Изобретение относится к аналитической химии и может быть использовано для определения концентрации азотсодержащих противомикробных препаратов (изониазида, этамбутола и др.) и антибиотиков (цефалоспоринового ряда - цефазолина, цефатоксима, цефуроксима, цефалексина, пеницилинового ряда, пиразинаммида и др.) в исследуемых жидких средах, например, для токсикологического и технического анализа лекарственных средств, в медицине для определения концентрации этих веществ в биосистемах (сыворотке крови, слюне и др.) с целью регулирования введения оптимальных их доз при лечении различных инфекционных заболеваний, при исследовании фармакокинетики и др. Изобретение может быть также применено для определения присутствия указанных противомикробных препаратов и антибиотиков в пищевых продуктах, сточных водах фармацевтических производств.
Притивомикробные препараты и антибиотики применяются в медицине, ветеринарии, пищевой промышленности при консервировании, для обработки пищевых продуктов при их транспортировке. В связи с этим требуется контроль их содержания в лекарственных формах, а также определение их содержания в биологических жидкостях организма человека и животных, продуктов питания, сточных водах фармацевтических предприятий и других объектах.
Известны различные способы количественного определения притивомикробных препаратов и антибиотиков: микробиологические, спектрофотометрические, флуориметрические, хемилюминесцентные, различные варианты хроматографических методов, в т.ч. высокоэффективная жидкостная хроматография (ВЭЖХ), хроматомасспектрометрия, инверсионная вольтамперометрия, электроаналитическое определение с модифицированными электродами.
Однако для проведения экспрессного анализа биоцидных азотсодержащих органических соединений (противомикробных препаратов и антибиотиков) предпочтительным является использование способов, основанных на использовании определенных свойств антибиотиков: вступать в реакции, сопровождающиеся образованием окрашенных соединений.
Известны спектрофотометрические методы определения антибиотиков, в основном, в лекарственных средах. В частности, известен спектрофотометрический способ определения антибиотиков пенициллиновой группы - ампициллина, амоксициллина, клоксациллина, сулбенициллина, карбенициллина, тикарциллина в готовых лекарственных формах [Amin A.S. Pyrocatechol violet in pharmaceutical analysis. Part I. A spectrophotometric method for the determination of some - 1Mactam antibiotics in pure and in pharmaceutical dosage forms // Farmaco. - 2001. - V.56, №3. - P.211-218], основанный на измерении поглощения (=323-346 нм) продуктом реакции пенициллинов с раствором 1,2,4-триазола, содержащим хлорид ртути (II). Данный способ применим преимущественно для определения вещества в лекарственных формах, составляющих его основу.
Известны спектрофотометрический и спектрофлуориметрический способы определения четырех пенициллинов (амоксициллина, бакампициллина, пиперациллина и сультамициллина) и десяти цефалоспориновых антибиотиков в фармацевтических препаратах, которые основаны на окислении антибиотиков церием (IV) в среде ОДМ H2SO4 при 100°C. Способы включают операцию измерения уменьшения светопоглощения церия (IV) при =317 нм или интенсивности флуоресценции образовавшегося церия (III) при длинах волн возбуждения и испускания 256 и 356 нм соответственно [El Walily М., Gazy A., Belal S. Use of cerium (IV) in the spectrophotometric and spectrofluorimetric determinations of penicillins and cephalosporins in their pharmaceutical preparations // Spectrosc Lett, 2000. - Vol.33. - №6. - P.931-948].
Известен спектрофотометрический способ определения ампициллина, амоксициллина и карбенициллина с применением фенольного реактива Фолина-Чокальтеу [Ахмад А.С., Рахман Н., Ислам Ф. Спектрофотометрическое определение ампициллина, амоксициллина и карбенициллина с применением фенольного реактива Фолина-Чокальтеу // Журн. аналит.химии, 2004. - Т.12. - №2. - С.138-142]. Смесь определяемых пенициллинов с реактивом при pH 2,25 нагревают в термостатируемой водяной бане при 95±2°C и возникающую синюю окраску образующихся гетерополисоединений измеряют спектрофотометрически при =750 нм для ампициллина и карбенициллина и при =770 нм для амоксициллина.
Однако фармакокинетические исследования, проводимые на биологических средах, требуют определения низких концентраций антибиотиков Cmin<10 мкг/мл, а, следовательно, для данных целей необходим более чувствительный и экспрессный метод.
Данные способы, предназначенные для определения антибиотиков в лекарственных средах, длительны и трудоемки и в силу недостаточной чувствительности не могут быть использованы для анализа биологических жидкостей организма человека и животных, продуктов питания, сточных вод фармацевтических предприятий и других объектов.
Известен способ определения натриевых солей цефотаксима и моногидрата цефадроксила в двух составляющих смесях методом производной спектрофотометрии [Morelli В. Derivative spectrophotometry in the analysis of mixtures of cefotaxime sodium and cefadroxil monohydrate // J Pharm and Biomed Anal., 2003. - Vol.32. - №2. - P.257-267]. Способ заключается в снятии спектров поглощения антибиотиков и оценке первой и второй производных спектров поглощения. Пределы чувствительности от 0,28 до 0,51 мг/мл.
Наиболее близким к предлагаемому техническому решению является спектрофотометрический способ количественного определения пенициллиновых антибиотиков в лекарственных средах, основанный на использовании гидроксамовой реакции при =475 нм [Красникова А.В., Иозеп А.А. Спектрофотометрическое определение пенициллиновых антибиотиков // Хим. фарм. журн., 2003. - Т.37. - №9. - С.49-51]. Ацильные соединения, реагируя с гидроксиламином, превращаются в гидроксамовые кислоты, которые с солями железа (III) образуют окрашенные комплексы. Способ заключается в приготовлении растворов антибиотиков, добавлении гидроксиламина для разрушения лактамного кольца в молекулах пенициллинов с образованием гидроксамовых кислот, добавление хлорида железа для образования окрашенного комплекса пенициллинов с ионами железа и измерением оптической плотности окрашенных соединений антибиотиков с ионами железа (III). Количественное содержание антибиотиков определяют по градуировочному графику. Для построения градуировочного графика используют следующую методику. Получают окрашенные комплексы к точной навеске (от 2 до 6·10-3 г) антибиотика добавлением 0,4 мл щелочного раствора гидроксиламина, который получают смешением 2 мл 2 М раствора гидроксиламина гидрохлорида с 1,2 мл 4 н. раствора гидроксида натрия и 0,8 мл 2 н. раствора карбоната калия. Смесь оставляют на 20 мин при 0°C, после чего к ней добавляют 0,5 мл 4 н. раствора соляной кислоты и 0,5 мл 10% раствора хлорида железа (III) в 0,1 н. соляной кислоте и доводят объем раствора дистиллированной водой до 20 мл. Оптическую плотность окрашенных растворов измеряют при 485 и 487 нм на фотоколориметре КФК-3 в кювете с толщиной рабочего слоя 10 мм. Раствор сравнения - те же компоненты без антибиотика.
Однако способ характеризуется длительностью процесса, многостадийностью, требует охлаждения раствора. Кроме того, данный способ предназначен только для определения ампициллина тригидрата и ампициллина натриевой соли, амоксициллина тригидрата и амоксициллина натриевой соли и не может быть распространен на другие группы антибиотиков и биологические среды.
Задачей изобретения является создание экспрессного и чувствительного способа количественного определения азотсодержащих противомикробных препаратов и антибиотиков, например изониозида, этамбутола и цефатоксима в водных и биологических средах (в том числе в жидкости ротовой полости, сыворотке крови и др.), а также их присутствия в продуктах питания, сточных водах фармацевтических предприятий и других объектах.
Техническим результатом является сокращение времени определения при снижении предела обнаружения антибиотиков и оптимизации метрологических характеристик способа (снижение предела обнаружения, увеличение точности определения, снижение погрешности определения результата).
Кроме того, одним из преимуществ заявляемого способа является возможность использования в полевых условиях, отсутствие необходимости в термической пробоподготовке и использования прибора для осуществления спектрометрического окончания при получении результатов.
Технический результат достигается способом определения содержания биоцидного азотсодержащего органического соединения в водном растворе, по которому воздействуют анализируемой пробой водного раствора на сорбент силикагель, модифицированный солью переходного металла, и по появлению окрашенной зоны судят о наличии в растворе биоцидного азотсодержащего органического соединения.
В частном случае используют сорбент, модифицированный солью переходного металла, размещенный в индикаторной трубке, воздействие на сорбент осуществляют путем пропускания анализируемой пробы через индикаторную трубку. При этом для количественного определения указанного соединения измеряют размер окрашенной зоны сорбента и определяют по нему концентрацию указанного соединения.
В другом частном случае используют структурированный материал, содержащий сорбент, модифицированный солью переходного металла, воздействие на сорбент осуществляют путем нанесения капли анализируемого водного раствора на указанный материал.
Предпочтительно в качестве соли переходного металла использовать соль меди.
Для повышения предела обнаружения в водной среде перед нанесением анализируемую пробу водного раствора пропускают через концентрирующий патрон.
Предложенный способ основан на свойстве образования азотсодержащих органических соединений образовывать окрашенные соединения с солями переходных металлов в водных растворах, но оно осуществляется в растворах в течение некоторого времени (10-15 мин) и при нагревании в течение не менее 5 мин при температуре свыше 60°C или требует добавления реактивов для осуществления предварительных реакций. Факт образования окрашенного соединения на твердом носителе - силикагеле (для алюмогелей, цеолитов и полимерных сорбентов изменение окраски не наблюдалось), практически моментально и при комнатной температуре установлен впервые.
Образование окраски на обработанном солями переходных металлов силикагеле для азотсодержащих органических соединений наблюдалось для различных металлов (медь, железо, кобальт, ртуть, свинец, никель и т.д.), но наиболее интенсивная окраска при более низкой концентрации в растворе определяемого вещества характерна для меди, при этом анионы в составе модифицирующего раствора не влияют на появление окраски, а определяют pH раствора, при котором данное соединение существует в растворенном состоянии при комнатной температуре, поэтому анализ осуществляют при определенном значении pH, сопровождающимся образованием окрашенной зоны сорбции анализируемого соединения.
При использовании индикаторной трубки, заполненной сорбентом, модифицированным солью переходного металла, определение концентрации анализируемого соединения осуществляют по градуировочному графику или калибровочному коэффициенту, построенному по эталонной среде, в качестве которой используют водный раствор соответствующего соединения в различных концентрациях. Градуировочный график строят по измеренным значениям длины окрашенной зоны от концентрации вещества в эталонном растворе.
При использовании структурированного гидрофильного материала, модифицированного солью переходного металла, определяют предельную концентрацию анализируемого соединения путем образования окрашенного пятна. При осуществлении способа путем нанесения капли раствора на структурированный материал, содержащий сорбент, модифицированный солями тяжелых металлов, фиксируют факт образования окрашенного пятна, соответствующего концентрации антибиотика, предварительно определенного в модельном растворе.
Предложенный способ осуществляют следующим образом.
Фракцию сорбента силикагеля с размером частиц 0.05-0.1 мм обрабатывают при температуре 50-70°C в течение 1-1,5 часов солями меди при величине pH от 3 до 5, помещают в стеклянную трубку или наносят на структурированный материал и высушивают при температуре 100-110°C в течение 60-90 мин, готовят водные растворы противомикробных препаратов и антибиотиков в дистиллированной воде с концентрацией 1,2 г/л, затем последовательным разбавлением готовят растворы меньших концентраций (0,12; 0,9; 0,7; 0,05; 0,01), пропускают через индикаторные трубки в объеме от 2 мл, строят градуировочный график или наносят на индикаторный материал, фиксируя образование окрашенного пятна. Объем исследуемого раствора пропускают через индикаторную трубку или наносят каплю на индикаторный материал. Определяемую концентрацию анализируемого вещества рассчитывают по калибровочному коэффициенту или определяют наличие при концентрации выше предельной по появлению окрашенного пятна на индикаторном материале. Для анализа сточных вод, концентрация определяемых компонентов которых может быть менее 0,01 г/л, применяется предварительное концентрирование пробы воды на концентрирующих патронах (например: патроны для твердофазной экстракции - Oasis HLB). Для этого 500 мл анализируемой пробы пропускают через концентрирующий патрон для твердофазной экстракции - Oasis HLB, затем десорбируют 5 мл водно-спиртового раствора. Пропускают от 2 мл десорбата анализируемого вещества через стеклянную трубку, содержащую модифицированный индикаторный сорбент, и фиксируют размер окрашенной зоны. Определяют концентрацию по калибровочному графику и по формуле рассчитывают концентрацию определяемого вещества в пробе: C=(Lc)/500, где
С - концентрация анализируемого вещества в пробе, г/л;
Lc - концентрация анализируемого вещества в по графику, г/л.
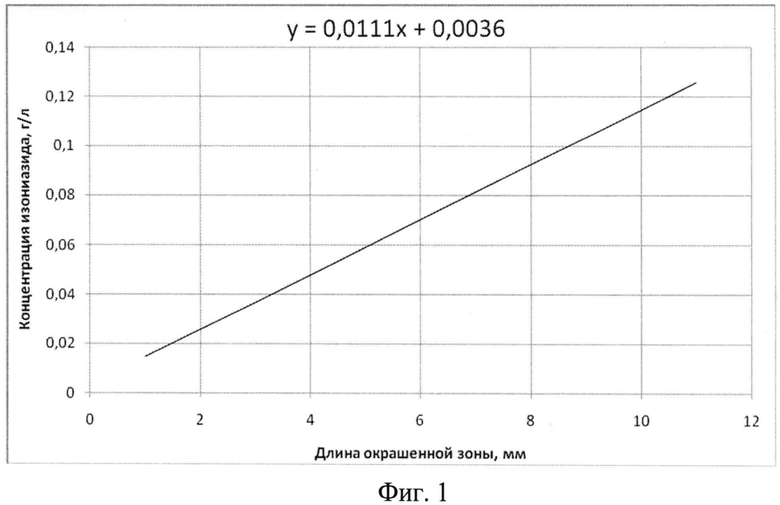
На фиг.1 показан график зависимости концентрации изониазида от длины окрашенной зоны индикаторной трубки.
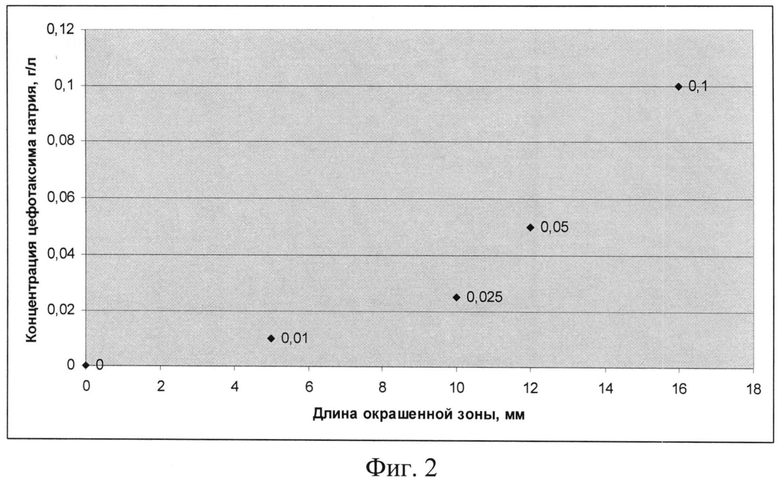
На фиг.2 показан график зависимости концентрации цефотаксима натрия от длины окрашенной зоны индикаторной трубки.
Пример 1. Определение изониазида в лекарственных формах, в биологических жидкостях организма человека и животных, продуктов питания. Пропускают от 0.1 до 5 мл водного раствора анализируемого вещества (инъекционные растворы, водные вытяжки из сиропов, микстур, таблеток, биологических жидкостей человека и животных, продуктов питания) через стеклянную трубку, содержащую модифицированный индикаторный сорбент, и фиксируют длину окрашенной зоны. Определяют концентрацию изониазида по калибровочному графику (фиг.1).
Пример 2. Определение изониазида в сточных водах. Проводят концентрирование пробы воды на концентрирующих патронах. Пропускают от 0.1 до 5 мл десорбата анализируемого вещества через стеклянную трубку, содержащую модифицированный индикаторный сорбент, и фиксируют длину окрашенной зоны. Определяют концентрацию изониазида по калибровочному графику (фиг.1).
Пример 3. Определение цефатоксима в лекарственных формах, в биологических жидкостях организма человека и животных, продуктах питания. Пропускают от 0.1 до 5 мл водного раствора анализируемого вещества (инъекционные растворы, водные вытяжки из сиропов, микстур, таблеток, биологических жидкостей человека и животных, продуктов питания) через стеклянную трубку, содержащую модифицированный индикаторный сорбент и фиксируют длину окрашенной зоны. Определяют концентрацию цефатоксима по калибровочному графику (фиг.2).
Пример 4. Определение цефатоксима натрия в сточных водах. Для определения цефотаксима натрия в водной среде с концентрацией 0,0005 г/л объем водного раствора цефотаксима натрия 500 мл пропускают через концентрирующий патрон для твердофазной экстракции - Oasis HLB, затем десорбируют 5 мл водно-спиртового раствора. Пропускают от 0.1 до 5 мл десорбата анализируемого вещества через стеклянную индикаторную трубку, содержащую модифицированный индикаторный сорбент, и фиксируют длину окрашенной зоны. Определяют концентрацию цефатоксима по калибровочному графику (фиг.2).
Индикаторная трубка содержала два слоя сорбента (нижний 2,5 см - силикагель КСКГ, модифицированный 5% водным ацетатом меди при 70°C в течение 1 часа, высушенный при 110°C в течение 1,5 часов, отобрана фракция сорбента 0,1-0,15 мм; верхний 0,5 см - Цеолит NaA (размер пор 4°А), фракция сорбента 0,1-0,15 мм, предварительно просушен при 150°C в течение 2,5 часов).
Пример 5. Определение азотсодержащих противомикробных препаратов и антибиотиков в водных средах. На индикаторный материал наносят одну каплю водного раствора анализируемого вещества или водной вытяжки, образование окрашенного пятна свидетельствует о присутствии вещества в концентрации более 0,1 мг/мл.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ обнаружения биоцидного азотсодержащего органического соединения в водном растворе этого соединения | 2015 |
|
RU2611047C2 |
| Способ количественного определения биоцидного азотсодержащего органического соединения гидразида изоникотиновой кислоты (изониазида) в водном растворе этого соединения | 2016 |
|
RU2633080C2 |
| Индикатор на носителе для определения содержания серосодержащих соединений в автомобильном топливе, способ определения содержания серосодержащих соединений в автомобильном топливе и способ получения индикатора на носителе | 2017 |
|
RU2649978C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ СОДЕРЖАНИЯ МОНОМЕТИЛАНИЛИНА В УГЛЕВОДОРОДНЫХ ТОПЛИВАХ ИНДИКАТОРНЫМ ТЕСТОВЫМ СРЕДСТВОМ И ИНДИКАТОРНОЕ ТЕСТОВОЕ СРЕДСТВО ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 2013 |
|
RU2548724C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ СОДЕРЖАНИЯ СВИНЦА В БЕНЗИНЕ, ИНДИКАТОРНЫЙ СОСТАВ ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ И СПОСОБ ПОЛУЧЕНИЯ ИНДИКАТОРА НА НОСИТЕЛЕ ДЛЯ ОПРЕДЕЛЕНИЯ СОДЕРЖАНИЯ СВИНЦА В БЕНЗИНЕ | 2003 |
|
RU2249814C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ СЕРО- И АЗОТСОДЕРЖАЩИХ ВЕЩЕСТВ В ЖИДКИХ УГЛЕВОДОРОДНЫХ ТОПЛИВАХ | 2018 |
|
RU2682570C1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ ЦЕФАЛОСПОРИНОВЫХ АНТИБИОТИКОВ В БИОСРЕДАХ | 2010 |
|
RU2445624C2 |
| СПОСОБ ОПРЕДЕЛЕНИЯ СОДЕРЖАНИЯ ЖЕЛЕЗА В АВТОМОБИЛЬНОМ БЕНЗИНЕ, ИНДИКАТОР НА НОСИТЕЛЕ ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ И СПОСОБ ПОЛУЧЕНИЯ ИНДИКАТОРА НА НОСИТЕЛЕ ДЛЯ ОПРЕДЕЛЕНИЯ СОДЕРЖАНИЯ ЖЕЛЕЗА В БЕНЗИНЕ | 2007 |
|
RU2339942C1 |
| СПОСОБ ПОЛУЧЕНИЯ КАРБОКСИЛЭСТЕРАЗЫ, МАТРИЦА ДЛЯ ОБНАРУЖЕНИЯ ФОСФОРОРГАНИЧЕСКИХ СОЕДИНЕНИЙ И БИОХИМИЧЕСКИЙ СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ ФОСФОРОРГАНИЧЕСКИХ СОЕДИНЕНИЙ | 1991 |
|
RU2057807C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ СОДЕРЖАНИЯ ЖЕЛЕЗА В АВТОМОБИЛЬНОМ БЕНЗИНЕ, ИНДИКАТОРНЫЙ ТЕСТ ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ И СПОСОБ ПОЛУЧЕНИЯ ИНДИКАТОРНОГО ТЕСТА | 2007 |
|
RU2339943C1 |
Изобретение относится к аналитической химии и может быть использовано для определения концентрации азотсодержащих противомикробных препаратов (изиниазида, этамбутола и др.) и антибиотиков (цефалоспоринового ряда - цефазолина, цефатоксима, цефуроксима, цефалексина и др.) в исследуемых жидких средах. Способ определения содержания биоцидного азотсодержащего органического соединения в водном растворе заключается в том, что модифицируют сорбент силикагель солью переходного металла путем обработки силикагеля водным раствором соли переходного металла при температуре 50-70°C и при величине pH от 3 до 5 в течение 1-1,5 часов, высушивают, помещают сорбент в стеклянную индикаторную трубку, затем пропускают анализируемую пробу через индикаторную трубку с размещенным в нем сорбентом, модифицированным солью переходного металла, измеряют длину окрашенной зоны сорбента и определяют по нему концентрацию указанного соединения. Способ позволяет сократить время определения антибиотиков при снижении предела их обнаружения, увеличить точность определения, снизить погрешность определяемого результата. 4 з.п. ф-лы, 4 пр., 2 ил.
1. Способ определения содержания биоцидного азотсодержащего органического соединения в водном растворе путем пропускания анализируемой пробы через индикаторную трубку с размещенным в ней сорбентом силикагелем, модифицированным обработкой водным раствором соли переходного металла при температуре 50-70°C и величине pH от 3 до 5 в течение 1-1,5 часов и высушенным, измеряют длину окрашенной зоны сорбента и определяют по нему концентрацию указанного соединения.
2. Способ по п. 1, отличающийся тем, что в качестве соли тяжелого металла используют соль меди.
3. Способ по п. 1, отличающийся тем, что для повышения чувствительности анализируемую пробу получают путем пропускания анализируемого водного раствора через концентрирующий патрон для твердофазной экстракции и десорбируют водно-спиртовым раствором.
4. Способ по п. 1, отличающийся тем, что определение концентрации анализируемого соединения осуществляют по градуировочному графику, построенному по измеренным значениям длины окрашенной зоны для различных концентраций анализируемого соединения в эталонном растворе.
5. Способ по п. 1, отличающийся тем, что определение концентрации анализируемого соединения осуществляют по калибровочному коэффициенту, определенному с помощью эталонного раствора анализируемого соединения в различных концентрациях.
| Красникова А.В., Иозеп А.А | |||
| Спектрофотометрическое определение пенициллиновых антибиотиков // Хим | |||
| фарм | |||
| журн., 2003 | |||
| Пишущая машина | 1922 |
|
SU37A1 |
| Разборный с внутренней печью кипятильник | 1922 |
|
SU9A1 |
| Способ смешанной растительной и животной проклейки бумаги | 1922 |
|
SU49A1 |
| Беклемишев и др | |||
| Сорбционно-каталитический метод определения азотсодержащих органических соединений | |||
| ВЕСТН | |||
| МОСК | |||
| УН-ТА | |||
| СЕР | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| ХИМИЯ | |||
| Способ и приспособление для нагревания хлебопекарных камер | 1923 |
|
SU2003A1 |
| Т | |||
| Приспособление для плетения проволочного каркаса для железобетонных пустотелых камней | 1920 |
|
SU44A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| с | |||
| Ударно-долбежная врубовая машина | 1921 |
|
SU115A1 |

