[1] Настоящая заявка испрашивает приоритет на основании патентной заявки Китая CN 201810323355.2, поданной 11 апреля 2018 г. Содержание патентной заявки Китая полностью включено в настоящее описание посредством ссылки.
Область техники
[2] Настоящее изобретение относится к полициклическим производным карбамоилпиридона, фармацевтическим композициям и их применению.
Уровень техники
[3] Грипп представляет собой острую вирусную инфекцию, которая легко распространяется среди людей. Инфекции гриппа достигают пика зимой в регионах с умеренным климатом. Вирус гриппа в настоящее время является серьезной проблемой для здравоохранения, поскольку этот вирус широко распространен во всем мире, им может заразиться любой человек любой возрастной группы, а в группах высокого риска часто возникают серьезные сопутствующие заболевания и наступает смерть. По статистике, пандемии гриппа ежегодно вызывают от 3 до 5 миллионов тяжелых случаев заболевания и от 250000 до 500000 смертей.
[4] Поскольку геном вируса гриппа очень мал, вирус гриппа полагается на систему трансляции клеток-хозяев для синтеза необходимых белков. Следовательно, матричная РНК (мРНК) вируса гриппа должна иметь как 5'кэп-структуру (CAP), так и структуру 3'-поли(A)-хвоста, которые могут распознаваться системой трансляции клетки-хозяина. 5'кэп-структура отщепляется с 5'-конца пре-мРНК клетки-хозяина за счет эндонуклеазной активности PA-субъединицы в полимеразном комплексе РНК вируса гриппа. В ходе этого процесса, называемого «отщеплением кэпа», кэп-структура CAP мРНК хозяина используется для транскрипции собственной мРНК вируса, что необходимо для инициации транскрипции вируса гриппа.
[5] Поскольку «отщепление кэпа» является ключевой частью цикла репликации вируса гриппа, и в клетке-хозяине нет аналогичного механизма и соответствующей протеазы, ингибиторы эндонуклеазы, ответственной за отщепление кэпа, могут избирательно блокировать процесс транскрипции вируса гриппа, не влияя одновременно на клетку-хозяина. Таким образом, этот механизм позволяет рассматривать эндонуклеазу как перспективную мишень для противогриппозных препаратов. Настоящее раскрытие удовлетворяет эти потребности и обеспечивает другие преимущества.
РАСКРЫТИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
[6] Настоящее изобретение относится к полициклическому производному карбамоилпиридона, представленному формулой (VII), стереоизомеру указанного соединения, таутомеру указанного соединения, гидрату указанного соединения, сольвату указанного соединения, активному метаболиту указанного соединения, кристаллической форме указанного соединения, фармацевтически приемлемой соли указанного соединения или пролекарству указанного соединения,
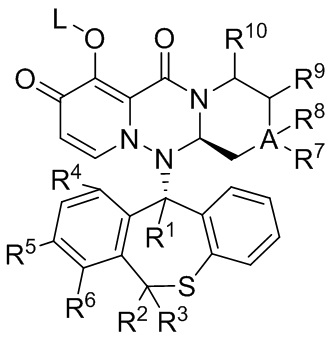
формула (VII)
[7] где
[8] R1 представляет собой водород или дейтерий;
[9] R2 представляет собой водород или дейтерий;
[10] R3 представляет собой водород или дейтерий;
[11] L представляет собой водород, алкил или (метоксикарбонил)оксиметил (т.е. -CH2OC(=O)OCH3);
[12] и по меньшей мере один из R1, R2 и R3 представляет собой дейтерий;
[13] R4 представляет собой водород или галоген (предпочтительно хлор);
[14] R5 представляет собой водород или галоген (предпочтительно фтор);
[15] R6 представляет собой водород или галоген (предпочтительно фтор);
[16] A представляет собой C или O, когда A представляет собой C, то R7 и R8 оба представляют собой водород или R7 и R8 оба представляют собой метил; когда A представляет собой O, тогда R7 и R8 отсутствуют;
[17] R9 представляет собой водород или метил; R10 представляет собой водород или метил;
[18] и R4, R5 и R6 одновременно не представляют собой водород.
[19] Предпочтительно, указанное полициклическое производное карбамоилпиридона, представленное формулой (VII), удовлетворяет любому одному из следующих условий:
[20] условие 1: R4 представляет собой водород; R5 представляет собой фтор; R6 представляет собой фтор; A представляет собой O; R9 представляет собой водород; R10 представляет собой водород;
[21] условие 2: R4 представляет собой водород; R5 представляет собой фтор; R6 представляет собой водород; A представляет собой O; R9 представляет собой водород; R10 представляет собой метил и при этом атом углерода, который непосредственно соединен с R10, имеет конфигурацию (S);
[22] условие 3: R4 представляет собой хлор; R5 представляет собой водород; R6 представляет собой водород; A представляет собой O; R9 представляет собой водород; R10 представляет собой водород;
[23] условие 4: R4 представляет собой водород; R5 представляет собой фтор; R6 представляет собой фтор; A представляет собой C; R7 и R8 оба представляют собой метил; R9 представляет собой водород; R10 представляет собой водород;
[24] условие 5: R4 представляет собой водород; R5 представляет собой фтор; R6 представляет собой фтор; A представляет собой C; R7 и R8 оба представляют собой водород; R9 представляет собой водород и при этом атом углерода, который непосредственно соединен с R9, имеет конфигурацию (R); R10 представляет собой водород;
[25] условие 6: R4 представляет собой водород; R5 представляет собой фтор; R6 представляет собой водород; A представляет собой C; R7 и R8 оба представляют собой водород; R9 представляет собой водород; R10 представляет собой водород.
[26] В настоящем описании, когда L представляет собой алкил, тогда указанный алкил предпочтительно представляет собой C1-4 алкил, а еще более предпочтительно метил, этил, пропил, изопропил, бутил, изобутил или трет-бутил.
[27] Настоящее изобретение относится к полициклическому производному карбамоилпиридона, представленному формулой (I), стереоизомеру указанного соединения, таутомеру указанного соединения, гидрату указанного соединения, сольвату указанного соединения, активному метаболиту указанного соединения, кристаллической форме указанного соединения, фармацевтически приемлемой соли указанного соединения или пролекарству указанного соединения,
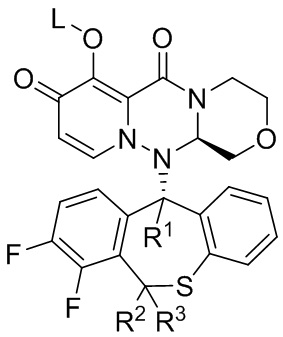
Формула (I)
[28] где:
[29] R1 представляет собой водород или дейтерий;
[30] R2 представляет собой водород или дейтерий;
[31] R3 представляет собой водород или дейтерий;
[32] L представляет собой водород, алкил или (метоксикарбонил)оксиметил (т.е. -CH2OC(=O)OCH3);
[33] и по меньшей мере один из R1, R2 и R3 представляет собой дейтерий.
[34] Указанное полициклическое производное карбамоилпиридона, представленное формулой (I), предпочтительно выбрано из следующих соединений:
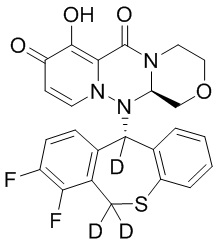
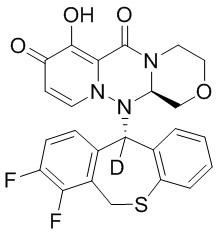
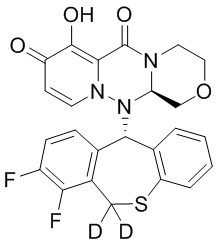
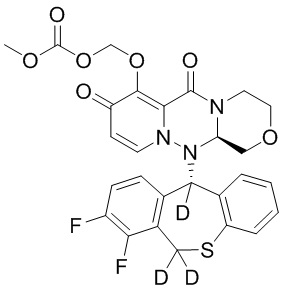
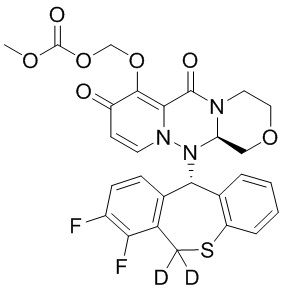
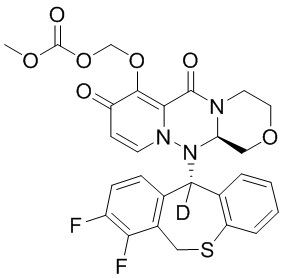 .
.
[35] Настоящее изобретение относится к полициклическому производному карбамоилпиридона, представленному формулой (II), стереоизомеру указанного соединения, таутомеру указанного соединения, гидрату указанного соединения, сольвату указанного соединения, активному метаболиту указанного соединения, кристаллической форме указанного соединения, фармацевтически приемлемой соли указанного соединения или пролекарству указанного соединения,
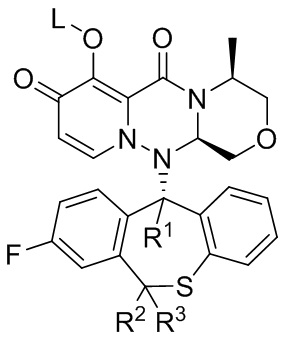
Формула (II)
[36] где:
[37] R1 представляет собой водород или дейтерий;
[38] R2 представляет собой водород или дейтерий;
[39] R3 представляет собой водород или дейтерий;
[40] L представляет собой водород, алкил или (метоксикарбонил)оксиметил (т.е. -CH2OC(=O)OCH3);
[41] и по меньшей мере один из R1, R2 и R3 представляет собой дейтерий.
[42] Указанное полициклическое производное карбамоилпиридона, представленное формулой (II), предпочтительно выбрано из следующих соединений:
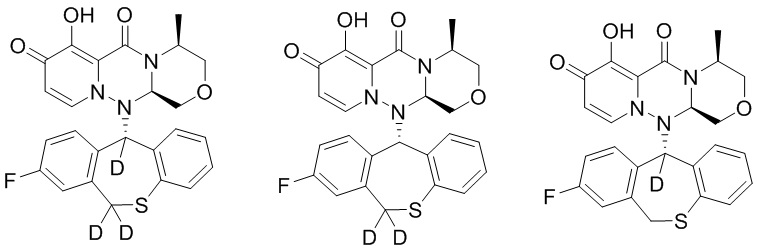 .
.
[43] Настоящее изобретение относится к полициклическому производному карбамоилпиридона, представленному формулой (III), стереоизомеру указанного соединения, таутомеру указанного соединения, гидрату указанного соединения, сольвату указанного соединения, активному метаболиту указанного соединения, кристаллической форме указанного соединения, фармацевтически приемлемой соли указанного соединения или пролекарству указанного соединения,
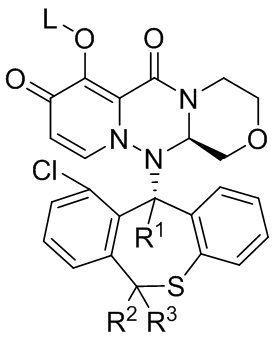
Формула (III),
[44] где:
[45] R1 представляет собой водород или дейтерий;
[46] R2 представляет собой водород или дейтерий;
[47] R3 представляет собой водород или дейтерий;
[48] L представляет собой водород, алкил или (метоксикарбонил)оксиметил (т.е. -CH2OC(=O)OCH3);
[49] и по меньшей мере один из R1, R2 и R3 представляет собой дейтерий.
[50] Указанное полициклическое производное карбамоилпиридона, представленное формулой (III), предпочтительно выбрано из следующих соединений:
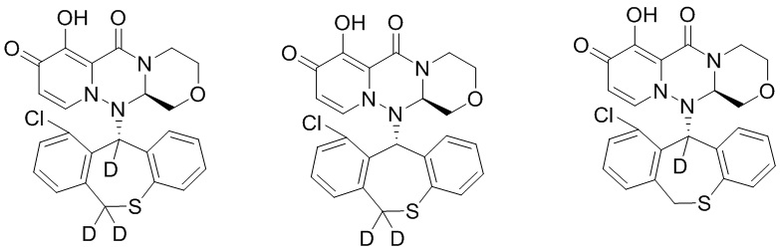 .
.
[51] Настоящее изобретение относится к полициклическому производному карбамоилпиридона, представленному формулой (IV), стереоизомеру указанного соединения, таутомеру указанного соединения, гидрату указанного соединения, сольвату указанного соединения, активному метаболиту указанного соединения, кристаллической форме указанного соединения, фармацевтически приемлемой соли указанного соединения или пролекарству указанного соединения,
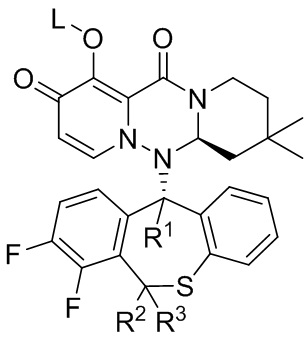
формула (IV),
[52] где:
[53] R1 представляет собой водород или дейтерий;
[24] R2 представляет собой водород или дейтерий;
[55] R3 представляет собой водород или дейтерий;
[56] L представляет собой водород, алкил или (метоксикарбонил)оксиметил (т.е. -CH2OC(=O)OCH3);
[57] и по меньшей мере один из R1, R2 и R3 представляет собой дейтерий.
[58] Указанное полициклическое производное карбамоилпиридона, представленное формулой (IV), предпочтительно выбрано из следующих соединений:
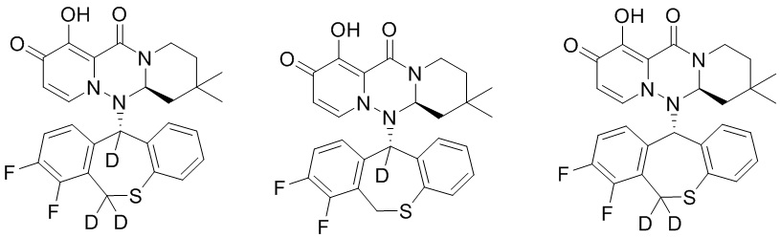 .
.
[59] Настоящее изобретение относится к полициклическому производному карбамоилпиридона, представленному формулой (V), стереоизомеру указанного соединения, таутомеру указанного соединения, гидрату указанного соединения, сольвату указанного соединения, активному метаболиту указанного соединения, кристаллической форме указанного соединения, фармацевтически приемлемой соли указанного соединения или пролекарству указанного соединения,
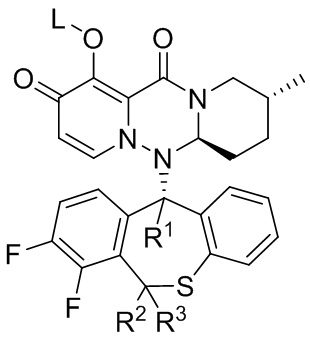
формула (V)
[60] где:
[61] R1 представляет собой водород или дейтерий;
[62] R2 представляет собой водород или дейтерий;
[63] R3 представляет собой водород или дейтерий;
[64] L представляет собой водород, алкил или (метоксикарбонил)оксиметил (т.е. -CH2OC(=O)OCH3);
[65] и по меньшей мере один из R1, R2 и R3 представляет собой дейтерий.
[66] Указанное полициклическое производное карбамоилпиридона, представленное формулой (V), предпочтительно выбрано из следующих соединений:
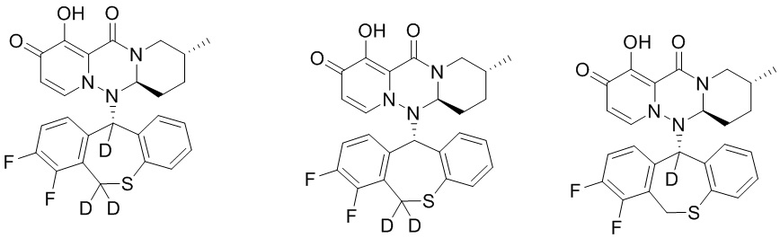 .
.
[67] Настоящее изобретение относится к полициклическому производному карбамоилпиридона, показанному формулой (VI), стереоизомеру указанного соединения, таутомеру указанного соединения, гидрату указанного соединения, сольвату указанного соединения, активному метаболиту указанного соединения, кристаллической форме указанного соединения, фармацевтически приемлемой соли указанного соединения или пролекарству указанного соединения,
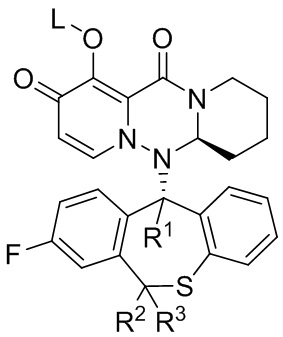
формула (VI)
[68] где:
[69] R1 представляет собой водород или дейтерий;
[70] R2 представляет собой водород или дейтерий;
[71] R3 представляет собой водород или дейтерий;
[72] L представляет собой водород, алкил или (метоксикарбонил)оксиметил (т.е. -CH2OC(=O)OCH3);
[73] и по меньшей мере один из R1, R2 и R3 представляет собой дейтерий.
[74] Указанное полициклическое производное карбамоилпиридона, представленное формулой (VI), предпочтительно выбрано из следующих соединений:
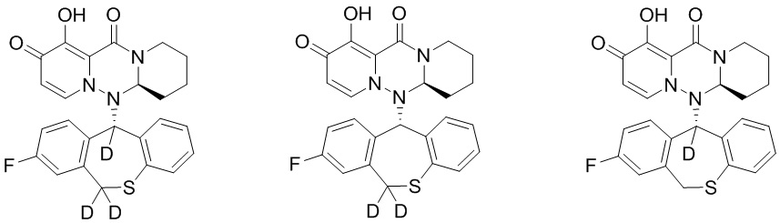 .
.
[75] Если не указано иное, термин «алкил» в настоящем документе используется для обозначения насыщенной углеводородной группы с линейной или разветвленной цепью, она может быть одновалентной (такой как метил), двухвалентной (такой как метилен) или многовалентной (такой как метин). Примеры алкила включают метил (Me), этил (Et), пропил (такой, как н-пропил и изопропил), бутил (такой, как н-бутил, изобутил, втор-бутил и трет-бутил), пентил (такой, как п-пентил, изопентил и неопентил) и тому подобное.
[76] Соединения согласно настоящему изобретению обычно применяют в форме свободной кислоты или свободного основания. Альтернативно, соединения согласно настоящему изобретению можно применять в форме солей присоединения кислоты или основания. Соль присоединения кислоты соединений согласно настоящему изобретению со свободной аминогруппой (аминогруппами) можно получить способами, хорошо известными в данной области техники, и ее можно получить из органических и неорганических кислот. Подходящие органические кислоты включают малеиновую кислоту, фумаровую кислоту, бензойную кислоту, аскорбиновую кислоту, янтарную кислоту, метансульфоновую кислоту, уксусную кислоту, трифторуксусную кислоту, щавелевую кислоту, пропионовую кислоту, винную кислоту, салициловую кислоту, лимонную кислоту, глюконовую кислоту, молочную кислоту, миндальную кислоту, фенилуксусную кислоту, аспарагиновую кислоту, стеариновую кислоту, пальмитиновую кислоту, гликолевую кислоту, глутаминовую кислоту и бензолсульфоновую кислоту. Подходящие неорганические кислоты включают соляную кислоту, бромистоводородную кислоту, серную кислоту, фосфорную кислоту и азотную кислоту. Основные соли включают соли, образованные с карбоксилат-анионами, и включают соли, образованные, например, с органическими и неорганическими катионами, выбранными из ионов щелочных металлов, ионов щелочноземельных металлов (например, лития, натрия, калия, магния, бария, кальция) и ионов аммония, и их замещенных производных (например, дибензиламмоний, бензиламмоний, 2-гидроксиэтиламмоний и так далее). Следовательно, термин «фармацевтически приемлемая соль» соединения формулы (I), формулы (II), формулы (III), формулы (IV), формулы (V), формулы (VI) или формулы (VII) должен включать все приемлемые формы солей.
[77] Кроме того, пролекарства также включены в объем настоящего изобретения. Пролекарство представляет собой любой ковалентно связанный носитель и высвобождает соединение, представленное формулой (I), формулой (II), формулой (III), формулой (IV), формулой (V), формулой (VI) или формулой (VII) in vivo при введении пациенту. Пролекарства обычно получают путем модификации функциональных групп таким образом, чтобы модификация была обратима обычным образом или при разложении in vivo с образованием исходного соединения. Пролекарства включают, например, соединения согласно настоящему изобретению, в которых гидроксильная группа, аминогруппа или сульфгидрильная группа объединена с любой группой, при этом указанная группа удаляется с образованием гидроксильной группы, аминогруппы или сульфгидрильной группы, когда соединение вводят пациенту.
[78] Следовательно, репрезентативные примеры пролекарств включают, но не ограничиваются ими, ацетатные, формиатные и бензоатные производные спиртовых и аминных функциональных групп соединений, представленных формулой (I), формулой (II), формулой (III), формулой (IV), формулой (V), формулой (VI) или формулой (VII). Кроме того, в случае карбоновой кислоты (-COOH) могут быть включены сложные эфиры, такие как метиловый эфир, этиловый эфир и т.п. В случае гидроксила могут быть включены смешанные ангидриды, такие как метокси, этокси, пропокси, трет-бутокси и т.п.
[79] Что касается стереоизомеров, соединения, представленные формулой (I), формулой (II), формулой (III), формулой (IV), формулой (V), формулой (VI) или формулой (VII), могут иметь хиральный центр и могут существуют в виде рацематов, рацемических смесей и индивидуальных энантиомеров или диастереомеров. Настоящее изобретение включает все изомерные формы, включая их смеси. Кроме того, определенные кристаллические формы соединений, представленных формулой (I), формулой (II), формулой (III), формулой (IV), формулой (V), формулой (VI) или формулой (VII), могут существовать в виде различных полиморфных форм, которые также включены в объем настоящего изобретения. Кроме того, некоторые из соединений, представленных формулой (I), формулой (II), формулой (III), формулой (IV), формулой (V), формулой (VI) или формулой (VII), также можно комбинировать с водой или другими органическими растворителями с получением сольвата. Такие сольваты аналогичным образом включены в объем настоящего изобретения.
[80] Специалисту в данной области техники будет понятно, что любое соединение может содержать изотопы атомов в пропорциях, отличных от существующих в природе, для одного или более атомов, которые составляют такие соединения. В полициклических производных карбамоилпиридона, включенных в объем настоящего изобретения, дейтерирование означает, что атомы в соответствующих узлах соединения содержат атомы дейтерия в пропорции, превышающей встречающиеся в природе (то есть превышающей естественное содержание дейтерия). Следовательно, любое полициклическое производное карбамоилпиридона, которое содержит атомы дейтерия в соотношении, превышающем встречающееся в природе содержание дейтерия в соответствующем положении, попадает в объем настоящего изобретения. Например, понятно, что соответствующие полициклические производные карбамоилпиридона с определенной степенью дейтерирования или содержанием дейтерия, которые получены введением атомов дейтерия при помощи коммерчески доступных дейтерированных реагентов с помощью способов химического синтеза, показанных в вариантах реализации настоящего изобретения, или аналогичных, все находятся в пределах объема настоящего изобретения. Для настоящего изобретения способы химического синтеза и дейтерированные реагенты не ограничиваются теми, которые приведены в примерах, но следует понимать, что они включают все методы или пути синтеза в данной области техники, которые могут быть использованы для получения соединений согласно настоящему изобретению, и все дейтерированные реагенты, при помощи которых можно ввести атом(ы) дейтерия в целевые молекулы с помощью вышеупомянутых методов или путей синтеза.
[81] В соответствии с определенными вариантами реализации настоящего изобретения, приведенными ниже, специалисты в данной области техники могут использовать те же или аналогичные принципы и методы для получения определенных полициклических производных карбамоилпиридона, представленных формулой (I), формулой (II), формулой (III), формулой (IV), формулой (V), формулой (VI) или формулой (VII).
[82] В настоящем изобретении дополнительно описано применение полициклического производного карбамоилпиридона, представленного формулой (VII) (предпочтительно формулой (I), формулой (II), формулой (III), формулой (IV), формулой (V) или формулой (VI)), стереоизомера указанного соединения, таутомера указанного соединения, гидрата указанного соединения, сольвата указанного соединения, активного метаболита указанного соединения, кристаллической формы указанного соединения, фармацевтически приемлемой соли указанного соединения или пролекарства указанного соединения в производстве ингибитора 5'кэп-зависимой эндонуклеазы.
[83] В настоящем изобретении дополнительно описано применение полициклического производного карбамоилпиридона, представленного формулой (VII) (предпочтительно формулой (I), формулой (II), формулой (III), формулой (IV), формулой (V) или формулой (VI)), стереоизомера указанного соединения, таутомера указанного соединения, гидрата указанного соединения, сольвата указанного соединения, активного метаболита указанного соединения, кристаллической формы указанного соединения, фармацевтически приемлемой соли указанного соединения или пролекарства указанного соединения в производстве лекарственного средства для предотвращения, лечения и/или облегчения заболевания, связанного с 5'кэп-зависимой эндонуклеазой. Заболевание, связанное с 5'кэп-зависимой эндонуклеазой, относится к симптомам и/или заболеваниям, которые можно предотвратить, лечить и/или облегчить путем ингибирования уровня 5'кэп-зависимой эндонуклеазы in vivo, особенно к симптомам и/или заболеваниям, вызванным заражением вирусом гриппа. Более конкретно, вирус гриппа включает, но не ограничивается ими, типы A, B и C. Симптомы включают, но не ограничиваются ими, например, симптомы простуды, такие как лихорадка, озноб, головная боль, мышечная боль, общая усталость и тому подобное, или воспаления дыхательных путей, такие как боль в горле, насморк, заложенность носа, кашель, мокрота и тому подобное; желудочно-кишечные симптомы, такие как боль в животе, рвота, диарея и тому подобное; дальнейшие вторичные инфекционные осложнения, такие как острая энцефалопатия, пневмония и тому подобное.
[84] Настоящее изобретение также относится к способу предотвращения, лечения и/или облегчения заболевания, связанного с 5'-кэп-зависимой эндонуклеазой, указанный способ включает введение терапевтически эффективного количества полициклического производного карбамоилпиридона, представленного формулой (VII) (предпочтительно формулой (I), формулой (II), формулой (III), формулой (IV), формулой (V) или формулой (VI)), стереоизомера указанного соединения, таутомера указанного соединения, гидрата указанного соединения, сольвата указанного соединения, активного метаболита указанного соединения, кристаллической формы указанного соединения, фармацевтически приемлемой соли указанного соединения или пролекарства указанного соединения субъекту, нуждающемуся в этом, для ингибирования in vivo уровня 5'-кэп-зависимой эндонуклеазы. При этом заболевание, связанное с 5'-кэп-зависимой эндонуклеазой, конкретно описано выше.
[85] В настоящем изобретении дополнительно описана фармацевтическая композиция, содержащая терапевтически эффективное количество полициклического производного карбамоилпиридона, представленного формулой (VII) (предпочтительно формулой (I), формулой (II), формулой (III), формулой (IV), формулой (V) или формулой (VI)), стереоизомера указанного соединения, таутомера указанного соединения, гидрата указанного соединения, сольвата указанного соединения, активного метаболита указанного соединения, кристаллической формы указанного соединения, фармацевтически приемлемой соли указанного соединения или пролекарства указанного соединения и фармацевтически приемлемый носитель.
[86] В настоящем изобретении дополнительно описана фармацевтическая композиция, которая содержит не только терапевтически эффективное количество полициклического производного карбамоилпиридона, представленного формулой (VII) (предпочтительно формулой (I), формулой (II), формулой (III), формулой (IV), формулой (V) или формулой (VI)), стереоизомера указанного соединения, таутомера указанного соединения, гидрата указанного соединения, сольвата указанного соединения, активного метаболита указанного соединения, кристаллической формы указанного соединения, фармацевтически приемлемой соли указанного соединения или пролекарства указанного соединения и фармацевтически приемлемый носитель, но и другие активный фармацевтический ингредиент. Указанный другой активный фармацевтический ингредиент выбран из ингибиторов нейраминидазы (таких как осельтамивир (oseltamivir), занамивир (zanamivir), перамивир (peramivir), инавир (inavir) и т.п.), ингибиторов РНК-зависимой РНК-полимеразы (таких как фавипиравир (favipiravir)), ингибиторов белка M2 (амантадин (amantadine)), ингибиторов связывания PB2 кэпа (таких как VX-787), антител против НА (MHAA4594A) или лекарственных средств иммунного действия (нитазоксанид (nitazoxanide)).
[87] Соединения согласно настоящему изобретению можно применять в комбинации с другими фармацевтическими препаратами, чтобы повысить эффективность указанных соединений или уменьшить их дозировку. Например, в случае гриппа их можно применять в сочетании с ингибиторами нейраминидазы (такими как осельтамивир (oseltamivir), занамивир (zanamivir), перамивир (peramivir), инавир (Inavir) и т.п.), ингибиторами РНК-зависимой РНК-полимеразы (такими как фавипиравир (favipiravir)), ингибиторами белка M2 (амантадин (amantadine)), ингибиторами связывания PB2 кэпа (VX-787), антителами против НА (MHAA4594A) или лекарственными средствами иммунного действия (нитазоксанид (nitazoxanide)) и т.д.
[88] Отдельные формы соединений согласно настоящему изобретению или их фармацевтически приемлемых солей или подходящие фармацевтические композиции можно вводить любым способом введения, приемлемым для лекарственного средства с подобным воздействием. Фармацевтическую композицию согласно настоящему изобретению можно приготовить путем объединения соединения согласно настоящему изобретению с подходящим фармацевтически приемлемым носителем, разбавителем или вспомогательным веществом, и ее можно приготовить в виде твердого, полутвердого, жидкого или газообразного состава, такого как таблетка, капсула, порошок, гранула, мазь, раствор, суппозиторий, состав для инъекции, средство для ингаляции, гель, микросферы и аэрозоль. Типичные пути введения указанной фармацевтической композиции включают, но не ограничиваются ими, пероральное, местное, трансдермальное, ингаляционное, парентеральное, сублингвальное, буккальное, ректальное, вагинальное и интраназальное введение. Используемый в настоящем документе термин «парентерально» включает подкожные инъекции, внутривенные, внутримышечные, внутригрудинные инъекции или инфузии. Фармацевтическая композиция согласно настоящему изобретению приготовлена так, чтобы активные ингредиенты, содержащиеся в ней, были биодоступными после введения указанной композиции пациенту. Композиция, предназначенная для введения субъекту или пациенту, может быть в виде форм, содержащих одну и более доз, где, например, таблетка может представлять собой лекарственную форму, содержащую одну дозу, а контейнер, содержащий соединение согласно настоящему изобретению в виде аэрозоля, содержат несколько единичных доз. Фактический способ приготовления лекарственной формы известен или будет известен специалистам в данной области техники. Вводимая композиция в любом случае будет содержать терапевтически эффективное количество соединения согласно настоящему изобретению или фармацевтически приемлемой соли указанного соединения для лечения рассматриваемого заболевания или состояния в соответствии с сущностью настоящего изобретения.
[89] Фармацевтическая композиция согласно настоящему изобретению может быть в твердой или жидкой форме. В одном аспекте носитель представляет собой микрочастицы, так что композиция находится, например, в форме таблетки или порошка. Носитель может представлять собой жидкостью, и указанная композиция представляет собой, например, сироп для перорального введения, жидкость для инъекций или аэрозоль, подходящий, например, для ингаляционного введения. Фармацевтическая композиция, предназначенная для перорального приема, предпочтительно находится в твердой или жидкой форме, при этом твердые или жидкие формы также включают полутвердые, полужидкие, суспензионные и гелевые формы. Для твердых композиций для перорального приема указанную фармацевтическую композицию можно приготовить в виде порошка, гранул, прессованной таблетки, пилюли, капсулы, жевательной таблетки, порошковой таблетки и т.п. Такие твердые композиции обычно содержат один или более инертных разбавителей или приемлемых с пищевой точки зрения носителей. Кроме того, также могут присутствовать одно или более из следующих веществ: связующие, такие как карбоксиметилцеллюлоза, этилцеллюлоза, микрокристаллическая целлюлоза, ксантановая камедь или желатин; вспомогательные вещества, такие как крахмал, лактоза или декстрин; разрыхлители, такие как альгиновая кислота, альгинат натрия, примогель, кукурузный крахмал и т.д.; смазывающие вещества, такие как стеарат магния или гидрогенизированное растительное масло (Sterotex); скользящие вещества, такие как коллоидный диоксид кремния; подсластители, например, сахароза или сахарин; ароматизаторы, такие как мята перечная, метилсалицилат или апельсиновые ароматизаторы; и красители.
[90] Когда указанная фармацевтическая композиция находится в форме капсулы, такой как желатиновая капсула, она может содержать жидкий носитель, такой как полиэтиленгликоль или масло, в дополнение к веществам упомянутых выше типов. Фармацевтическая композиция может быть в жидкой форме, такой как настойка, сироп, раствор, эмульсия или суспензия. Такие жидкие формы можно принимать перорально или вводить путем инъекции, в качестве двух примеров. Когда композиция предназначена для перорального приема, она предпочтительно содержит один или более из подсластителей, консервантов, красителей/окрашивающих веществ и усилителей вкуса в дополнение к соединению согласно настоящему изобретению. В композиции, предназначенной для введения путем инъекции, может содержаться одно или более из поверхностно-активных веществ, консервантов, смачивающих агентов, диспергаторов, суспендирующих агентов, буферов, стабилизаторов и изотонических агентов.
[91] Независимо от того, находится ли жидкая фармацевтическая композиция по настоящему изобретению в виде раствора, суспензии или в другой подобной форме, она может содержать один или более из следующих адъювантов: стерильный разбавитель, такой как вода для инъекций, физиологический раствор, предпочтительно физиологический раствор, раствор Рингера, изотонический раствор хлорида натрия, нелетучее масло (например, синтетические моноглицериды или диглицериды, которые можно использовать в качестве растворителей или суспензионных сред), растворители, такие как полиэтиленгликоль, глицерин, пропиленгликоль; антибактериальные агенты, такие как бензиловый спирт или метиловый эфир пара-гидроксибензойной кислоты; антиоксиданты, такие как аскорбиновая кислота или бисульфит натрия; хелатирующие агенты, такие как этилендиаминтетрауксусная кислота; буферы, такие как ацетатные соли, цитратные соли или фосфатные соли, и реагенты для регулирования тоничности, такие как оксид натрия или декстроза. Парентеральный состав может быть упакован в ампулу, одноразовый шприц или многодозовый флакон из стекла или пластика. Лучшим адъювантом является физиологический раствор. Фармацевтическая композиция для инъекций предпочтительно является стерильной.
[92] Жидкая фармацевтическая композиция согласно настоящему изобретению для парентерального или перорального введения должна содержать определенное количество соединения согласно настоящему изобретению, чтобы можно было получить подходящую дозировку. Фармацевтическая композиция согласно настоящему изобретению может быть предназначена для местного применения, и в этом случае носитель предпочтительно включает раствор, эмульсию, мазевую или гелевую основу. Например, такая основа может содержать одно или более из следующего: парафиновое масло, ланолин, полиэтиленгликоль, пчелиный воск, минеральное масло, разбавитель (такой как вода и спирт), а также эмульгатор и стабилизатор. В фармацевтической композиции для местного применения может содержаться загуститель. Если предполагается трансдермальное введение, композиция может включать трансдермальный пластырь или устройство для ионтофореза.
[93] Фармацевтическая композиция согласно настоящему изобретению может быть предназначена для ректального введения, например, в форме суппозитория, который тает в прямой кишке и высвобождает лекарственное средство. Композиция для ректального введения может содержать масляную основу в качестве подходящего нераздражающего наполнителя. Основа включает, но не ограничивается ими, ланолин, масло какао и полиэтиленгликоль.
[94] Фармацевтическая композиция согласно настоящему изобретению может содержать различные вещества, которые модифицируют физическую форму твердой или жидкой дозированной единицы. Например, указанная композиция может содержать вещество, которое образует оболочку вокруг активного ингредиента. Вещество, образующее оболочку, обычно инертно и может быть выбрано, например, из сахара, шеллака и других энтеросолюбильных покрытий. Альтернативно активный ингредиент может быть заключен в желатиновые капсулы.
[95] Фармацевтическая композиция согласно настоящему изобретению в твердой или жидкой форме может содержать агент, который связывает соединение согласно настоящему изобретению и тем самым способствует доставке соединения. Подходящие реагенты, обладающие такой способностью, включают моноклональные или поликлональные антитела, белки или липосомы.
[96] Фармацевтическая композиция согласно настоящему изобретению может состоять из дозированной формы, которую можно вводить в виде аэрозоля. Термин «аэрозоль» используется для обозначения множества систем от коллоидных типов до систем в упаковке под давлением. Доставка может осуществляться при помощи сжиженного или сжатого газа или при помощи подходящей насосной системы, которая дозирует активный ингредиент. Аэрозоли соединений согласно настоящему изобретению могут доставляться в однофазной, двухфазной или трехфазной системе для доставки активного ингредиента. Часть для вывода аэрозоля включает в себя необходимые контейнеры, стартеры, клапаны, суб-контейнеры и т. д., которые вместе могут образовывать набор. Специалист в данной области техники сможет определить предпочтительный аэрозоль без излишних экспериментов.
[97] Фармацевтическую композицию согласно настоящему изобретению можно приготовить способами, хорошо известными в области фармацевтики. Например, фармацевтическую композицию для введения путем инъекции можно приготовить путем объединения соединения согласно настоящему изобретению со стерильной дистиллированной водой с получением раствора. Для облегчения образования однородного раствора или суспензии могут быть добавлены поверхностно-активные вещества. Поверхностно-активные вещества представляют собой соединения, которые нековалентно взаимодействуют с соединениями согласно настоящему изобретению, тем самым способствуя растворению или однородному суспендированию соединений в водной системе доставки.
[98] Соединения согласно настоящему изобретению или фармацевтически приемлемые соли указанных соединений вводят в терапевтически эффективном количестве, и указанное терапевтически эффективное количество будет варьироваться в зависимости от различных факторов, включая активность конкретных используемых соединений, метаболическую стабильность соединений и продолжительность действия, а также возраст, массу, общее состояние здоровья, пол и диету пациента, способ и время введения, скорость выведения, комбинацию лекарственных средств, тяжесть конкретного заболевания или состояния индивидуума, подвергающегося терапии.
[99] Соединения согласно настоящему изобретению или фармацевтически приемлемые производные указанных соединений также можно вводить одновременно, до или после введения одного или более других терапевтических агентов. Такая комбинированная терапия включает введение одного фармацевтического состава, содержащего соединение согласно настоящему изобретению и один или более других активных агентов, и введение отдельных фармацевтических составов соединения согласно настоящему изобретению и каждого активного агента. Например, соединение согласно настоящему изобретению и другой активный агент можно вводить пациенту совместно в виде одной композиции для перорального введения (например, таблетке или капсуле), или каждый агент можно вводить в отдельной композиции для перорального введения. В случае применения составов для раздельного введения соединение согласно настоящему изобретению и один или более дополнительных активных агентов можно вводить по существу в одно и то же время (т.е., одновременно) или в моменты, разнесенные по времени (т.е., последовательно). Под комбинированной терапией следует понимать все эти схемы.
[100] Дозировку фармацевтической комбинации согласно настоящему изобретению можно регулировать в соответствии со стадией заболевания, путем введения, возрастом или массой пациента. Для перорального введения взрослым дозировка обычно составляет от 0,1 до 100 мг/кг/сутки, предпочтительно от 1 до 20 мг/кг/сутки. Соответствующую дозировку согласно настоящему изобретению необходимо определять с учетом возраста, массы, состояния пациента, способа введения и т.д., для перорального введения дозировка обычно находится в диапазоне от 0,05 до 100 мг/кг/сутки, предпочтительно от 0,1 до 10 мг/кг/сутки. В случае неперорального введения дозировка фармацевтической композиции согласно настоящему изобретению сильно варьируется в зависимости от различных путей введения, но обычно составляет от 0,005 до 10 мг/кг/сутки, предпочтительно от 0,01 до 1 мг/кг/сутки.
[101] Дозировка соединения согласно настоящему изобретению варьируется в зависимости от способа введения, возраста, массы, состояния пациента и типа заболевания, но обычно для перорального введения суточная доза для взрослых составляет от примерно 0,05 мг до 3000 мг, предпочтительно от примерно 0,1 до 1000 мг, и при необходимости их можно вводить не одновременно. Кроме того, в случае неперорального введения суточная доза для взрослых составляет примерно от 0,01 до 1000 мг, предпочтительно примерно от 0,05 до 500 мг. Следует понимать, что в настоящем изобретении разрешены только такие комбинации заместителей и/или переменных в химической формуле, которые позволяют получить стабильное соединение.
[102] Настоящее изобретение относится к замещенным полициклическим производным карбамоилпиридона, обладающим ингибирующей активностью в отношении 5'-кэп-зависимой (CAP) эндонуклеазы, пролекарствам указанных соединений, дейтерированным соединениям и фармацевтическим композициям, содержащим указанные соединения, а также способу применения композиции для ингибирования пролиферации вируса гриппа.
[103] Соединения согласно настоящему изобретению являются пролекарствами и, следовательно, имеют следующие преимущества: высокую пероральную абсорбцию, хорошую биодоступность, хороший клиренс, хороший метаболизм в легких и т.п. Таким образом, они могут составить превосходное лекарственное средство.
[104] Соединения, которые являются исходными для соединений согласно настоящему изобретению, обладают высокой ингибирующей активностью в отношении 5'-кэп-структурно-зависимой эндонуклеазы, и, поскольку эндонуклеаза является вирус-специфическим ферментом, соединения будут иметь высокую селективность и могут быть лекарственными средствами с уменьшенными побочными эффектами. Кроме того, соединения согласно настоящему изобретению и/или соединения, являющиеся исходными для соединений согласно настоящему изобретению, обеспечивают следующие преимущества, а именно: высокую метаболическую стабильность, высокую растворимость, высокую всасываемость при пероральном введении, хорошую биодоступность, хороший клиренс, хороший метаболизм в легких, длительный период полувыведения, высокую скорость небелкового связывания, низкое ингибирование в отношении канала hERG, низкое ингибирование в отношении ферментов печени, метаболизирующих лекарственные средства, подтвержденный эффект ингибирования цитопатического эффекта (CPE) и/или отрицательные результаты в тесте на фототоксичность, тесте Эймса и тесте на генотоксичность или отсутствие токсичности для печени и т.д., по этим причинам соединения согласно настоящему изобретению обладают лучшими фармакологическими свойствами.
[105] Соединения согласно настоящему изобретению и/или соединения согласно настоящему изобретению можно применять для лечения симптомов и/или заболеваний, вызванных вирусами гриппа, и они эффективны для лечения и/или предотвращения, например, симптомов простуды, таких как лихорадка, озноб, головная боль, боль в мышцах, общая усталость и тому подобное, или воспаления дыхательных путей, такие как боль в горле, насморк, заложенность носа, кашель, мокрота и тому подобное, желудочно-кишечные симптомы, такие как боль в животе, рвота, диарея и тому подобное, дальнейших вторичных инфекционных осложнений, таких как острая энцефалопатия, пневмония и т.п., и улучшают их симптомы.
[106] Если не указано иное, реагенты и материалы, используемые в настоящем изобретении, являются коммерчески доступными.
[107] Если не указано иное, названия соединениям согласно настоящему изобретению даны вручную или с помощью программного обеспечения ChemDraw®, а для коммерчески доступных соединений используются названия из каталогов поставщиков.
[108] В соответствии с общеизвестными знаниями в данной области техники, вышеупомянутые предпочтительные условия могут быть произвольно объединены для получения предпочтительных вариантов реализации настоящего изобретения.
ПОДРОБНОЕ ОПИСАНИЕ ВАРИАНТОВ РЕАЛИЗАЦИИ ИЗОБРЕТЕНИЯ
[109] Следующие ниже предпочтительные примеры дополнительно иллюстрируют настоящее изобретение, но настоящее изобретение ими не ограничивается. В следующих примерах экспериментальные способы, для которых не указаны конкретные условия, осуществляют в соответствии с общепринятыми способами и условиями или в соответствии со спецификацией продукта. Исходные материалы являются коммерчески доступными или получены в соответствии со способами, известными в данной области техники, или получены в соответствии со способами, описанными в настоящем документе.
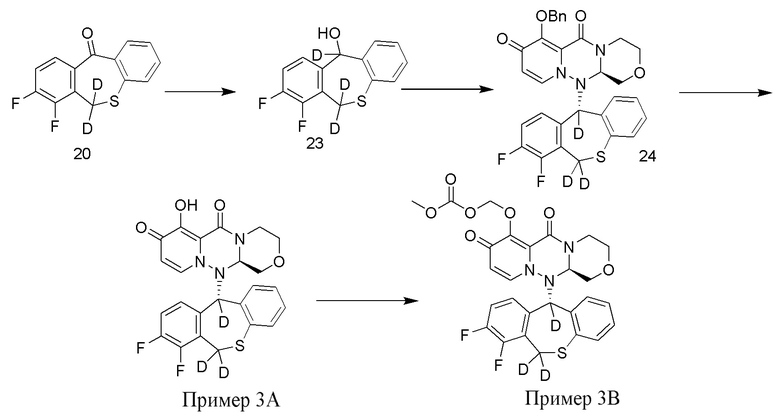
[110] Пример 1
[111] Путь синтезирования выглядит следующим образом:
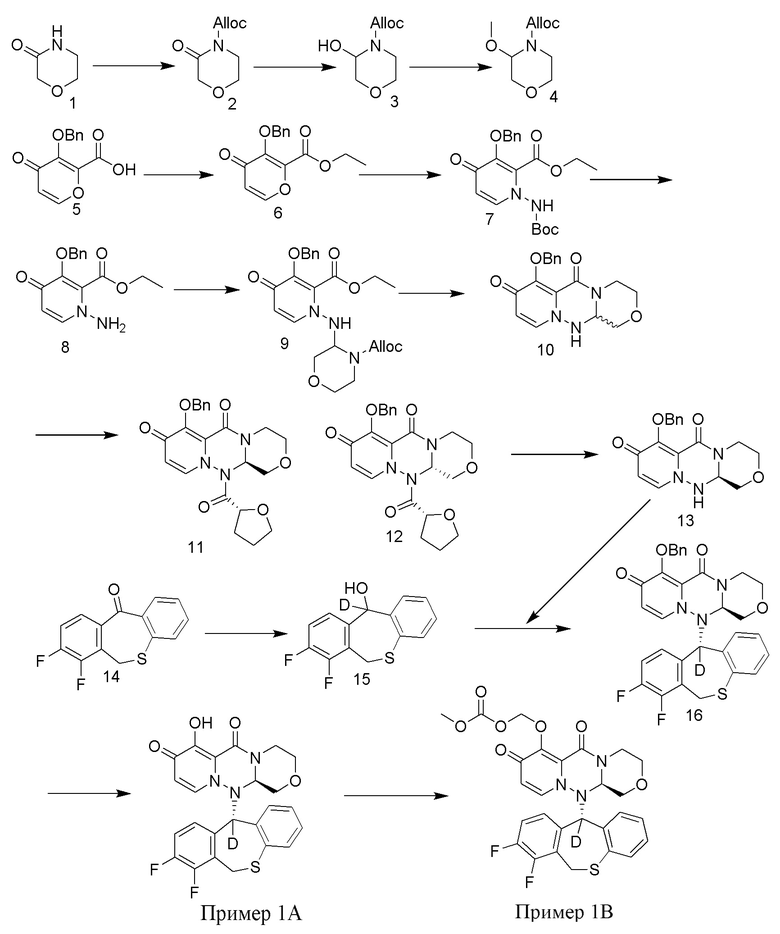
[112] Получение промежуточного соединения 15
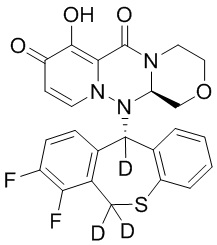
[113] 156 г промежуточного соединения 14 (приобретенного у Nanjing Leizheng Pharmaceutical Technology Co., Ltd., также можно получить согласно способу, приведенному в публикации Rajsner et al., Collection of Czechoslovak Chemical Communications, 1987, vol.47, #1, p.65-71) растворяли в 2,5 л ТГФ и медленно добавляли 14 г тетрагидроалюмината лития-D4 при температуре 0°C. Полученную смесь нагревали до комнатной температуры и перемешивали в течение 3 часов. После завершения реакции под контролем ТСХ к реакционной смеси добавляли 2 н. хлористоводородную кислоту. Смесь экстрагировали этилацетатом и концентрировали при пониженном давлении. Остаток суспендировали в циклогексане/дихлорметане с получением промежуточного соединения 15 в виде белого твердого вещества. МС: ESI 266,1[M+1]+.
[114] Получение промежуточного соединения 16
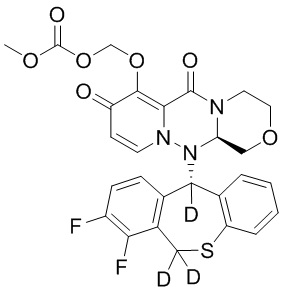
[115] 257 г промежуточного соединения 13 и 244,3 г промежуточного соединения 15 суспендировали в растворе (803 г) 50 мас.% пропилфосфорного ангидрида в этилацетате и добавляли 280 мл этилацетата. К указанной реакционной смеси при комнатной температуре добавляли метансульфоновую кислоту (109 г), и полученную смесь перемешивали при нагревании с обратным холодильником в течение 6 часов. К реакционной смеси добавляли воду при охлаждении на ледяной бане. После перемешивания при комнатной температуре в течение 1 часа добавляли ТГФ, и полученную смесь экстрагировали этилацетатом. Органический слой промывали водой, затем 8% водным раствором бикарбоната натрия, затем концентрировали при пониженном давлении. Полученное твердое вещество растворяли в 1,4 л ТГФ, добавляли 198 г карбоната калия. После повышения температуры до 50°C по каплям добавляли 60 мл бензилбромида, и указанную смесь перемешивали при температуре 60°C в течение 9 часов. К реакционной смеси по каплям добавляли 2 н. водный раствор хлористоводородной кислоты при охлаждении на ледяной бане, указанную смесь перемешивали при комнатной температуре, экстрагировали этилацетатом. Органический слой промывали водой, затем 8% водным раствором бикарбоната натрия, затем сушили над безводным сульфатом магния, добавляли активированный уголь для адсорбции в течение 1 часа, фильтровали через диатомовую земли и фильтрат концентрировали при пониженном давлении. Концентрированный раствор суспендировали в смеси этилацетат/гексан, при этом выпадал твердый осадок. Смесь фильтровали и полученное твердое вещество сушили с получением промежуточного соединения 16 с выходом 45%. МС: ESI 575,2[M+H]+.
[116] Иллюстративное соединение 1A
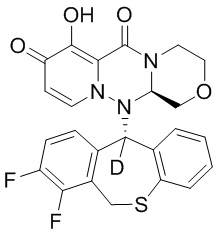
[117] 15 г хлорида лития добавляли к 120 мл раствора промежуточного соединения 16 (40 г) в DMA при температуре 25°C, и смесь перемешивали при температуре 80°C в течение 3 часов. К указанной реакционной смеси добавляли ацетон, 0,5 н. разбавленного раствора соляной кислоты и воду при температуре от 0 до 5°C и перемешивали в течение 1,5 часов, в течение которых выпадал осадок. Смесь фильтровали, и полученное твердое вещество перекристаллизовывали из смеси хлороформ/изопропиловый эфир, фильтровали и сушили с получением иллюстративного соединения 1A с выходом 79%. МС: ESI 485,1[M+H]+. 1H-ЯМР (CDCl3) δ: 3,00 - 3,51 (2H, m), 3,62 (1H, t), 3,83 (2H, m), 4,60 (2H, m), 5,28 - 5,33 (2H, m), 5,78 - 6,72 (2H, m), 6,86 - 6,90 (1H, m), 6,70 - 7,18 (5H, m). Дейтерирование: 99,6%.
[118] Иллюстративное соединение 1B
[119] К иллюстративному соединению 1A (5,0 г) добавляли DMA (25 мл) и перемешивали, затем добавляли метилхлорметилкарбонат (2,4 г), карбонат калия (2,9 г) и иодид калия (1,7 г). Полученную смесь нагревали до температуры 50°C и перемешивали в течение 6 часов. Добавляли DMA (5 мл) и перемешивание продолжали в течение 6 часов. Реакционную смесь охлаждали до комнатной температуры, добавляли DMA (25 мл), перемешивали при температуре 50°C в течение 5 минут и фильтровали. К фильтрату по каплям добавляли 1 моль/л водный раствор соляной кислоты (50 мл) и воду (15 мл) при температуре от 0 до 5°C и перемешивали в течение 1 часа. Смесь фильтровали и полученное твердое вещество сушили при пониженном давлении при температуре 60°C с получением иллюстративного соединения 1B с выходом 88%. МС: ESI 573,1[M+H]+. 1H-ЯМР (ДМСО-D6) δ: 2,89 - 3,28 (2H, m), 3,41 (1H, t), 3,68 (1H, m), 3,70 (3H, s), 3,97 - 4,39 (2H, m), 4,42 (1H, m), 5,39 - 5,68 (2H, m), 5,71 - 5,74 (3H, m), 6,81 - 7,00 (2H, J = 6,9 Гц), 7,08 - 7,45 (5H, m). Дейтерирование: 99,6%.
[120] Иллюстративное соединение 1С
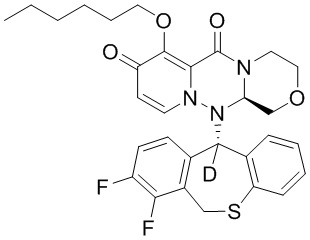
[121] К иллюстративному соединению 1A (4,8 г) добавляли DMA (30 мл) и перемешивали, а затем добавляли бромгексан (1,7 г), карбонат калия (2,9 г) и йодид калия (1,7 г). Указанную смесь нагревали до температуры 50°C и перемешивали в течение 6 часов. Добавляли DMA (5 мл) и перемешивание продолжали в течение 6 часов. Реакционную смесь охлаждали до комнатной температуры, добавляли DMA (30 мл), смесь перемешивали при температуре 50° C в течение 5 минут и фильтровали. К фильтрату по каплям добавляли 1 моль/л водный раствор соляной кислоты (50 мл) и воду (25 мл) при температуре от 0 до 5°C и перемешивали в течение 1 часа. Смесь фильтровали с получением твердого вещества. Затем полученное твердое вещество сушили при пониженном давлении при температуре 60°C с получением иллюстративного соединения 1C с выходом 79%. МС: ESI 569,2[M+H]+.
[122] Пример 2
[123] Путь синтезирования выглядит следующим образом:
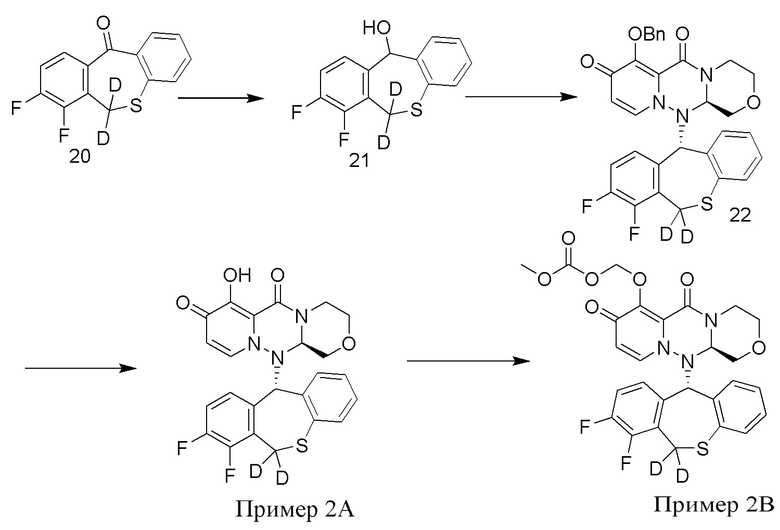
[124] Промежуточное соединение 20 было приобретено у Nanjing Leizheng Pharmaceutical Technology Co., Ltd., и его также можно получить согласно способу, приведенному в публикации Rajsner et al., Collection of Czechoslovak Chemical Communications, 1987, vol. 47, #1, p. 65-71. МС: ESI 265,5[M+H]+.
[125] Промежуточное соединение 21 получали согласно способу получения промежуточного соединения 15, за исключением того, что тетрагидроалюминат лития-D4 был заменен тетрагидроалюминатом лития, а промежуточное соединение 14 было заменено промежуточным соединением 20.
[126] Промежуточное соединение 22 получали согласно способу получения промежуточного соединения 16, за исключением того, что промежуточное соединение 15 было заменено промежуточным соединением 21.
[127] Получение иллюстративного соединения 2А
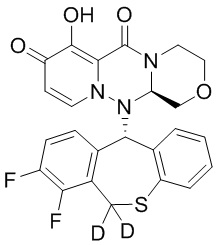
[128] 15 г хлорида лития добавляли к 120 мл раствора промежуточного соединения 22 (40 г) при температуре 25°C, и смесь перемешивали при температуре 80°C в течение 3 часов. К указанной реакционной смеси добавляли ацетон, 0,5 н. разбавленного раствора соляной кислоты и воду при температуре от 0 до 5°C и перемешивали в течение 1,5 часов, в течение которых выпадал осадок. Смесь фильтровали, и полученное твердое вещество перекристаллизовывали из смеси хлороформ/изопропиловый эфир, фильтровали и сушили с получением иллюстративного соединения 2A с выходом 65%. МС: ESI 486,1[M+H]+. 1H-ЯМР (CDCl3) δ: 3,01 - 3,50 (2H, m), 3,61 (1H, t), 3,80 (2H, m), 4,02 (1H, m), 4,58 (2H, m), 5,76 - 6,70 (2H, m), 6,85 - 6,8/8 (1H, m), 6,70 - 7,20 (5H, m). Дейтерирование: 98,6%.
[129] Иллюстративное соединение 2B
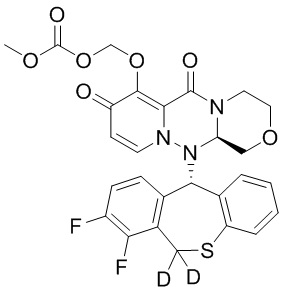
[130] DMA (50 мл) добавляли к иллюстративному соединению 2A (10,0 г) и перемешивали, а затем добавляли метилхлорметилкарбонат (4,8 г), карбонат калия (6,0 г), иодид калия (3,4 г). Указанную смесь нагревали до температуры 50°C и перемешивали в течение 6 часов. Добавляли DMA (10 мл) и перемешивание продолжали в течение 6 часов. Реакционную смесь охлаждали до комнатной температуры, добавляли DMA (50 мл), смесь перемешивали при температуре 50°C в течение 5 минут и фильтровали. К фильтрату по каплям добавляли 1 моль/л водный раствор соляной кислоты (100 мл) и воду (30 мл) при температуре от 0 до 5°C и перемешивали в течение 1 часа. Смесь фильтровали с получением твердого вещества. Затем полученное твердое вещество сушили при пониженном давлении при температуре 60°C с получением иллюстративного соединения 2B с выходом 90%. МС: ESI 574,2[M+H]+. 1H-ЯМР (ДМСО-D6) δ: 2,91 - 3,30 (2H, m), 3,44 (1H, t), 3,71 (1H, m), 3,73 (3H, s), 4,02 - 4,41 (3H, m), 4,43 (1H, m), 5,43 - 5,76 (2H, m), 5,79 (1H, m), 6,84 - 7,05 (2H, J = 6,9 Гц), 7,10 - 7,49 (5H, m). Дейтерирование: 99,6%.
[131] Пример 3
[132] Путь синтезирования выглядит следующим образом:
[133] Промежуточное соединение 23 получали согласно способу получения промежуточного соединения 15, за исключением того, что промежуточное соединение 14 было заменено промежуточным соединением 20.
[134] Промежуточное соединение 24 получали согласно способу получения промежуточного соединения 16, за исключением того, что промежуточное соединение 15 было заменено промежуточным соединением 23.
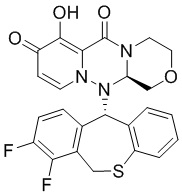
[135] Получение иллюстративного соединения 3А
[136] Промежуточное соединение 3А получали согласно способу получения соединения 1А, за исключением того, что промежуточное соединение 16 было заменено промежуточным соединением 24. МС: ESI 487,1[M+H]+. Дейтерирование: 99,5%.
[137] Получение иллюстративного соединения 3В
[138] Промежуточное соединение 3В получали согласно способу получения соединения 1В, за исключением того, что соединение 1А было заменено соединением 3А. МС: ESI 575,2[M+H]+. Дейтерирование: 99,6%.
[139] Получение промежуточного соединения 2
[140] 50 г промежуточного соединения 1 растворяли в 1 л ТГФ, добавляли 305 мл раствора н-бутиллития в гексане в атмосфере N2 при температуре -78°C, и указанную смесь перемешивали при температуре -78°C в течение 2 часов. К указанному реакционному раствору по каплям добавляли раствор 59,6 г аллилхлорформиата в ТГФ (200 мл), и смесь перемешивали при температуре -78°C в течение 2 часов. Указанный реакционный раствор гасили насыщенным водным раствором хлорида аммония, нагревали до комнатной температуры, экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили и концентрировали при пониженном давлении с удалением растворителя и с получением промежуточного соединения 2, выход 76%.
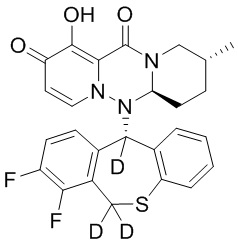
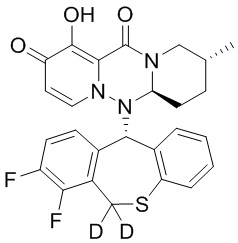
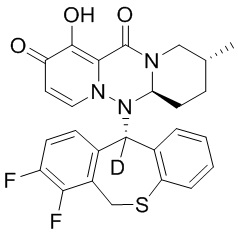
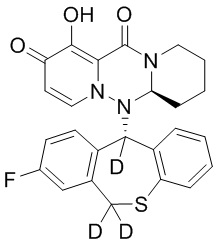
[141] Получение промежуточного соединения 3
[142] 20 г промежуточного соединения 2 растворяли в 200 мл ТГФ, в атмосфере N2 по каплям добавляли 138 мл раствора DIBAL-H в гексане при температуре -78°C, и указанную смесь перемешивали при температуре -78°C в течение 1 часа. Реакцию гасили добавлением водного раствора тартрата натрия-калия и указанную смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили и концентрировали при пониженном давлении с удалением растворителя и с получением промежуточного соединения 3.
[143] Получение промежуточного соединения 4
[144] К раствору промежуточного соединения 3 (12,4 г) в метаноле (130 мл) добавляли моногидрат п-толуолсульфоновой кислоты (1,3 г), и указанную смесь перемешивали при комнатной температуре в течение 8 часов. Реакцию гасили водным раствором бикарбоната натрия, концентрировали и экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили и концентрировали при пониженном давлении с удалением растворителя и с получением промежуточного соединения 4, выход 88%. МС: ESI 202,2[M+H]+.
[145] Получение промежуточного соединения 6
[146] 30 г промежуточного соединения 5 растворяли в 150 мл ДМФА, к раствору по каплям добавляли 15,2 г иодэтана и 27,6 мл диазабициклоундецена и перемешивали при комнатной температуре в течение ночи. Реакционную смесь гасили 10% водным раствором хлорида аммония и экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили и концентрировали при пониженном давлении с получением промежуточного соединения 6.
[147] Получение промежуточного соединения 7
[148] 2,5 г промежуточного соединения растворяли в 25 мл раствора DMA, добавляли 6,9 г тозилата пиридиния и 1,8 г boc-гидразина, и указанную смесь перемешивали при температуре 60°C в течение 15 часов. Добавляли воду и указанную смесь экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида аммония и насыщенным водным раствором хлорида натрия, сушили, концентрировали при пониженном давлении и подвергали колоночной флэш-хроматографии на силикагеле с получением промежуточного соединения 7.
[149] Получение промежуточного соединения 8
[150] 2,5 г соединения 8 растворяли в 4 н. растворе хлористого водорода в этилацетате (25 мл). Смесь перемешивали при комнатной температуре в течение 1 часа, концентрировали при пониженном давлении, промывали насыщенным водным раствором бикарбоната натрия и экстрагировали дихлорметаном. Органический слой промывали насыщенным солевым раствором, сушили и концентрировали при пониженном давлении с получением промежуточного соединения 8.
[151] Получение промежуточного соединения 10
[152] Соединение 4 (31 г) растворяли в 820 мл ацетонитрила при температуре -25°C в атмосфере азота, добавляли 40 г промежуточного соединения 8, по каплям добавляли тетрахлорид олова (230 мл) и смесь перемешивали при температуре -25°C в течение 45 минут. Реакционный раствор гасили водным раствором бикарбоната натрия, затем добавляли дихлорметан. Указанную смесь перемешивали при комнатной температуре, фильтровали через диатомовую землю и экстрагировали дихлорметаном. Органический слой промывали насыщенным солевым раствором, сушили и концентрировали при пониженном давлении с получением неочищенного промежуточного соединения 9. Неочищенное промежуточное соединение 9 растворяли в 800 мл ТГФ, добавляли морфолин (115 мл) и тетракис(трифенилфосфин)палладий (150 г), и указанную смесь перемешивали при комнатной температуре в течение 2,5 часов. К указанной реакционной смеси добавляли метил-трет-бутиловый эфир, смесь фильтровали с получением твердого вещества, и полученное твердое вещество сушили с получением промежуточного соединения 10.
[153] Получение промежуточного соединения 11
[154] (R)-тетрагидрофуран-2-карбоновую кислоту (855 мг, 7,36 ммоль) и 40 г промежуточного соединения 10 последовательно растворяли в 180 мл этилацетата при комнатной температуре, добавляли 80 мл пиридина и 50% пропилфосфорный ангидрид в этилацетате. Указанную смесь перемешивали в течение 8 часов и фильтровали с получением твердого вещества. Твердое вещество промывали этилацетатом и этанолом и суспендировали в 120 мл этанола при комнатной температуре в течение 7 часов. Суспензию фильтровали и осадок на фильтре дважды промывали этанолом с получением неочищенного промежуточного соединения 11.
[155] Получение промежуточного соединения 13
[156] 15 г неочищенного промежуточного соединения 11 суспендировали в этаноле (700 мл), добавляли DBU (10,5 мл) и смесь перемешивали в течение 30 минут. Добавляли 1,3 л диизопропилового эфира и перемешивали при комнатной температуре в течение 30 минут. Смесь фильтровали и дважды промывали этилацетатом с получением промежуточного соединения 13 с выходом 91%. МС: ESI 328,1[M+H]+.
[157] Получение соединения S-033188A
[158] S-033188A получали согласно способу получения иллюстративного соединения 1A.
[159] МС: ESI 485,1[M+H]+.
[160] Получение соединения S-033188B

[161] S-033188B получали согласно способу получения иллюстративного соединения 1В.
[162] МС: ESI 572,1[M+H]+.
[163] Соединения, представленные в таблице:
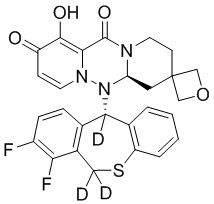
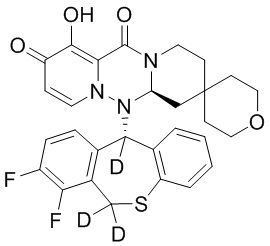
[164] Соединения, приведенные в следующей таблице, и их отдельные энантиомеры и пролекарства соответствующего сложного эфира соединений в следующей таблице были получены способами, аналогичными описанным выше. Такие сложные эфиры могут превращаться в активные метаболиты с гидроксильной группой (группами) in vivo, например, при пероральном введении под действием ферментов, метаболизирующих лекарственные средства, гидролитических ферментов, пищеварительного сока или бактерий в пищеварительном тракте и т.п. Соответствующие сложные эфиры соединений в таблице предпочтительно представляют собой метилметилкарбонат, но не ограничиваются этим.
Таблица 1: Структура соединений
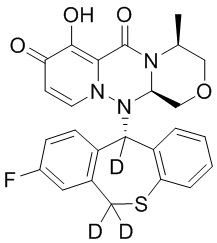
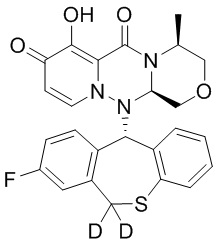
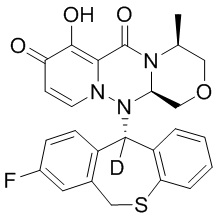

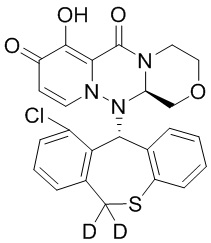
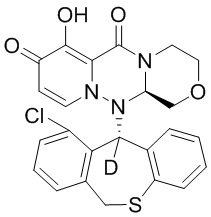
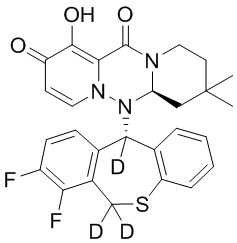
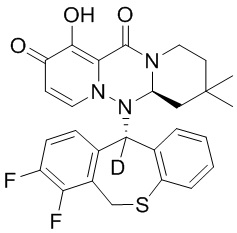
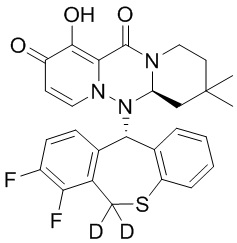
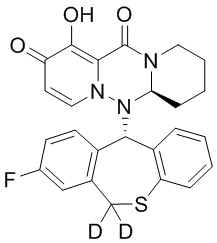
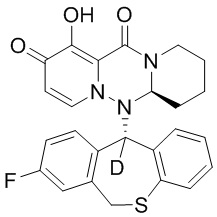
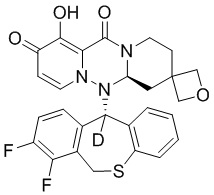
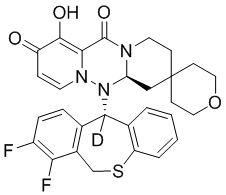
[165] Пример 4: Определение ингибирующей активности кэп-зависимой эндонуклеазы (CEN)
[166] 1) Подготовка субстрата
[167] Приобрели 30-мер РНК (5'-pp- [m2' -0]GAA UAU(-Cy3)GCA UCA CUA GUA AGCUUU GCU CUA-BHQ2-3', предоставлено фармацевтическим университетом Китая (China Pharmaceutical University)), в которой G на 5'-конце дифосфорилирован, гидроксил в 2'-положении метоксилирован, U в 6-м положении от 5'-конца помечен Cy3, а 3'-конец помечен BHQ2. При помощи системы ScriptCap добавили кэп-структуру с получением продукта m7G[5']-ppp-[5'] [m2' -0]GAA UAU(-Cy3)GCA UCA CUA GUA AGC UUU GCU CUA(-BHQ2)-3'. Его отделяли, очищали электрофорезом в модифицированном полиакриламидном геле и использовали в качестве субстрата.
[168] 2) Подготовка фермента
[169] РНП получали из вирусных частиц в соответствии с установленным способом (Ссылка: VIROLOGY (1976) 73, pages 327-338. LGA M.ROCHOVANSKY). В частности, 200 мкл 1×103 БОЕ/мл вируса A/WSN/33 инокулировали в развивающиеся куриные яйца возраста 10 дней, культивировали при 37°C в течение 2 дней, а затем выделяли аллантоисную жидкость из куриных яиц. Вирусные частицы очищали ультрацентрифугированием с 20% сахарозой, TritonX-100 и лизолецитин использовали для растворения вирусных частиц, а затем разделяли ультрацентрифугированием с градиентами плотности глицерина от 30% до 70%, фракцию РНП собирали (от 50% до 70% фракции глицерина), использовали в качестве раствора фермента (содержащего около 1 нМ комплекса PB1 • PB2 • PA).
[170] 3) Ферментативная реакция
[171] 2,5 мкл ферментативной реакционной смеси (состав: 53 мМ трис-гидрохлорида (pH 7,8), 1 мМ MgCl2, 1,25 мМ дитиотреитола, 80 мМ NaCl, 12,5% глицерина, 0,15 мкл раствора фермента) распределяли в 384-луночный планшет, сделанный из полипропилена. Затем 0,5 мкл ДМСО добавляли к 0,5 мкл раствора тестируемого соединения, серийно разбавленного ДМСО, к положительному контролю (ПК) и отрицательному контролю (ОК), и тщательно перемешивали. Затем добавляли 2 мкл раствора субстрата (1,4 нМ субстратной РНК, 0,05% Tween20) и инициировали реакцию. Реакционную смесь инкубировали при комнатной температуре в течение 60 минут, а затем 1 мкл реакционной смеси добавляли к 10 мкл раствора формамида высокой чистоты (содержащего стандарт GeneScan120LizSize в качестве калибровочного маркера, произведенный Applied Biosystem (ABI)), чтобы остановить реакцию. Реакцию ОК останавливали заранее, добавляя EDTA (4,5 мМ) перед началом реакции (все отмеченные концентрации были конечными концентрациями).
[172] 4) Определение ингибирующей концентрации (значение IC50)
[173] После прекращения реакции раствор нагревали при температуре 85°C в течение 5 минут, гасили на льду в течение 2 минут и затем анализировали с помощью генетического анализатора ABIPRIZM3730. Пик продукта кэп-зависимой эндонуклеазы количественно определяли с помощью аналитического программного обеспечения ABI Genemapper, интенсивность флуоресценции ПК и ОК использовали как 0% ингибирования и 100% ингибирования, соответственно, рассчитывали ингибирующую концентрацию реакции CEN (%) исследуемых соединений, и для расчета значения IC50 использовали программное обеспечение для построения кривой.
[174] Пример 5: Тест для подтверждения эффекта подавления CPE.
[175] Материалы:
[176] 1) 2% FCS E-MEM (приготовили путем добавления канамицина и FCS (фетальной бычьей сыворотки) к MEM (минимальная основная среда)).
[177] 2) 0,5% BSA E-MEM (приготовили путем добавления канамицина и BSA к MEM (минимальная основная среда)).
[178] 3) HBSS (сбалансированный солевой раствор Хэнкса).
[179] 4) Клетки MDBK: плотность клеток доводили до подходящего значения (3 × 105/мл) с помощью 2% FCS E-MEM.
[180] 5) Клетки MDCK: клетки дважды промывали HBSS, а затем плотность клеток доводили до подходящего значения (5 × 105/мл) с помощью 0,5% BSA E-MEM.
[181] 6) Раствор трипсина.
[182] Трипсин (SIGMA) из поджелудочной железы свиньи растворяли в PBS(-) и фильтровали через фильтр 0,45 мкм.
[183] 7) Считыватель микропланшетов.
[184] 8) Набор WST-8.
[185] 9) 10% раствор SDS.
[186] Осуществление процесса тестирования
[187] 1) Разведение и распределение образцов для теста
[188] 2% FCS E-MEM использовали в качестве культуральной среды для клеток MDBK, и 0,5% BSA E-MEM использовали в качестве культуральной среды для клеток MDCK. В дальнейшем ту же культуральную среду использовали для разведения вирусов, клеток и тестируемых образцов.
[189] Тестируемые образцы предварительно разбавляли до подходящей концентрации культуральным раствором и готовили 2-5-кратные серийные разведения (50 мкл/лунку) в 96-луночном планшете. Подготовили два планшета для измерения противогриппозной активности и измерения цитотоксичности. Определение выполняли в трех повторностях для каждого лекарственного средства.
[190] При использовании клеток MDCK к клеткам добавляли трипсин в конечной концентрации 3 мкг/мл только для определения противогриппозной активности.
[191] 2) Разведение и распределение вируса гриппа
[192] Вирус гриппа заранее разводили культуральной средой до подходящей концентрации, и каждое разведение вносили аликвотами по 50 мкл/лунку в 96-луночный планшет, содержащий тестируемый образец. Для измерения цитотоксичности культуральный раствор вносили аликвотами в планшет в количестве 50 мкл/лунку.
[193] 3) Разведение и распределение клеток
[194] Клетки с плотностью клеток, отрегулированной до подходящего значения, вносили аликвотами в количестве 100 мкл/лунку в 96-луночный планшет, содержащий тестируемый образец.
[195] Смесь перемешивали при помощи миксера для планшетов. Клетки культивировали в инкубаторе с СО2 в течение 3 дней как для теста противогриппозной активности, так для теста на цитотоксичность.
[196] 4) Распределение WST-8
[197] 96-луночный планшет культивировали в течение 3 дней и вели наблюдения невооруженным глазом и под микроскопом, и супернатант удаляли с планшета таким образом, чтобы не отсасывались клетки.
[198] Набор WST-8 разводили в 10 раз культуральной средой и аликвотировали по 100 мкл каждого раствора WST-8 в каждую лунку. Смесь перемешивали при помощи миксера для планшетов. Затем клетки инкубировали в инкубаторе с CO2 в течение 1-3 часов.
[199] Для планшета, на котором проводили измерения противогриппозной активности, после инкубации аликвоты 10 мкл 10% раствора SDS вносили в каждую лунку для инактивации вируса.
[200] 5) Определение оптической плотности
[201] Оптическую плотность 96-луночного планшета после перемешивания измеряли с помощью EnVision при двойной длине волны 450 нм/620 нм.
[202] Расчет значений каждого объекта измерения
[203] Для расчета использовали программу Microsoft Excel, расчет проводили на основе следующей формулы.
[204] Расчет ингибирующей концентрации 50% гибели клеток (ЕС50) для инфекции гриппа.
[205] EC50 =10Z
[206] Z = (50%-High%)/(High%-Low%)×{log(High conc.)-log(Low conc.)}+log(High conc.)
[207] Результаты измерений для примера 4 и примера 5 для тестируемого вещества (иллюстративные соединения) в качестве исходного соединения приведены в таблице 2.
[208] Таблица 2: Результаты теста на ингибирующую активность CEN и результаты теста на ингибирующий эффект CPE
нМ
нМ
[209] Из приведенных выше результатов можно видеть, что соединения 1A, 2A и 3A проявляют высокую ингибирующую активность кэп-зависимой эндонуклеазы и высокий ингибирующий эффект CPE, в частности, ингибирующий эффект CPE соединения 1A и соединения 3A в два раза выше, чем у S-033188A. Следовательно, иллюстративные соединения можно применять в качестве лекарственных средств для предотвращения/лечения симптома/заболевания, вызванного инфекцией вируса гриппа.
[210] Пример 6: Исследование биодоступности
[211] Экспериментальные материалы и способы для исследования пероральной абсорбции
[212] (1) Используемые животные: Использовали крыс линии Спрег-Доули (SD).
[213] (2) Условия кормления: Крысы SD имели твердый корм и очищенную воду в свободном доступе.
[214] (3) Дозирования и разбивка на группы: Проводили пероральное или внутривенное введение в предписанной дозировке. Группы были установлены следующим образом. (дозировка каждого соединения варьировалась)
[215] Пероральный прием от 1 до 30 мг/кг (n = от 5 до 6).
[216] Внутривенное введение от 0,5 до 10 мг/кг (n = от 5 до 6).
[217] (4) Приготовление дозируемого раствора: для перорального введения использовали суспензию внутрижелудочно. Для внутривенного введения через хвостовую вену использовали раствор.
[218] (5) Параметры оценки: Собирали образцы крови в разные моменты времени, и концентрации лекарств в плазме определяли с помощью ЖХ/МС/МС.
[219] (6) Статистический анализ: Для изменений концентрации в плазме рассчитывали площадь под кривой зависимости концентрации в плазме от времени (AUC) при помощи метода нелинейных наименьших квадратов, биодоступность (BA) рассчитывали на основе AUC в группе перорального введения и группе внутривенного введения, а период полувыведения концентрации лекарственного средства в плазме подсчитывали при внутривенном введении.
[220] Результаты исследования показаны в таблице 3.
[221] Таблица 3: Результаты измерения биодоступности иллюстративных соединений
[222] Из результатов в Таблице 3 можно увидеть, что пролекарства (1B, 2B, 3B) обладают улучшенной биодоступностью по сравнению с исходными соединениями (1A, 2A, 3A). Иллюстративные соединения 1A, 2A и 3A имеют значительно улучшенную биодоступность по сравнению с S-033188A; и биодоступность иллюстративных соединений 1B, 2B и 3B значительно улучшена по сравнению с S-033188B. Период полувыведения иллюстративных соединений при внутривенном введении увеличивался в разной степени.
[223] Следовательно, соединения согласно настоящему изобретению имеют лучшее пероральное всасывание, чем S-033188A/S-033188B, и их можно применять в качестве лекарственных средств для лечения и/или предотвращения симптомов и/или заболеваний, вызванных инфекцией вируса гриппа, и ожидается, что эти лекарственные средства будут требоваться в меньшей дозировке и оказывать меньше побочных эффектов.
[224] Пример 7: тест hERG
[225] Чтобы оценить риск удлинения интервала QT на электрокардиограмме, клетки HEK293, экспрессирующие hERG (ген специфических калиевых каналов сердца человека) каналы, были использованы для исследования калиевого тока замедленного выпрямления (IKr), который играет важную роль в процесс реполяризации желудочков.
[226] Применяли полностью автоматизированную систему пэтч-кламп для регистрации IKr, вызванного деполяризационной стимуляцией +50 мВ в течение 2 секунд и последующей реполяризационной стимуляцией -50 мВ в течение 2 секунд после удержания клеток при мембранном потенциале -80 мВ при помощи метода пэтч-клампа всей клетки. После стабилизации генерируемого тока клетки обрабатывали внеклеточной жидкостью с исследуемыми соединениями в целевых концентрациях (NaCl: 137 ммоль/л, KCl: 4 ммоль/л, CaCl2: 1,8 ммоль/л, MgCl2-6H2O: 1 ммоль/л, глюкоза: 10 ммоль/л, HEPES: 10 ммоль/л, pH 7,4) при комнатной температуре в течение 10 минут. Абсолютное значение максимального следового тока получали на основе текущего значения мембранного потенциала покоя при помощи программ анализа на основе полученного IKr. Кроме того, была рассчитана степень ингибирования по отношению к максимальному хвостовому току перед обработкой тестируемыми соединениями и по сравнению с группой, в которой применяли носитель (0,1% раствор ДМСО), и была проведена оценка влияния тестируемых соединений на IKr.
[227] Таблица 4: Степень ингибирования для иллюстративных соединений при 0,3-10 мкмоль/л
[228] Из результатов, приведенных в таблице 4, видно, что степень ингибирования у иллюстративных соединений 1А, 2А и 3А значительно ниже, чем у S-033188A, и особенно что степень ингибирования у соединений 1А и 3A составляет меньше половины от степени ингибирования S-033188A. Результаты испытаний показывают, что соединения 1A и 3A имеют более низкую кардиотоксичность, чем соединение S-033188A.
[229] Пример 8: Тест на летальное ингибирование у мышей, инфицированных вирусом гриппа
[230] Мышь
[231] В эксперименте использовали мышей BALB/cAnNCrlCrlj в возрасте от 6 до 7 недель.
[232] Приготовление раствора вируса
[233] Проводили пассажи A/WS/33, A/Victoria/3/75 или B/Maryland/1/59 (ATCC) в легких мышей с получением адаптированных к мышам вирусов. Криоконсервированные растворы адаптированных к мышам вирусов быстро размораживали и разбавляли DPBS до титра инфекционности (A/WS/33: 800-4000TC ID501 мышь, A/Victoria/3/75:750 TCID50/мышь, B/Maryland/1/59: 100 TCID50/мышь).
[234] Инфицирование
[235] Под анестезией смесью кетамин-ксилазин 100 мкл приготовленного раствора вируса вводили через нос для того, чтобы непосредственно заразить легкие мышей.
[236] Подготовка исследуемого образца
[237] Исследуемый образец суспендировали в 0,5% растворе метилцеллюлозы при подходящей концентрации.
[238] Введение инфицированным мышам
[239] Сразу после заражения вирусом или по прошествии определенного периода времени мышам перорально вводили 200 мкл разбавленного исследуемого образца.
[240] Оценка эффективности
[241] После заражения вирусом мышей содержали в течение 14 дней и рассчитывали количество ED50 для ежедневного введения (мг/кг/день), необходимое для 50% летального подавления, и сравнивали с контрольной группой для оценки эффекта подавления вируса.
[242] Результаты
[243] В таблице 5 приведены значения ED50 для однократного введения.
[244] Таблица 5: Значение ED50 в случае однократного введения иллюстративных соединений
[245] Из приведенных выше результатов можно видеть, что все тестируемые соединения демонстрируют различную степень ингибирующего действия вируса in vivo, в частности, что половинные эффективные дозы для соединения 1B и соединения 3B значительно меньше, чем для S-033188B. Это показывает, что дозировка иллюстративных соединений при клиническом применении будет меньше или интервал приема лекарств будет длиннее, а соответствующие побочные эффекты также будут меньше в сравнении с S-033188B.
[246] Пример 9: Обнаружение токсичности образцов для клеток MDCK
[247] В эксперименте использовали набор AlamarBlue® (Invitrogen) для обнаружения токсического действия лекарств на клетки.
[248] Принцип эксперимента: В эксперименте использовали набор AlamarBlue® (Invitrogen) для обнаружения токсического действия лекарств на клетки. AlamarBlue® - это окислительно-восстановительный индикатор, который изменяет поглощение и флуоресцентные сигналы в зависимости от клеточной метаболической активности. AlamarBlue® легко растворяется в воде. После того, как его окисленная форма попадает в клетки, она восстанавливается митохондриальными ферментами, вызывая измеримые изменения флуоресценции и цвета. Он используется для количественного анализа жизнеспособности и пролиферации клеток, а также для исследований цитотоксичности in vitro. Нормальные клетки с метаболической активностью могут преобразовывать реагенты с получением сильных изменений флуоресценции и цвета. Поврежденные и неактивные клетки имеют более низкую естественную метаболическую активность и более низкие соответствующие сигналы, поэтому сила сигнала флуоресценции может отражать уровень активности клетки.
[249] Процедура эксперимента: Клетки MDCK высевали в 96-луночный планшет для культивирования клеток и культивировали до прикрепления клеток для последующего использования. Лекарственное средство серийно разводили 3 раза от 2-кратной максимальной исследуемой концентрации до 8 градиентов. Лекарственное средство добавляли к клеткам и культивировали в инкубаторе с CO2 при температуре 37°C. После 48 часов дозирования и инкубации под микроскопом наблюдали цитопатический эффект (ЦПЭ), вызванный лекарственным средством. Затем добавляли среду AlamarBlue® для определения выживаемости клеток. Токсичность препарата для клеток обратно пропорциональна активности клеток и выражается как активность клеток.
[250] Формула расчета: активность клеток (%) = (группа препарата - пустой контроль)/(клеточный контроль - пустой контроль)*100.
[251] Лекарственное средство в очень высоких концентрациях оказывает токсическое действие на клетки MDCK. Концентрация, вызывающая 50% токсического действия, показана CC50. См. подробную информацию в таблице ниже.
[252] Таблица 6: Токсичность иллюстративных соединений для клеток
[253] Токсичность соединения 1A значительно ниже, чем токсичность S-033188A. Таким образом, соединение 1A имеет более высокую безопасность, чем S-033188A.
[254] Для специалистов в данной области техники настоящее изобретение не ограничивается приведенными выше иллюстративными примерами и может быть воплощено в других конкретных формах без отступления от его основных признаков. Следовательно, все аспекты следует рассматривать как иллюстративные примеры, относящиеся к прилагаемой формуле изобретения, а не как ограниченные приведенными примерами, и процитированные документы также предназначены для прилагаемой формулы изобретения, а не вышеизложенных примеров. Поэтому ожидается, что все модификации, имеющие эквивалентное значение и объем формулы изобретения, будут включены в данный документ.
[255] Все патенты, заявки на патенты и литературные ссылки, перечисленные в настоящем описании, полностью включены посредством ссылки. В случае несоответствия определения будут иметь значения, приведенные в настоящем документе.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЛЕКАРСТВЕННЫЙ ПРЕПАРАТ ДЛЯ ЛЕЧЕНИЯ ГРИППА, ХАРАКТЕРИЗУЮЩИЙСЯ ТЕМ, ЧТО В НЕМ ОБЪЕДИНЕНЫ ИНГИБИТОР КЭП-ЗАВИСИМОЙ ЭНДОНУКЛЕАЗЫ И ЛЕКАРСТВЕННОЕ СРЕДСТВО ПРОТИВ ГРИППА | 2016 |
|
RU2745071C2 |
| ЗАМЕЩЕННЫЕ ПОЛИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ПИРИДОНА И ИХ ПРОЛЕКАРСТВА | 2016 |
|
RU2712275C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ЗАМЕЩЕННЫЕ ПОЛИЦИКЛИЧЕСКИЕ ПИРИДОНОВЫЕ ПРОИЗВОДНЫЕ И ИХ ПРОЛЕКАРСТВО | 2017 |
|
RU2727962C1 |
| КЛАСС КОНДЕНСИРОВАННЫХ КОЛЬЦЕВЫХ СОЕДИНЕНИЙ И ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2021 |
|
RU2831125C1 |
| Противо-РНК вирусное, в том числе противокоронавирусное средство - замещенный хиноксалин, фармацевтическая композиция и применения | 2020 |
|
RU2744429C1 |
| БИВАЛЕНТНЫЕ ИНГИБИТОРЫ БРОМОДОМЕНОВ И ПУТИ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2742035C2 |
| ХИНОКСАЛИНСОДЕРЖАЩИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ ВИРУСА ГЕПАТИТА С | 2008 |
|
RU2493160C2 |
| ПРОИЗВОДНОЕ ПИРИДОПИРИМИДИНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2019 |
|
RU2778524C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ КАК ИНГИБИТОРЫ TRK | 2019 |
|
RU2803817C2 |
| ДЕЙТЕРИРОВАННЫЕ ПРОИЗВОДНЫЕ 2-(ИМИДАЗОЛ-4-ИЛ)-ЭТАНАМИД ПЕНТАНДИОВОЙ-1,5 КИСЛОТЫ | 2021 |
|
RU2783866C2 |
Группа изобретений относится к области фармацевтической промышленности и включает полициклическое производное карбамоилпиридона, представленное ниже, или его стереоизомер, или фармацевтически приемлемую соль, их применение, фармацевтическую композицию и способ лечения на их основе. Технический результат - полициклическое производное карбамоилпиридона, обладающее ингибирующей активностью в отношении 5'-кэп-зависимой эндонуклеазы. 5 н. и 7 з.п. ф-лы, 6 табл., 9 пр.
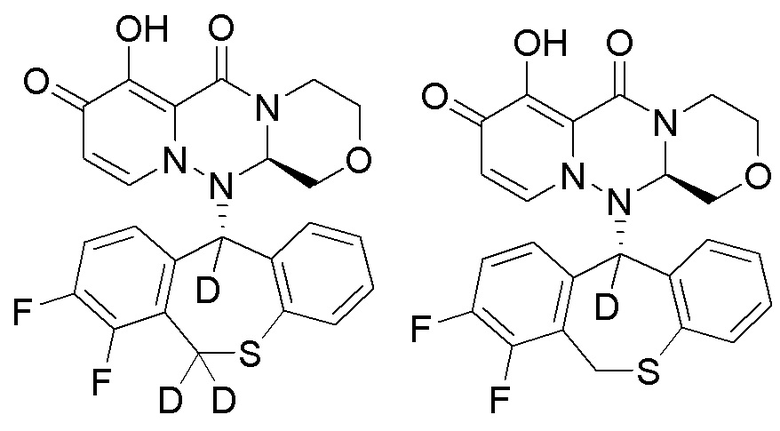
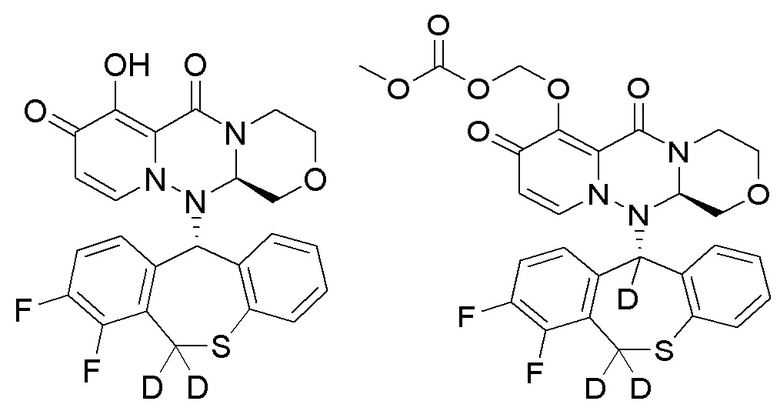
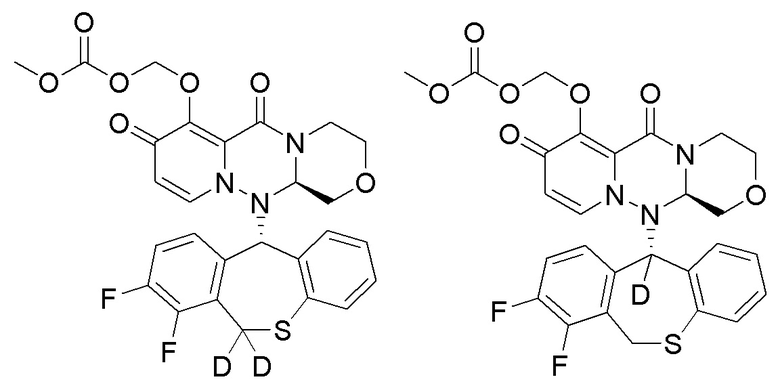
1. Полициклическое производное карбамоилпиридона, представленное ниже, стереоизомер указанного соединения или фармацевтически приемлемая соль указанного соединения
2. Полициклическое производное карбамоилпиридона, стереоизомер указанного соединения, фармацевтически приемлемая соль указанного соединения по п. 1, отличающееся тем, что
указанная фармацевтически приемлемая соль указанного соединения представляет собой соль присоединения кислоты, образованную указанным полициклическим производным карбамоилпиридона и органической кислотой или неорганической кислотой; где указанная органическая кислота представляет собой одну или более из кислот, выбранных из группы, состоящей из малеиновой кислоты, фумаровой кислоты, бензойной кислоты, аскорбиновой кислоты, янтарной кислоты, метансульфоновой кислоты, уксусной кислоты, трифторуксусной кислоты, щавелевой кислоты, пропионовой кислоты, винной кислоты, салициловой кислоты, лимонной кислоты, глюконовой кислоты, молочной кислоты, миндальной кислоты, фенилуксусной кислоты, аспарагиновой кислоты, стеариновой кислоты, пальмитиновой кислоты, гликолевой кислоты, глутаминовой кислоты и бензолсульфоновой кислоты; указанная неорганическая кислота представляет собой одну или более кислот, выбранных из группы, состоящей из хлористоводородной кислоты, бромистоводородной кислоты, серной кислоты, фосфорной кислоты и азотной кислоты;
или указанная фармацевтически приемлемая соль указанного соединения представляет собой соль, образованную полициклическим производным карбамоилпиридона и одним или более органическими и неорганическими катионами, выбранными из группы, состоящей из ионов щелочных металлов, ионов щелочноземельных металлов и ионов аммония; где указанный щелочной металл выбран из лития, натрия или калия и указанный щелочноземельный металл выбран из магния, бария или кальция.
3. Фармацевтическая композиция для ингибирования 5'-кэп-зависимой эндонуклеазы, содержащая терапевтически эффективное количество полициклического производного карбамоилпиридона, стереоизомера указанного соединения или фармацевтически приемлемой соли указанного соединения по п. 1 или 2 и фармацевтически приемлемый носитель.
4. Применение полициклического производного карбамоилпиридона, стереоизомера указанного соединения или фармацевтически приемлемой соли указанного соединения по п. 1 или 2 для получения ингибитора 5'-кэп-зависимой эндонуклеазы.
5. Применение полициклического производного карбамоилпиридона, стереоизомера указанного соединения или фармацевтически приемлемой соли указанного соединения по п. 1 или 2 для получения лекарственного средства для лечения и/или облегчения заболевания, представляющего собой грипп, связанный с 5'-кэп-зависимой эндонуклеазой.
6. Применение по п. 5, отличающееся тем, что заболевание, представляющее собой грипп, связанный с 5'-кэп-зависимой эндонуклеазой, представляет собой симптом и/или заболевание, вызванное инфекцией вируса гриппа.
7. Применение по п. 6, отличающееся тем, что указанный вирус гриппа выбран из типа A, типа B или типа C.
8. Применение по п. 7, отличающееся тем, что указанный симптом выбран из симптомов простуды, таких как лихорадка, озноб, головная боль, мышечная боль, общая усталость и тому подобное, или воспалений дыхательных путей, таких как боль в горле, насморк, заложенность носа, кашель, мокрота; желудочно-кишечных симптомов, таких как боль в животе, рвота, диарея; и дальнейших вторичных инфекционных осложнений, таких как острая энцефалопатия, пневмония.
9. Способ лечения и/или облегчения заболевания, представляющего собой грипп, связанный с 5'-кэп-зависимой эндонуклеазой, у индивидуума, нуждающегося в этом, включающий введение терапевтически эффективного количества указанного полициклического производного карбамоилпиридона, стереоизомера указанного соединения или фармацевтически приемлемой соли указанного соединения по п.1 или 2 указанному индивидууму.
10. Способ по п. 9, отличающийся тем, что указанное заболевание, представляющее собой грипп, связанный с 5'-кэп-зависимой эндонуклеазой, относится к симптому и/или заболеванию, вызванному инфекцией вируса гриппа.
11. Способ по п. 10, отличающийся тем, что вирус гриппа выбран из типа A, типа B или типа C.
12. Способ по п. 10, отличающийся тем, что симптом выбран из симптомов простуды, таких как лихорадка, озноб, головная боль, мышечная боль, общая усталость и тому подобное, или воспалений дыхательных путей, таких как боль в горле, насморк, заложенность носа, кашель, мокрота; желудочно-кишечных симптомов, таких как боль в животе, рвота, диарея; и дальнейших вторичных инфекционных осложнений, таких как острая энцефалопатия, пневмония.
| EP 3290424 A1, 07.03.2018 | |||
| R.D.TUNG, Deuterium medicinal chemistry comes of age | |||
| FUTURE MED | |||
| CHEM | |||
| Токарный резец | 1924 |
|
SU2016A1 |
| ПРОЛЕКАРСТВЕННАЯ ФОРМА ЗАМЕЩЕННОГО ПОЛИЦИКЛИЧЕСКОГО ПРОИЗВОДНОГО КАРБАМОИЛПИРИДОНА | 2011 |
|
RU2608519C2 |

