Изобретение относится к медицине, фармации, химии, а именно, к способу определения фармацевтических примесей. Заявленный способ может быть использован для контроля качества субстанций или различных лекарственных форм габапентина путем идентификации его примеси А с последующим ее количественным определением.
Уровень техники
Габапентин представляет собой химическое соединение 2-[1-(аминометил) циклогексил] уксусная кислота, являющееся синтетическим небензодиазепиновым аналогом γ-аминомасляной кислоты, и широко использующееся для лечения эпилепсии, а также для снижения симптомов периферической нейропатической боли, постгерпетической невралгии, диабетической периферической невропатии, острой алкогольной абстиненции, рассеянного склероза [1, 2, 3]. В растворе и в твердой фазе габапентин способен за счет внутримолекулярной циклизации образовывать примесь А, представляющую собой химическое соединение 2-азаспиро[4,5]декан-3-он. Примесь А обладает токсичностью [4], поэтому ее содержание в субстанциях габапентина и препаратах на его основе нормируют и определяют в ходе контроля качества [5, 6, 7].
Известен способ идентификации и определения содержания примеси А в лекарственных формах габапентина на основе метода высоко эффективной жидкостной хроматографии (далее - ВЭЖХ), включающий сравнение времен удерживания и площадей пиков на хроматограмме лекарственной формы габапентина со временами удерживания и площадями пиков на хроматограмме смеси стандартных образцов габапентина и примеси А [5, 6, 7]. Недостатком указанного способа является необходимость использования стандартного образца для построения градуировочной функции между измеряемой площадью пика на хроматограмме и содержанием примеси А в лекарственной форме габапентина (относительность измерения), а также зависимость неопределенности результата измерения содержания примеси А не только от неопределенности измерения площади пика на хроматограмме, но и от неопределенности взятия навесок испытуемого и стандартного образцов, объемов растворителя (косвенность измерения).
Наиболее близким прототипом к предлагаемому изобретению является способ определения содержания примесей А, В и С в фармацевтической субстанции салициловой кислоты методом спектроскопии ядерного магнитного резонанса на протонах (далее - 1Н ЯМР) [8], в котором количественное содержание примеси относительно основного компонента методом ЯМР определяют, измеряя отношение интегральных интенсивностей сигналов основного и примесного компонентов в спектре 1Н ЯМР. Неопределенность результата зависит только от неопределенности этого измерения.
Существует потребность в разработке эффективного и селективного способа идентификации примеси А с последующим ее количественным определением в субстанциях габапентина и его различных лекарственных формах, произведенных различными производителями.
Описание сущности изобретения
Задачей изобретения является разработка способа идентификации (определения) примеси А с последующим ее количественным определением в различных лекарственных формах габапентина, произведенных различными производителями, без применения стандартного образца. Поставленная задача решается путем использования метода спектроскопии ядерного магнитного резонанса на протонах (далее - 1Н ЯМР), что приводит к возможности идентификации (определения) примеси А с последующим ее количественным (весовым) определением содержания относительно габапентина без использования стандартного образца.
Техническим результатом предлагаемого технического решения для определения примеси А в субстанции габапентина и его лекарственных формах является эффективность и селективность.
Сущность предложенного способа заключается в эффективном выявлении характеристических сигналов метиленовых групп габапентина, где габапентин представляет собой химическое соединение 2-[1-(аминометил)циклогексил]уксусная кислота, которое является синтетическим небензодиазепиновым аналогом γ-аминомасляной кислоты, и имеет структурную формулу габапентина
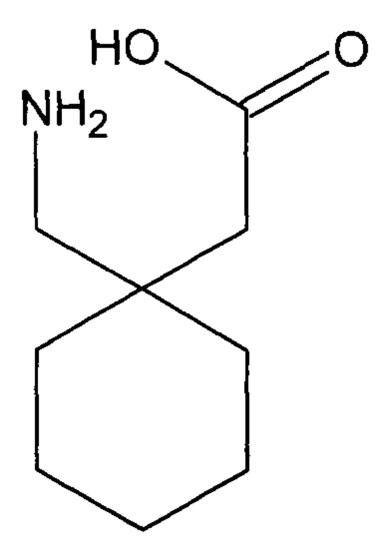
и примеси А, в виде химического соединения 2-азаспиро[4,5]декан-3-он, и имеет структурную формулу примеси А габапентина
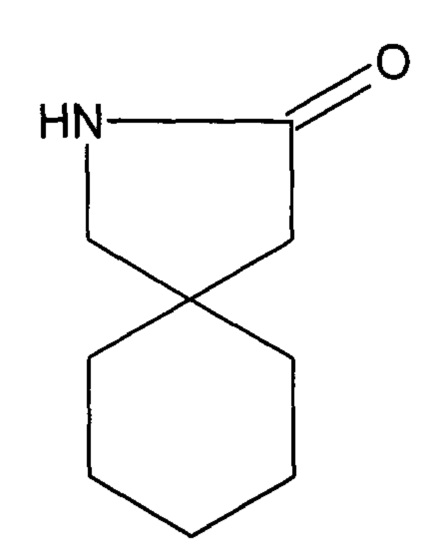
путем измерения значений их интегральных интенсивностей в спектрах 1Н ЯМР лекарственных форм габапентина.
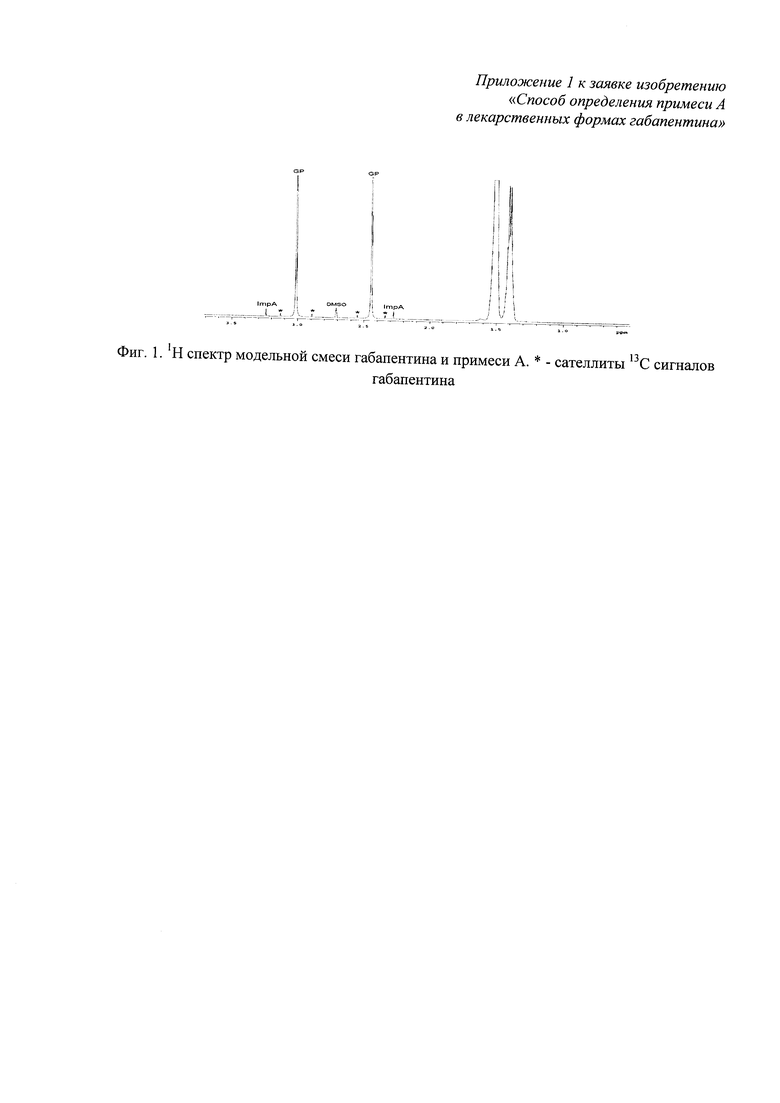
Несмотря на сходство структур габапентина и примеси А, перекрывание их сигналов на спектре 1Н наблюдается только в области циклогексанового фрагмента 1,25-1,70 м.д. (фиг. 1). Характеристическими сигналами идентификации габапентина и примеси А являются: 1Н (D2O), δ, м.д. габапентин: 2,45±0,01 (с, 2Н, СН2-С=O), 3,02±0,01 (с, 2Н, CH2-N); примесь А: 2,28±0,01 (с, 2Н, СН2-С=O), 3,24±0,01 (с, 2Н, CH2-N).
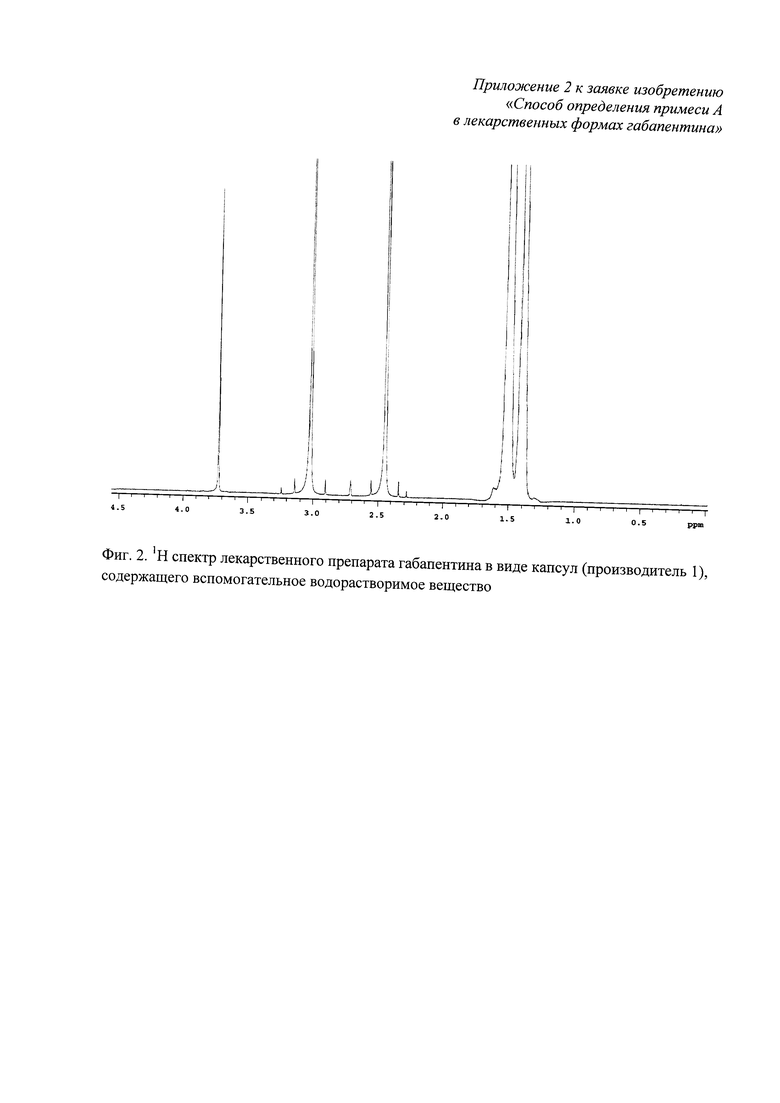
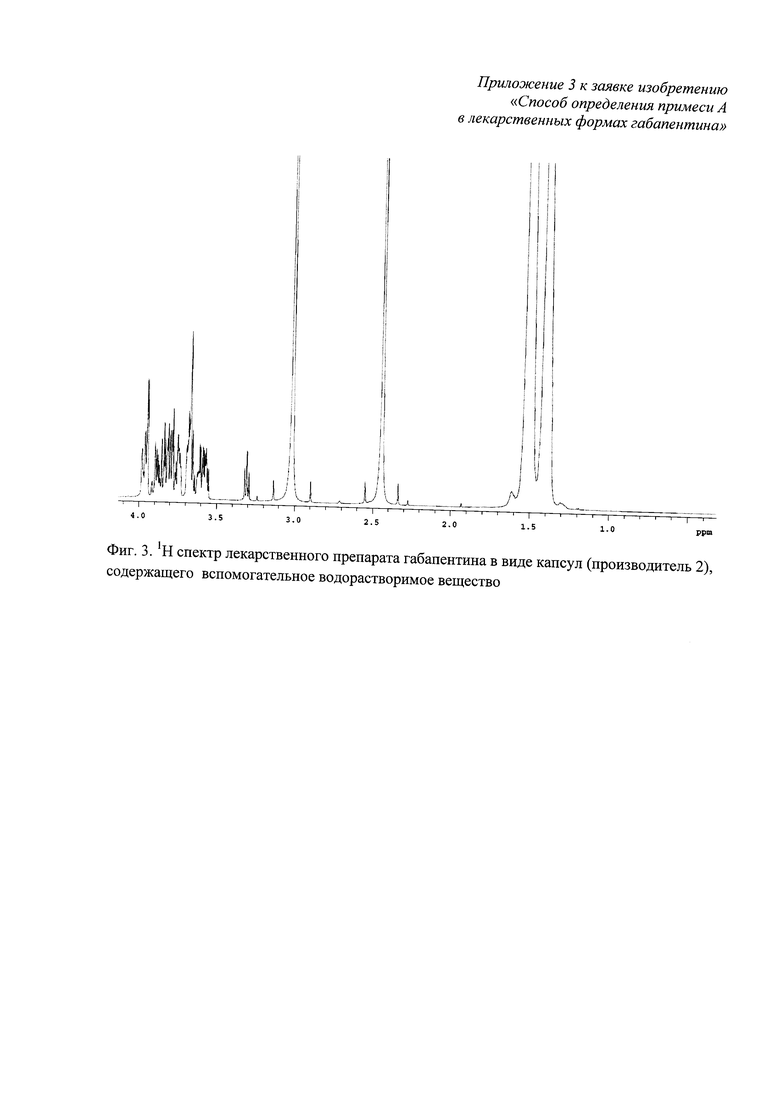
Характеристические сигналы примеси А не перекрываются с характеристическими сигналами 1Н габапентина и с их 13С сателлитами (фиг. 1), а также с сигналами водорастворимых вспомогательных веществ лекарственных форм габапентина - полиэтиленгликоля, лактозы моногидрата (фиг. 2, фиг. 3).
Неожиданно обнаружили, что эффективность заявленного способа заключается в том, что спектр 1Н регистрируют при температуре 300 К на ЯМР-спектрометре с рабочей частотой по протонам не менее 400 МГц; калибровку шкалы химических сдвигов 1Н проводят по сигналу метальной группы диметилсульфоксида - эталонного соединения, добавляемого в испытуемый образец (δ=2,71 м.д.). Примесь А в субстанции или лекарственной форме габапентина идентифицируют, наблюдая в спектре 1Н ЯМР характеристические синглетные сигналы 2,28±0,01 и 3,24±0,01 м.д. Положения характеристических сигналов габапентина и примеси А в спектре 1Н зависит от таких факторов, как температура эксперимента и значение химического сдвига сигнала эталонного соединения. Характеристические сигналы интегрируют с соблюдением правил прецизионного интегрирования: отдельно настраивают фазу каждого интегрируемого сигнала, диапазон интегрирования приравнивают к 64-кратной ширине интегрируемого сигнала на его полувысоте, углеродные сателлиты исключают из диапазона интегрирования [10]. Весовой % (w %) примеси А относительно габапентина рассчитывают по формуле (1):

где 0,901 - отношение молекулярных масс примеси А и габапентина;
IImpA - измеренная интегральная интенсивность любого из характеристических сигналов примеси А (2,28±0,01; 3,24±0,01 м.д.) или их усредненное значение;
Igp - измеренная интегральная интенсивность любого из характеристических сигналов габапентина (2,45±0,01; 3,02±0,01 м.д.) или их усредненное значение.
Существенным отличительным признаком заявляемого изобретения является идентификация с последующим количественным определением методом 1Н ЯМР примеси А, содержащейся в субстанциях или лекарственных формах габапентина без использования стандартных образцов и без построения градуировочной функции. Преимуществом заявленного технического решения является:
1) возможность идентификации с последующим количественным измерением содержания примеси А в субстанции габапентина и его лекарственных формах без использования фармакопейных и рабочих стандартных образцов;
2) повышение точности количественных измерений за счет ликвидации неопределенности измерения, связанной со взятием навесок испытуемого образца субстанции или лекарственной формы габапентина и стандартного образца примеси А, с измерением объемов растворителей, с построением градуировочной функции, с аттестованным значением содержания основного компонента в стандартном образце;
3) сокращение общих трудозатрат на стадиях пробоподготовки и анализа.
Заявленный способ позволяет идентифицировать примесь А с последующим ее количественным определением, не только в субстанциях габапентина, но и в различных лекарственных формах (препаратах) габапентина, произведенных различными производителями, где лекарственные формы могут содержать фармацевтически приемлемые вспомогательные вещества, выбранные из полиэтиленгликоля, лактозы моногидрат, кальция гидрофосфат дигидрат, крахмал картофельный, вода, магния стеарат и других приемлемых вспомогательных веществ.
Краткое описание чертежей и иных материалов (см. Приложения 1-6):
Фиг. 1. 1Н спектр модельной смеси габапентина и примеси А;
Фиг. 2. 1Н спектр лекарственного препарата габапентина в виде капсул (производитель 1), содержащего вспомогательное водорастворимое вещество;
Фиг. 3. 1Н спектр лекарственного препарата габапентина в виде капсул (производитель 2), содержащего вспомогательное водорастворимое вещество;
Фиг. 4. Фрагмент спектра 1Н субстанции габапентина с измеренными интегральными интенсивностями характеристических сигналов габапентина и примеси А;
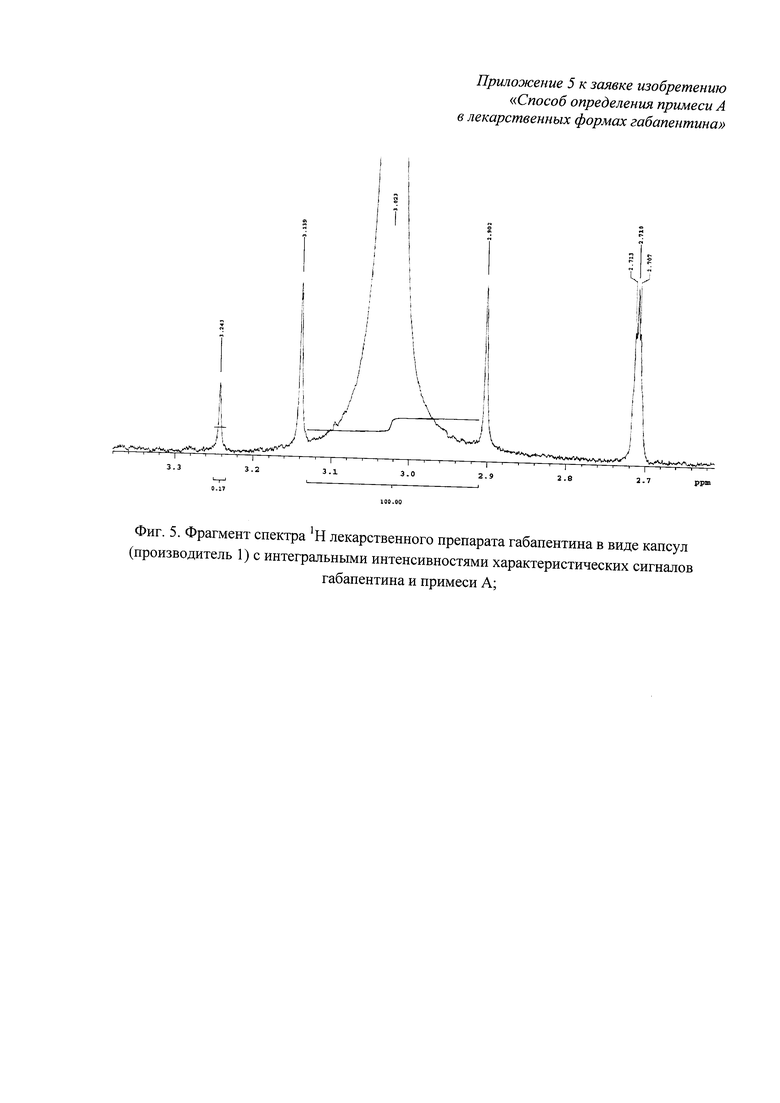
Фиг. 5. Фрагмент спектра 1Н лекарственного препарата габапентина в виде капсул (производитель 1) с измеренными интегральными интенсивностями характеристических сигналов габапентина и примеси А;
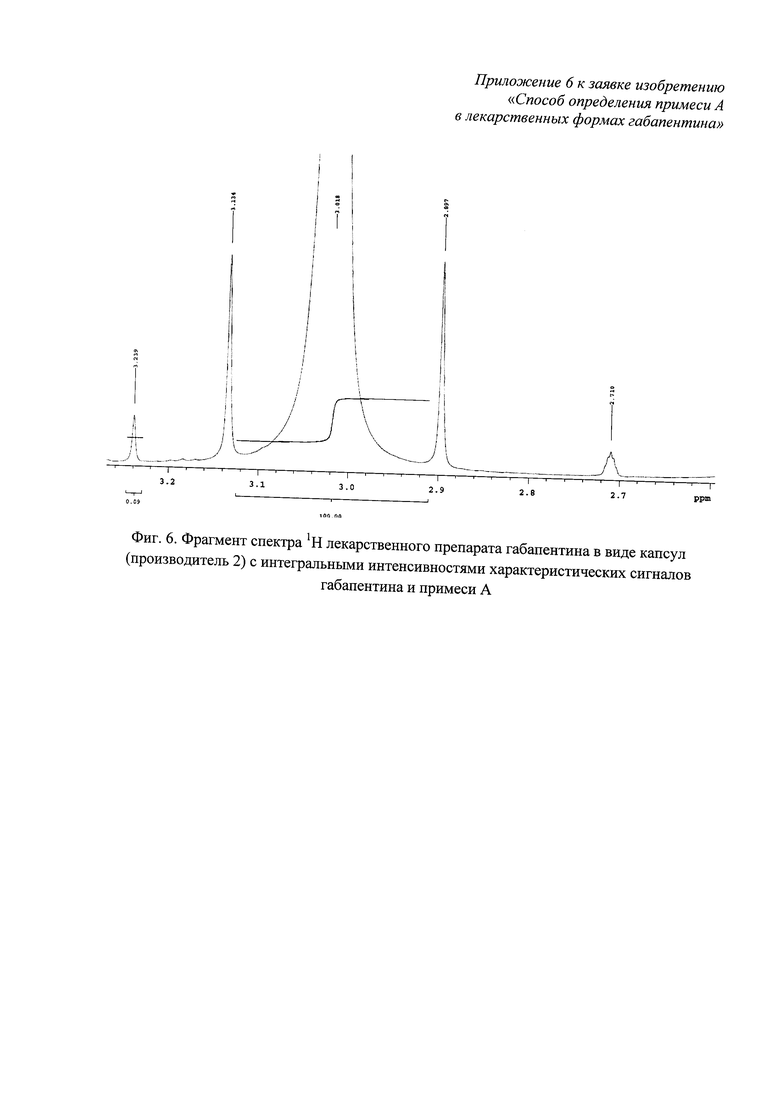
Фиг. 6. Фрагмент спектра 1Н лекарственного препарата габапентина в виде капсул (производитель 2) с измеренными интегральными интенсивностями характеристических сигналов габапентина и примеси А.
Возможность осуществления заявляемого изобретения раскрыта в следующих примерах.
Пример 1. Идентификация примеси А с последующим ее количественным определением содержания в субстанции габапентина методом 1Н ЯМР спектроскопии.
Эффективное количество субстанции в навеске, около 20 мг субстанции, помещают в ЯМР-ампулу, добавляют 0,5 мл дейтерированной воды (D2O) и 10 мкл диметилсульфоксида (ДМСО), интенсивно встряхивают до полного растворения навески образца субстанции; далее, идентифицируют характеристические сигналы габапентина и примеси А путем регистрации спектра 1Н на ЯМР-спектрометре с рабочей частотой по протонам не менее 400 МГц при температуре 300 К. Параметры эксперимента: ширина спектра -6009,6 Гц, угол поворота намагниченности - 90°, время релаксации - 10 с, количество накоплений сигнала свободной индукции - 512, число точек аналого-цифрового преобразования - 64к, экспоненциальное умножение - 0,3 Гц, автоматическая коррекция базовой линии спектра, ручная настройка фазы, калибровка шкалы δ под ДМСО в D2O (δ=2,71 м.д.).
Наблюдаемые в спектре 1Н характеристические синглетные сигналы габапентина δ 2,45; 3,02 м.д. и примеси А 8 2,28 и 3,24 м.д. подтверждают наличие в субстанции габапентина примеси А. Для расчета весовой доли примеси А относительно габапентина используют измеренные интегральные интенсивности характеристических сигналов δ 3,02 м.д. (IGp=1000) и 3,24 м.д. (IImPA=0,17) (фиг. 4) по формуле (1):
w %=0,901(0,17/1000)100=0,015%.
Пример 2. Идентификация примеси А с последующим ее количественным определением содержания в лекарственном препарате габапентина в виде капсул, производитель 1, методом 1Н ЯМР спектроскопии.
К эффективному количеству содержимого капсулы (около 1/2 капсулы), представляющего собой однородную смесь эффективных количеств габапентина и фармацевтических приемлемых вспомогательных веществ, добавляют 1,5 мл D2O и затем интенсивно встряхивают в течение 10 мин до получения однородной суспензии; полученную суспензию фильтруют через устройство для фильтрования (фильтр), известное специалисту из данной области техники, затем 0,5 мл фильтрата переносят в ЯМР-ампулу и добавляют 10 мкл ДМСО для калибровки шкалы химических сдвигов (δ=2,71 м.д.); затем идентифицируют характеристические сигналы габапентина и примеси А путем регистрации спектра 1Н на ЯМР-спектрометре с рабочей частотой по протонам не менее 400 МГц при температуре 300 К. Параметры эксперимента аналогичны примеру 1.
Наблюдаемые в спектре 1Н синглетные сигналы габапентина δ 2,45;3,02 м.д. и примеси А 8 2,28 и 3,24 м.д. подтверждают наличие в лекарственном препарате габапентина (капсулы) примеси А. Для расчета весовой доли примеси А относительно габапентина используют измеренные интегральные интенсивности характеристических сигналов δ 3,02 м.д. (Igp=100) и 3,24 м.д. (IImpA=0,17) (фиг. 5) по формуле (1):
w %=0,901(0,17/100)100=0,15%.
Пример 3. Идентификация примеси А с последующим ее количественным определением содержания в лекарственном препарате габапентина в виде капсул, производитель 2, методом 1Н ЯМР спектроскопии.
К эффективному количеству содержимого капсулы (около 1/2 капсулы), представляющего собой однородную смесь эффективных количеств габапентина и фармацевтических приемлемых вспомогательных веществ, добавляют 1,5 мл D2O и интенсивно встряхивают в течение 10 мин до получения однородной суспензии; далее полученную суспензию фильтруют через устройство для фильтрования (фильтр), известное специалисту из данной области техники, затем 0,5 мл фильтрата переносят в ЯМР-ампулу и добавляют 10 мкл ДМСО для калибровки шкалы химических сдвигов (δ=2,71 м.д.); затем идентифицируют характеристические сигналы габапентина и примеси А путем регистрации спектра 1Н на ЯМР-спектрометре с рабочей частотой по протонам не менее 400 МГц при температуре 300 К. Параметры эксперимента аналогичны примеру 1.
Наблюдаемые в спектре 1Н синглетные сигналы габапентина δ 2,45;3,02 м.д. и примеси А 8 2,28 и 3,24 м.д. подтверждают наличие в лекарственном препарате габапентина (капсулы) примеси А. Для расчета весовой доли примеси А относительно габапентина используют измеренные интегральные интенсивности характеристических сигналов: δ 3,02 м.д. (IGp=100) и 3,24 м.д. (IImpA=0,09) (фиг. 6) по формуле (1):
w %=0,901(0,09/100)100=0,008%.
Пример 4 Измерение весовых долей примеси А методом высокоэффективной жидкостной хроматографии (далее -ВЭЖХ).
В субстанции габапентина и его лекарственных формах (препаратах) в виде капсул различных производителей, весовые доли примеси А измеряли методом ВЭЖХ. Приготовление раствора для проверки пригодности системы, буферного раствора, испытуемых растворов субстанции габапентина и лекарственных препаратов габапентина в виде капсул, калибровочных растворов и подвижной фазы осуществляли по методикам [5, 6]. Условия хроматографирования: колонка мм × 4,6 мм × 5 мкм; температура колонки 40°С; режим элюирования изократический; скорость потока 1 мл / мин; детектор УФ 215 нм; объем инжекции 20 мкл; время хроматографирования не менее 50 мин. Весовые доли примеси А, измеренные методом ВЭЖХ, составили: 0,02 w % для субстанции габапентина; 0,13 w % для лекарственного препарата габапентина в виде капсул производитель 1); 0,07 w % для лекарственного препарата габапентина в виде капсул (производитель 2).

Результаты измерения методом спектроскопии 1Н ЯМР сопоставимы с результатами измерения методом ВЭЖХ (см. табл. 1). Приведенные примеры показывают возможность идентификации примеси А с последующим ее количественным определением методом 1Н ЯМР спектроскопии содержания в субстанции или различных лекарственных формах габапентина.
Представленные примеры не ограничивают объем притязаний настоящего изобретения и служат только для цели иллюстрации и раскрытия заявленного способа.
Промышленная применимость
Все приведенные примеры, подтверждают эффективность и селективность заявленного способа.
Таким образом, поставленная техническая задача, а именно, разработка эффективного и селективного способа для идентификации примеси А с последующим ее количественным определением в субстанции габапентина или его различных лекарственных формах, произведенных различными производителями, достигнута, что подтверждается приведенными примерами.
Применение в фармации, медицине, химии заявленного способа является эффективным и селективным.
Список литературы
1.1. K. Celikyurt, О. Mutlu, G. Ulak, F.Y. Akar, F. Erden. Neurosci. Lett. 2011, 492, 124-128.
2. S Hiom, G.K. Patel, R.G. Newcombe, S Khot, С Martin. Br. J. Dermatol. 2015, 173, 300-302.
3. M.L. Bums, E. Kinge, M.S. Opdal, S.I. Johannessen, C.J. Landmark. Acta Neurol Scand. 2019, 139, 446-454.
4. Z. Zong, J. Qiu, R. Tinmanee, L.E. Kirsch. J. Pharm. Set 2012, 101, 2123-2133.
5. USP43-NF38. Gabapentin. 2057. https://online.uspnf.com/
6. USP43-NF38. Gabapentin Capsules. 2058. https://online.uspnf.com/
7. USP43-NF38. Gabapentin Tablets. 2060. https://online.uspnf.comy
8. С.В. Моисеев, В.И. Крылов, Т.В. Мастеркова, В.А. Яшкир, Н.Д. Бунятян. Использование метода ЯМР-спектроскопии для подтверждения подлинности, идентификации и количественного определения посторонних примесей субстанции салициловой кислоты. Ведомости НЦЭСМП, 2014, 1, 15-19.
9. Н.Е. Gottlieb, V. Kotlyar and A. Nudelman. NMR Chemical Shifts of Common Laboratory Solvents as Trace Impurities. J. Org. Chem. 1997, 62(21), 7512-7515.
10. F. Malz and H.Jancke. Validation of quantitative nuclear magnetic resonance. J. Pharm. Biomed. 2005, 38, 813-823.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ идентификации и количественного определения эпимеров будесонида в его лекарственных формах | 2022 |
|
RU2802028C2 |
| Способ идентификации и количественного определения содержания олигопептидов в фармацевтической субстанции "Пептофорс" методом спектроскопии ЯМР | 2019 |
|
RU2709020C1 |
| Способ одновременного определения степеней окисления и алкилирования азоксимера бромида - действующего вещества полиоксидония - методом C спектроскопии ЯМР | 2019 |
|
RU2713805C1 |
| Способ определения летучих компонентов в лекарственных препаратах | 2022 |
|
RU2790000C1 |
| Способ количественного определения фтивазида | 2024 |
|
RU2828350C1 |
| Определение стабилизаторов углеводной природы в биологически активных препаратах | 2023 |
|
RU2816030C1 |
| Способ определения арбутина в листьях толокнянки | 2023 |
|
RU2802173C1 |
| Способ количественного определения аскорбиновой кислоты в лекарственных растительных препаратах | 2023 |
|
RU2801885C1 |
| Способ количественного определения ионов алюминия атомно-абсорбционной спектрометрией с электротермической атомизацией | 2022 |
|
RU2799235C1 |
| Определение полисорбата 80 в биологических лекарственных препаратах | 2023 |
|
RU2812788C1 |

Группа изобретений относится к медицине и фармацевтике и может быть использована для контроля качества субстанций или различных лекарственных форм габапентина путем идентификации его примеси А с последующим ее количественным определением. Способ определения примеси А в субстанциях габапентина спектроскопией ядерного магнитного резонанса на протонах (1Н ЯМР), где примесь А представляет собой 2-азаспиро[4,5]декан-3-он, характеризуется тем, что субстанцию габапентина растворяют в дейтерированной воде (D2O) и диметилсульфоксиде (ДМСО) при интенсивном встряхивании до полного ее растворения, идентифицируют характеристические сигналы габапентина и его примеси А путем регистрации спектра 1Н на ЯМР спектрометре с рабочей частотой по протонам не менее 400 МГц при температуре 300 К, калибруют шкалы химических сдвигов 1H под сигнал метильной группы ДМСО δ=2,71 м.д. и рассчитывают весовую долю примеси А относительно габапентина с использованием измеренных интегральных интенсивностей сигналов. Группа изобретений касается также варианта способа определения примеси А в лекарственных препаратах габапентина в виде капсул и применения указанных способов для определения примеси А в субстанции габапентина или ее лекарственных формах. Обеспечивается повышение точности определения примеси А без использования стандартных образцов и без построения градуировочной функции. 3 н. и 3 з.п. ф-лы, 6 ил., 1 табл., 4 пр.
1. Способ определения примеси А в субстанциях габапентина спектроскопией ядерного магнитного резонанса на протонах (1Н ЯМР), где примесь А представляет собой 2-азаспиро[4,5]декан-3-он, и характеризующийся тем, что субстанцию габапентина растворяют в дейтерированной воде (D2O) и диметилсульфоксиде (ДМСО) при интенсивном встряхивании до полного ее растворения, идентифицируют характеристические сигналы габапентина и его примеси А путем регистрации спектра 1Н на ЯМР спектрометре с рабочей частотой по протонам не менее 400 МГц при температуре 300 К, калибруют шкалы химических сдвигов 1H под сигнал метильной группы ДМСО δ=2,71 м.д., рассчитывают весовую долю примеси А относительно габапентина с использованием измеренных интегральных интенсивностей сигналов.
2. Способ определения примеси А в лекарственных препаратах габапентина в виде капсул спектроскопией ядерного магнитного резонанса на протонах (1Н ЯМР), где примесь А представляет собой 2-азаспиро[4,5]декан-3-он, и характеризующийся тем, что содержимое капсулы габапентина и дейтерированную воду (D2O) интенсивно встряхивают до получения однородной суспензии, фильтруют, добавляют диметилсульфоксид (ДМСО) для калибровки шкалы химических сдвигов, идентифицируют характеристические сигналы габапентина и примеси А путем регистрации спектра 1Н на ЯМР спектрометре с рабочей частотой по протонам не менее 400 МГц, при температуре 300 К, калибруют шкалы химических сдвигов 1H под сигнал метильной группы ДМСО δ=2,71 м.д., рассчитывают весовую долю примеси А относительно габапентина с использованием измеренных интегральных интенсивностей сигналов.
3. Способ по п. 1 или 2, дополнительно характеризующийся тем, что характеристические сигналы составляют для габапентина δ 2,45±0,01; 3,02±0,01 м.д. и для примеси А 2,28±0,01; 3,24±0,01 м.д.
4. Способ по п. 1 или 2, дополнительно характеризующийся тем, что для расчета весовой доли примеси А относительно габапентина используют измеренные интегральные интенсивности характеристических сигналов 3,02±0,01 м.д. для габапентина (IGP) и δ 3,24±0,01 м.д. примеси А (IImpA).
5. Способ по п. 2, дополнительно характеризующийся тем, что содержимое капсулы габапентина представляет собой однородную смесь габапентина и вспомогательных веществ.
6. Применение способа по п. 1 или 2 для определения примеси А в субстанции габапентина или ее лекарственных формах.
| Способ идентификации и количественного определения содержания олигопептидов в фармацевтической субстанции "Пептофорс" методом спектроскопии ЯМР | 2019 |
|
RU2709020C1 |
| CN 106053510 A, 26.10.2016 | |||
| КИМ Г.А | |||
| и др | |||
| Оптимизация метода ТСХ для обнаружения посторонних примесей в противоэпилептическом препарате | |||
| Вестник ВГУ, Серия: Химия | |||
| Биология | |||
| Фармация | |||
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| МОИСЕЕВ С.В | |||
| и др | |||
| Использование метода ЯМР-спектроскопии для подтверждения подлинности, идентификации и | |||

