Изобретение относится к способам контроля качества инъекционных лекарственных средств, а именно к идентификации и количественному определению основных компонентов в инъекционных лекарственных средствах методом спектрометрии комбинационного рассеяния света.
Спектрометрия комбинационного рассеяния (также называемая Рамановской спектрометрией) является неразрушающим аналитическим методом идентификации и контроля качества лекарственных средств, поскольку дает возможность получить индивидуальный спектр молекулы вещества.
Спектр комбинационного рассеяния (далее - Рамановский спектр) возникает при облучении вещества монохроматическим лазерным излучением ультрафиолетового или видимого диапазона (диапазон длин волн от ультрафиолетовой до ближней инфракрасной области). Под действием излучения молекулы вещества поляризуются и рассеивают свет в интервале от 2 до 4000 см-1. Если взаимодействие кванта падающего излучения с молекулой, находящейся в основном или возбужденном колебательном состоянии, является упругим, то энергетическое состояние молекулы не меняется, и частота рассеянного излучения будет такая же, как падающего (релеевская полоса Рамановского спектра). В случае неупругого взаимодействия происходит обмен энергией между квантом излучения и молекулой, за счет чего возникает рассеянное излучение, которое может быть большей или меньшей частоты (антистоксова и стоксова полоса соответственно). Таким образом, формируется Рамановский спектр. Спектры комбинационного рассеяния очень чувствительны к природе химических связей - как в органических молекулах и полимерных материалах, так и в кристаллических решетках и кластерах, что обуславливает индивидуальность спектра конкретного вещества.
В результате анализа можно идентифицировать молекулярные фрагменты - определять строение вещества или изучать внутримолекулярные взаимодействия, наблюдая положение и интенсивность полос в Рамановском спектре. При этом достаточно просто идентифицировать фрагменты, используя поиск по библиотекам спектров.
Идентификация соединения по спектру комбинационного рассеяния осуществляется сравнением его спектра со спектром образца оригинального (подлинного) лекарственного средства, полученного на одном и том же типе прибора в одних и тех же условиях.
Количественный анализ основан на прямо пропорциональной зависимости интенсивности (I) линий спектра числу молекул (N) в единице объема:
I=i*k*N,
где i - интенсивность рассеиваемого света на одну молекулу; k - коэффициент, зависящий от условий эксперимента, постоянная для данного прибора величина.
Таким образом, количественный анализ состоит из двух этапов: подтверждение подлинности (средний спектр образца сравнивается со спектром, хранящимся в библиотеке моделей) и количественная оценка концентрации действующего компонента.
Под термином модель следует понимать спектры оригинального лекарственного препарата, принимаемого за эталонной образец, калибровочных растворов, изготовленных с его помощью, и параметров математической обработки.
Модель является структурной составляющей библиотеки (базы данных), используемой для скрининга качества инъекционных медицинских препаратов без разрушения оригинальной упаковки в комплексе с портативными спектрометрами комбинационного рассеивания.
Эталонный образец - образец лекарственного средства, концентрация действующего вещества в котором определена согласно утвержденной нормативной документации, а за концентрацию вспомогательных компонентов принимается концентрация, указанная в нормативном документе, либо паспорте на лекарственный препарат. Эталонный образец должен иметь действительный срок годности и документальное подтверждение соблюдения условий хранения.
В КР-спектрометрии известна методология количественного определения растворов неорганических солей в воде (статья «Inorganic salts diluted in water probed by Raman spectrometry: Data processing and performance evaluation)), T.H. Kauffmann, M.D. Fontana, Sensors and Actuators B: Chemical, Volume 209, 31 March 2015, Pages 154-161). Процедура применяется к растворам нитрата натрия различной концентрации. Для количественного определения в работе применяют несколько подходов использования интенсивностей пиков. Показано, что интегральная интенсивность в отличие от абсолютной или относительной интенсивности пика практически не чувствительна к небольшим изменениям сдвига Рамановских линий вызванных, например, изменением температуры или дрейфом лазера, но требует дополнительных вычислений по сравнению с использованием интенсивности максимумов. Так же в статье приводят варианты нормализации. Нормализация по валентным колебаниям воды дает не только низкую чувствительность, но и ведет к ошибкам при определении концентрации. SNV-нормализация (Standard normal variate) при расчетах по площади дает высокую чувствительность и линейность, а также проста в вычислении. Несмотря на все достоинства, данная статья не отражает возможности переноса моделей между приборами, возможности многокомпонентного анализа, и анализа органических веществ не нарушая упаковки.
Известен метод количественного анализа комбинационного рассеяния света в жидких смесях (статья «Quantitative NIR Raman analysis in liquid mixtures", R. Ysacc  , Jorge Medina-Valtierra, Cirilo Medina-Gutierrez, Claudio Frausto-Reyes, Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, Август 2004). В этой статье показана возможность количественного анализа смесей воды с этанолом и родамина-6G с метанолом непосредственно в стеклянных сосудах с использованием простой математической модели смесей (линейная регрессия). Предполагая, что интенсивность КР-сигнала пропорциональна концентрации вещества, выводят уравнение, где представляют интенсивность спектра смеси в каждой точке как сумму соответствующих точек спектров ее компонентов (с учетом их концентрации). Спектры чистых компонентов при этом нормализуют по самому интенсивному пику. Для анализа используют не весь спектр, а интенсивности конкретных пиков. Авторами проведены процедуры и расчеты неопределенности согласно Руководству по выражению неопределенности в измерениях (Guide to the Expression of Uncertainty in Measurement, ISO, Geneva, 1995). Недостатками данного метода является использование интенсивности конкретных выделенных пиков, а также нормировка на самый интенсивный пик вещества. Данный подход не позволяет переносить модели между приборами, поскольку даже между двумя приборами может наблюдаться отклонение пиков по волновому числу, а также существенное различие в интенсивности пиков. Все это может привести к появлению ложных результатов количественного определения, а также не позволяет зафиксировать появление в смеси дополнительного компонента в небольшом количестве.
, Jorge Medina-Valtierra, Cirilo Medina-Gutierrez, Claudio Frausto-Reyes, Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, Август 2004). В этой статье показана возможность количественного анализа смесей воды с этанолом и родамина-6G с метанолом непосредственно в стеклянных сосудах с использованием простой математической модели смесей (линейная регрессия). Предполагая, что интенсивность КР-сигнала пропорциональна концентрации вещества, выводят уравнение, где представляют интенсивность спектра смеси в каждой точке как сумму соответствующих точек спектров ее компонентов (с учетом их концентрации). Спектры чистых компонентов при этом нормализуют по самому интенсивному пику. Для анализа используют не весь спектр, а интенсивности конкретных пиков. Авторами проведены процедуры и расчеты неопределенности согласно Руководству по выражению неопределенности в измерениях (Guide to the Expression of Uncertainty in Measurement, ISO, Geneva, 1995). Недостатками данного метода является использование интенсивности конкретных выделенных пиков, а также нормировка на самый интенсивный пик вещества. Данный подход не позволяет переносить модели между приборами, поскольку даже между двумя приборами может наблюдаться отклонение пиков по волновому числу, а также существенное различие в интенсивности пиков. Все это может привести к появлению ложных результатов количественного определения, а также не позволяет зафиксировать появление в смеси дополнительного компонента в небольшом количестве.
Популярна работа ((Quantitative Analysis Using Raman Spectrometry)) (APPLIED SPECTROSCOPY, Выпуск 1, 2003 г.). В данной статье рассматриваются изменения в позиции пиков, их ширины или формы (inhomogeneous broadening - неоднородное ущирение полос). Эти изменения могут быть эмпирически связаны с изменениями концентрации, однако фундаментальным механизмом такого количественного анализа является пропорциональная зависимость между интенсивностью Рамановского сигнала и концентрацией вещества. Рамановский спектр смеси равносилен взвешенной сумме спектров компонентов. Их вес определяется КР-активностью и количеством компонента в образце. При этом спектр компонента в образце может не быть идентичен спектру компонента в чистом виде, ввиду химических и физических взаимодействий. Принцип линейной суперпозиции рушится, если хотя бы одно вещество значительно поглощает КР-сигнал от аналита или излучение лазера. В этом случае вейвлет-преобразование схоже с преобразованием Фурье, но может лучше подходить под нужды анализа Рамановских спектров. Если из различных видов шума доминирующим является высокочастотный, то площадь пика может быть вычислена более точно, т.к. большее число фотонов включено в расчет. Вариации интенсивности сигнала в зависимости от положения образца могут быть снижены применением оптики с большей площадью сбора или использованием специальных приспособлений для точного позиционирования. В качестве внутреннего стандарта могут быть использованы несколько линий (в том числе с весами), их сумма, участки спектра или даже спектр целиком. Соотношение Рамановских пиков двух аналитов предоставляет такой же тип связи, что и соотношение сигналов аналита и внутреннего стандарта. Если известны соотношения площадей и относительная Рамановская активность для каждого компонента, то для вычисления искомой концентрации может быть применено уравнение массового баланса. Данная работа отражает общие принципы, которые можно применять для количественного определения в Рамановской спектроскопии, предлагает вероятные пути решения. Однако статья не описывает конкретных алгоритмов, позволяющих добиться решения всех поставленных задач.
В совокупности каждая из приведенных статей предлагает решение одной из возникающих проблем и задач количественного определения растворов и смесей (в том числе и лекарственных препаратов), но не отражает совокупного решения.
Основными преимуществами предложенного способа по сравнению с существующими являются:
- Низкая стоимость - для создания количественной модели не используются дорогостоящие стандарты чистых действующих веществ лекарственных препаратов. В случае двухкомпонентной модели достаточно лишь воды и оригинального препарата. В случае 3-х компонентной модели дополнительно необходимы дешевые и легкодоступные вспомогательные вещества;
- Надежность - спектроскопия комбинационного рассеяния крайне чувствительный метод. В традиционном варианте создания количественных моделей используются чистые вещества (стандартные образцы) отличные от тех, что мог использовать производитель при изготовлении оригинального препарата, либо рН раствора может отличаться. Такая разница может давать различные отклонения в спектрах калибровочных растворов по отношению к анализируемому образцу. В предложенном методе такая проблема отсутствует, так как в процедуре применяется сам эталонный образец оригинального лекарственного средства, предварительно проанализированный в лабораторных условиях;
- Точность - использование жидкого стандарта калибровки относительной интенсивности приборов взамен традиционно используемых подходов (вольфрамовая лампа, флуоресцентные стекла) позволяет в лучшей степени добиваться согласованности работы нескольких приборов. Относительная ошибка переноса моделей между приборами в среднем не превышает 5% в зависимости от образца.
Задачами, на решение которых направлено данное изобретение, являются количественное определение действующих веществ в растворах с низкой концентрацией, а именно инъекционных лекарственных препаратов, одновременное определение до двух компонентов смеси, возможность переноса моделей между приборами, нивелируя такие факторы, как разница в интенсивности и определении волновых чисел между приборами, разница температур, дрейф лазера, возможность количественного определения компонентов лекарственных препаратов без разрушения первичной упаковки с хорошей точностью, так же сам процесс приготовления моделей достаточно прост и не требует использования дополнительных реактивов и приготовления большого количества калибровочных растворов.
Данные задачи решаются за счет того, что способ идентификации и последующего количественного определения основных компонентов в инъекционных лекарственных средствах включает в себя измерение спектра лекарственного препарата с помощью спектрометра, в оригинальной упаковке сравнение полученного спектра препарата с эталонными качественной и количественной моделями и оценку полученных результатов. Идентификация заключается в определении корреляционного коэффициента для анализируемого спектра по отношению к библиотечному спектру, а количественное определение - расчет концентрационной доли анализируемого образца по отношению к количественной модели. При этом метод создания эталонных количественных моделей для применения в составе библиотек спектров, используемых для скрининга качества инъекционных лекарственных средств, включает в себя получение серий нормированных спектров шести растворов эталонного образца (оригинального лекарственного препарата): оригинального препарата, раствора оригинального препарата и воды в соотношении 1:1, раствора вспомогательных компонентов, раствора вспомогательных компонентов и воды в соотношении 1:1, раствора оригинального препарата и вспомогательных компонентов в соотношении 1:1, вода трех растворов для двухкомпонентных моделей в случае отсутствия вспомогательных веществ, - их усреднение и предобработка, вычисление по усредненным спектрам базовых коэффициентов, сохранение базовых спектров и вычисленных базовых коэффициентов в базе моделей.
Техническим результатом, обеспечиваемым приведенной совокупностью признаков, является получение более точных результатов и уменьшение ошибок во время проведения анализа, что приводит к подтверждению качества лекарственных средств.
Способ идентификации и последующего количественного определения основных компонентов в инъекционных лекарственных средствах включает в себя измерение спектра лекарственного препарата с помощью спектрометра в оригинальной упаковке, сравнение полученного спектра препарата с эталонной моделью и оценку полученных результатов.
При выполнении измерений применяют спектрометр комбинационного рассеяния, со спектральным диапазоном не хуже 100 - 4000 см-1 и разрешением не менее 22 см-1. Источник излучения - лазер с длиной волны 532 нм.
Спектрометры комбинационного рассеяния обладают различными характеристиками в разных частях матриц ПЗС, что в итоге приводит к получению существенной разницы при использовании одной и той же количественной модели на разных инструментах.
Для коррекции этого эффекта проводят процедуру калибровки. На базовом эталонном приборе измеряется спектр раствора пирацетама 200 мг/мл и сохраняется в энергонезависимую память каждого спектрометра (ХЭТпир).


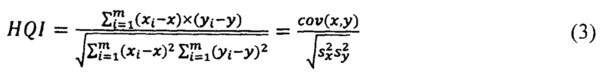
Далее любой спектр (X), полученный на второстепенных спектрометрах, нормируется на специально подобранную полиномиальную функцию (F(x)). Функция нормировки подбирается таким образом, чтобы коррелятор (HQI) спектра пирацетама, измеренный на эталонном и второстепенном приборе, был не ниже 0,99.
В качестве нормировочного спектра был выбран препарат пирацетама ввиду высокой стабильности препарата (его срок годности составляет 5 лет, препарат не имеет строгих условий хранения), и интенсивного сигнала на всем спектральном диапазоне измерений.
Все исходные спектры, используемые в расчетах, нормированы на спектр пирацетама с базового эталонного прибора.
Идентификация лекарственных препаратов осуществляется путем сравнения спектров комбинационного рассеяния испытуемого образца, с аналогичными спектрами, полученными от оригинальных медикаментов ранее и хранящимися в базе данных.
В качестве параметра соответствия испытуемого спектра модельному используется коэффициент корреляции Пирсона (HQI).
Для идентификации используют первую производную спектра, которая представляет собой распределение интенсивности комбинационного рассеяния света по длинам волн, предварительно обработанного с помощью фильтра Difference of Gaussians (DoG) (Разница по Гауссу) по формуле:
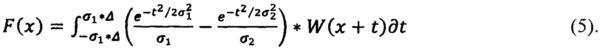
Сглаженная первая производная определяется по формуле:
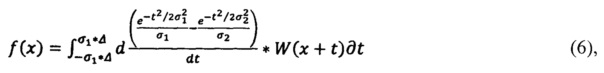
где W - сглаженный спектр, σ1 - щель (минимальное окно), σ2 - окно (максимальное окно), Δ - полуинтервал сглаживания (полуширина DoG фильтра).
Щель (σ1) - минимальная дисперсия Гаусса, которая обеспечивает сглаживание более узких пиков шума. Окно (σ2) - максимальная дисперсия Гаусса, которая обеспечивает удаление более широких пиков фона.
Таким образом для модельного спектра получают:
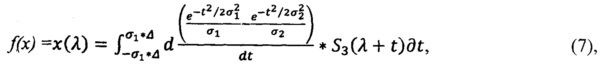
для спектра анализируемого препарата получают:
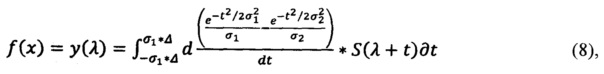
где S3 - спектр эталонного препарата, S - спектр анализируемого препарата, σ1 - щель, или минимальное окно сглаживания, σ2 - окно, или максимальное окно сглаживания, Δ - полуинтервал сглаживания, λ - волновое число.
Перед предобработкой спектр интерполяцией сбрасывается на равномерный шаг в 1,5 см-1, и выравнивается по волновому числу относительно модельного.
Корреляционный коэффициент HQI (индекс качества соответствия) для анализируемого спектра по отношению к опорному библиотечному спектру рассчитывается на выделенном диапазоне длин волн испытуемого и модельного спектров с использованием следующей формулы:
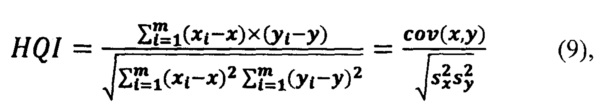
где х, у - выборочные средние, s - выборочная дисперсия.
При этом выборка х(λ)=(x1…,xm) - это интенсивности эталонного спектра, а у(λ)=(у1,…,ym) - интенсивности испытуемого спектра, a i=(1,…,m) - точки по оси абсцисс (волновое число); выделенный диапазон длин волн (1,…m) задается как один из параметров модели.
Значение HQI может находиться в диапазоне 0-1 (1 соответствует полному совпадению опорного и измеряемого спектров). Критерием получения удовлетворительных результатов идентификации образца в общем случае является коэффициент корреляции выше 0,90. В случае получения удовлетворительных результатов по HQI проводят количественное определение основных компонентов лекарственного препарата.
Состав инъекционных лекарственных средств условно можно разделить на следующие компоненты: действующее вещество (вещества), вспомогательные компоненты (стабилизаторы рН, консерванты и т.д.) и растворитель. Независимо от количества действующих веществ, вспомогательных компонентов и состава растворителя рассматривается каждая из этих трех составляющих, имеющая вполне определенный спектр. Спектр действующего вещества обозначен как Р (primary), спектр вспомогательных компонентов как A (additive), растворителя - как D (dissolvent). Считается, что спектр препарата можно разложить в сумму других спектров и определить коэффициенты этого разложения. Разложив спектр эталона и исследуемого вещества в суммы спектров Р, A, D становятся определены отклонения концентраций действующего вещества и добавок. Однако образцы действующего вещества и вспомогательных компонентов не всегда удается получить (необходимое количество может достигать десяток грамм), а процедура приготовления модельной смеси для калибровок в лаборатории далеко не всегда соответствует таковой в производственных условиях.
Считается, что доступен спектр вспомогательных компонентов без действующего вещества, растворенных в той же самой концентрации, что и в эталонном препарате. Сравнивая спектр такого раствора с эталонным можно определить спектр действующего вещества.
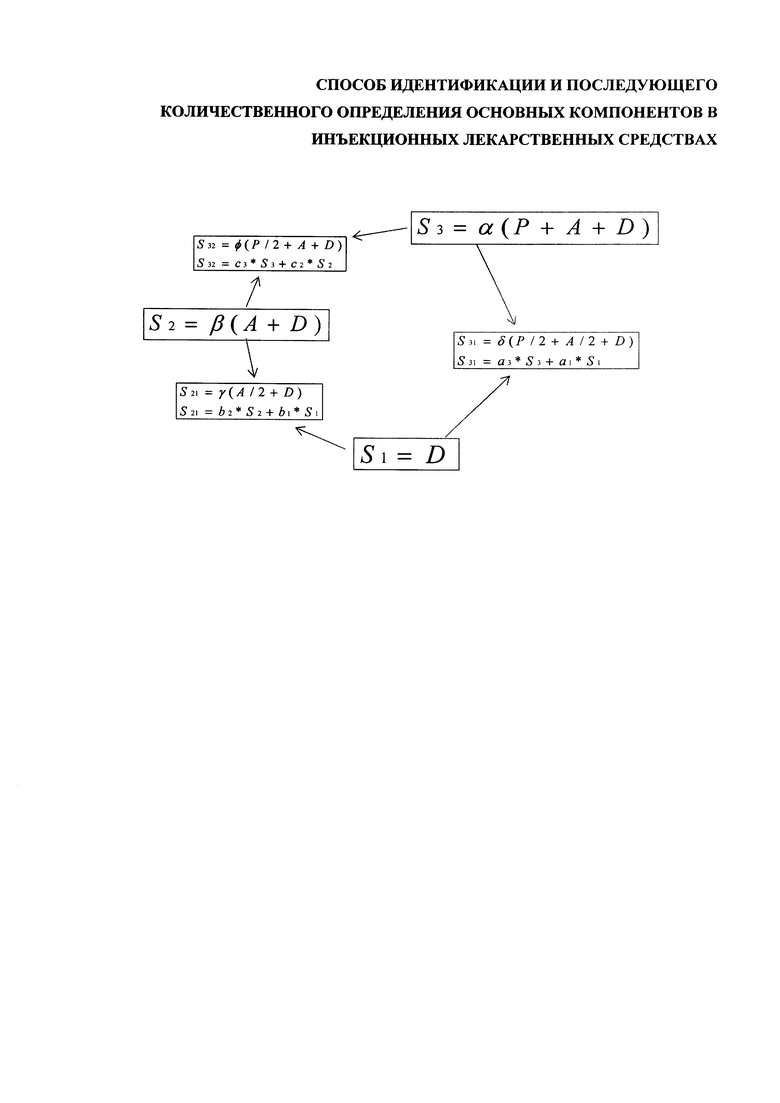
Спектры эталонного препарата, раствора вспомогательных компонентов и чистого растворителя обозначаются как S3, S2, S1 (фиг.). Их можно представить, как сумму спектров А, Р и D:
S1=D - растворитель;
S2=β(A+D) - раствора вспомогательных компонентов;
S3=α(A+D+P) - эталонный препарат.
Эти три спектра называют базовыми, а коэффициенты α, β - базовыми коэффициентами.
Помимо этих трех спектров измеряются также три спектра смесей, составленных из эталона, раствора вспомогательных компонентов и чистого растворителя, смешанных попарно в соотношении 1:1, которые так же можно разложить в суммы спектров А, Р и D:
S31=δ(P/2+A/2+D) - эталонный препарат + растворитель в соотношении 1:1;
S32=φ(P/2+A+D) - эталонный препарат + раствор вспомогательных компонентов в соотношении 1:1;
S21=γ(A/2+D) - раствор вспомогательных компонентов + растворитель в соотношении 1:1.
Множители, обозначенные буквами греческого алфавита (α, β, φ, δ, γ - коэффициенты разложения спектров), символизируют тот факт, что спектр всегда известен с точностью до произвольного множителя, φ, δ, γ в расчетах не участвуют.
Спектры S31, S32, S21 можно разложить в суммы базовых спектров, как показано на фиг.:

Коэффициенты a1, а3, b1, b2, c2, с3 - коэффициенты разложения, определяются стандартными математическими методами, например, методом наименьших квадратов.
Зная коэффициенты a1, а3, b1, b2, определяются базовые коэффициенты по формулам (13):
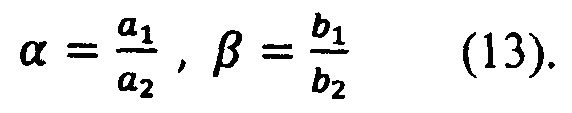
Для коэффициентов с2, с3 получается соотношение (14), которое может быть использовано для дополнительной проверки результата:
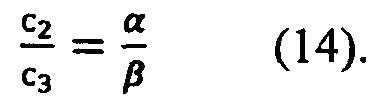
Спектр анализируемого препарата (S) так же можно разложить в сумму базовых спектров (15) с определением коэффициентов разложения k3, к2, k1:
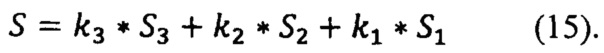
Зная базовые коэффициенты α и β, вычисляется концентрация действующего вещества и вспомогательных компонентов в анализируемом препарате (16) в единицах эталонной:
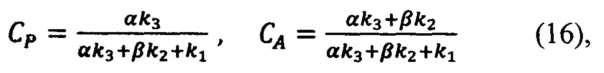
где CP - искомая концентрация действующего вещества в долях по отношению к концентрации действующего вещества в эталонном препарате, CA - искомая концентрация вспомогательного компонента в долях по отношению к концентрации вспомогательного вещества в эталонном препарате.
Для создания модели спектры шести растворов (S1, S2, S3, S31, S32, S21) (трех для двухкомпонентных моделей) измеряются несколько раз, после чего усредняются. По усредненным спектрам вычисляются базовые коэффициенты α и β. Базовые спектры и вычисленные базовые коэффициенты сохраняются в базе моделей.
Спектры S32 используются для контроля результата. Вычислив отношение αс3/βс2, полученное из усредненных спектров, проверяется, что оно находится в пределах допустимого отклонения от 1. Не усредненные спектры используются для дополнительного контроля разброса этой величины. Если результат для усредненных спектров отличается от 1 на величину большую, чем разброс результатов для не усредненных, это может свидетельствовать о систематических ошибках вроде нелинейного сложения спектров при смешивании компонент препарата.
Не усредненные спектры S3 используются для проверки полученной модели эталона. Вычислив концентрации действующего вещества и добавок для каждого из них, определяется разброс результатов. Этот разброс дает нижнюю оценку точности метода.
Таким образом, количественная модель состоит из шести наборов всех исходных спектров, а также базовых коэффициентов α и β. Каждый набор определяет усредненный спектр данного раствора Si.
Для количественного определения действующих и вспомогательных веществ исследуемого препарата его спектр раскладывают в сумму базовых спектров, затем, используя базовые коэффициенты, получают концентрации искомых компонентов, проверяют, что они находятся в допустимых пределах относительно эталонных.
Ниже приведен пример создания количественной модели двухкомпонентного лекарственного препарата (действующее вещество + растворитель).
В этом режиме отсутствует вспомогательный компонент, поэтому необходимы спектры 3 растворов - препарата, растворителя и их смеси в соотношении 1:1.
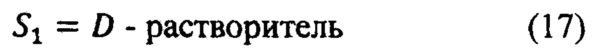

S21=γ(P/2+D) - эталонный препарат + растворитель в соотношении 1:1

Отсюда:

Таким образом, спектр испытуемого препарата можно разложить в сумму базовых:

Отсюда:
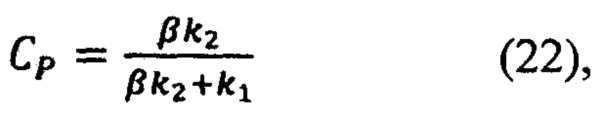
где, CP - искомая концентрация действующего вещества по отношению к концентрации эталонного спектра S2, а β, γ и φ - неизвестные множители, при этом значения γ и φ не интересует.
Модели, получаемые по разработанному способу, позволяют с высокой точностью определять количественное содержание до двух компонентов инъекционных лекарственных средств единовременно. После экспертизы образец может быть возвращен в реализацию, либо использован по своему прямому назначению.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ количественного определения действующих веществ в лекарственных препаратах методом ИК-спектроскопии | 2018 |
|
RU2698515C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ПОДЛИННОСТИ НЕФТЕПРОМЫСЛОВЫХ ХИМИЧЕСКИХ РЕАГЕНТОВ | 2020 |
|
RU2770161C1 |
| Способ качественного и количественного определения биологически активного действующего вещества в водорастворимых лекарственных препаратах | 2021 |
|
RU2774817C1 |
| СПОСОБ УВЕЛИЧЕНИЯ СООТНОШЕНИЯ СИГНАЛ/ШУМ ПРИ АНАЛИЗЕ ВОДНЫХ РАСТВОРОВ МЕТОДОМ КР-СПЕКТРОСКОПИИ | 2019 |
|
RU2719574C1 |
| СПОСОБ ЗАЩИТЫ ОТ ПОДДЕЛКИ И КОНТРОЛЯ ПОДЛИННОСТИ ИЗДЕЛИЙ | 2011 |
|
RU2450358C1 |
| ПРИМЕНЕНИЕ ГИБРИДНЫХ ПОДЛОЖЕК КРЕМНИЕВЫХ НАНОНИТЕЙ, ДЕКОРИРОВАННЫХ НАНОЧАСТИЦАМИ СЕРЕБРА И/ИЛИ ЗОЛОТА ДЛЯ ЭКСПРЕСС-ОБНАРУЖЕНИЯ ВЗРЫВЧАТЫХ ВЕЩЕСТВ | 2023 |
|
RU2821710C1 |
| Способ обнаружения наркотических веществ в отпечатках пальцев человека методом комбинационного (рамановского) рассеяния света | 2023 |
|
RU2833312C1 |
| Способ определения примеси А в лекарственных формах габапентина | 2021 |
|
RU2773851C1 |
| Способ определения давности выполнения печатных текстов на бумажной основе с помощью спектроскопии комбинационного рассеяния света | 2023 |
|
RU2808949C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ И ИДЕНТИФИКАЦИИ БИОЛОГИЧЕСКИХ МИКРООБЪЕКТОВ И ИХ НАНОКОМПОНЕНТОВ И УСТРОЙСТВО ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 2008 |
|
RU2406078C2 |
Группа изобретений относится к медицине, в частности к фармакологии, и может быть использована для идентификации и количественного определения основных компонентов в инъекционных лекарственных средствах методом спектрометрии комбинационного рассеяния света. Для этого проводят измерение спектра лекарственного препарата с помощью спектрометра в оригинальной упаковке. Сравнивают полученный спектр препарата с эталонными качественной и количественной моделями. При этом инъекционный лекарственный препарат рассматривают как смесь, состоящую из трех компонентов: действующего вещества, вспомогательных компонентов и растворителя. После чего проводят оценку полученных результатов. Идентификация заключается в определении корреляционного коэффициента для анализируемого спектра по отношению к библиотечному спектру. Также предложен способ идентификации и количественного определения основных компонентов в инъекционных лекарственных препаратах, где препарат рассматривают как смесь двух компонентов: действующего вещества и растворителя. Группа изобретений обеспечивает получение более точных результатов и уменьшение ошибок во время проведения анализа. 2 н. и 1 з.п. ф-лы, 1 ил.
1. Способ идентификации и последующего количественного определения основных компонентов в инъекционных лекарственных средствах, включающий в себя измерение спектра лекарственного препарата с помощью спектрометра в оригинальной упаковке, сравнение полученного спектра препарата с эталонной качественной и количественной моделями и оценку полученных результатов, отличающийся тем, что
метод создания эталонных количественных моделей для применения в составе библиотек спектров, используемых для скрининга качества инъекционных лекарственных средств, включает в себя получение серий нормированных спектров шести растворов исследуемого лекарственного препарата, их усреднение и предобработку, вычисление по усредненным спектрам базовых коэффициентов, сохранение базовых спектров и вычисленных базовых коэффициентов в базе моделей, при этом получение спектров растворов включает в себя
- рассмотрение инъекционного лекарственного препарата как смеси трех составляющих, включающей действующее вещество, вспомогательные компоненты и растворитель, каждая из которых имеет определенный спектр,
- получение базовых спектров: спектра действующего вещества (Р), спектра вспомогательных компонентов (А), спектра растворителя (D), а также трех спектров смесей, составленных из эталонного препарата, раствора добавок без действующего вещества и чистого растворителя, смешанных попарно в соотношении 1:1,
- разложение спектров трех смесей в суммы спектров A, D и Р согласно следующим формулам:
S1=D;
S2=β(A+D);
S3=α(A+D+P);
S31=δ(P/2+A/2+D)=a3*S3+a1*S1;
S32=φ(P/2+A+D)=c3*S3+c2*S2;
S21=γ(A/2+D)=b2*S2+b1*S1;
где S1 - спектр чистого растворителя, S2 - спектр раствора вспомогательных компонентов, S3 - спектр эталонного препарата, α, β - базовые коэффициенты, φ, δ, γ - произвольные множители, с точностью до которых известен спектр, а, b, с - коэффициенты разложения,
при этом базовые коэффициенты определяются по формулам
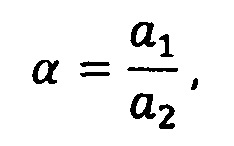
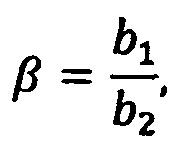
при этом спектр исследуемого инъекционного лекарственного средства раскладывают в сумму базовых спектров
S=k3*S3+k2*S2+k1*S1,
где k1, k2, k3 - коэффициенты разложения исследуемого спектра,
а концентрацию действующего вещества и добавок вычисляют в единицах эталонной модели по формулам
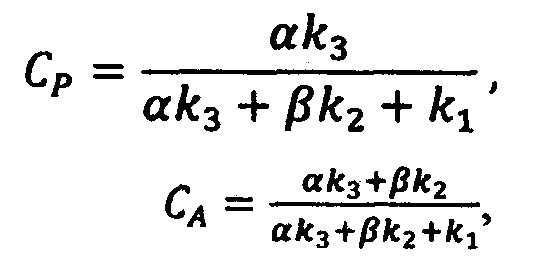
где CP - искомая концентрация действующего вещества по отношению к концентрации эталонного спектра, CA - искомая концентрация вспомогательных компонентов по отношению к концентрации эталонного спектра;
для идентификации основных компонентов в лекарственных средствах используют первую производную спектра, которая представляет собой распределение интенсивности комбинационного рассеяния света по длинам волн, предварительно обработанную с помощью фильтра разницы по Гауссу по формуле
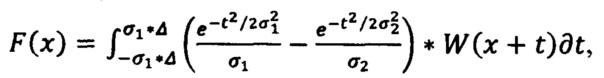
а сглаженная первая производная определяется по формуле
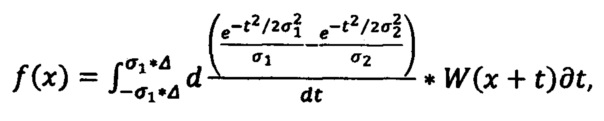
где W - сглаженный спектр, σ1 - щель (минимальное окно), σ2 - окно (максимальное окно), Δ - полуинтервал сглаживания (полуширина DoG фильтра);
а для определения соответствия испытуемого спектра модельному спектру используется коэффициент корреляции Пирсона по формуле
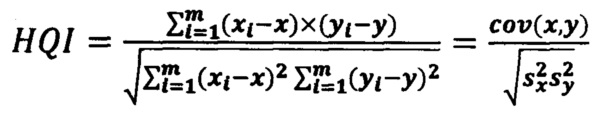
где х, у - выборочные средние, s - выборочная дисперсия, выборка xm=(x1, …, xm) - это интенсивности сравниваемого спектра, a ym=(у1, …, ym) - интенсивности эталонного спектра, a i=(1, …, m) - точки по оси абсцисс (волновое число); m определяется параметрами модели (диапазон волновых чисел);
при этом значение HQI может находиться в диапазоне 0-1, где 1 соответствует полному совпадению модельного и измеряемого спектров.
2. Способ идентификации и последующего количественного определения основных компонентов в инъекционных лекарственных средствах, включающий в себя измерение спектра лекарственного препарата с помощью спектрометра в оригинальной упаковке, сравнение полученного спектра препарата с эталонной качественной и количественной моделями и оценку полученных результатов, отличающийся тем, что метод создания эталонных количественных моделей для применения в составе библиотек спектров, используемых для скрининга качества инъекционных лекарственных средств, включает в себя получение серий нормированных спектров трех растворов исследуемого лекарственного препарата, их усреднение и предобработку, вычисление по усредненным спектрам базовых коэффициентов, сохранение базовых спектров и вычисленных базовых коэффициентов в базе моделей, при этом получение спектров растворов включает в себя
- рассмотрение инъекционного лекарственного препарата как смеси двух составляющих, включающей действующее вещество и растворитель, каждая из которых имеет определенный спектр,
- получение базовых спектров: спектра действующего вещества (Р), спектра растворителя (D) и их смеси в соотношении 1:1,
- разложение спектров согласно следующим формулам:
S1=D;
S2=β(P+D);
S21=γ(P/2+D)=b1S1+b2S2;
где S1 - спектр чистого растворителя, S2 - спектр эталонного препарата, β - базовый коэффициент, γ - произвольный множитель, с точностью до которого известен спектр, b - коэффициенты разложения,
при этом базовый коэффициент определяется по формуле
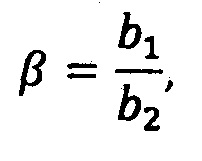
при этом спектр исследуемого инъекционного лекарственного средства раскладывают в сумму базовых спектров
S=k2*S2+k1*S1,
где k1, k2 - коэффициенты разложения исследуемого спектра,
а концентрацию действующего вещества вычисляют в единицах эталонной модели по формуле
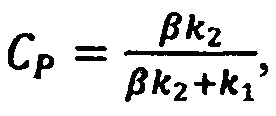
где CP - искомая концентрация действующего вещества по отношению к концентрации эталонного спектра;
для идентификации основных компонентов в лекарственных средствах используют первую производную спектра, которая представляет собой распределение интенсивности комбинационного рассеяния света по длинам волн, предварительно обработанную с помощью фильтра разницы по Гауссу по формуле
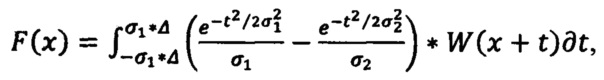
а сглаженная первая производная определяется по формуле
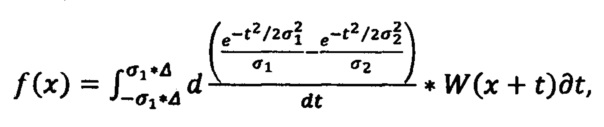
где W - сглаженный спектр, σ1 - щель (минимальное окно), σ2 - окно (максимальное окно), Δ - полуинтервал сглаживания (полуширина DoG фильтра);
а для определения соответствия испытуемого спектра модельному спектру используется коэффициент корреляции Пирсона по формуле
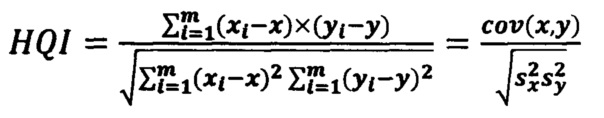
где х, у - выборочные средние, s - выборочная дисперсия, выборка xm=(x1, …, xm) - это интенсивности сравниваемого спектра, a ym=(у1, …, ym) - интенсивности эталонного спектра, a i=(1, …, m) - точки по оси абсцисс (волновое число); m определяется параметрами модели (диапазон волновых чисел);
при этом значение HQI может находиться в диапазоне 0-1, где 1 соответствует полному совпадению модельного и измеряемого спектров.
3. Способ идентификации и последующего количественного определения основных компонентов в инъекционных лекарственных средствах по п. 1 или 2, отличающийся тем, что при выполнении измерений применяют спектрометр комбинационного рассеяния со спектральным диапазоном не хуже 100-4000 см-1, разрешением не менее 22 см-1 и источником излучения - лазером с длиной волны 532 нм; а все исходные спектры, используемые в расчетах, нормированы на спектр раствора пирацетама 200 мг/мл с базового эталонного прибора, который сохраняется в энергонезависимую память каждого спектрометра (ХЭТпир)
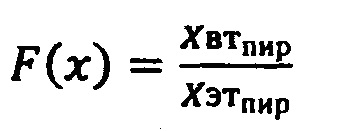
X=Хэт*F(x)
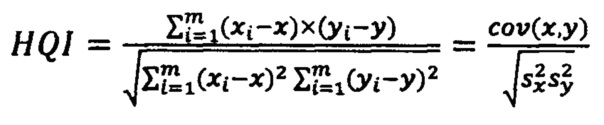
и любой спектр (X), полученный на второстепенных спектрометрах, нормируется на специально подобранную полиномиальную функцию (F(x)), причем функция нормировки подбирается таким образом, чтобы коррелятор (HQI) спектра пирацетама, измеренный на эталонном и второстепенном приборах, был не ниже 0,99.
| RU 2015109714 A, 20.08.2013 | |||
| Приспособление к зерноподъемникам для отбора проб зерна | 1927 |
|
SU18974A1 |
| SATO-BERRÚ R | |||
| Y | |||
| et al | |||
| Quantitative NIR Raman analysis in liquid mixtures, Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, 2004, Aug, V.60, (10), Р | |||
| Автоматический указатель остановок трамвайного вагона | 1924 |
|
SU2225A1 |
| PRATIWIA D | |||
| et al | |||
| Quantitative analysis of polymorphic mixtures of ranitidine hydrochloride by Raman spectroscopy and principal components analysis, Eur | |||
| J | |||
| Pharm | |||
| Biopharm., 2002, 54(3), P | |||
| Ленточный тормозной башмак | 1922 |
|
SU337A1 |

