТОПЛИВАХ ИНДИКАТОРНЫМ ТЕСТОВЫМ СРЕДСТВОМ И ИНДИКАТОРНОЕ ТЕСТОВОЕ СРЕДСТВО ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 2013 |
|
RU2548724C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ СЕРО- И АЗОТСОДЕРЖАЩИХ ВЕЩЕСТВ В ЖИДКИХ УГЛЕВОДОРОДНЫХ ТОПЛИВАХ | 2018 |
|
RU2682570C1 |
| Способ определения содержания монометиланилина в углеводородных топливах | 2016 |
|
RU2617053C1 |
| Способ определения монометиланилина в углеводородных топливах | 2015 |
|
RU2609864C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ СОДЕРЖАНИЯ ЖЕЛЕЗА В АВТОМОБИЛЬНОМ БЕНЗИНЕ, ИНДИКАТОРНЫЙ ТЕСТ ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ И СПОСОБ ПОЛУЧЕНИЯ ИНДИКАТОРНОГО ТЕСТА | 2007 |
|
RU2339943C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ПАЛЛАДИЯ (II) | 2008 |
|
RU2374640C1 |
| Способ определения простых сахаров в тонком слое сорбента | 2016 |
|
RU2642264C2 |
Изобретение относится к медицине для определения концентрации в биосистемах (сыворотке крови, слюне и др.) и может быть использовано для количественного определения биоцидного гидразида изоникотиновой кислоты (изониазида) в водных растворах этого соединения при токсикологическом и техническом анализе субстанции и лекарственных форм этого препарата. Для этого сорбенты (силикагель или оксид алюминия) предварительно обрабатывают водным раствором соли меди и высушивают. Затем анализируемую пробу водного раствора наносят на сорбент и после развития окраски регистрируют параметр окраски. Регистрацию оптического сигнала осуществляют при сканировании поверхности сорбента с помощью оптического сканера по каналам цветности RGB с получением графического изображения. Концентрацию изониазида определяют по интенсивности отклика сигнала по цветовому каналу В с использованием графического редактора. Изобретение обеспечивает регулирование введения оптимальных доз лекарств при лечении активных форм туберкулеза, а также при исследовании фармакокинетики лекарственного препарата. 2 табл., 8 пр.
Способ количественного определения гидразида изоникотиновой кислоты (изониазида) в водных растворах этого соединения (в том числе получаемого растворением таблетированных форм), лекарственных формах, содержащих изониазид, и биологических жидкостях, по которому воздействуют анализируемой пробой водного раствора на сорбент силикагель или оксид алюминия, модифицированный обработкой водным раствором соли меди и высушенный, после развития окраски на сорбенте регистрируют параметр окраски, отличающийся тем, что в качестве параметра окраски сорбента используют оптический сигнал, регистрацию которого осуществляют путем сканирования поверхности сорбента с помощью оптического сканера, обеспечивающего сканирование по каналам цветности RGB и обработки цветового сигнала образовавшегося окрашенного соединения изониазида с солью меди в графическом редакторе с получением графического изображения и по интенсивности отклика сигнала по цветовому каналу В графического изображения определяют концентрацию изониазида.
| СПОСОБ ОПРЕДЕЛЕНИЯ СОДЕРЖАНИЯ БИОЦИДНОГО АЗОТСОДЕРЖАЩЕГО ОРГАНИЧЕСКОГО СОЕДИНЕНИЯ В ВОДНОМ РАСТВОРЕ ЭТОГО СОЕДИНЕНИЯ | 2013 |
|
RU2567335C2 |
| Устройство для получения колебаний высокой частоты с помощью N-катодных ламп | 1927 |
|
SU25976A1 |
| Способ определения изониазида | 1981 |
|
SU989477A1 |
| Устройство для усиления микрофонного тока с применением самоиндукции | 1920 |
|
SU42A1 |
| Устройство двукратного усилителя с катодными лампами | 1920 |
|
SU55A1 |
| Синий свет и оптические покрытия, уменьшающие его пропускание, Оптические покрытия, 2014, стр | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| КомпьюАрт, 2006, 8, стр | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Полянская Е | |||
| М., Определение противотуберкулезных препаратов методом ВЭЖХ для оценки их фармакокинетики и анализ её соответствия генетическим предикторам у больных туберкулёзом лёгкого, МГ, Новосибирск - 2006, стр | |||
| Устройство для электрической сигнализации | 1918 |
|
SU16A1 |

