УРОВЕНЬ ТЕХНИКИ
Процессы, вовлеченные в прогрессирование роста и матастазирование опухоли, обусловлены сигнальным путями, которые активированы в раковых клетках. Ключевую роль в регуляции клеточного роста у млекопитающих играет сигнальный путь ERK (киназ, регулируемых внеклеточными сигналами), через который передаются внеклеточные сигналы от связанных с лигандами трансмембранных рецепторов с тирозинкиназной активностью (RTK), таких как семейство рецепторов с тирозинкиназной активностью ErbB, рецепторы с тирозинкиназной активностью PDGF (тромбоцитарных факторов роста), FGF (факторов роста фибробластов) и VEGF (факторов роста сосудистого эндотелия). Активация RTK индуцирует каскад процессов фосфорилирования, который начинается с активации Ras. Активация Ras ведет к рекрутингу и активации Raf, серин-треониновой киназы. Активированная Raf затем фосфорилирует и активирует MEK1/2 (киназу митоген-активируемых протеинкиназ), которая затем фосфорилирует и активирует ERK1/2. Активированная ERK1/2 фосфорилирует несколько следующих мишеней, вовлеченных в множество клеточных процессов, включая изменения цитоскелета и активацию транскрипции. Сигнальный путь ERK/MAPK (митоген-активируемая протеинкиназа) является одним из наиболее важных для пролиферации клеток, и считается, что сигнальный путь ERK/MAPK часто активирован во многих опухолях. Гены Ras, расположенные на хромосоме выше (upstream) генов ERK1/2, мутированы в клетках некоторых раковых опухолей, включая колоректальную опухоль, меланому, опухоль молочной железы и поджелудочной железы. Высокая активность Ras сопровождается повышенной активностью ERK во многих опухолях человека. Кроме того, мутации в BRAF, серин-треониновой киназе семейства Raf, ассоциированы с повышенной киназной активностью. Мутации в BRAF идентифицированы в меланомах (60%), в клетках рака щитовидной железы (более 40%) и клетках колоректального рака. Данные наблюдения показывают, что сигнальный путь ERK1/2 является перспективным для противораковой терапии в отношении широкого спектра опухолей человека (М. Hohno and J. Pouyssegur, Prog, in Cell Cycle Res. 2003 5:219).
Сигнальный путь ERK также представляет интерес в качестве терапевтической мишени для лечения боли и воспаления (Ma, Weiya and Remi, Quirion. "The ERK/MAPK Pathway, as a Target For The Treatment Of Neuropathic Pain" Expert Opin. Ther. Targets. 2005 9 (4): 699-713 и Sommer, Claudia and Frank Birklein "Resolvins and Inflammatory Pain" F1000 Medicine Reports 2011 3:19).
Поэтому низкомолекулярные ингибиторы активности ERK (а именно активности ERK1 и/или ERK2) могут быть использованы для лечения многих типов рака, таких как, например, меланома, рак поджелудочной железы, рак щитовидной железы, колоректальный рак, рак легкого, рак молочной железы и рак яичника, а также для лечения боли и воспаления, таких как артрит, боль внизу спины, воспалительное заболевание кишечника и ревматизм. В настоящем изобретении предложен способ и промежуточные соединения для получения (S)-1-(1-(4-хлор-3 -фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-она, фармацевтически приемлемых солей данного соединения и кристаллических форм этих солей. В настоящем изобретении также предложены фармацевтические композиции, содержащие указанные соли или кристаллические формы этих солей, и способы применения указанных солей и кристаллических форм этих солей. Синтез (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1//)-она описан в WO 2013/130976.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
В настоящем изобретении предложены способы получения соединения I, представляющего собой полезное промежуточное соединение, которое может быть использовано для синтеза соединения VIII (WO 2013/130976). Соединение VIII является ингибитором ERK и может быть использовано в качестве лекарства для лечения гиперпролиферативных расстройств. Предложенный способ представляет собой эффективный путь получения соединения VIII и полезных промежуточных соединений VI и VII. Алкилирование соединения VII с использованием соединения VI приводит к получению соединения I, которое затем конденсируют с 1-метил-1H-пиразол-5-амином (XIV) (СХЕМА А).
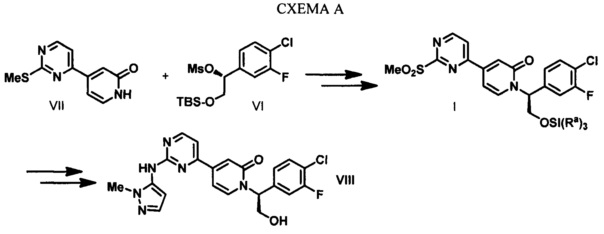
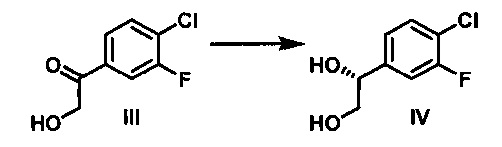
В настоящем изобретении дополнительно предложено асимметрическое ферментативное восстановление, которое позволяет осуществлять стереоспецифическое восстановление 1-(4-хлор-3-фторфенил)-2-гидроксиэтанона с получением (R)-1-(4-хлор-3-фторфенил)этан-1,2-диола (IV).
В настоящем изобретении также предложен улучшенный способ получения 4-(2-(метилтио)пиримидин-4-ил)пиридин-2(1H)-она (VII).
В настоящем изобретении предложена кристаллическая безилатная соль (VIIIb), обладающая желательными физическими свойствами, которые позволяют использовать эту соль в качестве препарата и обеспечивают хорошую биодоступность.
Согласно варианту осуществления 1, в настоящем изобретении предложены способы получения соединения Формулы VIII, включающие следующие стадии:

(а) контактирование 4-бром-1-хлор-2-фторбензола с металлирующим агентом в апротонном органическом растворителе с образованием магнийорганического соединения, которое подвергают взаимодействию с 2-хлор-N-метокси-N-метилацетамидом с получением 2-хлор-1-(4-хлор-3-фторфенил)этанона (II);
(b) контактирование соединения II с формиатом натрия и муравьиной кислотой в водном этаноле с образованием 1-(4-хлор-3-фторфенил)-2-гидроксиэтанона (III);
(c) контактирование соединения III с кеторедуктазой с образованием (R)-1-(4-хлор-3-фторфенил)этан-1,2-диола (IV);
(d) контактирование соединения IV с силилхлоридом (Ra)3SiCl и по меньшей мере с одним основанием в неполярном апротонном растворителе с образованием соединения V и последующее добавление сульфонилхлорида RbS(O)2Cl с получением соединения VI, где Ra в каждом случае независимо представляет собой С1-6 алкил или фенил и Rb выбран из См алкила или фенила, необязательно имеющих в качестве заместителей от 1 до 3 групп, независимо выбранных из C1-3 алкила, атома галогена, нитро, циано или C1-3 алкокси;
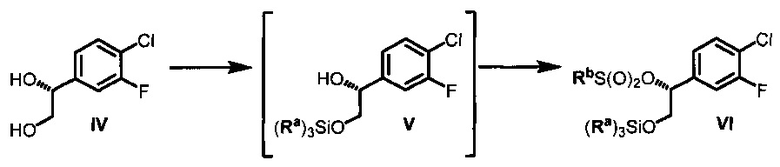
(e) контактирование 4-(2-(метилсульфонил)пиримидин-4-ил)пиридин-2(1Н)-она (VII) с сильным основанием в органическом растворителе и последующее добавление соединения VI с получением соединения XI;

(f) обработку соединения XI окисляющим агентом с получением соединения I;
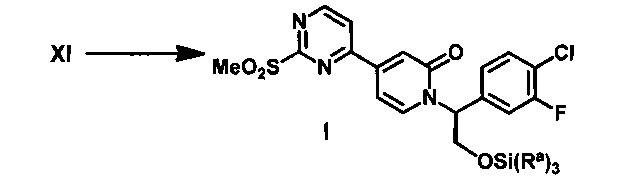
(g) обработку 1-метил-1H-пиразол-5-амина сильным основанием в апротонном растворителе при пониженной температуре и добавление соединения Формулы I с получением соединения IX; и

(h) контактирование соединения IX с десилилирующим агентом с образованием соединения VIII.
Согласно варианту осуществления 2, в настоящем изобретении предложены способы по варианту осуществления 1, отличающиеся тем, что кеторедуктаза на стадии (с) приводит к получению энантиомерного избытка по меньшей мере приблизительно 98%.
Согласно варианту осуществления 3, в настоящем изобретении предложены способы по варианту осуществления 2, отличающиеся тем, что кеторедуктаза на стадии (с) представляет собой KRED-NADH-112.
Согласно варианту осуществления 4, в настоящем изобретении предложены способы по варианту осуществления 2, отличающиеся тем, что стадия (с) дополнительно включает НАД(Н) (никотинамидадениндинуклеотид, восстановленный) или НАДФ(Н) (никотинамидадениндинуклеотидфосфат, восстановленный) в качестве кофактора.
Согласно варианту осуществления 5, в настоящем изобретении предложены способы по варианту осуществления 4, отличающиеся тем, что указанный кофактор регенерируют с использованием косубстрата, выбранного из вторичного спирта или из дополнительного фермента, выбранного из алкогольдегидрогеназы, глюкозодегидрогеназы, формиатдегидрогеназы, глюкозо-6-фосфат-дегидрогеназы, фосфитдегидрогеназы или гидрогеназы.
Согласно варианту осуществления 6, в настоящем изобретении предложены способы по любому из вариантов осуществления 2-5, отличающиеся тем, что стадию с участием кеторедуктазы выполняют в водной среде в присутствии органического сорастворителя при температуре от 1 до 50°С.
Согласно варианту осуществления 7, в настоящем изобретении предложены способы по варианту осуществления 6, отличающиеся тем, что стадия с участием кеторедуктазы приводит к получению гомогенной суспензии.
Согласно варианту осуществления 8, в настоящем изобретении предложены способы по варианту осуществления 1, отличающиеся тем, что силилхлорид представляет собой трет-бутил-хлор-диметилсилан, сульфонилхлорид представляет собой метансульфохлорид, основания, используемые на стадии (d), представляют собой DMAP и TEA, и неполярный апротонный растворитель представляет собой ДХМ, и органический растворитель, используемый на стадии (е), представляет собой диоксан.
Согласно варианту осуществления 9, в настоящем изобретении предложены способы по варианту осуществления 1, отличающиеся тем, что (Ra)3Si представляет собой трет-бутилдиметилсилил, Rb представляет собой метил и на стадии (е) сильное основание представляет собой гексаметилдисилазан калия и органический растворитель представляет собой диметиловый эфир диэтиленгликоля.
Согласно варианту осуществления 10, в настоящем изобретении предложены способы по варианту осуществления 1, отличающиеся тем, что на стадии (а) металлирующий агент представляет собой изо-PrMgCl и LiCl и растворитель представляет собой ТГФ, на стадии (с) кеторедуктаза представляет собой KRED-NADH-112, и стадия (с) дополнительно включает кофактор НАД и глюкозодегидрогеназу в качестве агента, регенерирующего данный кофактор, на стадии (d) (Ra)3Si представляет собой трет-бутилдиметилсилил, Rb представляет собой метил, основания представляют собой DMAP и TEA и неполярный апротонный растворитель представляет собой ДХМ, и на стадии (е) сильное основание представляет собой гексаметилдисилазан калия и органический растворитель представляет собой диметиловый эфир диэтиленгликоля.
Согласно варианту осуществления 11, в настоящем изобретении предложены способы по варианту осуществления 1, отличающиеся тем, что на стадии (а) металлирующий агент представляет собой изо-PrMgCl и LiCl и растворитель представляет собой ТГФ, на стадии (с) кеторедуктаза представляет собой KRED-NADH-112, и стадия (с) дополнительно включает кофактор НАД, и агент, регенерирующий данный кофактор, представляет собой глюкозодегидрогеназу, на стадии (d) (Ra)3Si представляет собой трет-бутилдиметилсилил, Rb представляет собой метил, основания представляют собой DMAP и TEA и неполярный апротонный растворитель представляет собой ДХМ, на стадии (е) сильное основание представляет собой гексаметилдисилазан калия и органический растворитель представляет собой диметиловый эфир диэтиленгликоля, и на стадии (g) сильное основание представляет собой гексаметилдисилазан калия и апротонный растворитель представляет собой ТГФ.
Согласно варианту осуществления 12, в настоящем изобретении предложены способы по варианту осуществления 1, отличающиеся тем, что на стадии (а) металлирующий агент представляет собой изо-PrMgCl и LiCl и растворитель представляет собой ТГФ, на стадии (с) кеторедуктаза представляет собой KRED-NADH-112, и стадия (с) дополнительно включает кофактор НАД, и агент, регенерирующий данный кофактор, представляет собой глюкозодегидрогеназу, на стадии (d) (Ra)3Si представляет собой трет-бутилдиметилсилил, Rb представляет собой метил, основания представляют собой DMAP и TEA и неполярный апротонный растворитель представляет собой ДХМ, на стадии (е) сильное основание представляет собой гексаметилдисилазан калия и органический растворитель представляет собой диметиловый эфир диэтиленгликоля, на стадии (g) сильное основание представляет собой гексаметилдисилазан калия и апротонный растворитель представляет собой ТГФ, и на стадии (h) десилилирующий агент представляет собой раствор HCl в метаноле.
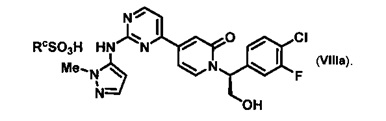
Согласно варианту осуществления 13, в настоящем изобретении предложены способы по варианту осуществления 1, отличающиеся тем, что соединение VIII, полученное на стадии h, контактирует с сульфоновой кислотой в органическом растворителе и воде с образованием соли VIIIa, где Rc представляет собой арилсульфоновую кислоту
 .
.
Согласно варианту осуществления 14, в настоящем изобретении предложены способы по варианту осуществления 13, отличающиеся тем, что RcSO3H представляет собой бензолсульфоновую кислоту и растворитель представляет собой метилэтилкетон и воду, что приводит к образованию желаемой безилатной соли VIIIb.
Согласно варианту осуществления 15, в настоящем изобретении предложены способы получения 4-(2-(метилсульфонил)пиримидин-4-ил)пиридин-2(1H)-она (VII), включающие следующие стадии:
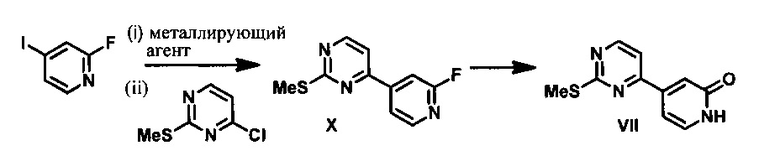
(a) контактирование 2-фтор-4-иодпиридина с металлирующим агентом в апротонном органическом растворителе с образованием магнийорганического соединения, которое подвергают взаимодействию с 4-хлор-2(метилтио)пиримидином в присутствии палладиевого катализатора с получением 4-(2-фторпиридин-4-ил)-2-(метилтио)пиримидина (X);
(b) обработку соединения X трет-бутилатом калия в ТГФ и затем водной кислотой с получением 4-(2-(метилсульфонил)пиримидин-4-ил)пиридин-2(1H)-она (VII).
Согласно варианту осуществления 16, в настоящем изобретении предложены способы по варианту осуществления 15, отличающиеся тем, что палладиевый катализатор представляет собой (1,3-диизопропилимидазол-2-илиден)(3-хлорпиридил)палладия (II) дихлорид, металлирующий агент представляет собой изо-PrMgCl и LiCl и апротонный растворитель представляет собой ТГФ.
Согласно варианту осуществления 17, в настоящем изобретении предложено соединение (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-она бензолсульфонат.
Согласно варианту осуществления 18, в настоящем изобретении предложены фармацевтические композиции, содержащие (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(H)-она бензолсульфонат и фармацевтически приемлемый эксципиент.
Согласно варианту осуществления 19, в настоящем изобретении предложен кристаллический (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-она бензолсульфонат.
Согласно варианту осуществления 20, в настоящем изобретении предложен кристаллический (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-она бензолсульфонат, который имеет порошковую рентгеновскую дифрактограмму, содержащую пики в положениях 6,16±0,2, 7,46±0,2, 16,36±0,2, 25,76±0,2 и 25,98±0,2 2θ
Согласно варианту осуществления 21, в настоящем изобретении предложен кристаллический (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-она бензолсульфонат, который имеет порошковую рентгеновскую дифрактограмму, по существу соответствующую дифрактограмме, приведенной на Фигуре 1.
Согласно варианту осуществления 22, в настоящем изобретении предложен кристаллический (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-она бензолсульфонат, который имеет 13С-ЯМР-спектр, по существу соответствующий 13С-ЯМР-спектру, приведенному на Фигуре 19.
Согласно варианту осуществления 23, в настоящем изобретении предложен кристаллический (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-она бензолсульфонат, который имеет 19F-ЯМР-спектр, по существу соответствующий 19F-ЯМР-спектру, приведенному на Фигуре 20.
Согласно варианту осуществления 24, в настоящем изобретении предложен кристаллический (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-она бензолсульфонат, который имеет 13С-ЯМР-спектр, по существу соответствующий 13С-ЯМР-спектру, приведенному на Фигуре 19, и 19Р-ЯМР-спектр, по существу соответствующий 19Р-ЯМР-спектру, приведенному на Фигуре 20.
Согласно варианту осуществления 25, в настоящем изобретении предложен кристаллический (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-она бензолсульфонат, который имеет 19F-ЯМР-спектр, содержащий пики в положениях -111,1±0,4 м.д. и -115,4±0,4 м.д. относительно CFCl3 (при 293°K).
Согласно варианту осуществления 26, в настоящем изобретении предложен кристаллический (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5 ил)амино)пиримидин-4-ил)пиридин-2(1H)-она бензолсульфонат, который имеет 13С-ЯМР-спектр, содержащий пики в положениях 157,7±0,2 м.д., 129,6±0,2 м.д., 125,8±0,2 м.д. и 117,0±0.2 м.д. относительно тетраметилсилана (при 293°K).
Согласно варианту осуществления 27, в настоящем изобретении предложен кристаллический (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-она бензолсульфонат, который имеет DSC-термограмму, по существу соответствующую DSC-термограмме, приведенной на Фигуре 2.
Согласно варианту осуществления 28, в настоящем изобретении предложены фармацевтические композиции, содержащие кристаллический (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-она бензолсульфонат по любому из вариантов осуществления 19-27 и фармацевтически приемлемый эксципиент.
Согласно варианту осуществления 29, в настоящем изобретении предложен (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-он в форме соли пара-толуолсульфоновой кислоты.
Согласно варианту осуществления 30, в настоящем изобретении предложены фармацевтические композиции, содержащие соль (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-она с пара-толуолсульфоновой кислотой и фармацевтически приемлемый эксципиент.
Согласно варианту осуществления 31, в настоящем изобретении предложена кристаллическая соль (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-она с пара-толуолсульфоновой кислотой.
Согласно варианту осуществления 32, в настоящем изобретении предложена кристаллическая Форма А соли (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-она с пара-толуолсульфоновой кислотой.
Согласно варианту осуществления 33, в настоящем изобретении предложена кристаллическая Форма А соли (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-она с пара-толуолсульфоновой кислотой, имеющая порошковую рентгеновскую дифрактограмму, содержащую пики в положениях 5,76±0,2, 13,44±0,2, 15,64±0,2, 19,40±0,2 2θ.
Согласно варианту осуществления 34, в настоящем изобретении предложена кристаллическая Форма А соли (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-она с пара-толуолсульфоновой кислотой, имеющая порошковую рентгеновскую дифрактограмму, по существу соответствующую дифрактограмме, приведенной на Фигуре 12.
Согласно варианту осуществления 35, в настоящем изобретении предложена кристаллическая Форма А соли (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-она с лора-толуолсульфоновой кислотой, которая имеет DSC-термограмму, по существу соответствующую DSC-термограмме, приведенной на Фигуре 13.
Согласно варианту осуществления 36, в настоящем изобретении предложены фармацевтические композиции, содержащие кристаллическую Форму А соли (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-она с пара-толуолсульфоновой кислотой по любому из пп. 31-35 и фармацевтически приемлемый эксципиент.
Согласно варианту осуществления 37, в настоящем изобретении предложена кристаллическая Форма В соли (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-она с пара-толуолсульфоновой кислотой.
Согласно варианту осуществления 38, в настоящем изобретении предложена кристаллическая Форма В соли (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-она с пара-толуолсульфоновой кислотой, имеющая порошковую рентгеновскую дифрактограмму, содержащую пики в положениях 7,02±0,2, 16,30±0,2, 17,30±0,2, 21,86±0,2 2θ.
Согласно варианту осуществления 39, в настоящем изобретении предложена кристаллическая Форма В соли (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-она с пара-толуолсульфоновой кислотой, имеющая порошковую рентгеновскую дифрактограмму, по существу соответствующую дифрактограмме, приведенной на Фигуре 15.
Согласно варианту осуществления 40, в настоящем изобретении предложена кристаллическая Форма В соли (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-она с пара-толуолсульфоновой кислотой, имеющая DSC-термограмму, по существу соответствующую DSC-термограмме, приведенной на Фигуре 16.
Согласно варианту осуществления 41, в настоящем изобретении предложены фармацевтические композиции, содержащие кристаллическую Форму В соли (S)-1-(1-(4-хлор-3 -фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-она с пара-толуолсульфоновой кислотой по любому из вариантов осуществления 37-40 и фармацевтически приемлемый эксципиент.
Согласно варианту осуществления 42, в настоящем изобретении предложен (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-он в форме соли нафталиндисульфоновой кислоты.
Согласно варианту осуществления 43, в настоящем изобретении предложены фармацевтические композиции, содержащие соль (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5 -ил)амино)пиримидин-4-ил)пиридин-2(1H)-она с нафталиндисульфоновой кислотой и фармацевтически приемлемый эксципиент.
Согласно варианту осуществления 44, в настоящем изобретении предложена кристаллическая соль (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-она с нафталиндисульфоновой кислотой.
Согласно варианту осуществления 45, в настоящем изобретении предложена кристаллическая Форма I соли (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-она с нафталиндисульфоновой кислотой.
Согласно варианту осуществления 46, в настоящем изобретении предложена кристаллическая Форма I соли (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-она с нафталиндисульфоновой кислотой, имеющая порошковую рентгеновскую дифрактограмму, содержащую пики в положениях 12,50±0,2, 13,86±0,2 2θ.
Согласно варианту осуществления 47, в настоящем изобретении предложена кристаллическая Форма I соли (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-она с нафталиндисульфоновой кислотой, имеющая порошковую рентгеновскую дифрактограмму, по существу соответствующую дифрактограмме, приведенной на Фигуре 6.
Согласно варианту осуществления 48, в настоящем изобретении предложена кристаллическая Форма I соли (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-она с нафталиндисульфоновой кислотой, имеющая DSC-термограмму, по существу соответствующую DSC-термограмме, приведенной на Фигуре 7.
Согласно варианту осуществления 49, в настоящем изобретении предложены фармацевтические композиции, содержащие кристаллическую Форму I соли (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-она с нафталиндисульфоновой кислотой по любому из вариантов осуществления 44-48 и фармацевтически приемлемый эксципиент.
Согласно варианту осуществления 50, в настоящем изобретении предложена кристаллическая Форма II соли (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-она с нафталиндисульфоновой кислотой.
Согласно варианту осуществления 51, в настоящем изобретении предложена кристаллическая Форма II соли (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-она с нафталиндисульфоновой кислотой, имеющая порошковую рентгеновскую дифрактограмму, содержащую пики в положениях 12,80±0,2, 22,42±0,2, 24,92±0,2 2θ.
Согласно варианту осуществления 52, в настоящем изобретении предложена кристаллическая Форма II соли (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5 -ил)амино)пиримидин-4-ил)пиридин-2(1H)-она с нафталиндисульфоновой кислотой, имеющая порошковую рентгеновскую дифрактограмму, по существу соответствующую дифрактограмме, приведенной на Фигуре 8.
Согласно варианту осуществления 53, в настоящем изобретении предложена кристаллическая Форма II соли (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-она с нафталиндисульфоновой кислотой, имеющая DSC-термограмму, по существу соответствующую DSC-термограмме, приведенной на Фигуре 9.
Согласно варианту осуществления 54, в настоящем изобретении предложены фармацевтические композиции, содержащие кристаллическую Форму II соли (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-она с нафталиндисульфоновой кислотой по любому из пп. 50-53 и фармацевтически приемлемый эксципиент.
Согласно варианту осуществления 55, в настоящем изобретении предложен аморфный (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-она бензолсульфонат.
Согласно варианту осуществления 56, в настоящем изобретении предложен аморфный (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-она бензолсульфонат, который имеет порошковую рентгеновскую дифрактограмму, по существу соответствующую дифрактограмме, приведенной на Фигуре 21.
Согласно варианту осуществления 57, в настоящем изобретении предложен аморфный (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-она бензолсульфонат, который имеет DSC-термограмму, по существу соответствующую DSC-термограмме, приведенной на Фигуре 22.
Согласно варианту осуществления 58, в настоящем изобретении предложены фармацевтические композиции, содержащие аморфный (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-она бензолсульфонат по любому из вариантов осуществления 55-57 и фармацевтически приемлемый эксципиент.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На Фигуре 1 представлена XRPD-дифрактограмма кристаллической формы А безилата соединения VIII.
На Фигуре 2 представлен DSC- и TGA-анализ кристаллической формы А безилата соединения VIII.
На Фигуре 3 представлен анализ структуры монокристалла кристаллической формы А безилата соединения VIII.
На Фигуре 4 представлена XRPD-дифрактограмма соединения VIII в форме свободного основания.
На Фигуре 5 представлен DSC-анализ соединения VIII в форме свободного основания.
На Фигуре 6 представлена XRPD-дифрактограмма формы I соли соединения VIII с нафталиндисульфоновой кислотой.
На Фигуре 7 представлен DSC-анализ формы I соли соединения VIII с нафталиндисульфоновой кислотой.
На Фигуре 8 представлена XRPD-дифрактограмма формы II соли соединения VIII с нафталиндисульфоновой кислотой, содержащей небольшое количество формы I.
На Фигуре 9 представлен DSC-анализ формы II соли соединения VIII с нафталиндисульфоновой кислотой.
На Фигуре 10 представлены данные анализа DVS (динамической сорбции паров) формы I соли соединения VIII с нафталиндисульфоновой кислотой.
На Фигуре 11 представлена XRPD-дифрактограмма IPA-сольвата соли соединения VIII с толуолсульфоновой кислотой.
На Фигуре 12 представлена XRPD-дифрактограмма формы А соли соединения VIII с толуолсульфоновой кислотой.
На Фигуре 13 представлен DSC-анализ формы А соли соединения VIII с толуолсульфоновой кислотой.
На Фигуре 14 представлен DVS-анализ формы А соли соединения VIII с толуолсульфоновой кислотой.
На Фигуре 15 представлена XRPD-дифрактограмма смеси аморфной формы и формы В соли соединения VIII с толуолсульфоновой кислотой.
На Фигуре 16 представлен DSC-анализ смеси аморфной формы и формы В соли соединения VIII с толуолсульфоновой кислотой.
На Фигуре 17 представлена XRPD-дифрактограмма аморфной соли соединения VIII с толуолсульфоновой кислотой.
На Фигуре 18 представлен DVS-анализ формы А безилатной соли соединения VIII.
На Фигуре 19 представлен 13С-спектр кристаллической формы А безилата соединения VIII, полученный с использованием твердотельной ЯМР-спектроскопии.
На Фигуре 20 представлен, 19F-спектр кристаллической формы А безилата соединения VIII, полученный с использованием твердотельной ЯМР-спектроскопии.
На Фигуре 21 представлена XRPD-дифрактограмма аморфного безилата соединения VIII.
На Фигуре 22 представлен DSC-анализ аморфного безилата соединения VIII.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Далее приведено подробное описание изобретения со ссылкой на конкретные варианты осуществления изобретения, примеры которых проиллюстрированы прилагаемыми структурами и формулами. Несмотря на то, что для описания данного изобретения использовано ограниченное количество конкретных вариантов осуществления изобретения, подразумевается, что изобретение не ограничено этими вариантами. Напротив, подразумевается, что изобретение включает все альтернативы, модификации и эквиваленты, которые могут быть включены в объем настоящего изобретения. Специалисту в данной области техники должны быть известны многие методики и вещества, аналогичные или эквивалентные методикам и веществам, приведенным в данном описании, которые можно было бы использовать на практике при осуществлении настоящего изобретения. Описанные методики и вещества не ограничивают настоящее изобретение каким-либо образом. В том случае, если в одном или более чем одном из включенных в заявку литературных источников, патентов и аналогичных материалов содержатся положения, отличающиеся от данной заявки или противоречащие ей, включая, без ограничения, определения терминов, использование терминов, описания методик и тому подобное, данная заявка имеет преимущественную силу.
В контексте данного описания, будь то переходная фраза или формула изобретения, термины "содержат(ит)" и "содержащий" должны быть интерпретированы как имеющие неограничивающее значение. То есть данные термины должны быть интерпретированы как синонимы фразам "имеющий по меньшей мере" или "включающий по меньшей мере". При описании способа термин "содержащий" означает, что данный способ включает по меньшей мере указанные стадии, но может включать и дополнительные стадии. При описании соединения или композиции термин "содержащий" означает, что данное соединение или композиция включает по меньшей мере указанные признаки или компоненты, но также может включать дополнительные признаки или компоненты. Кроме того, в контексте описания и формулы изобретения подразумевается, что слова "включают", "включающий" и "включает" определяют присутствие указанных признаков, чисел, компонентов или стадий, но не исключают присутствия или добавления одного или более чем одного другого признака, числа, компонента, стадии или их комбинации.
Термин "приблизительно" при использовании в отношении времени означает ±5 ч. Термин "приблизительно" при использовании в отношении температуры означает ±5°С. Термин "приблизительно" при использовании в отношении процентов или других единиц измерения означает ±10%.
Термин "хиральный" относится к молекулам, которые не совместимы со своим зеркальным отображением, термин "ахиральный" относится к молекулам, которые совместимы со своим зеркальным отображением.
Термин "изомер" относится к соединениям, которые представлены одной и той же Формулой, но отличаются расположением атомов в молекуле и обладают разными свойствами.
Термин "стереоизомеры" относится к соединениям, которые имеют одинаковое химическое строение, но различаются расположением атомов или групп в пространстве.
Термин "диастереомеры" относится к стереоизомерам, которые содержат два или более центра хиральности и молекулы которых не являются зеркальными изображениями друг друга. Диастереомеры имеют разные физические свойства, например точки плавления, точки кипения, спектральные свойства, и разную реакционную способность. Смеси диастереоизомеров могут быть разделены с использованием аналитических методик высокого разрешения, таких как электрофорез и хроматография.
Термин "энантиомеры" относятся к двум стереоизомерам соединения, которые являются неналагающимися зеркальными изображениями друг друга.
Стереохимические определения и условные обозначения, использованные в данном описании, обычно соответствуют S. P. Parker, Ed., McGraw-Hill Dictionary of Chemical Terms (1984) McGraw-Hill Book Company, New York; и Eliel, E. and Wilen, S., "Stereochemistry of Organic Compounds", John Wiley & Sons, Inc., New York, 1994. Соединения, описанные в данной заявке, могут содержать асимметричные или хиральные центры и поэтому могут существовать в разных стереоизомерных формах. Многие органические соединения существуют в оптически активных формах, то есть обладают способностью вращать плоскость плоскополяризованного света. При описании оптически активного соединения для обозначения абсолютной конфигурации молекулы относительно ее хирального(ых) центра(ов) используют префиксы D и L или R и S. Префиксы d и 1 или (+) и (-) используют для обозначения знака вращения плоскополяризованного света соединением, при этом (-) или 1 означает, что соединение является левовращающим. Соединение с префиксом (+) или d является правовращающим. Данные стереоизомеры с одинаковой химической структурой идентичны, за исключением того, что они являются зеркальными отражениями друг друга. Конкретный стереоизомер также может называться энантиомером, и смесь таких изомеров часто называют энантиомерной смесью. Смесь, содержащая энантиомеры в соотношении 50:50, называется рацемической смесью или рацематом, такая смесь может получаться в результате химической реакции или в результате использования методики, где отсутствует стереоселекция или стереоспецифичность. Термины "рацемическая смесь" и "рацемат" относятся к эквимолярной смеси двух энантиомеров, которая является оптически неактивной.
Способ по настоящему изобретению, описанный в данной заявке, также можно применять для получения меченых изотопами соединений согласно настоящему изобретению, которые идентичны соединениям, описанным в данной заявке, за тем исключением, что один или более чем один атом у этих соединений заменен атомом, имеющим атомную массу или массовое число, отличающиеся от атомной массы или массового числа соответствующего атома, обычно встречающегося в природе. Все изотопы любого конкретного атома или элемента, соответственно, включены в данное изобретение в объеме соединений согласно настоящему изобретению и их применений. Типичные изотопы, которые могут быть введены в соединения согласно настоящему изобретению, включают изотопы атомов водорода, углерода, азота, кислорода, фосфора, серы, фтора, хлора и иода, такие как 2Н, 3Н, 11С, 13С, 14С, 13N, 15N, 15O, 17O, 18O, 32P, 33P, 35S, 18F, 36Cl, 123I или 125I. Некоторые меченые изотопами соединения согласно настоящему изобретению (например соединения, меченные изотопами 3Н и 14С) могут быть использованы для анализа распределения соединения и/или субстрата в тканях. Тритиевые (3Н) и углеродные (14С) изотопы могут быть использованы ввиду легкости их получения и регистрации. Кроме того, замена атома более тяжелыми изотопами, таким как дейтерий (то есть 2Н), может давать некоторые терапевтические преимущества (например увеличение in vivo время полужизни или уменьшение требуемой дозы), обусловленные более высокой метаболической стабильностью, и поэтому в некоторых случаях такая замена может быть предпочтительна. Позитронно-активные изотопы, такие как 15O, 13N, 11С и 18F, могут быть использованы в позитронно-эмиссионной томографии (ПЭТ) для исследования степени занятости рецептора субстратом. Меченые изотопами соединения согласно настоящему изобретению обычно могут быть получены в соответствии с методиками, аналогичными методикам, описанным ниже в разделе Примеры, путем замены немеченого изотопом реагента на меченый изотопом реагент.
Термин "таутомер" или "таутомерная форма" относится к структурным изомерам, обладающим разной энергией, которые являются взаимопревращаемыми вследствие низкоэнергетического барьера этих превращений. Например, протонная таутомерия (также известная как прототропная таутомерия) включает взаимопревращения в результате миграции протона, примерами прототропной таутомерии являются кето-енольная и имин-енаминная изомеризации. Валентная таутомерия включает взаимопревращения в результате перераспределения некоторых электронных связей.
Термин "апротонный" (или неполярный) растворитель относится к таким органическим растворителям, как диэтиловый эфир, лигроин, пентан, гексан, циклогексан, гептан, хлороформ, бензол, толуол, диоксан, тетрагидрофуран, дихлорметан или этилацетат.
Термин "полярный апротонный растворитель" относится к таким органическим растворителям, как формамид, N,N-диметилформамид, диметилсульфоксид, N-метилпирролидон или гексаметилфосфорамид.
Термин "полярный протонный растворитель" относится к таким органическим растворителям, как низшие алканолы, муравьиная кислота или уксусная кислота.
Термин "эфирный растворитель" относится к таким растворителям, как тетрагидрофуран, диметоксиэтан, диоксан или диалкиловые простые эфиры, такие как диэтиловый эфир и метилтретбутиловый эфир.
Термин "производное" соединения в контексте данного описания означает соединение, которое может быть получено из исходного соединения с использованием простой химической методики.
Термин "защитная группа" в контексте данного описания относится к химической группе, которая (а) защищает реакционноспособную группу от участия в нежелательной химической реакции и (b) может быть легко удалена после того, как защита данной реакционноспособной группы больше не требуется. Например, бензильная группа является защитной группой для первичной гидроксильной функциональной группы.
Термин "гидроксизащитная группа" или "защитная группа для спиртовой функции" означает защитную группу, которая защищает гидроксильную группу, которая в противном случае была бы модифицирована в результате некоторых химических реакций. Гидроксизащитная группа может представлять собой простой эфир, сложный эфир или силан, которые могут быть легко удалены после завершения всех других реакционных стадий, такие как низшая ацильная группа (например ацетильная или пропионильная группа или диыетип-трет-бутилсилильная группа) или аралкильная группа (например бензильная группа, необязательно имеющая заместители у фенильного кольца). Термин "силилхлорид" в контексте данного описания относится к (Ra)3SiCl, где Ra в каждом случае независимо представляет собой C1-6 алкил или фенил.
Термин "реагент для снятия защитной группы" в контексте данного описания относится к реагентам, взаимодействующим с защищенной химической группировкой с целью удаления защитных групп. Реагенты и методики для снятия защитных групп хорошо известны и могут быть найдены в книге авторов Greene и Wuts или в книге авторов Harrison и Harrison (см. ниже). Специалисту в области химии должно быть понятно, что в некоторых случаях данные методики должны быть оптимизированы для конкретной молекулы, и такая оптимизация находится в рамках компетенции специалиста в данной области техники.
Термин "необязательный" или "необязательно" в контексте данного описания означает, что описываемое далее событие или условие может, но не обязано иметь место и что данное описание включает примеры, в которых указанное событие или условие имеет место, и примеры, в которых оно отсутствует. Например, выражение "арильная группа, необязательно имеющая в качестве заместителей одну или две алкильные группы" означает, что алкил может, но не обязан присутствовать и данное описание включает ситуации, когда арильная группа имеет в качестве заместителей одну или две алкильные группы, и ситуации, когда арильная группа не имеет в качестве заместителя алкильную группу.
В контексте данного описания термин "обработка", "контактирование" или "взаимодействие", используемый в отношении химической реакции, означает добавление или смешивание двух или более реагентов в подходящих условиях с целью получения указанного и/или желаемого продукта. Следует понимать, что реакция, в ходе которой образуется указанный и/или желаемый продукт, не всегда может являться прямым результатом комбинации двух реагентов, которые были добавлены на начальной стадии, то есть в реакционной смеси может иметь место образование одного или более чем одного промежуточного соединения, что, в конечном счете, приводит к получению указанного и/или желаемого продукта.
Термин "уходящая группа" имеет значение, которое обычно связано с ним в химии органического синтеза, то есть данный термин означает атом или группу, которые могут быть вытеснены нуклеофилом, и включает атом галогена (такой как атом хлора, брома и иода), алкансульфонилокси, аренсульфонилокси, алкилкарбонилокси (например ацетокси), арилкарбонилокси, мезилокси, тозилокси, трифторметансульфонилокси, арилокси (например 2,4-динитрофенокси), метокси, N,O-диметилгидроксиламино и тому подобное. Термин "сульфонилхлорид" относится к соединению RbS(O)2Cl, где Rb выбран из С1-4 алкила или фенила, необязательно имеющих в качестве заместителей от 1 до 3 групп, независимо выбранных из C1-3 алкила, атома галогена, нитро, циано, C1-3 алкокси.
Реагент Виттига может быть использован для получения алкена из альдегида. Реагент Виттига обычно получают из соли фосфония, которую, в свою очередь, получают в результате взаимодействия трифенилфосфина с алкилгалогенидом. Для получения реагента Виттига (илида) соль фосфония суспендируют в таком растворителе, как диэтиловый эфир или ТГФ и обрабатывают сильным основанием, таким как фениллития или н-бутиллития.
Дигидроксилирование, или бисгидроксилирование, по Шарплессу используют для энантиоселективного синтеза 1,2-диолов из прохиральных олефинов. Данную реакцию выполняют с использованием соединения осмия в качестве катализатора и стехиометрического окислителя [например K3Fe(CN)6 или оксида N-метилморфолина (NMO)]; реакцию выполняют в буферном растворе для поддержания стабильного рН, так как реакция проходит быстрее в слегка щелочной среде. Энантиоселективность реакции достигается путем добавления энантиомерно-обогащенных хиральных лигандов [(DHQD)2PHAL, (DHQ)2PHAL или их производных]. Данные реагенты также доступны в виде стабильных упакованных смесей (AD-mix α и AD-mix β AD = асимметрическое дигидроксилирование) для получения любых энантиомеров.
В методиках, описанных в данной заявке, может быть использовано титрование по Карлу Фишеру для определения следового количества воды в образце. Для названия данной методики может быть использована аббревиатура "KF."
В методиках получения соединений, приведенных в данном описании, бывает полезно отделить продукты реакции друг от друга и/или от исходных веществ. Желаемые продукты, полученные на каждой стадии или в результате нескольких стадий, разделяют и/или очищают (в дальнейшем разделяют) до желаемой степени гомогенности с использованием стандартных методик, известных в данной области техники. Обычно такое разделение включает многофазное экстрагирование, кристаллизацию из растворителя или смеси растворителей, дистилляцию, сублимацию или хроматографию. Хроматография может включать любое количество методик, в том числе, например, методики и соответствующее оборудование для обратнофазовой и нормальнофазовой хроматографии, эксклюзионной хроматографии, ионообменной хроматографии, жидкостной хроматографии высокого, среднего и низкого давления; методики аналитической хроматографии для маломасштабного разделения; хроматографии в псевдоподвижном слое (SMB) и препаративной тонкослойной или толстослойной хроматографии, а также методики тонкослойной хроматографии для маломасштабного разделения и флэш-хроматографии.
Другая группа методик разделения включает обработку реакционной смеси реагентом, который может избирательно связываться с желаемым продуктом, непрореагировавшим исходным веществом, побочным продуктом реакции и т.п. или обеспечивать разделение указанных веществ иным образом. Такие реагенты включают адсорбенты или абсорбенты, такие как активированный уголь, молекулярные сита, ионообменные среды и тому подобное. Альтернативно, данные реагенты могут представлять собой кислоты, в случае, если вещество является основанием, основания, в случае, если вещество является кислотой, связующие реагенты, такие как антитела, связывающие белки, селективные хелатирующиие агенты, такие как краун-эфиры, реагенты для жидкостной экстракции ионов (LIX) и тому подобное.
Выбор подходящих методик разделения зависит от природы присутствующих в реакционной смеси веществ. Например, температура кипения и молекулярная масса имеют значение при разделении путем дистилляции и сублимации, наличие или отсутствие полярных функциональных групп имеет значение при разделении путем хроматографии, стабильность веществ в кислой и щелочной среде имеет значение при мультифазовой экстракции и т.п. Специалист в данной области техники будет использовать методики, наиболее подходящие для достижения желаемого разделения.
В настоящем изобретении предложен способ получения (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-(1-метил-1H-пиразол-5-иламино)пиримидин-4-ил)пиридин-2(1H)-она (VIII), имеющего следующую структуру:
 ,
,
который является сильным ингибитором киназы ERK и может быть использован в качестве лекарства для лечения рака или других гиперпролиферативных расстройств. Конденсация соединения I с 1-метил-1H-пиразол-5-амином (XIV) в присутствии сильного основания приводит к получению соединения IX, которое легко превращается в соединение VIII в результате контактирования данного силильного эфира с водной кислотой. Полученное аморфное свободное основание может быть превращено в кристаллическую соль арилсульфоновой кислоты. Термин "арилсульфоновая кислота" в контексте данного описания относится к бензолсульфоновой кислоте или к нафталин моно- или дисульфоновой кислоте, у которых арильное кольцо необязательно имеет в качестве заместителей метил или атом галогена.
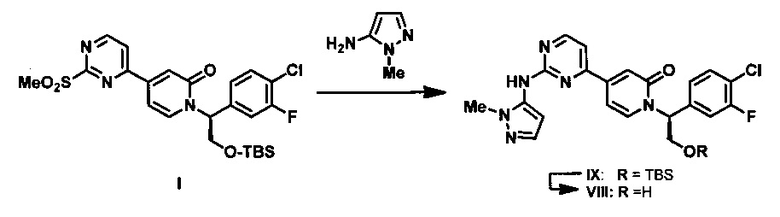
В настоящем изобретении дополнительно предложен способ получения промежуточного соединения I путем обработки 4-(2-(метилтио)пиримидин-4-ил)пиридин-2(1H)-она (VII) сильным основанием и последующего алкилирования полученного соединения (R)-2-((трет-бутилдиметилсилил)окси)-1-(4-хлор-3-фторфенил)этил метансульфонатом (VI).

N-Алкилирование амидов может быть выполнено в присутствии различных оснований, известных специалистам в данной области техники. Данную реакцию обычно проводят в апротонных растворителях, таких как ТГФ, ДМФА, ДМСО, NMP или их смеси, при температуре от -78°С до 100°С. Обычно в качестве основания используют реактивы Гриньяра, гидрид натрия, гидрид калия, метилат натрия, трет-бутилат калия, гексаметилдисилазид лития, гексаметилдисилазид натрия или гексаметилдисилазид калия. Соединение VII обрабатывают гексаметилдисилазидом калия в диметиловом эфире диэтиленгликоля при комнатной температуре с получением литиевой соли соединения VII, затем добавляют мезилат VI, и реакционную смесь нагревают при 90°С в течение 4 ч.
Тиоэфир обычно легко окисляется до сульфоксида или сульфона под действием множества известных реагентов, которые способны осуществить данное превращение. Окисление серы обычно выполняют с использованием водного раствора перекиси водорода, NaIO4, трет-бутилгипохлорита, ацил нитритов, пербората натрия, гидроперсульфата калия или перкислот, таких как перуксусная кислота и мета-хлорпербензойная кислота. Обычно для получения сульфоксида используют приблизительно один эквивалент окислителя. Использование двух или более эквивалентов окислителя приводит к окислению до сульфона. Окисление соединения XI с использованием МСРВА в МТВЕ при температуре окружающей среды приводит к получению соединения I.
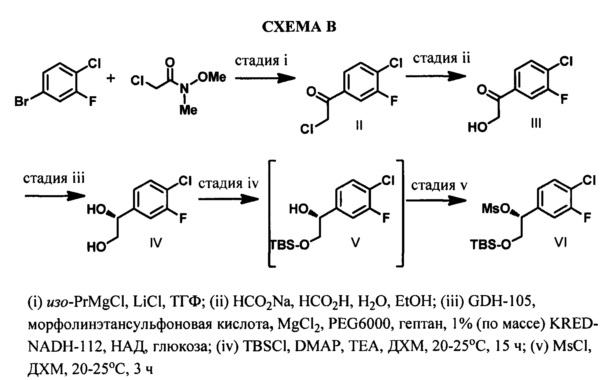
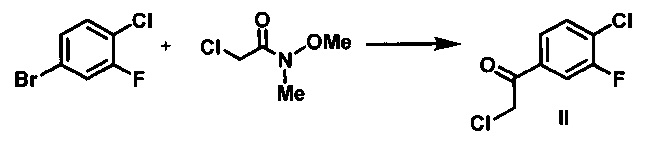
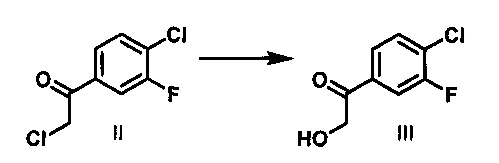
Мезилат VI получают с использованием пятистадийной методики из 1-бром-4-хлор-3-фторбензола, в качестве исходного вещества, который превращают в реактив Гриньяра и подвергают взаимодействию с 2-хлор-N-метокси-N-метилацетамидом с получением кетона II. Конденсация литийорганических и магнийорганических соединений с N,O-диметилгидроксиамидами приводит к получению соответствующих кетонов. (S. Nahm and D. M. Weinreb, S. M. Tetrahedron Lett. 1981, 22, 3815). Реактив Гриньяра получают путем обработки 1-бром-4-хлор-3-фторбензола изопропилмагния хлоридом в присутствии LiCl. Считается, что добавление солей повышает реакционную способность реактивов Гриньяра путем стимуляции дезинтеграции полимерных агрегатов, которые, как известно, существуют в классических растворах реактивов Гриньяра (А. Krasovskiy and P. Knochel, Angew. Chem. Int. Ed. 2004 43:3333). Реакцию Гриньяра останавливают путем добавления 1 н. раствора HCl, и затем органическую фазу промывают водой и концентрируют. Добавляют формиат натрия, муравьиную кислоту, этанол и воду, и данную смесь нагревают при 80-90°С с получением α-гидрокси кетона III.
Катализируемое ферментом восстановление кетонов, часто протекающее с высокой стереоселективностью, обычно проводят в присутствии НАД(Н) или НАДФ(Н), в качестве кофактора, который регенерируют in situ (J. С. Moore et al, Асе. Chem. Res, 2007 40 (12): 1412-19). Микробные оксидоредуктазы, найденные в дрожжах, бактериях, или оксидоредуктазы из клеток млекопитающих являются предпочтительными, данные оксидоредуктазы могут быть использованы в форме выделенного(ых) фермента(ов) или в виде целых клеток, необязательно в иммобилизованной форме, в соответствии с одной из многочисленных стандартных методик, описанных в литературе.
Окисленный кофактор, как правило, постоянно регенерируют с помощью вторичного спирта, используемого в качестве косубстрата. Типичные косубстраты может быть выбраны из 2-пропанола, 2-бутанола, пентан-1,4-диола, 2-пентанола, 4-метил-2-пентанола, 2-гептанола, гексан-1,5-диола, 2-гептанол или 2-октанола, предпочтительно из 2-пропанола. Предпочтительно, когда кофактор регенерируют с помощью агента, который является косубстратом для того же самого фермента, который катализирует основную реакцию. Согласно дополнительному предпочтительному варианту осуществления изобретения, ацетон, образующийся при использовании в качестве косубстрата 2-пропанола, постоянно удаляют из реакционной смеси.
Кофактор может быть регенерирован путем добавления дополнительного фермента, который восстанавливает кофактор, окисляя свой природный субстрат. Например, могут быть использованы следующие ферменты: дегидрогеназа, окисляющая вторичные спирты/спирт, глюкозодегидрогеназа/глюкоза, формиатдегидрогеназа/муравьиная кислота, глюкозо-6-фосфат-дегидрогеназа/глюкозо-6-фосфат, фосфитдегидрогеназа/фосфит или гидрогеназа/молекулярный водород и тому подобное. Кроме того, известны методики электрохимической регенерации, и также могут быть использованы химические методики регенерации кофактора, включающие металлический катализатор и восстанавливающий агент. Выбор предпочтительной системы катализатор/кофактор/косубстрат может зависеть от восстанавливаемых кетонов.
Ферментативное восстановление выполняют в водной среде в присутствии органического сорастворителя, который может быть выбран, например, из глицерина, 2-пропанола, диэтилового эфира, wpew-бутилметилового эфира, диизопропилового эфира, дибутилового эфира, этилацетата, бутилацетата, гептана, гексана или циклогексена, или их смесей. Присутствие органического сорастворителя является особенно предпочтительным в том случае, когда может образоваться гомогенная суспензия, что позволяет легко отделить желаемый спирт Формулы IV. Температуру реакционной смеси на стадии ферментативного восстановления обычно поддерживают в диапазоне от 1°С до 50°С, предпочтительно в диапазоне от 20°С до 40°С.
Концентрацию реакционной смеси (то есть концентрацию кетона и соответствующего спирта) обычно поддерживают в диапазоне от 1% до 25%, предпочтительно в диапазоне от 10 до 20%.
Согласно конкретному варианту осуществления способа по настоящему изобретению, каталитическое асимметрическое восстановление соединения III осуществляют с использованием KRED-NADH-112 (Codexis Inc., Redwood City, CA, USA) в присутствии окисляемого кофактора НАД, регенерирующего фермента GDH-105 (Codexis Inc., Redwood City, CA, USA) и глюкозы в качестве конечного восстановителя с получением (R)-1-(4-хлор-3-фторфенил)этан-1,2-диола (избыток энантиомера 99,5% в количественном химическом превращении).
Конечные стадии включают избирательную защиту первичной спиртовой группы с использованием трет-бутилхлордиметилсилана, 4-диметиламинопиридина (DMAP) и триэтиламина (TEA) в ДХМ и последующее получение метансульфонатного эфира с использованием метансульфонилхлорида, DMAP и TEA в ДХМ и могут быть выполнены последовательно в одном реакционном сосуде с получением (R)-2-((трет-бутилдиметилсилил)окси)-1-(4-хлор-3-фторфенил)этил метансульфоната (VI).
Специалисту в данной области техники должно быть понятно, что данный способ можно эффективно применять в отношении других замещенных бромбензольных производных.
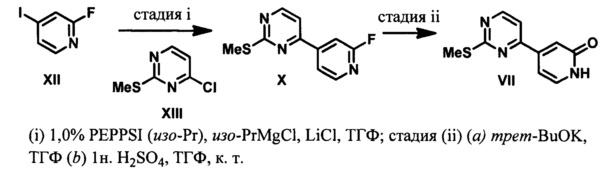
4-(2-(Метилтио)пиримидин-4-ил)пиридин-2(1H)-он (VII) получают в результате сочетания 4-хлор-2-тиометилпиримидина (XIII) с 2-фтор-4-иодпиридином (XII) в присутствии палладиевого катализатора. Реактив Гриньяра получают путем трансметаллирования с использованием изо-PrMgCl в присутствии LiCl (Krasovskiy, см. выше) и обработки полученного гетероарильного реактива Гриньяра соединением XIII в присутствии PEPPSI (изо-Pr) ([1,3-бис(2,6-диизопропилфенил)имидазол-2-илиден](3-хлорпиридил)палладия (И) дихлорида, CAS RN 905459-27-0). В результате взаимодействия соединения X с трет-бутлатои калия получают 4-(2-(трет-бутокси)пиридин-4-ил)-2-(метилтио)пиримидин, у которого удаляют трет-бутильную группу путем обработки H2SO4 с получением соединения VII.
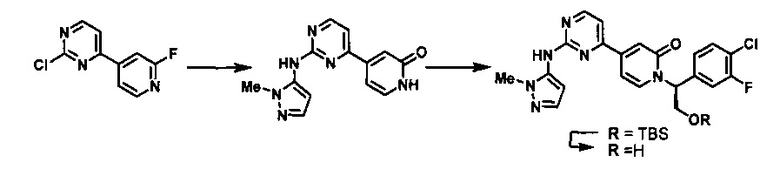
Данная последовательность стадий может быть модифицирована без отступления от изобретения, раскрытого в данном описании. Согласно одному из вариантов осуществления изобретения, 2,4-дизамещенное производное пиримидина, такое как 2,4-дихлор-пиримидин или 4-хлор-2-метилтиопиримидин, подвергают сочетанию с 2-фторпиридин-4-илбороновой кислотой (Pd(dppf)Cl2, K3PO4, диоксан) с получением 2-хлор-4-(2-фторпиридин-4-ил)пиримидина, который конденсируют с 1-метил-1H-пиразол-5-амином (LiHMDS, ТГФ) и гидролизуют с получением 4-(2-(1-метил-1H-пиразол-5-иламино)пиримидин-4-ил)пиридин-2(1H)-она, который может быть алкилирован, в соответствии с тем, как описано выше, с использованием двух эквивалентов основания.
В данной заявке могут быть использованы следующие общепринятые аббревиатуры: ацетил (Ас), водный (вод.), атмосфера (ат), трет-бутоксикарбонил (Boc), пироугольной кислоты ди-трет-бутиловый эфир или Вос-ангидрид (ВОС2О), бензил (Bn), бензотриазол-1-илокси-трис-(диметиламино)фосфонийгексафторфосфат (ВОР), бутил (Bu), бензоил (Bz), Chemical Abstracts Registration Number (CASRN), бензилоксикарбонил (CBZ или Z), карбонилдиимидазол (CDI), дибензилиденацетон (DBA), 1,5-диазабицикло[4.3.0]нон-5-ен (DBN), 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU), N,N'-дициклогексилкарбодиимид (DCC), 1,2-дихлорэтан (DCE), дихлорметан (ДХМ), азодикарбоновой кислоты диэтиловый эфир (ДЭАД), азодикарбоновой кислоты диизопропиловый эфир (DIAD), ди-изо-бутилалюминийгидрид (DIBAL или DIBAL-H), ди-изо-пропилэтиламин (DIPEA), N,N-диметилацетамид (DMA), 4-N,N-диметиламинопиридин (DMAP), N,N-диметилформамид (ДМФА), диметилсульфоксид (ДМСО), 1,1'-бис-(дифенилфосфино)этан (dppe), 1,1'-бис-(дифенилфосфино)ферроцен (dppf), 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (EDCI), этил (Et), диэтиловый эфир (Et2O), этилацетат (EtOAc), этанол (EtOH), 2-этокси-2H-хинолин-1-карбоновой кислоты этиловый эфир (EEDQ), диэтиловый эфир (Et2O), O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (HATU), O-(бензотриазол-1-ил)-МДД'Д'-тетраметилурония гексафторфосфат (HBTU), уксусная кислота (НОАс), 1-N-гидроксибензотриазол (HOBt), высокоэффективная жидкостная хроматография (ВЭЖХ), изо-пропанол (IPA), лития гексаметилдисилазид (LiHMDS), лития диизопропиламид (LDA), метанол (МеОН), температура плавления (т. пл.), MeSO2- (мезил или Ms), метил (Me), ацетонитрил (MeCN), мета-хлорпербензойная кислота (МСРВА), масс-спектр (МС), метилтретбутиловый эфир (МТВЕ), N-метилморфолин (NMM), N-метилпирролидон (NMP), пиридиния хлорхромат (РСС), петролейный эфир (петр. эфир, то есть смесь углеводородов), фенил (Ph), пропил (Pr), изо-пропил (изо-Pr), фунт-сила на квадратный дюйм (psi), бром-трис-пирролидинофосфония гексафторфосфат (PyBrOP), пиридин (pyr), комнатная температура (к. т. или К. Т.), насыщенный (нас), трет-бутилметиловый эфир (ТВМЕ), трет-бутилдиметилсилил или трет-BuMe2Si (TBDMS или TBS), триэтиламин (TEA или Et3N), трифлат или CF3SO2- (Tf), трифторуксусная кислота (ТФУ), О-бензотриазол-1-ил-N,N,N,'N'-тетраметилурония тетрафторборат (TBTU), тонкослойная хроматография (ТСХ), тетрагидрофуран (ТГФ), тетраметилэтилендиамин (TMEDA), триметилсилил или Me3Si (TMS), 2-(триметилсилил)этоксиметил (SEM), пара-толуолсульфоновой кислоты моногидрат (TsOH или pTsOH), 4-Me-C6H4SO2- или тозил (Ts), N-уретан-N-карбоксиангидрид (UNCA), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (Xantphos), киназа, регулируемая внеклеточными сигналами (ERK), тетрагидрофуран (ТГФ), час(ы) (ч), метахлорпероксибензойная кислота (МСРВА или mCPBA), никотинамидадениндинуклеотид (НАД), никотинамидадениндинуклеотидфосфат (НАДФ), 4-диметиламинопиридин (DMAP), фенил (Ph), метил (Me), этил (Et), трет-бутил (трет-Bu), трет-бутил-хлор-диметилсилан (TBSCl), мезил (Ms), этилацетат (EtOAc), газовая хроматография (ГХ), метилэтилкетон (MEK), высокоэффективная жидкостная хроматография (ВЭЖХ), дифракция рентгеновских лучей на порошке (XRPD), ядерный магнитный резонанс (ЯМР), температура стеклования (TG), термогравиметрический анализ (TGA), дифференциальная сканирующая калориметрия (DSC), политетрафторэтилен (PTFE). Префиксы, используемые с алкильной группировкой, включая префиксы нормальный (н-), изо (изо-), вторичный (втор-), третичный (трет- или -t) и нео-, имеют свои обычные значения в соответствии с общепринятой номенклатурой. (J. Rigaudy and D. P. Klesney, Nomenclature in Organic Chemistry, IUPAC 1979 Pergamon Press, Oxford.).
Следующие примеры включены в данное описание для иллюстрации настоящего изобретения. Однако необходимо понимать, что данные примеры не ограничивают изобретение и предложены в качестве иллюстрации способа осуществления настоящего изобретения. Специалисты в данной области техники должны понимать, что химические методики, приведенные в данном описании, могут быть адаптированы в соответствии с имеющимся в распоряжении оборудованием и имеющимся условиями. Кроме того, могут быть выбраны другие реагенты, например другие уходящие группы, активирующие группы, защитные группы, и, например, другие сильные основания и палладиевые катализаторы, без отклонения от настоящего изобретения.

Новый способ синтеза по настоящему изобретению включает восьмистадийный линейный синтез (в целом 10 стадий) с использованием трех имеющихся в продаже исходных веществ, 4-хлор-3-фторбензальдегида 1, 4-хлор-2-(метилтио)пиримидина XIII и (2-фторпиридин-4-ил)бороновой кислоты 6-2, (Схема 1). В результате реакции Виттига из соединения 1 получают олефиновое промежуточное соединение 2 с выходом 55%. Асимметрическое дигидроксилирование по Шарплессу стирола и последующая избирательная моно-защита диола IV с использованием TBSCl приводят к получению промежуточного соединения V с выходом после двух стадий 55%. Одно из промежуточных соединений VI получают путем мезилирования вторичного спирта. В то же время синтезируют пиридоновое промежуточное соединение VII с использованием реакции кросс-сочетания Сузуки между соединениями XIII и 6-2 и последующего гидролиза водным раствором HCl. Для хорошей очистки соединения VII от соединения 6 и его корректного превращения в следующей реакции требуется очистка соединения VII путем экстракции Сокслета с использованием EtOAc в течение 3 суток. Sn2-замещение с участием соединений VI и VII позволяет получить промежуточное соединение XI с выходом 50% (после двух стадий, начиная с промежуточного соединения V). В результате окисления с использованием мета-СРВА получают сульфоновое промежуточное соединение I, которое подвергают SnAr-замещению с использованием имеющегося в продаже аминопиразола, 2-метилпиразол-3-амина, с получением промежуточного соединения IX с выходом 60%. Снятие защитной TBS-группы в присутствии кислоты на заключительной стадии приводит к получению свободно основания соединения VIII с выходом 85%. Слабой стороной данной методики является низкий выход продукта на некоторых стадиях. Из-за довольно сложного реакционного профиля необходимо проводить множество трудоемких очисток, таких как дистилляция, флэш-хроматография и экстракция Сокслета. Использование не очень желательных растворителей и реагентов, таких как дихлорметан, гидрид натрия и оксид осмия, также не позволяет увеличить выход продукта.
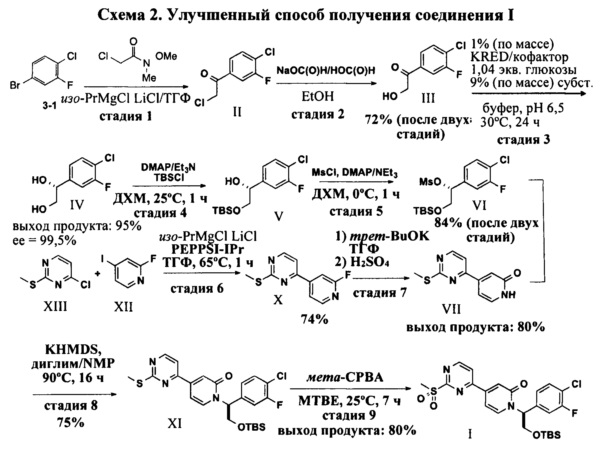
Предложен улучшенный путь синтеза соединения I. Промежуточное соединение III, гидроксил кетон, получают с выходом после двух стадий 72%. Реакция обмена между имеющимся в продаже ареном 3-1 и реактивом Гриньяра и последующее нуклеофильное присоединение к амиду Вайнреба приводят к получению промежуточного соединения II, которое затем гидролизуют с получением соединения III. Ферментативное асимметрическое восстановление кетона позволяет получить то же, что и на Схеме 1, диольное промежуточное соединение IV, с высоким выходом и высоким энантиомерным избытком. Для получения промежуточного соединения VI использованы те же методики избирательной TBS-защиты и мезилирования, как и на Схеме 1. Синтез пиридона VII улучшен. Для получения промежуточного соединения X с высоким выходом и более чистым профилем использовано сочетание Кумада, катализируемое PEPPSI-IPr. Чтобы избежать образования агрессивного HF, имеющего место в исходной методике, использован двухстадийный гидролиз. В результате замещения фторида трет-BuOK (трет-бутилатом калия) в ТГФ и последующего удаления трет-бутильной группы в кислых условиях получают пиридоновое промежуточное соединение VII с выходом 80%. Для улучшения стадии Sn2-замещения использованы другие, в сравнении с исходной методикой, основания и растворители. Промежуточное соединение XI окисляют в таких же условиях, как и в исходной методике, с получением соединения I.
Пример 1
2-((трет-Бутилдиметилсилил)окси)-1-(4-хлор-3-фторфенил)этил метансульфонат

Стадия 1. В стальной химический реактор объемом 2000 л в атмосфере азота вносили 4-бром-1-хлор-2-фторбензол (64 кг) и сухой толуол (170 кг). Из химического реактора откачивали газ, и затем реактор заполняли азотом, данную процедуру повторяли три раза, затем реактор охлаждали до -10-5°С в атмосфере азота. К полученному раствору добавляли по каплям изо-PrMgCl.LiCl (280 кг, 1,3 М в ТГФ) при -10-10°С. Данную реакционную смесь перемешивали дополнительно в течение 15-30 мин при -10-10°С и затем нагревали до приблизительно 20-25°С в течение 1 ч. Реакционную смесь перемешивали дополнительно в течение 6 ч для завершения обмена. Полученный раствор охлаждали до температуры в диапазоне от -50 до -40°С. К полученному раствору добавляли по каплям раствор 2-хлор-N-метокси-N-метилацетамида (44,5 кг) в сухом толуоле (289 кг), поддерживая температуру в диапазоне от -50 до -30°С. Данную реакционную смесь нагревали до 20-25°С в течение 1 ч и затем перемешивали в течение 3 ч для завершения реакции. Реакционную смесь гасили путем добавления 1 н. водного раствора HCl (808 л г) при температуре от -5 до 15°С. Водный слой отделяли, и органический слой фильтровали через подушку диатомовой земли. Органический слой дважды промывали 10% водным раствором NaCl (320 кг), затем концентрировали до объема приблизительно 300 л с получением (1-(4-хлор-3-фторфенил)-2-хлорэтанона (51,8 кг, выход: 81,9%), в виде продукта в толуоле.
Стадия 2. Раствор соединения II (51,7 кг) в толуоле концентрировали, и растворитель заменяли на EtOH с получением суспензии соединения II в EtOH (326 кг). Добавляли раствор HCOONa⋅2H2O (54,8 кг) и НСООН (44,5 кг) в воде (414 кг) при температуре 15-35°С в атмосфере азота. Полученную смесь нагревали до температуры дефлегмации и перемешивали в течение 4-5 ч. Раствор охлаждали до 20-30°С, имело место 95% превращение исходных веществ. Добавляли по каплям воду (450 кг) при 10-30°С в течение 2 ч. Полученную суспензию охлаждали до температуры в диапазоне от -10 до -3°С, и охлажденный раствор перемешивали в течение 1-2 ч. Твердое вещество отфильтровывали, и осадок на фильтре промывали водой (400 кг) для удаления остатков HCOONa и НСООН. Полученный 1-(4-хлор-3-фторфенил)-2-гидроксиэтанон суспендировали в смеси в EtOAc (41 кг) и н-гептана (64 кг), затем нагревали до 45-50°С, перемешивали в течение 2 ч, затем охлаждали до -2-5°С в течение 2 ч и перемешивали при этой же температуре в течение 2 ч. Твердое вещество отфильтровывали и сушили под вакуумом при 40-50°С в течение 12 ч с получением желаемого продукта в виде белого твердого вещества (40,0 кг, чистота: 99,3%, выход: 84,5%).
Стадия 3: В химический реактор объемом 500 л в атмосфере азота вносили очищенную воду (150 кг), 4-морфолинэтансульфоновую кислоту (0,90 кг), безводный MgCl2 (0,030 кг), н-гептан (37 кг), 1-(4-хлор-3-фторфенил)-2-гидроксиэтанон (30 кг), 0-(+)-глюкозы моногидрат (34,8 кг) и ПЭГ 6000 (30,0 кг). рН раствора доводили до 6,5-7,0 путем добавления 1 н. водного раствора NaOH при 28-32°С. Добавляли регенерирующий кофактор фермент, глюкозодегидрогеназу GDH-105 (0,300 кг) (Codexis Inc., Redwood City, CA, USA), кофактор, никотинамидадениндинуклеотид (НАД) (0,300 кг) (Roche) и оксидоредуктазу KRED-NADH-112 (0,300 кг) (Codexis Inc., Redwood City, CA, USA). Полученную суспензию перемешивали при 29-31°С в течение 10-12 ч, под держивая рН реакционной смеси в диапазоне от 6,5 до 7,0 путем добавления 1 н. водного раствора NaOH (160 кг). рН реакционной смеси доводили до 1-2 путем добавления 49% H2SO4 (20 кг), чтобы остановить реакцию. Добавляли EtOAc (271 кг), и данную смесь перемешивали при 20-30°С в течение 10-15 мин, затем фильтровали через подушку диатомовой земли. Осадок на фильтре промывали EtOAc (122 кг). Объединенные органические слои отделяли, и водный слой экстрагировали EtOAc (150 кг). К объединенным органическим слоям добавляли воду (237 кг). рН смеси доводили до 7,0-8,0 путем добавления твердого NaHCO3. Органический слой отделяли, концентрировали и затем разбавляли ДХМ (дихлорметаном) с получением (R)-1-(4-хлор-3-фторфенил)этан-1,2-диола (30,9 кг, выход: 100%) в виде продукта в ДХМ.
Стадия 4: В химический реактор объемом 1000 л в атмосфере азота вносили (R)-1-(4-хлор-3-фторфенил)этан-1,2-диол (29,5 кг) и сухой ДХМ (390 кг). Данный раствор охлаждали до -5-0°С. Добавляли порциями трет-бутилхлордиметилсилан (25,1 кг), поддерживая температуру в диапазоне от -5 до 2°С. К полученному раствору при -5-2°С добавляли по каплям раствор DMAP (0,95 кг) и TEA (41,0 кг) в сухом ДХМ (122 кг). Данный реакционный раствор перемешивали в течение 1 ч, затем нагревали до 20-25°С и перемешивали в течение 16 ч. Полученный раствор (R)-2-(трет-бутилдиметилсилил)окси)-1-(4-хлор-3-фторфенил)этанола снова охлаждали до температуры в диапазоне от -10 до -5°С. К данному раствору добавляли по каплям раствор метансульфонилхлорида (19,55 кг) в сухом ДХМ (122 кг), поддерживая температуру в диапазоне от -10 до 0°С. Данный реакционный раствор перемешивали при -10-0°С в течение 20-30 мин и затем нагревали до 0-5°С в течение 1 ч и перемешивали. Реакционный раствор промывали водой (210 кг), и затем 5% водным раствором лимонной кислоты (210 кг), 2% водным раствором NaHCO3 (210 кг) и в заключение водой (2×210 кг). Полученный ДХМ-раствор сушили (Na2SO4), фильтровали и концентрировали под вакуумом при температуре ниже 15°С (температура рубашки реактора ниже 35°С) с получением (R)-2-((тирет-бутилдиметилсилил)окси)-1-(4-хлор-3-фторфенил)этил метансульфоната (49,5 кг, выход: 83,5%, содержание воды по Карлу Фишеру = 0,01%) в виде продукта в ДХМ.
Пример 2
4-(2-(Метилсульфонил)пиримидин-4-ил)пиридин-2(1H)-он
Стадия 1. В химический реактор объемом 1000 л вносили 2-фтор-4-иодпиридин (82,2 кг) и сухой ТГФ (205 кг). Из химического реактора откачивали газ, и затем реактор заполняли азотом, данную процедуру повторяли три раза, затем реактор охлаждали до температуры в диапазоне от -30 до -20°С. К полученному раствору добавляли по каплям изо-PrMgCl.LiCl (319 кг, 1,3 М в ТГФ). Данную реакционную смесь нагревали до температуры в диапазоне от -20 до -10°С и перемешивали в течение 1,5 ч для завершения трансметаллирования.
В химический реактор объемом 2000 л вносили 4-хлор-2-метилтиопиримидин (45,6 кг), сухой ТГФ (205 кг) и [1,3-бис(2,6-диизопропилфенил)имидазол-2-илиден](3-хлорпиридил)палладия (II) дихлорид (PEPPSI™-IPr, 1,850 кг). Из химического реактора объемом 2000 л откачивали газ, и затем реактор заполняли азотом, данную процедуру повторяли три раза, затем реактор нагревали до 55-57°С. В течение 0,5-1 ч в химический реактор добавляли раствор (2-фторпиридин-4-ил)магния хлорида, поддерживая температуру в диапазоне от 50 до 62°С. Полученную реакционную смесь перемешивали при 50-62°С дополнительно в течение 2 ч. Реакцию останавливали путем добавления воды, в течение всего этого времени реакционную смесь охлаждали, поддерживая температуру в диапазоне от 5 до 25°С. рН смеси доводили до 8-9 путем добавления твердого моногидрата лимонной кислоты (7,3 кг). Органический слой отделяли, промывали 12,5% водным раствором NaCl (228 кг) и концентрировали под вакуумом при температуре ниже 50°С с получением 4-(2-фторпиридин-4-ил)-2-(метилтио)пиримидина (38,3 кг, выход: 61%) в виде продукта в ТГФ.
Стадия 2. Раствор 4-(2-фторпиридин-4-ил)-2-(метилтио)пиримидина (38,2 кг) в ТГФ концентрировали, и остатки воды удаляли путем выпаривали вместе с ТГФ. Полученную суспензию фильтровали через подушку диатомовой земли для удаления неорганических солей. К полученному раствору в ТГФ (510 кг) добавляли порциями трет-BuOK(39,7 кг), поддерживая температуру в диапазоне от 15 до 25°С. Данную смесь нагревали до 20-25°С и перемешивали в течение 5 ч. Добавляли NaHCO3 (14,9 кг), и затем добавляли раствор лимонной кислоты (5 кг) в ТГФ (15 кг) до получения рН 8-9. Добавляли воду (230 кг). Смесь фильтровали, и осадок на фильтре промывали ТГФ (100 кг). Объединенные порции ТГФ-раствора промывали 12,5% водным раствором NaCl (320 кг) и концентрировали до объема приблизительно 380 л с получением раствора 4-(2-(трет-бутокси)пиридин-4-ил)-2-(метилтио)пиримидина в ТГФ.
К данному ТГФ-раствору, охлажденному до 15-30°С, добавляли 1 н. водный раствор H2SO4 (311 кг). Полученную смесь перемешивали при этой же температуре в течение 4 ч. Вносили МТВЕ (280 кг), и рН реакционного раствора доводили до 14 путем добавления 30% водного раствора NaOH (120 кг). Водный слой отделяли, и органическую фазу фильтровали для удаления неорганических солей. Полученный водный слой промывали МТВЕ (2×280 кг). К водному раствору добавляли 2-MeTHF (1630 кг) и изо-РЮН (180 кг). Затем рН доводили до 8, медленно добавляя концентрированную HCl (19 кг). Органический слой отделяли, и водный слой экстрагировали 2-MeTHF (305 кг). Объединенные 2-MeTHF-экстракты промывали водой (300 кг) и концентрировали до объема приблизительно 100 л. Добавляли МТВЕ (230 кг), и перемешивали в течение 0,5 ч при 20-30°С. Твердое вещество отфильтровывали и суспендировали в смеси растворителей 2-MeTHF (68 кг) и МТВЕ (230 кг). Полученную суспензию перемешивали в течение 3 ч при 35-50°С и затем охлаждали до 0-10°С и дополнительно перемешивали в течение 2 ч. Твердое вещество отфильтровывали и сушили под вакуумом при 50-62°С в течение 20 ч с получением в качестве продукта 4-(2-(метилтио)пиримидин-4-ил)пиридин-2(1H)-она, в виде коричневого твердого вещества (33,55 кг, 89,6% анализ, выход: 19,4%).
Пример 3
(S)-1-(2-((трет-Бутилдиметилсилил)окси)-1-(4-хлор-3-фторфенил)этил)-4-(2-(метилтио)пиримидин-4-ил)пиридин-2(1Н)-он (XI)
Стадия 1. Из ТГФ-раствора 4-(2-(метилтио)пиримидин-4-ил)пиридин-2(1H)-она (25,5 кг) удаляли остатки воды путем выпаривали вместе с ТГФ. Добавляли сухой бис-(2-метоксиэтил)эфир (75 кг). Добавляли по каплям раствор KHMDS (131 кг, 1 М в ТГФ), поддерживая температуру в диапазоне от 25 до 40°С. Данную смесь нагревали до 75-80°С и перемешивали в течение 30-40 мин. Полученную смесь охлаждали до 20-30°С в атмосфере азота. В течение 30-60 мин добавляли раствор (R)-2-((трет-бутилдиметилсил ил)окси)-1-(4-хлор-3-фторфенил)этил метансульфоната (47,6 кг) в ТГФ (50 кг), поддерживая температуру в диапазоне от 20 до 40°С. Данный реакционный раствор нагревали до 80-85°С и перемешивали в течение 7 ч. Раствор охлаждали до 5-15°С, и добавляли воду (155 кг). рН раствора доводили до 7,5 путем добавления 30% водного раствора лимонной кислоты (30 кг). Добавляли EtOAc (460 кг), и данную смесь перемешивали в течение 20 мин. Органический слой отделяли и промывали 12,5% водным раствором NaCl (510 кг). Объединенные водные слои экстрагировали EtOAc (115 кг). Этилацетатные слои концентрировали до объема приблизительно 360 л с получением (S)-1-(2-((трет-бутилдиметилсилил)окси)-1-(4-хлор-3-фторфенил)этил)-4-(2-(метилтио)пиримидин-4-ил)пиридин-2(1 Н)-она (44,6 кг, выход: 75,7%) в виде продукта в EtOAc.
Стадия 2. К раствору (S)-1-(2-((трет-бутилдиметилсилил)окси)-1-(4-хлор-3-фторфенил)этил)-4-(2-(метилтио)пиримидин-4-ил)пиридин-2(1H)-она (44,6 кг) в EtOAc (401 кг, 10 об.), охлажденному до 5-10°С, добавляли порциями МСРВА (58 кг). К данной реакционной смеси добавляли раствор NaHCO3 (48,7 кг) в воде (304 кг) при температуре в диапазоне от 10 до -20°С. Для удаления остатков МСВРА добавляли по каплям раствор NaHCO3 (15 кг) в воде (150 кг). Органический слой отделяли, и водный слой экстрагировали EtOAc (130 кг). Объединенные органические слои промывали водой (301 кг), концентрировали, и растворитель заменяли на ДХМ с получением (S)-1-(2-((трет-бутилдиметилсилил)окси)-1-(4-хлор-3-фторфенил)этил)-4-(2-(метилсульфонил)пиримидин-4-ил)пиридин-2(1H)-она (45,0 кг, выход: 94,9%) в виде продукта в ДХМ. Данный ДХМ-раствор концентрировали до объема приблизительно 100 л, фильтровали через подушку SiO2 (60 кг) и элюировали градиентом EtOAc/ДХМ (0, 25 и 50% EtOAc). Фракции объединяли и концентрировали с получением продукта, который четыре раза ресуспендировали в смеси (ацетон: н-гептан = 1:3 об./об.) с получением конечного продукта (31,94 кг, выход: 71%).
Пример 4
(S)-1-(1-(4-Хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-он, бензолсульфонатная соль (VIIIb)
Стадия 1. В пустой цилиндрический химический реактор объемом 100 л вносили ТГФ (13 кг), затем добавляли при умеренном перемешивании последовательно (S)-1-(2-((трет-бутилдиметилсилил)окси)-1-(4-хлор-3-фторфенил)этил)-4-(2-(метилсульфонил)пиримидин-4-ил)пиридин-2(1 Н)-он (I, 5 кг) и 1-метил-1H-пиразол-5-амин (1,1 кг), и затем добавляли ТГФ (18 кг). Данную смесь охлаждали до -35°С, и к полученной жидкой суспензии медленно добавляли раствор LiHMDS (17,4 кг, 1,0 М) в ТГФ, со скоростью, которая позволяла поддерживать внутреннюю температуру ниже -25°С. После завершения добавления реакционную смесь выдерживали при температуре в диапазоне от -35 до -25°С в течение 20 мин и анализировали с использованием ВЭЖХ. Если согласно ВЭЖХ-анализу превращение составляло ≤98,5%, медленно добавляли еще одну порцию LiHMDS (0,34 кг, 1,0 М, 0,05 молярных %) при -35°С. Реакционную смесь медленно гасили при этой же температуре путем добавления раствора Н3РО4 (4,4 кг 85% Н3РО4 и 15 кг воды), и внутреннюю температуру поддерживали ниже 30°С. Реакционную смесь разбавляли EtOAc (18 кг), и фазы разделяли, органический слой промывали раствором Н3РО4 (1,1 кг 85% Н3РО4 и 12 кг воды) и затем второй раз промывали раствором Н3РО4 (0,55 кг 85% Н3РО4 и 12 кг воды). Если оставался 1-метил-1H-пиразол-5-амин, органический слой промывали еще раз раствором Н3РО4 (0,55 кг 85% Н3РО4 и 12 кг воды). В заключение органический слой промывали последовательно водой (20 кг) и раствором NaCl и NaHCO3 (2 кг NaCl, 0,35 кг NaHCO3 и 10 кг воды). После разделения фаз остаток воды в органическом растворе удаляли путем азеотропной дистилляции с EtOAc до ≤0,5% (согласно определению по Карлу Фишеру), и затем раствор концентрировали под вакуумом при температуре ниже 50°С до объема 20-30 л. Затем растворитель заменяли на МеОН с использованием 35 кг МеОН, и затем раствор концентрировали до объема 20-30 л и использовали на следующей стадии.
Стадия 2. К раствору (S)-1-(2-((трет-бутилдиметилсилил)окси)-1-(4-хлор-3-фторфенил)этил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-она (IX) в МеОН добавляли HCl (10,7 кг, 1,25 М в МеОН) при комнатной температуре. Наблюдали небольшое выделение тепла. После завершения добавления реакционную смесь нагревали до 45°С. Если реакция не завершалась через 14-16 ч, добавляли еще одну порцию HCl (1 кг, 1,25 М в МеОН) и перемешивание продолжали при 45°С в течение 2 ч. Реактор подключали к установке для перегонки с кислотным скруббером. Реакционную смесь концентрировали под вакуумом при температуре ниже 50°С до объема 20-30 л. К полученному раствору добавляли МеОН (35 кг), и данную реакционную смесь снова концентрировали под вакуумом при температуре ниже 50°С до объема 20-30 л. Затем растворитель заменяли на EtOAc с использованием 40 кг EtOAc. Соотношение исходного и нового растворителей определяли с использованием парофазной газовой хроматографии (Headspace GC), и процесс замены растворителя продолжали до тех пор, пока указанное отношение не становилось менее 1/5. Полученный раствор концентрировали под вакуумом при температуре ниже 50°С до объема 20-30 л. Раствор охлаждали до температуры ниже 30°С, затем медленно при умеренном перемешивании добавляли водный раствор NaHCO3 (1,2 кг NaHCO3 и 20 кг воды), и затем добавляли EtOAc (40 кг). Органический слой промывали водой (2×10 кг), затем концентрировали под вакуумом при температуре ниже 50°С до объема 20-30 л. Затем растворитель заменяли на MEK с использованием 35 кг MEK. Количество оставшегося МеОН определяли с использованием парофазной газовой хроматографии, и процесс замены растворителя продолжали до тех пор, пока количество МеОН не составляло <0,3%. Раствор, содержащий (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-он (VIII), концентрировали под вакуумом при температуре ниже 50°С до объема 20-30 л и использовали на следующей стадии.
Стадия 3. Раствор соединения VIII в MEK переносили во второй цилиндрический химический реактор объемом 100 л через встроенный фильтр с размером пор 1 мкм. В отдельном контейнере получали раствор бензолсульфоновой кислоты (1,3 кг бензолсульфоновой кислоты, 1,4 кг воды и 4,4 кг MEK). Отфильтрованный раствор соединения VIII нагревали до 75°С, и к полученному раствору добавляли через встроенный фильтр с размером пор 1 мкм 0,7 кг раствора бензолсульфоновой кислоты. В полученный прозрачный раствор вносили кристаллическую соль бензолсульфоновой кислоты соединения VIII (0,425 кг) в виде суспензии в MEK (0,025 кг кристаллической затравки соединения VIIIb и 0,4 кг MEK) с получением жидкой суспензии. Затем в течение 2 ч через встроенный фильтр с размером пор 1 мкм добавляли оставшийся раствор бензолсульфоновой кислоты. После добавления суспензию нагревали при 75°С дополнительно в течение 1 ч и затем охлаждали до 18°С минимум в течение
3 ч. Полученную густую суспензию перемешивали при 20°С в течение 14-16 ч. Твердое вещество отфильтровывали с использованием сушильного аппарата Aurora. Маточный раствор анализировали с использованием ВЭЖХ (потери составляли приблизительно 0,3%). Затем твердое вещество промывали с использованием 15,8 кг раствора MEK и воды (0,8 кг воды и 15 кг MEK), пропущенного через встроенный фильтр с размером пор 1 мкм, и затем с использованием 30 кг MEK, пропущенного через встроенный фильтр с размером пор 1 мкм. Промывочную жидкость анализировали с использованием ВЭЖХ (потери составляли<1%). Отфильтрованный осадок сушили под вакуумом и промывали азотом в течение минимум 12 ч, поддерживая температуру рубашки реактора при 45°С, с получением соли бензолсульфоновой кислоты соединения VIII, которая представляла собой желаемое соединения VIIIb.
Дополнительные Примеры
Стадия 1

В пустой цилиндрический химический реактор объемом 100 л вносили первую порцию ТГФ (13 кг). При умеренном перемешивании последовательно добавляли 5,0 кг соединения I и 1,1 кг 1-метил-1H-пиразол-5-амина и затем остаток ТГФ (18 кг). При -35°С к полученной жидкой суспензии медленно добавляли 17,4 кг LiHMDS (1,0 моль/л) в ТГФ, и внутреннюю температуру поддерживали ниже -25°С. После добавления реакционную смесь выдерживали при температуре в диапазоне от -35 до -25°С в течение 20 мин. За ходом реакции следили с использованием ВЭЖХ. Если согласно ВЭЖХ-анализу превращение составляло ≤98,5%, медленно добавляли еще 0,34 кг (0,05 молярных %) LiHMDS (1,0 моль/л) в ТГФ при -35°С. В противном случае, реакционную смесь гасили при этой же температуре, медленно добавляя 19,4 кг раствора Н3РО4 (4,4 кг 85% Н3РО4 и 15 кг воды), и внутреннюю температуру поддерживали ниже 30°С. Реакционную смесь разбавляли EtOAc (18 кг). Фазы разделяли, и органический слой промывали с использованием 13,1 кг раствора Н3РО4 (1,1 кг 85% Н3РО4 и 12 кг воды) и затем с использованием 12,6 кг раствора Н3РО4 (0,55 кг 85% Н3РО4 и 12 кг воды). Анализировали уровень 1-метил-1H-пиразол-5-амина в органическом слое с использованием ВЭЖХ. Если согласно ВЭЖХ-анализу уровень 1-метил-1H-пиразол-5-амина составлял ≥20 мкг/мл, органический слой дополнительно промывали с использованием 12,6 кг раствора Н3РО4 (0,55 кг 85% Н3РО4 и 12 кг воды). В противном случае, органический слой промывали с использованием 20 кг воды. Проводили повторный анализ содержания 1-метил-1H-пиразол-5-амина в органическом слое. Если согласно ВЭЖХ-анализу уровень 1-метил-1H-пиразол-5-амина составлял ≥2 мкг/мл, органический слой дополнительно промывали с использованием 20 кг воды. В противном случае, органический слой промывали с использованием 12,4 кг раствора NaCl и NaHCO3 (2 кг NaCl, 0,35 кг NaHCO3 и 10 кг воды). После разделения фаз остаток воды в органическом растворе удаляли путем азеотропной дистилляции с EtOAc до ≤0,5% (согласно определению по Карлу Фишеру), и затем раствор концентрировали под вакуумом при температуре ниже 50°С до объема 20-30 л. Затем растворитель заменяли на МеОН с использованием 35 кг МеОН, и затем раствор концентрировали до объема 20-30 л и использовали на следующей стадии.
Стадия 2
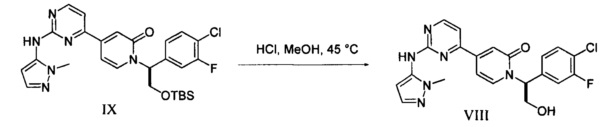
К раствору соединения IX в МеОН, полученному на предыдущей стадии, добавляли 10,7 кг HCl (1,25 М в МеОН) при комнатной температуре. Наблюдали небольшое выделение тепла. После добавления реакционную смесь нагревали до 45°С. Через 14-16 ч реакционную смесь анализировали с использованием ВЭЖХ. Если согласно ВЭЖХ-анализу превращение составляло ≤98%, добавляли еще 1 кг HCl (1,25 М в МеОН) и реакционную смесь перемешивали при 45°С дополнительно в течение 2 ч. В противном случае, реактор подключали к установке для перегонки с кислотным скруббером. Реакционную смесь концентрировали под вакуумом при температуре ниже 50°С до объема 20-30 л. К полученному раствору добавляли 35 кг МеОН, и реакционную смесь снова концентрировали под вакуумом при температуре ниже 50°С до объема 20-30. Затем растворитель заменяли на EtOAc с использованием 40 кг EtOAc. Соотношение исходного и нового растворителей определяли с использованием парофазной газовой хроматографии. Если отношение MeOH/EtOAc превышало значение 1/5, продолжали процесс замены растворителя. В противном случае, раствор концентрировали под вакуумом при температуре ниже 50°С до объема 20-30 л. После охлаждения раствора до температуры ниже 30°С медленно при умеренном перемешивании добавляли 21,2 кг раствора NaHCO3 (1,2 кг NaHCO3 и 20 кг воды), и затем добавляли 40 кг EtOAc. Фазы разделяли, и органический слой промывали водой (2×10 кг). Органический слой концентрировали под вакуумом при температуре ниже 50°С до объема 20-30 л. Затем растворитель заменяли на MEK с использованием 35 кг MEK. Количество оставшегося МеОН определяли с использованием парофазной газовой хроматографии. Если количество МеОН составляло ≥0,3%, продолжали процесс замены растворителя. В противном случае, раствор концентрировали под вакуумом при температуре ниже 50°С до объема 20-30 л и использовали на следующей стадии.
Стадия 3
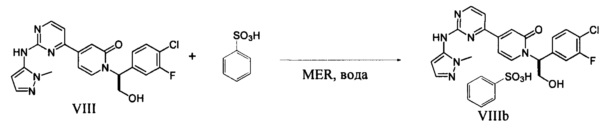
Раствор соединения VIII в MEK, полученный на предыдущей стадии, переносили во второй цилиндрический химический реактор объемом 100 л через встроенный фильтр с размером пор 3 мкм. В отдельном контейнере получали 7,1 кг раствора бензолсульфоновой кислоты (1,3 кг бензолсульфоновой кислоты, 1,4 кг воды и 4,4 кг MEK). Отфильтрованный раствор соединения G02584994 нагревали до 75°С, и к полученному раствору добавляли через встроенный фильтр с размером пор 3 мкм 0,7 кг раствора бензолсульфоновой кислоты (10%). В полученный прозрачный раствор вносили 0,425 кг кристаллической затравки соединения VIIIb в виде суспензии в MEK (0,025 кг кристаллической затравки соединения VIIIb и 0,4 кг MEK). Получали жидкую суспензию. Затем в течение 2 ч через встроенный фильтр с размером пор 3 мкм добавляли оставшийся раствор бензолсульфоновой кислоты. После добавления суспензию нагревали при 75°С дополнительно в течение 1 ч и затем охлаждали до 20°С в течение минимум 3 ч. Полученную густую суспензию перемешивали в течение 14-16 ч при 20°С. Твердое вещество отфильтровывали с использованием осушителя фильтра. Маточный раствор анализировали с использованием ВЭЖХ (потери составляли приблизительно 3%). Затем твердое вещество промывали с использованием 15,8 кг раствора MEK и воды (0,8 кг воды и 15 кг MEK), пропущенного через встроенный фильтр с размером пор 3 мкм, и затем с использованием 30 кг MEK, пропущенного через встроенный фильтр с размером пор 3 мкм. Промывочную жидкость анализировали с использованием ВЭЖХ (потери составляли <1%). Отфильтрованный осадок сушили под вакуумом и промывали азотом в течение минимум 12 ч, поддерживая температуру рубашки реактора при 45°С.
Перекристаллизация

В пустой цилиндрический химический реактор объемом 100 л вносили первую порцию EtOH (16 кг). При умеренном перемешивании добавляли 3,5 кг соединения VIIIb и затем остаток EtOH (8,5 кг). Полученную густую суспензию нагревали до 78°С, и добавляли воду (-1,1 кг) до тех пор, пока не получали прозрачный раствор. Горячий раствор фильтровали через встроенный фильтр с размером пор 3 мкм во второй пустой цилиндрический химический реактор объемом 100 л. Температура падала до 55-60°С, и отфильтрованный раствор оставался прозрачным. К полученному раствору добавляли 0,298 кг кристаллической затравки соединения VIIIb в виде суспензии в EtOH (0,018 кг кристаллической затравки соединения VIIIb и 0,28 кг EtOH). Полученную густую суспензию концентрировали под вакуумом при 60°С до объема 20-30 л и затем охлаждали до 20°С в течение 3 ч. Полученную суспензию перемешивали при 20°С в течение 14-16 ч. Твердое вещество отфильтровывали с использованием осушителя фильтра. Маточный раствор анализировали с использованием ВЭЖХ (потери составляли приблизительно 10%). Затем твердое вещество промывали с использованием 11,1 кг раствора ЕЮН и воды (0,56 кг воды и 11 кг EtOH), пропущенного через встроенный фильтр с размером пор 3 мкм, и затем с использованием 21 кг MEK, пропущенного через встроенный фильтр с размером пор 3 мкм. Промывочную жидкость анализировали с использованием ВЭЖХ (потери составляли 3%). Отфильтрованный осадок сушили под вакуумом и промывали азотом в течение минимум 12 ч, поддерживая температуру рубашки реактора при 45°С.
Дополнительный способ синтеза описан ниже.
Стадия 1

В пустой цилиндрический химический реактор объемом 100 л вносили первую порцию ТГФ (18 кг). При умеренном перемешивании последовательно добавляли 4,2 кг соединения I и 0,91 кг 1-метил-1H-пиразол-5-амина и затем остаток ТГФ (21 кг). При -40°С к полученной жидкой суспензии медленно добавляли 14,9 кг LiHMDS (1,0 моль/л) в ТГФ, и внутреннюю температуру поддерживали ниже -30°С. После добавления реакционную смесь выдерживали при температуре в диапазоне от -35 до -40°С в течение 20 мин. За ходом реакции следили с использованием ВЭЖХ. Согласно ВЭЖХ-анализу превращение составляло 99,1%. Реакционную смесь гасили при этой же температуре, медленно добавляя 16,7 кг раствора Н3РО4 (3,7 кг 85% Н3РО4 и 13 кг воды), и внутреннюю температуру поддерживали ниже 30°С. Реакционную смесь разбавляли EtOAc (17 кг). Фазы разделяли, и органический слой промывали с использованием 13,1 кг раствора Н3РО4 (1,1 кг 85% Н3РО4 и 12 кг воды) и затем с использованием 10,5 кг раствора Н3РО4 (0,46 кг 85% Н3РО4 и 10 кг воды). Анализировали уровень 1-метил- 1H-пиразол-5-амина в органическом слое с использованием ВЭЖХ. Согласно ВЭЖХ-анализу уровень 1-метил-1H-пиразол-5-амина составлял 2 мкг/мл. Органический слой промывали с использованием 15,8 кг раствора NaCl (0,3 кг NaCl и 15,5 кг воды). Проводили повторный анализ содержания соединения G02586778 в органическом слое. Согласно ВЭЖХ-анализу уровень 1-метил-1H-пиразол-5-амина составлял 0,5 мкг/мл. Органический слой промывали с использованием 10,3 кг раствор NaCl и NaHCO3 (1,7 кг NaCl, 0,6 кг NaHCO3 и 8 кг воды). После разделения фаз остаток воды в органическом растворе удаляли путем азеотропной дистилляции с EtOAc до ≤0,5% (согласно определению по Карлу Фишеру), и затем раствор концентрировали под вакуумом при температуре ниже 50°С до объема 20-30 л. Затем растворитель заменяли на МеОН с использованием 30 кг МеОН, и затем раствор концентрировали до объема 20-30 л и использовали на следующей стадии.
Стадия 2

К раствору соединения IX в МеОН, полученному на предыдущей стадии, добавляли 9,0 кг HCl (1,25 М в МеОН) при комнатной температуре. Наблюдали небольшое выделение тепла. После добавления реакционную смесь нагревали до 45°С. Через 16 ч реакционную смесь анализировали с использованием ВЭЖХ. Согласно ВЭЖХ-анализу превращение составляло 99,4%. Реактор подключали к установке для перегонки. Реакционную смесь концентрировали под вакуумом при температуре ниже 50°С до объема 20 л. К полученному раствору добавляли 35 кг МеОН, и реакционную смесь снова концентрировали под вакуумом при температуре ниже 50°С до объема 20 л. Затем растворитель заменяли на EtOAc с использованием 40 кг EtOAc. Соотношение исходного и нового растворителей определяли с использованием парофазной газовой хроматографии. Если отношение МеОН/EtOAc превышало значение 1/5, продолжали процесс замены растворителя. В противном случае, раствор концентрировали под вакуумом при температуре ниже 50°С до объема 20 л. После охлаждения раствора до температуры ниже 30°С медленно при умеренном перемешивании добавляли 18 кг раствора NaHCO3 (1 кг NaHCO3 и 17 кг воды), и затем добавляли 34 кг EtOAc. Фазы разделяли, и органический слой промывали водой (2×8 кг). Органический слой концентрировали под вакуумом при температуре ниже 50°С до объема 20 л. Затем растворитель заменяли на MEK с использованием 35 кг MEK. Количество оставшегося МеОН определяли с использованием парофазной газовой хроматографии. Если количество МеОН составляло ≥0,3%, продолжали процесс замены растворителя. В противном случае, раствор концентрировали под вакуумом при температуре ниже 50°С до объема 20 л и использовали на следующей стадии.
Стадия 3
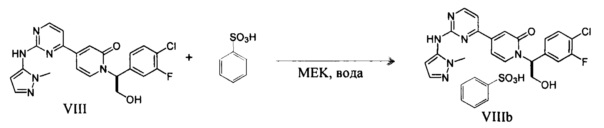
Раствор соединения VIII в MEK, полученный на предыдущей стадии, переносили во второй цилиндрический химический реактор объемом 100 л через фильтр тонкой очистки с размером пор 1 мкм. В отдельном контейнере получали 6,0 кг раствора бензолсульфоновой кислоты (1,1 кг бензолсульфоновой кислоты, 1,2 кг воды и 3,7 кг MEK). Отфильтрованный раствор соединения VIII нагревали до 75°С, и к полученному раствору добавляли через встроенный фильтр с размером пор 1 мкм 0,6 кг раствора бензолсульфоновой кислоты (10%). В полученный прозрачный раствор вносили 0,36 кг кристаллической затравки соединения VIIIb в виде суспензии в MEK (0,021 кг кристаллической затравки соединения VIIIb и 0,34 кг MEK). Получали жидкую суспензию. Затем в течение 2 ч через встроенный фильтр с размером пор 1 мкм добавляли оставшийся раствор бензолсульфоновой кислоты. После добавления суспензию нагревали при 75°С дополнительно в течение 1 ч и затем охлаждали до 18°С в течение минимум 3 ч. Полученную густую суспензию перемешивали в течение 14-16 ч при 18°С. Твердое вещество отфильтровывали с использованием сушильного аппарата Aurora. Затем твердое вещество промывали с использованием 8,15 кг раствора MEK и воды (0,35 кг воды и 7,8 кг MEK), пропущенного через встроенный фильтр с размером пор 1 мкм, и затем с использованием 12 кг MEK, пропущенного через встроенный фильтр с размером пор 1 мкм.
Перекристаллизация

В пустой цилиндрический химический реактор объемом 100 л вносили первую порцию EtOH (21 кг). При умеренном перемешивании добавляли 3,5 кг соединения VIIIb и затем остаток EtOH (9 кг). Полученную густую суспензию нагревали до 78°С, и добавляли воду (1,2 кг) до тех пор, пока не получали прозрачный раствор. Горячий раствор фильтровали через встроенный фильтр с размером пор 1 мкм во второй пустой цилиндрический химический реактор объемом 100 л. Температура падала до 69°С, и отфильтрованный раствор оставался прозрачным. К полученному раствору добавляли 0,37 кг кристаллической затравки соединения VIIIb в виде суспензии в EtOH (0,018 кг кристаллической затравки соединения VIIIb и 0,35 кг EtOH). Полученную жидкую суспензию концентрировали под вакуумом при 60-70°С до объема 20 л и затем охлаждали до 18°С в течение 3 ч. Полученную суспензию перемешивали в течение 14-16 ч при 18°С. Твердое вещество отфильтровывали с использованием осушителя фильтра. Затем твердое вещество промывали с использованием 8,6 кг раствора EtOH и воды (0,4 кг воды и 8,2 кг EtOH), пропущенного через встроенный фильтр с размером пор 1 мкм. Данный раствор вводили в два приема равными порциями. Затем твердое вещество промывали с использованием 6,7 кг MEK, пропущенного через встроенный фильтр с размером пор 1 мкм. Отфильтрованный осадок сушили под вакуумом и промывали азотом в течение минимум 12 ч, поддерживая температуру рубашки реактора при 35-40°С.
Альтернативный путь синтеза (стадии 1-10 ниже)

Стадия 1
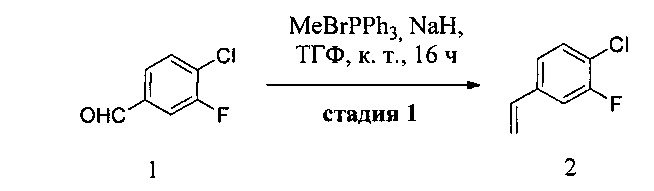
Методика
1. Соединение 1 и MeBrPPh3 вносили в атмосфере N2 в четырехгорлую колбу с теплозащитной рубашкой, оснащенную лопастной мешалкой.
2. В колбу добавляли ТГФ (5,0 V, KF<0,02%) (Примечание: V представляет собой отношение объема раствора к массе ключевого реагента, или л/кг).
3. Суспензию перемешивали при 0°С.
4. В колбу добавляли порциями NaH (60% суспензия в минеральном масле) при 0°С.
5. Перемешивали при 0°С в течение 30 мин.
6. Нагревали до 30°С, и перемешивали в течение 6 ч.
7. Охлаждали до 0°С.
8. В колбу добавляли РЕ (петролейный эфир) (5,0 V).
9. В колбу вносили затравочный кристалл ТРРО (трифенилфосфина оксида)(1 приблизительно 5% (по массе) от общего количества ТРРО).
10. Перемешивали при -10°С в течение 2 ч.
11. Фильтровали, и отфильтрованный осадок промывали с использованием РЕ (5,0 V).
12. Фильтрат концентрировали досуха.
13. В результате очистки полученного продукта путем перегонки при пониженном давлении получали соединение 2 в виде бесцветного масла.
Стадия 2

Методика
1. В колбу в атмосфере N2 вносили (DHQD)2PHAL, Na2CO3, K2Fe(CN)6, K2OsO2(OH)4 (Ad-mix beta, Aldrich, St. Louis, MO).
2. Охлаждали до 0°C.
3. Добавляли трет-BuOH (5 V) и H2O (5 V).
4. Добавляли соединение 2.
5. Смесь перемешивали в течение 6 ч при 0°С.
6. Охлаждали до 0°С.
7. Реакционную смесь гасили путем добавления Na2SO3.
8. Перемешивали при 0°С в течение 2 ч.
9. Фильтровали, и отфильтрованный осадок промывали ЕА (этилацетатом).
10. Органический слой отделяли.
11. Фильтровали, и концентрировали досуха.
Стадия 3
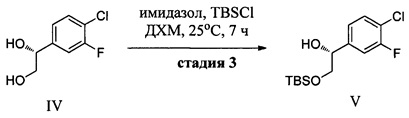
Методика
1. В колбу в атмосфере N2 вносили соединение IV (1 экв.) и ДХМ (5 V).
2. Охлаждали до 0°С.
3. Добавляли DMAP (0,1 экв.), затем TEA (1,5 экв.).
4. Добавляли по каплям при 0°С TBSC1 (1,05 экв.).
5. Смесь перемешивали в течение 1 ч при 0°С.
6. Реакционную смесь гасили путем добавления воды.
7. Слои разделяли.
8. Органический слой сушили над Na2SO4.
9. Фильтровали.
10. Фильтрат концентрировали досуха.
11. Использовали на следующей стадии без дополнительной очистки.
Стадия 4
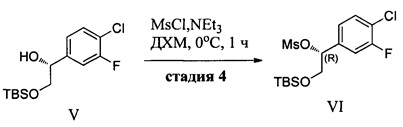
Методика
1. В колбу в атмосфере N2 вносили соединение V (1,0 экв.) и ДХМ (5 V).
2. Охлаждали до 0°С.
3. Добавляли TEA (1,51 экв.).
4. Добавляли по каплям при 0°С MsCl (1,05 экв.).
5. Смесь перемешивали в течение 1 ч при комнатной температуре.
6. Смесь разбавляли путем добавления ДХМ для лучшего перемешивания.
7. Реакционную смесь гасили путем добавления воды.
8. Слои разделяли.
9. Органический слой промывали NaHCO3.
10. Сушили над Na2SO4.
11. Фильтровали, и фильтрат концентрировали досуха.
12. Использовали на следующей стадии без дополнительной очистки.
Стадия 5
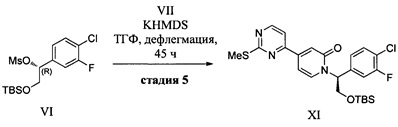
Методика
1. В колбу в атмосфере N2 вносили соединение VII (1 экв.) и DGME (20 V).
2. Охлаждали до 0°С.
3. Добавляли KHMDS (1 M в ТГФ, 1 экв.).
4. Добавляли соединение VI (1,2-1,5 экв.) в растворе DGME.
5. Перемешивали при 0°С в течение 5 мин.
6. Нагревали до температуры дефлегмации (температура рубашки 120°С), и перемешивали в течение 4 ч.
7. Охлаждали.
8. Гасили путем добавления воды, и экстрагировали МТВЕ.
9. Промывали с использованием 20% NaCl.
10. Сушили над Na2SO4.
11. Концентрировали досуха, и использовали на следующей стадии без дополнительной очистки.
Стадия 6
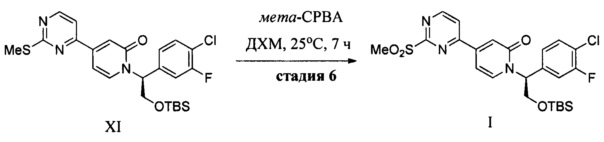
Методика
1. В колбу в атмосфере N2 вносили соединение XI (1 экв.), ДХМ (8 V).
2. Добавляли порциями мета-СРВА.
3. Перемешивали в течение 2 ч при комнатной температуре.
4. Промывали путем добавления 7% водного раствора NaHCO3.
5. Гасили путем добавления водного Na2S2O4.
6. Промывали 20% водным раствором NaCl.
7. Сушили над Na2SO4.
8. Фильтровали, и концентрировали досуха.
9. Суспендировали в МТВЕ (3 V) с получением соединения I.
Стадия 7
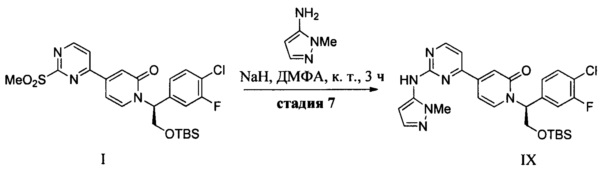
Методика
1. В колбу в атмосфере N2 вносили соединение I (1 экв.), 1-метил-1Н-пиразол-5-амин (4 экв.), Cs2CO3, ДМФА (4 V).
2. Перемешивали в течение 3 ч при комнатной температуре.
3. Выделяли продукт реакции с получением желаемого соединения.
Стадия 8

Методика
1. Соединение IX растворяли в МеОН.
2. Добавляли HCl (1,25 М в МеОН) при комнатной температуре.
3. После добавления реакционную смесь нагревали до 45°С в течение 16 ч.
4. Реакционную смесь охлаждали до комнатной температуры и гасили путем добавления водного NaHCO3, и разбавляли EtOAc (этилацетатом).
5. Фазы разделяли, и органический слой промывали водой. Органический слой концентрировали с получением неочищенного соединения VIII.
Стадия 9
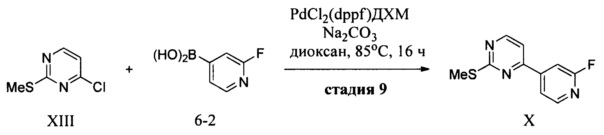
Методика
1. Соединение 6-2, соединение XIII, Pd-катализатор и бикарбонат натрия вносили в атмосфере N2 в четырехгорлую колбу с теплозащитной рубашкой, оснащенную лопастной мешалкой.
2. В колбу добавляли воду и 1,4-диоксан (5,0 V, KF<0,02%).
3. Суспензию перемешивали в течение 16 ч при 85°С.
4. Фильтровали через силикагель (2,0 X) и диатомовую землю (0,5 X)
5. Удаляли 1,4-диоксан путем перегонки под вакуумом.
6. Распределяли между водой (2,0 V) и EtOAc (5,0 V)
7. Органическую фазу отделяли и концентрировали.
8. Очищали путем перекристаллизации из РЕ и EtOAc.
Стадия 10
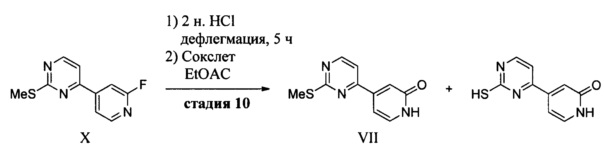
Методика
1. В колбу вносили соединение X.
2. Добавляли 2 М HCl (10-15 V).
3. Нагревали до 100°С, и перемешивали в течение 3 ч.
4. Охлаждали.
5. Нейтрализовали до рН 7-8 путем добавления 30% водного раствора NaOH.
6. Экстрагировали ТГФ.
7. Промывали 20% водным раствором NaCl.
8. Сушили над Na2SO4.
9. Фильтровали, и концентрировали досуха.
Синтез кристаллической (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1Н-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1Н)-она бензолсульфонатной соли
(S)-1-(1-(4-Хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-он (21,1 мг, 0,048 ммоль) растворяли в MEK (0,5 мл). Бензолсульфоновую кислоту (Fluka, 98%, 7,8 мг, 0,049 ммоль) растворяли в MEK (0,5 мл), и полученный раствор добавляли по каплям при перемешивании к приготовленному раствору соединения в форме свободного основания. Имело место выпадение осадка, который медленно растворялся по мере добавления дополнительной порции раствора бензолсульфоновой кислоты. На дне пробирки оставалось небольшое количество клейкого твердого вещества. Содержимое пробирки обрабатывали ультразвуком в течение 10 мин, при этом имело место дополнительное выпадение осадка. Твердое вещество выделяли путем центрифугирования и сушили под вакуумом при 40°С с использованием централизованной вакуумной системы (house vacuum).
Синтез (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1Н-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1Н)-она бензолсульфонатной соли кристаллической Формы А
(S)-1-(1-(4-Хлор-3-фторфенил)-2-гидроксиэтал)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-она бензолсульфонатную соль (23,1 мг) растворяли в горячем изопропаноле (5 мл) в термостате с установленной температурой 90°С. Нагревание термостата выключали, раствор оставляли охлаждаться до комнатной температуры и затем помещали в низкотемпературный шкаф (приблизительно -20°С). Твердое вещество собирали, пока раствор был холодным, согласно результатам XRPD-анализа полученное твердое вещество представляло собой (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-она бензолсульфонатную соль Формы А.
Синтез (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1Н-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1Н)-она бензолсульфонатной соли кристаллической Формы А в виде монокристаллов, подходящих для определения структуры монокристалла в соответствии с описанной ниже методикой
(S)-1-(1-(4-Хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-она бензолсульфонатная соль кристаллической Формы А. Кристаллы, подходящие по качеству для определения структуры, выращивали в метаноле с перемешиванием при температуре приблизительно 50°С, приблизительно через 1 сутки кристаллы выделяли с использованием масла Paratone-N и хранили в условиях окружающей среды.
Структурное решение. Бесцветный пластинчатый кристалл C27H23ClFN6O5S [C21H18ClFN6O2, C6H5O3S], имеющий приблизительные размеры 0,16×0,16×0,06 мм, монтировали на нити в произвольной ориентации. Предварительное тестирование и регистрацию данных проводили с использованием излучения Cu Kα (λ=1,54178Å) на дифрактометре Rigaku Rapid II, оснащенным конфокальной оптикой. Уточнения выполняли с использованием пакета программного обеспечения SHELX2013 [Sheldrick, G. М. Acta Cryst., 2008, А64, 112].
На основе полученного массива данных рассчитывали параметры решетки и матрицу ориентации путем уточнения методом наименьших квадратов с использованием установочных углов 24479 рефлексов в диапазоне 3°<θ<63°. Уточненная мозаичность, полученная с помощью DENZO/SCALEPACK, составляла 0,59°, что указывало на среднее качество кристаллов [Otwinowski, Z.; Minor, W. Methods Enzymol. 1997, 276, 307]. Пространственную группу определяли с использованием программы XPREP [Bruker, XPREP в пакете программного обеспечения SHELXTL v. 6.12., Bruker AXS Inc., Madison, WI, USA, 2002]. Систематические погасания рефлексов отсутствовали, пространственная группа кристалла была определена как P1 (№1).
Данные регистрировали при температуре 293±1 K, максимальный угол рассеяния 2θ составлял 126,9°.
Фреймы интегрировали с использованием программы HKL3000 [Flack, Н. D.; Bernardinelli, G., Acta Cryst. 1999, А55, 908]. В целом регистрировали 24479 рефлексов, из которых 6536 были уникальными. Данные корректировали с учетом фактора Лоренца и поляризационного фактора. Для излучения Си Ясс коэффициент линейного поглощения составлял 2,450 мм-1. Для коррекции эмпирической адсорбции использовали программу SCALEPACK [Otwinowski, Z.; Minor, W. Methods Enzymol. 1997, 276, 307]. Значения коэффициентов прохождения находились в диапазоне от 0,564 до 0,863. Также выполняли коррекцию вторичной экстинкции [Glusker, Jenny Pickworth; Trueblood, Kenneth N. Crystal Structure Analysis: A Primer, 2nd ed.; Oxford University press: New York, 1985; p.87]. Окончательное значение коэффициента, уточненное методом наименьших квадратов, составляло 0,00170 (в абсолютных единицах). Интенсивности эквивалентных рефлексов усредняли. Фактор согласия (agreement factor) для данного усреднения составлял 9,8% (на основе интенсивности).
Структура решали прямым методом с использованием SHELXT [Burla, М. С., Caliandro, R., Camalli, M. Carrozzini, В., Cascarano, G. L., De Caro, L., Giacovazzo, C, Polidori, G., and Spagna, R., J. Appl. Cryst. 2005, 38, 381]. Положения остальных атомов определяли из последующих разностных синтезов Фурье. Атомы водорода, принадлежащие атомам азота, уточняли независимо. Все другие атомы водорода включали в уточнение в модели "наездника" (атомы водорода уточняются в связке с атомом, с которым они связаны). Структуру уточняли методом наименьших квадратов в полноматричном приближении путем минимизации функции  .
.
Весовую функцию w определяли следующим образом: 1/[σ2(Fo2)+(0,2000Р)2+(0,0000Р)], где Р=(Fo2+2Fc2)/3.
Коэффициенты рассеяния были взяты из "International Tables for Crystallography" [International Tables for Crystallography, Vol. C, Kluwer Academic Publishers: Dordrecht, The Netherlands, 1992, Tables 4.2.6.8 и 6.1.1.4]. Из 6536 рефлексов, использованных для уточнений, только рефлексы с Fo2>2σ(Fo2) использовали для вычисления остаточного фактора R. Для вычисления было использовано 5796 рефлексов. Последний цикл уточнения включал 771 вариабельный параметр и сходился (самый большой сдвиг параметров составлял <0,01 от его рассчитанного стандартного отклонения) с невзвешенным и взвешенным факторами согласия

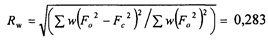 .
.
Стандартное отклонение наблюдаемой массы ячейки (добротность уточнения) составляло 1,385. Самый высокий пик в конечном разностном синтезе Фурье имел высоту 0,85 е/Å3. Это довольно большое значение, что указывало на плохое качество уточнения структуры. Минимальный негативный пик имел высоту -0,28 е/Å3. Фактор Флэка для определения абсолютной структуры [Flack, Н. D. Acta Cryst. 1983, А39, 876] был уточнен до -0,01(4).
Одну из гидроксильных групп одной из молекул в асимметрической ячейке уточняли с использованием разупорядочения. Это приводило к расщеплению атомов 022 и Н22 в парах атомных координат 022А, Н22А и 022 В, Н22 В.
Строение молекулы кристаллической Формы A (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-она бензолсульфонатной соли, определенное в результате анализа монокристалла, показано на Фигуре 3. Разупорядочение в гидроксиметильной группе можно наблюдать в верхнем правом углу Фигуры 3.
(S)-1-(1-(4-Хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-она бензолсульфонатная соль кристаллизуется в хиральную триклинную пространственную группу Р-1 с двумя симметрично независимыми катион-анионными парами. Хотя геометрические параметры указывают на то, что все межмолекулярные взаимодействия можно обрабатывать как относительно сильные, оба катиона характеризуется высокой степенью разупорядочения. Найдены две конформации хлорфторфенильных групп с относительным заполнением (occupancy ratio) приблизительно 60:40. Кроме того, гидроксиметильная группа также была разупорядочена с относительным заполнением приблизительно 50:50. Согласно установленной абсолютной стереохимии соединению была приписана S-конфигурация.
Синтез аморфной (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1Н-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1 Н)-она бензолсульфонатной соли
(S)-1-(1-(4-Хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-она бензолсульфонатную соль (39,6 мг) в трет-бутаноле (приблизительное 20 мл) нагревали до 60°С в термостате. Перед добавлением к безилатной соли трет-бутанол плавили приблизительно при 30°С. Добавляли воду (200 мкл), и нагревали до получения прозрачного раствора. Раствор охлаждали, фильтровали через фильтр с размером пор 0,2 мкм и помещали в а лиофилизатор. Соединение лиофилизировали с использованием сублимационной сушилки SP Scientific VirTis Advantage 2,0 Benchtop Freeze Dryer. Для удаления растворителя из соединения использовали 70-часовой режим сушки.
Вначале соединение замораживали под вакуумом в течение 1,5 ч при -70°С при давлении 500 мторр (66,65 Па). Данная процедура позволяла полностью заморозить внутренний раствор перед началом первичной сушки. Первичную сушку проводили с целью удаления основной массы растворителя путем сублимации. В течение 1 ч температуру увеличивали от -70°С до -35°С и давление уменьшали до 100 мторр (13,33 Па). После сушки в течение 1 ч при -35°С температуру увеличивали до 5°С и сушили дополнительно в течение 28 ч при том же давлении. Последнюю стадию первичной сушки проводили при температуре 15°С, которую поддерживали в течение 16 ч. Давление лиофилизации уменьшали до 50 мторр (6,665 Па), и температуру увеличивали до 35°С в течение 16 ч. Вторичную сушку продолжали в течение 6 ч понижая температуру до 30°С и давление до 10 мторр (1,333 Па). На последней стадии цикла лиофилизации температуру понижали до 25°С и давление увеличивали до 2500 мторр (333,25 Па) в течение 1 ч.
Синтез кристаллической формы I (5)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтш)-4-(2-((1-метил-1Н-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1Н)-она 1,5-нафталиндисульфоната
(S)-1-(1-(4-Хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-он (21,8 мг, 0,0494 ммоль) растворяли в MEK (0,5 мл). 1,5-Нафталиндисульфоновой кислоты тетрагидрат (25,1 мг, 0,0871 ммоль) растворяли в метаноле (1,0 мл), и приблизительно 0,36 мл полученного раствора добавляли по каплям при перемешивании к раствору свободного основания. Наблюдали выпадение осадка. Суспензию оставляли медленно упариваться, пока не оставалось следовое количество растворителя. Твердое вещество сушили под вакуумом при 40°С с использованием централизованной вакуумной системы.
Синтез кристаллической Формы II (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1Н-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1Н)-она 1,5-нафталиндисульфоната
(S)-1-(1-(4-Хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-он (103,3 мг, 0,234 ммоль) растворяли в MEK (2,5 мл). 1,5-Нафталиндисульфоновой кислоты тетрагидрат (110,4 мг, 0,383 ммоль) растворяли в метаноле (2,0 мл), и приблизительно 0,77 мл полученного раствора добавляли по каплям при перемешивании к раствору свободного основания. Наблюдали выпадение осадка, в том числе большого куска. Данный кусок разбивали шпателем, затем добавляли метанол (0,77 мл). Суспензию оставляли перемешиваться в течение 3 суток. Твердое вещество выделяли путем фильтрования и сушили при 60°С, используя централизованную вакуумную систему, с выходом желтого твердого вещества (57 мг).
Синтез IPA-солъвата (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1Н-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1Н)-она тозилата и Формы А (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1Н-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1Н)-она тозилата
(S)-1-(1-(4-Хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-он (105,1 мг, 0,239 ммоль) почти полностью растворяли в изопропаноле (1 мл) с использованием ультразвука. пара-Толуолсульфоновой кислоты моногидрат (чистота: 97,5%; 52,9 мг, 0,271 ммоль) растворяли в изопропаноле (1 мл). Данный раствор толуолсульфоновой кислоты добавляли по каплям при перемешивании к раствору свободного основания с получением желтого твердого вещества. Добавляли еще одну порцию изопропанола (1 мл). Полученное твердое вещество выделяли путем фильтрования, химический реактор и твердое вещество промывали 1 мл изопропанола. Твердое вещество анализировали с использованием XRPD, помещая его в держатель образца, который сообщался с атмосферой, пока твердое вещество оставалось влажным; получали разупорядоченный IPA-сольват (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-она тозилата. TG-анализ выполняли с использованием XRPD-образца.
Остальное твердое вещество сушили при 60°С под вакуумом в течение 4 суток с получением кристаллической Формы A (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидан-4-ил)пиридин-2(1H)-она тозилата.
Синтез аморфного (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1Н-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1Н)-она тозилата
(S-1-(1-(4-Хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-он (104,3 мг, 0,237 ммоль) растворяли в диэтиловом эфире (60 мл). пара-Толуолсульфоновой кислоты моногидрат (52,0 мг, 0,273 ммоль) растворяли в диэтиловом эфире (5 мл). Данный раствор толуолсульфоновой кислоты добавляли по каплям при перемешивании к раствору свободного основания, и полученную суспензию перемешивали в течение ночи. Полученный сложный эфир декантировали, и твердое вещество оставляли сушиться на воздухе с получением 103 мг желтого твердого вещества.
Синтез смеси аморфной формы и кристаллической Формы В (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1Н)-она тозилата
Аморфный (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-она тозилат (10,8 мг) помещали в пробирку с якорем для магнитной мешалки. Добавляли метилэтилкетон (MEK, 0,3 мл), и данную суспензию перемешивали в течение 4 суток. Растворитель выпаривали под вакуумом при 60°С с получением желтого твердого вещества.
Порошковые рентгеновские дифрактограммы образцов получали на порошковом рентгеновском дифрактометре Rigaku MiniFlexII с использованием геометрии отражения. Источник Cu-излучения использовали в режиме: напряжение = 30 кВ и ток = 15 мА. Каждый образец помещали в полость алюминиевого держателя образца, оснащенного кварцевой вставкой с нулевым уровнем шума, и разглаживали стеклянной пластиной для формирования хорошего рельефа поверхности, и помещали в держатель образца. Дифракцию образцов регистрировали в интервале значений угла 20 от 2° до 40° со скоростью сканирования 27 мин и шагом 0,02°.
Для измерения термических свойств порошковых образцов использовали дифференциальный сканирующий калориметр ТА Instruments (модель Q100 или модель Q2000) с механической системой охлаждения и стандартной ячейкой (форма которой соответствует форме тигля для образцов). Каждый образец вносили в алюминиевый тигель, закрываемый незапрессованной крышкой, без отверстий или содержащей одно отверстие, и помещали в ячейку дифференциального сканирующего калориметра (DSC). Ячейку продували азотом со скоростью приблизительно 50 см3/мин. Ячейку и образец уравновешивали при 20°С. Затем ячейку нагревали до 209°С или 250-350°С со скоростью 10,00°С/мин, регистрируя разницу в тепловых потоках пустого контрольного тигля и тигля с образцом.
Для анализа аморфного свободного основания (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-она использовали модулированную DSC. Для измерения термических свойств порошковых образцов использовали дифференциальный сканирующий калориметр ТА Instruments (модель Q2000) с механической системой охлаждения и стандартной ячейкой (форма которой соответствует форме тигля для образцов). Каждый образец вносили в алюминиевый тигель, закрываемый незапрессованной крышкой, без отверстий или содержащей одно отверстие, и помещали в ячейку дифференциального сканирующего калориметра (DSC). Ячейку продували азотом со скоростью приблизительно 50 см3/мин. Ячейку и образец уравновешивали при 25°С, температуру модулировали в режиме ±1°С каждые 60 секунд и поддерживали постоянной в течение 5 мин. Включали регистрацию данных, и температуру образца линейно увеличивали со скоростью 3°С/мин до 100°С. Затем температуру образца линейно уменьшали до 25°С со скоростью 3°С/мин. Затем образец нагревали со скоростью 3°С/мин до 200°С. Регистрировали инвертирующий сигнал.
Для автоматизированной регистрации данных при исследовании сорбции водяных паров использовали сорбционный анализатор водяных паров ТА Instruments Q5000SA. В качестве калибровочных стандартов использовали NaCl и PVP (поливинилпирролидон). Образцы перед анализом не сушили. Данные по адсорбции и десорбции регистрировали при 25°С при продувании азотом в диапазоне значений ОВВ (относительной влажности воздуха) от 5 до 95% с инкрементом 10% ОВВ. Образцы выдерживали при соответствующей ОВВ в течение 1 ч перед переходом к следующему диапазону ОВВ. Данные не корректировали на содержание начальной влажности в образцах.
Свободное основание S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-она представляет собой аморфное твердое вещество (XRPD, Фигура 4). Его температура стеклования (TG) изменяется в диапазоне приблизительно 74-96°С, в зависимости от степени чистоты и содержания растворителя, согласно данным, полученным с использованием дифференциальной сканирующей калориметрии (DSC, Фигура 5).
Выполнили приблизительно 200 безуспешных экспериментов по кристаллизации в попытке найти кристаллическую форму свободного основания (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-она. В нескольких экспериментах получали небольшое количество кристаллов, но впоследствии было установлено, что это кристаллы представляют собой загрязнения и образуются либо в процессе синтеза, либо из исходных веществ, и среди них не было кристаллов свободного основания (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-она. Исключение составляли эксперименты с использованием диффузии из паровой фазы, в которых в качестве растворителя присутствовал нитрометан и в качестве антирастворителя - гептан. Выделяли смесь аморфного и кристаллического вещества. Было установлено, что полученное кристаллическое вещество представляет собой ±1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-он.
Чтобы определить, можно ли получить подходящую форму соли, проводили скрининг солей. Определенное значение рКа свободного основания составляло менее 2, что ограничивало диапазон возможных солевых коформеров. Кроме того, соли, получаемые из свободного основания (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-она должны иметь pHmax менее 2 и, согласно ожиданиям, будут диспропорционированы в воде. Соответственно, оставалось неизвестным, может ли быть получена кристаллическая соль или будет ли соль подвергаться приемлемому воздействию in vivo (в водном окружении). Начальные попытки получения кристаллических солей с использованием соляной кислоты, серной кислоты, метансульфоновой кислоты, этансульфоновой кислоты, изэтионовой кислоты и этандисульфоновой кислоты провалились, и ни одну из указанных кристаллических солей не удалось получить. В конечном счете, получили кристаллические соли 1,5-нафталиндисульфоновой кислоты, пара-толуолсульфоновой кислоты и бензолсульфоновой кислоты.
Ниже приведены таблицы с первоначальными данными XRPD-пиков для солей, описанных в данной заявке. Специалистам в данной области техники хорошо известно, что эти пики могут быть сдвинуты вверх или вниз в зависимости от условий, в которых выполняли XRPD-анализ. В общем случае, сдвиг пиков может составлять ±0,2. Согласно другому аспекту настоящего изобретения, сдвиг пиков может составлять ±0,1.
Безилатная соль
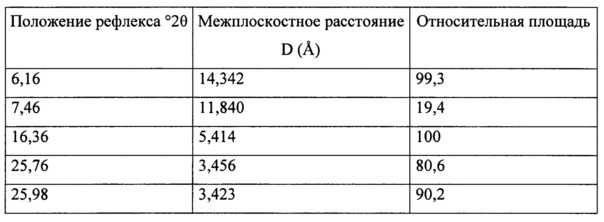
Тозилата IPA-сольват
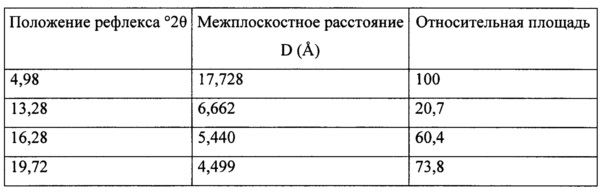
Тозилата Форма А

Тозилата Форма В

Нафталиндисульфоновой кислоты Форма I

Нафталиндисульфоновой кислоты Форма II

Содержит ли фармацевтический препарат (обычно таблетка или капсула) конкретную кристаллическую форму вещества, может быть определено, например, с использованием рентгеновской дифракции, спектроскопии рамановского рассеяния и/или методик твердотельного ЯМР. Например, 13С- и 19F-спектры кристаллической Формы А (S)-1-(1-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-она бензолсульфонатной соли, полученные с использованием твердотельной ЯМР-спектроскопии. приведены на Фигурах 19 и 20, соответственно. Методика получения данных ЯМР-спектров описана ниже.
Кристаллическую Форму А (S)-1-(1-(4-Хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-она бензолсульфонатной соли анализировали с использованием 13С и 19F твердотельной ЯМР-спектроскопии. Спектры регистрировали с использованием ЯМР-спектрометра Bruker Avance III в режиме 500,13 МГц для ядер 1Н, 125,77 МГц для ядер 13С и 470,55 МГц для ядер 19F. В 13С-экспериментах использовали датчик двойного резонанса (Bruker НХ) в диапазоне наблюдаемых ядер 1Н и 13С, с модулем вращения образца под магическим углом (MAS) диаметром 4 мм. В 19F-экспериментах использовали датчик тройного резонанса (Bruker HFC) в диапазоне наблюдаемых ядер 1Н, 19F и 13С, также оснащенный MAS-модулем диаметром 4 мм. Образцы упаковывали в изготовленные из ZrO2 роторы диаметром 4 мм и плотно закрывали крышками-приводами, изготовленными из полимера Kel-F. Все спектры регистрировали при 293 К. Спектры регистрировали, обрабатывали и анализировали с использованием программы Bruker TopSpin™ 3.2.
Импульсная последовательность при регистрации 13С-спектров включала кросс-поляризацию (CP) с линейным изменением амплитуды импульсов1-3, 5-π полное подавлением боковых полос (TOSS)4 и мощную развязку от 1Н по схеме SPINAL645 с использованием сильного поля 90 кГц. Вращение образца под магическим углом (MAS) выполняли при 8000±3 Гц. Ширина импульса 1Н 90° составляла 2,79 мс, и ширина импульсов 13С 180° в импульсной последовательности TOSS составляла 6,50 мс. Длительность контактного импульса CP составляла 3 мс, время ожидания восстановления намагниченности (recycle delay) составляло 18 с, всего 3888 накоплений сигнала усредняли для получения спектра. Химические сдвиги привязывали к внешнему стандарту, устанавливая пик метальной группы 3-метилглутаровой кислоты на 18,84 м.д. относительно тетраметилсилана.6
Импульсная последовательность при регистрации 19F-спектров включала кросс-поляризацию (CP) с линейным изменением амплитуды импульсов1-3 и мощную развязку от 1Н по схеме SPINAL645 с использованием сильного поля 71 кГц. Вращение образца под магическим углом (MAS) выполняли при 14000±5 Гц. Ширина импульса 1Н 90° составляла 3,54 мс, длительность контактного импульса CP составляла 3 мс, время ожидания восстановления намагниченности составляло 18 с, всего 16 накоплений сигнала усредняли для получения спектра. Химические сдвиги привязывали к внешнему стандарту, устанавливая пик фтора политетрафторэтилена (PTFE) на -122,38 м.д. относительно CFCl3 (определенному экспериментально по пику CFCl3 в PTFE-образце).
Список цитированной литературы по ЯМР
1. Pines, A.; Gibby, М. G.; Waugh, J. S., Proton-enhanced nuclear induction spectroscopy. Method for high-resolution NMR of dilute spins in solids. J. Chem. Phys. 1912, 56 (4), 1776-7.
2. Stejskal, E. O.; Schaefer, J.; Waugh, J. S., Magic-angle spinning and polarization transfer in proton-enhanced NMR. J. Magn. Reson. (1969-1992) 1977, 28 (1), 105-12.
3. Metz, G.; Wu, X.; Smith, S. O., Ramped-amplitude cross polarization in magic-angle-spinning NMR. J. Magn. Reson. Ser. A 1994,110 (2), 219-27.
4. Song, Z.; Antzutkin, O. N.; Feng, X.; Levitt, M. H., Sideband suppression in magic-angle-spinning NMR by a sequence of 5 pi pulses. Solid State Nucl. Magn. Reson. 1993, 2 (3), 143-6.
5. Fung, В. M.; Khitrin, A. K.; Ermolaev, K., An improved broadband decoupling sequence for liquid crystals and solids. J. Magn. Reson. 2000, 142 (1), 97-101.
6. Barich, D. H.; Gorman, E. M.; Zell, M. Т.; Munson, E. J., 3-Methylglutaric acid as a 13C solid-state NMR standard. Solid State Nucl. Magn. Reson. 2006, 30 (3-4), 125-129.
13С-спектр кристаллической Формы A (S)-1-(l-(4-хлор-3-фторфенил)-2-гидроксиэтил)-4-(2-((1-метил-1H-пиразол-5-ил)амино)пиримидин-4-ил)пиридин-2(1H)-она бензолсульфонатной соли, полученный с использованием твердотельной ЯМР-спектроскопии, характеризуется сильными пиками с химическими сдвигами 157,7±0,2 м.д., 129,6±0,2 м.д., 125,8±0,2 м.д. и 117,0±0,2 м.д. относительно тетраметилсилана (при 293 К). 19F-спектр характеризуется двумя изотропными пиками с химическими сдвигами -111,1±0,4 м.д.: и -115,4±0,4 м.д.: относительно CFCl3 (при 293 K).
Кристаллическая безилатная соль соединения VIII представляет собой высококристаллическое вещество с точкой плавления, которая приемлема для разработки фармацевтической лекарственной формы. Форма безилатной соли более предпочтительна, чем формы тозилатной соли и соли 1,5-нафталинсульфоновой кислоты, так как она имеет простой структурный ландшафт (идентифицирована только одна кристаллическая форма). Кроме того, более низкая гигроскопичность безилатной соли по сравнению с формами тозилатной соли и соли 1,5-нафталиндисульфоновой кислоты делает эту соль весьма привлекательной. Свободное основание соединения VIII имеет низкое значение pKa, менее 1,8, и поэтому можно ожидать, что любые полученные соли будут нестабильны в присутствии воды вследствие диспропорционирования с образование свободного основания и кислоты. Поэтому негигроскопичность безилатной соли была неожиданностью и позволяла повысить стабильность по сравнению с формами тозилатной соли и соли нафталинсульфоновой кислоты.
Согласно другому аспекту настоящего изобретения, предложены фармацевтические композиции, содержащие соединение по настоящему изобретению или соль или кристаллическую форму соли данного соединения и фармацевтически приемлемый эксципиент, такой как носитель, адъювант или наполнитель. Согласно некоторым вариантам осуществления настоящего изобретения указанная композиция представлена в виде препарата для введения нуждающемуся в этом пациенту.
Термин "пациент" или "индивидуум" в контексте данного описания относится к животному, такому как млекопитающее, например человек. Согласно одному из вариантов осуществления настоящего изобретения пациент или индивидуум относится к человеку.
Термин "фармацевтически приемлемый" означает, что указанное соединение или композиция совместима химически и/или токсикологически с другими ингредиентами (такими как эксципиенты), содержащимися в препарате, и/или с пациентом, который получает лечение, в особенности с человеком.
Термин "фармацевтически приемлемый носитель, адъювант или наполнитель " относится к нетоксичному носителю, адъюванту или наполнителю, который не нарушает фармакологическую активность соединения, вместе с которым он входит в состав препарата. Фармацевтически приемлемые носители, адъюванты или наполнители, которые могут быть использованы в композициях согласно настоящему изобретению, включают ионообменные вещества, оксид алюминия, стеарат алюминия, лецитин, сывороточные белки, такие как человеческий сывороточный альбумин, буферные вещества, такие как фосфаты, глицин, сорбиновую кислоту, сорбат калия, смеси неполных эфиров глицерина с насыщенными жирными кислотами растительного происхождения, воду, соли или электролиты, такие как протаминсульфат, гидроортофосфат натрия, гидроортофосфат калия, хлорид натрия, соли цинка, коллоидный кремнезем, трисиликат магния, поливинилпирролидон, вещества на основе целлюлозы, полиэтиленгликоль, натрий-карбоксиметилцеллюлозу, полиакрилаты, воски, полиэтилен-полиоксипропилен-блок-полимеры, полиэтиленгликоль и ланолин, но не ограничены ими.
Композиции, содержащие соединение согласно настоящему изобретению, могут быть введены перорально, парентерально, с использованием ингаляционного спрея, с использованием местного, трансдермального, ректального, назального, трансбуккального, сублингвального, вагинального, интраперитонеального, внутрилегочного, интрадермального, эпидурального пути введения или через имплантированный резервуар. Термин "парентеральный" в контексте данного описания включает методики подкожной, внутривенной, внутримышечной, интраартикулярной, интрасиновиальной, интрастернальной, интратекальной, внутрипеченочной, внутриочаговой и интракраниальной инъекции или инфузии.
Согласно одному из вариантов осуществления изобретения, композиция, содержащая соединение согласно настоящему изобретению, представлена в виде твердой лекарственной формы для перорального введения. Твердые лекарственные формы для перорального введения включают капсулы, таблетки, пилюли, порошки и гранулы. Согласно некоторым вариантам осуществления настоящего изобретения, твердая пероральная лекарственная форма, содержащая соединение Формулы (I) или его соль, дополнительно содержит один или более чем один ингредиент, выбранный из (i) инертного фармацевтически приемлемого эксципиента или носителя, таких как цитрат натрия или гидрофосфат кальция, и (ii) наполнителя или разбавителя, таких как крахмал, лактоза, сахароза, глюкоза, маннит или кремниевая кислота, (iii) связующих веществ, таких как карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидинон, сахароза или гуммиарабик, (iv) увлажнителей, таких как глицерин, (v) разрыхляющего агента, такого как агар, карбонат кальция, картофельный или маниоковый крахмал, альгиновая кислота, некоторые силикаты или карбонат натрия, (vi) агентов для замедления растворения, таких как парафин, (vii) ускорителей абсорбции, таких как четвертичные аммониевые соли, (viii) увлажняющего агента, такого как цетиловый спирт или моностеарат глицерина, (ix) абсорбента, такого как каолин или бентонитовая глина, и (х) смазывающего вещества, такого как тальк, стеарат кальция, стеарат магния, полиэтиленгликоль или лаурилсульфат натрия. Согласно некоторым вариантам осуществления настоящего изобретения, твердая пероральная лекарственная форма представлена препаратами в виде капсул, таблеток или пилюль. Согласно некоторым вариантам осуществления настоящего изобретения, твердая пероральная лекарственная форма дополнительно содержит буферные агенты. Согласно некоторым вариантам осуществления изобретения, такие композиции для твердых пероральных лекарственных форм могут быть представлены в виде наполнителей в мягких и твердых желатиновых капсулах, содержащих один или более чем один эксципиент, такой как лактоза, или молочный сахар, полиэтиленгликоль и тому подобное.
Согласно некоторым вариантам осуществления настоящего изобретения, композиции, содержащие соединение Формулы I или его соль, в форме таблеток, драже, капсул, пилюль и гранул необязательно содержат покрытия или оболочки, такие как кишечно-растворимые покрытия. Данные лекарственные формы могут необязательно содержать замутняющие агенты и также могут высвобождать только, или преимущественно, активный(ые) ингредиент(ы) композиции в определенной части кишечного тракта, необязательно в режиме отсроченного высвобождения. Примеры заливочных (embedding) композиций включают полимерные вещества и воски, которые также могут быть использованы в качестве наполнителей в мягких и твердых желатиновых капсулах с использованием таких эксципиентов, как лактоза, или молочный сахар, а также высокомолекулярные полиэтиленгликоли и тому подобное.
Согласно другому варианту осуществления изобретения, композиция содержит микроинкапсулированное соединение согласно настоящему изобретению и необязательно дополнительно содержит один или более чем один эксципиент.
Согласно другому варианту осуществления настоящего изобретения, композиции включают жидкие лекарственные препараты, содержащие соединение Формулы I или его соль, для перорального введения и необязательно дополнительно включают один или более чем один жидкий лекарственный препарат, выбранный из фармацевтически приемлемых эмульсий, микроэмульсий растворов, суспензий, сиропов и эликсиров. Согласно некоторым вариантам осуществления настоящего изобретения, жидкая лекарственная форма необязательно дополнительно содержит один или более чем один ингредиент, выбранный из инертного разбавителя, такого как вода или другой растворитель, солюбилизирующего агента и эмульгатора, такого как этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, диметилформамид, масла (в частности хлопковое, арахисовое, кукурузное масло, масло зародышей пшеницы, оливковое, касторовое и кунжутное масло), глицерин, тетрагидрофурфуриловый спирт, полиэтиленгликоли или эфиры сорбита и жирных кислот и их смеси. Согласно некоторым вариантам осуществления настоящего изобретения, жидкие пероральные композиции необязательно дополнительно содержат один или более чем один адьювант, такой как увлажняющий агент, суспендирующий агент, подслащивающий агент, корригент и ароматизирующий агент.
Инъекционные препараты, например, стерильные инъекционные водные или масляные суспензии, могут быть изготовлены в соответствии с известными в данной области техники методиками с использованием подходящих диспергирующих или увлажняющих агентов и суспендирующих агентов. Стерильный инъекционный препарат также может представлять собой стерильный инъекционный раствор, суспензию или эмульсию в нетоксичном и приемлемом для парентерального введения разбавителе или растворителе, например раствор в 1,3-бутандиоле. Приемлемые наполнители и растворители, которые могут быть использованы, включают воду, раствор Рингера, U.S.Р. и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или суспендирующей среды обычно используют стерильные нелетучие масла. Для этой цели может быть использовано любое безвкусное нелетучее масло, включая синтетические моно- или диглицериды. Кроме того, для получения инъекционных препаратов используют жирные кислоты, такие как олеиновая кислота.
Инъекционные препараты могут быть стерилизованы, например, путем фильтрования через фильтр, задерживающий бактерии, или путем введения стерилизующих агентов в твердую лекарственную форму стерильных композиций, которая перед использованием может быть растворена или диспергирована в стерильной воде или другой стерильной инъекционной среде.
Для пролонгирования действия соединения согласно настоящему изобретению часто желательно замедлить абсорбцию соединения после его введения путем подкожной или внутримышечной инъекции. Это может быть достигнуто путем использования жидкой суспензии кристаллического или аморфного вещества, обладающего плохой растворимостью в воде. Скорость абсорбции соединения зависит от скорости его растворения, которая, в свою очередь, может зависеть от размера кристаллов и кристаллической формы. Альтернативно, отсроченной абсорбции парентерально вводимой формы соединения можно добиться путем растворения или суспендирования соединения в масляном наполнителе. Инъекционные формы замедленного высвобождения получают путем формирования микроинкапсулированного матрикса соединения в биодеградируемых полимерах, таких как полилактид-полигликолид. Скорость высвобождения соединения можно контролировать, меняя соотношение соединения и полимера и используя полимеры определенной природы. Примеры других биодеградируемых полимеров включают поли(ортоэфиры) и поли(ангидриды). Инъекционные препараты замедленного высвобождения также получают путем заключения соединения в липосомы или микроэмульсии, которые совместимы с тканями организма.
Согласно некоторым вариантам осуществления настоящего изобретения, композиция для ректального или вагинального введения представлена в форме суппозиториев, которые могут быть изготовлены путем смешивания соединения согласно настоящему изобретению с подходящими не вызывающими раздражение эксципиентами или носителями, такими как масло какао, полиэтиленгликоль или воск для суппозиториев, например с такими, которые остаются твердыми при температуре окружающей среды, но становятся жидкими при температуре тела и поэтому плавятся при введении в прямую кишку или влагалище и высвобождают соединение согласно настоящему изобретению.
Примеры лекарственных форм для местного или трансдермального введения соединения согласно настоящему изобретению включают мази, пасты, кремы, лосьоны, гели, порошки, растворы, спреи, средства для ингаляции или пластыри. Соединение согласно настоящему изобретению смешивают в стерильных условиях с фармацевтически приемлемым носителем и необязательно с консервантами или буферными агентами. Дополнительные примеры препаратов включают офтальмологический препарат, ушные капли, глазные капли или чрезкожные пластыри. Трансдермальные лекарственные формы могут быть изготовлены путем растворения или распределения соединения согласно настоящему изобретению в среде, например в этаноле или диметилсульфоксиде. Для усиления потока соединения через кожу также могут быть использованы агенты, усиливающие абсорбцию. Скорость можно контролировать либо путем использования регулирующей скорость мембраны, либо путем диспергирования соединения в полимерном матриксе или геле.
Назальные аэрозоли или препараты для ингаляции соединения согласно настоящему изобретению могут быть изготовлены в форме растворов в физиологическом растворе, с использованием бензилового спирта или других подходящих консервантов, усилителей абсорбции для повышения биодоступности, фторуглеродов и/или других стандартных солюбилизирующих или диспергирующих агентов.
Согласно некоторым вариантам осуществления настоящего изобретения, фармацевтические композиции могут быть введены во время еды или отдельно от еды. Согласно некоторым вариантам осуществления настоящего изобретения, фармацевтически приемлемые композиции вводят отдельно от еды. Согласно некоторым вариантам осуществления изобретения, фармацевтически приемлемые композиции согласно настоящему изобретению вводят во время еды.
Конкретная доза и схема лечения, назначаемые конкретному пациенту, будут зависеть от ряда факторов, включая возраст, массу тела, общее состояние здоровья, пол, диету, время введения, скорость экскреции, комбинирование лекарств, мнение лечащего врача и тяжесть конкретного заболевания, которое лечат. Количество соединения согласно настоящему изобретению в композиции также будет зависеть от конкретного соединения, используемого в композиции.
Согласно одному из вариантов осуществления настоящего изобретения, терапевтически эффективная доза соединения согласно изобретению, предназначенная для парентерального введения, находится в диапазоне приблизительно 0,01-100 мг/кг, альтернативно в диапазоне приблизительно 0,1-20 мг/кг массы тела пациента в сутки, обычно начальная доза соединения находится в диапазоне 0,3-15 мг/кг/сутки. Согласно другому варианту осуществления настоящего изобретения, пероральные стандартные лекарственные формы, такие как таблетки и капсулы, содержат от приблизительно 5 до приблизительно 100 мг соединения согласно изобретению.
Например, таблетированная пероральная лекарственная форма содержит приблизительно 2 мг, 5 мг, 25 мг, 50 мг, 100 мг, 250 мг или 500 мг соединения Формулы (I) или его соли и дополнительно содержит приблизительно 5-30 мг безводной лактозы, приблизительно 5-40 мг кроскармелозы натрия, приблизительно 5-30 мг поливинилпирролидона (PVP) K30 и приблизительно 1-10 мг стеарата магния. Процесс изготовления данной таблетки включает смешивание всех порошковых ингредиентов вместе и последующее смешивание с раствором PVP. Полученная композиция может быть подвергнута сушке, грануляции, смешиванию со стеаратом магния и прессованию с получением таблетированной формы с использованием стандартного оборудования. Аэрозольный препарат, в качестве примера, может быть изготовлен путем растворения приблизительно 2-500 мг соединения Формулы I или его соли в подходящем буферном растворе, например в фосфатном буфере, и добавления, при желании, регулятора тоничности раствора (tonicifier), например соли, такой как хлорид натрия. Данный раствор может быть отфильтрован, например, с использованием 0,2-микронного фильтра, для удаления примесей и загрязняющих веществ.
Отличительные признаки изобретения, раскрытые выше в данном описании или в следующей далее формуле изобретения, выраженные в их конкретных формах или в терминах средств осуществления раскрываемой функции, или способа, или процесса, направленных на достижение раскрытого здесь результата, при необходимости, по отдельности или в виде любой комбинации таких признаков, могут применяться для осуществления настоящего изобретения в его разнообразных формах.
Для большей ясности и понимания некоторые детали настоящего изобретения описаны с использованием иллюстраций и примеров. Специалисту в данной области техники должно быть очевидно, что применение изобретения на практике допускает, что могут быть сделаны изменения и модификации, которые не выходят за пределы объема прилагаемой формулы изобретения. Поэтому необходимо понимать, что данное описание является иллюстративным и не ограничивает объем изобретения. Поэтому объем настоящего изобретения следует определять, руководствуясь не приведенным выше описанием, а прилагаемой формулой изобретения, вместе с полным объемом эквивалентов, на которые такая формула изобретения дает право.
Патенты, опубликованные заявки и научная литература, на которые ссылается данное описание, устанавливают уровень знаний специалистов в данной области техники и тем самым включены в данное описание посредством ссылки во всей своей полноте, как если бы для каждой из публикаций по отдельности было указано, что конкретно она включена посредством ссылки. Любое противоречие между любой цитированной здесь ссылкой и конкретными описаниями, приведенными в данной заявке, должно быть разрешено в пользу последних. Точно также любое противоречие между определением слова или фразы, принятым в данной области техники, и конкретным определением того же слова или фразы в данном описании должно быть разрешено в пользу последнего.
| название | год | авторы | номер документа |
|---|---|---|---|
| ГЕТЕРОЦИКЛИЧЕСКИЕ АНАЛОГИ ПИРИМИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ JAK | 2012 |
|
RU2564419C1 |
| ИНГИБИТОРЫ СЕРИН/ТРЕОНИНОВЫХ КИНАЗ | 2013 |
|
RU2650501C2 |
| КОНДЕНСИРОВАННЫЕ ТРИАЗОЛАМИНЫ В КАЧЕСТВЕ МОДУЛЯТОРОВ Р2Х7 | 2010 |
|
RU2533122C2 |
| ФЕНОКСИМЕТИЛЬНЫЕ ПРОИЗВОДНЫЕ | 2016 |
|
RU2746481C1 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ | 2005 |
|
RU2390522C2 |
| ИНГИБИТОРЫ СЕРИН/ТРЕОНИН КИНАЗЫ ДЛЯ ЛЕЧЕНИЯ ГИПЕРПРОЛИФЕРАТИВНЫХ ЗАБОЛЕВАНИЙ | 2013 |
|
RU2644947C2 |
| ПРОЛЕКАРСТВА СОПРЯЖЕННО-БИЦИКЛИЧЕСКИХ АНТАГОНИСТОВ C5aR | 2019 |
|
RU2794327C2 |
| ПРОИЗВОДНЫЕ АМИНОПИРАЗИНА И ЛЕКАРСТВЕННЫЕ СРЕДСТВА | 2010 |
|
RU2535217C2 |
| ЗАМЕЩЕННЫЕ ПИРИДИНОВЫЕ И ПИРАЗИНОВЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ PDE4 | 2014 |
|
RU2802185C2 |
| ТИАЗОЛОПИРИМИДИНОНЫ В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ РЕЦЕПТОРОВ NMDA | 2014 |
|
RU2703273C2 |
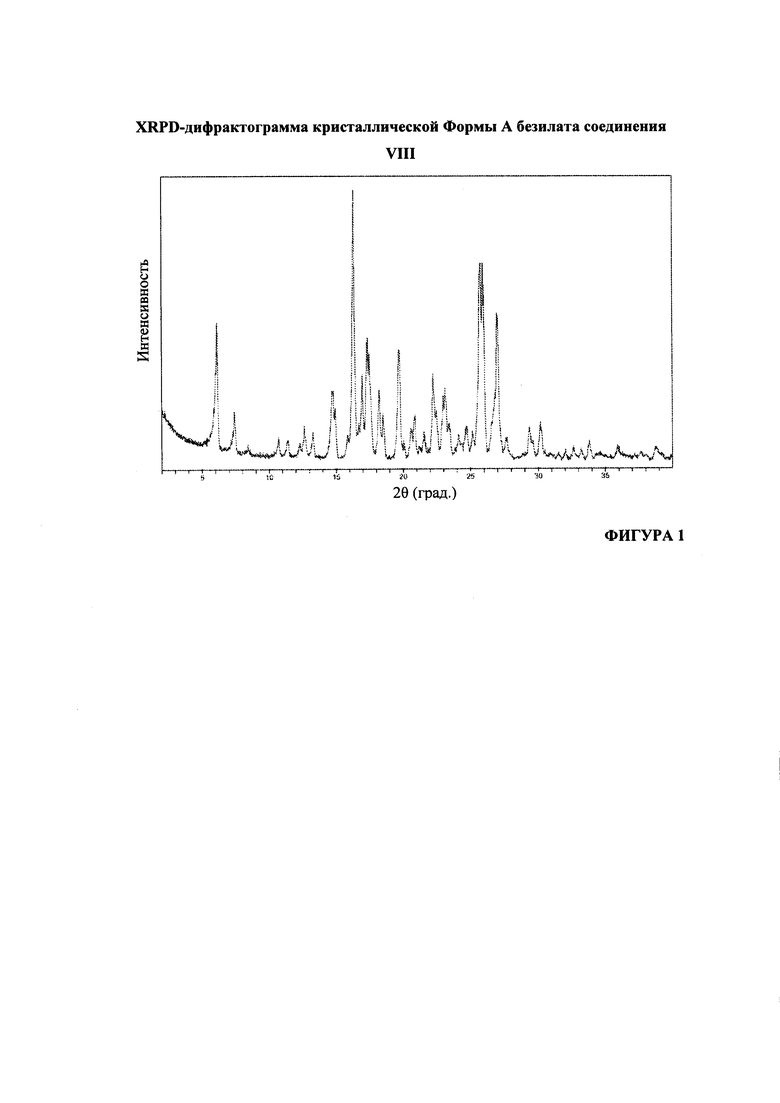
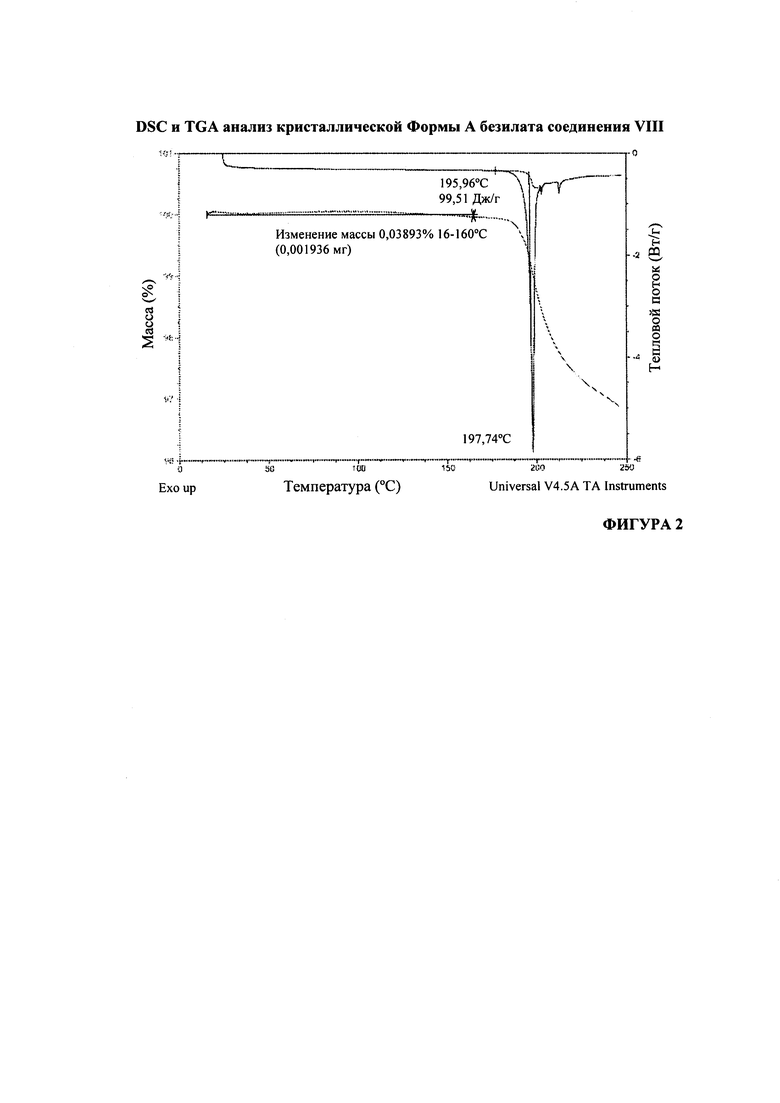

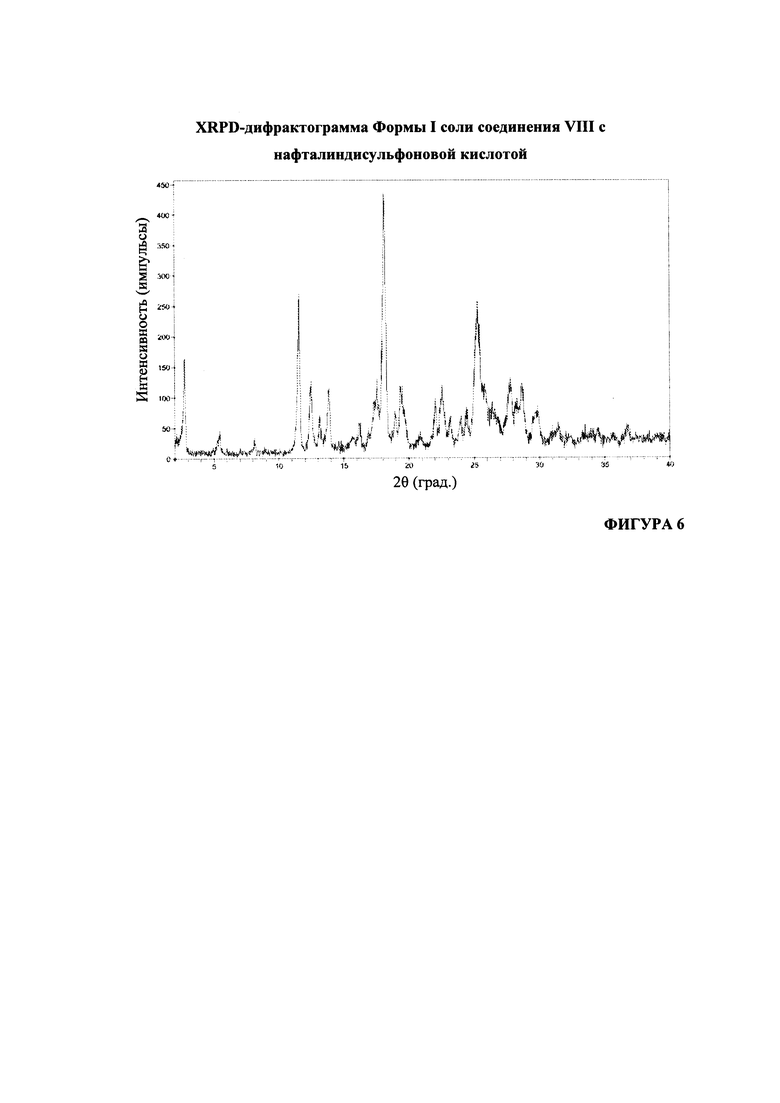

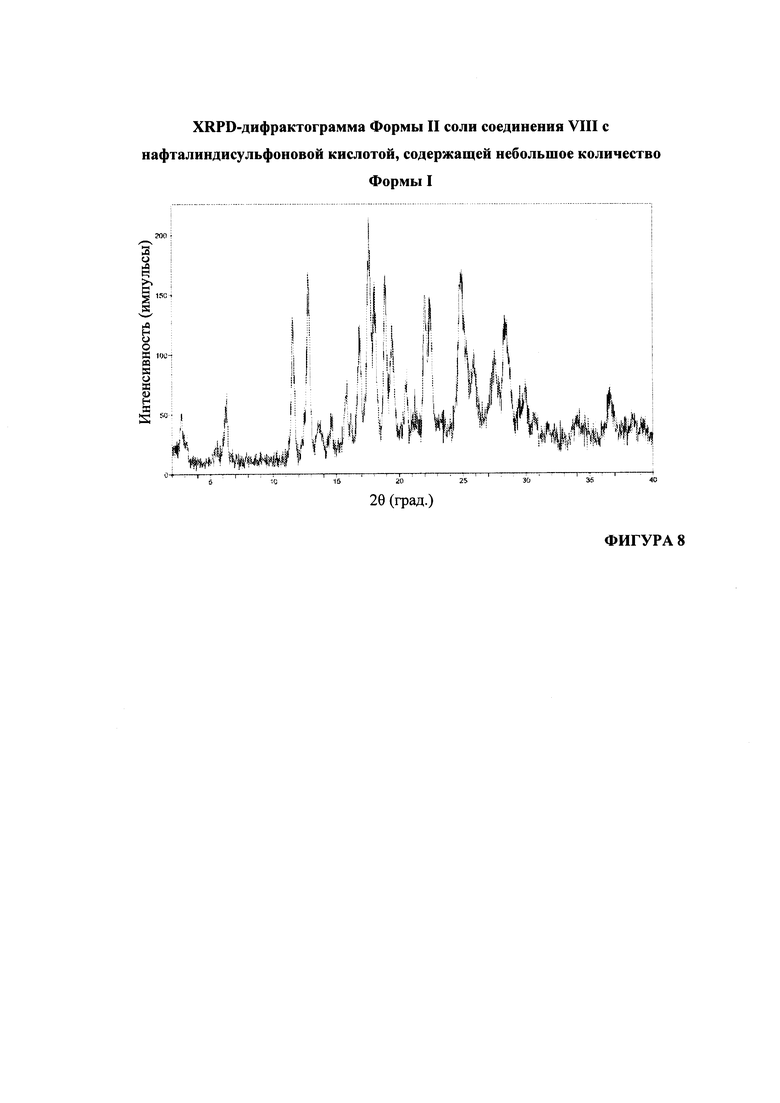
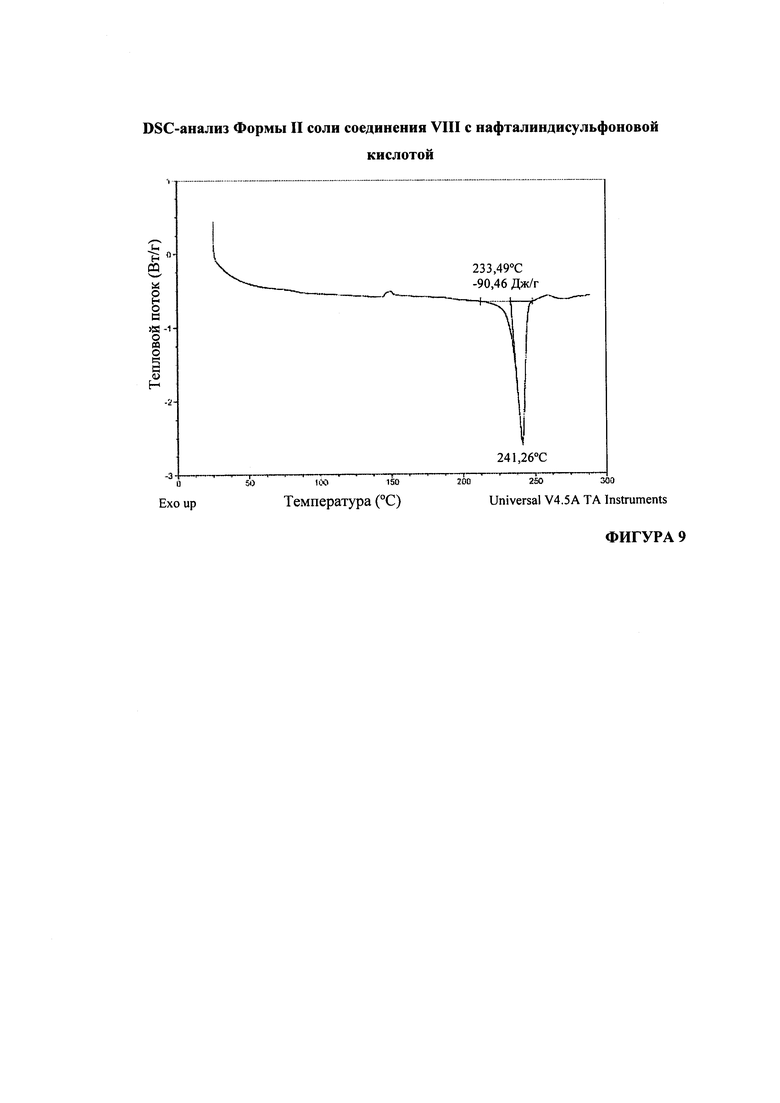
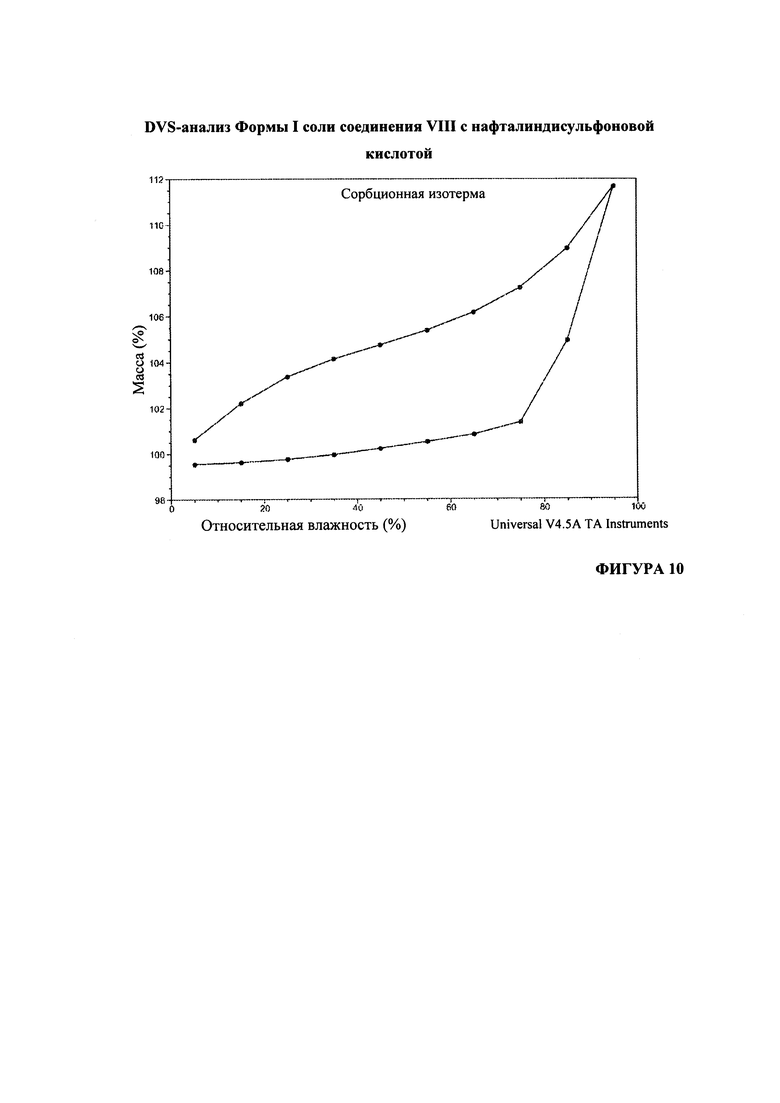
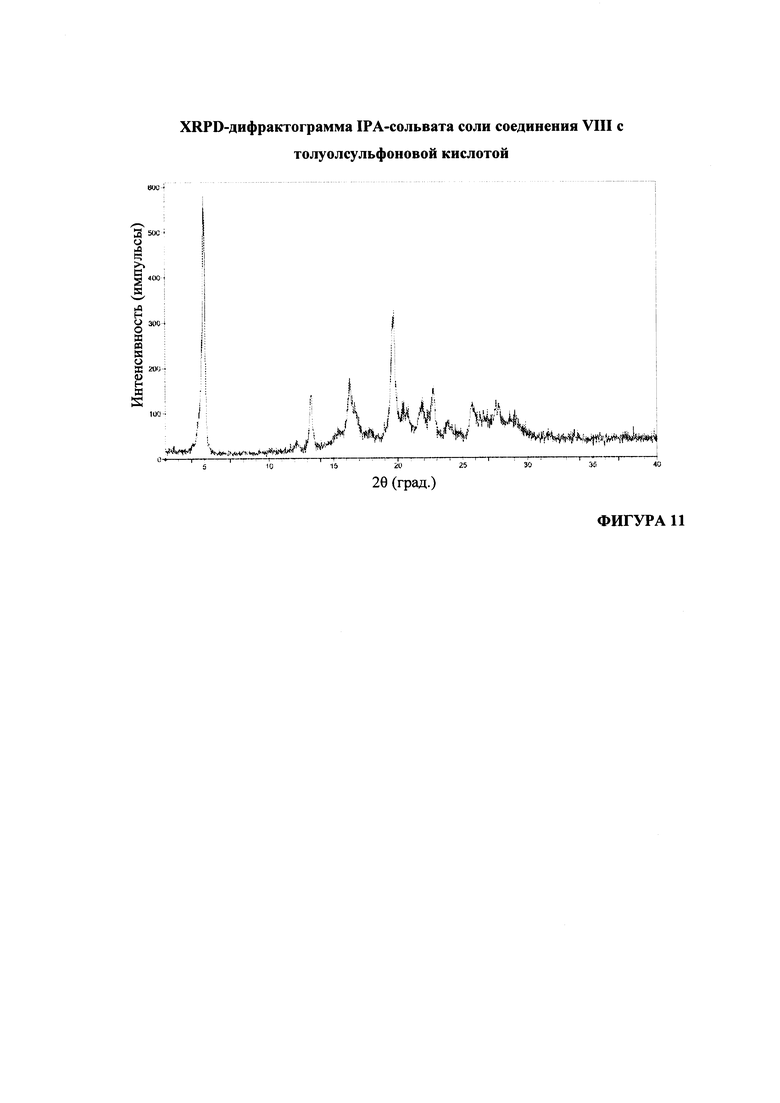

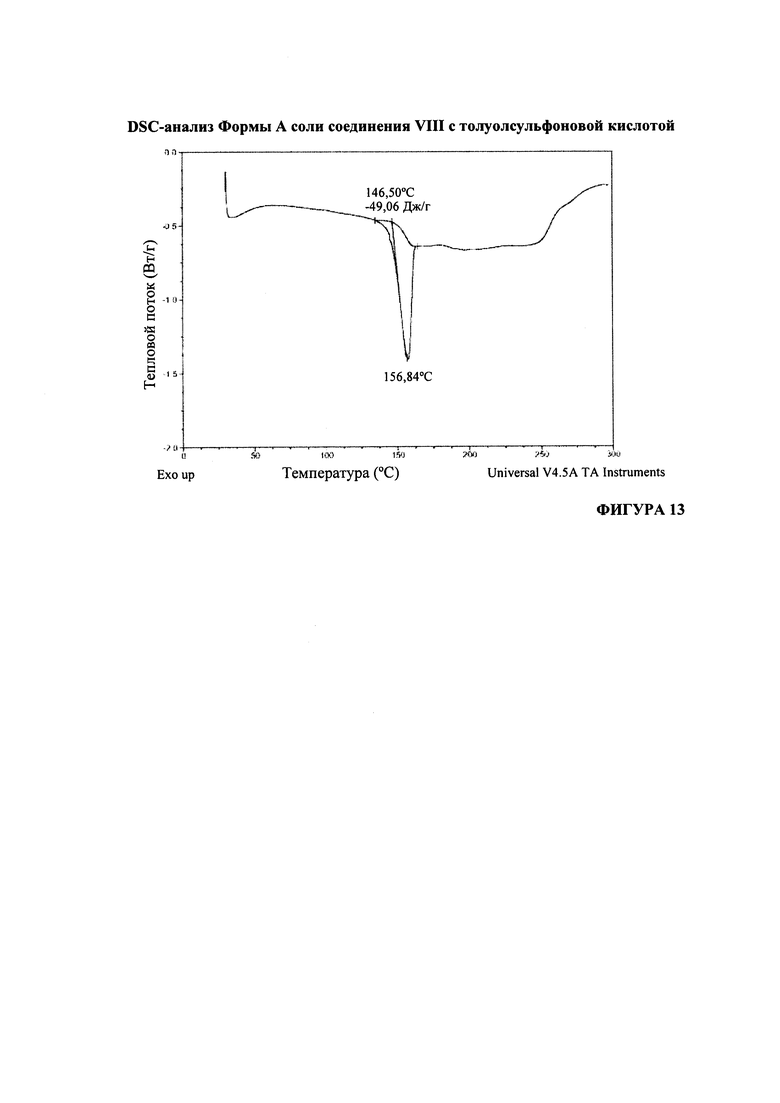
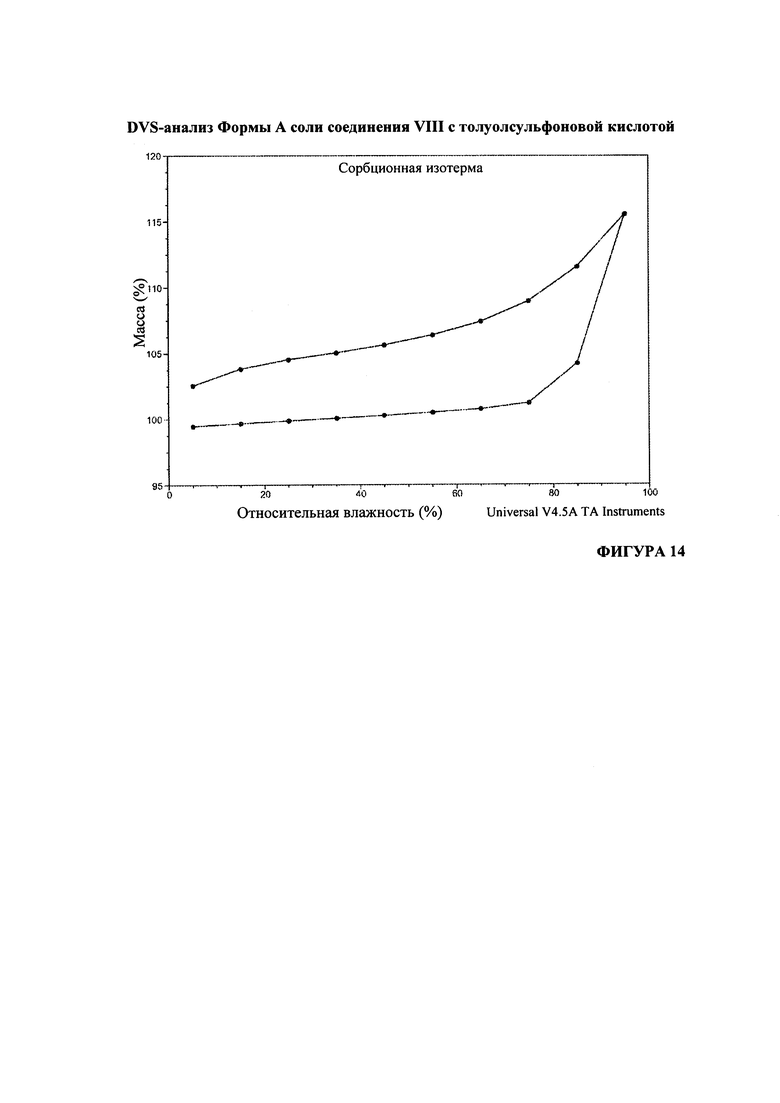
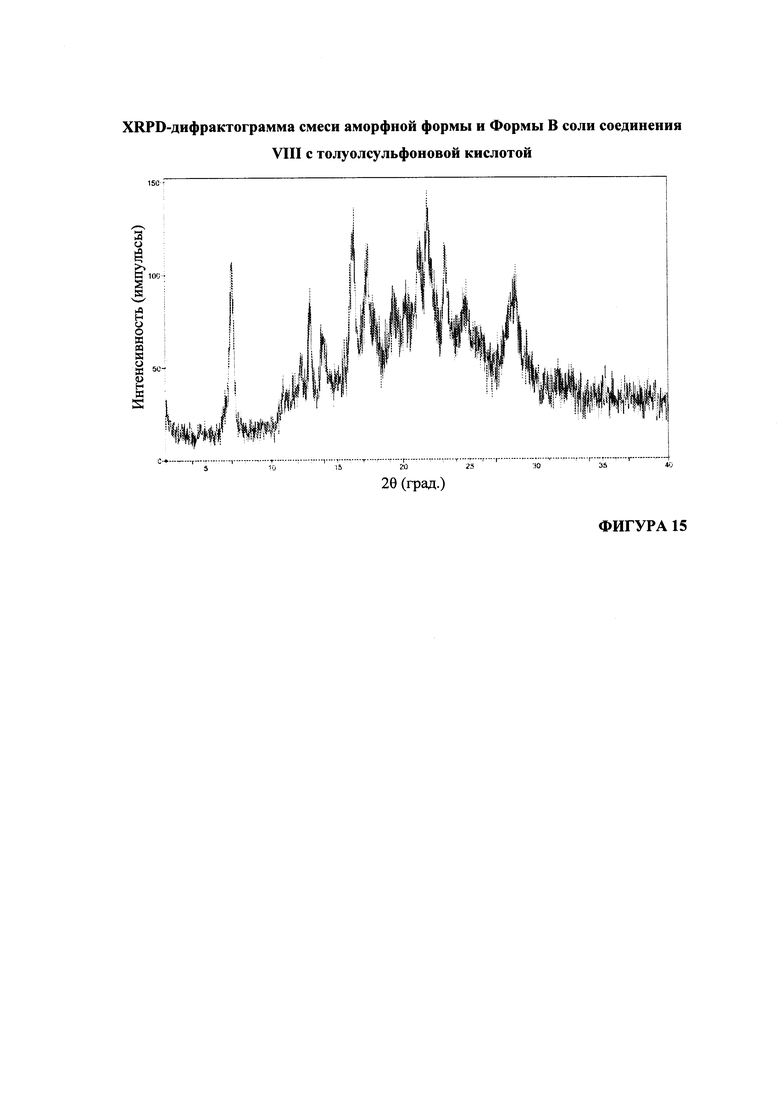

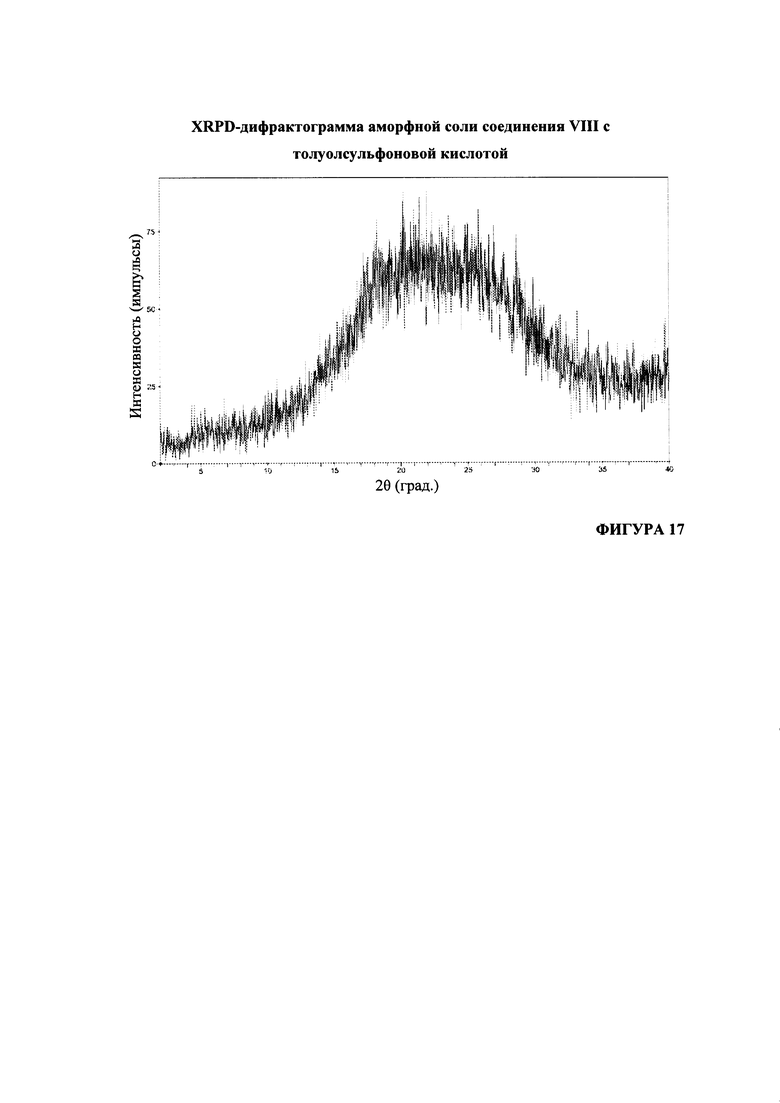


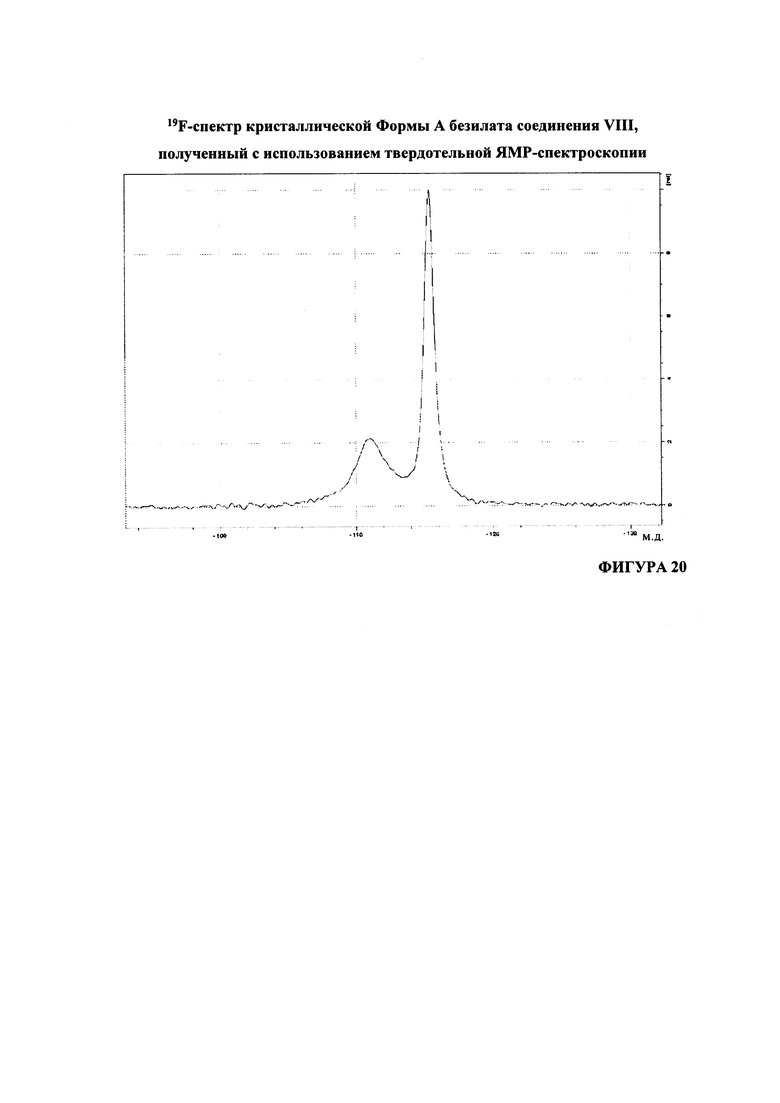

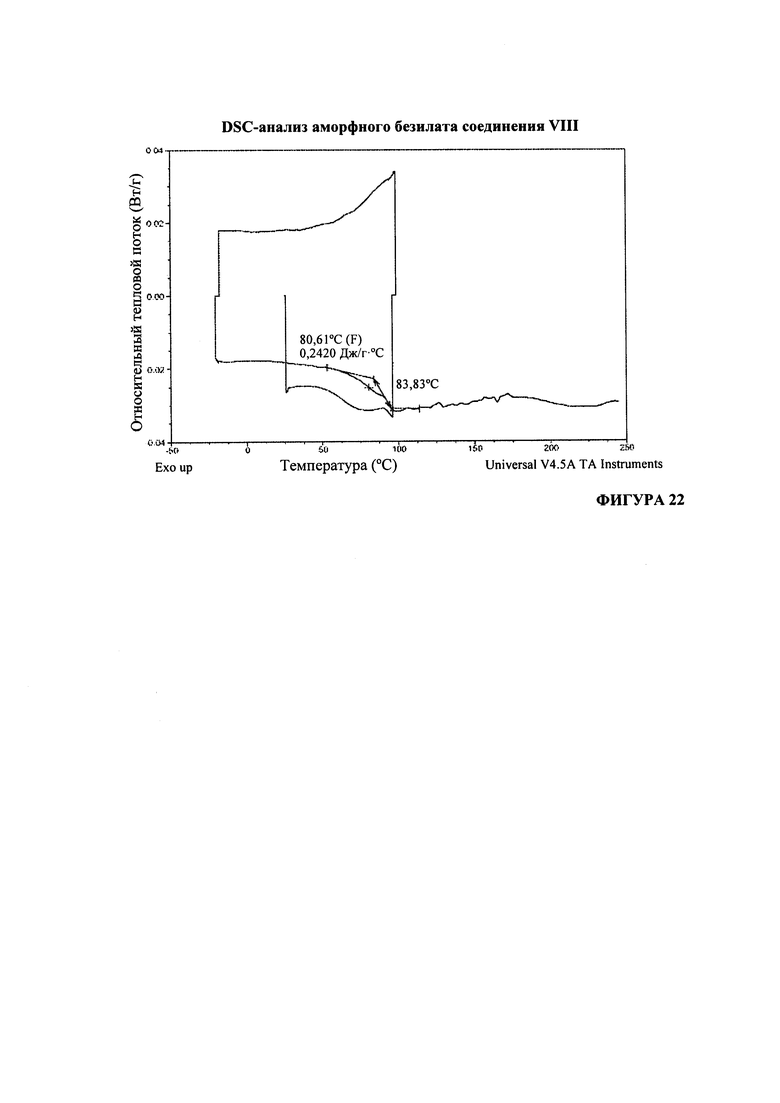
Описан многостадийный способ получения соединения Формулы VIII и его солевых форм формулы VIIIa, где RcSO3H представляет собой арилсульфоновую кислоту, выбранную из бензолсульфоновой или нафталин моно- или дисульфоновой кислоты, у которых арильное кольцо необязательно в качестве заместителя имеет метил или атом галогена. Технический результат - предложен улучшенный способ получения соединения формулы VIII и его арилсульфонатных солей, которые являются ингибиторами ERK и могут быть использованы для лечения гиперпролиферативных заболеваний. 11 з.п. ф-лы, 22 ил., 6 табл., 4 пр.

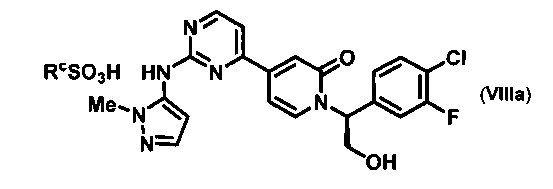 .
.
1. Способ получения соединения Формулы VIII или его соли с арилсульфоновой кислотой, включающий следующие стадии:
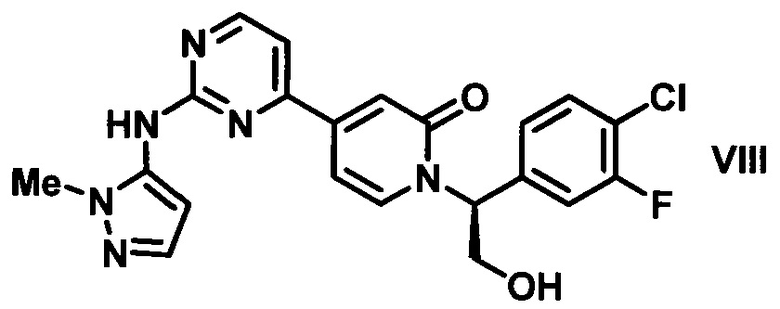
(a) контактирование 4-бром-1-хлор-2-фторбензола с металлирующим агентом в апротонном органическом растворителе с образованием магнийорганического соединения, которое подвергают взаимодействию с 2-хлор-N-метокси-N-метилацетамидом с получением 2-хлор-1-(4-хлор-3-фторфенил)этанона (II);
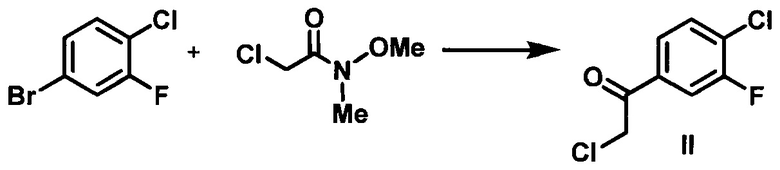
(b) контактирование соединения II с формиатом натрия и муравьиной кислотой в водном этаноле с образованием 1-(4-хлор-3-фторфенил)-2-гидроксиэтанона (III);
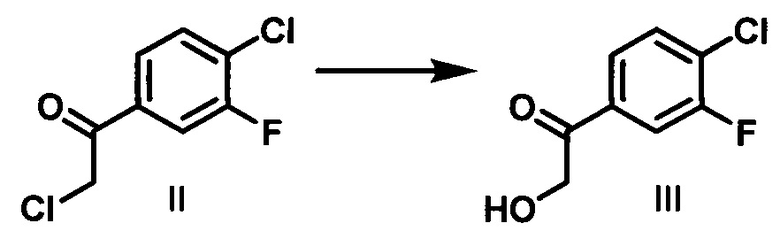
(c) контактирование соединения III с кеторедуктазой с образованием (R)-1-(4-хлор-3-фторфенил)этан-1,2-диола (IV);
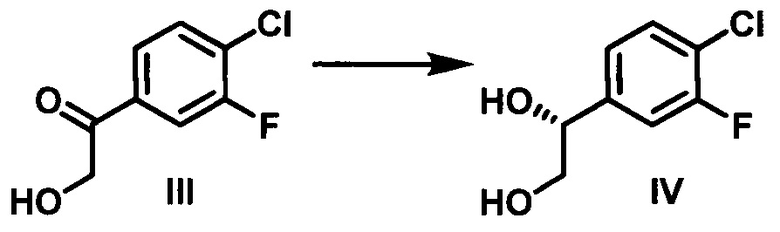
(d) контактирование соединения IV с силилхлоридом (Ra)3SiCl и по меньшей мере с одним основанием в неполярном апротонном растворителе с образованием соединения V и последующее добавление сульфонилхлорида RbS(O)2Cl с получением соединения VI, где Ra в каждом случае независимо представляет собой C1-6 алкил или фенил и Rb выбран из С1-4 алкила или фенила, необязательно имеющих в качестве заместителей от 1 до 3 групп, независимо выбранных из С1-3 алкила, галогена, нитро, циано или C1-3 алкокси;
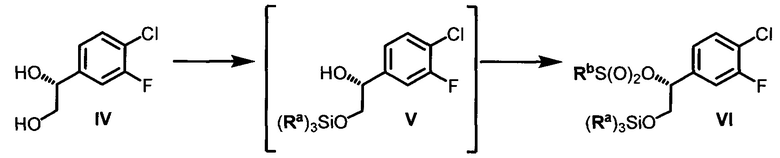
(e) контактирование 4-(2-(метилсульфонил)пиримидин-4-ил)пиридин-2(1H)-она (VII) с сильным основанием в органическом растворителе и последующее добавление соединения VI с получением соединения XI;
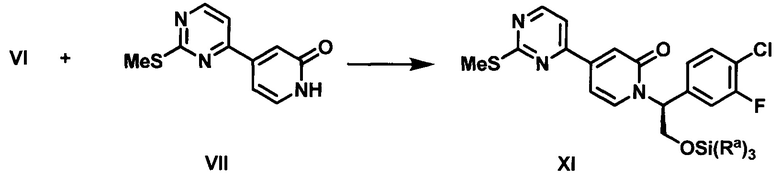
(f) обработку соединения XI окисляющим агентом с получением соединения I;
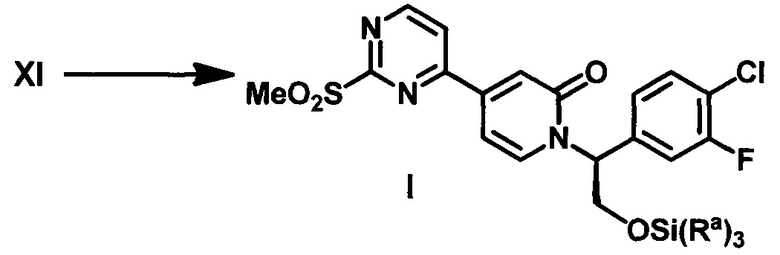
(g) обработку 1-метил-1H-пиразол-5-амина сильным основанием в апротонном растворителе при пониженной температуре и добавление соединения Формулы I с получением соединения IX; и

(h) контактирование соединения IX с десилилирующим агентом с образованием соединения VIII, в случае получения соли арилсульфоновой кислоты соединение VIII, полученное на стадии h, контактирует с арилсульфоновой кислотой в органическом растворителе и воде с образованием соли VIIIa:
где RcSO3H представляет собой арилсульфоновую кислоту, выбранную из бензолсульфоновой или нафталин моно- или дисульфоновой кислоты, у которых арильное кольцо необязательно в качестве заместителей имеет метил или атом галогена.
2. Способ по п. 1, отличающийся тем, что кеторедуктаза на стадии (с) приводит к получению энантиомерного избытка по меньшей мере приблизительно 98%.
3. Способ по п. 2, отличающийся тем, что кеторедуктаза на стадии (с) представляет собой KRED-NADH-112 и/или стадия (с) дополнительно включает НАД(Н) или НАДФ(Н) в качестве кофактора.
4. Способ по п. 3, отличающийся тем, что указанный кофактор регенерируют с использованием косубстрата, выбранного из вторичного спирта или из дополнительного фермента, выбранного из алкогольдегидрогеназы, глюкозодегидрогеназы, формиатдегидрогеназы, глюкозо-6-фосфат-дегидрогеназы, фосфитдегидрогеназы или гидрогеназы.
5. Способ по любому из пп. 2-4, отличающийся тем, что стадию с участием кеторедуктазы выполняют в водной среде в присутствии органического сорастворителя при температуре от 1 до 50°С.
6. Способ по п. 5, отличающийся тем, что стадия с участием кеторедуктазы приводит к получению гомогенной суспензии.
7. Способ по п. 1, отличающийся тем, что силилхлорид представляет собой трет-бутилхлордиметилсилан, сульфонилхлорид представляет собой метансульфохлорид, основания на стадии (d) представляют собой DMAP и TEA, и неполярный апротонный растворитель представляет собой ДХМ, и органический растворитель на стадии (е) представляет собой диоксан.
8. Способ по п. 1, отличающийся тем, что (Ra)3Si представляет собой трет-бутилдиметилсилил, Rb представляет собой метил и на стадии (е) сильное основание представляет собой гексаметилдисилазан калия и органический растворитель представляет собой диметиловый эфир диэтиленгликоля.
9. Способ по п. 1, отличающийся тем, что на стадии (а) металлирующий агент представляет собой изо-PrMgCl и LiCl и растворитель представляет собой ТГФ, на стадии (с) кеторедуктаза представляет собой KRED-NADH-112, и стадия (с) дополнительно включает кофактор НАД и глюкозодегидрогеназу в качестве агента, регенерирующего данный кофактор, на стадии (d) (Ra)3Si представляет собой трет-бутилдиметилсилил, Rb представляет собой метил, основания представляют собой DMAP и TEA и неполярный апротонный растворитель представляет собой ДХМ, и на стадии (е) сильное основание представляет собой гексаметилдисилазан калия и органический растворитель представляет собой диметиловый эфир диэтиленгликоля.
10. Способ по п. 1, отличающийся тем, что на стадии (а) металлирующий агент представляет собой изо-PrMgCl и LiCl и растворитель представляет собой ТГФ, на стадии (с) кеторедуктаза представляет собой KRED-NADH-112, и стадия (с) дополнительно включает кофактор НАД, и агент, регенерирующий данный кофактор, представляет собой глюкозодегидрогеназу, на стадии (d) (Ra)3Si представляет собой трет-бутилдиметилсилил, Rb представляет собой метил, основания представляют собой DMAP и TEA и неполярный апротонный растворитель представляет собой ДХМ, на стадии (е) сильное основание представляет собой гексаметилдисилазан калия и органический растворитель представляет собой диметиловый эфир диэтиленгликоля, и на стадии (g) сильное основание представляет собой гексаметилдисилазан калия и апротонный растворитель представляет собой ТГФ.
11. Способ по п. 1, отличающийся тем, что на стадии (а) металлирующий агент представляет собой изо-PrMgCl и LiCl и растворитель представляет собой ТГФ, на стадии (с) кеторедуктаза представляет собой KRED-NADH-112, и стадия (с) дополнительно включает кофактор НАД, и агент, регенерирующий данный кофактор, представляет собой глюкозодегидрогеназу, на стадии (d) (Ra)3Si представляет собой трет-бутилдиметилсилил, Rb представляет собой метил, основания представляют собой DMAP и TEA и неполярный апротонный растворитель представляет собой ДХМ, на стадии (е) сильное основание представляет собой гексаметилдисилазан калия и органический растворитель представляет собой диметиловый эфир диэтиленгликоля, на стадии (g) сильное основание представляет собой гексаметилдисилазан калия и апротонный растворитель представляет собой ТГФ, и на стадии (h) десилилирующий агент представляет собой раствор HCl в метаноле.
12. Способ по п. 1, отличающийся тем, что RcSO3H представляет собой бензолсульфоновую кислоту и растворитель представляет собой метилэтилкетон и воду, что приводит к образованию желаемой безилатной соли VIIIb.
| Устройство для закрепления лыж на раме мотоциклов и велосипедов взамен переднего колеса | 1924 |
|
SU2015A1 |
| RICHARD J | |||
| BASTIN ET AL." Salt Selection and Optimisation Procedures for Pharmaceutical Entities", Organic Process Research and Develop, vol.4, N5, 2000, pages 427-435 | |||
| ГЕТЕРОЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ ERK2 И ИХ ИСПОЛЬЗОВАНИЕ | 2002 |
|
RU2300377C2 |

