Область изобретения
[001] Настоящее изобретение относится к новым способам синтеза синтона для γ,δ-дигидроксиизолейцина 1 (CAS No. 55399-94-5] в качестве структурного блока для синтеза аматоксинов и для новых способов синтеза аматоксинов с использованием такого структурного блока.
Предпосылки изобретения
[002] Аматоксины представляют собой циклические пептиды, состоящие из 8 аминокислот, которые обнаружены в Amanita phalloides грибах (см. Фиг. 1). Аматоксины специфическим образом ингибируют ДНК-зависимую РНК полимеразу II клеток млекопитающих и, таким образом, также транскрипцию и биосинтез белка в пораженных клетках. Ингибирование транскрипции в клетке вызывает остановку роста и пролиферации. Хотя он не является ковалентно связанным, комплекс между аманитином и РНК-полимеразой II очень прочный (KD=3 нМ). Диссоциация аманитина из фермента является очень медленным процессом, делая, таким образом, маловероятным восстановление пораженной клетки. Когда ингибирование транскрипции длится слишком долго, клетка подвергается процессу запрограммированной клеточной гибели (апоптоз).
[003] Применение аматоксинов в качестве цитотоксичных компонентов для противоопухолевой терапии уже использовали в 1981 году путем связывания анти-Thy 1.2 антитела с α-аманитином с использованием линкера, присоединенного к индольному кольцу Trp (аминокислота 4; см. Фиг. 1) через диазотирование (Davis & Preston, Science 1981, 213, 1385-1388). Davis & Preston идентифицировали участок присоединения как положение 7’. Morris & Venton также продемонстрировали, что замещение в положении 7’ приводит к производному, которое поддерживает цитотоксическую активность (Morris & Venton, Int. J. Peptide Protein Res. 1983, 21 419-430).
[004] Патентная заявка EP 1 859 811 A1 (опубликована 28 ноября 2007 года) описывает конъюгаты, в которых γ C-атом аминокислоты 1 аматоксина β-аманитина был непосредственно связан, т.е. без линкерной структуры, с альбумином или с моноклональным антителом HEA125, OKT3 или PA-1. Кроме того, был показан ингибиторный эффект этих конъюгатов на пролиферацию раковых клеток молочной железы (MCF-7), клеток лимфомы Беркитта (Raji) и T-лимфомных клеток (Jurkat). Было предложено использовать линкеры, в том числе линкеры, включающие элементы, такие как амидные, сложноэфирные, простые эфирные, тиоэфирные, дисульфидные, мочевинные, тиомочевинные, углеводородные группы и т.п., но никакие подобные конструкции не были показаны на самом деле, и не представлены никакие более подробные сведения, такие как участки присоединения на аматоксинах.
[005] Патентные заявки WO 2010/115629 и WO 2010/115630 (обе опубликованы 14 октября 2010 года) описывают конъюгаты, где антитела, такие как анти-EpCAM антитела, такие как гуманизированное антитело huHEA125, связаны с аматоксинами через (i) γ C-атом аминокислоты 1 аматоксина, (ii) 6’ C-атом аминокислоты 4 аматоксина или (iii) через δ C-атом аминокислоты 3 аматоксина, в каждом случае либо непосредственно, либо через линкер между антителом и аматоксинами. Предложенные линкеры включают элементы, такие как амидные, сложноэфирные, простые эфирные, тиоэфирные, дисульфидные, мочевинные, тиомочевинные, углеводородные группы и т.п. Кроме того, были показаны ингибиторные эффекты этих конъюгатов на пролиферацию раковых клеток молочной железы (клеточная линия MCF-7), карциномы поджелудочной железы (клеточная линия Capan-1), рака толстой кишки (клеточная линия Colo205) и холангиокарциномы (клеточная линия OZ).
[006] Аматоксины можно выделить из собранных плодовых тел грибов Amanita phalloides или из чистых культур (Zhang P, Chen Z, Hu J, Wei B, Zhang Z, and Hu W, Production and characterization of Amanitin toxins from a pure culture of Amanita exitialis, FEMS Microbiol Lett. 2005 Nov 15;252(2):223-8. Epub 2005 Sep 15). Однако количества аматоксинов, которые можно получить, достаточно низкие (в пределах около 0,3 - 3 мг/г сухого вещества из природных плодовых тел и около 10% из чистых культур), и гибкость для дальнейшей модификации природных вариантов аматоксинов ограничена (см. ссылочные документы, обсуждаемые в [003]-[005], и указанные в них ссылочные документы).
[007] Альтернативно аматоксины можно получить путем ферментации с использованием базидиомицета (Muraoka S, and Shinozawa T., Effective production of amanitins by two-step cultivation of the basidiomycete, Galerina fasciculata GF-060, J Biosci Bioeng. 2000; 89(1):73-6; указанный выход около 5 мг/л культуры) или A. fissa (Guo XW, Wang GL, and Gong JH, Culture conditions and analysis of amantins on Amanita spissa, Wei Sheng Wu Xue Bao. 2006 Jun; 46(3):373-8; указанный выход около 30 мкг/л культуры). Снова выходы низкие и гибкость для дальнейшей модификации природных вариантов аматоксинов также ограничена.
[008] Наконец, аматоксины были получены путем частичного или общего синтеза (например, Zanotti G, Möhringer C, and Wieland T., Stnthesis of analogues of amaninamide, an amatoxin from the white Amanita virosa mushroom, Int J Pept Protein Res. 1987 Oct; 30(4):450-9; Zanotti G, Wieland T, Benedetti E, Di Blasio B, Pavone V, and Pedone C., Structure-toxicity relationships in the amatoxin series. Synthesis of S-deoxy[gamma(R)-hydroxy-Ile3]-amaninamide, its crystal and molecular structure and inhibitory efficiency, Int J Pept Protein Res. 1989 Sep; 34(3):222-8; Zanotti G, Petersen G, and Wieland T., Structure-toxicity relationships in the amatoxin series. Structural variations of side chain 3 and inhibition of RNA polymerase II, Int J Pept Protein Res. 1992 Dec; 40(6):551-8).
[009] Хотя использование полностью синтетических путей для получения аматоксинов может предложить получение бóльших количеств аматоксинов, требуемых для терапевтических применений, и может предложить создание различных новых вариантов аматоксинов с использованием подходящих исходных веществ в качестве структурных блоков, до сих пор не было никаких сообщений о полностью синтетическом подходе для получения наиболее релевантных аматоксинов, α-аманитина и β-аманитина, а также аманина и аманинамида. Это можно отнести, по меньшей мере частью, за счет того, что существенный структурный блок, γ,δ-дигидроксиизолейцин 1 или синтон для него до сих пор не является доступным в качестве чистого диастереомера.
Цель изобретения
[0010] Таким образом, в уровне техники существовала большая необходимость в получении γ,δ-дигидроксиизолейцина 1 или синтона для него в качестве структурного блока для синтеза аматоксинов и в определении способа получения γ,δ-дигидроксиизолейцина 1 или синтона для него. Кроме того, в уровне техники существовала большая необходимость определения альтернативного способа синтеза аматоксинов.
Краткое описание изобретения
[0011] Настоящее изобретение основано на неожиданном наблюдении, что γ,δ-дигидроксиизолейцин 1 или синтон для него можно получить многостадийным способом, где ключевой стадией является региоселективное метилирование подходяще защищенного производного аспарагиновой кислоты.
[0012] Таким образом, в одном аспекте настоящее изобретение относится к способу синтеза γ,δ-дигидроксиизолейцина 1 или синтона для соединения 1, включающему стадию метилирования соединения 3, в частности, при помощи метилиодида в присутствии бис(триметилсилил)амида лития (LHMDS).
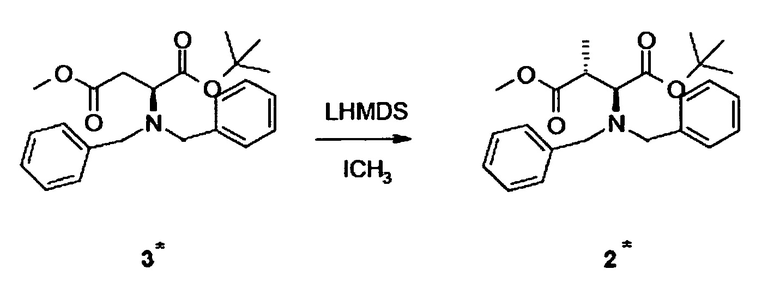
[0013] В альтернативном аспекте настоящее изобретение относится к способу синтеза γ,δ-дигидроксиизолейцина 1 или синтона для соединения 1, включающему стадию метилирования соединения 3* (содержащего вторую бензильную защитную группу по атому азота вместо фенилфлуоренильной группы в соединении 3), в частности, при помощи метилиодида в присутствии бис(триметилсилил)амида лития (LHMDS).
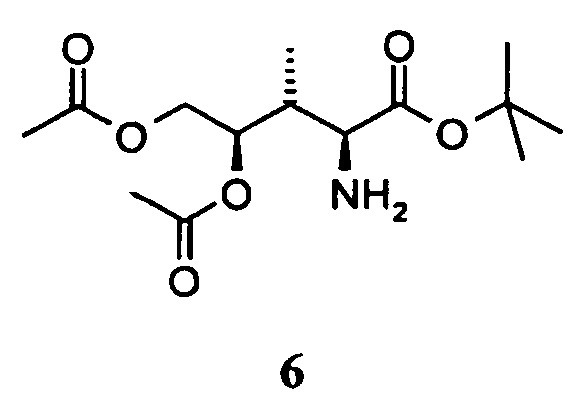
[0014] Во втором аспекте, настоящее изобретение относится к соединению 6.
[0015] В третьем аспекте, настоящее изобретение относится к набору, включающему соединение 6 и по меньшей мере один дополнительный реагент для синтеза аматоксинов или их предшественников.
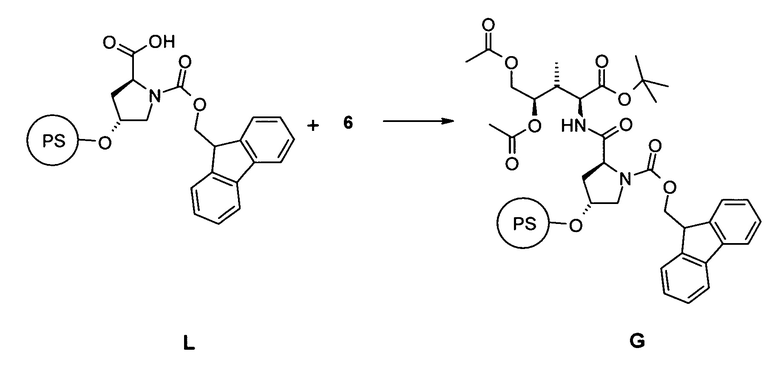
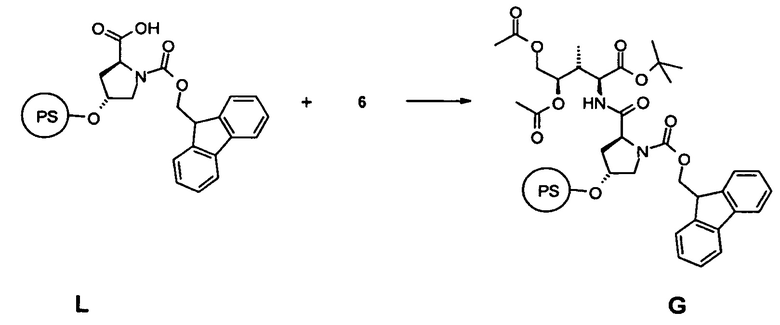
[0016] Еще в одном аспекте настоящее изобретение относится к способу синтеза аматоксина или молекулы, являющейся его предшественником, включающему стадию связывания соединения 6 с гидроксипролином, в частности, путем взаимодействия соединения 6 со смолой, содержащей предварительно нагруженный на нее гидроксипролин (PS) L.
Краткое описание чертежей
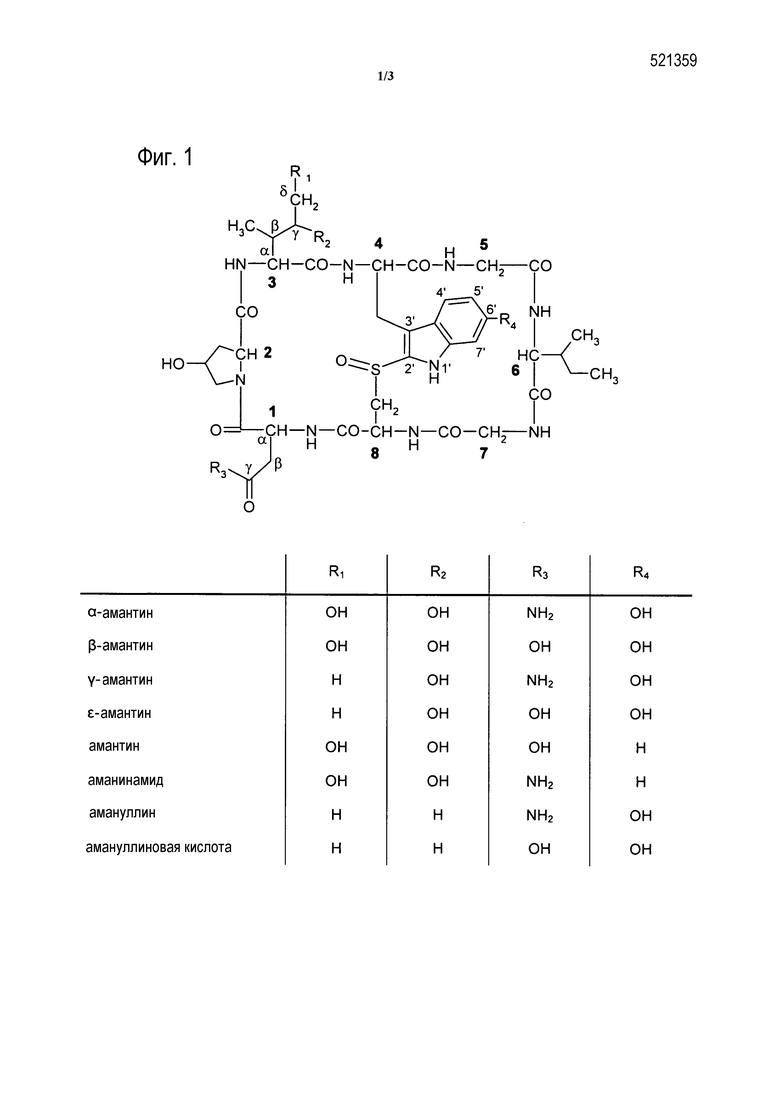
[0017] На Фиг. 1 показаны структурные формулы различных аматоксинов. Цифры, выделенные жирным шрифтом, (1-8) означают стандартную нумерацию восьми аминокислот, образующих аматоксин. Также показано стандартное обозначение атомов в аминокислотах 1, 3 и 4 (греческие буквы α-γ, греческие буквы α-δ и номера от 1’ до 7’, соответственно).
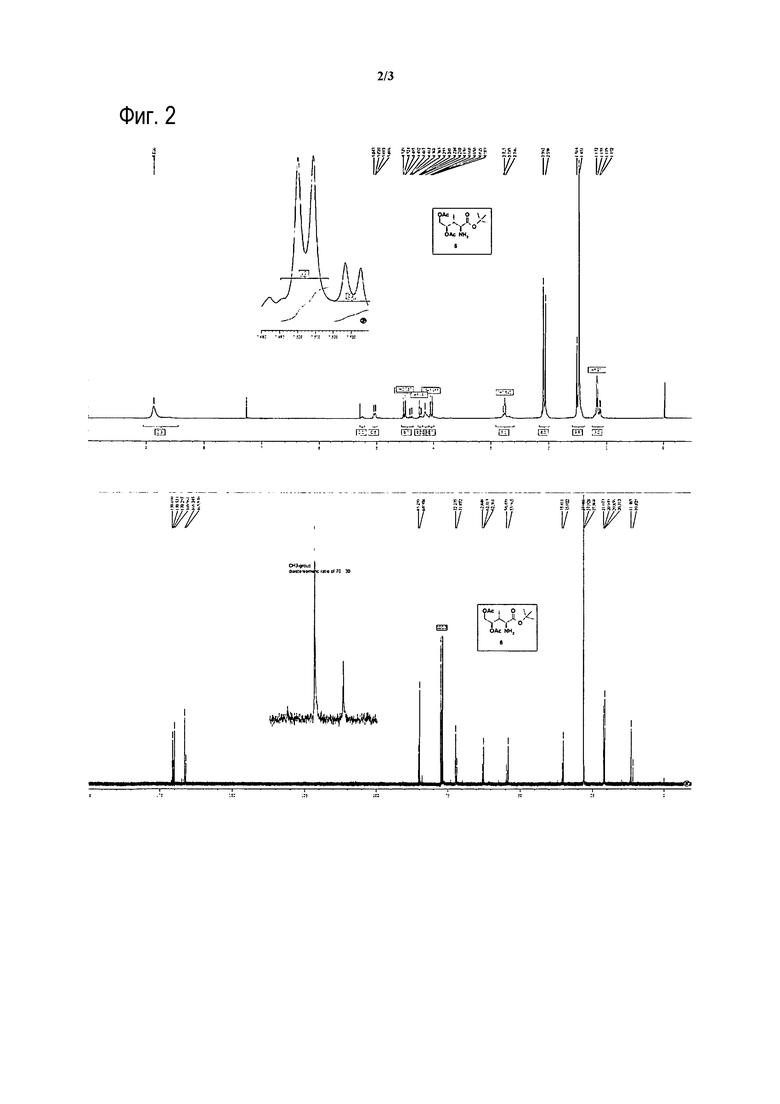
[0018] На фиг. 2 представлены 1H- и 13C-ЯМР спектры, иллюстрирующие диастереомерное отношение 70:30 соединения 6.
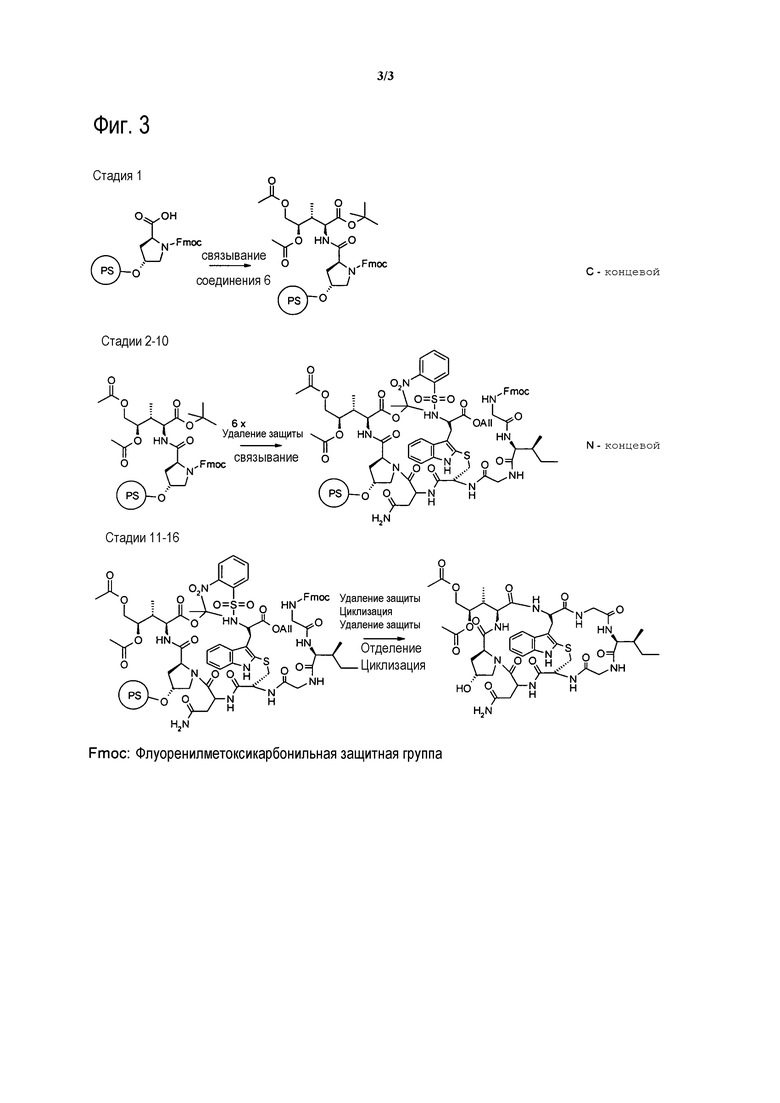
[0019] На фиг. 3 представлена общая схема синтеза аматоксинов.
Подробное описание изобретения
[0020] Перед тем как настоящее изобретение будет подробно описано ниже, следует отметить, что настоящее изобретение не ограничивается конкретными способами, протоколами и реагентами, описанными в настоящей заявке, поскольку они могут варьироваться. Также должно быть понятно, что терминология, используемая в настоящей заявке, предназначена для описания только конкретных вариантов воплощения и не предназначена для ограничения объема настоящего изобретения, который должен ограничиваться только прилагаемой формулой изобретения. Если не определено иначе, все технические и научные термины, используемые в настоящей заявке, имеют такие же значения, которые обычно известны рядовым специалистам в данной области.
[0021] Предпочтительно, термины, используемые в настоящей заявке, определяются так, как описано в "A multilingual glossary of biotechnological terms: (IUPAC Recommendations)", Leuenberger, H.G.W, Nagel, B. and Kölbl, H. eds. (1995), Helvetica Chimica Acta, CH-4010 Basel, Switzerland).
[0022] В тексте настоящего описания и в следующей далее формуле изобретения, если из контекста не следует иное, слово "включать" и варианты, такие как "включает" и "включающий", следует понимать как подразумевающие включение указанного целого, композиции или стадии или группы целых или стадий, хотя также, необязательно, могут присутствовать дополнительное целое, композиция, или стадия, или группа целых композиций или стадий, включая варианты воплощения, где не присутствует никакое дополнительное целое, композиция, или стадия, или группа целых композиций или стадий. В последних таких вариантах воплощения термин "включающий" используют как имеющий такое же значение как "состоящий из".
[0023] Некоторые документы повсеместно цитируются в тексте настоящего описания. Каждый из документов, цитируемых в настоящей заявке (включая все патенты, патентные заявки, научные публикации, спецификации изготовителей, инструкции, представление последовательностей в соответствии с GenBank Accession Number и т.п.), выше или ниже, включены в настоящую заявку посредством ссылки во всей полноте в той степени, которая является возможной согласно соответствующему патентному законодательству. Ничто в настоящей заявке не должно рассматриваться как допущение, что изобретение не имеет права предвосхищать такое раскрытие на основании предшествующего изобретения.
[0024] Настоящее изобретение далее описано более подробно. В следующих разделах различные аспекты настоящего изобретения определены более подробно. Каждый аспект, определенный таким образом, можно объединить с любым другим аспектом или аспектами, если только определенно не указано иное. В частности, любой признак, указанный как предпочтительный или выгодный, можно объединить с любым другим признаком или признаками, указанными как предпочтительные или выгодные.
[0025] Таким образом, в одном аспекте настоящее изобретение относится к способу синтеза γ,δ-дигидроксиизолейцина 1 или синтона для соединения 1, включающему стадию метилирования соединения 3, в частности, при помощи метилиодида в присутствии бис(триметилсилил)амида лития (LHMDS).
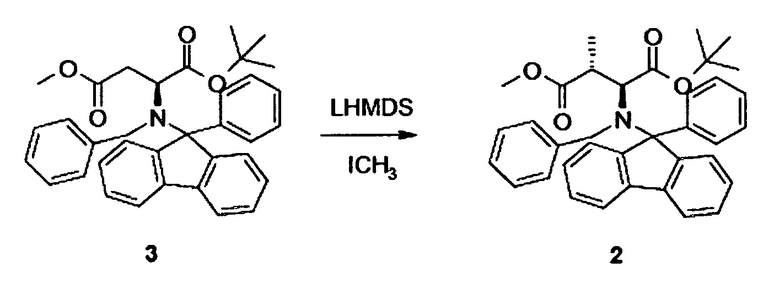
[0026] В альтернативном аспекте настоящее изобретение относится к способу синтеза γ,δ-дигидроксиизолейцина 1 или синтона для соединения 1, включающему стадию метилирования соединения 3* (содержащего вторую бензильную защитную группу по атому азота вместо фенилфлуоренильной группы в соединении 3), в частности, при помощи метилиодида в присутствии бис(триметилсилил)амида лития (LHMDS).
[0027] В контексте настоящего изобретения термин "синтон" относится к соединению, которое представляет собой или которое можно использовать как синтетический эквивалент для представляющего интерес конкретного соединения в химической реакции. Это определение включает соединения, где группу представляющего интерес соединения, которая может быть лабильной или реакционно-способной в условиях, используемых в данной химической реакции, защищают или маскируют при помощи подходящей защитной группы, которую можно отщепить после указанной химической реакции.
[0028] В конкретном варианте воплощения реакцию осуществляют при температуре в пределах от около -10°C до около -80°C в простом эфире в течение времени от около 12 до около 20 ч, в частности в течение около 16 ч. В конкретном варианте воплощения реакцию осуществляют при -20°C и в тетрагидрофуране в качестве растворителя. В конкретных вариантах воплощения получают диастереомерную чистоту больше чем 30:1, в частности больше чем 40:1, в частности около 50:1.
[0029] В контексте настоящего изобретения термин "около" или "приблизительно" означает от 90% до 110% данного значения или диапазона.
[0030] В конкретном варианте воплощения способ дополнительно включает одну или несколько из следующих стадий для синтеза соединения 3:
(а) взаимодействие L-аспарагиновой кислоты, монометилового эфира A, с 2-метилпропеном с образованием соединения B;

(b) взаимодействие соединения B с бензальдегидом с образованием соединения C;
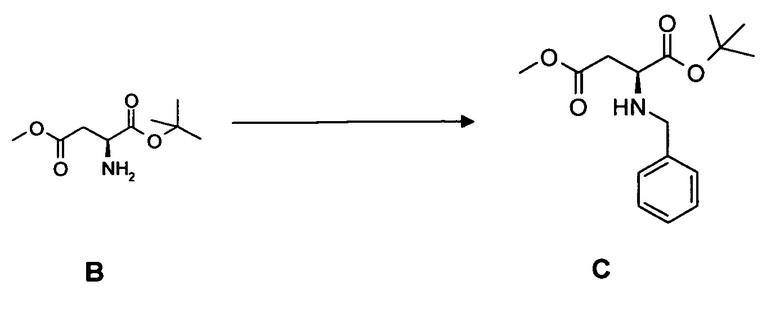
(c) взаимодействие соединения C с фенилфлуоренбромидом с образованием соединения 3.
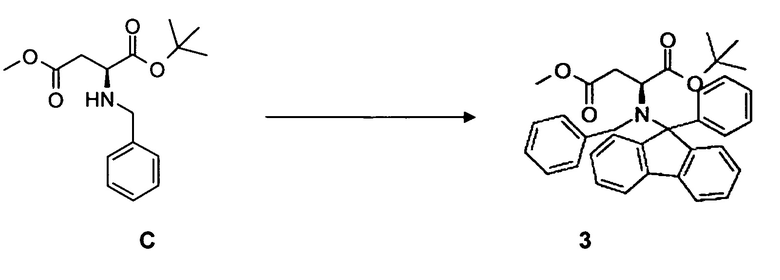
[0031] В одном варианте воплощения альтернативного аспекта способ дополнительно включает одну или несколько из следующих стадий для синтеза соединения 3*:
(а) взаимодействие L-аспарагиновой кислоты, монометилового эфира A, с 2-метилпропеном с образованием соединения B;
(b) взаимодействие соединения B с бензальдегидом с образованием соединения C;
(c) взаимодействие соединения C с бензилбромидом с образованием соединения 3*.
[0032] В конкретном варианте воплощения способ дополнительно включает одну или несколько из следующих стадий:
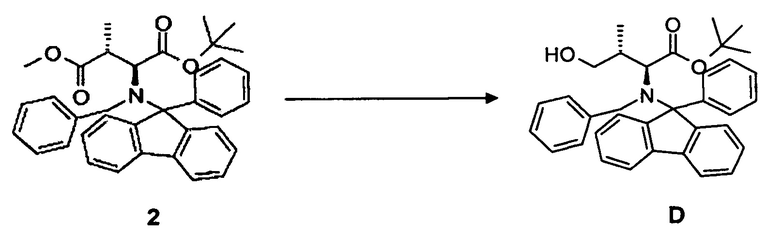
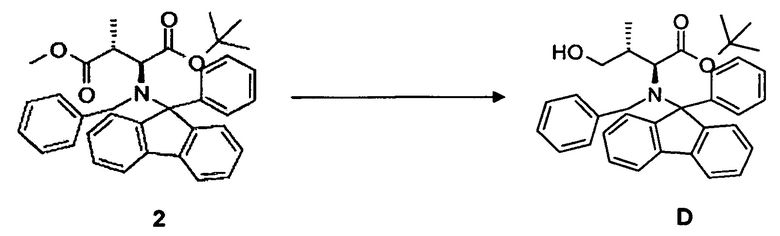
(а) восстановление соединения 2, в частности, при помощи диизобутилалюминийгидрида (DiBAl-H) с образованием соединения D;
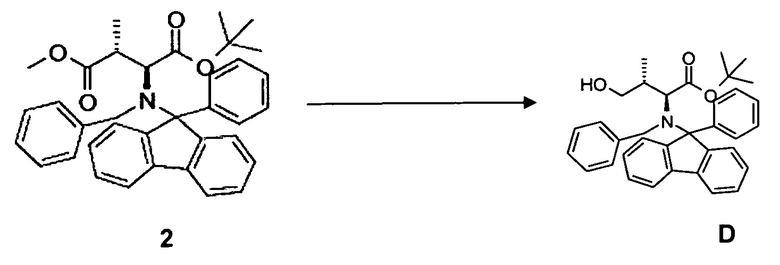
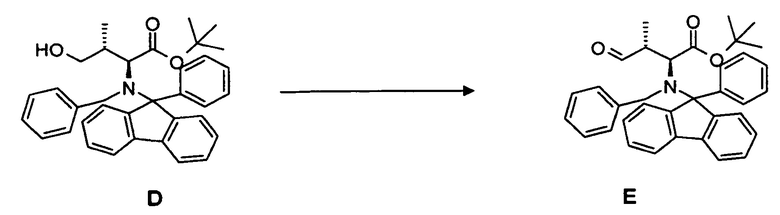
(b) окисление гидроксисоединения D, в частности, с использованием окисления по методу Сверна, с образованием соединения E;
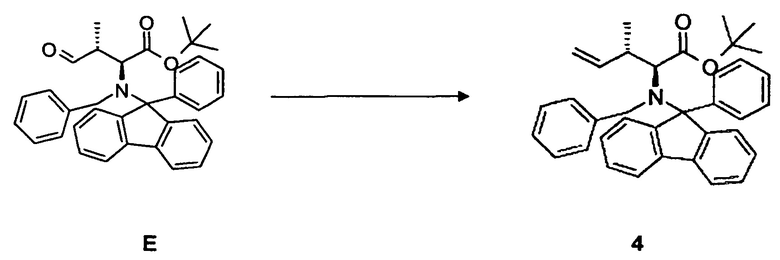
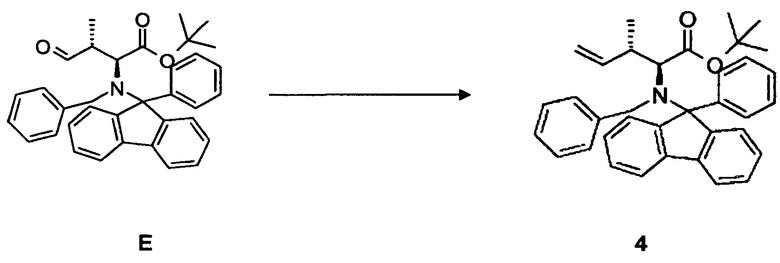
(c) преобразование соединения E, в частности, в условиях реакции Виттига, с образованием соединения 4;
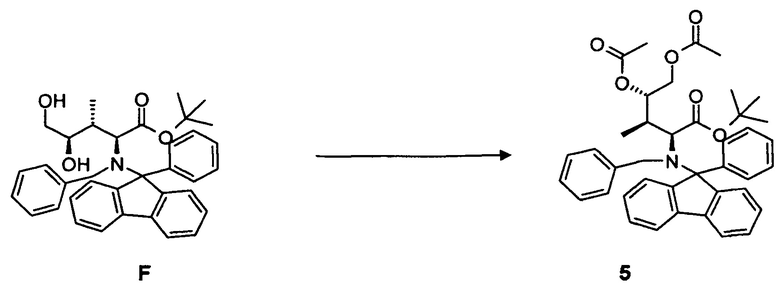
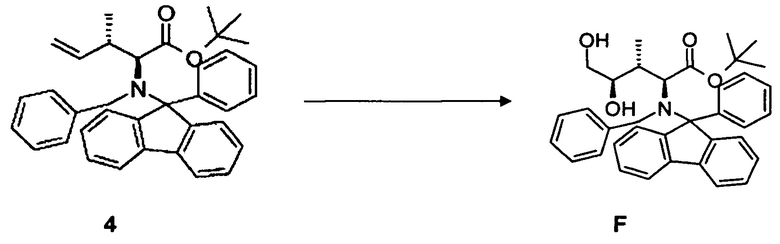
(d) преобразование соединения 4, в частности, в условиях окисления по методу Шарплесса, с образованием соединения F;
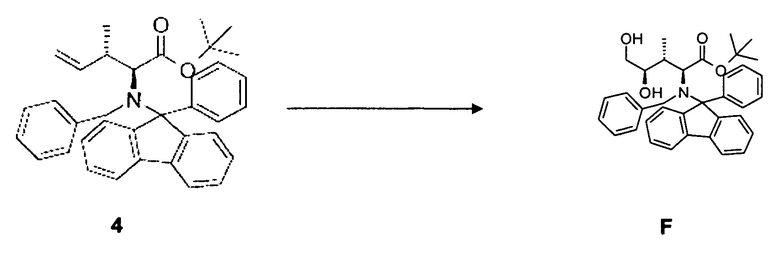
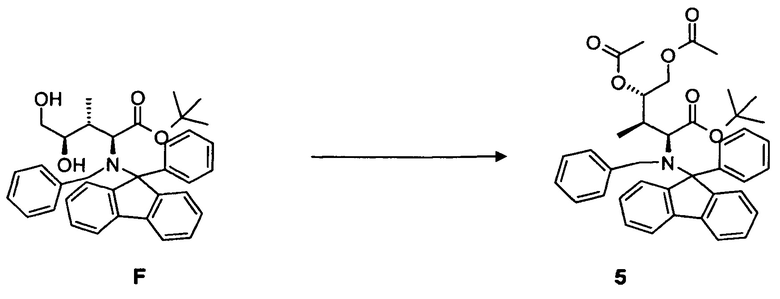
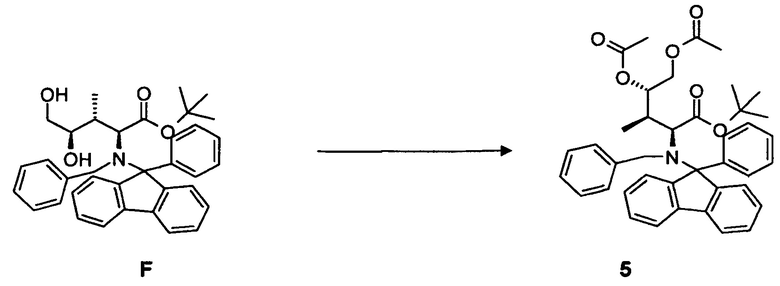
(e) преобразование соединения F, в частности, в условиях каталитической эстерификации, с образованием соединения 5;
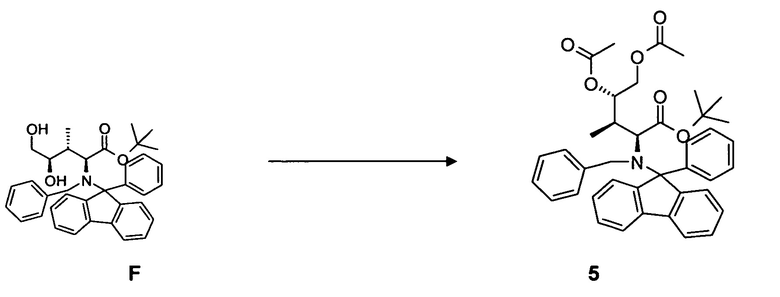
и
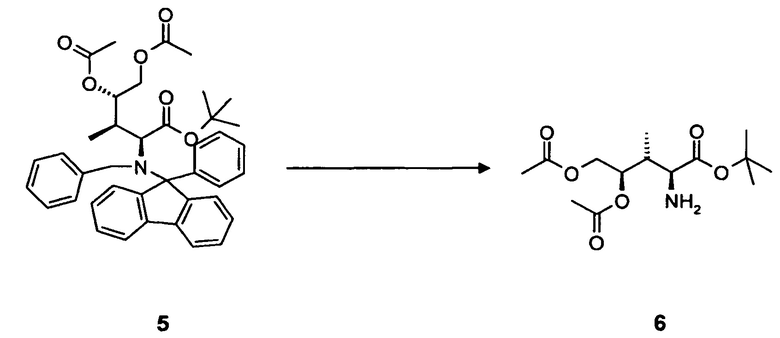
удаление N-защитной группы в соединении 5, в частности, с использованием палладийкатализируемой гидрогенизации, с образованием соединения 6.
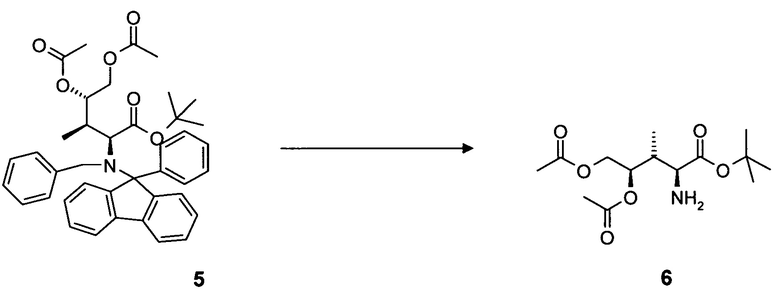
[0033] В конкретном варианте воплощения альтернативного аспекта способ дополнительно включает одну или несколько из следующих стадий:
(а) восстановление соединения 2*, в частности, при помощи диизобутилалюминийгидрида (DiBAl-H) с образованием соединения D* (содержащего вторую бензильную защитную группу по атому азота вместо фенилфлуоренильной группы в соединении D);
(b) окисление гидрокси-соединения D*, в частности, с использованием окисления по методу Сверна, с образованием соединения E* (содержащего вторую бензильную защитную группу по атому азота вместо фенилфлуоренильной группы в соединении E);
(c) преобразование соединения E*, в частности, в условиях реакции Виттига, с образованием соединения 4* (содержащего вторую бензильную защитную группу по атому азота вместо фенилфлуоренильной группы в соединении 4);
(d) преобразование соединения 4*, в частности, в условиях окисления по методу Шарплесса, с образованием соединения F* (содержащего вторую бензильную защитную группу по атому азота вместо фенилфлуоренильной группы в соединении F);
(e) преобразование соединения F*, в частности, в условиях каталитической эстерификации, с образованием соединения 5* (содержащего вторую бензильную защитную группу по атому азота вместо фенилфлуоренильной группы в соединении 5);
и
(f) удаление N-защитной группы в соединении 5*, в частности, с использованием палладийкатализируемой гидрогенизации, с образованием соединения 6.
[0034] В конкретном варианте воплощения, способ дополнительно включает стадию выделения и очистки соединения 6. В конкретном варианте воплощения соединение 6 очищают с использованием осаждения в виде гидрохлорида и/или хроматографической очистки.
[0035] Во втором аспекте настоящее изобретение относится к соединению 6.
[0036] В конкретном варианте воплощения соединение 6 имеет чистоту больше чем 90%, в частности больше чем 95%.
[0037] В контексте настоящего изобретения термин "чистота" относится к общему количеству соединения 6 и его диастереоизомеров, которые присутствуют. Чистота больше чем 90%, например, означает, что в 1 г композиции, включающей соединение 6, содержится больше чем 90%, т.е. больше чем 900 мг, соединения 6 и/или его стереоизомеров. Остальная часть, т.е. примеси, может включать непрореагировавшее исходное вещество и другие участвующие в реакции вещества, растворители, продукты расщепления и/или побочные продукты.
[0038] В конкретном варианте воплощения композиция, включающая соединение 6 с чистотой больше чем 90%, включает больше чем 100 мг соединения 6.
[0039] В конкретном варианте воплощения соединение 6 имеет диастереомерную чистоту больше чем 70:30.
[0040] В контексте настоящего изобретения термин "диастереомерная чистота" относится к отношению количества соединения 6, присутствующего в композиции, включающей соединение 6, к количеству его диастереоизомеров, присутствующих в указанной композиции. Диастереомерная чистота больше чем 70:30, например, означает, что больше чем 70% от общего количества защищенных дигидроксиизолейцинов в композиции, включающей соединение 6 и его диастереомеры, составляет соединение 6, тогда как общее количество всех диастереоизомеров соединения 6 соответственно меньше чем 30%.
[0041] В конкретном варианте воплощения композиция, включающая соединение 6 с диастереомерной чистотой больше чем 70:30, включает больше чем 100 мг соединения 6.
[0042] В третьем аспекте настоящее изобретение относится к набору, включающему соединение 6, в частности набору, включающему по меньшей мере 100 мг соединения 6 и по меньшей мере один дополнительный реагент для синтеза аматоксинов или их предшественников.
[0043] В конкретных вариантах воплощения соединение 6 в наборе имеет чистоту больше чем 90%, в частности больше чем 95%, и/или диастереомерную чистоту больше чем 70:30.
[0044] В конкретных вариантах воплощения указанный по меньшей мере один дополнительный реагент выбран из следующего перечня:
(i) смола, в частности, смола, выбранная из следующей группы: Merrifield смола; Rink-Amid смола; и THP-смола;
(ii) защищенный гидроксипролин, в частности флуоренилметилоксикарбонил-(Fmoc-)-защищенный O-аллил гидроксипролин (FmocHypOAll);
(iii) защищенный аспарагин, в частности Fmoc-защищенный N-тритиласпарагин (Fmoc(N-Tri)AsnOH);
(iv) защищенный Cys-Trp дипептид, в частности Fmoc-защищенный Cys-Trp дипептид с -SH и -OH защитными группами (FmocCys(S-2-((o-NO2Ph)SO2Trp-O-Аллил))]OH);
(v) защищенный глицин, в частности Fmoc-защищенный глицин (FmocGly);
(vi) защищенный изолейцин, в частности Fmoc-защищенный изолейцин (FmocIle);
(vii) агент пептидного связывания, в частности агент пептидного связывания, выбранный из следующей группы: O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилуроний тетрафторборат (TBTU); бензотриазол-1-ил-окситрипирролидинофосфоний гексафторфосфат (PyBOP); и o-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфат (HATU); и
(viii) третичный амин, в частности N,N-диизопропилэтиламин (DiPEA).
[0045] Еще в одном аспекте настоящее изобретение относится к способу синтеза аматоксина или молекулы, являющейся его предшественником, включающему стадию (a) связывания соединения 6 с гидроксипролином, в частности, путем взаимодействия соединения 6 со смолой, содержащей предварительно нагруженный на нее гидроксипролин, в частности, путем связывания соединения 6 со свободным C-концом FmocHypOH, который иммобилизован на смоле L, например, тетрагидропиранильной (THP) смоле.
[0046] В конкретных вариантах воплощения остальные аминокислоты затем связывают с использованием N-концевой стратегии синтеза. В таких конкретных вариантах воплощения способ по настоящему изобретению дополнительно включает одну или несколько из следующих стадий:
(b) повторное удаление N-защитной группы Fmoc и связывание соединения G с Fmoc-(N-Tri)Asn OH; FmocCys(S-2-((o-NO2Ph)SO2Trp-O-Аллил))]OH, Fmoc-Gly OH, Fmoc-Ile OH, Fmoc-Gly OH с образованием соединения H:
(c) удаление O-аллил- и N-Fmoc-защиты в соединении H с последующей циклизацией с образованием соединения I (замыкание B-кольца):
(d) Удаление 2-нитроарилсульфонамид-N-защиты и отделение соединения I от смолы с образованием соединения J:
(e) циклизация в фазе раствора соединения J с образованием Аманитинового производного K:
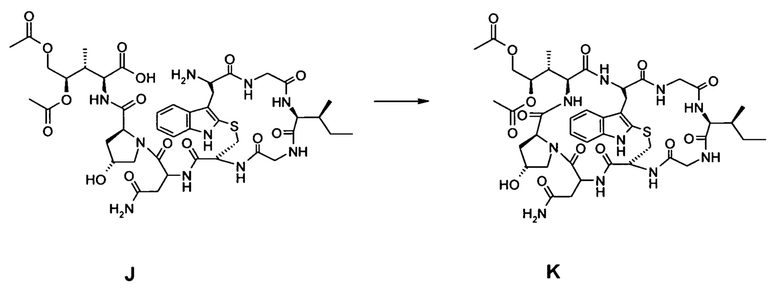
[0047] Еще в одном аспекте настоящее изобретение относится к способу синтеза аматоксина или молекулы, являющейся его предшественником, в растворе.
[0048] В некоторых вариантах воплощения такой способ включает одну или несколько из следующих стадий:
[0049] (a) связывание соединения 6 с гидроксипролином, в частности, путем взаимодействия соединения 6 с гидроксипролином, в частности, путем связывания соединения 6 со свободным C-концом Fmoc (OtBu) HypOH;
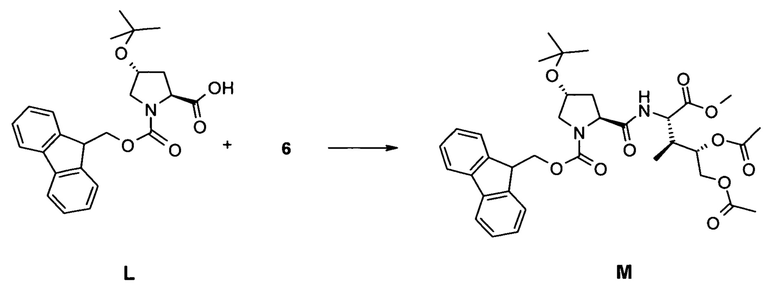
(b) повторное удаление N-защитной группы Fmoc и связывание соединения M с Fmoc-(N-Tri)Asn OH; Fmoc-(S-Tri)Cys OH, Fmoc-Gly OH, Fmoc-Ile OH, Fmoc-Gly OH и N-Boc-HPI OH с образованием соединения N;
(c) кислотный способ удаления N- и S-тритильной, O-трет-бутильной и N-трет-бутилоксикарбонильной защитных групп и in situ замыкание кольца при помощи реакции Savige-Fontana (Savige & Fontana, Int J Pept Protein Res. 15 (1980) 102-12) с получением соединения J.
[0050] Еще в одном аспекте настоящее изобретение относится к способу синтеза аматоксина или молекулы, являющейся его предшественником, в растворе, включающему стадию связывания соединения 6 с гидроксипролином, в частности, путем взаимодействия соединения 6 с гидроксипролином, в частности, путем связывания соединения 6 со свободным C-концом Fmoc (OtBu) HypOH.
[0051] В конкретных вариантах воплощения, аматоксин представляет собой аматоксин с дигидроксиизолейциновой группой в качестве аминокислоты 3 (см. Фиг. 1).
[0052] В контексте настоящего изобретения, термин “аматоксин” включает все циклические пептиды, состоящие из 8 аминокислот, выделенные из рода Amanita и описанные в Wieland, T. and Faulstich H. (Wieland T, Faulstich H., CRC Crit Rev Biochem. 1978 Dec;5(3):185-260), и, кроме того, включает все их химические производные; кроме того, все их полусинтетические аналоги; кроме того, все их синтетические аналоги, образованные из структурных блоков в соответствии с основной структурой природных соединений (циклические, 8 аминокислот), кроме того, все синтетические или полусинтетические аналоги, содержащие негидроксилированные аминокислоты вместо гидроксилированных аминокислот, кроме того, все синтетические или полусинтетические аналоги, в которых тиоэфирная сульфоксидная группа замещена сульфидом, сульфоном или атомами, отличными от серы, например атомом углерода, как в карбааналоге аманитина, при этом в каждом случае любое такое производное или аналог является функционально активным путем ингибирования РНК полимеразы II млекопитающего.
[0053] Функционально аматоксины определяются как пептиды или депсипептиды, которые ингибируют РНК полимеразу II млекопитающего. Предпочтительными аматоксинами являются такие, которые содержат функциональную группу (например, карбоксильную группу, аминогруппу, гидроксигруппу, тиольную или тиолзахватывающую группу), которая может взаимодействовать с линкерными молекулами или мишеньсвязывающими группами, определенными выше. Аматоксины, которые являются особенно подходящими для конъюгатов по настоящему изобретению, представляют собой α-аманитин, β-аманитин, γ-аманитин, ε-аманитин, аманин, аманинамид, амануллин и амануллиновую кислоту, как показано на Фиг. 1, а также их соли, химические производные, полусинтетические аналоги и синтетические аналоги. Особенно предпочтительными аматоксинами для использования в настоящем изобретении являются α-аманитин, β-аманитин и аманинамид.
[0054] Как используется в настоящей заявке, “химическое производное” (или кратко: “производное”) соединения относится к веществам, имеющим химическую структуру, которая подобна соединению, но при этом содержащим по меньшей мере одну химическую группу, которая не присутствует в соединении, и/или не содержащим по меньшей мере одну химическую группу, которая присутствует в соединении. Соединение, с которым сравнивают производное, известно как “исходное” соединение. Типично, "производное" можно получить из исходного соединения в одну или несколько химических реакционных стадий.
ПРИМЕРЫ
[0055] Далее настоящее изобретение объясняется более подробно при помощи неограничивающих примеров:
Пример 1
Реакция Манниха с использованием пропионового альдегида и N-PMP глиоксалимина
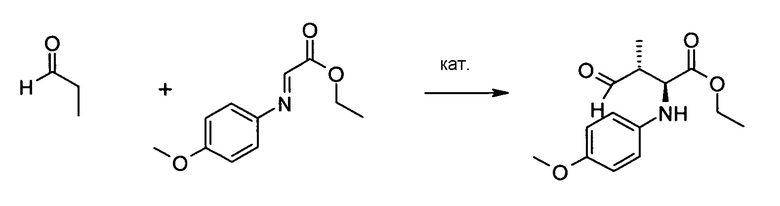
[0056] Одним из ключевых характерных структурных признаков соединения 1 является 2S,3R конформация амино- и метильной группы. Легкий доступ к конструкции смежных стереоцентров представляет собой реакцию Манниха. Однако оба стереоцентра предполагают тип реакции, противоположный реакции Манниха, с высоким диастерео- и энантиоконтролем. Это исходное условие описано с использованием специально разработанного органического катализатора, описание которого легко можно найти в J. Am. Chem. Soc., 2006, 128, 1040-1041. Однако различные подходы к удалению PMP-группы не были эффективными и давали реакционные смеси, таким образом, этот подход, в конечном счете, был отвергнут. Из литературы известно, что удаление может быть достаточно трудоемким и безуспешным, хотя PMP-группа необходима для реакции.
Пример 2
Алкилирование производного аспарагиновой кислоты
2.1 Введение
[0057] Альтернативный подход к синтезу γ,δ-дигидроксиизолейцин синтона с правильной стереоконфигурацией (2S,3R,4R) начинался с производного аспарагиновой кислоты 3. Подобные подходы описаны в литературе (см. Yoshida et al., A large scale production of (3S,4S)-3-(tert-Butoxycarbonyl)amino-4-methylpyrrolidine and its analogs from L-Aspartic acid, Chem Pharm Bull (1996), 44, 1128 - 1131; Wolf and Rapoport, Conformationally constrained Peptides. Chirospecific Synthesis of 4-Alkyl-Substituted g-Lactam-Bridged Dipeptides from L-Aspartic Acid’, J Org Chem (1989), 54, 3164-3173). Оказалось, что критически важным было (i) использование комбинации либо (ia) бензильной и фенилфлуоренильной группы, либо (ib) двух бензильных групп для защиты свободной амино группы, и (ii) использование литий гексаметилдисилазана (LHMDS) вместо соответствующей калиевой соли. Калий гексаметилдисилазан (KHMDS) приводил к противоположной конфигурации.
2.2 Синтез соединения 2
[0058] В техгорлую круглодонную колбу с магнитной мешалкой, капельной воронкой и низкотемпературным термометром загружали 16,3 ммоль, 8,7 г соединения 3 (см. Пример 3), растворенного в 150 мл безводного тетрагидрофурана, и охлаждали до -20°C. Добавляли гексаметилдисилазид лития, 40 мл 1,0M раствора в гексане, по каплям в течение 15 мин. Реакцию поддерживали при температуре при -20°C в течение 2 ч, затем охлаждали до -80°C. В завершение добавляли 12,2 мл, 19,6 ммоль, 1,2 экв. метилиодида. Реакционной смеси давали медленно нагреться и поддерживали еще в течение 4 ч при -20°C. Реакцию в конце гасили путем добавления 10 мл метанола, давали нагреться до комнатной температуры и выливали в 150 мл воды. Водную фазу экстрагировали трет-бутилметиловым эфиром, сушили над MgSO4 и концентрировали в вакууме. Неочищенный продукт: 9,0 г. 1H-ЯМР неочищенного продукта показал диастереоселективную чистоту лучше чем 5:1. Выход 7,2 г, 81%.
[0059] Неочищенный продукт очищали флэш-хроматографией, 330 г диоксида кремния, градиент н-гексан/трет-бутилметиловый эфир от 0% до 50%.
[0060] 1H-ЯМР: д 0,72 (д, 3H, J=5,6 Гц), 1,03 (с, 9H), 2,64 (дт, 1H, J=5,6, 8,4 Гц), 3,55 (с, 3H), 3,85 (д, 1H, J=8,4 Гц), 4,31 (д, 1H, J=11,2 Гц), 4,65 (д, 1H, J=11,2 Гц), 7,15-7,94 (м, 18H).
2.3 Синтез соединения 2*
[0061] Аналогично Примеру 2.2 соединение 2* можно синтезировать из соединения 3*.
Пример 3
Синтез соединения 3
3.1 Введение
[0062] Соединение 3 синтезировали в соответствии с протоколом, описанным Dunn et al. (Dunn et al., Stereoselective synthesis of 2,3-diamino acids. 2,3-Diamino-4-phenylbutanoic acid, J. Org. Chem. 55 (1990) 5017-25).
3.2 Синтез соединения B

[0063] В круглодонный стеклянный цилиндр с завинчивающейся крышкой и стержнем для перемешивания загружали 25 г, 136 ммоль 4-метил-L-аспартат гидрохлорида. Моноэфир A суспендировали в 100 мл смеси диоксан/тетрагидрофуран (1:1, об/об) и 25 мл серной кислоты и охлаждали до -30°C (криостат). К смеси добавляли 2-метилпропен, 200 г, 3,56 моль. Цилиндр закрывали и давали нагреться до комнатной температуры. Реакционную смесь выливали на 1000 мл насыщенного раствора бикарбоната натрия и экстрагировали этилацетатом (5 раз 400 мл). Объединенную органическую фазу сушили над MgSO4 и концентрировали в вакууме. Выход: 17,88 г, 64,6%.
3.3 Синтез соединения C
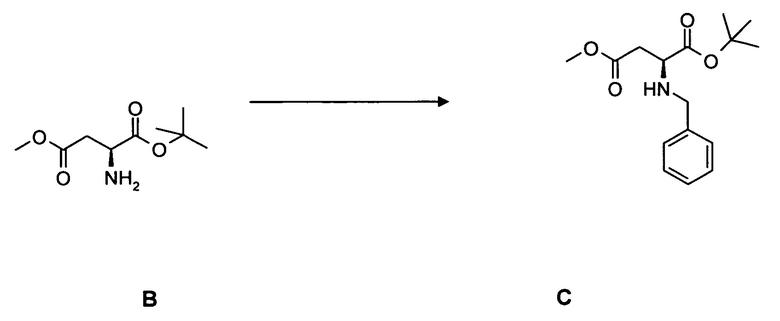
[0064] В круглодонную колбу с термометром и стержнем для перемешивания загружали соединение B, 17,9 г, 87,9 ммоль, растворенное в 500 мл метанола и 150 мл уксусной кислоты. Добавляли по каплям бензальдегид, 16,0 мл, 158 ммоль. Реакцию поддерживали при комнатной температуре в течение 2 ч и в завершение охлаждали до 0°C. Добавляли цианоборогидрид натрия, 10,0 г, 159 ммоль, в течение 45 мин и перемешивали еще в течение 15 мин при 0°C. Реакционную смесь в завершение выливали в 1000 мл бикарбоната натрия и перемешивали в течение 10 мин. После экстрагирования дихлорметаном (5 раз 250 мл) объединенные органические фазы сушили над MgSO4 и концентрировали в вакууме. Неочищенный продукт: 35 г. Выход 14,3 г, 55,4%.
[0065] Очистка методом флэш-хроматографии, 330 г SiO2, градиент н-гексан/этилацетат от 0 до 50%.
[0066] 1H-ЯМР: д 1,42 (с, 9H), 2,29 (с, 1H), 2,62 (т, 2H, J=9,2 Гц), 2,50 (т, 1H, J=8 Гц), 3,62 (с, 3H), 3,67 (д, 1H, 17,6 Гц), 3,83 (д, 1H, J=17,2 Гц), 7,19-7,30 (м, 5H).
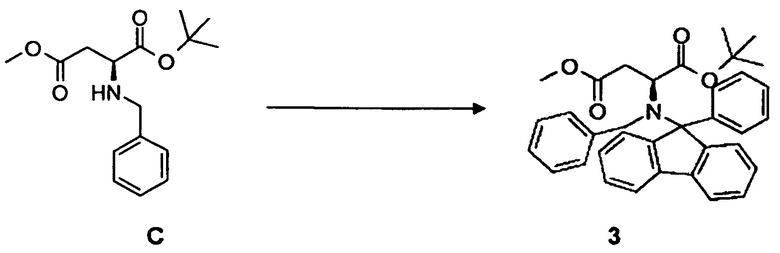
3.4 Синтез соединения 3
[0067] В круглодонную колбу со стержнем для перемешивания загружали соединение C, 21,3 г, 51,6 ммоль, в 500 мл ацетонитрила. Добавляли 9-бром-9-фенилфлуорен, 25,0 г, 77,8 ммоль, 20,0 г, 60,3 ммоль нитрата свинца, 31,7 г, 149,3 ммоль K3PO4. Реакционную смесь поддерживали при комнатной температуре в течение 2,0 ч. После завершения, реакционную смесь разбавляли при помощи 500 мл дихлорметана, сушили над Na2SO4 и фильтровали через Целит. Продукт концентрировали в вакууме. Выход: 16,2 г, 41,7%.
[0068] Очистка методом флэш-хроматографии, 330 г SiO2, н-гексан/трет-бутилметиловый эфир, градиент от 0 до 50%.
[0069] 1H-ЯМР: д 1,14 (с, 9H), 1,92 (дд, 1H, J=2,8, 16 Гц), 2,54 (дд, 1H, 10,8, 15,8 Гц), 3,40 (с, 3H), 3,86 (д, 1H, J=13,6 Гц), 4,21 (д, 1H, J=14 Гц), 7,17-7,83 (м, 18 H).
Пример 4
Синтез соединения 6
4.1 Синтез соединения D
[0070] В круглодонную колбу, снабженную стержнем для перемешивания, термометром и капельной воронкой, загружали соединение 2, 16,4 г, 29,9 ммоль, растворенное в 150 мл безводного тетрагидрофурана, и охлаждали до -30°C. Добавляли 1,0M раствор (150 мл) диизобутилалюминийгидрида в инертной атмосфере (аргон) в течение 1,0 часа и перемешивали еще в течение 16 ч. Реакционную смесь гидролизовали при помощи Na2SO4 декагидрата и давали нагреться до комнатной температуры. Осадок отфильтровывали и тщательно промывали трет-бутилметиловым эфиром. Органическую фазу концентрировали в вакууме. Выход: 14,3 г, 92%.
[0071] Очистка методом флэш-хроматографии, 330 г SiO2, н-гексан/этилацетат, градиент от 0 до 50%.
[0072] 1H-ЯМР: д 0,45 (д, 3H, J=6,8 Гц), 1,02 (с, 9H), 3,09 (д, 1H, J=10,8 Гц), 3,34 (дкв., 1H, J=7,2 Гц, J=14,8 Гц), 4,00 (д, 2H, J=10,8 Гц), 4,36 (д, 1H, 13,6 Гц), 4,73 (д, 1H, J=13,6 Гц), 7,20-7,76 (м, 18H).
4.2 Синтез соединения E
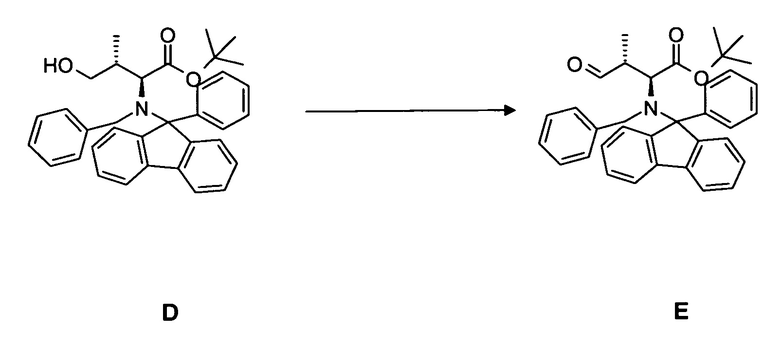
[0073] В круглодонную колбу, снабженную стержнем для перемешивания, термометром, капельной воронкой и вводом для аргона, загружали оксалилхлорид 3,28 мл, 35,5 ммоль в дихлорметане и охлаждали до -80°C. Медленно добавляли безводный диметилсульфоксид 5,47 мл, 71,1 ммоль, разбавленный 20 мл дихлорметана. Добавляли соединение D 13,67 г, 25,9 ммоль, растворенное в 30 мл дихлорметана, в течение 15 мин. По прошествии еще 15 мин при -80°C добавляли триэтиламин и реакционной смеси давали нагреться до комнатной температуры. Два слоя разделяли и водный слой экстрагировали дихлорметаном (4 раза 150 мл). Объединенные органические фазы сушили над MgSO4 и концентрировали в вакууме. Неочищенный продукт: 16,9 г. Неочищенный продукт непосредственно преобразовывали в олефин.
4.3 Синтез соединения 4
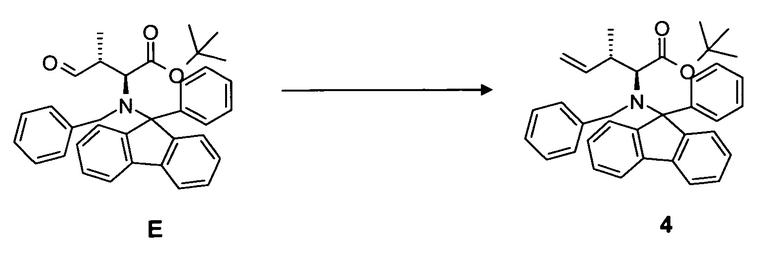
[0074] В круглодонную колбу со стержнем для перемешивания, термометром и вводом для аргона загружали гидрид натрия 2,32 г, 57,9 ммоль, суспендированный в 120 мл диметилсульфоксида. Суспензию нагревали до 50°C в течение 30 мин и охлаждали до комнатной температуры. Затем добавляли твердый метилтрифенилфосфонийбромид и перемешивали в течение 15 мин. Добавляли неочищенный продукт E, растворенный в 20 мл диметилсульфоксида, и перемешивали еще в течение 16 ч. После гидролиза (300 мл воды), экстрагирования этилацетатом (4 раза 150 мл), тщательной промывки объединенной органической фазы водой (3 раза 150 мл) и насыщенным солевым раствором раствор сушили при помощи MgSO4 и концентрировали в вакууме. Выход: 13,3 г, 99,2% (2 стадии).
[0075] Очистка методом флэш-хроматографии, 330 г SiO2, н-гексан/этилацетат, градиент от 0 до 80%.
[0076] 1H-ЯМР: д 0,51 (д, 3H, J=1,6 Гц), 1,05 (с, 9H), 2,14 (гепт., 1H, J=0,8 Гц), 3,15 (д, 1H, 10,8 Гц), 4,30 (д, 1H, J=14 Гц), 4,49 (дд, 1H, J=1,2, 17,6 Гц), 4,57 (д, 1H, 13,6 Гц), 5,03 (д, 1 H, J=0,8, 10,8 Гц), 5,95 (м, 1H), 7,26-7,60 (м, 18H).
4.4 Синтез соединения F
[0077] В круглодонную колбу загружали 100 г AD-смеси (бета, коммерческий источник) и растворяли в 60 мл смеси трет-бутанол/вода (1:1, об/об). Добавляли соединение 5, 3,75 г, 7,3 ммоль, растворенное в 17 мл диоксана, одной порцией. Реакционную смесь перемешивали при комнатной температуре в течение 4 дней вплоть до завершения реакции. Реакционную смесь гасили путем добавления Na2SO3 и экстрагировали этилацетатом (4 раза 50 мл), промывали насыщенным раствором NH4Cl и насыщенным солевым раствором, сушили над MgSO4 и концентрировали в вакууме. Выход: 1,19 г, 29%.
[0078] Очистка методом флэш-хроматографии, 330 г SiO2, дихлорметан/трет-бутилметиловый эфир, градиент от 0 до 80%.
[0079] 1H-ЯМР: д 0,38 (д, 3H, J=6,8 Гц), 1,03 (с, 9H), 1,84 (с+м, 2H, J=8,8 Гц), 3,24 (дд, 1H, J=4,8, 11,4 Гц), 3,38 (д, 2H, J=10,9 Гц), 3,76 (м, 1H), 4,40 (д, 1H, J=13,3 Гц), 4,95 (д, 1H, J=13,3 Гц), 5,55 (с, 1H), 7,26-7,91 (м, 18H).
4.4 Синтез соединения 5
[0080] В круглодонную колбу с магнитной мешалкой загружали соединение F, 2,1 г, 3,64 ммоль, растворенное в 75 мл дихлорметана. Добавляли избыточное количество ангидрида уксусной кислоты, 5,0 мл, 52,3 ммоль, и каталитическое количество диметиламинопиридина. Реакционную смесь оставляли для перемешивания при комнатной температуре в течение ночи. Выход: 1,83 г, 81%.
[0081] Очистка методом флэш-хроматографии, 330 г SiO2, гексан/трет-бутилметиловый эфир, градиент от 0 до 20%.
[0082] 1H-ЯМР: д 0,49 (д, 3H, J=7,2 Гц), 1,05 (с, 9H), 1,74 (м, 1H, J=2,90 Гц), 1,88 (с, 3H), 2,11 (с, 3H), 3,18 (д, 1H, J=10,5 Гц), 3,60 (дд, 1H, J=2,3, 12,2 Гц), 4,32 (д, 1H, J=13,9 Гц), 4,74 (д, 1H, J=13,9 Гц), 5,98 (дд, 1H, J=2,4, 84 Гц), 7,10-7,73 (м, 18H).
4.4 Синтез соединения 6

[0083] В круглодонную колбу, снабженную вводом для аргона и трубопроводом для создания вакуума, загружали соединение 5, 2,0 г, 3,1 ммоль, растворенное в 50 мл 0,1M хлористоводородной кислоты в этаноле, и 200 мг 10% палладия на угле. После продувки колбы водородом, реакционную смесь оставляли для перемешивания в течение 16 ч при комнатной температуре. Колбу продували аргоном, фильтровали и фильтрат концентрировали в вакууме. Прозрачное масло обрабатывали н-гексаном (3 раза) для удаления фенилфлуорена. Соединение 6 в форме соли хлористоводородной кислоты получали в виде белого твердого вещества. Выход: 0,92 г, 98%.
[0084] Очистка методом осаждения или хроматографии.
[0085] 1H-ЯМР: (основной изомер) д 1,16 (д, 3H, J=6,8 Гц), 1,47 (С, 9H), 2,06 (с, 3H), 2,09 (с, 3H), 2,78 (м, 1H, J=7,7 Гц), 4,04 (дд, 1H, J=3,9, 12,5 Гц), 4,15 (с, 1H), 4,52 (дд, 2H, J=2,08, 12,6 Гц), 5,02 (м, 2 H), 8,86 (с, 2H).
[0086] 13C-NMR: (основной изомер) д 11,5, 20,8, 21,7, 35,0, 53,9, 62,4, 72,2, 84,9, 166,2, 169,9, 170,7.
Пример 5
Синтез of α-Аматоксина
5.1 Синтез соединения G
[0087] В открытый сосуд из полипропилена в виде реакционной трубки, снабженной фриттой и дренажным клапаном, загружали 0,5 г, 0,5 ммоль FmocHypOH тетрагидропиранил полистирола и оставляли для набухания в 3,0 мл диметилформамида в течение 20 мин. После удаления растворителя через дренажный клапан в реакционный сосуд загружали 179 мг, 0,6 ммоль соединения 6, растворенного в 1,5 мл диметилформамида, 1,5 мл 1 мМ раствора гидроксибензотиазола в диметилформамиде, 1,5 мл 1 мМ раствора (бензотриазол-1-илокси)трипирролидинофосфоний гексафторфосфата в диметилформамиде и 259 мкл диизопропилэтиламина. После гомогенизации при помощи стеклянного стержня реакционную смесь нагревали с использованием микроволнового облучения до 70°C в течение 4,0 мин и в завершение промывали диметилформамидом (3 раза) и дихлорметаном (2 раза).
[0088] Аликвоту, приблизительно 20 мг полимера, отщепляли с использованием смеси трифторуксусная кислота/вода/триэтилсилан (8:2:10; об/об/об) для масс-спектроскопического анализа.
[0089] MS: 582,92; [M-tBu+H]+; Fmoc-Hyp-бис(O-ацетил)дигидрокси-Ile-OtBu
5.2 Синтез соединения H
[0090] Соединение G затем повторно подвергали процедуре удаления защитной группы Fmoc и связывали с остальными 6 аминокислотами следующим образом:
Удаление Fmoc- N-защитной группы:
[0091] Смолу два раза обрабатывали при помощи 4,5 мл 20% раствора пиперидина в диметилформамиде и нагревали до 70°C в течение 3 мин с использованием микроволнового облучения. Смолу затем промывали диметилформамидом (3 раза).
Связывание с аминокислотами:
[0092] Связанный со смолой после удаления защиты пептид в 1,5 мл диметилформамида последовательно подвергали взаимодействию с аминокислотами (см. перечень), растворенными в 1,5 мл диметилформамида, 1,5 мл 1 мМ раствора гидроксибензотиазола в диметилформамиде, 1,5 мл 1 мМ раствора (бензотриазол-1-илокси)трипирролидинофосфоний гексафторфосфата в диметилформамиде и 259 мкл диизопропилэтиламина. После гомогенизации с использованием стеклянного стержня реакционную смесь нагревали с использованием микроволнового облучения до 70°C в течение 4,0 мин и в завершение промывали диметилформамидом (3 раза) и дихлорметаном (2 раза).
[0093] Аликвоту, приблизительно 20 мг полимера, отщепляли с использованием смеси трифторуксусная кислота/вода/триэтилсилан (8:2:10; об/об/об) для масс-спектроскопического анализа.
Аминокислоты для связывания:
1. Fmoc-(N-Три)Asn OH
2. Fmoc-Cys(S-2-((o-NO2Ph)SO2Trp-O-Аллил))]OH
3. Fmoc-Gly OH
4. Fmoc-Ile OH
5. Fmoc-Gly OH
[0094] MS: 1227,14; [M-tBu+H]+; 1283,00; [M+H]+; FmocCys(S-2-((o-NO2Ph)SO2Trp-O-Аллил))]-Asn-Hyp-бис(O-ацетил)дигидрокси-Ile-OtBu.
[0095] MS: 1453,98; [M-tBu+H]+; [M+H]+; FmocGly-Ile-Gly-Cys(S-2-((o-NO2Ph)SO2Trp-O-Аллил))]-Asn-Hyp-бис(O-ацетил)дигидрокси-Ile-OtBu
5.3 Синтез соединения I; замыкание B-кольца
[0096] Смолу H последовательно подвергали удалению аллил- и Fmoc- защитных групп, затем B-кольцевой циклизации:
Удаление аллил-O-защиты
[0097] Смолу встряхивали в течение ночи при комнатной температуре с 874 мг, 5,6 ммоль N,N-диметилбарбитуровой кислоты, 258 мг, 0,224 ммоль Pd(PPh3)4 в дихлорметане. Через 16 ч смолу промывали дихлорметаном; диметилформамидом; ацетонитрилом; и трет-бутилметиловым эфиром.
Удаление N-защитной группы Fmoc:
[0098] Смолу два раза обрабатывали при помощи 4,5 мл 20% раствора пиперидина в диметилформамиде и нагревали до 70°C в течение 3 мин с использованием микроволнового облучения. Смолу затем промывали диметилформамидом (3 раза).
Образование B-кольца
[0099] Связанный со смолой после удаления защиты пептид в 3,0 мл диметилформамида подвергали взаимодействию с 1,5 мл 1 мМ раствора гидроксибензотиазола в диметилформамиде, 1,5 мл 1 мМ раствора (бензотриазол-1-илокси)трипирролидинофосфоний гексафторфосфата в диметилформамиде и 259 мкл диизопропилэтиламина. После гомогенизации с использованием стеклянного стержня реакционную смесь нагревали с использованием микроволнового облучения до 70°C в течение 4,0 мин и в завершение промывали диметилформамидом (3 раза) и дихлорметаном (2 раза).
[00100] Аликвоту, приблизительно 20 мг полимера, отщепляли с использованием смеси трифторуксусная кислота/вода/триэтилсилан (8:2:10; об/об/об) для масс-спектроскопического анализа.
[00101] MS: 1174,08; [M-tBu+H]+; 1229,96; [M+H]+; [Gly-Ile-Gly-Cys(S-2-((o-NO2Ph)SO2Trp))]кольцо-Asn-Hyp-бис(O-ацетил)дигидрокси-Ile-OtBu
5.4 Удаление 2-NO2-фенилсульфонильной N-защитной группы и отделение смолы, соединение J
[00102] Смолу I подвергали процедуре удаления 2-NO2-фенилсульфонил-защиты и в завершение отщепляли соединение от смолы
Удаление 2-NO2-фенилсульфонильной N-защитной группы:
[00103] Смолу повторно (3 раза) обрабатывали при помощи 500 мкл меркаптоэтанола и 500 мкл диазабициклоундецена в 4 мл диметилформамида в течение 2 ч. Смолу затем интенсивно промывали диметилформамидом, дихлорметаном, ацетонитрилом и трет-бутилметиловым эфиром.
Отделение от смолы:
[00104] Смолу затем обрабатывали при помощи 6 мл смеси трифторуксусная кислота/вода/триэтилсилан (8:2:10; об/об/об) при комнатной температуре в течение ночи и неочищенный белок концентрировали в вакууме.
[00105] MS: 989,18; [M-tBu+H]+; [Gly-Ile-Gly-Cys(S-2-(Trp))]кольцо-Asn-Hyp-бис(O-ацетил)дигидрокси-Ile-OtBu
5.5 Синтез соединения K; образование A-кольца
[00106] В реакционной колбе со стержнем для перемешивания неочищенный продукт J растворяли в 25 мл диметилформамида. К раствору добавляли 85 мкл, 2,5 ммоль диизопропилэтиламина и 135 мкл, 2,5 ммоль дифенилфосфазида. Реакционную смесь оставляли для перемешивания в течение ночи при комнатной температуре. Раствор в завершение концентрировали в вакууме, растворяли в 0,5 мл метанола и очищали. Выход: 4,9 мг
[00107] Неочищенную реакционную смесь очищали препаративной колоночной хроматографией.
[00108] MS: 858,94; [M+H]+
Пример 6
Синтез соединения 3*
[00109] Аналогично Примеру 3.4, соединение 3* можно синтезировать из соединения C с использованием бензилбромида вместо 9-бром-9-фенилфлуорена для N-защиты.
Пример 7
Альтернативный синтез соединения 6
[00110] Аналогично Примеру 4 соединение 6 можно синтезировать из соединения 2* через промежуточные соединения D*, E*, 4*, F* и 5* (каждое из которых содержит вторую бензильную защитную группу по атому азота вместо фенилфлуоренильной группы в соответствующем соединении D, E, 4, F и 5, соответственно).
| название | год | авторы | номер документа |
|---|---|---|---|
| КОНЪЮГАТ АНТИТЕЛА С АМАТОКСИНОМ НЕПРИРОДНОГО ТИПА | 2018 |
|
RU2798981C2 |
| КОНЪЮГАТ АНТИТЕЛА С АМАТОКСИНОМ НЕПРИРОДНОГО ТИПА | 2018 |
|
RU2840926C2 |
| НОВЫЙ СПОСОБ СИНТЕЗА АМАНИТИНОВ | 2018 |
|
RU2792210C2 |
| КОНЪЮГАТЫ АМАТОКСИНОВ С УЛУЧШЕННЫМИ ЛИНКЕРАМИ | 2011 |
|
RU2601411C2 |
| ПРОИЗВОДНЫЕ АМАТОКСИНА | 2014 |
|
RU2695370C2 |
| СИНТЕЗ (S)-6-ГИДРОКСИТРИПТОФАНА И ЕГО ПРОИЗВОДНЫХ | 2019 |
|
RU2810786C2 |
| УСОВЕРШЕНСТВОВАННЫЕ КОНЪЮГАТЫ N4 ХЕЛАТООБРАЗУЮЩИХ АГЕНТОВ | 2005 |
|
RU2360701C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ АНАЛОГОВ ОКСИТОЦИНА | 2015 |
|
RU2696276C2 |
| КОНЪЮГАТЫ АМАТОКСИН - АНТИТЕЛО | 2016 |
|
RU2724328C2 |
| КОНЪЮГАТЫ АМАТОКСИНА С УЛУЧШЕННЫМИ СВЯЗЯМИ | 2012 |
|
RU2575854C2 |
Изобретение относится к способу синтеза γ,δ-дигидроксиизолейцина 1 или его гидроксизащищенного производного (соединения 6) в качестве синтона для соединения 1, которые могут найти применение в качестве структурных блоков для получения аматоксинов. Способ включает стадию метилирования соединения 3 или соединения 3*, содержащего вторую бензильную защитную группу по атому азота вместо фенилфлуоренильной группы в соединении 3, при помощи метилиодида в присутствии бис(триметилсилил)амида лития (LHMDS). Предлагаемый способ позволяет получать соединение 6 с высокой диастереомерной чистотой. Изобретение относится также к соединению структуры 6, набору для синтеза аматоксинов или их предшественников и способу синтеза аматоксина или молекулы, являющейся его предшественником, с использованием соединения 6. 4 н. и 12 з.п. ф-лы, 3 ил., 7 пр.

1. Способ синтеза γ,δ-дигидроксиизолейцина 1
или его гидроксизащищенного производного (соединения 6) следующей формулы
в качестве синтона для соединения 1, включающий стадию метилирования соединения 3 или соединения 3*, содержащего вторую бензильную защитную группу по атому азота вместо фенилфлуоренильной группы в соединении 3, при помощи метилиодида в присутствии бис(триметилсилил)амида лития (LHMDS).
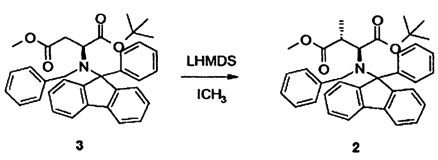
2. Способ по п.1, где реакцию осуществляют при температуре в пределах от около -10°С до около -80°С в простом эфире в течение времени от около 12 до около 20 ч.
3. Способ по п.1, дополнительно включающий одну или несколько из следующих стадий:
(a) взаимодействие L-аспарагиновой кислоты, монометилового эфира А, с 2-метилпропеном с образованием соединения В;
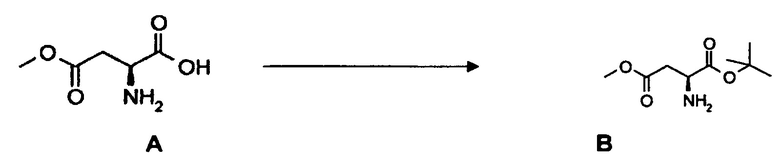
(b) взаимодействие соединения В с бензальдегидом с образованием соединения С;
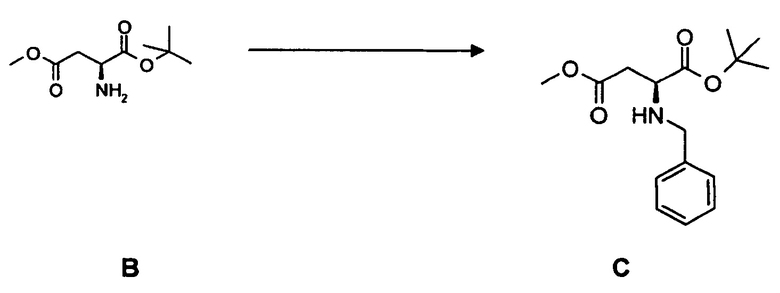
(c) взаимодействие соединения C с фенилфлуоренилбромидом с образованием соединения 3 или с бензилбромидом с образованием соединения 3*, содержащего вторую бензильную защитную группу по атому азота вместо фенилфлуоренильной группы в соединении 3.
4. Способ по п.1, дополнительно включающий одну или несколько из следующих стадий:
(а) восстановление соединения 2 или соединения 2*, содержащего вторую бензильную защитную группу по атому азота вместо фенилфлуоренильной группы в соединении 2, в частности, при помощи диизобутилалюминийгидрида, с образованием соединения D или соединения D*, содержащего вторую бензильную защитную группу по атому азота вместо фенилфлуоренильной группы в соединении D, соответственно;
(b) окисление гидрокси-соединения D или гидрокси-соединения D*, содержащего вторую бензильную защитную группу по атому азота вместо фенилфлуоренильной группы в соединении D, в частности, с использованием окисления по методу Сверна, с образованием соединения Е или соединения Е*, содержащего вторую бензильную защитную группу по атому азота вместо фенилфлуоренильной группы в соединении Е, соответственно;
(c) преобразование соединения Е или соединения Е*, содержащего вторую бензильную защитную группу по атому азота вместо фенилфлуоренильной группы в соединении Е, в частности, в условиях реакции Виттига, с образованием соединения 4 или соединения 4*, содержащего вторую бензильную защитную группу по атому азота вместо фенилфлуоренильной группы в соединении 4, соответственно;
(d) преобразование соединения 4 или соединения 4*, содержащего вторую бензильную защитную группу по атому азота вместо фенилфлуоренильной группы в соединении 4, в частности, в условиях окисления по методу Шарплесса, с образованием соединения F или соединения F*, содержащего вторую бензильную защитную группу по атому азота вместо фенилфлуоренильной группы в соединении F, соответственно;
(е) преобразование соединения F или соединения F*, содержащего вторую бензильную защитную группу по атому азота вместо фенилфлуоренильной группы в соединении F, в частности, в условиях каталитической эстерификации, с образованием соединения 5 или соединения 5*, содержащего вторую бензильную защитную группу по атому азота вместо фенилфлуоренильной группы в соединении 5, соответственно;
и
(f) удаление N-защиты в соединении 5 или соединении 5*, содержащем вторую бензильную защитную группу по атому азота вместо фенилфлуоренильной группы в соединении 5, в частности, с использованием палладий-катализируемой гидрогенизации, с образованием соединения 6.
5. Способ по п.2, дополнительно включающий одну или несколько из следующих стадий:
(а) восстановление соединения 2 или соединения 2*, содержащего вторую бензильную защитную группу по атому азота вместо фенилфлуоренильной группы в соединении 2, в частности, при помощи диизобутилалюминийгидрида, с образованием соединения D или соединения D*, содержащего вторую бензильную защитную группу по атому азота вместо фенилфлуоренильной группы в соединении D, соответственно;
(b) окисление гидрокси-соединения D или гидрокси-соединения D*, содержащего вторую бензильную защитную группу по атому азота вместо фенилфлуоренильной группы в соединении D, в частности, с использованием окисления по методу Сверна, с образованием соединения Е или соединения Е*, содержащего вторую бензильную защитную группу по атому азота вместо фенилфлуоренильной группы в соединении Е, соответственно;
(c) преобразование соединения Е или соединения Е*, содержащего вторую бензильную защитную группу по атому азота вместо фенилфлуоренильной группы в соединении Е, в частности, в условиях реакции Виттига, с образованием соединения 4 или соединения 4*, содержащего вторую бензильную защитную группу по атому азота вместо фенилфлуоренильной группы в соединении 4, соответственно;
(d) преобразование соединения 4 или соединения 4*, содержащего вторую бензильную защитную группу по атому азота вместо фенилфлуоренильной группы в соединении 4, в частности, в условиях окисления по методу Шарплесса, с образованием соединения F или соединения F*, содержащего вторую бензильную защитную группу по атому азота вместо фенилфлуоренильной группы в соединении F, соответственно;
(e) преобразование соединения F или соединения F*, содержащего вторую бензильную защитную группу по атому азота вместо фенилфлуоренильной группы в соединении F, в частности, в условиях каталитической эстерификации, с образованием соединения 5 или соединения 5*, содержащего вторую бензильную защитную группу по атому азота вместо фенилфлуоренильной группы в соединении 5, соответственно;
и
(f) удаление N-защиты в соединении 5 или соединении 5*, содержащем вторую бензильную защитную группу по атому азота вместо фенилфлуоренильной группы в соединении 5, в частности, с использованием палладий-катализируемой гидрогенизации, с образованием соединения 6.
6. Способ по п.3, дополнительно включающий одну или несколько из следующих стадий:
(a) восстановление соединения 2 или соединения 2*, содержащего вторую бензильную защитную группу по атому азота вместо фенилфлуоренильной группы в соединении 2, в частности, при помощи диизобутилалюминийгидрида, с образованием соединения D или соединения D*, содержащего вторую бензильную защитную группу по атому азота вместо фенилфлуоренильной группы в соединении D, соответственно;
(b) окисление гидроксисоединения D или гидроксисоединения D*, содержащего вторую бензильную защитную группу по атому азота вместо фенилфлуоренильной группы в соединении D, в частности, с использованием окисления по методу Сверна, с образованием соединения Е или соединения Е*, содержащего вторую бензильную защитную группу по атому азота вместо фенилфлуоренильной группы в соединении Е, соответственно;
(c) преобразование соединения Е или соединения Е*, содержащего вторую бензильную защитную группу по атому азота вместо фенилфлуоренильной группы в соединении Е, в частности, в условиях реакции Виттига, с образованием соединения 4 или соединения 4*, содержащего вторую бензильную защитную группу по атому азота вместо фенилфлуоренильной группы в соединении 4, соответственно;
(d) преобразование соединения 4 или соединения 4*, содержащего вторую бензильную защитную группу по атому азота вместо фенилфлуоренильной группы в соединении 4, в частности, в условиях окисления по методу Шарплесса, с образованием соединения F или соединения F*, содержащего вторую бензильную защитную группу по атому азота вместо фенилфлуоренильной группы в соединении F, соответственно;
(e) преобразование соединения F или соединения F*, содержащего вторую бензильную защитную группу по атому азота вместо фенилфлуоренильной группы в соединении F, в частности, в условиях каталитической эстерификации, с образованием соединения 5 или соединения 5*, содержащего вторую бензильную защитную группу по атому азота вместо фенилфлуоренильной группы в соединении 5, соответственно;
и
(f) удаление N-защиты в соединении 5 или соединении 5*, содержащем вторую бензильную защитную группу по атому азота вместо фенилфлуоренильной группы в соединении 5, в частности, с использованием палладий-катализируемой гидрогенизации, с образованием соединения 6.

7. Способ по любому из пп.1-6, дополнительно включающий стадию выделения и очистки соединения 6, в частности, где соединение 6 очищают с использованием осаждения в виде гидрохлорида и/или хроматографической очистки.
8. Соединение структуры 6:
 .
.
9. Соединение по п.8, где соединение имеет чистоту больше чем 90%, в частности больше чем 95%.
10. Соединение по п.8, где соединение 6 имеет диастереомерную чистоту больше чем 70:30.
11. Набор, включающий соединение 6, в частности включающий по меньшей мере 100 мг соединения 6 и один или несколько дополнительных реагентов для синтеза аматоксинов или их предшественников.
12. Набор по п.11, где указанные один или несколько дополнительных реагентов выбраны из следующего перечня:
(i) смола, в частности смола, выбранная из следующей группы: Merrifield смола; Rink-Amid смола и ТНР-смола;
(ii) защищенный гидроксипролин, в частности флуоренилметилоксикарбонил-(Fmoc-)-защищенный О-аллил гидроксипролин (FmocHypOAll);
(iii) защищенный аспарагин, в частности Fmoc-защищенный N-тритиласпарагин (Fmoc(N-Три)AsnOH);
(iv) защищенный Cys-Trp дипептид, в частности Fmoc-защищенный Cys-Trp дипептид с -SH и -ОН защитными группами (FmocCys(S-2-((o-NO2Ph)SO2Trp-О-Аллил))ОН);
(v) защищенный глицин, в частности Fmoc-защищенный глицин (FmocGly);
(vi) защищенный изолейцин, в частности Fmoc-защищенный изолейцин (FmocIle);
(vii) агент пептидного связывания, в частности агент пептидного связывания, выбранный из следующей группы: О-(бензотриазол-1-ил)-N,N,N',N'-тетраметилуроний тетрафторборат (TBTU); бензотриазол-1-ил-окситрипирролидинофосфоний гексафторфосфат (РуВОР) и о-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфат (HATU); и
(viii) третичный амин, в частности N,N-диизопропилэтиламин (DiPEA).
13. Способ синтеза аматоксина или молекулы, являющейся его предшественником, включающий стадию (а) связывания соединения 6 с гидроксипролином, в частности, путем взаимодействия соединения 6 со смолой, содержащей предварительно нагруженный на нее гидроксипролин, в частности, путем связывания соединения 6 со свободным С-концом FmocHypOH, иммобилизованного на смоле L, в частности тетрагидропиранильной (ТНР) смоле.
14. Способ по п.13, где остальные аминокислоты затем связывают с использованием N-концевой стратегии синтеза.
15. Способ по п.14, дополнительно включающий одну или несколько из следующих стадий:
(b) повторное удаление Fmoc-N-защиты и связывание соединения G с Fmoc-(N-Три)AsnОН; FmocCys(S-2-((o-NO2Ph)SO2Trp-О-Аллил))ОН, Fmoc-GlyОН, Fmoc-IleОН, Fmoc-GlyОН с образованием соединения Н:
(c) удаление О-аллил- и Fmoc-N-защиты в соединении Н с последующей циклизацией с образованием соединения I (замыкание В-кольца):
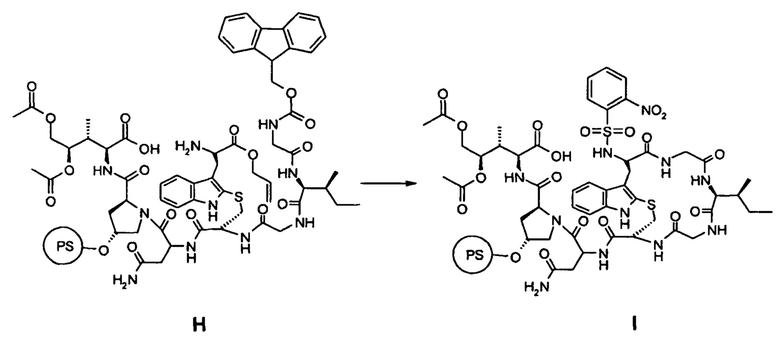
(d) удаление 2-нитроарилсульфонамид-N-защиты и отделение соединения I от смолы с образованием соединения J:
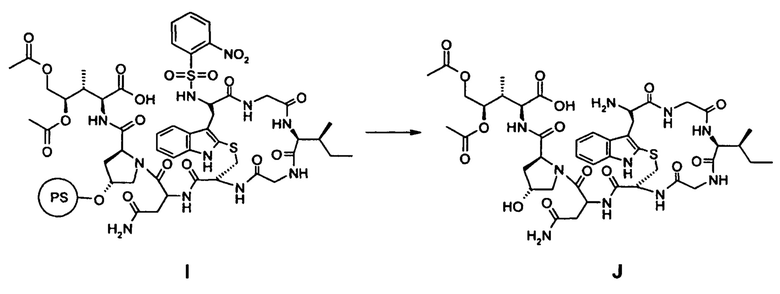
(е) циклизация в фазе раствора соединения J с образованием аманитинового производного K:
16. Способ по п.13, где аматоксин представляет собой аматоксин с дигидроксиизолейциновой группой в качестве аминокислоты 3.
| G | |||
| ZANOTTI et al | |||
| Synthesis of analogues of amaninamide, an amatoxin from the white Amanita virosa mushroom, INT | |||
| J | |||
| PEPTIDE PROTEIN RES., 1987, 30, p | |||
| Двигатель внутреннего горения | 1921 |
|
SU450A1 |
| T | |||
| WIELAND et al | |||
| Die absoluten Konfigurationen der in den Phytotoxinen enthaltenen γ -Hydroxyaminosauren und der γ -Hydroxynorvaline, LIEBIGS ANN | |||
| CHEM., 1968, vol | |||
| КОЛЬЦЕВОЙ ПОДПЯТНИК | 1923 |
|
SU717A1 |
| Автоматическая акустическая блокировка | 1921 |
|
SU205A1 |
| СПОСОБ ПОЛУЧЕНИЯ ОПТИЧЕСКИ АКТИВНЫХ ЭФИРОВ ЭРИТРО-3-АМИНО-2-ОКСИМАСЛЯНЫХ КИСЛОТ И СООТВЕТСТВУЮЩИХ ИСХОДНЫХ КИСЛОТ | 1997 |
|
RU2169138C2 |

