Область техники
Настоящее изобретение относится к 1,3,4-оксадиазолтиокарбонильным соединениям, обладающим ингибирующей активностью по отношению к гистондеацетилазе 6 (ГДАЦ6), их стереоизомерам, их фармацевтически приемлемым солям; их применению, их применению для получения терапевтического лекарственного средства, способу лечения заболеваний, применяя их; фармацевтической композиции, содержащей их; и способу их получения.
Уровень техники
В клетках посттрансляционная модификация, такая как ацетилирование, служит очень важным регуляторным модулем в центре биологических процессов и также строго контролируется несколькими ферментами. Как основной белок, составляющий хроматин, гистон действует как ось, вокруг которой накручивается ДНК, и, таким образом, способствует конденсации ДНК. Кроме того, баланс между ацетилированием и деацетилированием гистонов играет очень важную роль в экспрессии генов.
Известно, что гистондеацетилаза (ГДАЦ) как фермент для удаления ацетильной группы из остатка лизина белка-гистона, составляющего хроматин, связана с выключением генов и вызывает остановку клеточного цикла, ангиогенное ингибирование, иммунорегуляцию, апоптоз и т.д. (Hassig et al. al., Curr.Opin.Chem.Biol. 1, 300-308 (1997)). Также сообщается, что ингибирование функций фермента ГДАЦ побуждает раковые клетки входить в стадию апоптоза путем снижения активности факторов, связанных с выживанием раковых клеток, и активации факторов, связанных с гибелью раковых клеток в организме (Warrell et al., Natl. Cancer Inst.90, 1621-1625 (1998)).
Для человека известны 18 ГДАЦ, которые классифицируются на четыре класса по гомологии с ГДАЦ дрожжей. В данном случае одиннадцать ГДАЦ, применяющих цинк в качестве кофактора, можно разделить на три группы: класс I (ГДАЦ1, 2, 3, 8), класс II (IIa: ГДАЦ4, 5, 7, 9; IIb: ГДАЦ6, 10) и класс IV (ГДАЦ11). Кроме того, семь ГДАЦ класса III (SIRT 1-7) применяют НАД+ в качестве кофактора вместо цинка (Bolden et al., Nat. Rev. Drug Discov. 5(9), 769-784 (2006)).
Различные ингибиторы ГДАЦ в настоящее время находятся на стадии доклинической или клинической разработки, но в качестве противораковых средств пока известны только неселективные ингибиторы ГДАЦ. Вориностат (SAHA) и ромидепсин (FK228) получили одобрение в качестве терапевтического средства при кожной Т-клеточной лимфоме, а панобиностат (LBH-589) получил одобрение в качестве терапевтического средства при множественной миеломе. Однако известно, что неселективные ингибиторы ГДАЦ обычно вызывают побочные эффекты, такие как утомляемость, тошнота и т.п. при высоких дозах (Piekarz et al., Pharmaceuticals 3, 2751-2767 (2010)). Сообщается, что побочные эффекты вызваны ингибированием ГДАЦ I класса. Из-за побочных эффектов и т.д., разработка неселективных ингибиторов ГДАЦ была ограничена при разработке лекарственных средств в других областях, кроме противораковых средств (Witt et al., Cancer Letters 277, 8-21 (2009)).
Между тем, сообщается, что избирательное ингибирование ГДАЦ класса II не будет проявлять токсичности, которая имела место при ингибировании ГДАЦ класса I. В случае разработки селективных ингибиторов ГДАЦ, вероятно, удастся решить проблему побочных эффектов, таких как токсичность и т.д., вызванных неселективным ингибированием ГДАЦ. Соответственно, существует вероятность того, что селективные ингибиторы ГДАЦ могут быть разработаны в качестве эффективных терапевтических средств при различных заболеваниях (Matthias et al., Mol. Cell. Biol. 28, 1688-1701 (2008)).
ГДАЦ6, один из ГДАЦ класса IIb, как известно, в основном присутствует в цитоплазме и содержит белок тубулин, таким образом, участвуя в деацетилировании ряда негистоновых субстратов (HSP90, кортактин и т.д.) (Yao et al., Mol. Cell 18, 601-607 (2005)). ГДАЦ6 имеет два каталитических домена, в которых С-концевой домен цинкового пальца может связываться с убиквитинированным белком. Известно, что ГДАЦ6 имеет ряд негистоновых белков в качестве субстрата и, таким образом, играет важную роль в различных заболеваниях, таких как рак, воспалительные заболевания, аутоиммунные заболевания, неврологические заболевания, нейродегенеративные расстройства и подобные (Santo et al., Blood 119, 2579-2589 (2012); Vishwakarma et al., International Immunopharmacology 16, 72-78 (2013); Hu et al., J. Neurol. Sci. 304, 1-8 (2011)).
Структурная особенность, общая для различных ингибиторов ГДАЦ, состоит из кэп-группы, линкерной группы и цинксвязывающей группы (ZBG), как показано на следующей структуре вориностата. Многие исследователи провели исследование ингибирующей активности и селективности в отношении ферментов посредством структурной модификации кэп-группы и линкерной группы. Из групп известно, что цинксвязывающая группа играет более важную роль в ингибирующей активности и селективности фермента (Wiest et al., J. Org. Chem 78, 5051-5055 (2013); Methot et al., Bioorg. Med. Chem. Lett. 18, 973-978 (2008)).
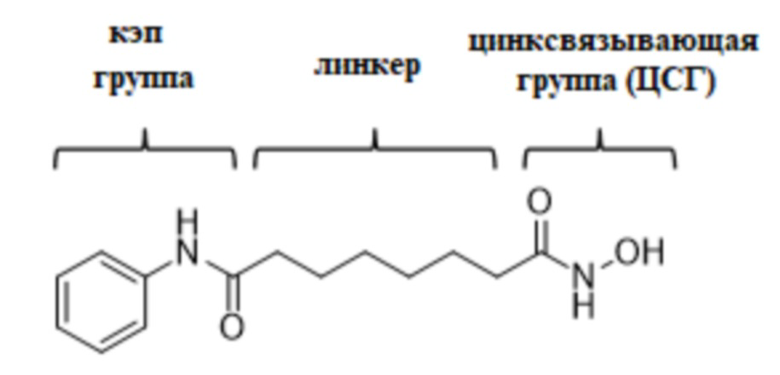
Большую часть цинксвязывающей группы составляет гидроксамовая кислота или бензамид. При этом производные гидроксамовой кислоты демонстрируют сильный ингибирующий ГДАЦ эффект, но имеют проблемы с низкой биодоступностью и серьезной нецелевой активностью. Производные бензамида включают анилин и поэтому имеют проблему, заключающуюся в том, что они могут давать токсичные метаболиты in vivo (Woster et al., Med. Chem. Commun., онлайн-публикация (2015)).
Соответственно, в отличие от неселективных ингибиторов, обладающих побочными эффектами, существует необходимость в разработке селективных ингибиторов ГДАЦ6, имеющих цинксвязывающую группу с улучшенной биодоступностью, но не вызывающих побочных эффектов, для лечения рака, воспалительных заболеваний, аутоиммунных заболеваний, неврологические заболевания, нейродегенеративные расстройства и подобных.
Ссылки родственного уровня техники
Патентные документы
Нерассмотренная международная патентная публикация No. WO 2011/091213 (опубл. 28 июля 2011): ACY-1215
Нерассмотренная международная патентная публикация No. WO 2011/011186 (опубл. 27 января 2011): Tubastatin
Нерассмотренная международная патентная публикация No. WO 2013/052110 (опубл. 11 апреля 2013): Sloan-K
Нерассмотренная международная патентная публикация No. WO 2013/041407 (опубл. 28 марта 2013): Cellzome
Нерассмотренная международная патентная публикация No. WO 2013/134467 (опубл. 12 сентября 2013): Kozi
Нерассмотренная международная патентная публикация No. WO 2013/008162 (опубл. 17 января 2013): Novartis
Нерассмотренная международная патентная публикация No. WO 2013/080120 (опубл. 06 июня 2013): Novartis
Нерассмотренная международная патентная публикация No. WO 2013/066835 (опубл. 10 мая 2013): Tempero
Нерассмотренная международная патентная публикация No. WO 2013/066838 (опубл. 10 мая 2013): Tempero
Нерассмотренная международная патентная публикация No. WO 2013/066833 (опубл. 10 мая 2013): Tempero
Нерассмотренная международная патентная публикация No. WO 2013/066839 (опубл. 10 мая 2013): Tempero.
Описание настоящего изобретения
Техническая проблема
Цель настоящего изобретения заключается в обеспечении 1,3,4-оксадиазолтиокарбонильных соединений, обладающих селективной ГДАЦ6 ингибирующей активностью, их стереоизомеров или их фармацевтически приемлемых солей.
Другая цель настоящего изобретения заключается в обеспечении фармацевтической композиции, содержащей 1,3,4-оксадиазолтиокарбонильные соединения, обладающие селективной ГДАЦ6 ингибирующей активностью, их стереоизомеры или их фармацевтически приемлемые соли.
Еще другая цель настоящего изобретения заключается в обеспечении способа их получения.
Еще другая цель настоящего изобретения заключается в обеспечении фармацевтической композиции, содержащей данные соединения.
Еще другая цель настоящего изобретения заключается в обеспечении фармацевтической композиции, содержащей соединения для предотвращения или лечения Заболеваний, связанных с активностью ГДАЦ6. В настоящем изобретении, заболевания, связанные с активностью ГДАЦ6, могут включать инфекционные заболевания, новообразования, эндокринопатии, болезни питания и обмена веществ, психические и поведенческие расстройства, неврологические заболевания, заболевания глаз и глазных придатков, болезни кровообращения, респираторные заболевания, проблемы с пищеварением, заболевания кожи и подкожной клетчатки, заболевания опорно-двигательного аппарата и соединительной ткани или тератоз, деформации и хромосомные аберрации.
Еще другая цель настоящего изобретения заключается в обеспечении их применения для получения лекарственного средства для предотвращения или лечения заболеваний, связанных с активностью ГДАЦ6.
Еще другая цель настоящего изобретения заключается в обеспечении способа лечения заболеваний, связанных с активностью ГДАЦ6, включающего введение терапевтически эффективного количества соединений или фармацевтической композиции, содержащей данные соединения.
Техническое решение проблемы
Изобретатели настоящего изобретения обнаружили оксадиазольное соединение, обладающее ингибирующей активностью по отношению к гистондеацетилазе 6 (ГДАЦ6) и применили его для ингибирования или лечения заболеваний, связанных с активностью ГДАЦ6, посредством этого завершив настоящее изобретение.
В настоящем изобретении далее, настоящее изобретение будет описано более подробно. Все комбинации различных элементов, описанных в настоящем изобретении, попадают в объем настоящего изобретения. Кроме того, нельзя не заметить, что объем настоящего изобретения не ограничивается конкретным описанием, приведенным ниже.
1,3,4-оксадиазолтиокарбонильные соединения
Согласно целям, соединения, обеспечиваемые в настоящем изобретении, могут представлять собой, как показано в (1)-(3) ниже.
(1) 1,3,4-оксадиазолтиокарбонильное соединение, представленное формулой I ниже, его стереоизомеры или его фармацевтически приемлемые соли:
<формула I>
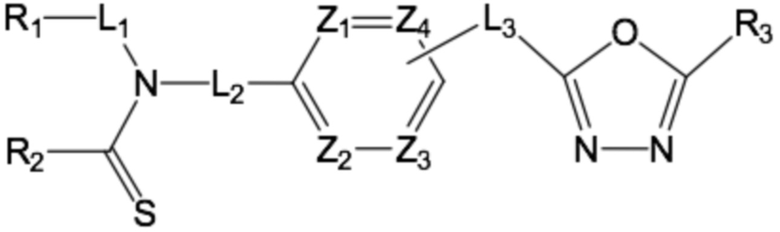
в формуле I,
каждый L1, L2 и L3 независимо представляет собой простую связь или -(C1-C4 алкилен)-;
R1 представляет собой -H, -(C1-C4 алкил), -(C1-C4 алкил)-O(C1-C4 алкил), -(C1-C4 алкил)-C(=O)-O(C1-C4 алкил), -(C3-C7 циклоалкил), -(C2-C6 циклогетероалкил), -арил, -гетероарил, -адамантил, 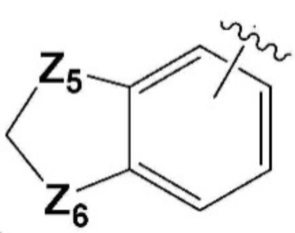 или
или 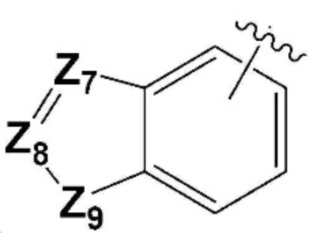 ,
,
в R1,
по меньшей мере один H -(C1-C4 алкила) может быть замещен -T или -OH,
по меньшей мере один H -арила или -гетероарила может каждый независимо быть замещен -T, -OH, -O(C1-C4 алкилом), -OCF3, -O-арилом, -NRDRE, -(C1-C4 алкилом), -CF3, -CF2H, -C(=O)-(C1-C4 алкилом), -C(=O)-O(C1-C4 алкилом), -C(=O)-NRDRE, -S(=O)2-(C1-C4 алкилом), -арилом, -гетероарилом, , или 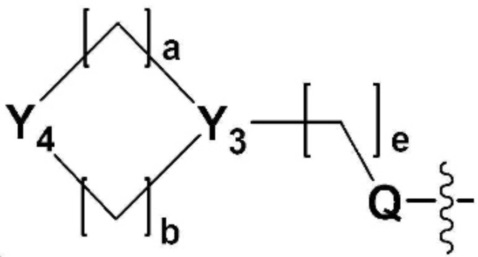 ,
,
в котором по меньшей мере один H может быть замещен -T, -(C1-C4 алкилом), -CF3 или -CF2H,
по меньшей мере один H -(C3-C7 циклоалкила), -(C2-C6 циклогетероалкила), -адамантила, или может каждый независимо быть замещен -T, -OH или -(C1-C4 алкилом);
R2 представляет собой -NRARB, -ORC, -гетероарил, 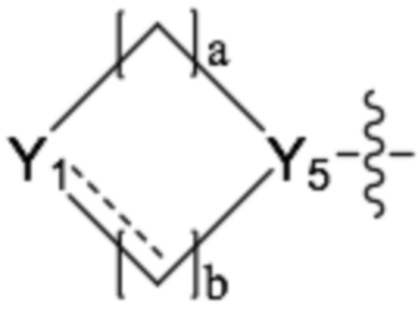 ,
,  или
или 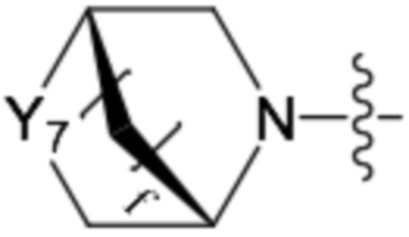 ,
,
в R2,
по меньшей мере один H или
может быть замещен -T, -OH, -O(C1-C4 алкилом), -NRDRE, -(C1-C4 алкилом), -CF3, -CF2H, -CN, -арилом, -гетероарилом, -(C1-C4 алкил)-арилом или -(C1-C4 алкил)-гетероарилом, в которых по меньшей мере один H -арила, -гетероарила, -(C1-C4 алкил)-арила или -(C1-C4 алкил)-гетероарила может быть замещен -T, -OH, -CF3 или -CF2H;
R3 представляет собой -CT3 или -CT2H;
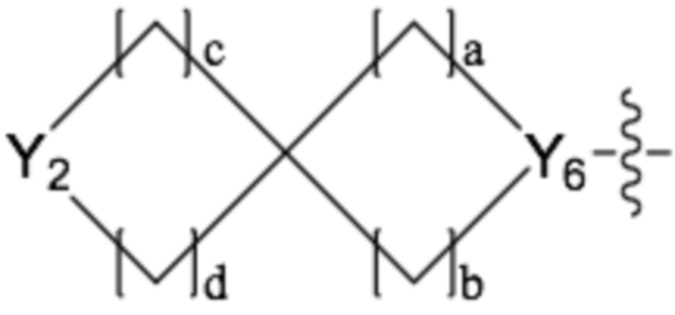
каждый Y1, Y2, Y4 и Y7 независимо представляет собой =CH-, -CHRF-, -NRF-, -O-, -C(=O)- или -S(=O)2-;
каждый Y3, Y5 и Y6 независимо представляет собой -CH- или -N-;
каждый Z1-Z4 независимо представляет собой N или CRZ,
в Z1-Z4,
по меньшей мере три Z1-Z4 могут не представлять собой N одновременно, и RZ представляет собой -H, -T или -O(C1-C4 алкил);
каждый Z5 и Z6 независимо представляет собой -CH2- или -O-;
каждый Z7 и Z8 независимо представляет собой =CH- или =N-;
Z9 представляет собой -NRG- или -S-;
каждый RA и RB независимо представляет собой -H, -(C1-C4 алкил), -(C1-C4 алкил)-OH, -(C1-C4 алкил)-NRDRE, -арил, -(C1-C4 алкил)-арил, -гетероарил, -(C1-C4 алкил)-гетероарил, -(C3-C7 циклоалкил), -(C2-C6 гетероциклоалкил) или ,
в RA и RB,
по меньшей мере один H -(C1-C4 алкила), -(C1-C4 алкил)-OH или -(C1-C4 алкил)-NRDRE может быть замещен -T,
по меньшей мере один H -арила, -(C1-C4 алкил)-арила, -гетероарила, -(C1-C4 алкил)-гетероарила, -(C3-C7 циклоалкила) или -(C2-C6 гетероциклоалкила) может быть замещен -T, -OH, -O(C1-C4 алкилом), -(C1-C4 алкилом), -CF3, -CF2H или -CN,
по меньшей мере один H может быть замещен -T, -OH, -O(C1-C4 алкилом), -(C1-C4 алкилом), -CF3, -CF2H, -CN, -(C2-C6 гетероциклоалкилом), -арилом, -(C1-C4 алкил)-арилом или -гетероарилом;
RC представляет собой -(C1-C4 алкил), -арил, -(C1-C4 алкил)-арил, -гетероарил или -(C1-C4 алкил)-гетероарил,
в RC,
по меньшей мере один H -(C1-C4 алкила) может быть замещен -T или -OH,
по меньшей мере один H -арила, -(C1-C4 алкил)-арила, -гетероарила или -(C1-C4 алкил)-гетероарила может быть замещен -T, -OH, -CF3 или -CF2H;
каждый RD и RE независимо представляет собой -H, -(C1-C4 алкил), -арил или -(C1-C4 алкил)-арил,
в RD и RE,
по меньшей мере один H -(C1-C4 алкила) может быть замещен -T или -OH,
по меньшей мере один H -арила или -(C1-C4 алкил)-арила может быть замещен -T, -OH, -CF3 или -CF2H;
RF представляет собой -H, -(C1-C6 алкил), -(C1-C4 алкил)-OH, -(C1-C4 алкил)-O-(C1-C4 алкил), -C(=O)-(C1-C4 алкил), -C(=O)-O(C1-C4 алкил), -(C1-C4 алкил)-C(=O)-O(C1-C4 алкил), -NRDRE, -(C1-C4 алкил)-NRDRE, -S(=O)2-(C1-C4 алкил), -арил, -(C1-C4 алкил)-арил, -(C2-C4 алкенил)-арил, -гетероарил, -(C1-C4 алкил)-гетероарил, -C(=O)-(C3-C7 циклоалкил), -(C2-C6 гетероциклоалкил) или -(C1-C4 алкил)-C(=O)-(C2-C6 гетероциклоалкил),
в RF,
по меньшей мере один H -(C1-C6 алкила), -(C1-C4 алкил)-OH, -(C1-C4 алкил)-O-(C1-C4 алкила), -C(=O)-(C1-C4 алкила), -C(=O)-O(C1-C4 алкила), -(C1-C4 алкил)-C(=O)-O(C1-C4 алкила), -NRDRE, -(C1-C4 алкил)-NRDRE или -S(=O)2-(C1-C4 алкила) может быть замещен -T,
по меньшей мере один H -арила, -(C1-C4 алкил)-арила, -(C2-C4 алкенил)-арила, -гетероарила, -(C1-C4 алкил)-гетероарила, -C(=O)-(C3-C7 циклоалкила), -(C2-C6 гетероциклоалкила) или -(C1-C4 алкил)-C(=O)-(C2-C6 гетероциклоалкила) может быть замещен -T, -OH, -(C1-C4 алкил), -CF3 или -CF2H;
RG представляет собой -H или -(C1-C4 алкил);
Q представляет собой -O- или простую связь;
 представляет собой простую связь или двойную связь, при условии, что когда представляет собой двойную связь, Y1 представляет собой =CH-;
представляет собой простую связь или двойную связь, при условии, что когда представляет собой двойную связь, Y1 представляет собой =CH-;
каждый a-e независимо представляет собой целое 0, 1, 2, 3 или 4, при условии, что a и b могут не представлять собой 0 вместе, и c и d могут не представлять собой 0 вместе;
f представляет собой целое 1 или 2; и
T представляет собой F, Cl, Br или I.
(2) 1,3,4-Оксадиазолтиокарбонильное соединение, его стереоизомеры или его фармацевтически приемлемые соли по (1) выше:
в формуле I,
каждый L1, L2 и L3 независимо представляет собой простую связь или -(C1-C2 алкилен)-;
R1 представляет собой -(C1-C4 алкил), -(C6-C12 арил) или -(C3-C10 гетероарил), включая по меньшей мере один гетероатом, выбранный из группы, состоящей из O, N и S,
в R1,
по меньшей мере один H -(C1-C4 алкила) может быть замещен -T или -OH,
по меньшей мере один H -(C6-C12 арила) или -(C3-C10 гетероарила), включая по меньшей мере один гетероатом, выбранный из группы, состоящей из O, N и S может каждый независимо быть замещен -T, -CF3 или -CF2H;
R2 представляет собой -(C3-C10 гетероарил), включая по меньшей мере один гетероатом, выбранный из группы, состоящей из O, N и S, , или ;
R3 представляет собой -CT3 или -CT2H;
каждый Y1, Y2, Y4 и Y7 независимо представляет собой =CH-, -CHRF-, -NRF-, -O-, -C(=O)- или -S(=O)2-;
каждый Y3, Y5 и Y6 независимо представляет собой -CH- или -N-;
каждый Z1-Z4 независимо представляет собой N или CRZ,
в Z1-Z4,
по меньшей мере три Z1-Z4 могут не представлять собой N одновременно,
Rz представляет собой -H, -T или -O(C1-C4 алкил);
RF представляет собой -H, -(C1-C6 алкил), -C(=O)-(C1-C4 алкил) или -(C2-C6 гетероциклоалкил);
представляет собой простую связь или двойную связь, при условии, что когда представляет собой двойную связь, Y1 представляет собой =CH-;
каждый a-e независимо представляет собой целое 0, 1, 2, 3 или 4, при условии, что a и b могут не представлять собой 0 вместе, и c и d могут не представлять собой 0 вместе;
f представляет собой целое 1 или 2; и
T представляет собой F, Cl, Br или I.
В настоящем изобретении, 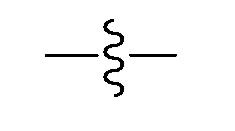 представляет собой соединенную часть формулы.
представляет собой соединенную часть формулы.
В настоящем изобретении, представляет собой простую связь или двойную связь. Другими словами, может представлять как простую связь или  как двойную связь.
как двойную связь.
В настоящем изобретении, «простая связь» относится к связи, в которой два атома имеют общую пару электронов с образованием связи
В настоящем изобретении, "Cm-Cn" (в котором каждый m и n независимо представляет собой целое 1 или более) может означать количество атомов углерода, например, "C1-C4 алкил" представляет собой алкил, содержащий 1-4 атомов углерода.
В настоящем изобретении, «алкил» означает линейную или разветвленную насыщенную углеводородную группу и, например, «C1-C4 алкил» может включать метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, трет-бутил, изобутил и т.д.
В настоящем изобретении, «алкилен» означает двухвалентную функциональную группу, полученную из указанного алкила (включая и линейный и разветвленный) и, например, «C1-C4 алкилен» может включать метилен (-CH2-), этилен (-CH2CH2-), н-пропилен (-CH2CH2CH2-), н-бутилен (-CH2CH2CH2CH2-) и т.д.
В настоящем изобретении, «гетероарил» означает ароматическую функциональную группу, содержащую по меньшей мере один гетероатом в кольце, и гетероатом может включать по меньшей мере гетероатом, выбранный из группы, состоящей из O, N и S. Гетероарил может включать гетероарил, который содержит 3-10 атомов углерода в кольце. Гетероарил может представлять собой 4- или более членное кольцо, например, 5-6-членное кольцо. Например, «гетероарил» может представлять собой фуран, тиофен, тиазол, тиадиазол, пиррол, пиразол, пиридин, пиримидин, имидазол, триазол, триазин, пиридазин, пиразин или подобные, но не ограничивается ими.
В настоящем изобретении, «гетероциклоалкил» означает циклический алкил, содержащий по меньшей мере один гетероатом в кольце. Гетероатом может включать по меньшей мере гетероатом, выбранный из группы, состоящей из O, N и S. Гетероциклоалкил может включать гетероциклоалкил, который содержит 3-10 атомов углерода в кольце. Гетероциклоалкил может представлять собой 3- или более членное кольцо, например, 3-6-членное кольцо. Например, «гетероциклоалкил» может представлять собой пропиленоксид, оксетан, тетрагидрофуран, тетрагидропиран, азетидин, морфолин, тиоморфолиндиоксид, пиперазин, пиперидин, оксадиазол, пирролидин и т.д., но не ограничивается ими.
В настоящем изобретении, T означает атом галогена и может представлять собой F, Cl, Br или I.
В настоящем изобретении, фармацевтически приемлемые соли могут относиться к солям, традиционно применяемым в фармацевтической промышленности, например, к солям неорганических ионов, полученным из кальция, калия, натрия, магния и т.п.; солям неорганических кислот, полученных из хлористоводородной кислоты, азотной кислоты, фосфорной кислоты, бромной кислоты, йодистой кислоты, хлорной кислоты, серной кислоты и т.д.; солям органических кислот, полученных из уксусной кислоты, трифторуксусной кислоты, лимонной кислоты, малеиновой кислоты, янтарной кислоты, щавелевой кислоты, бензойной кислоты, винной кислоты, фумаровой кислоты, миндальной кислоты, пропионовой кислоты, молочной кислоты, гликолевой кислоты, глюконовой кислоты, галактуроновой кислоты, глутаминовая кислота, глутаровая кислота, глюкуроновая кислота, аспарагиновая кислота, аскорбиновая кислота, угольная кислота, ванилиновая кислота, иодистоводородная кислота и т.д.; солям сульфокислот, полученным из метансульфокислоты, этансульфокислоты, бензолсульфокислоты, п-толуолсульфокислоты, нафталинсульфокислоты и т.д.; солям аминокислот, полученным из глицина, аргинина, лизина и т.д.; солям аминов, полученным из триметиламина, триэтиламина, аммиака, пиридина, пиколина и т.д.; и подобным, но типы солей, подразумеваемые в настоящем изобретении, не ограничиваются перечисленными солями.
«Стереоизомер» 1,3,4-оксадиазолтиокарбонильного соединения, представленного формулой I настоящего изобретения, может включать диастереомер и оптический изомер (энантиомер), в котором оптический изомер может включать не только энантиомер, но и смесь энантиомеров, и даже рацемат. Изомер можно отделить путем разделения согласно предшествующему уровню техники, например, с помощью колоночной хроматографии, ВЭЖХ или подобного. Альтернативно, каждый стереоизомер 1,3,4-оксадиазолтиокарбонильного соединения, представленного формулой I, можно стереоспецифически синтезировать с применением известного набора оптически чистых исходных материалов и/или реагентов.
(3) 1,3,4-оксадиазолтиокарбонильное соединение, его стереоизомер или его фармацевтически приемлемые соли по (1) или (2) выше,
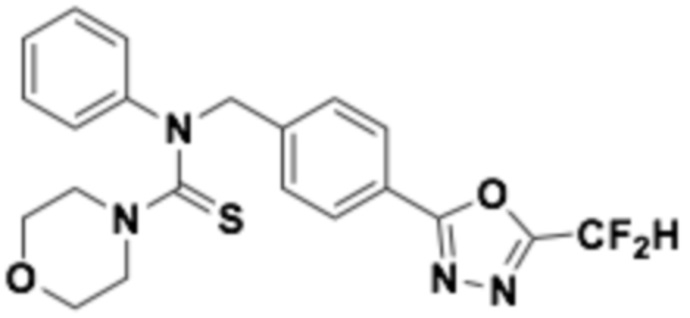
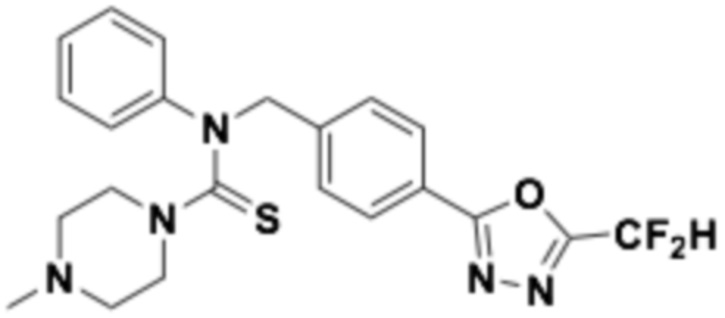
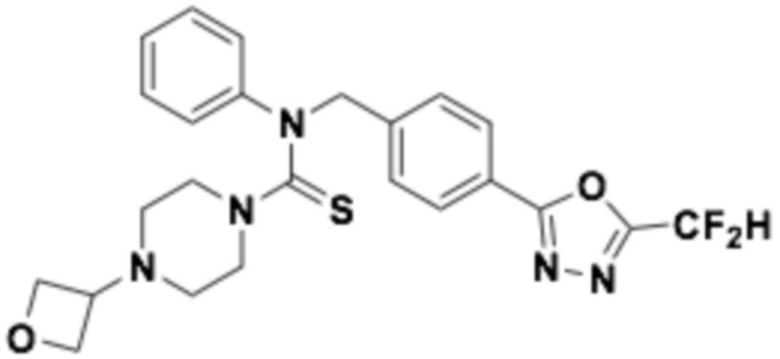
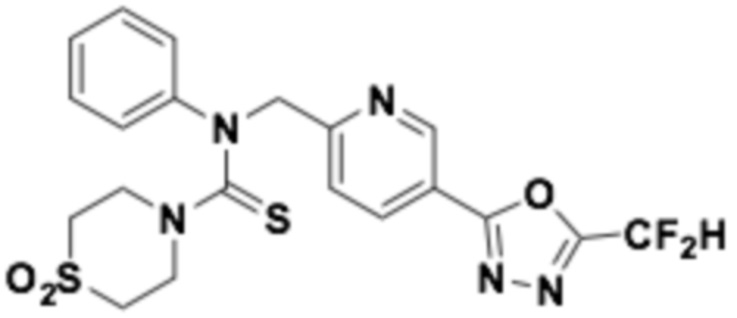
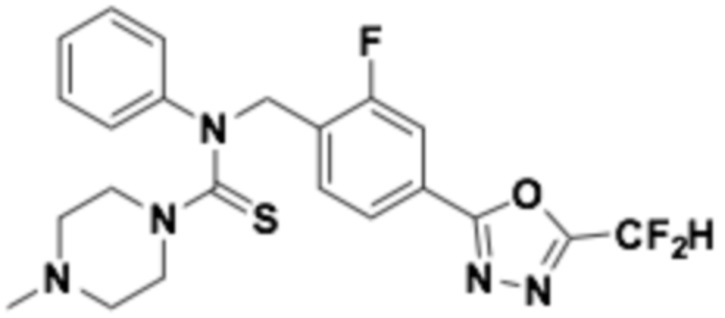
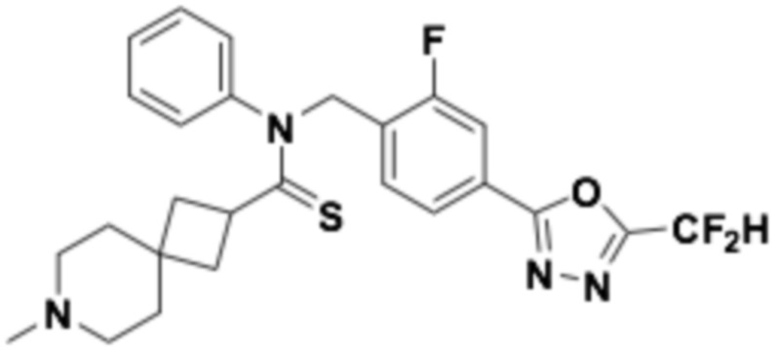
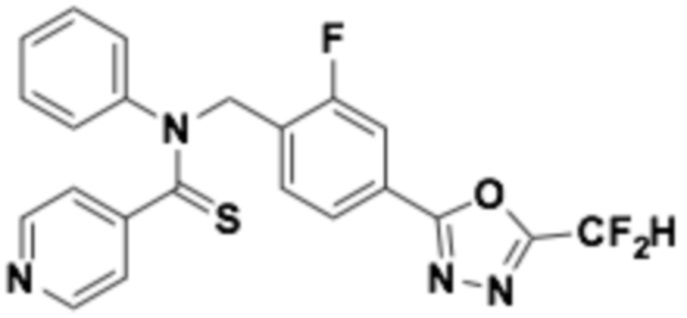
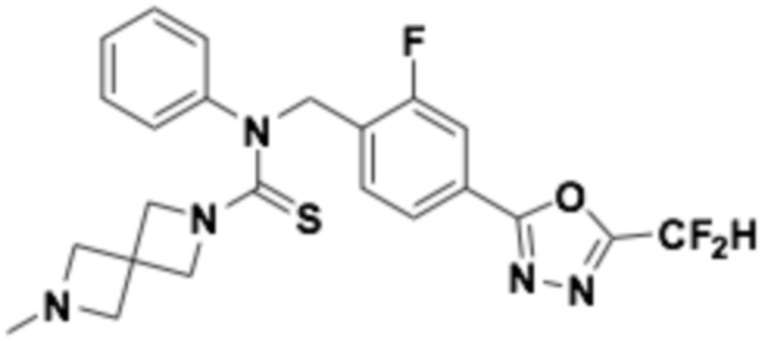
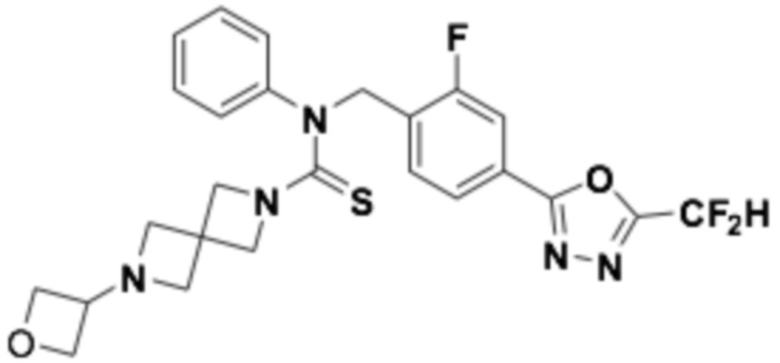
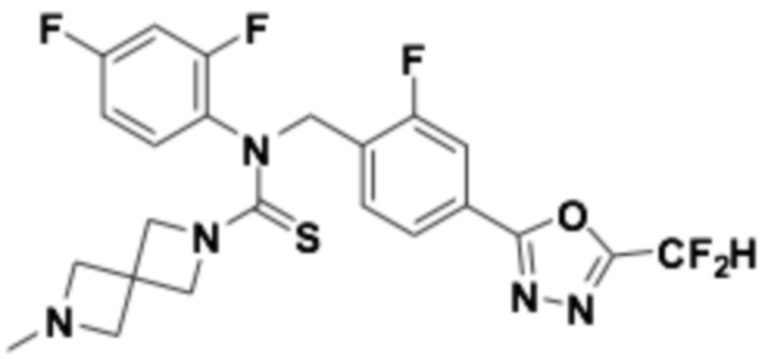
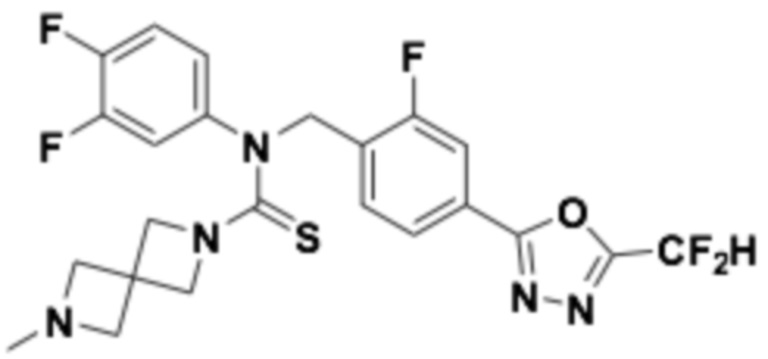
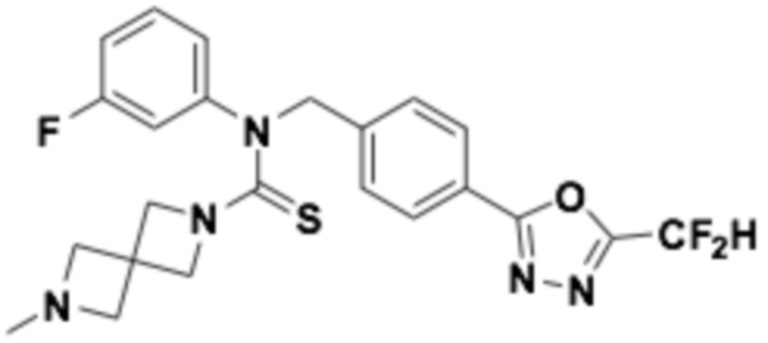
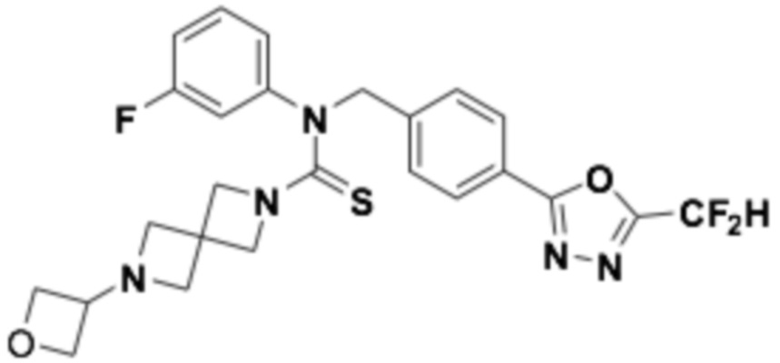
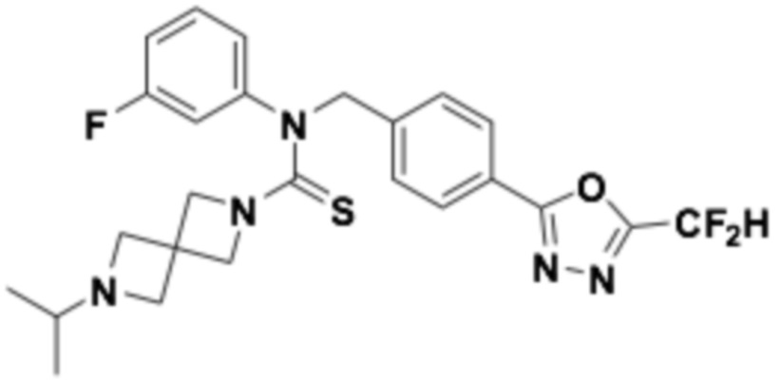
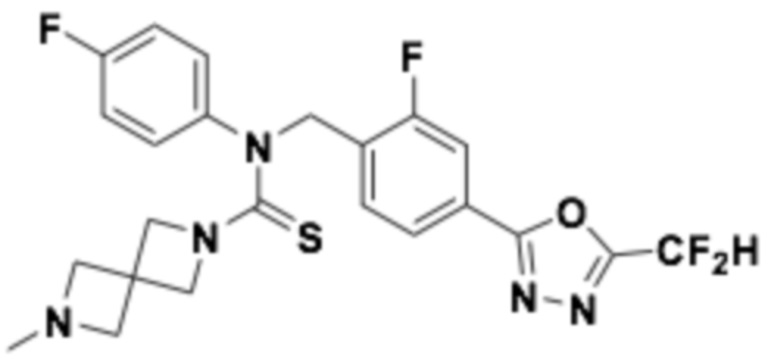
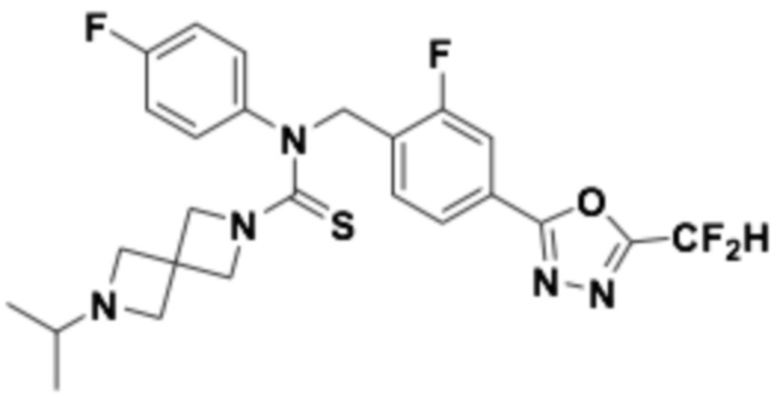
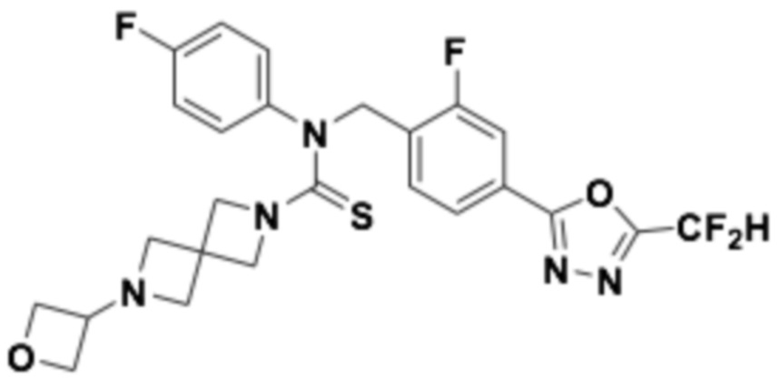
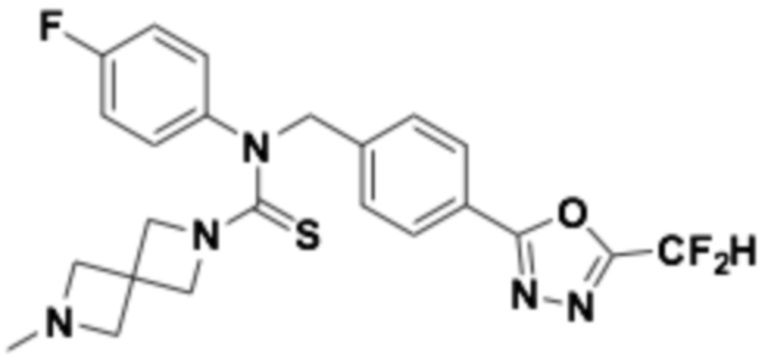
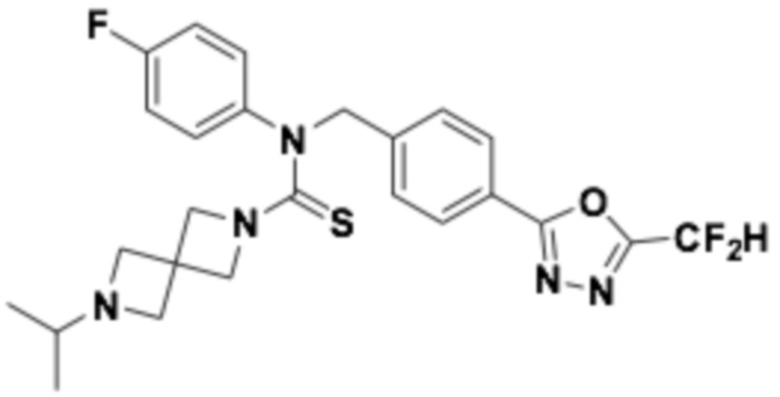
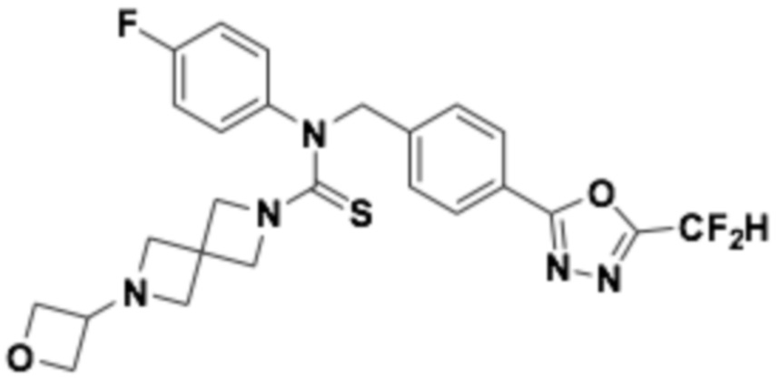
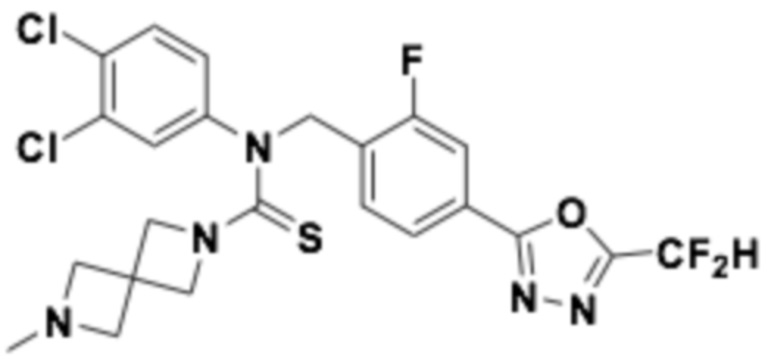
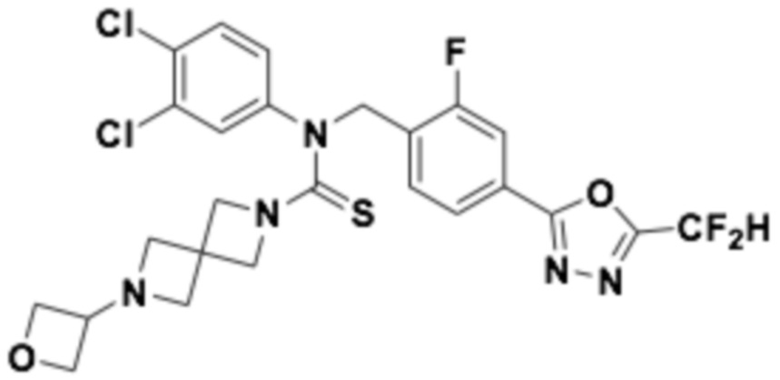
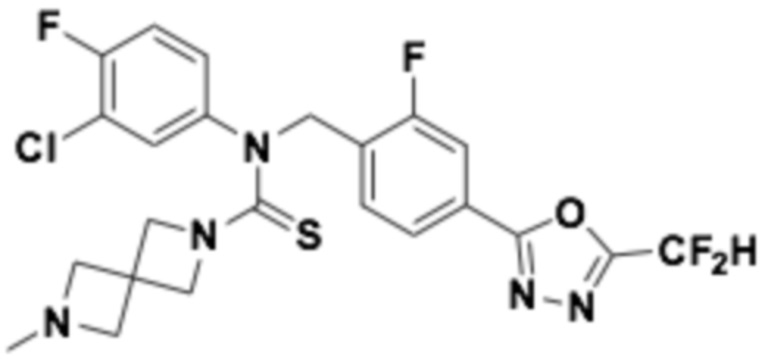
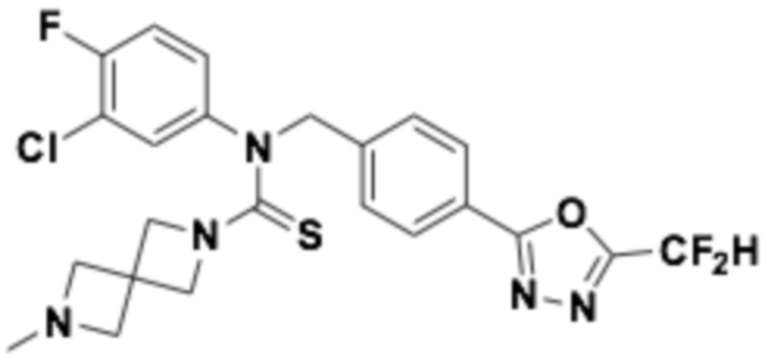
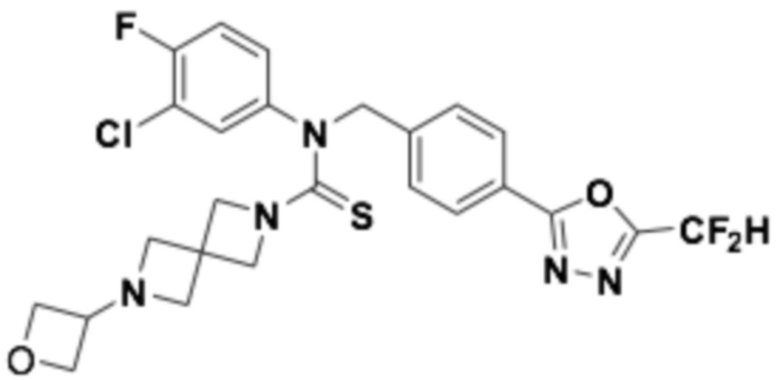
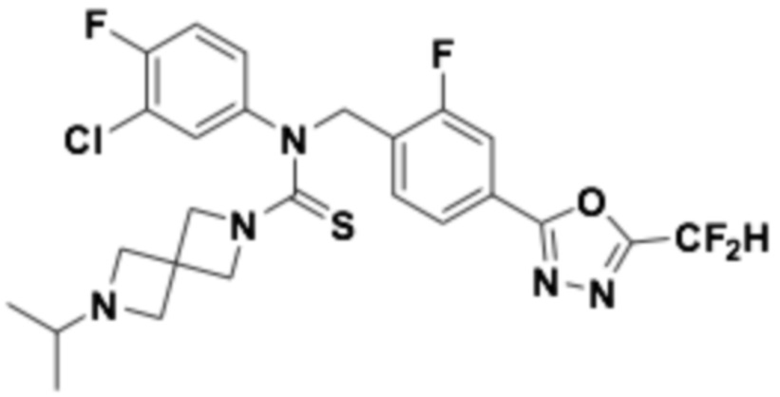
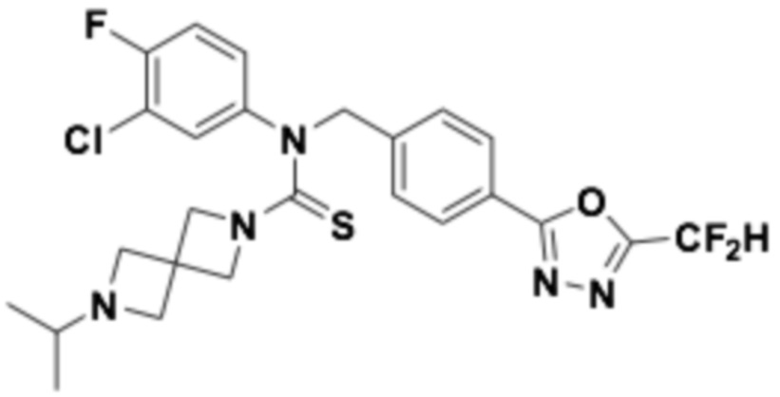
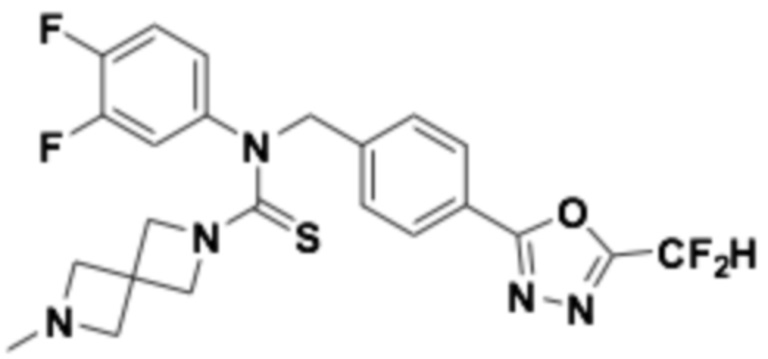
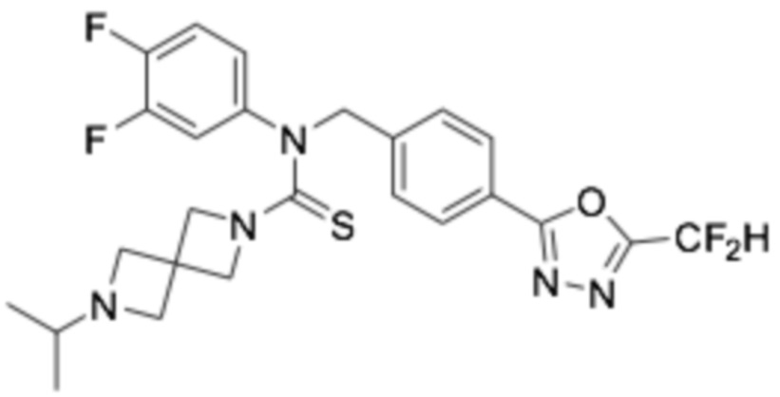
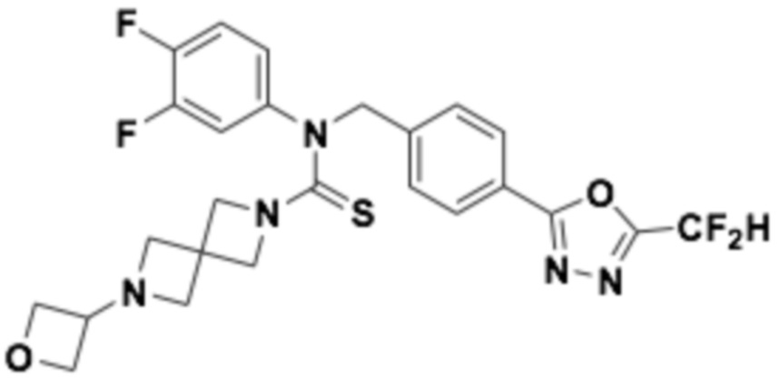
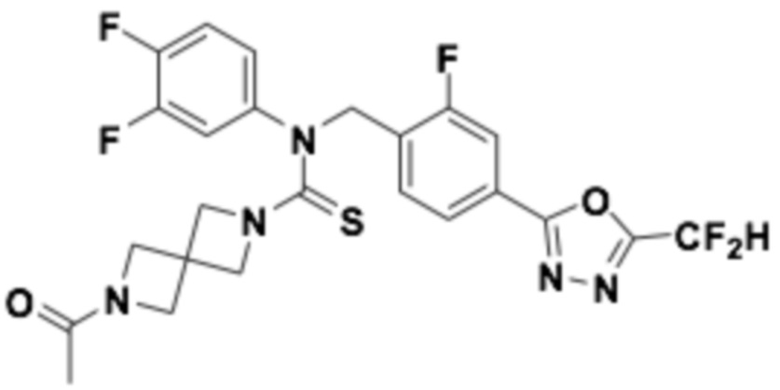
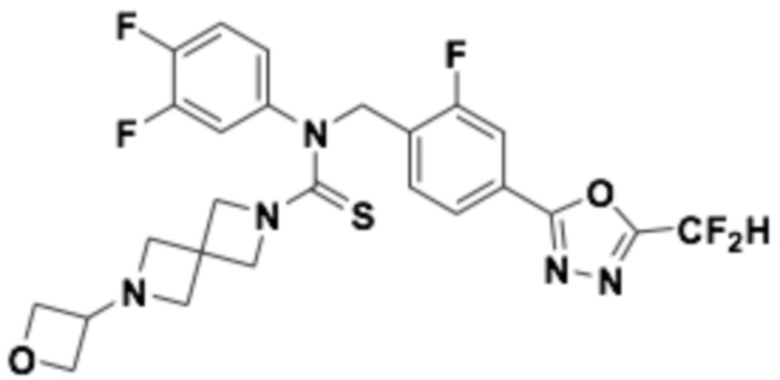
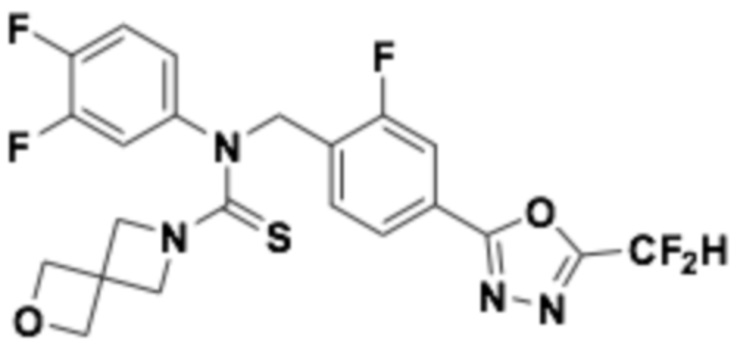
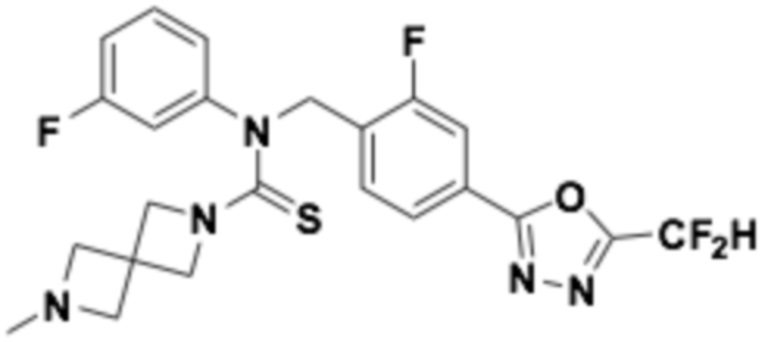
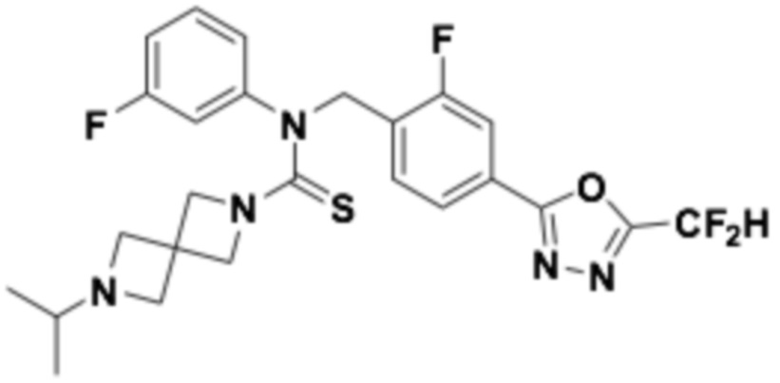
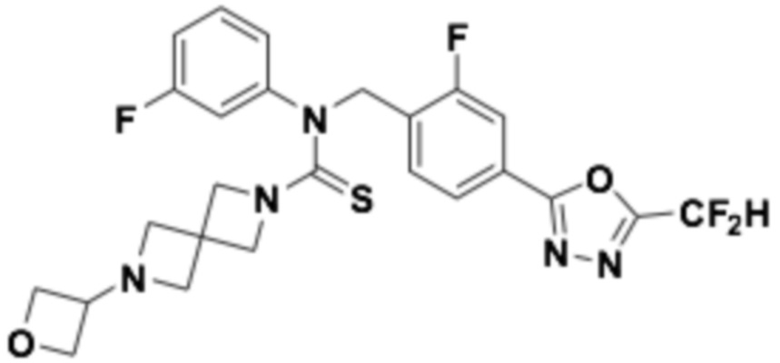
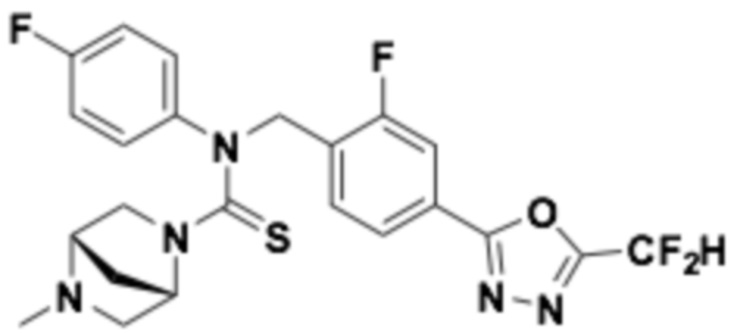
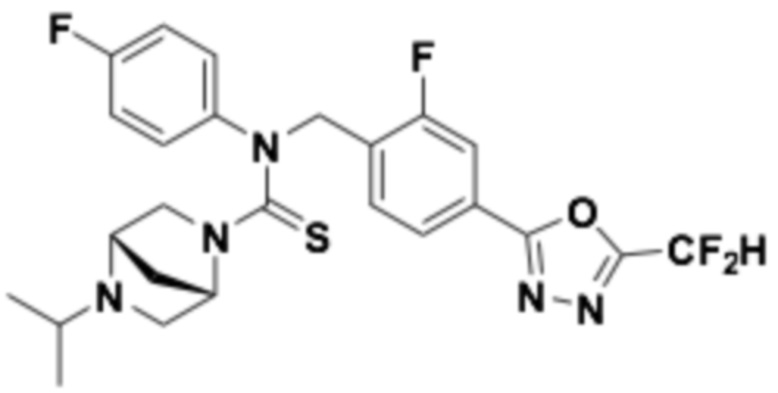
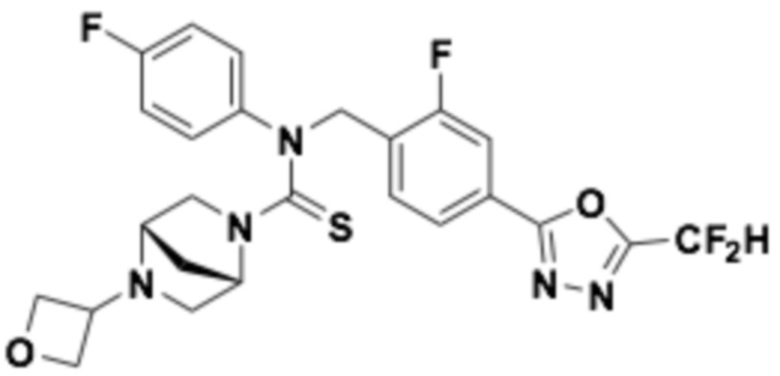
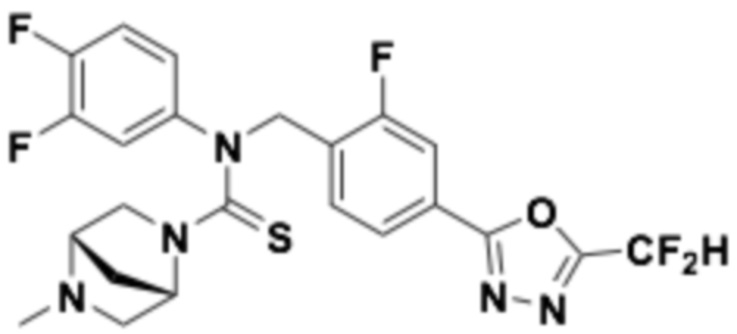
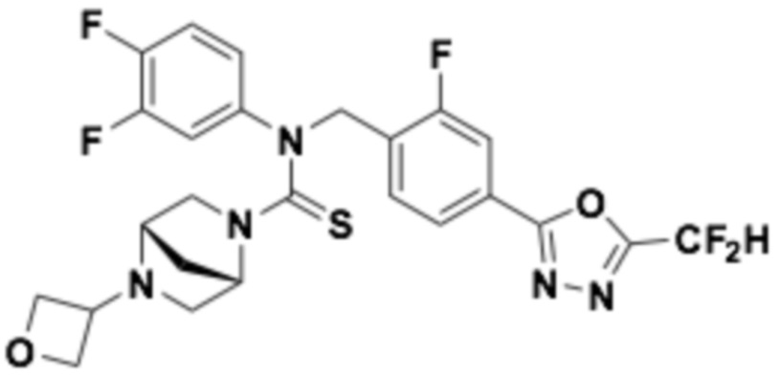
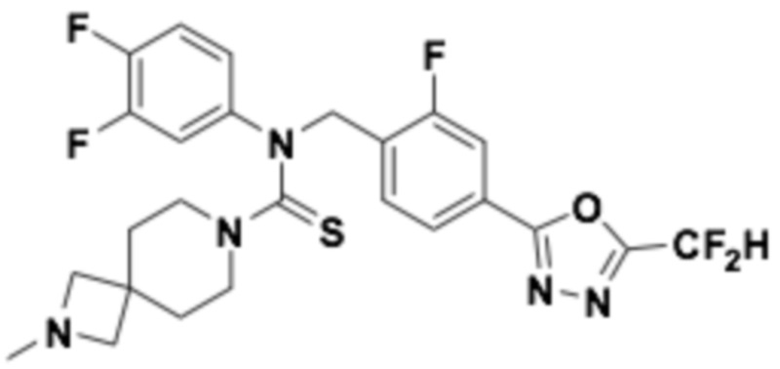
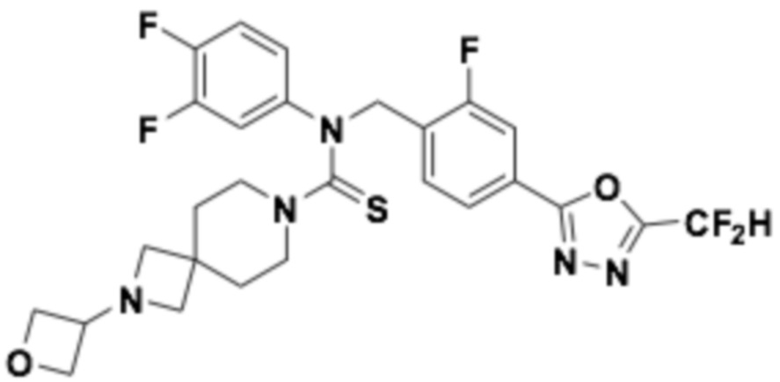
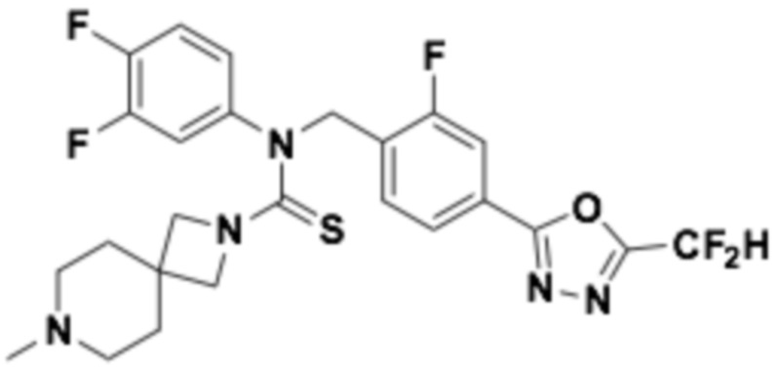
В которых соединение представляет собой соединение, выбранное из группы, состоящей из соединений 1-46, показанных в таблице 1.
Таблица 1
Способ получения 1,3,4-оксадиазолтиокарбонильных соединений формулы I
1,3,4-оксадиазолтиокарбонильное соединение, представленное формулой I, его стереоизомеры или его фармацевтически приемлемые соли можно получить в соответствии со способом получения, представленным реакционными формулами 1-4, и даже способ получения, модифицированный на уровне, понятном специалистам в данной области, также может быть включен в настоящее изобретение.
В настоящем изобретении далее в реакционных формулах X1-X4 может быть последовательно таким же, как Z1-Z4 формулы I, и другие символы могут быть представлены теми же символами, как символы формулы I в реакционных формулах, и символы, специально неописанные, могут быть такими же, как определено в формуле I. Таким образом, любое избыточное описание будет опущено.
В следующих реакционных формулах 1-4, заместитель, представленный «X», может означать уходящую группу.
В следующих реакционных формулах 1-4, «PG» может представлять собой защитную группу амина и, например, PG может представлять собой трет-бутилоксикарбонильную группу (BOC).
<Реакционная формула 1>
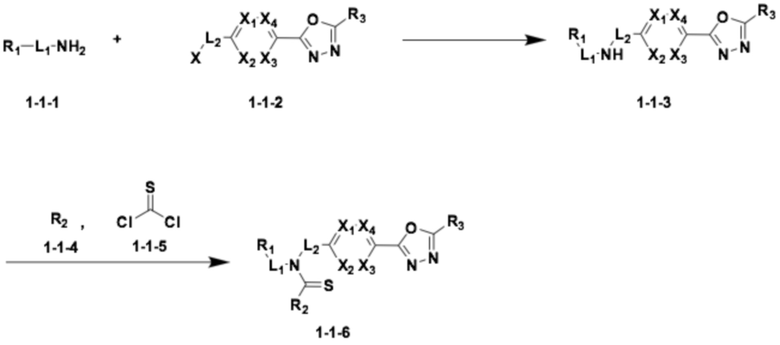 В реакционной формуле 1, соединение формулы 1-1-4, представленное «R2» может означать соединение, в которой первичная или вторичная аминогруппа введена в R2, которая представляет собой моновалентный заместитель, в определении формулы I.
В реакционной формуле 1, соединение формулы 1-1-4, представленное «R2» может означать соединение, в которой первичная или вторичная аминогруппа введена в R2, которая представляет собой моновалентный заместитель, в определении формулы I.
Согласно реакционной формуле 1, соединение формулы 1-1-3 можно получить реакцией замещения между соединением формулы 1-1-1 и соединением формулы 1-1-2, после чего соединение формулы 1-1-4 и соединение формулы 1-1-5 могут реагировать, давая соединение формулы 1-1-6.
Соединение, полученное реакционной формулой 1, может представлять собой соединения 1, 2, 3, 7, 35 и т.д.
<Реакционная формула 2>
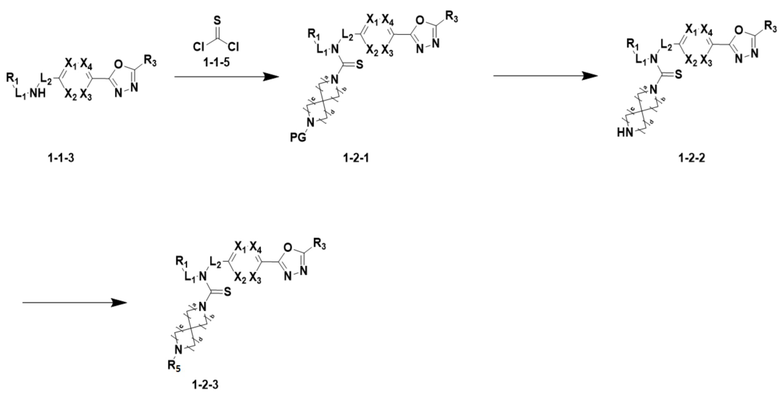
В реакционной формуле 2, R5 могут быть такими же, как определено как RF в формуле I.
Согласно реакционной формуле 2, соединение формулы 1-2-1 можно получить реакцией соединения формулы 1-1-3, соединения формулы 1-1-5 и спиросоединения, в которое введена аминогруппа, содержащая защитную группу (PG). После этого, защитную группу можно удалить, получая соединение формулы 1-2-2, и затем можно провести реакцию восстановительного аминирования или реакцию замещения для получения соединения формулы 1-2-3.
Соединение, полученное реакционной формулой 2, может представлять собой 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 36, 37, 38, 44, 45, 46 и т.д.
<Реакционная формула 3>
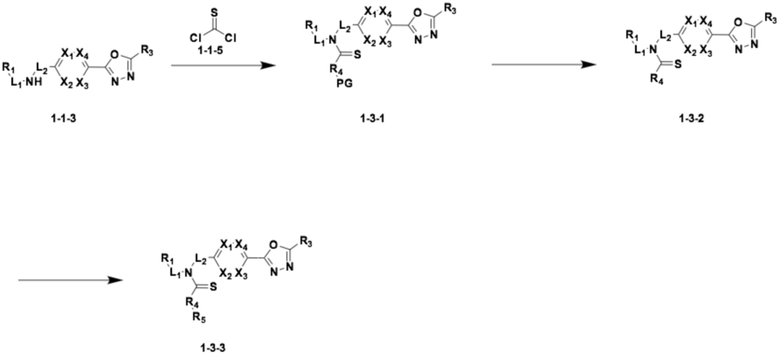
В реакционной формуле 3, R4 может представлять собой или (в которых каждый Y1 и Y7 может независимо представлять собой -N-), и R5 могут быть такими же, как определено как RF в формуле I.
Согласно реакционной формуле 3, соединение формулы 1-3-1 можно получить реакцией соединения формулы 1-1-3, соединения формулы 1-1-5, и R4 соединения, в которое введена аминогруппа, содержащая защитную группу (PG). После этого, защитную группу можно удалить, получая соединение формулы 1-3-2, и затем можно провести реакцию восстановительного аминирования или реакцию замещения для получения соединения формулы 1-3-3.
Соединение, полученное реакционной формулой 3, может представлять собой соединения , 5, 39, 40, 41, 42, 43 и т.д.
<Реакционная формула 4>
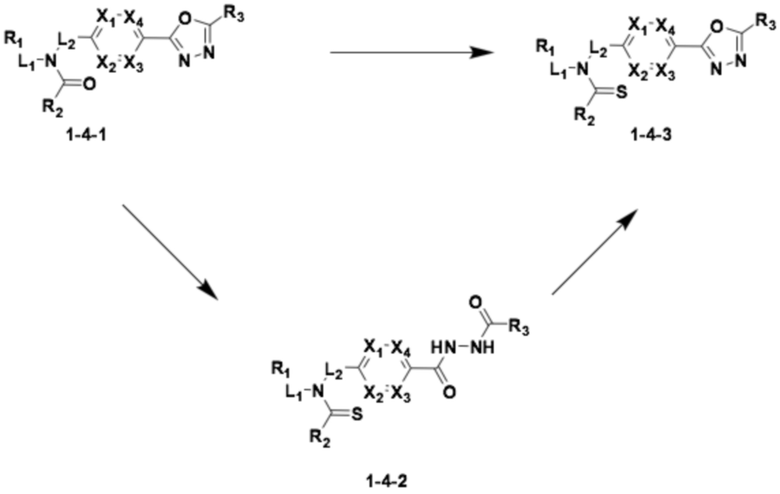 Согласно реакционной формуле 4, соединение формулы 1-4-1 может реагировать с 2,4-бис(4-метоксифенил)-1,3,2,4-дитиадифосфетан-2,4-дисульфидом (Реактив Ловессона), давая соединение формулы 1-4-2 или формулы 1-4-3.
Согласно реакционной формуле 4, соединение формулы 1-4-1 может реагировать с 2,4-бис(4-метоксифенил)-1,3,2,4-дитиадифосфетан-2,4-дисульфидом (Реактив Ловессона), давая соединение формулы 1-4-2 или формулы 1-4-3.
Альтернативно, соединение формулы 1-4-2 может реагировать с 1-метокси-N-триэтиламмониосульфонилметанимидатом (реагент Берджесса), давая соединение формулы 1-4-3.
Соединение, полученное реакционной формулой 4, может представлять собой соединения 6, 8, 9 и т.д.
Композиция, содержащая 1,3,4-оксадиазолтиокарбонильное соединение, представленное формулой I, ее применение и терапевтический способ с ее применением
Настоящее изобретение обеспечивает фармацевтическую композицию, содержащую 1,3,4-оксадиазолтиокарбонильное соединение, представленное формулой I, его стереоизомеры или его фармацевтически приемлемые соли в качестве активного ингредиента.
Кроме того, настоящее изобретение обеспечивает фармацевтическую композицию для предотвращения или лечения заболеваний, связанных с активностью гистондеацетилазы 6, содержащую 1,3,4-оксадиазолтиокарбонильное соединение, представленное формулой I, его стереоизомеры или его фармацевтически приемлемые соли в качестве активного ингредиента.
Фармацевтическая композиция настоящего изобретения селективно ингибирует гистондеацетилазу 6, посредством этого проявляя заметный эффект на предотвращение или лечение заболеваний, связанных с активностью гистондеацетилазы 6.
Заболевания, связанные, с активностью гистондеацетилазы 6 могут включать: инфекционные заболевания, такие как прионовая болезнь; новообразование, такое как доброкачественная опухоль (например, миелодиспластический синдром) или злокачественная опухоль (например, множественная миелома, лимфома, лейкемия, рак легких, колоректальный рак, рак толстой кишки, рак простаты, уротелиальная карцинома, рак молочной железы, меланома, рак кожи, рак печени, рак головного мозга, рак желудка, рак яичников, рак поджелудочной железы, рак головы и шеи, рак полости рта или глиома); эндокринопатию, алиментарные и метаболические заболевания, такие как болезнь Вильсона, амилоидоз или диабет; психические и поведенческие расстройства, такие как депрессия, синдром Ретта или подобный; неврологические заболевания, такие как атрофия центральной нервной системы (например, болезнь Хантингтона, спинальная мышечная атрофия (СМА), спиноцеребеллярная атаксия (СМА)), нейродегенеративное заболевание (например, болезнь Альцгеймера), двигательное расстройство (например, болезнь Паркинсона), невропатию (например, наследственную невропатию (болезнь Шарко-Мари-Тута), спорадическую невропатию, воспалительную невропатию, лекарственную невропатию), моторную невропатию (например, боковой амиотрофический склероз (БАС)), демиелинизирующее заболевание центральной нервной системы (например, рассеянный склероз (РС)) или подобный; заболевания глаз и глазных придатков, такие как увеит; заболевания системы кровообращения, такие как фибрилляция предсердий, инсульт или подобные; респираторные заболевания, такие как астма; расстройства пищеварения, такие как алкогольное заболевание печени, воспалительное заболевание кишечника, болезнь Крона, язвенная болезнь кишечника или подобные; заболевания кожи и подкожной клетчатки, такие как псориаз; заболевания опорно-двигательного аппарата и соединительной ткани, такие как ревматоидный артрит, остеоартрит, системная красная волчанка (СКВ) или подобные; или тератоз, деформации и хромосомные аберрации, такие как аутосомно-доминантный поликистоз почек, а также могут включать другие симптомы или заболевания, связанные с аномальными функциями гистондеацетилазы.
Для введения, фармацевтическая композиция настоящего изобретения может дополнительно содержать по меньшей мере один тип фармацевтически приемлемого носителя в добавление к 1,3,4-оксадиазолтиокарбонильному соединению, представленному формулой I, его стереоизомерам или его фармацевтически приемлемым солям. Фармацевтически приемлемый носитель, применяемый в настоящем изобретении, может включать физиологический раствор, стерилизованную воду, раствор Рингера, забуференный физиологический раствор, раствор декстрозы, раствор мальтодекстрина, глицерин, этанол и смесь одного из их компонентов, а также его можно применять с добавлением других обычных добавок, таких как антиоксиданты, буферные растворы, бактериостатики и т.д., при необходимости. Кроме того, для приготовления инъекционных лекарственных форм, таких как водные растворы, суспензии, эмульсии и т.д., пилюли, капсулы, гранулы или таблетки, могут быть добавлены разбавители, диспергаторы, поверхностно-активные вещества, связующие и смазочные вещества. Таким образом, композиция настоящего изобретения могут представлять собой пластыри, жидкие лекарственные средства, пилюли, капсулы, гранулы, таблетки, суппозитории и т.д. Данные препараты можно получить в соответствии с обычным способом, применяемым для формулирования в данной области техники, или способом, описанным в Remington's Pharmaceutical Science (последнее издание), Merck Publishing Company, Easton PA, и данную композицию можно составлять из различных препаратов в зависимости от каждого заболевания или компонента.
Композицию настоящего изобретения можно вводиться перорально или парентерально (например, применяться внутривенно, подкожно, внутрибрюшинно или местно) в соответствии с целевым способом, при котором ее дозировка варьируется в определенном диапазоне в зависимости от веса, возраста, пола, состояния здоровья и диеты пациента, время введения, способа введения, скорости выведения, тяжести заболевания и подобных. Суточная доза 1,3,4-оксадиазолтиокарбонильного соединения, представленного формулой I настоящего изобретения, может составлять от приблизительно 1 до приблизительно 1000 мг/кг, предпочтительно от приблизительно 5 до приблизительно 100 мг/кг, и ее можно вводить один раз в день или несколько раз в день путем деления суточной дозы соединения.
В добавление к 1,3,4-оксадиазолтиокарбонильному соединению, представленному формулой I, его стереоизомерам или его фармацевтически приемлемым солям, фармацевтическая композиция настоящего изобретения может дополнительно содержать по меньшей мере один активный ингредиент, который проявляет такие же или аналогничные медицинские эффекты.
Настоящее изобретение может обеспечивать способ предотвращения или лечения заболеваний, связанных с активностью гистондеацетилазы 6, включающий введение терапевтически эффективного количества 1,3,4-оксадиазолтиокарбонильного соединения, представленного формулой I, его стереоизомеров или его фармацевтически приемлемых солей.
Как применяют в настоящем изобретении, термин «терапевтически эффективное количество» может относиться к количеству 1,3,4-оксадиазолтиокарбонильного соединения, представленного формулой I, которое является эффективным для предотвращения или лечения заболеваний, связанных с активностью гистондеацетилазы 6.
Кроме того, настоящее изобретение может обеспечивать способ селективного ингибирования ГДАЦ6 введением 1,3,4-оксадиазолтиокарбонильного соединения, представленного формулой I, его стереоизомеров или его фармацевтически приемлемых солей млекопитающим, включая людей.
Способ предотвращения или лечения заболеваний, связанных с активностью гистондеацетилазы 6, согласно настоящему изобретению, может включать не только борьбу с заболеваниями как таковыми до проявления симптомов, но также подавление или избегание данных симптомов введением 1,3,4-оксадиазолтиокарбонильного соединения, представленного формулой I. При лечении заболевания профилактическая или терапевтическая доза определенного активного ингредиента может варьироваться в зависимости от природы и тяжести заболевания или состояния, а также пути введения активного ингредиента. Доза и частота могут варьироваться в зависимости от возраста, веса и реакции конкретного пациента. Подходящая доза и способ применения могут быть легко выбраны специалистами в данной области, естественно, с учетом данных факторов. Кроме того, способ предотвращения или лечения заболеваний, связанных с активностью гистондеацетилазы 6, настоящего изобретения может дополнительно включать введение эффективного количества дополнительного активного агента, который полезен при лечении заболеваний, вместе с 1,3,4-оксадиазолтиокарбонильным соединением, представленным формулой I, в которой дополнительный активный агент может проявлять синергический эффект или адъювантный эффект вместе с соединением формулы I.
Настоящее изобретение обеспечивает применение 1,3,4-оксадиазолтиокарбонильного соединения, представленного формулой I, его стереоизомеров или его фармацевтически приемлемых солей в получении лекарственного средства для лечения заболеваний, связанных с активностью гистондеацетилазы 6. 1,3,4-Оксадиазолтиокарбонильное соединение, представленное формулой I, для получения лекарственного средства можно комбинировать с приемлемым адъювантом, разбавителем, носителем и т.д., и можно получить в виде комплексного агента вместе другими активными агентами, таким образом, обладающими синергичным действием.
Положения, упомянутые в применении, композиции и терапевтическом способе настоящего изобретения, могут применяться в равной степени, если не противоречат друг другу.
Полезные эффекты настоящего изобретения
Согласно настоящему изобретению, 1,3,4-оксадиазолтиокарбонильное соединение, представленное формулой I, его стереоизомеры или его фармацевтически приемлемые соли могут селективно ингибировать ГДАЦ6, таким образом обладая значительным превосходным эффектом предотвращения или лечения заболеваний, связанных с активностью гистондеацетилазы 6.
Способ осуществления настоящего изобретения
В настоящем изобретении далее настоящее изобретение будут подробно описаны посредством предпочтительных примеров для лучшего понимания настоящего изобретения. Однако следующие примеры представлены только для иллюстрации настоящего изобретения, и, таким образом, настоящее изобретение не ограничивается ими.
Получение 1,3,4-оксадиазолтиокарбонильных соединений
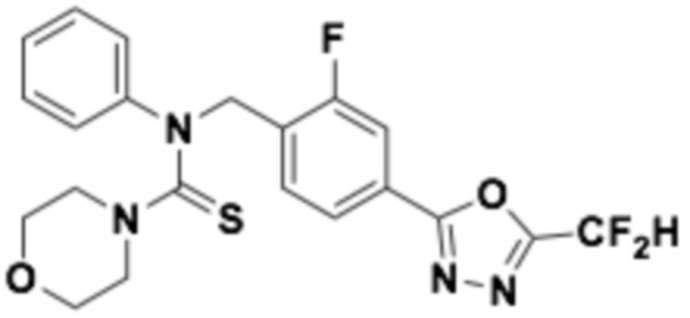
Пример 1: Получение соединения 1, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-фенилморфолин-4-карботиоамид
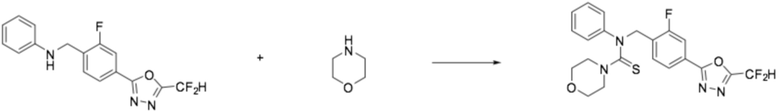
N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)анилин (0,500 г, 1,566 ммоль), N, N-диизопропилэтиламин (1,091 мл, 6,264 ммоль) и тиофосген (0,268 г, 2,349 ммоль) растворяли в дихлорметане (10 мл), после чего полученный в результате раствор перемешивали при 0°C в течение 30 минут и затем морфолин (0,135 мл, 1,566 ммоль) добавляли к нему и дополнительно перемешивали при комнатной температуре в течение 18 часов. Воду приливали в реакционную смесь, и органический слой экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрировали, получая заявленное в заголовке соединение (0,090 г, 12,8%) в виде желтого масла.
1H ЯМР (400 МГц, CDCl3) δ 7,86 (дд, J=8,1, 1,3 Гц, 1H), 7,80~7,76 (м, 2H), 7,35 (т, J=7,9 Гц, 2H), 7,17~7,11 (м, 3H), 7,05 (с, 0,25H), 6,92 (с, 0,5H), 6,79 (с, 0,25H), 5,51 (с, 2H), 3,67 (т, J=4,8 Гц, 4H), 3,51 (т, J=4,8 Гц, 4H); LRMS (ES) m/z 449,4 (M++1).
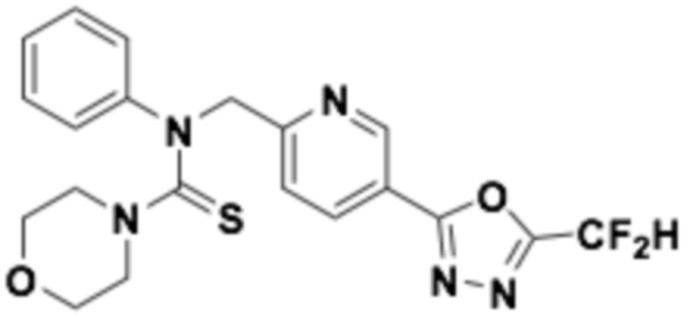
Пример 2: Получение соединения 2, N-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-N-фенилморфолин-4-карботиоамид
[Стадия 1] Получение N-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)анилина
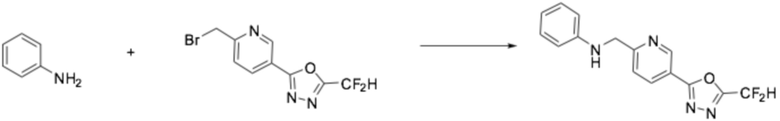 Анилин (0,294 мл, 3,221 ммоль) растворяли в N, N-диметилформамиде (20 мл) при 0°C, после чего гидрид натрия (60,00%, 0,193 г, 4,832 ммоль) добавляли к полученному в результате раствору и перемешивали при той же температуре в течение 30 минут. 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,934 г, 3,221 ммоль) добавляли в реакционную смесь и дополнительно перемешивали при комнатной температуре в течение трех часов. Растворитель удаляли из реакционной смеси под пониженным давлением, после чего воду выливали в полученный в результате концентрат, и органический слой экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрировали, получая требуемое заявленное в заголовке соединение (0,337 г, 34,6%) в виде желтого масла.
Анилин (0,294 мл, 3,221 ммоль) растворяли в N, N-диметилформамиде (20 мл) при 0°C, после чего гидрид натрия (60,00%, 0,193 г, 4,832 ммоль) добавляли к полученному в результате раствору и перемешивали при той же температуре в течение 30 минут. 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,934 г, 3,221 ммоль) добавляли в реакционную смесь и дополнительно перемешивали при комнатной температуре в течение трех часов. Растворитель удаляли из реакционной смеси под пониженным давлением, после чего воду выливали в полученный в результате концентрат, и органический слой экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрировали, получая требуемое заявленное в заголовке соединение (0,337 г, 34,6%) в виде желтого масла.
[Стадия 2] Получение соединения 2
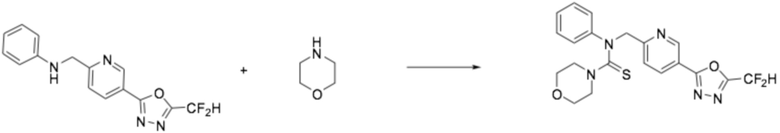 N-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)анилин (0,186 г, 0,615 ммоль), полученный на стадии 1, морфолин (0,053 мл, 0,615 ммоль) и N, N-диизопропилэтиламин (0,429 мл, 2,461 ммоль) растворяли в дихлорметане (10 мл), после чего тиофосген (0,106 г, 0,923 ммоль) добавляли к полученному в результате раствору при 0°C, перемешивали при той же температуре в течение 30 минут, и дополнительно перемешивали при комнатной температуре в течение 18 часов. Воду приливали в реакционную смесь, и органический слой экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрировали, получая требуемое заявленное в заголовке соединение (0,030 г, 11,3%) в виде бесцветного масла.
N-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)анилин (0,186 г, 0,615 ммоль), полученный на стадии 1, морфолин (0,053 мл, 0,615 ммоль) и N, N-диизопропилэтиламин (0,429 мл, 2,461 ммоль) растворяли в дихлорметане (10 мл), после чего тиофосген (0,106 г, 0,923 ммоль) добавляли к полученному в результате раствору при 0°C, перемешивали при той же температуре в течение 30 минут, и дополнительно перемешивали при комнатной температуре в течение 18 часов. Воду приливали в реакционную смесь, и органический слой экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрировали, получая требуемое заявленное в заголовке соединение (0,030 г, 11,3%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3) δ 9,26 (д, J=2,1 Гц, 1H), 8,34 (дд, J=8,2, 2,2 Гц, 1H), 7,69 (д, J=8,2 Гц, 1H), 7,35 (т, J=7,9 Гц, 2H), 7,19~7,12 (м, 3H), 7,07 (с, 0,25H), 6,94 (с, 0,5H), 6,81 (с, 0,25H), 5,65 (с, 2H), 3,68 (т, J=4,7 Гц, 4H), 3,55 (т, J=4,8 Гц, 4H); LRMS (ES) m/z 432,4 (M++1)
Пример 3: Получение соединения 3, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенилморфолин-4-карботиоамид
[Стадия 1] Получение N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)анилина
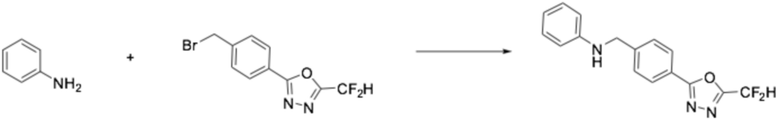 Анилин (0,490 мл, 5,369 ммоль) растворяли в N, N-диметилформамиде (20 мл) при 0°C, после чего гидрид натрия (60,00%, 0,322 г, 8,053 ммоль) добавляли к полученному в результате раствору и перемешивали при той же температуре в течение 30 минут. 2-(4-(бромметил)фенил)-5-(дифторметил)-1,3,4-оксадиазол (1,552 г, 5,369 ммоль) добавляли в реакционную смесь и дополнительно перемешивали при комнатной температуре в течение трех часов. Растворитель удаляли из реакционной смеси под пониженным давлением, после чего воду приливали в полученный в результате концентрат, и органический слой экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрировали, получая заявленное в заголовке соединение (0,550 г, 34,0%) в виде белого твердого остатка.
Анилин (0,490 мл, 5,369 ммоль) растворяли в N, N-диметилформамиде (20 мл) при 0°C, после чего гидрид натрия (60,00%, 0,322 г, 8,053 ммоль) добавляли к полученному в результате раствору и перемешивали при той же температуре в течение 30 минут. 2-(4-(бромметил)фенил)-5-(дифторметил)-1,3,4-оксадиазол (1,552 г, 5,369 ммоль) добавляли в реакционную смесь и дополнительно перемешивали при комнатной температуре в течение трех часов. Растворитель удаляли из реакционной смеси под пониженным давлением, после чего воду приливали в полученный в результате концентрат, и органический слой экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрировали, получая заявленное в заголовке соединение (0,550 г, 34,0%) в виде белого твердого остатка.
[Стадия 2] Получение соединения 3
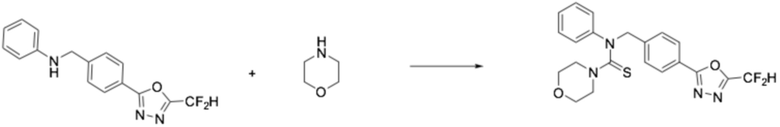 N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)анилин (0,300 г, 0,996 ммоль), полученный на стадии 1, и N, N-диизопропилэтиламин (0,694 мл, 3,983 ммоль) растворяли в дихлорметане (10 мл), после чего морфолин (0,086 мл, 0,996 ммоль) и тиофосген (0,172 г, 1,494 ммоль) добавляли к полученному в результате раствору при 0°C, перемешивали при той же температуре в течение 30 минут, и дополнительно перемешивали при комнатной температуре в течение 18 часов. Воду приливали в реакционную смесь, и органический слой экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрировали, получая заявленное в заголовке соединение (0,100 г, 23,3%) в виде бесцветного масла.
N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)анилин (0,300 г, 0,996 ммоль), полученный на стадии 1, и N, N-диизопропилэтиламин (0,694 мл, 3,983 ммоль) растворяли в дихлорметане (10 мл), после чего морфолин (0,086 мл, 0,996 ммоль) и тиофосген (0,172 г, 1,494 ммоль) добавляли к полученному в результате раствору при 0°C, перемешивали при той же температуре в течение 30 минут, и дополнительно перемешивали при комнатной температуре в течение 18 часов. Воду приливали в реакционную смесь, и органический слой экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрировали, получая заявленное в заголовке соединение (0,100 г, 23,3%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3) δ 8,03 (д, J=8,3 Гц, 2H), 7,55 (д, J=8,2 Гц, 2H), 7,33~7,28 (м, 2H), 7,12 (т, J=7,4 Гц, 1H), 7,06~7,04 (м, 2H), 7,06 (с, 0,25H), 6,91 (с, 0,5H), 6,78 (с, 0,25H), 3,65 (т, J=4,8 Гц, 4H), 3,50 (т, J=4,8 Гц, 4H); LRMS (ES) m/z 431,4 (M++1)
Пример 4: Получение соединения 4, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-4-метил-N-фенилпиперазин-1-карботиоамид
[Стадия 1] Получение трет-бутил 4-((4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)(фенил)карбамотиоил)пиперазин-1-карбоксилата
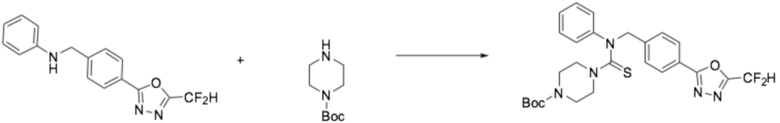 N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)анилин (0,677 г, 2,247 ммоль), полученный тем же способом, как описан на стадии 1 соединения 3, трет-бутил пиперазин-1-карбоксилат (0,419 г, 2,247 ммоль) и N, N-диизопропилэтиламин (1,565 мл, 8,988 ммоль) растворяли в дихлорметане (10 мл), после чего тиофосген (0,388 г, 3,370 ммоль) добавляли к полученному в результате раствору при 0°C, перемешивали при той же температуре в течение 30 минут, и дополнительно перемешивали при комнатной температуре в течение 18 часов. Воду приливали в реакционную смесь, и органический слой экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрировали, получая заявленное в заголовке соединение (0,600 г, 50,4%) в виде желтого масла.
N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)анилин (0,677 г, 2,247 ммоль), полученный тем же способом, как описан на стадии 1 соединения 3, трет-бутил пиперазин-1-карбоксилат (0,419 г, 2,247 ммоль) и N, N-диизопропилэтиламин (1,565 мл, 8,988 ммоль) растворяли в дихлорметане (10 мл), после чего тиофосген (0,388 г, 3,370 ммоль) добавляли к полученному в результате раствору при 0°C, перемешивали при той же температуре в течение 30 минут, и дополнительно перемешивали при комнатной температуре в течение 18 часов. Воду приливали в реакционную смесь, и органический слой экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрировали, получая заявленное в заголовке соединение (0,600 г, 50,4%) в виде желтого масла.
[Стадия 2] Получение N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенилпиперазин-1-карботиоамида
 Трет-бутил 4-((4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)(фенил)карбамотиоил)пиперазин-1-карбоксилат (0,600 г, 1,133 ммоль), полученный на стадии 1, и трифторуксусную кислоту (0,868 мл, 11,329 ммоль) растворяли в дихлорметане (20 мл) при комнатной температуре, после чего полученный в результате раствор перемешивали при той же температуре в течение пяти часов. Растворитель удаляли из реакционной смеси под пониженным давлением, после чего насыщенный водный раствор гидрокарбоната натрия выливали в полученный в результате концентрат, и органический слой экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный продукт применяли без дополнительной очистки (0,450 г, 92,5%, белый твердый остаток).
Трет-бутил 4-((4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)(фенил)карбамотиоил)пиперазин-1-карбоксилат (0,600 г, 1,133 ммоль), полученный на стадии 1, и трифторуксусную кислоту (0,868 мл, 11,329 ммоль) растворяли в дихлорметане (20 мл) при комнатной температуре, после чего полученный в результате раствор перемешивали при той же температуре в течение пяти часов. Растворитель удаляли из реакционной смеси под пониженным давлением, после чего насыщенный водный раствор гидрокарбоната натрия выливали в полученный в результате концентрат, и органический слой экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный продукт применяли без дополнительной очистки (0,450 г, 92,5%, белый твердый остаток).
[Стадия 3] Получение соединения 4
 N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенилпиперазин-1-карботиоамид (0,200 г, 0,466 ммоль), полученный на стадии 2, формальдегид (0,028 г, 0,931 ммоль) и триацетоксиборгидрид натрия (0,197 г, 0,931 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре, после чего полученный в результате раствор перемешивали при той же температуре в течение 18 часов. Воду приливали в реакционную смесь, и органический слой экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрировали, получая заявленное в заголовке соединение (0,050 г, 24,2%) в виде бесцветного масла.
N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенилпиперазин-1-карботиоамид (0,200 г, 0,466 ммоль), полученный на стадии 2, формальдегид (0,028 г, 0,931 ммоль) и триацетоксиборгидрид натрия (0,197 г, 0,931 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре, после чего полученный в результате раствор перемешивали при той же температуре в течение 18 часов. Воду приливали в реакционную смесь, и органический слой экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрировали, получая заявленное в заголовке соединение (0,050 г, 24,2%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3) δ 8,05 (д, J=8,3 Гц, 2H), 7,56 (д, J=8,3 Гц, 2H), 7,32~7,28 (м, 2H), 7,12 (т, J=7,4 Гц, 1H), 7,04 (д, J=7,9 Гц, 2H), 7,04 (с, 0,25H), 6,91 (с, 0,5H), 6,78 (с, 0,25H), 5,52 (с, 2H), 3,69 (т, J=4,9 Гц, 4H), 2,28 (т, J=5,0 Гц, 4H), 2,23 (с, 3H); LRMS (ES) m/z 444,3 (M++1).
Пример 5: Получение соединения 5, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-4-(оксетан-3-ил)-N-фенилпиперазин-1-карботиоамид
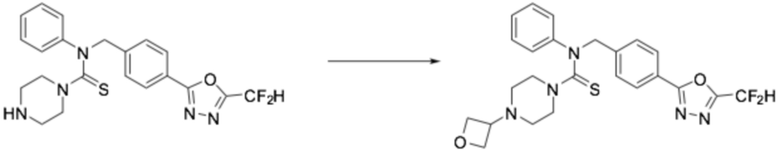 N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенилпиперазин-1-карботиоамид (0,200 г, 0,466 ммоль), полученный тем же способом, как описан на стадии 2 соединения 4, 3-оксетанон (0,055 мл, 0,931 ммоль) и триацетоксиборгидрид натрия (0,197 г, 0,931 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре, после чего полученный в результате раствор перемешивали при той же температуре в течение 18 часов. Воду приливали в реакционную смесь, и органический слой экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрировали, получая заявленное в заголовке соединение (0,100 г, 44,2%) в виде бесцветного масла.
N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенилпиперазин-1-карботиоамид (0,200 г, 0,466 ммоль), полученный тем же способом, как описан на стадии 2 соединения 4, 3-оксетанон (0,055 мл, 0,931 ммоль) и триацетоксиборгидрид натрия (0,197 г, 0,931 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре, после чего полученный в результате раствор перемешивали при той же температуре в течение 18 часов. Воду приливали в реакционную смесь, и органический слой экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрировали, получая заявленное в заголовке соединение (0,100 г, 44,2%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3) δ 8,04 (д, J=8,3 Гц, 2H), 7,55 (д, J=8,2 Гц, 2H), 7,32~7,28 (м, 2H), 7,14~7,10 (м, 1H), 7,04~7,02 (м, 2H), 7,04 (с, 0,25H), 6,91 (с, 0,5H), 6,78 (с, 0,25H), 5,51 (с, 2H), 4,62 (т, J=6,6 Гц, 2H), 4,52 (т, J=6,1 Гц, 2H), 3,70 (т, J=4,9 Гц, 4H), 3,44~3,38 (м, 1H), 2,19 (т, J=5,0 Гц, 4H); LRMS (ES) m/z 486,4 (M++1).
Пример 6: Получение соединения 6, N-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-N-фенилтиоморфолин-4-карботиоамид 1,1-диоксид

N-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-N-фенилтиоморфолин-4-карбоксамид 1,1-диоксид (0,200 г, 0,432 ммоль) и 2,4-бис(4-метоксифенил)-1,3,2,4-дитиадифосфетан-2,4-дисульфид (реактив Ловессона, 0,175 г, 0,432 ммоль) растворяли в толуоле (20 мл) при 110°C, после чего полученный в результате раствор перемешивали при той же температуре в течение 18 часов, завершая реакцию, понизив температуру до комнатной. Воду приливали в реакционную смесь, и органический слой экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрировали, получая заявленное в заголовке соединение (0,027 г, 13,0%) в виде желтого твердого остатка типа пена.
1H ЯМР (400 МГц, CDCl3) δ 9,27 (д, J=2,0 Гц, 1H), 8,41 (дд, J=8,2, 2,2 Гц, 1H), 7,62 (д, J=8,2 Гц, 1H), 7,41 (т, J=7,9 Гц, 2H), 7,28~7,21 (м, 3H), 7,09 (с, 0,25H), 6,96 (с, 0,5H), 6,83 (с, 0,25H), 5,62 (с, 2H), 4,11~4,06 (м, 4H), 2,97 (т, J=5,2 Гц, 4H); LRMS (ES) m/z 480,3 (M++1).
Пример 7: Получение соединения 7, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-4-метил-N-фенилпиперазин-1-карботиоамид
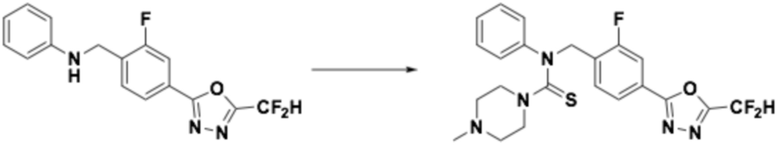
N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)анилин (0,200 г, 0,626 ммоль) и N, N-диизопропилэтиламин (0,218 мл, 1,253 ммоль) растворяли в дихлорметане (4 мл) при 0°C, после чего тиофосген (0,053 мл, 0,689 ммоль) добавляли к полученному в результате раствору и перемешивали при той же температуре. 1-метилпиперазин (0,084 мл, 0,752 ммоль) добавляли в реакционную смесь и дополнительно перемешивали при комнатной температуре в течение 18 часов. Насыщенный водный раствор хлорида натрия выливали в реакционную смесь, органический слой экстрагировали дихлорметаном, фильтровали через пластиковый фильтр и слой водного раствора из него, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-2,5%) и концентрировали, получая продукт, после чего полученный в результате продукт очищали снова хроматографией (SiO2 пластинка, 20×20×1 мм; метанол/дихлорметан=3%) и концентрировали, получая требуемое соединение (0,034 г, 11,8%) в виде желтого масла.
1H ЯМР (400 МГц, CDCl3) δ 7,87 (д, J=1,4 Гц, 1H), 7,85-7,76 (м, 2H), 7,35-7,28 (м, 2H), 7,15-7,11 (м, 3H), 6,89 (т, J=51,7 Гц, 1H), 5,52 (с, 2H), 3,68 (т, J=5,0 Гц, 4H), 2,26 (т, J=5,0 Гц, 4H), 2,07 (с, 3H); LRMS (ES) m/z 462,3 (M++1).
Пример 8: Получение соединения 8, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-7-метил-N-фенил-7-азаспиро[3,5]нонан-2-карботиоамид
[Стадия 1] Получение N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-фенил-7-азаспиро[3,5]нонан-2-карботиоамида
 Трет-бутил 2-((4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)(фенил)карбамоил)-7-азаспиро[3,5]нонан-7-карбоксилат (0,110 г, 0,193 ммоль) и 2,4-бис(4-метоксифенил)-1,3,2,4-дитиадифосфетан-2,4-дисульфид (реактив Ловессона, 0,117 г, 0,289 ммоль) растворяли в толуоле (10 мл) при 110°C, после чего полученный в результате раствор перемешивали при той же температуре в течение 18 часов завершая реакцию, понизив температуру до комнатной. Воду приливали в реакционную смесь, и органический слой экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрировали, получая заявленное в заголовке соединение (0,077 г, 82,1%) в виде коричневого масла.
Трет-бутил 2-((4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)(фенил)карбамоил)-7-азаспиро[3,5]нонан-7-карбоксилат (0,110 г, 0,193 ммоль) и 2,4-бис(4-метоксифенил)-1,3,2,4-дитиадифосфетан-2,4-дисульфид (реактив Ловессона, 0,117 г, 0,289 ммоль) растворяли в толуоле (10 мл) при 110°C, после чего полученный в результате раствор перемешивали при той же температуре в течение 18 часов завершая реакцию, понизив температуру до комнатной. Воду приливали в реакционную смесь, и органический слой экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрировали, получая заявленное в заголовке соединение (0,077 г, 82,1%) в виде коричневого масла.
[Стадия 2] Получение соединения 8
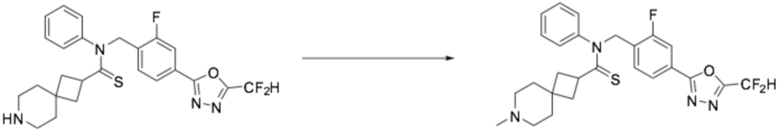 N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-фенил-7-азаспиро[3,5]нонан-2-карботиоамид (0,077 г, 0,158 ммоль), полученный на стадии 1, формальдегид (0,010 г, 0,317 ммоль) и триацетоксиборгидрид натрия (0,067 г, 0,317 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре, после чего полученный в результате раствор перемешивали при той же температуре в течение 18 часов. Воду приливали в реакционную смесь, и органический слой экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрировали, получая заявленное в заголовке соединение (0,035 г, 44,2%) в виде белого твердого остатка.
N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-фенил-7-азаспиро[3,5]нонан-2-карботиоамид (0,077 г, 0,158 ммоль), полученный на стадии 1, формальдегид (0,010 г, 0,317 ммоль) и триацетоксиборгидрид натрия (0,067 г, 0,317 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре, после чего полученный в результате раствор перемешивали при той же температуре в течение 18 часов. Воду приливали в реакционную смесь, и органический слой экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрировали, получая заявленное в заголовке соединение (0,035 г, 44,2%) в виде белого твердого остатка.
1H ЯМР (400 МГц, CDCl3) 7,88 (д, J=8,0 Гц, 1H), 7,73~7,72 (м, 2H), 7,39~7,38 (м, 3H), 7,05 (с, 0,25H), 6,98~6,97 (м, 2H), 6,92 (с, 0,5H), 6,79 (с, 0,25H), 5,72 (с, 2H), 3,26~3,22 (м, 1H), 3,10~2,90 (м, 2H), 2,67 (с, 3H), 2,40~2,24 (м, 2H), 2,06~2,02 (м, 4H), 1,76~1,74 (м, 4H); LRMS (ES) m/z 501,5 (M++1).
Пример 9: Получение соединения 9, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-фенилпиридин-4-карботиоамид
[Стадия 1] Получение N-(4-(2-(2,2-дифторацетил)гидразин-1-карбонил)-2-фторбензил)-N-фенилпиридин-4-карботиоамида
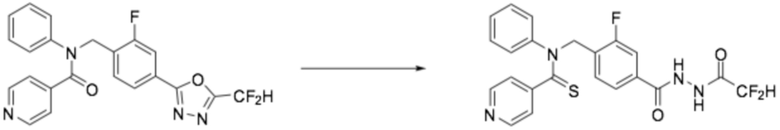
N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-фенилизоникотинамид (0,414 г, 0,976 ммоль) и 2,4-бис(4-метоксифенил)-1,3,2,4-дитиадифосфетан-2,4-дисульфид (реактив Ловессона, 0,592 г, 1,463 ммоль) растворяли в толуоле (10 мл) при 110°C, после чего полученный в результате раствор перемешивали при той же температуре в течение 18 часов завершая реакцию, понизив температуру до комнатной. Воду приливали в реакционную смесь, и органический слой экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрировали, получая заявленное в заголовке соединение (0,14 г, 31,3%) в виде коричневого масла.
[Стадия 2] Получение соединения 9

N-(4-(2-(2,2-дифторацетил)гидразин-1-карбонил)-2-фторбензил)-N-фенилпиридин-4-карботиоамид (0,140 г, 0,305 ммоль), полученный на стадии 1, и 1-метокси-N-триэтиламмониосульфонил-метанимидат (реагент Берджесса, 0,109 г, 0,458 ммоль) смешивали в тетрагидрофуране (10 мл), облучали микроволнами и нагревали при 150°C в течение 30 минут завершая реакцию, понизив температуру до комнатной. Воду приливали в реакционную смесь, и органический слой экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-40%) и концентрировали, получая заявленное в заголовке соединение (0,060 г, 44,6%) в виде коричневого масла.
1H ЯМР (400 МГц, CDCl3) δ 8,39 (д, J=5,8 Гц, 2H), 7,94~7,71 (м, 3H), 7,20~7,11 (м, 5H), 7,06 (с, 0,25H), 6,99~6,94 (м, 2H), 6,94 (с, 0,5H), 6,80 (с, 0,25H), 5,88 (с, 2H); LRMS (ES) m/z 441,4 (M++1).
Пример 10: Получение соединения 10, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-6-метил-N-фенил-2,6-диазаспиро[3,3]гептан-2-карботиоамид
[Стадия 1] Получение трет-бутил 6-((4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)(фенил)карбамотиоил)-2,6-диазаспиро[3,3]гептан-2-карбоксилата
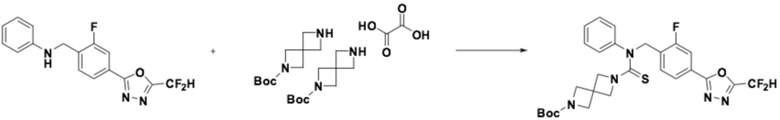 N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)анилин (0,500 г, 1,566 ммоль), гемиоксалат трет-бутил 2,6-диазаспиро[3,3]гептан-2-карбоксилата (0,457 г, 0,940 ммоль), тиофосген (0,132 мл, 1,723 ммоль) и N, N-диизопропилэтиламин (0,546 мл, 3,132 ммоль) растворяли в дихлорметане (5 мл) при комнатной температуре, после чего полученный в результате раствор перемешивали при той же температуре в течение 18 часов. Воду приливали в реакционную смесь, и органический слой экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=10-70%) и концентрировали, получая требуемое соединение (0,433 г, 49,4%) в виде оранжевого масла.
N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)анилин (0,500 г, 1,566 ммоль), гемиоксалат трет-бутил 2,6-диазаспиро[3,3]гептан-2-карбоксилата (0,457 г, 0,940 ммоль), тиофосген (0,132 мл, 1,723 ммоль) и N, N-диизопропилэтиламин (0,546 мл, 3,132 ммоль) растворяли в дихлорметане (5 мл) при комнатной температуре, после чего полученный в результате раствор перемешивали при той же температуре в течение 18 часов. Воду приливали в реакционную смесь, и органический слой экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=10-70%) и концентрировали, получая требуемое соединение (0,433 г, 49,4%) в виде оранжевого масла.
[Стадия 2] Получение N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-фенил-2,6-диазаспиро[3,3]гептан-2-карботиоамида
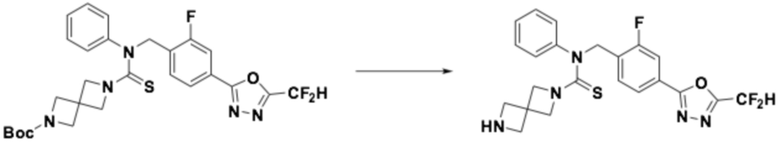
Трет-бутил 6-((4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)(фенил)карбамотиоил)-2,6-диазаспиро[3,3]гептан-2-карбоксилат (0,433 г, 0,774 ммоль), полученный на стадии 1, и трифторуксусную кислоту (0,415 мл, 5,416 ммоль) растворяли в дихлорметане (5 мл) при комнатной температуре, после чего полученный в результате раствор перемешивали при той же температуре в течение пяти часов. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь, органический слой экстрагировали дихлорметаном, фильтровали через пластиковый фильтр, удаляя твердый остаток и слой водного раствора из него, и концентрировали под пониженным давлением. Полученный продукт применяли без дополнительной очистки (0,340 г, 95,6%, желтый твердый остаток).
[Стадия 3] Получение соединения 10
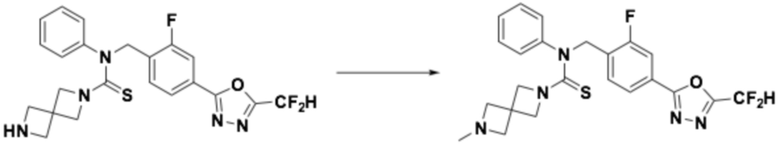
N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-фенил-2,6-диазаспиро[3,3]гептан-2-карботиоамид (0,150 г, 0,326 ммоль), полученный на стадии 2, и формальдегид (38,00% раствор, 0,036 мл, 0,490 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,138 г, 0,653 ммоль) добавляли к полученному в результате раствору и перемешивали при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь, органический слой экстрагировали дихлорметаном, фильтровали через пластиковый фильтр, удаляя твердый остаток и слой водного раствора из него, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрировали, получая требуемое соединение (0,107 г, 69,2%) в виде светло-желтого масла.
1H ЯМР (400 МГц, CDCl3) δ 7,95 (т, J=7,6 Гц, 1H), 7,87 (дд, J=8,1, 1,5 Гц, 1H), 7,68 (дд, J=9,9, 1,5 Гц, 1H), 7,34-7,32 (м, 2H), 7,28-7,24 (м, 1H), 7,13-7,10 (м, 2H), 6,91 (т, J=51,7 Гц, 1H), 5,63 (с, 2H), 3,74 (уш с, 4H), 3,18 (с, 4H), 2,22 (с, 3H); LRMS (ES) m/z 474,4 (M++1).
Пример 11: Получение соединения 11, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-6-(оксетан-3-ил)-N-фенил-2,6-диазаспиро[3,3]гептан-2-карботиоамид
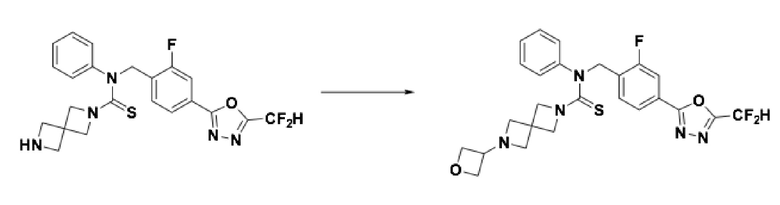
N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-фенил-2,6-диазаспиро[3,3]гептан-2-карботиоамид (0,150 г, 0,326 ммоль), полученный тем же способом, как описан на стадии 2 соединения 10, и 3-оксетанон (0,029 мл, 0,490 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,138 г, 0,653 ммоль) добавляли к полученному в результате раствору и перемешивали при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь, органический слой экстрагировали дихлорметаном, фильтровали через пластиковый фильтр, удаляя твердый остаток и слой водного раствора из него, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-2,5%) и концентрировали, получая продукт, после чего полученный в результате продукт очищали снова хроматографией (SiO2, 4 г картридж; этилацетат/гексан=50-100%) и концентрировали, получая требуемое соединение (0,062 г, 36,8%) в виде светло-желтого твердого остатка.
1H ЯМР (400 МГц, CDCl3) δ 7,94 (т, J=7,6 Гц, 1H), 7,87 (дд, J=8,1, 1,4 Гц, 1H), 7,67 (дд, J=9,9, 1,4 Гц, 1H), 7,35-7,31 (м, 2H), 7,29-7,26 (м, 1H), 7,13-7,11 (м, 2H), 6,91 (т, J=51,7 Гц, 1H), 5,63 (с, 2H), 4,63 (т, J=6,6 Гц, 2H), 4,37 (т, J=5,9 Гц, 2H), 3,84-3,80 (м, 5H), 3,26 (с, 4H); LRMS (ES) m/z 516,5 (M++1).
Пример 12: Получение соединения 12, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(2,4-дифторфенил)-6-метил-2,6-диазаспиро[3,3]гептан-2-карботиоамид
[Стадия 1] Получение N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-2,4-дифторанилина
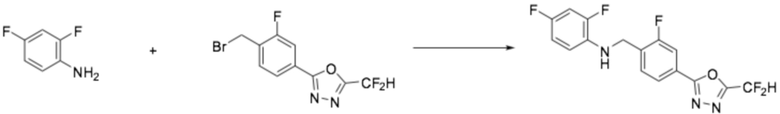 2,4-дифторанилин (0,500 г, 3,873 ммоль), 2-(4-(бромметил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (1,189 г, 3,873 ммоль) и карбонат калия (1,070 г, 7,745 ммоль) растворяли в ацетонитриле (20 мл) при 50°C, после чего полученный в результате раствор перемешивали при той же температуре в течение 18 часов завершая реакцию, понизив температуру до комнатной. Воду приливали в реакционную смесь, и органический слой экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-30%) и концентрировали, получая заявленное в заголовке соединение (1,100 г, 80,0%) в виде белого твердого остатка.
2,4-дифторанилин (0,500 г, 3,873 ммоль), 2-(4-(бромметил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (1,189 г, 3,873 ммоль) и карбонат калия (1,070 г, 7,745 ммоль) растворяли в ацетонитриле (20 мл) при 50°C, после чего полученный в результате раствор перемешивали при той же температуре в течение 18 часов завершая реакцию, понизив температуру до комнатной. Воду приливали в реакционную смесь, и органический слой экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-30%) и концентрировали, получая заявленное в заголовке соединение (1,100 г, 80,0%) в виде белого твердого остатка.
[Стадия 2] Получение трет-бутил 6-((4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)(2,4-дифторфенил)карбамотиоил)-2,6-диазаспиро[3,3]гептан-2-карбоксилата
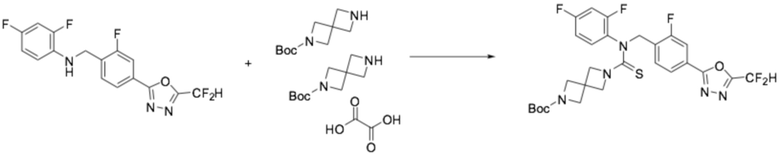 N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-2,4-дифторанилин (0,843 г, 2,373 ммоль), полученный на стадии 1, N, N-диизопропилэтиламин (1,653 мл, 9,491 ммоль) и тиофосген (0,704 г, 2,373 ммоль) растворяли в дихлорметане (20 мл), после чего полученный в результате раствор перемешивали при 0°C в течение 30 минут и затем гемиоксалат трет-бутил 2,6-диазаспиро[3,3]гептан-2-карбоксилата (0,577 г, 1,186 ммоль) добавляли к нему и дополнительно перемешивали при комнатной температуре в течение 18 часов. Воду приливали в реакционную смесь, и органический слой экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрировали, получая заявленное в заголовке соединение (0,200 г, 14,2%) в виде бесцветного масла.
N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-2,4-дифторанилин (0,843 г, 2,373 ммоль), полученный на стадии 1, N, N-диизопропилэтиламин (1,653 мл, 9,491 ммоль) и тиофосген (0,704 г, 2,373 ммоль) растворяли в дихлорметане (20 мл), после чего полученный в результате раствор перемешивали при 0°C в течение 30 минут и затем гемиоксалат трет-бутил 2,6-диазаспиро[3,3]гептан-2-карбоксилата (0,577 г, 1,186 ммоль) добавляли к нему и дополнительно перемешивали при комнатной температуре в течение 18 часов. Воду приливали в реакционную смесь, и органический слой экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрировали, получая заявленное в заголовке соединение (0,200 г, 14,2%) в виде бесцветного масла.
[Стадия 3] Получение N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(2,4-дифторфенил)-2,6-диазаспиро[3,3]гептан-2-карботиоамид 2,2,2-трифторацетата
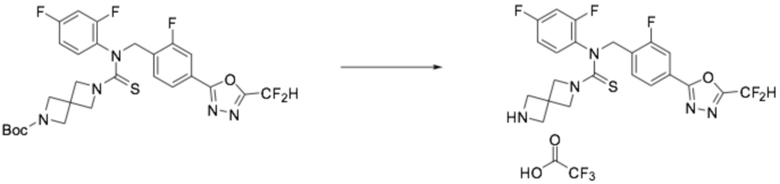 Трет-бутил 6-((4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)(2,4-дифторфенил)карбамотиоил)-2,6-диазаспиро[3,3]гептан-2-карбоксилат (0,084 г, 0,141 ммоль), полученный на стадии 2, растворяли в дихлорметане (10 мл) при комнатной температуре, после чего полученный в результате раствор перемешивали при той же температуре в течение 18 часов. Растворитель удаляли из реакционной смеси под пониженным давлением, после чего полученный продукт применяли без дополнительной очистки (0,084 г, 97,7%, желтое масло).
Трет-бутил 6-((4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)(2,4-дифторфенил)карбамотиоил)-2,6-диазаспиро[3,3]гептан-2-карбоксилат (0,084 г, 0,141 ммоль), полученный на стадии 2, растворяли в дихлорметане (10 мл) при комнатной температуре, после чего полученный в результате раствор перемешивали при той же температуре в течение 18 часов. Растворитель удаляли из реакционной смеси под пониженным давлением, после чего полученный продукт применяли без дополнительной очистки (0,084 г, 97,7%, желтое масло).
[Стадия 4] Получение соединения 12
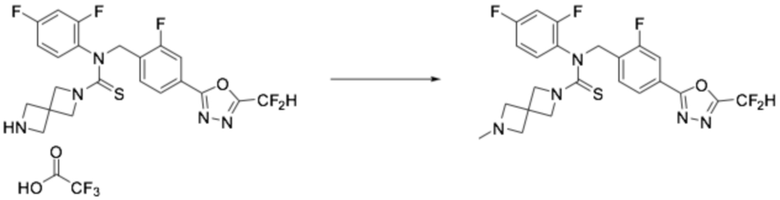
N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(2,4-дифторфенил)-2,6-диазаспиро[3,3]гептан-2-карботиоамид 2,2,2-трифторацетат (0,084 г, 0,138 ммоль), полученный на стадии 3, N, N-диизопропилэтиламин (0,024 мл, 0,138 ммоль), триацетоксиборгидрид натрия (0,058 г, 0,276 ммоль) и формальдегид (0,008 г, 0,276 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре, после чего полученный в результате раствор перемешивали при той же температуре в течение 18 часов. Воду приливали в реакционную смесь, и органический слой экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-30%) и концентрировали, получая заявленное в заголовке соединение (0,020 г, 28,5%) в виде желтого масла.
1H ЯМР (400 МГц, CDCl3) δ 8,01 (т, J=7,6 Гц, 1H), 7,87 (дд, J=8,1, 1,2 Гц, 1H), 7,67 (дд, J=9,9, 1,2 Гц, 1H), 7,07~7,01 (м, 1H), 7,04 (с, 0,25H), 6,92 (с, 0,5H), 6,92~6,82 (м, 2H), 6,79 (с, 0,25H), 5,55 (с, 2H), 3,84 (с, 4H), 3,41 (с, 4H), 2,34 (с, 3H); LRMS (ES) m/z 510,5 (M++1).
Пример 13: Получение соединения 13, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3,4-дифторфенил)-6-метил-2,6-диазаспиро[3,3]гептан-2-карботиоамид
[Стадия 1] Получение N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3,4-дифторанилина
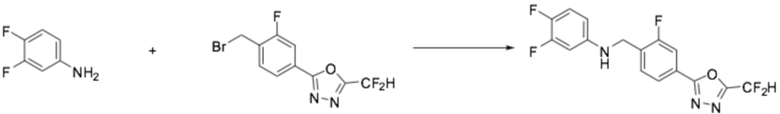 3,4-дифторанилин (0,500 г, 3,873 ммоль), 2-(4-(бромметил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (1,189 г, 3,873 ммоль) и карбонат калия (1,070 г, 7,745 ммоль) растворяли в ацетонитриле (20 мл) при 50°C, после чего полученный в результате раствор перемешивали при той же температуре в течение 18 часов завершая реакцию, понизив температуру до комнатной. Воду приливали в реакционную смесь, и органический слой экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-30%) и концентрировали, получая заявленное в заголовке соединение (0,880 г, 64,0%) в виде белого твердого остатка.
3,4-дифторанилин (0,500 г, 3,873 ммоль), 2-(4-(бромметил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (1,189 г, 3,873 ммоль) и карбонат калия (1,070 г, 7,745 ммоль) растворяли в ацетонитриле (20 мл) при 50°C, после чего полученный в результате раствор перемешивали при той же температуре в течение 18 часов завершая реакцию, понизив температуру до комнатной. Воду приливали в реакционную смесь, и органический слой экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-30%) и концентрировали, получая заявленное в заголовке соединение (0,880 г, 64,0%) в виде белого твердого остатка.
[Стадия 2] Получение трет-бутил 6-((4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)(3,4-дифторфенил)карбамотиоил)-2,6-диазаспиро[3,3]гептан-2-карбоксилата
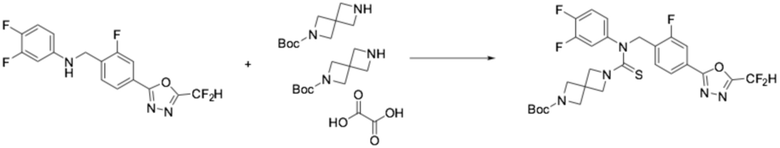 N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3,4-дифторанилин (0,756 г, 2,128 ммоль), полученный на стадии 1, N, N-диизопропилэтиламин (1,483 мл, 8,512 ммоль) и тиофосген (0,631 г, 2,128 ммоль) растворяли в дихлорметане (20 мл), после чего полученный в результате раствор перемешивали при 0°C в течение 30 минут и затем гемиоксалат трет-бутил 2,6-диазаспиро[3,3]гептан-2-карбоксилата (0,518 г, 1,064 ммоль) добавляли к нему и дополнительно перемешивали при комнатной температуре в течение 18 часов. Воду приливали в реакционную смесь, и органический слой экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрировали, получая заявленное в заголовке соединение (0,200 г, 15,8%) в виде бесцветного масла.
N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3,4-дифторанилин (0,756 г, 2,128 ммоль), полученный на стадии 1, N, N-диизопропилэтиламин (1,483 мл, 8,512 ммоль) и тиофосген (0,631 г, 2,128 ммоль) растворяли в дихлорметане (20 мл), после чего полученный в результате раствор перемешивали при 0°C в течение 30 минут и затем гемиоксалат трет-бутил 2,6-диазаспиро[3,3]гептан-2-карбоксилата (0,518 г, 1,064 ммоль) добавляли к нему и дополнительно перемешивали при комнатной температуре в течение 18 часов. Воду приливали в реакционную смесь, и органический слой экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрировали, получая заявленное в заголовке соединение (0,200 г, 15,8%) в виде бесцветного масла.
[Стадия 3] Получение 2,2,2-трифторацетата N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3,4-дифторфенил)-2,6-диазаспиро[3,3]гептан-2-карботиоамида
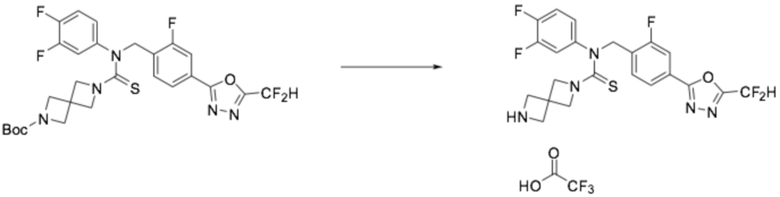
Трет-бутил 6-((4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)(3,4-дифторфенил)карбамотиоил)-2,6-диазаспиро[3,3]гептан-2-карбоксилат (0,140 г, 0,235 ммоль), полученный на стадии 2, и трифторуксусную кислоту (0,180 мл, 2,351 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре, после чего полученный в результате раствор перемешивали при той же температуре в течение 18 часов. Растворитель удаляли из реакционной смеси под пониженным давлением, после чего полученный продукт применяли без дополнительной очистки (0,140 г, 97,7%, желтое масло).
[Стадия 4] Получение соединения 13
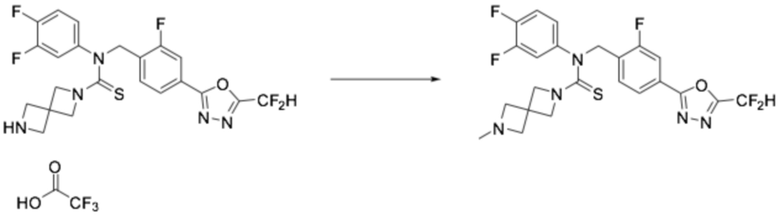
2,2,2-Трифторацетат N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3,4-дифторфенил)-2,6-диазаспиро[3,3]гептан-2-карботиоамида (0,140 г, 0,230 ммоль), полученный на стадии 3, N, N-диизопропилэтиламин (0,040 мл, 0,230 ммоль), триацетоксиборгидрид натрия (0,097 г, 0,459 ммоль) и формальдегид (0,014 г, 0,459 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре, после чего полученный в результате раствор перемешивали при той же температуре в течение 18 часов. Воду приливали в реакционную смесь, и органический слой экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-30%) и концентрировали, получая заявленное в заголовке соединение (0,060 г, 51,3%) в виде желтого масла.
1H ЯМР (400 МГц, CDCl3) δ 7,93~7,88 (м, 2H), 7,72 (д, J=10,2 Гц, 1H), 7,14 (дд, J=18,0, 8,9 Гц, 1H), 7,05 (с, 0,25H), 7,01~6,96 (м, 1H), 6,94 (с, 0,5H), 6,88~6,86 (м, 1H), 6,79 (с, 0,25H), 5,56 (с, 2H), 4,00~3,70 (м, 4H), 3,36 (с, 4H), 2,36 (с, 3H); LRMS (ES) m/z 510,5 (M++1).
Пример 14: Получение соединения 14, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-(3-фторфенил)-6-метил-2,6-диазаспиро[3,3]гептан-2-карботиоамид
[Стадия 1] Получение N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-(3-фторфенил)-2,6-диазаспиро[3,3]гептан-2-карботиоамида

Трет-бутил 6-((4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)(3-фторфенил)карбамотиоил)-2,6-диазаспиро[3,3]гептан-2-карбоксилат (0,500 г, 0,893 ммоль) и трифторуксусную кислоту (0,479 мл, 6,254 ммоль) растворяли в дихлорметане (5 мл) при комнатной температуре, после чего полученный в результате раствор перемешивали при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь, органический слой экстрагировали дихлорметаном, фильтровали через пластиковый фильтр, удаляя твердый остаток и слой водного раствора из него, и концентрировали под пониженным давлением. Полученный продукт применяли без дополнительной очистки (0,361 г, 93,7%, желтый твердый остаток).
[Стадия 2] Получение соединения 14
N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-(3-фторфенил)-2,6-диазаспиро[3,3]гептан-2-карботиоамид (0,100 г, 0,218 ммоль), полученный на стадии 1, и формальдегид (38,00% раствор, 0,024 мл, 0,326 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,092 г, 0,435 ммоль) добавляли к полученному в результате раствору и перемешивали при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь, органический слой экстрагировали дихлорметаном, фильтровали через пластиковый фильтр, удаляя твердый остаток и слой водного раствора из него, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрировали, получая требуемое соединение (0,038 г, 36,9%) в виде белого твердого остатка.
1H ЯМР (400 МГц, CDCl3) δ 8,04 (д, J=8,2 Гц, 2H), 7,54 (д, J=8,2 Гц, 2H), 7,32-7,26 (м, 1H), 7,05-6,79 (м, 4H), 5,55 (с, 2H), 3,83 (уш с, 4H), 3,25 (с, 4H), 2,27 (с, 3H); LRMS (ES) m/z 474,7 (M++1).
Пример 15: Получение соединения 15, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-(3-фторфенил)-6-(оксетан-3-ил)-2,6-диазаспиро[3,3]гептан-2-карботиоамид
N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-(3-фторфенил)-2,6-диазаспиро[3,3]гептан-2-карботиоамид (0,100 г, 0,218 ммоль), полученный тем же способом, как описан на стадии 1 соединения 14, и 3-оксетанон (0,021 мл, 0,326 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,092 г, 0,435 ммоль) добавляли к полученному в результате раствору и перемешивали при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь, органический слой экстрагировали дихлорметаном, фильтровали через пластиковый фильтр, удаляя твердый остаток и слой водного раствора из него, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-2,5%) и концентрировали, получая требуемое соединение (0,046 г, 41,0%) в виде светло-желтого твердого остатка.
1H ЯМР (400 МГц, CDCl3) δ 8,03 (д, J=8,2 Гц, 2H), 7,53 (д, J=8,2 Гц, 2H), 7,33-7,27 (м, 1H), 7,05-6,79 (м, 4H), 5,55 (с, 2H), 4,65 (т, J=6,7 Гц, 2H), 4,40 (т, J=5,9 Гц, 2H), 3,87 (уш с, 4H), 3,66-3,63 (м, 1H), 3,30 (с, 4H); LRMS (ES) m/z 516,7 (M++1).
Пример 16: Получение соединения 16, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-(3-фторфенил)-6-изопропил-2,6-диазаспиро[3,3]гептан-2-карботиоамид
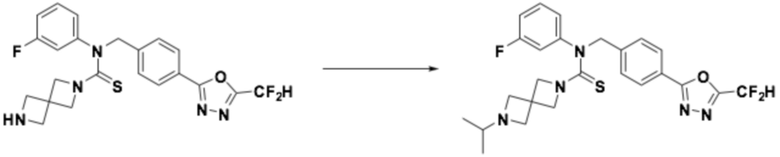
N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-(3-фторфенил)-2,6-диазаспиро[3,3]гептан-2-карботиоамид (0,100 г, 0,218 ммоль), полученный тем же способом, как описан на стадии 1 соединения 14, и ацетон (0,024 мл, 0,326 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,092 г, 0,435 ммоль) добавляли к полученному в результате раствору и перемешивали при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь, органический слой экстрагировали дихлорметаном, фильтровали через пластиковый фильтр, удаляя твердый остаток и слой водного раствора из него, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-5%) и концентрировали, получая требуемое соединение (0,028 г, 25,7%) в виде белого твердого остатка.
1H ЯМР (400 МГц, CDCl3) δ 8,03 (д, J=8,2 Гц, 2H), 7,54 (д, J=8,1 Гц, 2H), 7,31-7,26 (м, 1H), 7,05-6,79 (м, 4H), 5,55 (с, 2H), 3,83 (уш с, 4H), 3,22 (с, 4H), 2,23-2,15 (м, 1H), 0,90 (д, J=6,0 Гц, 6H); LRMS (ES) m/z 502,7 (M++1).
Пример 17: Получение соединения 17, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(4-фторфенил)-6-метил-2,6-диазаспиро[3,3]гептан-2-карботиоамид
[Стадия 1] Получение N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-4-фторанилина
4-фторанилин (1,000 г, 8,999 ммоль) и гидрид натрия (60,00%, 0,378 г, 9,449 ммоль) растворяли в N, N-диметилформамиде (30 мл) при 0°C, после чего 2-(4-(бромметил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (2,902 г, 9,449 ммоль) добавляли к полученному в результате раствору и перемешивали при комнатной температуре в течение 18 часов. Растворитель удаляли из реакционной смеси под пониженным давлением, после чего насыщенный водный раствор гидрокарбоната натрия выливали в полученный в результате концентрат, и органический слой экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом магния, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 24 г картридж; этилацетат/гексан=5-20%) и концентрировали, получая требуемое соединение (1,360 г, 44,8%) в виде желтого твердого остатка.
[Стадия 2] Получение трет-бутил 6-((4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)(4-фторфенил)карбамотиоил)-2,6-диазаспиро[3,3]гептан-2-карбоксилата
N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-4-фторанилин (1,000 г, 2,965 ммоль), полученный на стадии 1, и N, N-диизопропилэтиламин (1,549 мл, 8,895 ммоль) растворяли в дихлорметане (30 мл) при 0°C, после чего тиофосген (0,227 мл, 2,965 ммоль) добавляли к полученному в результате раствору и перемешивали при той же температуре. Гемиоксалат трет-бутил 2,6-диазаспиро[3,3]гептан-2-карбоксилата (0,866 г, 1,779 ммоль) добавляли в реакционную смесь и дополнительно перемешивали при комнатной температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь, органический слой экстрагировали дихлорметаном, фильтровали через пластиковый фильтр, удаляя твердый остаток и слой водного раствора из него, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=10-30%) и концентрировали, получая требуемое соединение (1,220 г, 71,2%) в виде светло-желтого твердого остатка.
[Стадия 3] Получение N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(4-фторфенил)-2,6-диазаспиро[3,3]гептан-2-карботиоамида

Трет-бутил 6-((4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)(4-фторфенил)карбамотиоил)-2,6-диазаспиро[3,3]гептан-2-карбоксилат (1,220 г, 2,112 ммоль), полученный на стадии 2, и трифторуксусную кислоту (1,132 мл, 14,785 ммоль) растворяли в дихлорметане (50 мл) при комнатной температуре, после чего полученный в результате раствор перемешивали при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь и органический слой экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом магния, фильтровали, и концентрировали под пониженным давлением. Полученный продукт применяли без дополнительной очистки (0,964 г, 95,6%, светло-желтый твердый остаток).
[Стадия 4] Получение соединения 17

N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(4-фторфенил)-2,6-диазаспиро[3,3]гептан-2-карботиоамид (0,100 г, 0,209 ммоль), полученный на стадии 3, и формальдегид (38,00% раствор, 0,023 мл, 0,314 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,089 г, 0,419 ммоль) добавляли к полученному в результате раствору и перемешивали при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь, органический слой экстрагировали дихлорметаном, фильтровали через пластиковый фильтр, удаляя твердый остаток и слой водного раствора из него, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрировали, получая требуемое соединение (0,037 г, 35,9%) в виде белого твердого остатка.
1H ЯМР (400 МГц, CDCl3) δ 7,95 (т, J=7,5 Гц, 1H), 7,88 (д, J=8,1 Гц, 1H), 7,68 (д, J=9,9 Гц, 1H), 7,10-7,08 (м, 2H), 7,07-6,79 (м, 3H), 5,60 (с, 2H), 3,78 (уш с, 4H), 3,20 (с, 4H), 2,23 (с, 3H); LRMS (ES) m/z 492,7 (M++1).
Пример 18: Получение соединения 18, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(4-фторфенил)-6-изопропил-2,6-диазаспиро[3,3]гептан-2-карботиоамид
N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(4-фторфенил)-2,6-диазаспиро[3,3]гептан-2-карботиоамид (0,100 г, 0,209 ммоль), полученный тем же способом, как описан на стадии 3 соединения 17, и ацетон (0,023 мл, 0,314 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,089 г, 0,419 ммоль) добавляли к полученному в результате раствору и перемешивали при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь, органический слой экстрагировали дихлорметаном, фильтровали через пластиковый фильтр, удаляя твердый остаток и слой водного раствора из него, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрировали, получая требуемое соединение (0,030 г, 27,6%) в виде светло-желтого твердого остатка.
1H ЯМР (400 МГц, CDCl3) δ 7,94 (т, J=7,6 Гц, 1H), 7,87 (д, J=8,1 Гц, 1H), 7,68 (д, J=9,9 Гц, 1H), 7,10-7,06 (м, 2H), 7,02-6,79 (м, 3H), 5,59 (с, 2H), 3,72 (уш с, 4H), 3,19 (с, 4H), 2,20-2,17 (м, 1H), 0,86 (д, J=6,2 Гц, 6H); LRMS (ES) m/z 520,7 (M++1).
Пример 19: Получение соединения 19, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(4-фторфенил)-6-(оксетан-3-ил)-2,6-диазаспиро[3,3]гептан-2-карботиоамид
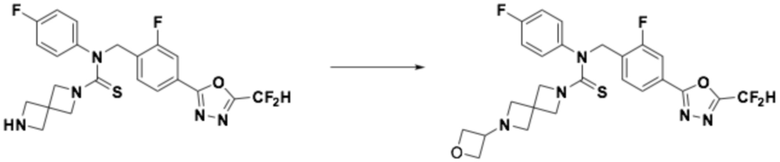
N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(4-фторфенил)-2,6-диазаспиро[3,3]гептан-2-карботиоамид (0,100 г, 0,209 ммоль), полученный тем же способом, как описан на стадии 3 соединения 17, и 3-оксетанон (0,020 мл, 0,314 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,089 г, 0,419 ммоль) добавляли к полученному в результате раствору и перемешивали при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь, органический слой экстрагировали дихлорметаном, фильтровали через пластиковый фильтр, удаляя твердый остаток и слой водного раствора из него, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-2,5%) и концентрировали, получая требуемое соединение (0,016 г, 14,3%) в виде белого твердого остатка.
1H ЯМР (400 МГц, CDCl3) δ 7,96 (т, J=7,6 Гц, 1H), 7,88 (д, J=8,1 Гц, 1H), 7,69 (д, J=9,9 Гц, 1H), 7,12-7,08 (м, 2H), 7,04 (д, J=8,1 Гц, 2H), 7,01-6,79 (м, 1H), 5,60 (с, 2H), 4,64 (т, J=6,6 Гц, 2H), 4,39 (т, J=5,9 Гц, 2H), 3,83 (уш с, 4H), 3,75-3,62 (м, 1H), 3,27 (с, 4H); LRMS (ES) m/z 534,6 (M++1).
Пример 20: Получение соединения 20, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-(4-фторфенил)-6-метил-2,6-диазаспиро[3,3]гептан-2-карботиоамид
[Стадия 1] Получение N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-4-фторанилина
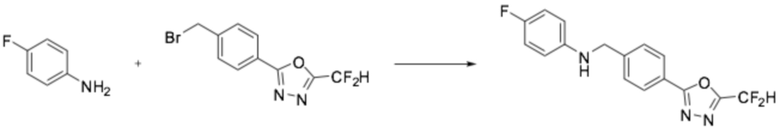
4-фторанилин (1,000 г, 8,999 ммоль) и гидрид натрия (60,00%, 0,378 г, 9,449 ммоль) растворяли в N, N-диметилформамиде (30 мл) при 0°C, после чего 2-(4-(бромметил)фенил)-5-(дифторметил)-1,3,4-оксадиазол (2,732 г, 9,449 ммоль) добавляли к полученному в результате раствору и перемешивали при комнатной температуре в течение 18 часов. Растворитель удаляли из реакционной смеси под пониженным давлением, после чего насыщенный водный раствор гидрокарбоната натрия выливали в полученный в результате концентрат, и органический слой экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом магния, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 24 г картридж; этилацетат/гексан=5-20%) и концентрировали, получая требуемое соединение (1,510 г, 52,6%) в виде розового твердого остатка.
[Стадия 2] Получение трет-бутил 6-((4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)(4-фторфенил)карбамотиоил)-2,6-диазаспиро[3,3]гептан-2-карбоксилата
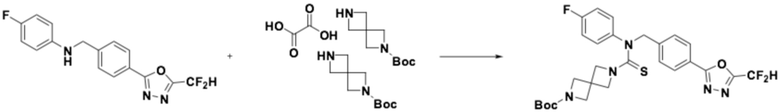
N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-4-фторанилин (1,000 г, 3,132 ммоль), полученный на стадии 1, и N, N-диизопропилэтиламин (1,637 мл, 9,396 ммоль) растворяли в дихлорметане (50 мл) при 0°C, после чего тиофосген (0,360 г, 3,132 ммоль) добавляли к полученному в результате раствору и перемешивали при той же температуре. Гемиоксалат трет-бутил 2,6-диазаспиро[3,3]гептан-2-карбоксилата (0,914 г, 1,879 ммоль) добавляли в реакционную смесь и дополнительно перемешивали при комнатной температуре в течение 18 часов. Водный раствор гидрокарбоната натрия выливали в реакционную смесь, органический слой экстрагировали дихлорметаном, фильтровали через пластиковый фильтр, удаляя твердый остаток и слой водного раствора из него, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=10-40%) и концентрировали, получая требуемое соединение (1,200 г, 68,5%) в виде желтого твердого остатка.
[Стадия 3] Получение N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-(4-фторфенил)-2,6-диазаспиро[3,3]гептан-2-карботиоамида
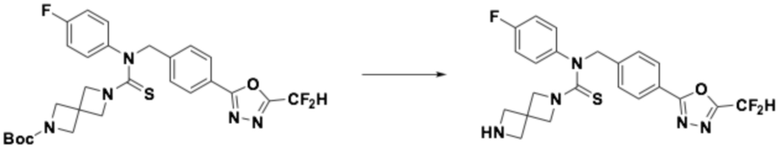
Трет-бутил 6-((4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)(4-фторфенил)карбамотиоил)-2,6-диазаспиро[3,3]гептан-2-карбоксилат (1,200 г, 2,144 ммоль), полученный тем же способом, как описан на стадии 2, и трифторуксусную кислоту (1,149 мл, 15,010 ммоль) растворяли в дихлорметане (15 мл) при комнатной температуре, после чего полученный в результате раствор перемешивали при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь, органический слой экстрагировали дихлорметаном, фильтровали через пластиковый фильтр, удаляя твердый остаток и слой водного раствора из него, и концентрировали под пониженным давлением. Полученный продукт применяли без дополнительной очистки (0,948 г, 96,2%, светло-желтый твердый остаток).
[Стадия 4] Получение соединения 20

N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-(4-фторфенил)-2,6-диазаспиро[3,3]гептан-2-карботиоамид (0,100 г, 0,218 ммоль), полученный на стадии 3, и формальдегид (38,00% раствор, 0,024 мл, 0,326 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,092 г, 0,435 ммоль) добавляли к полученному в результате раствору и перемешивали при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь, органический слой экстрагировали дихлорметаном, фильтровали через пластиковый фильтр, удаляя твердый остаток и слой водного раствора из него, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрировали, получая требуемое соединение (0,051 г, 49,5%) в виде белого твердого остатка.
1H ЯМР (400 МГц, CDCl3) δ 8,03 (д, J=8,1 Гц, 2H), 7,53 (д, J=8,1 Гц, 2H), 7,05-6,79 (м, 5H), 5,54 (с, 2H), 3,77 (уш с, 4H), 3,24 (с, 4H), 2,26 (с, 3H); LRMS (ES) m/z 474,6 (M++1).
Пример 21: Получение соединения 21, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-(4-фторфенил)-6-изопропил-2,6-диазаспиро[3,3]гептан-2-карботиоамид
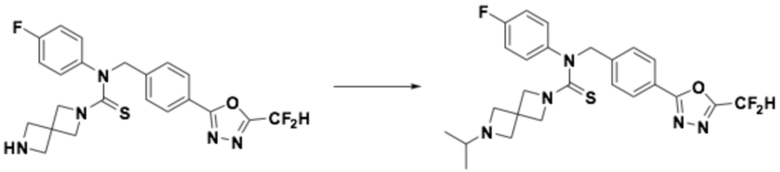
N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-(4-фторфенил)-2,6-диазаспиро[3,3]гептан-2-карботиоамид (0,100 г, 0,218 ммоль), полученный тем же способом, как описан на стадии 3 соединения 20, и ацетон (0,024 мл, 0,326 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,092 г, 0,435 ммоль) добавляли к полученному в результате раствору и перемешивали при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь, органический слой экстрагировали дихлорметаном, фильтровали через пластиковый фильтр, удаляя твердый остаток и слой водного раствора из него, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрировали, получая требуемое соединение (0,037 г, 33,9%) в виде белого твердого остатка.
1H ЯМР (400 МГц, CDCl3) δ 8,03 (д, J=8,1 Гц, 2H), 7,53 (д, J=8,2 Гц, 2H), 7,05-6,79 (м, 5H), 5,54 (с, 2H), 3,85 (уш с, 4H), 3,33 (уш с, 4H), 2,48-2,47 (м, 1H), 0,95-0,89 (м, 6H); LRMS (ES) m/z 502,7 (M++1).
Пример 22: Получение соединения 22, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-(4-фторфенил)-6-(оксетан-3-ил)-2,6-диазаспиро[3,3]гептан-2-карботиоамид
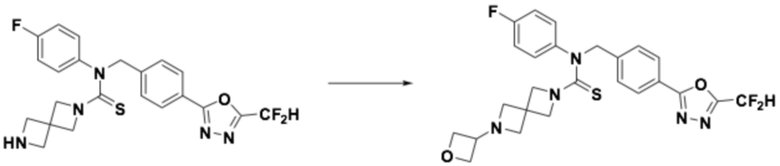
N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-(4-фторфенил)-2,6-диазаспиро[3,3]гептан-2-карботиоамид (0,100 г, 0,218 ммоль), полученный тем же способом, как описан на стадии 3 соединения 20, и 3-оксетанон (0,021 мл, 0,326 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,092 г, 0,435 ммоль) добавляли к полученному в результате раствору и перемешивали при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь, органический слой экстрагировали дихлорметаном, фильтровали через пластиковый фильтр, удаляя твердый остаток и слой водного раствора из него, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-2,5%) и концентрировали, получая требуемое соединение (0,069 г, 61,5%) в виде белого твердого остатка.
1H ЯМР (400 МГц, CDCl3) δ 8,03 (д, J=8,0 Гц, 2H), 7,53 (д, J=8,1 Гц, 2H), 7,05-6,79 (м, 5H), 5,55 (с, 2H), 4,68 (т, J=6,7 Гц, 2H), 4,42 (т, J=5,9 Гц, 2H), 3,85-3,72 (м, 5H), 3,38 (с, 4H); LRMS (ES) m/z 516,7 (M++1).
Пример 23: Получение соединения 23, N-(3,4-дихлорфенил)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-6-метил-2,6-диазаспиро[3,3]гептан-2-карботиоамид
[Стадия 1] Получение трет-бутил 6-((3,4-дихлорфенил)(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)карбамотиоил)-2,6-диазаспиро[3,3]гептан-2-карбоксилата
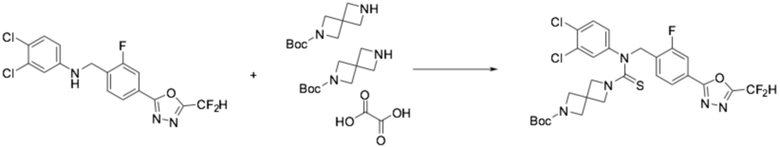 3,4-дихлор-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)анилин (0,930 г, 2,396 ммоль), тиофосген (0,184 мл, 2,396 ммоль) и N, N-диизопропилэтиламин (1,252 мл, 7,188 ммоль) растворяли в дихлорметане (20 мл), после чего полученный в результате раствор перемешивали при 0°C в течение 30 минут, и затем гемиоксалат трет-бутил 2,6-диазаспиро[3,3]гептан-2-карбоксилата (0,583 г, 1,198 ммоль) добавляли к нему и дополнительно перемешивали при комнатной температуре в течение 18 часов. Воду приливали в реакционную смесь, и органический слой экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-30%) и концентрировали, получая заявленное в заголовке соединение (0,280 г, 18,6%) в виде желтого масла.
3,4-дихлор-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)анилин (0,930 г, 2,396 ммоль), тиофосген (0,184 мл, 2,396 ммоль) и N, N-диизопропилэтиламин (1,252 мл, 7,188 ммоль) растворяли в дихлорметане (20 мл), после чего полученный в результате раствор перемешивали при 0°C в течение 30 минут, и затем гемиоксалат трет-бутил 2,6-диазаспиро[3,3]гептан-2-карбоксилата (0,583 г, 1,198 ммоль) добавляли к нему и дополнительно перемешивали при комнатной температуре в течение 18 часов. Воду приливали в реакционную смесь, и органический слой экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-30%) и концентрировали, получая заявленное в заголовке соединение (0,280 г, 18,6%) в виде желтого масла.
[Стадия 2] Получение 2,2,2-трифторацетата N-(3,4-дихлорфенил)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-2,6-диазаспиро[3,3]гептан-2-карботиоамида 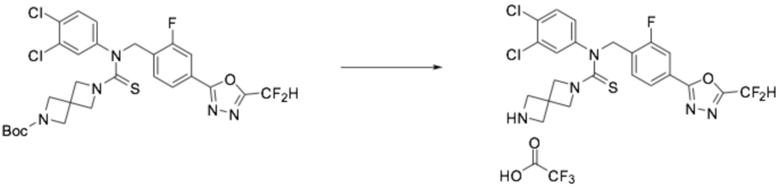
Трет-бутил 6-((3,4-дихлорфенил)(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)карбамотиоил)-2,6-диазаспиро[3,3]гептан-2-карбоксилат (0,275 г, 0,438 ммоль), полученный на стадии 1, и трифторуксусную кислоту (0,335 мл, 4,376 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре, после чего полученный в результате раствор перемешивали при той же температуре в течение 18 часов. Растворитель удаляли из реакционной смеси под пониженным давлением, после чего полученный продукт применяли без дополнительной очистки (0,275 г, 97,8%, желтое масло).
[Стадия 3] Получение соединения 23
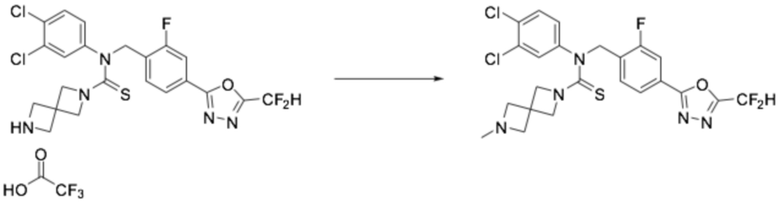
2,2,2-Трифторацетат N-(3,4-дихлорфенил)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-2,6-диазаспиро[3,3]гептан-2-карботиоамида (0,150 г, 0,233 ммоль), полученный на стадии 2, N, N-диизопропилэтиламин (0,041 мл, 0,233 ммоль), формальдегид (0,014 г, 0,467 ммоль) и триацетоксиборгидрид натрия (0,099 г, 0,467 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре, после чего полученный в результате раствор перемешивали при той же температуре в течение 12 часов. Воду приливали в реакционную смесь, и органический слой экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%), получая заявленное в заголовке соединение (0,100 г, 79,0%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3) δ 7,90~7,87 (м, 2H), 7,72 (д, J=9,8 Гц, 1H), 7,41 (д, J=8,6 Гц, 1H), 7,26 (д, J=2,3 Гц, 1H), 7,05 (с, 0,25H), 6,98 (дд, J=8,6, 2,4 Гц, 1H), 6,92 (с, 0,5H), 6,79 (с, 0,25H), 5,55 (с, 2H), 3,87~3,73 (м, 4H), 3,41 (с, 4H), 2,34 (с, 3H); LRMS (ES) m/z 542,2 (M++1).
Пример 24: Получение соединения 24, N-(3,4-дихлорфенил)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-6-(оксетан-3-ил)-2,6-диазаспиро[3,3]гептан-2-карботиоамид
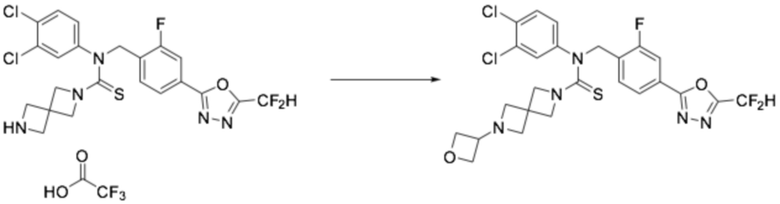
2,2,2-Трифторацетат N-(3,4-дихлорфенил)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-2,6-диазаспиро[3,3]гептан-2-карботиоамида (0,150 г, 0,233 ммоль), полученный тем же способом, как описан на стадии 2 соединения 23, N, N-диизопропилэтиламин (0,041 мл, 0,233 ммоль), 3-оксетанон (0,027 мл, 0,467 ммоль) и триацетоксиборгидрид натрия (0,099 г, 0,467 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре, после чего полученный в результате раствор перемешивали при той же температуре в течение 12 часов. Воду приливали в реакционную смесь, и органический слой экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%), получая заявленное в заголовке соединение (0,100 г, 73,3%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3) δ 7,90~7,89 (м, 2H), 7,73 (д, J=10,0 Гц, 1H), 7,42 (д, J=8,5 Гц, 1H), 7,28~7,27 (м, 1H), 7,05 (с, 0,25H), 6,99 (дд, J=8,5, 2,3 Гц, 1H), 6,92 (с, 0,5H), 6,79 (с, 0,25H), 5,57 (с, 2H), 4,69~4,63 (м, 2H), 4,48~4,45 (м, 2H), 3,94~3,89 (м, 4H), 3,67~3,61 (м, 1H), 3,29 (с, 4H); LRMS (ES) m/z 584,3 (M++1).
Пример 25: Получение соединения 25, N-(3-хлор-4-фторфенил)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-6-метил-2,6-диазаспиро[3,3]гептан-2-карботиоамид
[Стадия 1] Получение трет-бутил 6-((3-хлор-4-фторфенил)(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)карбамотиоил)-2,6-диазаспиро[3,3]гептан-2-карбоксилата
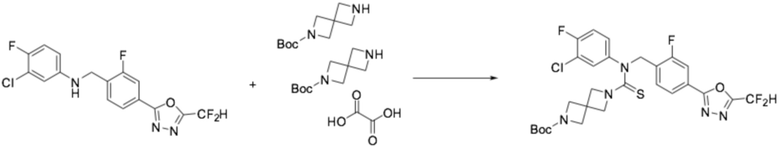 3-хлор-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-4-фторанилин (1,000 г, 2,690 ммоль), тиофосген (0,206 мл, 2,690 ммоль) и N, N-диизопропилэтиламин (1,406 мл, 8,071 ммоль) растворяли в дихлорметане (20 мл), после чего полученный в результате раствор перемешивали при 0°C в течение 30 минут и затем гемиоксалат трет-бутил 2,6-диазаспиро[3,3]гептан-2-карбоксилата (0,654 г, 1,345 ммоль) добавляли к нему и дополнительно перемешивали при комнатной температуре в течение 18 часов. Воду приливали в реакционную смесь, и органический слой экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-30%) и концентрировали, получая заявленное в заголовке соединение (0,650 г, 39,5%) в виде желтого масла.
3-хлор-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-4-фторанилин (1,000 г, 2,690 ммоль), тиофосген (0,206 мл, 2,690 ммоль) и N, N-диизопропилэтиламин (1,406 мл, 8,071 ммоль) растворяли в дихлорметане (20 мл), после чего полученный в результате раствор перемешивали при 0°C в течение 30 минут и затем гемиоксалат трет-бутил 2,6-диазаспиро[3,3]гептан-2-карбоксилата (0,654 г, 1,345 ммоль) добавляли к нему и дополнительно перемешивали при комнатной температуре в течение 18 часов. Воду приливали в реакционную смесь, и органический слой экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-30%) и концентрировали, получая заявленное в заголовке соединение (0,650 г, 39,5%) в виде желтого масла.
[Стадия 2] Получение 2,2,2-трифторацетата N-(3-хлор-4-фторфенил)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-2,6-диазаспиро[3,3]гептан-2-карботиоамида
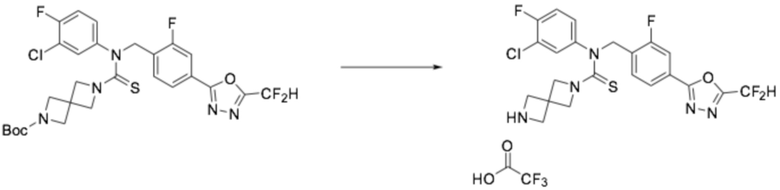
Трет-бутил 6-((3-хлор-4-фторфенил)(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)карбамотиоил)-2,6-диазаспиро[3,3]гептан-2-карбоксилат (0,680 г, 1,111 ммоль), полученный на стадии 1, и трифторуксусную кислоту (0,851 мл, 11,110 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре, после чего полученный в результате раствор перемешивали при той же температуре в течение 18 часов. Растворитель удаляли из реакционной смеси под пониженным давлением, после чего полученный продукт применяли без дополнительной очистки (0,680 г, 97,8%, желтое масло).
[Стадия 3] Получение соединения 25
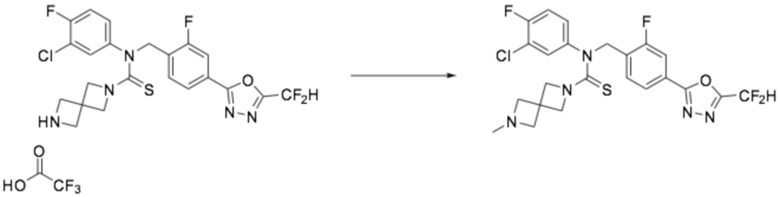
2,2,2-Трифторацетат N-(3-хлор-4-фторфенил)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-2,6-диазаспиро[3,3]гептан-2-карботиоамида (0,262 г, 0,419 ммоль), полученный на стадии 2, и N, N-диизопропилэтиламин (0,073 мл, 0,419 ммоль) растворяли в дихлорметане (10 мл), после чего полученный в результате раствор перемешивали при комнатной температуре в течение 30 минут и затем формальдегид (0,025 г, 0,837 ммоль) и триацетоксиборгидрид натрия (0,177 г, 0,837 ммоль) добавляли к нему и дополнительно перемешивали при той же температуре в течение 18 часов. Воду приливали в реакционную смесь, и органический слой экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрировали, получая заявленное в заголовке соединение (0,150 г, 68,1%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3) δ 7,93~7,88 (м, 2H), 7,72 (д, J=10,0 Гц, 1H), 7,22 (дд, J=6,3, 2,5 Гц, 1H), 7,12 (т, J=8,5 Гц, 1H), 7,05 (с, 0,25H), 7,01~6,97 (м, 1H), 6,92 (с, 0,5H), 6,79 (с, 0,25H), 5,55 (с, 2H), 3,92 (с, 4H), 3,39 (с, 4H), 2,32 (с, 3H); LRMS (ES) m/z 526,6 (M++1).
Пример 26: Получение соединения 26, N-(3-хлор-4-фторфенил)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-6-метил-2,6-диазаспиро[3,3]гептан-2-карботиоамид
[Стадия 1] Получение трет-бутил 6-((3-хлор-4-фторфенил)(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)карбамотиоил)-2,6-диазаспиро[3,3]гептан-2-карбоксилата
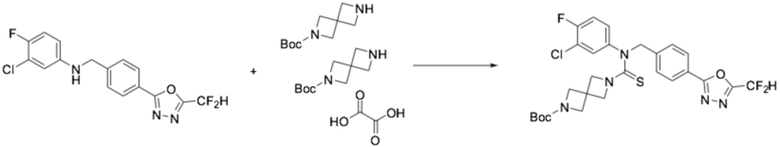 3-хлор-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-4-фторанилин (0,950 г, 2,686 ммоль), тиофосген (0,206 мл, 2,686 ммоль) и N, N-диизопропилэтиламин (1,403 мл, 8,057 ммоль) растворяли в дихлорметане (20 мл), после чего полученный в результате раствор перемешивали при 0°C в течение 30 минут и затем гемиоксалат трет-бутил 2,6-диазаспиро[3,3]гептан-2-карбоксилата (0,653 г, 1,343 ммоль) добавляли к нему и дополнительно перемешивали при комнатной температуре в течение 18 часов. Воду приливали в реакционную смесь, и органический слой экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-30%) и концентрировали, получая заявленное в заголовке соединение (0,853 г, 53,5%) в виде желтого масла.
3-хлор-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-4-фторанилин (0,950 г, 2,686 ммоль), тиофосген (0,206 мл, 2,686 ммоль) и N, N-диизопропилэтиламин (1,403 мл, 8,057 ммоль) растворяли в дихлорметане (20 мл), после чего полученный в результате раствор перемешивали при 0°C в течение 30 минут и затем гемиоксалат трет-бутил 2,6-диазаспиро[3,3]гептан-2-карбоксилата (0,653 г, 1,343 ммоль) добавляли к нему и дополнительно перемешивали при комнатной температуре в течение 18 часов. Воду приливали в реакционную смесь, и органический слой экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-30%) и концентрировали, получая заявленное в заголовке соединение (0,853 г, 53,5%) в виде желтого масла.
[Стадия 2] Получение 2,2,2-трифторацетата N-(3-хлор-4-фторфенил)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-2,6-диазаспиро[3,3]гептан-2-карботиоамида
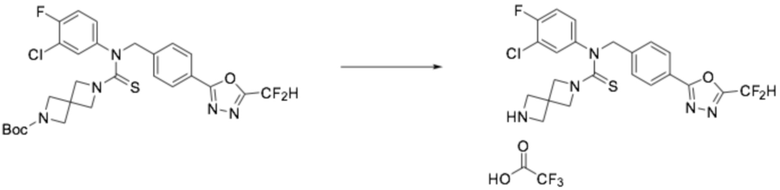
Трет-бутил 6-((3-хлор-4-фторфенил)(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)карбамотиоил)-2,6-диазаспиро[3,3]гептан-2-карбоксилат (0,853 г, 1,436 ммоль), полученный на стадии 1, и трифторуксусную кислоту (1,100 мл, 14,359 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре, после чего полученный в результате раствор перемешивали при той же температуре в течение 18 часов. Растворитель удаляли из реакционной смеси под пониженным давлением, после чего полученный продукт применяли без дополнительной очистки (0,853 г, 97,7%, желтое масло).
[Стадия 3] Получение соединения 26
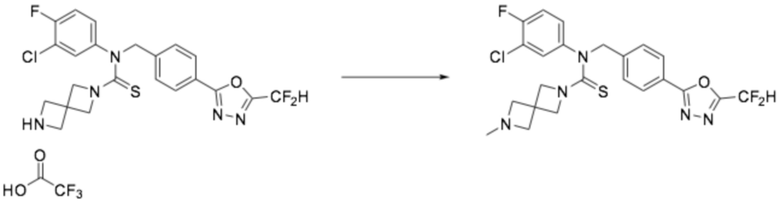
N-(3-хлор-4-фторфенил)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-2,6-диазаспиро[3,3]гептан-2-карботиоамид 2,2,2-трифторацетат, полученный на стадии 2, и N, N-диизопропилэтиламин (0,097 мл, 0,554 ммоль) растворяли в дихлорметане (10 мл), после чего полученный в результате раствор перемешивали при комнатной температуре в течение 30 минут и затем формальдегид (0,033 г, 1,109 ммоль) и триацетоксиборгидрид натрия (0,235 г, 1,109 ммоль) добавляли к нему и дополнительно перемешивали при той же температуре в течение 18 часов. Воду приливали в реакционную смесь, и органический слой экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрировали, получая заявленное в заголовке соединение (0,220 г, 78,1%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3) δ 8,04 (д, J=8,1 Гц, 2H), 7,51 (д, J=8,1 Гц, 2H), 7,16 (дд, J=6,3, 2,5 Гц, 1H), 7,10 (т, J=8,5 Гц, 1H), 7,05 (с, 0,25H), 6,95~6,91 (м, 1H), 6,92 (с, 0,5H), 6,79 (с, 0,25H), 5,50 (с, 2H), 3,86~3,73 (м, 4H), 3,51 (с, 4H), 2,40 (с, 3H); LRMS (ES) m/z 508,5 (M++1).
Пример 27: Получение соединения 27, N-(3-хлор-4-фторфенил)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-6-(оксетан-3-ил)-2,6-диазаспиро[3,3]гептан-2-карботиоамид
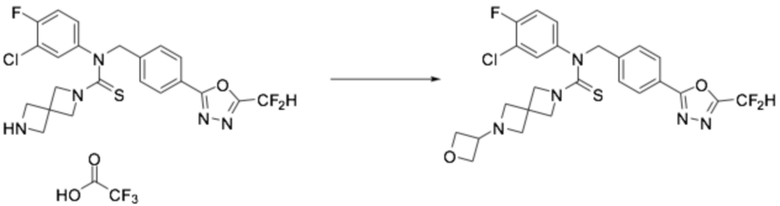
2,2,2-Трифторацетат N-(3-хлор-4-фторфенил)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-2,6-диазаспиро[3,3]гептан-2-карботиоамида (0,320 г, 0,526 ммоль), полученный тем же способом, как описан на стадии 2 соединения 26, и N, N-диизопропилэтиламин (0,092 мл, 0,526 ммоль) растворяли в дихлорметане (10 мл), после чего полученный в результате раствор перемешивали при комнатной температуре в течение 30 минут и затем 3-оксетанон (0,062 мл, 1,053 ммоль) и триацетоксиборгидрид натрия (0,223 г, 1,053 ммоль) добавляли к нему и дополнительно перемешивали при той же температуре в течение 18 часов. Воду приливали в реакционную смесь, и органический слой экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрировали, получая заявленное в заголовке соединение (0,188 г, 70,3%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3) δ 8,05 (д, J=8,1 Гц, 2H), 7,53 (д, J=8,1 Гц, 2H), 7,19 (дд, J=6,3, 2,4 Гц, 1H), 7,10 (т, J=8,5 Гц, 1H), 7,05 (с, 0,25H), 6,96~6,92 (м, 1H), 6,92 (с, 0,5H), 6,79 (с, 0,25H), 5,52 (с, 2H), 4,65 (т, J=6,6 Гц, 2H), 4,40 (т, J=5,8 Гц, 2H), 3,86~3,75 (м, 4H), 3,67~3,61 (м, 1H), 3,29 (с, 4H); LRMS (ES) m/z 550,4 (M++1).
Пример 28: Получение соединения 28, N-(3-хлор-4-фторфенил)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-6-изопропил-2,6-диазаспиро[3,3]гептан-2-карботиоамид
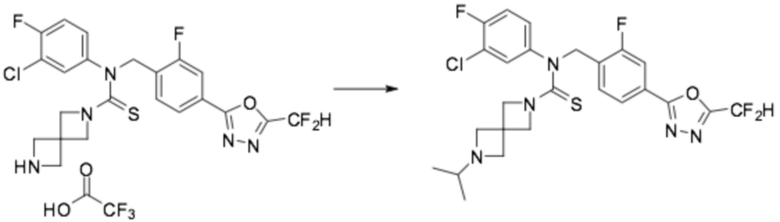
2,2,2-Трифторацетат N-(3-хлор-4-фторфенил)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-2,6-диазаспиро[3,3]гептан-2-карботиоамида (0,254 г, 0,406 ммоль), полученный тем же способом, как описан на стадии 2 соединения 25, и N, N-диизопропилэтиламин (0,071 мл, 0,406 ммоль) растворяли в дихлорметане (10 мл), после чего полученный в результате раствор перемешивали при комнатной температуре в течение 30 минут и затем ацетон (0,024 г, 0,812 ммоль) и триацетоксиборгидрид натрия (0,172 г, 0,812 ммоль) добавляли к нему и дополнительно перемешивали при той же температуре в течение 18 часов. Воду приливали в реакционную смесь, и экстракцию проводили дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрировали, получая заявленное в заголовке соединение (0,160 г, 71,2%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3) δ 7,93~7,88 (м, 2H), 7,72 (д, J=10,0 Гц, 1H), 7,22 (дд, J=6,3, 2,4 Гц, 1H), 7,14~7,09 (м, 1H), 7,05 (с, 0,25H), 7,01~6,97 (м, 1H), 6,92 (с, 0,5H), 6,79 (с, 0,25H), 5,50 (с, 2H), 3,95~3,84 (м, 4H), 3,42 (с, 4H), 2,49~2,42 (м, 1H), 0,98~0,96 (м, 6H); LRMS (ES) m/z 554,7 (M++1).
Пример 29: Получение соединения 29, N-(3-хлор-4-фторфенил)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-6-изопропил-2,6-диазаспиро[3,3]гептан-2-карботиоамид
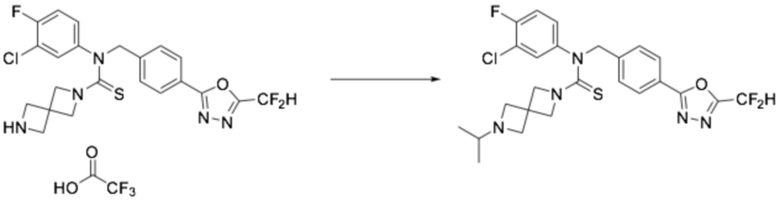
2,2,2-Трифторацетат N-(3-хлор-4-фторфенил)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-2,6-диазаспиро[3,3]гептан-2-карботиоамида (0,325 г, 0,535 ммоль), полученный тем же способом, как описан на стадии 2 соединения 26, и N, N-диизопропилэтиламин (0,093 мл, 0,535 ммоль) растворяли в дихлорметане (10 мл), после чего полученный в результате раствор перемешивали при комнатной температуре в течение 30 минут и затем ацетон (0,032 г, 1,069 ммоль) и триацетоксиборгидрид натрия (0,227 г, 1,069 ммоль) добавляли к нему и дополнительно перемешивали при той же температуре в течение 18 часов. Воду приливали в реакционную смесь, и органический слой экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрировали, получая заявленное в заголовке соединение (0,199 г, 69,4%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3) δ 8,04 (д, J=8,1 Гц, 2H), 7,53 (д, J=8,1 Гц, 2H), 7,17 (дд, J=6,2, 2,2 Гц, 1H), 7,08 (т, J=8,7 Гц, 1H), 7,05 (с, 0,25H), 6,94~6,92 (м, 1H), 6,92 (с, 0,5H), 6,79 (с, 0,25H), 5,52 (с, 2H), 3,91~3,74 (м, 4H), 3,18 (с, 4H), 2,20~2,16 (м, 1H), 0,88 (д, J=6,2 Гц, 6H); LRMS (ES) m/z 536,4 (M++1).
Пример 30: Получение соединения 30, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-(3,4-дифторфенил)-6-метил-2,6-диазаспиро[3,3]гептан-2-карботиоамид
[Стадия 1] Получение трет-бутил 6-((4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)(3,4-дифторфенил)карбамотиоил)-2,6-диазаспиро[3,3]гептан-2-карбоксилата
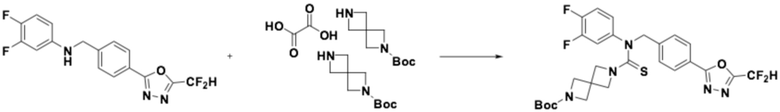 N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-3,4-дифторанилин (1,000 г, 2,965 ммоль), полученный на стадии 1, и N, N-диизопропилэтиламин (1,549 мл, 8,895 ммоль) растворяли в дихлорметане (50 мл) при 0°C, после чего тиофосген (0,341 г, 2,965 ммоль) добавляли к полученному в результате раствору и перемешивали при той же температуре. Гемиоксалат трет-бутил 2,6-диазаспиро[3,3]гептан-2-карбоксилата (0,866 г, 1,779 ммоль) добавляли в реакционную смесь и дополнительно перемешивали при комнатной температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь, органический слой экстрагировали дихлорметаном, фильтровали через пластиковый фильтр, удаляя твердый остаток и слой водного раствора из него, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=10-40%) и концентрировали, получая требуемое соединение (1,080 г, 63,1%) в виде желтого твердого остатка.
N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-3,4-дифторанилин (1,000 г, 2,965 ммоль), полученный на стадии 1, и N, N-диизопропилэтиламин (1,549 мл, 8,895 ммоль) растворяли в дихлорметане (50 мл) при 0°C, после чего тиофосген (0,341 г, 2,965 ммоль) добавляли к полученному в результате раствору и перемешивали при той же температуре. Гемиоксалат трет-бутил 2,6-диазаспиро[3,3]гептан-2-карбоксилата (0,866 г, 1,779 ммоль) добавляли в реакционную смесь и дополнительно перемешивали при комнатной температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь, органический слой экстрагировали дихлорметаном, фильтровали через пластиковый фильтр, удаляя твердый остаток и слой водного раствора из него, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=10-40%) и концентрировали, получая требуемое соединение (1,080 г, 63,1%) в виде желтого твердого остатка.
[Стадия 2] Получение N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-(3,4-дифторфенил)-2,6-диазаспиро[3,3]гептан-2-карботиоамида
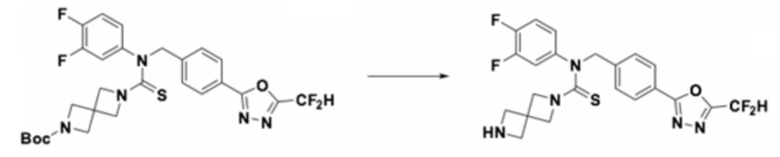
Трет-бутил 6-((4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)(3,4-дифторфенил)карбамотиоил)-2,6-диазаспиро[3,3]гептан-2-карбоксилат (1,080 г, 1,870 ммоль), полученный на стадии 1, и трифторуксусную кислоту (1,002 мл, 13,089 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре, после чего полученный в результате раствор перемешивали при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь, органический слой экстрагировали дихлорметаном, фильтровали через пластиковый фильтр, удаляя твердый остаток и слой водного раствора из него, и концентрировали под пониженным давлением. Полученный продукт применяли без дополнительной очистки (0,864 г, 96,8%, светло-желтый твердый остаток).
[Стадия 3] Получение соединения 30

N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-(3,4-дифторфенил)-2,6-диазаспиро[3,3]гептан-2-карботиоамид (0,100 г, 0,209 ммоль), полученный на стадии 2, и формальдегид (38,00% раствор в воде, 0,023 мл, 0,314 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,089 г, 0,419 ммоль) добавляли к полученному в результате раствору и перемешивали при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь, после чего экстракцию проводили дихлорметаном, затем фильтровали через пластиковый фильтр, удаляя твердый остаток и слой водного раствора из него, и затем концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрировали, получая требуемое соединение (0,030 г, 29,1%) в виде белого твердого остатка.
1H ЯМР (400 МГц, CDCl3) δ 8,02 (д, J=8,1 Гц, 2H), 7,51 (д, J=8,0 Гц, 2H), 7,08 (кв, J=9,3 Гц, 1H), 7,01-6,78 (м, 3H), 5,51 (с, 2H), 3,82 (уш с, 4H), 3,21 (с, 4H), 2,23 (с, 3H); LRMS (ES) m/z 492,7 (M++1)
Пример 31: Получение соединения 31, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-(3,4-дифторфенил)-6-изопропил-2,6-диазаспиро[3,3]гептан-2-карботиоамид
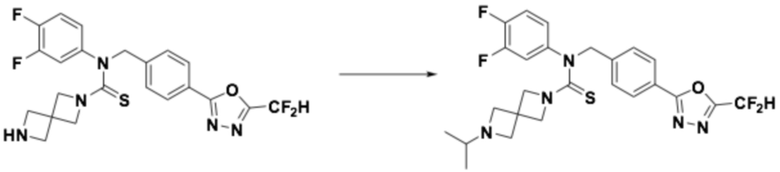
N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-(3,4-дифторфенил)-2,6-диазаспиро[3,3]гептан-2-карботиоамид (1,000 г, 2,094 ммоль), полученный тем же способом, как описан на стадии 2 соединения 30, и ацетон (0,234 мл, 3,141 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,888 г, 4,189 ммоль) добавляли к полученному в результате раствору и перемешивали при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь, органический слой экстрагировали дихлорметаном, фильтровали через пластиковый фильтр, удаляя твердый остаток и слой водного раствора из него, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-5%) и концентрировали, получая требуемое соединение (0,029 г, 2,7%) в виде белого твердого остатка.
1H ЯМР (400 МГц, CDCl3) δ 8,04 (д, J=8,4 Гц, 2H), 7,53 (д, J=8,4 Гц, 2H), 7,10 (кв, J=9,0 Гц, 1H), 7,05-6,80 (м, 3H), 5,52 (с, 2H), 3,84 (уш с, 4H), 3,18 (с, 4H), 2,20-2,15 (м, 1H), 0,89 (д, J=6,9 Гц, 6H); LRMS (ES) m/z 520,8 (M++1).
Пример 32: Получение соединения 32, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-(3,4-дифторфенил)-6-(оксетан-3-ил)-2,6-диазаспиро[3,3]гептан-2-карботиоамид
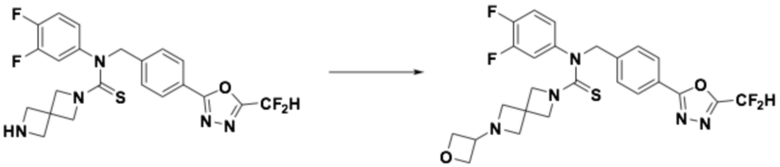
N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-(3,4-дифторфенил)-2,6-диазаспиро[3,3]гептан-2-карботиоамид (0,100 г, 0,209 ммоль), полученный тем же способом, как описан на стадии 2 соединения 30, и 3-оксетанон (0,020 мл, 0,314 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,089 г, 0,419 ммоль) добавляли к полученному в результате раствору и перемешивали при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь, органический слой экстрагировали дихлорметаном, фильтровали через пластиковый фильтр, удаляя твердый остаток и слой водного раствора из него, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-2,5%) и концентрировали, получая требуемое соединение (0,034 г, 30,4%) в виде белого твердого остатка.
1H ЯМР (400 МГц, CDCl3) δ 8,05 (д, J=8,4 Гц, 2H), 7,53 (д, J=8,4 Гц, 2H), 7,14 (кв, J=9,0 Гц, 1H), 7,06-6,80 (м, 3H), 5,53 (с, 2H), 4,68 (т, J=6,7 Гц, 2H), 3,89-3,70 (м, 5H), 3,38 (с, 4H); LRMS (ES) m/z 534,6 (M++1).
Пример 33: Получение соединения 33, 6-ацетил-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3,4-дифторфенил)-2,6-диазаспиро[3,3]гептан-2-карботиоамид
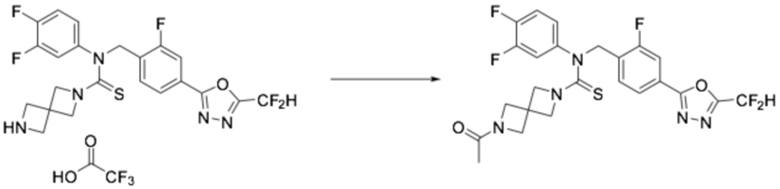
2,2,2-Трифторацетат N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3,4-дифторфенил)-2,6-диазаспиро[3,3]гептан-2-карботиоамида (0,159 г, 0,261 ммоль), полученный тем же способом, как описан на стадии 3 соединения 13, N, N-диизопропилэтиламин (0,091 мл, 0,522 ммоль) и ацетилхлорид (0,028 мл, 0,391 ммоль) растворяли в дихлорметане (20 мл) при комнатной температуре, после чего полученный в результате раствор перемешивали при той же температуре в течение 18 часов. Воду приливали в реакционную смесь, и органический слой экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-70%) и концентрировали, получая заявленное в заголовке соединение (0,100 г, 71,3%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3) δ 7,92~7,90 (м, 2H), 7,73~7,71 (м, 1H), 7,20~7,10 (м, 1H), 7,05 (с, 0,25H), 7,03~6,98 (м, 1H), 6,92 (с, 0,5H), 6,92~6,89 (м, 1H), 6,79 (с, 0,25H), 5,57 (с, 2H), 4,16~3,80 (м, 8H), 1,82 (с, 3H); LRMS (ES) m/z 538,5 (M++1).
Пример 34: Получение соединения 34, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3,4-дифторфенил)-6-(оксетан-3-ил)-2,6-диазаспиро[3,3]гептан-2-карботиоамид
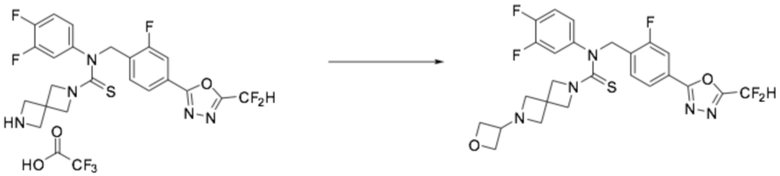
2,2,2-Трифторацетат N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3,4-дифторфенил)-2,6-диазаспиро[3,3]гептан-2-карботиоамида (0,186 г, 0,305 ммоль), полученный тем же способом, как описан на стадии 3 соединения 13, и N, N-диизопропилэтиламин (0,053 мл, 0,305 ммоль) растворяли в дихлорметане (10 мл), после чего полученный в результате раствор перемешивали при комнатной температуре в течение 30 минут и затем триацетоксиборгидрид натрия (0,129 г, 0,610 ммоль) и 3-оксетанон (0,044 г, 0,610 ммоль) добавляли к нему и дополнительно перемешивали при той же температуре в течение 18 часов. Воду приливали в реакционную смесь, и органический слой экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрировали, получая заявленное в заголовке соединение (0,100 г, 61,4%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3) δ 7,92~7,89 (м, 2H), 7,71 (дд, J=9,9, 1,4 Гц, 1H), 7,20~7,12 (м, 1H), 7,05 (с, 0,25H), 7,03~6,95 (м, 1H), 6,92 (с, 0,5H), 6,89~6,82 (м, 1H), 6,79 (с, 0,25H), 5,56 (с, 2H), 4,64 (т, J=6,7 Гц, 2H), 4,40 (дд, J=6,6, 5,2 Гц, 2H), 4,00~3,80 (м, 4H), 3,65~3,60 (м, 1H), 3,29 (с, 4H). LRMS (ES) m/z 552,5 (M++1).
Пример 35: Получение соединения 35, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3,4-дифторфенил)-2-окса-6-азаспиро[3,3]гептан-6-карботиоамид
N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3,4-дифторанилин (0,330 г, 0,929 ммоль), полученный тем же способом, как описан на стадии 1 соединения 13, N, N-диизопропилэтиламин (0,485 мл, 2,787 ммоль) и тиофосген (0,107 г, 0,929 ммоль) растворяли в дихлорметане (10 мл), после чего полученный в результате раствор перемешивали при 0°C в течение 30 минут и затем гемиоксалат 2-окса-6-азаспиро[3,3]гептана (0,134 г, 0,464 ммоль) добавляли к нему и дополнительно перемешивали при комнатной температуре в течение 18 часов. Воду приливали в реакционную смесь, и органический слой экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-70%) и концентрировали, получая заявленное в заголовке соединение (0,100 г, 21,7%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3) δ 7,94~7,88 (м, 2H), 7,74~7,71 (м, 1H), 7,17 (дд, J=18,2, 8,7 Гц, 1H), 7,05 (с, 0,25H), 7,02~6,97 (м, 1H), 6,93 (с, 0,5H), 6,91~6,87 (м, 1H), 6,80 (с, 0,25H), 5,57 (с, 2H), 4,67 (с, 4H), 3,92 (с, 4H); LRMS (ES) m/z 497,5 (M++1).
Пример 36: Получение соединения 36, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3-фторфенил)-6-метил-2,6-диазаспиро[3,3]гептан-2-карботиоамид
[Стадия 1] Получение трет-бутил 6-((4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)(3-фторфенил)карбамотиоил)-2,6-диазаспиро[3,3]гептан-2-карбоксилата
 N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-фторанилин (1,000 г, 2,965 ммоль) и N, N-диизопропилэтиламин (1,033 мл, 5,930 ммоль) растворяли в дихлорметане (30 мл) при 0°C, после чего тиофосген (0,309 мл, 3,261 ммоль) добавляли к полученному в результате раствору и перемешивали при той же температуре. Гемиоксалат трет-бутил 2,6-диазаспиро[3,3]гептан-2-карбоксилата (0,866 г, 1,779 ммоль) добавляли в реакционную смесь и дополнительно перемешивали при комнатной температуре в течение 18 часов. Реакционная смесь очищали колоночной хроматографией (SiO2, 24 г картридж; этилацетат/гексан=10-60%) и концентрировали, получая требуемое соединение (0,560 г, 32,7%) в виде светло-желтого масла.
N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-фторанилин (1,000 г, 2,965 ммоль) и N, N-диизопропилэтиламин (1,033 мл, 5,930 ммоль) растворяли в дихлорметане (30 мл) при 0°C, после чего тиофосген (0,309 мл, 3,261 ммоль) добавляли к полученному в результате раствору и перемешивали при той же температуре. Гемиоксалат трет-бутил 2,6-диазаспиро[3,3]гептан-2-карбоксилата (0,866 г, 1,779 ммоль) добавляли в реакционную смесь и дополнительно перемешивали при комнатной температуре в течение 18 часов. Реакционная смесь очищали колоночной хроматографией (SiO2, 24 г картридж; этилацетат/гексан=10-60%) и концентрировали, получая требуемое соединение (0,560 г, 32,7%) в виде светло-желтого масла.
[Стадия 2] Получение N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3-фторфенил)-2,6-диазаспиро[3,3]гептан-2-карботиоамида

Трет-бутил 6-((4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)(3-фторфенил)карбамотиоил)-2,6-диазаспиро[3,3]гептан-2-карбоксилат (0,560 г, 0,970 ммоль), полученный на стадии 1, и трифторуксусную кислоту (0,520 мл, 6,787 ммоль) растворяли в дихлорметане (6 мл) при комнатной температуре, после чего полученный в результате раствор перемешивали при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь, органический слой экстрагировали дихлорметаном, фильтровали через пластиковый фильтр, удаляя твердый остаток и слой водного раствора из него, и концентрировали под пониженным давлением. Полученный продукт применяли без дополнительной очистки (0,420 г, 90,7%, желтый твердый остаток).
[Стадия 3] Получение соединения 36

N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3-фторфенил)-2,6-диазаспиро[3,3]гептан-2-карботиоамид (0,100 г, 0,209 ммоль), полученный на стадии 2, и формальдегид (38,00% раствор в воде, 0,023 мл, 0,314 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,089 г, 0,419 ммоль) добавляли к полученному в результате раствору и перемешивали при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь, органический слой экстрагировали дихлорметаном, фильтровали через пластиковый фильтр, удаляя твердый остаток и слой водного раствора из него, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрировали, получая требуемое соединение (0,008 г, 7,8%) в виде светло-желтого твердого остатка.
1H ЯМР (400 МГц, CDCl3) δ 7,94~7,88 (м, 2H), 7,71 (д, J=10,2 Гц, 1H), 7,34~7,29 (м, 1H), 7,05~6,79 (м, 4H), 5,61 (с, 2H), 3,84 (уш с, 4H), 3,23 (с, 4H), 2,26 (с, 3H); LRMS (ES) m/z 492,2 (M++1).
Пример 37: Получение соединения 37, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3-фторфенил)-6-изопропил-2,6-диазаспиро[3,3]гептан-2-карботиоамид
N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3-фторфенил)-2,6-диазаспиро[3,3]гептан-2-карботиоамид (0,100 г, 0,209 ммоль), полученный тем же способом, как описан на стадии 2 соединения 36, и ацетон (0,023 мл, 0,314 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,089 г, 0,419 ммоль) добавляли к полученному в результате раствору и перемешивали при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь, органический слой экстрагировали дихлорметаном, фильтровали через пластиковый фильтр, удаляя твердый остаток и слой водного раствора из него, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрировали, получая требуемое соединение (0,006 г, 5,5%) в виде светло-желтого твердого остатка.
1H ЯМР (400 МГц, CDCl3) δ 7,94~7,87 (м, 2H), 7,71 (дд, J=9,9, 1,3 Гц, 1H), 7,33~7,27 (м, 1H), 7,05~6,79 (м, 4H), 5,61 (с, 2H), 3,80 (уш с, 4H), 3,20 (с, 4H), 2,22~2,19 (м, 1H), 0,88 (д, J=4,8 Гц, 6H); LRMS (ES) m/z 520,4 (M++1).
Пример 38: Получение соединения 38, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3-фторфенил)-6-(оксетан-3-ил)-2,6-диазаспиро[3,3]гептан-2-карботиоамид
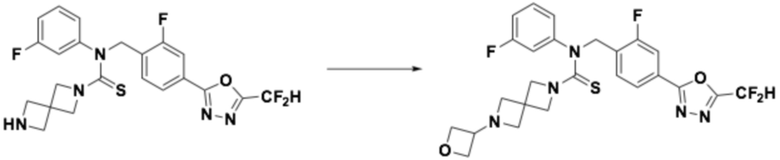
N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3-фторфенил)-2,6-диазаспиро[3,3]гептан-2-карботиоамид (0,100 г, 0,209 ммоль), полученный тем же способом, как описан на стадии 2 соединения 36, и 3-оксетанон (0,020 мл, 0,314 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,089 г, 0,419 ммоль) добавляли к полученному в результате раствору и перемешивали при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь, органический слой экстрагировали дихлорметаном, фильтровали через пластиковый фильтр, удаляя твердый остаток и слой водного раствора из него, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-2,5%) и концентрировали, получая требуемое соединение (0,004 г, 3,6%) в виде светло-желтого твердого остатка.
1H ЯМР (400 МГц, CDCl3) δ 7,94~7,88 (м, 2H), 7,72 (дд, J=10,0, 1,3 Гц, 1H), 7,35~7,29 (м, 1H), 7,05~6,79 (м, 4H), 4,66 (т, J=6,7 Гц, 2H), 4,42~4,41 (м, 2H), 3,88~3,67 (м, 5H), 3,32 (с, 4H); LRMS (ES) m/z 534,3 (M++1).
Пример 39: Получение соединения 39, (1S,4S)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(4-фторфенил)-5-метил-2,5-диазабицикло[2,2,1]гептан-2-карботиоамид
[Стадия 1] Получение трет-бутил (1S,4S)-5-((4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)(4-фторфенил)карбамотиоил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилата

N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-4-фторанилин (1,000 г, 2,965 ммоль), полученный на стадии 1 соединения 17, и N, N-диизопропилэтиламин (1,549 мл, 8,895 ммоль) растворяли в дихлорметане (30 мл) при 0°C, после чего тиофосген (0,227 мл, 2,965 ммоль) добавляли к полученному в результате раствору и перемешивали при той же температуре. Трет-бутил (1S,4S)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилат (0,705 г, 3,558 ммоль) добавляли в реакционную смесь и дополнительно перемешивали при комнатной температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь и органический слой экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом магния, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 24 г картридж; этилацетат/гексан=10-40%) и концентрировали, получая требуемое соединение (1,120 г, 65,4%) в виде светло-желтого твердого остатка.
[Стадия 2] Получение (1S,4S)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(4-фторфенил)-2,5-диазабицикло[2,2,1]гептан-2-карботиоамида
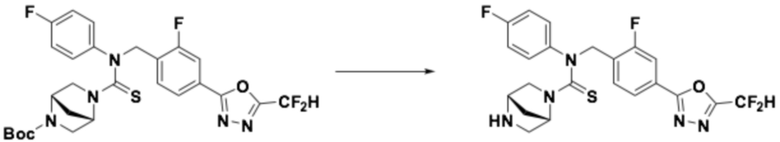
Трет-бутил (1S,4S)-5-((4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)(4-фторфенил)карбамотиоил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилат (1,120 г, 1,939 ммоль), полученный на стадии 1, и трифторуксусную кислоту (1,039 мл, 13,573 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре, после чего полученный в результате раствор перемешивали при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь, органический слой экстрагировали дихлорметаном, фильтровали через пластиковый фильтр, удаляя твердый остаток и слой водного раствора из него, и концентрировали под пониженным давлением. Полученный продукт применяли без дополнительной очистки (0,780 г, 84,2%, желтый твердый остаток).
[Стадия 3] Получение соединения 39

(1S,4S)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(4-фторфенил)-2,5-диазабицикло[2,2,1]гептан-2-карботиоамид (0,150 г, 0,314 ммоль), полученный на стадии 2, и формальдегид (38,00% раствор в воде, 0,034 мл, 0,471 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,133 г, 0,628 ммоль) добавляли к полученному в результате раствору и перемешивали при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь, органический слой экстрагировали дихлорметаном, фильтровали через пластиковый фильтр, удаляя твердый остаток и слой водного раствора из него, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрировали, получая требуемое соединение (0,070 г, 45,3%) в виде белого твердого остатка.
1H ЯМР (400 МГц, CDCl3) δ 7,89~7,82 (м, 2H), 7,75 (дд, J=10,2, 1,3 Гц, 1H), 7,13~7,08 (м, 2H), 7,13~6,79 (м, 3H), 5,64 (д, J=15,9 Гц, 1H), 5,31 (д, J=3,4 Гц, 1H), 4,94 (с, 1H), 3,35~3,30 (м, 2H), 2,79~2,74 (м, 3H), 2,33 (с, 3H), 1,85 (д, J=10,0 Гц, 1H), 1,57 (дд, J=10,0, 1,5 Гц, 1H); LRMS (ES) m/z 492,4 (M++1).
Пример 40: Получение соединения 40, (1S,4S)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(4-фторфенил)-5-изопропил-2,5-диазабицикло[2,2,1]гептан-2-карботиоамид
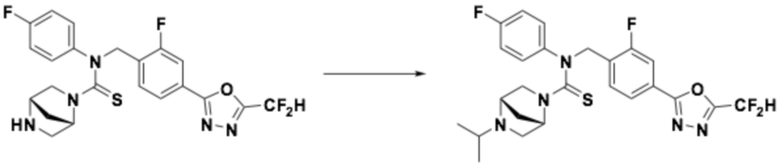 (1S,4S)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(4-фторфенил)-2,5-диазабицикло[2,2,1]гептан-2-карботиоамид (0,150 г, 0,314 ммоль), полученный тем же способом, как описан на стадии 2 соединения 39, и ацетон (0,035 мл, 0,471 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,133 г, 0,628 ммоль) добавляли к полученному в результате раствору и перемешивали при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь, органический слой экстрагировали дихлорметаном, фильтровали через пластиковый фильтр, удаляя твердый остаток и слой водного раствора из него, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрировали, получая требуемое соединение (0,087 г, 53,3%) в виде светло-желтого твердого остатка.
(1S,4S)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(4-фторфенил)-2,5-диазабицикло[2,2,1]гептан-2-карботиоамид (0,150 г, 0,314 ммоль), полученный тем же способом, как описан на стадии 2 соединения 39, и ацетон (0,035 мл, 0,471 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,133 г, 0,628 ммоль) добавляли к полученному в результате раствору и перемешивали при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь, органический слой экстрагировали дихлорметаном, фильтровали через пластиковый фильтр, удаляя твердый остаток и слой водного раствора из него, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрировали, получая требуемое соединение (0,087 г, 53,3%) в виде светло-желтого твердого остатка.
1H ЯМР (400 МГц, CDCl3) δ 7,89~7,82 (м, 2H), 7,76 (д, J=9,6 Гц, 1H), 7,13~7,09 (м, 2H), 7,06~6,80 (м, 3H), 5,61 (д, J=15,9 Гц, 1H), 5,33 (д, J=15,8 Гц, 1H), 4,91 (с, 1H), 3,64 (с, 1H), 3,37 (с, 1H), 3,04~3,02 (м, 1H), 2,72~2,70 (м, 2H), 2,49 (с, 1H), 1,87 (д, J=9,1 Гц, 1H), 1,60 (д, J=10,1 Гц, 1H), 0,92~0,88 (м, 6H); LRMS (ES) m/z 520,4 (M++1).
Пример 41: Получение соединения 41, (1S,4S)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(4-фторфенил)-5-(оксетан-3-ил)-2,5-диазабицикло[2,2,1]гептан-2-карботиоамид
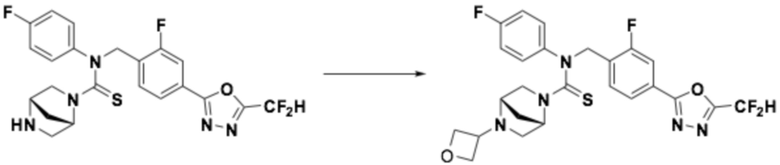
(1S,4S)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(4-фторфенил)-2,5-диазабицикло[2,2,1]гептан-2-карботиоамид (0,100 г, 0,209 ммоль), полученный тем же способом, как описан на стадии 2 соединения 39, и 3-оксетанон (0,020 мл, 0,314 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,089 г, 0,419 ммоль) добавляли к полученному в результате раствору и перемешивали при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь, органический слой экстрагировали дихлорметаном, фильтровали через пластиковый фильтр, удаляя твердый остаток и слой водного раствора из него, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=50-90%) и концентрировали, получая требуемое соединение (0,068 г, 60,9%) в виде белого твердого остатка.
1H ЯМР (400 МГц, CDCl3) δ 7,89~7,85 (м, 2H), 7,75 (д, J=10,5 Гц, 1H), 7,12~7,08 (м, 2H), 7,05~6,79 (м, 3H), 5,58 (д, J=15,7 Гц, 1H), 5,34 (д, J=15,7 Гц, 1H), 4,97 (с, 1H), 4,67~4,63 (м, 2H), 4,49~4,44 (м, 2H), 3,87~3,81 (м, 1H), 3,32 (с, 1H), 3,12~3,09 (м, 2H), 2,75 (д, J=8,4 Гц, 1H), 2,70~2,69 (м, 1H), 1,80 (д, J=10,0 Гц, 1H), 1,57 (д, J=10,0 Гц, 1H); LRMS (ES) m/z 534,4 (M++1).
Пример 42: Получение соединения 42, (1S,4S)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3,4-дифторфенил)-5-метил-2,5-диазабицикло[2,2,1]гептан-2-карботиоамид
[Стадия 1] Получение трет-бутил (1S,4S)-5-((4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)(3,4-дифторфенил)карбамотиоил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилата
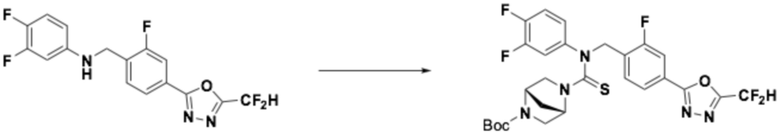 N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3,4-дифторанилин (1,000 г, 2,815 ммоль), полученный тем же способом, как описан на стадии 1 примера 13, тиофосген (0,216 мл, 2,815 ммоль) и N, N-диизопропилэтиламин (1,716 мл, 9,852 ммоль) растворяли в дихлорметане (30 мл), после чего полученный в результате раствор перемешивали при 0°C в течение 30 минут и затем трет-бутил (1S,4S)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилат (0,558 г, 2,815 ммоль) добавляли к нему и дополнительно перемешивали при комнатной температуре в течение 18 часов. Воду приливали в реакционную смесь, и органический слой экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-30%) и концентрировали, получая заявленное в заголовке соединение (0,460 г, 27,4%) в виде желтого масла.
N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3,4-дифторанилин (1,000 г, 2,815 ммоль), полученный тем же способом, как описан на стадии 1 примера 13, тиофосген (0,216 мл, 2,815 ммоль) и N, N-диизопропилэтиламин (1,716 мл, 9,852 ммоль) растворяли в дихлорметане (30 мл), после чего полученный в результате раствор перемешивали при 0°C в течение 30 минут и затем трет-бутил (1S,4S)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилат (0,558 г, 2,815 ммоль) добавляли к нему и дополнительно перемешивали при комнатной температуре в течение 18 часов. Воду приливали в реакционную смесь, и органический слой экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-30%) и концентрировали, получая заявленное в заголовке соединение (0,460 г, 27,4%) в виде желтого масла.
[Стадия 2] Получение (1S,4S)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3,4-дифторфенил)-2,5-диазабицикло[2,2,1]гептан-2-карботиоамида
 Трет-бутил (1S,4S)-5-((4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)(3,4-дифторфенил)карбамотиоил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилат (0,460 г, 0,772 ммоль), полученный на стадии 1, и трифторуксусную кислоту (0,591 мл, 7,723 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре, после чего полученный в результате раствор перемешивали при той же температуре в течение 18 часов. Растворитель удаляли из реакционной смеси под пониженным давлением, после чего насыщенный водный раствор гидрокарбоната натрия выливали в полученный в результате концентрат, и органический слой экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Заявленное в заголовке соединение применяли без дополнительной очистки (0,350 г, 91,5%, бесцветное масло).
Трет-бутил (1S,4S)-5-((4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)(3,4-дифторфенил)карбамотиоил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилат (0,460 г, 0,772 ммоль), полученный на стадии 1, и трифторуксусную кислоту (0,591 мл, 7,723 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре, после чего полученный в результате раствор перемешивали при той же температуре в течение 18 часов. Растворитель удаляли из реакционной смеси под пониженным давлением, после чего насыщенный водный раствор гидрокарбоната натрия выливали в полученный в результате концентрат, и органический слой экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Заявленное в заголовке соединение применяли без дополнительной очистки (0,350 г, 91,5%, бесцветное масло).
[Стадия 3] Получение соединения 42

(1S,4S)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3,4-дифторфенил)-2,5-диазабицикло[2,2,1]гептан-2-карботиоамид (0,168 г, 0,339 ммоль), полученный на стадии 2, формальдегид (0,020 г, 0,678 ммоль) и N, N-диизопропилэтиламин (0,118 мл, 0,678 ммоль) растворяли в дихлорметане (20 мл) при комнатной температуре, после чего полученный в результате раствор перемешивали при той же температуре в течение 18 часов. Воду приливали в реакционную смесь, и органический слой экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрировали, получая заявленное в заголовке соединение (0,110 г, 63,7%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3) δ 7,88 (дд, J=8,0, 1,6 Гц, 1H), 7,81~7,75 (м, 2H), 7,15~7,05 (м, 1H), 7,02 (с, 0,25H), 7,01~6,97 (м, 1H), 6,92 (с, 0,5H), 6,91~689,00 (м, 1H), 6,79 (с, 0,25H), 5,62 (д, J=15,9 Гц, 1H), 5,21 (д, J=16,0 Гц, 1H), 4,96 (с, 1H), 3,47~3,45 (м, 2H), 2,88~2,80 (м, 3H), 2,38 (с, 3H), 1,94 (д, J=10,4 Гц, 1H), 1,64 (д, J=10,2 Гц, 1H); LRMS (ES) m/z 510,8 (M++1).
Пример 43: Получение соединения 43, (1S,4S)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3,4-дифторфенил)-5-(оксетан-3-ил)-2,5-диазабицикло[2,2,1]гептан-2-карботиоамид
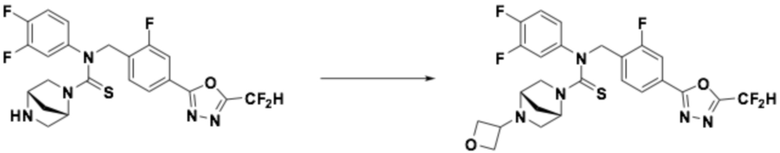
(1S,4S)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3,4-дифторфенил)-2,5-диазабицикло[2,2,1]гептан-2-карботиоамид (0,126 г, 0,254 ммоль), полученный тем же способом, как описан на стадии 2 соединения 42, 3-оксетанон (0,030 мл, 0,509 ммоль) и N, N-диизопропилэтиламин (0,089 мл, 0,509 ммоль) растворяли в дихлорметане (20 мл) при комнатной температуре, после чего полученный в результате раствор перемешивали при той же температуре в течение 18 часов. Воду приливали в реакционную смесь, и органический слой экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрировали, получая заявленное в заголовке соединение (0,088 г, 62,7%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3) δ 7,89~7,76 (м, 3H), 7,15~7,05 (м, 1H), 7,02 (с, 0,25H), 7,00~6,97 (м, 1H), 6,92 (с, 0,5H), 6,91~6,87 (м, 1H), 6,79 (с, 0,25H), 5,53 (д, J=15,8 Гц, 1H), 5,29 (д, J=15,8 Гц, 1H), 4,96 (с, 1H), 4,65 (дд, J=13,8, 6,7 Гц, 2H), 4,48~4,41 (м, 2H), 3,84~3,81 (м, 1H), 3,81 (с, 1H), 3,25~3,00 (м, 2H), 2,78~2,75 (м, 2H), 1,82 (д, J=10,1 Гц, 1H), 1,61 (д, J=27,1 Гц, 1H); LRMS (ES) m/z 552,8 (M++1).
Пример 44: Получение соединения 44, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3,4-дифторфенил)-2-метил-2,7-диазаспиро[3,5]нонан-7-карботиоамид
[Стадия 1] Получение трет-бутил 7-((4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)(3,4-дифторфенил)карбамотиоил)-2,7-диазаспиро[3,5]нонан-2-карбоксилата
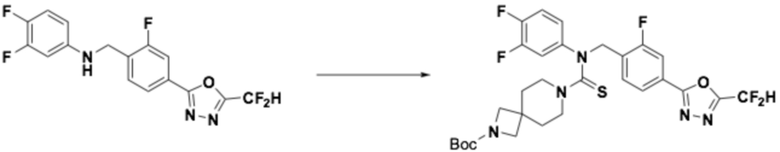
N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3,4-дифторанилин (1,000 г, 2,815 ммоль), полученный тем же способом, как описан на стадии 1 соединения 13, тиофосген (0,216 мл, 2,815 ммоль) и N, N-диизопропилэтиламин (1,716 мл, 9,852 ммоль) растворяли в дихлорметане (30 мл), после чего полученный в результате раствор перемешивали при 0°C в течение 30 минут и затем трет-бутил 2,7-диазаспиро[3,5]нонан-2-карбоксилат (0,637 г, 2,815 ммоль) добавляли к нему и дополнительно перемешивали при комнатной температуре в течение 18 часов. Воду приливали в реакционную смесь, и органический слой экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-30%) и концентрировали, получая заявленное в заголовке соединение (0,600 г, 34,2%) в виде желтого масла.
[Стадия 2] Получение N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3,4-дифторфенил)-2,7-диазаспиро[3,5]нонан-7-карботиоамида

Трет-бутил 7-((4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)(3,4-дифторфенил)карбамотиоил)-2,7-диазаспиро[3,5]нонан-2-карбоксилат (0,600 г, 0,962 ммоль), полученный на стадии 1, и трифторуксусную кислоту (0,737 мл, 9,621 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре, после чего полученный в результате раствор перемешивали при той же температуре в течение 18 часов. Растворитель удаляли из реакционной смеси под пониженным давлением, после чего насыщенный водный раствор гидрокарбоната натрия выливали в полученный в результате концентрат, и органический слой экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный продукт применяли без дополнительной очистки (0,500 г, 99,3%, бесцветное масло).
[Стадия 3] Получение соединения 44

N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3,4-дифторфенил)-2,7-диазаспиро[3,5]нонан-7-карботиоамид (0,216 г, 0,413 ммоль), полученный на стадии 2, формальдегид (0,025 г, 0,825 ммоль) и N, N-диизопропилэтиламин (0,144 мл, 0,825 ммоль) растворяли в дихлорметане (20 мл) при комнатной температуре, после чего полученный в результате раствор перемешивали при той же температуре в течение 18 часов. Воду приливали в реакционную смесь, и органический слой экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрировали, получая заявленное в заголовке соединение (0,100 г, 45,1%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3) δ 7,87 (дд, J=8,0, 1,4 Гц, 1H), 7,81 (дд, J=10,3, 1,4 Гц, 1H), 7,73 (т, J=7,7 Гц, 1H), 7,14~7,10 (м, 1H), 7,05 (с, 0,25H), 6,97~6,93 (м, 1H), 6,93 (с, 0,5H), 6,85~6,83 (м, 1H), 6,80 (с, 0,25H), 5,38 (с, 2H), 3,75~3,55 (м, 4H), 3,36 (с, 4H), 2,56 (с, 3H), 1,72~1,69 (м, 4H); LRMS (ES) m/z 538,7 (M++1).
Пример 45: Получение соединения 45, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3,4-дифторфенил)-2-(оксетан-3-ил)-2,7-диазаспиро[3,5]нонан-7-карботиоамид

N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3,4-дифторфенил)-2,7-диазаспиро[3,5]нонан-7-карботиоамид (0,185 г, 0,353 ммоль), полученный тем же способом, как описан на стадии 2 соединения 44, 3-оксетанон (0,041 мл, 0,707 ммоль) и N, N-диизопропилэтиламин (0,123 мл, 0,707 ммоль) растворяли в дихлорметане (20 мл) при комнатной температуре, после чего полученный в результате раствор перемешивали при той же температуре в течение 18 часов. Воду приливали в реакционную смесь, и органический слой экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрировали, получая заявленное в заголовке соединение (0,035 г, 17,1%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3) δ 7,89~7,87 (м, 1H), 7,82~7,80 (м, 1H), 7,75~7,71 (м, 1H), 7,17~7,12 (м, 1H), 7,06 (с, 0,25H), 7,02~6,94 (м, 1H), 6,93 (с, 0,5H), 6,89~6,87 (м, 1H), 6,80 (с, 0,25H), 5,38 (с, 2H), 4,97~4,93 (м, 2H), 4,70~4,67 (м, 2H), 4,35~4,25 (м, 1H), 3,80~3,40 (м, 8H), 1,72~1,69 (м, 4H); LRMS (ES) m/z 580,9 (M++1).
Пример 46: Получение соединения 46, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3,4-дифторфенил)-7-метил-2,7-диазаспиро[3,5]нонан-2-карботиоамид
[Стадия 1] Получение трет-бутил 2-((4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)(3,4-дифторфенил)карбамотиоил)-2,7-диазаспиро[3,5]нонан-7-карбоксилата
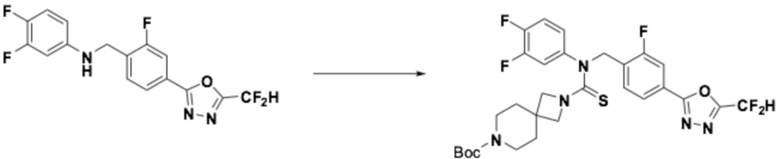
N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3,4-дифторанилин (1,000 г, 2,815 ммоль), полученный тем же способом, как описан на стадии 1 соединения 13, тиофосген (0,216 мл, 2,815 ммоль) и N, N-диизопропилэтиламин (1,716 мл, 9,852 ммоль) растворяли в дихлорметане (30 мл), после чего полученный в результате раствор перемешивали при 0°C в течение 30 минут, добавляли и дополнительно перемешивали при комнатной температуре в течение 18 часов. Воду приливали в реакционную смесь, и органический слой экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-30%) и концентрировали, получая заявленное в заголовке соединение (0,230 г, 13,1%) в виде желтого масла.
[Стадия 2] Получение N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3,4-дифторфенил)-2,7-диазаспиро[3,5]нонан-2-карботиоамида

Трет-бутил 2-((4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)(3,4-дифторфенил)карбамотиоил)-2,7-диазаспиро[3,5]нонан-7-карбоксилат (0,230 г, 0,369 ммоль), полученный на стадии 1 и трифторуксусная кислота (0,282 мл, 3,688 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре, после чего полученный в результате раствор перемешивали при той же температуре в течение 18 часов. Растворитель удаляли из реакционной смеси под пониженным давлением, после чего насыщенный водный раствор гидрокарбоната натрия выливали в полученный в результате концентрат, и органический слой экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный продукт применяли без дополнительной очистки (0,150 г, 77,7%, бесцветное масло).
[Стадия 3] Получение соединения 46
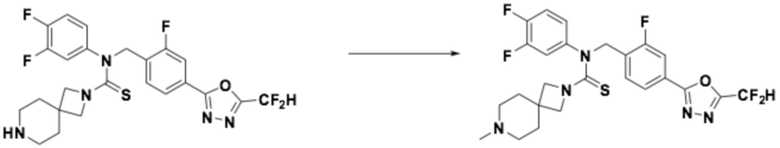
N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3,4-дифторфенил)-2,7-диазаспиро[3,5]нонан-2-карботиоамид (0,139 г, 0,266 ммоль), полученный на стадии 2, формальдегид (0,016 г, 0,531 ммоль) и N, N-диизопропилэтиламин (0,092 мл, 0,531 ммоль) растворяли в дихлорметане (20 мл) при комнатной температуре, после чего полученный в результате раствор перемешивали при той же температуре в течение 18 часов. Воду приливали в реакционную смесь, и органический слой экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали безводным сульфатом натрия, фильтровали, и концентрировали под пониженным давлением. Полученный в результате концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-10%) и концентрировали, получая заявленное в заголовке соединение (0,060 г, 42,0%) в виде черного масла.
1H ЯМР (400 МГц, CDCl3) δ 7,95~7,88 (м, 2H), 7,72 (дд, J=10,0, 1,4 Гц, 1H), 7,13 (дд, J=18,2, 8,8 Гц, 1H), 7,05 (с, 0,25H), 7,02~6,97 (м, 1H), 6,92 (с, 0,5H), 6,90~6,87 (м, 1H), 6,79 (с, 0,25H), 5,57 (с, 2H), 3,80~3,20 (м, 4H), 2,60~2,40 (м, 4H), 2,32 (с, 3H), 1,74 (т, J=5,4 Гц, 4H); LRMS (ES) m/z 538,7 (M++1).
Протокол измерения и анализа активности соединений настоящего изобретения
Экспериментальный пример 1. Подтверждение ингибирования активности ГДАЦ фермента (in vitro)
1. Экспериментальный способ
Способность ингибировать фермент ГДАЦ тестируемого материала измеряли с применением набора для флуориметрического анализа лекарственных средств ГДАЦ1 (Enzolifesciences: BML-AK511) и человеческого рекомбинантного препарата ГДАЦ6 (Calbiochem: 382180). Для анализа ГДАЦ1 образцы обрабатывали в концентрации 100, 1000 и 10000 нМ. Для анализа ГДАЦ6 образцы обрабатывали в концентрации 0,1 нМ, 1 нМ, 10 нМ, 100 нМ и 1000 нМ. После обработки образца реакцию продолжали при 37°С в течение 60 минут, обрабатывали проявителем и подвергали реакции при 37°С в течение 30 минут, после чего измеряли интенсивность флуоресценции (Ex 390 нм, Em 460 нм), применяя FlexStation3 (Molecular device). Для получения окончательных результатов каждое значение IC50 рассчитывали с помощью программы GraphPad Prism 4.0.
2. Экспериментлаьные результаты
Результаты поиска ингибирования активности фермента ГДАЦ, полученные экспериментальным способом, представлены в таблице 2.
Таблица 2
Как описано в таблице 2, результаты тестирования ингибирования активности ГДАЦ1 и ГДАЦ6 подтвердили, что тиокарбонильное соединение настоящего изобретения, его стереоизомеры или его фармацевтически приемлемые соли демонстрируют превосходную селективную ГДАЦ6 ингибирующую активность в отношении ГДАЦ1.
Изобретение относится к 1,3,4-оксадиазолтиокарбонильным соединениям формулы I, где каждый L1, L2 и L3 независимо представляет собой простую связь или -(C1-C4 алкилен)-; R1 представляет собой -(C6-C12 арил), в R1, по меньшей мере один H -(C6-C12 арила) может независимо быть замещен –T; R2 представляет собой структуру (а), где каждый a и b независимо представляют собой целое число 2 и Y1 представляет собой O или NRF, и Y5 представляет собой N, структуру (b), где a=b=c=d=1, когда Y2 представляет собой NRF и Y6 представляет собой N, или a=b=1 и c=d=2, когда Y6=CH и Y2=NRF, или структуру (с), где f=1 и Y7 представляет собой NRF; R3 представляет собой -CT2H; каждый Z1-Z4 независимо представляет собой CRZ; RZ представляет собой –H или Т; RF представляет собой -H, -(C1-C6 алкил), -C(=O)-(C1-C4 алкил), или -(C2-C6 гетероциклоалкил), содержащий 1 гетероатом О; фрагмент (d) представляет собой простую связь; и каждый T представляет собой F или Cl. Также изобретение относится к фармацевтической композиции на основе соединения формулы I или его фармацевтически приемлемых солей, способу ингибирования ГДАЦ6 путем введения соединения формулы I или его фармацевтически приемлемых солей и применению соединения формулы I или его фармацевтически приемлемых солей в получении лекарственного средства для ингибирования ГДАЦ6. Технический результат - получение соединений формулы I, обладающих ингибирующей активностью в отношении гистондеацетилазы 6 (ГДАЦ6). 5 н. и 5 з.п. ф-лы, 2 табл., 47 пр.
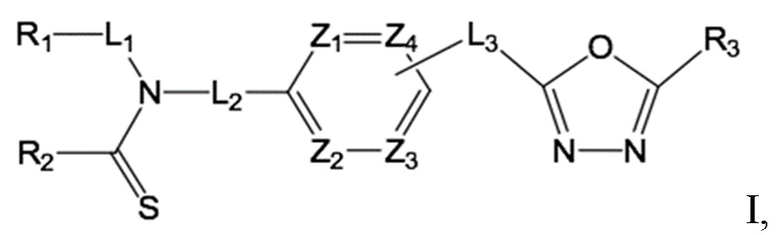
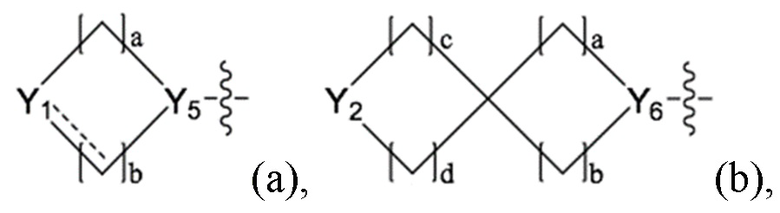
1. 1,3,4-Оксадиазолтиокарбонильное соединение, представленное формулой I, или его фармацевтически приемлемые соли:
<формула I>
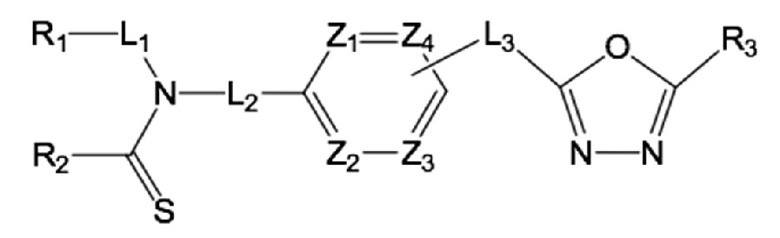
где
каждый L1, L2 и L3 независимо представляет собой простую связь или -(C1-C4 алкилен)-;
R1 представляет собой -(C6-C12 арил),
в R1,
по меньшей мере один H -(C6-C12 арила) может независимо быть замещен -T;
R2 представляет собой 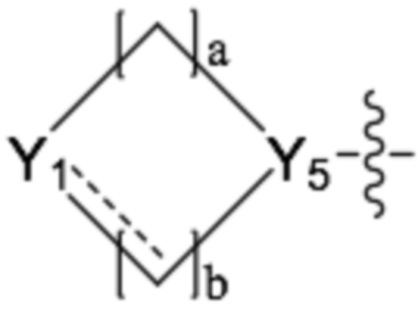 (каждый a и b независимо представляют собой целое число 2 и Y1 представляет собой O или NRF, и Y5 представляет собой N),
(каждый a и b независимо представляют собой целое число 2 и Y1 представляет собой O или NRF, и Y5 представляет собой N),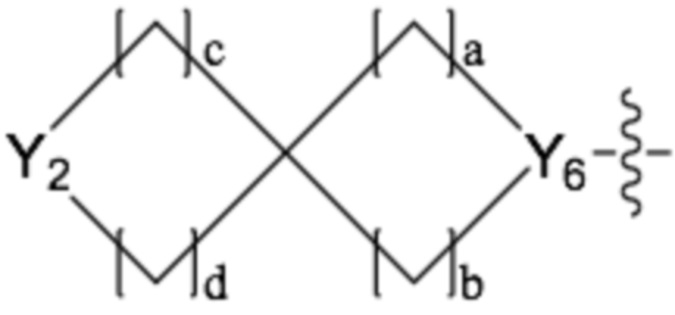 (a=b=c=d=1, когда Y2 представляет собой NRF и Y6 представляет собой N, или a=b=1 и c=d=2, когда Y6=CH и Y2=NRF) или
(a=b=c=d=1, когда Y2 представляет собой NRF и Y6 представляет собой N, или a=b=1 и c=d=2, когда Y6=CH и Y2=NRF) или  (f=1 и Y7 представляет собой NRF),
(f=1 и Y7 представляет собой NRF),
R3 представляет собой -CT2H;
каждый Z1-Z4 независимо представляет собой CRZ;
RZ представляет собой -H или Т;
RF представляет собой -H, -(C1-C6 алкил), -C(=O)-(C1-C4 алкил), или -(C2-C6 гетероциклоалкил), содержащий 1 гетероатом О,
 представляет собой простую связь; и
представляет собой простую связь; и
каждый T представляет собой F или Cl.
2. 1,3,4-Оксадиазолтиокарбонильное соединение, представленное формулой I, или его фармацевтически приемлемые соли по п. 1,
где в формуле I,
каждый L1, L2 и L3 независимо представляет собой простую связь или -(C1-C2 алкилен)-;
R1 представляет собой -С6 арил,
в R1,
по меньшей мере один H -С6 арила может независимо быть замещен -T;
R2 представляет собой (каждый a и b независимо представляют собой целое число 2 и Y1 представляет собой O или NRF, и Y5 представляет собой N), (a=b=c=d=1, когда Y2 представляет собой NRF и Y6 представляет собой N, или a=b=1 и c=d=2, когда Y6=CH и Y2=NRF) или (f=1 и Y7 представляет собой NRF);
R3 представляет собой -CT2H;
каждый Z1-Z4 независимо представляет собой CRZ,
Rz представляет собой -H или-T;
RF представляет собой -H, -(C1-C6 алкил), -C(=O)-(C1-C4 алкил) или -(C4 гетероциклоалкил), содержащий 1 гетероатом О;
представляет собой простую связь; и
каждый T представляет собой F или Cl.
3. 1,3,4-Оксадиазолтиокарбонильное соединение, представленное формулой I, или его фармацевтически приемлемые соли по п. 1, где соединение, представленное формулой I, представляет собой любое соединение, выбранное из следующих соединений:
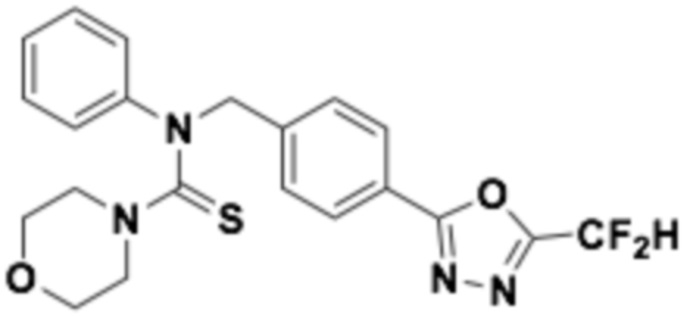
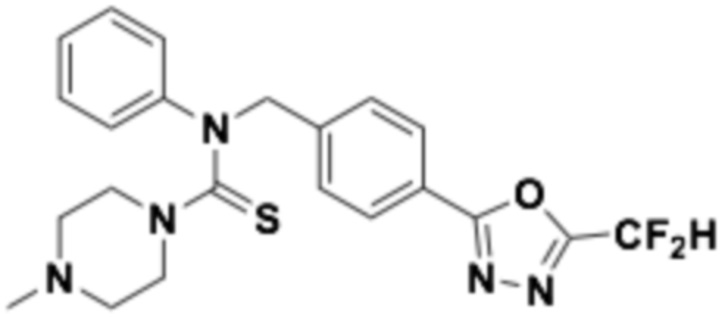
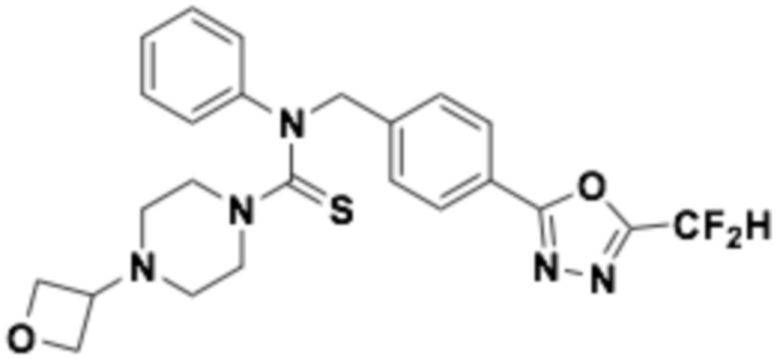
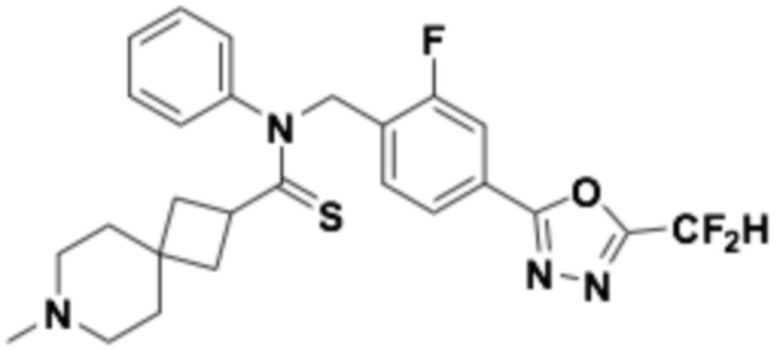
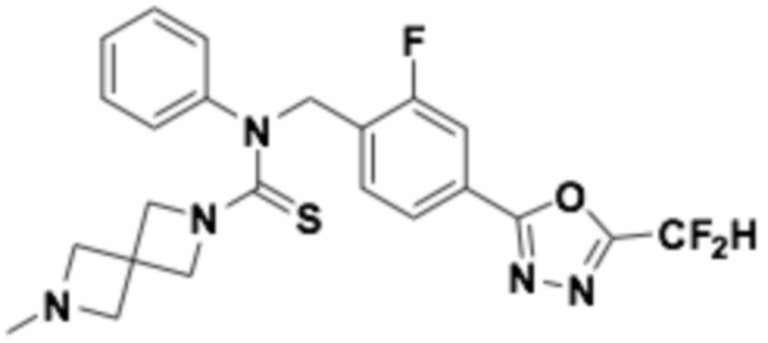
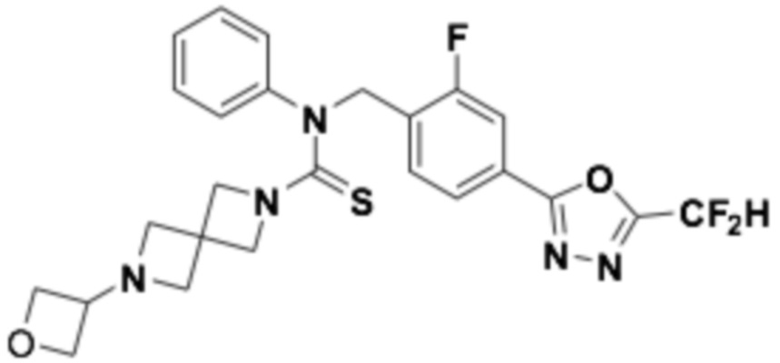
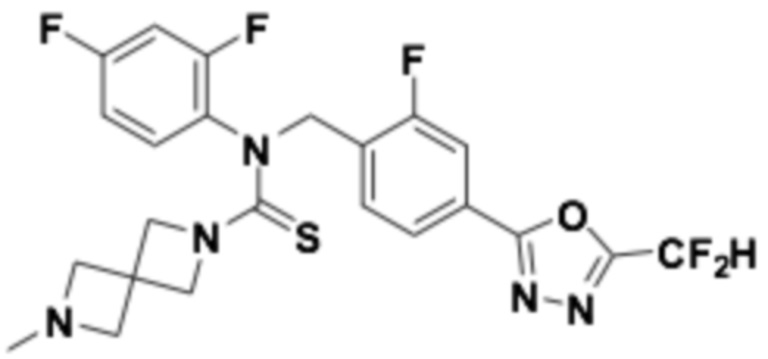
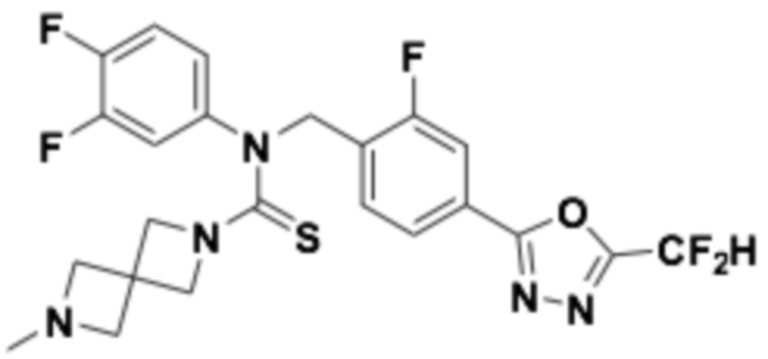
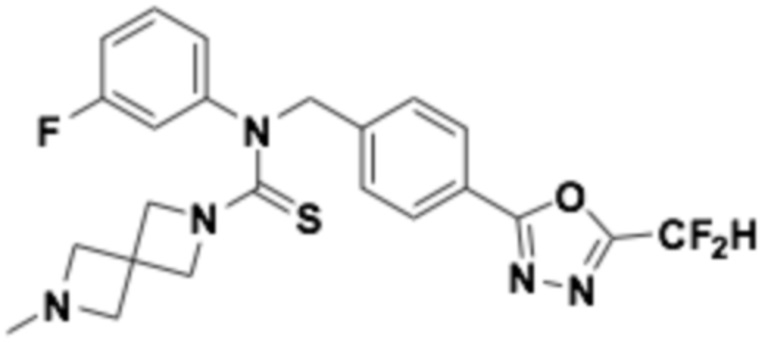
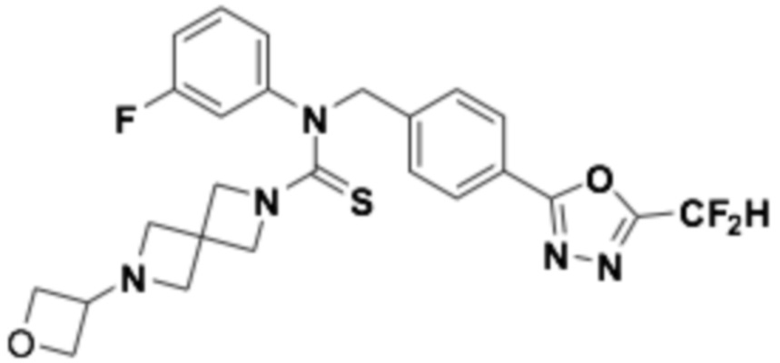
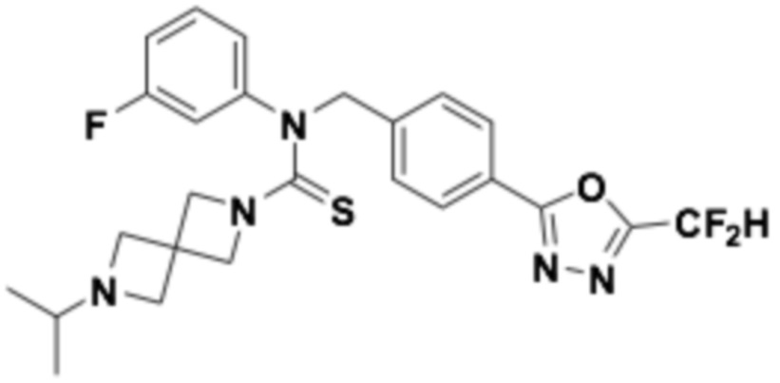
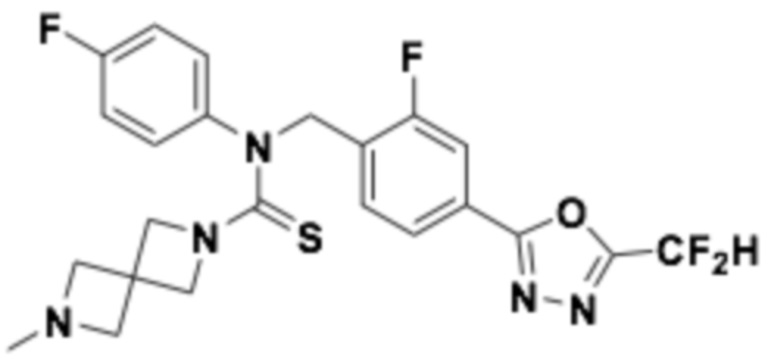
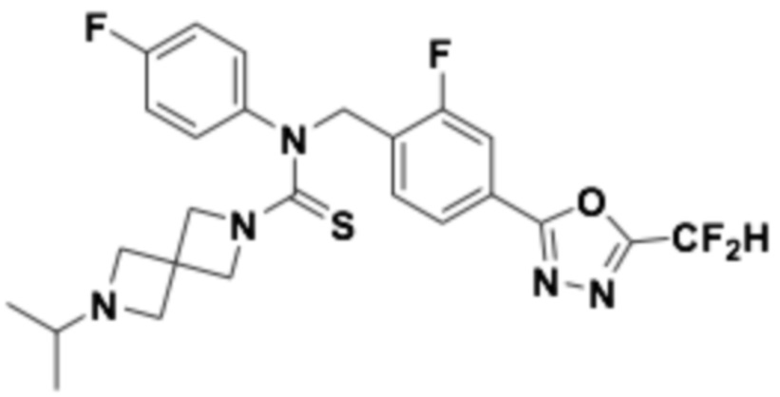
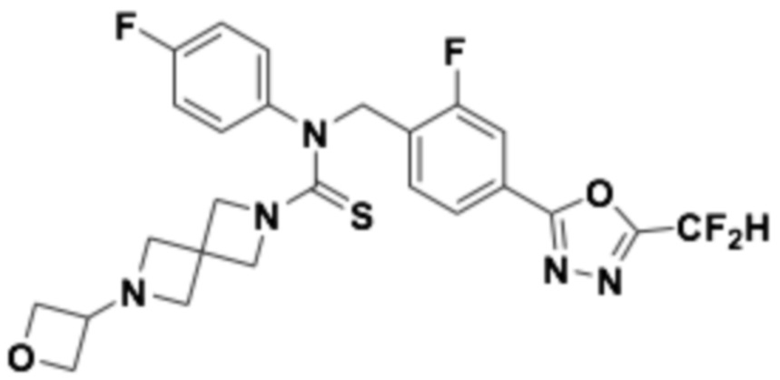
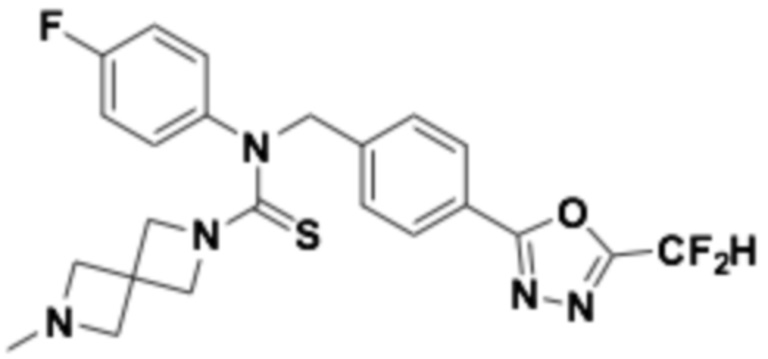
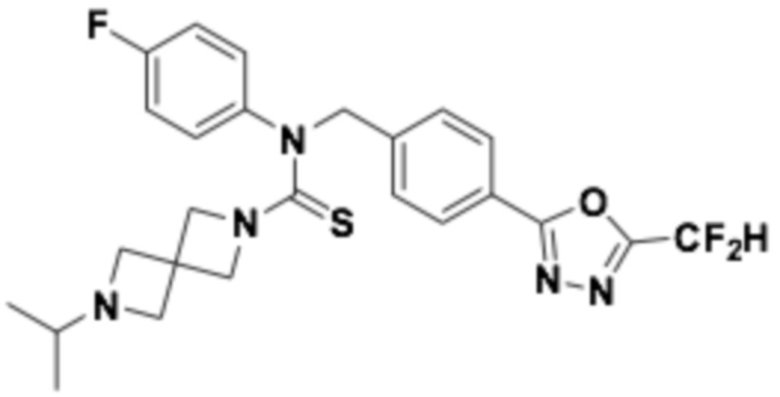
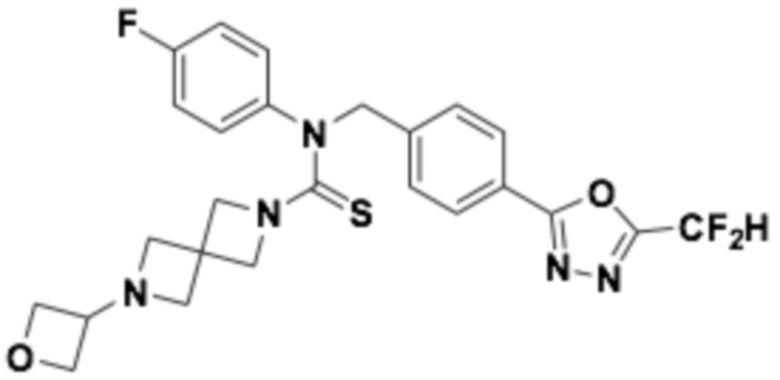
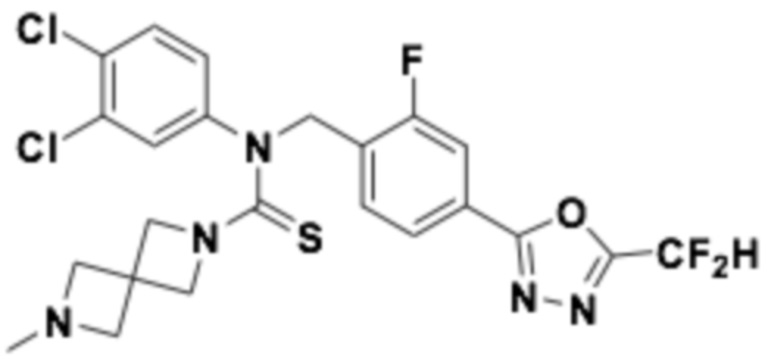
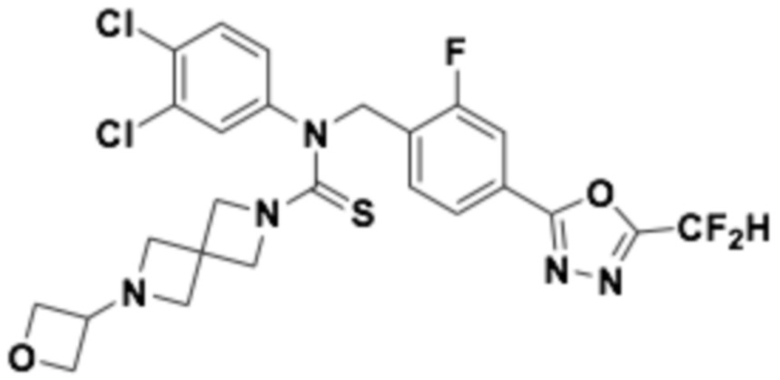
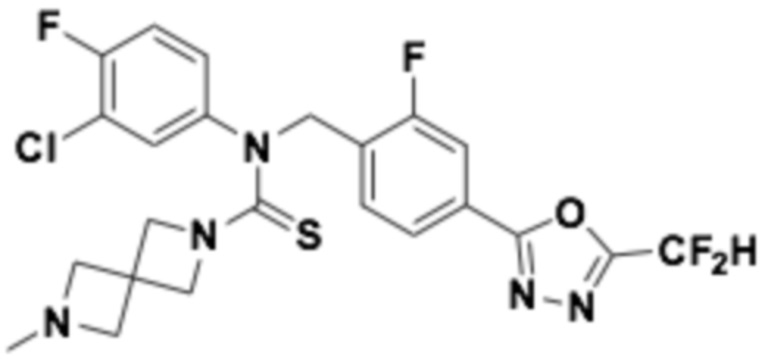
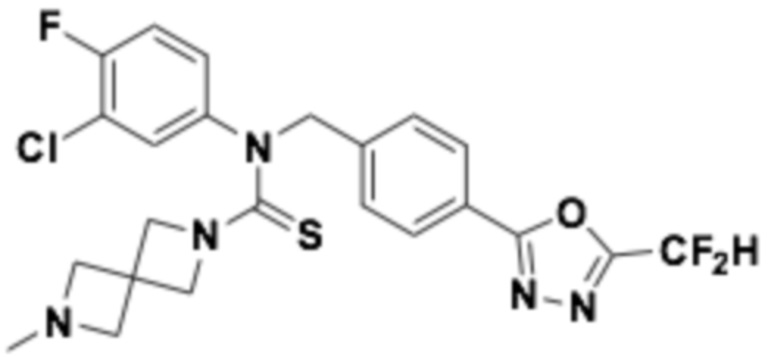
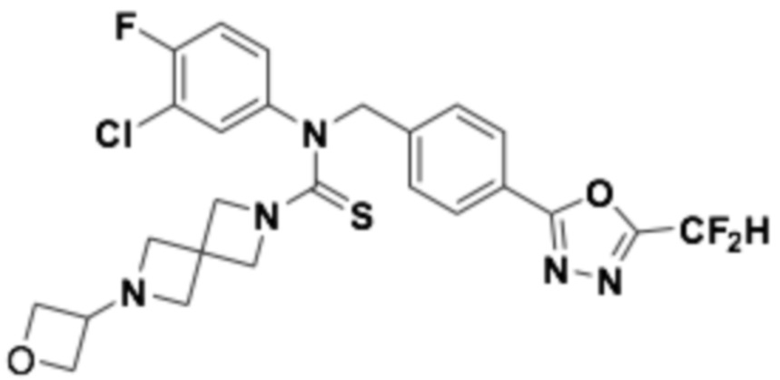
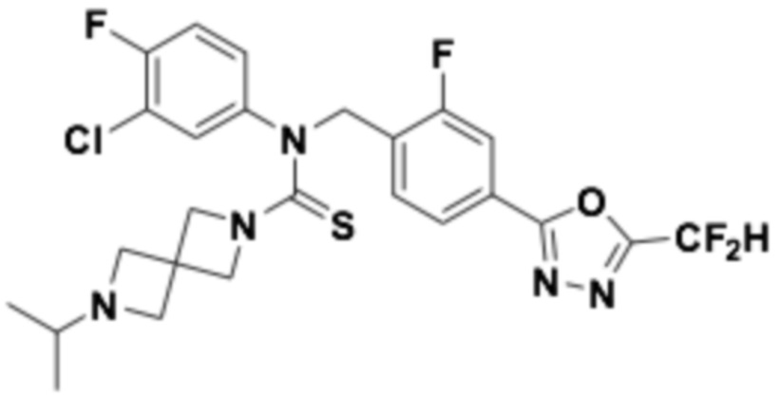
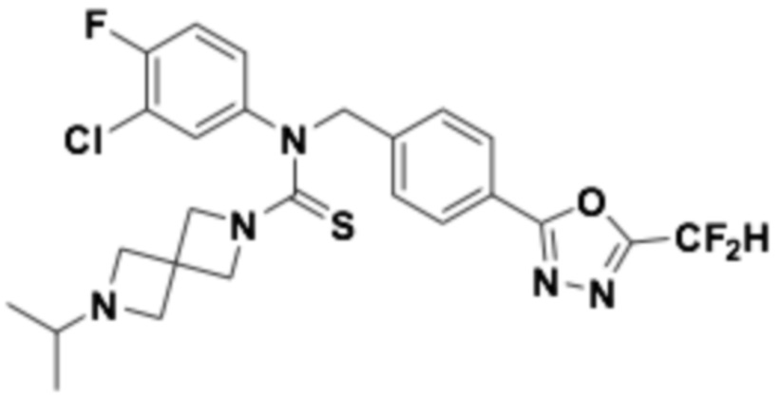
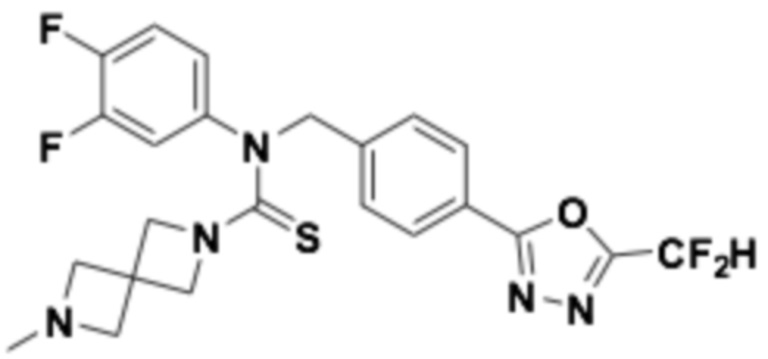
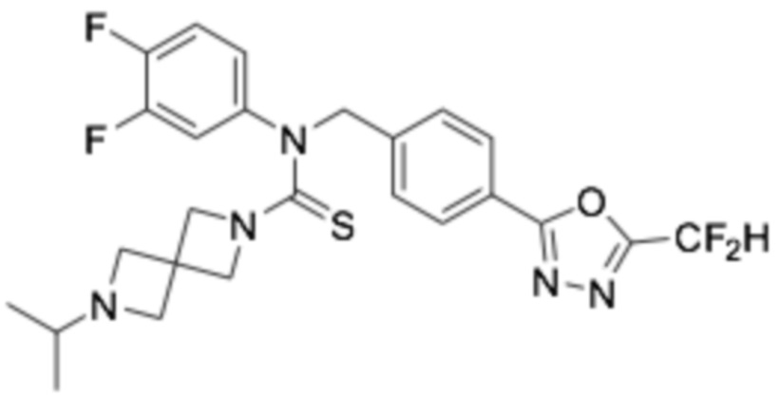
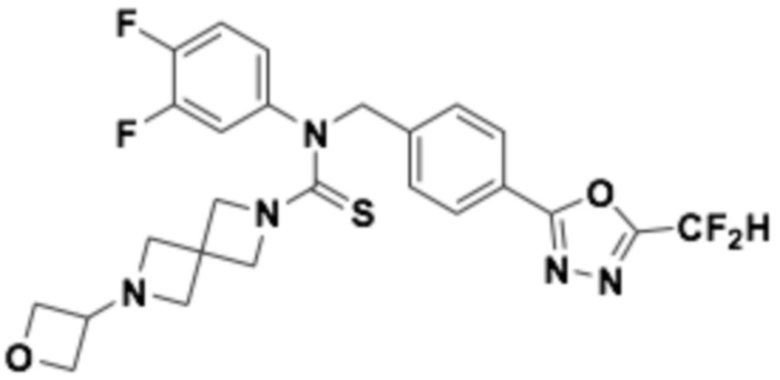
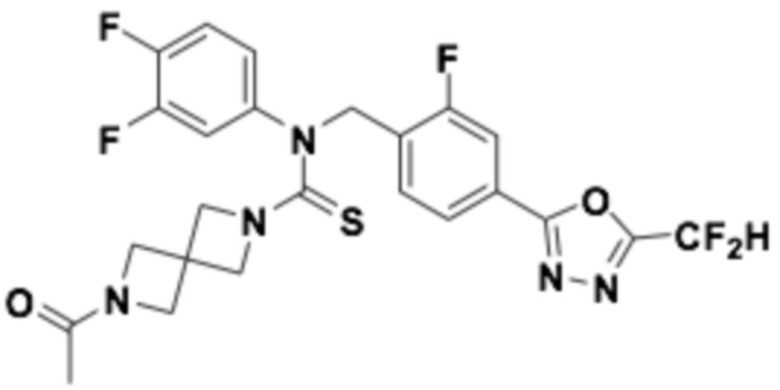
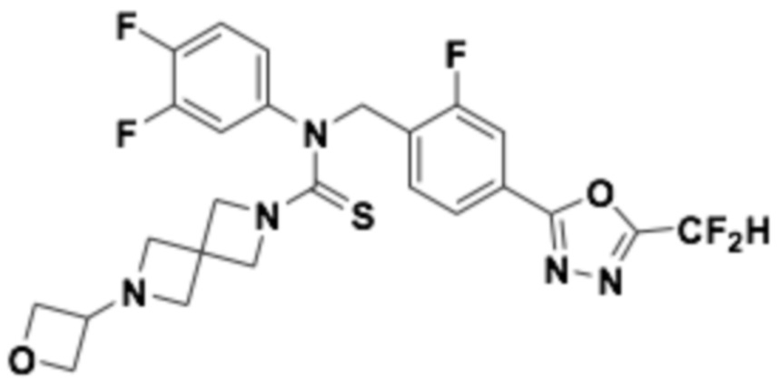
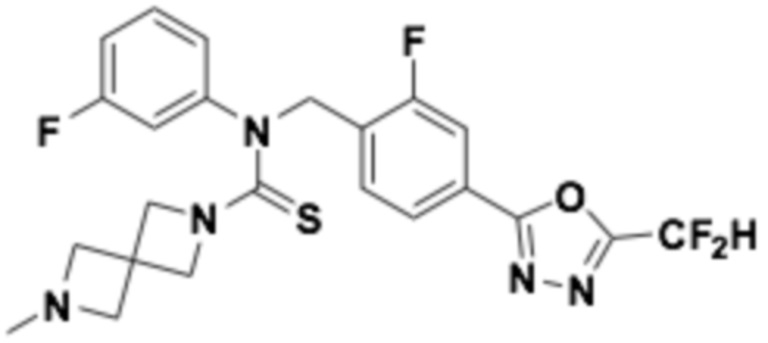
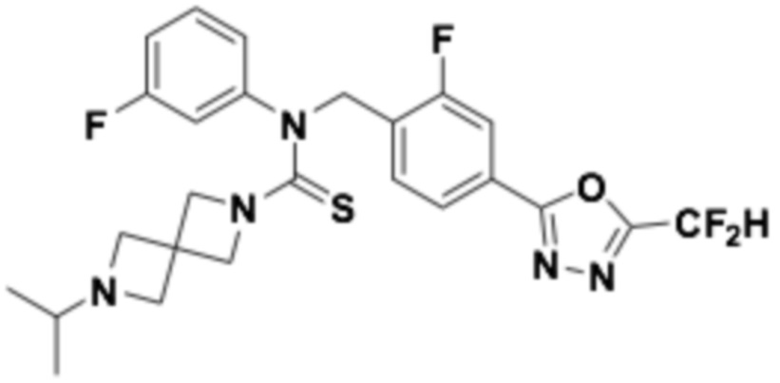
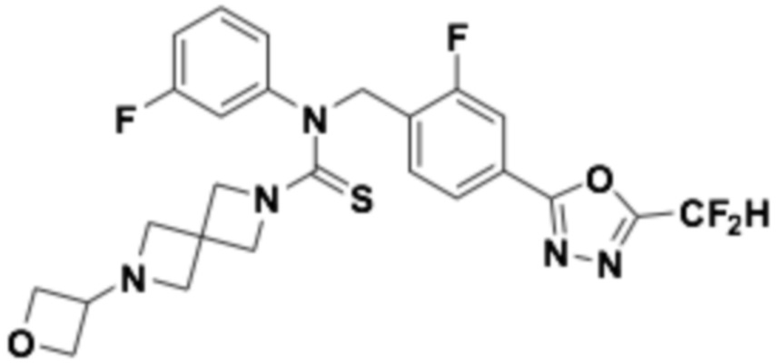
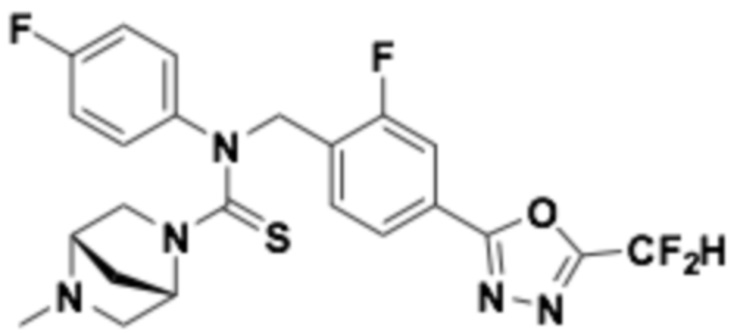
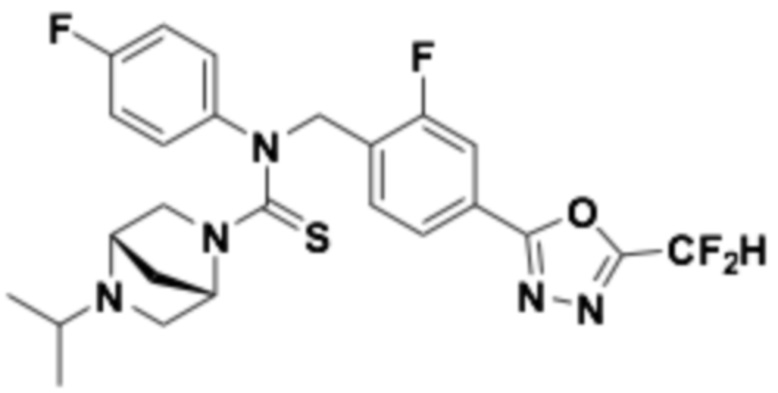
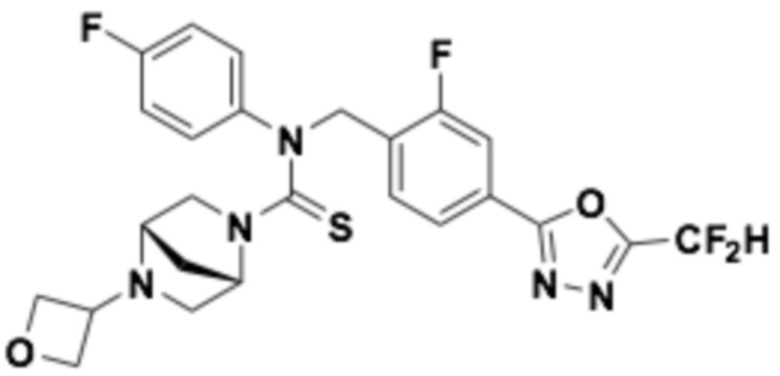
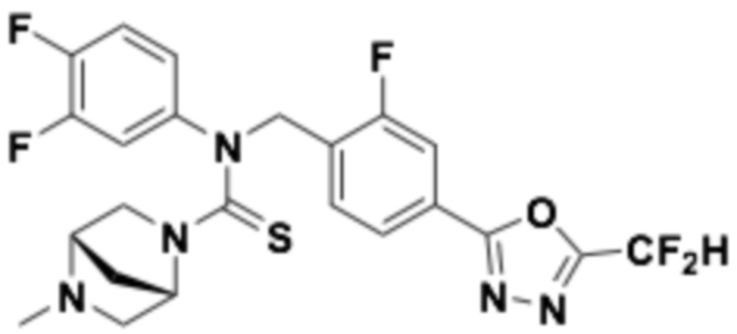
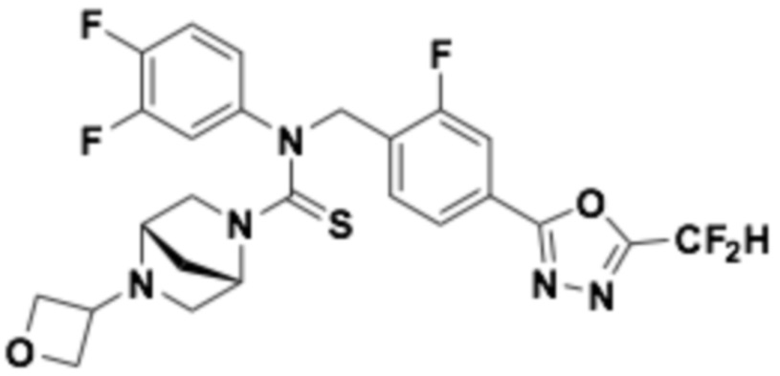
4. Фармацевтическая композиция для ингибирования гистондеацетилазы 6 (ГДАЦ6), содержащая терапевтически эффективное количество 1,3,4-оксадиазолтиокарбонильного соединения по любому из пп. 1-3 или его фармацевтически приемлемых солей в качестве активного ингредиента, и фармацевтически приемлемый носитель.
5. Фармацевтическая композиция по п. 4, где фармацевтическая композиция применяется для профилактики или лечения заболеваний, опосредованных гистондеацетилазой 6 (ГДАЦ6).
6. Фармацевтическая композиция по п. 5, где заболевания, опосредованные гистондеацетилазой 6 (ГДАЦ6), представляют собой инфекционные заболевания; новообразование; эндокринопатию, алиментарные и метаболические заболевания; психические и поведенческие расстройства; неврологические заболевания; заболевания глаз и глазных придатков; респираторные заболевания; проблемы с пищеварением; заболевания кожи и подкожной клетчатки; заболевания опорно-двигательного аппарата и соединительной ткани; или тератоз, деформации и хромосомные аберрации.
7. Фармацевтическая композиция по п. 6, где
инфекционные заболевания представляют собой прионное заболевание;
новообразование представляет собой доброкачественную опухоль или злокачественную опухоль;
эндокринопатия, алиментарные и метаболические заболевания представляют собой болезнь Вильсона, амилоидоз или диабет;
психические и поведенческие расстройства представляют собой депрессию или синдром Ретта;
неврологические заболевания представляют собой атрофию центральной нервной системы, нейродегенеративное заболевание, двигательное расстройство, невропатию, заболевание двигательных нейронов или демиелинизирующее заболевание центральной нервной системы;
заболевания глаз и глазных придатков представляют собой увеиты;
заболевания кожи и подкожной клетчатки представляют собой псориаз;
респираторные заболевания представляют собой астму;
расстройства пищеварения представляют собой алкогольные заболевания печени, воспалительные заболевания кишечника, болезнь Крона или язвенную болезнь кишечника;
заболевания опорно-двигательного аппарата и соединительной ткани представляют собой ревматоидный артрит, остеоартрит или системную красную волчанку;
и тератоз, деформации и хромосомные аберрации представляют собой аутосомно-доминантный поликистоз почек.
8. Способ ингибирования гистондеацетилазы 6 (ГДАЦ6), причем способ включает введение терапевтически эффективного количества 1,3,4-оксадиазолтиокарбонильного соединения по любому из пп. 1-3 или его фармацевтически приемлемых солей.
9. Применение 1,3,4-оксадиазолтиокарбонильного соединения по любому из пп. 1-3 или его фармацевтически приемлемых солей для ингибирования гистондеацетилазы 6 (ГДАЦ6).
10. Применение 1,3,4-оксадиазолтиокарбонильного соединения по любому из пп. 1-3 или его фармацевтически приемлемых солей в получении лекарственного средства для ингибирования гистондеацетилазы 6 (ГДАЦ6), причем лекарственное средство находится в виде пластыря, жидкого лекарственного средства, пилюли, капсулы, гранулы, таблетки, суппозитория.
| EP 3330259 A1, 06.06.2018 | |||
| WO 2017023133 A2, 09.02.2017 | |||
| US 20190185462 A1, 20.06.2019 | |||
| ЗЕФИРОВА О.Н | |||
| и др | |||
| Об истории возникновения и развития концепции биоизостеризма | |||
| Вестник Московского Университета, 2002, 43(4), с.251-256 | |||
| HEIMANN, D | |||
| et al | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |

