ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к классу конъюгатов лиганд-цитотоксическое лекарственное средство, имеющих новую структуру. Говоря конкретнее, настоящее изобретение относится к конъюгатам лиганд-цитотоксическое лекарственное средство, а также способу их получения, содержащей их фармацевтической композиции и применению конъюгата или фармацевтической композиции.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Химиотерапия все еще является одной из наиболее важных противоопухолевых стратегий, включающих хирургическое лечение, метод лучевой терапии и таргетной терапии. Несмотря на то, что существует много типов высокоэффективных цитотоксинов, небольшое различие между раковыми клетками и нормальными клетками ограничивает широкое применение данных противоопухолевых соединений клинически вследствие токсичных и побочных эффектов. Кроме того, специфичность противоопухолевых моноклональных антител против антигена поверхности опухолевых клеток приводит к тому, что лекарственные средства на основе антител становятся лекарственными средствами противоопухолевой терапии первой линии, однако, эффективность действия часто является неудовлетворительной при применении в качестве противоопухолевого лекарственного средства только антитела.
Под конъюгатом антитело-лекарственное средство (ADC) подразумевается соединение моноклонального антитела или фрагмента антитела с биологически активным цитотоксином через стабильное химическое линкерное соединение, использующее все преимущества специфичности связывания антитела с антигеном поверхности нормальных и опухолевых клеток и высокой эффективности цитотоксинов, в то же время позволяющее избежать низкой эффективности первого и избыточных побочных эффектов второго. Это означает, что по сравнению с общепринятыми традиционными химиотерапевтическими лекарственными средствами, конъюгат антитело-лекарственное средство способен связываться именно с опухолевыми клетками и уменьшать воздействие на нормальные клетки (Milliard А, (2013) Nature Reviews Drug Discovery, 12: 329-332; DiJoseph JF, Armellino DC, (2004) Blood, 103: 1807-1814).
Первые лекарственные средства ADC прежде всего используют мышиные моноклональные антитела, некоторым из них сложно достигать мишени в результате иммунного ответа человека. Во-вторых, эффекторные молекулы, включающие доксорубицин, используемый на ранней стадии, демонстрировали более низкую биологическую активность, которая ограничивала эффективность первого поколения конъюгатов антитело-лекарственное средство. Кроме того, источник антител, способ связывания и число линкеров еще не оптимизированы.
В 2000 г. первый конъюгат антитело-лекарственное средство Mylotarg® (гемтузумаб озогамицин, Wyeth Pharmaceuticals) был одобрен US FDA (Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США) для лечения острого миелоидного лейкоза (Drugs of the Future (2000) 25(7): 686; US 4970198; US 5079233; US 5585089; US 5606040; US 5693762; US 5739116; US 5767285; US 5773001). Mylotarg® представляет собой конъюгат гуманизированное антитело против CD33 - калихеамицин, который в 2010 г был отозван с рынка самой компанией Pfizer из-за ограниченной эффективности и высокой токсичности.
В августе 2011 г Adcetris® (брентуксимаб ведотин, Seattle Genetics Inc.) был одобрен в результате ускоренной процедуры рассмотрения US FDA для лечения лимфомы Ходжкина и рецидивирующей анапластической крупноклеточной лимфомы (Nat. Biotechnol (2003) 21(7): 778-784; WO 2004010957; WO 2005001038; US 7090843 A; US 7659241; WO 2008025020). Adcetris® представляет собой новое специфическое лекарственное средство ADC, которое может приводить к прямому действию лекарственного средства на мишень CD30 в клетках лимфомы, запускать эндоцитоз и, следовательно, вызывать апоптоз опухолевых клеток.
В феврале 2013 г Kadcyla® (адо-трастузумаб эмтанзин, T-DM1) получил одобрение у FDA для лечения пациентов с распространенным или метастатическим раком молочной железы, которые являются HER2-положительными, с устойчивостью к трастузумабу (торговое наименование: Herceptin®) и паклитакселю (WO 2005037992; US 8088387). Как Mylotarg®, так и Adcetris® представляют собой таргетные терапии для гематологических опухолей, структура организации которых является относительно простой по сравнению со структурой организации солидной опухоли. Kadcyla® представляет собой первое лекарственное средство ADC, одобренное FDA для лечения солидной опухоли.
Kadcyla®, с использованием технологии ImmunoGen, получают путем конъюгированйя высокоактивного ингибитора митоза DM1 с трастузумабом от Roche посредством устойчивого тиоэфирного линкера, где средняя загрузка лекарственным средством одного трастузумаба составляет примерно 3,5 DM1. Трастузумаб у пациента специфично связывается с клетками рака молочной железы и расщепляется с внутриклеточным высвобождением DM1 после эндоцитоза. Внутриклеточная концентрация агрегирования DM1 является достаточной для того, чтобы вызывать гибель клеток вследствие нарушения митоза с последующей регрессией опухолевого очага (в отличие от mAb (моноклональное антитело) монотерапии Herceptin®, которая часто приводит к замедлению опухолевого роста). T-DM1 не только поддерживает антитело-зависимое ингибирование пролиферации клеток, подобно Herceptin®, но также усиливает потенциальный эффект цитотоксического лекарственного средства. И поскольку его токсины высвобождаются в опухолевых клетках-мишенях, его побочный эффект одновременно не усиливается с усилением его лечебного эффекта.
Пертузумаб (также известен, как 2С4, торговое наименование Perjeta) представляет собой рекомбинантное гуманизированное моноклональное антитело, которое прежде всего называется "ингибитором димеризации HER". Пертузумаб блокирует димеризацию HER2 и других рецепторов HER посредством связывания с HER2 (Agus DB, (2002) Cancer Cell (2): 127-137; Schaefer G, (1997) Oncogene (15): 1385-1394; Mendoza N, (2002) Cancer Res (62): 5485-5488; Takai N, (2005) Cancer (104): 2701-2708; Jackson JG, (2004) Cancer Res (64): 2601-2609). Было подтверждено, что пертузумаб оказывает ингибирующее действие на рост опухоли как в моделях рака предстательной железы с высокой экспрессией HER2, так и моделях рака предстательной железы с низкой экспрессией HER2 (Craft N, (1999) Nat Med (5):280-285; Oxley JD, (2002) J Clin Pathol (55): 118-120; Reese DM, (2001) Am J Clin Pathol (116): 234-239; Agus DB, (2002) Cancer Cell (2): 127-137).
Отличаясь от Трастузумаба (торговое наименование Герцептин), который ингибирует прямые сигнальные пути через сайт связывания, локализованный на околомембранной области субдомена IV внеклеточного домена HER2, пертузумаб эффективно ингибирует гетерологичную димеризацию HER2 посредством связывания с доменом II (домен димеризации). Таким образом, трастузумаб только оказывает некоторое действие на пациентов с раком со сверхэкспрессией HER2, особенно на пациентов с раком молочной железы. Хотя и имея с трастузумабом одну и ту же мишень и эндоцитоз, вследствие его отличного механизма действия пертузумаб может блокировать сигнальный путь, опосредованный рецептором семейства ЕгbВ после ингибирования димеризации, и может иметь более широкое применение, чем одно только блокирование сигнального пути HER2 (Franklin МС, (2001) Cancer Cell (5): 317-328).
В настоящее время главным образом существует две методики конъюгирования лекарственного средства ADC: в отношении T-DM1 принято случайное конъюгирование цитотоксического лекарственного средства со свободной аминогруппой в антителе (WO 2005037992); в то время как в отношении Adcetris® принято конъюгирование цитотоксического лекарственного средства со свободной тиольной группой в антителе после восстановления шарнирной области (WO 2004010957). Оба способа конъюгирования приводят к получению смеси с изменчивым числовым значением соотношения лекарственное средство - антитело. Например, среднее соотношение лекарственное средство - антитело Т-DM1 составляет 3,5, однако распределение загрузки лекарственного средства составляет от 0 до 8. Низкое значение соотношения лекарственное средство - антитело влияет на эффективность ADC, в то время как высокое значение соотношения лекарственное средство - антитело легче приводит к избыточной модификации антитела, приводя к распознаванию и разрушению лекарственного средства ADC системой тканевых макрофагов. Это не только укорачивает период полувыведения ADC, но также усиливает токсичные побочные эффекты вследствие аккумуляции токсинов в тканях, не являющихся мишенью; и в отношении Adcetris® дисульфидную связь шарнирной области антитела восстанавливают восстанавливающим агентом, что оказывало бы определенное влияние на стабильность самого антитела.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Для улучшения эффекта конъюгирования лиганда, особенно антитела, и лекарственного средства настоящее изобретение предлагает улучшенное соединительное звено для соединения лиганда и лекарственного средства.
В настоящем изобретении раскрыт конъюгат лиганд-цитотоксическое лекарственное средство или его фармацевтически приемлемая соль или его сольват, где конъюгат лиганд-цитотоксическое лекарственное средство содержит соединительное звено X, имеющее следующую структуру:

X1 выбран из группы, состоящей из Н, алкила, циклоалкила, гетероциклила, арила и гетероарила, где каждый из алкила, циклоалкила, гетероциклила, арила и гетероарила независимо и необязательно дополнительно замещен одной или более чем одной группой, выбранной из группы, состоящей из галогена, гидрокси, циано, алкила, алкокси, циклоалкила, гетероциклила, арила и гетероарила,
Х2 выбран из группы, состоящей из алкила, циклоалкила, алкил-циклоалкила, циклоалкил-алкила, алкил-циклоалкил-алкила, гетероциклила, алкил-гетероциклила, гетероциклил-алкила, алкил-гетероциклил-алкила, арила, алкил-арила, арил-алкила, алкил-арил-алкила, гетероарила, алкил-гетероарила, гетероарил-алкила, алкил-гетероарил-алкила, (СН2)p(OСН2СН2)р, (СН2СН2О)p(СН2)p, каждый р представляет собой целое число, независимо выбранное из 1-10, где алкил, циклоалкил, гетероциклил, арил и гетероарил независимо и необязательно дополнительно замещены одной или более чем одной группой, выбранной из группы, состоящей из гало, гидрокси, циано, алкила, алкокси, циклоалкила, гетероциклила, арила и гетероарила;
Или когда X1 не является Н, X1 и X2 с атомом углерода, соединяющим X1 и X2, в своей совокупности образуют циклоалкильную группу, где циклоалкил независимо и необязательно дополнительно замещен одной или более чем одной группой, выбранной из группы, состоящей из гало, гидрокси, циано, алкила, алкокси, циклоалкила, гетероциклила, арила и гетероарила;
S представляет собой атом серы.
В предпочтительном воплощении настоящего изобретения предложен конъюгат лиганд-цитотоксическое лекарственное средство или его фармацевтически приемлемая соль или его сольват, упомянутые выше, где X1 представляет собой Н или алкил, предпочтительно Н.
В другом предпочтительном воплощении настоящего изобретения предложен конъюгат лиганд-цитотоксическое лекарственное средство или его фармацевтически приемлемая соль или его сольват, упомянутые выше, где Х2 представляет собой алкил или циклоалкил, предпочтительно алкил, более предпочтительно линейный алкил.
В другом предпочтительном воплощении настоящего изобретения предложен конъюгат лиганд-цитотоксическое лекарственное средство или его фармацевтически приемлемая соль или сольват, упомянутые выше, содержащие структуру формулы (I):
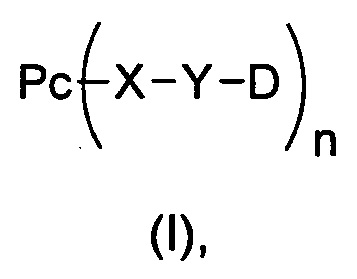
где:
Рс представляет собой лиганд;
X является таким, как определено в п. 1;
Y представляет собой промежуточное звено;
D представляет собой цитотоксическое лекарственное средство;
n представляет собой соотношение лекарственное средство-антитело, n выбран из 1-8.
В другом предпочтительном воплощении настоящего изобретения предложен конъюгат лиганд-цитотоксическое лекарственное средство или его фармацевтически приемлемая соль или его сольват, упомянутые выше, где соединительное звено X связано с N-концевой аминогруппой полипептидной цепи Рс или ε-аминогруппой остатка лизина, n выбран из 1-4.
В другом предпочтительном воплощении настоящего изобретения предложен конъюгат лиганд-цитотоксическое лекарственное средство или его фармацевтически приемлемая соль или его сольват, упомянутые выше, где лиганд представляет собой антитело.
В другом предпочтительном воплощении настоящего изобретения предложен конъюгат лиганд-цитотоксическое лекарственное средство или его фармацевтически приемлемая соль или его сольват, упомянутые выше, где антиген указанного антитела представляет собой антиген поверхности клеток, экспрессирующийся в клетке-мишени и/или ткани пролиферативного заболевания; пролиферативное заболевание предпочтительно представляет собой рак; антиген поверхности клеток предпочтительно представляет собой рецептор поверхности клеток.
В другом предпочтительном воплощении настоящего изобретения предложен конъюгат лиганд-цитотоксическое лекарственное средство или его фармацевтически приемлемая соль или его сольват, упомянутые выше, где рецептор поверхности клеток выбран из группы, состоящей из:
1) HER2 (ErbB2),
2) HER3 (ErbB3),
3) HER4 (ErbB4),
4) CD20,
5) CD22,
6) CD30,
7) CD33,
8) CD44,
9) Lewis Y,
10) CD56,
11) CD105,
12) VEGFR и
13) GPNMB.
В другом предпочтительном воплощении настоящего изобретения предложен конъюгат лиганд-цитотоксическое лекарственное средство или его фармацевтически приемлемая соль или его сольват, упомянутые выше, где рецептор поверхности клеток выбран из группы, состоящей из:
1) HER2 (ErbB2),
2) CD 22,
3) CD30
4) CD33,
5) CD44
6) CD56,
7) Lewis Y и
8) GPNMB.
В другом предпочтительном воплощении настоящего изобретения предложен конъюгат лиганд-цитотоксическое лекарственное средство или его фармацевтически приемлемая соль или его сольват, упомянутые выше, где антитело выбрано из группы, состоящей из:
1) Трастузумаб (HER2),
2) Инотузумаб (CD22),
3) Пинатузумаб (CD22),
4) Брентуксимаб (CD30),
5) Гемтузумаб (CD33),
6) Биватузумаб (CD44),
7) Лорвотузумаб (CD56),
8) cBR96 (Lewis Y),
9) Глематумамаб (GPNMB) и
10) Пертузумаб
В другом предпочтительном воплощении настоящего изобретения предложен конъюгат лиганд-цитотоксическое лекарственное средство или его фармацевтически приемлемая соль или его сольват, упомянутые выше, где антитело способно связываться с белком HER2, причем данное антитело содержит:
1) легкую цепь, содержащую по меньшей мере одну CDR (гипервариабельная область), выбранную из данных трех CDR-L1, CDR-L2 и CDR-L3, определенных в соответствии с системой нумерации Kabat, где:
i) CDR-L1 представляет собой CDR SEQ ID NO: 1 или по меньшей мере одной последовательности, обладающей по меньшей мере 80% идентичностью с SEQ ID NO: 1 после оптимального выравнивания;
ii) CDR-L2 представляет собой CDR SEQ ID NO: 2 или по меньшей мере одной последовательности, обладающей по меньшей мере 80% идентичностью с SEQ ID NO: 2 после оптимального выравнивания;
iii) CDR-L3 представляет собой CDR SEQ ID NO: 3 или по меньшей мере одной последовательности, обладающей по меньшей мере 80% идентичностью с SEQ ID NO: 3 после оптимального выравнивания;
2) тяжелую цепь, содержащую по меньшей мере одну CDR, выбранную из данных трех CDR-H1, CDR-H2 and CDR-H3, определенных в соответствии с системой нумерации Kabat, где:
iv) CDR-H1 представляет собой CDR SEQ ID NO: 4 или по меньшей мере одной последовательности, обладающей по меньшей мере 80% идентичностью с SEQ ID NO: 4 после оптимального выравнивания;
v) CDR-H2 представляет собой CDR SEQ ID NO: 5 или по меньшей мере одной последовательности, обладающей по меньшей мере 80% идентичностью с SEQ ID NO: 5 после оптимального выравнивания;
vi) CDR-H3 представляет собой CDR SEQ ID NO: 6 или по меньшей мере одной последовательности, обладающей по меньшей мере 80% идентичностью с SEQ ID NO: 6 после оптимального выравнивания.
В другом предпочтительном воплощении настоящего изобретения предложен конъюгат лиганд-цитотоксическое лекарственное средство или его фармацевтически приемлемая соль или его сольват, упомянутые выше, где антитело, способное связываться с белком HER2, содержит легкую цепь и/или тяжелую цепь, причем легкая цепь содержит аминокислотную последовательность SEQ ID NO: 7, тяжелая цепь содержит аминокислотную последовательность SEQ ID NO: 8.
В другом предпочтительном воплощении настоящего изобретения предложен конъюгат лиганд-цитотоксическое лекарственное средство или его фармацевтически приемлемая соль или его сольват, упомянутые выше, где цитотоксическое лекарственное средство выбрано из группы, состоящей из: ингибиторов тубулина, ДНК-алкилирующих агентов, ингибиторов тирозинкиназы, ингибиторов топоизомеразы и ингибиторов синтеза ДНК, предпочтительно ингибиторов тубулина.
В другом предпочтительном воплощении настоящего изобретения предложен конъюгат лиганд-цитотоксическое лекарственное средство или его фармацевтически приемлемая соль или его сольват, упомянутые выше, где ингибитор топоизомеразы выбран из камптотецина, иринотекана, актиномицина, адриамицина, доксорубицина, даунорубицина, эпирубицина; ингибитор синтеза ДНК выбран из фторурацила, цитарабина, азацитидина, анцитабина, гемцитабина, капецитабина, метотрексата, блеомицина, комплексов платины; ДНК-алкилирующий агент выбран из азотистых ипритов (циклофосфамид), этилиденгидразоноаминов (тиотепа, митомицин), сложных эфиров метансульфоновой кислоты (бусульфан), полиолов (дибромманнит), нитрозомочевин (кармустин), триазенимидазола (дакарбазин) и гидразинов (прокарбазин); ингибитор тирозинкиназы выбран из иматиниба, гефитиниба, эрлотиниба, сунитиниба, сорафениба, лапатиниба, дазатиниба, нилотиниба.
В другом предпочтительном воплощении настоящего изобретения предложен конъюгат лиганд-цитотоксическое лекарственное средство или его фармацевтически приемлемая соль или его сольват, упомянутые выше, где цитотоксическое лекарственное средство, ингибитор тубулина, выбрано из группы, состоящей из: майтанзиноидов, калихеамицина, таксанов, винкристина, колхицина и доластатинов/ауристатинов, предпочтительно майтанзиноида или доластатинов/ауристатинов.
В еще одном предпочтительном воплощении настоящего изобретения предложен конъюгат лиганд-цитотоксическое лекарственное средство или его фармацевтически приемлемая соль или его сольват, упомянутые выше, где D выбран из доластатинов/ауристатинов со структурой формулы (D1):
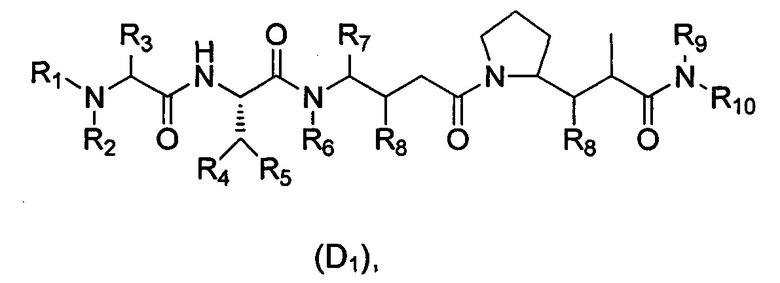
где:
R1 выбран из связи, Н, алкила или циклоалкила, предпочтительно связи; когда R1 выбран из Н, алкила или циклоалкила, D связан с Y через R10 в формуле (I); когда R1 предпочтительно представляет собой связь, D связан с Y через R10 в формуле (I);
R2 выбран из Н или алкила;
Или R1 и R2 с шарнирным атомом N в своей совокупности образуют гетероциклил, где гетероциклил дополнительно независимо и необязательно замещен одной или более чем одной группой, выбранной из группы, состоящей из гало, гидрокси, циано, алкила, алкокси, циклоалкила, гетероциклила, арила и гетероарила; или образуют структуру -(CRaRb)e-, Ra и Rb независимо выбраны из Н, алкила или гетероциклила, е представляет собой целое число, выбранное из 2-6;
R3 выбран из группы, состоящей из Н, алкила, циклоалкила, арила, алкил-арила, алкил-циклоалкила, гетероциклила и алкил-гетероциклила;
R4 выбран из группы, состоящей из Н, алкила, циклоалкила, арила, алкил-арила, алкил-циклоалкила, гетероциклила и алкил-гетероциклила;
R5 выбран из Н или метила;
R6 выбран из Н или алкила;
R7 выбран из группы, состоящей из Н, алкила, циклоалкила, арила, алкил-арила, алкил-циклоалкила, гетероциклила и алкил-гетероциклила;
R8 выбран из группы, состоящей из Н, гидрокси, алкила, циклоалкила и алкокси;
R9 выбран из Н или алкила;
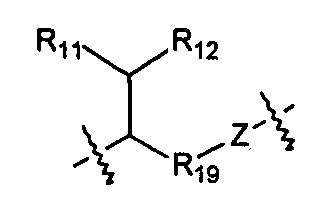
Когда R1 выбран из алкила или циклоалкила, или R1 и R2 с шарнирным атомом N в своей совокупности образуют гетероциклил, где гетероциклил независимо и необязательно дополнительно замещен одной или более чем одной группой, выбранной из группы, состоящей из гало, гидрокси, циано, алкила, алкокси, циклоалкила, гетероциклила, арила и гетероарила, R10 выбран из следующих структур:
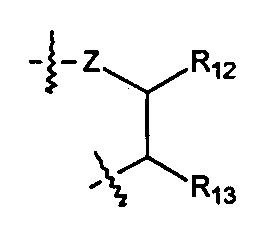 ,
,  ,
, 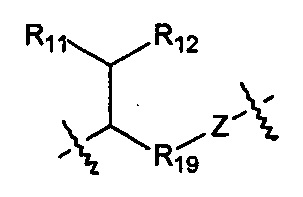 ,
, 
и 
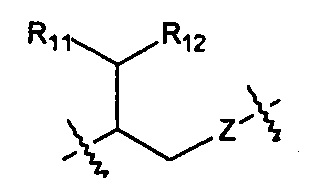
Когда R1 представляет собой Н, R10 выбран из следующих структур:
 ,
,  ,
,  и
и 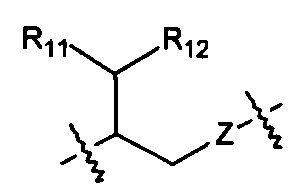 .
.
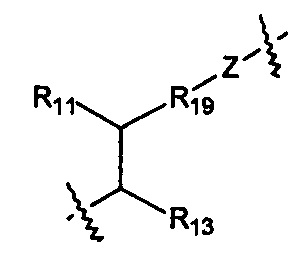
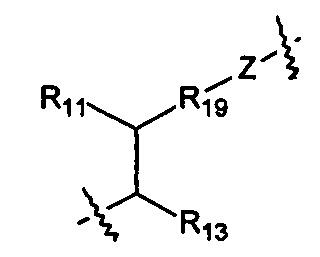
Когда R1 представляет собой связь, он связан с промежуточным звеном Y, где R10 выбран из следующих структур:
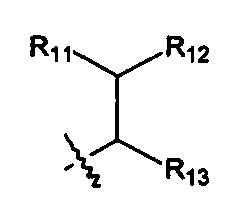 и
и 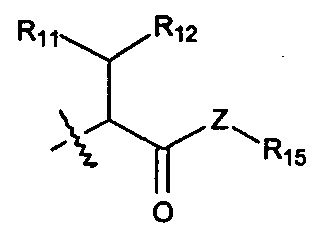
Z выбран из О, S, NH и N(R14);
R11 выбран из группы, состоящей из Н, гидрокси, амино, NHR14, N(R14)2 алкокси, алкила, циклоалкила, арила, гетероциклила, алкил-арила, алкил-циклоалкила и алкил-гетероциклила; или когда R11 представляет собой О, он может замещать Н, присоединенный к шарнирному атому углерода, и образовывать карбонильную группу (С=О) с данным атомом углерода;
R12 выбран из арила и гетероциклила, арил или гетероциклил необязательно дополнительно замещен одной или более чем одной группой, выбранной из группы, состоящей из гидрокси, алкокси, алкила и галогена;
R13 выбран из группы, состоящей из Н, гидрокси, амино, NHR14, N(R14)2, COOR14, алкокси, алкила, циклоалкила, арила, гетероциклила, алкил-арила, алкил-циклоалкила, алкил-гетероциклила и алкокси-алкокси-алкокси;
R14 выбран из Н или алкила;
R15 выбран из группы, состоящей из Н, алкила, арила, гетероциклила, (R16O)m-R14 и (R16O)m-CH(R17)2;
m представляет собой целое число, выбранное из 1-1000;
R16 представляет собой С2-С8 алкил;
R17 выбран из группы, состоящей из Н, карбоксила, -(CH2)t-N(R-18)2 и -(СН2)t-SO3R14;
R18 выбран из группы, состоящей из Н, алкила и -(CH2)t-COOH;
t представляет собой целое число, выбранное из 0-6;
R19 выбран из группы, состоящей из арила, циклоалкила и гетероциклила.
В еще одном предпочтительном воплощении настоящего изобретения предложен конъюгат лиганд-цитотоксическое лекарственное средство или его фармацевтически приемлемая соль или его сольват, формулы (D1),
где:
R1 выбран из связи или алкила;
R2 выбран из Н или алкила;
R3 выбран из Н, алкила или циклоалкила;
Каждый из R4, R5, R6, R7 независимо и необязательно выбран из Н, алкила, циклоалкила или гетероциклила;
R8 выбран из Н, алкила, циклоалкила или алкокси;
R9 выбран из Н или алкила;
Когда R1 выбран из -алкила, R10 выбран из следующей структуры:
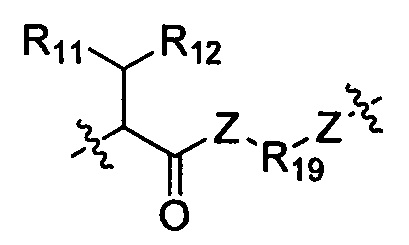
Когда R1 выбран из связи, он связан с промежуточным звеном Y, где R10 выбран из следующей структуры:
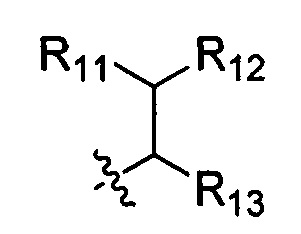
Z выбран из NH;
R11 выбран из Н, гидрокси или алкила;
R12 выбран из арила, арил необязательно дополнительно замещен одной или более чем одной группой, выбранной из группы, состоящей из гидрокси, алкокси, алкила и галогена;
R13 выбран из Н, алкила или COOR14;
R14 выбран из Н или алкила, алкил необязательно дополнительно замещен алкокси или алкокси-алкокси-алкокси;
R19 выбран из арила.
В еще одном предпочтительном воплощении настоящего изобретения R3 в формуле (D1) выбран из Н или изопропила.
В другом предпочтительном воплощении настоящего изобретения предложен конъюгат лиганд-цитотоксическое лекарственное средство или его фармацевтически приемлемая соль или его сольват, упомянутые выше, где D выбран из майтанзина со структурой формулы (DM):
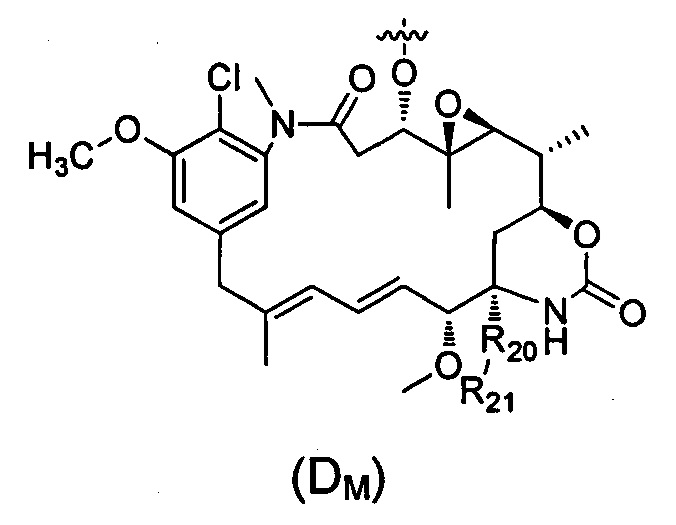
где:
R20 выбран из О или S;
R21 выбран из группы, состоящей из Н, алкила, циклоалкила, гетероциклила, арила и гетероарила, где алкил, циклоалкил, гетероциклил, арил и гетероарил независимо и необязательно дополнительно замещены одной или более чем одной группой, выбранной из группы, состоящей из галогена, гидрокси, циано, нитро, алкила, алкокси, циклоалкила, гетероциклила, арила и гетероарила.
В другом предпочтительном воплощении настоящего изобретения предложен конъюгат лиганд-цитотоксическое лекарственное средство или его фармацевтически приемлемая соль или его сольват, упомянутые выше, где D представляет собой D1, где промежуточное звено Y имеет структуру следующей формулы:
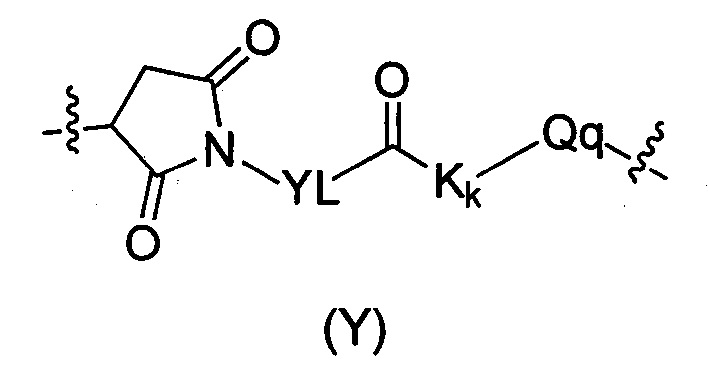
где:
YL выбран из группы, состоящей из алкила, циклоалкила, О-алкила, O-алкокси, арила, алкил-циклоалкила, циклоалкил-алкила, алкил-арила, алкил-циклоалкил-алкила, гетероциклила, алкил-гетероциклила, гетероциклил-алкила, алкил-гетероциклил-алкила, арила, алкил-арила, арил-алкила, алкил-арил-алкила, гетероарила, алкил-гетероарила, гетероарил-алкила, алкил-гетероарил-алкила, CH2(OCH2CH2)t, (CH2CH2O)tCH2 и (CH2CH2O)t, t представляет собой целое число, выбранное из 1-10, предпочтительно алкильной группы, более предпочтительно C2-C8 линейного алкила;
Kk представляет собой аминокислотное звено, где K представляет собой аминокислоту, k представляет собой целое число, выбранное из 0-10, предпочтительно 2, Kk предпочтительно представляет собой валин-цитруллин;
Qq представляет собой протяженное звено, где q представляет собой 0, 1 или 2.
Настоящее изобретение также относится к соединению, имеющему формулу (II):
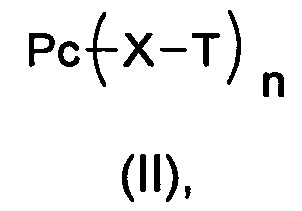
которое используют для получения промежуточного соединения, имеющего формулу (III),
где:
Рс является таким, как определено в формуле (I);
X определен, как соединительное звено X;
Т выбран из группы, состоящей из Н, трет-бутила, ацетила, н-пропионила, изопропионила, трифенилметила, метоксиметила и 2-(триметилсилил)этоксиметила, предпочтительно Н или ацетила;
n выбран из 1-4.
В другом предпочтительном воплощении настоящего изобретения предложен конъюгат лиганд-цитотоксическое лекарственное средство или его фармацевтически приемлемая соль или его сольват, упомянутые выше, содержащие структуру формулы (III):
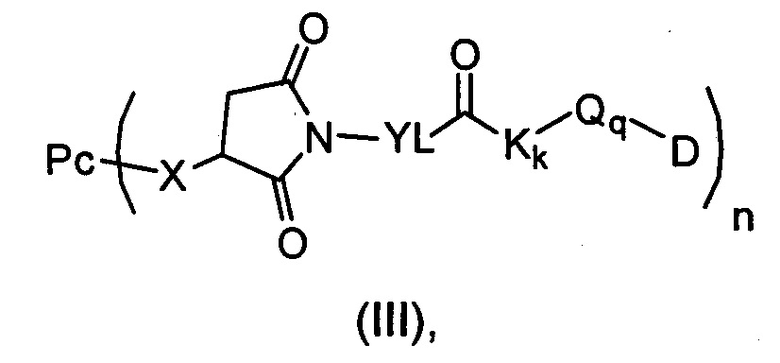
где:
Рс представляет собой антитело;
X определен, как соединительное звено X;
YL, Kk, Qq определены в формуле (Y);
n выбран из 1-4;
D представляет собой цитотоксическое лекарственное средство.
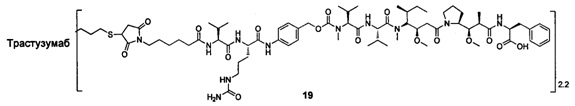
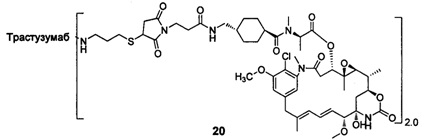
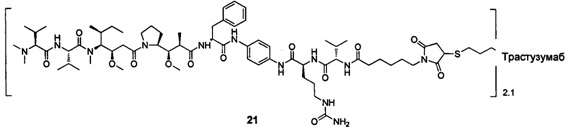
В другом предпочтительном воплощении настоящего изобретения предложен конъюгат лиганд-цитотоксическое лекарственное средство или его фармацевтически приемлемая соль или его сольват, упомянутые выше, содержащие любую из следующих структур:
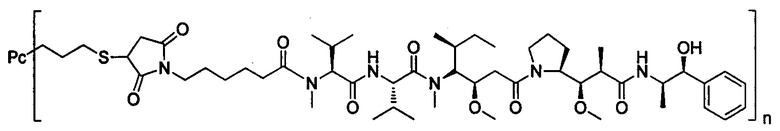 ,
,
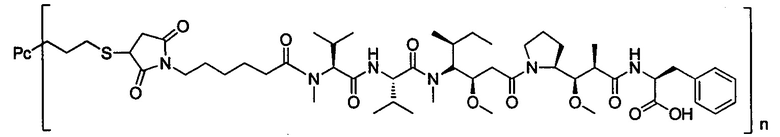 ,
,
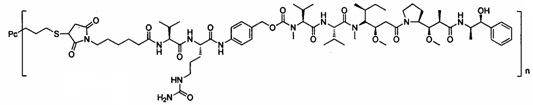 ,
,
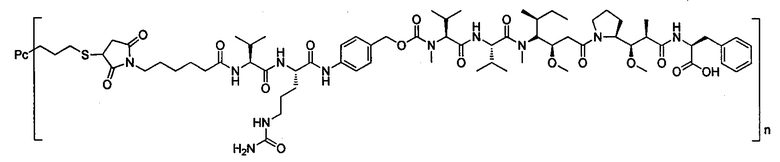 ,
,
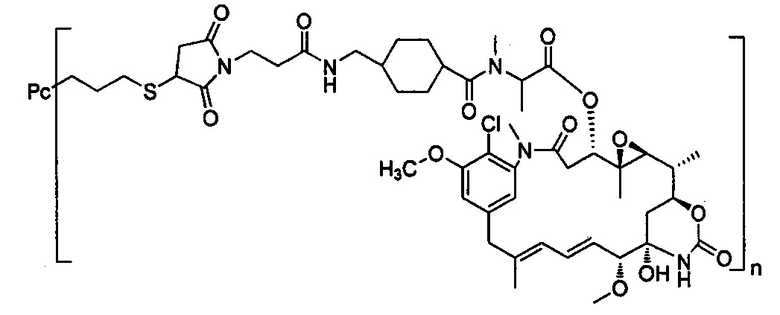 ,
,
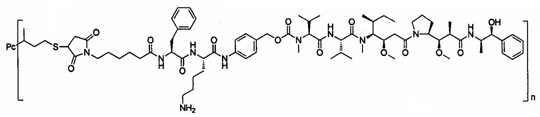 ,
,
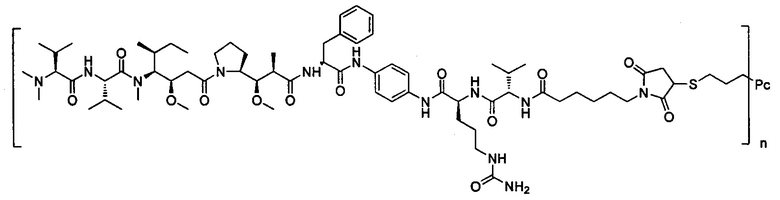 ,
,
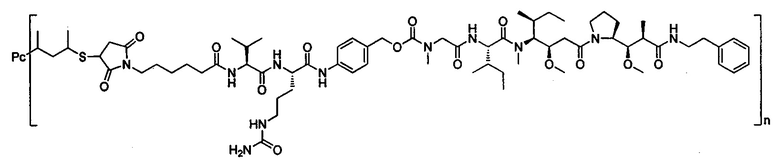 ,
,
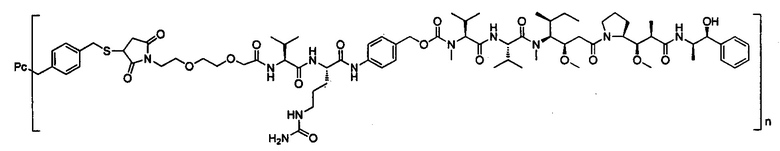 ,
,
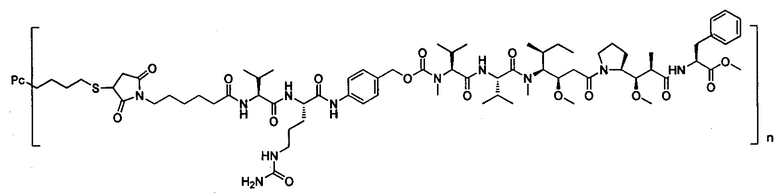 ,
,
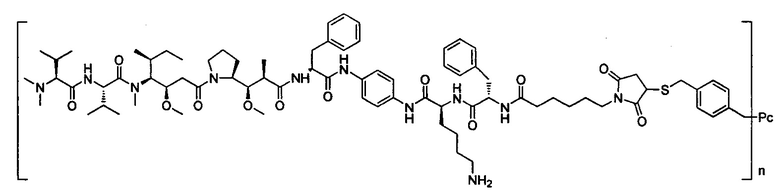 ,
,
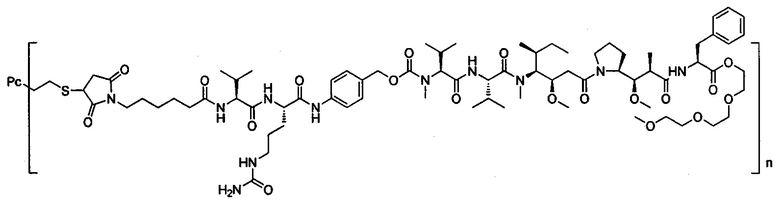 ,
,
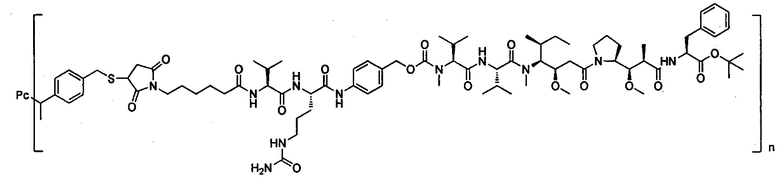 ,
,
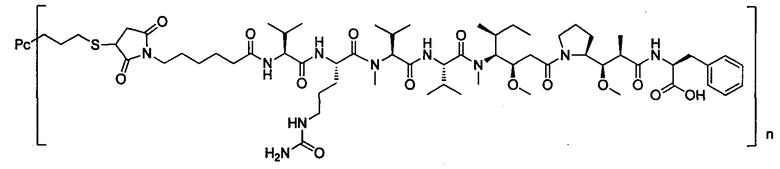 ,
,
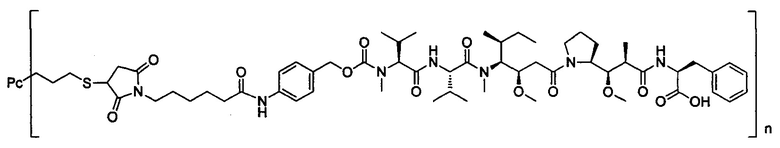 ;
;
где лиганд Рс представляет собой лиганд, n представляет собой соотношение лекарственное средство-антитело и n выбран из 1-8.
В другом конкретном воплощении настоящего изобретения предложен конъюгат лиганд-цитотоксическое лекарственное средство или его фармацевтически приемлемая соль или его сольват, упомянутые выше, где Рс выбран из Трастузумаба, Инотузумаба и Брентуксимаба, предпочтительно Трастузумаба или Пертузумаба, более предпочтительно Пертузумаба.
В другом конкретном воплощении настоящего изобретения предложен конъюгат лиганд-цитотоксическое лекарственное средство или его фармацевтически приемлемая соль или его сольват, упомянутые выше, выбранные из группы, состоящей из:
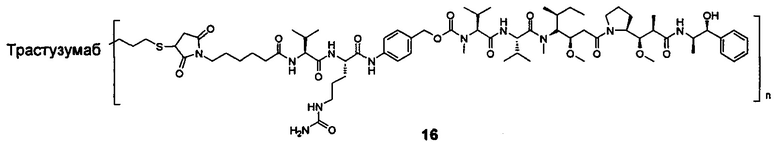
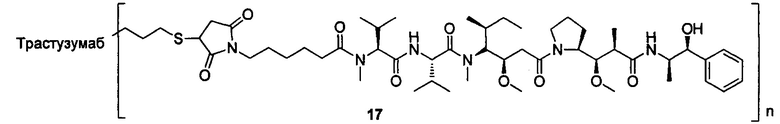
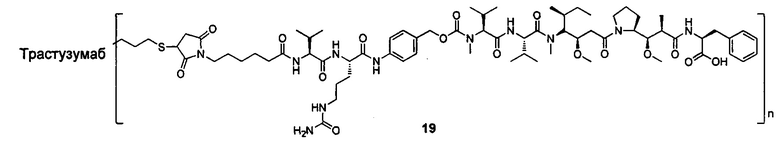
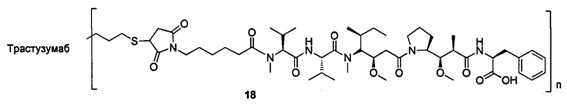
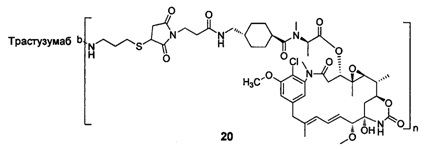
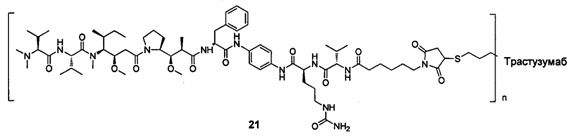
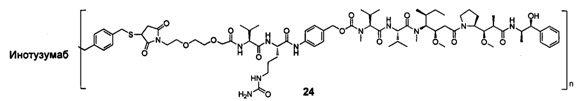

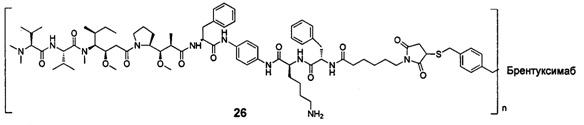
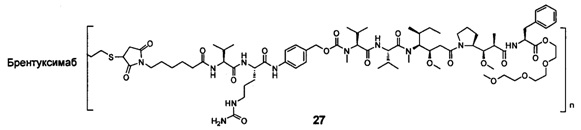
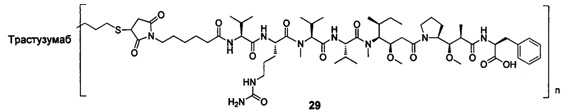
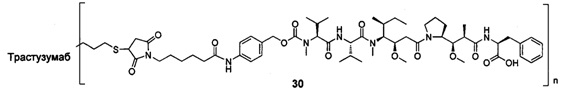
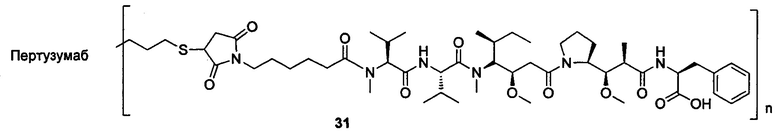

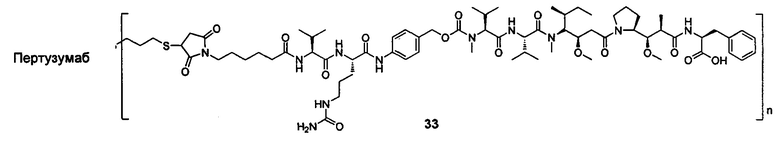
и
n выбран из 1-8, предпочтительно 1-4.
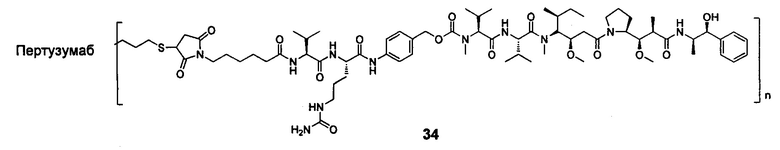
В другом конкретном воплощении настоящего изобретения предложен конъюгат лиганд-цитотоксическое лекарственное средство или его фармацевтически приемлемая соль или его сольват, упомянутые выше, выбранные из группы, состоящей из:

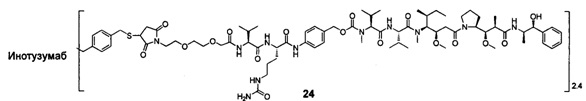
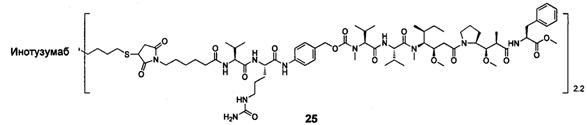
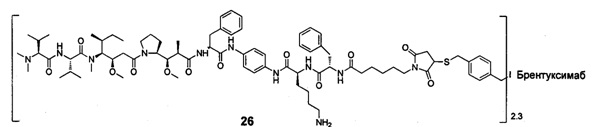
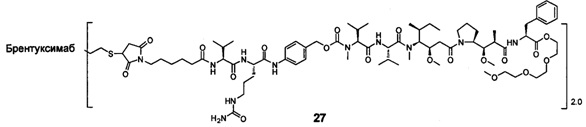

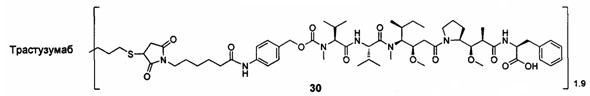
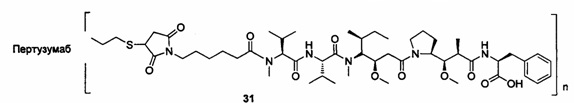
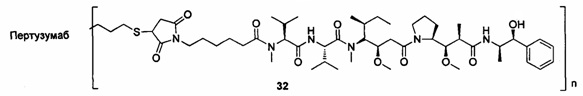
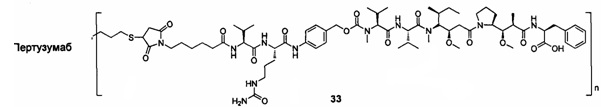
 ;
;
и
n выбран из 1-8, предпочтительно 1-4.
Настоящее изобретение дополнительно относится к способу получения конъюгата антитело-цитотоксическое лекарственное средство формулы (III),
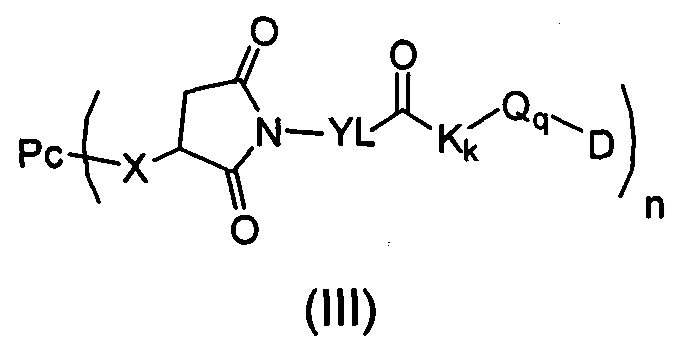
причем способ содержит следующие стадии:
1) добавление восстанавливающего агента RA к соединению формулы IA и соединению формулы IB, проведение реакции в условиях рН реакционной системы 3-6 и температуры реакции 0-40°С и получение соединения формулы IC:
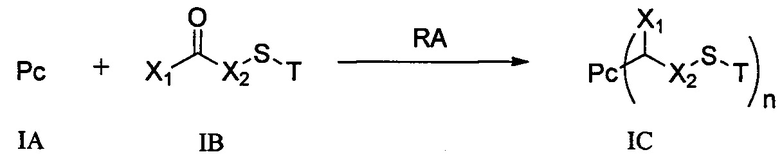 ;
;
где Т выбран из группы, состоящей из третичного бутила, ацетила, н-пропионила, изопропионила, трифенилметила, метоксиметила и 2-(триметилсилил)этоксиметила, предпочтительно ацетила;
2) в условиях температуры реакции 0-40°С добавление агента для снятия защиты к соединению формулы IC для удаления защитной группы Т тиольной группы и получение соединения формулы ID:
 ;
;
3) в условиях температуры реакции 0-40°С проведение реакции присоединения по Михаэлю между соединением формулы ID и соединением формулы IE и получение соединения формулы (III):
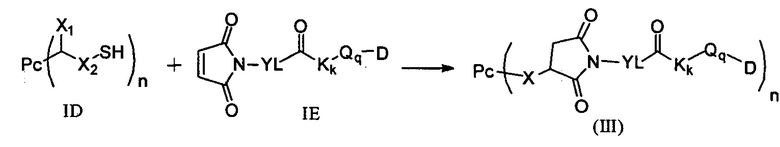 ;
;
где температура реакции предпочтительно составляет 15-30°С, наиболее предпочтительно 20-25°С; агент для снятия защиты предпочтительно представляет собой гидрохлорид гидроксиламина; восстанавливающий агент RA предпочтительно представляет собой цианоборогидрид натрия или триацетоксиборогидрид натрия;
где X1, Х2 являются такими, как определено в формуле X; Рс представляет собой лиганд; Т, n являются такими, как определено в формуле (II); YL, Kk, Qq являются такими, как определено в формуле (Y), D представляет собой цитотоксическое лекарственное средство.
Настоящее изобретение дополнительно относится к фармацевтической композиции, отличающейся тем, что фармацевтическая композиция содержит терапевтически эффективное количество конъюгата лиганд-цитотоксическое лекарственное средство или его фармацевтически приемлемой соли или его сольвата, упомянутых выше, и один или более чем один фармацевтически приемлемый носитель, разбавитель или эксципиент.
Настоящее изобретение дополнительно относится к применению конъюгата лиганд-цитотоксическое лекарственное средство или его фармацевтически приемлемой соли или его сольвата, или фармацевтической композиции, описанной выше, для получения лекарственного препарата для лечения рака, где рак представляет собой рак со сверхэкспрессией опухолеассоциированного рецептора, где опухолассоциированный рецептор представляет собой один или более чем один, выбранный из группы, состоящей из (1)-(8):
1) HER2 (ErbB2),
2) CD22,
3) CD30,
4) CD33,
5) CD44,
6) CD56,
7) Lewis Y и
8) GPNMB.
Настоящее изобретение дополнительно относится к способу модулирования рецептора in vitro, причем способ включает введение субъекту, подлежащему тестированию, эффективного количества конъюгата лиганд-цитотоксическое лекарственное средство или его фармацевтически приемлемой соли или его сольвата, или фармацевтической композиции, упомянутой выше, рецептор выбран из группы, состоящей из:
1) HER2 (ErbB2),
2) CD22,
3) CD30,
4) CD33,
5) CD44,
6) CD56,
7) Lewis Y и
8) GPNMB.
Настоящее изобретение дополнительно относится к способу лечения рака у млекопитающих, данный способ включает введение млекопитающему терапевтически эффективного количества конъюгата лиганд-цитотоксическое лекарственное средство или его фармацевтически приемлемой соли или его сольвата, или фармацевтической композиции, упомянутой выше, где млекопитающее представляет собой человека, рак выбран из группы, состоящей из рака молочной железы, рака яичника, рака желудка, рака эндометрия, рака слюнной железы, рака легкого, рака толстой кишки, рака почки, рака толстой и прямой кишки, рака щитовидной железы, рака поджелудочной железы, рака предстательной железы, рака мочевого пузыря, острого лимфоцитарного лейкоза, острого миелоидного лейкоза, острого промиелоцитарного лейкоза, хронического миелогенного лейкоза, хронического лимфоцитарного лейкоза, лимфомы Ходжкина, неходжкинской лимфомы и рецидивирующей анапластической крупноклеточной лимфомы, предпочтительно рака молочной железы, лимфомы Ходжкина или рецидивирующей анапластической крупноклеточной лимфомы; более предпочтительно рака молочной железы со сверхэкспрессией HER2 уровня 2+ или более высокого уровня, наиболее предпочтительно рака молочной железы, ассоциированного с экспрессией HER2.
Настоящее изобретение дополнительно относится к применению соединения формулы (IV), таутомеру, мезомеру, рацемату, энантиомеру, диастереомеру или их смеси, или его фармацевтически приемлемой соли в получении лекарственного препарата для лечения рака:
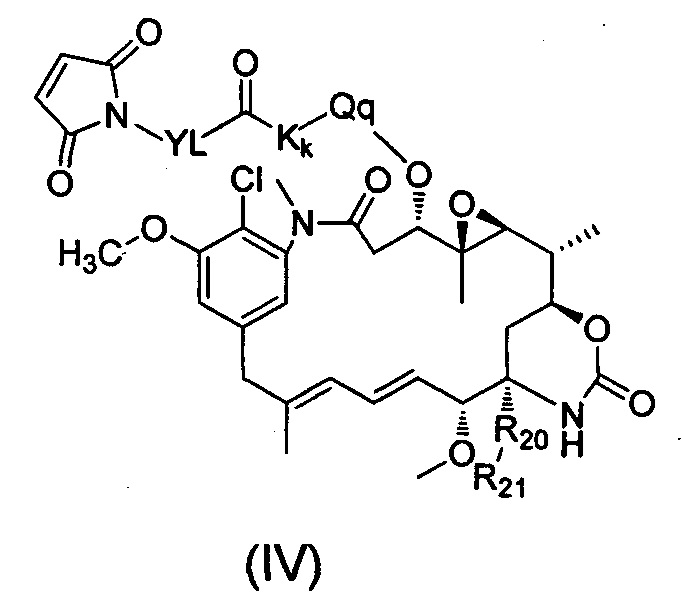
где:
YL выбран из алкила, циклоалкила, О-алкила, О-алкокси, арила, алкил-циклоалкила, циклоалкил-алкила, алкил-арила, алкил-циклоалкил-алкила, гетероциклила, алкил-гетероциклила, гетероциклил-алкила, алкил-гетероциклил-алкила, арила, алкил-арила, арил-алкила, алкил-арил-алкила, гетероарила, алкил-гетероарила, гетероарил-алкила, алкил-гетероарил-алкила, CH2(OCH2CH2)t, (CH2CH2O)tCH2 и (CH2CH2O)t, t представляет собой целое число, выбранное из 1-10, предпочтительно алкильной группы, более предпочтительно С2-С8 линейного алкила;
Kk представляет собой аминокислотное звено, где K представляет собой аминокислоту, k представляет собой целое число, выбранное из 0-10, предпочтительно k представляет собой 2, Kk предпочтительно представляет собой валин-цитруллин;
Qq представляет собой протяженное звено, где q представляет собой 0, 1 или 2;
R20 выбран из О или S;
R21 выбран из группы, состоящей из Н, алкила, циклоалкила, гетероциклила, арила и гетероарила, где алкил, циклоалкил, гетероциклил, арил и гетероарил независимо и необязательно дополнительно замещены одной или более чем одной группой, выбранной из группы, состоящей из галогена, гидрокси, циано, нитро, алкила, алкокси, циклоалкила, гетероциклила, арила и гетероарила, После связывания N-концевой аминогруппы и/или аминогруппы остатка лизина антитела согласно настоящему изобретению с соединительным звеном X, имеющим свободную тиольную группу, реакции восстановления избегают в шарнирной области антитела, таким образом, воздействие на структуру самого антитела снижается. Кроме того, введенная структура связи углерод-азот является стабильной, нелегко поддающейся разрушению при циркуляции в организме, соотношение лекарственное средство-антитело можно контролировать в пределах нормального распределения 0-5 посредством дополнительного контроля условий реакции.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Если не указано иное, все технические и научные термины, используемые в данном документе, согласуются с теми, которые обычно понятны среднему специалисту в данной области. Несмотря на то, что похожие или эквивалентные способы и материалы могут быть также использованы для осуществления или испытания настоящего изобретения, в настоящем описании описываются предпочтительные способы и материалы. Следующие термины используются для описания и заявления настоящего изобретения в соответствии со следующими определениями.
При использовании в данном изобретении торговых наименований, предполагается, что они включают препараты, непатентованные лекарственные средства и активные группировки лекарственных средств под торговыми наименованиями.
Если не указано иное, используемые термины в данном описании и формуле изобретения, имеют следующие значения.
Термин «алкил» относится к насыщенной алифатической углеводородной группе, включающей С1-С20 линейные или разветвленные группы, предпочтительно алкилу, имеющему 1-12 атомов углерода, более предпочтительно алкилу, имеющему 1-10 атомов углерода, наиболее предпочтительно алкилу, имеющему 1-6 атомов углерода. Типичные примеры включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, 1,1-диметил пропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1-этил-2-метилпропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2-этилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2,3-диметилбутил, н-гептил, 2-метилгексил, 3-метилгексил, 4-метилгексил, 5-метилгексил, 2,3-диметилпентил, 2,4-диметилпентил, 2,2-диметилпентил, 3,3-диметилпентил, 2-этилпентил, 3-этилпентил, н-октил, 2,3-диметилгексил, 2,4-диметилгексил, 2,5-диметилгексил, 2,2-диметилгексил, 3,3-диметилгексил, 4,4-диметилгексил, 2-этилгексил, 3-этилгексил, 4-этилгексил, 2-метил-2-этилпентил, 2-метил-3-этилпентил, н-нонил, 2-метил-2-этилгексил, 2-метил-3-этилгексил, 2,2-диэтилпентил, н-децил, 3,3-диэтилгексил, 2,2-диэтилгексил и их разные разветвленные изомеры, но не ограничиваются ими. Низший алкил, имеющий от 1 до 6 атомов углерода, является более предпочтительным. Типичные примеры включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, emop-бутил, н-пентил, 1,1-диметил пропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1-этил-2-метил пропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2-этилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2,3-диметилбутил и тому подобное, но не ограничиваются ими. Алкильная группа может быть замещенной или незамещенной. Будучи замещенной, замещающая(ие) группа(ы) может(гут) быть замещена(ны) в любой доступной точке присоединения, и замещающая(ие) группа(пы) предпочтительно представляет(ют) собой одну или более чем одну группу, независимо выбранную из группы, состоящей из алкила, алкенила, алкинила, алкилоксила, алкилтиола, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклического алкила, арила, гетероарила, циклоалкокси, гетероциклического алкокси, циклоалкилтио, гетероциклоалкилтио и оксо.
Термин «Циклоалкил» относится к насыщенной или частично ненасыщенной моноциклической или полициклической гидрокарбильной группе. Циклоалкил имеет от 3 до 20 атомов углерода, предпочтительно от 3 до 12 атомов углерода, более предпочтительно от 3 до 10 атомов углерода, наиболее предпочтительно от 3 до 8 атомов углерода. Типичные примеры моноциклического циклоалкила включают циклопропил, циклобутил, циклопентил, циклопентенил, циклогексил, циклогексенил, циклогексадиенил, циклогептил, циклогептатриенил, циклооктил и т.д., но не ограничиваются ими. Полициклический циклоалкил включает циклоалкил, имеющий спирокольцо, конденсированное кольцо или кольцо с внутренним мостиком.
Термин «Гетероциклил» относится к насыщенному или частично ненасыщенному моноциклическому или полициклическому углеводородному заместителю, имеющему от 3 до 20 атомов цикла, где один или более чем один атом цикла представляет собой гетероатом, выбранный из группы, состоящей из N, О и S(O)m (где m представляет собой целое число от 0 до 2), но исключая -О-О-, -O-S- или -S-S- в кольце, остальные атомы цикла представляют собой атомы С. От 3 до 12 атомов цикла является предпочтительным, где от 1 до 4 атомов представляют собой гетероатомы; От 3 до 10 атомов цикла является более предпочтительным. Типичные примеры моноциклического гетероциклила включают пирролидинил, пиперидил, пиперазинил, морфолинил, тиоморфолинил, гомопиперазинил и тому подобное, но не ограничиваются ими. Полициклический гетероциклил включает гетероциклил, имеющий спирокольцо, конденсированное кольцо или кольцо с внутренним мостиком.
Кольцо указанного гетероциклила может быть конденсировано с кольцом арила, гетероарила или циклоалкила, где кольцо, связанное с исходной структурой, представляет собой гетероциклил. Типичные примеры включают следующие группы, но не ограничиваются ими:
 и
и  и т.д.
и т.д.
Гетероциклил может быть необязательно замещен или не замещен. Будучи замещенным, замещающая(ие) группа(ы) предпочтительно представляет(ют) собой одну или более чем одну группу(ы), независимо выбранную(ые) из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилсульфо, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклического алкила, арила, гетероарила, циклоалкокси, гетероциклического алкокси, циклоалкилтио, гетероциклического алкилтио и оксогруппы.
Термин «арил» относится к 6-14-членному цельноуглеродному моноциклическому кольцу или конденсированному полициклическому кольцу (то есть кольца имеют общую пару смежных атомов углерода), которое имеет систему сопряженных связей с 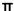 -электронами. Арил предпочтительно является 6-10-членным, таким как фенил и нафтил, предпочтительно фенил. Арильное кольцо может быть конденсировано гетероарильным, гетероциклильным или циклоалкильным кольцом, где кольцо, связанное с исходной структурой, представляет собой арил. Типичные примеры включают следующие группы, но не ограничиваются ими:
-электронами. Арил предпочтительно является 6-10-членным, таким как фенил и нафтил, предпочтительно фенил. Арильное кольцо может быть конденсировано гетероарильным, гетероциклильным или циклоалкильным кольцом, где кольцо, связанное с исходной структурой, представляет собой арил. Типичные примеры включают следующие группы, но не ограничиваются ими:
 и
и  .
.
Арильная группа может быть замещенной или незамещенной. Будучи замещенной, замещающая(ие) группа(ы) предпочтительно представляет(ют) собой одну или более чем одну группу, независимо выбранную из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтиола, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклоалкила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио и гетероциклоалкилтио.
Термин «гетероарил» относится к гетероароматической системе, имеющей от 1 до 4 гетероатомов и от 5 до 14 атомов цикла, где гетероатомы выбраны из группы, состоящей из О, S и N. Гетероарил предпочтительно является 5-10-членным, более предпочтительно 5- или 6-членным, таким как фурил, тиенил, пиридинил, пирролил, N-алкилпирролил, пиримидинил, пиразинил, имидазолил, тетразолил и тому подобное. Гетероарил может быть конденсирован с арильным кольцом, гетероциклильным или циклоалкильным кольцом, где кольцо, связанное с исходной структурой, представляет собой гетероарил. Типичные примеры включают следующие группы, но не ограничиваются ими:
 и
и  .
.
Гетероарильная группа может быть необязательно замещенной или незамещенной. Будучи замещенной, замещающая(ие) группа(ы) предпочтительно представляет(ют) собой одну или более чем одну группу, независимо выбранную из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтиола, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклоалкила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио и гетероциклоалкилтио.
Термин «алкокси» относится как к группе -О-(алкил), так и к группе -О-(ненасыщенный циклоалкил), где алкил является таким, как определено выше. Типичные примеры алкокси включают метокси, этокси, пропокси, бутокси, циклопропилокси, циклобутилокси, циклопентилокси и циклогексилокси, но не ограничиваются ими. Алкокси может быть необязательно замещенным или незамещенным. Будучи замещенным, заместитель предпочтительно представляет собой одну или более чем одну группу, независимо выбранную из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилсульфо, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклоалкила, арила, гетероарила, циклоалкокси, гетероциклического алкокси, циклоалкилтио и гетероциклического алкилтио.
Термин «связь» относится к ковалентной связи, представленной как «-»
Термин «Гидрокси» относится к -ОН группе.
Термин «Галоген» относится к атомам фтора, хлора, брома или йода.
Термин «Амино» относится к -NH2 группе.
Термин «Циано» относится к -CN группе.
Термин «Нитро» относится к -NO2 группе.
Термин «Оксогруппа» относится к =O группе.
Термин «Карбоксил» относится к -С(O)ОН группе.
Термин «Алкоксикарбонил» относится к -С(O)O(алкильной) или (циклоалкильной) группе, где алкил является таким, как определено выше.
Термин «бензил» относится к метилбензольной группе:  .
.
Термин «необязательный» или «необязательно» означает, что событие или обстоятельство, описанное далее, может происходить, но не обязательно происходит, и описание включает примеры, в которых событие или обстоятельство происходит или не происходит. Например, фраза «гетероциклическая группа, необязательно замещенная алкилом» означает, что алкильная группа может присутствовать, но не обязательно присутствует, и описание включает случай, когда гетероциклическая группа замещена алкилом и случай, когда гетероциклическая группа не замещена алкилом.
«Замещенный» относится к одному или более чем одному атому водорода в группе, предпочтительно вплоть до 5, более предпочтительно от 1 до 3 атомов водорода, причем каждый независимо замещен соответствующим числом заместителей. Излишне говорить о том, что заместители существуют только в своем единственно возможном химическом положении. Специалист в данной области способен определять, возможно ли замещение или невозможно, не прилагая излишних усилий в экспериментальном исследовании или теории. Например, комбинация амино- или гидроксигруппы, имеющей свободный водород, и атомов углерода, имеющих ненасыщенные связи (такие как олефиновые), может быть нестабильной.
Термин «фармацевтическая композиция» относится к смеси одного или более чем одного соединения, описанного в настоящем изобретении, или его физиологически/фармацевтически приемлемых солей или пролекарств и других химических компонентов, таких как физиологически/фармацевтически приемлемые носители и эксципиенты. Назначение фармацевтической композиции заключается в облегчении введения соединения в организм, помощи в абсорбции активного ингредиента, демонстрируя, таким образом, биологическую активность.
Термин «фармацевтически приемлемая соль» относится к солевой форме конъюгата лиганд-цитотоксическое лекарственное средство по настоящему изобретению, такой вид солей является безопасным и эффективным и обладает биологической активностью, которая необходима для млекопитающих in vivo. Соединение конъюгата антитело-лекарственное средство по настоящему изобретению содержит по меньшей мере одну аминогруппу, которая может образовывать соль с кислотой. Неограничивающие примеры фармацевтически приемлемых солей включают: гидрохлорид, гидробромид, гидройодид, сульфат, бисульфат, цитрат, ацетат, сукцинат, аскорбат, оксалат, нитрат, грушевые соли, гидрофосфат, дигидрофосфат, салицилат, гидроцитрат, тартрат, малеат, фумарат, формиат, бензоат, метансульфонат, этансульфонат, бензолсульфонат, п-толуолсульфонат.
Термин «сольват» относится к фармацевтически приемлемому растворителю, образованному соединениями конъюгата лиганд-лекарственное средство по настоящему изобретению и одной или более чем одной молекулой сольвата. Неограничивающие примеры молекул сольвата включают: воду, этанол, ацетонитрил, изопропиловый спирт, DMSO (диметилсульфоксид), этилацетат.
Термин «лиганд» представляет собой макромолекулярное соединение, способное распознавать и связываться с антигенами или рецепторами, ассоциированными с клеткой-мишенью. Роль лиганда заключается в доставке лекарственного средства, связанного с лигандом, к популяции клеток-мишеней, лиганд включает гормоны белковой природы, пектины, факторы роста, антитела или другие молекулы, способные связываться с клетками, но не ограничивается ими. В воплощении настоящего изобретения лиганд выражен в виде Рс. Соединительная связь может быть образована между гетероатомом в лиганде и соединительным звеном.
«Антиген или рецептор» используется лигандом для распознавания и связывания с клетками-мишенями. В настоящем изобретении лиганды против антигенов или рецепторов поверхности клеток, экспрессирующихся в клетках-мишенях и/или ткани пролиферативных заболеваний, таких как рак, являются предпочтительными; неограничивающие воплощения рецепторов поверхности клеток выбраны из рецепторов поверхности клеток HER2, HER3, HER4, CD20, CD22, CD30, CD33, CD44, Lewis Y, CD56, CD105, VEGFR или GPNMB. Наиболее предпочтительными являются рецепторы поверхности клеток, выбранные из группы рецепторов поверхности клеток HER2, CD22, CD30, CD33, CD44, CD56, Lewis Y и GPNMB. Конкретно, предпочтительные неограничивающие воплощения включают: Трастузумаб (HER2), Инотузумаб (CD22), Пинатузумаб (CD22), Брантуксимаб (CD30), Гемтузумаб (CD33), Биватузумаб (CD44), Лорвотузумаб (CD56), cBR96 (Lewis Y) или Глематумамаб (GPNMB).
Термин «антитело», в том виде, в котором он используется в данном документе, относится к любой форме антитела, демонстрирующей желательную биологическую активность. Таким образом, он используется в самом широком смысле, в частности, включая полноразмерные антитела, связывающие фрагменты антитела или их производные, но не ограничиваясь ими. Источники антител включают моноклональные антитела, поликлональные антитела, генетически сконструированные антитела (например биспецифичные антитела), но не ограничиваются ими.
Термин «полноразмерное антитело» относится к полимеру иммуноглобулина (такому как IgM), содержащему 4 полипептидные цепи (то есть 2 тяжелые цепи и 2 легкие цепи), поперечно связанные дисульфидными связями. Каждая тяжелая цепь содержит фрагмент вариабельной области тяжелой цепи (сокращенно VH) и фрагмент константной области тяжелой цепи, константная область тяжелой цепи содержит три домена: СН1, СН2 и СН3. Каждая легкая цепь содержит фрагмент вариабельной области легкой цепи (сокращенно VL) и фрагмент константной области легкой цепи, константная область легкой цепи содержит один домен (CL1). Области VH и VL могут быть дополнительно разделены на гипервариабельные области, которые называются областями, определяющими комплементарность (CDR), более консервативные домены, называемые каркасной областью (FR), располагаются у каждой области, определяющей комплементарность.
Термин «связывающий фрагмент антитела или производное» включает любой встречающийся в природе, полученный ферментативным путем, синтезированный или генетически сконструированный полипептид или гликопротеин, который может связываться с антигеном и образовывать комплекс; обычно он содержит по меньшей мере часть антигенсвязывающей области или вариабельной области (например одну или более чем одну CDR) исходного антитела и сохраняет по меньшей мере некоторую специфичность связывания исходного антитела. «Связывающий фрагмент антитела или производное» может происходить от антитела, например, полученного посредством преобразования полноразмерного антитела с помощью соответствующих стандартных методик, включая методики протеолиза или рекомбинантных ДНК (включая манипуляции с и экспрессию ДНК, экспрессирующую вариабельную область антитела и часть константной области). Термин «связывающий фрагмент антитела или производное» включает: (i) Fab фрагменты; (ii) F(ab')2 фрагменты; (iii) Fd фрагменты; (iv) Fv фрагменты; (V) одноцепочечный Fv (scFv); (vi) dAb фрагменты; и (vii) минимальную единицу распознавания имитирующих аминокислотных остатков гипервариабельной области антитела (например выделенная область, определяющая комплементарность (CDR)), но не ограничивается ими. Другие конструирующие молекулы, такие как бивалентные антитела, трехвалентные антитела, четырехвалентные антитела и микроантитела подпадают под термин «связывающий фрагмент антитела или производное».
«Fab фрагмент» состоит из полноразмерной легкой цепи и VH тяжелой цепи и функциональных доменов СН1. Тяжелая цепь молекулы Fab не может образовывать дисульфидную связь с другой молекулой тяжелой цепи.
«Fc» область содержит два фрагмента тяжелой цепи, содержащих домены СН1 и СН2 антитела. Данные два фрагмента тяжелой цепи соединены с помощью двух или более чем двух дисульфидных связей и гидрофобного эффекта домена СН3.
«Fab' фрагмент» содержит легкую цепь и функциональные области VH и СН1 тяжелой цепи и дополнительно содержит область между доменом СН1 и доменом СН2, так что между двумя данными тяжелыми цепями двух Fab' фрагментов может образовываться дисульфидная связь с образованием молекулы F(ab')2.
«F(ab')2 фрагмент» содержит две легкие цепи и две тяжелые цепи, содержащие неполную константную область между доменом СН1 и доменом СН2, так что между двумя данными тяжелыми цепями может образовываться межцепочечная дисульфидная связь. Таким образом, F(ab')2 фрагмент состоит из двух Fab' фрагментов, удерживаемых вместе посредством дисульфидной связи между двумя данными тяжелыми цепями.
«Fv фрагмент» содержит легкую цепь или/и функциональный домен вариабельной области тяжелой цепи (VH).
«Область Fc» соответствует функциональным доменам СН2 и СН3 IgG без какой-либо антигенсвязывающей активности, она представляет собой часть молекулы антитела, взаимодействующую с эффекторными молекулами и клетками.
«Шарнирная область» используется для связи Fab фрагмента антитела с Fc фрагментом. В настоящем изобретении с Fc фрагментом может быть связан биспецифичный слитый белок.
Антитело по настоящему изобретению предпочтительно представляет собой специфичное антитело, связывающееся с антигеном поверхности клеток-мишеней; неограничивающие воплощения включают следующие антитела:
Антитела против антигена поверхности клеток HER2 (в большей степени существующий на поверхности клеток рака молочной железы); антитела против большинства В-клеточных лимфом со сверхэкспрессией антигенов CD20 или CD22; антитела против антигена поверхности клеток CD33 (антиген поверхности клеток преобладает в некоторых миеломах человека, особенно при остром миелоидном лейкозе); антитела против антигенов поверхности клеток CD30, CD44, Lewis Y, CD56, CD105, VEGFR или GPNMB; кроме того, другие коммерчески доступные антитела, такие как трастузумаб (торговое наименование Herceptin®), можно также использовать в качестве лиганда. Трастузумаб представляет собой гуманизированное антитело против HER2, используемое для лечения рака молочной железы, для лечения метастатического рака молочной железы со сверхэкспрессией HER2.
Термин «идентичность» относится к сходству последовательностей у двух полинуклеотидных последовательностей или двух полипептидов. Когда положения двух сравниваемых последовательностей занимает одно и то же основная или аминокислотная мономерная субъединица, например, если каждое положение двух молекул ДНК занимает аденин, тогда две данные молекулы являются идентичными в данном положении. Выраженная в процентах идентичность двух последовательностей представлена в виде функции: число общих совпадающих или идентичных положений двух последовательностей, разделенное на число сравниваемых положений х 100. Например, при оптимальном выравнивании последовательностей, если шесть положений совпадают или являются идентичными в 10 положениях двух последовательностей, тогда идентичность двух данных последовательностей составляет 60%. В общем, сравните две последовательности и получите самый высокий уровень идентичности, выраженный в процентах.
Термин «промежуточное звено» (Y) представляет собой бифункциональное соединение, используемое для соединения лиганда по настоящему изобретению и цитотоксического лекарственного средства с образованием конъюгата лиганд-промежуточное звено-лекарственное средство, или используемое для образования иммуноконъюгата против опухолеассоциированного антигена. Такой иммуноконъюгат может селективно доставлять цитотоксические лекарственные средства к опухолевым клеткам.
Под термином «цитотоксическое лекарственное средство» подразумевается химическая молекула, способная сильно нарушать нормальный рост в опухолевых клетках. По существу, цитотоксические лекарственные средства могут уничтожать опухолевые клетки в достаточно высоких концентрациях, но из-за недостатка специфичности, при уничтожении опухолевых клеток, это будет также приводить к апоптозу в нормальных клетках, приводя к серьезным побочным эффектам. В воплощении настоящего изобретения цитотоксическое лекарственное средство представлено как D, неограничивающие примеры включают: ингибиторы тубулина, ДНК-алкилирующие агенты, ингибиторы тирозинкиназы, ингибиторы топоизомеразы или ингибиторы синтеза ДНК, предпочтительно ингибиторы тубулина.
Ауристатины представляют собой полностью синтетические лекарственные средства с относительно легко преобразованной химической структурой, которая облегчает оптимизацию физических свойств и способности поддаваться воздействию лекарственных средств. Производные ауристатинов, используемые для конъюгирования с антителами, включают монометилауристатин Е (ММАЕ) и монометилауристатин F (MMAF), причем первый представляет собой синтетический пентапептид, происходящий от встречающегося в природе ингибитора полимеразы тубулина доластатина-10, синтезированный посредством добавления 2-амино-1-ол-фенилпропила на С-конец. Ингибирующие активности ММАЕ против множества линий опухолевых клеток человека являются меньше, чем однонаномолярными. Для снижения цитотоксической активности самого ММАЕ, в случае MMAF, на С-конец долостатина-10 вводят фенилаланин, в результате введения карбоксильной группы в структуру, MMAF обладает плохой способностью к прохождению через мембрану, и, вследствие этого, биологическая активность против клеток значительно снижена, но ингибирующая активность против клеток после конъюгирования с антителом существенно повышается (US 7750116).
Термин «ингибитор тубулина» относится к классу соединений, которые оказывают противоопухолевое действие посредством ингибирования или стимулирования полимеризации тубулина и, следовательно, препятствуя процессу митоза в клетках. Неограничивающие примеры включают: майтанзиноиды, калихеамицины, таксаны, винкристины, колхицины, доластатины/ауристатины, предпочтительно выбранные из майтанзиноидов или доластатинов/ауристатинов; более предпочтительно выбранные из соединений формулы D1 или DM.
Термин «ДНК-алкилирующий агент» относится к классу соединений, которые могут образовывать карбокатионы или другие положительно заряженные ионы или другие активные электрофильные группы в организме и дополнительно ковалентно связываются с группами, содержащими избыточное количество электронов в ДНК в клетках (например, амино, меркапто, гидрокси, карбоксил, фосфорил и т.д.), вызывать структурные изменения или разрыв ДНК и, следовательно, приводить к гибели опухолевых клеток. Неограничивающие примеры включают: азотистые иприты (циклофосфамид), этилиденгидразоноамины (тиотепа, митомицин), метансульфонаты (бусульфан), полиолы (дибромманнит), нитрозомочевины (кармустин), триазенимидазол (дакарбазин) и гидразины (прокарбазин).
Термин «ингибитор тирозинкиназы» относится к классу конкурентных ингибиторов, которые могут связываться с тирозинкиназой в виде аденозинтрифосфата (АТФ), также относится к классу соединений, которые блокируют активность тирозинкиназ и ингибируют пролиферацию клеток в виде тирозиновых аналогов. Неограничивающие примеры включают: иматиниб, гефитиниб, эрлотиниб, сунитиниб, сорафениб, лапатиниб, дазатиниб, нилотиниб и тому подобное.
Термин «ингибитор синтеза белков» относится к классу соединений, которые могут воздействовать на белок, синтезируемый клеткой-мишенью. Ингибиторы синтеза белков могут действовать на каждом этапе синтеза белков, таком как репликация ДНК, транскрипция РНК, и играют роль путем ингибирования факторов инициации, факторов элонгации и рибосом. Неограничивающие примеры включают: аминогликозиды, тетрациклины, макролиды и хлорамфениколы.
Термин «соотношение лекарственное средство-антитело» обозначает среднее число цитотоксических лекарственных средств, загруженных на каждом лиганде приведенной выше формулы (I), также представлено в виде соотношения количества лекарственного средства и количества антитела, интервал загрузок лекарственного средства в отношении каждого лиганда (Рс) может составлять 1-8 цитотоксических лекарственных средств (D), в воплощении настоящего изобретения соотношение лекарственное средство-антитело представлено как n, среднее число лекарственных средств в каждой молекуле ADC после реакции связывания может быть идентифицировано общепринятыми способами, такими как спектроскопия в УФ- и видимой области, масс-спектрометрия, анализ ELISA (твердофазный иммуноферментный анализ) и идентификация характеристических ионов посредством ВЭЖХ (высокоэффективная жидкостная хроматография).
В настоящем изобретении n может ограничиваться числом сайтов соединения, в одном воплощении настоящего изобретения цитотоксическое лекарственное средство конъюгировали на N-концевой аминогруппе и/или ε-аминогруппе остатка лизина через соединительное звено, в общем, число молекул лекарственного средства, которые конъюгированы с антителом в реакции связывания, будет меньше, чем теоретический максимум.
Для контроля количества загрузки конъюгата лиганд-цитотоксическое лекарственное средство можно использовать следующие неограничивающие способы, включающие:
(1) контроль молярного соотношения соединительных реагентов и MAb,
(2) контроль времени и температуры реакции
(3) выбор разных реагентов реакции.
Термин «соединительное звено» относится к фрагменту химической структуры, который ковалентно связан с лигандом через атом углерода на одном конце и ковалентно связан с цитотоксическими лекарственными средствами через атом серы на другом конце. В настоящем изобретении соединительное звено определено в виде общей формулы (X). Соединительное звено связано с аминогруппой антитела способом восстановительного аминирования, предпочтительно с N-концом антитела и/или с ε-амино остатков лизина.
Термин «промежуточное звено» представляет собой бифункциональный фрагмент соединения, используемый для связи соединительного звена с цитотоксическим лекарственным средством и, в конечном итоге, образования конъюгата лиганд-цитотоксическое лекарственное средство; такой способ соединения может селективно доставлять цитотоксические лекарственные средства к соединительному звену. В настоящем изобретении промежуточное звено предпочтительно показано в виде общей формулы (Y).
Термин «аминокислотное звено» относится к тому случаю, когда существует протяженное звено, карбонильная группа следующей структурной формулы YR может быть соединена с протяженным звеном. При отсутствии протяженного звена YR непосредственно связан с аминокислотой на цитотоксических лекарственных средствах. В воплощениях настоящего изобретения аминокислотное звено представлено как -Kk-:
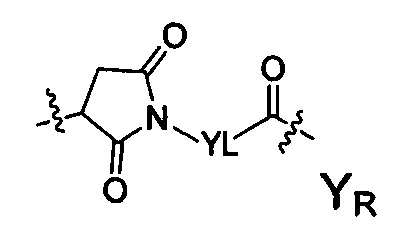
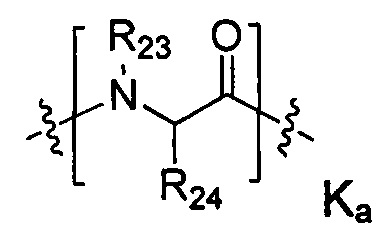
-Kk- представляет собой дипептид, трипептид, тетрапептид, пентапептид, гексапептид, гептапептид, октапептид, нонапептид или декапептид, каждое -K- звено независимо содержит следующую структурную формулу Ka или Kb, k представляет собой целое число из 0-10:
 или
или 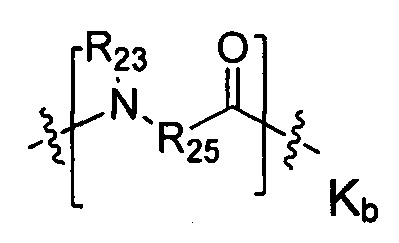
где:
R23 представляет собой -Н или метил;
R24 представляет собой Н, метил, изопропил, изобутил, втор-бутил, бензил, п-гидроксибензил, -СН2ОН, -СН(ОН)СН3, -CH2CH2SCH3, -CH2CONH2, -СН2СООН, -CH2CH2CONH2, -СН2СН2СООН, -(CH2)3NHC(=NH)NH2, -(CH2)3NH2, (CH2)3NHCOCH3, -(CH2)3NHCHO, -(CH2)4NHC(=NH)NH2, -(CH2)4NH2, (CH2)4NHCOCH3, -(CH2)4NHCHO, -(CH2)3NHCONH2, -(CH2)4NHCONH2, -CH2CH2CH(OH)CH2NH2, 2-пиридилметил-, 3-пиридилметил-, 4-пиридилметил-, фенил или циклогексил,
 ,
,  ,
,  ,
,
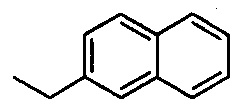 ,
, 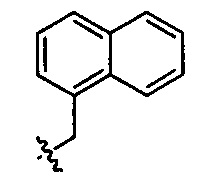 ,
,  ,
,
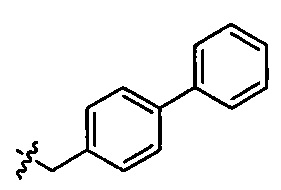 ,
, 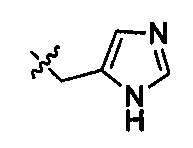 ,
, 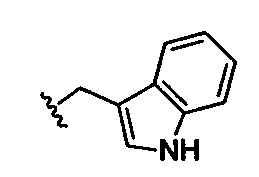 ;
;
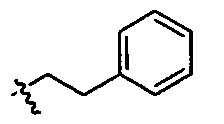
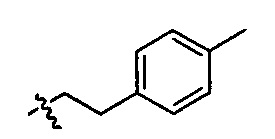
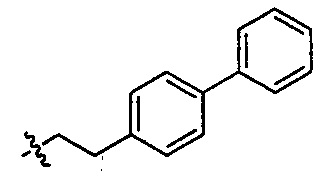
R25 представляет собой -арил-, -алкил-арил-, -циклоалкил-, -алкил-циклоалкил-, -циклоалкил-алкил-, -алкил-циклоалкил-алкил-, -гетероциклил-, -алкил-гетероциклил-, -гетероцикл-алкил-, -алкил-гетероцикл-алкил-, -арил-, -алкил-арил-, -арил-алкил-, -алкил-арил-алкил-, -гетероарил-, -алкил-гетероарил-, -гетероарил-алкил-, -алкил-гетероарил-алкил-.
В одном воплощении -Kk- представляет собой дипептид, предпочтительно -валин-цитруллин-, -фенилаланин-лизин- или -N-метилвалин-цитруллин- и более предпочтительно -валин-цитруллин-.
В другом воплощении -Kk- представляет собой дипептид, предпочтительно 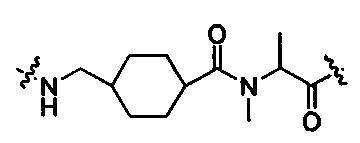 и
и 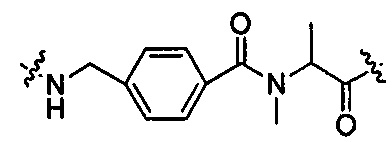 .
.
Термин «аминокислота» относится к органическому соединению, структура молекулы которого содержит амино- и карбоксильную группы, и амино- и карбоксильная группы непосредственно связаны с -СН- структурой. Формула представляет собой H2NCHRCOOH. В соответствии с положением аминогруппы относительно атома углерода их можно подразделять на α, β, γ, δ, ε … - аминокислоты. В области биологии аминокислоты, которые составляют структуру нативного белка, имеют свои специфические характеристики, то есть: аминогруппа присоединяется непосредственно к α-атому углерода, а именно α-аминокислоты, включая Gly(Глицин), Ala(Аланин), Val(Валин), Leu(лейцин), Ile(изолейцин), Phe(фенилаланин), Trp(триптофан), Tyr(тирозин), Asp(аспарагиновая кислота), His(гистидин), Asn(аспарагин), Glu (глутаминовая кислота), Lys (лизин), Gln(глутамин), Met(метионин), Arg(аргинин), Ser(серин), Thr(треонин), Cys (цистеин), Pro(пролин) и тому подобное.
В одном воплощении настоящего изобретения аминокислота выбрана из  и
и 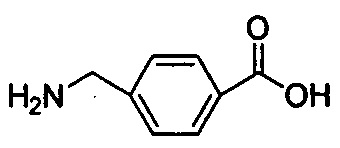 .
.
Термин «протяженное звено» относится к случаю, когда присутствует аминокислотное звено, оно может связывать данное аминокислотное звено с цитотоксическим лекарственным средством, или когда аминокислотное звено отсутствует, протяженное звено может представлять собой химическую структуру, конъюгированную с цитотоксическим и лекарственными средствами посредством карбонильной группы YR. В воплощении настоящего изобретения протяженное звено представлено в виде -Qq-, q выбран из 0, 1, 2.
В одном предпочтительном воплощении Q представляет собой структуру парааминобензилового спирта. В данном воплощении возможный механизм высвобождения лекарственного средства in vivo показан на Схеме 1 (Toki BE, Cerveny CG, J Org. Chem. 2002 (67) 1866-1872):
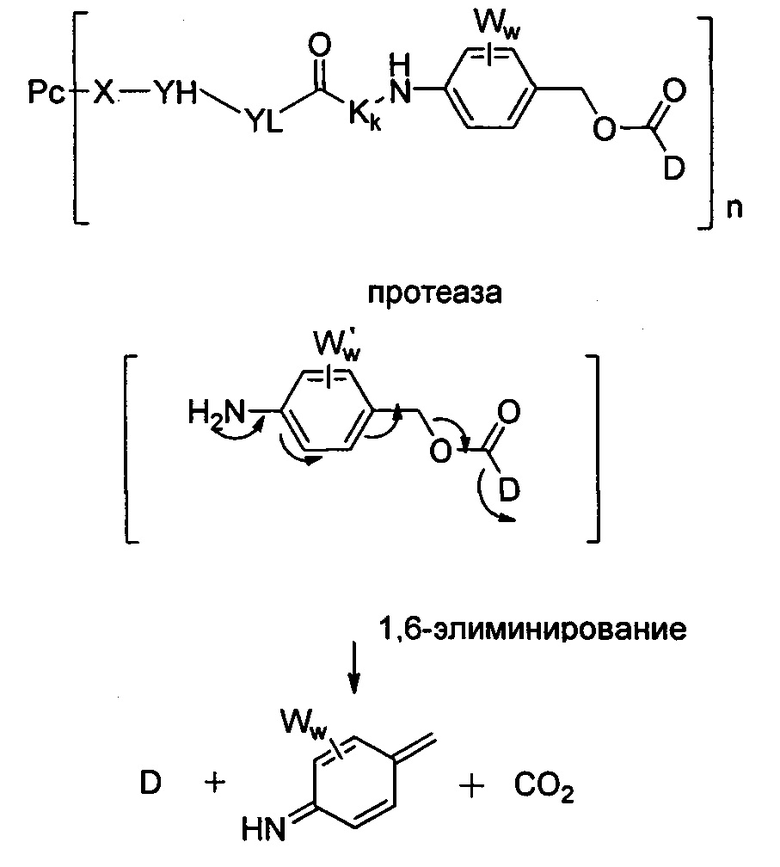
Схема 1
где:
W выбран из C1-C8 алкила, галогена, нитро или циано;
w представляет собой целое число, выбранное из 0-4.
Получение традиционных фармацевтических композиций показано в Китайской фармакопее.
Термин «носитель» применяют в отношении лекарственного средства по настоящему изобретению, относится к системе, которая может менять способ поступления лекарственного средства в организм человека и менять распространение in vivo, контролировать скорость высвобождения лекарственного средства и доставку лекарственное средства к органу-мишени. Высвобождение носителя лекарственного средства и система нацеливания способно уменьшать деградацию и потерю лекарственного средства, уменьшая побочные эффекты и улучшая биодоступность. Например, макромолекулярное поверхностно-активное вещество, используемое в качестве носителя, может самостоятельно собираться с образованием агрегатов в разных формах благодаря своей уникальной амфифильной структуре, предпочтительные примеры включают мицеллы, эмульсии, гели, жидкие кристаллы, везикулы и т.д. Данные агрегаты не только обладают способностью к захвату молекул лекарственных средств, но также демонстрируют хорошую проницаемость в отношении мембраны, могут быть использованы в качестве превосходных носителей лекарственных средств.
Термин «эксципиент» представляет собой дополнительный компонент, отличный от основного лекарственного средства в фармацевтических композициях, также называемый вспомогательным. Например, связывающие агенты, наполнители, дезинтегрирующие агенты, смазывающие вещества в таблетке; матричные элементы в полутвердых композициях, такие как мази, кремы; консерванты, антиоксиданты, вкусовые добавки, ароматизирующие агенты, сорастворители, эмульгаторы, солюбилизаторы, агенты, регулирующие тоничность, красители в жидкой композиции и тому подобное могут быть названы эксципиентами.
Термин «разбавитель» также известен как наполнитель, его основное назначение заключается в повышении массы и объема таблетки. Добавление разбавителя не только должно обеспечивать получение определенного объема, но также снижать отклонение дозы основных компонентов и улучшать формуемость лекарственного средства. Когда фармацевтические таблетки содержат масляный компонент, для абсорбции данного масляного вещества и поддержания «сухого» состояния, которое облегчает формование таблеток, нужно добавлять абсорбент. Например, крахмал, лактозу, соли кальция, микрокристаллическую целлюлозу и тому подобное.
Фармацевтическая композиция может представлять собой форму стерильного инъецируемого водного раствора. Применяемые на практике приемлемые носители и растворители включают воду, раствор Рингера и изотонический раствор хлорида натрия. Стерильный инъецируемый препарат может представлять собой стерильную инъецируемую микроэмульсию типа «масло в воде», где активный ингредиент растворен в масляной фазе. Например, активный ингредиент растворен в смеси соевого масла и лецитина. Затем масляный раствор добавляют в смесь воды и раствора глицерина и оставляют для образования микроэмульсии. Раствор для инъекции или микроэмульсию можно инъецировать в кровоток пациента посредством обильной местной инъекции. Или, предпочтительно, раствор и микроэмульсию вводят способом поддержания постоянной концентрации соединения по настоящему изобретению в системе кровообращения. Для поддержания такой постоянной концентрации можно использовать устройство для непрерывной внутривенной доставки. Примером такого устройства является внутривенный насос Deltec CADD-PLUS. ТМ. 5400.
Фармацевтические композиции могут находиться в форме стерильной инъецируемой водной или масляной суспензии для внутримышечного и подкожного введения. Согласно известным методикам данные подходящие диспергирующие агенты или увлажнители, описанные выше, можно применять вместе со суспендирующими агентами для получения суспензии. Стерильный инъецируемый препарат может также представлять собой стерильный инъецируемый раствор или суспензию, полученную в нетоксичном парентерально приемлемом разбавителе или растворителе, например, растворе, полученном с помощью 1,3-бутандиола. Кроме того, в качестве растворителя или суспендирующей среды можно беспрепятственно использовать стерильные нелетучие масла. В данных целях можно использовать любое нелетучее масло, включая синтезированный глицерид или диглицерид. Кроме того, жирные кислоты, такие как олеиновая кислота, можно также использовать для получения инъецируемого раствора.
Термин «восстанавливающий агент» представляет собой вещество, теряющее электрон или перенаправляющее электрон в оксилительно-восстановительной реакции. Сам восстанавливающий агент в широком смысле также представляет собой антиоксидант со способностью к восстановлению, будучи окисленным, его продукт называется продуктами окисления. В воплощении настоящего изобретения восстанавливающий агент представлен в виде RA, неограничивающие примеры восстанавливающих агентов включают: Н2, углерод (С), монооксид углерода (СО), восстановленный железный порошок (Fe), цинк (Zn), щелочной металл (обычно используют Li, Na, K), другие активные металлы (например, Mg, Al, Са, La и т.д.), хлорид олова (SnCl2), щавелевую кислоту, борогидрид калия (KBH4), борогидрид натрия (NaBH4), цианоборогидрид натрия (NaCNBH3), триацетоксиборогидрид натрия ((CH3COO)3BHNa), литийалюминийгидрид (LiAlH4), гипофосфорную кислоту, гипофосфит натрия, тиосульфат натрия (Na2S2O3), восстанавливающий агент по настоящему изобретению предпочтительно представляет собой цианоборогидрид натрия или триацетоксиборогидрид натрия.
Термин «меркапто-защитная группа» относится к случаю, когда как тиольная группа, так и другая химическая группа принимают участие в реакции для обеспечения того, чтобы реакция протекала только по конкретной группе, и предотвращения воздействия на тиольную группу, тиол защищают до завершения реакции, затем защитную группу удаляют. В воплощении настоящего изобретения меркапто-защитная группа представлена в виде Т. Неограничивающие примеры меркапто-защитных групп включают -трет-бутил, -ацетил, -н-пропионил, -изопропионил, -трифенилметил, -метоксиметил, -2-(триметилсилил)этоксиметил, меркапто-защитная группа по настоящему изобретению предпочтительно представляет собой ацетил.
Термин «рак», также известный как злокачественная опухоль, относится к расстройствам и заболеваниям, вызванным неконтролируемым ростом клеток и механизмом пролиферации. В соответствии с разными встречающимися локализациями патологические характеристики раковых заболеваний различны, неограничивающие примеры включают следующие раковые заболевания: фибросаркома, саркома слизистой, липосаркома, хондросаркома, остеосаркома, хордома, ангиосаркома, эндотелиальная саркома, лимфосаркома, лимфоэндотелиальная саркома, синовиома, мезотелиома, лейомиосаркома, рабдомиосаркома, карцинома толстой кишки, рак толстой и прямой кишки, рак почки, рак поджелудочной железы, рак костей, рак молочной железы, рак яичника, рак предстательной железы, рак пищевода, рак желудка, рак ротовой полости, рак полости носа, карцинома гортани, карцинома потовой железы, карцинома сальной железы, папиллярная карцинома, папиллярная аденокарцинома, цистаденокарцинома, медуллярная карцинома, бронхогенная карцинома, гепатоклеточная карцинома, холангиокарцинома, хориокарцинома, рак шейки матки, рак матки, рак яичка, мелкоклеточный рак легкого, немелкоклеточный рак легкого, рак мочевого пузыря, эпителиальная карцинома, глиома, глиобластома, астроцитома, медуллобластома, краниофарингиома, эпендимома, опухоли пинеальной области, раковые заболевания клеток крови, олигодендроглиома, менингиома, рак кожи, меланома, нейробластома, ретинобластома, острый лимфобластный лейкоз «ALL», В-клеточный острый лимфобластный лейкоз, острый лимфоцитарный Т-клеточный лейкоз, острый миелогенный лейкоз «AML», острый промиелоцитарный лейкоз «APL», острый моноцитарный лейкоз, острый лейкоз, острый первичный мегакариоцитарный лейкоз, острый миеломоноцитарный лейкоз, острый нелимфоидный лейкоз, острый недифференцированный лейкоз, хронический миелолейкоз «CML», хронический лимфоцитарный лейкоз «CLL», лейкоз ворсистых клеток, множественная миелома, лимфома Ходжкина, неходжкинская лимфома.
Следующие сокращения соединительных фрагментов, в том виде, в котором они используются в данном документе, соответствуют соответствующей структуре:
МС представляет собой фрагмент, показанный в виде формулы (V):
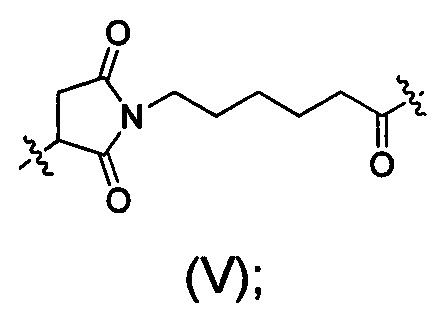
Val представляет собой фрагмент валина;
Cit представляет собой фрагмент цитруллина;
РАВ представляет собой фрагмент 1,4-аминобензилкарбамоила, который связан с D, со структурой, показанной в виде формулы (VI):
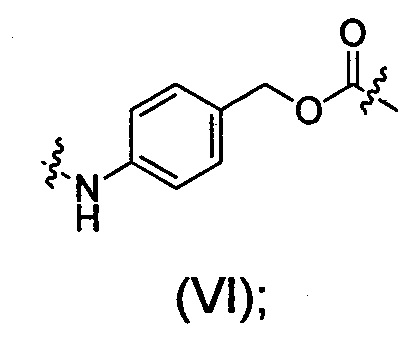
Сокращения следующих цитотоксических лекарственных средств, в том виде, в котором они используются в данном документе, имеют определения, приведенные ниже:
ММАЕ представляет собой монометил-ауристатин Е (молекулярная масса: 718), структура показана в виде формулы (VII):
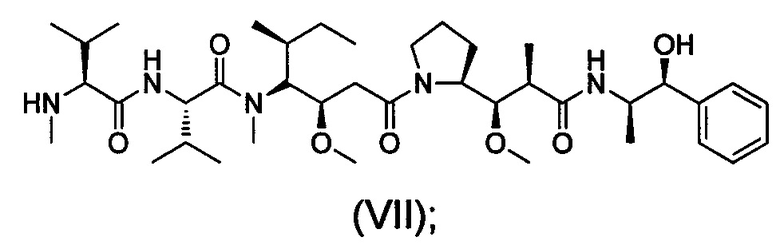
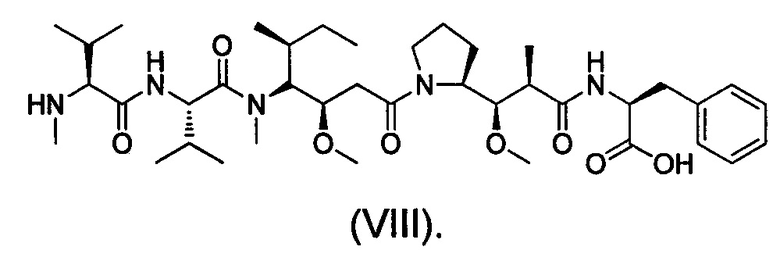
MMAF представляет собой N-метил валин-валин-долаизолейцин (Dil)-долапроин (Dap)-фенилаланин (молекулярная масса: 731,5), структура показана в виде формулы (VIII):
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
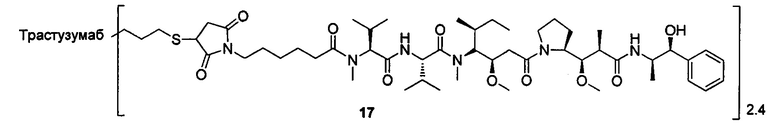
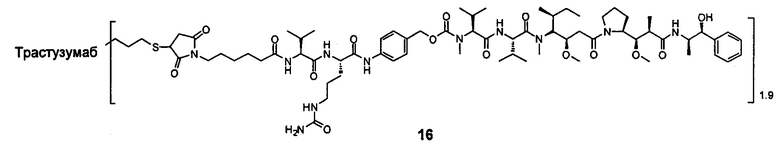
На Фиг. 1 показана эффективность Соединения 16, Соединения 17, Соединения 18 и Соединения 35 положительного контроля на ксенотрансплантаты рака желудка человека NCI-N87 у голых мышей;
На Фиг. 2 показано воздействие Соединения 16, Соединения 17, Соединения 18 и Соединения 35 положительного контроля на массу голых мышей, несущих опухоль.
ПРЕДПОЧТИТЕЛЬНЫЕ ВОПЛОЩЕНИЯ
Изобретение будет дополнительно проиллюстрировано со ссылкой на следующие конкретные примеры. Следует понимать, что данные примеры предназначены только для демонстрации изобретения без ограничения объема изобретения.
Структуры соединений идентифицировали посредством Ядерного Магнитного Резонанса (NMR) и/или Масс-Спектрометрии (MS). NMR определяли с помощью прибора Bruker AVANCE-400. Растворители представляли собой дейтерированный диметилсульфоксид (DMSO-d6), дейтерированный хлороформ (CDCl3) и дейтерированный метанол (CD3OD) с тетраметилсиланом (TMS) в качестве внутреннего стандарта. Химические сдвиги ЯМР (δ) даны в 10-6 (млн-1).
МС определяли посредством масс-спектрометра FINNIGAN LCQAd ((ESI) (электрораспылительная ионизация)) (производитель: Thermo, тип: Finnigan LCQ advantage MAX).
Высокоэффективную жидкостную хроматографию (ВЭЖХ) осуществляли на спектрометре для жидкостной хроматографии высокого давления Agilent 1200DAD (хроматографическая колонка Sunfire С18 150×4,6 мм) и спектрометре для жидкостной хроматографии высокого давления Waters 2695-2996 (хроматографическая колонка Gimini С18 150×4,6 мм).
Для тонкослойной хроматографии на силикагеле (TLC) использовали силикагелевую пластину Yantai Huanghai HSGF254 или Qingdao GF254. Размеры пластин, используемых в TLC, составляли 0,15 мм - 0,2 мм, и размеры пластин, используемых в очистке продукта, составляли 0,4 мм - 0,5 мм.
Для колоночной хроматографии в качестве носителя обычно использовали силикагель Yantai Huanghai 200-300 меш.
Известные исходные вещества по изобретению могут быть получены посредством способов синтеза, общепринятых в предшествующем уровне технике, или могут быть приобретены у ABCR GmbH & Co. KG, Acros Organics, Aldrich Chemical Company, Accela ChemBio Inc. или Dari Chemical Company и т.д.
Если не указано иное, следующие реакции проводили в атмосфере азота или атмосфере аргона.
Термин «атмосфера аргона» или «атмосфера азота» означает, что реакционная колба снабжена баллоном аргона или азота 1 л.
Если не указано иное, раствор, используемый в примерах, относится к водному раствору.
Если не указано иное, температура реакции в примерах представляла собой комнатную температуру в диапазоне 20°С - 30°С.
Реакционный процесс контролировали посредством тонкослойной хроматографии (TLC), и система проявляющего растворителя включала следующее: А: система дихлорметан и метанол, Б: система н-гексан и этил ацетат, В: система петролейный эфир и этилацетат, Г: ацетон. Соотношение объема растворителя регулировали в соответствии с полярностью соединений.
Система элюирования для очистки соединений посредством колоночной хроматографии и проявляющего растворителя посредством тонкослойной хроматографии включала следующее: А: система дихлорметан и метанол, Б: система н-гексан и этилацетат, В: система н-гексан и ацетон, Г: н-гексан, Д: этилацетат. Объем растворителя регулировали в соответствии с полярностью соединений, и иногда также можно добавлять немного триэтиламина и кислотного или щелочного реагента.
Структуры соединений по настоящему изобретению определяли посредством Q-TOF LC/MS (квадрупольно-времяпролетная система жидкостная хроматография-масс-спектрометрия). Для Q-TOF LC/MS использовали квадруполь-времяпролетный масс-спектрометр с точными измеряемыми массами Agilent 6530 и Agilent 1290-Infinity UHPLC (колонка Agilent Poroshell 300SB-C8 5 мкм, 2,1×75 мм).
Известные исходные вещества по настоящему изобретению синтезировали путем адаптирования или использования способов, известных в данной области, экспериментальные способы в следующих примерах, для которых не были указаны конкретные условия, осуществляли согласно общепринятым условиям или условиям, рекомендованным производителями продукта. Реагенты, используемые в экспериментах, для которых не указаны конкретные источники, представляют собой традиционные реагенты, обычно приобретаемые на рынке.
Пример 1: Получение промежуточных соединений
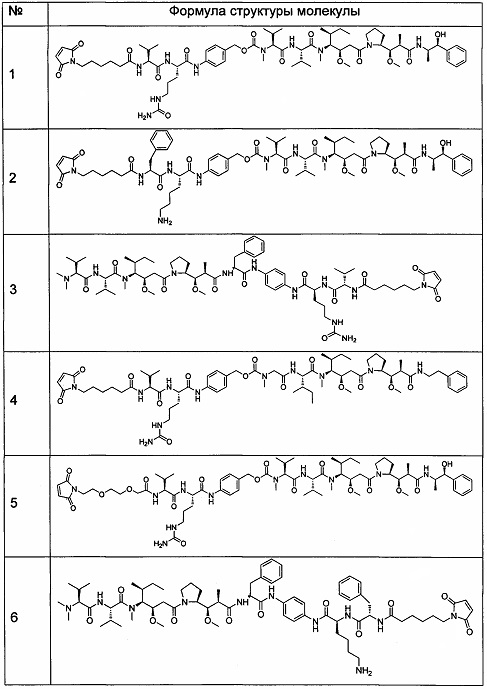
Получение промежуточных соединений в качестве лекарственных средств 1. Следующие промежуточные соединения 1-6 получали посредством способа, описанного в патентной заявке РСТ WO 2004010957.
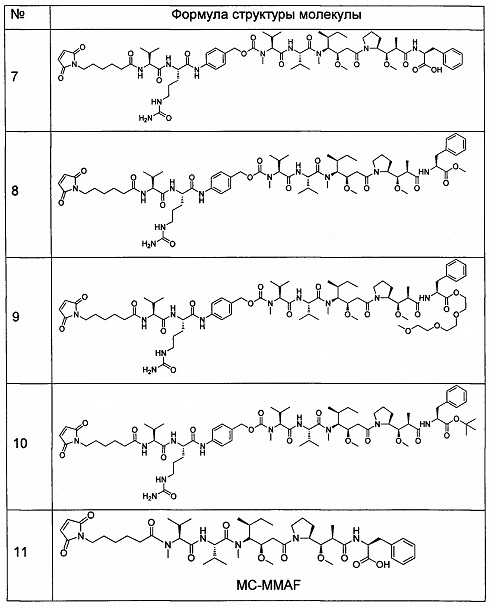
2. Следующие промежуточные соединения 7-11 получали способом, описанным в патентной заявке РСТ WO 2005081711.
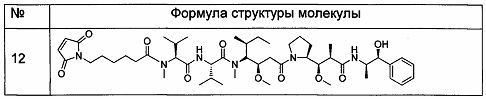
3. Следующие промежуточные соединения 12-14 получали способом, описанным в патенте США US 7750116.
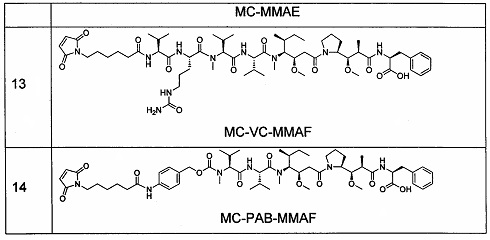
4. Получение промежуточного соединения 15
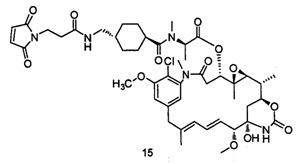
Конкретный путь синтеза выглядел следующим образом:
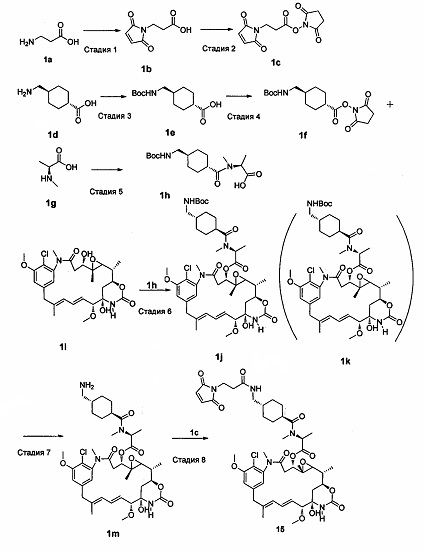
Стадия 1
3-(малеимид)пропионовая кислота
β-аланин 1а (2,29 г, 25,8 ммоль) и малеиновый ангидрид (2,52 г, 25,8 ммоль) растворяли в 20 мл уксусной кислоты, нагревали с обратным холодильником и перемешивали в течение 2 часов. Реакционный раствор концентрировали при пониженном давлении, остаток обрабатывали толуолом для азеотропной дистилляции, сушили безводным сульфатом натрия и концентрировали при пониженном давлении, и остаток перекристаллизовывали посредством этилацетата, фильтровали и сушили с получением продукта 3-(малеимид)пропионовая кислота 1b (2,80 г, выход 64,3%) в виде бесцветного кристалла.
MS m/z (ESI): 170,04 [М+1].
Стадия 2
Сложный эфир сукцинимида и 3-(малеимид)пропионовой кислоты 3-(малеимид)пропионовую кислоту 1b (2,80 г, 16,6 ммоль) и N-гидроксисукцинимид (2,25 г, 19,9 ммоль) растворяли в 30 мл DMF (диметилформамид) на водяной бане и охлаждали до 0°С, перемешивали в течение 10 минут с добавлением N,N'-дициклогексилкарбодиимида (6,85 г, 33,2 ммоль), затем реакционную смесь нагревали до комнатной температуры и перемешивали всю ночь. После фильтрования фильтрат смешивали с 80 мл дихлорметана, промывали водой (60 мл×3), 5% водным раствором бикарбоната натрия (60 мл×3) и насыщенным физиологическим раствором (50 мл×3) соответственно. Органический слой сушили безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении с получением сложного эфира 3-(малеимид)пропионовой кислоты и сукцинимида 1 с (2,76 г, выход 62,5%) в виде белого твердого вещества.
MS m/z (ESI): 267,06 [М+1].
Стадия 3
(1r,4r)-4-(трет-бутоксикарбониламинометил)циклогексилметановая кислота
(1r,4r)-4-(аминометил)циклогексилметановую кислоту 1d (4,72 г, 30,0 ммоль) и гидроксид натрия (1,28 г, 32,0 ммоль) растворяли в смеси растворителей 20 мл воды и 44 мл трет-бутанола с последующим добавлением трет-бутилдикарбоната (6,99 г, 32,0 ммоль), реакционную смесь перемешивали в течение 18 часов при комнатной температуре. В реакционный раствор добавляли 100 мл воды, его промывалит н-гексаном (100 мл×3), водный слой охлаждали до 4°С, рН доводили до 3 насыщенным водным раствором лимонной кислоты, подкисленный раствор экстрагировали этилацетатом (50 мл×3), органические слои объединяли, сушили безводным сульфатом натрия, фильтровали, и фильтрат концентрировали при пониженном давлении с получением (1r,4r)-4-(трет-бутоксикарбониламинометил)циклогексилметановой кислоты 1е (7,33 г, выход 95%) в виде бесцветного кристалла.
MS m/z (ESI): 258,17 [М+1].
Стадия 4
Сложный эфир (1r,4r)-4-((трет-бутоксикарбониламино)метил)циклогексилметановой кислоты и сукцинимида
(1r,4r)-4-(трет-бутоксикарбониламинометил)циклогексилметановую кислоту 1е (7,33 г, 28,5 ммоль) и N-гидроксисукцинимид (3,87 г, 34,2 ммоль) растворяли в 35 мл DMF на водяной бане и охлаждали до 0°С, перемешивали в течение 10 минут с добавлением N,N'-дициклогексилкарбодиимида (11,76 г, 57,0 ммоль), затем реакционную смесь нагревали до комнатной температуры и перемешивали всю ночь. После фильтрования в фильтрат добавляли 90 мл дихлорметана, промывали водой (60 мл×3), 5% водным раствором NaHCO3 (60 мл×3), насыщенным физиологическим раствором (50 мл×3) соответственно. Органический слой сушили безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении с получением сложного эфира (1r,4r)-4-((трет-бутоксикарбониламинометил)циклогексилметановой кислоты и сукцинимида 1f (6,75 г, выход 66,8%) в виде почти белого твердого вещества.
MS m/z (ESI): 355,18 [М+1].
Стадия 5
5-2-((1r,4s)-4-((трет-бутоксикарбониламино)метил)-N-метил-N-циклогексилформил)пропановая кислота
Сложный эфир (1r,4r)-4-((трет-бутоксикарбониламинометил)циклогексилметановой кислоты и сукцинимида 1f (6,75 г, 19,0 ммоль) и N-метил-L-аланин 1g (1,96 г, 19,0 ммоль) растворяли в 90 мл смеси растворителей диметиловый эфир этиленгликоля/вода с объемным соотношением 1:1 с последующим добавлением триэтиламина (4,05 г, 40 ммоль), реакция протекала в смеси в течение 6 ч при комнатной температуре, смесь концентрировали при пониженном давлении, остаток растворяли 100 мл этилацетата, промывали насыщенным физиологическим раствором (80 мл×3), органический слой сушили безводным сульфатом натрия, фильтровали, и фильтрат концентрировали при пониженном давлении, остаток очищали посредством тонкослойной хроматографии с проявляющим растворителем дихлорметан/метанол (50:1) с получением S-2-((1r,4s)-4-((трет-бутоксикарбониламино)метил)-N-метил-N-циклогексилформил)пропановой кислоты 1h (4,78 г, выход 73,5%) в виде почти белого твердого вещества.
MS m/z (ESI): 343,22 [М+1].
Стадия 6
Майтанзинол 1i (565,5 мг, 1,0 ммоль, полученный хорошо известным способом синтеза, опубликованным Wayne CW, Sharon DW, Emily EC, et al., J. Med. Chem, 2006, 49, 4392-4408) и S-2-((1r,4s)-4-((трет-бутоксикарбониламино)метил)-N-метил-N-циклогексилформил)пропановую кислоту 1h (2,05 г, 6,0 ммоль) растворяли в 20 мл дихлорметана. N,N'-дициклогексилкарбодиимид (1,30 г, 6,3 ммоль) растворяли в 5 мл дихлорметана и добавляли в указанный выше реакционный раствор, затем смесь по каплям медленно титровали 1 М хлоридом цинка в раствор диэтилового эфира (1,2 мл, 1,2 ммоль), перемешивали при комнатной температуре в течение 2 часов. После того, как реакционный раствор разводили в 30 мл этилацетата, фильтровали, и фильтрат промывали насыщенным раствором бикарбоната натрия (15 мл×2) и насыщенным физиологическим раствором (10 мл×2), соответственно, органический слой сушили безводным сульфатом натрия, фильтровали, и фильтрат концентрировали при пониженном давлении, остаток очищали посредством тонкослойной хроматографии с проявляющим растворителем дихлорметан/метанол (50:1) с получением соединения 1j (201,8 мг, выход 22,7%) в виде почти белого твердого вещества.
MS m/z (ESI): 889,43 [М+1].
Стадия 7
Соединение 1j (88,9 мг, 0,1 ммоль) растворяли в 8 мл дихлорметана с последующим добавлением трифторуксусной кислоты (12,6 мг, 0,11 ммоль), реакционную смесь перемешивали при комнатной температуре в течение 1 часа, концентрировали при пониженном давлении, остаток растворяли в 20 мл этилацетата, промывали 5% водным раствором карбоната натрия (6 мл×3), органический слой сушили безводным сульфатом натрия, фильтровали, и фильтрат концентрировали при пониженном давлении с получением соединения 1m (76,8 мг, выход 97,3%) в виде почти белого твердого вещества.
MS m/z (ESI): 789,38 [М+1].
Стадия 8
Соединение 1m (76,8 мг, 0,097 ммоль) и сложный эфир 3-(малеимид)пропионовой кислоты и сукцинимида 1 с (29,3 мг, 0,11 ммоль) растворяли в 10,0 мл N,N'-диметилформамида с последующим добавлением триэтиламина (30,6 мг, 0,3 ммоль), реакция в смеси протекала при комнатной температуре в течение 8 ч, смесь концентрировали при пониженном давлении, остаток растворяли 20 мл этилацетата, промывали насыщенным физиологическим раствором (10 мл×3), органический слой сушили безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении, остаток очищали посредством тонкослойной хроматографии с проявляющим растворителем дихлорметан/метанол (40:1) с получением соединения 15 (40,6 мг, выход 44,5%) в виде почти белого твердого вещества.
MS m/z (ESI): 940,41 [М+1].
Получение промежуточных соединений в качестве антител
Следующие антитела получали согласно традиционным способам: например, конструирование вектора, трансфекция клеток HEK293 (Life Technologies, кат. №11625019), очистка и экспрессия.
1. Последовательности антител
(1) Трастузумаб, способный специфично связываться с мишенью HER2:
Последовательность легкой цепи:

Последовательность тяжелой цепи:

(2) Инотузумаб, способный специфично связываться с мишенью CD22:
Последовательность легкой цепи:

Последовательность тяжелой цепи:

(3) Брентуксимаб, способный специфично связываться с мишенью CD30:
Последовательность легкой цепи:

Последовательность тяжелой цепи:


(4) Пертузумаб, способный специфично связываться с мишенью HER2:
Последовательность CDR-L1 легкой цепи: KASQDVSIGVA SEQ ID NO: 7
Последовательность CDR-L2 легкой цепи: SASYRYT SEQ ID NO: 8
Последовательность CDR-L3 легкой цепи: QQYYIYPYT SEQ ID NO: 9
Последовательность CDR-H1 тяжелой цепи: DYTMD SEQ ID NO: 10
Последовательность CDR-H2 тяжелой цепи: DVNPNSGGSIYNQRFKG SEQ ID NO: 11
Последовательность CDR-H3 тяжелой цепи: NLGPSFYFDY SEQ ID NO: 12
Последовательность легкой цепи:

Последовательность тяжелой цепи:

Пример 2
Получение соединения 16 конъюгата антитело-лекарственное средство
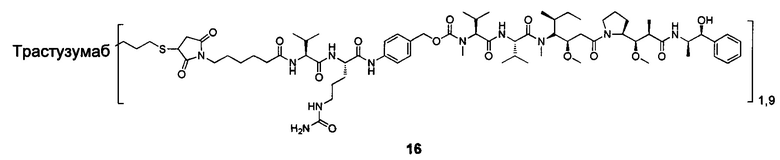
Путь синтеза выглядит следующим образом:
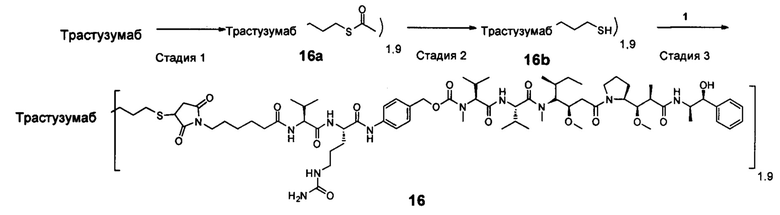
Стадия 1
Сложный этиловый эфир трастузумаб-пропантиола
32,0 мл (1,49 мкмоль) стокового раствора трастузумаба (6,9 мг/мл, рН=6,3 в растворе PBS (фосфатно-солевой буферный раствор) заменяли равным объемом буфера, состоящего из 0,1 М уксусной кислоты и ацетата натрия, с рН 5,0. 3-ацетил-меркаптопропиональдегид (0,79 мг, 5,98 мкмоль) растворяли в 3,0 мл ацетонитрила и затем по каплям титровали в указанный выше буферный раствор, по каплям добавляли цианоборогидрид натрия (0,92 мг, 14,6 мкмоль), растворенный в 1,0 мл воды, перемешивали в течение 3 часов при 25°С. После остановки реакции проводили обессоливание и очистку с помощью колонки с гелем Sephadex G25 (фаза элюирования: 0,05 М раствора PBS, содержащего 2 мМ EDTA (этилендиаминтетрауксусная кислота), рН 6,2) с получением 45,1 мл раствора сложного этилового эфира трастузумаб-пропантиола (16а) в концентрации 4,8 мг/мл.
Стадия 2
Трастузумаб-пропантиол
Раствор сложного этилового эфира трастузумаб-пропантиола (16а) (полученный на стадии 1) смешивали с 73 мкл 2 М гидрохлорида гидроксиламина, перемешивали при 25°С в течение 1 часа, затем обессоливание и очистку проводили с помощью колонки с гелем Sephadex G25 (фаза элюирования: 0,05 М раствора PBS, содержащего 2 мМ EDTA, рН 6,2) с получением 71,3 мл раствора трастузумаб-пропантиола (16б) в концентрации 3,0 мг/мл.
Стадия 3
Трастузумаб-пропил-1-cepa-MC-Val-Cit-PAB-MMAE (соединение 16)
Соединение 1 (MC-Val-Cit-PAB-MMAE, 19,1 мг, 14,5 мкмоль) растворяли в 7 мл ацетонитрила с последующим добавлением раствора трастузумаб-пропантиола (16b) (полученного на стадии 2), реакционный раствор перемешивали при 25°С в течение 4 часов, реакционную смесь обессоливали и очищали с помощью колонки с гелем Sephadex G25 (фаза элюирования: 0,05 М раствора PBS, содержащего 2 мМ EDTA, рН 6,2) и фильтровали в стерильных условиях через 0,2 мкм фильтр с получением 101,0 мл раствора Трастузумаб-пропил-1-cepa-MC-Val-Cit-PAB-MMAE (16) в концентрации 2,03 мг/мл, раствор фильтровали в стерильных условиях через 0,2 мкм фильтр и хранили при -20°С в замороженном состоянии.
Q-TOF LC/MS: 148381,5(MAb+0D), 149613,2(MAb+1D), 151169,8(MAb+2D), 152587,7(MAb+3D), 153868,1(MAb+4D), 155484,4(MAb+5D). n=1,9.
Пример 3
Получение соединения 22 конъюгата антитело-лекарственное средство
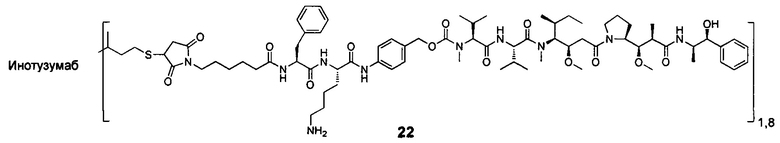
Путь синтеза выглядит следующим образом: (согласуется с путями синтеза, описанными выше)
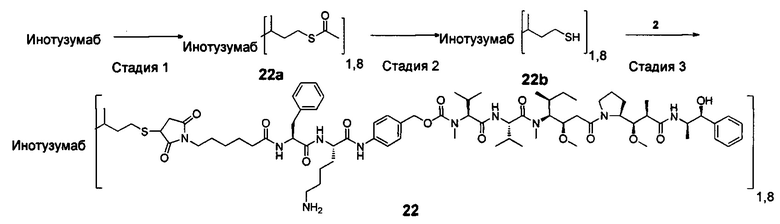
Стадия 1
Сложный этиловый эфир инотузумаб-1-метилпропантиола
8-(3-оксибутил) тиоэтилацетат (0,70 мг, 4,32 мкмоль) растворяли в 3,0 мл ацетонитрила, затем по каплям титровали в 20,0 мл (1,08 мкмоль) стокового раствора Инотузумаба (8,1 мг/мл, рН=6,0 в растворе PBS). Триметоксиборогидрид натрия (2,29 мг, 10,8 мкмоль) растворяли в 2,0 мл воды и по каплям титровали в указанный выше реакционный раствор, перемешивали в течение 48 часов при 30°С. После этого реакционный раствор обессоливали и очищали с помощью колонки с гелем Sephadex G25 (фаза элюирования: 0,05 М раствора PBS, содержащего 2 мМ EDTA, рН 6,2) с получением 29,2 мл раствора сложного этилового эфира Инотузумаб-1-метилпропантиола (22а) в концентрации 5,4 мг/мл.
Соединение 22 получали согласно стадиям 2 и 3 в Примере 2, кроме того, соединение 2 использовали на стадии 3.
Q-TOF LC/MS: 149611,1(MAb+0D), 151039,9(MAb+1D), 152470,6(MAb+2D), 153904,4(MAb+3D), 155317,0(MAb+4D), 156757,9(MAb+5D).
n=1,8.

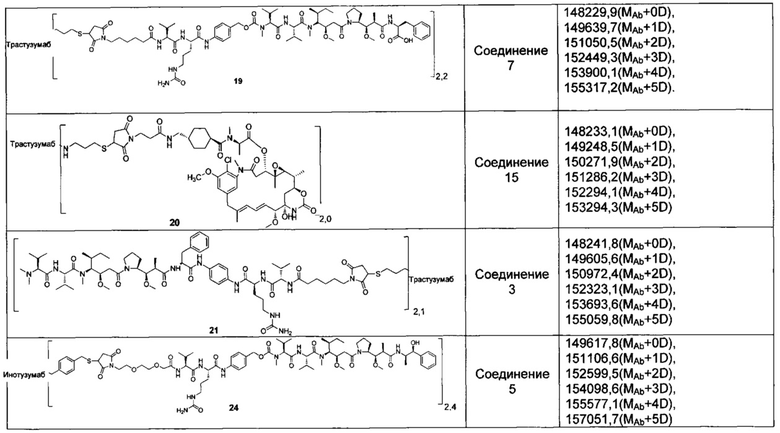
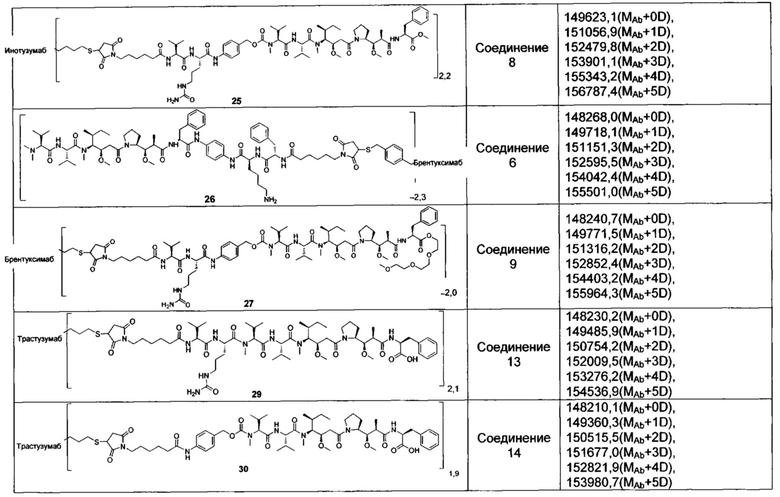
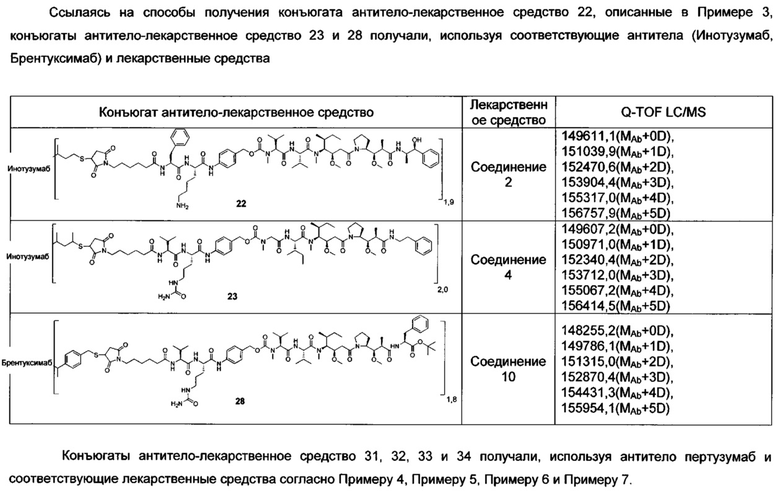

Пример 4
Получение соединения 31 конъюгата антитело-лекарственное средство
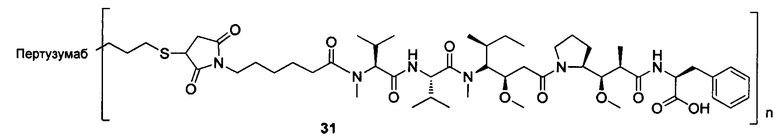
Путь синтеза выглядит следующим образом:
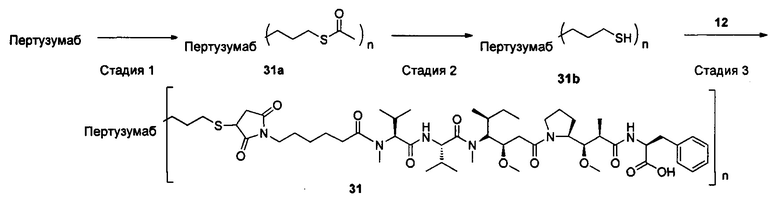
Стадия 1
Сложный этиловый эфир Пертузумаб-пропантиола
Маточный раствор Пертузумаба (сохраненный в буферной системе с 20 мМ L-гистидин-ацетата, 120 мМ сахарозы, РН=5,7) заменяли на 100 мМ буфер, содержащий уксусную кислоту и ацетат натрия (РН=4,3-4,5) посредством использования колонки для эксклюзионной хроматографии G-25, концентрировали до концентрации примерно 10,0 мг/мл с получением 200 мл буфера P-mAb, содержащего уксусную кислоту и ацетат натрия (13,5 ммоль). 3-ацетилмеркаптопропиональдегид (14,3 мг, 0,108 ммоль) растворяли в 20 мл ацетонитрила и затем по каплям титровали в указанный выше буфер, цианоборогидрид натрия (173 мг, 2,7 ммоль) растворяли в 10 мл воды и по каплям титровали в указанный выше реакционный раствор и перемешивали при 25°С в течение 3 часов. После этого реакционный раствор обессоливали и очищали с помощью колонки с гелем Sephadex G25 (фаза элюирования: 0,05 М раствора PBS, содержащего 2 мМ EDTA, рН 6,3) с получением 300 мл раствора сложного этилового эфира пертузумаб-пропантиола (31а) в концентрации 6,5 мг/мл.
Стадия 2
Пертузумаб-пропантиол
6,0 мл 2 М гидрохлорида гидроксиламина добавляли в раствор сложного этилового эфира Пертузумаб-пропантиола (31а) (полученного на стадии 1), перемешивали при 25°С в течение 1 часа, затем реакционный раствор обессоливали и очищали с помощью колонки с гелем Sephadex G25 (фаза элюирования: 0,05 М раствора PBS, содержащего 2 мМ EDTA, рН 6,3) с получением 450 мл раствора пертузумаб-пропантиола (31b) в концентрации 4,3 мг/мл.
Стадия 3
Пертузумаб-пропил-1-cepa-MC-MMAF
Соединение 12 (MC-MMAF, 125 мг, 13,5 ммоль) растворяли в 45 мл ацетонитрила и добавляли в раствор пертузумаб-пропантиола (31b) (полученный на стадии 2). После перемешивания при 25°С в течение 4 часов реакционный раствор обессоливали и очищали с помощью колонки с гелем Sephadex G25 (фаза элюирования: 0,05 М раствор PBS, содержащий 2 мМ EDTA, рН 6,3), концентрировали и фильтровали в стерильных условиях через 0,2 мкм фильтр с получением 240 мл раствора пертузумаб-пропил-1-cepa-MC-MMAF (соединение 31) в концентрации 8,02 мг/мл и затем хранили при -20°С в замороженном состоянии.
Пример 5
Получение соединения 32 конъюгата антитело-лекарственное средство
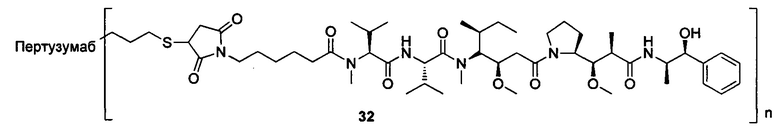
Путь синтеза выглядит следующим образом:
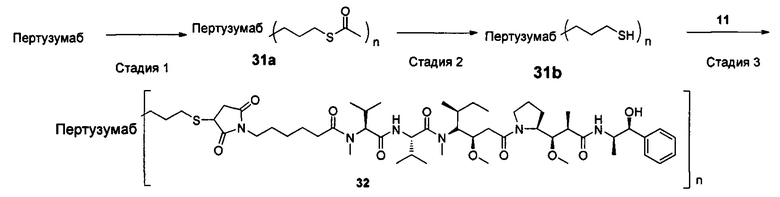
Стадия 1
Сложный этиловый эфир Пертузумаб-пропантиола
Маточный раствор Пертузумаба (сохраненный в буферной системе с 20 мМ L-гистидин-ацетата, 120 мМ сахарозы, РН=5,7) заменяли 100 мМ буфером, содержащим уксусную кислоту и ацетат натрия (РН=4,3-4,5), посредством использования колонки для эксклюзионной хроматографии G-25 и концентрировали до концентрации примерно 10,0 мг/мл с получением 2,0 мл буфера P-mAb, содержащего уксусную кислоту и ацетат натрия (0,135 ммоль). 3-ацетил-меркапто-пропиональдегид (0,15 мг, 1,1 мкмоль) растворяли в 0,2 мл ацетонитрила и затем по каплям титровали в указанный выше буферный раствор. Цианоборогидрид натрия (173 мг, 2,7 ммоль) растворяли в 0,2 мл воды и по каплям титровали в указанный выше реакционный раствор и перемешивали при 25°С в течение 3 часов. После этого реакционный раствор обессоливали и очищали с помощью колонки с гелем Sephadex G25 (фаза элюирования: 0,05 М раствор PBS, содержащий 2 мМ EDTA, рН 6,3) с получением 3,0 мл раствора сложного этилового эфира пертузумаб-пропантиола (31а) в концентрации 6,5 мг/мл.
Стадия 2
Пертузумаб-пропантиол
0,06 мл 2 М гидрохлорида гидроксиламина добавляли в раствор сложного этилового эфира Пертузумаб-пропантиола (31а) (полученного на стадии 1), перемешивали при 25°С в течение 1 часа, затем реакционный раствор обессоливали и очищали с помощью колонки с гелем Sephadex G25 (фаза элюирования: 0,05 М раствор PBS, содержащий 2 мМ EDTA, рН 6,3) с получением 5,0 мл раствора пертузумаб-пропантиола (31b) в концентрации 3,8 мг/мл.
Стадия 3
Пертузумаб-пропил-1-сера-МС-ММАЕ
Соединение 11 (МС-ММАЕ, 1,3 мг, 1,4 мкмоль) растворяли в 0,05 мл ацетонитрила и добавляли в раствор пертузумаб-пропантиола (31b) (полученный на стадии 2). После перемешивания при 25°С в течение 4 часов реакционный раствор обессоливали и очищали с помощью колонки с гелем Sephadex G25 (фаза элюирования: 0,05 М раствор PBS, содержащий 2 мМ EDTA, рН 6,3), фильтровали в стерильных условиях через 0,2 мкм фильтр с получением 8,0 мл раствора пертузумаб-пропил-1-сера-МС-ММАЕ (соединение 32) в концентрации 2,4 мг/мл и затем хранили при -20°С в замороженном состоянии.
Пример 6
Получение соединения 33 конъюгата антитело-лекарственное средство

Путь синтеза выглядит следующим образом:
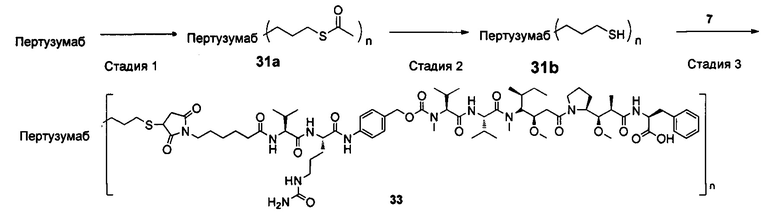
Стадия 1
Сложный этиловый эфир Пертузумаб-пропантиола
Маточный раствор Пертузумаба (сохраненный в буферной системе с 20 мМ L-гистидин-ацетата, 120 мМ сахарозы, РН=5,7) заменяли 100 мМ буфером, содержащим уксусную кислоту и ацетат натрия (РН=4,3-4,5), с использованием колонки для эксклюзионной хроматографии G-25, концентрировали до концентрации примерно 10,0 мг/мл с получением 200 мл буфера P-mAb, содержащего уксусную кислоту и ацетат натрия (13,5 ммоль). 3-ацетил-меркапто-пропиональдегид (14,3 мг, 0,108 ммоль) растворяли в 20 мл ацетонитрила и затем по каплям титровали в указанный выше буфер, цианоборогидрид натрия (173 мг, 2,7 ммоль) растворяли в 10 мл воды и по каплям титровали в реакционный раствор и перемешивали при 25°С в течение 3 часов. После этого реакционный раствор обессоливали и очищали с помощью колонки с гелем Sephadex G25 (фаза элюирования: 0,05 М раствор PBS, содержащий 2 мМ EDTA, рН 6,3) с получением 300 мл раствора сложного этилового эфира пертузумаб-пропантиола (31а) в концентрации 6,5 мг/мл.
Стадия 2
Пертузумаб-пропантиол
6,0 мл 2 М гидрохлорида гидроксиламина добавляли в раствор сложного этилового эфира Пертузумаб-пропантиола (31а) (полученный на стадии 1), перемешивали при 25°С в течение 1 часа, затем реакционный раствор обессоливали и очищали с помощью колонки с гелем Sephadex G25 (фаза элюирования: 0,05 М раствор PBS, содержащий 2 мМ EDTA, рН 6,3) с получением 450 мл раствора пертузумаб-пропантиола (31b) в концентрации 4,3 мг/мл.
Стадия 3
Пертузумаб-пропил-1-сера-МС-VC-PAB-MMAF
Соединение 7 (MC-VC-PAB-MMAF, 180 мг, 13,5 ммоль) растворяли в 45 мл ацетонитрила и добавляли в раствор пертузумаб-пропантиола (31b) (полученный на стадии 2). После перемешивания при 25°С в течение 4 часов реакционный раствор обессоливали и очищали с помощью колонки с гелем Sephadex G25 (фаза элюирования: 0,05 М раствор PBS, содержащий 2 мМ EDTA, рН 6,3), концентрировали и фильтровали в стерильных условиях через 0,2 мкм фильтр с получением 240 мл раствора пертузумаб-пропил-1-сера-МС-VC-PAB-MMAF (соединение 33) в концентрации 8,0 мг/мл и затем хранили при -20°С в замороженном состоянии.
Пример 7
Получение соединения 34 конъюгата антитело-лекарственное средство
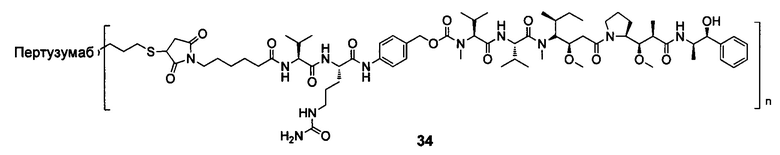
Путь синтеза выглядит следующим образом:
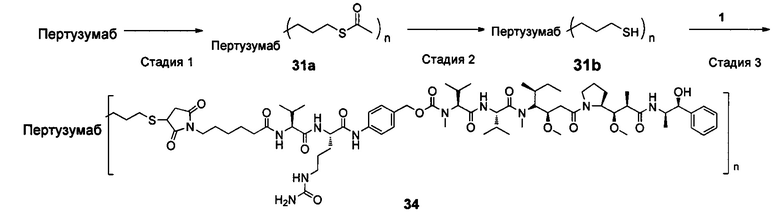
Стадия 1
Сложный этиловый эфир Пертузумаб-пропантиола
Маточный раствор Пертузумаба (сохраненный в буферной системе с 20 мМ L-гистидин-ацетата, 120 мМ сахарозы, РН=5,7) заменяли 100 мМ буфером, содержащим уксусную кислоту и ацетат натрия (РН=4,3-4,5), с использованием колонки для эксклюзионной хроматографии G-25 и концентрировали до концентрации примерно 10,0 мг/мл с получением 2,0 мл буфера P-mAb, содержащего уксусную кислоту и ацетат натрия (0,135 ммоль). 3-ацетилмеркаптопропиональдегид (0,15 мг, 1,1 мкмоль) растворяли в 0,2 мл ацетонитрила и затем по каплям титровали в указанный выше буферный раствор. Цианоборогидрид натрия (173 мг, 2,7 ммоль) растворяли в 0,2 мл воды и по каплям титровали в указанный выше реакционный раствор и перемешивали при 25°С в течение 3 часов. После этого реакционный раствор обессоливали и очищали с помощью колонки с гелем Sephadex G25 (фаза элюирования: 0,05 М раствор PBS, содержащий 2 мМ EDTA, рН 6,3) с получением 3,0 мл раствора сложного этилового эфира пертузумаб-пропантиола (31а) в концентрации 6,5 мг/мл.
Стадия 2
Пертузумаб-пропантиол
0,06 мл 2 М гидрохлорида гидроксиламина добавляли в раствор сложного этилового эфира Пертузумаб-пропантиола (31а) (полученный на стадии 1) и перемешивали при 25°С в течение 1 часа, затем реакционный раствор обессоливали и очищали с помощью колонки с гелем Sephadex G25 (фаза элюирования: 0,05 М раствор PBS, содержащий 2 мМ EDTA, рН 6,3) с получением 5,0 мл раствора пертузумаб-пропантиола (31b) в концентрации 3,8 мг/мл.
Стадия 3
Пертузумаб-пропил-1-сера-МС-VC-PAB-MMAE
Соединение 1 (MC-VC-PAB-MMAE, 1,7 мг, 1,4 мкмоль) растворяли в 0,55 мл ацетонитрила и добавляли в раствор пертузумаб-пропантиола (31b) (полученный на стадии 2). После перемешивания при 25°С в течение 4 часов реакционный раствор обессоливали и очищали с помощью колонки с гелем Sephadex G25 (фаза элюирования: 0,05 М раствор PBS, содержащий 2 мМ EDTA, рН 6,3), концентрировали и фильтровали в стерильных условиях через 0,2 мкм фильтр с получением 8,0 мл пертузумаб-пропил-1-сера-МС-VC-РАВ-ММАЕ (соединение 34) в концентрации 2,4 мг/мл и затем хранили при -20°С в замороженном состоянии.
Пример 8
Получение соединения 35 конъюгата антитело-лекарственное средство в качестве положительного контроля
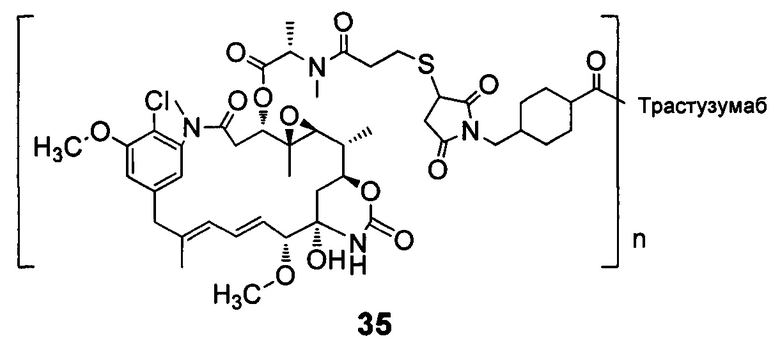
Путь синтеза выглядит следующим образом:
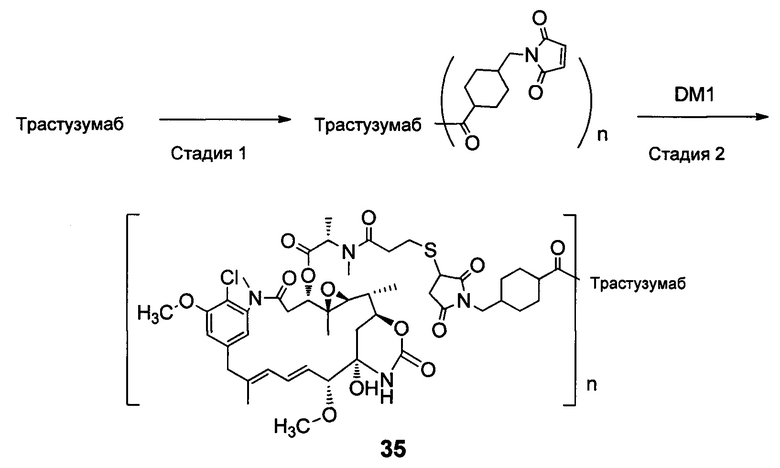
Соединение 35 конъюгат антитело-лекарственное средство получали посредством способа, описанного в патенте US 20050169933.
Следующие тестовые примеры служат для иллюстрации изобретения, но данные примеры не следует рассматривать как ограничивающие объем изобретения.
ТЕСТОВЫЕ ПРИМЕРЫ
Оценка биологического действия
Тестовый пример 1. Анализ пролиферации клеток ВТ474
Цель
Протестировать ингибирующие эффекты образцов по настоящему изобретению, оказываемые на пролиферацию клеток ВТ474
Материалы:
Образцы по настоящему изобретению: соединение 16 конъюгат антитело-лекарственное средство (называемое далее в данном документе соединением), соединение 17, соединение 18, соединения 31-34
Лекарственное средство - положительный контроль: соединение 35;
Клетки ВТ474: приобретены в Банке клеток Китайской Академии Наук, кат. №: TCHuH3;
CCK-8: набор для подсчета клеток-8, доступный от Dojindo, кат. №: CK04;
FBS: фетальная бычья сыворотка, доступная от Gibco, кат. №: 10099-141;
RPMI1640: доступна от Hyclone, кат. №: SH30809.01B;
Многофункциональный микропланшетный ридер NOVOSTAR (BMG).
Способ:
1. 100 мкл среды RPMI1640, содержащей 10% FBS и 15000 клеток ВТ474, добавляли в каждую лунку 96-луночного планшета и культивировали в инкубаторе при 37°С, 5% CO2.
2. Осуществляли последовательное двухкратное разведение образца средой RPMI1640, содержащей 10% FBS, всего 9 разведений, исходная концентрация составляла 5 мкг/мл.
3. Разведенные лекарственные средства переносили в 96-луночный планшет, в который предварительно высевали клетки ВТ474, 50 мкл/лунка. Каждую концентрацию добавляли в трехкратной повторности. Между тем лунки без какого-либо лекарственного средства использовали в качестве контроля в трехкратной повторности. Далее, клетки непрерывно культивировали в условиях 37°С, 5% CO2.
4. Спустя 96 часов, в каждую лунку добавляли 10 мкл раствора CCK-8 для развития окраски, помещали в инкубатор при 37°С, 5% CO2. Спустя 4 часа развития окраски считывали значение OD450 (оптическая плотность) на микропланшеном ридере ELISA, значение IC50 (концентрация полумаксимального ингибирования) получали в результате использования программного обеспечения Graphpad Prism 5.
Результаты:
Биологическую активность соединений по настоящему изобретению получали с помощью указанных выше способов; расчетные значения IC50 перечислены ниже в Таблице 1:

Тестовый пример 2: анализ пролиферации клеток Дауди
Цель:
Протестировать ингибирующие эффекты образцов по настоящему изобретению, оказываемые на пролиферацию клеток Дауди
Материалы и оборудование:
Образцы по настоящему изобретению: Соединение 22, Соединение 23, Соединение 24, Соединение 25;
RPMI1640: доступна от Hyclone, кат. №: SH30809.01B;
Pen Strep (P/S) (пенициллин/стрептомицин), приобретенный у Gibco, кат. №15140;
CCK-8: набор для подсчета клеток -8, имеющийся у Dojindo, кат. №: CK04;
75 см ТС(культура клеток)-обработанная колба для культуры, доступная от Corning Incorporated, товар: 430641;
PBS: приобретенный у Gibco, Catalog No: 20012-027;
Клетки лимфомы Беркитта человека Дауди, приобретенные в банке клеток Китайской Академии Наук, кат. №: TcHu140;
Многофункциональный микропланшетный ридер NOVOSTAR (BMG).
Антитело Инотузумаб: положительный контроль
Способ:
1. Клетки лимфомы Беркитта человека Дауди инкубировали в среде RPMI-1640, содержащей 10% FBS и 1% P/S. В день эксперимента плотность клеток доводили до 5×104 клеток/мл, в каждую лунку 96-луночного планшета добавляли 90 мкл среды.
2. Осуществляли последовательное четырехкратное разведение образца PBS, всего 9 разведений; исходная концентрация составляла 2,5 мкг/мл.
3. Разведенные лекарственные средства переносили в 96-луночный планшет, в который предварительно высевали клетки лимфомы Беркитта человека Дауди, 10 мкл/лунка. В контрольные лунки добавляли 10 мкл PBS. Далее клетки непрерывно культивировали в инкубаторе при 37°С, 5% CO2.
4. 72 часа спустя в каждую лунку добавляли 10 мкл проявляющего раствора CCK-8, помещали в инкубатор при 37°С, 5% CO2. Спустя 4 часа проявления считывали значение OD450 на микропланшетном ридере ELISA, значение IC50 получали в результате использования программного обеспечения Graphpad Prism 5.
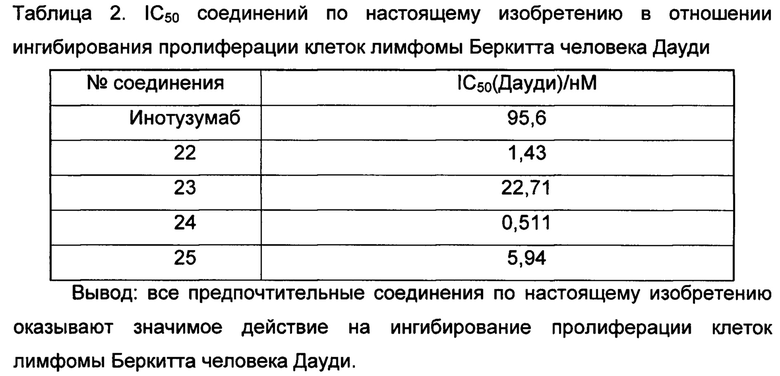
Тестовый пример 3: Тестирование степени ингибирования на NCI-N87
Цель
Оценить и сравнить эффективность действия цитотоксических конъюгатов антител по настоящему изобретению на ингибирование роста ксенотрансплантата клеток рака желудка человека NCI-N87 (АТСС, CRL-5822) у голых мышей.
Тестируемые лекарственные средства
Образцы по настоящему изобретению: Соединение 16; Соединение 17; Соединение 18; Соединение 31.
Лекарственное средство-положительный контроль: Соединение 35;
Способ получения: приготовлены с физиологическим раствором.
Животные
Голые мыши BALB/cA, 6-7 недель, самки, приобретенные в Shanghai SLAC laboratory Animal Co., Ltd. №свидетельства: SCXK (Шанхай) 2012-0002. Среда размещения: категория SPF (свободные от специфической патогенной микрофлоры)
Способ:
Голых мышей подкожно инокулировали клетками рака желудка человека NCI-N87. Когда объем опухоли достигал 100-200 мм3, животных случайным образом разделяли на группы (D0). Дозировка и режим показаны в Таблице 1. Объем и массу опухоли измеряли 2-3 раза в неделю, данные записывали. Объем опухоли (V) рассчитывали следующим образом:
V=1/2×a×b2
Где: а и b представляют собой длину и ширину, соответственно.
Т/С(%)=(Т-Т0)/(С-С0)×100%, где Т и С измеряли в конце эксперимента; Т0 и С0 измеряли в начале эксперимента
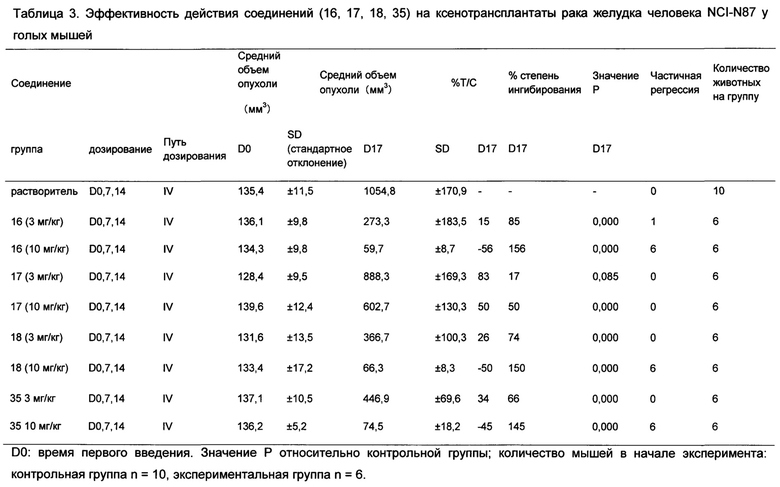
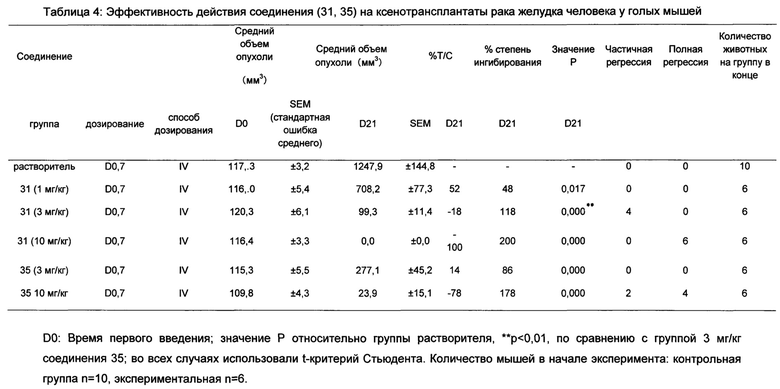
Результаты
В первой экспериментальной группе соединение 16 (3, 10 мг/кг, IV, D0, 7, 14) значимо ингибировало рост раковой опухоли с высоким уровнем экспрессии HER2 NCI-N87, подкожно пересаженной голым мышам, степень ингибирования составляла 85% и 156% соответственно, и частичная регрессия опухоли была вызвана у 1/6 и 6/6 мышей; Для одной и той же дозировки и режима введения соединения 18 степень ингибирования в отношении NCI-N87 составляла 74% и 150%, соответственно, где более высокая доза вызывала частичную регрессию опухоли у 6/6 мышей; Степень ингибирования контрольного соединения 35 в отношении NCI-N87 составляла 66% и 145% соответственно, где более высокая доза вызывала частичную регрессию опухоли у 6/6 мышей; Степень ингибирования контрольного соединения 17 в отношении NCI-N87 составляла 17% и 50% соответственно. Мыши, несущие опухоль, хорошо переносили данные лекарственные средства, указанные выше. Ингибирующее действие тестируемых лекарственных средств, оказываемое на рост опухоли, показано в Таблице 1 и на Фиг. 1. Во время введения не наступало смертей, и массы тела каждой группы мышей не были значимо снижены во время введения, как показано на Фиг. 2, давая основания предполагать, что существующая на данный момент доза не вызывала значительных побочных эффектов.
Во второй экспериментальной группе соединение 31 (1, 3, 10 мг/кг, IV, один раз в неделю, всего дважды) дозозависимым образом ингибировало рост раковой опухоли желудка с высоким уровнем экспрессии HER2, подкожно пересаженной голым мышам, степень ингибирования составляла 48%, 118% и 200% соответственно; для группы 3 мг/кг частичная регрессия опухоли была показана у 4/6, для группы 10 мг/кг полная регрессия опухоли была показана у 6/6; Для контрольного соединения 35 (3, 10 мг/кг, IV, один раз в неделю, всего дважды) степень ингибирования в отношении NCI-N87 составляла 86% и 178% соответственно; для группы 10 мг/кг частичную регрессию опухоли наблюдали у 2/6, и полную регрессию опухоли наблюдали у 4/6. Мыши, несущие опухоль, хорошо переносили данные соединения. Эффективность действия соединения 31 в отношении NCI-N87 была выше, чем эффективность действия соединения - положительного контроля 35 (Р<0,01, по сравнению с группой 3 мг/кг) (Таблица 4).
Тестовый пример 4: анализ пролиферации клеток SK-BR-3
Цель:
Протестировать ингибирующее действие образцов на пролиферацию клеток Дауди посредством использования способа CCK и определить активность образцов in vitro в соответствии с IC50.
Материалы:
Клетки SK-BR-3: АТСС, кат. №: НТВ-30;
Среда McCoy's 5А: приобретенная у Gibco, кат. №: 16600-108;
CCK-8: набор для подсчета клеток-8, доступный от Dojindo, кат. №: CK04;
PBS: приобретенный у Gibco, кат №: 20012-027;
Способ:
1. Клетки SK-BR-3 культивировали в среде McCoy 5А, содержащей 10% FBS, пересевали дважды-трижды в неделю при соотношении пересевания 1:3 или 1:6. Для пересевания клеток среду отсасывали, адгезивный слой клеток промывали 5 мл 0,25% трипсина, затем трипсин отсасывали, проводили расщепление клеток в течение 3-5 минут в инкубаторе и их ресуспендировали с добавлением свежей среды.
2. 100 мкл суспензии клеток добавляли в каждую лунку 96-луночного планшета при плотности клеток 5×104 клеток/мл с культуральной средой McCoy 5А, содержащей 10% FBS, по периферии 96-луночного планшета добавляли только 100 мкл среды McCoy 5А, содержащей 10% FBS.
3. Спустя 24 часа адгезии клеток, среду удаляли, в каждую лунку добавляли 90 мкл среды McCoy 5А, содержащей 2% FBS.
4. Осуществляли последовательное разведение образца PBS до разных концентраций, 10 мкл образца с разными концентрациями добавляли в каждую лунку 96-луночного планшета. Каждую концентрацию повторяли в двухкратной повторности.
5. Планшет инкубировали в течение 3 суток в инкубаторе (37°С, 5% СО2).
6. В каждую лунку добавляли 10 мкл раствора CCK-8 (будьте осторожными, чтобы не допустить образования пузырьков в лунках, так как это может влиять на считывание значений OD)
7. После 3 часов инкубации в инкубаторе значение поглощения считывали на микропланшетном ридере ELISA при 450 нм.
Результаты:
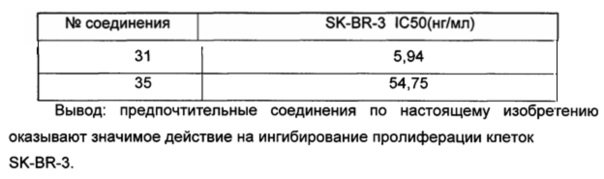






























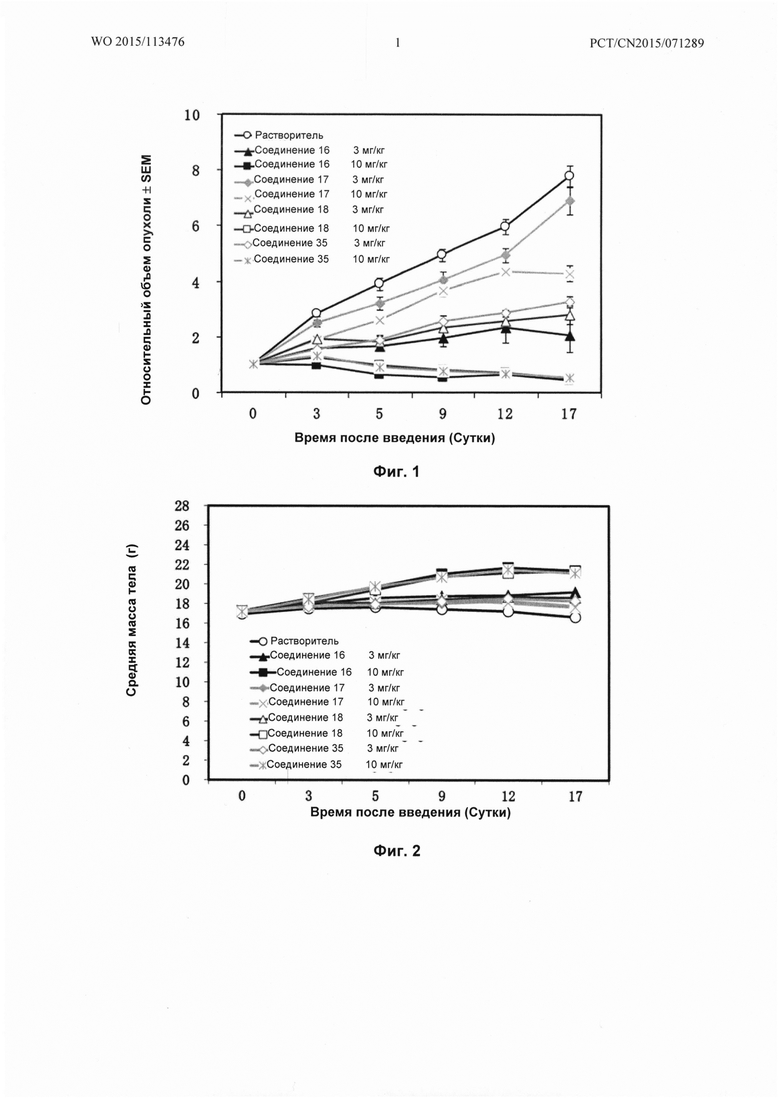
Группа изобретений относится к области фармацевтики. Первое изобретение представляет собой конъюгат лиганд-цитотоксическое лекарственное средство со структурной формулой
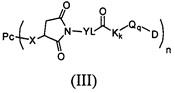 .
.
Также группа изобретений включает способ получения указанного конъюгата, содержащего его фармацевтическую композицию, применение указанного конъюгата или композиции для получения лекарственного препарата для лечения рака, способ модулирования рецептора in vitro и способ лечения рака. 6 н. и 26 з.п. ф-лы, 12 пр., 4 табл., 2 ил.
1. Конъюгат лиганд-цитотоксическое лекарственное средство, или его фармацевтически приемлемая соль, или его сольват со структурой формулы (III):
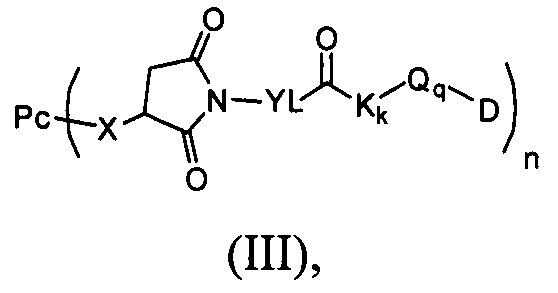
где конъюгат лиганд-цитотоксическое лекарственное средство содержит соединительное звено X, имеющее следующую структуру:
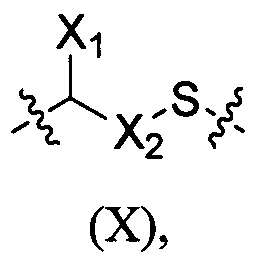
где соединительное звено X связано с N-концевой аминогруппой полипептидной цепи Рс и/или с ε-аминогруппой остатка лизина Рс;
X1 представляет собой Н или алкил,
Х2 выбран из группы, состоящей из алкила, арила и арил-алкила;
S представляет собой атом серы;
Рс представляет собой антитело;
YL выбран из алкила или (CH2CH2O)tCH2, где t представляет собой целое число, выбранное из 1-10;
Kk представляет собой аминокислотное звено, где K представляет собой аминокислоту, k представляет собой целое число, выбранное из 0-10;
Qq представляет собой протяженное звено, где q представляет собой 0, 1 или 2, Q представляет собой структуру 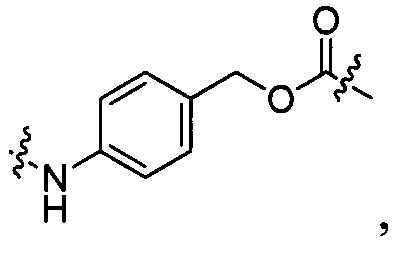
где D выбран из доластатинов/ауристатинов со структурой формулы (D1):
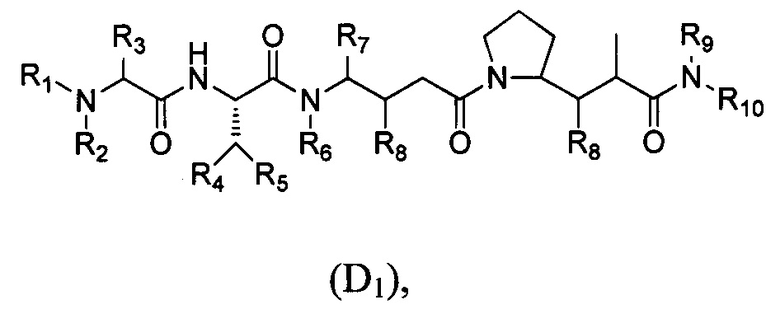
где:
R1 выбран из связи, Н или алкила;
R2 выбран из Н или алкила;
R3 выбран из Н или алкила;
R4 выбран из Н или алкила;
R5 выбран из Н или метила;
R6 выбран из Н или алкила;
R7 выбран из Н или алкила;
R8 выбран из алкила или алкокси;
R9 выбран из Н или алкила;
когда R1 представляет собой алкил,
R10 выбран из следующей структуры:
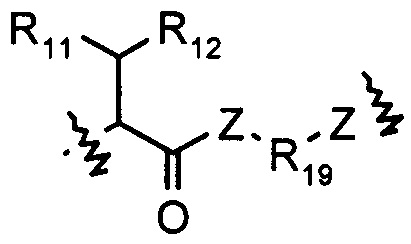 ,
,
когда R1 представляет собой связь, он связан с промежуточным звеном Y, где R10 выбран из следующих структур:
 и
и  ,
,
Z выбран из О, NH и N(R14);
R11 выбран из группы, состоящей из Н, гидрокси и алкила;
R12 выбран из арила;
R13 выбран из группы, состоящей из Н, COOR14 и алкила;
R14 выбран из Н или алкила;
R15 выбран из группы, состоящей из Н, алкила и (R16O)m-R14;
m представляет собой целое число, выбранное из 1-10;
R16 представляет собой С2-С8 алкил;
R19 представляет собой арил; или
где D выбран из майтанзина со структурой формулы (DM):
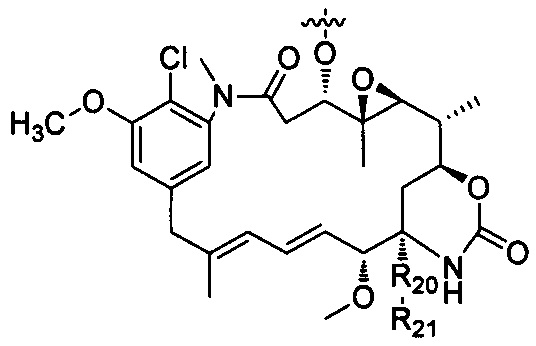
(DM),
где:
R20 выбран из О или S;
R21 выбран из Н или алкила;
n представляет собой соотношение лекарственное средство-антитело, n выбран из 1-8.
2. Конъюгат лиганд-цитотоксическое лекарственное средство, или его фармацевтически приемлемая соль, или его сольват по п. 1, где X1 представляет собой Н.
3. Конъюгат лиганд-цитотоксическое лекарственное средство, или его фармацевтически приемлемая соль, или его сольват по п. 1, где Х2 представляет собой алкил.
4. Конъюгат лиганд-цитотоксическое лекарственное средство, или его фармацевтически приемлемая соль, или его сольват по п. 1, где Х2 представляет собой линейный алкил.
5. Конъюгат лиганд-цитотоксическое лекарственное средство, или его фармацевтически приемлемая соль, или его сольват по п. 1, где n выбран из 1-4.
6. Конъюгат лиганд-цитотоксическое лекарственное средство, или его фармацевтически приемлемая соль, или его сольват по п. 1, где антиген указанного антитела представляет собой антиген поверхности клеток, экспрессирующийся в клетке-мишени и/или в ткани пролиферативного заболевания.
7. Конъюгат лиганд-цитотоксическое лекарственное средство, или его фармацевтически приемлемая соль, или его сольват по п. 6, где пролиферативное заболевание представляет собой рак.
8. Конъюгат лиганд-цитотоксическое лекарственное средство, или его фармацевтически приемлемая соль, или его сольват по п. 6, где антиген поверхности клеток представляет собой рецептор поверхности клеток.
9. Конъюгат лиганд-цитотоксическое лекарственное средство, или его фармацевтически приемлемая соль, или его сольват по п. 8, отличающийся тем, что рецептор поверхности клеток выбран из группы, состоящей из:
1) HER2 (ErbB2),
2) HER3 (ErbB3),
3) HER4 (ErbB4),
4) CD20,
5) CD22,
6) CD30,
7) CD33,
8) CD44,
9) Lewis Y,
10) CD56,
11) CD105,
12) VEGFR и
13) GPNMB.
10. Конъюгат лиганд-цитотоксическое лекарственное средство, или его фармацевтически приемлемая соль, или его сольват по п. 8, отличающийся тем, что рецептор поверхности клеток выбран из группы, состоящей из:
1) HER2 (ErbB2),
2) CD22,
3) CD30
4) CD33,
5) CD44
6) CD56,
7) Lewis Y и
8) GPNMB.
11. Конъюгат лиганд-цитотоксическое лекарственное средство, или его фармацевтически приемлемая соль, или его сольват по п. 1, отличающийся тем, что антитело выбрано из группы, состоящей из следующих антител:
1) Трастузумаб (HER2),
2) Инотузумаб (CD22),
3) Пинатузумаб (CD22),
4) Брентуксимаб (CD30),
5) Гемтузумаб (CD33),
6) Биватузумаб (CD44),
7) Лорвотузумаб (CD56),
8) cBR96 (Lewis Y),
9) Глематумамаб (GPNMB) и
10) Пертузумаб.
12. Конъюгат лиганд-цитотоксическое лекарственное средство, или его фармацевтически приемлемая соль, или его сольват по п. 1, где антитело способно связываться с белком HER2, где антитело содержит:
1) легкую цепь, содержащую по меньшей мере одну CDR (область, определяющую комплементарность), выбранную из трех CDR-L1, CDR-L2 и CDR-L3, определенных в соответствии с системой нумерации Kabat, где
i) CDR-L1 представляет собой CDR SEQ ID NO: 7 или по меньшей мере одной последовательности, обладающей по меньшей мере 80% идентичностью с SEQ ID NO: 7 после оптимального выравнивания;
ii) CDR-L2 представляет собой CDR SEQ ID NO: 8 или по меньшей мере одной последовательности, обладающей по меньшей мере 80% идентичностью с SEQ ID NO: 8 после оптимального выравнивания;
iii) CDR-L3 представляет собой CDR SEQ ID NO: 9 или по меньшей мере одной последовательности, обладающей по меньшей мере 80% идентичностью с SEQ ID NO: 9 после оптимального выравнивания;
2) тяжелую цепь, содержащую по меньшей мере одну CDR, выбранную из трех CDR-H1, CDR-H2 и CDR-H3, определенных в соответствии с системой нумерации Kabat, где
iv) CDR-H1 представляет собой CDR SEQ ID NO: 10 или по меньшей мере одной последовательности, обладающей по меньшей мере 80% идентичностью с SEQ ID NO: 10 после оптимального выравнивания;
v) CDR-H2 представляет собой CDR SEQ ID NO: 11 или по меньшей мере одной последовательности, обладающей по меньшей мере 80% идентичностью с SEQ ID NO: 11 после оптимального выравнивания;
vi) CDR-H3 представляет собой CDR SEQ ID NO: 12 или по меньшей мере одной последовательности, обладающей по меньшей мере 80% идентичностью с SEQ ID NO: 12 после оптимального выравнивания.
13. Конъюгат лиганд-цитотоксическое лекарственное средство, или его фармацевтически приемлемая соль, или его сольват по п. 1, отличающийся тем, что антитело, способное связываться с белком HER2, содержит легкую цепь и/или тяжелую цепь, легкая цепь содержит аминокислотную последовательность SEQ ID NO: 13, тяжелая цепь содержит аминокислотную последовательность SEQ ID NO: 14.
14. Конъюгат лиганд-цитотоксическое лекарственное средство, или его фармацевтически приемлемая соль, или его сольват по п. 1, где R1 представляет собой связь.
15. Конъюгат лиганд-цитотоксическое лекарственное средство, или его фармацевтически приемлемая соль, или его сольват по п. 1, где Kk представляет собой валин-цитруллин.
16. Конъюгат лиганд-цитотоксическое лекарственное средство, или его фармацевтически приемлемая соль, или его сольват по п. 1, где YL представляет собой С2-С8 линейный алкил.
17. Конъюгат лиганд-цитотоксическое лекарственное средство, или его фармацевтически приемлемая соль, или его сольват по п. 1, где структура выбрана из группы, состоящей из:
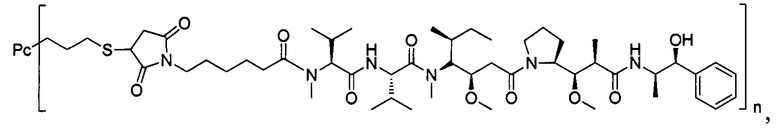
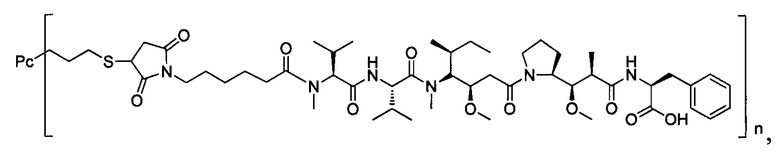
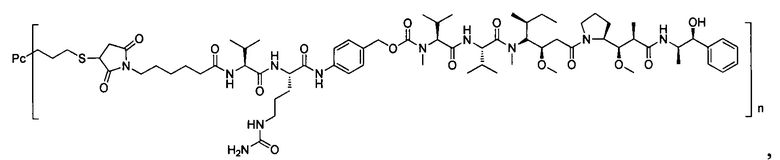

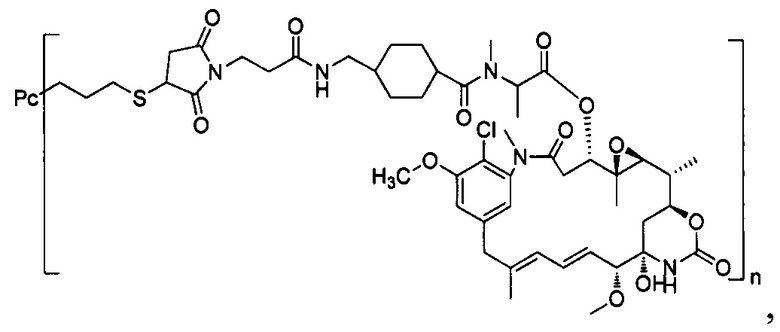
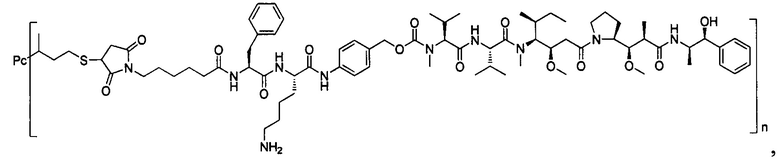
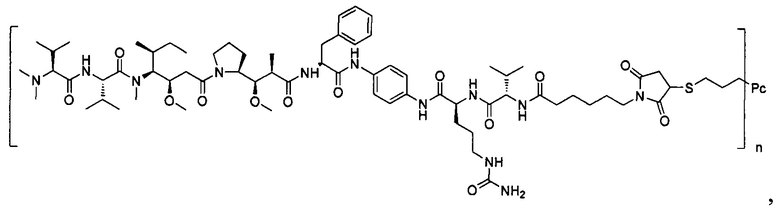
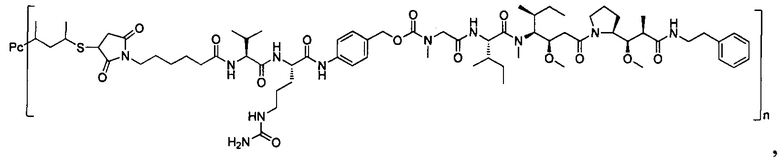
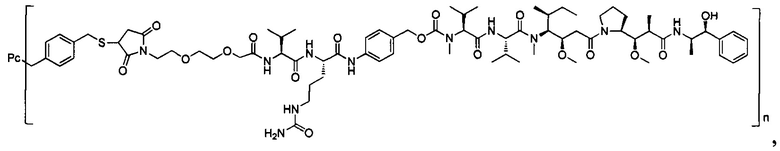
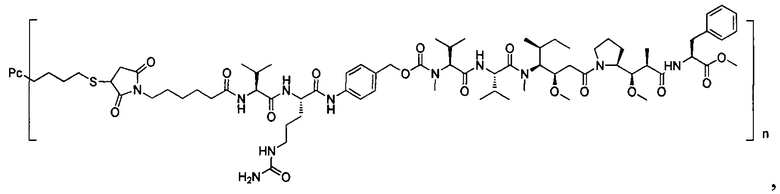
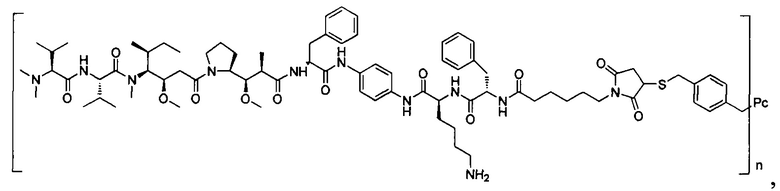
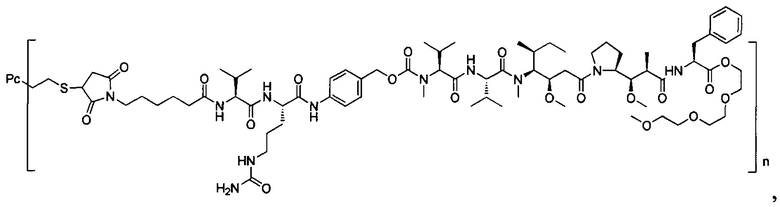
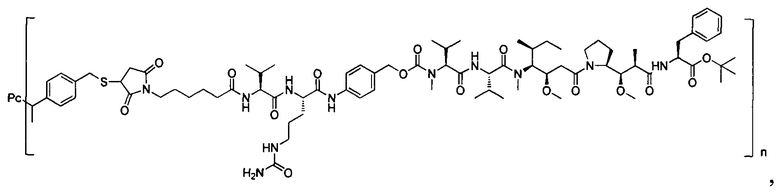
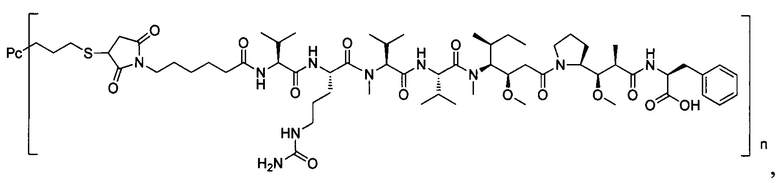
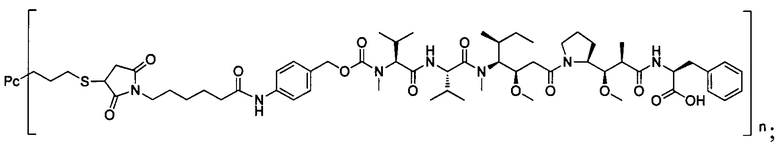
где лиганд Рс и n являются такими, как определено в п. 1.
18. Конъюгат лиганд-цитотоксическое лекарственное средство, или его фармацевтически приемлемая соль, или его сольват по п. 1, где Рс выбран из Трастузумаба, Инотузумаба, Брентуксимаба и Пертузумаба.
19. Конъюгат лиганд-цитотоксическое лекарственное средство, или его фармацевтически приемлемая соль, или его сольват по п. 1, где Рс представляет собой Трастузумаб или Пертузумаб.
20. Конъюгат лиганд-цитотоксическое лекарственное средство, или его фармацевтически приемлемая соль, или его сольват по п. 1, где Рс представляет собой Пертузумаб.
21. Конъюгат лиганд-цитотоксическое лекарственное средство, или его фармацевтически приемлемая соль, или его сольват по п. 1, выбранный из группы, состоящей из:
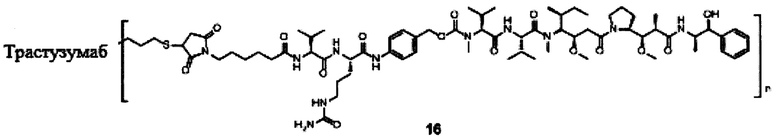
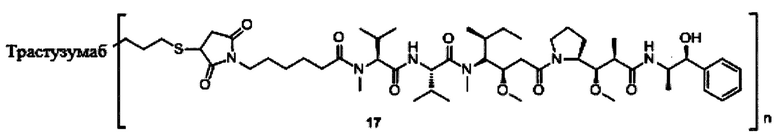
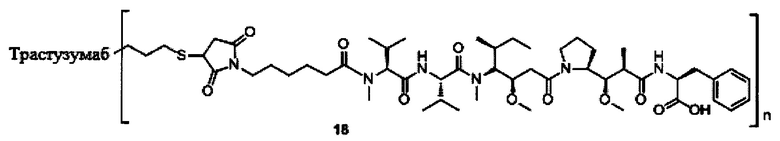
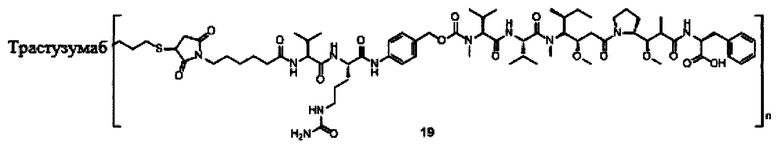

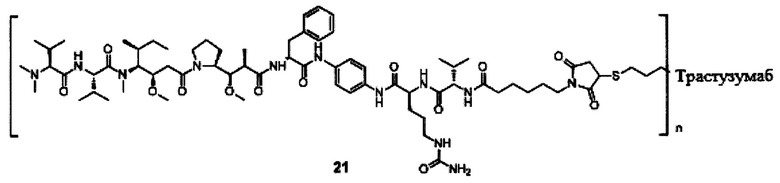
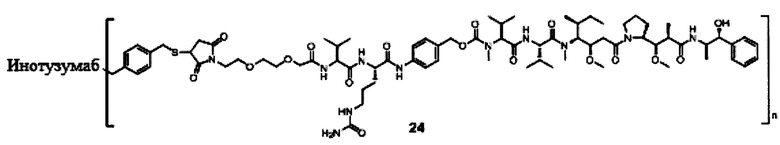
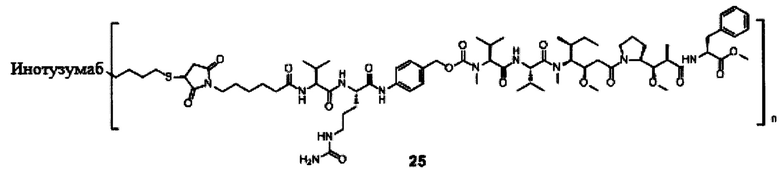
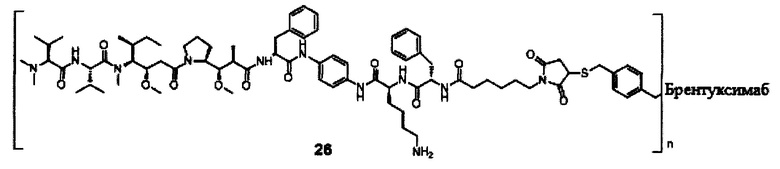
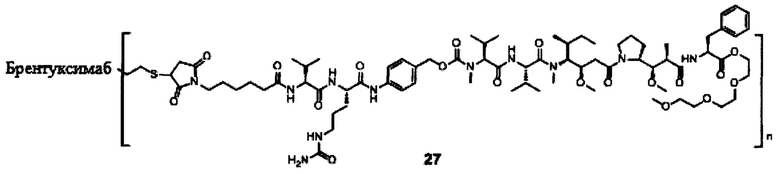
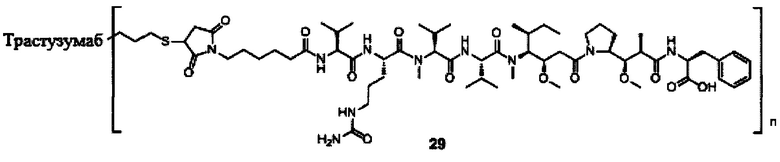
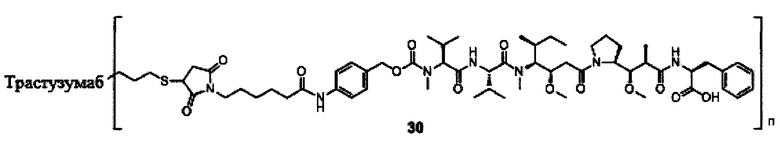
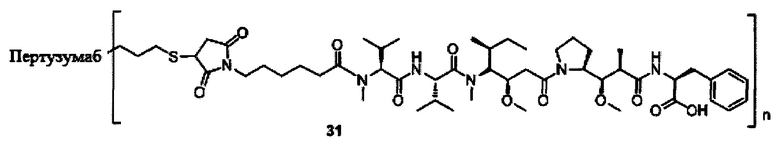
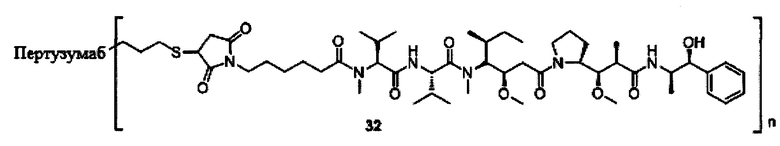
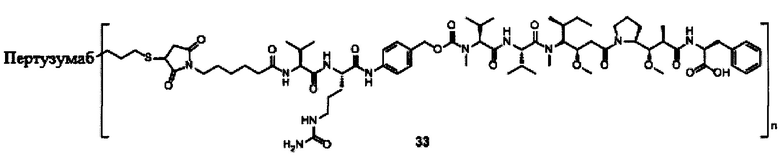
и
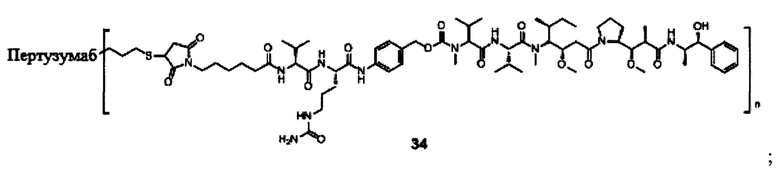
n является таким, как определено в п. 1.
22. Способ получения конъюгата антитело-цитотоксическое лекарственное средство формулы (III)
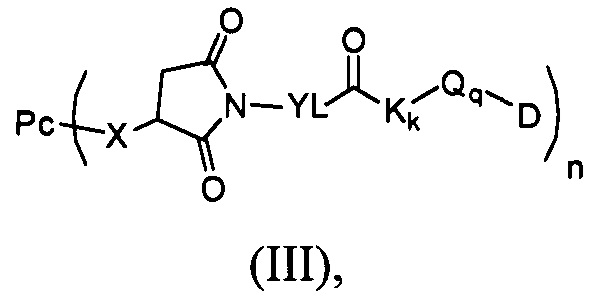
где способ включает стадии:
1) добавление восстанавливающего агента RA к соединению формулы IA и соединению формулы IB, проведение реакции и получение соединения формулы IC; рН реакционной системы составляет 3-6,
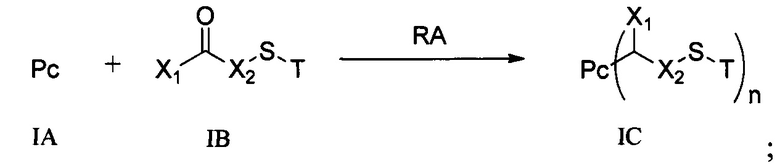
2) добавление агента для снятия защиты к соединению формулы IC для удаления защитной группы Т тиольной группы и получение соединения формулы ID,

3) проведение реакции присоединения по Михаэлю между соединением формулы ID и соединением формулы IE и получение соединения формулы (III),
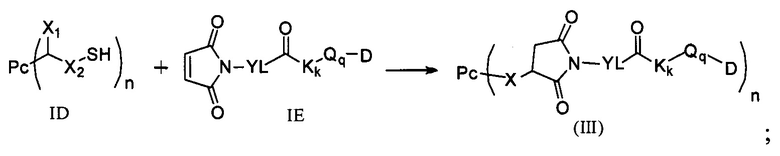
где Т представляет собой Н или ацетил; и
Рс, YL, Kk, Qq, D, n, X1 и Х2 являются такими, как определено в п. 1.
23. Способ получения конъюгата антитело-цитотоксическое лекарственное средство формулы (III) по п. 22, где температура реакции составляет 0-40°С.
24. Способ получения конъюгата антитело-цитотоксическое лекарственное средство формулы (III) по п. 22, где температура реакции составляет 15-30°С.
25. Способ получения конъюгата антитело-цитотоксическое лекарственное средство формулы (III) по п. 22, где температура реакции составляет 20-25°С.
26. Способ получения конъюгата антитело-цитотоксическое лекарственное средство формулы (III) по п. 22, где агент для снятия защиты представляет собой гидрохлорид гидроксиламина; восстанавливающий агент RA представляет собой цианоборогидрид натрия или триацетоксиборогидрид натрия.
27. Фармацевтическая композиция для лечения рака со сверхэкспрессией опухолеассоциированного рецептора, отличающаяся тем, что фармацевтическая композиция содержит терапевтически эффективное количество конъюгата лиганд-цитотоксическое лекарственное средство, или его фармацевтически приемлемой соли, или его сольвата по любому из пп. 1-21, и один или более чем один фармацевтически приемлемый носитель, разбавитель или эксципиент.
28. Применение конъюгата лиганд-цитотоксическое лекарственное средство, или его фармацевтически приемлемой соли, или его сольвата по любому из пп. 1-21 или фармацевтической композиции по п. 27 для получения лекарственного препарата для лечения рака, где рак представляет собой рак со сверхэкспрессией опухолеассоциированного рецептора, где опухолеассоциированный рецептор представляет собой один или более чем один, выбранный из группы, состоящей из (1)-(8):
1) HER2(ErbB2),
2) CD22,
3) CD30,
4) CD33,
5) CD44,
6) CD56,
7) Lewis Y и
8) GPNMB.
29. Способ модулирования рецептора in vitro, включающий введение субъекту, подлежащему тестированию, эффективного количества конъюгата лиганд-цитотоксическое лекарственное средство, или его фармацевтически приемлемой соли, или его сольвата по любому из пп. 1-21 или фармацевтической композиции по п. 27, рецептор выбран из группы, состоящей из:
1) HER2(ErbB2),
2) CD22,
3) CD30,
4) CD33,
5) CD44,
6) CD56,
7) Lewis Y и
8) GPNMB.
30. Способ лечения рака у млекопитающих, включающий введение млекопитающему терапевтически эффективного количества конъюгата лиганд-цитотоксическое лекарственное средство, или его фармацевтически приемлемой соли, или сольвата по любому из пп. 1-21 или фармацевтической композиции по п. 27, где млекопитающее представляет собой человека, рак выбран из группы, состоящей из рака молочной железы, рака яичника, рака желудка, рака эндометрия, рака слюнной железы, рака легкого, рака толстой кишки, рака почки, рака толстой и прямой кишки, рака щитовидной железы, рака поджелудочной железы, рака предстательной железы, рака мочевого пузыря, острого лимфоцитарного лейкоза, острого миелоидного лейкоза, острого промиелоцитарного лейкоза, хронического миелогенного лейкоза, хронического лимфоцитарного лейкоза, лимфомы Ходжкина, неходжкинской лимфомы и рецидивирующей анапластической крупноклеточной лимфомы.
31. Способ по п. 30, где рак выбран из группы, состоящей из рака молочной железы, лимфомы Ходжкина или рецидивирующей анапластической крупноклеточной лимфомы.
32. Способ по п. 30, где рак представляет собой рак молочной железы, ассоциированный с экспрессией HER2.
| ГАЗОГЕНЕРАТОР | 2004 |
|
RU2286844C2 |
| WO 2005081711 A2, 09.09.2005 | |||
| US 2013156693 A1, 20.06.2013. | |||

