Область техники, к которой относится изобретение
Настоящее изобретение относится к хиназолинам, которые ингибируют киназу RIP2, и к способам их получения и их применению.
Уровень техники
Киназа взаимодействующего с рецептором белка-2 (RIP2), которую также называют CARD3, RICK, CARDIAK или RIPK2, представляет собой сериновую/треониновую протеинкиназу семейства TKL, вовлеченную во врожденный иммунный сигнальный путь. Киназа RIP2 состоит из N-терминального киназного домена и C-терминального домена рекрутирования каспазы (CARD), связанных через промежуточную (IM) область ((1998) J. Biol. Chem. 273, 12296-12300; (1998) Current Biology 8, 885-889; и (1998) J. Biol. Chem. 273, 16968-16975). CARD-домен киназы RIP2 опосредует взаимодействие с другими белками, содержащими CARD, такими как NOD1 и NOD2 ((2000) J. Biol. Chem. 275, 27823-27831 и (2000) EMBO reports 2, 736-742). NOD1 и NOD2 являются цитоплазматическими рецепторами, которые играют ключевую роль во врожденном иммунном надзоре. Они распознают как грамположительные, так и грамотрицательные бактериальные патогены и активируются особыми пептидогликановыми мотивами - диаминопимелиновой кислотой (то есть DAP) и мурамилдипептидом (MDP), соответственно ((2007) J. Immunol. 178, 2380-2386).
Вслед за активацией, киназа RIP2 связывается с NOD1 или NOD2 и, по-видимому, функционирует главным образом как молекулярный каркас, сближающий другие киназы (TAK1, IKKα/β/γ), вовлеченные в активацию NF-κB и митоген-активируемой протеинкиназы ((2006) Nature Reviews Immunology 6, 9-20). Киназа RIP2 подвергается K63-сопряженному полиубиквитинированию по лизину-209, которое облегчает рекрутирование TAK1 ((2008) EMBO Journal 27, 373-383). Данная посттрансляционная модификация необходима для передачи сигнала, так как мутация данного остатка предотвращает опосредованную NOD1/2 активацию NF-κB. Киназа RIP2 также подвергается аутофосфорилированию по серину-176 и, возможно, другим остаткам ((2006) Cellular Signaling 18, 2223-2229). Исследования с использованием мутантов (K47A) с инактивированной киназой и неселективных низкомолекулярных ингибиторов продемонстрировали, что активность киназы RIP2 важна для регуляции стабильности экспрессии киназы RIP2 и передачи сигнала ((2007) Biochem. J. 404, 179-190 и (2009) J. Biol. Chem. 284, 19183-19188).
Нарушение регуляции RIP2-зависимого сигнального пути связывали с аутовоспалительными заболеваниями. Мутации приобретения функции в NACHT-домене NOD2 вызывают синдром Блау, ранний саркоидоз, детскую гранулематозную болезнь, характеризующуюся увеитом, дерматитом и артритом ((2001) Nature Genetics 29, 19-20; (2005) Journal of Rheumatology 32, 373-375; (2005) Current Rheumatology Reports 7, 427-433; (2005) Blood 105, 1195-1197; (2005) European Journal of Human Genetics 13, 742-747; (2006) American Journal of Ophthalmology 142, 1089-1092; (2006) Arthritis & Rheumatism 54, 3337-3344; (2009) Arthritis & Rheumatism 60, 1797-1803 и (2010) Rheumatology 49, 194-196). Мутации в LRR-домене NOD2 определенно связывали с предрасположенностью к болезни Крона ((2002) Am. J. Hum. Genet. 70, 845-857; (2004) European Journal of Human Genetics 12, 206-212; (2008) Mucosal Immunology (2008) 1 (Suppl. 1), S5-S9. 1, S5-S9; (2008) Inflammatory Bowel Diseases 14, 295-302; (2008) Experimental Dermatology 17, 1057-1058; (2008) British Medical Bulletin 87, 17-30; (2009) Inflammatory Bowel Diseases 15, 1145-1154 и (2009) Microbes and Infection 11, 912-918). Мутации в NOD1 связывали с астмой ((2005) Hum. Mol. Genet. 14, 935-941) и ранним и имеющим внекишечные проявления воспалительным заболеванием кишечника ((2005) Hum. Mol. Genet. 14, 1245-1250). Генетические и функциональные исследования также указывали на роль RIP2-зависимого сигнального пути в различных других гранулематозных нарушениях, таких как саркоидоз ((2009) Journal of Clinical Immunology 29, 78-89 и (2006) Sarcoidosis Vasculitis and Diffuse Lung Diseases 23, 23-29) и гранулематоз Вегенера ((2009) Diagnostic Pathology 4, 23).
Активный, селективный, низкомолекулярный ингибитор активности киназы RIP2 мог бы блокировать RIP2-зависимый провоспалительный сигнальный путь и тем самым обеспечивать терапевтически полезный эффект в аутовоспалительных и/или аутоиммунных заболеваниях, характеризующихся повышенной и/или разрегулированной активностью киназы RIP2.
Сущность изобретения
Изобретение относится к соединению, выбираемому из:
6-(трет-бутилсульфонил)-N-(4,5-диметил-1H-пиразол-3-ил)-7-(2-метоксиэтокси)хиназолин-4-амина, имеющего формулу:
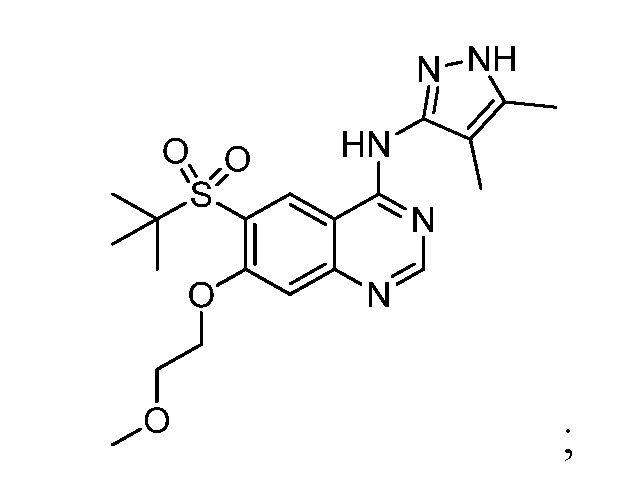
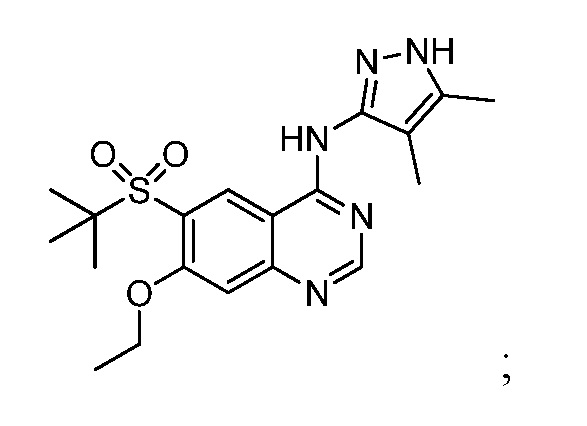
6-(трет-бутилсульфонил)-N-(4,5-диметил-1H-пиразол-3-ил)-7-этоксихиназолин-4-амина, имеющего формулу:
6-(трет-бутилсульфонил)-N-(4,5-диметил-1H-пиразол-3-ил)-7-пропоксихиназолин-4-амина, имеющего формулу:

и 6-(трет-бутилсульфонил)-N-(4,5-диметил-1H-пиразол-3-ил)-7-((тетрагидрофуран-2-ил)метокси)хиназолин-4-амина, имеющего формулу:
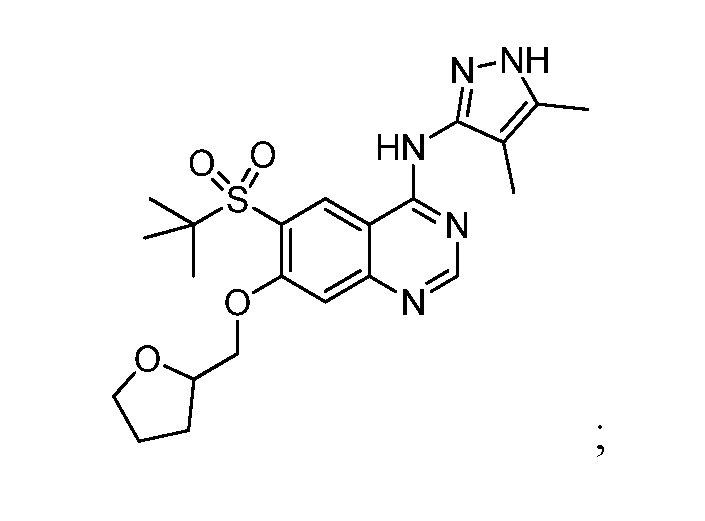
или его соли, в частности фармацевтически приемлемой соли.
Соответственно, настоящее изобретение относится к способу ингибирования киназы RIP2, где способ включает в себя приведение в контакт клетки с соединением по изобретению или с его солью, в частности фармацевтически приемлемой солью.
Изобретение дополнительно относится к способу лечения опосредованного киназой RIP2 заболевания или нарушения, который включает в себя введение терапевтически эффективного количества соединения по изобретению или его соли, в частности фармацевтически приемлемой соли, пациенту (человеку или другому млекопитающему, в частности человеку), нуждающемуся в нем. Изобретение также дополнительно относится к применению соединения по изобретению или фармацевтической композиции, содержащей соединение по изобретению, для ингибирования киназы RIP2 и/или лечения опосредованного киназой RIP2 заболевания или нарушения.
Примеры опосредованных киназой RIP2 заболеваний или нарушений включают в себя увеит, болезнь Крона, язвенный колит, раннее и имеющее внекишечные проявления воспалительное заболевание кишечника и гранулематозные нарушения, такие как саркоидоз, синдром Блау, ранний саркоидоз и гранулематоз Вегенера.
Настоящее изобретение дополнительно относится к фармацевтической композиции, содержащей соединение по изобретению или его соль, в частности фармацевтически приемлемую соль, и фармацевтически приемлемое вспомогательное вещество. В частности, данное изобретение относится к фармацевтической композиции для лечения опосредованного киназой RIP2 заболевания или нарушения, где композиция содержит соединение по изобретению или его соль, в частности фармацевтически приемлемую соль, и одно или более фармацевтически приемлемых вспомогательных веществ.
Краткое описание чертежей
На Фиг. 1 представлена порошковая рентгенодифрактограмма (PXRD) кристаллической формы 6-(трет-бутилсульфонил)-N-(4,5-диметил-1H-пиразол-3-ил)-7-этоксихиназолин-4-амина (свободное основание).
На Фиг. 2 показан объединенный IL8-цитокиновый ответ в образцах крысиной цельной крови, полученных после предварительного введения крысам доз соединения 6-(трет-бутилсульфонил)-N-(4,5-диметил-1H-пиразол-3-ил)-7-этоксихиназолин-4-амина с последующим введением доз L18-MDP.
Подробное описание изобретения
Использованные здесь термины “соединение(я) по изобретению” и “соединение(я) по данному изобретению” означают любое из соединений, определенных здесь, в любой форме, то есть в любой солевой или несолевой форме (например, в виде формы свободной кислоты или свободного основания, или в виде соли, в частности его фармацевтически приемлемой соли) и его любой физической форме (например, включая нетвердые формы (например, жидкие или полутвердые формы) и твердые формы (например, аморфные или кристаллические формы, особые полиморфные формы, сольватные формы, включая гидратные формы (например, моно-, ди- и полугидраты)), и в виде смесей различных форм (гидрат соли). А именно, будет понятно, что настоящее изобретение охватывает соединения по изобретению в виде свободного основания и в виде его солей, например в виде его фармацевтически приемлемой соли. В одном варианте осуществления изобретение относится к соединениям по изобретению в форме свободного основания. В другом варианте осуществления изобретение относится к соединениям по изобретению в форме соли, в частности фармацевтически приемлемой соли.
Также специалистам в данной области будет понятно, что пиразолильный фрагмент, присутствующий в соединениях по данному изобретению, может существовать в виде таутомерных пиразолильных изомеров, представляемых формулой (I-A) и формулой (I-B):
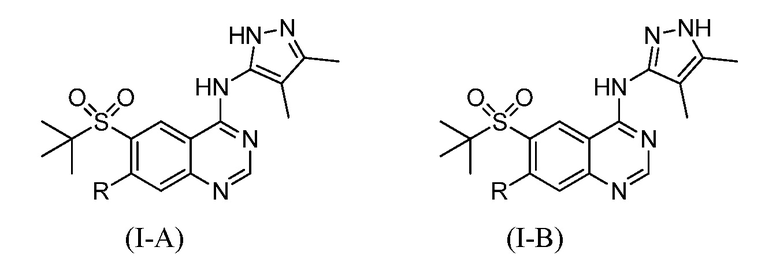
Будет понятно, что получающийся в результате пиразолильный фрагмент может быть назван либо как 3,4-диметил-1H-пиразол-5-ильный фрагмент, либо как 4,5-диметил-1H-пиразол-3-ильный фрагмент. Следует также понимать, что любая ссылка на названное соединение по данному изобретению подразумевается как охватывающая все таутомеры названного соединения и любые смеси таутомеров названного соединения. Например, название соединения 6-(трет-бутилсульфонил)-N-(4,5-диметил-1H-пиразол-3-ил)-7-этоксихиназолин-4-амин подразумевается как охватывающее соединения 6-(трет-бутилсульфонил)-N-(4,5-диметил-1H-пиразол-3-ил)-7-этоксихиназолин-4-амин и 6-(трет-бутилсульфонил)-N-(3,4-диметил-1H-пиразол-5-ил)-7-этоксихиназолин-4-амин и их смеси. Подразумевается, что все таутомерные формы описанных здесь соединений охватываются объемом настоящего изобретения.
Соединения по изобретению могут содержать один или более асимметрических центров (также именуемых хиральным центром) и, следовательно, могут существовать в виде индивидуальных энантиомеров, диастереомеров или других стереоизомерных форм, или в виде их смесей. Хиральные центры, такие как хиральный углерод, могут также присутствовать в соединениях по данному изобретению. Если стереохимия хирального центра, присутствующего в соединении по данному изобретению (например, относясь к названию соединения) или в любой проиллюстрированной здесь химической структуре, конкретно не указана, то подразумевается, что соединение, название соединения или структура охватывает все индивидуальные стереоизомеры и все их смеси. Таким образом, соединения по изобретению, содержащие один или более хиральных центров, могут присутствовать в виде рацемических смесей, энантиомерно обогащенных смесей или в виде энантиомерно чистых индивидуальных стереоизомеров.
Индивидуальные стереоизомеры соединения по изобретению, которые содержат один или более асимметрических центров, могут быть разделены способами, известными специалистам в данной области. Например, такое разделение может быть выполнено (1) формированием диастереомерных солей, комплексов или других производных; (2) селективной реакцией со специфичным к стереоизомеру реагентом, например ферментативным окислением или восстановлением; или (3) газожидкостной или жидкостной хроматографией в хиральной среде, например на хиральном носителе, таком как диоксид кремния с закрепленным хиральным лигандом, или в присутствии хирального растворителя. Квалифицированному специалисту будет понятно, что если желаемый стереоизомер превращается в другую химическую единицу в результате одной из описанных выше процедур разделения, для высвобождения желаемой формы требуется дополнительная стадия. Альтернативно, конкретные стереоизомеры могут быть синтезированы асимметрическим синтезом, используя оптически активные реагенты, субстраты, катализаторы или растворители, или путем превращения одного энантиомера в другой путем асимметрической трансформации.
Следует понимать, что твердая форма соединения по изобретению может существовать в кристаллических формах, некристаллических формах или в виде их смеси. Такие кристаллические формы также могут обнаруживать полиморфизм (то есть способность встречаться в разных кристаллических формах). Данные разные кристаллические формы обычно известны как “полиморфы”. Полиморфы имеют одинаковую химическую композицию, но отличаются упаковкой, геометрическим расположением и другими описательными свойствами кристаллического твердого состояния. Полиморфы, следовательно, могут иметь разные физические свойства, такие как форма, плотность, твердость, деформируемость, стабильность и растворимость. Полиморфы обычно обнаруживают разные температуры плавления, ИК-спектры и порошковые рентгенодифрактограммы, которые можно использовать для идентификации. Специалисту в данной области будет понятно, что разные полиморфы могут быть получены, например, изменением или регулированием условий, используемых при кристаллизации/перекристаллизации соединения.
Специалистам в данной области хорошо известно и понятно, что используемая установка, влажность, температура, ориентация кристаллов в порошке и другие параметры, вовлеченные в получение порошковой рентгенодифрактограммы (PXRD), могут вызывать некоторую вариабельность во внешнем виде, интенсивностях и положениях линий на дифрактограмме. Порошковая рентгенодифрактограмма, которая “по существу соответствует” таковой, приведенной здесь на Фигуре, представляет собой дифрактограмму PXRD, которая рассматривалась бы специалистом в данной области как представляющая соединение, обладающее той же кристаллической формой, что и соединение, которое давало дифрактограмму PXRD, показанную на Фигуре. Например, дифрактограмма PXRD может быть идентичной таковой на Фигуре 1 или, что более вероятно, она может несколько отличаться. Такая дифрактограмма PXRD необязательно может показывать каждую из линий представленных здесь дифракционных картин и/или может показывать слабое изменение во внешнем виде, интенсивности или сдвиг в положении указанных линий в результате различий в условиях, вовлеченных в получение данных. Специалист в данной области способен определить, имеет ли образец кристаллического соединения ту же форму, что и раскрытая здесь форма, или форму, отличную от нее, путем сравнения их дифрактограмм PXRD. Например, специалист в данной области может наложить дифрактограмму PXRD образца кристаллической формы соединения примера 2, то есть 6-(трет-бутилсульфонил)-N-(4,5-диметил-1H-пиразол-3-ил)-7-этоксихиназолин-4-амина (свободное основание), на дифрактограмму PXRD, представленную на Фиг. 1, и, используя навыки и знания в данной области, без труда определить, соответствует ли по существу дифрактограмма PXRD образца дифрактограмме PXRD, приведенной на Фигуре 1. Если дифрактограмма PXRD по существу соответствует таковой на Фиг. 1, то форма образца может быть без труда и точно идентифицирована как имеющая ту же форму, что и описанная здесь кристаллическая форма 6-(трет-бутилсульфонил)-N-(4,5-диметил-1H-пиразол-3-ил)-7-этоксихиназолин-4-амина (свободное основание). Подобно этому специалист в данной области способен определить, расположен ли данный дифракционный угол (выраженный в °2θ), полученный из дифрактограммы PXRD, примерно в том же положении, что и цитированное значение.
По причине их потенциального применения в медицине соли соединений по данному изобретению представляют собой предпочтительно фармацевтически приемлемые соли. Подходящие фармацевтически приемлемые соли включают соли, полученные присоединением кислоты или основания, такие как соли, описанные в статье Berge, Bighley и Monkhouse, J. Pharm. Sci. (1977) 66, стр. 1-19, и монографии “Pharmaceutical Salts: Properties, Selection, and Use”, издание 2ое, исправленное, под редакцией P.H. Stahl и C.G. Wermuth, Wiley, Hoboken, NJ, US (2011).
Термин “фармацевтически приемлемый” относится к тем соединениям, материалам, композициям и лекарственным формам, которые в рамках здравого медицинского суждения являются подходящими для применения в контакте с тканями человеческих существ и животных без избыточной токсичности, раздражения или другой проблемы или осложнения, соразмерно с разумным соотношением пользы/риска.
Термин “фармацевтически приемлемая(ые) соль(и)” относится к соединению, которое подходит для фармацевтического применения. Солевые и сольватные (например, гидраты и гидраты солей) формы соединений по изобретению, которые подходят для применения в медицине, представляют собой таковые, в которых противоион или связанный растворитель является фармацевтически приемлемым. Однако соли и сольваты, содержащие не являющиеся фармацевтически приемлемыми противоионы или связанные растворители, входят в объем настоящего изобретения, например, для применения в качестве промежуточных реагентов при получении других соединений по изобретению и их солей и сольватов.
Когда соединение по изобретению представляет собой основание (содержит основный фрагмент), желаемая солевая форма может быть получена любым подходящим способом, известным в данной области, включая обработку свободного основания кислотой. Примеры фармацевтически приемлемых солей, полученных присоединением кислоты, включают в себя ацетат, адипат, аскорбат, аспартат, бензолсульфонат, бензоат, камфорат, камфорсульфонат (камзилат), капрат (деканоат), капроат (гексаноат), каприлат (октаноат), карбонат, бикарбонат, циннамат, цитрат, цикламат, додецилсульфат (эстолат), этан-1,2-дисульфонат (эдизилат), этансульфонат (эзилат), формиат, фумарат, галактарат (мукат), гентизат (2,5-дигидроксибензоат), глюкогептонат (глюкептат), глюконат, глюкуронат, глутамат, глутарат, глицерофосфорат, гликолят, гиппурат, гидробромид, гидрохлорид, гидроиодид, изобутират, лактат, лактобионат, лаурат, малеат, малат, малонат, манделат, метансульфонат (мезилат), нафталин-1,5-дисульфонат (нападизилат), нафталинсульфонат (напзилат), никотинат, нитрат, олеат, оксалат, пальмитат, памоат, фосфат, дифосфат, пропионат, пироглутамат, салицилат, себацинат, стеарат, сукцинат, сульфат, тартрат, тиоцианат, тозилат, ундециленат, 1-гидрокси-2-нафтоат, 2,2-дихлорацетат, 2-гидроксиэтансульфонат (изетионат), 2-оксоглутарат, 4-ацетамидобензоат и 4-аминосалицилат. Не являющиеся фармацевтически приемлемыми соли, например трифторацетат, могут быть использованы, например, при выделении соединения изобретения и включены в объем данного изобретения.
Когда соединение по изобретению представляет собой кислоту (содержит кислотный фрагмент), желаемая солевая форма может быть получена любым подходящим способом, известным в данной области, включая обработку свободной кислоты неорганическим или органическим основанием. Примеры фармацевтически приемлемых солей, полученных присоединением основания, включают в себя аммонийные, литиевые, натриевые, калиевые, кальциевые, магниевые, алюминиевые соли, цинковые соли, соли триметиламина, триэтиламина, морфолина, пиридина, пиперидина, пиколина, дициклогексиламина, N,N'-дибензилэтилендиамина, 2-гидроксиэтиламина, бис(2-гидроксиэтил)амина, три(2-гидроксиэтил)амина, прокаина, дибензилпиперидина, дегидроабиетиламина, глюкамина, N-метилглюкамина, коллидина, хинина, хинолина, лизина и аргинина. В одном варианте осуществления фармацевтически приемлемая соль, полученная присоединением основания, представляет собой натриевую соль.
Определенные соединения по изобретению могут образовывать соли с одним или более эквивалентами кислоты (если соединение содержит основный фрагмент) или основания (если соединение содержит кислотный фрагмент). Настоящее изобретение включает в свой объем все возможные стехиометрические и нестехиометрические солевые формы.
Данное изобретение также предусматривает превращение одной фармацевтически приемлемой соли соединения по данному изобретению в другую фармацевтически приемлемую соль соединения по данному изобретению.
Если основное соединение выделяют в виде соли, соответствующая форма свободной кислоты или свободного основания такого соединения может быть получена любым подходящим способом, известным в данной области.
Что касается сольватов соединений по изобретению, включая сольваты солей соединений по изобретению, которые находятся в кристаллической форме, специалисту в данной области будет понятно, что могут быть сформированы фармацевтически приемлемые сольваты, в которых молекулы растворителя включаются в кристаллическую решетку в ходе кристаллизации. Сольваты могут включать в себя неводные растворители, такие как этанол, изопропанол, ДМСО, уксусная кислота, этаноламин и EtOAc, или они могут включать в себя воду в качестве растворителя, который включен в кристаллическую решетку. Сольваты, в которых вода представляет собой растворитель, который включен в кристаллическую решетку, обычно называют “гидратами”. Гидраты включают в себя стехиометрические гидраты, а также композиции, содержащие переменные количества воды. Изобретение включает в себя все такие сольваты, в частности гидраты.
Поскольку соединения по изобретению предназначены для применения в фармацевтических композициях, будет совершенно понятно, что их в каждом случае предпочтительно предоставляют в по существу чистой форме, например с чистотой по меньшей мере 60%, предпочтительнее с чистотой по меньшей мере 75% и предпочтительно с чистотой по меньшей мере 85%, в особенности с чистотой по меньшей мере 98% (% приведены по массе в расчете на массу). Соединения, полученные не в чистой форме, могут быть использованы для получения более чистых форм, используемых в фармацевтических композициях.
Общие синтетические способы
Соединения по изобретению могут быть получены с использованием синтетических методик, проиллюстрированных на приведенных ниже схемах, или опираясь на знания квалифицированного специалиста в области органической химии. Показанные на данных схемах синтезы применимы для получения соединений по изобретению, содержащих разнообразные замещающие группы, с использованием надлежащих предшественников, которые при необходимости подходящим образом защищены для того, чтобы добиться совместимости с изложенными здесь реакциями. Последующее удаление защит, где это необходимо, дает соединения раскрытой в общем виде природы. Хотя на схемах показаны соединения лишь общей формулы, они иллюстрируют способы, которые можно использовать для получения соединений по изобретению.
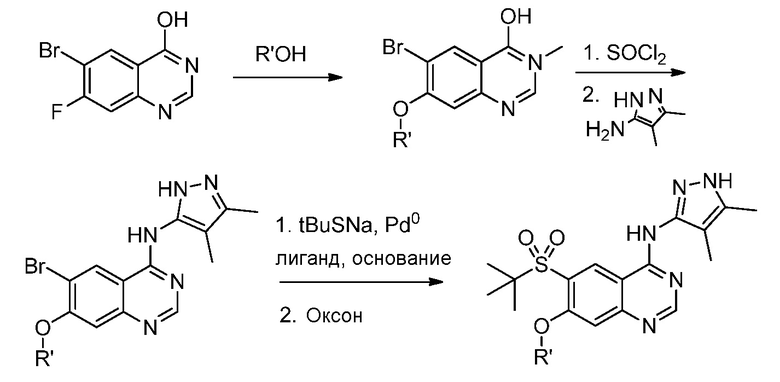
Перед введением пиразолильного фрагмента можно было бы ввести заместитель в положение C6. Катализируемое палладием сочетание тиола с 6-иодхиназолиноном может давать сульфид, который впоследствии может быть окислен до сульфона. Хлорирование с помощью POCl3 или SOCl2 может давать 4-хлорхиназолин.
Схема 1

Анилины/амины можно было бы вводить в реакцию с 4-хлорхиназолинами в основных или кислых условиях с получением 4-аминохиназолинов, которые могли бы представлять собой конечные соединения или можно было бы использовать в качестве промежуточных реагентов для дальнейшего синтеза.
Схема 2

Альтернативно, некоторые соединения по изобретению можно получать из 6-бром-7-фторхиназолин-4-ола реакцией с подходящими спиртами в присутствии основания при нагревании, что дает соответствующий 6-бром-7-алкоксихиназолин-4-ол. Последующее хлорирование и замещение аминами/анилинами будет давать 4-амино-6-бром-алкоксихиназолины. Дальнейшая реакция данных соединений с тиолами или тиолятами в присутствии подходящего сочетания палладиевого катализатора, лиганда и основания при нагревании будет давать 4-амино-6-алкилтио-7-алкоксихиназолины. Окисление будет приводить к 4-амино-6-сульфонил-7-алкоксихиназолинам, которые могут представлять собой конечные соединения или можно использовать в качестве промежуточных реагентов в дальнейших химических превращениях.
Схема 3
Получение некоторых из соединений по изобретению можно осуществлять из 7-фтор-6-сульфонил-4-хиназолинона. Синтез данного промежуточного реагента начинается с бромирования 4-фтор-2-аминобензойной кислоты с последующей конденсацией с ацетатом формамидина, проводимой in situ. Катализируемое палладием сочетание с тиолом дает сульфид, который впоследствии окисляют до сульфона.
Схема 4

Замещение фторного заместителя на алкоксильную группу может быть достигнуто обработкой соответствующим спиртом и т-бутоксидом калия.
Схема 5

Конкретным соединением по изобретению является 6-(трет-бутилсульфонил)-N-(4,5-диметил-1H-пиразол-3-ил)-7-этоксихиназолин-4-амин (в виде свободного основания). В другом варианте осуществления конкретным соединением по изобретению является 6-(трет-бутилсульфонил)-N-(4,5-диметил-1H-пиразол-3-ил)-7-этоксихиназолин-4-амин или его соль. В другом варианте осуществления конкретным соединением по изобретению является 6-(трет-бутилсульфонил)-N-(4,5-диметил-1H-пиразол-3-ил)-7-этоксихиназолин-4-амин или его фармацевтически приемлемая соль. В другом варианте осуществления конкретным соединением по изобретению является кристаллическая форма 6-(трет-бутилсульфонил)-N-(4,5-диметил-1H-пиразол-3-ил)-7-этоксихиназолин-4-амина, отличающаяся дифрактограммой PXRD, представленной на Фигуре 1. В другом варианте осуществления конкретным соединением по изобретению является кристаллическая форма 6-(трет-бутилсульфонил)-N-(4,5-диметил-1H-пиразол-3-ил)-7-этоксихиназолин-4-амина, характеризующаяся дифракционными данными, представленными в Таблице 1.
Соединения по данному изобретению являются ингибиторами киназы RIP2. Соответственно, в одном варианте осуществления изобретение относится к способу ингибирования киназы RIP2, включающему в себя приведение в контакт клетки с соединением по изобретению. В другом варианте осуществления изобретение относится к способу лечения опосредованного киназой RIP2 заболевания или нарушения, включающему в себя введение терапевтически эффективного количества соединения по изобретению человеку, нуждающемуся в нем.
В другом конкретном варианте осуществления изобретение относится к способу лечения опосредованного киназой RIP2 заболевания или нарушения, включающему в себя введение терапевтически эффективного количества 6-(трет-бутилсульфонил)-N-(4,5-диметил-1H-пиразол-3-ил)-7-этоксихиназолин-4-амина или его фармацевтически приемлемой соли человеку, нуждающемуся в нем.
Соединения по изобретению могут быть полезны, в частности, для лечения опосредованных киназой RIP2 заболеваний или нарушений, в частности заболеваний или нарушений, при которых ингибирование киназы RIP2 обеспечило бы полезный эффект. Примеры таких опосредованных киназой RIP2 заболеваний или нарушений включают в себя увеит, лихорадочный синдром (ИПФ-лихорадка), связанный с интерлейкин-1-превращающим ферментом (ИПФ, также известен как каспаза-1), дерматит, острое повреждение легких, сахарный диабет типа 2, артрит (в особенности ревматоидный артрит), воспалительные кишечные нарушения (такие как язвенный колит и болезнь Крона), раннее воспалительное заболевание кишечника, имеющее внекишечные проявления воспалительное заболевание кишечника, предотвращение ишемического реперфузионного повреждения паренхиматозных органов (в особенности почки) в ответ на ишемию, вызванную операциями на сердце, состояние после трансплантации органов, сепсис и другие поражения, заболевания печени (неалкогольный стеатогепатит, алкогольный стеатогепатит и аутоиммунный гепатит), аллергические заболевания (такие как астма), реакции на трансплантат (такие как реакция “трансплантат против хозяина”), аутоиммунные заболевания (такие как системная красная волчанка и множественный склероз) и гранулематозные нарушения (такие как саркоидоз, синдром Блау, ранний саркоидоз, гранулематоз Вегенера и интерстициальное заболевание легких).
Соединения по данному изобретению могут быть полезны, в частности, для лечения увеита, ИПФ-лихорадки, синдрома Блау, раннего саркоидоза, язвенного колита, болезни Крона, гранулематоза Вегенера и саркоидоза. Лечение опосредованных киназой RIP2 заболеваний или нарушений или, в более широком смысле, лечение иммуноопосредованных заболеваний, включая следующие, но без ограничения ими: аллергические заболевания, аутоиммунные заболевания, предотвращение отторжения трансплантата и тому подобное, может быть достигнуто с применением соединения по данному изобретению в виде монотерапии или в двухкомпонентной или многокомпонентной комбинированной терапии, в частности для лечения упорных случаев, как, например, в сочетании с другими противовоспалительными агентами и/или агентами анти-ФНО (ингибиторы фактора некроза опухолей), которые могут быть введены в терапевтически эффективных количествах, как известно в данной области.
Соединения по данному изобретению можно использовать в одиночку или в сочетании с другими терапевтическими агентами. Комбинированная терапия по настоящему изобретению включает в себя, таким образом, введение по меньшей мере одного соединения по изобретению и применение по меньшей мере одного другого терапевтически активного агента. Предпочтительно, комбинированная терапия по настоящему изобретению включает в себя введение по меньшей мере одного соединения по изобретению и по меньшей мере одного другого терапевтически активного агента. Соединение(я) по изобретению и другой(ие) терапевтически активный(е) агент(ы) можно вводить совместно в единственной фармацевтической композиции или по отдельности, и при введении по отдельности введение может происходить одновременно или последовательно в любом порядке. Количества соединения(й) по изобретению и другого(их) терапевтически активного(ых) агента(ов) и относительные времена введения будут выбираться в целях достижения желаемого комбинированного терапевтического эффекта. Таким образом, в дополнительном аспекте предоставляется сочетание, содержащее соединение по изобретению вместе с одним или более другими терапевтически активными агентами. В дополнительном аспекте предоставляется сочетание, содержащее 6-(трет-бутилсульфонил)-N-(4,5-диметил-1H-пиразол-3-ил)-7-этоксихиназолин-4-амин или его фармацевтически приемлемую соль вместе с одним или более другими терапевтически активными агентами.
Таким образом, в одном аспекте данного изобретения соединение по изобретению и фармацевтические композиции, содержащие соединение по данному изобретению, могут быть использованы в сочетании с одним или более другими терапевтическими агентами или включать в себя один или более других терапевтических агентов, например противовоспалительный агент и/или агент анти-ФНО.
Соединения по данному изобретению могут быть введены в сочетании с кортикостероидами и/или агентами анти-ФНО для лечения синдрома Блау, раннего саркоидоза; или в сочетании с биопрепаратами анти-ФНО или другими противовоспалительными биопрепаратами для лечения болезни Крона; или в сочетании с 5-АСК (месаламином) или сульфасалазином для лечения язвенного колита; или в сочетании с низкодозовыми кортикостероидами и/или метотрексатом для лечения гранулематоза Вегенера или саркоидоза или интерстициального заболевания легких; или в сочетании с биопрепаратом (например, анти-ФНО, анти-ИЛ-6 и так далее) для лечения ревматоидного артрита; или в сочетании с анти-ИЛ-6 и/или метотрексатом для лечения ИПФ-лихорадки.
Примеры подходящих противовоспалительных агентов включают в себя 5-аминосалициловую кислоту и препараты месаламина, сульфасалазин, гидроксихлорохин, тиопурины (азатиоприн, меркаптопурин), метотрексат, циклофосфамид, циклоспорин, ингибиторы JAK (тофацитиниб), кортикостероиды, в частности низкодозовые кортикостероиды (такие как преднизон (Deltasone®) и бундесонид) и противовоспалительные биопрепараты, такие как моноклональные антитела анти-ИЛ6Р (Actemra® (тоцилизумаб)), биопрепараты анти-ИЛ6, биопрепараты анти-ИЛ1 или анти-ИЛ12 или анти-ИЛ23 (устекинумаб (Stelara®)), антиинтегриновые агенты (натализумаб (Tysabri®)), моноклональные антитела анти-CD20 (ритуксимаб (Rituxan®) и офатумумаб (Arzerra®)) и другие агенты, такие как абатацепт (Orencia®), анакинра (Kineret®) и белимумаб (Benlysta®), биопрепараты CD4 и другие ингибиторы цитокинов или биопрепараты, активные по отношению к рецепторам T-клеток или B-клеток или интерлейкинам. Примеры подходящих агентов анти-ФНО включают в себя биопрепараты анти-ФНО, такие как Enbrel® (этанерцепт), Humira® (адалимумаб), Remicade® (инфликсимаб), Cimzia® (цертолизумаб) и Simponi® (голимумаб).
Другие примеры подходящих противовоспалительных агентов включают в себя 5-аминосалициловую кислоту и препараты месаламина, сульфасалазин, гидроксихлорохин, тиопурины (азатиоприн, меркаптопурин), метотрексат, циклофосфамид, циклоспорин, ингибиторы кальциневрина (циклоспорин, пимекролимус, такролимус), микофеноловую кислоту (CellCept®), ингибиторы mTOR (темсиролимус, эверолимус), ингибиторы JAK (тофацитиниб (Xeljan®)), ингибиторы Syk (фостаматиниб), кортикостероиды, в частности низкодозовые кортикостероиды (такие как преднизон (Deltasone®) и бундесонид) и противовоспалительные биопрепараты, такие как моноклональные антитела анти-ИЛ6Р (Actemra® (тоцилизумаб)), биопрепараты анти-ИЛ6, биопрепараты анти-ИЛ1 (анакинра (Kineret®), канакинумаб (Ilaris®), рилонацепт (Arcalyst®)), анти-ИЛ12 или анти-ИЛ23 (устекинумаб (Stelara®)), биопрепараты анти-ИЛ17 (секукинумаб), анти-CD22 (эпратузумаб), антиинтегриновые агенты (натализумаб (Tysabri®)), ведолизумаб (Entyvio®), анти-IFNa (сифалимумаб), моноклональные антитела анти-CD20 (ритуксимаб (Rituxan®) и офатумумаб (Arzerra®)) и другие агенты, такие как абатацепт (Orencia®), анакинра (Kineret®), канакинумаб (Ilaris®), рилонацепт (Arcalyst®), секукинумаб, эпратузумаб, сифалимумаб и белимумаб (Benlysta®), биопрепараты CD4 и другие ингибиторы цитокинов или биопрепараты, активные по отношению к рецепторам T-клеток или B-клеток или интерлейкинам. Примеры подходящих агентов анти-ФНО включают в себя биопрепараты анти-ФНО, такие как Enbrel® (этанерцепт), Humira® (адалимумаб), Remicade® (инфликсимаб), Cimzia® (цертолизумаб) и Simponi® (голимумаб). Данное изобретение также предоставляет соединение по изобретению для применения в терапии. А именно, данное изобретение предоставляет описанные здесь соединения или их фармацевтически приемлемую соль для применения в терапии. Конкретнее, данное изобретение предоставляет 6-(трет-бутилсульфонил)-N-(4,5-диметил-1H-пиразол-3-ил)-7-этоксихиназолин-4-амин или его фармацевтически приемлемую соль для применения в терапии.
В другом варианте осуществления данное изобретение предоставляет соединение по изобретению для применения в лечении опосредованного киназой RIP2 заболевания или нарушения. А именно, данное изобретение предоставляет описанные здесь соединения или их фармацевтически приемлемую соль для применения в лечении опосредованного киназой RIP2 заболевания или нарушения.
В другом варианте осуществления данное изобретение предоставляет описанные здесь соединения или их фармацевтически приемлемую соль для применения в лечении увеита, лихорадочного синдрома, связанного с интерлейкин-1-превращающим ферментом, дерматита, острого повреждения легких, сахарного диабета типа 2, артрита (в особенности ревматоидного артрита), воспалительных кишечных нарушений (таких как язвенный колит и болезнь Крона), раннего воспалительного заболевания кишечника, имеющего внекишечные проявления воспалительного заболевания кишечника, для предотвращения ишемического реперфузионного повреждения паренхиматозных органов (в особенности почки) в ответ на ишемию, вызванную операциями на сердце, в лечении состояния после трансплантации органов, сепсиса и других поражений, заболеваний печени (неалкогольного стеатогепатита, алкогольного стеатогепатита и аутоиммунного гепатита), аллергических заболеваний (таких как астма), реакций на трансплантат (таких как реакция “трансплантат против хозяина”), аутоиммунных заболеваний (таких как системная красная волчанка и множественный склероз) и гранулематозных нарушений (таких как саркоидоз, синдром Блау, ранний саркоидоз, гранулематоз Вегенера и интерстициальное заболевание легких).
В другом варианте осуществления данное изобретение предоставляет описанные здесь соединения или их фармацевтически приемлемую соль для применения в лечении увеита. В другом варианте осуществления данное изобретение предоставляет описанные здесь соединения или их фармацевтически приемлемую соль для применения в лечении лихорадочного синдрома, связанного с интерлейкин-1-превращающим ферментом. В другом варианте осуществления данное изобретение предоставляет описанные здесь соединения или их фармацевтически приемлемую соль для применения в лечении синдрома Блау. В другом варианте осуществления данное изобретение предоставляет описанные здесь соединения или их фармацевтически приемлемую соль для применения в лечении раннего саркоидоза. В другом варианте осуществления данное изобретение предоставляет описанные здесь соединения или их фармацевтически приемлемую соль для применения в лечении язвенного колита. В другом варианте осуществления данное изобретение предоставляет описанные здесь соединения или их фармацевтически приемлемую соль для применения в лечении болезни Крона. В другом варианте осуществления данное изобретение предоставляет описанные здесь соединения или их фармацевтически приемлемую соль для применения в лечении раннего воспалительного заболевания кишечника. В другом варианте осуществления данное изобретение предоставляет описанные здесь соединения или их фармацевтически приемлемую соль для применения в лечении имеющего внекишечные проявления воспалительного заболевания кишечника. В другом варианте осуществления данное изобретение предоставляет описанные здесь соединения или их фармацевтически приемлемую соль для применения в лечении гранулематоза Вегенера. В другом варианте осуществления данное изобретение предоставляет описанные здесь соединения или их фармацевтически приемлемую соль для применения в лечении саркоидоза.
Изобретение также предусматривает применение соединения по изобретению в производстве медикамента для применения в лечении опосредованного киназой RIP2 заболевания или нарушения, например, каждого из заболеваний и нарушений, перечисленных здесь. А именно, данное изобретение предусматривает применение описанных здесь соединений или их фармацевтически приемлемой соли в производстве медикамента для лечения опосредованного киназой RIP2 заболевания или нарушения. Конкретнее, данное изобретение предусматривает применение 6-(трет-бутилсульфонил)-N-(4,5-диметил-1H-пиразол-3-ил)-7-этоксихиназолин-4-амина или его фармацевтически приемлемой соли в производстве медикамента для лечения опосредованного киназой RIP2 заболевания или нарушения.
Соответственно, изобретение предусматривает применение описанных здесь соединений или их фармацевтической соли в производстве медикамента для применения в лечении человека, нуждающегося в нем, имеющего заболевание или нарушение, опосредованное киназой RIP2. Подразумевается, что терапевтически “эффективное количество” означает такое количество соединения, которое при введении пациенту, нуждающемуся в таком лечении, является достаточным для осуществления лечения, как определено здесь. Таким образом, например, терапевтически эффективное количество соединения по изобретению или его фармацевтически приемлемой соли представляет собой количество агента по изобретению, которое при введении человеку, нуждающемуся в нем, является достаточным для модуляции или ингибирования активности киназы RIP2, так что патологическое состояние, которое опосредовано такой активностью, ослабляется, облегчается или предотвращается. Количество данного соединения, которое будет соответствовать такому количеству, будет изменяться в зависимости от таких факторов, как конкретное соединение (например, активность (pIC50), эффективность (EC50) и биологический период полувыведения конкретного соединения), патологическое состояние и его тяжесть, личность (например, возраст, рост и масса) пациента, нуждающегося в лечении, но, тем не менее, может быть определено установленным образом специалистом в данной области. Аналогично, продолжительность лечения и период времени введения (период времени между введениями доз и время введения доз, например до/во время/после еды) соединения будут изменяться в зависимости от индивидуальных характеристик млекопитающего (например, массы), нуждающегося в лечении, конкретного соединения и его свойств (например, фармацевтических характеристик), заболевания или нарушения и его тяжести и конкретной композиции и используемого способа лечения, но, тем не менее, могут быть определены специалистом в данной области.
Подразумевается, что “осуществление лечения” или “лечение” означает, по меньшей мере, смягчение заболевания или нарушения, наблюдающегося у пациента. Способы лечения для смягчения заболевания или нарушения включают в себя применение соединений, описанных в данном изобретении, любым общепринятым образом, например для предотвращения, замедления, профилактики, терапии или лечения опосредованного заболевания или нарушения. Конкретные заболевания и нарушения, которые могут быть в особенности чувствительны к лечению с использованием соединения по данному изобретению, описаны здесь.
Соединения по изобретению могут быть введены любым подходящим путем введения, включая как системное введение, так и местное введение. Системное введение включает в себя пероральное введение, парентеральное введение, трансдермальное введение, ректальное введение и введение ингаляцией. Парентеральное введение относится к путям введения, отличным от энтерального, трансдермального или посредством ингаляции, и обычно осуществляется инъекцией или инфузией. Парентеральное введение включает в себя внутривенную, внутримышечную и подкожную инъекцию или инфузию. Ингаляция относится к введению в легкие пациента, осуществляемому через рот или через носовые проходы. Местное введение включает в себя нанесение на кожу.
Соединения изобретения можно вводить один раз или согласно режиму дозирования, в котором ряд доз вводят в разные интервалы времени в течение заданного периода времени. Например, дозы можно вводить один, два, три или четыре раза в день. Дозы можно вводить до достижения желаемого терапевтического эффекта или в течение неопределенного периода времени для поддержания желаемого терапевтического эффекта. Подходящие режимы дозирования для соединения изобретения зависят от фармакокинетических свойств такого соединения, таких как абсорбция, распределение и период полувыведения, которые может определить специалист в данной области. Кроме того, подходящие режимы дозирования, включая продолжительность таких режимов, для соединения по изобретению зависят от подвергаемого лечению заболевания или нарушения, тяжести подвергаемого лечению заболевания или нарушения, возраста и физического состояния подвергаемого лечению пациента, истории болезни подвергаемого лечению пациента, природы сопутствующей терапии, желаемого терапевтического эффекта и тому подобных факторов в пределах знаний и опыта специалиста в данной области. Кроме того, специалистам в данной области будет понятно, что подходящие режимы дозирования могут требовать корректирования, принимая во внимание ответную реакцию пациента на режим дозирования, или с течением времени по мере изменения потребностей пациента.
Для применения в терапии из соединения по изобретению будут обычно, но не обязательно, составлять фармацевтическую композицию перед введением пациенту. Соответственно, изобретение также относится к фармацевтическим композициям, содержащим соединение по изобретению и одно или более фармацевтически приемлемых вспомогательных веществ.
В одном варианте осуществления предоставлена фармацевтическая композиция, содержащая 6-(трет-бутилсульфонил)-N-(4,5-диметил-1H-пиразол-3-ил)-7-этоксихиназолин-4-амин или его фармацевтически приемлемую соль и одно или более фармацевтических приемлемых вспомогательных веществ. В другом варианте осуществления предоставлена фармацевтическая композиция, содержащая 6-(трет-бутилсульфонил)-N-(4,5-диметил-1H-пиразол-3-ил)-7-этоксихиназолин-4-амин (в виде свободного основания) и одно или более фармацевтических приемлемых вспомогательных веществ. В другом варианте осуществления предоставлена фармацевтическая композиция, содержащая кристаллическую форму 6-(трет-бутилсульфонил)-N-(4,5-диметил-1H-пиразол-3-ил)-7-этоксихиназолин-4-амина, характеризующуюся дифрактограммой PXRD, представленной на Фигуре 1, и одно или более фармацевтических приемлемых вспомогательных веществ. Фармацевтические композиции по изобретению могут быть получены и упакованы в нефасованной форме, из которой эффективное количество соединения изобретения может быть отобрано, а затем дано пациенту, как, например, в порошках, сиропах и растворов для инъекции. Альтернативно, фармацевтические композиции по изобретению могут быть получены и упакованы в виде единичных лекарственных форм. Например, в случае перорального введения можно вводить одну или более таблеток или капсул. Доза фармацевтической композиции содержит, по меньшей мере, терапевтически эффективное количество соединения по данному изобретению. При получении в виде единичной лекарственной формы фармацевтические композиции могут содержать от 1 мг до 1000 мг соединения по данному изобретению.
Предусмотренные здесь единичные лекарственные формы (фармацевтические композиции), содержащие от 1 мг до 1000 мг соединения по изобретению, можно вводить один, два, три или четыре раза в день, предпочтительно один, два или три раза в день и предпочтительнее один или два раза в день для проведения лечения опосредованного киназой RIP2 заболевания или нарушения.
Фармацевтические композиции изобретения обычно содержат одно соединение по изобретению. Однако в определенных вариантах осуществления фармацевтические композиции по изобретению содержат более одного соединения по изобретению. Кроме того, фармацевтические композиции по изобретению необязательно могут дополнительно содержать одно или более дополнительных фармацевтически активных соединений.
В контексте данного изобретения “фармацевтически приемлемое вспомогательное вещество” означает материал, композицию или наполнитель, участвующие в придании композиции формы или консистенции. Каждое вспомогательное вещество должно быть совместимо с другими ингредиентами фармацевтической композиции при смешении так, чтобы избежать взаимодействий, которые существенно снижали бы эффективность соединения по изобретению при введении пациенту, и взаимодействий, которые приводили бы к фармацевтическим композициям, которые не являются фармацевтически приемлемыми. Кроме того, каждое вспомогательное вещество должно иметь, несомненно, достаточно высокую чистоту, сообщающую ему фармацевтическую приемлемость.
Из соединений по изобретению и фармацевтически приемлемого вспомогательного вещества или вспомогательных веществ будут обычно составлять лекарственную форму, выполненную с возможностью введения пациенту желаемым путем введения. Общепринятые лекарственные формы включают в себя таковые, выполненные с возможностью (1) перорального введения, такие как таблетки, капсулы, покрытые таблетки, пилюли, пастилки, порошки, сиропы, эликсиры, суспензии, растворы, эмульсии, саше и облатки; (2) парентерального введения, такие как стерильные растворы, суспензии и порошки для растворения; (3) трансдермального введения, такие как трансдермальные пластыри; (4) ректального введения, такие как суппозитории; (5) ингаляции, такие как аэрозоли и растворы; и (6) местного введения, такие как крема, мази, лосьоны, растворы, пасты, спреи, пены и гели.
Подходящие фармацевтически приемлемые вспомогательные вещества будут изменяться в зависимости от конкретной выбранной лекарственной формы. Кроме того, подходящие фармацевтически приемлемые вспомогательные вещества могут быть выбраны для конкретной функции, которой они могут служить в композиции. Например, определенные фармацевтически приемлемые вспомогательные вещества могут быть выбраны в связи с их способностью облегчать получение однородных лекарственных форм. Определенные фармацевтически приемлемые вспомогательные вещества могут быть выбраны в связи с их способностью облегчать получение стабильных лекарственных форм. Определенные фармацевтически приемлемые вспомогательные вещества могут быть выбраны в связи с их способностью облегчать перенос или транспорт соединения или соединений по изобретению после введении пациенту из одного органа или одной части тела в другой орган или другую часть тела. Определенные фармацевтически приемлемые вспомогательные вещества могут быть выбраны в связи с их способностью улучшать соблюдение пациентом режима лечения.
Подходящие фармацевтически приемлемые вспомогательные вещества включают в себя следующие типы вспомогательных веществ: разбавители, наполнители, связующие, разрыхлители, смазывающие вещества, глиданты, агенты для гранулирования, покрывающие агенты, смачивающие агенты, растворители, сорастворители, суспендирующие агенты, эмульгаторы, подсластители, ароматизаторы, агенты маскирования запахов, красители, противослеживатели, увлажнители, хелатирующие агенты, пластификаторы, агенты увеличения вязкости, антиоксиданты, консерванты, стабилизаторы, поверхностно-активные вещества и буферные агенты. Специалисту в данной области будет понятно, что определенные фармацевтически приемлемые вспомогательные вещества могут выполнять более одной функции и могут выполнять альтернативные функции в зависимости от того, какое количество вспомогательного вещества присутствует в рецептуре и какие другие ингредиенты присутствуют в рецептуре.
Квалифицированные специалисты обладают знаниями и навыками в данной области, позволяющими им выбрать подходящие фармацевтически приемлемые вспомогательные вещества в надлежащих количествах для применения в изобретении. Кроме того, существует ряд источников, которые доступны специалисту в данной области, в которых описаны фармацевтически приемлемые вспомогательные вещества, и такие источники могут быть полезны при выборе подходящих фармацевтически приемлемых вспомогательных веществ. Примеры включают в себя Remington’s Pharmaceutical Sciences (Mack Publishing Company), The Handbook of Pharmaceutical Additives (Gower Publishing Limited) и The Handbook of Pharmaceutical Excipients (the American Pharmaceutical Association and the Pharmaceutical Press).
Фармацевтические композиции по изобретению получают, применяя методики и способы, известные специалистам в данной области. Некоторые из способов, обычно применяемых в данной области, описаны в Remington’s Pharmaceutical Sciences (Mack Publishing Company).
В одном аспекте изобретение относится к твердой пероральной лекарственной форме, такой как таблетка или капсула, содержащей эффективное количество соединения по изобретению и разбавитель или наполнитель. Подходящие разбавители и наполнители включают в себя лактозу, сахарозу, декстрозу, маннит, сорбит, крахмал (например, кукурузный крахмал, картофельный крахмал и предварительно клейстеризованный крахмал), целлюлозу и ее производные (например, микрокристаллическую целлюлозу), сульфат кальция и гидрофосфат кальция. Пероральная твердая лекарственная форма может дополнительно содержать связующее. Подходящие связующие включают в себя крахмал (например, кукурузный крахмал, картофельный крахмал и предварительно клейстеризованный крахмал), желатин, камедь акации, альгинат натрия, альгиновую кислоту, трагакант, гуаровую камедь, повидон, целлюлозу и ее производные (например, микрокристаллическую целлюлозу). Пероральная твердая лекарственная форма может дополнительно содержать разрыхлитель. Подходящие разрыхлители включают в себя кросповидон, натриевый гликолят крахмала, кроскармеллозу, альгиновую кислоту и натриевую карбоксиметилцеллюлозу. Пероральная твердая лекарственная форма может дополнительно содержать смазывающее вещество. Подходящие смазывающие вещества включают в себя стеариновую кислоту, стеарат магния, стеарат кальция и тальк.
Примеры
Следующие примеры иллюстрируют изобретение. Подразумевается, что данные примеры не ограничивают объем настоящего изобретения, но скорее предназначены служить для специалиста в данной области руководством по получению и применению соединений, композиций и способов по настоящему изобретению. Хотя описаны конкретные варианты осуществления настоящего изобретения, специалисту в данной области будет понятно, что в пределах сущности и объема изобретения могут быть произведены различные изменения и модификации.
Изобретение также включает в себя различные дейтерированные формы соединений по изобретению. Любой доступный атом водорода, соединенный с атомом углерода, может быть независимым образом заменен на атом дейтерия. Специалисту обычной квалификации в данной области будет известно, как синтезировать дейтерированные формы соединений по изобретению.
Названия описанных здесь промежуточных и конечных соединений генерировали, используя программное обеспечение для создания названий ACD/Name Pro V6.02, доступное от Advanced Chemistry Development, Inc., 110 Yonge Street, 14th Floor, Торонто, Онтарио, Канада, M5C 1T4 (http://www.acdlabs.com/), или программу для создания названий, входящую в ChemDraw, Struct=Name Pro 12.0, как часть ChemBioDraw Ultra, доступную от CambridgeSoft, 100 CambridgePark Drive, Кембридж, Массачусетс, 02140 США (www.cambridgesoft.com).
В следующих описаниях экспериментов могут быть использованы следующие сокращения:
Пример получения 1
6-(трет-Бутилсульфонил)-7-фторхиназолин-4-ол
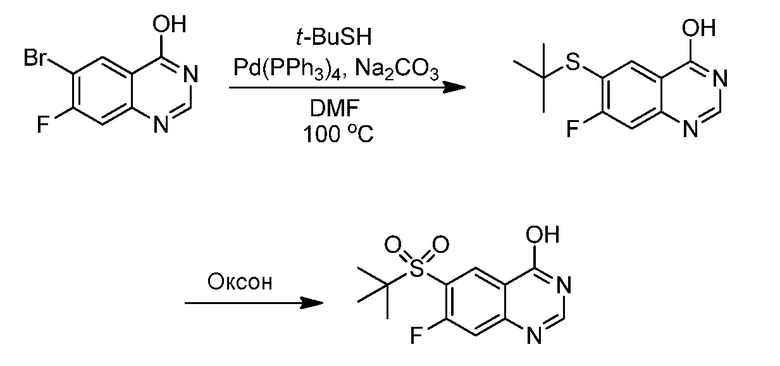
Стадия 1: 6-(трет-бутилтио)-7-фторхиназолин-4-ол: Смесь 6-бром-7-фторхиназолин-4-ола (69 г, 285 ммоль), тетракис(трифенилфосфин)палладия(0) (20 г, 17 ммоль) и карбоната натрия (60 г, 570 ммоль) перемешивали в DMF (1 л), продувая газообразным азотом в течение 5 минут. Прибавляли 2-метилпропан-2-тиол (64 мл, 570 ммоль) и реакционную смесь нагревали с обратным холодильником при 100°C в течение 6 часов. Реакционную смесь охлаждали и фильтровали через фильтровальную бумагу, а затем медленно вливали в 1500 мл перемешиваемой воды. Образовавшийся красный осадок фильтровали и затирали с 200 мл EtOAc. Твердое вещество фильтровали и последовательно промывали 110 мл смеси гексанов, 150 мл смеси, состоящей из смеси гексанов:EtOAc в соотношении 90:10, что давало 6-(трет-бутилтио)-7-фторхиназолин-4-ол (44,5 г, выход 61,9%) в виде желтовато-коричневого твердого вещества. LC/MS: M+H=253,2. 1H ЯМР (400 МГц, DMSO-d6) δ м.д. 12,23-12,72 (м, 1H), 8,24 (д, J=8,1 Гц, 1H), 8,19 (с, 1H), 7,58 (д, J=9,6 Гц, 1H), 1,28 (с, 9H).
Стадия 2: 6-(трет-бутилсульфонил)-7-фторхиназолин-4-ол: Суспензию 6-(трет-бутилтио)-7-фторхиназолин-4-ола (45 г, 124 ммоль) и оксона (191 г, 311 ммоль) в этилацетате (1220 мл), метаноле (1220 мл) и воде (1220 мл) перемешивали в течение 4 ч при 25°C, после чего добавили дополнительно 28 г (всего 2,8 экв.) оксона. Реакционную смесь перемешивали механической мешалкой в течение 12 ч. Реакционную смесь фильтровали и фильтрат медленно подщелачивали насыщенным водным раствором бикарбоната натрия, затем твердым бикарбонатом натрия, до pH~7,5. Смесь экстрагировали дополнительным 1,25 л EtOAc, а затем 500 мл EtOAc. Объединенные органические растворы промывали рассолом, затем сушили над MgSO4, фильтровали и концентрировали в вакууме. Небольшую примесь удаляли растиранием с 200 мл EtOAc. Желаемый 6-(трет-бутилсульфонил)-7-фторхиназолин-4-ол (33,2 г, выход 94%) отфильтровывали в виде желтого твердого вещества. LC/MS: M+H=285,2. 1H ЯМР (400 МГц, DMSO-d6) δ м.д. 12,48-13,03 (м, уш. с, 1H), 8,47 (д, J=7,8 Гц, 1H), 8,32 (с, 1H), 7,73 (д, J=11,1 Гц, 1H), 1,17-1,40 (с, 9H).
Пример получения 2
6-(трет-Бутилсульфонил)-N-(4,5-диметил-1H-пиразол-3-ил)-7-фторхиназолин-4-амин

К раствору 6-(трет-бутилсульфонил)-7-фторхиназолин-4-ола (4,14 г, 14,56 ммоль) в ацетонитриле (42,7 мл) прибавляли POCl3 (2,036 мл, 21,84 ммоль) и DIEA (3,81 мл, 21,84 ммоль). Реакционную смесь нагревали при 80°C в течение ночи в течение 16 ч. Добавляли дополнительное количество POCl3 (500 мкл) и реакционную смесь перемешивали при 80°C в течение 18 ч. Полное превращение в хлорид наблюдали посредством LCMS. Добавляли 4,5-диметил-1H-пиразол-3-амин (1,942 г, 17,47 ммоль) и реакционную смесь перемешивали в течение 1 ч при 80°C. Осадок отфильтровывали, промывали ацетонитрилом и сушили, что давало гидрохлорид 6-(трет-бутилсульфонил)-N-(4,5-диметил-1H-пиразол-3-ил)-7-фторхиназолин-4-амина (4,15 г, 9,93 ммоль, выход 68,2%). 1H ЯМР (400 МГц, DMSO-d6) δ м.д. 9,10-9,44 (м, 1H), 8,88 (уш. с, 1H), 7,94 (д, J=10,36 Гц, 1H), 2,23 (с, 3H), 1,82 (с, 3H), 1,24-1,45 (м, 9H). MS (m/z) 378 (M+H)+.
Пример 1
6-(трет-Бутилсульфонил)-N-(4,5-диметил-1H-пиразол-3-ил)-7-(2-метоксиэтокси)хиназолин-4-амин
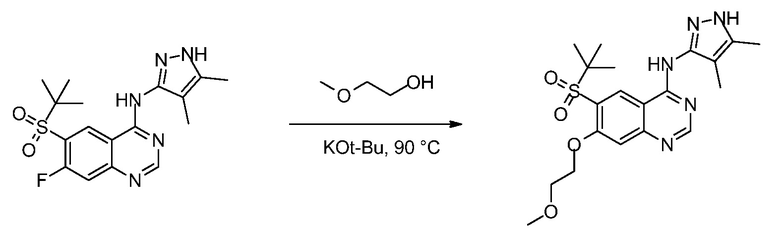
Смесь гидрохлорида 6-(трет-бутилсульфонил)-N-(4,5-диметил-1H-пиразол-3-ил)-7-фторхиназолин-4-амина (300 мг, 0,73 ммоль), 2-метоксиэтанола (5,7 мл, 73 ммоль) и KOtBu (410 мг, 3,6 ммоль) нагревали при 90°C в течение 4 д. Реакционную смесь концентрировали досуха, наносили в сухом виде на силикагель и очищали колоночной хроматографией (ISCO-Rf, 0-25% метанола (с 1% NH4OH)/этилацетат), что давало 6-(трет-бутилсульфонил)-N-(4,5-диметил-1H-пиразол-3-ил)-7-(2-метоксиэтокси)хиназолин-4-амин (230 мг, 0,531 ммоль, выход 73,2%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ м.д. 12,19 (с, 1H), 10,36 (с, 1H), 8,99 (с, 1H), 8,45 (с, 1H), 7,34 (с, 1H), 4,26-4,42 (м, 2H), 3,73 (т, J=4,4 Гц, 2H), 3,34 (с, 3H), 2,18 (с, 3H), 1,74 (с, 3H), 1,33 (с, 9H). MS (m/z) 434.
Пример 2
6-(трет-Бутилсульфонил)-N-(4,5-диметил-1H-пиразол-3-ил)-7-этоксихиназолин-4-амин

Стадия 1: 6-(трет-бутилсульфонил)-N-(4,5-диметил-1H-пиразол-3-ил)-7-фторхиназолин-4-амин: К суспензии 6-(трет-бутилсульфонил)-7-фторхиназолин-4-ола (5,50 г, 19,35 ммоль) в ацетонитриле (48 мл) прибавляли POCl3 (2,70 мл, 29,0 ммоль) и TEA (4,0 мл, 29 ммоль). Реакционную смесь перемешивали при 80°C в течение ночи. К раствору добавляли 4,5-диметил-1H-пиразол-3-амин (2,58 г, 23,2 ммоль) и реакционную смесь продолжили перемешивать при 80°C в течение 1 ч. Начинало осаждаться твердое вещество. Реакционной смеси позволяли охладиться до комнатной температуры. Твердое вещество отфильтровывали и промывали холодным ацетонитрилом. Твердое вещество сушили в вакуумной печи, что давало гидрохлорид 6-(трет-бутилсульфонил)-N-(4,5-диметил-1H-пиразол-3-ил)-7-фторхиназолин-4-амина (4,91 г, 11,86 ммоль, выход 61,3%). (M+H)+ 378,2.
Стадия 2: 6-(трет-бутилсульфонил)-N-(4,5-диметил-1H-пиразол-3-ил)-7-этоксихиназолин-4-амин: Смешивали этоксид натрия (24 мл, 65,6 ммоль, 21% в EtOH) и гидрохлорид 6-(трет-бутилсульфонил)-N-(4,5-диметил-1H-пиразол-3-ил)-7-фторхиназолин-4-амина (4,80 г, 11,60 ммоль) и суспензию нагревали при 80°C в течение 2 часов. Реакционной смеси позволяли охладиться до комнатной температуры. EtOH упаривали, а остаток растворяли в 25 мл воды. Раствор нейтрализовали до pH~9 прибавлением 1 н. HCl. Осаждалось светло-желтое твердое вещество. Твердое вещество отфильтровывали, промывали водой и сушили в вакуумной печи в течение ночи, что давало 6-(трет-бутилсульфонил)-N-(4,5-диметил-1H-пиразол-3-ил)-7-этоксихиназолин-4-амин (3,90 г, 9,67 ммоль, выход 83%). (M+H)+ 404,1; 1H ЯМР (DMSO-d6, 400 МГц): δ = 12,20 (с, 1H), 10,36 (с, 1H), 8,99 (с, 1H), 8,46 (с, 1H), 7,30 (с, 1H), 4,13-4,34 (м, 2H), 2,18 (с, 3H), 1,74 (с, 3H), 1,40 (т, J=6,9 Гц, 3H), 1,33 (с, 9H) м.д.
Образец 6-(трет-бутилсульфонил)-N-(4,5-диметил-1H-пиразол-3-ил)-7-этоксихиназолин-4-амина (120 г) суспендировали в EtOH (2000 мл), затем нагревали до 70°C. Прибавляли дополнительное количество EtOH (2000 мл) и полученную смесь нагревали до кипения с обратным холодильником. Большая часть твердого вещества растворялась в растворителе. Горячую суспензию фильтровали, и раствор вливали в 12 л холодной воды. Данную смесь перемешивали в течение приблизительно 60 мин, затем позволяли осесть в течение ночи по мере нагревания бани до RT. Светло-желтый осадок отделяли фильтрованием и сушили в вакуумной печи, что давало 105,9 г (261 ммоль, извлечение 88%) кристаллического 6-(трет-бутилсульфонил)-N-(4,5-диметил-1H-пиразол-3-ил)-7-этоксихиназолин-4-амина, который характеризуется дифрактограммой PXRD, представленной на Фигуре 1, и дифракционными данными, приведенными в Таблице 1.
Анализ PXRD проводили на настольном рентгеновском дифрактометре Rigaku, модель Miniflex II, серийный номер DD02652, используя сцинтилляционный детектор NaI(TI). Условия сбора данных включали в себя: излучение Cu Kα (λ=1,54059  ), напряжение генератора: 30 кВ, ток генератора: 15 мА, начальный угол °2θ: 3,0, конечный угол °2θ: 40,0, шаг сканирования °2θ: 0,04, время на шаг: 0,5 секунды. Образец получали, используя методику нулевого фона (фронтальное заполнение).
), напряжение генератора: 30 кВ, ток генератора: 15 мА, начальный угол °2θ: 3,0, конечный угол °2θ: 40,0, шаг сканирования °2θ: 0,04, время на шаг: 0,5 секунды. Образец получали, используя методику нулевого фона (фронтальное заполнение).
]
Пример 3
6-(трет-Бутилсульфонил)-N-(4,5-диметил-1H-пиразол-3-ил)-7-пропоксихиназолин-4-амин
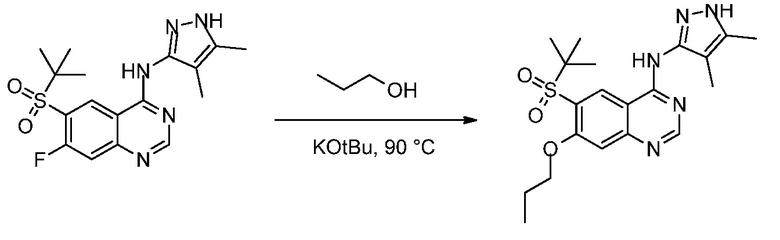
Смесь 6-(трет-бутилсульфонил)-N-(4,5-диметил-1H-пиразол-3-ил)-7-фторхиназолин-4-амина (1,5 г, 3,97 ммоль), пропан-1-ола (17,85 мл, 238 ммоль) и KOtBu (2,230 г, 19,87 ммоль) нагревали при 90°C в течение 21 ч. Реакционную смесь выливали в эфир, раствор становился мутным, осадок не образовывался. Смесь нейтрализовали лимонной кислотой и экстрагировали EtOAc (1×) и 2-MeTHF (1×). Объединенные органические растворы промывали рассолом, сушили над Na2SO4 и концентрировали досуха, что давало сырой продукт, который очищали посредством HPLC (10-50% ACN/вода, 0,1% TFA). Содержащие продукт фракции распределяли между EtOAc и нас. раствором бикарбоната натрия, промывали рассолом, сушили над Na2SO4 и концентрировали досуха. Полученный остаток растирали с EtOAc и фильтровали, что давало 6-(трет-бутилсульфонил)-N-(4,5-диметил-1H-пиразол-3-ил)-7-пропоксихиназолин-4-амин (280 мг, 0,671 ммоль, выход 16,87%) в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ м.д. 12,19 (с, 1H), 10,36 (с, 1H), 8,99 (с, 1H), 8,45 (с, 1H), 7,29 (с, 1H), 4,17 (т, J=6,19 Гц, 2H), 2,18 (с, 3H), 1,76-1,84 (м, 2H), 1,74 (с, 3H), 1,26-1,37 (м, 9H), 1,07 (т, J=7,45 Гц, 3H). MS (m/z) 418,3 (M+H)+.
Пример 4
6-(трет-Бутилсульфонил)-N-(4,5-диметил-1H-пиразол-3-ил)-7-((тетрагидрофуран-2-ил)метокси)хиназолин-4-амин

К раствору (тетрагидрофуран-2-ил)метанола (148 мг, 1,45 ммоль) в DMF (1 мл) прибавляли KOtBu (163 мг, 1,45 ммоль). Раствор перемешивали при комнатной температуре в течение 5 мин. Затем прибавляли 6-(трет-бутилсульфонил)-7-хлор-N-(4,5-диметил-1H-пиразол-3-ил)хиназолин-4-амин (30 мг, 0,076 ммоль) и в течение ночи перемешивали реакционную смесь при 80°C. Большую часть DMF удаляли в вакууме. Сырой материал очищали на колонке от Biotage (от 0 до 16% MeOH/DCM), что давало 6-(трет-бутилсульфонил)-N-(4,5-диметил-1H-пиразол-3-ил)-7-((тетрагидрофуран-2-ил)метокси)хиназолин-4-амин (40 мг, 0,084 ммоль, выход 35%). 1H ЯМР (DMSO-d6) δ 12,19 (уш. с, 1H), 10,36 (уш. с, 1H), 8,99 (с, 1H), 8,45 (с, 1H), 7,34 (с, 1H), 4,21 (м, 3H), 3,77-3,87 (м, 1H), 3,65-3,76 (м, 1H), 2,18 (с, 3H), 2,00 (м, 2H), 1,79-1,90 (м, 2H), 1,75 (с, 3H), 1,32 (с, 9H). MS (m/z) 460.
Фармацевтические композиции
Пример A
Получают таблетки, используя традиционные способы, и они имеют следующий состав:
Пример B
Получают капсулы, используя традиционные способы, и они имеют следующий состав:
Биологический анализ:
Для количественного описания взаимодействия новых тестируемых соединений в АТФ-связывающем кармане RIPK2 разработали основанный на поляризации флуоресценции анализ связывания, в основе которого лежит конкуренция с флуоресцентно меченным лигандом, конкурирующим с АТФ. Непроцессированную RIPK2, меченную FLAG His, получали очисткой из бакуловирусной экспрессирующей системы и использовали при конечной концентрации анализа, равной удвоенному значению кажущейся константы Kd. Флуоресцентно меченный лиганд (5-({[2-({[3-({4-[(5-гидрокси-2-метилфенил)амино]-2-пиримидинил}амино)фенил]карбонил}амино)этил]амино}карбонил)-2-(6-гидрокси-3-оксо-3H-ксантен-9-ил)бензойная кислота, полученная как описано в WO 2011/120025) использовали при конечной концентрации анализа, равной 5 нМ. Как из фермента, так и из лиганда готовили растворы, содержащие 50 мМ HEPES pH 7,5, 150 мМ NaCl, 10 мМ MgCl2, 1 мМ DDT и 1 мМ CHAPS. Растворы тестируемых соединений готовили в 100% DMSO и распределяли по 100 нл в отдельные лунки многолуночного планшета. Затем 5 мкл RIPK2 прибавляли к тестируемым соединениям в концентрации в два раза превышающей конечную концентрацию анализа и инкубировали при rt в течение 10 мин. После инкубации 5 мкл раствора флуоресцентно меченного лиганда прибавляли в каждую реакцию в концентрации в два раза превышающей конечную концентрацию анализа и инкубировали при rt в течение по меньшей мере 10 мин. Наконец, данные образцов считывали на приборе, способном измерять поляризацию флуоресценции. Ингибирование тестируемыми соединениями выражали как процент (%) ингибирования внутренних контролей анализа.
Для экспериментов концентрация/дозовый ответ, нормированные данные аппроксимировали и определяли значения pIC50, используя общепринятые методики. Значения pIC50 усредняли для определения среднего значения минимум для 2 экспериментов.
Длительное тестирование приводило к слабому изменению в приведенных средних значениях pIC50 для соединения примера 1 (7,5) и примера 3 (8,1).
Получение RIPK2, меченной FLAG His:
Непроцессированную человеческую кДНК RIPK2 (взаимодействующая с рецептором серин-треониновая киназа 2) приобретали у Invitrogen (Карлсбад, Калифорния, США, идентификатор клона: IOH6368, RIPK2-pENTR 221). Клонирование Gateway® LR использовали для сайт-специфической рекомбинации RIPK2 за N-концевой меткой FLAG-6His, содержащейся в векторе доставки pDEST8-FLAG-His6, согласно протоколу, описанному Invitrogen. Трансфекцию клеток насекомого Spodoptera frugiperda (Sf9) проводили с использованием Cellfectin® (Invitrogen) согласно протоколу производителя.
Клетки Sf9 выращивали в питательных средах Excell 420 (SAFC Bioscience, Ленекса, Канзас, США; Андовер, Хэмпшир, Великобритания) при 27°C, 80 об/мин во встряхиваемой колбе до достижения достаточного объема для засевания биореактора. Клетки выращивали в биореакторе рабочим объемом 50 литров (Applikon, Фостер Сити, Калифорния, США; Схидам, Нидерланды) при 27°C, 30% растворенного кислорода и скорости перемешивания 60-140 об/мин до достижения требуемого объема с концентрацией клеток приблизительно 3,7×106 клеток/мл. Клетки насекомого инфицировали бакуловирусом с множественностью заражения (MOI), равной 12,7. Культивирование продолжали в течение 43 часов фазы экспрессии. Инфицированные клетки извлекали из питательных сред центрифугированием при 2500 г, используя центрифугу непрерывного действия Viafuge (Carr) при скорости потока 80 литров/час. Сгусток клеток немедленно замораживали и впоследствии направляли на очистку.
Методика очистки I: 9,83×105 клеток насекомого ресуспендировали в 1,4 л буфера для лизиса (50 мМ Tris (pH 8,0), 150 мМ NaCl, 0,5 мМ NaF, 0,1% Triton X-100, 1 мл/литр смеси ингибиторов протеаз, комплект III (доступен от EMD Group; CalBiochem/Merck Bioscience, Гиббстаун, Нью-Джерси, США; Дармштадт, Германия)) и гомогенизировали в гомогенизаторе Даунса при охлаждении льдом. Затем суспензию осветляли центрифугированием при 47900 г в течение 2 ч при 4°C. Лизат декантировали с нерастворимого сгустка и подавали с линейной скоростью потока 16 см/ч на колонку для аффинной хроматографии FLAG-M2 объемом 55 мл (2,6×10,4 см), которую предварительно приводили в равновесное состояние с помощью буфера A (50 мМ Tris (pH 8,0), 150 мМ NaCl, 0,5 мМ NaF, 1 мл/литр смеси ингибиторов протеаз, комплект III), взятого в объеме, равном 10 объемам колонки. Затем колонку промывали буфером Α в объеме 15 объемов колонки и элюировали буфером B (буфер Α+150 мкг/мл пептида 3X FLAG) в объеме 6 объемов колонки с линейной скоростью потока 57 см/ч. Фракции, которые с помощью SDS-PAGE идентифицировали как содержащие интересующий белок, подвергали диализу для удаления из препарата пептида 3X FLAG против 5 л буфера Α (не содержащего смеси ингибиторов протеаз) в течение ночи, используя гофрированную диализную трубку SnakeSkin c MWCO 10 кДа. Данный способ очистки дал в итоге 11,3 мг белка, в котором RIPK2 присутствовала с чистотой 40% согласно гель-денситометрическому сканированию, а ее идентичность была подтверждена методом отпечатков пептидных масс. В качестве основных примесных белков в препарате были идентифицированы низкомолекулярные распавшиеся формы RIPK2.
Методика очистки II: 100 г клеток (культивирование в масштабе 10 л) замораживали, размораживали и ресуспендировали в 1 л буфера для лизиса (50 мМ Tris HCl pH 7,5, 250 мМ NaCl, 0,1 мМ TCEP, 3 мл смеси ингибиторов протеаз) и единожды подвергали лизису гомогенизацией при высоком давлении при 10000 фунтах на кв. дюйм (Avestin)(68,95 МПа). Затем суспензию осветляли центрифугированием при 35000 g в течение 45 минут при 4°C. Надосадочную жидкость отделяли центрифугированием и инкубировали с 5 мл смолы анти-FLAG-M2, которую предварительно приводили в равновесное состояние с помощью буфера A (50 мМ Tris HCl pH 7,5, 250 мМ NaCl, 0,1 мМ TCEP). После связывания белка при 4°C в течение 1 часа смолу набивали в две одноразовые колонки объемом 25 мл. Каждую колонку промывали 25 мл буфера A и элюировали 10 мл (буфер Α+200 мкг/мл пептида Flag). Элюированный пул концентрировали до 1 мл и наносили на колонку Superdex 200 (16/60) для эксклюзионной хроматографии. Фракции, содержащие непроцессированную RIPK2, собирали в соответствии с результатами анализа методом SDS-PAGE. Способ очистки дал 1,36 мг/л белка с чистой RIPK2 80%, идентичность которой была подтверждена методом отпечатков пептидных масс.
Биологический анализ:
Для оценки клеточной активности и эффективности новых тестируемых соединений был разработан анализ на стимулированное мурамилдипептидом (MDP) продуцирование цитокинов в человеческой цельной крови. Гепаризированную кровь (160 мкл), полученную от здоровых людей-добровольцев распределяли в отдельные лунки многолуночного планшета. Тестируемые соединения растворяли в 100% DMSO и разводили в не содержащем кальция и магния растворе D-PBS для получения рабочих исходных растворов 10× (10-кратной концентрации). В каждую лунку прибавляли двадцать микролитров разведенного раствора тестируемого соединения, и планшеты помещали на встряхиватель для планшетов (500 об/мин) и инкубировали в течение 30 мин во влажном инкубаторе (37°C, 5%CO2). Исходный раствор MDP 10× готовили в стерильной воде, в которой отсутствовали эндотоксины, содержащей 1% DMSO. Двадцать микролитров исходного раствора MDP прибавляли в каждую лунку (конечная концентрация=100 нг/мл), чтобы стимулировать зависимое от киназы RIP2 продуцирование цитоксинов. Во всех лунках конечная концентрация DMSO составляла 0,1% (об./об.). Планшеты инкубировали в течение дополнительных 6 ч (как отмечено выше). Затем в каждую лунку прибавляли дополнительные 100 мкл D-PBS (забуференный фосфатом солевой раствор Дульбекко), планшеты центрифугировали и отделяли надосадочные жидкости. Содержания TNFα в надосадочных жидкостях количественно определяли, используя коммерческий иммуноанализ (MesoScale Discovery). Ингибирование тестируемыми соединениями выражали как процент (%) ингибирования внутренних контролей анализа. Для экспериментов концентрация/дозовый ответ, нормированные данные аппроксимировали и определяли значения pIC50, используя общепринятые методики. Значения pIC50 усредняют для определения среднего значения минимум для 2 экспериментов.
Биологический анализ in vivo - Ингибирование индуцированного воспалительного ответа
Эффективность ингибиторов RIP2 может быть также оценена in vivo на примере грызунов. Было показано, что интраперитонеальное (i.p.) или внутривенное (i.v.) введение L18-MDP мышам индуцировало воспалительный ответ через активацию сигнального пути NOD2 (Rosenweig, H. L., et al. 2008. Journal of Leukocyte Biology 84:529-536). Уровень воспалительного ответа у крыс, подвергнутых воздействию L18-MDP, отслеживали с использованием общепринятых методик измерением возрастания уровней одного или более цитокинов (IL8, TNFα, IL6 и IL-1β) в сыворотке и/или жидкости перитонеального смыва и/или измерением инфлюкса нейтрофилов в перитонеальное пространство (когда дозу L18-MDP вводят i.p.). Ингибирование воспалительного ответа, индуцированного L18-MDP, у подвергнутых такому воздействию крыс может быть продемонстрировано пероральным предварительным введением доз тестируемого соединения, а затем измерением и сравнением уровней одного или более цитокинов (IL8, TNFα, IL6 и IL-1β) в сыворотке для контроля результатов, полученных для подвергнутых такому воздействию животных, используя общепринятые методики.
Например, крысам перорально предварительно вводили дозы соединения примера 2, то есть 6-(трет-бутилсульфонил)-N-(4,5-диметил-1H-пиразол-3-ил)-7-этоксихиназолин-4-амина, составляющие 0, 0,04, 0,4 и 4 мг/кг, с последующим введением дозы L18-MDP (50 мкг/крысу) спустя 0,25 ч после предварительного введения. Уровни цитокина IL8 в образцах цельной крови, взятых у крыс в данном исследовании, измеряли с использованием детектирования на основе антител (Meso-Scale Discovery platform). IL8-Цитокиновый ответ рассчитывали как усредненный ответ для каждого уровня дозы, выраженный относительно ответа, наблюдавшегося у крыс, которым вводили носитель, и результаты представлены на Фигуре 2 как среднее ± стандартная ошибка среднего (n=8 крыс/группа).
Биологический анализ in vivo - Клиновидный препарат кроличьего сердца
Самкам кроликов массой 2,2-3 кг вводили гепарин для предотвращения коагуляции и анестезировали фенобарбиталом (50 мг/кг, i.v.). Грудь вскрывали левосторонней торакотомией, и сердце иссекали и помещали в кардиоплегический раствор, состоящий из холодного (4°C) нормального раствора Тироде. Трансмуральный клин с размерами приблизительно 1,5 см шириной и 2-3 см длиной вырезали из левого желудочка.
Ткань клина канюлировали через левую переднюю нисходящую артерию или огибающую артерию и проводили перфузию кардиоплегическим раствором. Затем препарат помещали в малую ванну для тканей и проводили его артериальную перфузию раствором Тироде (T: 35,7±0,1°C, давление перфузии: 30-45 мм рт. ст.). Желудочковому клину предоставляли возможность прийти в равновесное состояние в ванне для тканей до достижения электрической стабильности, обычно в течение одного часа. Препараты стимулировали при длине сердечного цикла (BCL) от 1000 до 2000 мсек, используя биполярные серебряные электроды, изолированные за исключением концов, и приложенные к эндокардиальной поверхности.
Трансмуральную электрокардиограмму (ЭКГ) регистрировали во всех экспериментах, используя внеклеточные электроды из серебра/хлорида серебра, помещенные в раствор Тироде, омывающий препарат, на расстоянии от 1,0 до 1,5 см от эпикардиальной и эндокардиальной поверхностей вдоль того же вектора, что и трансмембранные данные регистрации (Epi: полюс “+”). На ЭКГ трансмуральную дисперсию реполяризации (TDR) определяли по интервалу между концом и пиком T-зубца (Tp-e). Интервал QT определяли как время от начала QRS до точки, в которой конечный склон T-зубца пересекал изоэлектрическую линию. Длительность QRS, QT и Tp-e измеряют для 10 разверток и усредняют для каждой обработки. Данные из всей популяции животных усредняют для каждой обработки и сравнивают со средними контрольными значениями.
Генерацию изометрической сократительной силы (%ICF) измеряют для 10 разверток и усредняют для каждой обработки. Данные из всей популяции животных усредняют для каждой обработки и сравнивают со средними контрольными значениями.
Для каждого тестируемого соединения готовили раствор в 100% DMSO с исходной концентрацией 30 мМ. Раствор соединения разводили до наивысшей тестируемой концентрации в буфере Тироде (содержащем в мМ: 129 NaCl, 4 KCl, 0,9 NaH2PO4, 20 NaHCO3, 1,8 CaCl2, 0,5 MgSO4 и 5,5 глюкозы, pH 7,4, забуференном при 95% Ο2 и 5% CO2), и из такого раствора впоследствии готовили последовательные разведения.
Каждое тестируемое соединение тестировали в 4 концентрациях от 1 до 30 мкМ. После того, как клиновидные препараты подвергали перфузии нормальным раствором Тироде и стимулировали при BCL 1000 мсек в течение одного часа, частоту стимуляции снижали до BCL 2000 мсек для 5-минутного периода стабилизации, после которого регистрировали базисную ЭКГ и изометрическую сократительную силу (ICF). Затем на препараты снова воздействовали BCL 1000 мсек и подвергали перфузии раствором Тироде, содержащим тестируемое соединение. Для каждой концентрации тестируемого соединения проводили перфузию клиновидных препаратов в течение 20 минут при BCL 1000 мсек, после чего следовали 5 минут при BCL 2000 мсек, в ходе которых регистрировали ЭКГ и ICF. В анализе с использованием клиновидного препарата кроличьего сердца оценивали соединение примера 2 (6-(трет-бутилсульфонил)-N-(4,5-диметил-1H-пиразол-3-ил)-7-этоксихиназолин-4-амин). Четыре главных регистрируемых показателя, полученные для клиновидного препарата включают в себя удлинение QT, пируэтную желудочковую тахикардию (индекс TdP, полученный из QT, Tp-e и вскоре после деполяризаций), проведение импульса (с которым связан QRS) и сократительная способность, которые представлены в Таблице 2.
Для оценки обусловленного соединением риска TdP использовали систему баллов, основанную на эксперименте с клиновидным препаратом извлеченного левого желудочка: баллы для интервала QT, соотношения Tp-e/QT. Индекс TdP получали сначала переводя интервал QT и соотношение Tp-e/QT в значение % изменения относительно базовой линии. Данным значениям по отдельности приписывали индекс TdP на основе следующей системы: <-5%=-1, от -5% до 10%=0, от 10% до 20%=1, от 20% до 30%=2, >30%=3. Итоговый диапазон системы балов представляет собой диапазон от -2 до 14 при BCL=2000 мсек.
Сводка данных (среднее, n=2).
Селективность кином
Селективность кином (исследование проведено Reaction Biology Corporation, One Great Valley Parkway, Малверн, Пенсильвания, США, 19355, http://www.reactionbiology.com) для соединения примера 2 (6-(трет-бутилсульфонил)-N-(4,5-диметил-1H-пиразол-3-ил)-7-этоксихиназолин-4-амин) определяли in vitro анализом по отношению к панели 337 киназ. При концентрации 1 мкМ соединение примера 2 продемонстрировало >70% ингибирование 1 из 337 киназ и >50% ингибирование 4 из 337 протестированных киназ.
Ссылки: WO 2011/120025, WO 2011/120026, WO 2011/123609, WO 2011/140442, WO 2012/021580, WO 2012/122011, WO 2013/025958.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФАРМАЦЕВТИЧЕСКИ АКТИВНЫЕ СОЕДИНЕНИЯ | 2013 |
|
RU2654942C2 |
| N-(2-ЦИАНОГЕТЕРОЦИКЛИЛ)ПИРАЗОЛОПИРИДОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ЯНУС-КИНАЗЫ | 2014 |
|
RU2669922C2 |
| ИНГИБИТОРЫ ТИРОЗИНКИНАЗЫ | 2006 |
|
RU2408584C2 |
| МОДУЛИРУЮЩИЕ JAK КИНАЗУ ХИНАЗОЛИНОВЫЕ ПРОИЗВОДНЫЕ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2010 |
|
RU2529019C2 |
| ХИМИЧЕСКИЕ СОЕДИНЕНИЯ - 759 | 2008 |
|
RU2481348C2 |
| ИМИДАЗОЛОНИЛХИНОЛИНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ АТМ | 2016 |
|
RU2743343C2 |
| ПИРАЗОЛОПИРИМИДИНОВЫЕ СОЕДИНЕНИЯ-ИНГИБИТОРЫ JAK И СПОСОБЫ | 2009 |
|
RU2539568C2 |
| N-ЗАМЕЩЕННЫЕ ДИОКСОЦИКЛОБУТЕНИЛАМИНО-3-ГИДРОКСИПИКОЛИНАМИДЫ, ПРИГОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ CCR6 | 2019 |
|
RU2784831C2 |
| СОЕДИНЕНИЯ ПИРАЗОЛО[3,4-b]ПИРИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ ТАМ И МЕТ | 2019 |
|
RU2773611C1 |
| ПРОИЗВОДНЫЕ 2-ПИРАЗИНОНА ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЯ ИЛИ СОСТОЯНИЯ, ПРИ КОТОРЫХ ПОЛЕЗНО ИНГИБИРОВАНИЕ АКТИВНОСТИ НЕЙТРОФИЛЬНОЙ ЭЛАСТАЗЫ | 2007 |
|
RU2448098C2 |

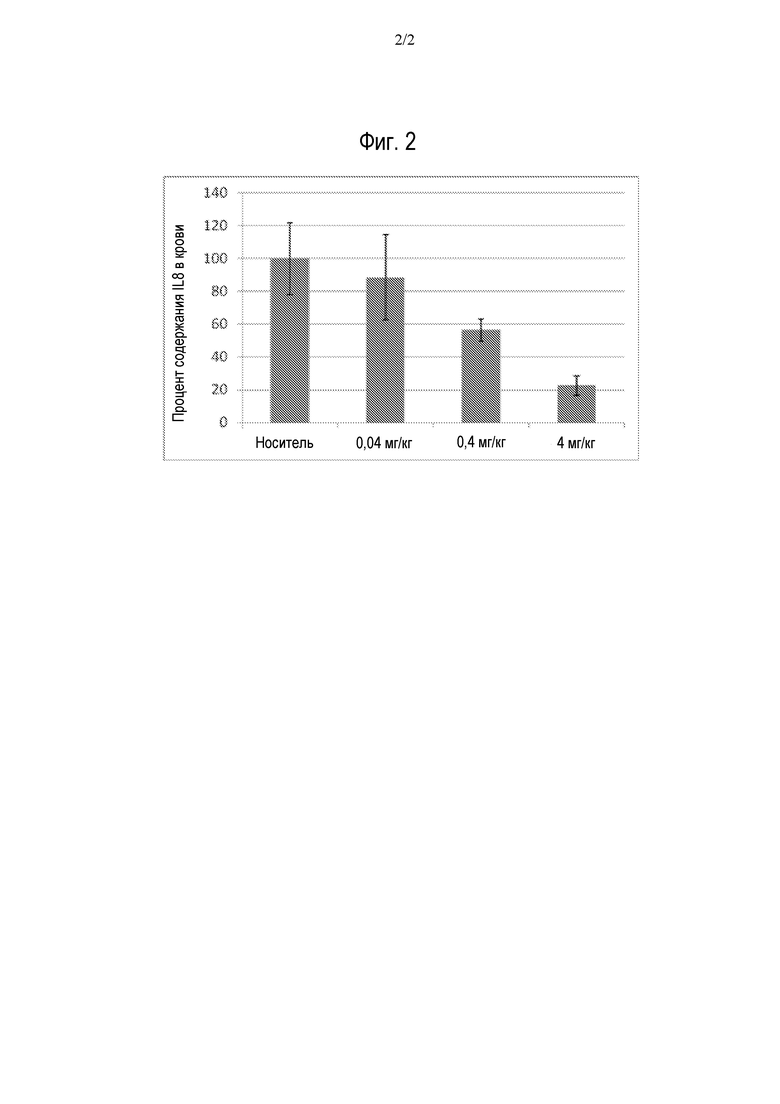
Изобретение относится к новому соединению, выбранному из нижеуказанной группы, или его фармацевтически приемлемой соли, которые обладают свойствами ингибиторов киназы RIP21. Соединения могут найти применение для лечения заболевания или нарушения, выбранного из увеита, лихорадочного синдрома, связанного с интерлейкин-1-превращающим ферментом, дерматита, острого повреждения легких, сахарного диабета типа 2, артрита, ревматоидного артрита, язвенного колита, болезни Крона, раннего воспалительного заболевания кишечника, имеющего внекишечные проявления воспалительного заболевания кишечника, предотвращения ишемического реперфузионного повреждения трансплантата паренхиматозного органа, неалкогольного стеатогепатита, алкогольного стеатогепатита, аутоиммунного гепатита, астмы, реакции “трансплантат против хозяина”, системной красной волчанки, множественного склероза, саркоидоза, синдрома Блау, раннего саркоидоза, гранулематоза Вегенера и интерстициального заболевания легких. Новые соединения имеют формулу
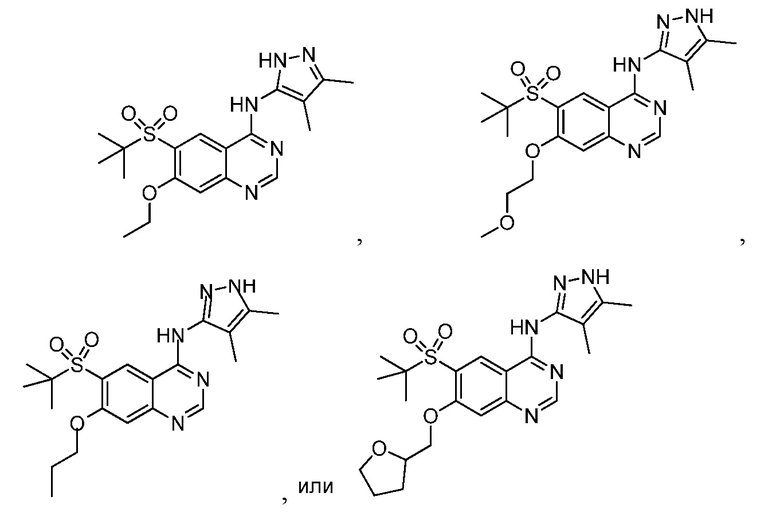
7 н. и 11 з.п. ф-лы,2 ил., 2 табл., 4 пр.
1. Соединение, имеющее формулу:
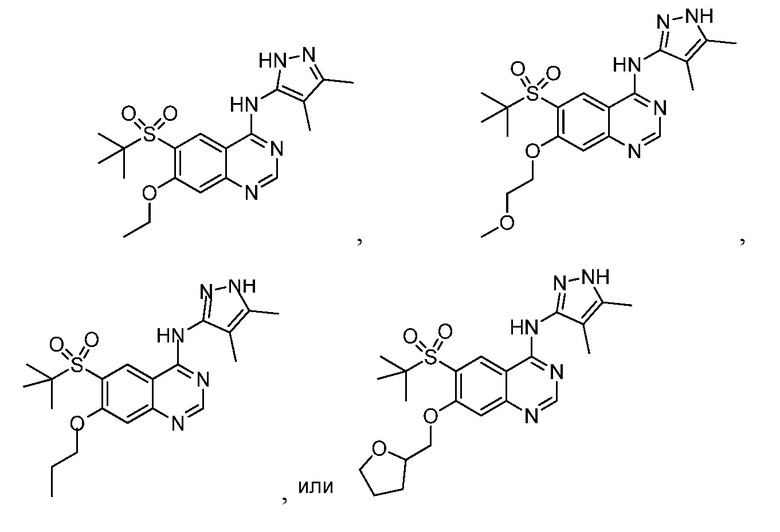
или его фармацевтически приемлемая соль.
2. Соединение, которое представляет собой 6-(трет-бутилсульфонил)-N-(4,5-диметил-1H-пиразол-3-ил)-7-этоксихиназолин-4-амин.
3. Соединение, которое представляет собой 6-(трет-бутилсульфонил)-N-(4,5-диметил-1H-пиразол-3-ил)-7-этоксихиназолин-4-амин или его фармацевтически приемлемую соль.
4. Соединение или его фармацевтически приемлемая соль по п.1, оказывающая ингибирующее действие на киназу RIP2, для применения в терапии.
5. Соединение по п. 4, которое представляет собой 6-(трет-бутилсульфонил)-N-(4,5-диметил-1H-пиразол-3-ил)-7-этоксихиназолин-4-амин или его фармацевтически приемлемую соль.
6. Соединение или его фармацевтически приемлемая соль по п.1 для применения в лечении опосредованного киназой RIP2 заболевания или нарушения, где заболевание или нарушение выбрано из увеита, лихорадочного синдрома, связанного с интерлейкин-1-превращающим ферментом, дерматита, острого повреждения легких, сахарного диабета типа 2, артрита, ревматоидного артрита, язвенного колита, болезни Крона, раннего воспалительного заболевания кишечника, имеющего внекишечные проявления воспалительного заболевания кишечника, предотвращения ишемического реперфузионного повреждения трансплантата паренхиматозного органа, неалкогольного стеатогепатита, алкогольного стеатогепатита, аутоиммунного гепатита, астмы, реакции “трансплантат против хозяина”, системной красной волчанки, множественного склероза, саркоидоза, синдрома Блау, раннего саркоидоза, гранулематоза Вегенера и интерстициального заболевания легких.
7. Соединение по п. 6, которое представляет собой 6-(трет-бутилсульфонил)-N-(4,5-диметил-1H-пиразол-3-ил)-7-этоксихиназолин-4-амин или его фармацевтически приемлемую соль.
8. Соединение или его фармацевтически приемлемая соль по п.6, где заболевание или нарушение выбрано из увеита, лихорадочного синдрома, связанного с интерлейкин-1-превращающим ферментом, синдрома Блау, раннего саркоидоза, язвенного колита, болезни Крона, гранулематоза Вегенера и саркоидоза.
9. Соединение по п. 8, которое представляет собой 6-(трет-бутилсульфонил)-N-(4,5-диметил-1H-пиразол-3-ил)-7-этоксихиназолин-4-амин или его фармацевтически приемлемую соль.
10. Фармацевтическая композиция, оказывающая ингибирующее действие на киназу RIP2, содержащая 6-(трет-бутилсульфонил)-N-(4,5-диметил-1H-пиразол-3-ил)-7-этоксихиназолин-4-амин или его фармацевтически приемлемую соль и одно или более фармацевтически приемлемых вспомогательных веществ.
11. Фармацевтическая композиция, оказывающая ингибирующее действие на киназу RIP2, содержащая 6-(трет-бутилсульфонил)-N-(4,5-диметил-1H-пиразол-3-ил)-7-этоксихиназолин-4-амин и одно или более фармацевтически приемлемых вспомогательных веществ.
12. Способ лечения опосредованного киназой RIP2 заболевания или нарушения, который включает в себя введение терапевтически эффективного количества соединения или его фармацевтически приемлемой соли по п.1 субъекту, нуждающемуся в нем, где заболевание или нарушение выбрано из увеита, лихорадочного синдрома, связанного с интерлейкин-1-превращающим ферментом, дерматита, острого повреждения легких, сахарного диабета типа 2, артрита, ревматоидного артрита, язвенного колита, болезни Крона, раннего воспалительного заболевания кишечника, имеющего внекишечные проявления воспалительного заболевания кишечника, предотвращения ишемического реперфузионного повреждения трансплантата паренхиматозного органа, неалкогольного стеатогепатита, алкогольного стеатогепатита, аутоиммунного гепатита, астмы, реакции “трансплантат против хозяина”, системной красной волчанки, множественного склероза, саркоидоза, синдрома Блау, раннего саркоидоза, гранулематоза Вегенера и интерстициального заболевания легких.
13. Способ по п. 12, где соединение представляет собой 6-(трет-бутилсульфонил)-N-(4,5-диметил-1H-пиразол-3-ил)-7-этоксихиназолин-4-амин или его фармацевтически приемлемую соль.
14. Способ по п.12, в котором заболевание или нарушение выбрано из увеита, лихорадочного синдрома, связанного с интерлейкин-1-превращающим ферментом, синдрома Блау, раннего саркоидоза, язвенного колита, болезни Крона, гранулематоза Вегенера и саркоидоза.
15. Способ по п. 14, где соединение представляет собой 6-(трет-бутилсульфонил)-N-(4,5-диметил-1H-пиразол-3-ил)-7-этоксихиназолин-4-амин или его фармацевтически приемлемую соль.
16. Применение соединения или его фармацевтически приемлемой соли по п.1 в производстве медикамента для лечения опосредованного киназой RIP2 заболевания или нарушения, где заболевание или нарушение выбрано из увеита, лихорадочного синдрома, связанного с интерлейкин-1-превращающим ферментом, дерматита, острого повреждения легких, сахарного диабета типа 2, артрита, ревматоидного артрита, язвенного колита, болезни Крона, раннего воспалительного заболевания кишечника, имеющего внекишечные проявления воспалительного заболевания кишечника, предотвращения ишемического реперфузионного повреждения трансплантата паренхиматозного органа, неалкогольного стеатогепатита, алкогольного стеатогепатита, аутоиммунного гепатита, астмы, реакции “трансплантат против хозяина”, системной красной волчанки, множественного склероза, саркоидоза, синдрома Блау, раннего саркоидоза, гранулематоза Вегенера и интерстициального заболевания легких.
17. Применение по п. 16, где соединение представляет собой 6-(трет-бутилсульфонил)-N-(4,5-диметил-1H-пиразол-3-ил)-7-этоксихиназолин-4-амин или его фармацевтически приемлемую соль.
18. Применение по п.16, в котором заболевание или нарушение выбрано из увеита, лихорадочного синдрома, связанного с интерлейкин-1-превращающим ферментом, синдрома Блау, раннего саркоидоза, язвенного колита, болезни Крона, гранулематоза Вегенера и саркоидоза.
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Изложница с суживающимся книзу сечением и с вертикально перемещающимся днищем | 1924 |
|
SU2012A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Sheth P R; Ramanathan L et al | |||
| Expression, purification, stability optimization and characterization of human Aurora B kinase domain from E | |||
| coli; ARCHIVES OF BIOCHEMISTRY AND BIOPHYSICS, 2010, Vol:503, Nr:2, Page(s):191 - 201 | |||
| Способ приготовления лака | 1924 |
|
SU2011A1 |
| СОЕДИНЕНИЯ АМИНОХИНАЗОЛИНОВ | 2005 |
|
RU2382034C2 |

