ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
По настоящей заявке испрашивается приоритет согласно разделу 35 Свода законов США §119 (e) предварительной заявки на патент США № 61/883919, озаглавленной: STRUCTURE BASED PREDICTIVE MODELING, зарегистрированной 27 сентября 2013, которая включена в настоящее раскрытие посредством ссылки во всей своей полноте для всех целей.
УРОВЕНЬ ТЕХНИКИ
Конструирование белка, как было давно известно, является трудной задачей по причине комбинаторного взрыва возможных молекул, которые составляют доступное для поиска пространство последовательностей. Пространство последовательностей белков является огромным и в нем невозможно выполнить исчерпывающий поиск с применением способов, известных в технике в настоящий момент. Одна из частей проблемы возникает вследствие большого числа полипептидных вариантов, которые должны быть секвенированы, подвергнуты скринингу и анализу. Способы направленной эволюции повышают эффективность путем усовершенствования биомолекул-кандидатов, обладающих выгодными свойствами. Сегодня, в направленной эволюции белков доминируют форматы высокопроизводительного скрининга и рекомбинации, часто выполняемые итеративно.
Также были предложены различные вычислительные методики для исследования пространства последовательности-активности. Собственно говоря, эти методики находятся в своей ранней стадии развития и все еще требуют существенных улучшений. Соответственно, новые способы для улучшения эффективности скрининга, секвенирования и анализа биомолекул-кандидатов являются очень востребованными.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее раскрытие относится к областям молекулярной биологии, молекулярной эволюции, биоинформатики и цифровых систем.
Способы по настоящему раскрытию являются целесообразными для оптимизации белков для промышленного и терапевтического применения. Способы и системы являются особенно полезными для проектирования и разработки ферментов, имеющих выгодные свойства или активности.
Определенные аспекты настоящего раскрытия относятся к способам для разработки белков, обладающих выгодными свойствами и/или для управления программами направленной эволюции. Раскрытие предоставляет способы для идентификации биомолекул с требуемыми свойствами (или которые являются наиболее подходящими для направленной эволюции в направлении таких свойств) в комплексных библиотеках биомолекул или множествах таких библиотек. Некоторые варианты осуществления настоящего раскрытия предоставляют способы для создания модели активности последовательности в отношении структурных данных, и эта модель может применяться для управления направленной эволюцией белков, имеющих выгодные свойства. Некоторые варианты осуществления применяют генетический(-ие) алгоритм(-ы) и структурные данные для отфильтровывания неинформативных данных. Некоторые варианты осуществления используют машины опорных векторов для обучения модели активности последовательности. Способы фильтрации и обучения могут генерировать модели активности последовательности, обладающие более высокой прогнозирующей способностью, чем стандартные способы моделирования.
Некоторые варианты осуществления настоящего раскрытия предоставляют способы для проведения направленной эволюции. В некоторых вариантах осуществления способ реализован с применением компьютерной системы, которая содержит один или более процессоров и системную память. Способ включает в себя:
(a) получение множества данных, имеющего информацию физических измерений молекул, при этом множество данных содержит следующую информацию для каждой из множества вариантных биомолекул: (i) активность вариантной биомолекулы с лигандом в сайте связывания вариантной биомолекулы, (ii) последовательность вариантной биомолекулы, и (iii) один или несколько геометрических параметров, характеризующих геометрию лиганда в сайте связывания; (b) фильтрацию множества данных для создания отфильтрованного подмножества данных путем удаления информации для одной или более вариантных биомолекул, при этом фильтрация включает в себя тестирование прогнозирующей способности моделей активности последовательности, обученных на множестве выбранных подмножеств данных, при этом каждое выбранное подмножество данных имеет информацию для определенного множества вариантных биомолекул, удаленных из множества данных (a); и (c) обучение улучшенной модели активности последовательности с использованием отфильтрованного подмножества данных. В некоторых вариантах осуществления, информация для каждого множества вариантных биомолекул также содержит (iv) энергию взаимодействия, характеризующую взаимодействие лиганда в сайте связывания. В некоторых вариантах осуществления, вариантные биомолекулы представляют собой ферменты.
В некоторых вариантах осуществления улучшенную модель активности последовательности получают посредством машины опорных векторов, множественной линейной регрессии, регрессии главных компонент, регрессии методом дробных наименьших квадратов или нейронной сети.
В некоторых вариантах осуществления фильтрация множества данных включает в себя удаление по меньшей мере одного из геометрических параметров из множества данных. В некоторых вариантах осуществления фильтрацию множества данных выполняют с помощью генетического алгоритма. В некоторых вариантах осуществления генетический алгоритм изменяет пороги для удаления информации, ассоциированной с геометрическими параметрами для одной или более вариантных биомолекул.
В некоторых вариантах осуществления способ для направленной эволюции дополнительно включает в себя применение улучшенной модели активности последовательности для идентификации одного или более новых вариантов биомолекулы, предсказанных улучшенной моделью активности последовательности как обладающих активностью, соответствующей определенным критериям. Каждый из одного или более новых вариантов биомолекул имеет последовательность, отличающуюся от последовательностей вариантов биомолекулы, предоставляющих информацию для множества данных (a). В некоторых вариантах осуществления применение улучшенной модели активности последовательности для идентификации одного или более новых вариантов биомолекул включает в себя выполнение генетического алгоритма, в котором потенциальные новые варианты биомолекул оценивают с применением улучшенной модели активности последовательности в качестве функции пригодности.
В некоторых вариантах осуществления способ для направленной эволюции дополнительно включает в себя анализ новых вариантов биомолекул в отношении активности. В некоторых вариантах осуществления способ также включает в себя измерение активности вариантных биомолекул посредством анализа in vitro.
В некоторых вариантах осуществления способ также включает в себя создание структурной модели для каждого из новых вариантов биомолекул. Способ также применяет структурные модели для генерации геометрических параметров для сайтов связывания новых вариантов биомолекул. Геометрические параметры характеризуют геометрию лиганда в сайтах связывания новых вариантов биомолекул. В некоторых вариантах осуществления способ также включает в себя получение структурных моделей вариантов биомолекул и определение одного или более геометрических параметров с применением структурных моделей. В некоторых вариантах осуществления, структурные модели представляют собой модели на основе гомологии. В некоторых вариантах осуществления модели на основе гомологии подготавливают с использованием деталей измерений физической структуры биомолекул. детали измерений физической структуры биомолекул могут включать в себя трехмерные позиции атомов, полученные посредством NMR или рентгеноструктурной кристаллографии.
В некоторых вариантах осуществления способ также включает в себя применение докера для определения одного или более геометрических параметров. В некоторых вариантах осуществления, способ также применяет докер для определения энергии взаимодействия.
В некоторых вариантах осуществления обрабатываемые вариантные биомолекулы являются множеством ферментов. В некоторых вариантах осуществления, активность вариантной биомолекулы на лиганде представляет собой активность фермента на субстрате. В некоторых вариантах осуществления, активность фермента на субстрате включает в себя одну или более функций каталитического преобразования субстрата ферментом.
В некоторых вариантах осуществления способ для направленной эволюции также включает в себя применение улучшенной модели активности последовательности для идентификации одной или более биомолекул, обладающих требуемой активностью. В некоторых вариантах осуществления способ также включает в себя синтезирование биомолекул, обладающих требуемой активностью.
В некоторых вариантах осуществления также предоставлены компьютерные программные продукты и компьютерные системы, реализующие способы для направленной эволюции биомолекул.
Эти и другие характеристики будут представлены ниже в отношении соответствующих чертежей.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Фигура 1A представляет собой блок-схему, изображающую поток операций направленной эволюции согласно некоторым вариантам осуществления настоящего раскрытия.
Фигура 1B представляет собой блок-схему, иллюстрирующую один из подходов к фильтрации необработанных данных согласно некоторым вариантам осуществления настоящего раскрытия.
Фигура 1C представляет собой блок-схему, представляющую процесс фильтрации данных согласно некоторым вариантам осуществления, на которой этап выбора характеристики не выполняется или объединен с этапом выбора расстояния.
Фигура 2 показывает три табличных представления множества данных активности последовательности для иллюстрации примера фильтрации данных согласно некоторым вариантам осуществления настоящего раскрытия.
Фигура 3A представляет собой блок-схему, показывающую генетический алгоритм для фильтрации необработанных данных с целью удаления одного или более типов энергии и/или геометрических типов согласно некоторым вариантам осуществления настоящего раскрытия.
Фигура 3B представляет собой блок-схему, показывающую генетический алгоритм для фильтрации необработанных данных с целью удаления данных для вариантов, имеющих значения энергии и/или геометрические значения вне заданных диапазонов согласно некоторым вариантам осуществления настоящего раскрытия.
Фигура 3C представляет собой блок-схему, показывающую генетический алгоритм для идентификации новых вариантов биомолекул с применением модели активности последовательности с высокой прогнозирующей способностью согласно некоторым вариантам осуществления настоящего раскрытия.
На фигуре 4 показано типовое цифровое устройство, которое может быть реализовано согласно некоторым вариантам осуществления.
ПОДРОБНОЕ ОПИСАНИЕ
Способы для разработки моделей активности последовательности в отношении структурных данных раскрыты в настоящем раскрытии. Модели активности последовательности могут применяться для управления направленной эволюцией белков, имеющих выгодные свойства. Некоторые варианты осуществления могут помочь в исследовании большого пространства последовательностей и быстро оптимизировать молекулы с выгодными свойствами. Материалы и/или ресурсы могут также быть сэкономлены в процессах нахождения или разработки белков с требуемыми свойствами. Некоторые варианты осуществления являются особенно полезными для проектирования и разработки ферментов, обладающих требуемой активностью и/или селективностью для каталитических реакций, включающих конкретные субстраты.
I. ОПРЕДЕЛЕНИЯ
Если в настоящем раскрытии не определено иное, все технические и научные термины, используемые в настоящем раскрытии, имеют то же самое значение, которое обычно понимается специалистом в данной области техники. Различные научные словари, которые содержат термины, содержащиеся в настоящем раскрытии, известны и доступны специалистам в данной области техники. Любые способы и материалы, аналогичные или эквивалентные описанным в настоящем раскрытии, применяются при практическом применении вариантов осуществления, раскрытых в настоящем раскрытии.
Термины, определенные ниже, более полно понимаются в отношении спецификации в целом. Определения приведены только с целью описания конкретных вариантов осуществления и способствования пониманию сложных концепций, описанных в данной спецификации. Не предполагается, что они ограничивают полный объем раскрытия. Конкретно, следует понимать, что настоящее раскрытие не ограничено конкретными последовательностями, композициями, алгоритмами, системами, методологиями, протоколами, и/или реагентами, описанными в настоящем раскрытии, поскольку они могут меняться в зависимости от контекста, в котором они применяются специалистами в данной области техники.
При использовании в данной спецификации и прилагаемой формуле изобретения, формы единственного числа включают в себя ссылки на множественные элементы, если содержание и контекст явно не указывают противоположное. Таким образом, например, ссылка на «устройство» включает в себя комбинацию двух или более таких устройств, и т.п. Если не указано другое, предполагается, что конъюнкция «или» используется в ее корректном смысле в качестве Булева логического оператора, охватывая как выбор характеристик в виде альтернативы (или B, где выбор является взаимно исключающим относительно B) и выбор характеристик совместно (или B, где выбраны и A, и B).
Машины опорных векторов (SVM) являются инструментами машинного обучения с соответствующими алгоритмами обучения для классификации и регрессионного анализа. Базовая SVM берет множество входных данных и предсказывает, для каждых входных данных, какой из двух возможных классов формирует выходные данные. Получив множество обучающих примеров, каждый из которых отмечен как принадлежащий к одной из двух категорий, обучающий алгоритм SVM создает модель, относящую новые примеры в одну из двух категорий. SVM является представлением примеров в виде точек в пространстве, отображенном таким образом, чтобы примеры различных категорий были разделены с явным разрывом, который является максимально широким, что реализуется посредством максимизации расстояния между точками данных и гиперплоскостью, разделяющей эти две категории. В дополнение к выполнению линейной классификации SVM может эффективно выполнять нелинейную классификацию с помощью функции ядра для неявного отображения входных данных в имеющие высокую размерность пространства признаков.
При использовании для оптимизации моделей активности последовательности, SVM берут в качестве входных данных множества данных обучения для последовательностей, которые были классифицированы по двум или более группам на основании активностей. Методы опорных векторов функционируют посредством взвешивания элементов обучающего множества по-разному в зависимости от того, насколько они близко к границе гиперплоскости, разделяющей «активные» и «неактивные» элементы обучающего множества. В данной методике требуется, чтобы ученый сначала решил, какие элементы обучающего множества поместить в «активную» группу и какие элементы обучающего множества поместить в «неактивную» группе. Это может быть достигнуто посредством выбора соответствующего числового значения активности, которое будет служить границей между «активными» и «неактивными» элементами обучающего множества. По этой классификации метод опорных векторов будет генерировать вектор, W, который может предоставить значения коэффициентов для индивидуальных независимых переменных, определяющих последовательности для членов активных и неактивных групп в обучающем множестве. Эти коэффициенты могут использоваться, чтобы «упорядочить» индивидуальные остатки, как описано в другом месте настоящего раскрытия. Методика пытается идентифицировать гиперплоскость, которая максимизирует интервал между самыми близкими элементами обучающего множества на противоположных сторонах этой плоскости. В другой вариации выполняется регрессионное моделирование на основе опорных векторов.
В этом случае, зависимая переменная представляет собой вектор непрерывных значений активности. Регрессионная модель на основе опорных векторов будет генерировать вектор коэффициентов, W, который может применяться для ранжирования индивидуальных остатков.
SVM применялись для изучения больших множеств данных во многих исследованиях и были достаточно популярными в области микропанелей ДНК. Их потенциальные преимущества включают в себя возможность точного различения (посредством взвешивания) факторов, которые отделяют выборки друг от друга. До такой степени, в которой SVM могут точно выделить, какие остатки вносят вклад в функцию, они могут являться особенно полезным инструментом для ранжирования остатков в соответствии с настоящим изобретением. SVM описаны в работе S. Gunn (1998) «Support Vector Machines for Classification and Regressions», Технический отчет, факультет инженерии и прикладных наук, отдел электроники и информатики, университет Саутгемптона, которая включена в настоящее раскрытие посредством ссылки для всех целей.
Докер (программное обеспечение для докинга или программа для докинга) – «докер» представляет собой компьютерную программу, вычислительно предсказывающую, будет ли лиганд связываться или стыковаться с представляющим интерес сайтом связывания в белке или другой биологической молекуле. Процесс, посредством которого лиганд приближается и в конечном итоге связывается с сайтом связывания, иногда называется «докингом» («стыковкой»). Понятие докинга может быть понято как взаимодействие, заставляющее лиганд связываться с биомолекулой таким образом, чтобы лиганд не мог быть легко перемещен. При успешном докинге лиганд и биомолекула формируют стабильный комплекс. Состыкованный лиганд может действовать как агонист или антагонист. Докер может моделировать и/или характеризовать докинг.
Докеры обычно реализованы как программное обеспечение, которое может быть временно или постоянно храниться в связи с аппаратными средствами, такими как процессор или процессоры. Коммерчески доступные программы для докинга включают CDocker (Accelrys), DOCK (Калифорнийский университет, Сан-Франциско), AutoDock (Исследовательский институт Scripps), FlexX (tripos.com), GOLD (ccdc.cam.ac.uk) и GLIDE (schrodinger.com).
Различные докеры выводят показатель докинга или другую меру связывания между лигандом и биомолекулой. Для некоторых комбинаций лиганд-биомолекула программа докинга будет определять, что связывание является маловероятным. В таких случаях программа докинга будет выдавать заключение, что лиганд не связывается с биомолекулой.
Докеры могут генерировать «положения» лигандов относительно сайтов связывания. Некоторые из этих положений могут использоваться в генерации показателя докинга или в другой оценке докинга. В некоторых вариантах осуществления докер разрешает пользователю задавать множество положений (n) для использования в оценке докинга. Только лучшие n положений с наилучшими показателями докинга рассматриваются в оценке докинга.
Докер может быть запрограммирован на вывод оценки вероятности того, что лиганд состыкуется с сайтом связывания биомолекулы, или качества такой стыковки, если она произойдет. На одном уровне докер определяет, является ли вероятными связывание лиганда с сайтом связывания биомолекулы. Если логический блок докера заключает, что связывание является маловероятным или является очень неблагоприятным, то он может выдать результат «не было найдено уточненных положений». Это может произойти, когда все конформации, сгенерированные программой докинга, имеют неблагоприятные Ван-дер-Ваальсовы наложения и/или электростатические отталкивания с сайтом связывания. В приведенном выше примере процедуры докинга в случае, если вторая операция не находит положения с мягкой энергией, меньшей порога, докер может возвратить такой результат, как «не было найдено уточненных положений». Поскольку в мягкой энергии прежде всего рассматриваются невалентные взаимодействия, включая ван-дер-ваальсовские и электростатические силы, результат «не было найдено уточненных положений» означает, что лиганд имеет серьезные пространственные наложения и/или электростатические отталкивания с рецептором биомолекулы для заданного числа положений.
В определенных вариантах осуществления, докер выдает показатель докинга, который представляет взаимодействие между лигандом и сайтом связывания биомолекулы. Докеры могут вычислять различные характеристики взаимодействия лиганда-биомолекулы. В одном из примеров результат представляет собой просто энергию взаимодействия между лигандом и биомолекулой. В другом варианте осуществления полная энергия является результатом. Полная энергия может пониматься как комбинация энергии взаимодействия лиганда-биомолекулы и деформации лиганда. В определенных реализациях такая энергия может быть вычислена с использованием силового поля, такого как CHARMm.
В различных вариантах осуществления программы докинга генерируют такие результаты путем рассмотрения множества положений лиганда в сайте связывания биомолекулы. Каждое положение будет иметь свои собственные ассоциированные с ним значения энергии. В некоторых вариантах осуществления программа докинга ранжирует положения и рассматривает энергию, ассоциированную с одним или более положениями, имеющими высокий ранг. В некоторых случаях она может усреднять энергии определенных положений с высоким рангом или выполнять статистический анализ положений с высоким рангом другим способом. В других вариантах осуществления она просто выбирает значение, ассоциированное с имеющим наибольший ранг положением, и выдает его в качестве полученной в результате энергии для докинга.
«Положение» представляет собой положение или ориентацию лиганда относительно сайта связывания биологической молекулы. В положении трехмерные позиции некоторых или всех атомов лиганда определены относительно некоторых или всех положений атомов в сайте связывания. Несмотря на то, что конформация лиганда не является его положением – потому что конформация не рассматривает сайт связывания – конформация может использоваться в определении положения. В некоторых вариантах осуществления ориентация лиганда и конформация совместно определяют положение. В некоторых вариантах осуществления положение существует, только если комбинация ориентации/конформации лиганда соответствует определенному уровню энергии в опорном сайте связывания.
Различные вычислительные механизмы могут применяться для генерации положений для докинга. Примеры включают систематические или стохастические поиски вращений вокруг поворотных связей, моделирование молекулярной динамики и генетические алгоритмы для “развития” новых низкоэнергетических конформаций. Эти методики применяются для изменения вычислительных представлений лиганда и/или сайта связывания в целях исследования «пространства положений».
Докеры оценивают положения с целью определения того, как лиганд взаимодействует с сайтом связывания. В некоторых вариантах осуществления они делают это посредством вычисления энергии взаимодействия на основании одного или более указанных выше типов взаимодействия (например, ван-дер-ваальсовских сил). Эта информация используется для того, чтобы охарактеризовать докинг и, в некоторых случаях, для получения показателя докинга. В некоторых реализациях докеры упорядочивают положения на основании показателя докинга. В некоторых реализациях докеры удаляют из рассмотрения положения с неблагоприятными показателями докинга.
В определенных вариантах осуществления система виртуального скрининга белков оценивает положения с целью определения того, является ли положение активным. Положение считается активным, если оно соответствует заданным ограничениям, которые, как известно, являются важными для рассматриваемой требуемой активности. Например, система виртуального скрининга белков может определять, поддерживает ли положение каталитическое превращение лиганда в сайте связывания.
«Лиганд» представляет собой молекулу или комплекс, который взаимодействует с сайтом связывания биомолекулы с формированием стабильного комплекса, содержащего, по меньшей мере, лиганд и биомолекулу. В дополнение к лиганду и биомолекуле, стабильный комплекс может содержать (иногда обязательно) другие химические сущности, такие как органические и неорганические кофакторы (например, коэнзимы и простетические группы), ионы металлов, и т.п. Лиганды могут являться агонистами или антагонистами.
В случае, когда биомолекула представляет собой фермент, сайт связывания является каталитическим сайтом и лиганд является субстратом, промежуточным продуктом реакции субстрата или переходным состоянием субстрата. «Промежуточный продукт реакции» представляет собой химическую сущность, сгенерированную из субстрата в процессе превращения субстрата в продукт реакции. «Переходное состояние» субстрата представляет собой субстрат в состоянии, соответствующем самой высокой потенциальной энергии вдоль пути реакции. В переходном состоянии, которое имеет тенденцию быть кратковременным, соударяющиеся молекулы реагента переходят к формированию продуктов. В настоящем раскрытии иногда, когда субстрат описан в процессе, промежуточный продукт и переходное состояние могут также быть подходящими для процесса. В таких ситуациях субстрат, промежуточный продукт и переходное состояние могут совместно называться «лигандами». В некоторых случаях, множество промежуточных продуктов генерируется при каталитическом превращении субстрата. В определенных вариантах осуществления соединение лиганда (субстрат, или промежуточный продукт, или переходное состояние), выбранное для анализа, является соединением, о котором известно, что оно ассоциировано с этапом ограничения скорости в каталитическом превращении. Например, субстрат, ковалентно связанный с кофактором фермента, может быть химически изменен на ограничивающем скорость этапе. В таком случае соединение субстрат-кофактор используется в моделировании взаимодействия.
Как должно быть ясно, концепция лиганда является более общей, чем концепция «субстрата». Некоторые лиганды связываются с сайтом связывания, но не подвергаются каталитическому превращению. Примеры включают лиганды, оцениваемые в области разработки лекарств. Такие лиганды могут представлять собой малые молекулы, выбранные вследствие их способности к нековалентному связыванию с целевой биомолекулой для фармакологических целей. В некоторых случаях лиганд оценивают по его способности стимулировать, активировать или ингибировать естественное поведение биомолекулы.
При использовании в настоящем раскрытии, «биомолекула» или «биологическая молекула» относятся к молекуле, которая обычно находится в биологическом организме. В некоторых вариантах осуществления биологические молекулы содержат полимерные биологические макромолекулы, имеющие множественные субъединицы (то есть, «биополимеры»). Типичные биомолекулы включают в себя, но не ограничиваются указанным, молекулы, которые имеют некоторые общие структурные характеристики с естественными полимерами, такими как РНК (сформированные из нуклеотидных субъединиц), ДНК (сформированные из нуклеотидных субъединиц) и пептиды или полипептиды (сформированные из аминокислотных субъединиц), включая, например, РНК, аналоги РНК, ДНК, аналоги ДНК, полипептиды, аналоги полипептидов, пептидные нуклеиновые кислоты (PNA), комбинации РНК и ДНК (например, химерапласты), и т.п. Не предполагается, что биомолекулы ограничены какой-либо конкретной молекулой, поскольку любая соответствующая биологическая молекула находит применение в настоящем изобретении, включая, но не ограничиваясь, например, липиды, углеводы или другие органические молекулы, которые сделаны из одной или более генетически кодируемых молекул (например, один или более ферментов или ферментных путей), и т.п. Особенно интересными для некоторых аспектов настоящего раскрытия являются биомолекулы, имеющие сайты связывания, которые взаимодействуют с лигандом с целью осуществления химического или биологического превращения, например, катализа субстрата, активации биомолекулы или инактивации биомолекулы.
В некоторых вариантах осуществления, «выгодное свойство» или «активность» заключается в увеличении или уменьшении одного или более из следующего: скорость каталитической реакции (kcat), связывающая способность субстрата (KM), эффективность каталитической реакции (kcat/KM), специфичность субстрата, хемоселективность, региоселективность, стереоселективность, стереоспецифичность, специфичность лиганда, агонизм рецептора, антагонизм рецептора, преобразование кофактора, кислородная стабильность, уровень экспрессии белка, растворимость, термоактивность, теплоустойчивость, активность pH фактор, стабильность pH фактор (например, при щелочном или кислотном pH фактор), ингибирование глюкозы и/или устойчивость к ингибиторам (например, уксусной кислоте, лектинам, дубильным кислотам и производным фенола) и протеазам. Другие требуемые активности могут включать в себя изменение профиля в ответ на конкретный стимул (например, изменение температуры и/или профилей pH фактор). В контексте рационального проектирования лиганда оптимизация нацеленного ковалентного ингибирования (TCI) является типом активности. В некоторых вариантах осуществления два или более вариантов, подвергнутых скринингу в соответствии с описанным в настоящем раскрытии, действуют на один и тот же субстрат, но отличаются в отношении одной или более следующих активностей: скорости образования продукта, процента преобразования субстрата в продукт, селективности и/или процента преобразования кофактора. Не предполагается, что настоящее раскрытие ограничено каким-либо конкретным выгодным свойством и/или требуемой активностью.
В некоторых вариантах осуществления «активность» используется для описания более ограниченной концепции возможности фермента катализировать оборот субстрата в продукт. Соответствующей характеристикой фермента является его «селективность» для конкретного продукта, такого как зеркальный изомер или региоселективный продукт. Широкое определение «активности», представленное в настоящем раскрытии, включает в себя селективность, хотя традиционно селективность иногда рассматривается как отличное от активности фермента понятие.
Термины «белок», «полипептид» и «пептид» используются взаимозаменяемо для обозначения полимера из по меньшей мере двух аминокислот, ковалентно связанных посредством амидной связи, независимо от длины или посттрансляционной модификации (например, гликозилирование, фосфорилирование, липидизация, миристиолирование, убиквитинирование, и т.д.). В некоторых случаях, полимер имеет по меньшей мере около 30 аминокислотных остатков, и обычно по меньшей мере около 50 аминокислотных остатков. В большинстве случаев, они содержат по меньшей мере около 100 аминокислотных остатков. Не предполагается, что настоящее изобретение ограничено аминокислотными последовательностями какой-либо конкретной длины. Термины включают композиции, которые, как обычно полагают, являются фрагментами полноразмерных белков или пептидов. В пределы этого определения включены D-и L-аминокислоты, и смеси D-и L-аминокислот. Полипептиды, описанные в настоящем раскрытии, не ограничены генетически закодированным аминокислотами. Действительно, в дополнение к генетически закодированным аминокислотам, полипептиды, описанные в настоящем раскрытии, могут быть составлены из, полностью или частично, естественных и/или синтетических незакодированных аминокислот. В некоторых вариантах осуществления полипептид представляет собой часть полноразмерного предкового или родительского полипептида, содержащую аминокислотные добавления или удаления (например, гэпы) и/или замены по сравнению с аминокислотной последовательностью полноразмерного родительского полипептида, при этом все еще сохраняя функциональную активность (например, каталитическую активность).
При использовании в настоящем раскрытии, термин «дикий тип» или «дикого типа» (WT) относится к встречающимся в природе ферментам и/или другим продуктам (например, нерекомбинантным ферментам). Субстрат или лиганд, который реагирует с биомолекулой дикого типа, иногда считают «нативным» субстратом или лигандом.
При использовании в настоящем раскрытии, термины «вариант», «мутант», «последовательность мутанта» и «последовательность варианта» относятся к биологической последовательности, которая отличается в некотором отношении от стандартной или опорной последовательности (например, в некоторых вариантах осуществления, родительской последовательности). Отличие может называться «мутацией». В некоторых вариантах осуществления мутант представляет собой полипептидную или полинуклеотидную последовательность, которая была изменена посредством по меньшей мере одной замены, вставки, перехода, удаления и/или другой генетической операции. Для целей настоящего раскрытия мутанты и варианты не ограничиваются конкретным способом, посредством которого они были созданы. В некоторых вариантах осуществления, мутант или вариант последовательности имеет повышенные, пониженные или по существу аналогичные активности или свойства по сравнению с родительской последовательностью. В некоторых вариантах осуществления вариант полипептида содержит один или более аминокислотных остатков, которые были мутированы по сравнению с аминокислотной последовательностью полипептида дикого типа (например, родительского полипептида). В некоторых вариантах осуществления один или более аминокислотных остатков полипептида оставляются неизменными, являются инвариантными или не подвергаются мутации по сравнению с родительским полипептидом в вариантах полипептидах, составляющих множество полипептидов. В некоторых вариантах осуществления родительский полипептид используется в качестве основы для генерации вариантов с улучшенной стабильностью, активностью или любым другим требуемым свойством.
При использовании в настоящем раскрытии, термины «вариант фермента» и «вариантный фермент» используются в отношении ферментов, которые аналогичны опорному ферменту, в частности, по своей функции, но имеют мутации в своей аминокислотной последовательности, которые делают их отличающимися по последовательности от фермента дикого типа или другого опорного фермента. Варианты фермента могут быть получены посредством широкого ряда различных методик мутагенеза, известных специалистам в данной области техники. Кроме того, наборы для мутагенеза также поставляются многими коммерческими поставщиками в области молекулярной биологии. Существуют способы для выполнения конкретных замен в заданных аминокислотах (сайт-направленных), специфичных или случайных мутаций в локализованной области гена (регион-специфичных) или неспецифического мутагенеза по всему гену (например, насыщающий мутагенез). Специалистам в технике известны многочисленные соответствующие способы для генерации вариантов фермента, включая, но не ограничиваясь, сайт-направленный мутагенез одноцепочечной ДНК или двухцепочечной ДНК с применением ПЦР, кассетного мутагенеза, синтез генов, ПЦР с внесением ошибок, перетасовку и химический насыщающий мутагенез, или любой другой соответствующий способ, известный в технике. После того, как варианты были продуцированы, они могут подвергаться скринингу в отношении требуемого свойства (например, высокой или увеличенной; или низкой или пониженной активности, повышенной тепловой и/или щелочной стабильности, и т.д.).
«Панель ферментов» представляет собой группу ферментов, выбранных таким образом, что каждый элемент панели катализирует одну и ту же химическую реакцию. В некоторых вариантах осуществления элементы панели могут коллективно осуществлять оборот множества субстратов, каждый из которых подвергается одной и той же реакции. Часто элементы панели выбирают для эффективного осуществления оборота множества субстратов. В некоторых случаях панели являются коммерчески доступными. В других случаях они являются индивидуально изготавливаемыми для субъекта. Например, панель может содержать различные ферменты, идентифицированные как «попадания» (хиты) в процедуре скрининга. В определенных вариантах осуществления один или более элементов панели существуют только как вычислительное представление. Другими словами, фермент представляет собой виртуальный фермент.
«Модель» является представлением структуры биомолекулы или лиганда. Иногда она предоставляется как совокупность трехмерных позиций для атомов или функциональных составляющих представляемой сущности. Модели часто содержат полученные вычислительно представления сайтов связывания или другие аспекты вариантов фермента. Примеры моделей, относящихся к вариантам осуществления настоящего раскрытия, получены посредством моделирования на основе гомологии, нарезания резьбы белка (нарезание резьбы белка), или моделирования белка с нуля с применением такой процедуры, как Розетта (rosettacommons.org/software/) или моделирования молекулярной динамики.
«Модель на основе гомологии» представляет собой трехмерную модель белка или части белка, содержащую, по меньшей мере, сайт связывания рассматриваемого лиганда. Моделирование на основе гомологии полагается на наблюдение, что структуры белка имеют тенденцию сохраняться среди гомологичных белков. Модель на основе гомологии предоставляет трехмерные позиции остатков, включая остов и боковые цепи. Модель генерируется по матрице структуры гомологичного белка, который, вероятно, имеет структуру, аналогичную структуре моделируемой последовательности. В некоторых вариантах осуществления матрица структуры используется на двух этапах: «выравнивание последовательности с матрицами» и «построение моделей на основе гомологии».
На этапе «выравнивания последовательности с матрицами» выполняется выравнивание последовательности модели с одной или более последовательностям матрицы структуры и подготовка выравнивания входной последовательности для построения модели на основе гомологии. Выравнивание идентифицирует пропуски и другие области расхождения между последовательностью модели и последовательностью(-ями) матрицы структуры.
На этапе «построения модели на основе гомологии» используются структурные характеристики для выведения пространственных ограничений, которые, в свою очередь, используются для генерации, например, модельных структур белка с использованием процедур сопряженного градиента и оптимизации посредством имитации отжига. Конструктивные характеристики матрицы могут быть получены с помощью таких методик, как NMR или рентгеноструктурная кристаллография. Примеры таких методик можно найти в обзорной статье “A Guide to Template Based Structure Prediction,” by Qu X, Swanson R, Day R, Tsai J. Curr Protein Pept Sci. 2009 Jun;10(3):270-85.
Термин «активная конформация» используется в отношении конформации белка (например, фермента), которая позволяет белку вызывать подвергание субстрата химическому превращению (например, каталитической реакции).
«Активное положение» представляет собой положение, в котором лиганд, вероятно, будет подвергаться каталитическому превращению или выполнять некоторую требуемую роль, такую как ковалентное связывание с сайтом связывания.
Термин «последовательность» используется в настоящем раскрытии для обозначения порядка и идентичности произвольных биологических последовательностей, включая, но не ограничиваясь, весь геном, целую хромосому, сегмент хромосомы, совокупность последовательностей генов для взаимодействующих генов, ген, последовательность нуклеиновой кислоты, белок, пептид, полипептид, полисахарид, и т.д. В некоторых контекстах «последовательность» относится к порядку и идентичности аминокислотных остатков в белке (то есть, последовательности белка или символьной строке белка) или к порядку и идентичности нуклеотидов в нуклеиновой кислоте (то есть, последовательности нуклеиновой кислоты или символьной строке нуклеиновой кислоты). Последовательность может быть представлена символьной строкой. «Последовательность нуклеиновой кислоты» относится к порядку и идентичности нуклеотидов, включая нуклеиновую кислоту. «Последовательность белка» относится к порядку и идентичности аминокислот, включая белок или пептид.
«Кодон» относится к конкретной последовательности трех последовательных нуклеотидов, которая является частью генетического кода, и которая определяет конкретную аминокислоту в белке или запускает или останавливает синтез белка.
Термин «ген» используется в широком смысле для обозначения произвольного сегмента ДНК или другой нуклеиновой кислоты, ассоциированного с биологической функцией. Таким образом, гены включают в себя кодирующие последовательности и, необязательно, регуляторные последовательности, требующиеся для их экспрессии. Гены также, необязательно, включают в себя неэкспрессируемые сегменты нуклеиновых кислот, которые, например, формируют последовательности распознавания для других белков. Гены могут быть получены из множества источников, включая клонирование из интересующего источника или синтезирование по известной или предсказанной информации о последовательности, и могут включать последовательности, спроектированные как имеющие требуемые параметры.
«Функциональная составляющая» представляет собой часть молекулы, которая может включать в себя или целые функциональные группы, или части функциональных групп в качестве подструктур, тогда как функциональные группы представляют собой группы атомов или связей в пределах молекул, которые ответственны за характеристические химические реакции этих молекул.
«Скрининг» относится к процессу, в котором определяют одно или более свойств одной или более биомолекул. Например, типичные процессы скрининга включают процессы, в которых определяют одно или более свойств одного или более элементов одной или более библиотек. Скрининг может быть выполнен вычислительно с применением вычислительных моделей биомолекул и виртуальной среды биомолекул. В некоторых вариантах осуществления системы виртуального скрининга белка предоставлены для отобранных ферментов с требуемой активностью и селективностью.
«Экспрессионная система» представляет собой систему для экспрессирования белка или пептида, закодированного геном или другой нуклеиновой кислотой.
«Направленная эволюция», «управляемая эволюция» или «искусственная эволюция» относятся к in silico, in vitro или в естественных условиях процессам искусственного изменения одной или более последовательностей биомолекул (или строки символов, представляющей последовательность) посредством искусственного отбора, мутации, рекомбинации или другой манипуляции. В некоторых вариантах осуществления направленная эволюция происходит в репродуктивной популяции, в которой (1) присутствуют изменчивости индивидов, (2) некоторые изменчивости имеют наследуемую генетическую информацию и (3) некоторые изменчивости отличаются по приспособляемости. Репродуктивный успех определяется результатом отбора для предварительно заданного свойства, такого как благоприобретенное свойство. Репродуктивная популяция может представлять собой, например, физическую популяцию в процессе in vitro или виртуальную популяцию в компьютерной системе в процессе in silico.
Способы направленной эволюции могут быть без труда применены к полинуклеотидам для создания библиотек вариантов, которые могут подвергаться экспрессии, скринингу или анализу. Способы мутагенеза и направленной эволюции известны в технике (см. например, патенты США № 5605793, 5830721, 6132970, 6420175, 6277638, 6365408, 6602986, 7288375, 6287861, 6297053, 6576467, 6444468, 5811238, 6117679, 6165793, 6180406, 6291242, 6995017, 6395547, 6506602, 6519065, 6506603, 6413774, 6573098, 6323030, 6344356, 6372497, 7868138, 5834252, 5928905, 6489146, 6096548, 6387702, 6391552, 6358742, 6482647, 6335160, 6653072, 6355484, 603344, 6319713, 6613514, 6455253, 6579678, 6586182, 6406855, 6946296, 7534564, 7776598, 5837458, 6391640, 6309883, 7105297, 7795030, 6326204, 6251674, 6716631, 6528311, 6287862, 6335198, 6352859, 6379964, 7148054, 7629170, 7620500, 6365377, 6358740, 6406910, 6413745, 6436675, 6961664, 7430477, 7873499, 7702464, 7783428, 7747391, 7747393, 7751986, 6376246, 6426224, 6423542, 6479652, 6319714, 6521453, 6368861, 7421347, 7058515, 7024312, 7620502, 7853410, 7957912, 7904249, и все родственные неамериканские экземпляры; Ling и соавт., Anal. Biochem., 254(2):157-78 [1997]; Dale и соавт., Meth. Mol. Biol., 57:369-74 [1996]; Smith, Ann. Rev. Genet., 19:423-462 [1985]; Botstein и соавт., Science, 229:1193-1201 [1985]; Carter, Biochem. J., 237:1-7 [1986]; Kramer и соавт., Cell, 38:879-887 [1984]; Wells и соавт., Gene, 34:315-323 [1985]; Minshull и соавт., Curr. Op. Chem. Biol., 3:284-290 [1999]; Christians и соавт., Nat. Biotechnol., 17:259-264 [1999]; Crameri и соавт., Nature, 391:288-291 [1998]; Crameri, и соавт., Nat. Biotechnol., 15:436-438 [1997]; Zhang и соавт., Proc. Nat. Acad. Sci. U.S.A., 94:4504-4509 [1997]; Crameri и соавт., Nat. Biotechnol., 14:315-319 [1996]; Stemmer, Nature, 370:389-391 [1994]; Stemmer, Proc. Nat. Acad. Sci. USA, 91:10747-10751 [1994]; WO 95/22625; WO 97/0078; WO 97/35966; WO 98/27230; WO 00/42651; WO 01/75767; и WO 2009/152336, все из которых включены в настоящее раскрытие посредством ссылки).
В определенных вариантах осуществления способы направленной эволюции генерируют библиотеки вариантов белка посредством рекомбинации генов, кодирующих варианты, полученные из родительского белка, а также посредством рекомбинации генов, кодирующих варианты в библиотеке вариантов родительского белка. В способах могут использоваться олигонуклеотиды, содержащие последовательности или подпоследовательности, кодирующие по меньшей мере один белок родительской библиотеки вариантов. Некоторые из олигонуклеотидов родительской библиотеки вариантов могут быть очень близкими, отличаясь только выбором кодонов для чередующихся аминокислот, выбранных для изменения посредством рекомбинации с другими вариантами. Способ может быть выполнен в течение одного или множества циклов, пока требуемые результаты не будут достигнуты. Если используется множество циклов, каждый из них обычно включает в себя этап скрининга с целью идентификации тех вариантов, которые имеют приемлемую или улучшенную производительность и являются кандидатами на использование по меньшей мере в одном последующем цикле рекомбинации. В некоторых вариантах осуществления этап скрининга включает в себя систему виртуального скрининга белка для определения каталитической активности и селективности ферментов для требуемых субстратов.
В некоторых вариантах осуществления способы направленной эволюции генерируют варианты белка посредством сайт-направленного мутагенеза в заданных остатках. Эти заданные остатки обычно идентифицируют посредством структурного анализа сайтом связывания, анализа квантовой химии, анализа гомологии последовательностей, моделей активности последовательности, и т.д. В некоторых вариантах осуществления используется насыщающий мутагенез, в котором производится попытка сгенерировать все возможные (или как можно ближе ко всем возможным) мутации в активном центре или узкой области гена.
«Перетасовка» и «генная перетасовка» являются типами направленной эволюции, в которых рекомбинируется совокупность фрагментов родительских полинуклеотидов через ряд циклов удлинения цепи. В определенных вариантах осуществления, один или более циклов удлинения цепи является самоинициирующимися; то есть, выполняются без добавления праймеров, помимо самих фрагментов. Каждый цикл включает в себя отжиг одноцепочечных фрагментов через гибридизацию, последовательное удлинение отожженных фрагментов через удлинение цепи и денатурацию. В течение перетасовки растущая цепь нуклеиновой кислоты обычно подвергается воздействию множества различных партнеров по отжигу в процессе, иногда называемом «обменом матрицами», который включает в себя обмен одного домена нуклеиновой кислоты из одной нуклеиновой кислоты на второй домен из второй нуклеиновой кислоты (то есть, первые и вторые нуклеиновые кислоты служат матрицами в процедуре перетасовки).
Обмен матрицами часто приводит к образованию химерных последовательностей, которые возникают вследствие внесения перекрытий между фрагментами из различных источников. Перекрытия создаются через рекомбинации посредством обмена матрицами в течение множества циклов отжига, удлинения и денатурации. Таким образом, перетасовка обычно приводит к продуцированию вариантов полинуклеотидных последовательностей. В некоторых вариантах осуществления вариантные последовательности составляют «библиотеку» вариантов (то есть, группу, содержащую множество вариантов). В некоторых вариантах осуществления этих библиотек варианты содержат сегменты последовательности из двух или более из родительских полинуклеотидов.
Когда используется два или более родительских полинуклеотидов, индивидуальные родительские полинуклеотиды являются достаточно гомологичными для того, чтобы фрагменты от различных родителей гибридизировались в условиях отжига, используемых в циклах перетасовки. В некоторых вариантах осуществления перетасовка допускает рекомбинацию родительских полинуклеотидов, имеющих относительно ограниченные/низкие уровни гомологии. Часто, индивидуальные родительские полинуклеотиды имеют отличительные и/или уникальные домены и/или другие интересующие характеристики последовательности. При использовании родительских полинуклеотидов, имеющих отличительные характеристики последовательности, перетасовка может произвести имеющие высокое разнообразие варианты полинуклеотидов.
Различные методики перетасовки известны в технике. См. например, патенты США № 6917882, 7776598, 8029988, 7024312 и 7795030, все из которых включены в настоящее раскрытие посредством ссылки во всех их полноте.
В некоторых методиках направленной эволюции используется «сплайсинг генов путем удлинения перекрытия» или «SOE генов», который представляет собой основанный на ПЦР способ рекомбинации последовательностей ДНК без зависимости от сайтов рестрикции и непосредственной генерации фрагментов ДНК in vitro. В некоторых реализациях методики начальные ПЦР генерируют перекрывающиеся сегменты гена, которые используются в качестве матричной ДНК для второй ПЦР для создания полноразмерного продукта. Внутренние праймеры ПЦР создают перекрывающиеся комплементарные 3'-концы на промежуточных сегментах и вносят нуклеотидные замены, вставки или удаления для сплайсинга генов. Перекрывающиеся цепочки этих промежуточных сегментов гибридизуют в 3'-областях во второй ПЦР и удлиняют с целью генерации полноразмерного продукта. В различных приложениях полный продукт усилен путем обрамления учебников для начинающих, которые могут включать сайты фермента ограничения для вставки продукта в вектор экспрессии для клонирования целей. См., например, Horton, и соавт., Biotechniques, 8 (5): 528-35 [1990]). «Мутагенез» представляет собой процесс внесения по меньшей мере одной мутации в стандартную или опорную последовательность, такую как родительская нуклеиновая кислота или родительский полипептид.
Сайт-направленный мутагенез является одним из примеров полезной методики для внесения мутаций, хотя любой соответствующий способ может быть применен. Таким образом, альтернативно или в дополнении, мутанты могут быть получены посредством генного синтеза, насыщающего неспецифического мутагенеза, полусинтетических комбинаторных библиотек остатков, рекурсивной рекомбинации последовательностей («RSR») (см. например, публикацию заявки на патент США № 2006/0223143, включенную в настоящее раскрытие посредством ссылки во всей ее полноте), перетасовки генов, ПЦР с внесением ошибок и/или любого другого соответствующего способа.
Один из примеров соответствующей процедуры насыщающего мутагенеза описан в публикации заявки на патент № 2010/0093560, которая включена в настоящее раскрытие посредством ссылки во всей ее полноте.
«Фрагмент» представляет собой произвольную часть последовательности нуклеотидов или аминокислот. Фрагменты могут быть произведены с применением произвольного подходящего способа, известного в технике, включая, но не ограничиваясь, расщепление полипептидной или полинуклеотидной последовательности. В некоторых вариантах осуществления фрагменты производят посредством применения нуклеаз, которые расщепляют полинуклеотиды. В некоторых дополнительных вариантах осуществления фрагменты создаются с применением методик химического и/или биологического синтеза. В некоторых вариантах осуществления фрагменты включают в себя последовательности по меньшей мере из одной родительской последовательности, созданные с применением частичного удлинения цепи комплементарной(-ых) нуклеиновой(-ых) кислоты(-т). В некоторых вариантах осуществления, в которых применяются методики in silico, виртуальные фрагменты генерируются вычислительно с целью имитации результатов генерации фрагментов посредством химических и/или биологических методик. В некоторых вариантах осуществления фрагменты полипептида демонстрируют активность полноразмерного полипептида, тогда как в некоторых других вариантах осуществления фрагменты полипептида не обладают активностью полноразмерного полипептида.
«Родительский полипептид», «родительский полинуклеотид», «родительская нуклеиновая кислота» и «родитель» обычно используются для обозначения полипептида дикого типа, полинуклеотида дикого типа, или варианта, используемого в качестве исходной точки в процедуре создания разнообразия, такой как направленная эволюция. В некоторых вариантах осуществления сам родитель продуцируется через перетасовку или другую(-ие) процедуру(-ы) создания разнообразия. В некоторых вариантах осуществления мутанты, используемые в направленной эволюции, прямо связаны с родительским полипептидом. В некоторых вариантах осуществления родительский полипептид является стабильным, когда подвергается действию экстремальных условий температуры, pH фактор и/или растворителя, и может служить основой для создания вариантов для перетасовки. В некоторых вариантах осуществления родительский полипептид не является устойчивым к экстремальным условиям температуры, pH фактор и/или растворителя, и родительский полипептид изменяется для создания устойчивых вариантов.
«Родительская нуклеиновая кислота» кодирует родительский полипептид.
«Библиотека» или «популяция» относятся к совокупности по меньшей мере из двух различных молекул, символьных строк и/или моделей, таких как последовательности нуклеиновых кислот (например, гены, олигонуклеотиды, и т.д.) или их продукты экспрессии (например, ферменты или другие белки). Библиотека или популяция обычно содержит ряд различных молекул. Например, библиотека или популяция обычно содержит по меньшей мере около 10 различных молекул. Большие библиотеки обычно содержат по меньшей мере около 100 различных молекул, и, как правило, по меньшей мере около 1000 различных молекул. Для некоторых приложений библиотека содержит по меньшей мере приблизительно 10000 или более различных молекул. Однако, не предполагается, что настоящее изобретение ограничивается конкретным числом различных молекул. В определенных вариантах осуществления библиотека содержит ряд различных или фантастические нуклеиновые кислоты или белки, произведенные направленной процедурой эволюции.
Две нуклеиновых кислоты «рекомбинируются», когда последовательности от каждой из этих двух нуклеиновых кислот комбинируются с образованием нуклеиновой(-ых) кислоты(-т)-потомка(-ов). Две последовательности рекомбинируются «прямо», когда обе нуклеиновые кислоты являются субстратами для рекомбинации.
Термин «отбор» относится к процессу, в котором одна или более биомолекул идентифицируются как имеющие одно или более интересующих свойств. Таким образом, например, можно провести скрининг библиотеки с целью определения одного или более свойств одного или более элементов библиотеки. Если один или более элементов библиотеки идентифицированы как обладающие интересующим свойством, они отбираются. Отбор может включать в себя выделение элемента библиотеки, но это не является необходимым. Кроме того, отбор и скрининг могут выполняться, и часто выполняются, одновременно. Некоторые варианты осуществления, раскрытые в настоящем раскрытии, предоставляют системы и способы для скрининга и отбора ферментов с требуемой активностью и/или селективностью.
«Секвенирование следующего поколения» и «высокопроизводительное секвенирование» являются методиками секвенирования, которые распараллеливают процесс секвенирования, производя тысячи или миллионы последовательностей единовременно.
Примеры соответствующих методов секвенирования следующего поколения включают в себя, но не ограничиваются указанным, секвенирование единичной молекулы в реальном времени (например, Pacific Biosciences, Менло-Парк, Калифорния), ионное полупроводниковое секвенирование (например, Ion Torrent, Южный Сан-Франциско, Калифорния), пиросеквенирование (например, 454, Брэнфорд, Коннектикут), секвенирование посредством лигирования (например, секвенирование SOLiD, которым владеет Life Technologies, Карлсбад, Калифорния), секвенирование посредством синтеза и обратимого обрывателя цепи (например, Illumina, Сан-Диего, Калифорния), технологии визуализации нуклеиновой кислоты, такие как просвечивающая электронная микроскопия, и т.п.
«Зависимая переменная» («DV») представляет результат или эффект, или тестируется на предмет определения, является ли она эффектом. «Независимые переменные» («IV») представляют входные данные или причины, или тестируется на предмет определения того, являются ли они причиной. Зависимая переменная может исследоваться с целью установления того, меняется ли она и насколько сильно при изменении независимых переменных.
В простой стохастической линейной модели
yi=+bxi+ei
где элемент yi представляет собой i-ое значение зависимой переменной, и xi представляет собой i-ое значение независимой переменной (IV). Элемент ei известен как «ошибка» и содержит изменчивость зависимой переменной, не объясняемую независимой переменной.
Независимая переменная (IV) также известна как «предикторная переменная», «регрессор», «управляемая переменная», «регулируемая переменная», «объясняющая переменная» или «входная переменная».
Термин «коэффициент» относится к скалярной величине, умноженной на зависимую переменную или выражение, содержащее зависимую переменную.
Термины «ортогональный» и ортогональность» относятся к независимой переменной, которая не коррелирует с другими независимыми переменными в модели или другой функциональной зависимости.
Термин «модель активности последовательности» относится к любым математическим моделям, которые описывают зависимость между активностями, характеристиками или свойствами биологических молекул с одной стороны, и различными биологическими последовательностями с другой стороны.
Термин «символьная строка» относится к представлению биологической молекулы, которое сохраняет информации о последовательности/структурную информацию для этой молекулы. В некоторых вариантах осуществления символьная строка содержит информацию о мутациях последовательности в библиотеке вариантов. Символьные строки биомолекул и информация об активности для биомолекул могут использоваться в качестве обучающего множества для модели активности последовательности. Не относящиеся к последовательности свойства биомолекул могут сохранены или иначе ассоциированы с символьными строками для биомолекул.
«Опорная последовательность» представляет собой последовательность, относительно которой производится изменение последовательности. В некоторых случаях, «опорная последовательность» используется для задания изменений. Такая последовательность может являться последовательностью, предсказанной посредством модели как имеющая самое высокое значение (или одно из самых высоких значений) требуемой активности. В другом случае опорная последовательность может являться последовательностью элемента исходной библиотеки вариантов белка. В определенных вариантов осуществления опорная последовательность представляет собой последовательность родительского белка или нуклеиновой кислоты.
Фраза «обучающее множество» относится к множеству данных или наблюдений последовательности-активности, под которое подогнаны или на основании которого построены одна или более моделей. Например, для модели активности последовательности белка, обучающее множество содержит последовательности остатков для исходной или улучшенной библиотеки вариантов белка. Как правило, эти данные включают в себя полную или частичную информацию о последовательности остатков вместе со значением активности для каждого белка в библиотеке. В некоторых случаях, множество типов активностей (например, данные константы скорости реакции и данные тепловой стабильности) представлены вместе в обучающем множестве. Активность иногда является выгодным свойством.
Термин «наблюдение» представляет собой информацию о белке или другом биологическом объекте, который может использоваться в обучающем множестве для создания модели, такой как модель активности последовательности. Термин «наблюдение» может относиться к любым отсеквенированным и/или проанализированным биологическим молекулам, включая варианты белка. В определенных вариантах осуществления каждое наблюдение представляет собой значение активности и ассоциированную последовательность для варианта в библиотеке. Обычно, чем больше наблюдений используется для создания модели активности последовательности, тем лучше предсказательная способность этой модели активности последовательности.
Фраза «прогнозирующая способность» относится к возможности модели правильно предсказывать значения зависимой переменной для данных при различных условиях. Например, прогнозирующая способность модели активности последовательности относится к возможности модели предсказывать активность по информации о последовательности.
Фраза «перекрестная проверка» относится к способу для проверки обобщаемости способности модели предсказывать значение зависимой переменной. Способ подготавливает модель с использованием одного множества данных и проверяет ошибки модели, используя другое множество данных. Первое множество данных рассматривается как обучающее множество, и второе множество данных представляет собой контрольное множество.
Фраза «систематическое изменение» относится к различным дескрипторам элемента или множества элементов, изменяемых в различных комбинациях.
Фраза «систематически изменяющиеся данные» относится к данным, произведенным, выведенным или следующим из различных дескрипторов элемента или множества элементов, изменяемых в различных комбинациях. Множество различных дескрипторов могут быть изменены одновременно, но в различных комбинациях. Например, данные активности, собранные для полипептидов, в которых были изменены комбинации аминокислот, являются систематически изменяющимися данными.
Фраза «систематически изменяющиеся последовательности» относится к множеству последовательностей, в которых каждый остаток можно видеть во множестве контекстов. В принципе, уровень систематического изменения может быть определен количественно по степени, в которой последовательности являются ортогональными друг относительно друга (то есть, максимально отличаются по сравнению со средним значением).
Термин «переключение» относится к введению множества типов аминокислотных остатков в конкретное положение в последовательностях вариантов белка в оптимизированной библиотеке.
Термины «регрессия» и «регрессионный анализ» относятся к методикам, применяемым для выяснения того, какие из независимых переменных связаны с зависимой переменной, и исследования форм таких связей. При ограниченных обстоятельствах регрессионный анализ может применяться для выведения причинно-следственных связей между независимыми и зависимыми переменными. Он представляет собой статистическую методику для оценки связей между переменными. Он включает в себя множество методик для моделирования и анализа нескольких переменных, в которых внимание сосредоточено на связи между зависимой переменной и одной или более независимых переменных. Более конкретно, регрессионный анализ помогает в понимании того, каким образом типичное значение зависимой переменной изменяется, когда изменяется любая из независимых переменных, в то время как другие независимые переменные остаются зафиксированными. Методики регрессии могут применяться для создания моделей активности последовательности по обучающим множествам, содержащим множество наблюдений, которые могут содержать информацию о последовательность и активности.
Методы дробных наименьших квадратов (PLS) составляют семейство методов, которые строят линейную регрессионную модель посредством отображения предсказанных переменных (например, активностей) и наблюдаемых переменных (например, последовательностей) в новое пространство. PLS также называют проекцией на скрытые структуры. Данные как для X (независимые переменные), так и для Y (зависимые переменные) проецируются в новые пространства. PLS применяется для нахождения фундаментальных соотношений между двумя матрицами (X и Y). Подход, основанный на скрытых переменных, применяется для моделирования структур ковариации в пространствах Y и X. PLS-модель будет пытаться найти многомерное направление в пространстве X, которое объясняет направление максимальной многомерной изменчивости в пространстве Y. Регрессия является особенно подходящей, когда матрица предикторов имеет больше переменных, чем наблюдений, и когда присутствует мультиколлинеарность среди значений X.
Скрытые переменные (в отличие от наблюдаемых переменных) представляют собой переменные, которые не наблюдают непосредственно, а выводят из непосредственно измеренных переменных. Математические модели, цель которых состоит в объяснении наблюдаемых переменных через скрытые переменные, называются моделями со скрытыми переменными.
«Дескриптор» относится к чему-либо, что служит для описания или идентификации элемента. Например, символы в символьной строке могут быть дескрипторами аминокислот в полипептиде, представленном символьной строкой.
В регрессионной модели зависимая переменная связана с независимыми переменными посредством суммы членов. Каждый член содержит произведение независимой переменной и ассоциированного коэффициента регрессии. В случае чисто линейной регрессионной модели коэффициенты регрессии задаются β в форме следующего выражения:
yi
=

где yi представляет собой зависимую переменную, кси представляют собой независимые переменные, εi представляет собой переменную ошибки, и T обозначает транспонирование, то есть, скалярное произведение векторов xi и β.
Фраза «регрессия главных компонентов» (PCR) относится к регрессионному анализу, в котором применяется анализ главных компонентов при оценке коэффициентов регрессии. Вместо того, чтобы регрессировать зависимую переменную по независимой переменной напрямую, используются главные компоненты независимых переменных. PCR обычно использует только подмножество главных компонентов в регрессионном анализе.
Фраза «анализ главных компонентов» (PCA) относится к математической процедуре, в которой применяется ортогональное преобразование для преобразования множества наблюдений, возможно, скоррелированных переменных, в множество значений линейно нескоррелированных переменных, называемых главными компонентами. Число главных компонентов меньше либо равно числу исходных переменных. Это преобразование определено таким образом, что первый главный компонент имеет наибольшую возможную изменчивость (то есть, учитывает столько изменчивости в данных, сколько возможно), и каждый последующий компонент, в свою очередь, имеет самую высокую изменчивость, которая возможна при условии его ортогональности (то есть, отсутствии корреляции) с предшествующими компонентами.
«Нейронная сеть» представляет собой модель, содержащую взаимосвязанную группу обрабатывающих элементов, или «нейронов», которые обрабатывают информацию с применением коннекционного подхода к вычислениям. Нейронные сети применяются для моделирования сложных взаимосвязей между входными и выходными данными и/или для нахождения шаблонов в данных. Большинство нейронных сетей обрабатывает данные в нелинейным, распределенным, параллельным образом. В большинстве случаев нейронные сети являются адаптивными системами, которые изменяют свою структуру во время фазы обучения. Функции выполняются совместно и параллельно всеми обрабатывающими элементами, вместо того, чтобы использовать четкое разделение на подзадачи, которые назначаются различным элементам.
Обычно, нейронная сеть включает в себя сеть простых обрабатывающих элементов, которые демонстрируют сложное глобальное поведение, определяемое связями между обрабатывающими элементами и параметрами элементов. Нейронные сети применяются с алгоритмами, спроектированными для изменения силы связей в сети с целью обеспечения требуемого потока сигнала. Мощность изменяется во время обучения или изучения.
«Генетический алгоритм» («GA») представляет собой процесс, который имитирует процесс эволюции. Генетические алгоритмы (GA) применяются во многих областях для решения задач, которые не являются полностью охарактеризованным или являются слишком сложными для того, чтобы обеспечить возможность полной характеризации, но для которых доступна некоторая аналитическая оценка. Таким образом, GA применяются для решения задач, которые могут быть оценены посредством некоторой количественно оцениваемой меры для относительной ценности решения (или, по меньшей мере, относительной ценности одного потенциального решения по сравнению с другим). В контексте настоящего раскрытия генетический алгоритм представляет собой процесс для отбора или манипулирования символьными строками в компьютере, при этом как правило символьная строка соответствует одной или более биологическим молекулам (например, нуклеиновым кислотам, белкам, и т.п.) или данным, использованным для обучения модели, такой как модель активности последовательности или метод опорных векторов.
В одном из примеров генетический алгоритм предоставляет и оценивает популяцию моделей в первом поколении алгоритма. Каждая модель содержит множество параметров, описывающих связь между по меньшей мере между одной независимой переменной (IV) и зависимой переменной (DV). «Функция приспособленности» оценивает члены популяции моделей и ранжирует их на основании одного или более критериев, таких как требуемая активность или низкий уровень ошибки предсказания модели. Члены популяции моделей также иногда называются индивидами или хромосомами в контексте генетических алгоритмов. В некоторых вариантах осуществления приспособленность модели оценивают с помощью информационного критерия Акаике (AIC) или байесовского информационного критерия (BIC), при этом индивидов, имеющих наименьшие значения AIC или BIC, выбирают в качестве наиболее приспособленных индивидов. Имеющие высокий ранг модели отбирают для продвижения во второе поколение и/или спаривания с целью продуцирования популяции «дочерних моделей» для второго поколения алгоритма. Популяция во втором поколении оценивается аналогично с помощью функцией приспособленности, и имеющие высокий ранг элементы продвигаются и/или спариваются, как и в случае с первым поколением. Генетический алгоритм продолжается таким образом для последующих поколений, пока не будет выполняться «критерий сходимости», после чего алгоритм завершается с одним или более имеющими высокий ранг индивидами (моделями).
В другом примере «индивиды» представляют собой вариантные пептидные последовательности, и функция приспособленности представляет собой предсказанную активность этих индивидов. Каждое поколение содержит популяцию пептидных последовательностей индивидов, которые оценивают в отношении их приспособленности. Самые приспособленные в поколении выбирают для продвижения и/или спаривания для создания популяции следующего поколения. После множества поколений генетический алгоритм может сходиться к популяции высокопроизводительных пептидных последовательностей.
Как в примере выше, генетический алгоритм часто проходит через множество итераций для поиска оптимальных параметров в пространстве параметров. Каждая итерация генетического алгоритма также называется «поколением» генетического алгоритма. Модели в поколении генетического алгоритма формируют «популяцию» для поколения. В контексте генетических алгоритмов термины «хромосома» и «индивид» иногда используются в качестве обозначений для модели или множества параметров модели в популяции. Такие обозначения используются потому, что модель из родительского поколения передает свои параметры (или «гены») модели дочернего поколения, что напоминает биологический процесс, в котором родительская хромосома передает свои гены хромосоме потомка.
Термин «генетическая операция» («GO») относится к биологическим и/или вычислительным генетическим операциям, в которых все изменения в любой популяции любого типа символьных строк (и, таким образом, в любых физических свойствах физических объектов, закодированных такими рядами), могут быть описаны как результат случайного и/или предварительно определенного применения конечного множества логических алгебраических функций. Примеры ИДУТ включают в себя, но не ограничиваются, размножение, перекрестное соединение, рекомбинацию, мутацию, лигирование, фрагментацию, и т.д.
«Информационный критерий Акаике» (AIC) является мерой относительной степени согласия статистической модели, и он часто используется в качестве критерия для выбора модели из конечного множества моделей. AIC основан в понятии информационной энтропии, фактически предлагая относительную меру информации, которая теряется, когда данная модель применяется для описания действительности. Можно сказать, что он описывает компромисс между погрешностью измерения и дисперсией при создании модели, или, грубо говоря, между точностью и сложностью модели.
AIC может быть вычислен как:
AIC= -2logeL+2k,
где L представляет собой максимальное правдоподобие функции, и k является числом свободных параметров модели, которая должна быть оценена.
«Байесов информационный критерий» («BIC») представляет собой критерий для выбора модели из конечного множества моделей, и тесно связан с AIC. BIC может быть вычислен как: BIC= -2logeL+kloge(n), где n является количеством наблюдений данных. При увеличении числа наблюдений BIC часто штрафует дополнительное количество свободных параметров в большей степени, чем AIC.
«Функция правдоподобия» или «правдоподобие» модели является функцией параметров статистической модели.
Вероятность множества значений параметров при наличии некоторых наблюдаемых исходов равняется вероятности этих наблюдаемых исходов при этих заданных значениях параметров, то есть, L(θ|x)=P(x|θ).
«Ансамбль моделей» представляет собой модель, члены которой включают в себя все члены группы моделей, при этом коэффициенты ансамбля моделей для членов основаны на взвешенных коэффициентах соответствующих членов для индивидуальных моделей группы. Взвешивание коэффициентов основано на предсказательной способности и/или приспособленности индивидуальных моделей.
«Моделирование методом Монте-Карло» представляет собой моделирование, которое основано на использовании большого количества случайных выборок для получения численных результатов, которые моделируют реальное явление.
«Моделирование Монте-Карло» представляет собой моделирование, которое основывается на большом количестве случайных выборок для получения числовых результатов, моделирующих реальное явление. Например, «вытягивание» большого числа псевдослучайных универсальных переменных из интервала (0,1) и отнесение значений, меньших либо равных 0,50, к орлам, и значений, больших 0,50, к решкам, представляет собой моделирование Монте-Карло поведения многократно бросаемой монеты.
II. ОБЩЕЕ ОПИСАНИЕ ПОТОКА ОПЕРАЦИЙ
A. Поток операций для цикла направленной эволюции
В определенных вариантах осуществления, в полном потоке операций применяются и методики in vitro, и вычислительные методики для управления процессом направленной эволюции. Вычислительная сторона процесса задействует структурные модели и моделей активности последовательности.
На каждом цикле направленной эволюции используется новый набор структурных моделей и новая модель активности последовательности. Кроме того, в каждом цикле варианты биомолекул, идентифицированные для дальнейшего анализа, оценивают с применением трехмерных структурных моделей вариантов. Информацию из структурных моделей объединяют с последовательностями вариантов и данными анализа (активности) с целью генерации большого нефильтрованного множества данных. Как правило, часть множества данных используют в качестве обучающего множества. Для текущего цикла направленной эволюции обучающее множество обучает модель активности последовательности, которая затем идентифицирует варианты биомолекул для следующего цикла направленной эволюции.
В определенных вариантах осуществления, один или несколько генетических алгоритмов (GA) применяются для оценки объединенных нефильтрованных данных, предоставленных в начале каждого цикла направленной эволюции. GA идентифицируют подмножество информации, содержащейся в нефильтрованном множестве данных, и это подмножество используют в качестве независимых переменных для обучения новой модели активности последовательности. Активность является зависимой переменной; модель активности последовательности предоставляет активность как функцию независимых переменных, идентифицированных во время фильтрации. В различных вариантах осуществления модель активности последовательности является нелинейной моделью. В определенных вариантах осуществления модель активности последовательности является гиперплоскостью в n-мерном пространстве, которое может быть сгенерировано машиной опорных векторов.
В примере, изображенном на фигуре 1А, поток операций направленной эволюции разворачивается следующим образом. Первоначально, собирают информацию для множества вариантов биомолекул. Каждый из этих вариантов мог быть идентифицирован на предыдущем цикле направленной эволюции. Если проект только начинается (т.е. отсутствуют предыдущие циклы направленной эволюции), варианты получают из другого источника, такого как панель биомолекул, которые, как известно, обладают потенциально интересующими свойствами. Иногда, выбирают варианты первого цикла, охватывающие относительно широкий диапазон пространства последовательности и/или активности.
После того, как варианты были идентифицированы, система оценки получает различные типы информации для каждого варианта. Следует отметить, что определяют по меньшей мере одну интересующую активность и последовательность каждого варианта. В некоторых вариантах осуществления последовательность представлена как совокупность мутаций относительно последовательности дикого типа или другой опорной последовательности. В некоторых вариантах осуществления активность хранится как численное значение, имеющее заданные единицы измерения. В некоторых вариантах осуществления значения активности являются нормализованными. Если последовательность заданного варианта неизвестна, она может быть получена путем секвенирования физического образца варианта.
В дополнение к данным активности последовательности структурная модель генерируется для каждой вариантной биомолекулы. В определенных вариантах осуществления структурные модели являются моделями на основе гомологии. Структурные модели оценивают вычислительно с целью получения дополнительных данных, которые объединяют с данными последовательности и активности для каждого варианта. В некоторых реализациях структурная модель каждого варианта применяется для идентификации энергии взаимодействия лиганда с сайтом рецептора биомолекулы и/или одного или более параметров, описывающих геометрию лиганда в сайте рецептора. Такая геометрия может включать в себя расстояния между атомами лиганда и атомами функциональной составляющей остатка в сайте связывания и/или атомами функциональной составляющей кофактора в сайте связывания. Определенные примеры представлены ниже.
Нефильтрованное множество данных содержит данные последовательности и активности для каждого варианта и обычно содержит множество дополнительных данных для каждого варианта. Как описано в настоящем раскрытии, эти дополнительные данные выведены из структурных моделей для каждого варианта. Кроме того, эти дополнительные данные обычно содержат (i) энергии взаимодействия или энергии связывания между рассматриваемым лигандом и сайтом связывания каждого варианта и/или (ii) структурные/геометрические дескрипторы, характеризующие взаимодействие лиганда с рецептором. См. блок 103 с фигуры 1A.
Было найдено, что множество необработанных нефильтрованных данных не всегда является оптимальным для обучения новой модели активности последовательности. Напротив, отфильтрованное подмножество объединенного множества необработанных данных обычно обеспечивает более полезную модель активности последовательности. Следовательно, множество необработанных данных из блока 103 фильтруется в соответствии с проиллюстрированным в блоках 105 и 107.
Фильтрация может быть выполнена с помощью любой(-ых) подходящей(-их) методики(-к). Как более полно описано ниже, одна необязательная методика удаляет определенные типы параметров, полученных из структурных моделей вариантов (например, определенные расстояния от атома субстрата до атома остатка). Блок 105. Например, нефильтрованное множество данных может содержать десять доступных геометрических характеристик лиганда в сайте связывания рецептора, но фильтрация устраняет три из них, в результате чего подмножество только из семи таких параметров используется в обучающем множестве. Эти параметры, вместе с последовательностью, служат независимыми переменными в модели активности последовательности, обученной на обучающем множестве. Альтернативно или дополнительно, фильтрация может удалять варианты, имеющие значения одной или более независимых переменных, выходящие за пределы диапазона, или ниже порога, определенного в качестве целесообразного для создания модели активности последовательности. Блок 107. В определенных вариантах осуществления, независимые переменные, фильтруемые таким образом, выводят из структурной модели.
Как проиллюстрировано в блоке 109, после того, как набор необработанных данных был должным образом отфильтрован, он используется для генерации модели активности последовательности. Как упомянуто, модель активности последовательности может быть нелинейной моделью, такой как гиперплоскость в n-мерном пространстве, определенном машиной опорных векторов. После того, как модель активности последовательности была сгенерирована, она используется для помощи в идентификации высокопроизводительных вариантов для следующего цикла направленной эволюции. См. блок 111. В одном из вариантов осуществления обученная модель активности последовательности используется в генетическом алгоритме (GA) для выбора множества различных последовательностей, которые, вероятно, обладают выгодными свойствами. Выбранные варианты используются в следующем цикле направленной эволюции. В таком следующем цикле варианты, выбранные с помощью модели активности последовательности, обрабатываются в соответствии с описанным выше (блоки 103, необязательно 105, 107 и 109). Однако они сначала анализируются для создания нового множества необработанных данных. См. блок 113. В определенных вариантах осуществления, варианты продуцируют физически и анализируют в отношении активности. Это обеспечивает некоторые необработанные данные. Варианты также структурно моделируются с целью определения значений энергии взаимодействия и геометрических значений связывания лиганда для каждого из типов энергий и типов геометрии, использованных в более раннем цикле направленной эволюции. Докер может быть применен для генерации значений для этих типов данных. Если необходимо, один или более вариантов секвенируются для дополнения необработанных данных.
Циклы направленной эволюции продолжаются таким образом, пока один или несколько циклов не покажут ограниченное улучшение или не будут соответствовать другим критериям сходимости. Проект направленной эволюции после этого завершается. На фигуре 1А проверка критериев сходимости проиллюстрирована блоком решения 115.
B. Поток операций генерации модели
Как указано выше, некоторые реализации фильтруют множество необработанных данных до обучения модели активности последовательности. Фильтрация может удалять определенные типы переменных из необработанных данных. Каждый тип переменной является потенциальной независимой переменной для модели активности последовательности. Альтернативно, или в дополнение, фильтрация может удалять определенные варианты, имеющие значения параметров вне заданных диапазонов. Было обнаружено, что такая фильтрация уменьшает шум, производимый моделями, обученными с использованием данных. В некоторых реализациях фильтрация выполняется с применением одного или более GA. В определенных вариантах осуществления, типы данных, фильтруемые в необработанных данных, ограничиваются энергией взаимодействия между лигандом и биомолекулой и/или геометрическими характеристиками лиганда в сайте связывания биомолекулы.
На фигуре 1B представлен один из подходов к фильтрации необработанных данных. В изображенном варианте осуществления данные из трех источников объединены с формированием множества 153 необработанных данных. Каждый вариант вносит свои собственные данные из всех трех источников. Объединенные данные включают в себя данные активности для взаимодействия лиганд-вариант. Данные активности, представленные блоком 141, могут быть сгенерированы с применением стандартных инструментов анализа, таких как жидкостная хроматография, газовая хроматография, и т.д. Кроме того, данные последовательности предоставлены для индивидуальных вариантов, имеющих требуемые данные активности (блок 141). Данные последовательности, представленные блоком 143, могут быть известны заранее или могут быть определены посредством секвенирования аминокислот или кодирующих нуклеиновых кислот вариантов. Секвенирование может быть выполнено с применением любой из множества доступных технологий секвенирования. Секвенирование с массовым распараллеливанием применяется в некоторых вариантах осуществления. Наконец, структурные данные могут быть сгенерированы из структурных моделей вариантов. Такая информация может быть получена с использованием не только структурных моделей, но и программы докинга (докера), которая оценивает положения лиганда в сайте связывания структурной модели рассматриваемого варианта. Необработанные структурные данные содержат данные для множества типов параметров, включая конкретные типы энергии взаимодействия и межатомные расстояния между лигандами и остатками кофактора и/или сайта связывания. Необработанные структурные данные представлены блоком 145 на фигуре 1B.
Все три источника данных объединены, как изображено на фигуре 1B, с получением объединенных необработанных данных 153. В определенных вариантах осуществления объединенные необработанные данные предоставлены в форме читаемого компьютером файла или группы файлов, которые доступны для дальнейшей обработки посредством инструментом фильтрации или реализованного компьютером алгоритма.
В изображенном варианте осуществления показаны два отдельных этапа фильтрации: выбор характеристики на этапе 155 и выбор расстояния на этапе 157. В изображенном варианте осуществления каждая из этих операций фильтрации выполняется с помощью своего собственного генетического алгоритма, использующего свою собственную модель активности последовательности в качестве целевой функции. В определенном варианте осуществления модели активности последовательности сгенерированы с помощью машин 159 и 161 опорных векторов, как изображено на фигуре 1B. Фильтр выбора характеристики идентифицирует конкретные типы энергии взаимодействия и/или межатомного расстояния для удаления из объединенного множества необработанных данных. В этом варианте осуществления понятие «расстояние» включает в себя другие геометрические параметры, такие как угловые, торсионные и общие позиционные характеристики атомов лиганда относительно атомов кофактора и/или биомолекулы. Идентифицированные типы данных удаляют для всех вариантов, вносящих вклад во множество данных. При использовании генетического алгоритма процесс удаления может быть плавным. Другими словами, один или более удаленных типов данных могут быть удалены только временно, для одного или более поколений, во время выполнения генетического алгоритма выбора характеристики. Примеры подходящих методик для выполнения этого описаны ниже. Фильтр выбора расстояния удаляет данные для определенных вариантов, которые вносят вклад в необработанные данные. Этот фильтр выбирает определенные значения энергии и/или расстояния, которые находятся вне обозначенных численных диапазонов. Для любого варианта, имеющего значения энергии и/или расстояния за пределами этих диапазонов, его данные полностью удаляются из множества необработанных данных. Когда фильтрация реализована с помощью генетического алгоритма, различные данные, удаленные в один из моментов в процессе, могут быть внесены повторно, при необходимости, во время последующего выполнения генетического алгоритма. Например, данные вариантов, удаленные во время одного поколения генетического алгоритма, могут быть внесены в более позднем поколении. Процесс будет описан более подробно ниже.
После того, как фильтрация завершена, как описано в отношении блоков 155 и 157, модель активности последовательности обучают с помощью отфильтрованных данных. В некоторых реализациях обучение выполняется с помощью машины опорных векторов. Полученная в результате модель активности последовательности изображена как блок 165. Она используется в качестве целевой функции в другом генетическом алгоритме, рассматривающем и оценивающем последовательности вариантов на основании предсказанных значений активности. Рассматриваемый генетический алгоритм изображен в блоке 167 фигуры 1B.
В некоторых других вариантах осуществления не выполняется этап выбора 155 характеристики. Поэтому ни одна из характеристик не отфильтровывается. Другими словами, все доступные характеристики используются в обучении модели активности последовательности 165 для генетического алгоритма предсказания 167. Фильтрация удаляет только варианты, имеющие энергию или геометрические значения вне идентифицированных диапазонов. В некоторых других вариантах осуществления этап 155 выбора характеристики и этап 157 выбора расстояния объединяют в один этап выбора, который может быть реализован с применением генетического алгоритма. В этих вариантах осуществления типы характеристик и значения характеристик одновременно изменяются в данных обучающего множества, оцениваемых с применением генетического алгоритма.
На фигуре 1C представлен процесс, в котором этап выбора характеристики не выполняется или объединяется с этапом 157 выбора расстояния. Как показано, необработанные данные 171 фильтруются с применением единственного генетического алгоритма 173, который выбирает варианты, имеющие один или несколько геометрических параметров, ограниченных в пределах выбранных диапазонов. В одном из примеров геометрические параметры представляют собой расстояния между атомами субстрата и атомами остатка или кофактора в сайте связывания. Например, один параметр может являться расстоянием между атомом азота на кофакторе и атомом кислорода на остатке тирозина в сайте связывания, другой параметр может являться расстоянием между карбонильным углеродом на субстрате и атомом фосфора на кофакторе, и т.д. Каждое из этих расстояний может быть установлено в пределах произвольных порогов (например, может требоваться, чтобы первое расстояние составляло менее 5 ангстрем, и второе расстояние составляло менее 7,5 ангстрем).
Функция приспособленности алгоритма 173 представляет собой точность прогнозирования моделей 175 активности последовательности, обученных с использованием различных комбинаций ограничений параметров. Таким образом, комбинации ограниченных геометрических параметров оценивают в отношении их способности обучать точные модели 175 активности последовательности. В определенных вариантах осуществления модели активности последовательности обучают с применением машин опорных векторов.
Варианты, не выбранные генетическим алгоритмом 173, удаляют из рассмотрения, в результате чего получают отфильтрованное по вариантам множество данных 177. Другими словами, результатом фильтрации посредством единственного генетического алгоритма 173 является подмножество 171 необработанных данных, содержащее только данные для подмножества вариантов в данных 171. Это подмножество используется для обучения высокоточной модели активности последовательности, которая, в свою очередь, используется в другом генетическом алгоритме, алгоритме 179 предсказания. В определенных вариантах осуществления, алгоритм 179 предсказания идентифицирует новые последовательности вариантов, предсказанные как обладающие высокой активностью. Это может быть сделано путем применения альтернативных аминокислотных (или нуклеотидных) последовательностей к обученной модели активности последовательности и определения, какие из них, вероятно, будут иметь высокие значения для выгодного свойства (например, активности для модели активности последовательности). Генетический алгоритм 179 генерирует альтернативные последовательности, которые обученная модель активности последовательности оценивает в отношении приспособленности. В итоге высокопроизводительные последовательности вариантов идентифицируют для дальнейшего исследования и/или продуцирования.
III. ОБЩЕЕ ОПИСАНИЕ ПРИМЕНЕНИЙ ГЕНЕТИЧЕСКОГО АЛГОРИТМА
Некоторые варианты осуществления предоставляют способы применения генетических алгоритмов для генерации фильтрованного множества данных для обучения модели активности последовательности, такой как модель, оптимизированная посредством машины опорных векторов (например, первый и второй генетические алгоритмы, описанные ниже). Другие варианты осуществления предоставляют способы применения генетических алгоритмов для настройки значений коэффициентов моделей активности последовательности с целью подгонки моделей к фильтрованному обучающему множеству данных. Другие варианты осуществления применяют генетический алгоритм для исследования пространства последовательностей и идентификации вариантов белка, имеющих выгодные свойства (например, третий генетический алгоритм, описанный ниже).
В генетическом алгоритме определяются подходящая функция приспособленности и подходящая процедура спаривания. Функция приспособленности предоставляет критерий определения, какие «индивиды» (модели в некоторых вариантах осуществления) являются «самыми приспособленными» в отношении наблюдаемых данных или имеют самую высокую прогнозирующую способность (т.е., модели, вероятно, предоставят наилучшие результаты). В некоторых вариантах осуществления, модель задается соотношением между одной или более независимыми переменными (IV) и зависимой переменной (DV), и соотношение описывается одним или более параметрами. Генетический алгоритм предоставляет механизм для поиска в пространствах параметров с целью нахождения комбинаций параметров или диапазонов значений параметров, генерирующих самые успешные модели.
Многие процессы в генетических алгоритмах основаны на биологических генетических операциях. Поэтому термины, используемые в генетических алгоритмах, заимствованы из биологических терминов, относящихся к генетическим операциям. В этих вариантах осуществления каждый из «индивидов» (иногда называемых элементами или хромосомами) популяции содержит «гены», представляющие все параметры, тестируемые в модели, и гены, имеющие выбранные значения для параметров в заданных диапазонах. Например, хромосома может иметь ген, представляющий наличие Gly в позиции 131.
В некоторых вариантах осуществления, генетический алгоритм может применяться для выбора подходящих IV для моделей (например, первый генетический алгоритм, описанный ниже, для фильтрации столбцов). Один из примеров такого алгоритма включает в себя гены/параметры с двоичными значениями 1 и 0, при этом каждый параметр ассоциирован с одной IV. Если параметр сходится к 0 для IV среди самых приспособленных индивидов в конце алгоритма, то IV отбрасывается из модели. Этот элемент сохраняется в обратном случае.
В некоторых вариантах осуществления, приспособленность модели измеряется с помощью прогнозирующей способности модели. В некоторых вариантах осуществления приспособленность измеряется коэффициентами попаданий на основании матрицы несоответствий, описанной ниже. В некоторых вариантах осуществления, приспособленность измеряется по AIC или BIC. Модели в этом примере могут в некоторых случаях фактически являться базовыми множествами данных, использованными для создания этих моделей.
После того, как для каждой «модели» в определенном поколении была оценена ее прогнозирующая способность, генетический алгоритм проверяется на сходимость или другие критерии (такие как фиксированное число поколений), чтобы определить, должен ли процесс продолжаться в следующем поколении. Если предположить, что генетический алгоритм еще не соответствует критерию для остановки, то производится ранжирование моделей текущего поколения. Модели, имеющие самую высокую прогнозирующую способность, могут сохраняться и использоваться в следующем поколении. Например, может использоваться уровень элитизма, составляющий 10%. Другими словами, лучшие 10% моделей (в соответствии с определенным с использованием функции приспособленности и измеренным посредством, например, точности или AIC) переносятся, чтобы стать элементами следующего поколения. Оставшиеся 90% элементов в следующем поколении получают путем спаривания «родителей» из предыдущего поколения.
Как указано, «родители» являются моделями, выбранными из предыдущего поколения. Обычно выбор взвешивается в направлении более приспособленных элементов предыдущего поколения, хотя в их выборе может присутствовать случайный компонент. Например, родительские модели могут быть выбраны с использованием линейного взвешивания (например, модель, производительность которой в 1,2 раза выше, чем у другой модели, будет выбрана с вероятностью, большей на 20%) или геометрического взвешивания (то есть, различия в предсказаниях моделей возводятся в степень для получения вероятности выбора). В некоторых вариантах осуществления, родителей выбирают путем простого выбора имеющих наилучшую производительность двух или более моделей в соответствии с ранжированием моделей в предыдущем поколении, и никакие другие модели не выбирают. В этих вариантах осуществления спариваются все выбранные модели из предыдущего поколения. В других вариантах осуществления некоторые модели из предыдущего поколения выбирают для включения в модель следующего поколения без спаривания, и другие имеющие худшую производительности модели из предыдущего поколения случайным образом выбирают в качестве родителей. Эти родители могут спариваться друг с другом и/или с имеющими лучшую производительность моделями, выбранными для включения без спаривания в следующее поколение.
После того, как было выбрано множество родительских моделей, пары таких моделей спариваются с получением моделей-потомков путем предоставления некоторых генов (значений параметров) от одного родителя и других генов (значений параметров) от другого родителя. В одном из подходов коэффициенты двух родителей выравнивают, и каждое значение рассматривают последовательно с целью определения, должен ли потомок взять элемент от родителя A или от родителя B. В одной из реализаций процесс спаривания начинается с родителя A и случайным образом определяет, должно ли событие «кроссинговера» происходить с первым рассматриваемым элементом. Если да, то элемент берется от родителя B. В противном случае элемент берется от родителя A. Следующий элемент последовательно рассматривают в отношении кроссинговера, и т.д. Элементы продолжают прибывать от родителя, отдавшего предыдущий рассмотренный элемент, пока не произойдет событие кроссинговера. В этой точке следующий элемент передается от другого родителя, и все последующие элементы будут передаваться от этого родителя, пока не произойдет другое событие кроссинговера. Для того, чтобы гарантировать, что один и тот же элемент не будет выбран в двух различных местоположениях в модели-потомке, могут применяться различные методики, например, методика кроссинговера с частичным соответствием. В некоторых вариантах осуществления, вместо того, чтобы использовать значения генов от одного из родителей, среднее значений генов может быть присвоено хромосоме-потомку.
В некоторых вариантах осуществления, генетический алгоритм также использует один или несколько механизмов мутации для генерации дальнейшего разнообразия моделей, что помогает исследовать области пространства параметров, не покрытые какими-либо существующими генами в родительском поколении. С другой стороны, механизмы мутации влияют на сходимость таким образом, что чем выше частота мутаций или больше диапазон мутаций, тем дольше алгоритм будет сходиться (если вообще сойдется). В некоторых вариантах осуществления мутация реализована посредством случайного выбора хромосомы/модели и случайного выбора параметра/гена указанной хромосомы, который затем изменяется случайным образом. В некоторых вариантах осуществления случайным образом измененные значения параметров/генов выбирают из случайного равномерного распределения с заданным диапазоном. В других вариантах осуществления случайным образом измененные значения параметров/генов выбирают из случайного нормального распределения с заданным диапазоном.
После того, как был рассмотрен каждый параметр, «модель»-потомок задается для следующего поколения. Затем другие два родителя могут быть выбраны для создания другой модели-потомка и т.д. В итоге популяция потомков в новом поколении будет готова к оценке посредством функции приспособленности способами, описанными выше.
Процесс продолжается от поколения к поколению, пока не будет выполнен критерий остановки, такой как сходимость значений. В этой точке по меньшей мере одну из имеющих самый высокий ранг моделей выбирают из текущего поколения в качестве общей наилучшей модели. Сходимость может быть протестирована посредством многих стандартных методик. В некоторых вариантах осуществления, это включает определение того, что производительность наилучшей модели не изменяется существенно в течение ряда последовательных поколений. Примеры критериев остановки включают, но не ограничиваются указанным, число поколений, сгенерированных до настоящего момента, активность лучших белков из текущей библиотеки, значения требуемой активности, и уровень улучшения, наблюдаемого в последнем поколении моделей.
IV. ВАРИАНТЫ ОСУЩЕСТВЛЕНИЯ, ПРИМЕНЯЮЩИЕ ГЕНЕТИЧЕСКИЕ АЛГОРИТМЫ ДЛЯ ФИЛЬТРАЦИИ ДАННЫХ
В некоторых вариантах осуществления присутствует два или три этапа для получения и использования модели активности последовательности из доступной информации. На каждом из этих этапов применяется генетический алгоритм. В трехэтапном процессе первый генетический алгоритм функционирует на данных из множества необработанных данных с целью выбора независимых переменных для использования в модели активности последовательности. Эти независимые переменные выбирают из пула имеющихся независимых переменных (иногда называемых параметрами). Не все имеющиеся независимые переменные используются в итоговой модели. В одном из вариантов осуществления, информация последовательности или мутации всегда используется в качестве независимой переменной, но другие типы независимых переменных выбирают посредством генетического алгоритма. Выбирают конкретную комбинацию независимых переменных, которая сделала очень хорошую работу (или, в некоторых вариантах осуществления, лучшую работу) по точному предсказанию активности. Например, может иметься от пяти до десяти доступных независимых переменных для использования в дополнение к информации последовательности, но только три из этих переменных выбирают для использования в модели активности последовательности. Генетический алгоритм идентифицирует, какая из множества альтернативных комбинаций независимых переменных делает лучшую работу по обучению модели активности последовательности для предсказания активности.
Другой генетический алгоритм идентифицирует подходящие диапазоны некоторых или всех независимых переменных в множестве данных. Диапазоны могут быть заданы порогами или значениями отсечения для независимых переменных. Этот генетический алгоритм применяется в двух- и трехэтапных процессах.
Заключительный генетический алгоритм идентифицирует последовательности биомолекулы (например, варианта белка), заслуживающие отбора или дальнейшего анализа. Данный генетический алгоритм предоставляет различные последовательности и тестирует их приспособленность с использованием модели активности последовательности, обученной с использованием фильтрованных данных, выбранных с применением одного или двух предыдущих генетических алгоритмов. Следует отметить различие между этим генетическим алгоритмом и другими генетическими алгоритмами, обсужденными в настоящем раскрытии. Этот алгоритм предоставляет последовательность нуклеиновых кислот, аминокислот или другие последовательности биомолекул в качестве индивидов в популяции. Напротив, в другом генетическом алгоритме, обсужденном в настоящем раскрытии, индивиды представляют собой модели или множества параметров модели.
В некоторых вариантах осуществления модель активности последовательности является нелинейной моделью. В других вариантах осуществления она является линейной моделью.
Как проиллюстрировано на фигуре 2, доступные данные для обучающего множества модели активности последовательности включают в себя информацию для каждой из множества вариантных биомолекул, использованных для подготовки обучающего множества. Информация для каждого варианта включает его последовательность и его активность. В различных примерах, представленных в настоящем раскрытии, активность представляет собой скорость и/или стереоселективность биомолекулы фермента в обороте субстрата. Могут использоваться другие типы активности или выгодного свойства, и некоторые из этих типов описаны в другом месте в настоящем раскрытии. Данные активности определяют с помощью анализа in vitro и/или вычислительной методики, такой как виртуальный скрининг, описанный в заявке на патент США № 61/883838 [реестр поверенного № CDXSP020P], поданной в той же день и включенной в настоящее раскрытие посредством ссылки во всей ее полноте.
В определенных вариантах осуществления, информация о последовательности может быть предоставлена как группа мутаций в исходной основе, при этом основа может представлять собой последовательность дикого типа или некоторую другую последовательностью, такую как консенсусная последовательность. Информация о последовательности, относящаяся к мутациям, может быть представлена в форме исходного остатка и остатка, на который производится замена в данной позиции. Другая альтернатива просто идентифицирует конечный остаток в определенной позиции. В различных вариантах осуществления, информация о последовательности предоставляется генетическим алгоритмом или другой вычислительной методикой, и поэтому она известна без необходимости секвенирования нуклеиновой кислоты или другой композиции. Если требуется секвенирование, может применяться любой из множества типов секвенирования. Некоторые из этих типов описаны в другом месте в настоящем раскрытии. Например, в некоторых вариантах осуществления, применяются высокопроизводительные методики для секвенирования нуклеиновых кислот.
В дополнение к данным последовательности и активности, необработанные данные содержат различные типы дополнительной информации, которая может быть включена, или не включена, в итоговое обучающее множество для модели активности последовательности. Дополнительная информация может относиться к множеству различных типов. Каждый тип потенциально служит в качестве независимой переменной для модели активности последовательности. Как объяснено в настоящем раскрытии, генетический алгоритм или другая методика оценивает целесообразность каждого типа информации.
В различных вариантах осуществления, дополнительная информация описывает характеристики связывания лиганда-рецептора. Такая информация может быть получена из измерений и/или вычисления. Как упомянуто, структурные модели вариантов могут идентифицировать значения для этих других типов информации. В одном из примеров структурная модель представляет собой модель на основе гомологии. Докер или аналогичный инструмент могут применяться для получения дополнительной информации из структурной модели. Примеры информации, сгенерированной докером, включают энергии взаимодействия и/или полные энергии, в соответствии с вычисленным посредством программы докинга, такой как программа Accelrys CDocker. Другие примеры касаются геометрических параметров, характеризующих относительную позицию лиганда или его активных функциональных составляющих или атомов относительно кофактора, остатка сайта связывания и/или другой характеристики, ассоциированной с сайтом связывания рассматриваемого варианта. Как упомянуто, часть этой информации может относиться к расстояниям, углам и/или торсионной информации об относительных позициях субстрата или промежуточного продукта и кофактора или остатка в сайте связывания. В качестве примера, значения энергии взаимодействия могут основываться на силе Ван-дер-Ваальса и/или электростатическом взаимодействии. Также может рассматриваться внутренняя энергия лиганда.
Фигура 2A-2C иллюстрирует пример фильтрации необработанного множества данных активности последовательности согласно некоторым вариантам осуществления настоящего раскрытия. На фигуре 2A показано необработанное множество данных активности последовательности для n вариантов семейства трансаминазы. Каждый вариант ассоциирован с данными активности, данными последовательности, энергетическими данными и геометрическими данными. В некоторых вариантах осуществления данные активности могут представлять собой скорость каталитической реакции, энантиоспецифичность и т.д., которые могут быть проанализированы посредством различных способов, описанных в другом месте в настоящем раскрытии. Три позиции последовательности для каждого варианта, P1, P2 и P3, предоставлены во множестве необработанных данных для включения в модель активности последовательности. Кроме того, два значения энергии, полная энергия и энергия взаимодействия, в соответствии с определенным системой виртуального докинга в другом месте в настоящем раскрытии, предоставляются для потенциального включения в модель. Наконец, пять геометрических значений предоставляются системой виртуального докинга для потенциального включения в модель. В этом примере, включающем лиганд, каждое из этих геометрических значений представляет собой расстояние между ключевым атомом лиганда, когда он состыкован с вариантом фермента, по сравнению со случаем, когда он состыкован с ферментом дикого типа. В частности, N1 обозначает атом азота, P представляет собой фосфор или фосфатную группу, C(O) представляет собой атом углерода карбоксильной группы, C(H3) представляет собой атом углерода метильной группы, и O(H) представляет собой атом кислорода гидроксильной группы.
Согласно некоторым вариантам осуществления, необработанные данные активности последовательности могут быть отфильтрованы посредством генетического алгоритма с целью исключения столбцов данных, которые являются неинформативными для обучения модели активности последовательности с высокой прогнозирующей способностью. На фигуре 2B показан пример столбцов данных, отфильтрованных генетическим алгоритмом. В этой реализации генетический алгоритм генерирует популяцию индивидов, в которой каждый индивид имеет множество «генов» или коэффициентов с двоичным значением (например, 0 и 1), указывающих, должны ли значения энергии и геометрические значения быть включены в модель активности последовательности. В примере на фигуре 2B показан результат для индивида из популяции GA, при этом индивид имеет следующие параметры: E Total=1, E Interact=1, N1=1, P=1, C(O)=0, C(H3)=1, O(H)=0. Поскольку параметр имеет значение 0, характеристика, ассоциированная с параметром, фактически исключена из модели. Этот индивид GA отфильтровывает геометрические данные C(O) и O(H), предоставляя в результате подмножество данных для обучения модели активности последовательности. В некоторых вариантах осуществления, модель активности последовательности обучают с помощью подмножества данных, включающего в себя три IV последовательности, две IV энергии и три геометрических IV. Следует отметить, что имеющие двоичные значения коэффициенты или гены GA могут быть реализованы отдельно от модели активности последовательности, в результате чего модель активности последовательности не будет содержать значения коэффициентов. В некоторых вариантах осуществления модель активности последовательности оптимизируют с помощью SVM, которая выдает попадания и непопадания для предсказанной активности. Функция приспособленности GA, определенная для каждого индивида, основывается на точности предсказания. Множество индивидов в популяции поколения GA тестируются таким же образом, как описано выше. Каждый индивид имеет ряд параметров со значениями 0 или 1, при этом параметры со значением 0 фактически отфильтровывают множество характеристик, в результате чего выдается подмножество данных для обучения модели активности последовательности. Индивиды сравниваются и ранжируются на основании их функций приспособленности. Затем один или более «самых подходящих» индивидов выбираются как родители для следующего поколения популяции с применением по меньшей мере одного механизма разнообразия, как описано в другом месте в настоящем раскрытии. В некоторых вариантах осуществления сравнение приспособленности реализовано с помощью информационного критерия Акаике (AIC) или байесовского информационного критерия (BIC), при этом индивиды, имеющие самые маленькие значения AIC или BIC, выбирают в качестве самых приспособленных индивидов. Как правило, GA повторяется для двух или более поколений, пока не будет выполнен критерий сходимости.
Следует отметить, что фильтрация столбцов является необязательной в некоторых вариантах осуществления. Согласно некоторым вариантам осуществления, необработанные данные активности последовательности могут быть отфильтрованы посредством генетического алгоритма с целью исключения строк данных вместо, или в дополнение, к фильтрации столбцов. На фигуре 2C показан пример строк данных (вариантов фермента), фильтруемых посредством генетического алгоритма. В этой реализации генетический алгоритм предоставляет популяцию индивидов, при этом каждый индивид имеет множество «генов» или коэффициентов с непрерывными значениями, показывающими пороговое значение исключения. Если значения энергии и геометрические значения выше порога для варианта, то вариант исключают из модели активности последовательности. В примере на фигуре 2C показан индивид GA, имеющий следующие пороговые значения: E Total>1,5, E Interaction>1,5, N1>3,3, P>2,8, C(O)>3,6, C(H3)>6 и O(H)>6. Эти пороговые значения приведены только в целях иллюстрации и не показывают оптимальные пороги для фактических реализаций. В этом примере данный индивид GA отфильтровывает вариант 1 и вариант 5, предоставляя подмножество данных для обучения модели активности последовательности. Следует отметить, что пороговые значения GA могут быть реализованы отдельно от модели активности последовательности, в результате чего модель активности последовательности не будет содержать пороговые значения. Как и при фильтрации столбцов, в некоторых вариантах осуществления модель активности последовательности оптимизируют с применением SVM, которая выдает попадания и непопадания для предсказанной активности. Функция приспособленности основывается на точности предсказания. Множество индивидов в GA тестируются таким же образом, как описано в примере выше. Индивиды сравниваются и оцениваются на основании их функций приспособленности. Затем один или более самых приспособленных индивидов отбирают для генерации следующего поколения популяции с применением по меньшей мере одного механизма разнообразия, как описано в другом месте в настоящем раскрытии.
В некоторых вариантах осуществления самые приспособленные индивиды, полученные из GA, показанного в примерах на фигуре 2, предоставляют подмножества данных и обучают машину опорных векторов с целью задания параметров модели активности последовательности, имеющей высокую прогнозирующую способность. В некоторых вариантах осуществления эта модель активности последовательности может управлять проектированием новых вариантов для нового цикла направленной эволюции, как подробнее описано ниже. После того, как один или несколько «наилучших моделей активности последовательности» были получены, некоторые варианты осуществления используют эти модели для управления синтезом фактических белков, которые могут быть далее разработаны посредством направленной эволюции. Некоторые варианты осуществления предоставляют способы для разработки белков с требуемой активностью посредством изменения предсказанных моделью последовательностей, как описано в другом месте в настоящем раскрытии.
A. Первый генетический алгоритм - выбор параметров
В определенных вариантах осуществления, таких как вариант осуществления, изображенный на фигуре 3А, генетический алгоритм выбирает определенные параметры из пула доступных параметров, а также информацию активности для множества вариантов. Вариант осуществления, показанный на фигуре 3А, является одним из способов реализации этапа 105 фильтрации необработанных данных с целью удаления одного или более типов энергии и/или геометрических типов в процессе, изображенном на фигуре 1А. Данные для этих параметров предоставлены в нефильтрованном множестве данных. См. блок 303 с фигуры 3A. Все данные могут быть объединены в одном или более читаемых компьютеров файлах для удобного доступа во время выполнения первого генетического алгоритма.
Для реализации первого генетического алгоритма случайным образом выбранная группа параметров из пула доступных параметров используется для предоставления первого поколения подмножеств данных. См. блок 305. Каждая совокупность параметров, которые служат совокупностями независимых переменных, задает уникальное подмножество данных. Различные случайным образом выбранные группы независимых переменных (т.е., множество отдельных подмножеств данных) применяются для обучения моделей активности последовательности. В некоторых вариантах осуществления одно и то же число независимых переменных используется для создания каждого подмножества данных. Во многих реализациях информация последовательности или мутации используется в качестве дополнительной независимой переменной в каждом подмножестве данных. Коллективно, подмножества данных составляют «индивидов» в популяции поколения генетического алгоритма.
В первом поколении генетического алгоритма моделей активности последовательности предоставлены для каждого из подмножеств данных, при этом каждая модель ассоциирована с различной случайным образом выбранной комбинацией независимых переменных. Они затем используются для предсказания активности. См. блок 307. В определенных вариантах осуществления, предсказание выполняется на последовательностях, не использовавшихся для фактического обучения модели, с тестированием прогнозирующей способности модели перекрестной проверкой. Например, неотфильтрованные данные могут быть доступны для 100 вариантов, но данные только для 70 из них используются для обучения моделей активности последовательности. Оставшиеся 30 вариантов, или, точнее, данные для этих оставшихся 30 вариантов, используются в качестве тестового множества для тестирования эффективности моделей активности последовательности, обеспечивая перекрестную проверку прогнозирующей способности модели.
Получающиеся подмножества данных, полученные во время этого первого поколения первого генетического алгоритма, ранжируют на основании их способности обучать модели, точно предсказывающие активность. См. блок 311. Ранжирование выполняется с помощью функции приспособленности, которая может рассматриваться как производительность обученных моделей. Другими словами, процесс выводит модели из необработанных данных, отфильтрованных различными способами с целью удаления различных комбинаций переменных. Модели оценивают приспособленность подмножеств данных (т.е., индивидов), которые использовались для их обучения.
Имеющие самый низкий ранг подмножества данных отражают имеющие самый низкий ранг совокупности независимых переменных, и они отклоняются до перемещения во второе поколение генетического алгоритма. Отклоненные подмножества данных заменяются подмножествами данных, полученными путем спаривания наиболее эффективных типов моделей первого поколения. См. блок 313.
Спаривание подмножеств данных может быть выполнено посредством различных методик. В основном, некоторые из выбранных независимых переменных для каждого из двух родительских подмножеств данных используются в спаривании, с тем чтобы они могли быть перенесены в подмножество данных потомков. В одном из примеров два родительских подмножества данных представлены как последовательность единиц и нулей, указывающих, используются ли определенные параметры из пула доступных независимых переменных в качестве независимых переменных в подмножествах данных. Эти двоичные представления подмножеств данных сокращаются в точке кроссинговера, и полученные в результате сегменты объединяются с комплементарными сегментами подмножества данных от другого родителя.
Функция приспособленности, или, точнее, способ оценки точности конкретной модели активности последовательности, может быть реализован различными способами. В одном из подходов функция приспособленности оценивает точность модели с помощью матрицы несоответствий. В такой методике каждый из вариантов, используемых в тестовом множестве, считается активным или неактивным в зависимости от того, больше ли его измеренная активность или меньше, чем определенный порог. Аналогично, модель активности последовательности характеризуется как предсказывающая вариант из тестового множества как являющийся активным или неактивным на основании того, предсказывает ли она значение активности как находящееся выше или ниже определенного порогового значения. Для каждого элемента тестового множества сравниваются фактические и предсказанные состояния активности элемента. Модель активности последовательности получает баллы, когда она правильно характеризует тестовый вариант как активный или как неактивный. Она теряет баллы, когда она предсказывает, что тестовый вариант является неактивным, когда в соответствии с измерениями он является активным, или когда она предсказывает, что тестовый вариант является активным, когда в соответствии с измерениями он является неактивным. Эти четыре альтернативы составляют матрицу несоответствий. Частота, с которой определенная модель правильно предсказывает активность или неактивность, используется для ранжирования подмножества данных, использованного для обучения модели. Другая опция для характеризации точности модели полагается на ошибку или различие между предсказанной ей активностью (или ее амплитудой) и фактической измеренной активностью. Это расстояние может быть просуммировано или усреднено по всем элементам тестового множества.
В конце генетического алгоритма первого поколения выбираются несколько групп независимых переменных (т.е., подмножеств данных) для моделей активности последовательности. Как упомянуто, имеющие высокий ранг подмножества данных выбирают для спаривания и/или продвижения в следующее поколение. Эти подмножества содержат выбранные структурные (например, расстояние) и/или энергетические независимые переменные в дополнение к независимой переменной последовательности.
Второе поколение подмножеств данных оценивают в отношении прогнозирующей способности моделей, обученных с их использованием. Процесс повторяется для множества поколений, пока выбор независимых переменных не будет сходиться. См. блок сходимости 309. В определенных вариантах осуществления критерий сходимости определяет, является ли улучшение текущего поколения, по сравнению с предыдущим поколением, меньшим, чем пороговый уровень для одного или более последовательных поколений. В некоторых вариантах осуществления другие способы тестирования сходимости включают, но не ограничиваются, тестирование на максимальное или минимальное значение приспособленности, такое как 100%-я приспособленность, выполнение в течение установленного числа поколений, выполнение в течение установленного временного интервала, или комбинацию указанного выше. В определенных вариантах осуществления около 5-100 подмножеств данных производятся и оцениваются в каждом поколении. В определенных вариантах осуществления, около 30-70 подмножеств данных производятся и оцениваются в каждом поколении. Не предполагается, что настоящее изобретение ограничено каким-либо конкретным числом подмножеств данных и/или поколений.
B. Второй генетический алгоритм
Во втором генетическом алгоритме, как иллюстрируется на фигуре 3B, предоставлен процесс для реализации этапа 107 с фигуры 1A для фильтрации необработанных данных, посредством чего удаляются данные для вариантов, имеющих значения энергии и/или геометрические значения вне заданных диапазонов. На фигуре 3B независимые переменные, идентифицированные в первом генетическом алгоритме, фиксируются. Невыбранные независимые переменные больше не считаются релевантными, и второй генетический алгоритм начинается с получения множества данных, отфильтрованного первым генетическим алгоритмом. См. блок 323. Можно предположить, что независимые переменные, выбранные первым генетическим алгоритмом, вероятно, вносят наибольший вклад в точное предсказание активности, по меньшей мере, с использованием рассматриваемой формы модели активности последовательности (например, n-мерная плоскость, сгенерированная машиной опорных векторов). В альтернативных вариантах осуществления первый генетический алгоритм не выполняется, и используются все независимые переменные из множества необработанных данных.
Следует понимать, что последовательности вариантов обязательно устанавливают значения дополнительных независимых переменных – переменных энергетических и структурных ограничений. Например, комбинация мутаций, присутствующая в связывающем кармане, будет задавать определенные геометрические структурные характеристики связывания и значения энергии взаимодействия, служащие доступными независимыми переменными. Тем не менее, одна только информация о последовательности может являться неподходящей для эффективного обучения модели активности последовательности для точного предсказания активности.
Во втором генетическом алгоритме каждая независимая переменная (кроме последовательности) оптимизирована таким образом, что только варианты, соответствующие пороговому значению независимой переменной, отбирают для использования в подмножестве данных. Эта оптимизации может быть применена к множеству независимых переменных, не относящихся к последовательности. Другими словами, второй генетический алгоритм выбирает поддиапазон в пределах общего доступного диапазона амплитуд для одной или более выбранных независимых переменных, не относящихся к последовательности. В качестве примера одного из подходов, заданная независимая переменная может иметь динамический диапазон от около 0 до около 20 Å, который представляет расстояние между двумя атомами или между двумя позициями стыковки одного и того же атома. Более оптимизированная версия этой независимой переменной рассматривает только варианты, имеющие значения около 12 Å или меньше. Другим примером диапазона значений может быть около 5 Å или меньше. Цель второго генетического алгоритма состоит в том, чтобы сконцентрироваться на части полного диапазона амплитуд переменных, которая полезна для предсказания активности. Это, как оказывается, уменьшает шум в прогнозирующей способности обученных моделей.
В первом поколении этого второго типа генетического алгоритма каждая из независимых переменных (кроме переменной последовательности) разделяется на части. Разделение выполняется случайным образом. См. блок 325. Например, конкретные значения амплитуды для каждой из независимых переменных выбирают случайным образом. Рассматриваются только варианты, имеющие значения, меньшие, чем данная точка разделения. Это фактически чистит независимые переменные, используемые в обучающем множестве для модели активности последовательности.
В первом поколении подмножества данных индивидов имеют случайным образом выбранные точки для каждой не относящейся к последовательности независимой переменной. Блок 325. Каждое подмножество данных индивидов в первом поколении производит обучение с использованием своей собственной уникальной модели активности последовательности. См. блок 327. Полученные в результате модели используются для предсказания активности для каждого элемента тестового множества. Блок 327. Каждое подмножество данных индивида оценивается в отношении его способности обучить точную модель посредством, например, матрицы несоответствий, как описано выше. См. блок 331. Такая оценка представляет собой функцию приспособленности. Возможны альтернативные функции приспособленности. Они включают функции, использующие величину разности между предсказанным и фактическим значением. Приспособленность может также основываться на типах независимых переменных, используемых в моделях и/или части полного диапазона используемых значений независимой переменной.
В определенных вариантах осуществления подмножество данных содержит данные для подмножества вариантов во множестве необработанных данных. Данные для части этих вариантов используются для обучения модели активности последовательности. Данные для оставшихся вариантов используются для тестирования полученной в результате модели активности последовательности. Другими словами, каждое подмножество данных разделено на обучающее множество и тестовое множество. Разделение может быть проведено посредством случайного выбора. В некоторых вариантах осуществления обучающее множество содержит от около 20 до около 90% (или от около 50 до около 80%) вариантов в подмножестве. Не предполагается, что настоящее изобретение ограничено каким-либо конкретным числом вариантов в подмножествах и/или обучающих множествах.
Имеющие высокие показатели подмножества в первом поколении выбирают для использования во втором поколении и/или в качестве родителей для спаривания с целью получения потомков для второго поколения. См. блок 333. Спаривание может проводиться с применением любой(-ых) подходящей(-их) методики(-к). В одном из вариантов осуществления схема, взвешивающая затраты, такая как взвешенная сумма различий, применяется с использованием значения отсечения (т.е., порога) для каждого из двух спаривающихся родителей для заданной независимой переменной. Во взвешивающей затраты схеме выбор спаривания смещен в направлении индивидов (т.е., подмножества данных), имеющих относительно более высокую приспособленность. Наиболее приспособленные индивиды спариваются больше, чем менее приспособленные индивиды. Другие схемы выбора спаривания включают пропорциональный выбор с помощью колеса рулетки, основанный на рангах выбор с помощью колеса рулетки и турнирный выбор.
Фактический процесс спаривания может принимать множество форм. Одним из примеров является спаривание непрерывных параметров. В этом подходе значение отсечения для заданного параметра в подмножестве данных потомка представляет собой значение, которое находится между значениями отсечения для этого же параметра в двух родительских подмножествах данных. Например, один родитель может иметь значение отсечения, равное 0,1 ангстрем, для первого параметра (расстояние X), в то время как другой родитель может иметь значение отсечения, равное 0,6 ангстрем для расстояния X. Значение отсечения потомка для расстояния X будет находиться между 0,1 и 0,6 ангстремами. Различные функции могут быть заданы для определения промежуточного значения отсечения потомка для расстояния X. В схеме спаривания с непрерывными параметрами значение «бета» выбирают случайным образом и применяют для определения дробного расстояния между двумя значениями отсечения родителей. В приведенном выше примере, если было выбрано значение бета, равное 0,7, и создается два потомка, то значения потомков могут быть вычислены следующим образом:
Расстояние потомка 1=0,1-(0,7)*0,1+(0,7)*0,6=0,45
Расстояние потомка 2=0,6+(0,7)*0,1-(0,7)*0,6=0,25
Потомок 1=a+бета*(b-a)
Потомок 2=b+бета*(a-b)
Во втором поколении индивиды (заданные подмножества данных), отобранные и/или произведенные путем спаривания в первом цикле, оценивают путем применения функции приспособленности к каждому из них. Другими словами, процесс в блоках 327, 331 и 333 применяется ко второму поколению. Как и в случае первого поколения, подмножества данных могут быть ранжированы на основании их способности обучать модели, точно предсказывающие активность в тестовом множестве вариантов. Имеющие высокий ранг подмножества могут быть перенесены в следующее поколение и/или спарены, как описано выше.
Дальнейшие поколения продолжаются таким же образом, как и в случае со вторым поколением, пока сходимость не будет достигнута. Как изображено на фигуре 3B, каждое поколение подвергается проверке сходимости. См. блок 329. В определенных вариантах осуществления критерий сходимости определяет, является ли улучшение текущего поколения, по сравнению с предыдущим поколением, меньшим, чем пороговый уровень для одного или более последовательных поколений. Другие способы тестирования на сходимость включают тестирование на максимальное/минимальное значение приспособленности, такое как 100%-я приспособленность, выполнение в течение установленного числа поколений, выполнение в течение установленного временного интервала, или комбинацию указанного выше.
В определенных вариантах осуществления около 5-100 подмножеств данных производятся и оцениваются в каждом поколении. В определенных вариантах осуществления, около 30-70 подмножеств данных производятся и оцениваются в каждом поколении. В конкретном примере около 45 подмножеств данных индивидов присутствует в каждом поколении второго генетического алгоритма. Не предполагается, что настоящее изобретение ограничено каким-либо конкретным числом подмножеств данных и/или поколений.
В некоторых аспектах этот процесс фильтрации множества данных может быть охарактеризован следующим образом. Первоначально, система использует нефильтрованное множество данных для создания популяции подмножеств данных. Каждое из этих подмножеств является «индивидом» в популяции поколения генетического алгоритма. Каждое подмножество данных идентифицируется с использованием порогов (значений отсечения) для значений параметров для геометрических параметров, характеризующих связывание лиганда с сайтом связывания биомолекулы. Когда система применяет пороги значений параметров, она фактически удаляет определенные варианты из нефильтрованного множества данных. Другими словами, каждое подмножество данных содержит данные только для некоторых вариантов, включенных в нефильтрованное множество данных.
Для каждого подмножества данных (т.е., индивида), система разделяет составляющие его варианты на те, которые принадлежат к обучающему множеству и те, которые принадлежат к тестовому множеству. Варианты, принадлежащие к обучающему множеству, используются для обучения модели активности последовательности. Обучение может быть выполнено с помощью такой методики, как машина опорных векторов или дробные наименьшие квадраты. Полученная в результате обученная модель активности последовательности применяется к вариантам тестового множества. Модель предсказывает активность для каждого варианта тестового множества, и система, таким образом, оценивает точность модели активности последовательности и, следовательно, ассоциированного с ней подмножества данных. Каждое подмножество данных (т.е., индивид) в популяции поколения генетического алгоритма оценивают в отношении точности таким же образом.
Для заданного поколения генетического алгоритма каждое из подмножеств данных и ассоциированных моделей активности последовательности оценивают на основании их способности точно предсказывать активность для вариантов в ассоциированном тестовом множестве. В пределах поколения, процесс выбирает имеющие высокий ранг подмножества для продвижения в следующее поколение. Кроме того, процесс спаривает некоторые имеющие высокий ранг подмножества для создания подмножеств-потомков, которые также предоставлены для следующего поколения. Подмножества данных следующего поколения (т.е., индивиды) обрабатываются, как описано выше. Множество поколений обрабатывается и оценивается до тех пор, пока сходимость не будет достигнута.
C. Третий генетический алгоритм
В описанном потоке операций подмножество данных, отобранное путем фильтрации необработанных данных последовательности, активности и структуры, обучает высокоточную модель активности последовательности. Машина опорных векторов может применяться для выполнения обучения. Полученная в результате модель активности последовательности идентифицирует новые вариантные биомолекулы. В некоторых вариантах осуществления, эти новые вариантные биомолекулы используются по меньшей мере в одном цикле направленной эволюции. В определенных вариантах осуществления заключительный генетический алгоритм применяется для идентификации новых вариантов биомолекул, описанной в блоке 111 с фигуры 1A. Пример подходящего генетического алгоритма изображен на фигуре 3C. Как показано там, процесс начинается с модели активности последовательности, выбранной после завершения второго генетического алгоритма. Блок 353.
Как подчеркнуто выше, существует различие между этим генетическим алгоритмом и другими генетическими алгоритмами, обсужденными в настоящем раскрытии. Этот алгоритм предоставляет последовательности нуклеиновых кислот, аминокислот или других биомолекул в качестве индивидов в популяции. Напротив, в другом генетическом алгоритме, обсужденном в настоящем раскрытии, индивиды представляют собой модели или множества параметров модели. В первом поколении этого GA генетический алгоритм предоставляет случайную популяцию индивидов, каждая из которых представляет различную последовательность белка (или другой биомолекулы). Блок 355. Отдельные белки отличаются друг от друга мутациями в заданных позициях. В некоторых реализациях мутации генерируют случайным образом, по меньшей мере, в первом поколении. Мутации могут быть сгенерированы относительно единственного основного белка, такого как основа белка дикого типа или опорная основа, идентифицированная во время цикла направленной эволюции.
Индивиды в первом поколении оценивают или выбирают с использованием функции приспособленности, которая представляет собой модель активности последовательности, обученную на подмножестве данных, полученном в конце второго генетического алгоритма (т.е., модель, переданную в блоке 353). См. блоки 357 и 359. Идентифицирующая информация последовательности для каждой биомолекулы индивида вводится в модель активности последовательности. Эта информация может представлять собой список мутаций, необязательно, идентифицирующий и начальный, и конечный остатки для каждой из позиций, в которых находятся мутации. Модель воздействует на эти входные данные путем присвоения предсказанной активности каждому индивиду. Блок 357. Биомолекулы индивидов, имеющие высокий ранг значения активности (как предсказано моделью), выбирают для спаривания и/или для передачи в следующее поколение. Блоки 359 и 363. Спаренные индивиды обеспечивают новые комбинации мутаций, при этом каждая новая комбинация является элементом следующего поколения. В определенных вариантах осуществления спаривание выполняется посредством операции кроссинговера. Пример операции кроссинговера в этом генетическом алгоритме может быть понят следующим образом. Родитель 1 имеет мутации в позициях 12 и 25, и родитель 2 имеет мутации в позициях 15 и 30. Первый потомок может иметь мутации в позиции 12 от родителя 1 и в позиции 30 от родителя 2, и второй потомок будет иметь мутации в позициях 25 от родителя 1 и позиции 12 от родителя 2.
В некоторых случаях некоторые потомки, произведенные путем спаривания (например, 20% из них) подвергаются дополнительным мутациям с помощью любого подходящего способа, включая точечные мутации, но не ограничиваясь ими. Такие мутации могут быть выполнены случайным образом.
Последующие поколения популяций различных биомолекул получают таким образом, как описано для второго поколения. Создание новых поколений повторяется до тех пор, пока активность, предсказанная моделью, не перестанет значительно улучшаться в течение определенного числа поколений. В этой точке популяцию биомолекул считают сходящейся к итоговому списку ранжированных индивидов, которые идентифицируются по множеству мутаций и предсказанной активности. Условие сходимости показано в блоке 361 на фигуре 3C.
В определенных вариантах осуществления биомолекулы индивидов из заключительного списка синтезируют и подвергают скринингу in vitro. Кроме того, отдельные биомолекулы могут быть проанализированы с целью предоставления геометрических ограничений или других структурных данных и/или энергии взаимодействия посредством применения программного обеспечения докинга или других инструментов. Полученные в результате данные последовательности, активности, и структурные/энергетические данные затем объединяют, и они служат входными данными для потока операций следующего цикла направленной эволюции. Другими словами, белки, подвергнутые скринингу после генетического алгоритма, предоставляют данные, которые могут служить новым обучающим множеством для второго цикла анализа. Таким образом, фильтрующий данные генетический алгоритм выполняется снова, но с полностью новым обучающим множеством. В некоторых вариантах осуществления множество данных и модель активности последовательности от одного цикла направленной эволюции не сохраняются в следующем цикле. Т.е., следующий цикл запускается заново и ищет новое множество независимых переменных с использованием нового нефильтрованного множества данных.
В некоторых вариантах осуществления, модель активности последовательности, используемую в третьем генетическом алгоритме, обучают с использованием энергетических и/или структурных (геометрических) параметров, а также информации последовательности. В определенных реализациях, однако, заключительный генетический алгоритм вводит в модель только информацию последовательности, но не энергетическую и/или структурную информацию. Другими словами, в то время как модель была разработана с использованием независимых переменных последовательности и энергии и/или структуры, модель не получает энергетические и/или структурные независимые переменные при оценке новых последовательностей в третьем генетическом алгоритме.
В определенных вариантах осуществления, от около 10 до около 10000 биомолекул оценивают в каждом поколении. В определенных вариантах осуществления от около 100 до около 1000 биомолекул оценивают в каждом поколении. В конкретном примере присутствует около 500 биомолекул индивидов в каждом поколении третьего генетического алгоритма. Не предполагается, что настоящее изобретение ограничено каким-либо конкретным числом оцениваемых биомолекул.
В некоторый момент вышеописанный процесс завершается, и один или несколько вариантов из текущего поколения выбирают для дальнейшего исследования, синтеза, разработки, продуцирования, и т.д. В одном из примеров выбранный вариант биомолекулы используется для запуска одного или более циклов направленной эволюции in vitro. Например, цикл направленной эволюции in vitro может включать в себя (i) подготовку множества олигонуклеотидов, содержащих или кодирующих, по меньшей мере, часть выбранного варианта белка, и (ii) выполнение цикла направленной эволюции in vitro с использованием множества олигонуклеотидов. Олигонуклеотиды могут быть подготовлены посредством синтеза генов, фрагментации нуклеиновой кислоты, кодирующей часть или весь выбранный вариант белка, и т.д. В определенных вариантах осуществления, цикл направленной эволюции in vitro включает в себя фрагментацию и повторное объединение множества олигонуклеотидов. В определенных вариантах осуществления, цикл направленной эволюции in vitro включает в себя выполнение насыщающего мутагенеза на множестве олигонуклеотидов.
V. МОДЕЛИ АКТИВНОСТИ ПОСЛЕДОВАТЕЛЬНОСТИ
Способы и системы, раскрытые в настоящем раскрытии, предоставляют модель активности последовательности с высокой прогнозирующей способностью. В некоторых вариантах осуществления модель активности последовательности является нелинейной моделью. В других вариантах осуществления она является линейной моделью. Примеры линейных и нелинейных моделей активности последовательности описаны в патенте США № 7747391, публикации заявки на патент США № 2005/0084907, предварительной заявке на патент США № 61/759276 и предварительной заявке на патент США № 61/799377, каждая из которых включена в настоящее раскрытие посредством ссылки во всей своей полноте. В различных вариантах осуществления, описанных в настоящем раскрытии, модель активности последовательности реализуется как n-мерная гиперплоскость, которая может быть сгенерирована посредством машины опорных векторов. В приведенном ниже описании в случае, когда модель активности последовательности иллюстрируется как n-мерная гиперплоскость, сгенерированная машиной опорных векторов, предполагается, что эта форма или модель могут быть заменены на другие типы линейных и нелинейных моделей, такие как модели наименьших квадратов, модели дробных наименьших квадратов, многократная линейная регрессия, регрессия главных компонент, регрессия дробных наименьших квадратов, машина опорных векторов, нейронная сеть, Байесовская линейная регрессия или бутстреп, и совокупности указанного выше.
Как обозначено выше, в некоторых вариантах осуществления, модель активности последовательности, используемая в вариантах осуществления по настоящему раскрытию, сопоставляет информацию последовательности белка с активностью белка. Информация последовательности белка, используемая моделью, может принимать множество форм. В некоторых вариантах осуществления она представляет собой полную последовательность аминокислотных остатков в белке. Однако в некоторых вариантах осуществления полная аминокислотная последовательность не является необходимой. Например, в некоторых вариантах осуществления достаточно предоставить только те остатки, которые должны изменяются в конкретной научно-исследовательской работе. В некоторых вариантах осуществления, включающих в себя более поздние этапы исследования, многие остатки фиксируются, и только ограниченные области пространства последовательностей остаются для исследования. В некоторых таких ситуациях удобно предоставить модели активности последовательности, которые требуют, в качестве входных данных, только идентификацию этих остатков в областях белка, в которых продолжается исследование. В некоторых дополнительных вариантах осуществления модели не требуют, чтобы точные идентификаторы остатков в представляющих интерес позициях остатков были известны. В некоторых таких вариантах осуществления идентифицируют одно или более физических или химических свойств, характеризующих аминокислоту в определенной позиции остатка. В некоторых вариантах осуществления, геометрические параметры, описывающие структурную информацию, например, расстояния между функциональными составляющими, включают в модель. Несмотря на то, что структурная информация может быть реализована в структурной модели, она может также быть реализована как часть модели активности последовательности. Альтернативно, структурная информация может использоваться для фильтрации данных с целью выбора подмножества данных активности последовательности для обучения модели активности последовательности.
Кроме того, в некоторых моделях используются комбинации таких свойств. Фактически, не предполагается, что настоящее изобретение ограничено каким-либо конкретным подходом, поскольку модели находят применение в различных конфигурациях информации о последовательности, информации об активности, информации о структуре и/или других физических свойств (например, гидрофобности, и т.д.).
В некоторых вариантах осуществления, описанных выше, аминокислотные последовательности предоставляют информацию для независимых переменных для моделей активности последовательности. В других вариантах осуществления последовательности нуклеиновых кислот, в противоположность аминокислотным последовательностям, предоставляют информацию для независимых переменных. В последних вариантах осуществления IV, представляющие наличие или отсутствие нуклеотидов определенных типов в определенных позициях нуклеотидных последовательностей, используется в качестве входных данных для модели. Белки, выведенные из нуклеотидных последовательностей, предоставляют данные активности в качестве выходных данных модели. Специалисту в данной области техники будет понятно, что различные нуклеотидные последовательности могут быть оттранслированы в одну и ту же аминокислотную последовательность вследствие вырожденности кодонов, при которой два или более различных кодонов (т.е., триплеты нуклеотидов) кодируют одну и ту же аминокислоту. Поэтому различные нуклеотидные последовательности могут потенциально относиться к одному и тому же белку и активности белка. Однако модель активности последовательности, берущая информацию нуклеотидной последовательности в качестве входных данных, и выдающая активность белка в качестве выходных данных, не обязательно должна рассматривать эту вырожденность. Практически, отсутствие взаимно-однозначного соответствия между входными и выходными данными может внести шум в модель в некоторых вариантах осуществления, но такой шум не нивелирует полезность модели. В некоторых вариантах осуществления, такой шум может даже улучшить прогнозирующую способность модели, поскольку, например, вероятность переобучения модели на данных будет меньше. В некоторых вариантах осуществления, модели обычно обрабатывают активность как зависимую переменную и значения последовательности/остатка как независимые переменные. Данные активности могут быть получены с помощью любых подходящих средств, известных в технике, включая, но не ограничиваясь указанным, анализы и/или скрининги, разработанные соответствующим образом для измерения амплитуд представляющей(-их) интерес активности(-ей). Такие способы известны специалистам в данной области техники и не являются существенными для настоящего изобретения. Действительно, принципы для разработки соответствующего анализа или скрининга широко известны и понятны в технике. Способы для получения последовательностей белка также являются хорошо известными и не являются ключевыми для настоящего изобретения. Как упомянуто, могут применяться технологии секвенирования следующего поколения. В некоторых вариантах осуществления представляющая интерес активность может являться стабильностью белка (например, тепловой стабильностью). Однако во многих важных вариантах осуществления рассматриваются другие активности, такие как каталитическая активность, устойчивость к патогенам и/или токсинам, терапевтическая активность, токсичность, и т.п. Действительно, не предполагается, что настоящее изобретение ограничено каким-либо конкретным способом анализа/скрининга и/или методом(-ами) секвенирования, поскольку любой подходящий способ, известный в технике, находит применение в настоящем изобретении.
В различных вариантах осуществления форма модели активности последовательности может значительно изменяться, пока она предоставляет механизм для корректной аппроксимации относительной активности белков на основании информации последовательности, в соответствии с требованиями. Примеры математической/логической формы моделей включают, но не ограничиваются, аддитивные, мультипликативные, линейные/без взаимодействия и нелинейные/с взаимодействием математические выражения различных порядков, нейронные сети, классификационные и регрессионные деревья/графы, подходы кластеризации, рекурсивное разделение, машины опорных векторов, и т.п.
Различные методики для создания моделей доступны и находят применение в настоящем изобретении. В некоторых вариантах осуществления методики включают в себя оптимизацию моделей или минимизацию ошибок модели. Конкретные примеры включают дробные наименьшие квадраты, множественную регрессию, случайный лес, различные другие методы регрессии, а также методики нейронных сетей, рекурсивное разделение, методики опорных векторов, CART (деревья классификации и регрессии), и/или т.п. Обычно, методика должна произвести модель, которая может отличить остатки, которые оказывают значительное влияние на активность от тех, которые не оказывают. В некоторых вариантах осуществления модели также упорядочивают индивидуальные остатки или положения остатков на основании их воздействия на активность. Не предполагается, что настоящее изобретение ограничено каким-либо конкретным способом для создания модели, поскольку любой соответствующий способ, известный в технике, находит применение в настоящем изобретении.
В некоторых вариантах осуществления модели создаются посредством методики регрессии, которая идентифицирует ковариацию независимых и зависимых переменных в обучающем множестве. Различные методики регрессии известны и широко применяются. Примеры включают множественную линейную регрессию (MLR), регрессию главных компонент (PCR) и регрессия дробных наименьших квадратов (PLS). В некоторых вариантах осуществления модели создаются с применением методик, которые включают в себя множество составных частей, включая, множественную регрессию и случайный лес, но не ограничиваясь указанным. Эти и любые другие соответствующие методики находят применение в настоящем изобретении. Не предполагается, что настоящее изобретение ограничено какой-либо конкретной методикой.
MLR является самой простой из этих методик. Она применяется просто для решения множества уравнений относительно коэффициентов обучающего множества. Каждое уравнение относится к активности элемента обучающего множества (то есть, зависимым переменным) при наличии или отсутствии конкретного остатка в конкретном положении (то есть, независимые переменные). В зависимости от количества возможных вариантов остатка в обучающем множестве число этих уравнений может быть довольно большим.
Как и MLR, и PCR создают модели по уравнениям, связывающим активность последовательности со значениями остатков. Однако данные методики делают это другим образом. Они сначала выполняют преобразование координат с целью сокращения числа независимых переменных. Затем они выполняют регрессию на преобразованных переменных. В MLR имеется потенциально очень большое количество независимых переменных: две или более для каждого положения остатка, которое изменяется в пределах обучающего множества. С учетом того, что белки и интересующие пептиды часто являются достаточно большими, и обучающее множество может предоставлять множество различных последовательностей, количество независимых переменных может быстро стать очень большим. В результате сокращения количества с целью сосредоточения на тех, которые обеспечивают большую часть изменчивости в множестве данных, и PCR обычно требуют меньшего количества выборок и упрощают этапы, включенные в создание моделей.
PCR аналогична регрессии PLEASE в том, что фактическая регрессия выполняется на относительно небольшом количестве скрытых переменных, полученных посредством преобразования координат необработанных независимых переменных (то есть, значений остатков). Различие между PLEASE и PCR заключается в том, что скрытые переменные в PCR создаются посредством максимизации ковариации между независимыми переменными (то есть, значениями остатка). В регрессии PLEASE скрытые переменные создаются таким образом, чтобы максимизировать ковариацию между независимыми переменными и зависимыми переменными (то есть, значениями активности). Регрессия дробных наименьших квадратов описана в Hand, D.J., и соавт. (2001) Principles of Data Mining (Adaptive Computation and Machine Learning), Boston, MA, MIT Press, and in Geladi, et al. (1986) «Partial Least-Squares Regression: a Tutorial», Analytica Chimica Acta, 198:1-17. Обе эти ссылки включены в настоящее раскрытие посредством ссылки для всех целей.
В PCR и непосредственным результатом регрессионного анализа является выражение для активности, которое представляет собой функцию взвешенных скрытых переменных. Это выражение может быть преобразовано к выражению для активности как функции исходных независимых переменных посредством выполнения преобразования координат, которое преобразует скрытые переменные обратно в исходные независимые переменные.
По существу, и PCR, и сначала понижают размерность информации, содержавшейся в обучающем множестве, и затем выполняют регрессионный анализ преобразованного множества данных, которое было преобразовано для создания новых независимых переменных, но при этом сохраняет исходные значения зависимой переменной. Преобразованные версии множеств данных могут давать в результате только относительно немного выражений для выполнения регрессионного анализа. В протоколах, в которых какое-либо снижение размерности не выполнялось, необходимо рассмотреть каждый отдельный остаток, который может быть изменен. В результате может быть получено очень большое множество коэффициентов (например, 2 Н коэффициентов для двусторонних взаимодействий, где N представляет собой количество положений остатка, которые могут измениться в обучающем множестве). В типичном анализе главных компонентов используется только 3, 4, 5, 6 главных компонентов. Однако, не предполагается, что настоящее изобретение ограничено каким-либо конкретным числом главных компонентов.
Способность методов машинного обучения подгонять обучающие данные часто называют «подгонкой модели» и в методиках регрессии, таких как MLR, PCR и, подгонку модели обычно измеряют по сумме квадратов разностей между измеренными и предсказанными значениями. Для данного обучающего множества оптимальная подгонка модели будет достигнута с применением MLR, при этом PCR и часто дают худшую подгонку модели (выше сумма квадратичной ошибки между измерениями и предсказаниями). Однако, главное преимущество использования методик регрессии скрытой переменной, таких как PCR и, заключается в прогнозирующей способности таких моделей. Получение подгонки модели с очень небольшой суммой квадратичной ошибки никоим образом не гарантирует, что модель будет в состоянии точно предсказывать новые образцы, не присутствующие в обучающем множестве - фактически, часто происходит противоположное, особенно когда имеется много переменных и только несколько наблюдений (то есть, образцов). Таким образом, методики регрессии скрытой переменной (например, PCR,), хотя и часто имеют худшую подгонку модели на обучающих данных, обычно являются более гибкими и способными предсказывать новые образцы вне обучающего множества более точно.
Метод опорных векторов (SVM) также может применяться для генерации моделей по настоящему изобретению. Как объяснено выше, SVM берут множества данных обучения для последовательностей, которые были классифицированы по двум или более группам на основании активностей в качестве входных данных. Методы опорных векторов функционируют посредством взвешивания элементов обучающего множества по-разному в зависимости от того, насколько они близко к границе гиперплоскости, разделяющей «активные» и «неактивные» элементы обучающего множества. В данной методике требуется, чтобы ученый сначала решил, какие элементы обучающего множества поместить в «активную» группу и какие элементы обучающего множества поместить в «неактивную» группе. В некоторых вариантах осуществления это выполняется посредством выбора соответствующего числового значения для уровня активности, которое служит границей между «активными» и «неактивными» элементами обучающего множества. По этой классификации метод опорных векторов генерирует вектор, W, который может предоставить значения коэффициентов для индивидуальных независимых переменных, определяющих последовательности для членов активных и неактивных групп в обучающем множестве. Эти коэффициенты могут использоваться, чтобы «упорядочить» индивидуальные остатки, как описано в другом месте настоящего раскрытия. Методика применяется для идентификации гиперплоскости, которая максимизирует интервал между самыми близкими элементами обучающего множества на противоположных сторонах этой плоскости.
VI. ДОКИНГ БЕЛКОВ
В некоторых вариантах осуществления система виртуального докинга или скрининга белков сконфигурирована для выполнения различных операций, связанных с вычислительной идентификацией вариантов биомолекулы, которые, вероятно, будут обладать требуемой активностью, например, эффективно и селективно катализировать реакцию при определенной температуре. Система виртуального докинга белков может получать в качестве входных данных представления по меньшей мере одного лиганда, который предназначен для взаимодействия с вариантами. Система может получать в качестве других входных данных представления вариантов биомолекулы или, по меньшей мере, сайтов связывания этих вариантов. Представления могут содержать трехмерные позиции атомов и/или функциональных составляющих лигандов и/или вариантов. Модели на основе гомологии являются примерами представлений вариантов биомолекулы. В некоторых вариантах осуществления система виртуального скрининга белков может применять информацию докинга и ограничения активности для оценки функционирования этих вариантов.
В определенных вариантах осуществления, система виртуального докинга и скрининга белков определяет одно или более значений энергии и одно или более геометрических значений в отношении взаимосвязей между функциональными составляющими на двух различных молекулах. В некоторых вариантах осуществления, значения энергий могут включать в себя энергию взаимодействия между субстратом и ферментом, при этом субстрат в одном или более положениях состыковался с ферментом. В некоторых вариантах осуществления значения энергии могут включать в себя общую энергию стыковки, включая энергию взаимодействия и внутреннюю энергию участников связывающего взаимодействия. В некоторых вариантах осуществления, геометрические значения могут включать в себя расстояние, угол или торсионные значения между функциональными составляющими двух молекул. В некоторых вариантах осуществления, геометрические значения включают в себя расстояние между соответствующими функциональными составляющими на нативном и требуемом субстрате, когда они оба состыкованы с одним и тем же ферментом. В других вариантах осуществления геометрические значения включают в себя расстояние между субстратом и ферментом, состыкованными друг с другом.
При рассмотрении каталитического оборота субстрата в качестве активности, система виртуального скрининга белков может быть сконфигурирована для идентификации положений, о которых, известно, что они ассоциированы с конкретной реакцией. В некоторых вариантах осуществления это включает в себя рассмотрение промежуточного продукта реакции или переходного состояния, а не субстрата непосредственно. В дополнение к обороту, положения могут быть оценены для других типов активности, таких как синтез стереоселективных зеркальных изомеров, связывание с рецептором целевой биомолекулы, идентифицированной как важной для поиска новых лекарственных средств, и т.д. В некоторых случаях, активность представляет собой необратимую или обратимую ковалентную связь, такую как нацеленное ковалентное ингибирование (TCI).
В определенных вариантах осуществления протокол для вычисления энергии связывания выполняется для оценки энергетики каждого активного положения варианта. В некоторых реализациях протокол может рассматривать силу Ван-дер-Ваальса, электростатическое взаимодействие и энергию сольватации. Сольватацию обычно не рассматривают в вычислениях, выполняемых докерами. Имеются различные модели сольватации для вычисления энергий связывания. Они включают в себя, но не ограничиваются указанным, зависимые от расстояния диэлектрики, обобщенную модель Борна с попарным суммированием (GenBorn), обобщенную модель Борна с неявной мембраной (GBIM), обобщенную модель Борна с интегрированием молекулярного объема (GBMV), обобщенную модель Борна с простым переключением (GBSW) и уравнение Пуассона-Больцмана с площадью неполярной поверхности (PBSA). Протоколы для вычисления энергии связывания отличаются или являются отдельными от программ докера. Они обычно выдают результаты, которые являются более точными, чем показатели докинга, отчасти вследствие включения эффектов сольватации в их вычисления. В различных реализациях энергии связывания вычисляют только для положений, которые считаются активными.
A. Структурные модели биомолекул и их сайтов связывания
В определенных вариантах осуществления компьютерная система предоставляет трехмерные модели для вариантов белка (или других биомолекул). Трехмерные модели являются вычислительными представлениями некоторых или всех полноразмерных последовательностей вариантов белка. Обычно, как минимум, представления вычисления покрывают, по меньшей мере, сайты связывания вариантов белка.
Как описано в настоящем раскрытии, трехмерные модели могут представлять собой модели на основе гомологии, подготовленные с применением соответствующим образом спроектированной вычислительной системы. Трехмерные модели используют шаблон структуры, в которой варианты белка отличаются друг друга по своим аминокислотным последовательностям. Обычно, шаблон структуры представляет собой структуру, ранее определенную посредством рентгеноструктурной кристаллографии или NMR для последовательности, которая гомологична последовательности модели. Качество модели на основе гомологии зависит от идентичности последовательности и разрешения шаблона структуры. В определенных вариантах осуществления трехмерные модели могут храниться в базе данных для использования в случае необходимости для текущих или будущих проектов.
Трехмерные модели вариантов белка могут быть получены посредством методик, отличных от моделирования на основе гомологии. Одним из примеров является протягивание белка, для которого также требуется шаблон структуры. Другим примером является моделирование белка с нуля, или моделирование de novo, для которого не требуется шаблон структуры, и которое основано на базовых физических принципах. Примеры методик с нуля включают моделирование молекулярной динамики и моделирования с применением пакета программного обеспечения Розетта.
В некоторых вариантах осуществления варианты белка отличаются друг от друга в своих сайта связывания. В некоторых случаях, сайты связывания отличаются от друг друга по меньшей мере одной мутацией в аминокислотной последовательности сайта связывания. Мутация может быть сделана в последовательности белка дикого типа или некоторой другой опорной последовательности белка. В некоторых случаях, два или более вариантов белка имеют одну и ту же аминокислотную последовательность для сайта связывания, но отличаются по аминокислотной последовательности для другой области белка. В некоторых случаях, два варианта белка отличаются от друг друга по меньшей мере приблизительно 2 аминокислотами, или, по меньшей мере, приблизительно 3 аминокислотами, или, по меньшей мере, приблизительно 4 аминокислотами. Однако, не предполагается, что настоящее изобретение ограничено конкретным числом различий в аминокислотах между вариантами белка.
В определенных вариантах осуществления множество вариантов включает в себя элементы библиотеки, произведенной посредством одного или более циклов направленной эволюции. Способы генерации разнообразия, применяемые в направленной эволюции, включают в себя перетасовку генов, мутагенез, рекомбинацию и т.п. Примеры методик направленной эволюции описаны в патенте США № 7024312, публикации заявки на патент США № 2012/0040871, патенте США № 7981614, WO2013/003290, заявке на патент № PCT/US2013/030526, каждая из которых включена в настоящее раскрытие посредством ссылки во всей ее полноте.
B. Докинг лиганда с вариантами белка
Как объясняется в настоящем раскрытии, докинг может применяться для идентификации энергии взаимодействия и/или геометрических параметров для использования в обучении моделей активности последовательности. Обычно докинг проводится посредством соответствующим образом запрограммированной компьютерной системой, которая использует вычислительное представление лиганда и вычислительные представления сайтов связывания сгенерированного множества вариантов.
Например, докер может быть сконфигурирован для выполнения некоторых или всех следующих операций:
1. генерации множества конформаций лиганда с использованием высокотемпературной молекулярной динамики со случайными точками запуска. Докер может генерировать такие конформации без рассмотрения среды лиганда. Следовательно, докер может идентифицировать благоприятные конформации посредством рассмотрения только силы сцепления или других соображений, специфичных только для лиганда. Число конформаций, которые будут сгенерированы, может быть задано произвольно. В одном из вариантов осуществления генерируется по меньшей мере около 10 конформаций. В другом варианте осуществления генерируется по меньшей мере около 20 конформаций, или, по меньшей мере, около 50 конформаций, или, по меньшей мере, около 100 конформаций. Однако, не предполагается, что настоящее изобретение ограничено конкретным числом конформаций.
2. генерации случайных ориентаций конформаций посредством переноса центра лиганда в заданное местоположение в пределах активного центра рецептора и выполнения ряда случайных вращений. Число ориентаций, которые будут оптимизироваться, может быть задано произвольно. В одном из вариантов осуществления генерируется по меньшей мере около 10 ориентаций. В другом варианте осуществления генерируется по меньшей мере около 20 ориентаций, или, по меньшей мере, около 50 ориентаций, или, по меньшей мере, около 100 ориентаций. Однако, не предполагается, что настоящее изобретение ограничено каким-либо конкретным числом ориентаций. В определенных вариантах осуществления докер вычисляет «менее жесткую» энергию с целью генерации дальнейших комбинаций ориентации и конформации. Докер вычисляет менее жесткую энергию с использованием физически нереалистичных предположений о допустимости определенных ориентаций в сайте связывания. Например, докер может предположить, что атомы лиганда и атомы сайта связывания могут занимать, по существу, одно и то же место, что является невозможным на основании отталкивания Паули и стерических ограничений. Это менее жесткое предположение может быть реализовано, например, с применением ослабленной формы потенциала Леннарда-Джонса при исследовании пространства конформаций. При использовании вычисления менее жесткой энергии докер позволяет более полно исследовать конформации, чем при использовании только физически реалистичных энергетических соображений. Если менее жесткая энергия конформации в конкретной ориентации меньше, чем указанный порог, то конформация-ориентация сохраняется. Эти низкоэнергетические конформации сохраняются как «положения». В определенных реализациях данный процесс продолжается, пока не будет найдено требуемое число низкоэнергетических положений, или пока не будет найдено максимальное число плохих положений.
3. подвергания каждого сохраненного положения с этапа 2 процедуре молекулярной динамики с имитацией отжига в целях оптимизации положения. Температуру увеличивают до высокого значения, и затем охлаждают до целевой температуры. Докер может сделать это в целях обеспечения физически более реалистичной ориентации и/или конформации, чем предоставляемая посредством вычисления менее жесткой энергии.
4. выполнения итоговой минимизации лиганда в жестком рецепторе с использованием несмягченного потенциала. Это обеспечивает более точное значение энергии для сохраненных положений. Однако, вычисление может предоставлять только частичную информацию об энергиях положений.
5. для каждого конечного положения, вычисления полной энергии (энергия взаимодействия рецептор-лиганд плюс сила сцепления лиганда) и только энергии взаимодействия. Вычисление может быть выполнено с использованием CHARMm. Положения сортируются по энергии CHARMm и сохраняются положения, имеющие наилучшие значения (имеющие наибольшие отрицательные значения, то есть, благоприятные для связывания). В некоторых вариантах осуществления на этом этапе (и/или этапе 4) удаляются положения, которые являются энергетически неблагоприятными.
По следующей ссылке предоставлен пример функционирования докера: Wu и соавт., Detailed Analysis of Grid-Based Molecular Docking: A Case Study of CDOCKER – A CHARMm-Based MD Docking Algorithm, J. Computational Chem., Vol. 24, No. 13, pp 1549-62 (2003), которая включена в настоящее раскрытие посредством ссылки во всей ее полноте.
Докер, такой как описан в настоящем раскрытии, может предоставлять такую информацию, как идентификаторы вариантов, для которых стыковка с требуемым субстратом маловероятна, множества положений (одно множество для каждого варианта), которые можно рассмотреть в отношении активности, и энергии взаимодействия для положений в множествах.
C. Определение геометрических параметров состыкованного лиганда
Для варианта белка, которые успешно состыковался с лигандом, геометрические параметры связывания могут идентифицировать одно или более активных положений. Активное положение представляет собой положение, соответствующее одному или более ограничениям, обеспечивающим связывание лиганда при заданных условиях (а не при произвольных условиях связывания). Если лиганд является субстратом, и белок является ферментом, то активное связывание может представлять собой связывание, которое позволяет субстрату подвергаться катализируемому химическому превращению, в частности, стереоспецифическому превращению. В некоторых реализациях геометрические характеристики связывания задают относительные позиции одного или более атомов в лиганде и одного или более атомов в белке и/или кофакторе, ассоциированном с белком.
В некоторых случаях геометрические параметры идентифицируют по одной или более конформациям нативного субстрата и/или последующего промежуточного продукта, когда он подвергается катализируемому химическому превращению посредством фермента дикого типа. В определенных вариантах осуществления геометрические параметры включают в себя (i) расстояние между конкретной функциональной составляющей на субстрате и/или последующим промежуточным продуктом и конкретным остатком или функциональной составляющей остатка в каталитическом центре, (ii) расстояние между конкретной функциональной составляющей на субстрате и/или последующем промежуточном продукте и конкретным кофактором в каталитическом центре, и/или (iii) расстояние между конкретной функциональной составляющей на субстрате и/или последующем промежуточном продукте и конкретной функциональной составляющей на идеально размещенном нативном субстрате, и/или последующем промежуточном продукте в каталитическом центре. Альтернативы расстоянию включают углы между связями или расположениями атомов между соединениями, торсионные позиции вокруг общей оси, и т.д. Примеры этих геометрических параметров описаны в заявке на патент США № 61/883838 США [реестр поверенного № CDXSP020P], поданной в той же день и включенной в настоящее раскрытие посредством ссылки во всей ее полноте.
Множество положений вычислительного представления субстрата и/или последующего промежуточного продукта может быть сгенерировано в отношении вычислительного представления рассматриваемого варианта белка. Множество положений может быть сгенерировано с помощью различных методик. Общие примеры таких методик включают в себя, но не ограничиваются указанным, систематические или стохастические торсионные поиски вокруг поворотных связей, моделирование молекулярной динамики и генетические алгоритмы, разработанные для обнаружения низкоэнергетических конформаций. В одном из примеров положения генерируют с применением высокотемпературной молекулярной динамики, после которой проводится случайное вращение, оптимизация посредством имитации отжига с регулярным шагом и/или конечная минимизация с регулярным шагом или минимизации силового поля с целью генерации конформации и/или ориентации субстрата и/или последующего промежуточного продукта в каталитическом центре вычислительного представления. Некоторые из этих операций являются необязательными, например, оптимизация посредством имитации отжига с регулярным шагом и минимизация с регулярным шагом или минимизация силового поля.
В определенных вариантах осуществления число рассматриваемых положений составляет, по меньшей мере около 10, или, по меньшей мере, около 20, или, по меньшей мере, около 50, или, по меньшей мере, около 100, или, по меньшей мере, около 200, или, по меньшей мере, около 500. Однако не предполагается, что настоящее изобретение ограничено конкретным числом рассматриваемых положений.
VII. ГЕНЕРАЦИЯ БЕЛКОВ С ТРЕБУЕМОЙ АКТИВНОСТЬЮ ПУТЕМ МОДИФИКАЦИИ ПРЕДСКАЗАННЫХ МОДЕЛЬЮ ПОСЛЕДОВАТЕЛЬНОСТЕЙ
Одна из целей изобретения состоит в создании оптимизированной библиотеки вариантов белка через направленную эволюцию. Некоторые варианты осуществления изобретения предоставляют способы для ведения направленной эволюции вариантов белка с применением созданных моделей последовательности-активности. Различные модели активности последовательности, подготовленные и настроенные в соответствии со способами, описанными выше, являются подходящими для ведения направленной эволюции белков или биологических молекул. В качестве части процесса, способы могут идентифицировать последовательности, которые будут использоваться для генерации новых вариантов белка для следующего цикла направленной эволюции, как показано блоком 111 с фигуры 1 А. Такие последовательности содержат изменения в определенных остатках, идентифицированных выше, или являются предшественниками, используемыми для последующего внесения таких изменений. Последовательности могут быть модифицированы посредством выполнения мутагенеза и/или основанного на рекомбинации механизма создания разнообразия для создания новой библиотеки вариантов белка. В некоторых вариантах осуществления новые варианты могут анализироваться в отношении представляющей интерес активности. См. блок 113 с фигуры 1А. В некоторых применениях структурные модели могут быть сгенерированы для новых вариантов, и эти структурные модели могут предоставлять значения энергии и геометрические значения для вариантов. См. блок 113 с фигуры 1А. В некоторых вариантах осуществления эти данные могут затем использоваться в разработке новой модели активности последовательности в новом цикле направленной эволюции. См. блок 115 с фигуры 1А.
В некоторых вариантах осуществления подготовка олигонуклеотидов или последовательностей нуклеиновых кислот достигается посредством синтеза олигонуклеотидов или последовательностей нуклеиновых кислот с применением синтезатора нуклеиновой кислоты. Некоторые варианты осуществления изобретения включают выполнение цикла направленной эволюции с использованием готовых олигонуклеотидов или последовательности белка в качестве элементарных звеньев для направленной эволюции. Различные варианты осуществления изобретения могут применять рекомбинацию и/или мутагенез к этим элементарным звеньям для создания разнообразия.
В некоторых вариантах осуществления процесс идентифицирует одну или более последовательностей, обладающих выгодными свойствами. Затем из идентифицированных последовательностей генерируют варианты в качестве обучающего множества для модели активности последовательности в новом цикле направленной эволюции. См. блоки 355 и 357 с фигуры 3C.
Для генерации вариантов, в качестве одного конкретного примера, некоторые варианты осуществления применяют методики рекомбинации к олигонуклеотидам. В этих вариантах осуществления способ включают в себя выбор одной или более мутаций для цикла направленной эволюции посредством оценки коэффициентов членов модели активности последовательности. Мутации выбирают из комбинаций заданных аминокислот или нуклеотидов определенных типов остатков в определенных позициях на основании их вклада в активность белков в соответствии с предсказанным моделями. В некоторых вариантах осуществления выбор мутаций включает в себя идентификацию одного или более коэффициентов, для которых было определено, что они больше, чем другие коэффициенты. Каждый из коэффициентов относится к вкладу остатка в активность белка, и остаток задается как имеющий конкретный тип в конкретном местоположении. Выбор мутаций включает в себя выбор остатков, ассоциированных с одним или более коэффициентами, идентифицированными таким образом. В некоторых вариантах осуществления после выбора мутаций согласно моделям активности последовательности, способы включают в себя подготовку множества олигонуклеотидов, содержащих или кодирующих по меньшей мере одну мутацию, и выполнение цикла направленной эволюции. В некоторых вариантах осуществления методики направленной эволюции включают в себя объединение и/или повторное объединение олигонуклеотидов.
В других вариантах осуществления методики рекомбинации применяются к последовательностям белков. В некоторых вариантах осуществления способы включают в себя идентификацию нового белка или новой последовательности нуклеиновой кислоты, и подготовку и исследование нового белка или белка, закодированного новой последовательностью нуклеиновой кислоты. В некоторых вариантах осуществления способы также включают в себя использование нового белка или белка, закодированного новой последовательностью нуклеиновой кислоты, в качестве исходной точки для дальнейшей направленной эволюции. В некоторых вариантах осуществления процесс направленной эволюции включает в себя фрагментацию и повторное объединение последовательности белка, которая имела требуемый уровень активности в соответствии с предсказанием модели.
В некоторых вариантах осуществления способы идентифицируют и/или подготавливают новый белок или новую последовательность нуклеиновой кислоты на основании индивидуальных мутаций, которые, как было предсказано моделью, являются важными. Эти способы включают в себя: выбор одной или более мутаций путем оценки коэффициентов членов модели активности последовательности с целью идентификации одной или более заданных аминокислот или нуклеотидов в заданных позициях, которые вносят вклад в активность; идентификацию нового белка или новой последовательности нуклеиновой кислоты, содержащий одну или более мутаций, выбранных выше, и приготовление и анализ нового белка или белка, закодированного новой последовательностью нуклеиновой кислоты.
В других вариантах осуществления способы идентифицируют и/или подготавливают новый белок или новую последовательность нуклеиновой кислоты на основании предсказанной активности всей последовательности вместо индивидуальных мутаций. В некоторых из этих вариантов осуществления способы включают в себя применение множественных последовательностей белка или множественных последовательностей аминокислот к модели активности последовательности и определение значений активности, предсказанных моделью активности последовательности, для каждой из множества последовательностей белка или последовательностей нуклеиновых кислот. Способы также включают в себя выбор новой последовательности белка или новой последовательности нуклеиновой кислоты из числа множества последовательностей белка или множества последовательностей аминокислот, примененных выше, посредством оценки значений активности, предсказанных моделью активности последовательности для множества последовательностей. Способы также включают в себя подготовку и исследование белка, имеющего новую последовательность белка, или белка, закодированного новой последовательностью нуклеиновой кислоты.
В некоторых вариантах осуществления, вместо того, чтобы просто синтезировать единственный предсказанный как лучший белок, комбинаторная библиотека белков создается на основании анализа чувствительности лучших изменений в выборе остатка в каждом положении в белке. В этом варианте осуществления, чем более чувствительным является заданный выбор остатка для предсказанного белка, тем больше будет предсказанное изменение пригодности. В некоторых вариантах осуществления эти чувствительности сортируются от самого высокой до самой низкой, и показатели чувствительности используются для создания комбинаторных библиотек белка в последующих циклах (то есть, посредством включения этих остатков на основании чувствительности). В некоторых вариантах осуществления, в которых применяется линейная модель/модель без взаимодействия, чувствительность идентифицируют путем простого рассмотрения размера коэффициента, ассоциированного с членом данного остатка в модели. Однако это невозможно для нелинейных моделей/моделей с взаимодействием. Вместо этого в вариантах осуществления, применяющих нелинейные модели/модели с взаимодействием, чувствительность остатка определяют посредством применения модели для вычисления изменений в активности, когда единственный остаток изменяют в «лучшей» предсказанной последовательности.
Некоторые варианты осуществления изобретения включают в себя выбор одного или более положений в последовательности белка или последовательности нуклеиновой кислоты и проведение насыщающего мутагенеза в одном или более положений, идентифицированных таким образом. В некоторых вариантах осуществления положения выбирают посредством оценки коэффициентов членов модели активности последовательности с целью идентификации одной или более определенных аминокислот или нуклеотидов в определенных положениях, которые вносят вклад в активность. Соответственно, в некоторых вариантах осуществления, цикл направленной эволюции включает в себя выполнение насыщающего мутагенеза на последовательности белка в положениях, выбранных с применением моделей активности последовательности. В некоторых вариантах осуществления, включающих модели, содержащие один или более членов взаимодействия, каждый член взаимодействия относится к двум или более взаимодействующим остаткам. Способы включают в себя одновременное применение мутагенеза к двум или более взаимодействующим остаткам.
В некоторых вариантах осуществления остатки рассматривают в порядке их ранжирования. В некоторых вариантах осуществления для каждого рассматриваемого остатка процесс определяет, переключить ли этот остаток. Термин «переключение» относится к включению или исключению конкретного аминокислотного остатка в конкретной позиции в последовательностях вариантов белка в оптимизированной библиотеке. Например, серин может появляться в положении 166 в одном из вариантов белка, тогда как фенилаланин может появляться в положении 166 в другом варианте белка в той же самой ZAJQ3 5X. Аминокислотные остатки, которые не изменяются между последовательностями вариантов белка в обучающем множестве, обычно остаются фиксированными в оптимизированной библиотеке. Однако, это происходит не всегда, поскольку может присутствовать изменчивость в оптимизированных библиотеках.
В некоторых вариантах осуществления оптимизированная библиотека вариантов белка спроектирована таким образом, что все идентифицированные остатки, имеющие «высокий» ранг коэффициента регрессии, фиксируются, и остальные остатки, имеющие более низкий ранг коэффициента регрессии, переключаются. Объяснение для данного варианта осуществления состоит в том, что поиск должен осуществляться в локальном пространстве, окружающем 'наилучший' предсказанный белок. Отмечено, что исходная «основа», в которую вносятся переключения, может являться лучшим белком, предсказанным моделью, и/или уже проверенным 'наилучшим' белком из подвергнутой скринингу библиотеки. Фактически, не предполагается, что исходная основа ограничена каким-либо конкретным белком.
В альтернативном варианте осуществления по меньшей мере один или более, но не все, идентифицированные остатки с высоким рангом коэффициента регрессии фиксируют в оптимизированной библиотеке, и другие переключают. Этот подход является рекомендуемым в некоторых вариантах осуществления, если имеется требование не изменяться сильно контекст других аминокислотных остатков путем единовременного внесения слишком большого числа изменений. Снова, исходная точка для переключения может являться наилучшим множеством остатков в соответствии с предсказанным моделью, наилучшим проверенным белком из существующей библиотеки, или «средним» клоном, который хорошо моделируется. В последнем случае может требоваться переключение остатков, которые были предсказаны как имеющие более высокую важность, поскольку большее пространство должно исследоваться при поиске пиков активности, ранее пропущенных при осуществлении выборки. Этот тип библиотеки обычно является более подходящим на ранних циклах продуцирования библиотеки, поскольку он создает более усовершенствованную картину последующих циклов. Также не предполагается, что исходная основа ограничена каким-либо конкретным белком.
Некоторые альтернативы для вышеупомянутых вариантов осуществления включают в себя различные процедуры для использования значимости остатков (то есть, рангов) в определении того, какие остатки будут переключаться. В одном таком альтернативном варианте осуществления имеющие более высокий ранг положения остатков более активно предпочитаются для переключения. Информация, необходимая в данном подходе, включает в себя последовательность наилучшего белка из обучающего множества, предсказанную или PCR наилучшую последовательность и ранжирование остатков из модели или PCR. В некоторых вариантах осуществления «наилучший» белок представляет собой проверенный в практической лаборатории «наилучший» клон в множестве данных (то есть, клон с самым высоким значением взвешенной функцией, который все еще хорошо моделируется, то есть, попадает относительно близко к заранее определенному значению при перекрестной проверке). Способ сравнивает каждый остаток из этого белка с соответствующим остатком из «наилучшей предсказанной» последовательности, имеющей самое высокое значение требуемой активности. Если остаток с самым высоким коэффициентом нагрузки или регрессии не присутствует в 'наилучшем' клоне, способ вносит это положение в качестве положения переключения для последующей библиотеки. Если остаток присутствует в наилучшем клоне аналоге, способ не обрабатывает положение как положение переключения, и он будет последовательно переходить в следующее положение. Процесс повторяется для различных остатков с перемещением через последовательно уменьшающиеся значения нагрузки, пока не будет создана библиотека достаточного размера.
В некоторых дополнительных вариантах осуществления проверенный в практической лаборатории 'наилучший' (или один из лучших) белок в текущей оптимизированной библиотеке (то есть, белок с самым высоким значением, или одним из самых высоких значений, взвешенной функции, который все еще хорошо моделируется, то есть, попадает относительно близко к заранее определенному значению при перекрестной проверке) служит основой, в которую вносят различные изменения. В другом подходе практическая лаборатория проверила 'наилучший' (или один из лучших) белок в текущей библиотеке, который может плохо моделироваться, и который служит основой, в которую вносятся различные изменения. В некоторых других подходах последовательность, предсказанная моделью последовательности-активности как имеющая самое высокое значение (или одно из самых высоких значений) требуемой активности, служит основой. В этих подходах множество данных для библиотеки «следующего поколения» (и, возможно, соответствующую модель) получают посредством изменения остатков по меньшей мере в одном лучших белков. В одном из вариантов осуществления эти изменения включают в себя систематическое изменение остатков в остове. В некоторых случаях, изменения включают в себя различные методики мутагенеза, рекомбинации и/или выбора последовательности. Каждый из них может быть выполнен in vitro, в естественных условиях и/или in silico. Фактически, не предполагается, что настоящее изобретение ограничено каким-либо конкретным форматом, поскольку любой соответствующий формат находит применение.
В некоторых вариантах осуществления оптимизированные библиотеки вариантов белка создаются с применением способов рекомбинации, описанных в настоящем раскрытии, или, альтернативно, посредством способов генного синтеза с последующей экспрессией в естественных условиях или in vitro. В некоторых вариантах осуществления, после того, как был проведен скрининг оптимизированных библиотек вариантов белка относительно требуемой активности, их секвенируют. Как указано выше, информация об активности и последовательности из оптимизированной библиотеки вариантов белка может применяться для создания другой модели активности последовательности, из которой может быть спроектирована последующая оптимизированная библиотека с применением способов, описанных в настоящем раскрытии.
В одном из вариантов осуществления все белки из этой новой библиотеки используются в качестве части множества данных.
VIII. СЕКВЕНИРОВАНИЕ ПОЛИНУКЛЕОТИДОВ И ПОЛИПЕПТИДОВ
В некоторых вариантах осуществления, информация полинуклеотидной и полипептидной последовательности используется для генерации моделей последовательности-активности или вычислительных представлений активных центров вариантов белка. В некоторых вариантах осуществления, информация полинуклеотидной и полипептидной последовательности используется в процессах направленной эволюции для получения вариантов белка с требуемыми свойствами.
В различных вариантах осуществления последовательности вариантов белка устанавливают из физических биомолекул с помощью способов белкового секвенирования, и некоторые из этих способов подробнее описаны ниже. Секвенирование белка включает в себя определение аминокислотной последовательности белка. Некоторые методики секвенирования белка также определяет конформацию, которую белок принимает, и степень, до которой он образует комплекс с произвольными непептидными молекулами. Масс-спектрометрия и реакция расщепления по Эдману могут применяться для непосредственного определения аминокислотной последовательности белка.
Реакция расщепления по Эдману позволяет обнаружить упорядоченный аминокислотный состав белка. В некоторых вариантах осуществления автоматизированные секвенаторы Эдмана могут применяться для определения последовательности вариантов белка. Автоматизированные секвенаторы Эдмана способны упорядочить пептиды все более длинных последовательностей, например, длиной вплоть до приблизительно 50 аминокислот. В некоторых вариантах осуществления процесс секвенирования белка, реализующий расщепление по Эдману, включает в себя одно или более из следующего:
- разрыв дисульфидных связей в белке с помощью восстановителя, например, 2-меркаптоэтанола.
Блокирующая группа, такая как йодоуксусная кислота, может применяться для предотвращения повторного образования связей.
- отделение и очистка индивидуальных цепей комплекса белка, если их больше одной
- определение аминокислотного состава каждой цепи
- определение концевых аминокислот каждой цепи
- разделение каждой цепи на фрагменты, например, фрагменты длиной до 50 аминокислот
- разделение и очистка фрагментов
- определение последовательности каждого фрагмента с применением реакции расщепления по Эдману
- повторение указанных выше шагов с применением различных профилей расщепления с целью предоставления дополнительного (-ых) прочтения (-ий) аминокислотных последовательностей
- конструирование последовательности полного белка их прочтений аминокислотной последовательности
В различных реализациях пептиды, превышающие приблизительно 50-70 аминокислот по длине, должны быть разбиты на небольшие фрагменты, чтобы облегчить реакции секвенирования Эдмана. Расщепление более длинных последовательностей может быть выполнено посредством эндопептидаз, таких как трипсин или пепсин, или посредством химических реагентов, таких как бромистый циан. Различные ферменты дают различные профили расщепления, и перекрытие между фрагментами может использоваться для конструирования полной последовательности.
Во время реакции расщепления по Эдману пептид, который будет секвенироваться, адсорбируется на твердую поверхность субстрата. В некоторых вариантах осуществления одним из подходящих субстратов является стекловолокно, покрытое полибреном, катионным полимером. Реагент Эдмана, фенилизотиоцианат (PITC), добавляют к адсорбированному пептиду, вместе со слабоосновным буферным раствором триметиламина. Этот реакционный раствор реагирует с аминной группой N-концевой аминокислоты. Концевая аминокислота может затем селективно отсоединяться посредством добавления безводной кислоты. Производная затем изомеризуется с целью получения замещенного фенилтиогидантоина, который может быть смыт и идентифицирован посредством хроматографии. Затем цикл может быть повторен.
В некоторых вариантах осуществления масс-спектрометрия может применяться для определения аминокислотной последовательности путем определения отношений массы к заряду для фрагментов аминокислотной последовательности. Масс-спектр, содержащий пики, соответствующие фрагментам с различными зарядами, может быть определен, при этом расстояние между пиками, соответствующее различным изотопам, является обратно пропорциональным заряду на фрагменте. Масс-спектр анализируют, например, путем сравнения с базой данных ранее отсеквенированных белков, чтобы определить последовательности фрагментов. Этот процесс затем повторяют с другим расщепляющим ферментом, и перекрытия в последовательностях используются для конструирования полной аминокислотной последовательности.
Пептиды часто легче подготовить и проанализировать для масс-спектрометрии, чем целые белки. В некоторых вариантах осуществления, ионизация электрораспылением применяется для доставки пептидов в спектрометр. Белок расщепляется с помощью эндопротеазы, и полученный в результате раствор передают через колонку жидкостной хроматографии высокого давления. В конце этой колонки раствор распыляют в масс-спектрометр, при этом раствор заряжен положительным потенциалом. Заряд на каплях раствора вызывает их фрагментацию на единичные ионы. Пептиды затем фрагментируют, и измеряют отношение массы к заряду для фрагментов.
Также можно косвенно определить аминокислотную последовательность по последовательности ДНК или мРНК, кодирующей белок. Способы секвенирования нуклеиновых кислот, например, различные способы секвенирования следующего поколения, могут применяться для определения последовательностей РНК или ДНК. В некоторых реализациях последовательность белка выделяют впервые, не имея информации о нуклеотидах, кодирующих белок. В таких реализациях можно сначала определить короткую последовательность полипептида с применением одного из прямых способов секвенирования белка. Комплементарный маркер для РНК этого белка может быть определен по этой короткой последовательности. Это может затем использоваться для выделения мРНК, кодирующей белок, которая затем может быть реплицирована в полимеразной цепной реакции с получением значительного количества ДНК, которая затем может быть отсеквенирована с применением методов секвенирования ДНК. Аминокислотная последовательность белка может затем быть выведена из последовательности ДНК. При выведении необходимо принять во внимание аминокислоты, удаленные после того, как мРНК была оттранслирована.
В различных вариантах осуществления информация о последовательности полинуклеотидов используется для генерации моделей последовательности-активности или вычислительного представления сайтов активности белка. Информация нуклеотидной последовательности может быть установлена из физических биомолекул с помощью способов секвенирования нуклеиновых кислот, и некоторые из этих способов подробнее описаны ниже.
В одном или более вариантах осуществления данные последовательностей нуклеиновых кислот могут быть использованы на различных этапах в процессе направленной эволюции белков. В одном или более вариантах осуществления данные последовательностей могут быть получены с применением методов массового секвенирования, включая, например, секвенирование по Сэнгеру или секвенирование методом Максама-Гилберта, которые считают методами секвенирования первого поколения. Секвенирование по Сэнгеру, которое включает в себя использование помеченных дидезокси-элементов обрыва цепи, известно в технике; см., например, Sanger и соавт., Proceedings of The National Academy of Sciences of the United States of America 74, 5463-5467 (1997). Секвенирование Максама-Гилберта, которое включает в себя выполнение множества частичных химических реакций разложения на частях образца нуклеиновой кислоты, после которых следует обнаружение и анализ фрагментов с целью выведения последовательности, также известно в техике; см., например, Maxam и соавт., Proceedings of The National Academy of Sciences of the United States of America 74, 560-564 (1977). Другим методом массового секвенирования является секвенирование посредством гибридизации, в котором последовательность образца выводят на основании ее свойств гибридизации со множеством последовательностей, например, на микрочипе или ДНК-чипе; см., например, Drmanac, и соавт., Nature Biotechnology 16, 54-58 (1998).
В одном или более вариантах осуществления данные последовательностей нуклеиновых кислот получают с применением методов секвенирования следующего поколения. Секвенирование следующего поколения также называется "высокопроизводительным секвенированием". Методики распараллеливают процесс секвенирования, выдавая тысячи или миллионы последовательностей единовременно. Примеры соответствующих методов секвенирования следующего поколения включают в себя, но не ограничиваются указанным, секвенирование единичной молекулы в реальном времени {например, Pacific Biosciences, Менло-Парк, Калифорния), ионное полупроводниковое секвенирование (например, Ion Torrent, Южный Сан-Франциско, Калифорния), пиросеквенирование (например, 454, Брэнфорд, Коннектикут), секвенирование посредством лигирования (например, секвенирование SOLiD, которым владеет Life Technologies, Карлсбад, Калифорния), секвенирование посредством синтеза и обратимого обрывателя цепи (например, Illumina, Сан-Диего, Калифорния), технологии визуализации нуклеиновой кислоты, такие как просвечивающий электронный микроскоп, и т.п.
Как правило, методы секвенирования следующего поколения обычно применяют в качестве этапа клонирования in vitro для амплификации индивидуальных молекул ДНК. Эмульсионная ПЦР (emПЦР) изолирует индивидуальные молекулы ДНК на покрытых праймером гранулах в водных каплях в пределах масляной фазы. ПЦР производит копии молекулы ДНК, которые связываются с праймерами на грануле, после чего производится фиксация для последующего секвенирования. emПЦР применяется в способах, приведенных в работах Marguilis и др. (коммерциализированных 454 Life Sciences, Брэнфорд, Коннектикут), Shendure и Porreca и др. (также известных как "секвенирование молекулярных колоний") и секвенировании SOLiD, (Applied Biosystems Inc., Фостер-Сити, Калифорния). См. M. Margulies, и соавт. (2005) "Genome sequencing in microfabricated high-density picolitre reactors" Nature 437: 376-380; J. Shendure и соавт. (2005) "Accurate Multiplex Polony Sequencing of an Evolved Bacterial Genome" Science 309 (5741): 1728-1732. Амплификация клонов in vitro также может быть выполнена посредством "бридж-ПЦР", в которой фрагменты амплифицируют после докинг праймеров к твердой поверхности. Braslavsky и др. разработали метод единичной молекулы (коммерциализированный Helicos Biosciences Corp., Кембридж, Массачусетс), который пропускает данный этап амплификации, напрямую фиксируя молекулы ДНК на поверхности. I. Braslavsky, и соавт. (2003) "Sequence information can be obtained from single ДНК molecules" Proceedings of the National Academy of Sciences of the United States of America 100: 3960-3964.
Молекулы ДНК, которые физически связаны с поверхностью, могут быть отсеквенированы параллельно. В «секвенировании посредством синтеза» комплементарная нить строится на основании последовательности матричной нити с применением ДНК-полимеразы, как в электрофоретическом секвенирование с окрашенным прерывателем. Методы обратимого прерывания (коммерциализированные Illumina, Inc., Сан-Диего, Калифорния и Helicos Biosciences Corp., Кембридж, Массачусетс) используют обратимые версии окрашенных прерывателей, добавляя один нуклеотид за один раз, и детектируют флуоресценцию в каждом положении в режиме реального времени, посредством циклического удаления блокирующей группы с целью обеспечения полимеризации другого нуклеотида. В «пиросеквенировании» также применяется полимеризация ДНК, добавление одного нуклеотид за один раз и детектирование и количественное определение числа нуклеотидов, добавленных к данному положению, через свет, излучаемый при высвобождении присоединенных пирофосфатов (коммерциализировано 454 Life Sciences, Брэнфорд, Коннектикут). См. M. Ronaghi и соавт. (1996). «Real-time DNA sequencing using detection of pyrophosphate release» Analytical Biochemistry 242: 84-89.
Конкретные примеры методов секвенирования следующего поколения подробнее описаны ниже. Одна или более реализаций настоящего изобретения могут применять один или более из приведенных ниже методов секвенирования, не отклоняясь от принципов изобретения.
Одномолекулярное секвенирование в реальном времени (также известное как SMRT) представляет собой технологию распараллеленного секвенирования одиночных молекул ДНК посредством синтеза, разработанную Тихоокеанские Биологические науки. Одномолекулярное секвенирование в реальном времени использует нулевой волновод (ZMW). Единственный фермент ДНК-полимеразы прикреплен на дне ZMW к единственной молекуле ДНК в качестве матрицы матрица. ZMW представляет собой структуру, которая создает освещенный объем наблюдения, который является достаточно малым для наблюдения только одного нуклеотида ДНК (также известного как основание), присоединяемого ДНК-полимеразой. Каждое из четырех оснований ДНК присоединено к одному из четырех различных флуоресцентных красителей. Когда нуклеотид присоединяется ДНК-полимеразой, флуоресцентная метка отщепляется и диффундирует из области наблюдения ZMW, где ее флуоресценция больше не наблюдается. Детектор обнаруживает флуоресцентный сигнал присоединения нуклеотида, и определение основания выполняется согласно соответствующей флуоресценции красителя.
Другой подходящей технологией одномолекулярного секвенирования является технология истинного одномолекулярного секвенирования Helicos (tSMS) (например, как описано в работе Harris T.D. и соавт., Nature 320: 106-109 [2008]). В методике tSMS образец ДНК расщепляется на нити длиной около 100-200 нуклеотидов, и последовательности полиА добавляется к 3 '-концам каждой нити ДНК. Каждая нить помечается посредством добавления флуоресцентно помеченного аденозинового нуклеотида. Нити ДНК затем гибридизуются в проточной ячейке, которая содержит миллионы сайтов захвата олиго-T, которые зафиксированы на поверхности проточной ячейки. В определенных вариантах осуществления матрицы могут присутствовать с плотностью около 100 миллионов матриц/см2. Проточная ячейка затем загружается в прибор, например, секвенатор HeliScope™, и лазер освещает поверхность проточной ячейки, показывая положение каждой матрицы. Камера CCD может отобразить положение матриц на поверхности клеток потока. Матричная флуоресцентная метка затем отщепляется и смывается. Реакция секвенирования начинается посредством внесения ДНК-полимеразы и флуоресцентно помеченного нуклеотида. Нуклеиновая кислота олиго-T служит праймером. Полимераза присоединяет помеченные нуклеотиды к праймеру на основании матрицы. Полимераза и неприсоединенные нуклеотиды удаляются. Матрицы, которые направляли присоединение флуоресцентно помеченного нуклеотида, различают посредством визуализации поверхности проточной ячейки. После визуализации на этапе расщепления удаляется флуоресцентная метка, и процесс повторяется с другими флуоресцентно помеченными нуклеотидами, пока требуемая длина чтения не будет достигнута. Информация последовательности собирается на каждом шаге добавления нуклеотида. Секвенирование всего генома посредством технологий одномолекулярного секвенирования исключает или обычно устраняет основанную на ПЦР амплификацию при подготовке библиотек секвенирования, и способы позволяют осуществлять прямое измерение образца, а не измерение копий этого образца.
Ионное полупроводниковое секвенирование представляет собой метод секвенирования ДНК, основанный на обнаружении ионов водорода, которые высвобождаются во время полимеризации ДНК. Данный метод является методом «секвенирования посредством синтеза», во время которого комплементарная нить строится на основании последовательности матричной нити. Микролунка, содержащая матричную нить ДНК, которая будет секвенироваться, заливаются единичными молекулами дезоксирибонуклеотидтрифосфата, (dNTP). Если внесенный dNTP является комплементарным находящемуся в начале матричному нуклеотиду, он включается в растущую комплементарную нить. Это вызывает высвобождение иона водорода, который переключает ионный датчик ISFET, который показывает, что реакция произошла. Если гомополимерные повторы будут присутствовать в матричной последовательности, то множество молекул dNTP будут включены в одном цикле. Это приводит к соответствующему количеству высвобожденных водородов и пропорционально более высокому электронному сигналу. Эта технология отличается от других технологий секвенирования, в том, что в ней не используются какие-либо измененные нуклеотиды или оптика. Ионное полупроводниковое секвенирование может также называться как поточным ионным секвенированием, pH-фактор-опосредованным секвенированием, кремниевым секвенированием или полупроводниковым секвенирование.
В пиросеквенировании пирофосфат-ион, высвобожденный в результате реакции полимеризации, реагирует с 5'-фосфосульфатом аденозина посредством АТФ-сульфурилазы с образованием АТФ; АТФ затем управляет преобразованием люциферина в оксилюциферин плюс свет от люциферазы. Поскольку флуоресценция являеся временной, то в данном методе отсутствует необходимость в отдельном шаге удаления флуоресценции. Один тип дезоксирибонуклеотитрифосфата (dNTP) добавляется за один раз, и информацию о последовательности различают по тому, какой dNTP генерирует значительный сигнал в месте реакции. Имеющийся в продаже прибор Roche GS FLX получает последовательность с применением данного метода. Эта методика и ее применения подробно обсуждаются, например, в Ronaghi и соавт., Analytical Biochemistry 242, 84-89 (1996) и Margulies и соавт., Nature 437, 376-380 (2005) (поправки в Nature 441, 120 (2006)). Коммерчески доступной технологией пиросеквенирования является 454-секвенирование (Roche) (например, как описано в Margulies и соавт., Nature 437:376-380 [2005]).
В секвенировании посредством лигирования фермент лигаза применяется для соединения являющего частично двухцепочечным олигонуклеотида с «липким» концом с секвенируемой нуклеиновой кислотой, которая имеет «липкий» конец; для того, чтобы происходило лигирование, липкие концы должны быть комплементарными. Основания в липком конце являющего частично двухцепочечным олигонуклеотида могут быть идентифицированы согласно флуорофору, конъюгированному с частично двухцепочечным олигонуклеотидом и/или для вторичным олигонуклеотидом, который гибридизуется с другой частью частично двухцепочечного олигонуклеотида. После получения флуоресцентных данных лигированный комплекс расщепляется в точке, находящейся ближе к 5 '-концу, чем сайт лигирования, например, рестрикционным ферментом типа IIs, например, Bbvl, который разрезает в сайте, находящемся на фиксированном расстоянии от сайта распознавания (который был включен в частично двухцепочечный олигонуклеотид). Данная реакция расщепления открывает новый липкий конец, находящийся в 5 '-направлении непосредственно рядом с предыдущим липким концом, и процесс повторяется. Эта методика и ее применения подробно обсуждаются, например, в работе Brenner и соавт., Nature Biotechnology 18, 630-634 (2000). В некоторых вариантах осуществления секвенирование посредством лигирования адаптировано для способов по изобретению посредством получения продукта амплификации по типу катящегося кольца круговой молекулы нуклеиновой кислоты и использования данного продукта амплификации по типу катящегося кольца в качестве матрицы для секвенирования посредством лигирования.
Коммерчески доступным примером технологии секвенирования посредством лигирования является технология SOLiD™ (Прикладные Биосистемы). В секвенировании посредством лигирования SOLiD™ геномная ДНК разрезается на фрагменты, и адаптеры присоединяются к 5'- и 3'-концам фрагментов для создания библиотеки фрагментов. Альтернативно, внутренние адаптеры могут быть введены посредством лигирования адаптеров с 5'- и 3 '-концами фрагментов, расщепления скругленного фрагмента для создания внутреннего адаптера и присоединения адаптеров к 5'- и 3'-концам полученных в результате фрагментов для создания парно сопряженной (mate-paired) библиотеки. Затем, популяции клональных гранул подготавливаются в микрореакторах, содержащих гранулы, праймеры, матрицу и компоненты ПЦР. После ПЦР матрицы денатурируются, и гранулы обогащают с целью выделения гранул с удлиненными матрицами. Матрицы на отобранных гранулах подвергаются 3 '-модификациям, которые позволяют связываться со стеклянной пластинкой. Последовательность может быть определена посредством последовательной гибридизации и лигирования частично случайных олигонуклеотидов с центральным определенным основанием (или парой оснований), которое идентифицируют по конкретном флуророфору. После того, как цвет был записан, лигированный олигонуклеотид расщепляется и удаляется, и процесс затем повторяют.
В секвенировании с применением обратимого обрывателя цепи флуоресцентный помеченный красителем аналог нуклеотида, который является обратимым обрывателем цепи вследствие наличия блокирующей группы, вносят в реакцию удлинения на одно основание. Идентификация основания производится согласно флуорофору; другими словами, каждая основа соединена с различным флуорофору. После того, как данные флуоресценции/последовательности были получены, флуорофор и блокирующая группа химически удаляют, и цикл повторяют для получения информации о следующем основании из последовательности. Прибор Illumina GA функционирует в соответствии с данным методом. Данная методика и ее применения подробно обсуждены, например, в Ruparel и соавт., Proceedings of the National Academy of Sciences of the United States of America 102, 5932-5937 (2005), и Harris и соавт., Science 320, 106-109 (2008).
Коммерчески доступным примером секвенирования с применением обратимого обрывателя цепи является секвенирование посредством синтеза и основанное на обратимом обрывателе цепи секвенирование Illumina с (например, как описано в Bentley и соавт., Nature 6:53-59 [2009]). Технология секвенирования Illumina основана на прикреплении фрагментированной геномной ДНК к плоской оптически прозрачной поверхности, на которой связаны фиксаторы олигонуклеотидов. Матричная ДНК подвергается восстановлению конца с целью получения 5 '-фосфорилированных тупых концов, и полимеразная активность фрагмента Кленова используется для добавления единичного основания к 3 '-концу тупых фосфорилированных фрагментов ДНК. Это добавление подготавливает фрагменты ДНК к лигированию с олигонуклеотидными адаптерами, которые имеют липкий конец из одного основания T на своих 3 '-концах, с целью повышения эффективности лигирования. Олигонуклеотиды адаптера являются комплементарными к фиксаторам проточной ячейки. В условиях предельного разведения модифицированная адаптером одноцепочечная матричная ДНК добавляется к проточной ячейке и фиксируется посредством гибридизации с фиксаторами. Присоединенные фрагменты ДНК удлиняются и бридж-амплифицируются для создания проточной ячейки секвенирования со сверхвысокой плотностью с сотнями миллионов кластеров, каждый из которых содержит ~1000 копий одной и той же матрицы. Матрицы секвенируют с применением гибкой четырехцветной технологии секвенирования ДНК посредством синтеза, в которой используют обратимые обрыватели цепи с удаляемыми флуоресцентными красителями. Обнаружение флуоресценции с высокой чувствительностью достигается с применением возбуждения лазером и оптики полного внутреннего отражения. Короткие прочтения последовательности около 20-40 н.п. например, 36 н.п. выравнивают с опорным геномом с маскированными повторами, и уникальное отображение коротких прочтений последовательности на опорный геном идентифицируют с применением специально разработанного конвейерного программного обеспечения анализа данных. Также могут использоваться опорные геномы без маскирования повторов. Независимо от того, используются ли опорные геномы с маскированными повторами или без маскирования повторов, подсчитывают только прочтения, которые уникально отображаются на опорный геном. После завершения первого прочтения матрицы могут быть восстановлены на месте с целью обеспечения второго прочтения с противоположного конца фрагментов. Таким образом, может быть применено или одностороннее секвенирование, или секвенирование парных концов фрагментов ДНК. Выполняется частичное секвенирование фрагментов ДНК, присутствующих в образце, и маркерные последовательности, содержащие прочтения с предварительно заданной длиной, например, 36 н.п. отображают на известный опорный геном и подсчитывают.
В нанопорном секвенировании одноцепочечная молекула нуклеиновой кислоты продевается через пору, например, с применением электрофоретической движущей силы, и последовательность выводят посредством анализа данных, полученных при прохождении одноцепочечной молекулы нуклеиновой кислоты через пору. Данные могут быть данными ионного тока, при этом каждое основание изменяет ток, например, посредством частичного блокирования тока, проходящего через пору, в различной различимой степени.
В другом иллюстративном, но неограничивающем, варианте осуществления способы, описанные в настоящем раскрытии, включают в себя получение информации о последовательности с применением просвечивающей электронной микроскопии (TEM). Способ включает в себя применением визуализации посредством трансмиссионного электронного микроскопа с разрешением в один атом имеющей высокий молекулярный вес ДНК (150 т.п.н. или более), выборочно помеченной маркерами из тяжелых атомов, и размещение этих молекул на ультратонких пленках в сверхплотных (3 нм между цепочками) параллельных множествах с согласованным интервалом от между основаниями. Электронный микроскоп применяется для визуализации молекул на пленках с целью определения положения маркеров из тяжелых атомов и извлечения информации о последовательности оснований из ДНК. Способ дополнительно описан в патентной публикации WO 2009/046445.
В другом иллюстративном, но неограничивающем, варианте осуществления способы, описанные в настоящем раскрытии, включают в себя получение информацию о последовательности с применением секвенирования третьего поколения. В секвенировании третьего поколения пластинка с алюминиевым покрытием с множеством маленьких отверстий (~50 нм) используется в качестве нулевого волновода (см., например, Levene и соавт., Science 299, 682-686 (2003)). Алюминиевая поверхность предохраняется от докинг ДНК-полимеразы посредством полифосфонатной химии, например, поливинилфосфонатной химии (см., например, Korlach и соавт., Proceedings of the National Academy of Sciences of the United States of America 105, 1176-1181 (2008)). Это приводит к преимущественному прикреплению молекул ДНК-полимеразы к открытому кремнию в отверстиях алюминиевого покрытия. Такая конструкция позволяет использовать явление нераспространяющейся волны для снижения фонового уровня флуоресценции, что позволяет использовать более высокие концентрации флуоресцентно помеченных dNTP. Флуорофор присоединен к концевому фосфату dNTP таким образом, что флуоресценция высвобождается после внесения dNTP, но флуорофор не остается присоединенным к недавно внесенному нуклеотиду, что означает, что комплекс сразу готов к еще одному циклу включения. Посредством этого метода, включение dNTP в индивидуальные комплексы праймер-матрица, присутствующие в отверстиях алюминиевого покрытия, может быть обнаружено. См., например, Eid и соавт., Nature 323, 133-138 (2009).
IX. Анализ вариантов генов и белков
В некоторых вариантах осуществления полинуклеотиды, сгенерированные в связи со способами по настоящему изобретению, дополнительно клонируются в клетки с целью экспрессии вариантов белков для выполнения скрининга активности (или используются в реакциях транскрипции in vitro с целью получения продуктов, которые будут подвергаться скринингу). Кроме того, нуклеиновые кислоты, кодирующие варианты белка, могут быть обогащены, отсеквенированы, экспрессированы, амплифицированы in vitro или обработаны любым другим рекомбинантным способом.
Общие тексты, в которых описаны методики молекулярной биологии, полезные для настоящего раскрытия, включая клонирование, мутагенез, построение библиотек, анализы скрининга, культивацию клеток и т.п., включают работы Berger and Kimmel, Guide to Molecular Cloning Techniques, Methods in Enzymology volume 152 Academic Press, Inc., San Diego, CA (Berger); Sambrook и соавт., Molecular Cloning A Laboratory Manual (2nd Ed.), Vol. 1-3, Cold Spring Harbor Laboratory, Cold Spring Harbor, New York, 1989 (Sambrook) and Current Protocols in Molecular Biology, F.M. Ausubel и соавт., eds., Current Protocols, a joint venture between Greene Publishing Associates, Inc. and John Wiley & Sons, Inc., New York (supplemented through 2000) (Ausubel). Способы трансфекции клеток, включая клетки растений и животных, нуклеиновыми кислотами, являются общедоступными, так же как и способы экспрессии белков, закодированных такими нуклеиновыми кислотами. В дополнение к работам Berger, Ausubel и Sambrook, полезные общие ссылки для культивации животных клеток включают Freshney (Culture of Animal Cells, a Manual of Basic Technique, third edition Wiley Liss, New York (1994)) и ссылки, процитированные в этой работе, Humason (Animal Tissue Techniques, fourth edition W.H. Freeman and Company (1979)) and Ricciardelli, и соавт., In Vitro Cell Dev. Biol. 25:1016 1024 (1989). Ссылки для клонирования, культивации и регенерации растительных клеток включают Payne и соавт. (1992) Plant Cell and Tissue Culture in Liquid Systems John Wiley & Sons, Inc. New York, NY (Payne); и Gamborg and Phillips (eds) (1995) Plant Cell, Tissue and Organ Culture; Fundamental Methods Springer Lab Manual, Springer-Verlag (Berlin Heidelberg New York) (Gamborg). Множество сред для культивирования клеток описано в Atlas and Parks (eds) The Handbook of Microbiological Media (1993) CRC Press, Boca Raton, FL (Atlas). Дополнительную информацию для культивации растительных клеток можно найти в доступной коммерческой литературе, такой как Life Science Research Cell Culture Catalogue (1998), компании Sigma Aldrich, Inc (St Louis, MO) (Sigma-LSRCCC) и, например, the Plant Culture Catalogue и приложения (1997), также от компании Sigma Aldrich, Inc (St Louis, MO) (Sigma-PCCS).
Примеры методик, достаточных для специалистов, в области способов амплификации in vitro, полезных, например, для амплификации олигонуклеотидов рекомбинированных нуклеиновых кислот, включают в себя полимеразные цепные реакции (ПЦР), лигазные цепные реакции (LCR), амплификации с помощью Q-репликазы и β другие опосредованные РНК-полимеразой методики (например, NASBA). Эти методики можно найти в Berger, Sambrook, и Ausubel, см. выше, а также в Mullis и соавт., (1987) патент США № 4683202; PCR Protocols A Guide to Methods and Applications (Innis и соавт. eds) Academic Press Inc. San Diego, CA (1990) (Innis); Arnheim & Levinson (October 1, 1990) C&EN 36-47; The Journal Of NIH Research (1991) 3, 81-94; Kwoh и соавт. (1989) Proc. Natl. Acad. Sci. USA 86, 1173; Guatelli и соавт. (1990) Proc. Natl. Acad. Sci. USA 87, 1874; Lomell и соавт. (1989) J. Clin. Chem 35, 1826; Landegren и соавт., (1988) Science 241, 1077-1080; Van Brunt (1990) Biotechnology 8, 291-294; Wu and Wallace, (1989) Gene 4, 560; Barringer и соавт. (1990) Gene 89, 117, and Sooknanan and Malek (1995) Biotechnology 13: 563-564. Улучшенные способы клонирования in vitro амплифицированных нуклеиновых кислот описаны в Wallace и соавт., патент США. № 5426039. Улучшенные способы амплификации больших нуклеиновых кислот посредством ПЦР резюмированы в работе Cheng и соавт. (1994) Nature 369: 684-685 и приведенных в ней ссылках, в которой генерируются ПЦР-ампликоны вплоть до 40 т.н.п. Специалисту в данной области техники будет понятно, что, по существу, любая РНК может быть преобразована в двухцепочечную ДНК, подходящую для рестрикционного расщепления, ПЦР-наработки и секвенирования с применением обратной транскриптазы и В одном предпочтительном варианте осуществления заново собранные последовательности проверяют относительно включения основанных на семействе олигонуклеотидов рекомбинации. Это может быть сделано посредством клонирования и секвенирования нуклеиновых кислот и/или посредством рестрикционного расщепления, например, как по существу изложено в Sambrook, Berger и Ausubel, см. выше. Кроме того, последовательности могут быть амплифицированы посредством ПЦР и отсеквенированы напрямую. Таким образом, в дополнение, например, к Sambrook, Berger, Ausubel и Innis (см. выше), дополнительные методологии ПЦР-секвенирования также являются особенно полезными. Например, выполняется прямое секвенирование сгенерированных посредством ПЦР ампликонов посредством селективного внедрения борсодержащих устойчивых к нуклеазе нуклеотидов в ампликоны во время ПЦР и расщепления ампликонов с помощью нуклеазы с получением отсортированных по размеру фрагментов шаблонов (Porter и соавт. (1997) Nucleic Acids Research 25 (8):1611-1617). В этих способах выполняются четыре реакции ПЦР на шаблоне, в каждой из которых один из трифосфатов нуклеотида в реакционной смеси ПЦР частично замещается на 2'-дезоксинуклеозид 5'-[P-борано]-трифосфат. Борсодержащий нуклеотид стохастически включается в ПЦР-продукты в изменяющихся позициях вдоль ПЦР-ампликона во вложенном множестве ПЦР-фрагментов шаблона. Экзонуклеаза, которая блокируется внесенными борсодержащими нуклеотидами, используется для расщепления ПЦР-ампликонов. Расщепленные ампликоны затем разделяют по размеру с применением электрофореза в полиакриламидном геле, предоставляя последовательность ампликона. Преимущество этого способа состоит в том, что в нем применяется меньше биохимических манипуляций, чем при выполнении стандартного секвенирования ПЦР-ампликонов по Сэнгеру.
Синтетические гены могут подвергаться обычным подходам клонирования и экспрессии; таким образом, свойства генов и белков, которые они кодируют, могут быть легко исследованы после их экспрессии в клетке-хозяине. Синтетические гены могут также использоваться для генерации полипептидных продуктов посредством in vitro (бесклеточной) транскрипции и трансляции. Полинуклеотиды и полипептиды могут, таким образом, быть исследованы в отношении их способности связывать множество предварительно определенных лигандов, малых молекул и ионов, или полимерных и гетерополимерных веществ, включая другие белки и антигенные детерминанты полипептида, а также микробные клеточные стенки, вирусные частицы, поверхности и мембраны.
Например, многие физические способы могут применяться для обнаружения полинуклеотидов, кодирующих фенотипы, ассоциированные с катализом химических реакций непосредственно с помощью полинуклеотидов или посредством закодированных полипептидов. Исключительно с целью иллюстрации и в зависимости от специфики конкретных предварительно заданных интересующих химических реакций, эти способы могут включать в себя множество методик, известных в технике, которые учитывают физические различия между субстратом (-ами) и продуктом (-ами), или учитывают изменения в реакционных средах, ассоциированных с химической реакцией (например, изменения в электромагнитных излучениях, адсорбции, рассеянии и флюоресценции, в ультрафиолетовой, видимой или инфракрасной (тепло) области). Эти способы также могут быть выбраны из любой комбинации следующего: масс-спектрометрия; ядерный магнитный резонанс; вещества, меченные радиоактивными атомами, способы разделения и спектральные способы, учитывающие распределение изотопов или образование меченных продуктов; спектральные и химические способы для обнаружения сопутствующих изменений в композициях ионов или элементов в продукте (-ах) реакции (включая изменения в pH-фактор-факторе, неорганических и органических ионах и т.п.). Другие способы физических анализов, подходящие для применения в способах по настоящему раскрытию, могут быть основаны на использовании биосенсоров, специфичных для продукта (-ов) реакции, включая те, которые содержат антитела с репортерными свойствами, или могут быть основаны на распознавании аффинности в естественных условиях вместе с экспрессией и активностью репортерного гена. Соединенные с ферментами анализы для обнаружения продукта реакции и выбора жизни-смерти-роста клеток в естественных условиях могут также применяться там, где это необходимо. Независимо от конкретной природы физических анализов, все они применяются для выбора требуемой активности, или комбинации требуемых активностей, предоставленных или закодированных интересующей биомолекулой.
Конкретный анализ, применяемый для отбора, будет зависеть от применения. Известно множество анализов для белков, рецепторов, лигандов, ферментов, субстратов и т.п. Форматы включают связывание с иммобилизованными компонентами, жизнеспособность клеток или организмов, продуцирование репортерных композиций, и т.п.
Высокопроизводительные анализы являются особенно подходящими для скрининга библиотек, используемых в настоящем изобретении. В высокопроизводительных анализах можно выполнить скрининг вплоть до нескольких тысяч различных вариантов за один день. Например, каждая лунка микротитрационного планшета может применяться для выполнения отдельного анализа, или, если должны наблюдаться концентрация или эффекты времени инкубации, в каждых 5-10 лунках может проверяться единственный вариант (например, в различных концентрациях). Таким образом, в одном стандартном микротитрационном планшете может анализироваться около 100 (например, 96) реакций. Если используются планшеты с 1536 лунками, то в единственном планшете может легко анализироваться от около 100 до около 1500 различных реакций. Можно проанализировать несколько различных планшетов в сутки; в анализ скрининга, включающий в себя вплоть до около 6000-20000 различных анализов (то есть, включающих различные нуклеиновые кислоты, закодированные белки, концентрации, и т.д.), является возможным с применением интегрированных систем по изобретению. В последнее время были разработаны микрожидкостные подходы к манипулированию реагентами, например, Caliper Technologies (Маунтин-Вью, Калифорния), которые могут предоставить обладающие очень высокой производительностью способы микрожидкостного анализа.
Системы высокопроизводительного скрининга имеются в продаже (см., например, Zymark Corp., Hopkinton, MA; Air Technical Industries, Mentor, OH; Beckman Instruments, Inc. Fullerton, CA; Precision Systems, Inc., Natick, MA, и т.д.). Эти системы обычно автоматизируют все процедуры, включая раскапывание всего образца и реагента, распределение жидкости, рассчитанное по времени инкубирование и итоговое считывание микропланшета в детекторе(-ах), подходящие для анализа. Эти конфигурируемые системы обеспечивают высокую производительность и быстрый запуск, а также высокую степень гибкости и возможностей по настройке.
Производители таких систем предоставляют подробные протоколы для различных высокопроизводительных анализов скрининга. Таким образом, например, Zymark Corp. предоставляет технические бюллетени, описывающие системы скрининга для обнаружения модуляции транскрипции генов, связывания лиганда, и т.п.
Множество коммерчески доступного периферийного оборудования и программного обеспечения доступно для оцифровки, хранения и анализа оцифрованного видео или оцифрованных изображений оптических или других анализов, например, с использованием ПК (совместимые с Intel x86 или чипом пентиум MAC OS, семейство WINDOWS™, или основанные на UNIX (например, рабочее место SUN™) компьютеры.
Системы для анализа обычно включают в себя цифровой компьютер, специально запрограммированный для выполнения специализированных алгоритмов с применением программного обеспечения для направления одного или более этапов одного или более способов по настоящему раскрытию, и, дополнительно, также включают в себя, например, программное обеспечение для управления платформой секвенирования следующего поколения, программное обеспечение для управления жидкостной высокопроизводительной системой, программное обеспечение для анализа изображений, программное обеспечение для обработки экспериментальных данных, автоматизированную арматуру для контроля жидкости для передачи растворов от источника к месту назначения, функционально связанную с цифровым компьютером, устройством ввода данных (например, клавиатурой компьютера) для ввода данных в цифровой компьютер с целью управления операциями или высокопроизводительным переносом жидкости посредством автоматизированной арматуры для контроля жидкости и, необязательно, сканер изображений для оцифровки сигналов меток от меченных компонентов анализа. Сканер изображений может взаимодействовать с программным обеспечением анализа изображений с целью предоставления измерения интенсивности метки зонда. Как правило, измерение интенсивности метки зонда интерпретируется посредством программного обеспечения интерпретации данных, чтобы показать, гибридизируется ли помеченный зонд с ДНК на твердой субстрате.
В некоторых вариантах осуществления клетки, вирусные пятна, споры и т.п., содержащие продукты опосредованной олигонуклеотидом рекомбинации in vitro или физические варианты осуществления рекомбинированных in silico нуклеиновых кислот, могут быть разделены на твердых средах с получением индивидуальных колоний (или пятен). С применением автоматизированного средства отбора колоний (например, Qbot, Genetix, Великобритания), колонии или пятна идентифицируют, отбирают, и инокулируют вплоть до 10000 различных мутантов в 96-луночных микротитрационных планшетах, содержащих два 3-миллиметровых стеклянных шарика на лунку. Qbot не отбирает всю колонию, а вставляет иглу через центр колонии и выходит с небольшой выборкой клеток (или мицелий) и спор (или вирусов в приложениях с пятнами). Время, в течение которого игла находится в колонии, число погружений для инокуляции культуральной среды и время, в течение которого игла находится в этой среде, влияют на размер инокулята, и каждым параметром можно управлять и можно его оптимизировать.
Равномерный процесс автоматизированного отбора колоний, такой как Qbot, снижает ошибки обработки вручную и повышает скорость установления культур (примерно 10000/4 часа). Эти культуры, необязательно, встряхиваются в термостате с контролируемой температурой и влажностью. Дополнительные стеклянные шарики в микротитрационных планшетах способствуют равномерной аэрации клеток и рассеиванию клеточных (например, мицелярных) фрагментов аналогично лопастям ферментатора. Клоны из представляющих интерес культур могут быть изолированы предельным разведением. Как также описано выше, пятна или клетки, составляющие библиотеки, также могут подвергаться непосредственному скринингу с целью продуцирования белков, посредством обнаружения гибридизации, активности белка, связывания белка с антителами, и т.п. Для того, чтобы повысить вероятность идентификации пула достаточного размера, может применяться предварительный скрининг, который увеличивает число обработанных мутантов в 10 раз. Цель предварительного скрининга состоит в том, чтобы быстро идентифицировать мутанты, имеющие равные или лучшие титры продукта, чем родительский(-ие) штамм(-ы), и перемещать только эти мутанты в жидкую клеточную культуру для последующего анализа.
Один из подходов к скринингу разнообразных библиотек заключается в применении твердофазной процедуры с массовым распараллеливанием для скрининга клеток, экспрессирующих варианты полинуклеотида, например, полинуклеотиды, которые кодируют варианты фермента. Имеются устройства для твердофазного скрининга с массовым распараллеливанием с применением абсорбции, флуоресценции, или FRET. См., например, патент США № 5914245 Bylina и соавт. (1999); см. также, http://www.kairos-scientific.com/; Youvan и соавт. (1999) «Fluorescence Imaging Micro-Spectrophotometer (FIMS)» Biotechnology et alia, <www.et-al.com> 1:1-16; Yang и соавт. (1998) «High Resolution Imaging Microscope (HIRIM)» Biotechnology et alia, <www.et-al.com> 4:1-20; и Youvan и соавт. (1999) «Calibration of Fluorescence Resonance Energy Transfer in Microscopy Using Genetically Engineered GFP Derivatives on Nickel Chelating Beads», выложенные на www.kairos-scientific.com. После выполнения скрининга посредством этих методик, интересующие молекулы обычно выделяют, и, необязательно, секвенируют с применением способов, известных в технике. Информация о последовательности затем используется в соответствии с изложенным в настоящем раскрытии для проектирования новой библиотеки вариантов белка.
Аналогично, ряд известных автоматизированных систем также был разработан для химических сред фазы раствора, полезных в системах анализа. Эти системы включают в себя автоматизированные рабочие станции, такие как как автоматизированный прибор синтеза, разработанный Takeda Chemical Industries, LTD. (Осака, Япония) и множество автоматизированных систем, использующих роботизированные руки-манипуляторы (Zymate II, Zymark Corporation, Hopkinton, Массачусетс; Orca, Beckman Coulter, Inc. (Фуллертон, Калифорния)), которые имитируют ручные синтетические операции, выполняемые ученым. Любое из вышеупомянутых устройств является подходящим для применения с настоящим изобретением, например, для высокопроизводительного скрининга молекул, закодированных нуклеиновыми кислотами, полученными в соответствии с описанным в настоящем раскрытии. Природа и реализация модификаций (при их наличии) в этих устройствах таким образом, чтобы они могли функционировать в соответствии с обсуждаемым в настоящем раскрытии, будут очевидны специалистам в соответствующей области техники.
X. ЦИФРОВОЙ ПРИБОР И СИСТЕМЫ
Как должно быть очевидным, в вариантах осуществления, описанных в настоящем раскрытии, применяются процессы, выполняемые под управлением инструкций и/или данных, хранящихся или передаваемых через одну или более вычислительных систем. Варианты осуществления, раскрытые в настоящем раскрытии, также относятся к устройству для выполнения этих операций. В некоторых вариантах осуществления устройство специально спроектировано и/или сконструировано для заданных целей, или оно может представлять собой универсальный компьютер, выборочно активированный или реконфигурированный посредством компьютерной программы и/или структуры данных, хранящейся в компьютере. Процессы, предоставленные настоящим раскрытием, по своей сути не связаны с каким-либо конкретным компьютером или другим конкретным устройством. В частности, различные универсальные машины находят применение с программами, написанными в соответствии с изложенным в настоящем раскрытии. Однако, в некоторых вариантах осуществления, специализированное устройство конструируется для выполнения требуемых операций способа. Один из вариантов осуществления конкретной структуры для множества таких машин описан ниже.
Кроме того, определенные варианты осуществления настоящего раскрытия относятся к машиночитаемым носителями или компьютерным программным продуктам, которые содержат инструкции программы и/или данные (включая структуры данных) для выполнения различных реализованных компьютером операций. Примеры машиночитаемых носителей включают в себя, но не ограничиваются указанным, магнитные носители, такие как жесткие диски; оптические носители, такие как устройства CD-ROM и голографические устройства; магнитооптические носители; и полупроводниковые запоминающие устройства такие как флэш-память. Аппаратные устройства, такие как постоянные запоминающие устройства (ROM) и запоминающее устройство с произвольной выборкой (RAM), могут быть сконфигурированы для хранения инструкций программ. Аппаратные устройства, такие как специализированные интегральные схемы (ASIC) и программируемые логические устройства (PLD) могут быть сконфигурированы для хранения инструкций программ. Не предполагается, что настоящее раскрытие ограничено какими-либо конкретными машиночитаемыми носителями или любыми другими компьютерными программными продуктами, которые содержат инструкции и/или данные для выполнения реализуемых компьютером операций.
Примеры инструкций программы включают, но не ограничены, коды низкого уровня, такие как выдаваемый компилятором код, и файлы, содержащие код более высокого уровня, которые могут быть выполнены компьютером с применением интерпретатора. Кроме того, инструкции программы включают, но не ограничены, машинный код, исходный кодом и любой другой код, который прямо или косвенно управляет работой вычислительной машины в соответствии с настоящим раскрытием. Код может определять входные данные, выходные данные, вычисления, условные выражения, ветвления, итерационные циклы, и т.д.
В одном иллюстративном примере воплощающие код способы, изложенные в настоящем раскрытии, воплощены в фиксированных носителях или передаваемом программном компоненте, содержащем логические инструкции и/или данных, которые при загрузке в соответствующим образом сконфигурированное вычислительное устройство вызывает выполнение устройством моделирования генетической операции (GO) на одной или более символьных строках. На фигуре 4 показан пример цифрового устройства 800, которое представляет собой логическое устройство, которое может считывать инструкции с носителя 817, сетевого порта 819, клавиатуры 809 для ввода данных пользователем, устройства 811 пользовательского ввода, или других средств ввода. Устройство 800 может затем применять эти инструкции для направления статистических операций в пространстве данных, например, создания одного или более множеств данных (например, определения множества репрезентативных элементов пространства данных). Одним из типов логического устройства, которое может воплотить раскрытые варианты осуществления, является вычислительная система, такая как вычислительная система 800, содержащая ЦП 807, необязательную клавиатуру - устройство ввода данных пользователем 809, и манипулятор GUI 811, а также периферийные компоненты, такие как дисковые накопители 815 и монитор 805 (какие отображает модифицированные посредством ИДУТ символьные строки и обеспечивает упрощенный выбор подмножеств таких символьных строк пользователем). Несъемные носители 817, необязательно, применяются для программирования системы в целом и могут включать, например, оптические или магнитные носители данных дискового типа или другие электронный запоминающие элементы памяти. Коммуникационный порт 819 может использоваться для программирования системы и может представлять любой тип коммуникационного соединения.
Определенные варианты осуществления могут также быть воплощены в пределах электрической схемы специализированной интегральной схемы (ASIC) или программируемого логического устройства (PLD). В таком случае варианты осуществления реализуют на читаемом компьютером дескрипторном языке, который может применяться для создания ASIC или PLD. Некоторые варианты осуществления настоящего раскрытия реализованы в пределах электрической схемы или логических процессоров множества других цифровых устройств, таких как PDA, системы ноутбука, дисплеи, оборудование для редактирования изображений, и т.д.
В некоторых вариантах осуществления настоящее раскрытие относится к компьютерному программному продукту, содержащему один или более читаемых компьютером носителей, которые, при их выполнении одним или более процессорами вычислительной системы, вызывают выполнение вычислительной системой реализации способа для виртуального скрининга вариантов белков и/или направленной эволюции in silico белков, обладающих требуемой активностью. Такой способ может представлять собой любой способ, описанный в настоящем раскрытии, такой как способы, охваченные фигурами и псевдокодом. В некоторых вариантах осуществления, например, способ получает данные последовательности для множества ферментов, создает трехмерные модели на основе гомологии для биологических молекул, стыкует модели ферментов на основе гомологии с одним или более вычислительными представлениями субстратов, и выводит структурные данные, относящиеся к геометрическим параметрам, в отношении ферментов и субстратов. В некоторых вариантах осуществления способ может дополнительно разрабатывать модели активности последовательности посредством фильтрации данных в отношении смоделированных структурных данных. Библиотеки вариантов могут пользоваться в повторяющейся направленной эволюции, в результате чего могут быть получены ферменты с требуемыми выгодными свойствами.
В некоторых вариантах осуществления стыковка моделей ферментов на основе гомологии с одним или более вычислительными представлениями субстратов проводится программой докинга в компьютерной системе, которая использует вычислительное представление лиганда и вычислительные представления сайтов связывания множества вариантов в соответствии с описанным в настоящем раскрытии. В различных вариантах осуществления программа докинга оценивает энергию связывания между положением субстрата и ферментом. Для варианта белка, который успешно стыкуется с лигандом, система определяет геометрические значения в отношении участвующих лиганда и белка. В различных вариантах осуществления, компьютерная система конструирует модель активности последовательности посредством обучения соответствующих опорных векторов. В различных вариантах осуществления, компьютерная система применяет генетические алгоритмы для отфильтровывания неинформативных данных, в результате чего предоставляется подмножество данных для обучения опорных векторов.
XI. ВАРИАНТЫ ОСУЩЕСТВЛЕНИЯ В ВЕБ-САЙТАХ И ОБЛАЧНЫХ ВЫЧИСЛЕНИЯХ
Интернет включает в себя компьютеры, информационные устройства и компьютерные сети, которые соединены через линии связи. Соединенные компьютеры обмениваются информацией с использованием различных служб, таких как электронная почта, протокол передачи файлов, всемирная паутина («WWW») и другие службы, включая безопасные службы. Службу WWW можно понимать как разрешение вычислительной системе сервера (например, веб-сервера или Веб-сайта) посылать веб-страницы с информацией удаленному клиентскому информационному устройству или вычислительной системе. Удаленная клиентская вычислительная система может затем отобразить веб-страницы. Обычно, каждый ресурс (например, компьютер или веб-страница) WWW является уникально идентифицируемым посредством унифицированного локатора ресурса («URL»). Для того, чтобы просмотреть или взаимодействовать с конкретной веб-страницей, клиентская компьютерная система задает URL для этой веб-страницы в запросе. Запрос перенаправляется серверу, который поддерживает эту веб-страницу. Когда сервер получает запрос, он отправляет эту веб-страницу клиентской информационной системе. Когда клиентская компьютерная система получает эту веб-страницу, она может отобразить веб-страницу с использованием браузера, или может взаимодействовать с веб-страницей или интерфейсом иным способом. Браузер представляет собой логический модуль, который осуществляет запрос веб-страниц и отображение или взаимодействие с веб-страницами.
В настоящий момент, визуализируемые веб-страницы обычно задают с применением языка разметки гипертекста («HTML»). HTML предоставляет стандартный набор тегов, которые задают, каким образом веб-страница должна быть отображена. HTML Документ содержит различные теги, которые управляют отображением текста, графики, элементов управления и других особенностей. HTML Документ может содержать URL других веб-страниц, доступных в этой серверной компьютерной системе или других серверных компьютерных системах. URL могут также указывать другие типы интерфейсов, включая такие аспекты, как CGI-скрипты или исполняемые интерфейсы, которые информационные устройства используют для взаимодействия с удаленными информационными устройствами или серверами, не обязательно отображая информацию пользователю.
Интернет главным образом способствует предоставлению информационных услуг одному или более удаленным заказчикам. Услуги могут включать элементы (например, музыка или биржевые котировки), которые доставляют электронно покупателю через Интернет. Услуги могут также включать обработку заказов для элементов (например, продукты, книги, или химические или биологические соединения, и т.д.) которые могут быть доставлены через стандартные каналы распространения (например, общественные транспортные предприятия). Услуги могут также включать обработку заказов на элементы, такие как бронирование авиабилетов или билетов в театры, к которым покупатель будет осуществлять доступ позднее. Серверная компьютерная система может предоставлять электронную версию интерфейса, которая приводит список доступных элементов или услуг. Пользователь или потенциальный покупатель могут получить доступ к интерфейсам, используя браузер, и выбрать различные интересующие элементы. Когда пользователь завершил выбор требуемых элементов, серверная компьютерная система может затем запросить у пользователя информацию, необходимую для завершения услуги. Эта операционно-специфичная информация для заказа может включать в себя имя покупателя или другую идентификацию, идентификацию для оплаты (такую как корпоративный номер заказа на поставку или номер счета), или дополнительную информацию, необходимую для завершения услуги, такую как информация о рейсе.
Среди представляющих особенный интерес услуг, которые могут быть предоставлены через Интернет и по другим сетям, находятся биологические данные и биологические базы данных. Такие услуги включают множество услуг, предоставленных Национальным центром информации по биотехнологии (NCBI) Национальных институтов здравоохранения (NIH). NCBI отвечает за создание автоматизированных систем для хранения и анализа знаний о молекулярной биологии, биохимии и генетике; способствование использованию таких баз данных и программного обеспечения исследовательским и медицинским сообществом; координирование усилий по сбору биотехнологической информации из национальных и международных источников; и выполнение исследований относительно передовых способов компьютерной обработки информации для анализа структуры и функции биологически важных молекул.
NCBI отвечает за базу данных последовательностей ДНК GenBank®. База данных была создана из последовательностей, предоставленных индивидуальными лабораториями, и путем обмена данными с международными базами данных нуклеотидных последовательностей, Европейской лаборатории молекулярной биологии (EMBL) и базы данных ДНК Японии (DDBJ) и содержит данные по патентованным последовательностям, представленным Бюро США по патентам и товарным знакам. В дополнение к GenBank® NCBI поддерживает и распространяет множество баз данных для медицинских и научных сообществ. Они включают Онлайновую Менделевскую наследственность у индивида (OMIM), базу данных молекулярного моделирования (MMDB) трехмерных структур белка, совокупность уникальных последовательностей человеческих генов (UniGene), генную карту человеческого генома, браузер таксономии и проект по анатомии генома рака (CGAP), в сотрудничестве с Национальным онкологическим институтом. Entrez представляет собой систему поиска и извлечения данных NCBI, которая предоставляет пользователям интегрированный доступ к последовательностям, картированию, таксономии и структурным данным. Entrez также предоставляет графические представления последовательностей и хромосомных карт. Функцией Entrez является возможность извлечения родственных последовательностей, структур и ссылок. BLAST, как описано в настоящем раскрытии, является программой для поиска подобия последовательности, разработанной в NCBI для идентификации генов и генетических характеристик, которая может выполнять поиск последовательности во всей базе данных ДНК. Дополнительные программные инструменты, предоставляемые NCBI, включают в себя: средство обнаружения открытых рамок считывания (ORF finder), электронная ПЦР, и средства загрузки последовательностей, Sequin и BankIt. Различные базы данных и программные инструменты NCBI доступны по WWW или FTP или почтовым серверам. Дополнительная информация доступна по адресу www.ncbi.nlm.nih.gov.
Некоторые биологические данные, доступные по Интернету, представляют собой данные, которые обычно просматривают с помощью специального «плагина» к браузеру или другого исполнимого кода. Одним из примеров такой системы является CHIME, плагин к браузеру, который позволяет получать интерактивное виртуальное 3-мерное отображение молекулярных структур, включая биологические молекулярные структуры. Дополнительная информация относительно CHIME доступна по адресу www.mdlchime.com/chime/.
Множество компаний и учреждений предоставляют онлайновые системы для заказа биологических соединений.
Множество компаний и учреждений предоставляют онлайновые системы для заказа биологических соединений. Примеры таких систем можно найти по адресу www.genosys.com/oligo_custinfo.cfm или www.genomictechnologies.com/Qbrowser2_FP.html. Как правило, эти системы принимают некоторый дескриптор требуемого биологического соединения (такого как олигонуклеотид, цепь ДНК, цепь РНК, аминокислотную последовательность и т.д.), и затем запрашиваемое соединение производится и отправляется заказчику. Поскольку способы, представленные в настоящем раскрытии, могут быть реализованы на веб-сайте, как подробнее описано ниже, вычислительные результаты или физические результаты, включающие в себя полипептиды, или полинуклеотиды, продуцированные некоторыми вариантами осуществления раскрытия, могут быть предоставлены через Интернет способами, аналогичными биологической информации и соединениям, описанным выше.
В качестве дополнительной иллюстрации, способы по настоящему изобретению могут быть реализованы в локализованной или распределенной вычислительной среде. В распределенной среде способы могут быть реализованы на единственном компьютере, содержащем множество процессоров, или на множестве компьютеров. Компьютеры могут быть соединены, например, через общую шину, но, более предпочтительно, компьютер (-ы) представляют собой узлы в сети. Сеть может представлять собой обобщенную или выделенную локальную или глобальную сеть, и, в определенных предпочтительных вариантах осуществления, компьютеры могут являться компонентами сети Интранет или Интернет.
В одном из Интернет-вариантов осуществления клиентская система обычно выполняет веб-браузер и соединена с серверным компьютером, выполняющим веб-сервер. Веб-браузер обычно представляет собой программу, такую как IBM Web Explorer, Microsoft Internet Explorer, NetScape, Opera или Mosaic. Веб-сервер обычно, но не обязательно, представляет собой программу, такую как IBM HTTP Daemon или другой www-процесс-демон (например, формы программы для LINUX). Клиентский компьютер имеет двустороннее соединение с серверным компьютером по линии или через беспроводную систему. В свою очередь, серверный компьютер имеет двустороннее соединение с веб-сайтом (сервером, на котором размещен веб-сайт), предоставляющим доступ к программному обеспечению, реализующему способы по настоящему изобретению.
Как указано, пользователь клиента, соединенного с сетью Интранет или Интернет, может вызывать выполнение клиентом запроса ресурсов, которые являются частью веб-сайта(-ов), на котором(-ых) размещено(-ы) приложение(-я), обеспечивающее(-ие) реализацию способов по настоящему изобретению. Серверная(-ые) программа(-ы) затем обрабатывает(-ют) запрос с возвращением указанных ресурсов (если предположить, что они в настоящий момент доступны). Стандартное соглашение о присвоении имен (то есть, унифицированный локатор ресурса («URL»)) охватывает несколько типов названий размещений, в настоящий момент включающих в себя такие подклассы, как протокол передачи гипертекста («http»), протокол передачи файлов («ftp»), протокол gopher и глобальная служба информации («WAIS»). Когда ресурс скачан, он может содержать URL дополнительных ресурсов. Таким образом, пользователь клиента может легко узнать о существования новых ресурсов, которые он или она не запрашивали в явном виде.
Программное обеспечение, реализующее способ(-ы) по настоящему изобретению, может работать локально на сервере, на котором размещен веб-сайт, в истинной клиент-серверной архитектуре. Таким образом, клиентский компьютер направляет запрос хост-серверу, который выполняет требуемый(-е) процесс(-ы) локально и затем выгружает результаты обратно клиенту. Альтернативно, способы по настоящему изобретению могут быть реализованы в «многоуровневом» формате, в котором компоненты способа(-ов) выполняются локально клиентом. Это может быть реализовано посредством программного обеспечения, загружаемого с сервера по запросу клиентом (например, Java-приложение), или это может быть реализовано посредством программного обеспечения, «постоянно» установленного на клиенте.
В одном из вариантов осуществления приложение(-я), реализующее(-ие) способы по настоящему изобретению разделено(-ы) на группы. В этой парадигме полезно рассматривать приложение не столько как совокупность характеристик или как функциональность, но, вместо этого, как совокупность дискретных групп или представлений. Типичное приложение, например, обычно включает в себя множество пунктов меню, каждый из которых вызывает конкретную группу, то есть форму, которая демонстрирует определенную функциональность приложения. С этой перспективы приложение рассматривается не как монолитное тело кода, но как совокупность апплетов, или пакетов функциональности. Таким образом, в браузере пользователь выбирал бы ссылку на веб-страницу, которая, в свою очередь, вызовет конкретную группу приложения (то есть, подприложение). Таким образом, например, одна или более групп могут предоставлять функциональность для ввода и/или кодирования биологической(-их) молекулы(-л) в одно или более пространств данных, в то время как другая группа предоставляет инструменты для оптимизации модели пространства данных.
В определенных вариантах осуществления способы по настоящему изобретению реализованы как одна или более групп, предоставляющих, например, следующие функциональности: функцию(-и) для кодирования двух или более биологических молекул в символьные строки с целью предоставления совокупности двух или более различных исходных символьных строк, в которой каждая из указанных биологических молекул содержит выбранное множество субъединиц; функции для выбора по меньшей мере двух подстрок из символьных строк; функции для соединения подстрок с целью формирования одной или более строк продукта, имеющих приблизительно такую же длину, как одна или более исходных символьных строк; функции для добавления (помещения) строк продуктов к совокупности строк; функции для создания и манипулирования вычислительным представлением/моделями ферментов и субстратов, функции для докинга вычислительного представления субстрата (например, лиганда) с вычислительным представлением фермента (например, белка); функции для применения молекулярной динамики к молекулярным моделям; функции для вычисления различных ограничений между молекулами, которые влияют на химические реакции, в которые вовлекаются молекулы (например, расстояние или угол между функциональной составляющей субстрата и активным центром фермента); и функции для реализации любого множества характеристик, изложенных в настоящем раскрытии.
Одна или более из этих функциональностей может также быть реализована исключительно на сервере или на клиентском компьютере. Эти функции, например, функции для создания или манипулирования вычислительными моделями биологических молекул, могут предоставлять одно или более окон, в которые пользователь может вставлять представление(-я) или манипулировать представлением(-ями) биологических молекул. Кроме того, функции также, дополнительно, предоставляют доступ к частным и/или общедоступным базам данных, доступным через локальную сеть и/или сеть интранет, посредством чего одна или более последовательностей, содержащихся в базах данных, могут быть введены в способы по настоящему изобретению. Таким образом, например, в одном из вариантов осуществления, пользователь может, необязательно, иметь возможность запросить поиск в GenBank® и ввести одну или более последовательностей, возвращенных таким поиском, в функцию кодирования и/или генерации разнообразия.
Способы реализации сети Интранет и/или Интранет-вариантов осуществления вычислительных процессов и/или процессов доступа к данным известны специалистам в данной области техники и очень подробно задокументированы (см., например, Cluer и соавт. (1992) «A General Framework for the Optimization of Object-Oriented Queries», Proc SIGMOD International Conference on Management of Data, San Diego, California, Jun. 2-5, 1992, SIGMOD Record, vol. 21, Issue 2, Jun., 1992; Stonebraker, M., Editor; ACM Press, pp. 383-392; ISO-ANSI, Working Draft, «Information Technology-Database Language SQL», Jim Melton, Editor, International Organization for Standardization and American National Standards Institute, Jul. 1992; Microsoft Corporation, «ODBC 2.0 Programmer's Reference and SDK Guide. The Microsoft Open Database Standard for Microsoft Windows.TM and Windows NTTM, Microsoft Open Database Connectivity.TM. Software Development Kit», 1992, 1993, 1994 Microsoft Press, pp. 3-30 and 41-56; ISO Working Draft, «Database Language SQL-Part 2: Foundation (SQL/Foundation)», CD9075-2:199.chi.SQL, Sep. 11, 1997, и т.п.). Дополнительные соответствующие подробности относительно веб-приложений можно найти в WO 00/42559, озаглавленной «METHODS OF POPULATING DATA STRUCTURES FOR USE IN EVOLUTIONARY SIMULATIONS», авторы Selifonov и Stemmer.
В некоторых вариантах осуществления способы для исследования, скрининга и/или разработки полинуклеотида или последовательностей полипептида могут быть реализованы как многопользовательская система на компьютерной системе со множеством процессорных элементов и модулей памяти, распределенных по компьютерной сети, при этом сеть может включать сеть Интранет на LAN и/или Интернет. В некоторых вариантах осуществления распределенная вычислительная архитектура включает в себя «облако», которое представляет собой совокупность вычислительных систем, доступных по компьютерной сети для вычисления и хранения данных. Вычислительная среда, включающая в себя облако, называется облачной вычислительной средой. В некоторых вариантах осуществления один или более пользователей могут получить доступ к компьютерам облака, распределенным по сети Интранет и/или Интернет. В некоторых вариантах осуществления пользователь может удаленно получить доступ, через веб-клиент, к компьютерам сервера, которые реализуют способы для скрининга и/или разработки вариантов белка, описанных выше.
В некоторых вариантах осуществления, включающих в себя облачную вычислительную среду, виртуальные машины (VM) настроены на компьютерах сервера, и результаты виртуальных машин могут быть отправлены назад пользователю. Виртуальная машина (VM) является основанной на программном обеспечении эмуляцией компьютера. Виртуальные машины могут быть основаны на спецификациях гипотетического компьютера или эмулировать архитектуру ЭВМ и функции компьютера реального мира. Структура и функции VM известны в технике. Как правило, VM установлена на хост-платформе, которая включает в себя системные аппаратные средства, и сама VM включает в себя виртуальные системные аппаратные средства и гостевое программное обеспечение.
Аппаратные средства хост-системы для VM включают в себя один или более центральных процессоров (ЦП), память, один или более жестких дисков и различные другие устройства. Виртуальные системные аппаратные средства VM включают в себя один или более виртуальных ЦП, виртуальную память, один или более виртуальных жестких дисков и одно или более виртуальных устройств. Гостевое программное обеспечение VM включает в себя гостевое системное программное обеспечение и гостевые приложения. В некоторых реализациях гостевое системное программное обеспечение включает в себя гостевую операционную систему с драйверами для виртуальных устройств. В некоторых реализациях гостевые приложения VM включают в себя по меньшей мере один экземпляр системы виртуального скрининга белка в соответствии с описанным выше.
В некоторых вариантах осуществления число предусмотренных VM может масштабироваться в зависимости от вычислительной нагрузки задачи, которая будет решаться. В некоторых вариантах осуществления пользователь может запросить виртуальную машину в облаке, при этом VM включает в себя систему виртуального скрининга. В некоторых вариантах осуществления облачная вычислительная среда может предоставить VM на основании пользовательского запроса. В некоторых вариантах осуществления VM может существовать в виде ранее сохраненного образа VM, который может храниться в хранилище образов. Облачная вычислительная среда может искать и передавать образ на сервер или в пользовательскую систему. Облачная вычислительная среда может затем загружать образ на сервере или в пользовательской системе.
Несмотря на то что приведенное выше было описано с некоторыми подробностями для целей ясности и понимания, специалисту в данной области техники после прочтения настоящего раскрытия будет ясно, что различные изменения в форме и деталях могут быть произведены без отклонения от фактического объема изобретения. Например, все методики и устройство, описанные выше, могут применяться в различных комбинациях. Все публикации, патенты, заявки на патент или другие документы, процитированные в настоящей заявке, включены посредством ссылки во всей их полноте для всех целей до той же самой степени, как если бы каждая индивидуальная публикация, патент, заявка на патент или другой документ были бы индивидуально указаны как включенные посредством ссылки для всех целей.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБЫ, СИСТЕМЫ И ПРОГРАММНОЕ ОБЕСПЕЧЕНИЕ ДЛЯ ИДЕНТИФИКАЦИИ БИОМОЛЕКУЛ С ПОМОЩЬЮ МОДЕЛЕЙ МУЛЬТИПЛИКАТИВНОЙ ФОРМЫ | 2014 |
|
RU2664695C2 |
| СПОСОБЫ, СИСТЕМЫ И ПРОГРАММНОЕ ОБЕСПЕЧЕНИЕ ДЛЯ ИДЕНТИФИКАЦИИ БИОМОЛЕКУЛ СО ВЗАИМОДЕЙСТВУЮЩИМИ КОМПОНЕНТАМИ | 2014 |
|
RU2695146C2 |
| СПОСОБЫ И СРЕДСТВА ПОЛУЧЕНИЯ БИБЛИОТЕКИ ДЛЯ СЕКВЕНИРОВАНИЯ | 2019 |
|
RU2815513C2 |
| СИСТЕМА АНАЛИЗА ДЛЯ ОРТОГОНАЛЬНОГО ДОСТУПА К БИОМОЛЕКУЛАМ И ИХ МЕЧЕНИЯ В КЛЕТОЧНЫХ КОМПАРТМЕНТАХ | 2017 |
|
RU2771892C2 |
| ПЛАТФОРМА ДЛЯ ОБНАРУЖЕНИЯ И АНАЛИЗА ТЕРАПЕВТИЧЕСКИХ АГЕНТОВ | 2016 |
|
RU2724998C2 |
| КЛАССИФИКАЦИЯ САЙТОВ СПЛАЙСИНГА НА ОСНОВЕ ГЛУБОКОГО ОБУЧЕНИЯ | 2018 |
|
RU2780442C2 |
| АМПЛИФИКАЦИЯ БИБЛИОТЕК НУКЛЕИНОВЫХ КИСЛОТ С ПРИМЕНЕНИЕМ КИНЕТИЧЕСКОГО ИСКЛЮЧЕНИЯ | 2017 |
|
RU2759690C2 |
| СПОСОБ И СИСТЕМЫ ДЛЯ ПРОГНОЗИРОВАНИЯ СПЕЦИФИЧЕСКИХ ДЛЯ HLA КЛАССА II ЭПИТОПОВ И ОХАРАКТЕРИЗАЦИИ CD4+ T-КЛЕТОК | 2019 |
|
RU2826261C2 |
| СПОСОБЫ И ПРИБОРЫ ДЛЯ ОБНАРУЖЕНИЯ И ИДЕНТИФИКАЦИИ ЗАКОДИРОВАННЫХ ГРАНУЛ И БИОЛОГИЧЕСКИХ МОЛЕКУЛ | 2007 |
|
RU2487169C2 |
| ИДЕНТИФИКАЦИЯ, ПРОИЗВОДСТВО И ПРИМЕНЕНИЕ НЕОАНТИГЕНОВ | 2016 |
|
RU2729116C2 |
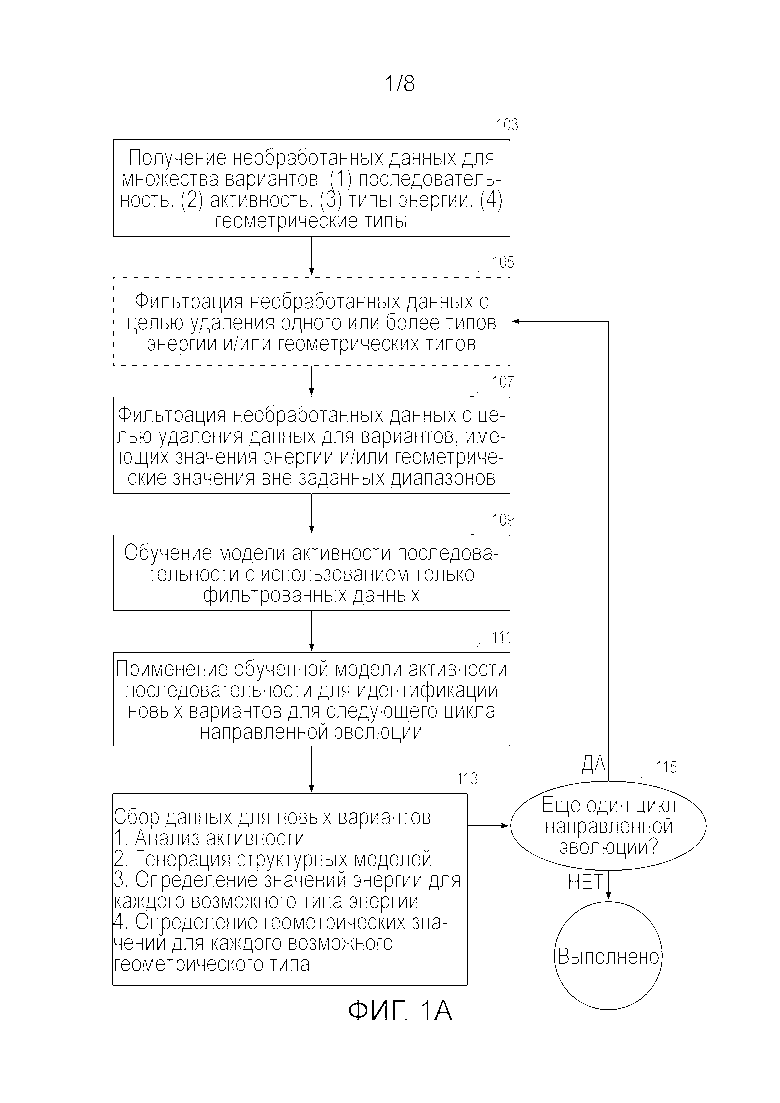
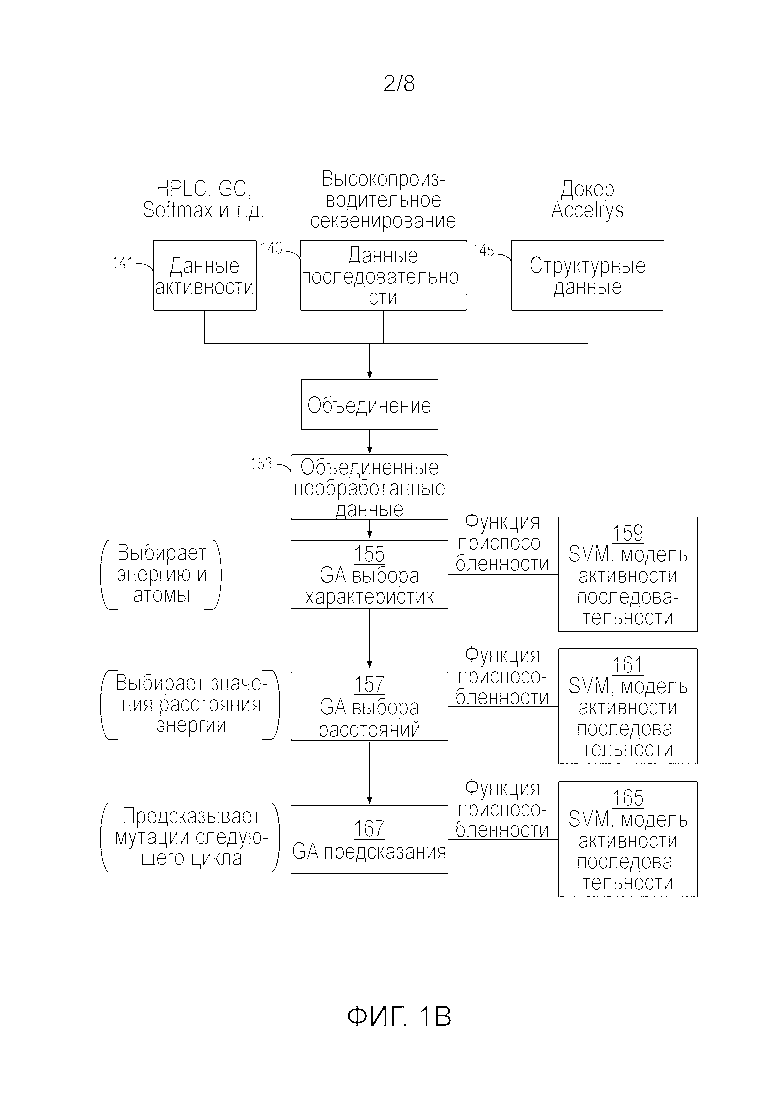
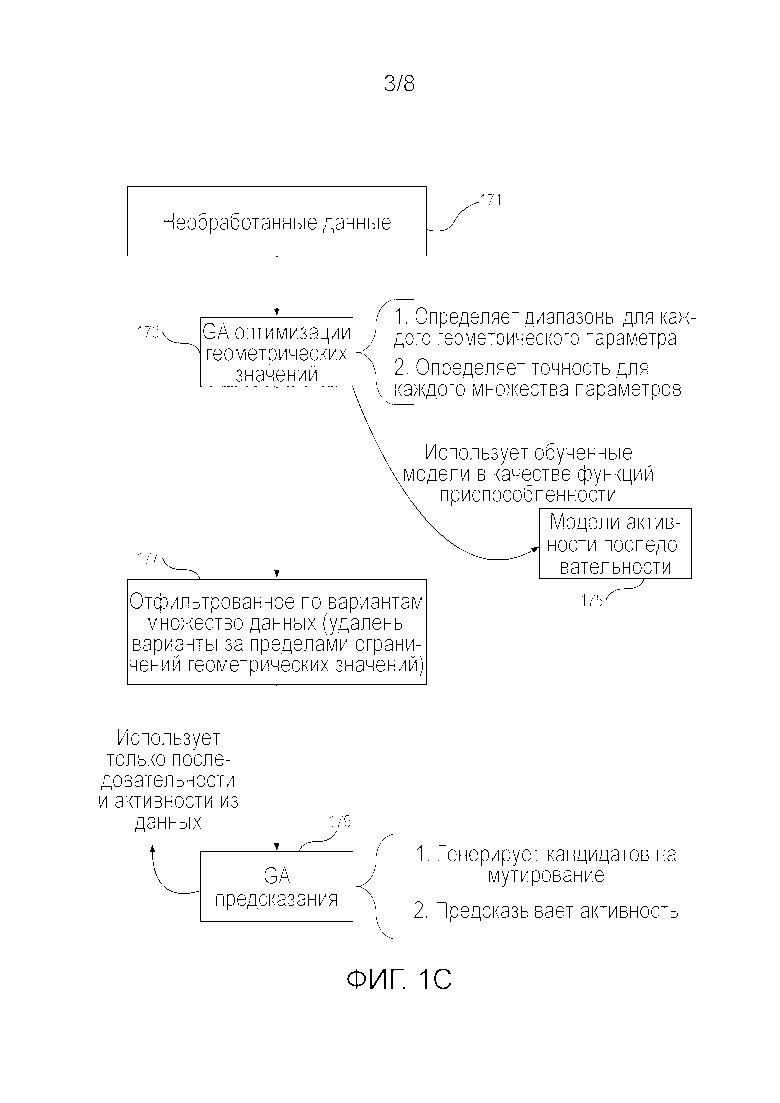

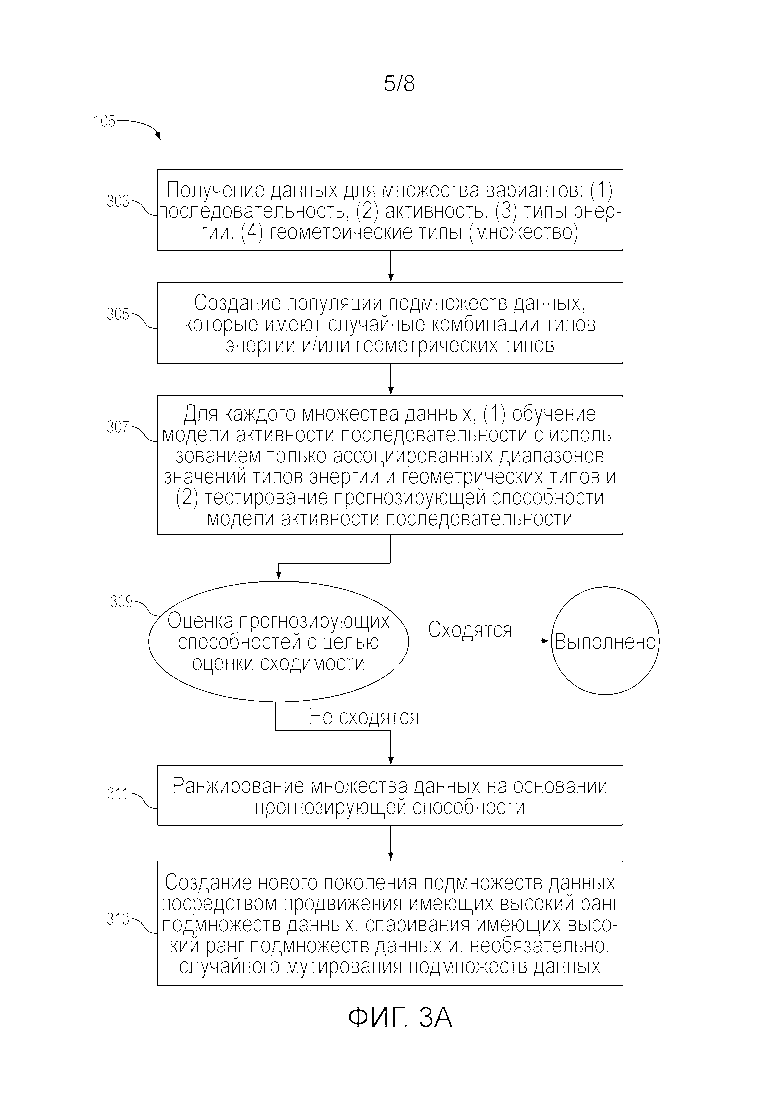

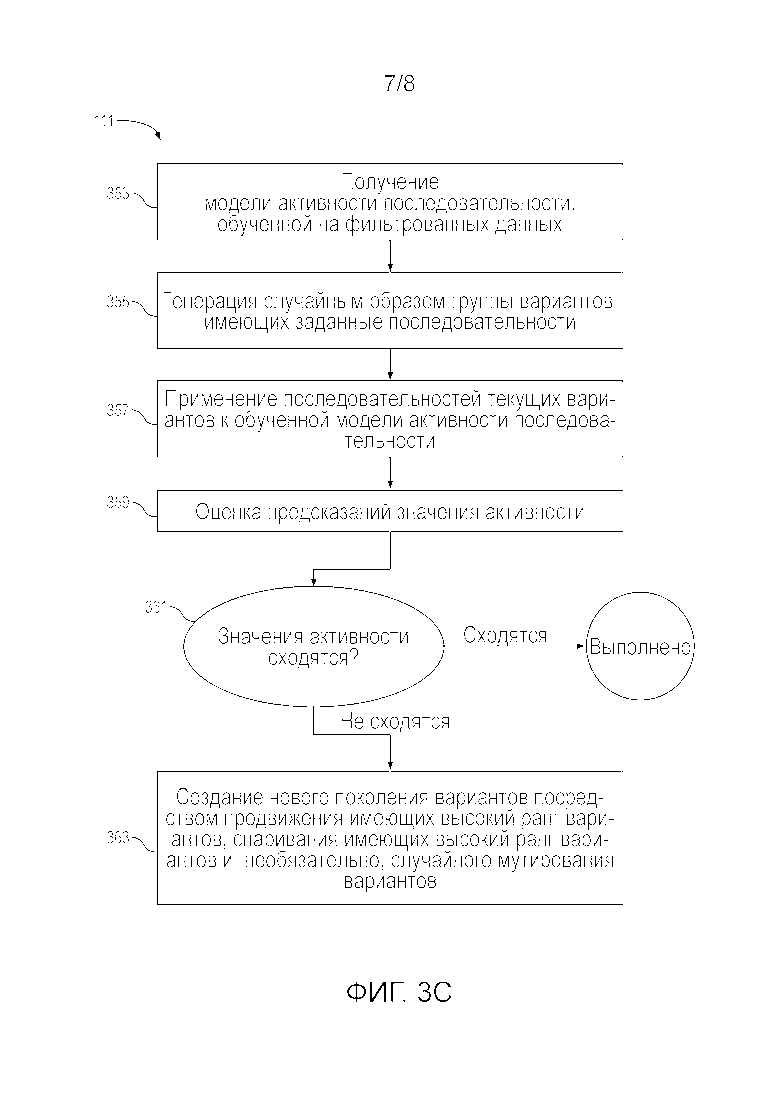

Настоящее изобретение относится к биоинформатике. Предложен способ проведения направленной эволюции одной или более полипептидной или полинуклеотидной молекулы, а также считываемый компьютером невременный носитель и система для выполнения упомянутого способа. Данное изобретение обеспечивает получение полипептидных и полинуклеотидных молекул, имеющих улучшенную активность. 3 н. и 27 з.п. ф-лы, 4 ил.
1. Способ проведения направленной эволюции одной или более полипептидной или полинуклеотидной молекул с использованием компьютера, при этом способ содержит:
(a) получение посредством компьютера набора данных, содержащих следующую информацию для каждой из множества вариантных биомолекул: (i) величину активности вариантной биомолекулы с лигандом в сайте связывания вариантной биомолекулы, (ii) последовательность вариантной биомолекулы, и (iii) величину геометрического параметра для каждого параметра из одного или более геометрических параметров, характеризующих геометрию лиганда в сайте связывания вариантной биомолекулы, при этом вариантная биомолекула представляет собой полипептидную молекулу или полинуклеотидную молекулу;
(b) получение посредством компьютера множества поднаборов данных из набора данных путем удаления, из каждого поднабора данных, данных любой из по меньшей мере одной вариантной биомолекулы из множества вариантных биомолекул, при этом по меньшей мере одна вариантная биомолекула имеет по меньшей мере одну величину геометрического параметра из по меньшей мере одного геометрического параметра лиганда в сайте связывания по меньшей мере одной вариантной биомолекулы, при этом значение по меньшей мере одного геометрического параметра находится вне одного или более пределов, определенных поднабором данных;
(c) обучение посредством одного или более процессоров множества моделей, причем каждый использует различные поднаборы данных из множества поднаборов данных, при этом каждая модель из множества моделей содержит последовательность данных и по меньшей мере один из одного или более геометрических параметров в качестве независимых переменных и прогнозируемое значение активности в качестве зависимой переменной;
(d) определение посредством одного или более процессоров прогнозируемой способности каждой модели из множества моделей для точного прогноза активности биомолекул;
(е) выбор одним или более процессорами по меньшей мере одной модели из множества моделей на основе по меньшей мере частично прогнозируемой способности по меньшей мере одной модели;
(е’) выбор одного или более поднаборов данных, которые использовались для обучения одной или более моделей, выбранных на этапе (е);
(e’’) формирование одного или более новых поднаборов данных, используя один или более поднаборов данных, выбранных на этапе (e’);
(e’’’) повтор этапов (b-e) или (b-e’’) по меньшей мере один раз, используя один или более новых поднаборов данных, сформированных на этапе (e’’) в качестве одного или более поднаборов данных из множества поднаборов данных этапа (b);
(f) определение одним или более процессорами одной или более последовательностей аминокислот или одной или более последовательностей нуклеиновых кислот, используя выбранную по меньшей мере одну модель или новую модель, обученную поднабором данных, который использовался для обучения указанной по меньшей мере одной выбранной модели на этапе (е), при этом этапы (a-е) выполняют на компьютере, содержащем один или более процессоров и системную память;
(g) синтез или экспрессию одной или более молекул аминокислоты или одной или более молекул нуклеиновой кислоты на основе одной или более последовательностей аминокислот или нуклеиновых кислот; и
(h) рекомбинацию или осуществление мутагенеза одной или более молекул аминокислоты или одной или более молекул нуклеиновой кислоты, полученных на шаге (g), для получения одной или более полипептидной или полинуклеотидной молекул.
2. Способ по п.1, в котором обучение на шаге (с) множества данных выполняется с помощью генетического алгоритма.
3. Способ по п.1, в котором генетический алгоритм изменяет пороги для удаления информации, ассоциированной с геометрическими параметрами для одной или более вариантных биомолекул, при этом один или более пределов являются различными для множества поднаборов данных.
4. Способ по п.1, в котором прогнозируют, что одна или более молекул аминокислоты или одна или более молекул нуклеиновой кислоты имеет активность, удовлетворяющую одному или более критериям в соответствии с выбранной одной моделью или новой моделью, при этом каждая из одной или более молекул аминокислоты или одной или более молекул нуклеиновой кислоты имеет последовательность, отличающуюся от последовательностей множества вариантов биомолекул, предоставляющих информацию для множества данных (a).
5. Способ по п.4, в котором шаг (f) содержит выполнение генетического алгоритма, в котором потенциальные новые варианты биомолекул оценивают с применением выбранной по меньшей мере одной модели или новой модели в качестве функции пригодности.
6. Способ по п.4, дополнительно включающий в себя анализ активности одной или более молекул аминокислоты или одной или более молекул нуклеиновой кислоты.
7. Способ по п.4, дополнительно включающий в себя
создание структурной модели для каждой одной или более молекул аминокислоты или одной или более молекул нуклеиновой кислоты; и
применение структурных моделей для генерации геометрических параметров для сайтов связывания одной или более молекул аминокислоты или одной или более молекул нуклеиновой кислоты, при этом геометрические параметры характеризуют геометрию лиганда в сайтах связывания одной или более молекул аминокислоты или одной или более молекул нуклеиновой кислоты.
8. Способ по п.1, дополнительно включающий в себя получение структурных моделей вариантов биомолекул и определение одного или более геометрических параметров с использованием структурных моделей.
9. Способ по п. 8, в котором структурные модели представляют собой модели на основе гомологии.
10. Способ по п.8, в котором модели на основе гомологии подготавливают с использованием деталей измерений физической структуры биомолекул.
11. Способ по п.10, в котором детали измерений физической структуры биомолекул включают в себя трехмерные позиции атомов, полученные посредством ядерно-магнитного резонанса (NMR) или рентгеноструктурной кристаллографии.
12. Способ по п.8, дополнительно содержащий применение докера для определения одного или более геометрических параметров.
13. Способ по п.1, в котором информация для каждой из множества вариантных биомолекул дополнительно содержит (iv) энергию взаимодействия, характеризующую взаимодействие лиганда в сайте связывания.
14. Способ по п.13, дополнительно включающий в себя применение докера для определения энергии взаимодействия.
15. Способ по п.1, в котором выбранную по меньшей мере одну модель или новую модель получают посредством машины опорных векторов, множественной линейной регрессии, регрессии главных компонент, регрессии методом дробных наименьших квадратов или нейронной сети.
16. Способ по п.15, в котором выбранную по меньшей мере одну модель или новую модель получают посредством машины опорных векторов.
17. Способ по п.1, в котором множество вариантных биомолекул содержит множество ферментов.
18. Способ по п.17, в котором активность вариантной биомолекулы на лиганде является активностью фермента на субстрате.
19. Способ по п.18, в котором активность фермента на субстрате включает в себя одну или более характеристик каталитического преобразования субстрата ферментом.
20. Способ по п. 1, в котором множество наборов данных на шаге (b) получают путем удаления, для каждого набора данных, данных по меньшей мере одного параметра из одного или более геометрических параметров, а также данные любого из по меньшей мере одного варианта биомолекулы, имеющей любое значение геометрического параметра, находящееся вне одного или более пределов, определенных для набора данных.
21. Способ по п. 1, дополнительно содержащий шаги, выполняемые до шага (а):
обеспечивают множество наборов данных, содержащих набор данных, используемый на шаге (а);
обучают множество предварительных моделей с использованием различных наборов данных из множества наборов данных;
выбирают по меньшей мере одну предварительную модель из множества предварительных моделей на основе по меньшей мере частично прогнозируемой способности по меньшей мере одной предварительной модели; и
получают набор данных для шага (а) из по меньшей мере одного набора данных, который использовался для обучения по меньшей мере одной выбранной предварительной модели.
22. Способ по п. 1, в котором каждый набор данных из множества наборов данных имеет: (i) величину активности вариантной биомолекулы, (ii) последовательность вариантной биомолекулы, и (iii) величину геометрического параметра для каждого параметра из набора геометрических параметров, при этом каждый набор данных из множества наборов данных имеет набор геометрических параметров, отличающийся от каждого другого набора данных из множества наборов данных.
23. Способ по п. 1, в котором получение набора данных на шаге (a) из по меньшей мере одного набора данных, который использовался для обучения по меньшей мере одной выбранной предварительной модели, содержит объединение двух или более наборов данных, которые использовались для обучения двух или более выбранных предварительных моделей.
24. Способ по п. 1, в котором по меньшей мере одна модель, выбранная на шаге (е), имеет прогнозируемую способность, которая соответствует критерию.
25. Способ по п. 1, в котором выбранная по меньшей мере одна модель содержит две или более моделей, и обучают новую модель с использованием данных, полученных путем объединения двух или более поднаборов данных, которые использовались для обучения двух или более моделей.
26. Способ по п. 1, в котором определяют одну или более последовательностей аминокислот или одной или более последовательностей нуклеиновых кислот с использованием новой модели.
27. Способ по п. 1, в котором обучают новую модель с использованием данных, полученных путем мутации данных поднабора, который использовался для обучения по меньшей мере одной выбранной модели.
28. Способ по п. 1, в котором в новой модели исключен по меньшей мере один из одного или более геометрических параметров.
29. Считываемый компьютером невременный носитель информации, на котором хранятся исполняемые компьютером инструкции, которые, при их выполнении одним или более процессорами системы, содержащей систему синтеза, один или более процессоров и системную память, обеспечивают исполнение системой способа для проведения направленной эволюции одной или более полипептидной или полинуклеотидной молекул по п. 1, при этом способ включает в себя:
(a) получение посредством компьютера набора данных, содержащих следующую информацию для каждой из множества вариантных биомолекул: (i) величину активности вариантной биомолекулы с лигандом в сайте связывания вариантной биомолекулы, (ii) последовательность вариантной биомолекулы, и (iii) величину геометрического параметра для каждого параметра из одного или более геометрических параметров, характеризующих геометрию лиганда в сайте связывания вариантной биомолекулы, при этом вариантная биомолекула представляет собой полипептидную молекулу или полинуклеотидную молекулу;
(b) получение посредством компьютера множества поднаборов данных из набора данных путем удаления, из каждого поднабора данных, данных любой из по меньшей мере одной вариантной биомолекулы из множества вариантных биомолекул, при этом по меньшей мере одна вариантная биомолекула имеет по меньшей мере одну величину геометрического параметра из по меньшей мере одного геометрического параметра лиганда в сайте связывания по меньшей мере одной вариантной биомолекулы, при этом значение по меньшей мере одного геометрического параметра находится вне одного или более пределов, определенных поднабором данных; и
(c) обучение посредством одного или более процессоров множества моделей, причем каждый использует различные поднаборы данных из множества поднаборов данных, при этом каждая модель из множества моделей содержит последовательность данных и по меньшей мере один из одного или более геометрических параметров в качестве независимых переменных и прогнозируемое значение активности в качестве зависимой переменной;
(d) определение посредством одного или более процессоров прогнозируемой способности каждой модели из множества моделей для точного прогноза активности биомолекул,
(е) выбор одним или более процессорами по меньшей мере одной модели из множества моделей на основе по меньшей мере частично прогнозируемой способности по меньшей мере одной модели;
(е’) выбор одного или более поднаборов данных, которые использовались для обучения одной или более моделей, выбранных на этапе (е);
(e’’) формирование одного или более новых поднаборов данных, используя один или более поднаборов данных, выбранных на этапе (e’);
(e’’’) повтор этапов (b-e) или (b-e’’) по меньшей мере один раз, используя один или более новых поднаборов данных, сформированных на этапе (e’’) в качестве одного или более поднаборов данных из множества поднаборов данных этапа (b);
(f) определение одной или более последовательностей аминокислот или одной или более последовательностей нуклеиновых кислот, используя выбранную по меньшей мере одну модель или новую модель, обученную поднабором данных, который использовался для обучения указанной по меньшей мере одной выбранной модели на этапе (е);
(g) синтез или экспрессию одной или более молекул аминокислоты или одной или более молекул нуклеиновой кислоты на основе одной или более последовательностей аминокислот или нуклеиновых кислот; и
(h) рекомбинацию или осуществление мутагенеза одной или более молекул аминокислоты или одной или более молекул нуклеиновой кислоты, полученных на шаге (g), для получения одной или более полипептидной или полинуклеотидной молекул;
при этом шаги (g) и (h) выполняют системой синтеза.
30. Система для проведения направленной эволюции одной или более полипептидной или полинуклеотидной молекул, содержащая:
систему синтеза;
один или более процессоров;
системную память; и
один или более читаемых компьютером носителей, на которых хранятся исполняемые компьютером инструкции, которые при их выполнении одним или более процессорами обеспечивают выполнение системой способа для проведения направленной эволюции одной или более полипептидной или полинуклеотидной молекул по п. 1, при этом способ включает в себя:
(a) получение посредством компьютера набора данных, содержащих следующую информацию для каждой из множества вариантных биомолекул: (i) величину активности вариантной биомолекулы с лигандом в сайте связывания вариантной биомолекулы, (ii) последовательность вариантной биомолекулы, и (iii) величину геометрического параметра для каждого параметра из одного или более геометрических параметров, характеризующих геометрию лиганда в сайте связывания вариантной биомолекулы, при этом вариантная биомолекула представляет собой полипептидную молекулу или полинуклеотидную молекулу;
(b) получение посредством компьютера множества поднаборов данных из набора данных путем удаления, из каждого поднабора данных, данных любой из по меньшей мере одной вариантной биомолекулы из множества вариантных биомолекул, при этом по меньшей мере одна вариантная биомолекула имеет по меньшей мере одну величину геометрического параметра из по меньшей мере одного геометрического параметра лиганда в сайте связывания по меньшей мере одной вариантной биомолекулы, при этом значение по меньшей мере одного геометрического параметра находится вне одного или более пределов, определенных поднабором данных; и
(c) обучение посредством одного или более процессоров множества моделей, причем каждый использует различные поднаборы данных из множества поднаборов данных, при этом каждая модель из множества моделей содержит последовательность данных и по меньшей мере один из одного или более геометрических параметров в качестве независимых переменных и прогнозируемое значение активности в качестве зависимой переменной;
(d) определение посредством одного или более процессоров прогнозируемой способности каждой модели из множества моделей для точного прогноза активности биомолекул,
(е) выбор одним или более процессорами по меньшей мере одной модели из множества моделей на основе по меньшей мере частично прогнозируемой способности по меньшей мере одной модели;
(е’) выбор одного или более поднаборов данных, которые использовались для обучения одной или более моделей, выбранных на этапе (е);
(e’’) формирование одного или более новых поднаборов данных, используя один или более поднаборов данных, выбранных на этапе (e’);
(e’’’) повтор этапов (b-e) или (b-e’’) по меньшей мере один раз, используя один или более новых поднаборов данных, сформированных на этапе (e’’) в качестве одного или более поднаборов данных из множества поднаборов данных этапа (b);
(f) определение одним или более процессорами одной или более последовательностей аминокислот или одной или более последовательностей нуклеиновых кислот, используя выбранную по меньшей мере одну модель или новую модель, обученную поднабором данных, который использовался для обучения указанной по меньшей мере одной выбранной модели на этапе (е), при этом этапы (a-е) выполняют на компьютере, содержащем один или более процессоров и системную память;
(g) синтез или экспрессию одной или более молекул аминокислоты или одной или более молекул нуклеиновой кислоты на основе одной или более последовательностей аминокислот или нуклеиновых кислот; и
(h) рекомбинацию или осуществление мутагенеза одной или более молекул аминокислоты или одной или более молекул нуклеиновой кислоты, полученных на шаге (g), для получения одной или более молекул полипептидов или полинуклеотидов;
при этом шаги (g) и (h) выполняют системой синтеза.
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| УСТАНОВКА ДЛЯ НЕПРЕРЫВНОЙ РАЗЛИВКИ СТАЛЬНЫХ СЛИТКОВ | 1998 |
|
RU2216429C2 |
| CHAPARRO-RIGGERS, Javier F., et al., "Better library design: data-driven protein engineering." Biotechnology journal, 2007, 2(2): 180-191 | |||
| YANG, JINN-MOON, CHUN-CHEN CHEN | |||
| "GEMDOCK: a generic evolutionary method for molecular docking." Proteins: Structure, Function, and Bioinformatics, 2004, 55(2): 288-304 | |||
| VILAR S, COSTANZI S | |||
| "Predicting the biological activities through QSAR analysis and docking-based scoring." Methods Mol Biol., 2012, 914:271-84. | |||

