[Область техники]
Настоящее изобретение относится к производным 1,3,4-оксадиазолсульфамида, имеющим ингибирующую активность в отношении гистондеацетилазы 6 (HDAC6), их стереоизомерам или их фармацевтически приемлемым солям; к их применению для получения терапевтических лекарственных средств; к способам лечения заболеваний с их использованием; к содержащим их фармацевтическим композициям; и к способам их получения.
[Уровень техники]
Посттрансляционные модификации, такие как ацетилирование, являются очень важными регулирующими модулями в основе биологических процессов в клетках и жестко регулируются множеством ферментов. Гистоны являются основными белковыми компонентами хроматина и действуют как катушки, вокруг которых цепи ДНК. Также, баланс ацетилирования и деацетилирования гистонов является важной ролью в регуляции экспрессии генов.
Гистондеацетилазы (HDAC) представляют собой ферменты, которые удаляют ацетильные группы из лизиновых остатков на гистоновых белках хроматина и, как известно, связаны с сайленсингом гена и индукцией остановки клеточного цикла, ингибированием ангиогенеза, иммунной регуляцией, гибелью клетки и тому подобное (Hassig et al., Curr. Opin. Chem. Biol. 1997, 1, 300-308). Кроме того, сообщалось, что ингибирование ферментативной функции HDAC индуцирует апоптоз раковых клеток in vivo за счет уменьшения активности факторов, связанных с выживаемостью раковых клеток, и активации факторов, связанных с апоптозом раковых клеток (Warrell et al, J. Natl. Cancer Inst. 1998, 90, 1621-1625).
У людей было идентифицировано 18 HDAC и разделены на четыре класса, основанные на их гомологии с дрожжевыми HDAC. Среди них 11 HDAC имеют цинк в качестве кофактора и могут быть разделены на три группы: I класс (HDAC1, 2, 3 и 8), II класс (IIa: HDAC4, 5, 7 и 9; IIb: HDAC6 и 10), IV класс (HDAC 11). Кроме того, 7 HDAC III класса (SIRT 1-7) требуют NAD+ вместо цинка в качестве кофактора (Bolden et al., Nat. Rev. Drug Discov. 2006, 5(9), 769-784).
Различные ингибиторы HDAC находятся в доклиническом или клиническом испытании, но до настоящего времени только неселективные ингибиторы HDAC были идентифицированы как противоопухолевые средства, и были одобрены только вориностат (SAHA) и ромидепсин (FK228) для лечения кожной Т-клеточной лимфомы. Однако неселективные ингибиторы HDAC, как известно, вызывают побочные эффекты, такие как усталость и тошнота, обычно, при высоких дозах (Piekarz et al., Pharmaceuticals 2010, 3, 2751-2767). Сообщается, что такие побочные эффекты обусловлены ингибированием HDAC класса I. Из-за таких побочных эффектов использование неселективных ингибиторов HDAC при разработке лекарственных средств, отличных от противоопухолевых лекарственных средств, было ограничено (Witt et al., Cancer Letters, 2009, 277, 8-21).
Между тем было сообщено, что селективное ингибирование HDAC класса II не проявляет токсичности, показанной при ингибировании HDAC класса I. Кроме того, когда селективные ингибиторы HDAC разрабатывают, можно преодолеть побочные эффекты, такие как токсичность, которые вызваны неселективным ингибированием HDAC. Таким образом, селективные ингибиторы HDAC обладают потенциалом для разработки в качестве терапевтических средств, эффективных для лечения различных заболеваний (Matthias et al., Mol. Cell. Biol. 2008, 28, 1688-1701).
Известно, что HDAC6, являющийся членом HDAC класса IIb, присутствует в основном в цитоплазме и участвует в деацетилировании ряда негистонных субстратов (HSP90, кортактин и т.п.), включая тубулин, (Yao et al., Mol. Cell 2005, 18, 601-607). HDAC6 имеет два каталитических домена, и домен цинкового пальца С-конца может связываться с убиквитинированными белками. Известно, что HDAC6 имеет множество негистоновых белков в качестве субстратов и, таким образом, играет важную роль в различных заболеваниях, включая рак, воспалительные заболевания, аутоиммунные заболевания, неврологические заболевания и нейродегенеративные нарушения (Santo et al., Blood 2012 119: 2579-258; Vishwakarma et al., International Immunopharmacology 2013, 16, 72-78; Hu et al., J. Neurol. Sci. 2011, 304, 1-8).
Общей структурной характеристикой различных ингибиторов HDAC является структура, состоящая из кэп-группы, линкера и цинк-связывающей группы (ZBG), как показано в следующей структуре вориностата. Многие исследователи провели исследования на ингибирующую активность и селективность фермента путем структурной модификации кэп-группы и линкера. Известно, что среди этих групп цинк-связывающая группа играет более важную роль в ингибирующей активности и селективности ферментов (Wiest et al., J. Org. Chem. 2013 78: 5051-5065; Methot et al., Bioorg. Med. Chem. Lett. 2008, 18, 973-978).
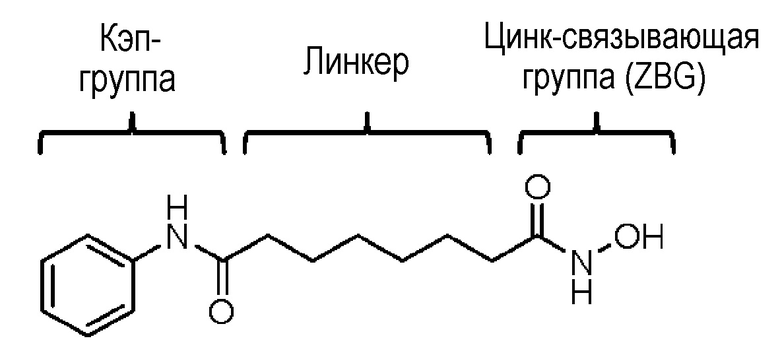
Цинк-связывающая группа обычно представляет собой производное гидроксамовой кислоты или бензамида. В этом случае производное гидроксамовой кислоты обладает мощным ингибирующим действием в отношении HDAC, но имеет проблемы низкой биодоступности и сильной нецелевой активности. В дополнение, производное бензамида имеет проблему в том, что оно может продуцировать токсичные метаболиты in vivo, так как оно содержит анилин (Woster et al., Med. Chem. Commun. 2015, online publication).
Соответственно, существует потребность в разработке селективных ингибиторов HDAC 6 для лечения таких заболеваний, как рак, воспалительные заболевания, аутоиммунные заболевания, неврологические заболевания и нейродегенеративные нарушения, которые имеют цинк-связывающую группу с улучшенной биодоступностью и, в то же время, не вызывают побочных эффектов, в отличие от неселективных ингибиторов, которые вызывают побочные эффекты.
[Раскрытие сущности изобретения]
[Техническая задача]
Задачей настоящего изобретения является предоставление производных 1,3,4-оксадиазолсульфамида, имеющих селективную ингибирующую активность в отношении HDAC6, их стереоизомеров или их фармацевтически приемлемых солей.
Другой задачей настоящего изобретения является предоставление фармацевтических композиций, содержащих производные 1,3,4-оксадиазолсульфамида, имеющих селективную ингибирующую активность в отношении HDAC6, их стереоизомеры или их фармацевтически приемлемые соли.
Еще одной задачей настоящего изобретения является предоставление способов получения новых соединений.
Еще одной задачей настоящего изобретения является предоставление фармацевтических композиций для профилактики или лечения заболеваний, связанных с активностью HDAC6, включая инфекционные заболевания; новообразования; болезни эндокринной системы, расстройства питания и нарушения обмена веществ; психические и поведенческие расстройства; неврологические заболевания; болезни глаза и его придаточного аппарата; заболевания сердечно-сосудистой системы; респираторные заболевания; заболевания пищеварительного тракта; заболевания кожи и подкожной клетчатки; заболевания костно-мышечной системы и соединительной ткани; или врожденные аномалии, деформации и хромосомные нарушения, которые содержат вышеуказанное соединение.
Еще одной задачей настоящего изобретения является обеспечение использования соединений для получения терапевтических лекарственных средств против заболеваний, связанных с активностью HDAC6.
Еще одной задачей настоящего изобретения является предоставление способов лечения заболеваний, связанных с активностью HDAC6, которые включают введение терапевтически эффективного количества фармацевтических композиций, содержащих соединения.
[Решение задачи]
Авторы настоящего изобретения обнаружили производные 1,3,4-оксадиазолсульфамида, обладающие ингибирующей активностью гистондеацетилазы 6 (HDAC6), и обнаружили, что эти соединения могут быть использованы для ингибирования или лечения заболеваний, связанных с активностью гистондеацетилазы 6 (HDAC6), тем самым завершая настоящее изобретение.
Производные 1,3,4-оксадиазолсульфамида
Для достижения вышеуказанных задач настоящее изобретение предоставляет производное 1,3,4-оксадиазолсульфамида, представленное следующей формулой I, его стереоизомер или его фармацевтически приемлемую соль:
[формула I]
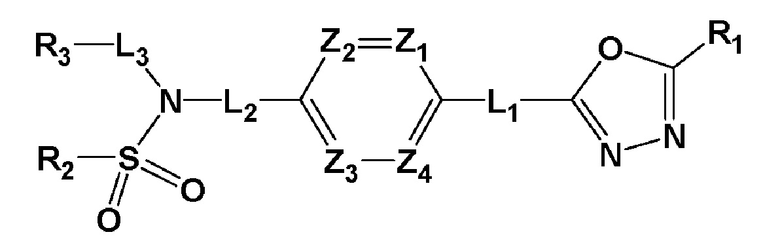
где L1, L2 или L3 каждый независимо представляют собой -(C0-C2 алкил)-;
Z1-Z4 каждый независимо представляют собой N или CRZ, где RZ представляет собой -H или -X;
R1 представляет собой -CX2H или -CX3;
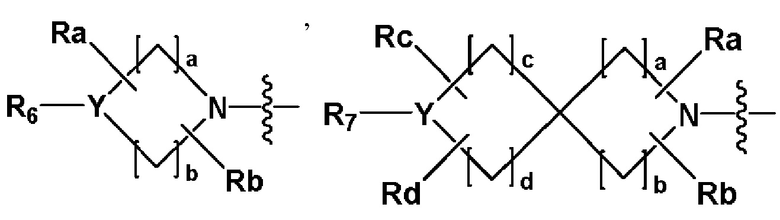
R2 представляет собой
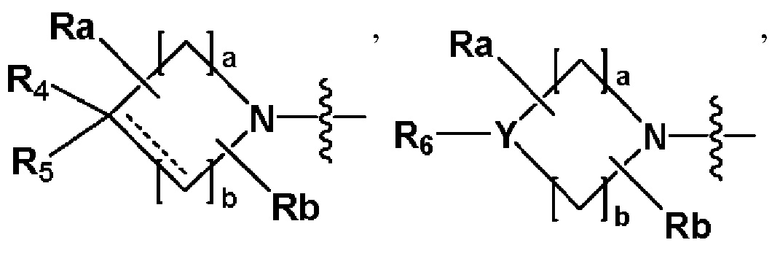 ,
,  или -NR8R9,
или -NR8R9,
где Y представляет собой -N-, -O- или -S(=O)2-,
a-d каждый независимо представляют собой целое число 1, 2 или 3,
Ra-Rd каждый независимо представляет собой -H или -(C1-C4 алкил),
пунктирная линия представляет собой одинарную связь или двойную связь,
R4 и R5 каждый независимо представляют собой -H, -X, -(C1-C4 алкил), арил или -NReRf, при условии, что пунктирная линия представляет собой двойную связь, R5 отсутствует,
Re и Rf каждый независимо представляют собой -H или -(C1-C4 алкил),
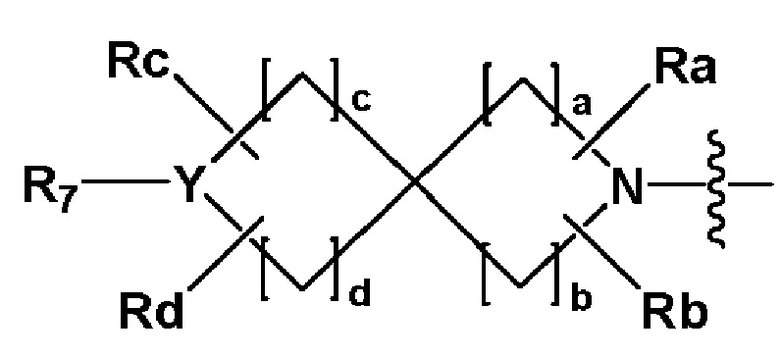
когда Y представляет собой -N-, R6 и R7, каждый независимо представляет собой -H, -(C1-C4 алкил), -(C1-C4 алкил)-O-(C1-C4 алкил), -C(=O)-(C1-C4 алкил), -C(=O)-O(C1-C4 алкил), -C(=O)-CF3, -(C1-C4 алкил)-C(=O)-O(C1-C4 алкил), -S(=O)2-(C1-C4 алкил), -(C3-C7 циклоалкил), -(C2-C6 гетероциклоалкил), -арил, -(C1-C4 алкил)-арил, -гетероарил или аминозащитную группу, где по меньшей мере один H из -(C1-C4 алкил) может быть замещен -X или -OH, по меньшей мере один H из -арила, -(C1-C4 алкил)-арила или -гетероарила может быть замещен -X, -OH или -CF3, и -(C2-C6 гетероциклоалкил) может содержать атом N, O или S в кольце,
и когда Y представляет собой -O- или -S(=O)2-, R6 и R7 отсутствуют,
R8 и R9 каждый независимо представляют собой -H, -(C1-C4 алкил), -(C3-C7 циклоалкил), -(C2-C6 гетероциклоалкил), -(C1-C4 алкил)-(C2-C6 гетероциклоалкил), -арил, -гетероарил или -(C1-C4 алкил)-арил, где по меньшей мере один H из -(C3-C7 циклоалкил), -(C2-C6 гетероциклоалкил), -(C1-C4 алкил)-(C2-C6 гетероциклоалкил), -арила, -гетероарила или -(C1-C4 алкил)-арила может быть замещен -(C1-C4 алкил), -C(=O)-(C1-C4 алкил), -S(=O)2-(C1-C4 алкил) или -(C2-C6 гетероциклоалкил); и
R3 представляет собой -H, -(C1-C4 алкил), -(C1-C4 алкил)-O(C1-C4 алкил), -(C1-C4 алкил)-C(=O)-O(C1-C4 алкил), -(C3-C7 циклоалкил), -арил, -гетероарил или 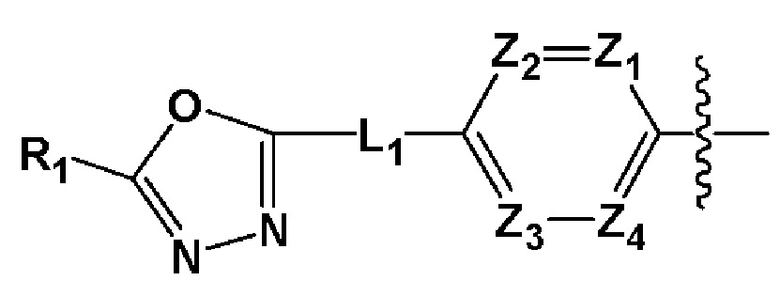 , где по меньшей мере один H из -(C3-C7 циклоалкил), -арила или -гетероарила может быть замещен -X, -OH, -(C1-C4 алкил), -CF3, -(C1-C4 алкил)-(C2-C6 гетероциклоалкил)-(C1-C4 алкил), -C(=O)-(C1-C4 алкил), -C(=O)-O(C1-C4 алкил), -O(C1-C4 алкил), -OCF3, -S(=O)2-(C1-C4 алкил), -арилом, -гетероарилом или -NR11R12,
, где по меньшей мере один H из -(C3-C7 циклоалкил), -арила или -гетероарила может быть замещен -X, -OH, -(C1-C4 алкил), -CF3, -(C1-C4 алкил)-(C2-C6 гетероциклоалкил)-(C1-C4 алкил), -C(=O)-(C1-C4 алкил), -C(=O)-O(C1-C4 алкил), -O(C1-C4 алкил), -OCF3, -S(=O)2-(C1-C4 алкил), -арилом, -гетероарилом или -NR11R12,
R11 и R12 каждый независимо представляют собой -H или -(C1-C4 алкил),
R1, L1, Z1, Z2, Z3 и Z4 являются такими как определено выше; и
X представляет собой F, Cl, Br или I.
В соответствии с предпочтительным вариантом осуществления настоящего изобретения,
L1 или L3 каждый независимо представляют собой -(C0 алкил)-;
L2 представляет собой -(C1 алкил)-;
Z1-Z4 каждый независимо представляют собой N или CRZ, где RZ представляет собой -H или -X;
R1 представляет собой -CX2H или -CX3;
R2 представляет собой
 , или -NR8R9,
, или -NR8R9,
где Y представляет собой -N-, -O- или -S(=O)2-,
a-d каждый независимо представляют собой целое число 1 или 2,
Ra-Rd каждый независимо представляет собой -H или -(C1-C4 алкил),
когда Y представляет собой -N-, R6 и R7, каждый независимо представляет собой -H, -(C1-C4 алкил), -C(=O)-(C1-C4 алкил), -S(=O)2-(C1-C4 алкил), -(C3-C7 циклоалкил) или -(C2-C6 гетероциклоалкил), где по меньшей мере один H из -(C1-C4 алкил) может быть замещен -X или -OH, и -(C2-C6 гетероциклоалкил) может содержать атом N, О или S,
и когда Y представляет собой -O- или -S(=O)2-, R6 и R7 отсутствуют,
R8 и R9 каждый независимо представляют собой -H, -(C1-C4 алкил) или -(C1-C4 алкил)-(C2-C6 гетероциклоалкил), где по меньшей мере один H из -(C1-C4 алкил)-(C2-C6 гетероциклоалкил) может быть замещен -(C1-C4 алкил), -C(=O)-(C1-C4 алкил), -S(=O)2-(C1-C4 алкил) или -(C2-C6 гетероциклоалкил);
R3 представляет собой -арил или -гетероарил, где по меньшей мере один Н из -арила или -гетероарила может быть замещен -X; и
X представляет собой F, Cl, Br или I.
В соответствии с более предпочтительным вариантом осуществления настоящего изобретения,
L1 или L3 каждый независимо представляют собой -(C0 алкил)-;
L2 представляет собой -(C1 алкил)-;
Z1-Z4 каждый независимо представляют собой N или CRZ, где RZ представляет собой -H или -X;
R1 представляет собой -CF2H или -CF3;
R2 представляет собой 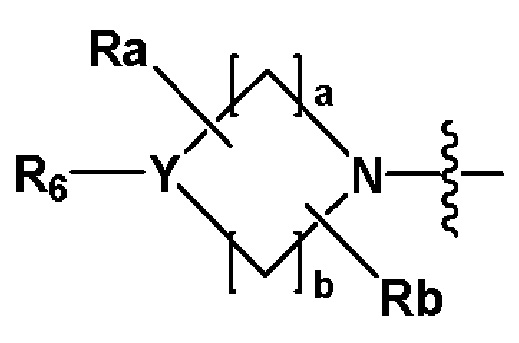 или
или 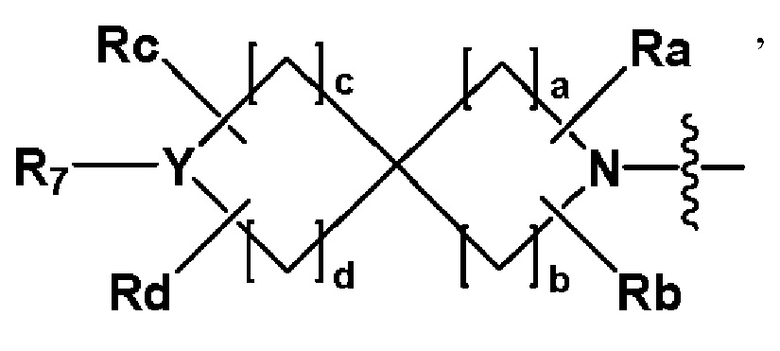
где Y представляет собой -N- или -S(=O)2-,
a-d каждый независимо представляют собой целое число 1 или 2,
Ra-Rd каждый независимо представляют собой -H или -(C1-C4 алкил),
когда Y представляет собой -N-, R6 и R7 каждый независимо представляют собой -H, -(C1-C4 алкил), -C(=O)-(C1-C4 алкил), -S(=O)2-(C1-C4 алкил), -(C3-C7 циклоалкил) или -(C2-C6 гетероциклоалкил), где по меньшей мере один H из -(C1-C4 алкил) может быть замещен -X или -OH, и -(C2-C6 гетероциклоалкил) может содержать атом N, О или S,
и когда Y представляет собой -S(=O)2-, R6 и R7 отсутствуют;
R3 представляет собой -арил или -гетероарил, где по меньшей мере один H -арила или -гетероарила может быть замещен -X; и
X представляет собой F, Cl, Br или I.
В соответствии с особенно предпочтительным вариантом осуществления настоящего изобретения,
L1 или L3 каждый независимо представляют собой -(C0 алкил)-;
L2 представляет собой -(C1 алкил)-;
Z1-Z4 каждый независимо представляют собой N или CRZ, где RZ представляет собой -H или -X;
R1 представляет собой -CF2H или -CF3;
R2 представляет собой 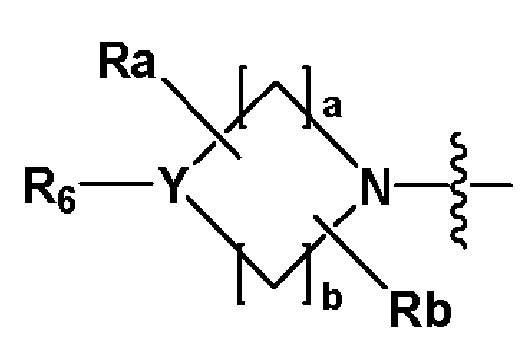 или
или 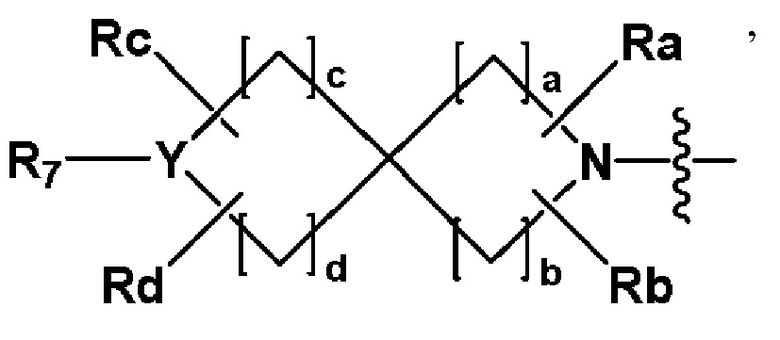
где Y представляет собой -N- или -S(=O)2-,
a и b равны 2, c и d равны 1,
Ra-Rd каждый независимо представляет собой -H или -(C1-C4 алкил),
когда Y представляет собой -N-, R6 и R7, каждый независимо представляет собой -H, -(C1-C4 алкил), -C(=O)-(C1-C4 алкил), -S(=O)2-(C1-C4 алкил), -(C3-C7 циклоалкил) или -(C2-C6 гетероциклоалкил), где по меньшей мере один H из -(C1-C4 алкил) может быть замещен -X или -OH, и -(C2-C6 гетероциклоалкил) может содержать атом N, О или S,
и когда Y представляет собой -S(=O)2-, R6 и R7 отсутствуют;
R3 представляет собой -арил или -гетероарил, где по меньшей мере один H -арила или -гетероарила может быть замещен -F; и
X представляет собой F или Cl.
Конкретные соединения, представленные формулой I, показаны в таблице 1 ниже:
[Таблица 1]
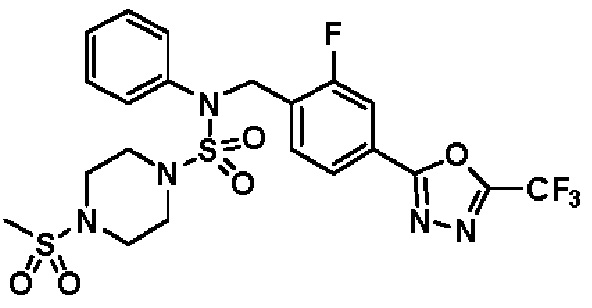
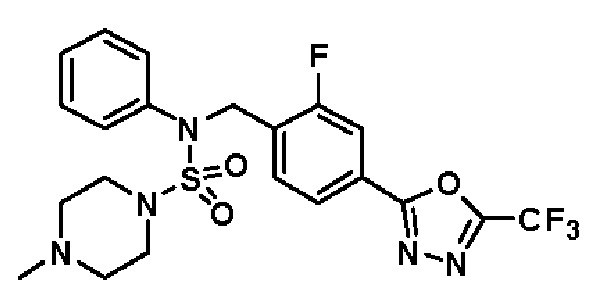
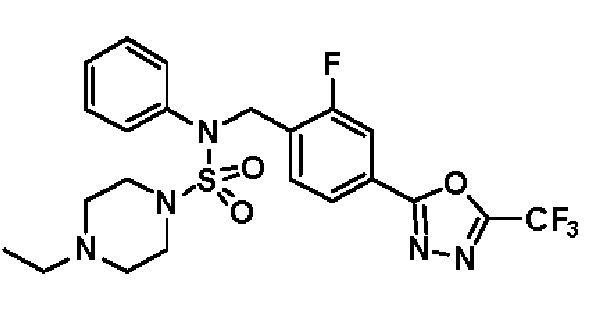
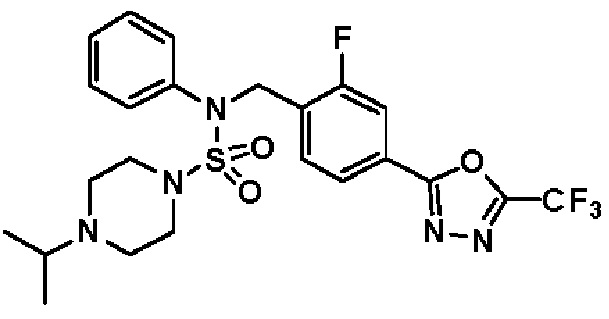
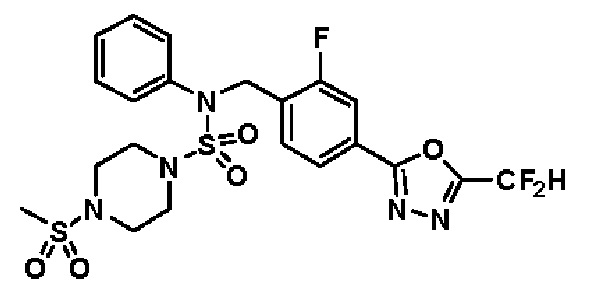
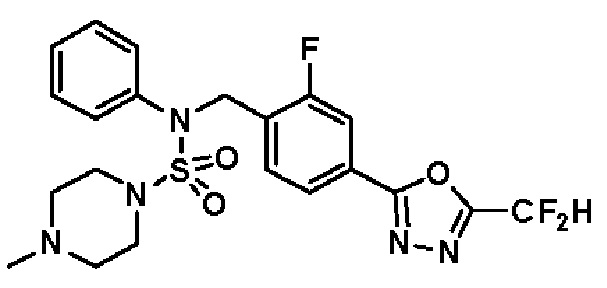
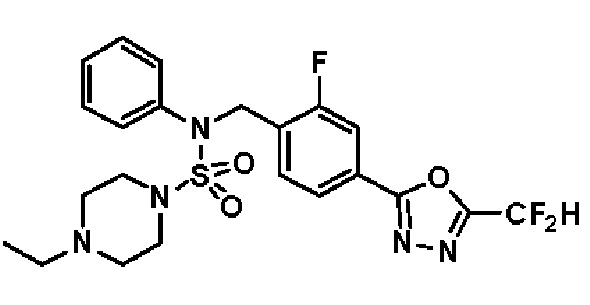
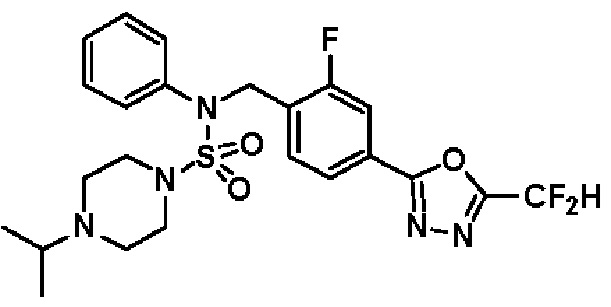
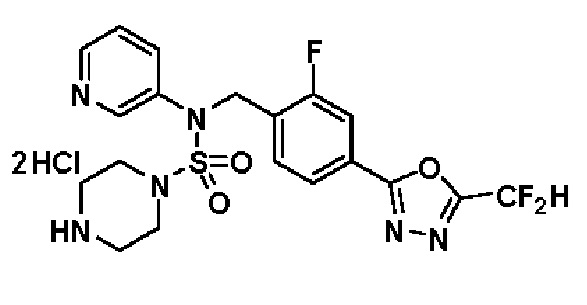
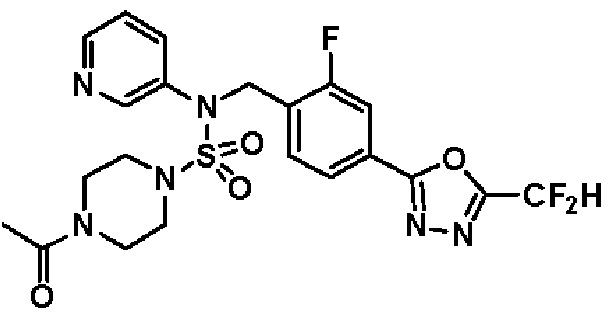
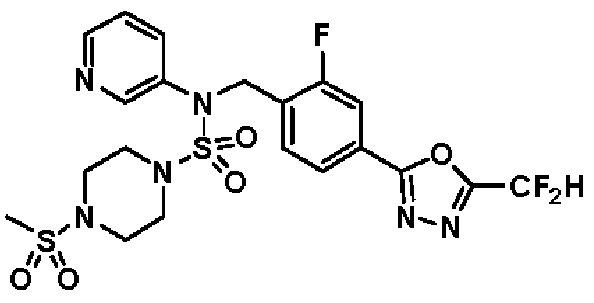
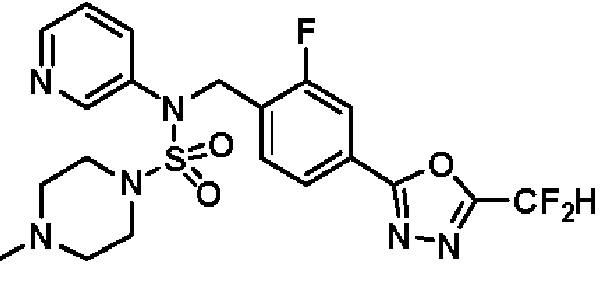
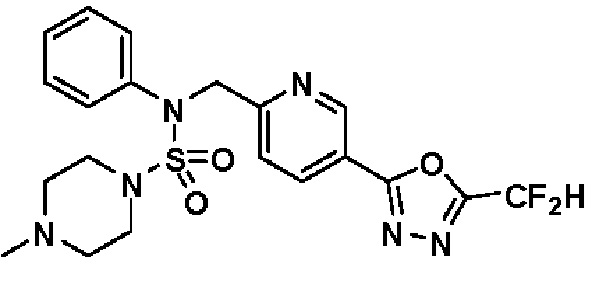
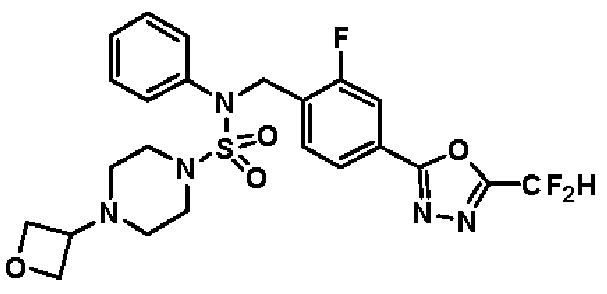
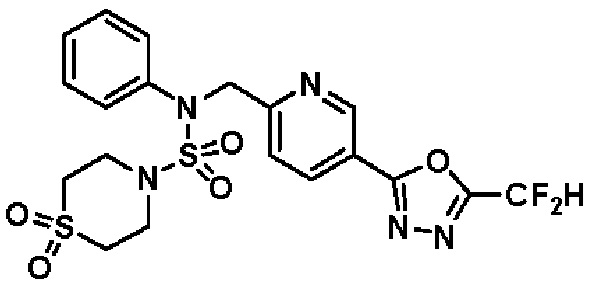
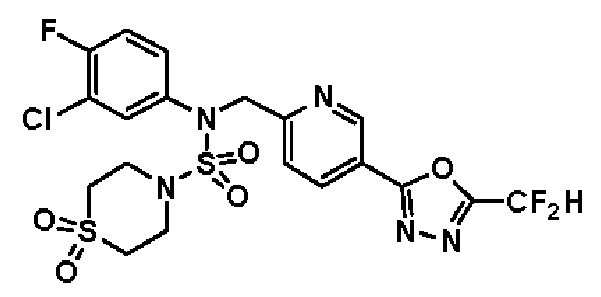
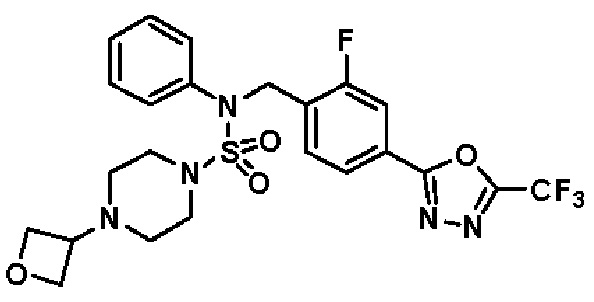
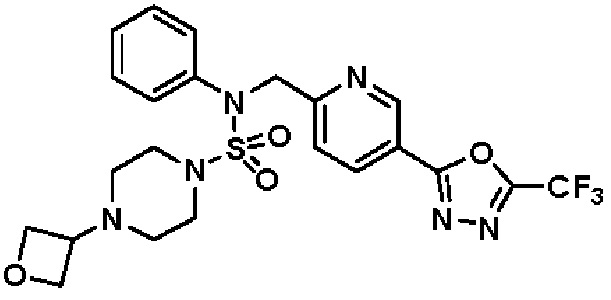
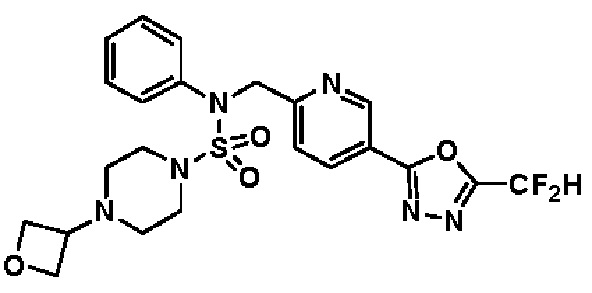
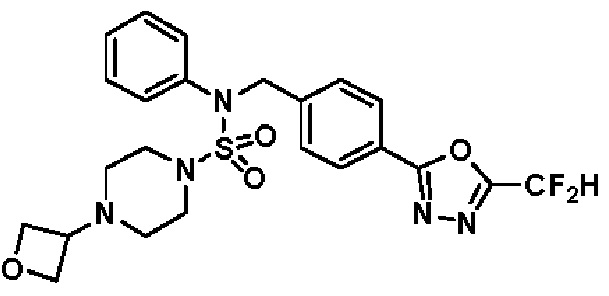
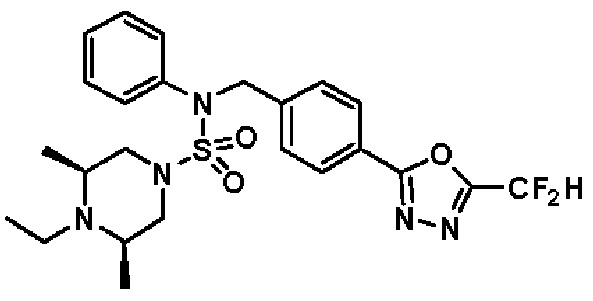
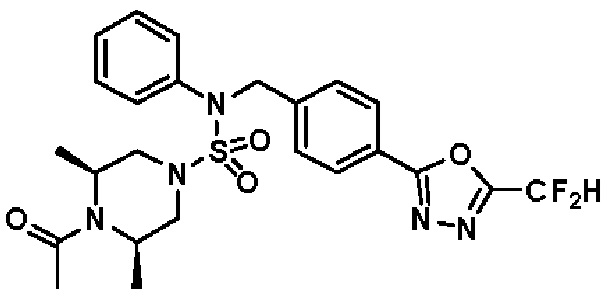
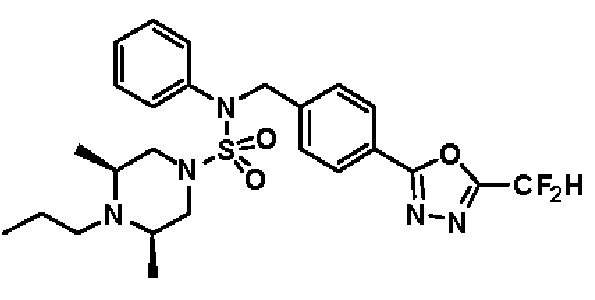
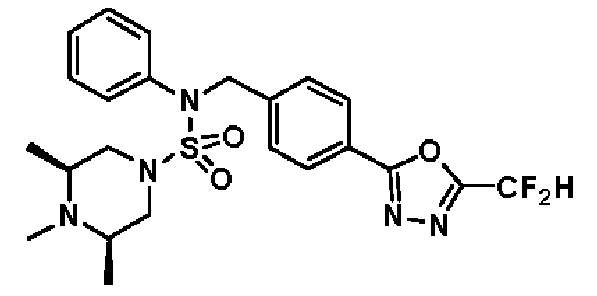
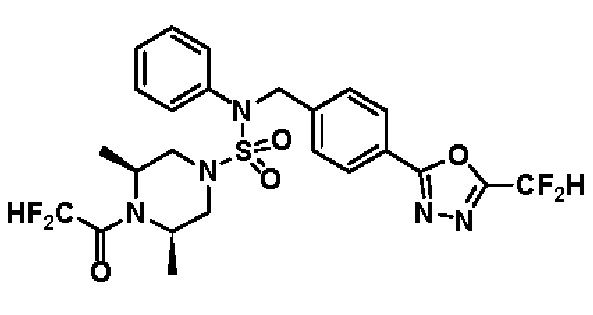
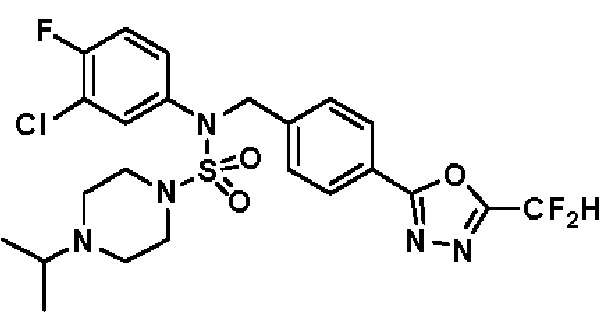
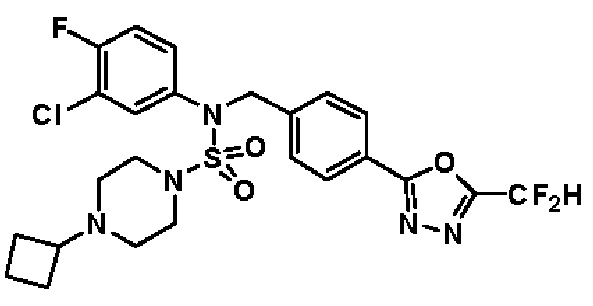
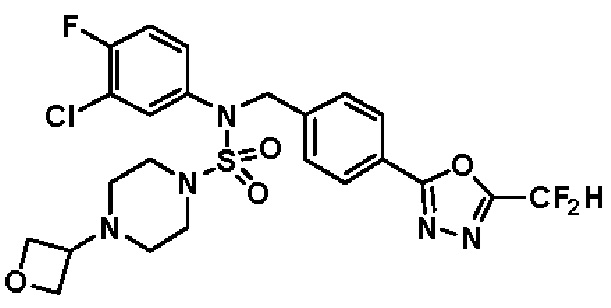
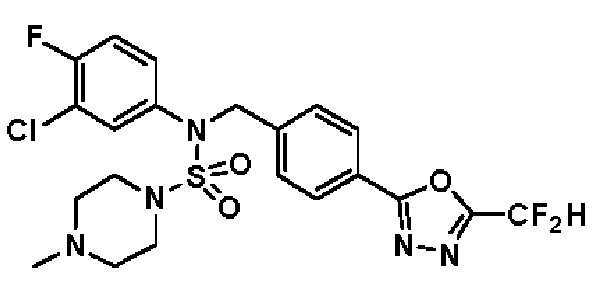
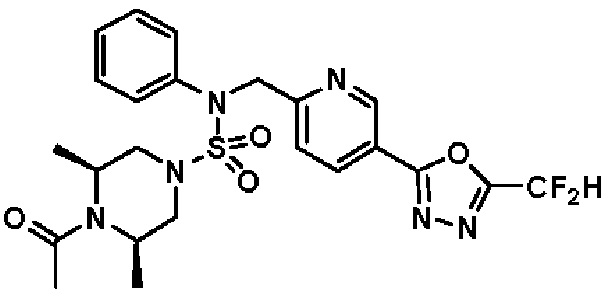
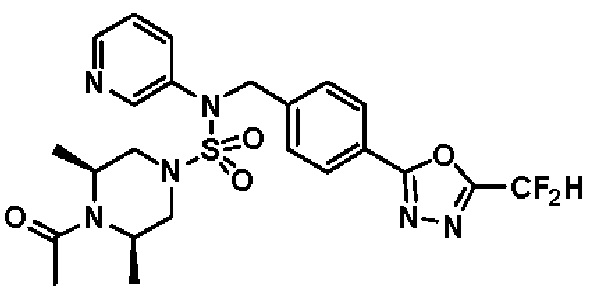
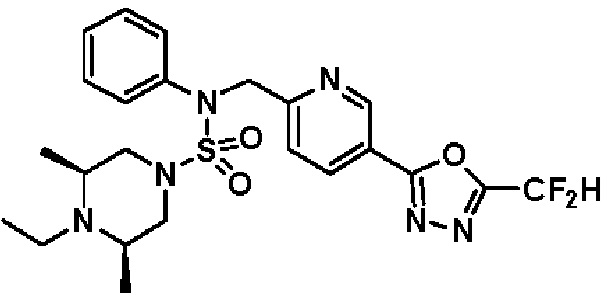
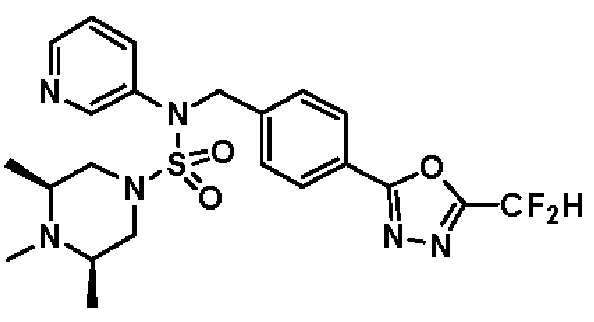
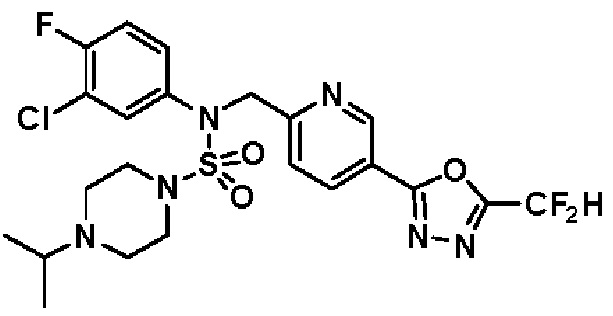
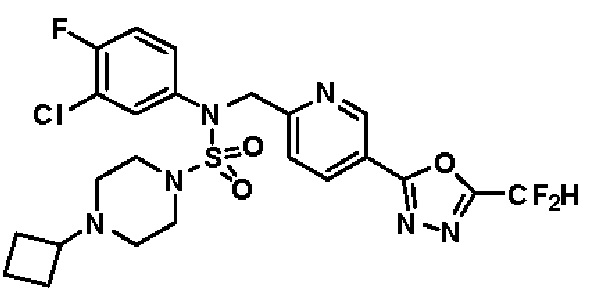
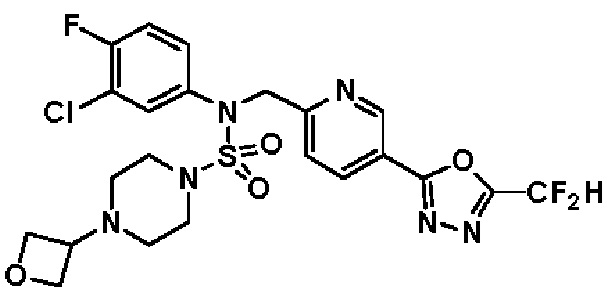
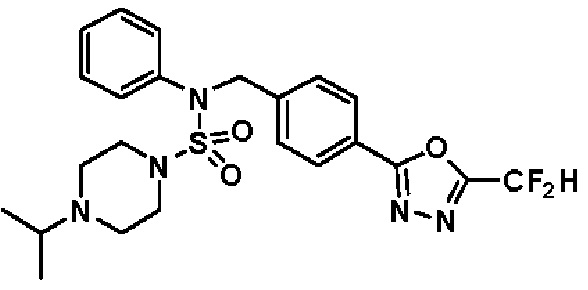
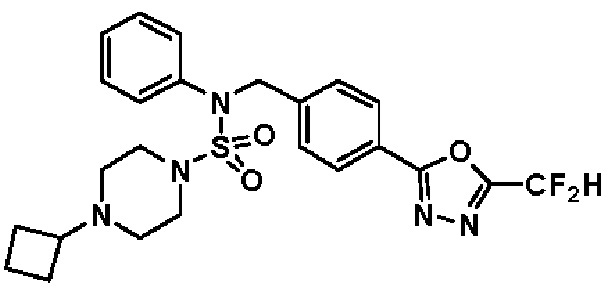
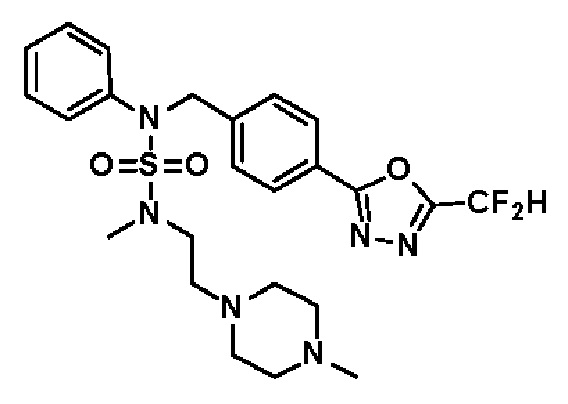
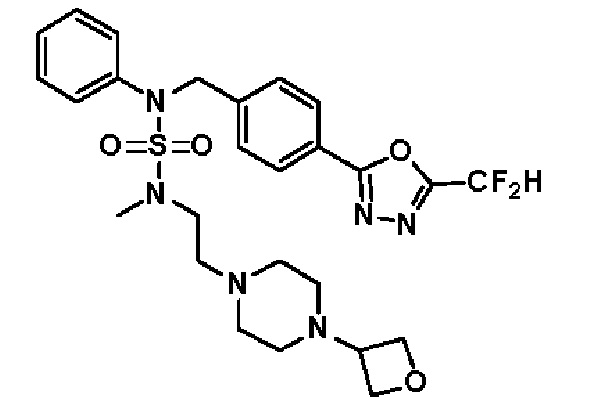
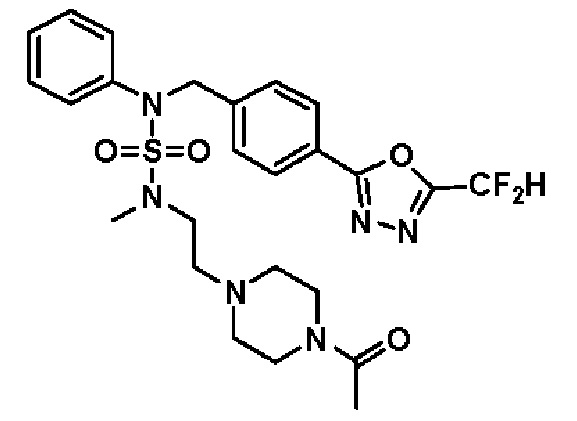
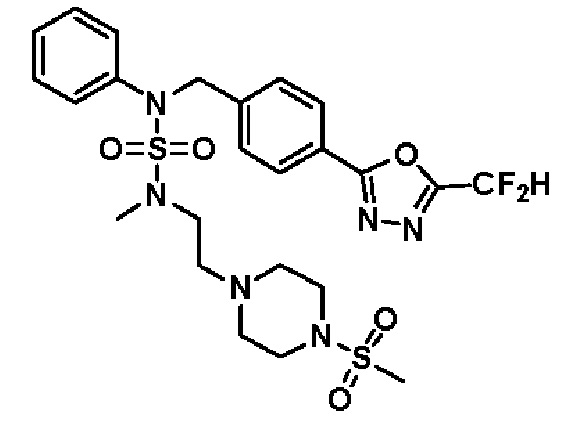
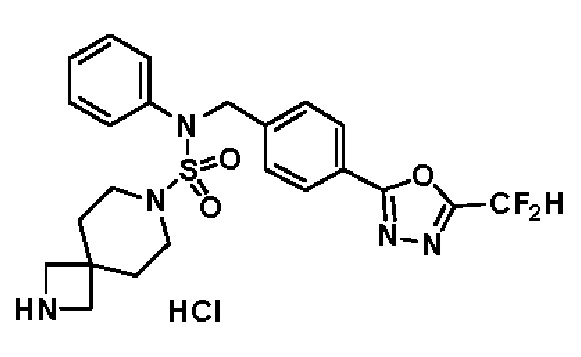
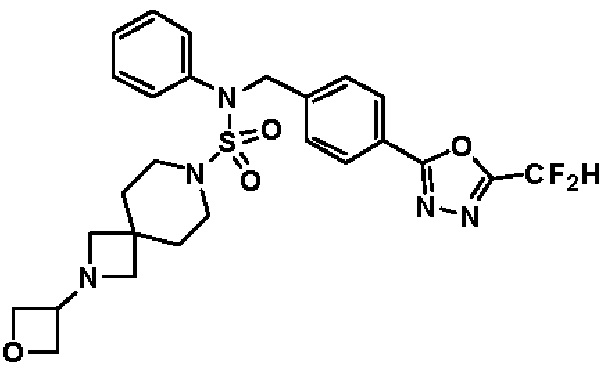
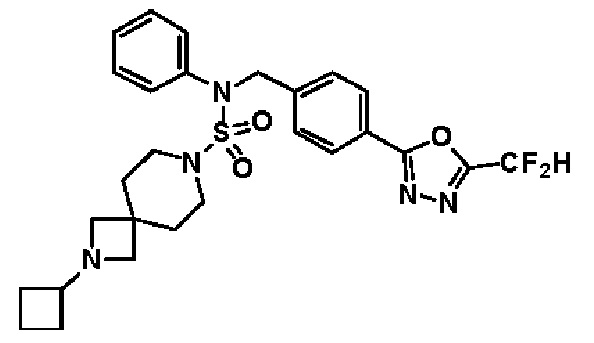
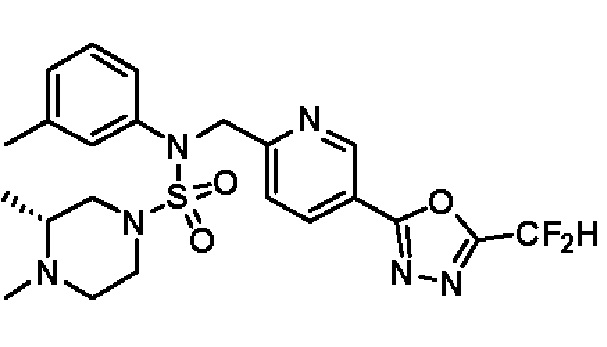
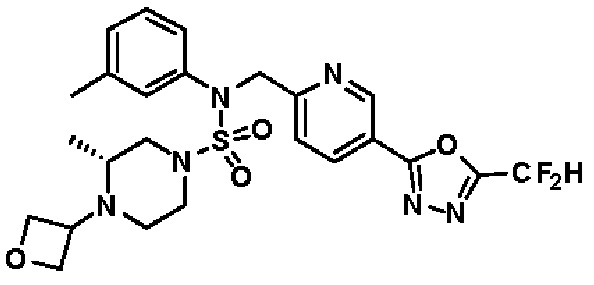
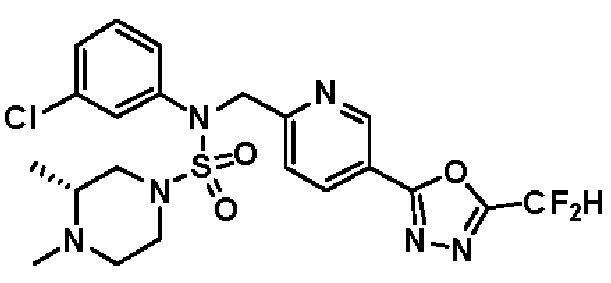
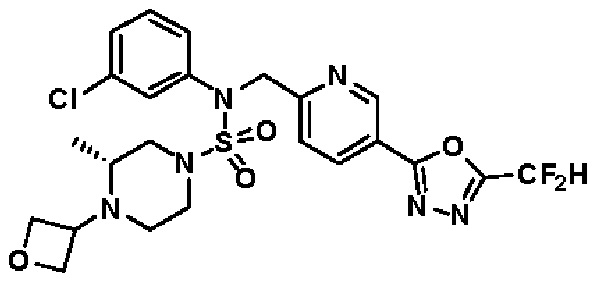
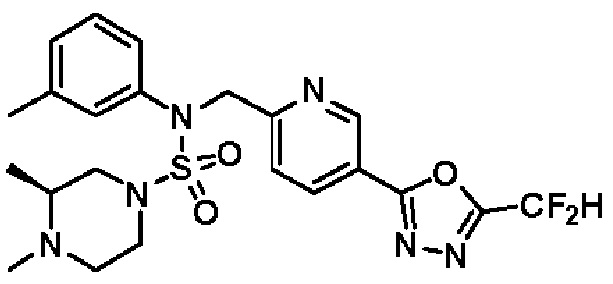
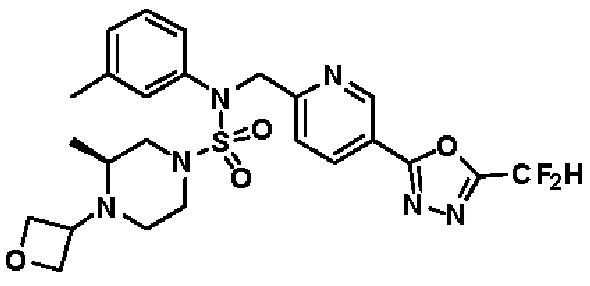
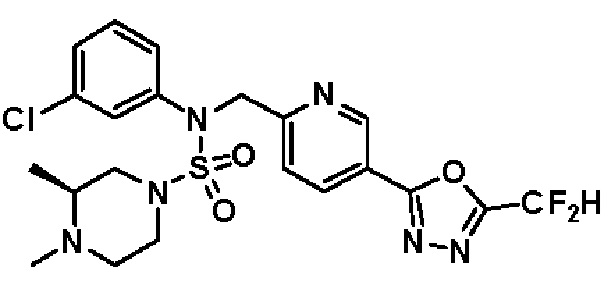
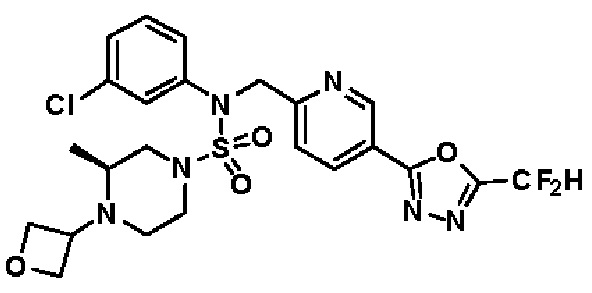
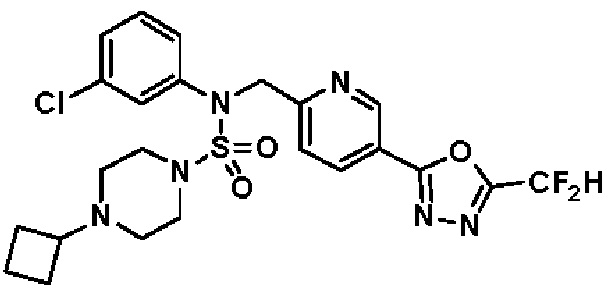
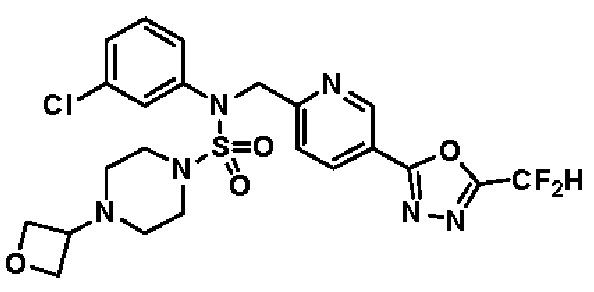
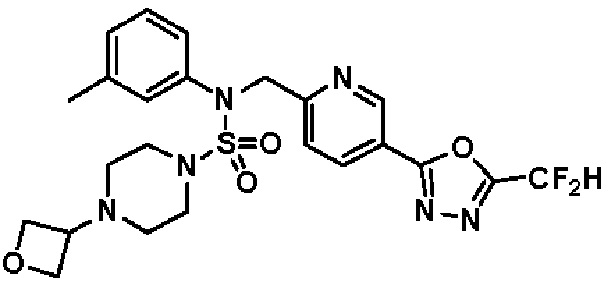
Предпочтительно, соединения, представленные формулой I, их стереоизомеры или их фармацевтически приемлемые соли, могут быть выбраны из группы, состоящей из соединений 11301, 11302, 11303, 11305, 11306, 11307, 11308, 11313, 11314, 11315, 11316, 11318, 11321, 11363, 11379, 11440, 11498, 11574, 11575, 11641, 11653, 11654, 11659, 11662, 11670, 11671, 11672, 11823, 11824, 11825, 11826, 11827, 11828, 11829, 11830, 11831, 11832 и 11833. Более предпочтительно, соединения, представленные формулой I, их стереоизомеры или их фармацевтически приемлемые соли, могут быть выбраны из группы, состоящей из соединений 11301, 11302, 11303, 11305, 11306, 11313, 11314, 11315, 11316, 11363, 11379, 11440, 11498, 11574, 11641, 11654, 11659, 11670, 11671, 11672, 11825, 11829, 11830, 11831 и 11832.
Как используется в настоящем описании, термин «фармацевтически приемлемая соль» означает любую соль, которая обычно используется в фармацевтической области. Примеры фармацевтически приемлемой соли включают, но ими не ограничиваются, соли с неорганическими ионами, такими как ионы кальция, калия, натрия или магния, соли с неорганическими кислотами, такими как хлористоводородная кислота, азотная кислота, фосфорная кислота, бромноватая кислота, иодноватая кислота, перхлорная кислота или серная кислота, соли с органическими кислотами, такими как уксусная кислота, трифторуксусная кислота, лимонная кислота, малеиновая кислота, янтарная кислота, щавелевая кислота, бензойная кислота, винная кислота, фумаровая кислота, миндальная кислота, пропионовая кислота, молочная кислота, гликолевая кислота, глюконовая кислота, галактуроновая кислота, глютаминовая кислота, глутаровая кислота, глюкуроновая кислота, аспарагиновая кислота, аскорбиновая кислота, угольная кислота, ванилиновая кислота, иодистоводородная кислота или тому подобное, соли с сульфоновыми кислотами, такими как метансульфоновая кислота, этансульфоновая кислота, бензолсульфоновая кислота, п-толуолсульфоновая кислота или нафталинсульфоновая кислота, соли с аминокислотами, такими как глицин, аргинин или лизин, и соли с аминами, такими как триметиламин, триэтиламин, аммиак, пиридин или пиколин.
В настоящем изобретении предпочтительные соли включают соли с хлористоводородной кислотой, фосфорной кислотой, серной кислотой, трифторуксусной кислотой, лимонной кислотой, бромноватой кислотой, малеиновой кислотой, винной кислотой или тому подобное, и предпочтительные примеры таких соединений включают соединения 11293, 11301, 11306, 11309, 11313, 11317, 11321 и 11787, как раскрыто в настоящем описании.
Соединения, представленные формулой I, могут содержать один или несколько асимметричных атомов углерода и, таким образом, могут существовать в форме рацематов, рацемических смесей, одиночных энантиомеров, диастереомерных смесей и индивидуальных диастереомеров. Соединения формулы I могут быть разделены на такие изомеры с помощью способов, известных в данной области, например, колоночной хроматографией или ВЭЖХ. Альтернативно, стереоизомеры соединений формулы I могут быть синтезированы стереоспецифическим синтезом с использованием оптически чистых исходных материалов и/или реагентов известной конфигурации.
Способы получения производных 1,3,4-оксадиазолсульфамида
Настоящее изобретение относится к способам получения производных 1,3,4-оксадиазолсульфамида, представленных формулой I, их изомеров или их фармацевтически приемлемых солей.
Предпочтительные способы получения производных 1,3,4-оксадиазолсульфамида, представленных формулой I, их стереоизомеров или их фармацевтически приемлемых солей, показаны на схемах реакций 1-6 ниже, и также включают модификации, очевидные для специалистов в данной области.
[Схема реакции 1]
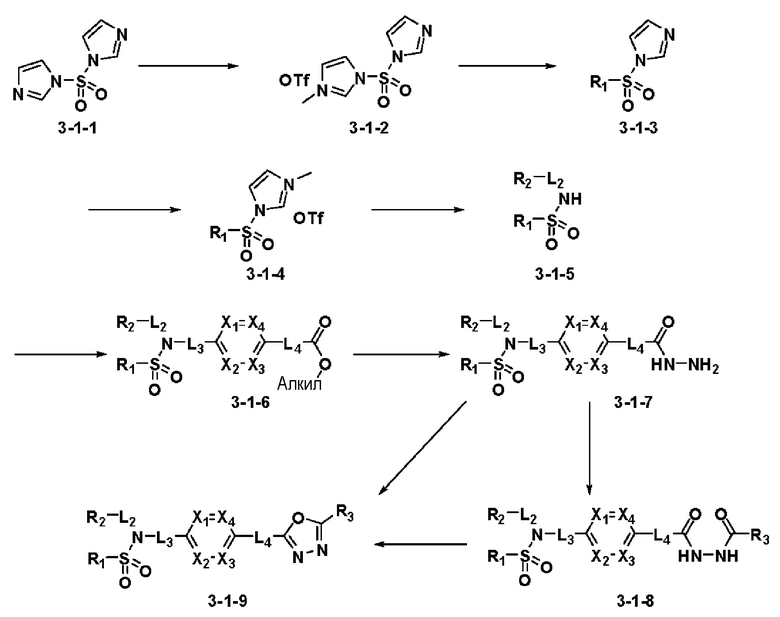
Схема реакции 1, выше, показывает общий способ синтеза соединений, имеющих сульфамидную структуру [Journal of Organic Chemistry, 2003, vol. 68, 115-119]. Как показано на схеме реакции 1, метильную группу вводят в 1,1′-сульфонилдиимидазол для повышения реакционной способности с последующим замещением амином. Этот процесс проводят дважды, получая таким образом соединение формулы 3-1-5. Затем алкильную группу вводят в соединение формулы 3-1-5 в присутствии гидрида натрия и сложноэфирный фрагмент замещают гидразином, получая таким образом соединение формулы 3-1-7. Затем соединение формулы 3-1-7 подвергают взаимодействию с трифторуксусным ангидридом или дифторуксусным ангидридом для синтеза соединения формулы 3-1-9 или формулы 3-1-8, которое затем подвергают взаимодействию с 1-метокси-N-триэтиламмонийсульфонил-метанимидатом (реагент Бургесса), синтезируя таким образом соединения 11198, 11199, 11440 и 11498, которые имеют оксадиазольную структуру.
[Схема реакции 2]
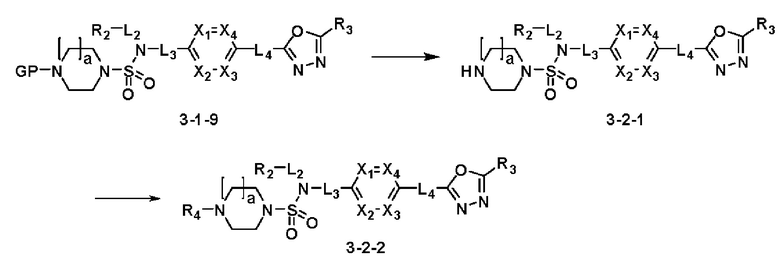
Схема реакции 2, выше, показывает способ введения заместителя во вторичный амин. Как показано на схеме, защитную группу удаляют из соединения формулы 3-1-9, синтезированного в соответствии со схемой реакции 1, таким образом синтезируя соединения 11293, 11301, 11309, 11313, 11317 и 11321. Затем заместитель вводят в соединение формулы 3-2-1 реакцией с ацилхлоридом или сульфонилхлоридом, или алкильную группу вводят в соединение формулы 3-2-1 с помощью восстановительного аминирования, тем самым синтезируя соединения 11294, 11295, 11296, 11297, 11298, 11299, 11300, 11302, 11303, 11304, 11305, 11306, 11307, 11308, 11310, 11311, 11312, 11314, 11315, 11316, 11318, 11319, 11320 и 11322.
[Схема реакции 3]
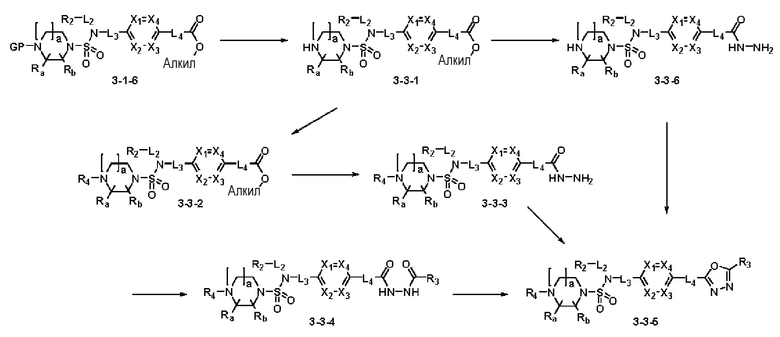
Схема реакции 3 выше показывает другой способ введения заместителя во вторичный амин. Как показано на схеме, защитную группу соединения формулы 3-1-6, синтезированного в соответствии со схемой реакции 1, удаляют для синтеза соединения формулы 3-3-1 и алкильную группу вводят в соединение формулы 3-3-1 путем восстановительного аминирования, тем самым синтезируя соединение формулы 3-3-2. Затем, согласно тому же способу синтеза, который показан на схеме реакции 1, синтезируют соединения 11363, 11379, 11527, 11528, 11574, 11575, 11640, 11641, 11642, 11643, 11644, 11651, 11652, 11653, 11654, 11659, 11660, 11661, 11662, 11670, 11671, 11672, 11673, 11674, 11823, 11824, 11825, 11826, 11827, 11828, 11829, 11830, 11831, 11832 и 11833.
[Схема реакции 4]
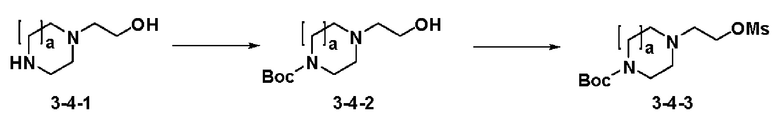
Схема реакции 4 выше показывает реакцию для синтеза соединения для замещения в сульфамидной структуре. Как показано на схеме, защитную группу вводят во вторичный амин, который затем подвергают взаимодействию с метансульфонилхлоридом для получения соединения формулы 3-4-3.
[Схема реакции 5]
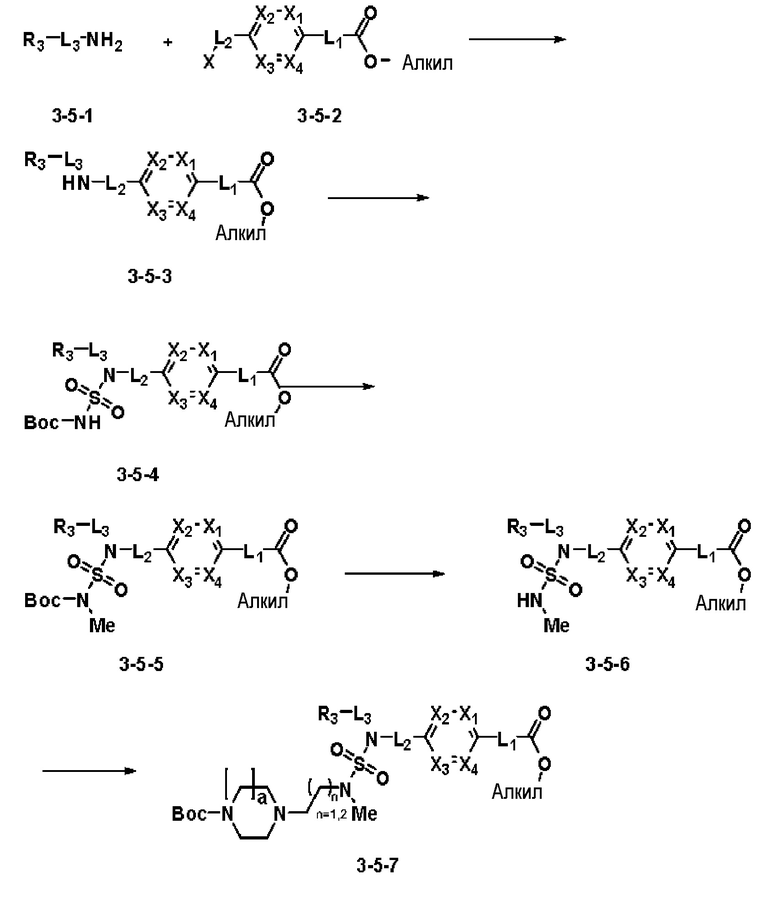
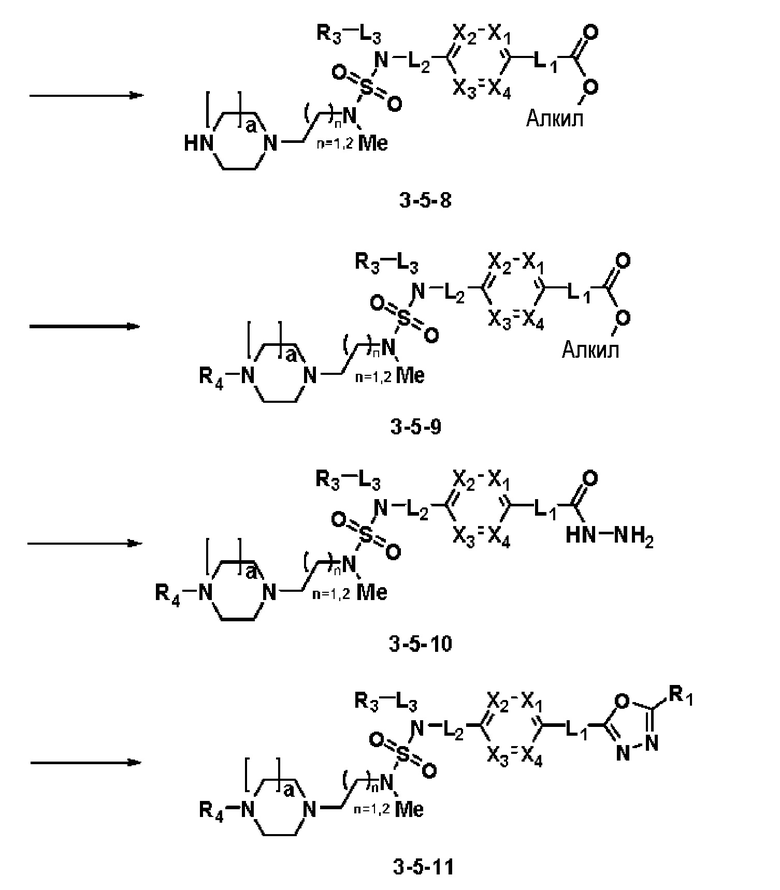
Схема реакции 5 выше показывает общий способ синтеза соединений, имеющих сульфамидную структуру. Как показано на схеме, соединение формулы 3-5-1 подвергают взаимодействию с соединением формулы 3-5-2 с получением соединения формулы 3-5-3, и хлорсульфонилизоцианат и трет-бутанол подвергают взаимодействию друг с другом, а затем подвергают взаимодействию с соединением формулы 3-5-3, получая таким образом соединение формулы 3-5-4. Затем метильную группу вводят во вторичный амин с получением соединения формулы 3-5-5, которое затем депротектируют (удаление защитной группы), получая таким образом соединение формулы 3-5-6. Соединение формулы 3-5-6 подвергают реакции замещения с соединением формулы 3-4-3, полученным в соответствии со схемой реакции 4, получая таким образом соединение формулы 3-5-7. Соединение формулы 3-5-7 депротектируют, получая таким образом соединение формулы 3-5-8. Затем соединение формулы 3-5-8 подвергают реакции замещения или реакции восстановительного аминирования с получением соединения формулы 3-5-9, которое затем подвергают взаимодействию с гидразином с получением соединения формулы 3-5-10. Затем соединение формулы 3-5-10 подвергают взаимодействию с трифторуксусным ангидридом или дифторуксусным ангидридом, получая таким образом соединение формулы 3-5-11, которое имеет оксадиазольную структуру.
В соответствии со схемой реакции 5, выше, получали соединения 11702, 11704, 11713 и 11714.
[Схема реакции 6]
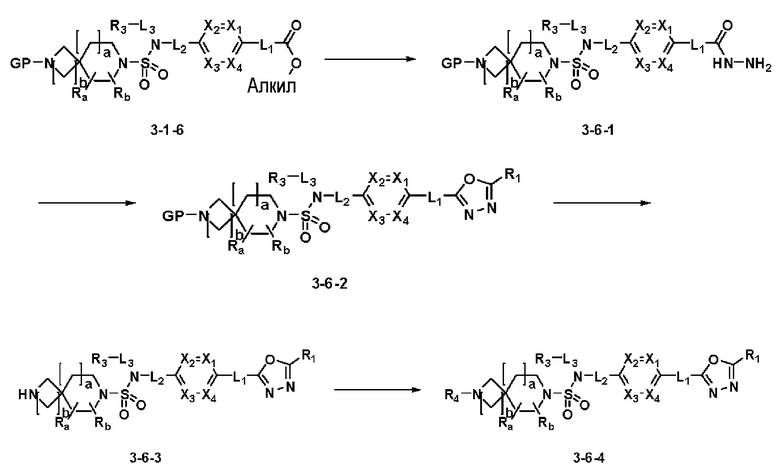
Схема реакции 6, выше, показывает способ введения заместителя во вторичный амин. Как показано здесь, соединение формулы 3-1-6, синтезированное в соответствии со схемой реакции 1, подвергают взаимодействию с гидразином для синтеза соединения формулы 3-6-1. Затем соединение формулы 3-6-1 подвергают взаимодействию с трифторуксусным ангидридом или дифторуксусным ангидридом с получением соединения формулы 3-6-2, имеющим оксадиазольную структуру. Затем соединение формулы 3-6-2 депротектируют, тем самым синтезируя соединение 11787. Алкильную группу вводят в соединение 11787 или соединение формулы 3-6-3 путем восстановительного аминирования, тем самым синтезируя соединения 11788 и 11789.
Композиции, содержащие производные 1,3,4-оксадиазолсульфамида, их применение и способ лечения заболеваний
Настоящее изобретение обеспечивает фармацевтическую композицию для профилактики или лечения заболеваний, связанных с активностью гистондеацетилазы 6 (HDAC6), которая содержит в качестве активного ингредиента соединение, представленное следующей формулой I, его стереоизомер или его фармацевтически приемлемую соль:
[Формула I]
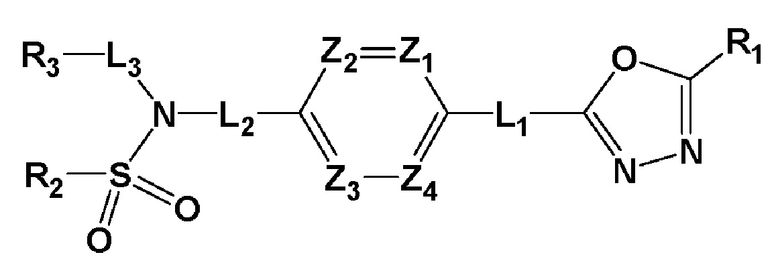
где формула I является такой, как определено выше.
Фармацевтическая композиция по настоящему изобретению проявляет заметный эффект на профилактику или лечение заболеваний, связанных с активностью гистондеацетилазы 6 (HDAC6), путем селективного ингибирования гистондеацетилазы 6 (HDAC6).
Заболевания, связанные с активностью гистондеацетилазы 6 (HDAC6), включают инфекционные заболевания, такие как прионное заболевание; новообразования, такие как доброкачественная опухоль (например, миелодиспластический синдром) или злокачественная опухоль (например, множественная миелома, лимфома, лейкоз, рак легкого, рак прямой кишки, рак толстой кишки, рак предстательной железы, уротелиальная карцинома, рак молочной железы, меланома, рак кожи, рак печени, рак мозга, рак желудка, рак яичников, рак поджелудочной железы, рак головы и шеи, рак полости рта или глиома); болезни эндокринной системы, расстройства питания и нарушения обмена веществ, такие как болезнь Вильсона, амилоидоз или диабет; психические и поведенческие нарушения, такие как депрессия или синдром Ретта, и тому подобное; неврологические заболевания, такие как атрофия центральной нервной системы (например, болезнь Хантингтона, спинальная мышечная атрофия (SMA), спиноцеребеллярная атаксия (SCA)), нейродегенеративное заболевание (например, болезнь Альцгеймера), нарушение движений (например, болезнь Паркинсона), невропатию (например, наследственная невропатия (болезнь Шарко-Мари-Тута), спорадическая невропатия, воспалительная невропатия, невропатия, вызванные лекарственными средствами), болезни двигательного нейрона (боковой амиотрофический склероз (ALS)) или демиелинизирующие заболевания центральной нервной системы (например, рассеянный склероз (MS)), и тому подобное; болезни глаза и его придаточного аппарата, такие как увеит; сердечно-сосудистые заболевания, такие как фибрилляция предсердий или инсульт и тому подобное; респираторные заболевания, такие как астма; заболевания пищеварительного тракта, такие как алкогольная болезнь печени, воспалительное заболевание кишечника, болезнь Крона или язвенная болезнь кишечника и тому подобное; заболевания кожи и подкожной клетчатки, такие как псориаз; заболевания костно-мышечной системы и соединительной ткани, такие как ревматоидный артрит, остеоартрит или системная красная волчанка (SLE) и тому подобное; или врожденные аномалии, деформации и хромосомные нарушения, такие как аутосомно-доминантное поликистозное заболевание почек, а также нарушения или заболевания, связанные с аномальной функцией гистондеацетилазы.
Фармацевтически приемлемая соль является такой, как описано выше, в отношении фармацевтически приемлемой соли соединения, представленного формулой I, по настоящему изобретению.
Для введения фармацевтическая композиция по настоящему изобретению может дополнительно содержать по меньшей мере один фармацевтически приемлемый носитель в дополнение к соединению формулы I, его изомеру или его фармацевтически приемлемой соли. Фармацевтически приемлемый носитель, который используется в настоящем изобретении, может быть по меньшей мере одним из физиологического раствора, стерильной воды, раствора Рингера, забуференного физиологического раствора, раствора декстрозы, раствора мальтодекстрина, глицерина, этанола и смеси двух или более из них. При необходимости, композиция может содержать другие обычные добавки, такие как антиоксидант, буфер или бактериостатическое средство. Кроме того, композиция может быть составлена в виде инъекционных составов, таких как растворы, суспензии, густая жидкость и т.п., пилюль, капсул, гранул или таблеток с использованием разбавителя, диспергирующего средства, поверхностно-активного вещества, связующего вещества и лубриканта. Таким образом, композиция по настоящему изобретению может быть в виде патчей, жидкостей, пилюль, капсул, гранул, таблеток, суппозиториев и т.п. Эти составы могут быть получены либо обычными способами, которые используют для получения в данной области техники, либо способом, описанным в Remington's Pharmaceutical Science (последнее издание), Mack Publishing Company, Easton PA.
Фармацевтическая композиция по настоящему изобретению может вводиться перорально или парентерально (например, внутривенно, подкожно, внутрибрюшинно или местно) в зависимости от предполагаемого использования. Доза фармацевтической композиции варьируется в зависимости от массы, возраста, пола, состояния здоровья и режима питания пациента, режима введения, способа введения, скорости экскреции, тяжести заболевания и тому подобного. Суточная доза соединения формулы I по настоящему изобретению может составлять приблизительно 1-1000 мг/кг, предпочтительно 5-100 мг/кг и может вводиться от одного раза до нескольких раз в день.
Фармацевтическая композиция по настоящему изобретению может дополнительно содержать в дополнение к соединению, представленному формулой I, его стереоизомер или его фармацевтически приемлемую соль, один или несколько активных ингредиентов, которые проявляют медицинскую эффективность, идентичную или сходную с ним.
Настоящее изобретение также относится к способу профилактики или лечения заболевания, опосредованного гистондеацетилазой, который включает введение терапевтически эффективного количества соединения, представленного формулой I, его стереоизомера или его фармацевтически приемлемой соли.
Как используется в настоящем описании термин «терапевтически эффективное количество» относится к количеству соединения, представленного формулой I, которое является эффективным для профилактики или лечения заболеваний, связанных с активностью гистондеацетилазы 6.
Настоящее изобретение также относится к способу селективного ингибирования HDAC6, который включает введение соединения формулы I, его стереоизомера или его фармацевтически приемлемой соли млекопитающим, включая человека.
Способ профилактики или лечения заболевания, связанного с активностью гистондеацетилазой 6, по настоящему изобретению включает ингибирование или предотвращение заболевания, а также устранение самого заболевания до появления симптомов путем введения соединения, представленного формулой I. При лечении заболеваний величина профилактической или терапевтической дозы конкретного активного ингредиента будет варьироваться в зависимости от характера и тяжести заболевания или состояния и может также варьироваться в зависимости от пути введения активного ингредиента. Доза и частота приема будут варьировать в зависимости от возраста, массы тела и реакции конкретного пациента. Подходящие режимы дозирования могут быть легко выбраны специалистами в данной области техники с учетом таких факторов. Кроме того, способ профилактики или лечения заболевания, связанного с активностью гистондеацетилазы 6, по настоящему изобретению, может дополнительно включать введение терапевтически эффективного количества дополнительного активного средства, полезного для лечения заболевания, вместе с соединением, представленным формулой I, в котором дополнительное активное средство может проявлять синергический эффект с соединением формулы I или вспомогательный эффект.
Настоящее изобретение также предназначено для обеспечения использования соединения, представленного формулой I, его стереоизомера или его фармацевтически приемлемой соли, для получения лекарственного средства для лечения заболевания, связанного с активностью гистондеацетилазы 6. Для получения лекарственного средства соединение, представленное формулой I, может быть смешано с фармацевтически приемлемым адъювантом, разбавителем, носителем или т.п. и объединено с другими активными средствами такими, что активные ингредиенты могут обладать синергическими эффектами.
Подробные сведения, упомянутые в использовании, композиции и способе лечения по настоящему изобретению, могут быть соответствующим образом объединены, если они не противоречат друг другу.
[Полезные эффекты изобретения]
Соединения, представленные формулой I, их стереоизомеры или их фармацевтически приемлемые соли, могут селективно ингибировать HDAC6 и, таким образом, демонстрируют превосходные эффекты на профилактику или лечение заболеваний, связанных с активностью гистондеацетилазы 6.
[Вариант осуществления изобретения]
Далее будут представлены предпочтительные примеры, которые помогут понять настоящее изобретение. Однако эти примеры приведены только для лучшего понимания настоящего изобретения и не предназначены для ограничения объема настоящего изобретения.
Получение производных 1,3,4-оксадиазолсульфамида
Конкретные способы получения соединений формулы I являются следующими.
Пример 1: Соединение 11198, N-(4-фторфенил)-N-(4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)морфолин-4-сульфонамид
[Стадия 1] N-(4-фторфенил)-N-(4-(гидразинкарбонил)бензил)морфолин-4-сульфонамид
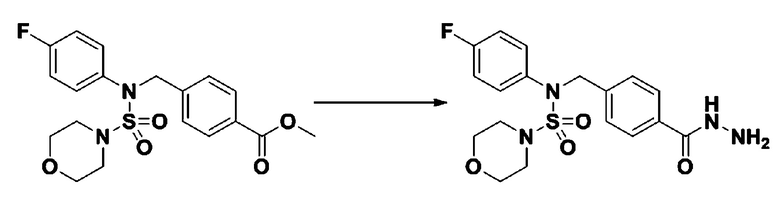
Смесь метил 4-((N-(4-фторфенил)морфолин-4-сульфонамидо)метил)бензоата (0,150 г, 0,367 ммоль) и гидразин моногидрата (0,347 мл, 7,345 ммоль) в этаноле (3 мл) нагревали при 120°С в течение 2 часов под действием микроволнового излучения, и охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Указанное в заголовке соединение использовали без дополнительной очистки (0,136 г, 90,5%, бесцветное масло).
[Стадия 2] Соединение 11198
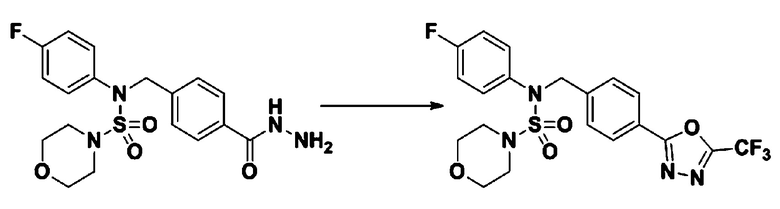
Раствор N-(4-фторфенил)-N-(4-(гидразинкарбонил)бензил)морфолин-4-сульфонамида (0,067 г, 0,164 ммоль) и триэтиламина (0,045 мл, 0,328 ммоль) в N,N-диметилформамиде (2 мл) перемешивали при 0°C, и смешивали с трифторуксусным ангидридом (0,028 мл, 0,197 ммоль). Реакционную смесь перемешивали при 80°С в течение дополнительных 18 часов, охлаждали до комнатной температуры для прекращения реакции, и концентрировали при пониженном давлении для удаления растворителя. Затем к концентрату добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; этилацетат/гексан=10%-40%) с получением указанного в заголовке соединения в виде желтого твердого вещества (0,047 г, 59,0%).
1H ЯМР (400 МГц, CDCl3) δ 8,04~8,02 (м, 2 H), 7,42 (д, 2 H, J=8,4 Гц), 7,25~7,22 (м, 2 H), 7,02~6,98 (м, 2 H), 4,85 (с, 2 H), 3,64 (т, 4 H, J=4,7 Гц), 3,17 (т, 4 H, J=4,8 Гц); LRMS (МС низкого разрешения) (ES) m/z 487,4 (M++1).
Пример 2: Соединение 11199, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-(4-фторфенил)морфолин-4-сульфонамид
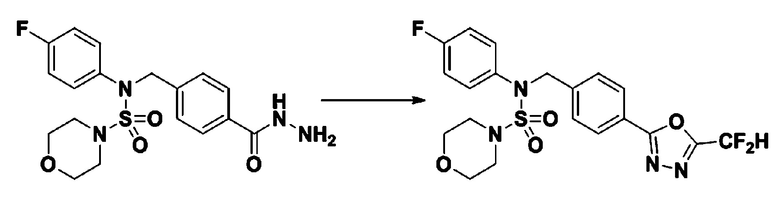
Раствор N-(4-фторфенил)-N-(4-(гидразинкарбонил)бензил)морфолин-4-сульфонамида (0,067 г, 0,164 ммоль) и триэтиламина (0,045 мл, 0,328 ммоль) в N,N-диметилформамиде (3 мл) перемешивали при 0°C, и смешивали с 2,2-дифторуксусным ангидридом (0,021 мл, 0,197 ммоль). Реакционную смесь перемешивали при 80°С в течение дополнительных 18 часов, охлаждали до комнатной температуры для прекращения реакции, и концентрировали при пониженном давлении для удаления растворителя, и концентрировали при пониженном давлении для удаления растворителя. Затем к концентрату добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; этилацетат/гексан=10%-40%) с получением указанного в заголовке соединения в виде желтого твердого вещества (0,051 г, 65,8%).
1H ЯМР (400 МГц, CDCl3) δ 8,04~8,02 (м, 2 H), 7,40 (д, 2 H, J=8,3 Гц), 7,25~7,21 (м, 2 H), 7,02~6,98 (м, 2 H), 6,90 (т, 1 H, J=51,7 Гц), 4,85 (с, 2 H), 3,64 (т, 4 H, J=4,7 Гц), 3,17 (т, 4 H, J=4,7 Гц); LRMS (ES) m/z 469,3 (M++1).
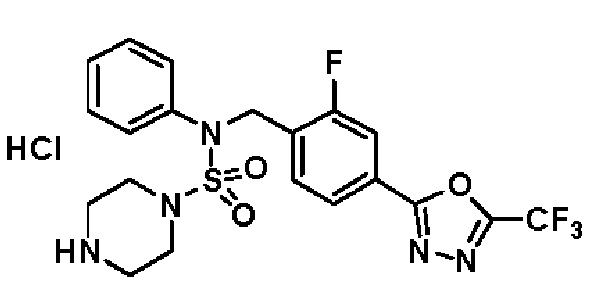
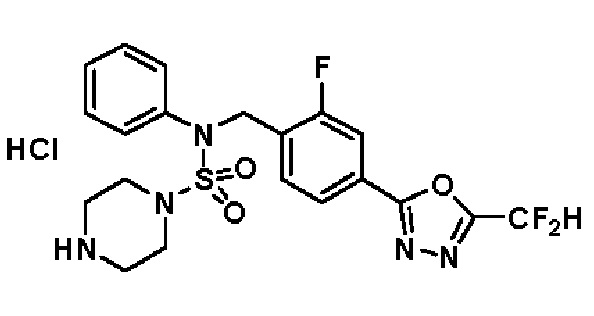
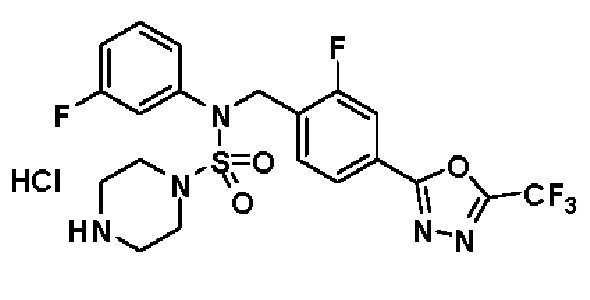
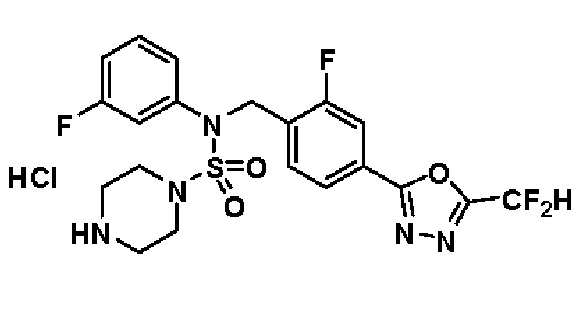
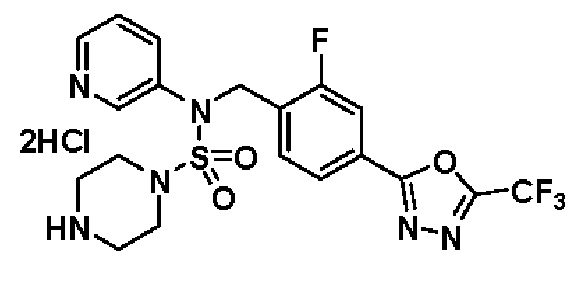
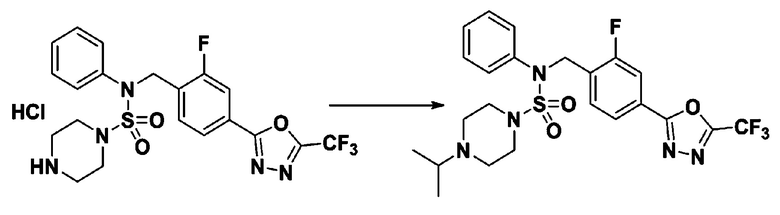
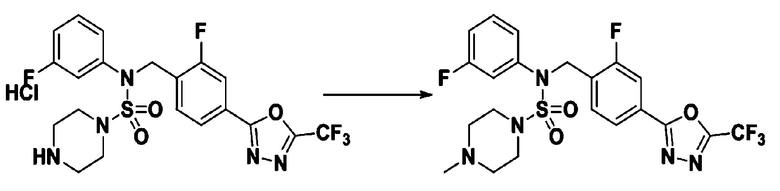
Пример 3: Соединение 11293, N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенилпиперазин-1-сульфонамид гидрохлорид
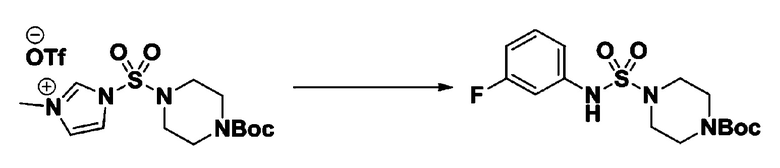
[Стадия 1] 1-((1H-имидазол-1-ил)сульфонил)-3-метил-1H-имидазол-3-ий трифторметансульфонат
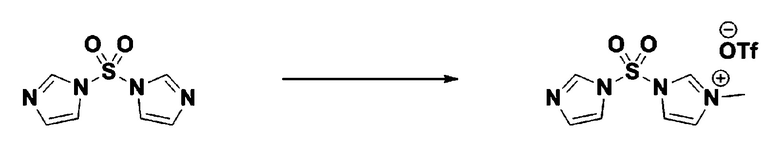
Раствор 1,1'-сульфонилбис(1Н-имидазол) (10,000 г, 50,454 ммоль) в дихлорметане (120 мл) смешивали при 0°С с трифторметансульфонатом (MeOTf, 5,710 мл, 50,454 ммоль), и перемешивали при той же температуре в течение 3 часов. Осадки собирали фильтрованием, промывали дихлорметаном и сушили с получением 1-((1H-имидазол-1-ил)сульфонил)-3-метил-1H-имидазол-3-ий трифторметансульфоната в виде белого твердого вещества (16,720 г, 91,5%).
[Стадия 2] трет-бутил 4-((1H-имидазол-1-ил)сульфонил)пиперазин-1-карбоксилат
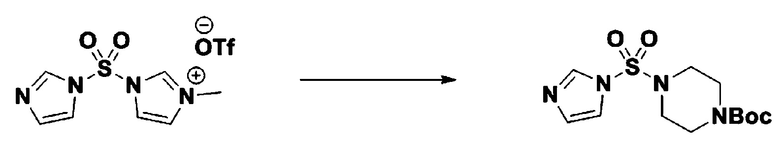
Смесь трет-бутил пиперазин-1-карбоксилата (2,500 г, 13,422 ммоль) и 1-((1H-имидазол-1-ил)сульфонил)-3-метил-1H-имидазол-3-ий трифторметансульфоната (5,835 г, 16,107 ммоль) в ацетонитриле (50 мл) перемешивали при комнатной температуре в течение 16 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 80 г; этилацетат/гексан=20%-50%) с получением трет-бутил 4-((1H-имидазол-1-ил)сульфонил)пиперазин-1-карбоксилата в виде бежевого твердого вещества (2,417 г, 56,9%).
[Стадия 3] 1-((4-(трет-бутоксикарбонил)пиперазин-1-ил)сульфонил)-3-метил-1H-имидазол-3-ий трифторметансульфонат
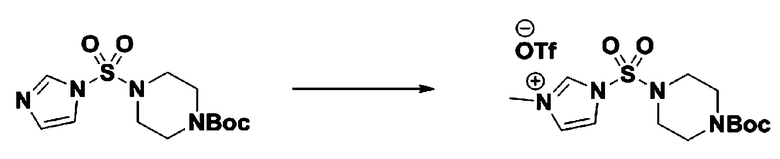
Раствор трет-бутил 4-((1H-имидазол-1-ил)сульфонил)пиперазин-1-карбоксилата (2,417 г, 7,640 ммоль) в дихлорметане (30 мл) смешивали при 0°C с MeOTf (0,908 мл, 8,022 ммоль), и перемешивали при комнатной температуре в течение 5 часов. Реакционную смесь разбавляли гексаном (30 мл), и перемешивали. Полученные осадки собирали фильтрованием, промывали гексаном и сушили с получением 1-((4-(трет-бутоксикарбонил)пиперазин-1-ил)сульфонил)-3-метил-1H-имидазол-3-ий трифторметансульфоната в виде белого твердого вещества (3,510 г, 95,6%).
[Стадия 4] трет-бутил 4-(N-фенилсульфамоил)пиперазин-1-карбоксилат
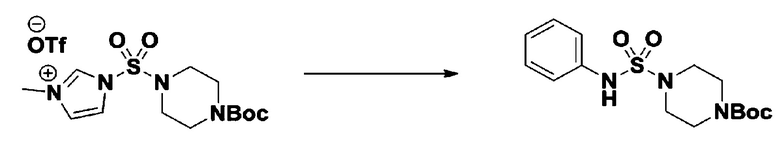
Смесь 1-((4-(трет-бутоксикарбонил)пиперазин-1-ил)сульфонил)-3-метил-1H-имидазол-3-ий трифторметансульфоната (3,510 г, 7,305 ммоль) и анилина (0,734 мл, 8,036 ммоль) в ацетонитриле (40 мл), полученную при температуре окружающей среды, нагревали с обратным холодильником в течение 16 часов, и охлаждали до температуры окружающей среды. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 40 г; этилацетат/гексан=20%-50%) с получением неочищенного продукта, который растворяли в этилацетате (20 мл) и гексане (100 мл), и перемешивали. Полученные осадки собирали фильтрованием, промывали гексаном и сушили с получением трет-бутил 4-(N-фенилсульфамоил)пиперазин-1-карбоксилата в виде белого твердого вещества (2,440 г, 97,8%).
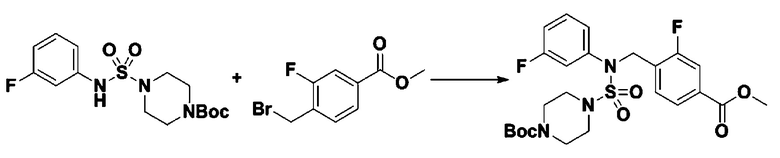
[Стадия 5] трет-бутил 4-(N-(2-фтор-4-(метоксикарбонил)бензил)-N-фенилсульфамоил)пиперазин-1-карбоксилат
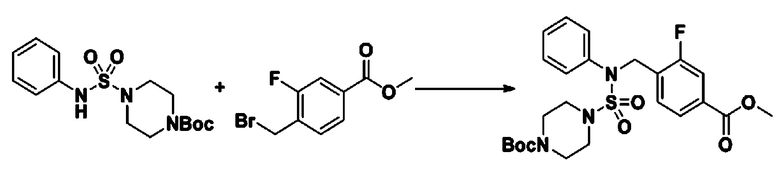
К раствору трет-бутил 4-(N-фенилсульфамоил)пиперазин-1-карбоксилата (2,440 г, 7,146 ммоль) в N,N-диметилформамиде (50 мл) добавляли NaH (60,00%, 0,372 г, 9,290 ммоль) при 0°C, и смесь перемешивали при той же температуре в течение 10 минут. Реакционную смесь обрабатывали метил 4-(бромметил)-3-фторбензоатом (1,942 г, 7,861 ммоль), и перемешивали в течение дополнительного 1 часа при комнатной температуре. Затем к реакционной смеси добавляли насыщенный водный раствор хлорида аммония с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 40 г; этилацетат/гексан=10%-30%) с получением трет-бутил 4-(N-(2-фтор-4-(метоксикарбонил)бензил)-N-фенилсульфамоил)пиперазин-1-карбоксилата в виде белого твердого вещества (3,070 г, 84,6%).
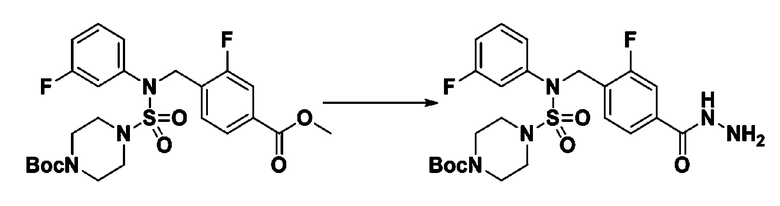
[Стадия 6] трет-бутил 4-(N-(2-фтор-4-(гидразинкарбонил)бензил)-N-фенилсульфамоил)пиперазин-1-карбоксилат
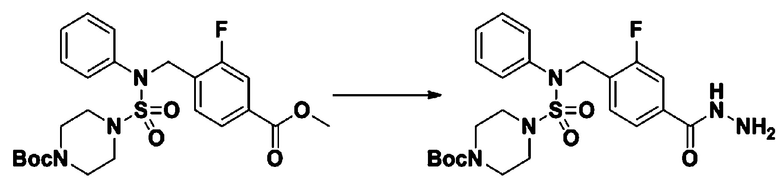
Смесь трет-бутил 4-(N-(2-фтор-4-(метоксикарбонил)бензил)-N-фенилсульфамоил)пиперазин-1-карбоксилата (2,570 г, 5,063 ммоль) и гидразина моногидрата (4,782 мл, 101,265 ммоль) в этаноле (20 мл) нагревали при 120°С в течение 2 часов под действием микроволнового излучения, и охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Указанное в заголовке соединение использовали без дополнительной очистки (2,462 г, 95,8%, белое твердое вещество).
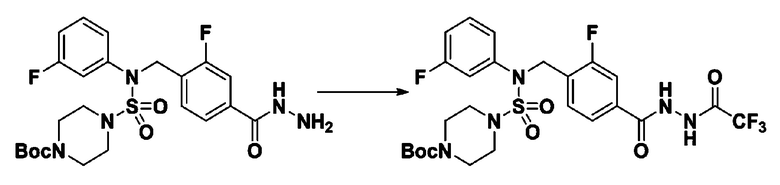
[Стадия 7] трет-бутил 4-(N-(2-фтор-4-(2-(2,2,2-трифторацетил)гидразин-1-карбонил)бензил)-N-фенилсульфамоил)пиперазин-1-карбоксилат
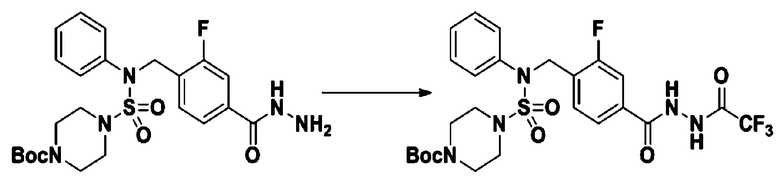
Раствор трет-бутил 4-(N-(2-фтор-4-(гидразинкарбонил)бензил)-N-фенилсульфамоил)пиперазин-1-карбоксилата (1,458 г, 2,872 ммоль) и триэтиламина (0,796 мл, 5,745 ммоль) в N,N-диметилформамиде (10 мл) перемешивали при 0°C, и смешивали с трифторуксусным ангидридом (0,486 мл, 3,447 ммоль). Реакционную смесь перемешивали при 80°С в течение дополнительных 18 часов, охлаждали до комнатной температуры для прекращения реакции, и концентрировали при пониженном давлении для удаления растворителя. Затем к концентрату добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 24 г; этилацетат/гексан=10%-70%) с получением указанного в заголовке соединения в виде белого твердого вещества (0,393 г, 23,3%).
[Стадия 8] трет-бутил 4-(N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенилсульфамоил)пиперазин-1-карбоксилат
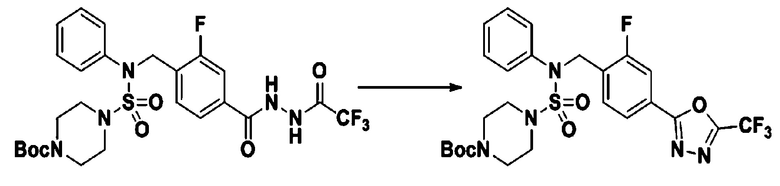
Смесь трет-бутил 4-(N-(2-фтор-4-(2-(2,2,2-трифторацетил)гидразин-1-карбонил)бензил)-N-фенилсульфамоил)пиперазин-1-карбоксилата (0,614 г, 1,017 ммоль) и 1-метокси-N-триэтиламмонийсульфонил-метанимидата (реагент Бургесса, 0,364 г, 1,526 ммоль) в тетрагидрофуране (4 мл) нагревали при 150°С в течение 30 минут под действием микроволнового излучения, охлаждали до комнатной температуры для прекращения реакции. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Затем к концентрату добавляли воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 12 г; этилацетат/гексан=0%-25%) с получением указанного в заголовке соединения в виде белого твердого вещества (0,576 г, 96,7%).
[Стадия 9] Соединение 11293
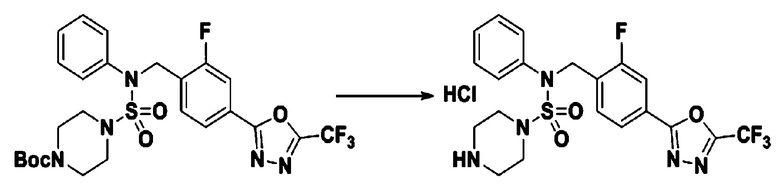
Раствор трет-бутил 4-(N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенилсульфамоил)пиперазин-1-карбоксилата (0,969 г, 1,655 ммоль) в 1,4-диоксане (10 мл) смешивали при комнатной температуре с хлористоводородной кислотой (4,00 M раствор в 1,4-диоксане, 8,274 мл, 33,096 ммоль), и перемешивали при той же температуре в течение 2 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Остаток разбавляли диэтиловым эфиром (5 мл) и гексаном (50 мл), и перемешивали. Полученные осадки собирали фильтрованием, промывали гексаном и сушили с получением N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенилпиперазин-1-сульфонамид гидрохлорида в виде белого твердого вещества (0,860 г, 99,6%).
1H ЯМР (400 МГц, ДМСО-d6) δ 8,96 (с, 2H), 7,86 (дд, 1H, J=8,0, 1,6 Гц), 7,83 (д, 1H, J=10,2 Гц), 7,62 (т, 1H, J=7,8 Гц), 7,43 (д, 2H, J=8,1 Гц), 7,37 (т, 2H, J=7,6 Гц), 7,29 (м, 1H), 5,05 (с, 2H), 3,40-3,37 (м, 4H), 3,13-3,11 (м, 4H); LRMS (ES) m/z 486,0 (M++1).
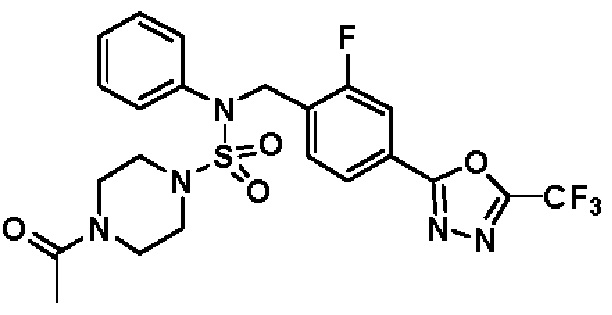
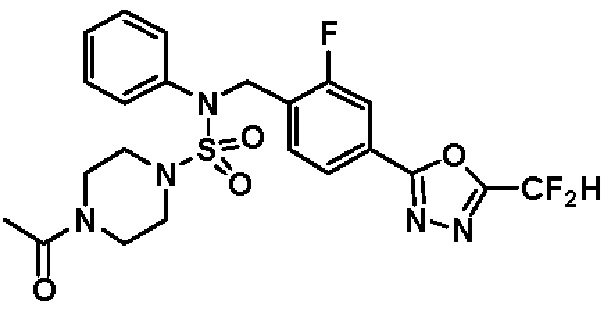
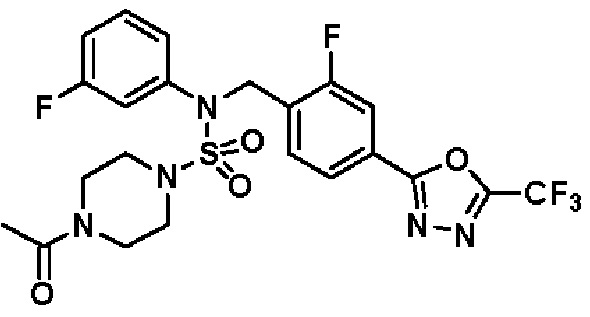
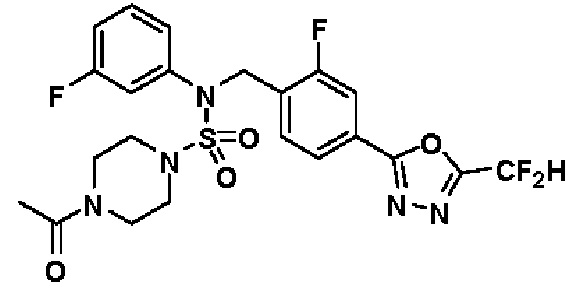
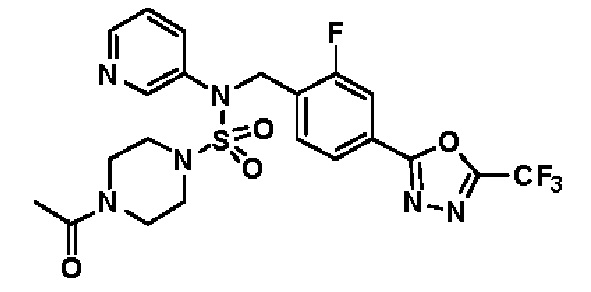
Пример 4: Соединение 11294, 4-ацетил-N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенилпиперазин-1-сульфонамид
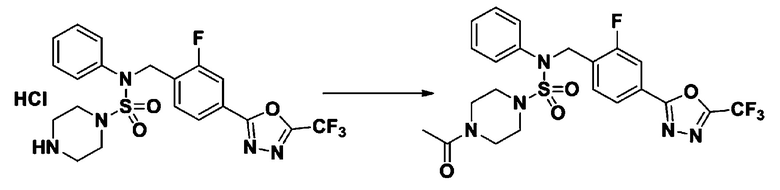
Суспензию N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенилпиперазин-1-сульфонамид гидрохлорида (0,030 г, 0,057 ммоль) в дихлорметане (3 мл) смешивали при комнатной температуре с ацетилхлоридом (0,008 мл, 0,115 ммоль) и N,N-диизопропилэтиламином (0,030 мл, 0,172 ммоль), и перемешивали при той же температуре в течение 1 часа. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 12 г; этилацетат/гексан=30%-70%) с получением 4-ацетил-N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенилпиперазин-1-сульфонамида в виде ярко-желтого масла (0,020 г, 66,0%).
1H ЯМР (400 МГц, ДМСО-d6) δ 7,86 (д, 1H, J=8,2 Гц), 7,81 (д, 1H, J=10,0 Гц), 7,63 (т, 1H, J=7,7 Гц), 7,44 (д, 2H, J=7,6 Гц), 7,35 (т, 2H, J=7,6 Гц), 7,26 (т, 1H, J=7,3 Гц), 5,04 (с, 2H), 3,44 (с, 4H), 3,20 (м, 2H), 3,14 (м, 2H), 2,00 (с, 3H); LRMS (ES) m/z 528,3 (M++1).
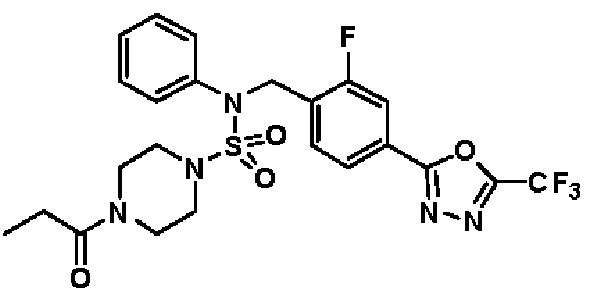
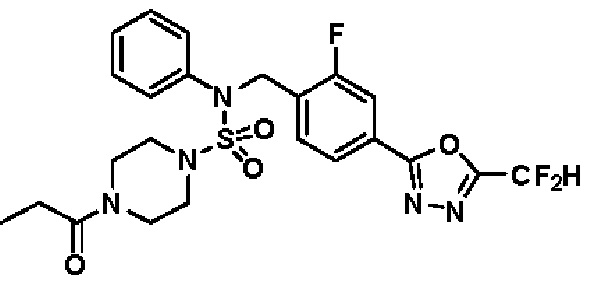
Пример 5: Соединение 11295, N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенил-4-пропионилпиперазин-1-сульфонамид
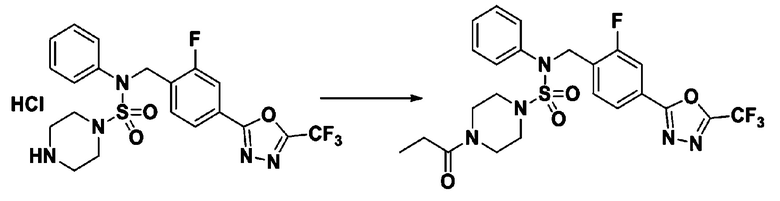
Суспензию N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенилпиперазин-1-сульфонамид гидрохлорида (0,030 г, 0,057 ммоль) в дихлорметане (3 мл) смешивали при комнатной температуре с пропионилхлоридом (0,010 мл, 0,115 ммоль) и N,N-диизопропилэтиламином (0,030 мл, 0,172 ммоль), и перемешивали при той же температуре в течение 1 часа. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 12 г; этилацетат/гексан=30%-70%) с получением N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенил-4-пропионилпиперазин-1-сульфонамида в виде бесцветной жидкости (0,024 г, 77,1%).
1H ЯМР (400 МГц, ДМСО-d6) δ 7,86 (дд, 1H, J=8,0, 1,6 Гц), 7,83-7,77 (м, 1H), 7,63 (т, 1H, J=7,7 Гц), 7,44 (д, 2H, J=7,3 Гц), 7,34 (т, 2H, J=7,6 Гц), 7,26 (т, 1H, J=7,3 Гц), 5,04 (с, 2H), 3,45 (м, 4H), 3,18 (м, 4H), 2,32 (кв, 2H, J=7,4 Гц), 0,98 (т, 3H, J=7,4 Гц); LRMS (ES) m/z 542,3 (M++1).
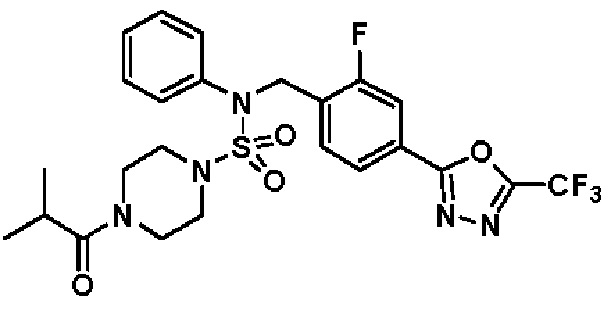
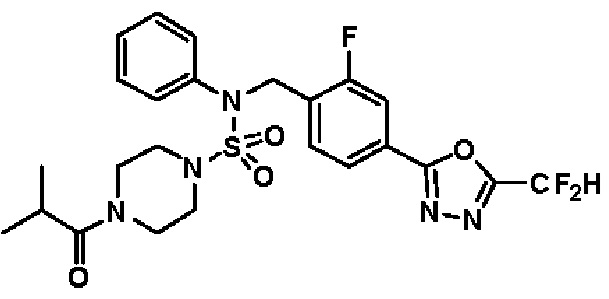
Пример 6: Соединение 11296, N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-4-изобутирил-N-фенилпиперазин-1-сульфонамид
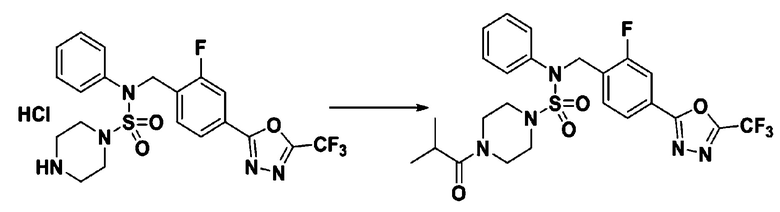
Суспензию N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенилпиперазин-1-сульфонамид гидрохлорида (0,030 г, 0,057 ммоль) в дихлорметане (3 мл) смешивали при комнатной температуре с изобутирилхлоридом (0,012 мл, 0,115 ммоль) и N,N-диизопропилэтиламином (0,030 мл, 0,172 ммоль), и перемешивали при той же температуре в течение 1 часа. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 12 г; этилацетат/гексан=20%-50%) с получением N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-4-изобутирил-N-фенилпиперазин-1-сульфонамида в виде белого твердого вещества (0,025 г, 78,3%).
1H ЯМР (400 МГц, ДМСО-d6) δ 7,86 (дд, 1H, J=8,0, 1,6 Гц), 7,83-7,77 (м, 1H), 7,63 (т, 1H, J=7,6 Гц), 7,44 (д, 2H, J=7,9 Гц), 7,34 (т, 2H, J=7,7 Гц), 7,26 (т, 1H, J=7,3 Гц), 5,05 (с, 2H), 3,49 (м, 4H), 3,16 (с, 4H), 2,84 (дт, 1H, J=13,6, 6,8 Гц), 0,98 (д, 6H, J=6,7 Гц); LRMS (ES) m/z 556,3 (M++1).
Пример 7: Соединение 11297, N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-4-(метилсульфонил)-N-фенилпиперазин-1-сульфонамид
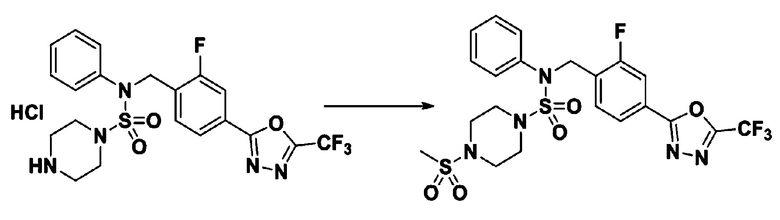
Суспензию N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенилпиперазин-1-сульфонамид гидрохлорида (0,030 г, 0,057 ммоль) в дихлорметане (3 мл) смешивали при комнатной температуре с метансульфонилхлоридом (0,009 мл, 0,115 ммоль) и N,N-диизопропилэтиламином (0,030 мл, 0,172 ммоль), и перемешивали при той же температуре в течение 1 часа. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 12 г; этилацетат/гексан=20%-50%) с получением N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-4-(метилсульфонил)-N-фенилпиперазин-1-сульфонамида в виде белого твердого вещества (0,029 г, 89,5%).
1H ЯМР (400 МГц, ДМСО-d6) δ 7,86 (дд, 1H, J=8,1, 1,6 Гц), 7,84-7,78 (м, 1H), 7,63 (т, 1H, J=7,6 Гц), 7,45 (д, 2H, J=8,1 Гц), 7,36 (т, 2H, J=7,7 Гц), 7,27 (т, 1H, J=7,3 Гц), 5,05 (с, 2H), 3,31-3,25 (м, 4H), 3,19-3,07 (м, 4H), 2,90 (с, 3H); LRMS (ES) m/z 564,2 (M++1).
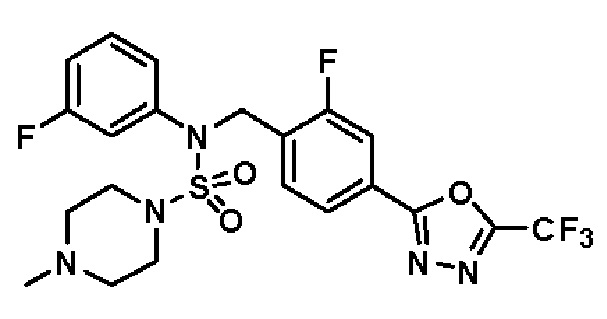
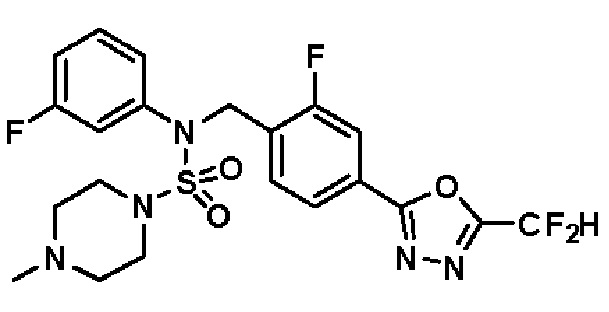
Пример 8: Соединение 11298, N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-4-метил-N-фенилпиперазин-1-сульфонамид
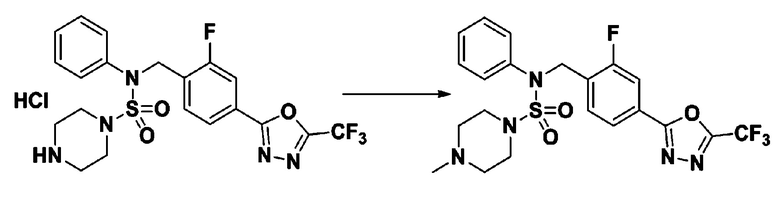
Смесь N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенилпиперазин-1-сульфонамид гидрохлорида (0,050 г, 0,096 ммоль) и формальдегида (37,00% раствор в воде, 0,071 мл, 0,958 ммоль) в дихлорметане (4 мл) обрабатывали при комнатной температуре триацетоксиборгидридом натрия (0,061 г, 0,287 ммоль), и перемешивали при той же температуре в течение 16 часов. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; этилацетат/гексан=70%-100%) с получением N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-4-метил-N-фенилпиперазин-1-сульфонамида в виде светло-желтого твердого вещества (0,031 г, 64,8%).
1H ЯМР (400 МГц, ДМСО-d6) δ 7,86 (дд, 1H, J=7,9, 1,6 Гц), 7,81 (д, 1H, J=10,1 Гц), 7,62 (т, 1H, J=7,8 Гц), 7,43 (д, 2H, J=7,5 Гц), 7,35 (т, 2H, J=7,7 Гц), 7,26 (т, 1H, J=7,2 Гц), 5,03 (с, 2H), 3,15 (м, 4H), 2,30 (с, 4H), 2,16 (с, 3H); LRMS (ES) m/z 500,3 (M++1).
Пример 9: Соединение 11299, 4-этил-N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенилпиперазин-1-сульфонамид
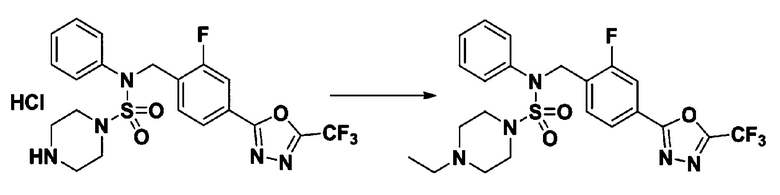
Смесь N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенилпиперазин-1-сульфонамид гидрохлорида (0,050 г, 0,096 ммоль) и ацетальдегида (0,027 мл, 0,479 ммоль) в дихлорметане (4 мл) обрабатывали при комнатной температуре триацетоксиборгидридом натрия (0,061 г, 0,287 ммоль), и перемешивали при той же температуре в течение 16 часов. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; этилацетат/гексан=70%-100%) с получением 4-этил-N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенилпиперазин-1-сульфонамида в виде коричневого твердого вещества (0,023 г, 46,8%).
1H ЯМР (400 МГц, ДМСО-d6) δ 7,86 (дд, 1H, J=8,0, 1,6 Гц), 7,83-7,77 (м, 1H), 7,62 (т, 1H, J=7,6 Гц), 7,43 (д, 2H, J=8,1 Гц), 7,35 (т, 2H, J=7,7 Гц), 7,26 (т, 1H, J=7,3 Гц), 5,03 (с, 2H), 3,15 (м, 4H), 2,48-2,08 (м, 6H), 0,97 (т, 3H, J=7,2 Гц); LRMS (ES) m/z 514,1 (M++1).
Пример 10: Соединение 11300, N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-4-изопропил-N-фенилпиперазин-1-сульфонамид
Смесь N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенилпиперазин-1-сульфонамид гидрохлорида (0,050 г, 0,096 ммоль) и ацетона (0,035 мл, 0,479 ммоль) в дихлорметане (4 мл) обрабатывали при комнатной температуре триацетоксиборгидридом натрия (0,061 г, 0,287 ммоль), и перемешивали при той же температуре в течение 16 часов. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; этилацетат/гексан=60%-90%) с получением N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-4-изопропил-N-фенилпиперазин-1-сульфонамида в виде белого твердого вещества (0,020 г, 39,6%).
1H ЯМР (400 МГц, ДМСО-d6) δ 7,86 (дд, 1H, J=8,0, 1,7 Гц), 7,81 (дд, 1H, J=10,0, 1,6 Гц), 7,63 (т, 1H, J=7,7 Гц), 7,48-7,41 (м, 2H), 7,35 (т, 2H, J=7,7 Гц), 7,25 (т, 1H, J=7,3 Гц), 5,03 (с, 2H), 3,12 (м, 4H), 2,69-2,60 (м, 1H), 2,36 (м, 4H), 0,93 (д, 6H, J=6,6 Гц); LRMS (ES) m/z 528,1 (M++1).
Пример 11: Соединение 11301, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-фенилпиперазин-1-сульфонамид гидрохлорид
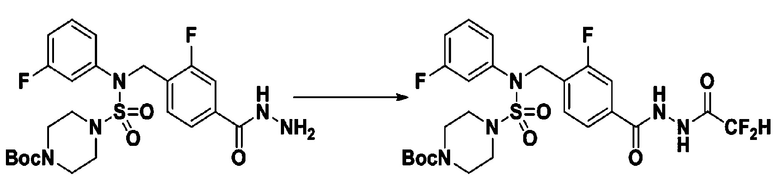
[Стадия 1] трет-бутил 4-(N-(4-(2-(2,2-дифторацетил)гидразин-1-карбонил)-2-фторбензил)-N-фенилсульфамоил)пиперазин-1-карбоксилат
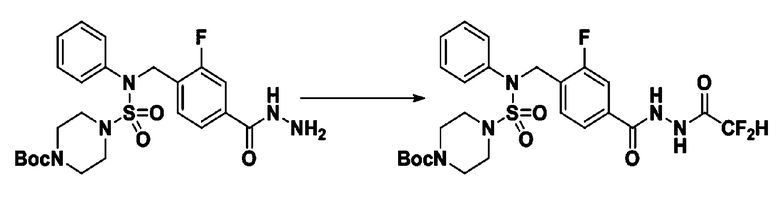
Раствор трет-бутил 4-(N-(2-фтор-4-(гидразинкарбонил)бензил)-N-фенилсульфамоил)пиперазин-1-карбоксилата (1,458 г, 2,872 ммоль) и триэтиламина (0,796 мл, 5,745 ммоль) в N,N-диметилформамиде (10 мл) перемешивали при 0°C, и смешивали с 2,2-дифторуксусным ангидридом (0,375 мл, 3,447 ммоль). Реакционную смесь перемешивали при 80°С в течение дополнительных 18 ч, охлаждали до комнатной температуры для прекращения реакции, и концентрировали при пониженном давлении для удаления растворителя. Затем к концентрату добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 24 г; этилацетат/гексан=10%-70%) с получением указанного в заголовке соединения в виде бесцветного масла (0,078 г, 4,8%).
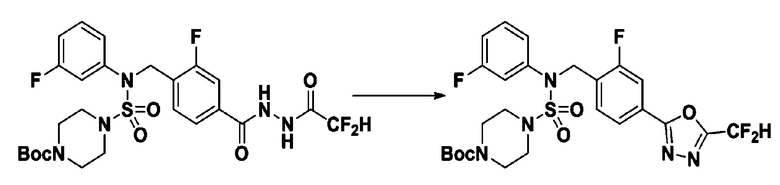
[Стадия 2] трет-бутил 4-(N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-фенилсульфамоил)пиперазин-1-карбоксилат
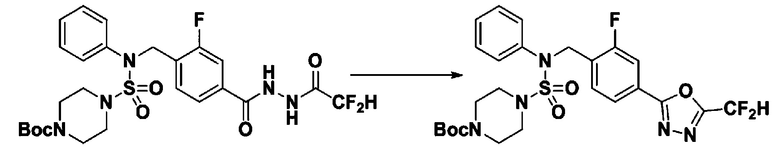
Смесь трет-бутил 4-(N-(4-(2-(2,2-дифторацетил)гидразин-1-карбонил)-2-фторбензил)-N-фенилсульфамоил)пиперазин-1-карбоксилата (0,671 г, 1,146 ммоль) и 1-метокси-N-триэтиламмонийсульфонил-метанимидата (реагент Бургесса, 0,410 г, 1,719 ммоль) в тетрагидрофуране (4 мл) нагревали при 150°С в течение 30 минут под действием микроволнового излучения, и охлаждали до комнатной температуры для прекращения реакции. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Затем к концентрату добавляли воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 12 г; этилацетат/гексан=0%-25%) с получением указанного в заголовке соединения в виде белого твердого вещества (0,382 г, 58,7%).
[Стадия 3] Соединение 11301
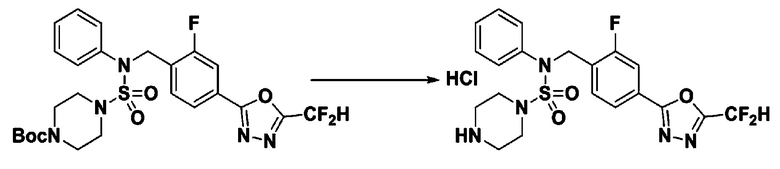
Раствор трет-бутил 4-(N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-фенилсульфамоил)пиперазин-1-карбоксилата (0,460 г, 0,810 ммоль) в 1,4-диоксане (10 мл) смешивали при комнатной температуре с хлористоводородной кислотой (4,00 M раствор в 1,4-диоксане, 4,052 мл, 16,209 ммоль), и перемешивали при той же температуре в течение 2 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток разбавляли диэтиловым эфиром (5 мл) и гексаном (50 мл), и перемешивали. Полученные осадки собирали фильтрованием, промывали гексаном и сушили с получением N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-фенилпиперазин-1-сульфонамид гидрохлорида в виде белого твердого вещества (0,400 г, 97,9%).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,04 (с, 2H), 7,84 (м, 1H), 7,78 (м, 1H), 7,67 (с, 0,25H), 7,61 (т, 1H, J=7,8 Гц), 7,54 (с, 0,5H), 7,44-7,38 (м, 2,25H), 7,37 (т, 2H, J=7,6 Гц), 7,30 (т, 1H, J=7,2 Гц), 5,04 (с, 2H), 3,41-3,37 (м, 4H), 3,14-3,09 (м, 4H); LRMS (ES) m/z 468,2 (M++1).
Пример 12: Соединение 11302, 4-ацетил-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-фенилпиперазин-1-сульфонамид
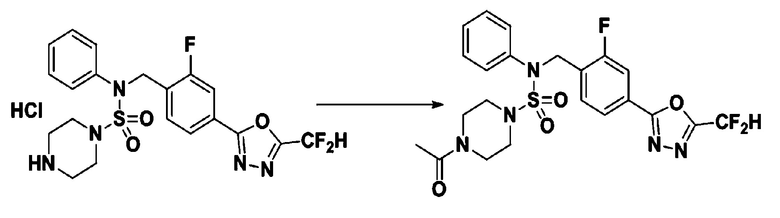
Суспензию N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-фенилпиперазин-1-сульфонамид гидрохлорида (0,030 г, 0,060 ммоль) в дихлорметане (3 мл) смешивали при комнатной температуре с ацетилхлоридом (0,008 мл, 0,119 ммоль) и N,N-диизопропилэтиламином (0,031 мл, 0,179 ммоль), и перемешивали при той же температуре в течение 1 часа. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 12 г; этилацетат/гексан=30%-70%) с получением 4-ацетил-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-фенилпиперазин-1-сульфонамида в виде светло-желтой жидкости (0,017 г, 56,0%).
1H ЯМР (400 МГц, ДМСО-d6) δ 7,83 (м, 1H), 7,77 (м, 1H), 7,66 (с, 0,25H), 7,61 (т, 1H, J=7,7 Гц), 7,53 (с, 0,5H), 7,44 (д, 2H, J=7,5 Гц), 7,40 (с, 0,25H), 7,35 (т, 2H, J=7,7 Гц), 7,26 (т, 1H, J=7,3 Гц), 5,03 (с, 2H), 3,43 (с, 4H), 3,19 (м, 2H), 3,14 (м, 2H), 2,00 (с, 3H); LRMS (ES) m/z 510,3 (M++1).
Пример 13: Соединение 11303, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-фенил-4-пропионилпиперазин-1-сульфонамид
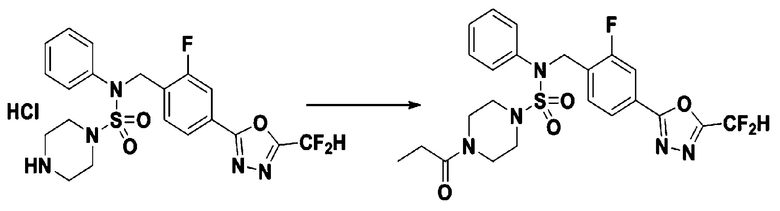
Суспензию N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-фенилпиперазин-1-сульфонамид гидрохлорида (0,030 г, 0,060 ммоль) в дихлорметане (3 мл) смешивали при комнатной температуре с пропионилхлоридом (0,010 мл, 0,119 ммоль) и N,N-диизопропилэтиламином (0,031 мл, 0,179 ммоль), и перемешивали при той же температуре в течение 1 часа. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 12 г; этилацетат/гексан=30%-70%) с получением N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-фенил-4-пропионилпиперазин-1-сульфонамида в виде бесцветной жидкости (0,021 г, 67,4%).
1H ЯМР (400 МГц, ДМСО-d6) δ 7,83 (дд, 1H, J=8,0, 1,7 Гц), 7,77 (дд, 1H, J=10,1, 1,5 Гц), 7,66 (с, 0,25H), 7,62 (т, 1H, J=7,8 Гц), 7,53 (с, 0,5H), 7,44 (д, 2H, J=7,4 Гц), 7,41 (с, 0,25H), 7,35 (т, 2H, J=7,7 Гц), 7,26 (т, 1H, J=7,3 Гц), 5,03 (с, 2H), 3,44 (с, 4H), 3,16 (м, 4H), 2,32 (кв, 2H, J=7,4 Гц), 0,98 (т, 3H, J=7,4 Гц); LRMS (ES) m/z 524,3 (M++1).
Пример 14: Соединение 11304, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-4-изобутирил-N-фенилпиперазин-1-сульфонамид
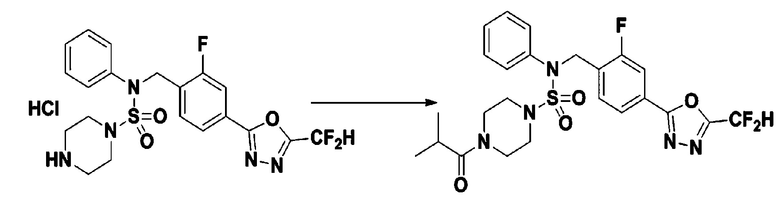
Суспензию N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-фенилпиперазин-1-сульфонамид гидрохлорида (0,030 г, 0,060 ммоль) в дихлорметане (3 мл) смешивали при комнатной температуре с изобутирилхлоридом (0,012 мл, 0,119 ммоль) и N,N-диизопропилэтиламином (0,031 мл, 0,179 ммоль), и перемешивали при той же температуре в течение 1 часа. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 12 г; этилацетат/гексан=20%-50%) с получением N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-4-изобутирил-N-фенилпиперазин-1-сульфонамида в виде бесцветного масла (0,023 г, 71,9%).
1H ЯМР (400 МГц, ДМСО-d6) δ 7,83 (дд, 1H, J=8,0, 1,6 Гц), 7,77 (дд, 1H, J=10,1, 1,6 Гц), 7,66 (с, 0,25H), 7,62 (т, 1H, J=7,8 Гц), 7,53 (с, 0,5H), 7,47-7,42 (м, 2H), 7,41 (с, 0,25H), 7,35 (т, 2H, J=7,6 Гц), 7,26 (т, 1H, J=7,3 Гц), 5,03 (с, 2H), 3,50 (м, 4H), 3,16 (с, 4H), 2,84 (дт, 1H, J=13,6, 6,7 Гц), 0,98 (д, 6H, J=6,7 Гц); LRMS (ES) m/z 538,1 (M++1).
Пример 15: Соединение 11305, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-4-(метилсульфонил)-N-фенилпиперазин-1-сульфонамид
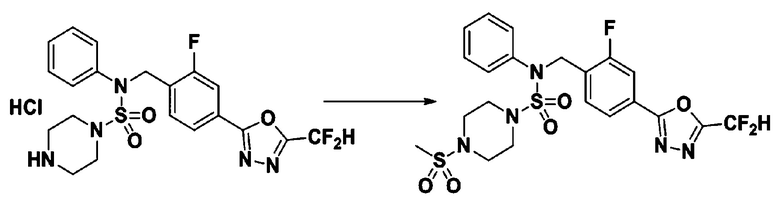
Суспензию N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-фенилпиперазин-1-сульфонамид гидрохлорида (0,030 г, 0,060 ммоль) в дихлорметане (3 мл) смешивали при комнатной температуре с метансульфонилхлоридом (0,009 мл, 0,119 ммоль) и N,N-диизопропилэтиламином (0,031 мл, 0,179 ммоль), и перемешивали при той же температуре в течение 1 часа. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 12 г; этилацетат/гексан=20%-50%) с получением N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-4-(метилсульфонил)-N-фенилпиперазин-1-сульфонамида в виде белого твердого вещества (0,028 г, 86,2%).
1H ЯМР (400 МГц, ДМСО-d6) δ 7,83 (м, 1H), 7,78 (д, 1H, J=10,1 Гц), 7,66 (с, 0,25H), 7,62 (т, 1H, J=7,8 Гц), 7,53 (с, 0,5H), 7,46 (д, 2H, J=7,4 Гц), 7,41 (с, 0,25H), 7,36 (т, 2H, J=7,7 Гц), 7,28 (т, 1H, J=7,3 Гц), 5,04 (с, 2H), 3,31-3,21 (м, 4H), 3,19-3,06 (м, 4H), 2,90 (с, 3H); LRMS (ES) m/z 546,2 (M++1).
Пример 16: Соединение 11306 гидрохлорид, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-4-метил-N-фенилпиперазин-1-сульфонамид гидрохлорид
[Стадия 1] Соединение 11306, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-4-метил-N-фенилпиперазин-1-сульфонамид
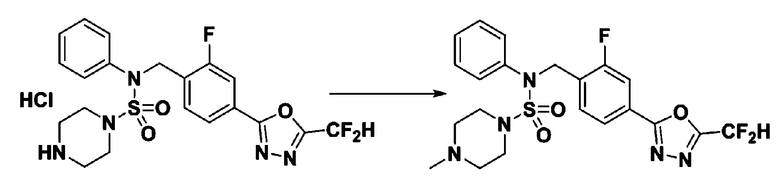
Смесь N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-фенилпиперазин-1-сульфонамид гидрохлорида (0,050 г, 0,099 ммоль) и формальдегида (37,00% раствор в воде, 0,074 мл, 0,992 ммоль) в дихлорметане (4 мл) обрабатывали при комнатной температуре триацетоксиборгидридом натрия (0,063 г, 0,298 ммоль), и перемешивали при той же температуре в течение 16 часов. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; этилацетат/гексан=70%-100%) с получением N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-4-метил-N-фенилпиперазин-1-сульфонамида в виде белого твердого вещества (0,020 г, 41,9%).
1H ЯМР (400 МГц, ДМСО-d6) δ 7,83 (дд, 1H, J=8,1, 1,6 Гц), 7,80-7,74 (м, 1H), 7,66 (с, 0,25H), 7,61 (т, 1H, J=7,7 Гц), 7,53 (с, 0,5H), 7,46-7,42 (м, 2H), 7,41 (с, 0,25H), 7,35 (т, 2H, J=7,6 Гц), 7,26 (т, 1H, J=7,3 Гц), 5,00 (с, 2H), 3,20-3,07 (м, 4H), 2,36-2,20 (м, 4H), 2,15 (с, 3H); LRMS (ES) m/z 482,5 (M++1).
[Стадия 2] Соединение 11306 гидрохлорид
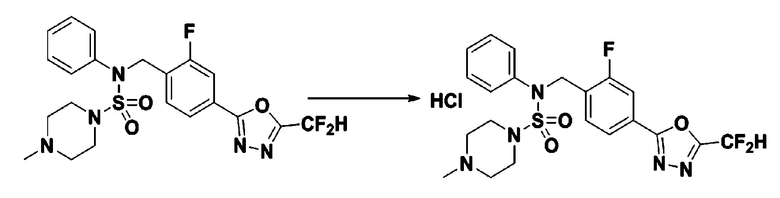
Раствор N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-4-метил-N-фенилпиперазин-1-сульфонамида (0,040 г, 0,083 ммоль) в этилацетате (5 мл) смешивали при комнатной температуре с хлористоводородной кислотой (1,00 M раствор в EtOAc, 0,249 мл, 0,249 ммоль), и перемешивали при той же температуре в течение 10 мин. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Остаток разбавляли этилацетатом (2 мл) и гексаном (20 мл) и перемешивали. Полученные осадки собирали фильтрованием, промывали гексаном и сушили с получением N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-4-метил-N-фенилпиперазин-1-сульфонамид гидрохлорида в виде белого твердого вещества (0,036 г, 83,7%).
Пример 17: Соединение 11307, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-4-этил-N-фенилпиперазин-1-сульфонамид
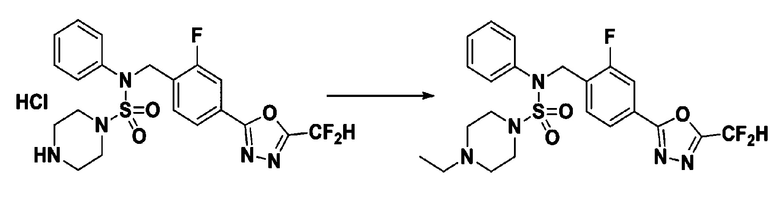
Смесь N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-фенилпиперазин-1-сульфонамид гидрохлорида (0,050 г, 0,099 ммоль) и ацетальдегида (0,028 мл, 0,496 ммоль) в дихлорметане (4 мл) обрабатывали при комнатной температуре триацетоксиборгидридом натрия (0,063 г, 0,298 ммоль), и перемешивали при той же температуре в течение 16 часов. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; этилацетат/гексан=70%-100%) с получением N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-4-этил-N-фенилпиперазин-1-сульфонамида в виде коричневой жидкости (0,025 г, 50,8%).
1H ЯМР (400 МГц, ДМСО-d6) δ 7,83 (д, 1H, J=8,0 Гц), 7,78 (д, 1H, J=10,1 Гц), 7,66 (с, 0,25H), 7,61 (т, 1H, J=7,8 Гц), 7,53 (с, 0,5H), 7,44 (д, 2H, J=8,0 Гц), 7,41 (с, 0,25H), 7,35 (т, 2H, J=7,7 Гц), 7,26 (т, 1H, J=7,3 Гц), 5,03 (с, 2H), 3,15 (м, 4H), 2,33 (м, 6H), 0,97 (т, 3H, J=7,1 Гц); LRMS (ES) m/z 496,1 (M++1).
Пример 18: Соединение 11308, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-4-изопропил-N-фенилпиперазин-1-сульфонамид
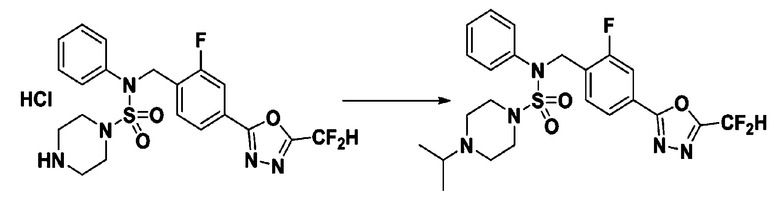
Смесь N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-фенилпиперазин-1-сульфонамид гидрохлорида (0,050 г, 0,099 ммоль) и ацетона (0,036 мл, 0,496 ммоль) в дихлорметане (4 мл) обрабатывали при комнатной температуре триацетоксиборгидридом натрия (0,063 г, 0,298 ммоль), и перемешивали при той же температуре в течение 16 часов. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; этилацетат/гексан=60%-90%) с получением N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-4-изопропил-N-фенилпиперазин-1-сульфонамида в виде белого твердого вещества (0,019 г, 37,6%).
1H ЯМР (400 МГц, ДМСО-d6) δ 7,83 (дд,1H, J=7,9, 1,6 Гц), 7,78 (дд, 1H, J=10,2, 1,6 Гц), 7,66 (с, 0,25H), 7,62 (т, 1H, J=7,7 Гц), 7,54 (с, 0,5H), 7,44 (д, 2H, J=8,0 Гц), 7,41 (с, 0,25H), 7,35 (т, 2H, J=7,7 Гц), 7,26 (т, 1H, J=7,3 Гц), 5,00 (с, 2H), 3,19-3,07 (м, 4H), 2,70-2,60 (м, 1H), 2,43-2,34 (м, 4H), 0,92 (д, 6H, J=6,5 Гц); LRMS (ES) m/z 510,4 (M++1).
Пример 19: Соединение 11309, N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-(3-фторфенил)пиперазин-1-сульфонамид гидрохлорид
[Стадия 1] трет-бутил 4-(N-(3-фторфенил)сульфамоил)пиперазин-1-карбоксилат
Смесь 1-((4-(трет-бутоксикарбонил)пиперазин-1-ил)сульфонил)-3-метил-1H-имидазол-3-ий трифторметансульфоната (3,340 г, 6,952 ммоль) и 3-фторанилина (0,732 мл, 7,647 ммоль) в ацетонитриле (30 мл), полученную при температуре окружающей среды, нагревали с обратным холодильником в течение 16 часов, и охлаждали до температуры окружающей среды. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 40 г; этилацетат/гексан=20%-50%) с получением неочищенного продукта, который растворяли в этилацетате (10 мл) и гексане (100 мл), и перемешивали. Полученные осадки собирали фильтрованием, промывали диэтиловым эфиром и сушили с получением трет-бутил 4-(N-(3-фторфенил)сульфамоил)пиперазин-1-карбоксилата в виде белого твердого вещества (1,950 г, 78,0%).
[Стадия 2] трет-бутил 4-(N-(2-фтор-4-(метоксикарбонил)бензил)-N-(3-фторфенил)сульфамоил)пиперазин-1-карбоксилат
К раствору трет-бутил 4-(N-(3-фторфенил)сульфамоил)пиперазин-1-карбоксилата (1,950 г, 5,425 ммоль) в N,N-диметилформамиде (20 мл) добавляли гидрид натрия (60,00%, 0,282 г, 7,053 ммоль) при комнатной температуре и смесь перемешивали при той же температуре в течение 10 минут. Реакционную смесь обрабатывали метил 4-(бромметил)-3-фторбензоатом (1,474 г, 5,968 ммоль), и перемешивали в течение дополнительного 1 ч при той же температуре. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 40 г; этилацетат/гексан=10%-30%) с получением трет-бутил 4-(N-(2-фтор-4-(метоксикарбонил)бензил)-N-(3-фторфенил)сульфамоил)пиперазин-1-карбоксилата в виде белого твердого вещества (2,810 г, 98,5%).
[Стадия 3] трет-бутил 4-(N-(2-фтор-4-(гидразинкарбонил)бензил)-N-(3-фторфенил)сульфамоил)пиперазин-1-карбоксилат
трет-бутил 4-(N-(2-фтор-4-(метоксикарбонил)бензил)-N-(3-фторфенил)сульфамоил)пиперазин-1-карбоксилат (2,810 г, 5,347 ммоль) и гидрат гидразина (5,197 мл, 106,932 ммоль) смешивали при комнатной температуре в этаноле (70 мл), и затем смесь перемешивали при 100°C в течение 16 часов. Реакционную смесь охлаждали до комнатной температуры для прекращения реакции, и концентрировали при пониженном давлении для удаления растворителя. Затем к концентрату добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Указанное в заголовке соединение использовали без дополнительной очистки (трет-бутил 4-(N-(2-фтор-4-(гидразинкарбонил)бензил)-N-(3-фторфенил)сульфамоил)пиперазин-1-карбоксилат, 2,376 г, 84,6%, белое твердое вещество).
[Стадия 4] трет-бутил 4-(N-(2-фтор-4-(2-(2,2,2-трифторацетил)гидразин-1-карбонил)бензил)-N-(3-фторфенил)сульфамоил)пиперазин-1-карбоксилат
Раствор трет-бутил 4-(N-(2-фтор-4-(гидразинкарбонил)бензил)-N-(3-фторфенил)сульфамоил)пиперазин-1-карбоксилата (1,000 г, 1,903 ммоль) в 1,4-диоксане (15 мл) смешивали при комнатной температуре с 2,2,2-трифторуксусным ангидридом (0,265 мл, 1,903 ммоль) и триэтиламином (0,659 мл, 4,757 ммоль). Реакционную смесь нагревали с обратным холодильником в течение 3 часов, и охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 40 г; этилацетат/гексан=20%-50%) с получением трет-бутил 4-(N-(2-фтор-4-(2-(2,2,2-трифторацетил)гидразин-1-карбонил)бензил)-N-(3-фторфенил)сульфамоил)пиперазин-1-карбоксилата в виде белого твердого вещества (0,855 г, 72,3%).
[Стадия 5] трет-бутил 4-(N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-(3-фторфенил)сульфамоил)пиперазин-1-карбоксилат
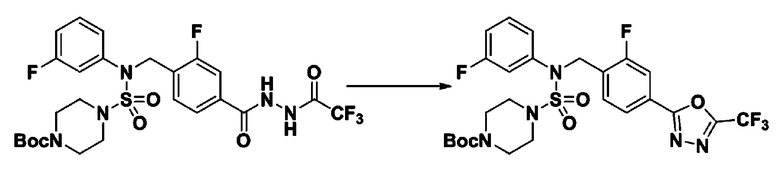
трет-бутил 4-(N-(2-фтор-4-(2-(2,2,2-трифторацетил)гидразин-1-карбонил)бензил)-N-(3-фторфенил)сульфамоил)пиперазин-1-карбоксилат (0,855 г, 1,376 ммоль) и 1-метокси-N-триэтиламмонийсульфонил-метанимидат (реагент Бургесса, 0,492 г, 2,063 ммоль) смешивали при комнатной температуре в тетрагидрофуране (10 мл), и затем смесь нагревали при 150°С в течение 30 минут под действием микроволнового излучения. Реакционную смесь охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 40 г; этилацетат/гексан=5%-30%) с получением трет-бутил 4-(N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-(3-фторфенил)сульфамоил)пиперазин-1-карбоксилата в виде белого твердого вещества (0,525 г, 63,2%).
[Стадия 6] Соединение 11309
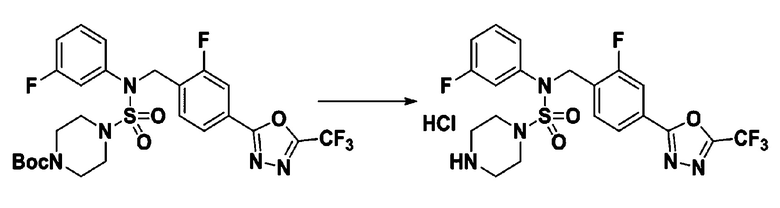
Раствор трет-бутил 4-(N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-(3-фторфенил)сульфамоил)пиперазин-1-карбоксилата (0,525 г, 0,870 ммоль) в 1,4-диоксане (10 мл) смешивали при комнатной температуре с хлористоводородной кислотой (4,00 M раствор в 1,4-диоксане, 4,349 мл, 17,396 ммоль), и перемешивали при той же температуре в течение 2 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток разбавляли диэтиловым эфиром (5 мл) и гексаном (50 мл), и перемешивали. Полученные осадки собирали фильтрованием, промывали гексаном и сушили с получением N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-(3-фторфенил)пиперазин-1-сульфонамид гидрохлорида в виде бежевого твердого вещества (0,460 г, 98,0%).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,17 (с, 2H), 7,88-7,85 (м, 2H), 7,64 (т, 1H, J=7,7 Гц), 7,43-7,38 (м, 2H), 7,29 (д, 1H, J=8,0 Гц), 7,16 (т, 1H, J=8,5 Гц), 5,07 (с, 2H), 3,53-3,38 (м, 4H), 3,22-3,05 (м, 4H); LRMS (ES) m/z 504,2 (M++1).
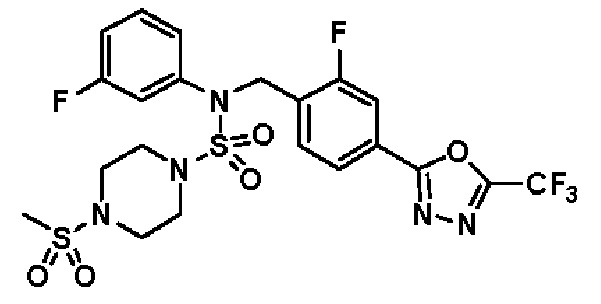
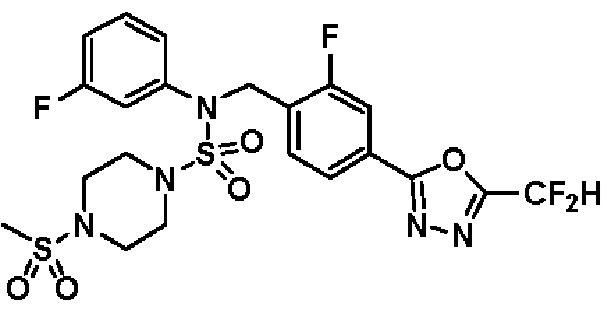
Пример 20: Соединение 11310, N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-(3-фторфенил)-4-(метилсульфонил)пиперазин-1-сульфонамид
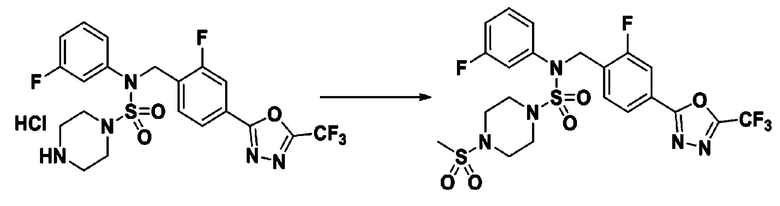
Суспензию N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-(3-фторфенил)пиперазин-1-сульфонамид гидрохлорида (0,050 г, 0,093 ммоль) в дихлорметане (3 мл) смешивали при комнатной температуре с метансульфонилхлоридом (0,014 мл, 0,185 ммоль) и N,N-диизопропилэтиламином (0,049 мл, 0,278 ммоль), и перемешивали при той же температуре в течение 1 часа. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 12 г; этилацетат/гексан=30%-70%) с получением N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-(3-фторфенил)-4-(метилсульфонил)пиперазин-1-сульфонамида в виде белого твердого вещества (0,044 г, 81,7%).
1H ЯМР (400 МГц, ДМСО-d6) δ 7,87 (д, 1H, J=8,1 Гц), 7,83 (д, 1H, J=10,2 Гц), 7,63 (т, 1H, J=7,6 Гц), 7,43-7,37 (м, 2H), 7,32 (д, 1H, J=7,9 Гц), 7,15 (м, 1H), 5,08 (с, 2H), 3,32 (м, 4H), 3,15 (м, 4H), 2,91 (с, 3H); LRMS (ES) m/z 582,2 (M++1).
Пример 21: Соединение 11311, 4-ацетил-N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-(3-фторфенил)пиперазин-1-сульфонамид
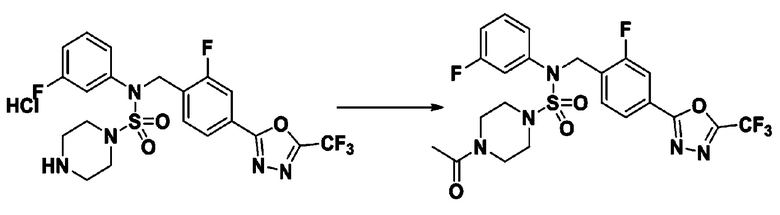
Суспензию N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-(3-фторфенил)пиперазин-1-сульфонамид гидрохлорида (0,050 г, 0,093 ммоль) в дихлорметане (3 мл) смешивали при комнатной температуре с ацетилхлоридом (0,013 мл, 0,185 ммоль) и N,N-диизопропилэтиламином (0,049 мл, 0,278 ммоль), и перемешивали при той же температуре в течение 1 часа. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 12 г; этилацетат/гексан=40%-80%) с получением 4-ацетил-N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-(3-фторфенил)пиперазин-1-сульфонамида в виде коричневой жидкости (0,035 г, 69,3%).
1H ЯМР (400 МГц, ДМСО-d6) δ 7,87 (дд, 1H, J=8,0, 1,6 Гц), 7,84-7,79 (м, 1H), 7,63 (т, 1H, J=7,7 Гц), 7,44-7,34 (м, 2H), 7,30 (м, 1H), 7,12 (м, 1H), 5,07 (с, 2H), 3,45 (м, 4H), 3,23 (м, 2H), 3,19 (м, 2H), 2,01 (с, 3H); LRMS (ES) m/z 546,1 (M++1).
Пример 22: Соединение 11312, N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-(3-фторфенил)-4-метилпиперазин-1-сульфонамид
Смесь N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-(3-фторфенил)пиперазин-1-сульфонамид гидрохлорида (0,050 г, 0,093 ммоль) и формальдегида (37,00% раствор в воде, 0,069 мл, 0,926 ммоль) в дихлорметане (4 мл) обрабатывали при комнатной температуре триацетоксиборгидридом натрия (0,059 г, 0,278 ммоль), и перемешивали при той же температуре в течение 16 часов. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; этилацетат/гексан=70%-100%) с получением N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-(3-фторфенил)-4-метилпиперазин-1-сульфонамида в виде белого твердого вещества (0,040 г, 83,5%).
1H ЯМР (400 МГц, ДМСО-d6) δ 7,87 (дд, 1H, J=7,9, 1,7 Гц), 7,84 (дд, 1H, J=10,1, 1,6 Гц), 7,64 (т, 1H, J=7,8 Гц), 7,43-7,37 (м, 2H), 7,30 (м, 1H), 7,16 (м, 1H), 5,07 (с, 2H), 3,41 (м, 4H), 3,02 (м, 4H), 2,68 (с, 3H); LRMS (ES) m/z 518,3 (M++1).
Пример 23: Соединение 11313, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3-фторфенил)пиперазин-1-сульфонамид гидрохлорид
[Стадия 1] трет-бутил 4-(N-(4-(2-(2,2-дифторацетил)гидразин-1-карбонил)-2-фторбензил)-N-(3-фторфенил)сульфамоил)пиперазин-1-карбоксилат
Раствор трет-бутил 4-(N-(2-фтор-4-(гидразинкарбонил)бензил)-N-(3-фторфенил)сульфамоил)пиперазин-1-карбоксилата (1,370 г, 2,607 ммоль) в 1,4-диоксане (15 мл) смешивали при комнатной температуре с 2,2-дифторуксусным ангидридом (0,324 мл, 2,607 ммоль) и триэтиламином (0,903 мл, 6,517 ммоль). Реакционную смесь нагревали с обратным холодильником в течение 3 часов, и охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 40 г; этилацетат/гексан=20%-50%) с получением трет-бутил 4-(N-(4-(2-(2,2-дифторацетил)гидразин-1-карбонил)-2-фторбензил)-N-(3-фторфенил)сульфамоил)пиперазин-1-карбоксилата в виде бежевого твердого вещества (1,230 г, 78,2%).
[Стадия 2] трет-бутил 4-(N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3-фторфенил)сульфамоил)пиперазин-1-карбоксилат
трет-бутил 4-(N-(4-(2-(2,2-дифторацетил)гидразин-1-карбонил)-2-фторбензил)-N-(3-фторфенил)сульфамоил)пиперазин-1-карбоксилат (1,230 г, 2,038 ммоль) и 1-метокси-N-триэтиламмонийсульфонил-метанимидат (реагент Бургесса, 0,728 г, 3,057 ммоль) смешивали при комнатной температуре в тетрагидрофуране (10 мл), и затем смесь нагревали при 150°С в течение 30 минут под действием микроволнового излучения. Реакционную смесь охлаждали до комнатной температуры для прекращения реакции. Затем к концентрату добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 40 г; этилацетат/гексан=5%-30%) с получением трет-бутил 4-(N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3-фторфенил)сульфамоил)пиперазин-1-карбоксилата в виде белого твердого вещества (0,941 г, 78,9%).
[Стадия 3] Соединение 11313
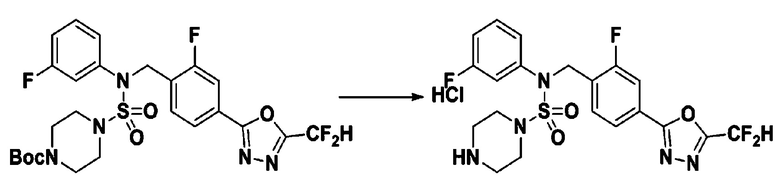
Раствор трет-бутил 4-(N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3-фторфенил)сульфамоил)пиперазин-1-карбоксилата (0,941 г, 1,607 ммоль) в 1,4-диоксане (10 мл) смешивали при комнатной температуре с хлористоводородной кислотой (4,00 M раствор в 1,4-диоксане, 8,035 мл, 32,140 ммоль), и перемешивали при той же температуре в течение 2 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток разбавляли диэтиловым эфиром (5 мл) и гексаном (50 мл), и перемешивали. Полученные осадки собирали фильтрованием, промывали гексаном и сушили с получением N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3-фторфенил)пиперазин-1-сульфонамида гидрохлорида в виде белого твердого вещества (0,785 г, 93,6%).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,02 (с, 2H), 7,85 (дд, 1H, J=7,9, 1,8 Гц), 7,81 (д, 1H, J=10,2 Гц), 7,67 (с, 0,25H), 7,62 (т, 1H, J=7,8 Гц), 7,54 (с, 0,5H), 7,46-7,36 (м, 2,25H), 7,29 (д, 1H, J=7,1 Гц), 7,17 (т, 1H, J=8,4 Гц), 5,07 (с, 2H), 3,44-3,37 (м, 4H), 3,14-3,12 (м, 4H); LRMS (ES) m/z 486,2 (M++1).
Пример 24: Соединение 11314, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3-фторфенил)-4-(метилсульфонил)пиперазин-1-сульфонамид
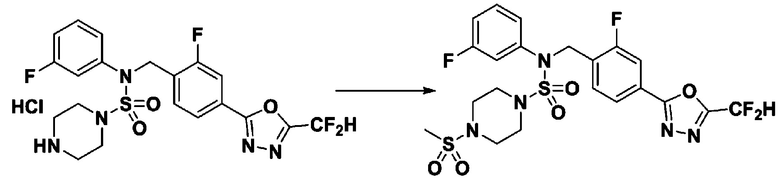
Суспензию N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3-фторфенил)пиперазин-1-сульфонамид гидрохлорида (0,050 г, 0,096 ммоль) в дихлорметане (3 мл) смешивали при комнатной температуре с метансульфонилхлоридом (0,015 мл, 0,192 ммоль) и N,N-диизопропилэтиламином (0,050 мл, 0,287 ммоль), и перемешивали при той же температуре в течение 1 часа. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 12 г; этилацетат/гексан=20%-50%) с получением N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3-фторфенил)-4-(метилсульфонил)пиперазин-1-сульфонамида в виде белого твердого вещества (0,047 г, 87,1%).
1H ЯМР (400 МГц, ДМСО-d6) δ 7,84 (дд, 1H, J=8,0, 1,7 Гц), 7,80 (дд, 1H, J=10,2, 1,5 Гц), 7,66 (с, 0,25H), 7,62 (т, 1H, J=7,7 Гц), 7,54 (с, 0,5H), 7,46-7,36 (м, 2,25H), 7,32 (д, 1H, J=8,9 Гц), 7,19-7,10 (м, 1H), 5,07 (с, 2H), 3,31 (м, 4H), 3,20-3,11 (м, 4H), 2,91 (с, 3H); LRMS (ES) m/z 564,3 (M++1).
Пример 25: Соединение 11315, 4-ацетил-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3-фторфенил)пиперазин-1-сульфонамид
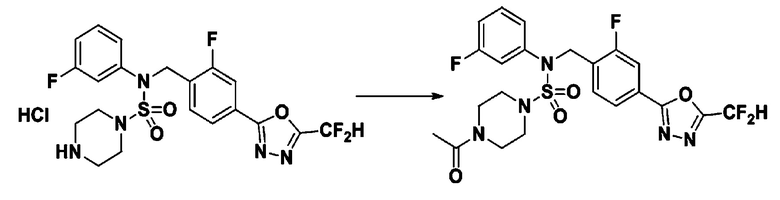
Суспензию N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3-фторфенил)пиперазин-1-сульфонамид гидрохлорида (0,050 г, 0,096 ммоль) в дихлорметане (3 мл) смешивали при комнатной температуре с ацетилхлоридом (0,014 мл, 0,192 ммоль) и N,N-диизопропилэтиламином (0,050 мл, 0,287 ммоль), и перемешивали при той же температуре в течение 1 часа. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 12 г; этилацетат/гексан=40%-80%) с получением 4-ацетил-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3-фторфенил)пиперазин-1-сульфонамида в виде желтой жидкости (0,036 г, 71,2%).
1H ЯМР (400 МГц, ДМСО-d6) δ 7,84 (дд, 1H, J=8,0, 1,5 Гц), 7,81-7,75 (м, 1H), 7,66 (с, 0,25H), 7,62 (т, 1H, J=7,8 Гц), 7,53 (с, 0,5H), 7,43-7,37 (м, 2H), 7,36 (с, 0,25H), 7,30 (д, 1H, J=8,0 Гц), 7,12 (т, 1H, J=7,3 Гц), 5,07 (с, 2H), 3,44 (м, 4H), 3,23 (м, 4H), 3,17 (м, 2H), 2,01 (с, 3H); LRMS (ES) m/z 528,2 (M++1).
Пример 26: Соединение 11316, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3-фторфенил)-4-метилпиперазин-1-сульфонамид
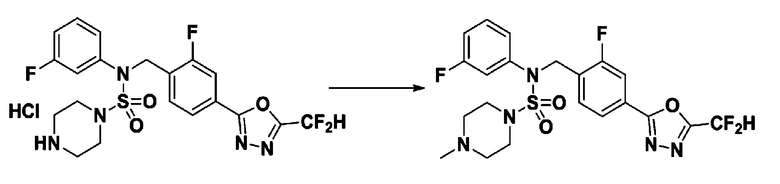
Смесь N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3-фторфенил)пиперазин-1-сульфонамид гидрохлорида (0,050 г, 0,096 ммоль) и формальдегида (37,00% раствор в воде, 0,071 мл, 0,958 ммоль) в дихлорметане (4 мл) обрабатывали при комнатной температуре триацетоксиборгидридом натрия (0,061 г, 0,287 ммоль), и перемешивали при той же температуре в течение 16 часов. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; этилацетат/гексан=70%-100%) с получением N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3-фторфенил)-4-метилпиперазин-1-сульфонамида в виде светло-желтой жидкости (0,038 г, 79,4%).
1H ЯМР (400 МГц, ДМСО-d6) δ 7,84 (д, 1H, J=8,0 Гц), 7,80 (д, 1H, J=10,1 Гц), 7,66 (с, 0,25H), 7,62 (т, 1H, J=7,7 Гц), 7,54 (с, 0,5H), 7,45-7,35 (м, 2,25H), 7,30 (д, 1H, J=8,2 Гц), 7,12 (т, 1H, J=7,1 Гц), 5,06 (с, 2H), 3,17 (с, 4H), 2,36 (м, 4H), 2,17 (с, 3H); LRMS (ES) m/z 500,2 (M++1).
Пример 27: Соединение 11317, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(пиридин-3-ил)пиперазин-1-сульфонамид дигидрохлорид
[Стадия 1] трет-бутил 4-(N-(пиридин-3-ил)сульфамоил)пиперазин-1-карбоксилат
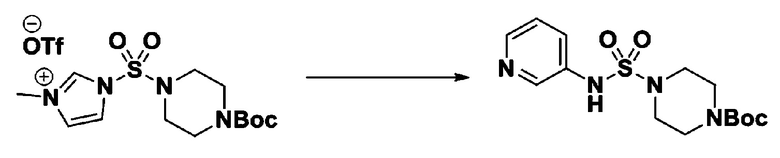
Смесь 1-((4-(трет-бутоксикарбонил)пиперазин-1-ил)сульфонил)-3-метил-1H-имидазол-3-ий трифторметансульфоната (3,340 г, 6,952 ммоль) и пиридин-3-амина (0,720 г, 7,647 ммоль) в ацетонитриле (30 мл), полученную при температуре окружающей среды, нагревали с обратным холодильником в течение 16 часов, и охлаждали до температуры окружающей среды. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 40 г; этилацетат/гексан=50%-80%) с получением неочищенного продукта, который растворяли в этилацетате (10 мл) и гексане (100 мл), и перемешивали. Полученные осадки собирали фильтрованием, промывали гексаном и сушили с получением трет-бутил 4-(N-(пиридин-3-ил)сульфамоил)пиперазин-1-карбоксилата в виде белого твердого вещества (1,745 г, 73,3%).
[Стадия 2] трет-бутил 4-(N-(2-фтор-4-(метоксикарбонил)бензил)-N-(пиридин-3-ил)сульфамоил)пиперазин-1-карбоксилат
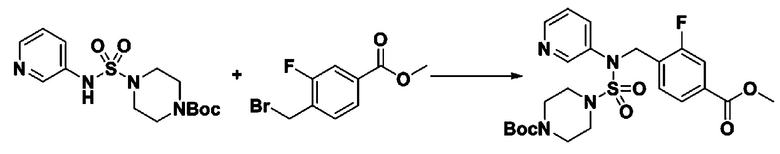
К раствору трет-бутил 4-(N-(пиридин-3-ил)сульфамоил)пиперазин-1-карбоксилата (1,745 г, 5,096 ммоль) в N,N-диметилформамиде (20 мл) добавляли гидрид натрия (60,00%, 0,265 г, 6,625 ммоль) при комнатной температуре и смесь перемешивали при той же температуре в течение 10 минут. Реакционную смесь обрабатывали метил 4-(бромметил)-3-фторбензоатом (1,385 г, 5,606 ммоль), и перемешивали в течение дополнительного 1 часа при той же температуре. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 40 г; этилацетат/гексан=20%-50%) с получением трет-бутил 4-(N-(2-фтор-4-(метоксикарбонил)бензил)-N-(пиридин-3-ил)сульфамоил)пиперазин-1-карбоксилата в виде бежевого твердого вещества (1,220 г, 47,1%).
[Стадия 3] трет-бутил 4-(N-(2-фтор-4-(гидразинкарбонил)бензил)-N-(пиридин-3-ил)сульфамоил)пиперазин-1-карбоксилат
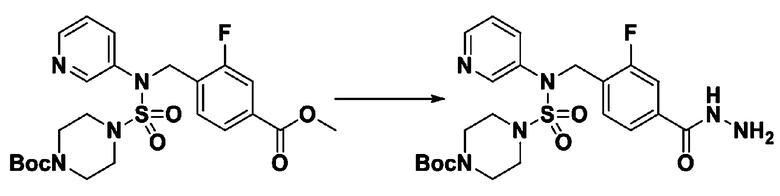
трет-бутил 4-(N-(2-фтор-4-(метоксикарбонил)бензил)-N-(пиридин-3-ил)сульфамоил)пиперазин-1-карбоксилат (1,220 г, 2,399 ммоль) и гидрат гидразина (2,332 мл, 47,978 ммоль) смешивали при комнатной температуре в этаноле (50 мл), и затем смесь перемешивали при 100°C в течение 16 часов. Реакционную смесь охлаждали до комнатной температуры для прекращения реакции, и концентрировали при пониженном давлении для удаления растворителя. Затем к концентрату добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Указанное в заголовке соединение использовали без дополнительной очистки (трет-бутил 4-(N-(2-фтор-4-(гидразинкарбонил)бензил)-N-(пиридин-3-ил)сульфамоил)пиперазин-1-карбоксилат, 0,702 г, 57,5%, белое твердое вещество).
[Стадия 4] трет-бутил 4-(N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(пиридин-3-ил)сульфамоил)пиперазин-1-карбоксилат
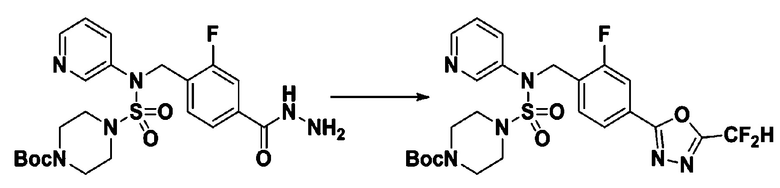
Раствор трет-бутил 4-(N-(2-фтор-4-(гидразинкарбонил)бензил)-N-(пиридин-3-ил)сульфамоил)пиперазин-1-карбоксилата (0,350 г, 0,688 ммоль) в 1,4-диоксане (15 мл) смешивали при комнатной температуре с 2,2-дифторуксусным ангидридом (0,086 мл, 0,688 ммоль) и триэтиламином (0,238 мл, 1,721 ммоль). Реакционную смесь нагревали с обратным холодильником в течение 3 часов, и охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 40 г; этилацетат/гексан=10%-40%) с получением трет-бутил 4-(N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(пиридин-3-ил)сульфамоил)пиперазин-1-карбоксилата в виде белого твердого вещества (0,233 г, 59,5%).
[Стадия 5] Соединение 11317
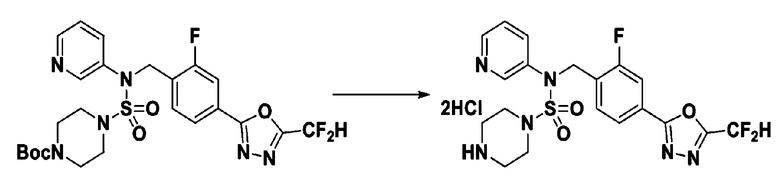
Раствор трет-бутил 4-(N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(пиридин-3-ил)сульфамоил)пиперазин-1-карбоксилата (0,233 г, 0,410 ммоль) в 1,4-диоксане (5 мл) смешивали при комнатной температуре с хлористоводородной кислотой (4,00 M раствор в 1,4-диоксане, 2,049 мл, 8,196 ммоль), и перемешивали при той же температуре в течение 2 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток разбавляли гексаном (30 мл) и перемешивали. Полученные осадки собирали фильтрованием, промывали гексаном и сушили с получением N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(пиридин-3-ил)пиперазин-1-сульфонамид дигидрохлорида в виде ярко-желтого твердого вещества (0,210 г, 94,8%).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,61 (с, 2H), 8,81 (м, 1H), 8,60 (м, 1H), 8,20 (д, 1H, J=8,0 Гц), 7,86-7,79 (м, 1,25H), 7,75-7,37 (м, 3,75H), 5,11 (2, 2H), 3,49 (м, 4H), 3,14 (м, 4H); LRMS (ES) m/z 469,3 (M++1).
Пример 28: Соединение 11318, 4-ацетил-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(пиридин-3-ил)пиперазин-1-сульфонамид
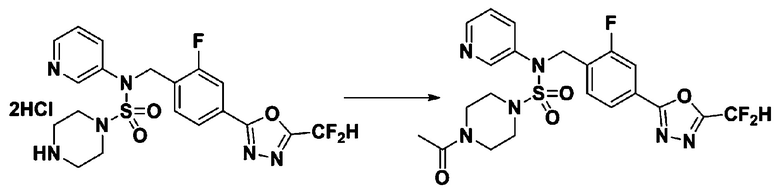
Суспензию N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(пиридин-3-ил)пиперазин-1-сульфонамид дигидрохлорида (0,050 г, 0,092 ммоль) в дихлорметане (3 мл) смешивали при комнатной температуре с ацетилхлоридом (0,013 мл, 0,185 ммоль) и N,N-диизопропилэтиламином (0,065 мл, 0,369 ммоль), и перемешивали при той же температуре в течение 1 часа. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 12 г; этилацетат/гексан=60%-90%) с получением 4-ацетил-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(пиридин-3-ил)пиперазин-1-сульфонамида в виде коричневой жидкости (0,018 г, 38,2%).
1H ЯМР (400 МГц, ДМСО-d6) δ 8,60 (м, 1H), 8,45 (дд, 1H, J=4,7, 1,4 Гц), 7,97-7,89 (м, 1H), 7,85 (дд, 1H, J=8,0, 1,6 Гц), 7,79 (дд, 1H, J=10,1, 1,5 Гц), 7,65 (д, 1H, J=8,9 Гц), 7,52 (с, 0,25H), 7,53 (с, 0,5H), 7,43 (с, 0,25H), 7,41 (т, 1H, J=4,1 Гц), 5,08 (с, 2H), 3,46 (с, 4H), 3,28-3,16 (м, 4H), 2,02 (с, 3H); LRMS (ES) m/z 511,2 (M++1).
Пример 29: Соединение 11319, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-4-(метилсульфонил)-N-(пиридин-3-ил)пиперазин-1-сульфонамид
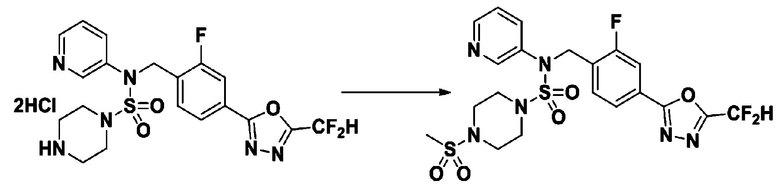
Суспензию N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(пиридин-3-ил)пиперазин-1-сульфонамид дигидрохлорида (0,050 г, 0,092 ммоль) в дихлорметане (3 мл) смешивали при комнатной температуре с метансульфонилхлоридом (0,014 мл, 0,185 ммоль) и N,N-диизопропилэтиламином (0,065 мл, 0,369 ммоль), и перемешивали при той же температуре в течение 1 часа. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 12 г; этилацетат/гексан=30%-70%) с получением N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-4-(метилсульфонил)-N-(пиридин-3-ил)пиперазин-1-сульфонамида в виде бежевого твердого вещества (0,041 г, 81,2%).
1H ЯМР (400 МГц, ДМСО-d6) δ 8,61 (с, 1H), 8,46 (д, 1H, J=4,5 Гц), 7,95 (д, 1H, J=8,7 Гц), 7,85 (д, 1H, J=8,2 Гц), 7,79 (д, 1H, J=9,8 Гц), 7,69-7,60 (м, 1,25H), 7,53 (с, 0,5H), 7,42 (м, 1,25H), 5,08 (с, 2H), 3,34 (м, 4H), 3,16 (с, 4H), 2,91 (с, 3H); LRMS (ES) m/z 547,0 (M++1).
Пример 30: Соединение 11320, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-4-метил-N-(пиридин-3-ил)пиперазин-1-сульфонамид
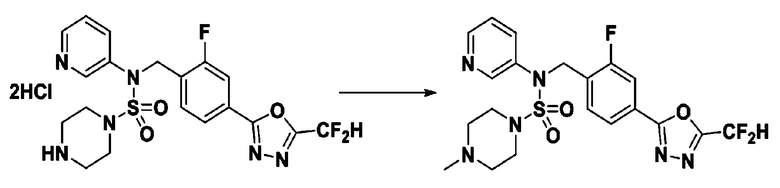
Смесь N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(пиридин-3-ил)пиперазин-1-сульфонамид дигидрохлорида (0,050 г, 0,092 ммоль) и формальдегида (37,00% раствор в воде, 0,069 мл, 0,924 ммоль) в дихлорметане (4 мл) обрабатывали при комнатной температуре триацетоксиборгидридом натрия (0,059 г, 0,277 ммоль), и перемешивали при той же температуре в течение 16 часов. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 12 г; метанол/дихлорметан=0%-10%) с получением N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-4-метил-N-(пиридин-3-ил)пиперазин-1-сульфонамида в виде бесцветной жидкости (0,011 г, 24,7%).
1H ЯМР (400 МГц, ДМСО-d6) δ 8,59 (д, 1H, J=2,6 Гц), 8,44 (дд, 1H, J=4,7, 1,5 Гц), 7,96-7,89 (м, 1H), 7,85 (дд, 1H, J=8,0, 1,6 Гц), 7,79 (дд, 1H, J=10,1, 1,6 Гц), 7,66 (с, 0,25H), 7,63 (т, 1H, J=7,8 Гц), 7,54 (с, 0,5H), 7,43 (с, 0,25H), 7,41 (т, 1H, J=4,1 Гц), 5,07 (с, 2H), 3,26-3,13 (м, 4H), 2,36-2,22 (м, 4H), 2,17 (с, 3H); LRMS (ES) m/z 483,3 (M++1).
Пример 31: Соединение 11321, N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-(пиридин-3-ил)пиперазин-1-сульфонамид дигидрохлорид
[Стадия 1] трет-бутил 4-(N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-(пиридин-3-ил)сульфамоил)пиперазин-1-карбоксилат
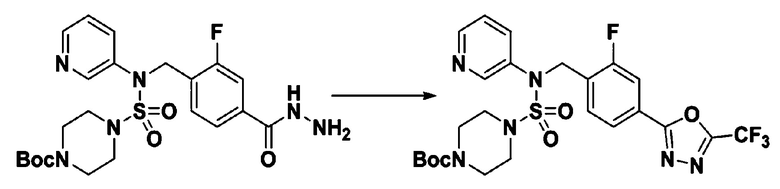
Раствор трет-бутил 4-(N-(2-фтор-4-(гидразинкарбонил)бензил)-N-(пиридин-3-ил)сульфамоил)пиперазин-1-карбоксилата (0,350 г, 0,688 ммоль) в 1,4-диоксане (15 мл) смешивали при комнатной температуре с 2,2,2-трифторуксусным ангидридом (0,096 мл, 0,688 ммоль) и триэтиламином (0,238 мл, 1,721 ммоль). Реакционную смесь нагревали с обратным холодильником в течение 3 часов, и охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 40 г; этилацетат/гексан=10%-40%) с получением трет-бутил 4-(N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-(пиридин-3-ил)сульфамоил)пиперазин-1-карбоксилата в виде белого твердого вещества (0,097 г, 24,0%).
[Стадия 2] Соединение 11321
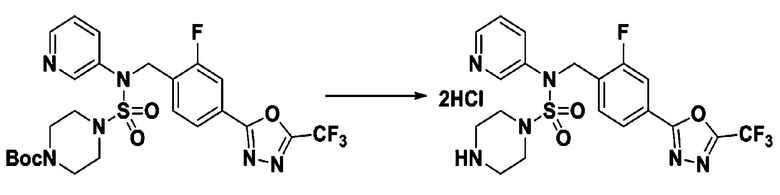
Раствор трет-бутил 4-(N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-(пиридин-3-ил)сульфамоил)пиперазин-1-карбоксилата (0,097 г, 0,165 ммоль) в 1,4-диоксане (5 мл) смешивали при комнатной температуре с хлористоводородной кислотой (4,00 M раствор в 1,4-диоксане, 0,827 мл, 3,307 ммоль), и перемешивали при той же температуре в течение 2 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток разбавляли гексаном (30 мл) и перемешивали. Полученные осадки собирали фильтрованием, промывали гексаном и сушили с получением N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-(пиридин-3-ил)пиперазин-1-сульфонамид дигидрохлорида в виде белого твердого вещества (0,082 г, 88,6%).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,57 (с, 2H), 8,76 (д, 1H, J=2,3 Гц), 8,57 (д, 1H, J=4,1 Гц), 8,16 (д, 1H, J=8,3 Гц), 7,91-7,80 (м, 2H), 7,68 (т, 1H, J=7,7 Гц), 7,60 (дд, 1H, J=8,3, 4,9 Гц), 5,13 (с, 2H), 3,53-3,45 (м, 4H), 3,14 (с, 4H); LRMS (ES) m/z 487,3 (M++1).
Пример 32: Соединение 11322, 4-ацетил-N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-(пиридин-3-ил)пиперазин-1-сульфонамид
Суспензию N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-(пиридин-3-ил)пиперазин-1-сульфонамид гидрохлорида (0,070 г, 0,125 ммоль) в дихлорметане (3 мл) смешивали при комнатной температуре с ацетилхлоридом (0,018 мл, 0,250 ммоль) и N,N-диизопропилэтиламином (0,087 мл, 0,501 ммоль), и перемешивали при той же температуре в течение 1 часа. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 12 г; метанол/дихлорметан=0%-5%) с получением 4-ацетил-N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-(пиридин-3-ил)пиперазин-1-сульфонамида в виде желтой жидкости (0,046 г, 69,6%).
1H ЯМР (400 МГц, ДМСО-d6) δ 8,60 (д, 1H, J=2,2 Гц), 8,44 (дд, 1H, J=4,7, 1,4 Гц), 7,93 (м, 1H), 7,87 (дд, 1H, J=8,0, 1,7 Гц), 7,82 (дд, 1H, J=10,1, 1,7 Гц), 7,65 (т, 1H, J=7,8 Гц), 7,41 (м, 1H), 5,09 (с, 2H), 3,48-3,45 (м, 4H), 3,25 (м, 2H), 3,20 (м, 2H), 2,02 (с, 3H); LRMS (ES) m/z 529,3 (M++1).
Пример 33: Соединение 11363, N-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4-метил-N-фенилпиперазин-1-сульфонамид
[Стадия 1] трет-бутил 4-(N-((5-(метоксикарбонил)пиридин-2-ил)метил)-N-фенилсульфамоил)пиперазин-1-карбоксилат
К раствору трет-бутил 4-(N-фенилсульфамоил)пиперазин-1-карбоксилата (0,600 г, 1,757 ммоль) в N,N-диметилформамиде (10 мл) медленно добавляли гидрид натрия (60,00%, 0,091 г, 2,285 ммоль) при комнатной температуре, и смесь перемешивали в течение 5 минут при той же температуре. Реакционную смесь обрабатывали метил 6-(бромметил)никотинатом (0,485 г, 2,109 ммоль), и перемешивали при той же температуре в течение дополнительных 16 часов. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 40 г; этилацетат/гексан=10%-50%) с получением неочищенного продукта, который растворяли в гексане (100 мл), и перемешивали. Полученные осадки собирали фильтрованием, промывали гексаном и сушили с получением трет-бутил 4-(N-((5-(метоксикарбонил)пиридин-2-ил)метил)-N-фенилсульфамоил)пиперазин-1-карбоксилата в виде белого твердого вещества (0,585 г, 67,9%).
[Стадия 2] метил 6-((N-фенилпиперазин-1-сульфонамидо)метил)никотинат дигидрохлорид
Раствор трет-бутил 4-(N-((5-(метоксикарбонил)пиридин-2-ил)метил)-N-фенилсульфамоил)пиперазин-1-карбоксилата (0,585 г, 1,192 ммоль) в 1,4-диоксане (5 мл) смешивали при комнатной температуре с хлористоводородной кислотой (4,00 M раствор в 1,4-диоксане, 4,472 мл, 17,887 ммоль), и перемешивали при той же температуре в течение 2 часов. Реакционную смесь и концентрировали при пониженном давлении для удаления растворителя. Остаток разбавляли этилацетатом (10 мл) и гексаном (20 мл), и перемешивали. Полученные осадки собирали фильтрованием, промывали гексаном и сушили с получением метил 6-((N-фенилпиперазин-1-сульфонамидо)метил)никотинат дигидрохлорида в виде бежевого твердого вещества (0,550 г, 99,5%).
[Стадия 3] метил 6-(((4-метил-N-фенилпиперазин)-1-сульфонамидо)метил)никотинат
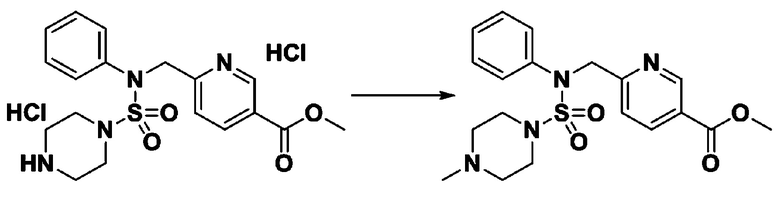
Смесь метил 6-((N-фенилпиперазин-1-сульфонамидо)метил)никотинат дигидрохлорида (0,550 г, 1,187 ммоль) и формальдегида (37,00% раствор в воде, 0,884 мл, 11,870 ммоль) в дихлорметане (15 мл) обрабатывали при комнатной температуре триацетоксиборгидридом натрия (0,755 г, 3,561 ммоль), и перемешивали при той же температуре в течение 16 часов. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 40 г; метанол/дихлорметан=0%-10%) с получением неочищенного продукта, который растворяли в дихлорметане (3 мл) и гексане (50 мл), и перемешивали. Полученные осадки собирали фильтрованием, промывали гексаном и сушили с получением метил 6-(((4-метил-N-фенилпиперазин)-1-сульфонамидо)метил)никотината в виде белого твердого вещества (0,420 г, 87,5%).
[Стадия 4] N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-4-метил-N-фенилпиперазин-1-сульфонамид
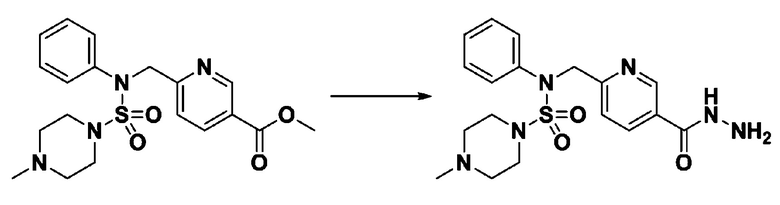
Суспензию метил 6-(((4-метил-N-фенилпиперазин)-1-сульфонамидо)метил)никотината (0,420 г, 1,038 ммоль) в этаноле (10 мл) смешивали при комнатной температуре с моногидратом гидразина (1,009 мл, 20,767 ммоль), и перемешивали при 110°С в течение 16 часов. Реакционную смесь охлаждали до комнатной температуры для прекращения реакции, и концентрировали при пониженном давлении для удаления растворителя. Остаток разбавляли дихлорметаном (10 мл) и диэтиловым эфиром (70 мл), и перемешивали. Полученные осадки собирали фильтрованием, промывали диэтиловым эфиром и сушили с получением N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-4-метил-N-фенилпиперазин-1-сульфонамида в виде белого твердого вещества (0,299 г, 71,2%).
[Стадия 5] N-((5-(2-(2,2-дифторацетил)гидразин-1-карбонил)пиридин-2-ил)метил)-4-метил-N-фенилпиперазин-1-сульфонамид
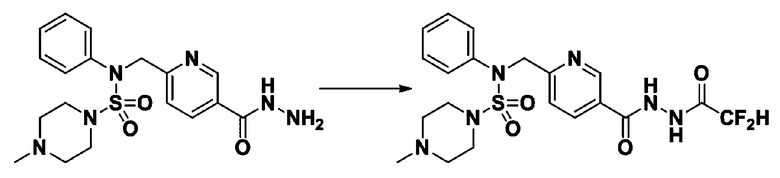
Раствор N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-4-метил-N-фенилпиперазин-1-сульфонамида (0,299 г, 0,739 ммоль) в тетрагидрофуране (20 мл) смешивали при 70°C с 2,2-дифторуксусным ангидридом (0,088 мл, 0,813 ммоль), и перемешивали при той же температуре в течение 1 часа. Реакционную смесь охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 12 г; метанол/дихлорметан=0%-10%) с получением неочищенного продукта, который растворяли в дихлорметане (5 мл) и гексане (100 мл), и перемешивали. Полученные осадки собирали фильтрованием, промывали гексаном и сушили с получением N-((5-(2-(2,2-дифторацетил)гидразин-1-карбонил)пиридин-2-ил)метил)-4-метил-N-фенилпиперазин-1-сульфонамида в виде ярко-желтого твердого вещества (0,256 г, 71,8%).
[Стадия 6] Соединение 11363
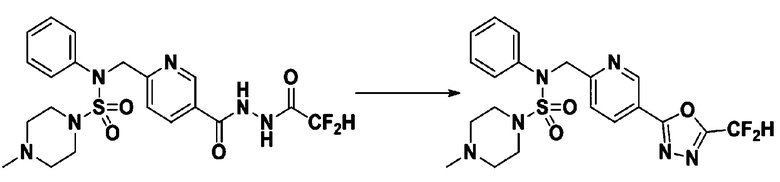
N-((5-(2-(2,2-дифторацетил)гидразин-1-карбонил)пиридин-2-ил)метил)-4-метил-N-фенилпиперазин-1-сульфонамид (0,256 г, 0,531 ммоль) и 1-метокси-N-триэтиламмонийсульфонил-метанимидат (реагент Бургесса, 0,632 г, 2,653 ммоль) смешивали при комнатной температуре в тетрагидрофуране (10 мл), и затем смесь нагревали при 150°С в течение 30 минут под действием микроволнового излучения. Реакционную смесь охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 12 г; метанол/дихлорметан=0%-10%) с получением неочищенного продукта, который растворяли в гексане (20 мл), и перемешивали. Полученные осадки собирали фильтрованием, промывали гексаном и сушили с получением N-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4-метил-N-фенилпиперазин-1-сульфонамида в виде бежевого твердого вещества (0,082 г, 33,3%).
1H ЯМР (400 МГц, ДМСО-d6) δ9,10 (д, 1H, J=2,2 Гц), 8,40 (дд, 1H, J=8,3, 2,3 Гц), 7,70-7,68 (м, 1,25H), 7,56 (с, 0,5H), 7,51 (дд, 2H, J=8,3, 1,0 Гц), 7,43 (с, 0,25H), 7,38-7,34 (м, 2H), 7,24 (м, 1H), 5,11 (с, 2H), 3,17 (т, 4H, J=5,0 Гц), 2,28 (т, 4H, J=4,7 Гц), 2,15 (с, 3H); LRMS (ES) m/z 465,4 (M++1).
Пример 34: Соединение 11379, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-4-(оксетан-3-ил)-N-фенилпиперазин-1-сульфонамид
[Стадия 1] метил 3-фтор-4-((N-фенилпиперазин-1-сульфонамидо)метил)бензоат гидрохлорид
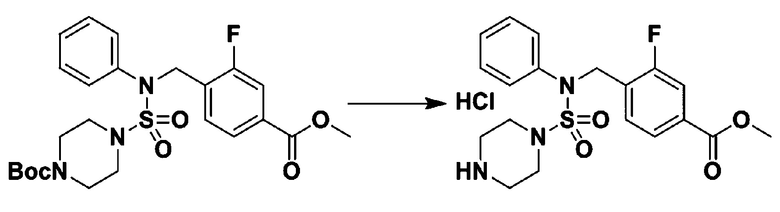
Суспензию трет-бутил 4-(N-(2-фтор-4-(метоксикарбонил)бензил)-N-фенилсульфамоил)пиперазин-1-карбоксилата (2,640 г, 5,201 ммоль) в этилацетате (20 мл) смешивали при комнатной температуре с хлористоводородной кислотой (4,00 M раствор в 1,4-диоксане, 13,003 мл, 52,012 ммоль), и перемешивали при той же температуре в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Остаток разбавляли этилацетатом (20 мл) и гексаном (20 мл), и перемешивали. Полученные осадки собирали фильтрованием, промывали гексаном и сушили с получением метил 3-фтор-4-((N-фенилпиперазин-1-сульфонамидо)метил)бензоат гидрохлорида в виде бежевого твердого вещества (2,280 г, 98,7%).
[Стадия 2] метил 3-фтор-4-(((4-(оксетан-3-ил)-N-фенилпиперазин)-1-сульфонамидо)метил)бензоат
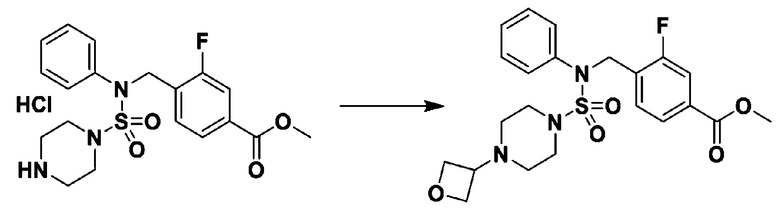
Метил 3-фтор-4-((N-фенилпиперазин-1-сульфонамидо)метил)бензоат гидрохлорид (0,440 г, 0,991 ммоль), оксетан-3-она (0,214 г, 2,974 ммоль) и триацетоксиборгидрид натрия (0,630 г, 2,974 ммоль) смешивали при комнатной температуре в дихлорметане (10 мл), и затем смесь перемешивали при той же температуре в течение 16 часов. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 12 г; метанол/дихлорметан=0%-10%) с получением неочищенного продукта, который растворяли в дихлорметане (3 мл) и гексане (50 мл), и перемешивали. Полученные осадки собирали фильтрованием, промывали гексаном и сушили с получением метил 3-фтор-4-(((4-(оксетан-3-ил)-N-фенилпиперазин)-1-сульфонамидо)метил)бензоата в виде бежевого твердого вещества (0,286 г, 62,3%).
[Стадия 3] N-(2-фтор-4-(гидразинкарбонил)бензил)-4-(оксетан-3-ил)-N-фенилпиперазин-1-сульфонамид
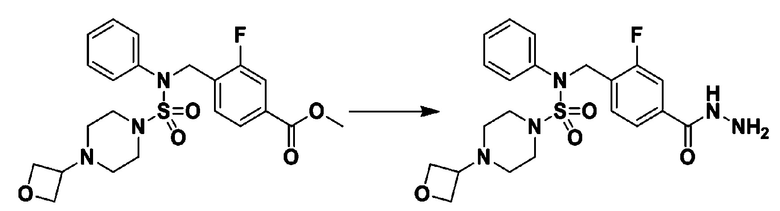
Метил 3-фтор-4-(((4-(оксетан-3-ил)-N-фенилпиперазин)-1-сульфонамидо)метил)бензоат (0,286 г, 0,617 ммоль) и гидразин моногидрат (0,300 мл, 6,170 ммоль) смешивали при комнатной температуре в этаноле (10 мл), и затем смесь перемешивали при 100°C в течение 16 часов. Реакционную смесь охлаждали до комнатной температуры для прекращения реакции, и концентрировали при пониженном давлении для удаления растворителя. Остаток разбавляли дихлорметаном (5 мл) и гексаном (30 мл), и перемешивали. Полученные осадки собирали фильтрованием, промывали гексаном и сушили с получением N-(2-фтор-4-(гидразинкарбонил)бензил)-4-(оксетан-3-ил)-N-фенилпиперазин-1-сульфонамида в виде белого твердого вещества (0,241 г, 84,3%).
[Стадия 4] N-(4-(2-(2,2-дифторацетил)гидразин-1-карбонил)-2-фторбензил)-4-(оксетан-3-ил)-N-фенилпиперазин-1-сульфонамид
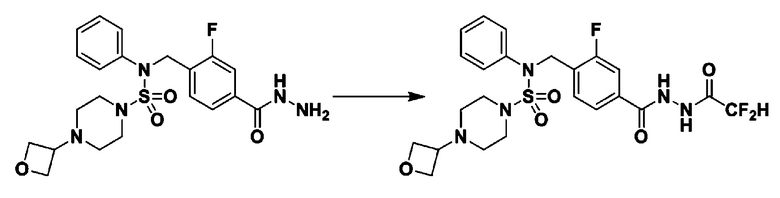
Раствор N-(2-фтор-4-(гидразинкарбонил)бензил)-4-(оксетан-3-ил)-N-фенилпиперазин-1-сульфонамида (0,241 г, 0,520 ммоль) в тетрагидрофуране (10 мл) смешивали при 70°C с 2,2-дифторуксусным ангидридом (0,068 мл, 0,624 ммоль) и N,N-диизопропилэтиламином (0,136 мл, 0,780 ммоль), и перемешивали при той же температуре в течение 1 часа. Реакционную смесь охлаждали до комнатной температуры для прекращения реакции, и концентрировали при пониженном давлении для удаления растворителя. Неочищенный продукт использовали без дополнительной очистки (N-(4-(2-(2,2-дифторацетил)гидразин-1-карбонил)-2-фторбензил)-4-(оксетан-3-ил)-N-фенилпиперазин-1-сульфонамид, 0,270 г, 95,9%, коричневое твердое вещество).
[Стадия 5] Соединение 11379
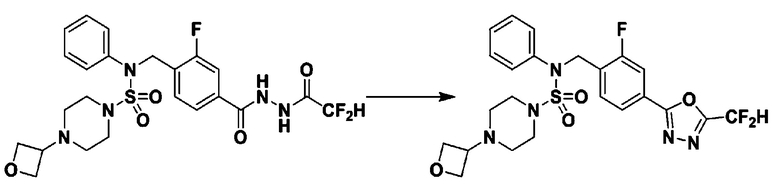
N-(4-(2-(2,2-дифторацетил)гидразин-1-карбонил)-2-фторбензил)-4-(оксетан-3-ил)-N-фенилпиперазин-1-сульфонамид (0,270 г, 0,499 ммоль) и 1-метокси-N-триэтиламмонийсульфонил-метанимидат (реагент Бургесса, 0,238 г, 0,997 ммоль) смешивали при комнатной температуре в тетрагидрофуране (10 мл), и затем смесь нагревали при 150°С в течение 30 минут под действием микроволнового излучения. Реакционную смесь охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 12 г; этилацетат/гексан=60%-90%) с получением N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-4-(оксетан-3-ил)-N-фенилпиперазин-1-сульфонамид в виде белого твердого вещества (0,042 г, 16,1%).
1H ЯМР (400 МГц, ДМСО-d6) δ 7,83 (дд, 1H, J=7,9, 1,7 Гц), 7,78 (дд, 1H, J=10,2, 1,7 Гц), 7,69-7,57 (м, 1,25H), 7,54 (с, 0,5H), 7,48-7,40 (м, 2,25H), 7,40-7,31 (м, 2H), 7,31-7,22 (м, 1H), 5,03 (с, 2H), 4,52 (т, 2H, J=6,6 Гц), 4,40 (т, 2H, J=6,1 Гц), 3,41 (p, 1H, J=6,2 Гц), 3,20-3,17 (м, 4H), 2,25 (т, 4H, J=4,9 Гц); LRMS (ES) m/z 524,4 (M++1).
Пример 35: Соединение 11440, N-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-N-фенилтиоморфолин-4-сульфонамид 1,1-диоксид
[Стадия 1] 4-((1H-имидазол-1-ил)сульфонил)тиоморфолин 1,1-диоксид
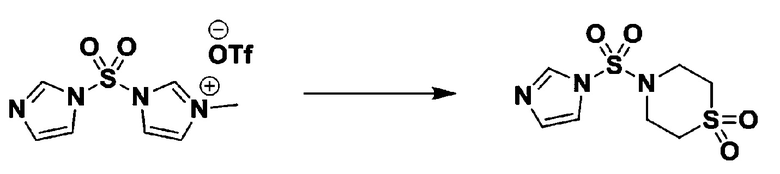
Смесь 1-((1H-имидазол-1-ил)сульфонил)-3-метил-1H-имидазол-3-ий трифторметансульфоната (4,400 г, 12,145 ммоль) и тиоморфолин 1,1-диоксида (1,970 г, 14,574 ммоль) в ацетонитриле (50 мл) перемешивали при комнатной температуре в течение 16 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 40 г; этилацетат/гексан=40%-70%) с получением неочищенного продукта, который растворяли в дихлорметане (10 мл) и гексане (200 мл), и перемешивали. Полученные осадки собирали фильтрованием, промывали гексаном и сушили с получением 4-((1H-имидазол-1-ил)сульфонил)тиоморфолин 1,1-диоксида в виде белого твердого вещества (1,635 г, 50,7%).
[Стадия 2] 1-((1,1-диоксидотиоморфолино)сульфонил)-3-метил-1H-имидазол-3-ий трифторметансульфонат
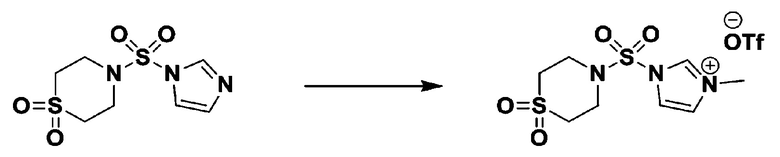
Раствор 4-((1H-имидазол-1-ил)сульфонил)тиоморфолин 1,1-диоксида (1,635 г, 6,163 ммоль) в дихлорметане (20 мл) смешивали при 0°С с метил трифторметансульфонатом (0,744 мл, 6,779 ммоль), и перемешивали при комнатной температуре в течение 5 часов. Реакционную смесь разбавляли гексаном (40 мл) и перемешивали. Полученные осадки собирали фильтрованием, промывали гексаном и сушили с получением 1-((1,1-диоксидотиоморфолино)сульфонил)-3-метил-1H-имидазол-3-ий трифторметансульфоната в виде белого твердого вещества (2,600 г, 98,2%).
[Стадия 3] N-фенилтиоморфолин-4-сульфонамид 1,1-диоксид
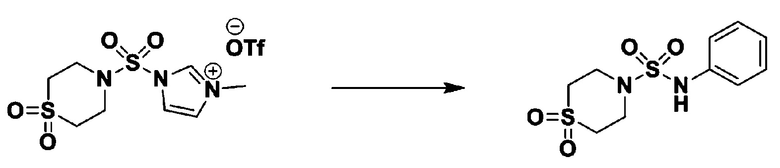
Раствор 1-((1,1-диоксидотиоморфолино)сульфонил)-3-метил-1H-имидазол-3-ий трифторметансульфоната (1,300 г, 3,027 ммоль) в ацетонитриле (10 мл) смешивали при комнатной температуре с анилином (0,415 мл, 4,541 ммоль). Реакционную смесь нагревали с обратным холодильником в течение 16 часов, и охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли насыщенный водный раствор хлорида аммония с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 12 г; этилацетат/гексан=30%-70%) с получением N-фенилтиоморфолин-4-сульфонамид 1,1-диоксида в виде коричневого твердого вещества (0,220 г, 25,0%).
[Стадия 4] метил 6-(((1,1-диоксидо-N-фенилтиоморфолин)-4-сульфонамидо)метил)никотинат
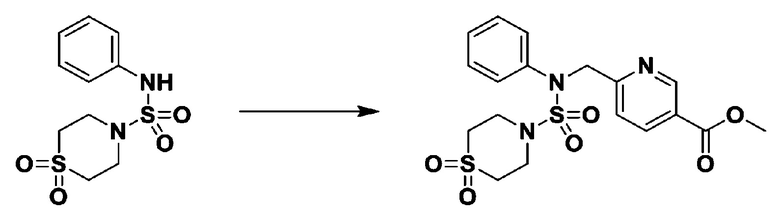
К раствору N-фенилтиоморфолин-4-сульфонамид 1,1-диоксида (0,220 г, 0,758 ммоль) в N,N-диметилформамиде (10 мл) добавляли гидрид натрия (60,00%, 0,036 г, 0,909 ммоль) при комнатной температуре и смесь перемешивали при той же температуре в течение 5 минут. Реакционную смесь обрабатывали метил 6-(бромметил)никотинатом (0,227 г, 0,985 ммоль), и перемешивали в течение дополнительных 3 часов при той же температуре. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 12 г; этилацетат/гексан=40%-70%) с получением неочищенного продукта, который растворяли в дихлорметане (5 мл) и гексане (50 мл), и перемешивали. Полученные осадки собирали фильтрованием, промывали гексаном и сушили с получением метил 6-(((1,1-диоксидо-N-фенилтиоморфолин)-4-сульфонамидо)метил)никотината в виде бежевого твердого вещества (0,250 г, 75,1%).
[Стадия 5] N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-N-фенилтиоморфолин-4-сульфонамид 1,1-диоксид
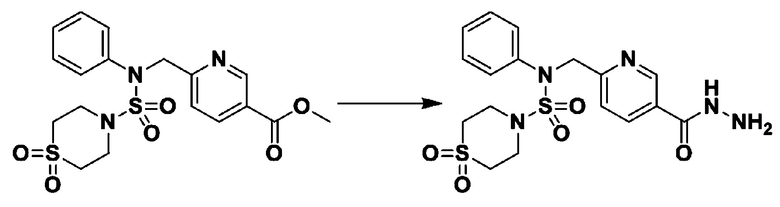
метил 6-(((1,1-диоксидо-N-фенилтиоморфолин)-4-сульфонамидо)метил)никотинат (0,250 г, 0,569 ммоль) и гидразин моногидрат (0,036 мл, 0,739 ммоль) смешивали при комнатной температуре в этаноле (10 мл), и затем смесь перемешивали при 100°C в течение 16 часов. Реакционную смесь охлаждали до комнатной температуры для прекращения реакции, и концентрировали при пониженном давлении для удаления растворителя. Остаток разбавляли этилацетатом (10 мл) и гексаном (20 мл), и перемешивали. Полученные осадки собирали фильтрованием, промывали гексаном и сушили с получением N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-N-фенилтиоморфолин-4-сульфонамид 1,1-диоксида в виде белого твердого вещества (0,212 г, 84,8%).
[Стадия 6] N-((5-(2-(2,2-дифторацетил)гидразин-1-карбонил)пиридин-2-ил)метил)-N-фенилтиоморфолин-4-сульфонамид 1,1-диоксид
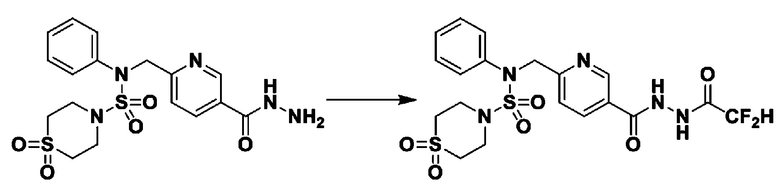
Раствор N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-N-фенилтиоморфолин-4-сульфонамид 1,1-диоксида (0,212 г, 0,482 ммоль) в тетрагидрофуране (10 мл) смешивали при 70°C с 2,2- дифторуксусным ангидридом (0,063 мл, 0,579 ммоль) и N,N-диизопропилэтиламином (0,126 мл, 0,724 ммоль), и перемешивали при той же температуре в течение 1 часа. Реакционную смесь охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Неочищенный продукт использовали без дополнительной очистки (N-((5-(2-(2,2-дифторацетил)гидразин-1-карбонил)пиридин-2-ил)метил)-N-фенилтиоморфолин-4-сульфонамид 1,1-диоксид, 0,220 г, 88,1%, желтая жидкость).
[Стадия 7] Соединение 11440
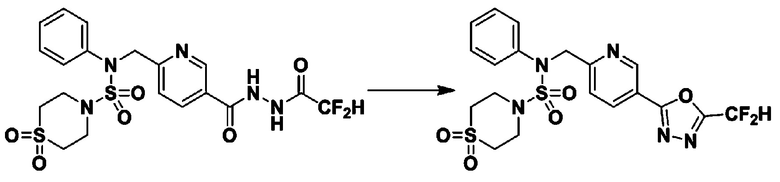
N-((5-(2-(2,2-дифторацетил)гидразин-1-карбонил)пиридин-2-ил)метил)-N-фенилтиоморфолин-4-сульфонамид 1,1-диоксид (0,220 г, 0,425 ммоль) и 1-метокси-N-триэтиламмонийсульфонил-метанимидат (реагент Бургесса, 0,203 г, 0,850 ммоль) смешивали при комнатной температуре в тетрагидрофуране (10 мл), и затем смесь нагревали при 150°С в течение 30 минут под действием микроволнового излучения. Реакционную смесь охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 12 г; этилацетат/гексан=40%-70%) с получением N-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-N-фенилтиоморфолин-4-сульфонамид 1,1-диоксида в виде бежевого твердого вещества (0,043 г, 20,3%).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,13 (д, 1H, J=2,2 Гц), 8,39 (дд, 1H, J=8,3, 2,3 Гц), 7,72-7,63 (м, 1,25H), 7,59-7,47 (м, 2,5H), 7,46-7,34 (м, 2,25H), 7,29 (т, 1H, J=7,3 Гц), 5,11 (с, 2H), 3,69-3,68 (м, 4H), 3,22-3,20 (м, 4H); LRMS (ES) m/z 500,3 (M++1).
Пример 36: Соединение 11498, N-(3-хлор-4-фторфенил)-N-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)тиоморфолин-4-сульфонамид 1,1-диоксид
[Стадия 1] N-(3-хлор-4-фторфенил)тиоморфолин-4-сульфонамид 1,1-диоксид
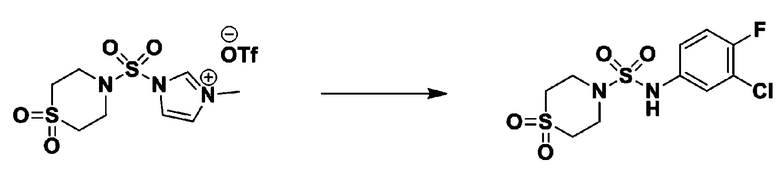
Смесь 1-((1,1-диоксидотиоморфолино)сульфонил)-3-метил-1H-имидазол-3-ий трифторметансульфоната (1,460 г, 3,400 ммоль) и 3-хлор-4-фторанилина (0,594 г, 4,080 ммоль) в ацетонитриле (20 мл), полученную при температуре окружающей среды, нагревали с обратным холодильником в течение 16 часов, и охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 40 г; этилацетат/гексан=20%-50%) с получением N-(3-хлор-4-фторфенил)тиоморфолин-4-сульфонамид 1,1-диоксида в виде бежевого твердого вещества (0,709 г, 60,8%).
[Стадия 2] метил 6-(((N-(3-хлор-4-фторфенил)-1,1-диоксидиотиоморфолин)-4-сульфонамидо)метил)никотинат
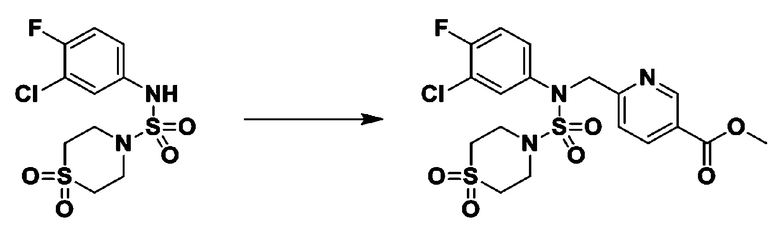
К раствору N-(3-хлор-4-фторфенил)тиоморфолин-4-сульфонамид 1,1-диоксида (0,400 г, 1,167 ммоль) в N,N-диметилформамиде (10 мл) добавляли гидрид натрия (60,00%, 0,061 г, 1,517 ммоль) при комнатной температуре и смесь перемешивали при той же температуре в течение 5 минут. Реакционную смесь обрабатывали метил 6-(бромметил)никотинатом (0,322 г, 1,400 ммоль), и перемешивали в течение дополнительных 3 часов при той же температуре. Затем к реакционной смеси добавляли насыщенный водный раствор хлорида аммония с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 12 г; этилацетат/гексан=30%-60%) с получением метил 6-(((N-(3-хлор-4-фторфенил)-1,1-диоксидиотиоморфолин)-4-сульфонамидо)метил)никотината в виде бежевого твердого вещества (0,348 г, 60,6%).
[Стадия 3] N-(3-хлор-4-фторфенил)-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)тиоморфолин-4-сульфонамид 1,1-диоксид
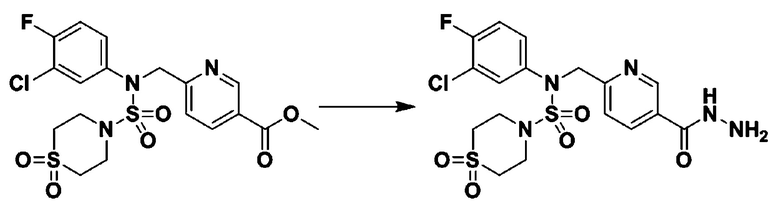
Суспензию метил 6-(((N-(3-хлор-4-фторфенил)-1,1-диоксидиотиоморфолин)-4-сульфонамидо)метил)никотината (0,250 г, 0,508 ммоль) в этаноле (10 мл) смешивали при комнатной температуре с моногидратом гидразина (0,494 мл, 10,164 ммоль), и перемешивали при 100°С в течение 16 часов. Реакционную смесь охлаждали до комнатной температуры для прекращения реакции, и концентрировали при пониженном давлении для удаления растворителя. Остаток разбавляли этилацетатом (5 мл) и гексаном (20 мл), и перемешивали. Полученные осадки собирали фильтрованием, промывали гексаном и сушили с получением N-(3-хлор-4-фторфенил)-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)тиоморфолин-4-сульфонамид 1,1-диоксида в виде светло-желтого твердого вещества (0,207 г, 82,8%).
[Стадия 4] N-(3-хлор-4-фторфенил)-N-((5-(2-(2,2-дифторацетил)гидразин-1-карбонил)пиридин-2-ил)метил)тиоморфолин-4-сульфонамид 1,1-диоксид
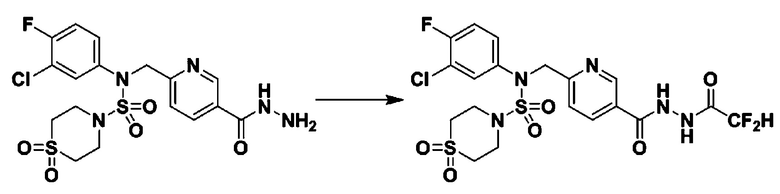
Раствор N-(3-хлор-4-фторфенил)-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)тиоморфолин-4-сульфонамид 1,1-диоксида (0,207 г, 0,421 ммоль) в тетрагидрофуране (10 мл) смешивали при 70°C с 2,2-дифторуксусным ангидридом (0,115 мл, 0,926 ммоль) и N,N-диизопропилэтиламином (0,220 мл, 1,262 ммоль). Реакционную смесь перемешивали при той же температуре в течение 1 часа, и охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 12 г; этилацетат/гексан=60%-90%) с получением N-(3-хлор-4-фторфенил)-N-((5-(2-(2,2-дифторацетил)гидразин-1-карбонил)пиридин-2-ил)метил)тиоморфолин-4-сульфонамид 1,1-диоксида в виде светло-желтого твердого вещества (0,148 г, 61,7%).
[Стадия 5] Соединение 11498
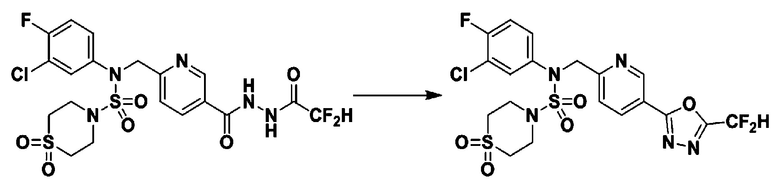
N-(3-хлор-4-фторфенил)-N-((5-(2-(2,2-дифторацетил)гидразин-1-карбонил)пиридин-2-ил)метил)тиоморфолин-4-сульфонамид 1,1-диоксид (0,148 г, 0,260 ммоль) и 1-метокси-N-триэтиламмонийсульфонил-метанимидат (реагент Бургесса, 0,093 г, 0,390 ммоль) смешивали при комнатной температуре в тетрагидрофуране (5 мл), и затем смесь нагревали при 150°С в течение 30 минут под действием микроволнового излучения. Реакционную смесь охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 12 г; этилацетат/гексан=30%-60%) с получением N-(3-хлор-4-фторфенил)-N-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)тиоморфолин-4-сульфонамид 1,1-диоксида в виде коричневого твердого вещества (0,051 г, 35,6%).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,14 (дд, 1H, J=2,2, 0,8 Гц), 8,40 (дд, 1H, J=8,2, 2,3 Гц), 7,86 (дд, 1H, J=6,6, 2,7 Гц), 7,71-7,64 (м, 1,25H), 7,62-7,51 (м, 1,5H), 7,47-7,37 (м, 1,25H), 5,11 (с, 2H), 3,79-3,71 (м, 4H), 3,26 (м, 4H); LRMS (ES) m/z 552,5 (M++1).
Пример 37: Соединение 11527, N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-4-(оксетан-3-ил)-N-фенилпиперазин-1-сульфонамид
[Стадия 1] N-(2-фтор-4-(2-(2,2,2-трифторацетил)гидразин-1-карбонил)бензил)-4-(оксетан-3-ил)-N-фенилпиперазин-1-сульфонамид
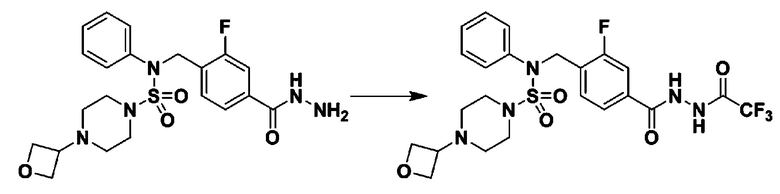
Раствор N-(2-фтор-4-(гидразинкарбонил)бензил)-4-(оксетан-3-ил)-N-фенилпиперазин-1-сульфонамида (0,500 г, 1,079 ммоль) в тетрагидрофуране (20 мл) смешивали при 70°C с трифторуксусным ангидридом (0,168 мл, 1,187 ммоль) и N,N-диизопропилэтиламином (0,225 мл, 1,294 ммоль), и перемешивали при той же температуре в течение 30 мин. Реакционную смесь охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 40 г; этилацетат/гексан=70%-100%) с получением неочищенного продукта, который растворяли в этилацетате (5 мл) и гексане (30 мл), и перемешивали. Полученные осадки собирали фильтрованием, промывали гексаном и сушили с получением N-(2-фтор-4-(2-(2,2,2-трифторацетил)гидразин-1-карбонил)бензил)-4-(оксетан-3-ил)-N-фенилпиперазин-1-сульфонамида в виде белого твердого вещества (0,589 г, 97,6%).
[Стадия 2] Соединение 11527
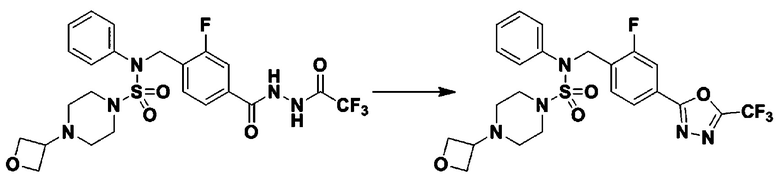
Суспензию N-(2-фтор-4-(2-(2,2,2-трифторацетил)гидразин-1-карбонил)бензил)-4-(оксетан-3-ил)-N-фенилпиперазин-1-сульфонамида (0,569 г, 1,017 ммоль) в дихлорметане (10 мл) смешивали при комнатной температуре с метансульфонилхлоридом (0,157 мл, 2,034 ммоль) и N,N-диизопропилэтиламином (0,354 мл, 2,034 ммоль), и перемешивали при той же температуре в течение 3 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 40 г; этилацетат/гексан=40%-70%) с получением неочищенного продукта, который растворяли в этилацетате (5 мл) и гексане (30 мл), и перемешивали. Полученные осадки собирали фильтрованием, промывали гексаном и сушили с получением N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-4-(оксетан-3-ил)-N-фенилпиперазин-1-сульфонамида в виде белого твердого вещества (0,186 г, 33,8%).
1H ЯМР (400 МГц, ДМСО-d6) δ 7,86 (дд, 1H, J=8,0, 1,7 Гц), 7,84-7,77 (м, 1H), 7,63 (т, 1H, J=7,7 Гц), 7,48-7,41 (м, 2H), 7,36 (т, 2H, J=7,6 Гц), 7,28 (т, 1H, J=7,2 Гц), 5,05 (с, 2H), 4,53 (с, 2H), 4,39 (с, 2H), 3,40 (м, 1H), 3,19 (с, 4H), 2,26 (с, 4H); LRMS (ES) m/z 542,5 (M++1).
Пример 38: Соединение 11528, 4-(оксетан-3-ил)-N-фенил-N-((5-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)пиперазин-1-сульфонамид
[Стадия 1] метил 6-((N-фенилпиперазин-1-сульфонамидо)метил)никотинат дигидрохлорид
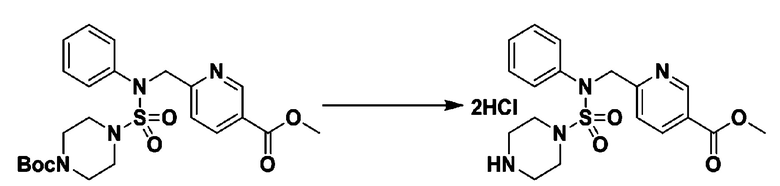
Суспензию трет-бутил 4-(N-((5-(метоксикарбонил)пиридин-2-ил)метил)-N-фенилсульфамоил)пиперазин-1-карбоксилата (0,760 г, 1,549 ммоль) в 1,4-диоксане (10 мл) смешивали при комнатной температуре с хлоридом водорода (4,00 M раствор в 1,4-диоксане, 5,809 мл, 23,238 ммоль), и перемешивали при той же температуре в течение 6 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Остаток разбавляли этилацетатом (20 мл) и гексаном (20 мл), и перемешивали. Полученные осадки собирали фильтрованием, промывали гексаном и сушили с получением метил 6-((N-фенилпиперазин-1-сульфонамидо)метил)никотинат дигидрохлорида в виде коричневого твердого вещества (0,710 г, 98,9%).
[Стадия 2] метил 6-(((4-(оксетан-3-ил)-N-фенилпиперазин)-1-сульфонамидо)метил)никотинат
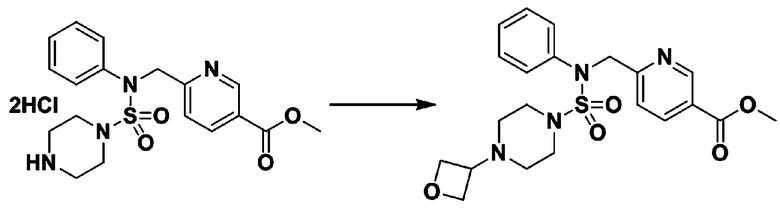
Смесь метил 6-((N-фенилпиперазин-1-сульфонамидо)метил)никотинат дигидрохлорида (0,710 г, 1,532 ммоль), оксетан-3-она (0,269 мл, 4,597 ммоль) и N,N-диизопропилэтиламина (0,667 мл, 3,831 ммоль) в дихлорметане (20 мл) обрабатывали при комнатной температуре триацетоксиборгидридом натрия (0,649 г, 3,065 ммоль), и перемешивали при той же температуре в течение 6 часов. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 40 г; метанол/дихлорметан=0%-10%) с получением метил 6-(((4-(оксетан-3-ил)-N-фенилпиперазин)-1-сульфонамидо)метил)никотината в виде бежевого твердого вещества (0,621 г, 90,8%).
[Стадия 3] N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-4-(оксетан-3-ил)-N-фенилпиперазин-1-сульфонамид
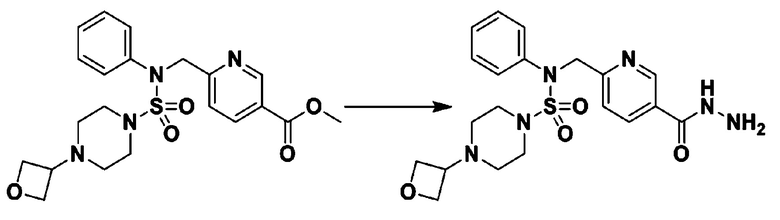
Суспензию метил 6-(((4-(оксетан-3-ил)-N-фенилпиперазин)-1-сульфонамидо)метил)никотината (0,621 г, 1,391 ммоль) в этаноле (10 мл) смешивали при комнатной температуре с моногидратом гидразина (1,352 мл, 27,815 ммоль), и нагревали с обратным холодильником в течение 16 часов. Реакционную смесь охлаждали до комнатной температуры для прекращения реакции, и концентрировали при пониженном давлении для удаления растворителя. Остаток хроматографировали (SiO2, картридж 80 г; метанол/дихлорметан=0%-5%) с получением N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-4-(оксетан-3-ил)-N-фенилпиперазин-1-сульфонамида в виде бежевого твердого вещества (0,420 г, 67,6%).
[Стадия 4] 4-(оксетан-3-ил)-N-фенил-N-((5-(2-(2,2,2-трифторацетил)гидразин-1-карбонил)пиридин-2-ил)метил)пиперазин-1-сульфонамид
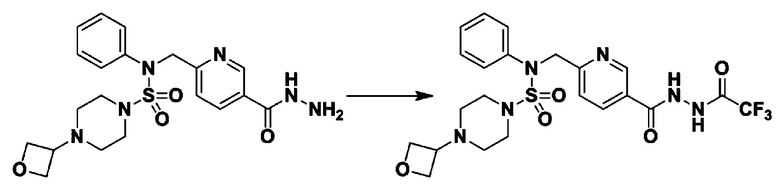
Раствор N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-4-(оксетан-3-ил)-N-фенилпиперазин-1-сульфонамида (0,200 г, 0,448 ммоль) в тетрагидрофуране (10 мл) смешивали при 70°C с трифторуксусным ангидридом (0,070 мл, 0,493 ммоль) и N,N-диизопропилэтиламином (0,094 мл, 0,537 ммоль), и перемешивали при той же температуре в течение 30 мин. Реакционную смесь охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 12 г; этилацетат/гексан=80%-100%) с получением неочищенного продукта, который растворяли в этилацетате (3 мл) и гексане (30 мл), и перемешивали. Полученные осадки собирали фильтрованием, промывали гексаном и сушили с получением 4-(оксетан-3-ил)-N-фенил-N-((5-(2-(2,2,2-трифторацетил)гидразин-1-карбонил)пиридин-2-ил)метил)пиперазин-1-сульфонамида в виде белого твердого вещества (0,202 г, 83,1%).
[Стадия 5] Соединение 11528
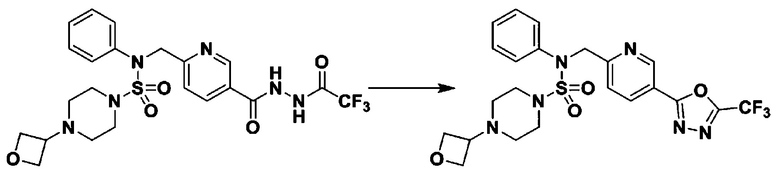
Суспензию 4-(оксетан-3-ил)-N-фенил-N-((5-(2-(2,2,2-трифторацетил)гидразин-1-карбонил)пиридин-2-ил)метил)пиперазин-1-сульфонамида (0,202 г, 0,372 ммоль) в дихлорметане (10 мл) смешивали при комнатной температуре с метансульфонилхлоридом (0,058 мл, 0,745 ммоль) и N,N-диизопропилэтиламином (0,130 мл, 0,745 ммоль), и перемешивали при той же температуре в течение 3 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 12 г; этилацетат/гексан=50%-80%) с получением неочищенного продукта, который растворяли в этилацетате (3 мл) и гексане (15 мл), и перемешивали. Полученные осадки собирали фильтрованием, промывали гексаном и сушили с получением 4-(оксетан-3-ил)-N-фенил-N-((5-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)пиперазин-1-сульфонамида в виде бежевого твердого вещества (0,092 г, 47,1%).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,13 (д, 1H, J=2,1 Гц), 8,43 (дд, 1H, J=8,2, 2,3 Гц), 7,70 (д, 1H, J=8,2 Гц), 7,56-7,48 (м, 2H), 7,37 (т, 2H, J=7,7 Гц), 7,26 (т, 1H, J=7,4 Гц), 5,13 (с, 2H), 4,54 (с, 2H), 4,43 (с, 2H), 3,37 (м, 1H), 3,23 (с, 4H), 2,34-2,28 (с, 4H); LRMS (ES) m/z 525,4 (M++1).
Пример 39: Соединение 11574, N-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4-(оксетан-3-ил)-N-фенилпиперазин-1-сульфонамид
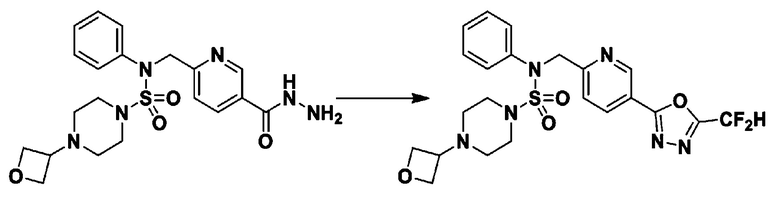
Раствор N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-4-(оксетан-3-ил)-N-фенилпиперазин-1-сульфонамида (0,600 г, 1,344 ммоль) в тетрагидрофуране (15 мл), полученный при температуре окружающей среды, смешивали при кипячении с обратным холодильником с 2,2-дифторуксусным ангидридом (0,501 мл, 4,031 ммоль) и N,N-диизопропилэтиламином (0,936 мл, 5,375 ммоль). Реакционную смесь нагревали с обратным холодильником в течение дополнительного 1 часа, и охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 40 г; этилацетат/гексан=70%-100%) с получением неочищенного продукта, который растворяли в этилацетате (5 мл) и гексане (30 мл), и перемешивали. Полученные осадки собирали фильтрованием, промывали гексаном и сушили с получением N-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4-(оксетан-3-ил)-N-фенилпиперазин-1-сульфонамида в виде белого твердого вещества (0,480 г, 70,5%).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,10 (с, 1H), 8,43-8,36 (м, 1H), 7,70-7,68 (м, 1,25H), 7,59-7,43 (м, 2,75H), 7,36 (т, 2H, J=7,7 Гц), 7,25 (т, 1H, J=7,3 Гц), 5,12 (с, 2H), 4,52 (т, 2H, J=6,6 Гц), 4,40 (т, 2H, J=6,1 Гц), 3,40 (т, 1H, J=6,4 Гц), 3,21 (с, 4H), 2,25 (с, 4H); LRMS (ES) m/z 507,2 (M++1).
Пример 40: Соединение 11575, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-4-(оксетан-3-ил)-N-фенилпиперазин-1-сульфонамид
[Стадия 1] метил 4-((N-фенилпиперазин-1-сульфонамидо)метил)бензоат гидрохлорид
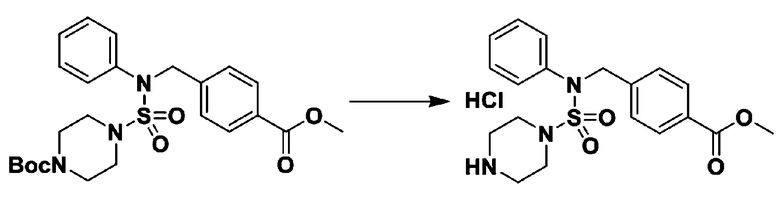
Суспензию трет-бутил 4-(N-(4-(метоксикарбонил)бензил)-N-фенилсульфамоил)пиперазин-1-карбоксилата (3,060 г, 6,250 ммоль) в 1,4-диоксане (30 мл) смешивали при комнатной температуре с хлористоводородной кислотой (4,00 M раствор в 1,4-диоксане, 15,625 мл, 62,501 ммоль), и перемешивали при той же температуре в течение 6 часов. Реакционную смесь разбавляли этилацетатом (20 мл) и гексаном (100 мл), и перемешивали. Полученные осадки собирали фильтрованием, промывали гексаном и сушили с получением метил 4-((N-фенилпиперазин-1-сульфонамидо)метил)бензоат гидрохлорида в виде белого твердого вещества (2,560 г, 96,2%).
[Стадия 2] метил 4-(((4-(оксетан-3-ил)-N-фенилпиперазин)-1-сульфонамидо)метил)бензоат
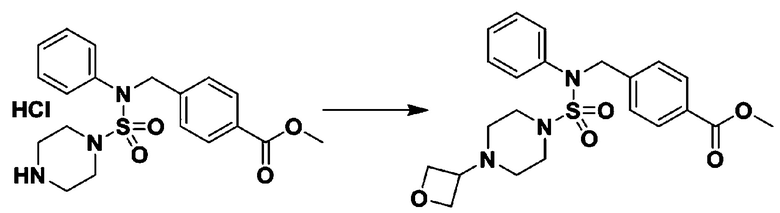
Смесь метил 4-((N-фенилпиперазин-1-сульфонамидо)метил)бензоат гидрохлорида (2,560 г, 6,010 ммоль), оксетан-3-она (0,704 мл, 12,021 ммоль) и N,N-диизопропилэтиламина (1,570 мл, 9,016 ммоль) в дихлорметане (50 мл) перемешивали при комнатной температуре в течение 30 минут, и обрабатывали триацетоксиборгидридом натрия (2,548 г, 12,021 ммоль). Реакционную смесь перемешивали при той же температуре в течение дополнительных 6 часов. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 40 г; метанол/дихлорметан=0%-10%) с получением метил 4-(((4-(оксетан-3-ил)-N-фенилпиперазин)-1-сульфонамидо)метил)бензоата в виде белого твердого вещества (2,450 г, 91,5%).
[Стадия 3] N-(4-(гидразинкарбонил)бензил)-4-(оксетан-3-ил)-N-фенилпиперазин-1-сульфонамид
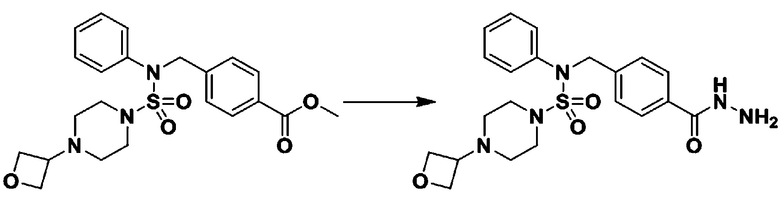
Смесь метил 4-(((4-(оксетан-3-ил)-N-фенилпиперазин)-1-сульфонамидо)метил)бензоата (1,850 г, 4,152 ммоль) и карбоната калия (11,477 г, 83,047 ммоль) в воде (5 мл)/метаноле (50 мл) обрабатывали при комнатной температуре моногидратом гидразина (6,054 мл, 124,571 ммоль), и перемешивали при той же температуре в течение 3 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток разбавляли этилацетатом (50 мл), и перемешивали. Полученные осадки собирали фильтрованием, промывали этилацетатом и сушили с получением N-(4-(гидразинкарбонил)бензил)-4-(оксетан-3-ил)-N-фенилпиперазин-1-сульфонамида в виде белого твердого вещества (1,120 г, 60,5%).
[Стадия 4] Соединение 11575
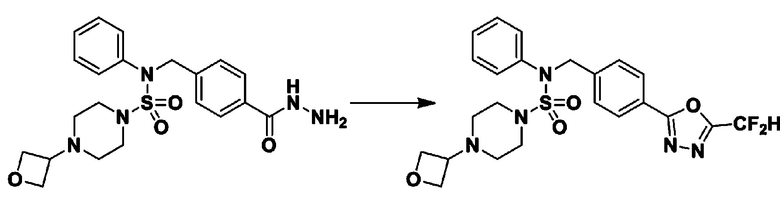
Перемешиваемый раствор N-(4-(гидразинкарбонил)бензил)-4-(оксетан-3-ил)-N-фенилпиперазин-1-сульфонамида (0,500 г, 1,122 ммоль) в тетрагидрофуране (15 мл), полученный при температуре окружающей среды, смешивали при кипячении с обратным холодильником с 2,2-дифторуксусным ангидридом (0,419 мл, 3,367 ммоль) и N,N-диизопропилэтиламином (0,586 мл, 3,367 ммоль). Реакционную смесь нагревали с обратным холодильником в течение дополнительного 1 часа, и охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 40 г; этилацетат/гексан=70%-100%) с получением неочищенного продукта, который растворяли в этилацетате (5 мл) и гексане (30 мл), и перемешивали. Полученные осадки собирали фильтрованием, промывали гексаном и сушили с получением N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-4-(оксетан-3-ил)-N-фенилпиперазин-1-сульфонамида в виде белого твердого вещества (0,397 г, 70,0%).
1H ЯМР (400 МГц, ДМСО-d6) δ 7,97 (д, 2H, J=8,0 Гц), 7,66 (с, 0,25H), 7,52-7,50 (м, 2,5H), 7,45-7,36 (м, 2,25H), 7,34 (т, 2H, J=7,6 Гц), 7,24 (м, 1H), 5,03 (с, 2H), 4,52 (т, 2H, J=6,5 Гц), 4,40 (т, 2H, J=6,1 Гц), 3,41 (м, 1H), 3,20 (с, 4H), 2,26 (с, 4H); LRMS (ES) m/z 506,3 (M++1).
Пример 41: Соединение 11640, (3S,5R)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-4-этил-3,5-диметил-N-фенилпиперазин-1-сульфонамид
[Стадия 1] трет-бутил (2S,6R)-4-((1H-имидазол-1-ил)сульфонил)-2,6-диметилпиперазин-1-карбоксилат
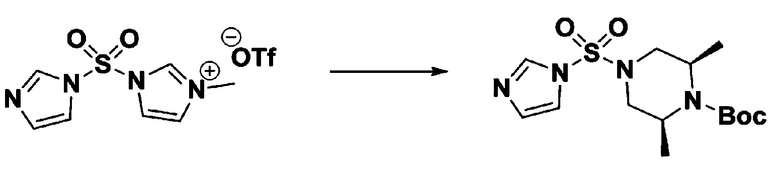
Раствор 1-((1H-имидазол-1-ил)сульфонил)-3-метил-1H-имидазол-3-ий трифторметансульфоната (4,000 г, 11,041 ммоль) и трет-бутил (2S,6R)-2,6-диметилпиперазин-1-карбоксилата (2,603 г, 12,145 ммоль) в ацетонитриле (100 мл) перемешивали при комнатной температуре в течение 16 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 80 г; этилацетат/гексан=0%-40%) с получением трет-бутил (2S,6R)-4-((1H-имидазол-1-ил)сульфонил)-2,6-диметилпиперазин-1-карбоксилата в виде желтого твердого вещества (2,800 г, 73,6%).
[Стадия 2] 1-(((3S,5R)-4-(трет-бутоксикарбонил)-3,5-диметилпиперазин-1-ил)сульфонил)-3-метил-1H-имидазол-3-ий трифторметансульфонат
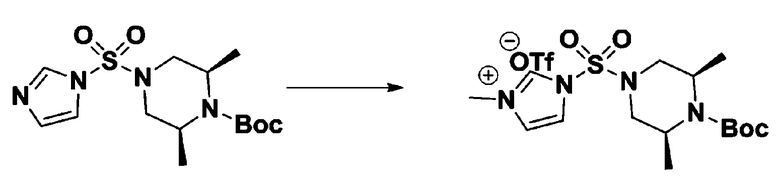
Раствор трет-бутил (2S,6R)-4-((1H-имидазол-1-ил)сульфонил)-2,6-диметилпиперазин-1-карбоксилата (2,800 г, 8,129 ммоль) и метил трифторметансульфоната (0,892 мл, 8,129 ммоль) в дихлорметане (80 мл) перемешивали при 0°C в течение 3 часов, и концентрировали при пониженном давлении для удаления растворителя. Указанное в заголовке соединение использовали без дополнительной очистки (1-(((3S,5R)-4-(трет-бутоксикарбонил)-3,5-диметилпиперазин-1-ил)сульфонил)-3-метил-1H-имидазол-3-ий трифторметансульфонат, 4,000 г, 96,8%, белое твердое вещество).
[Стадия 3] трет-бутил (2S,6R)-2,6-диметил-4-(N-фенилсульфамоил)пиперазин-1-карбоксилат
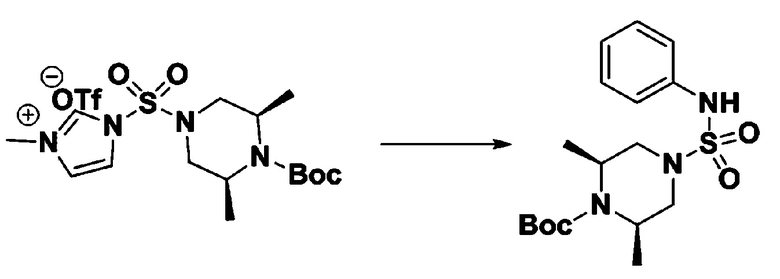
Смесь 1-(((3S,5R)-4-(трет-бутоксикарбонил)-3,5-диметилпиперазин-1-ил)сульфонил)-3-метил-1H-имидазол-3-ий трифторметансульфоната (4,000 г, 7,866 ммоль) и анилина (0,790 мл, 8,652 ммоль) в ацетонитриле (100 мл), полученную при температуре окружающей среды, нагревали с обратным холодильником в течение 16 часов, и охлаждали до температуры окружающей среды. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида аммония, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 40 г; этилацетат/гексан=0%-30%) с получением трет-бутил (2S,6R)-2,6-диметил-4-(N-фенилсульфамоил)пиперазин-1-карбоксилата в виде белого твердого вещества (1,800 г, 61,9%).
[Стадия 4] трет-бутил (2S,6R)-4-(N-(4-(метоксикарбонил)бензил)-N-фенилсульфамоил)-2,6-диметилпиперазин-1-карбоксилат
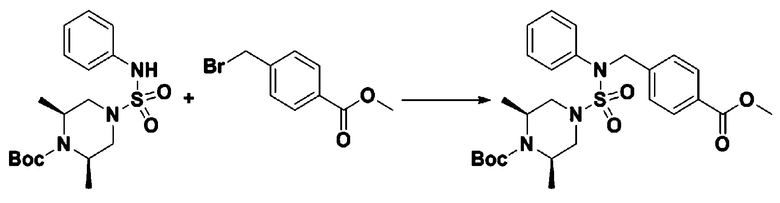
Раствор трет-бутил (2S,6R)-2,6-диметил-4-(N-фенилсульфамоил)пиперазин-1-карбоксилата (1,700 г, 4,601 ммоль) и карбоната калия (0,954 г, 6,902 ммоль) в N,N-диметилформамиде (5 мл) перемешивали при комнатной температуре в течение 30 мин и смешивали с метил 4-(бромметил)бензоатом (1,159 г, 5,061 ммоль) и иодидом калия (0,382 г, 2,301 ммоль). Реакционную смесь перемешивали при той же температуре в течение дополнительных 12 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида аммония, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 24 г; этилацетат/гексан=0%-40%) с получением трет-бутил (2S,6R)-4-(N-(4-(метоксикарбонил)бензил)-N-фенилсульфамоил)-2,6-диметилпиперазин-1-карбоксилата в виде белого твердого вещества (2,100 г, 88,2%).
[Стадия 5] метил 4-((((3S,5R)-3,5-диметил-N-фенилпиперазин)-1-сульфонамидо)метил)бензоат гидрохлорид
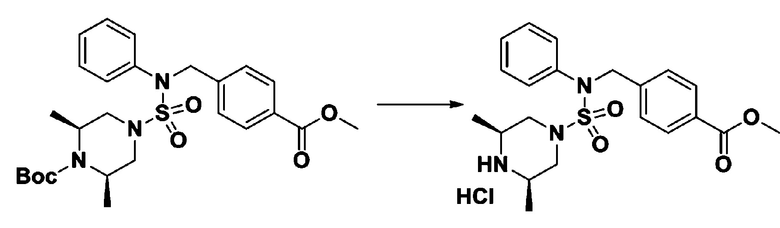
Раствор трет-бутил (2S,6R)-4-(N-(4-(метоксикарбонил)бензил)-N-фенилсульфамоил)-2,6-диметилпиперазин-1-карбоксилата (1,500 г, 2,898 ммоль) и хлорида водорода (4,00 M раствор в 1,4-диоксане, 1,449 мл, 5,796 ммоль) в дихлорметане (50 мл) перемешивали при комнатной температуре в течение 5 часов, и концентрировали при пониженном давлении для удаления растворителя. Неочищенный продукт использовали без дополнительной очистки (метил 4-((((3S,5R)-3,5-диметил-N-фенилпиперазин)-1-сульфонамидо)метил)бензоат гидрохлорид, 1,300 г, 98,8%, белое твердое вещество).
[Стадия 6] метил 4-((((3S,5R)-4-этил-3,5-диметил-N-фенилпиперазин)-1-сульфонамидо)метил)бензоат
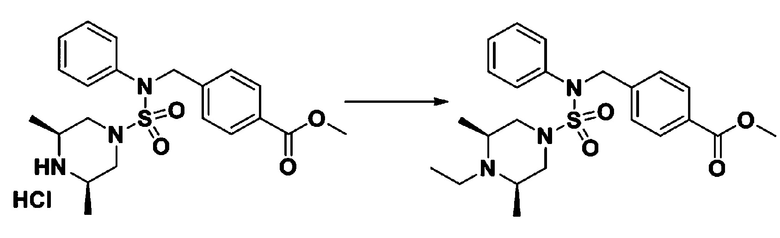
Раствор метил 4-((((3S,5R)-3,5-диметил-N-фенилпиперазин)-1-сульфонамидо)метил)бензоат гидрохлорида (0,200 г, 0,441 ммоль), ацетальдегида (0,039 г, 0,881 ммоль) и уксусной кислоты (0,028 мл, 0,485 ммоль) в дихлорметане (12 мл) перемешивали при комнатной температуре в течение 30 мин и смешивали с триацетоксиборгидридом натрия (0,187 г, 0,881 ммоль). Реакционную смесь перемешивали при той же температуре в течение дополнительных 12 часов. Затем, к реакционной смеси добавляли воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором бикарбоната натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; метанол/дихлорметан=0%-5%) с получением метил 4-((((3S,5R)-4-этил-3,5-диметил-N-фенилпиперазин)-1-сульфонамидо)метил)бензоата в виде желтого масла (0,130 г, 66,2%).
[Стадия 7] (3S,5R)-4-этил-N-(4-(гидразинкарбонил)бензил)-3,5-диметил-N-фенилпиперазин-1-сульфонамид
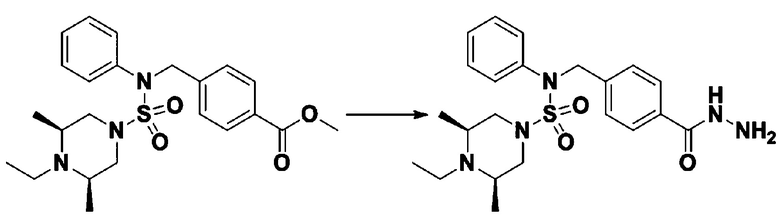
Раствор метил 4-((((3S,5R)-4-этил-3,5-диметил-N-фенилпиперазин)-1-сульфонамидо)метил)бензоата (0,130 г, 0,292 ммоль) и гидразина моногидрата (0,142 мл, 2,918 ммоль) в этаноле (10 мл) перемешивали при 90°C в течение 12 часов, и охлаждали до комнатной температуры для прекращения реакции. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Остаток разбавляли водой (10 мл) и бикарбонатом натрия (5 мл) и перемешивали. Полученные осадки собирали фильтрованием и сушили с получением (3S,5R)-4-этил-N-(4-(гидразинкарбонил)бензил)-3,5-диметил-N-фенилпиперазин-1-сульфонамида в виде белого твердого вещества (0,100 г, 76,9%).
[Стадия 8] Соединение 11640
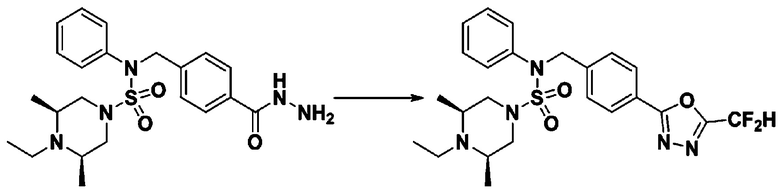
Раствор (3S,5R)-4-этил-N-(4-(гидразинкарбонил)бензил)-3,5-диметил-N-фенилпиперазин-1-сульфонамида (0,050 г, 0,112 ммоль), триэтиламина (0,078 мл, 0,561 ммоль) и 2,2-дифторуксусного ангидрида (0,042 мл, 0,337 ммоль) в тетрагидрофуране (5 мл) перемешивали при 70°С в течение 5 часов, и охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида аммония, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; этилацетат/гексан=0%-80%) с получением (3S,5R)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-4-этил-3,5-диметил-N-фенилпиперазин-1-сульфонамида в виде белого твердого вещества (0,035 г, 61,7%).
1H ЯМР (400 МГц, CDCl3) δ 8,02-8,01 (м, 2 H), 7,44-7,42 (м, 2 H), 7,34-7,24 (м, 5 H), 7,04 (с, 0,3 H), 6,91 (с, 0,5 H), 6,78 (с, 0,3 H), 4,90 (с, 2 H), 3,55-3,53 (м, 2 H), 3,04-3,01 (м, 2 H), 2,97-2,49 (м, 4 H), 1,16-1,00 (м, 6 H), 0,98-0,82 (м, 3 H); LRMS (ES) m/z 506,4 (M++1).
Пример 42: Соединение 11641, (3S,5R)-4-ацетил-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-3,5-диметил-N-фенилпиперазин-1-сульфонамид
[Стадия 1] метил 4-((((3S,5R)-4-ацетил-3,5-диметил-N- фенилпиперазин)-1-сульфонамидо)метил)бензоат
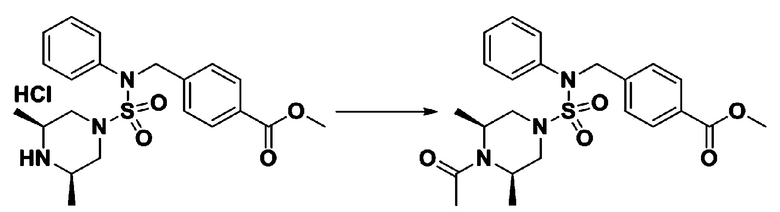
Раствор метил 4-((((3S,5R)-3,5-диметил-N-фенилпиперазин)-1-сульфонамидо)метил)бензоат гидрохлорида (0,200 г, 0,441 ммоль), триэтиламина (0,092 мл, 0,661 ммоль) и уксусного ангидрида (0,083 мл, 0,881 ммоль) в дихлорметане (10 мл) перемешивали при комнатной температуре в течение 12 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида аммония, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; метанол/дихлорметан=0%-5%) с получением метил 4-((((3S,5R)-4-ацетил-3,5-диметил-N-фенилпиперазин)-1-сульфонамидо)метил)бензоата в виде желтого масла (0,140 г, 69,1%).
[Стадия 2] (3S,5R)-4-ацетил-N-(4-(гидразинкарбонил)бензил)-3,5-диметил-N-фенилпиперазин-1-сульфонамид
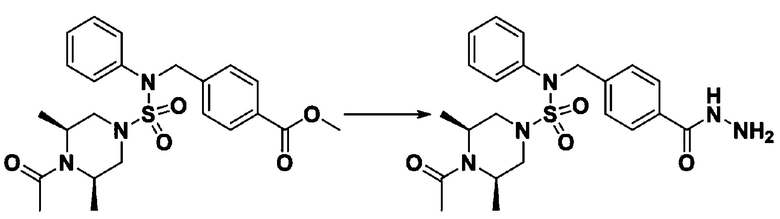
Раствор метил 4-((((3S,5R)-4-ацетил-3,5-диметил-N-фенилпиперазин)-1-сульфонамидо)метил)бензоата (0,140 г, 0,305 ммоль) и гидразина моногидрата (0,148 мл, 3,046 ммоль) в этаноле (10 мл) перемешивали при 90°C в течение 12 часов, и охлаждали до комнатной температуры для прекращения реакции. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Остаток разбавляли водой (10 мл) и бикарбонатом натрия (5 мл), и перемешивали. Полученные осадки собирали фильтрованием, промывали водой и сушили с получением (3S,5R)-4-ацетил-N-(4-(гидразинкарбонил)бензил)-3,5-диметил-N-фенилпиперазин-1-сульфонамида в виде белого твердого вещества (0,098 г, 70,0%).
[Стадия 3] Соединение 11641
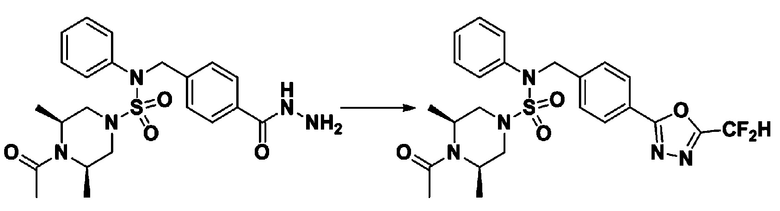
Раствор (3S,5R)-4-ацетил-N-(4-(гидразинкарбонил)бензил)-3,5-диметил-N-фенилпиперазин-1-сульфонамида (0,050 г, 0,109 ммоль), триэтиламина (0,076 мл, 0,544 ммоль) и 2,2-дифторуксусного ангидрида (0,041 мл, 0,326 ммоль) в тетрагидрофуране (10 мл) перемешивали при 70°С в течение 5 часов, и охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида аммония, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; этилацетат/гексан=0%-80%) с получением (3S,5R)-4-ацетил-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-3,5-диметил-N-фенилпиперазин-1-сульфонамида в виде белого твердого вещества (0,039 г, 69,0%).
1H ЯМР (400 МГц, CDCl3) δ 8,05-8,03 (м, 2 H), 7,45-7,43 (м, 2 H), 7,36-7,16 (м, 5 H), 7,04 (с, 0,2 H), 6,91 (с, 0,4 H), 6,79 (с, 0,2 H), 4,94 (с, 2 H), 3,97 (ушир.с, 2 H), 3,53-3,45 (м, 2 H), 2,70-2,66 (м, 2 H), 2,06 (с, 3 H), 1,31-1,22 (м, 6 H); LRMS (ES) m/z 520,3 (M++1).
Пример 43: Соединение 11642, (3S,5R)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-3,5-диметил-N-фенил-4-пропилпиперазин-1-сульфонамид
[Стадия 1] метил 4-((((3S,5R)-3,5-диметил-N-фенил-4-пропилпиперазин)-1-сульфонамидо)метил)бензоат
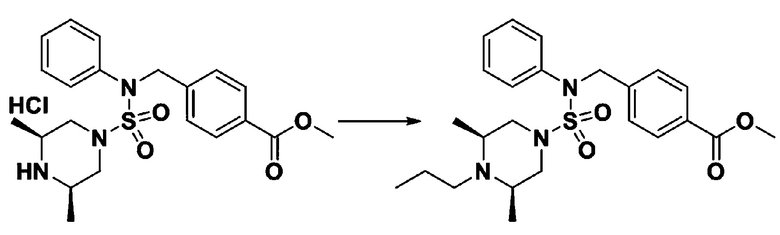
Раствор метил 4-((((3S,5R)-3,5-диметил-N-фенилпиперазин)-1-сульфонамидо)метил)бензоат гидрохлорида (0,200 г, 0,441 ммоль), пропиональдегида (0,051 г, 0,881 ммоль) и уксусной кислоты (0,028 мл, 0,485 ммоль) в дихлорметане (10 мл) перемешивали при комнатной температуре в течение 30 мин и смешивали с триацетоксиборгидридом натрия (0,187 г, 0,881 ммоль). Реакционную смесь перемешивали при той же температуре в течение дополнительных 12 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором бикарбоната натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; метанол/дихлорметан=0%-5%) с получением метил 4-((((3S,5R)-3,5-диметил-N-фенил-4-пропилпиперазин)-1-сульфонамидо)метил)бензоата в виде желтого масла (0,150 г, 74,1%).
[Стадия 2] (3S,5R)-N-(4-(гидразинкарбонил)бензил)-3,5-диметил-N-фенил-4-пропилпиперазин-1-сульфонамид
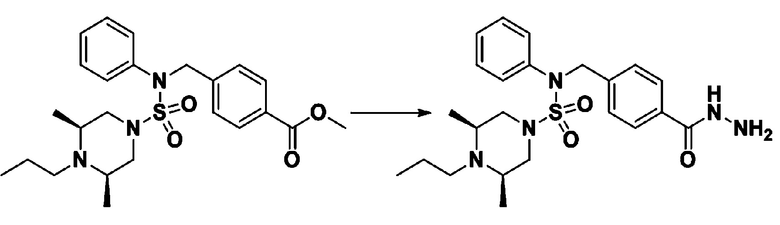
Раствор метил 4-((((3S,5R)-3,5-диметил-N-фенил-4-пропилпиперазин)-1-сульфонамидо)метил)бензоата (0,150 г, 0,326 ммоль) и гидразина моногидрата (0,016 мл, 0,573 ммоль) перемешивали при 90°C в течение 12 часов, и охлаждали до комнатной температуры для прекращения реакции. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Остаток разбавляли водой (10 мл) и бикарбонатом натрия (5 мл) и перемешивали. Полученные осадки собирали фильтрованием, промывали гексаном и сушили с получением (3S,5R)-N-(4-(гидразинкарбонил)бензил)-3,5-диметил-N-фенил-4-пропилпиперазин-1-сульфонамида в виде белого твердого вещества (0,110 г, 73,3%).
[Стадия 3] Соединение 11642
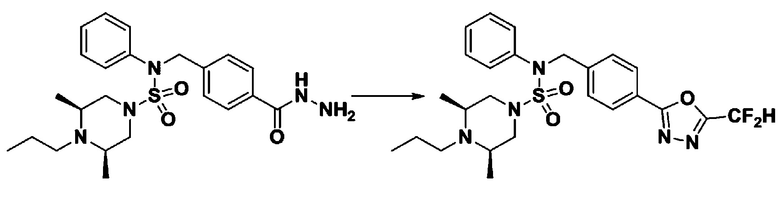
Раствор (3S,5R)-N-(4-(гидразинкарбонил)бензил)-3,5-диметил-N-фенил-4-пропилпиперазин-1-сульфонамида (0,050 г, 0,109 ммоль), триэтиламина (0,076 мл, 0,544 ммоль) и 2,2-дифторуксусного ангидрида (0,041 мл, 0,326 ммоль) в тетрагидрофуране (10 мл) перемешивали при 70°С в течение 5 часов, и охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида аммония, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; метанол/дихлорметан=0%-5%) с получением (3S,5R)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-3,5-диметил-N-фенил-4-пропилпиперазин-1-сульфонамида в виде белого твердого вещества (0,034 г, 60,1%).
1H ЯМР (400 МГц, CDCl3) δ 8,04-8,01 (м, 2 H), 7,43-7,41 (м, 2 H), 7,37-7,27 (м, 5 H), 7,04 (с, 0,3 H), 6,91 (с, 0,5 H), 6,78 (с, 0,3 H), 4,88 (с, 2 H), 3,65 (ушир.с, 4 H), 3,29-3,21 (м, 4 H), 1,64-1,60 (м, 2 H), 1,45-1,40 (м, 6 H), 1,29-1,26 (м, 3 H); LRMS (ES) m/z 520,3(M++1).
Пример 44: Соединение 11643, (3S,5R)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-3,4,5-триметил-N-фенилпиперазин-1-сульфонамид
[Стадия 1] метил 4-((((3S,5R)-3,4,5-триметил-N-фенилпиперазин)-1-сульфонамидо)метил)бензоат
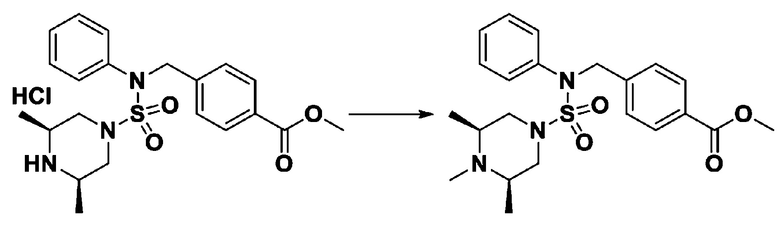
Смесь метил 4-((((3S,5R)-3,5-диметил-N-фенилпиперазин)-1-сульфонамидо)метил)бензоат гидрохлорида (0,200 г, 0,441 ммоль), N,N-диизопропилэтиламина (0,077 мл, 0,441 ммоль), параформальдегида (0,066 г, 2,203 ммоль) и триацетоксиборгидрида натрия (0,187 г, 0,881 ммоль) в дихлорметане (10 мл), полученную при температуре окружающей среды, кипятили с обратным холодильником в течение 12 ч, и охлаждали до температуры окружающей среды для прекращения реакции. Затем к реакционной смеси добавляли воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором бикарбоната натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; метанол/дихлорметан=0%-5%) с получением метил 4-((((3S,5R)-3,4,5-триметил-N-фенилпиперазин)-1-сульфонамидо)метил)бензоата в виде белого твердого вещества (0,160 г, 84,2%).
[Стадия 2] (3S,5R)-N-(4-(гидразинкарбонил)бензил)-3,4,5-триметил-N-фенилпиперазин-1-сульфонамид
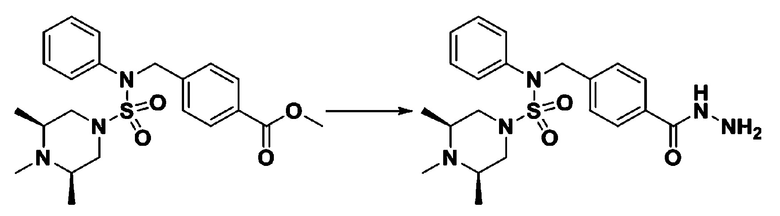
Раствор метил 4-((((3S,5R)-3,4,5-триметил-N-фенилпиперазин)-1-сульфонамидо)метил)бензоата (0,160 г, 0,371 ммоль) и гидразина моногидрата (0,180 мл, 3,708 ммоль) в этаноле (10 мл) перемешивали при 90°C в течение 12 часов, и охлаждали до комнатной температуры для прекращения реакции. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Остаток разбавляли водой (10 мл) и бикарбонатом натрия (5 мл), и перемешивали. Полученные осадки собирали фильтрованием, промывали гексаном и сушили с получением (3S,5R)-N-(4-(гидразинкарбонил)бензил)-3,4,5-триметил-N-фенилпиперазин-1-сульфонамида в виде белого твердого вещества (0,130 г, 81,2%).
[Стадия 3] Соединение 11643
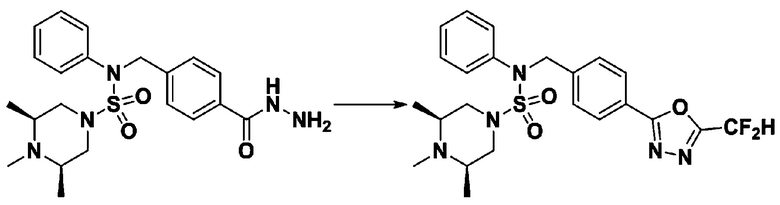
Раствор (3S,5R)-N-(4-(гидразинкарбонил)бензил)-3,4,5-триметил-N-фенилпиперазин-1-сульфонамида (0,050 г, 0,116 ммоль), триэтиламина (0,081 мл, 0,579 ммоль) и 2,2-дифторуксусного ангидрида (0,043 мл, 0,348 ммоль) в тетрагидрофуране (5 мл) перемешивали при 70°С в течение 5 часов, и охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида аммония, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; этилацетат/гексан=0%-80%) с получением (3S,5R)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-3,4,5-триметил-N-фенилпиперазин-1-сульфонамида в виде белого твердого вещества (0,036 г, 63,2%).
1H ЯМР (400 МГц, CDCl3) δ 8,04-8,01 (м, 2 H), 7,44-7,42 (м, 2 H), 7,35-7,25 (м, 5 H), 7,04 (с, 0,3 H), 6,91 (с, 0,5 H), 6,78 (с, 0,3 H), 4,90 (с, 2 H), 3,56-3,53 (м, 2 H), 2,50 (ушир.с, 2 H), 2,41-2,37 (м, 5 H), 1,29-1,26 (ушир.с, 6 H); LRMS (ES) m/z 492,1 (M++1).
Пример 45: Соединение 11644, (3S,5R)-4-(2,2-дифторацетил)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-3,5-диметил-N-фенилпиперазин-1-сульфонамид
[Стадия 1] (3S,5R)-N-(4-(гидразинкарбонил)бензил)-3,5-диметил-N-фенилпиперазин-1-сульфонамид
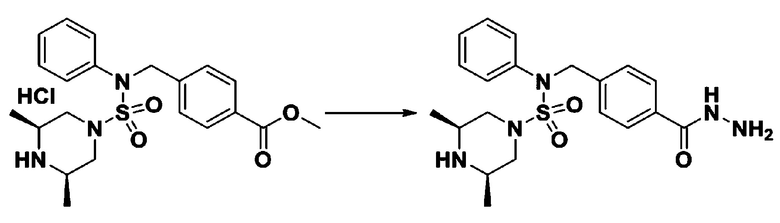
Раствор метил 4-((((3S,5R)-3,5-диметил-N-фенилпиперазин)-1-сульфонамидо)метил)бензоат гидрохлорида (0,150 г, 0,359 ммоль) и гидразина моногидрата (0,175 мл, 3,593 ммоль) в этаноле (10 мл) перемешивали при 90°C в течение 12 часов, и охлаждали до комнатной температуры для прекращения реакции. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Остаток разбавляли водой (10 мл) и бикарбонатом натрия (5 мл) и перемешивали. Полученные осадки собирали фильтрованием и сушили с получением (3S,5R)-N-(4-(гидразинкарбонил)бензил)-3,5-диметил-N-фенилпиперазин-1-сульфонамида в виде белого твердого вещества (0,110 г, 73,3%).
[Стадия 2] Соединение 11644
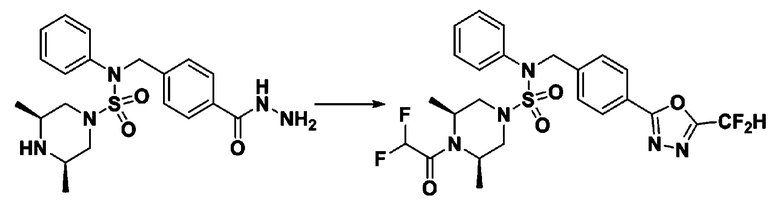
Раствор (3S,5R)-N-(4-(гидразинкарбонил)бензил)-3,5-диметил-N-фенилпиперазин-1-сульфонамида (0,050 г, 0,120 ммоль), триэтиламина (0,083 мл, 0,599 ммоль) и 2,2-дифторуксусного ангидрида (0,045 мл, 0,359 ммоль) в тетрагидрофуране (10 мл) перемешивали при 70°С в течение 5 часов, и охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида аммония, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; этилацетат/гексан=0%-50%) с получением (3S,5R)-4-(2,2-дифторацетил)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-3,5-диметил-N-фенилпиперазин-1-сульфонамида в виде белого твердого вещества (0,031 г, 46,6%).
1H ЯМР (400 МГц, CDCl3) δ 8,05-8,01 (м, 2 H), 7,44-7,42 (м, 2 H), 7,36-7,05 (м, 5 H), 6,92 (с, 0,3 H), 6,92 (с, 0,5 H), 6,79 (с, 0,3 H), 6,23 (с, 0,3 H), 6,09 (с, 0,5 H), 5,96 (с, 0,3 H), 4,93 (с, 2 H), 4,55 (ушир.с, 1 H), 4,24-4,22 (м, 1 H), 3,56 (д, 2 H, J=12,4 Гц), 2,76-2,72 (м, 2 H), 1,42-1,37 (м, 6 H); LRMS (ES) m/z 555,9 (M++1).
Пример 46: Соединение 11651, N-(3-хлор-4-фторфенил)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-4-изопропилпиперазин-1-сульфонамид
[Стадия 1] трет-бутил 4-(N-(3-хлор-4-фторфенил)сульфамоил)пиперазин-1-карбоксилат
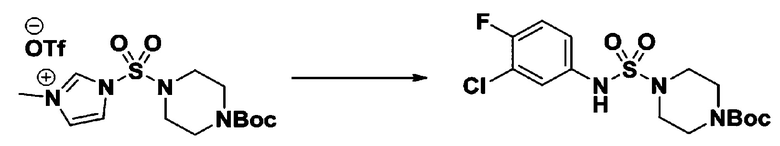
1-((4-(трет-бутоксикарбонил)пиперазин-1-ил)сульфонил)-3-метил-1H-имидазол-3-ий трифторметансульфонат (1,500 г, 3,122 ммоль) и 3-хлор-4-фторанилин (0,682 г, 4,683 ммоль) смешивали при комнатной температуре в ацетонитриле (100 мл), и затем смесь перемешивали при 80°C в течение 18 часов. Реакционную смесь охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 12 г; этилацетат/гексан=0%-30%) с получением трет-бутил 4-(N-(3-хлор-4-фторфенил)сульфамоил)пиперазин-1-карбоксилата в виде пурпурного твердого вещества (0,710 г, 57,7%).
[Стадия 2] трет-бутил 4-(N-(3-хлор-4-фторфенил)-N-(4-(метоксикарбонил)бензил)сульфамоил)пиперазин-1-карбоксилат
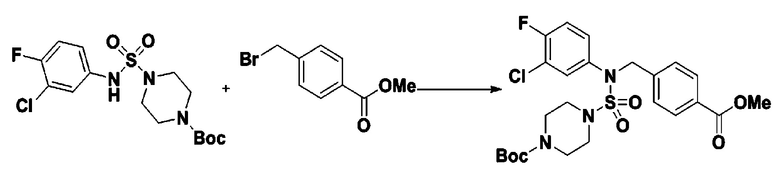
Раствор трет-бутил 4-(N-(3-хлор-4-фторфенил)сульфамоил)пиперазин-1-карбоксилата (0,710 г, 1,803 ммоль) и гидрида натрия (60,00%, 0,216 г, 5,408 ммоль) в N,N-диметилформамиде (80 мл) смешивали при комнатной температуре с метил 4-(бромметил)бензоатом (0,619 г, 2,704 ммоль), и перемешивали при 50°С в течение 18 часов. Реакционную смесь охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 12 г; этилацетат/гексан=0%-80%) с получением трет-бутил 4-(N-(3-хлор-4-фторфенил)-N-(4-(метоксикарбонил)бензил)сульфамоил)пиперазин-1-карбоксилата в виде белого твердого вещества (0,616 г, 63,0%).
[Стадия 3] метил 4-((N-(3-хлор-4-фторфенил)пиперазин-1-сульфонамидо)метил)бензоат гидрохлорид
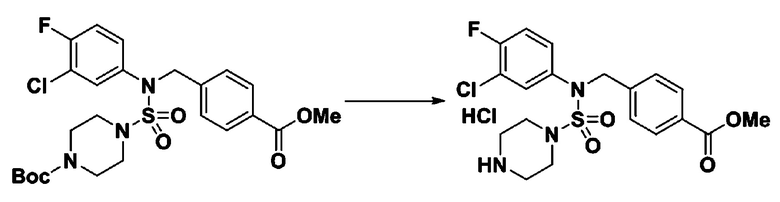
Раствор трет-бутил 4-(N-(3-хлор-4-фторфенил)-N-(4-(метоксикарбонил)бензил)сульфамоил)пиперазин-1-карбоксилата (0,616 г, 1,136 ммоль) в дихлорметане (15 мл) смешивали при комнатной температуре с хлористоводородной кислотой (4,00 M раствор в диоксане, 1,136 мл, 4,546 ммоль). Реакционную смесь перемешивали при той же температуре в течение 18 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Осадки собирали фильтрованием, промывали дихлорметаном и сушили с получением метил 4-((N-(3-хлор-4-фторфенил)пиперазин-1-сульфонамидо)метил)бензоат гидрохлорида в виде белого твердого вещества (0,330 г, 60,7%).
[Стадия 4] метил 4-(((N-(3-хлор-4-фторфенил)-4-изопропилпиперазин)-1-сульфонамидо)метил)бензоат
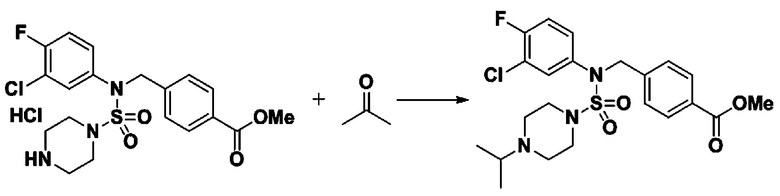
Раствор метил 4-((N-(3-хлор-4-фторфенил)пиперазин-1-сульфонамидо)метил)бензоат гидрохлорида (0,100 г, 0,209 ммоль) и пропан-2-она (0,023 мл, 0,314 ммоль) в дихлорметане (10 мл) смешивали при комнатной температуре с триацетоксиборгидридом натрия (0,133 г, 0,627 ммоль). Реакционную смесь перемешивали при той же температуре в течение 18 ч. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; метанол/дихлорметан=0%-15%) с получением метил 4-(((N-(3-хлор-4-фторфенил)-4-изопропилпиперазин)-1-сульфонамидо)метил)бензоата в виде белого твердого вещества (0,042 г, 41,5%).
[Стадия 5] N-(3-хлор-4-фторфенил)-N-(4-(гидразинкарбонил)бензил)-4-изопропилпиперазин-1-сульфонамид
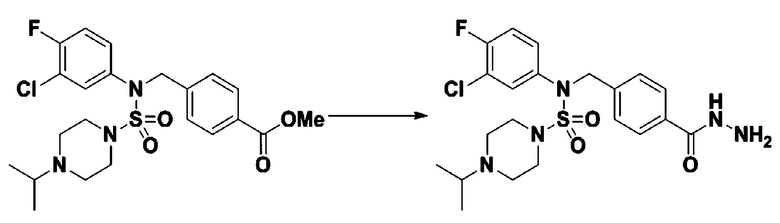
метил 4-(((N-(3-хлор-4-фторфенил)-4-изопропилпиперазин)-1-сульфонамидо)метил)бензоат (0,042 г, 0,087 ммоль) и гидразин моногидрат (0,084 мл, 1,736 ммоль) смешивали при комнатной температуре в этаноле (8 мл)/воде (2 мл), и затем смесь перемешивали при 80°C в течение 18 часов. Реакционную смесь охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Указанное в заголовке соединение использовали без дополнительной очистки (N-(3-хлор-4-фторфенил)-N-(4-(гидразинкарбонил)бензил)-4-изопропилпиперазин-1-сульфонамид, 0,038 г, 90,5%, бесцветное масло).
[Стадия 6] Соединение 11651
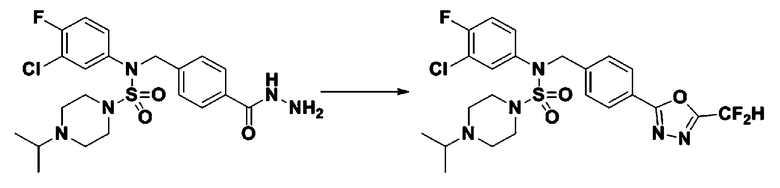
Раствор N-(3-хлор-4-фторфенил)-N-(4-(гидразинкарбонил)бензил)-4-изопропилпиперазин-1-сульфонамида (0,042 г, 0,087 ммоль) и триэтиламина (0,024 мл, 0,174 ммоль) в тетрагидрофуране (10 мл) смешивали при комнатной температуре с 2,2-дифторуксусным ангидридом (0,032 мл, 0,260 ммоль), и перемешивали при 80°С в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2 пластинка, 20×20×1 мм; метанол/дихлорметан=10%) с получением N-(3-хлор-4-фторфенил)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-4-изопропилпиперазин-1-сульфонамида в виде белого твердого вещества (0,009 г, 19,1%).
1H ЯМР (400 МГц, CDCl3) δ 8,04 ~ 8,02 (м, 2 H), 7,42 (д, 2 H, J=8,4 Гц), 7,25 ~ 7,22 (м, 2 H), 7,02 ~ 6,98 (м, 2 H), 4,85 (с, 2 H), 3,64 (т, 4 H, J=4,7 Гц), 3,17 (т, 4 H, J=4,8 Гц); LRMS (ES) m/z 544,0 (M++1).
Пример 47: Соединение 11652, N-(3-хлор-4-фторфенил)-4-циклобутил-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)пиперазин-1-сульфонамид
[Стадия 1] метил 4-(((N-(3-хлор-4-фторфенил)-4-циклобутилпиперазин)-1-сульфонамидо)метил)бензоат
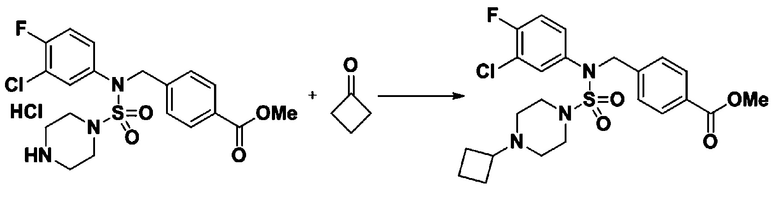
Раствор метил 4-((N-(3-хлор-4-фторфенил)пиперазин-1-сульфонамидо)метил)бензоат гидрохлорида (0,100 г, 0,209 ммоль) и циклобутанона (0,023 мл, 0,314 ммоль) в дихлорметане (10 мл) смешивали при комнатной температуре с триацетоксиборгидридом натрия (0,133 г, 0,627 ммоль). Реакционную смесь перемешивали при той же температуре в течение 18 ч. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; метанол/дихлорметан=0%-30%) с получением метил 4-(((N-(3-хлор-4-фторфенил)-4-циклобутилпиперазин)-1-сульфонамидо)метил)бензоата в виде белого твердого вещества (0,098 г, 94,5%).
[Стадия 2] N-(3-хлор-4-фторфенил)-4-циклобутил-N-(4-(гидразинкарбонил)бензил)пиперазин-1-сульфонамид
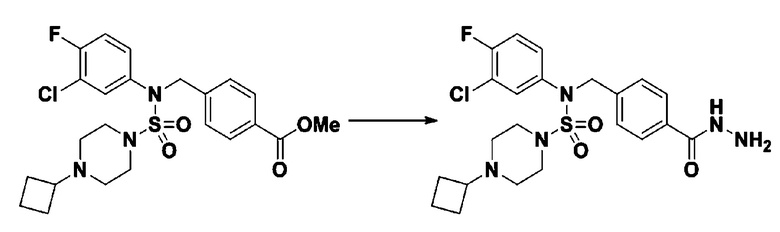
Метил 4-(((N-(3-хлор-4-фторфенил)-4-циклобутилпиперазин)-1-сульфонамидо)метил)бензоат (0,098 г, 0,198 ммоль) и гидразин моногидрат (0,192 мл, 3,952 ммоль) смешивали при комнатной температуре в этаноле (8 мл)/воде (2 мл), и затем смесь перемешивали при 80°C в течение 18 часов. Реакционную смесь охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Указанное в заголовке соединение использовали без дополнительной очистки (N-(3-хлор-4-фторфенил)-4-циклобутил-N-(4-(гидразинкарбонил)бензил)пиперазин-1-сульфонамид, 0,087 г, 88,8%, бесцветное масло).
[Стадия 3] Соединение 11652
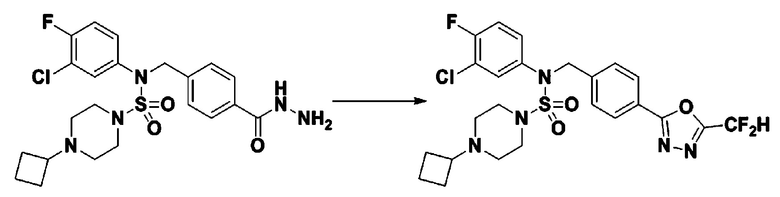
Раствор N-(3-хлор-4-фторфенил)-4-циклобутил-N-(4-(гидразинкарбонил)бензил)пиперазин-1-сульфонамида (0,098 г, 0,198 ммоль) и триэтиламина (0,055 мл, 0,395 ммоль) в тетрагидрофуране (10 мл) смешивали при комнатной температуре с 2,2-дифторуксусным ангидридом (0,074 мл, 0,593 ммоль), и перемешивали при 80°С в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2 пластинка, 20×20×1 мм; метанол/дихлорметан=10%) с получением N-(3-хлор-4-фторфенил)-4-циклобутил-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)пиперазин-1-сульфонамида в виде белого твердого вещества (0,007 г, 6,4%).
1H ЯМР (400 МГц, CDCl3) δ 8,05~8,00 (м, 2H), 7,40~7,35 (м, 2 H), 7,15~7,04 (м, 2 H), 7,03 (с, 0,25 H), 6,90 (с, 0,5 H), 6,77 (с, 0,25 H), 4,82 (с, 2 H), 4,67~4,60 (м, 4 H), 3,60 ~ 3,45 (м, 1 H), 3,40~3,20 (м, 4 H), 2,45~2,23 (м, 4 H); LRMS (ES) m/z 556,0 (M++1).
Пример 48: Соединение 11653, N-(3-хлор-4-фторфенил)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-4-(оксетан-3-ил)пиперазин-1-сульфонамид
[Стадия 1] метил 4-(((N-(3-хлор-4-фторфенил)-4-(оксетан-3-ил)пиперазин)-1-сульфонамидо)метил)бензоат
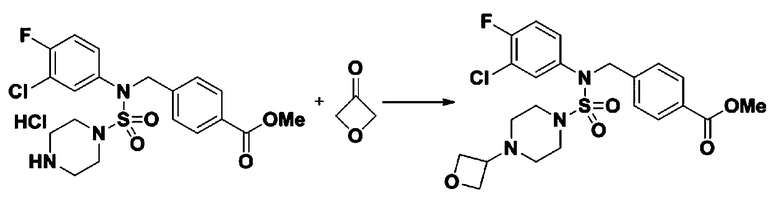
Раствор метил 4-((N-(3-хлор-4-фторфенил)пиперазин-1-сульфонамидо)метил)бензоат гидрохлорида (0,100 г, 0,209 ммоль) и оксетан-3-она (0,020 мл, 0,314 ммоль) в дихлорметане (10 мл) смешивали при комнатной температуре с триацетоксиборгидридом натрия (0,133 г, 0,627 ммоль). Реакционную смесь перемешивали при той же температуре в течение 18 ч. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; метанол/дихлорметан=0%-15%) с получением метил 4-(((N-(3-хлор-4-фторфенил)-4-(оксетан-3-ил)пиперазин)-1-сульфонамидо)метил)бензоата в виде белого твердого вещества (0,079 г, 75,9%).
[Стадия 2] N-(3-хлор-4-фторфенил)-N-(4-(гидразинкарбонил)бензил)-4-(оксетан-3-ил)пиперазин-1-сульфонамид
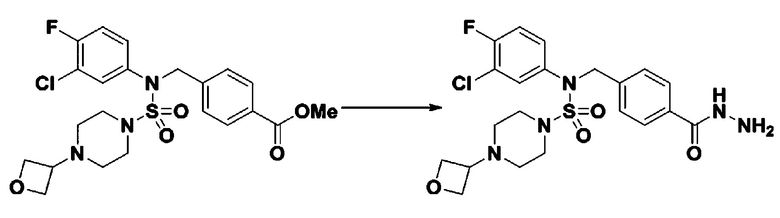
Метил 4-(((N-(3-хлор-4-фторфенил)-4-(оксетан-3-ил)пиперазин)-1-сульфонамидо)метил)бензоат (0,079 г, 0,159 ммоль) и гидразин моногидрат (0,154 мл, 3,173 ммоль) смешивали при комнатной температуре в этаноле (8 мл)/воде (2 мл), и затем смесь перемешивали при 80°C в течение 18 часов. Реакционную смесь охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Указанное в заголовке соединение использовали без дополнительной очистки (N-(3-хлор-4-фторфенил)-N-(4-(гидразинкарбонил)бензил)-4-(оксетан-3-ил)пиперазин-1-сульфонамид, 0,068 г, 86,1%, бесцветное масло).
[Стадия 3] Соединение 11653
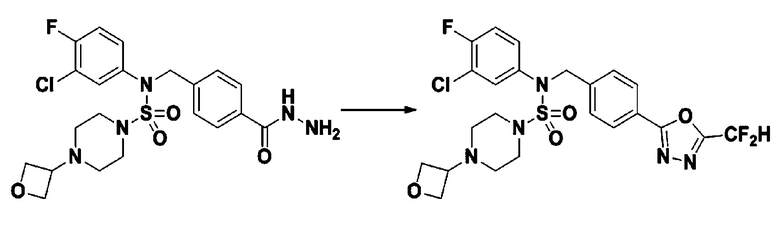
Раствор N-(3-хлор-4-фторфенил)-N-(4-(гидразинкарбонил)бензил)-4-(оксетан-3-ил)пиперазин-1-сульфонамида (0,076 г, 0,153 ммоль) и триэтиламина (0,043 мл, 0,305 ммоль) в тетрагидрофуране (10 мл) смешивали при комнатной температуре с 2,2-дифторуксусным ангидридом (0,057 мл, 0,458 ммоль), и перемешивали при 80°С в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2 пластинка, 20×20×1 мм; метанол/дихлорметан=10%) с получением N-(3-хлор-4-фторфенил)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-4-(оксетан-3-ил)пиперазин-1-сульфонамида в виде белого твердого вещества (0,008 г, 9,4%).
1H ЯМР (400 МГц, CDCl3) δ 8,05~8,02 (м, 2 H), 7,40~7,35 (м, 3 H), 7,16~7,12 (м, 1 H), 7,07~7,03 (м, 1 H), 7,03 (с, 0,25 H), 6,90 (с, 0,5 H), 6,77 (с, 0,25 H), 4,82 (с, 2 H), 3,43~3,22 (м, 4 H), 2,78~2,82 (м, 1 H), 2,62~2,21 (м, 4 H), 2,20~1,81 (м, 4 H), 1,80~1,61 (м, 2 H); LRMS (ES) m/z 558,0 (M++1).
Пример 49: Соединение 11654, N-(3-хлор-4-фторфенил)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-4-метилпиперазин-1-сульфонамид
[Стадия 1] метил 4-(((N-(3-хлор-4-фторфенил)-4-метилпиперазин)-1-сульфонамидо)метил)бензоат
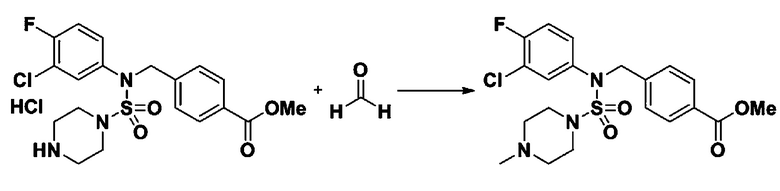
Раствор метил 4-((N-(3-хлор-4-фторфенил)пиперазин-1-сульфонамидо)метил)бензоат гидрохлорида (0,060 г, 0,125 ммоль) и параформальдегида (0,006 г, 0,188 ммоль) в дихлорметане (10 мл) смешивали при комнатной температуре с триацетоксиборгидридом натрия (0,080 г, 0,376 ммоль). Реакционную смесь перемешивали при той же температуре в течение 18 ч. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; метанол/дихлорметан=0%-15%) с получением метил 4-(((N-(3-хлор-4-фторфенил)-4-метилпиперазин)-1-сульфонамидо)метил)бензоата в виде белого твердого вещества (0,038 г, 66,4%).
[Стадия 2] N-(3-хлор-4-фторфенил)-N-(4-(гидразинкарбонил)бензил)-4-метилпиперазин-1-сульфонамид
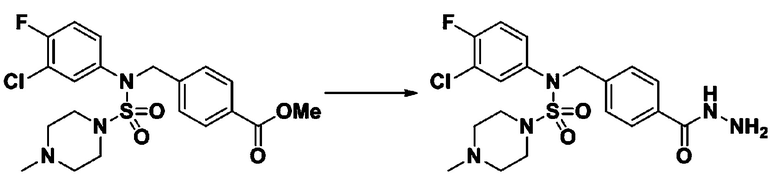
метил 4-(((N-(3-хлор-4-фторфенил)-4-метилпиперазин)-1-сульфонамидо)метил)бензоат (0,038 г, 0,083 ммоль) и гидразин моногидрат (0,081 мл, 1,667 ммоль) смешивали при комнатной температуре в этаноле (8 мл)/воде(2 мл), и затем смесь перемешивали при 80°C в течение 18 часов. Реакционную смесь охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Указанное в заголовке соединение использовали без дополнительной очистки (N-(3-хлор-4-фторфенил)-N-(4-(гидразинкарбонил)бензил)-4-метилпиперазин-1-сульфонамид, 0,033 г, 86,8%, бесцветное масло).
[Стадия 3] Соединение 11654
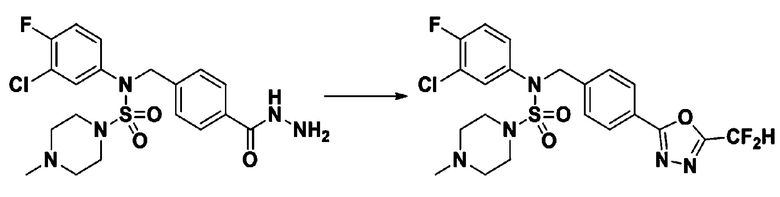
Раствор N-(3-хлор-4-фторфенил)-N-(4-(гидразинкарбонил)бензил)-4-метилпиперазин-1-сульфонамида (0,038 г, 0,083 ммоль) и триэтиламина (0,023 мл, 0,167 ммоль) в тетрагидрофуране (10 мл) смешивали при комнатной температуре с 2,2-дифторуксусным ангидридом (0,031 мл, 0,250 ммоль), и перемешивали при 80°С в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2 пластинка, 20×20×1 мм; метанол/дихлорметан=10%) с получением N-(3-хлор-4-фторфенил)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-4-метилпиперазин-1-сульфонамида в виде белого твердого вещества (0,008 г, 18,6%).
1H ЯМР (400 МГц, CDCl3) δ 8,05 ~ 8,03 (м, 2 H), 7,40 ~ 7,36 (м, 3 H), 7,20 ~ 7,08 (м, 2 H), 7,03 (с, 0,25 H), 6,90 (с, 0,5 H), 6,77 (с, 0,25 H), 4,83 (с, 2 H), 3,80 ~ 3,57 (м, 4 H), 3,18 ~ 2,80 (м, 4 H), 2,64 (с, 3 H); LRMS (ES) m/z 516,0 (M++1).
Пример 50: Соединение 11659, (3S,5R)-4-ацетил-N-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3,5-диметил-N-фенилпиперазин-1-сульфонамид
[Стадия 1] трет-бутил (2S,6R)-4-(N-((5-(метоксикарбонил)пиридин-2-ил)метил)-N-фенилсульфамоил)-2,6-диметилпиперазин-1-карбоксилат
Раствор трет-бутил (2S,6R)-2,6-диметил-4-(N-фенилсульфамоил)пиперазин-1-карбоксилата (1,110 г, 3,004 ммоль) и карбоната калия (0,623 г, 4,506 ммоль) в N,N-диметилформамиде (20 мл) перемешивали при комнатной температуре в течение 30 мин и смешивали с метил-6-(бромметил)никотинатом (0,760 г, 3,305 ммоль) и иодидом калия (0,249 г, 1,502 ммоль). Реакционную смесь перемешивали при той же температуре в течение дополнительных 12 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида аммония, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 24 г; этилацетат/гексан=0%-50%) с получением трет-бутил (2S,6R)-4-(N-((5-(метоксикарбонил)пиридин-2-ил)метил)-N-фенилсульфамоил)-2,6-диметилпиперазин-1-карбоксилата в виде желтого твердого вещества (1,440 г, 92,4%).
[Стадия 2] метил 6-((((3S,5R)-3,5-диметил-N-фенилпиперазин)-1-сульфонамидо)метил)никотинат гидрохлорид
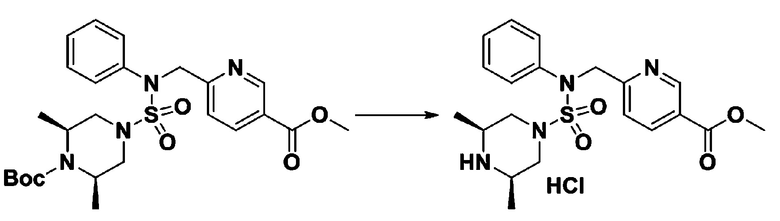
Раствор трет-бутил (2S,6R)-4-(N-((5-(метоксикарбонил)пиридин-2-ил)метил)-N-фенилсульфамоил)-2,6-диметилпиперазин-1-карбоксилата (1,440 г, 2,777 ммоль) и хлорида водорода (4,00 M раствор в 1,4-диоксане, 1,388 мл, 5,553 ммоль) в дихлорметане (80 мл) перемешивали при комнатной температуре в течение 5 часов, и концентрировали при пониженном давлении для удаления растворителя. Неочищенный продукт использовали без дополнительной очистки (метил 6-((((3S,5R)-3,5-диметил-N-фенилпиперазин)-1-сульфонамидо)метил)никотинат гидрохлорид, 0,900 г, 71,2%, белое твердое вещество).
[Стадия 3] метил 6-((((3S,5R)-4-ацетил-3,5-диметил-N-фенилпиперазин)-1-сульфонамидо)метил)никотинат
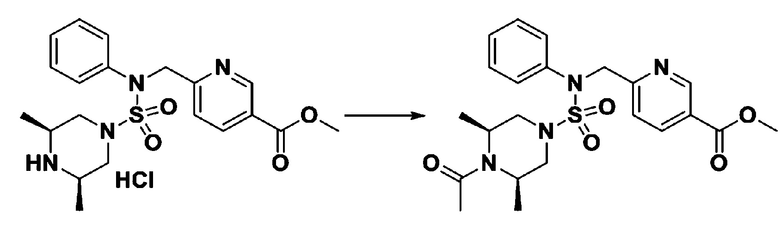
Раствор метил 6-((((3S,5R)-3,5-диметил-N-фенилпиперазин)-1-сульфонамидо)метил)никотинат гидрохлорида (0,200 г, 0,440 ммоль), триэтиламина (0,032 г, 0,319 ммоль) и уксусного ангидрида (0,062 мл, 0,659 ммоль) в дихлорметане (10 мл) перемешивали при комнатной температуре в течение 12 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида аммония, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; метанол/дихлорметан=0%-5%) с получением метил 6-((((3S,5R)-4-ацетил-3,5-диметил-N-фенилпиперазин)-1-сульфонамидо)метил)никотината в виде бесцветного масла (0,130 г, 64,2%).
[Стадия 4] (3S,5R)-4-ацетил-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-3,5-диметил-N-фенилпиперазин-1-сульфонамид
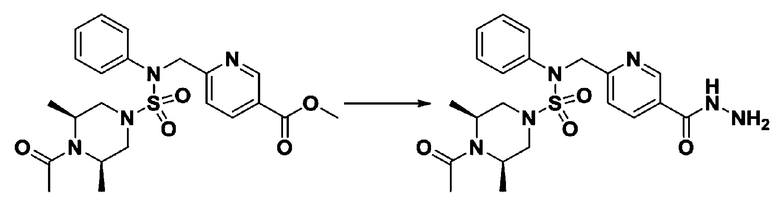
Раствор метил 6-((((3S,5R)-4-ацетил-3,5-диметил-N-фенилпиперазин)-1-сульфонамидо)метил)никотината (0,130 г, 0,282 ммоль) и гидразина моногидрата (0,137 мл, 2,823 ммоль) в этаноле (10 мл) перемешивали при 90°C в течение 12 часов. Реакционную смесь охлаждали до комнатной температуры для прекращения реакции, и концентрировали при пониженном давлении для удаления растворителя. Остаток разбавляли водой (5 мл) и бикарбонатом натрия (10 мл) и перемешивали. Полученные осадки собирали фильтрованием, промывали водой и сушили с получением (3S,5R)-4-ацетил-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-3,5-диметил-N-фенилпиперазин-1-сульфонамида в виде белого твердого вещества (0,085 г, 65,4%).
[Стадия 5] Соединение 11659
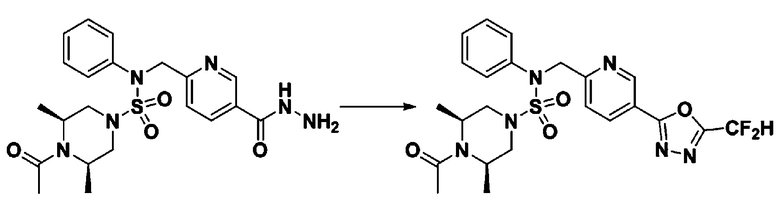
Раствор (3S,5R)-4-ацетил-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-3,5-диметил-N-фенилпиперазин-1-сульфонамида (0,085 г, 0,185 ммоль), триэтиламина (0,129 мл, 0,923 ммоль) и дифторуксусного ангидрида (0,069 мл, 0,554 ммоль) в тетрагидрофуране (10 мл) перемешивали при 70°С в течение 12 часов, и охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида аммония, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; этилацетат/гексан=0%-60%) с получением (3S,5R)-4-ацетил-N-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3,5-диметил-N-фенилпиперазин-1-сульфонамида в виде белого твердого вещества (0,015 г, 15,6%).
1H ЯМР (400 МГц, CDCl3) δ 9,20 (с, 1 H), 8,37-8,35 (м, 1 H), 7,69 (д, 1 H, J=8,2 Гц), 7,46-7,44 (м, 2 H), 7,35-7,31 (м, 2 H), 7,28-7,23 (м, 2 H), 7,06 (с, 0,3 H), 6,93 (с, 0,5 H), 6,81 (с, 0,3 H), 5,15 (с, 2 H), 4,63 (ушир.с, 1 H), 3,95 (ушир.с, 1 H), 3,53 (д, 2 H, J=12,1 Гц), 2,79-2,76 (м, 2 H), 2,08 (с, 3 H), 1,28 (с, 6 H); LRMS (ES) m/z 521,2(M++1).
Пример 51: Соединение 11660, (3S,5R)-4-ацетил-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-3,5-диметил-N-(пиридин-3-ил)пиперазин-1-сульфонамид
[Стадия 1] трет-бутил (2S,6R)-2,6-диметил-4-(N-(пиридин-3-ил)сульфамоил)пиперазин-1-карбоксилат
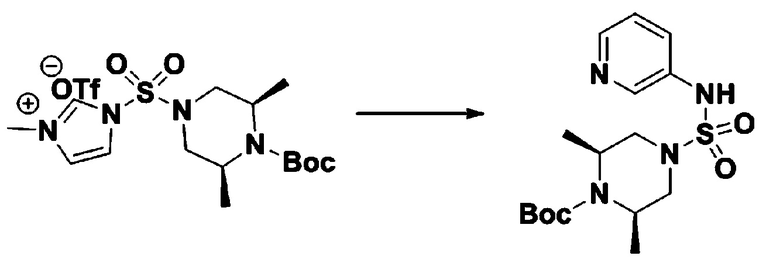
Раствор 1-(((3S,5R)-4-(трет-бутоксикарбонил)-3,5-диметилпиперазин-1-ил)сульфонил)-3-метил-1H-имидазол-3-ий трифторметансульфоната (2,000 г, 3,933 ммоль) и пиридин-3-амина (0,407 г, 4,326 ммоль) в ацетонитриле (5 мл) перемешивали при комнатной температуре в течение 16 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 24 г; этилацетат/гексан=0%-20%) с получением трет-бутил (2S,6R)-2,6-диметил-4-(N-(пиридин-3-ил)сульфамоил)пиперазин-1-карбоксилата в виде желтого твердого вещества (1,100 г, 75,5%).
[Стадия 2] трет-бутил (2S,6R)-4-(N-(4-(метоксикарбонил)бензил)-N-(пиридин-3-ил)сульфамоил)-2,6-диметилпиперазин-1-карбоксилат
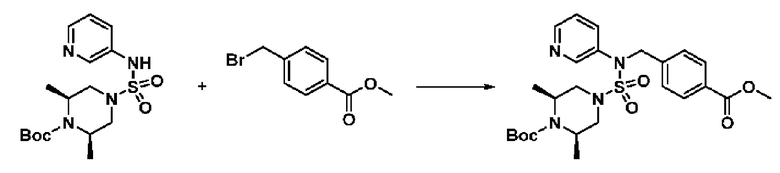
Раствор трет-бутил (2S,6R)-2,6-диметил-4-(N-(пиридин-3-ил)сульфамоил)пиперазин-1-карбоксилата (0,510 г, 1,377 ммоль) и карбоната калия (0,285 г, 2,065 ммоль) в N,N-диметилформамиде (20 мл) перемешивали при комнатной температуре в течение 30 мин и смешивали с метил 4-(бромметил)бензоатом (0,347 г, 1,514 ммоль) и иодидом калия (0,114 г, 0,688 ммоль). Реакционную смесь перемешивали при той же температуре в течение дополнительных 12 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида аммония, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 12 г; этилацетат/гексан=0%-30%) с получением трет-бутил (2S,6R)-4-(N-(4-(метоксикарбонил)бензил)-N-(пиридин-3-ил)сульфамоил)-2,6-диметилпиперазин-1-карбоксилата в виде белого твердого вещества (0,300 г, 42,0%).
[Стадия 3] метил 4-((((3S,5R)-3,5-диметил-N-(пиридин-3-ил)пиперазин)-1-сульфонамидо)метил)бензоат гидрохлорид
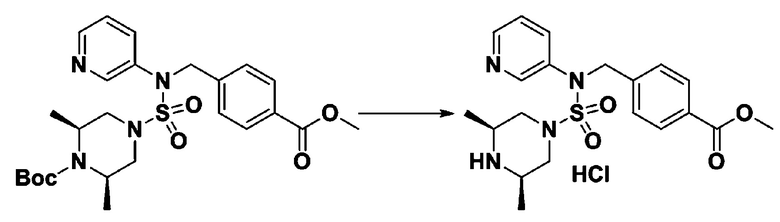
Раствор трет-бутил (2S,6R)-4-(N-(4-(метоксикарбонил)бензил)-N-(пиридин-3-ил)сульфамоил)-2,6-диметилпиперазин-1-карбоксилата (0,280 г, 0,540 ммоль) и хлористоводородной кислоты (4,00 M раствор в 1,4-диоксане, 0,270 мл, 1,080 ммоль) в дихлорметане (20 мл) перемешивали при комнатной температуре в течение 5 часов, и концентрировали при пониженном давлении для удаления растворителя. Неочищенный продукт использовали без дополнительной очистки (метил 4-((((3S,5R)-3,5-диметил-N-(пиридин-3-ил)пиперазин)-1-сульфонамидо)метил)бензоат гидрохлорид, 0,150 г, 61,1%, желтое твердое вещество).
[Стадия 4] метил 4-((((3S,5R)-4-ацетил-3,5-диметил-N-(пиридин-3-ил)пиперазин)-1-сульфонамидо)метил)бензоат
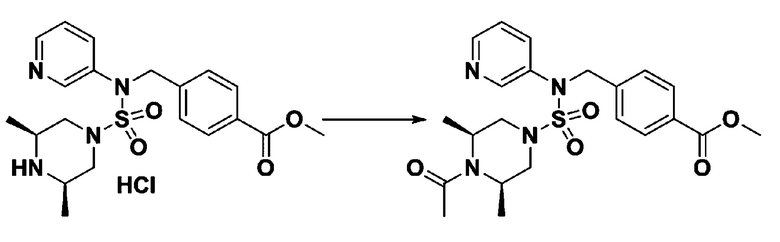
Раствор метил 4-((((3S,5R)-3,5-диметил-N-(пиридин-3-ил)пиперазин)-1-сульфонамидо)метил)бензоат гидрохлорида (0,120 г, 0,264 ммоль), триэтиламина (0,032 г, 0,317 ммоль) и уксусного ангидрида (0,037 мл, 0,396 ммоль) в дихлорметане (5 мл) перемешивали при комнатной температуре в течение 12 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида аммония, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; метанол/дихлорметан=0%-5%) с получением метил 4-((((3S,5R)-4-ацетил-3,5-диметил-N-(пиридин-3-ил)пиперазин)-1-сульфонамидо)метил)бензоата в виде желтого масла (0,081 г, 66,7%).
[Стадия 5] (3S,5R)-4-ацетил-N-(4-(гидразинкарбонил)бензил)-3,5-диметил-N-(пиридин-3-ил)пиперазин-1-сульфонамид
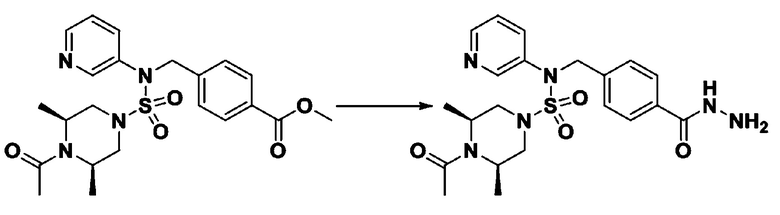
Раствор метил 4-((((3S,5R)-4-ацетил-3,5-диметил-N-(пиридин-3-ил)пиперазин)-1-сульфонамидо)метил)бензоата (0,081 г, 0,176 ммоль) и гидразина моногидрата (0,085 мл, 1,759 ммоль) в этаноле (10 мл) перемешивали при 90°C в течение 12 часов. Реакционную смесь охлаждали до комнатной температуры для прекращения реакции, и концентрировали при пониженном давлении для удаления растворителя. Остаток разбавляли водой (5 мл) и бикарбонатом натрия (10 мл) и перемешивали. Полученные осадки собирали фильтрованием, промывали водой и сушили с получением (3S,5R)-4-ацетил-N-(4-(гидразинкарбонил)бензил)-3,5-диметил-N-(пиридин-3-ил)пиперазин-1-сульфонамида в виде белого твердого вещества (0,039 г, 48,1%).
[Стадия 6] Соединение 11660
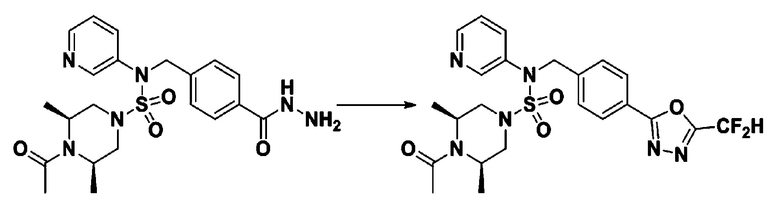
Раствор (3S,5R)-4-ацетил-N-(4-(гидразинкарбонил)бензил)-3,5-диметил-N-(пиридин-3-ил)пиперазин-1-сульфонамида (0,039 г, 0,085 ммоль), триэтиламина (0,043 г, 0,423 ммоль) и дифторуксусного ангидрида (0,044 г, 0,254 ммоль) в тетрагидрофуране (10 мл) перемешивали при 70°С в течение 12 часов, и охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида аммония, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; этилацетат/гексан=0%-60%) с получением (3S,5R)-4-ацетил-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-3,5-диметил-N-(пиридин-3-ил)пиперазин-1-сульфонамида в виде белого твердого вещества (0,020 г, 45,4%).
1H ЯМР (400 МГц, CDCl3) δ 8,69 (ушир.с, 1 H), 8,53-8,52 (м, 1 H), 8,06 (д, 2 H, J=8,1 Гц), 7,79-7,77 (м, 1 H), 7,46 (д, 2 H, J=7,6 Гц), 7,38-7,36 (м, 1 H), 7,05 (0,3 H), 6,92 (с, 0,5 H), 6,79 (с, 0,3 H), 5,02 (с, 2 H), 3,59-3,51 (м, 2 H), 3,50 (с, 2 H), 2,88-2,85 (м, 2 H), 2,10 (с, 3 H), 1,32-1,25 (м, 6 H); LRMS (ES) m/z 521,3 (M++1).
Пример 52: Соединение 11661, (3S,5R)-N-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4-этил-3,5-диметил-N-фенилпиперазин-1-сульфонамид
[Стадия 1] метил 6-((((3S,5R)-4-этил-3,5-диметил-N-фенилпиперазин)-1-сульфонамидо)метил)никотинат
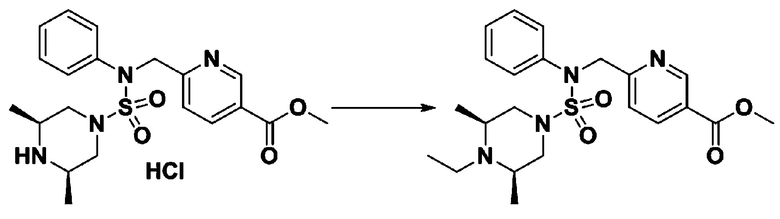
Раствор метил 6-((((3S,5R)-3,5-диметил-N-фенилпиперазин)-1-сульфонамидо)метил)никотинат гидрохлорида (0,200 г, 0,440 ммоль), ацетальдегида (0,039 г, 0,879 ммоль) и уксусной кислоты (0,028 мл, 0,484 ммоль) в дихлорметане (5 мл) перемешивали при комнатной температуре в течение 30 мин и смешивали с триацетоксиборгидридом натрия (0,186 г, 0,879 ммоль). Реакционную смесь перемешивали при той же температуре в течение дополнительных 12 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором бикарбоната натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; метанол/дихлорметан=0%-5%) с получением метил 6-((((3S,5R)-4-этил-3,5-диметил-N-фенилпиперазин)-1-сульфонамидо)метил)никотината в виде желтого масла (0,110 г, 56,0%).
[Стадия 2] (3S,5R)-4-этил-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-3,5-диметил-N-фенилпиперазин-1-сульфонамид
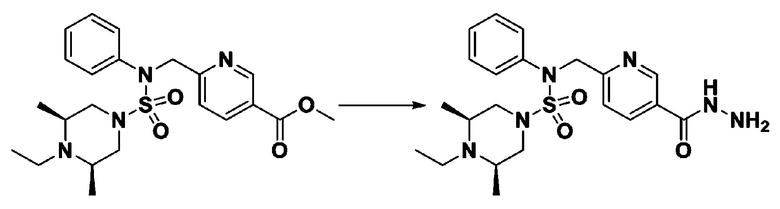
Раствор метил 6-((((3S,5R)-4-этил-3,5-диметил-N-фенилпиперазин)-1-сульфонамидо)метил)никотината (0,110 г, 0,246 ммоль) и гидразина моногидрата (0,120 мл, 2,463 ммоль) в этаноле (10 мл) перемешивали при 90°C в течение 12 часов, и охлаждали до комнатной температуры для прекращения реакции. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Остаток разбавляли водой (5 мл) и бикарбонатом натрия (10 мл) и перемешивали. Полученные осадки собирали фильтрованием и сушили с получением (3S,5R)-4-этил-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-3,5-диметил-N-фенилпиперазин-1-сульфонамида в виде белого твердого вещества (0,055 г, 50,0%).
[Стадия 3] Соединение 11661
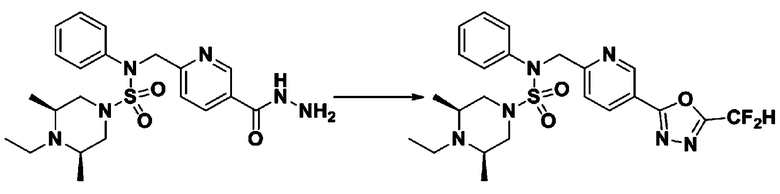
Раствор (3S,5R)-4-этил-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-3,5-диметил-N-фенилпиперазин-1-сульфонамида (0,055 г, 0,123 ммоль), триэтиламина (0,062 г, 0,616 ммоль) и дифторуксусного ангидрида (0,064 г, 0,369 ммоль) в тетрагидрофуране (10 мл) перемешивали при 70°С в течение 12 часов, и охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида аммония, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; этилацетат/гексан=0%-70%) с получением (3S,5R)-N-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4-этил-3,5-диметил-N-фенилпиперазин-1-сульфонамида в виде белого твердого вещества (0,021 г, 33,7%).
1H ЯМР (400 МГц, CDCl3) δ 9,21 (с, 1 H), 8,37-8,35 (м, 1 H), 7,72-7,71 (м, 1 H), 7,45-7,42 (м, 2 H), 7,36-7,31 (м, 2 H), 7,27-7,23 (м, 1 H), 7,07 (с, 0,3 H), 6,94 (с, 0,5 H), 6,81 (с, 0,3 H), 5,13 (с, 2 H), 3,55-3,53 (м, 2 H), 2,97-2,86 (м, 2 H), 2,60-2,46 (м, 3 H), 1,62 (ушир.с, 1 H), 1,19-1,01 (м, 6 H), 0,99-0,82 (м, 3 H); LRMS (ES) m/z 507,1 (M++1).
Пример 53: Соединение 11662, (3S,5R)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-3,4,5-триметил-N-(пиридин-3-ил)пиперазин-1-сульфонамид
[Стадия 1] метил 4-((((3S,5R)-3,4,5-триметил-N-(пиридин-3-ил)пиперазин)-1-сульфонамидо)метил)бензоат
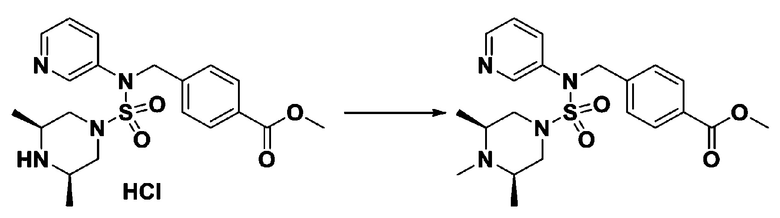
Раствор метил 4-((((3S,5R)-3,5-диметил-N-(пиридин-3-ил)пиперазин)-1-сульфонамидо)метил)бензоат гидрохлорида (0,120 г, 0,287 ммоль), параформальдегида (0,017 г, 0,573 ммоль), уксусной кислоты (0,020 мл, 0,344 ммоль) и триацетоксиборгидрида натрия (0,122 г, 0,573 ммоль) в дихлорметане (5 мл) перемешивали при комнатной температуре в течение 12 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором бикарбоната натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; метанол/дихлорметан=0%-5%) с получением метил 4-((((3S,5R)-3,4,5-триметил-N-(пиридин-3-ил)пиперазин)-1-сульфонамидо)метил)бензоата в виде бесцветного масла (0,095 г, 76,6%).
[Стадия 2] (3S,5R)-N-(4-(гидразинкарбонил)бензил)-3,4,5-триметил-N-(пиридин-3-ил)пиперазин-1-сульфонамид
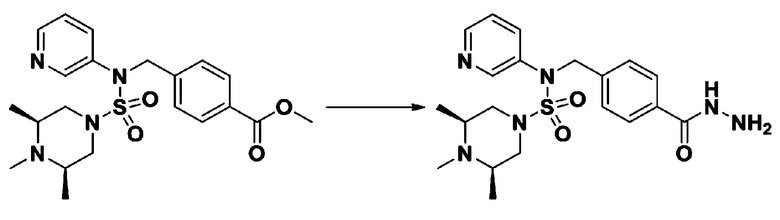
Раствор метил 4-((((3S,5R)-3,4,5-триметил-N-(пиридин-3-ил)пиперазин)-1-сульфонамидо)метил)бензоата (0,095 г, 0,220 ммоль) и гидразина моногидрата (0,107 мл, 2,196 ммоль) в этаноле (10 мл) перемешивали при 90°C в течение 12 часов, и охлаждали до комнатной температуры для прекращения реакции. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Остаток разбавляли водой (5 мл) и бикарбонатом натрия (10 мл), и перемешивали. Полученные осадки собирали фильтрованием, промывали водой и сушили с получением (3S,5R)-N-(4-(гидразинкарбонил)бензил)-3,4,5-триметил-N-(пиридин-3-ил)пиперазин-1-сульфонамида в виде белого твердого вещества (0,048 г, 50,5%).
[Стадия 3] Соединение 11662
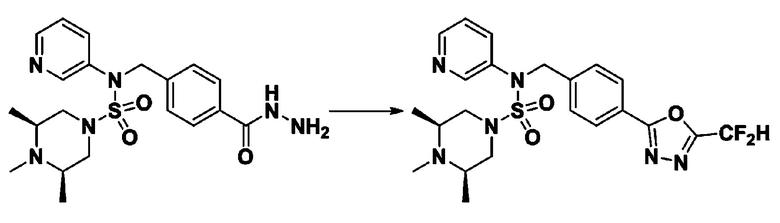
Раствор (3S,5R)-N-(4-(гидразинкарбонил)бензил)-3,4,5-триметил-N-(пиридин-3-ил)пиперазин-1-сульфонамида (0,048 г, 0,111 ммоль), триэтиламина (0,077 мл, 0,555 ммоль) и 2,2-дифторуксусного ангидрида (0,041 мл, 0,333 ммоль) в тетрагидрофуране (10 мл) перемешивали при 70°С в течение 12 часов, и охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида аммония, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; этилацетат/гексан=0%-70%) с получением (3S,5R)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-3,4,5-триметил-N-(пиридин-3-ил)пиперазин-1-сульфонамида в виде белого твердого вещества (0,019 г, 34,8%).
1H ЯМР (400 МГц, CDCl3) δ 8,53 (д, 1 H, J=2,2 Гц), 8,51-8,50 (м, 1 H), 8,06-8,03 (м, 2 H), 7,66-7,63 (м, 1 H), 7,44-7,42 (м, 2 H), 7,30-7,26 (м, 1 H), 7,05 (с, 0,2 H), 6,92 (с, 0,5 H), 6,79 (с, 0,2 H), 4,93 (с, 2 H), 3,56-3,53 (м, 2 H), 2,85 (ушир.с, 2 H), 2,46-2,32 (м, 5 H), 1,29-1,23 (м, 6 H); LRMS (ES) m/z 493,1(M++1).
Пример 54: Соединение 11670, N-(3-хлор-4-фторфенил)-N-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4-изопропилпиперазин-1-сульфонамид
[Стадия 1] трет-бутил 4-(N-(3-хлор-4-фторфенил)-N-((5-(метоксикарбонил)пиридин-2-ил)метил)сульфамоил)пиперазин-1-карбоксилат
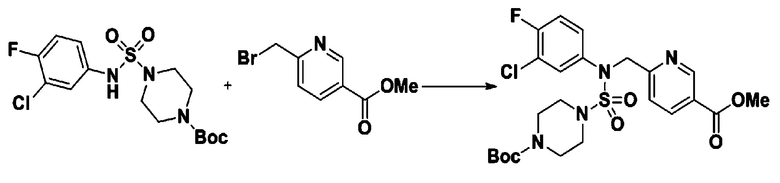
Раствор трет-бутил 4-(N-(3-хлор-4-фторфенил)сульфамоил)пиперазин-1-карбоксилата (1,800 г, 4,570 ммоль) и гидрида натрия (60,00%, 0,548 г, 13,710 ммоль) в N,N-диметилформамиде (50 мл) перемешивали при комнатной температуре в течение 10 минут, и перемешивали с метил 6-(бромметил)никотинатом (1,262 г, 5,484 ммоль). Реакционную смесь перемешивали при 50°С в течение дополнительных 18 часов, и охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 40 г; этилацетат/гексан=0%-80%) с получением трет-бутил 4-(N-(3-хлор-4-фторфенил)-N-((5-(метоксикарбонил)пиридин-2-ил)метил)сульфамоил)пиперазин-1-карбоксилата в виде белого твердого вещества (1,140 г, 45,9%).
[Стадия 2] метил 6-((N-(3-хлор-4-фторфенил)пиперазин-1-сульфонамидо)метил)никотинат гидрохлорид
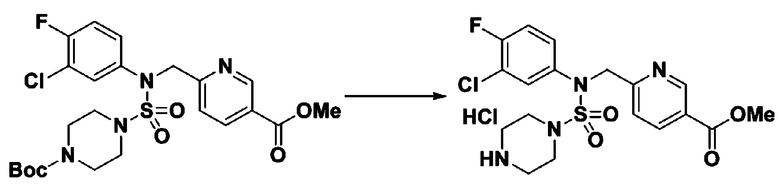
Раствор трет-бутил 4-(N-(3-хлор-4-фторфенил)-N-((5-(метоксикарбонил)пиридин-2-ил)метил)сульфамоил)пиперазин-1-карбоксилата (1,140 г, 2,099 ммоль) в дихлорметане (80 мл) смешивали при комнатной температуре с хлористоводородной кислотой (4,00 M раствор в диоксане, 2,099 мл, 8,398 ммоль). Реакционную смесь перемешивали при той же температуре в течение 18 часов, и концентрировали при пониженном давлении для удаления растворителя. Указанное в заголовке соединение использовали без дополнительной очистки (метил 6-((N-(3-хлор-4-фторфенил)пиперазин-1-сульфонамидо)метил)никотинат гидрохлорид, 0,980 г, 97,4%, красное твердое вещество).
[Стадия 3] метил 6-(((N-(3-хлор-4-фторфенил)-4-изопропилпиперазин)-1-сульфонамидо)метил)никотинат
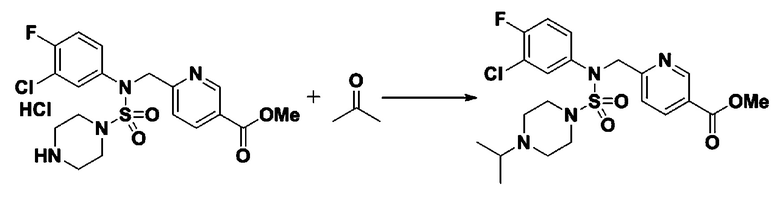
Раствор метил 6-((N-(3-хлор-4-фторфенил)пиперазин-1-сульфонамидо)метил)никотинат гидрохлорида (0,150 г, 0,313 ммоль) и триацетоксиборгидрид натрия (0,133 г, 0,626 ммоль) в дихлорметане (10 мл) смешивали при комнатной температуре с пропан-2-оном (0,034 мл, 0,469 ммоль). Реакционную смесь перемешивали при той же температуре в течение 18 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 12 г; метанол/дихлорметан=0%-15%) с получением метил 6-(((N-(3-хлор-4-фторфенил)-4-изопропилпиперазин)-1-сульфонамидо)метил)никотината в виде желтого твердого вещества (0,112 г, 73,8%).
[Стадия 4] N-(3-хлор-4-фторфенил)-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-4-изопропилпиперазин-1-сульфонамид
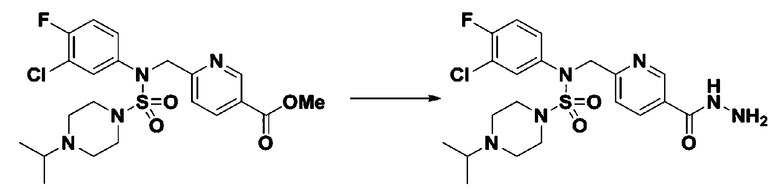
метил 6-(((N-(3-хлор-4-фторфенил)-4-изопропилпиперазин)-1-сульфонамидо)метил)никотинат (0,153 г, 0,315 ммоль) и гидразин моногидрат (0,460 мл, 9,465 ммоль) смешивали при комнатной температуре в этаноле (8 мл)/воде (2 мл), и затем смесь перемешивали при 80°C в течение 18 часов. Реакционную смесь охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Указанное в заголовке соединение использовали без дополнительной очистки (N-(3-хлор-4-фторфенил)-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-4-изопропилпиперазин-1-сульфонамид, 0,147 г, 96,1%, белое твердое вещество).
[Стадия 5] Соединение 11670
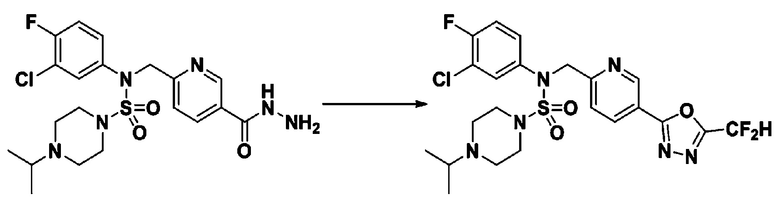
Раствор N-(3-хлор-4-фторфенил)-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-4-изопропилпиперазин-1-сульфонамида (0,147 г, 0,303 ммоль) и триэтиламина (0,084 мл, 0,606 ммоль) в тетрагидрофуране (15 мл) смешивали при комнатной температуре с 2,2-дифторуксусным ангидридом (0,113 мл, 0,909 ммоль), и перемешивали при 80°С в течение 1 часа. Реакционную смесь охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 12 г; метанол/дихлорметан=0%-30%) с получением N-(3-хлор-4-фторфенил)-N-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4-изопропилпиперазин-1-сульфонамида в виде бесцветного масла (0,085 г, 51,5%).
1H ЯМР (400 МГц, CDCl3) δ 9,24 (с, 1 H), 8,37 (дд, 1 H, J=8,2, 2,2 Гц), 7,62 ~ 7,60 (м, 1 H), 7,55 ~ 7,52 (м, 1 H), 7,35 ~ 7,31 (м, 1 H), 7,11 ~ 7,06 (м, 1 H), 7,05 (с, 0,25 H), 6,92 (с, 0,5 H), 6,79 (с, 0,25 H), 5,01 (с, 2 H), 3,43 ~ 3,24 (м, 4 H), 2,67 ~ 2,47 (м, 4 H), 1,62 ~ 1,59 (м, 2 H), 1,10 ~ 0,97 (м, 5 H); LRMS (ES) m/z 545,2 (M++1).
Пример 55: Соединение 11671, N-(3-хлор-4-фторфенил)-4-циклобутил-N-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)пиперазин-1-сульфонамид
[Стадия 1] метил 6-(((N-(3-хлор-4-фторфенил)-4-циклобутилпиперазин)-1-сульфонамидо)метил)никотинат
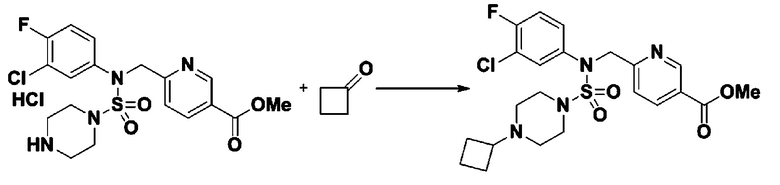
Раствор метил 6-((N-(3-хлор-4-фторфенил)пиперазин-1-сульфонамидо)метил)никотинат гидрохлорида (0,150 г, 0,313 ммоль) и циклобутанона (0,035 мл, 0,469 ммоль) в дихлорметане (10 мл) смешивали при комнатной температуре с триацетоксиборгидридом натрия (0,133 г, 0,626 ммоль). Реакционную смесь перемешивали при той же температуре в течение 18 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 12 г; метанол/дихлорметан=0%-15%) с получением метил 6-(((N-(3-хлор-4-фторфенил)-4-циклобутилпиперазин)-1-сульфонамидо)метил)никотината в виде желтого твердого вещества (0,093 г, 59,8%).
[Стадия 2] N-(3-хлор-4-фторфенил)-4-циклобутил-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)пиперазин-1-сульфонамид
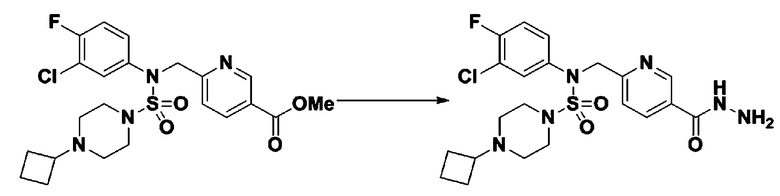
метил 6-(((N-(3-хлор-4-фторфенил)-4-циклобутилпиперазин)-1-сульфонамидо)метил)никотинат (0,100 г, 0,201 ммоль) и гидразин моногидрат (0,293 мл, 6,036 ммоль) смешивали при комнатной температуре в этаноле (8 мл)/воде (2 мл), и затем смесь перемешивали при 80°C в течение 18 часов. Реакционную смесь охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Неочищенный продукт использовали без дополнительной очистки (N-(3-хлор-4-фторфенил)-4-циклобутил-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)пиперазин-1-сульфонамид, 0,098 г, 98,0%, белое твердое вещество).
[Стадия 3] Соединение 11671
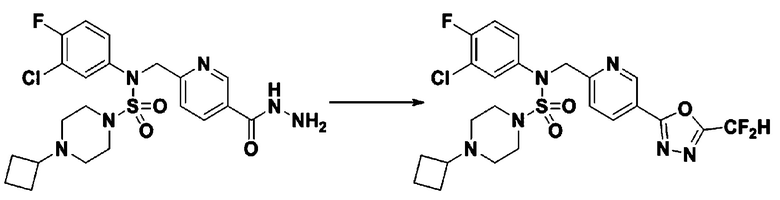
Раствор N-(3-хлор-4-фторфенил)-4-циклобутил-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)пиперазин-1-сульфонамида (0,100 г, 0,201 ммоль) и триэтиламина (0,056 мл, 0,402 ммоль) в тетрагидрофуране (15 мл) смешивали при комнатной температуре с 2,2-дифторуксусным ангидридом (0,075 мл, 0,604 ммоль), и перемешивали при 80°С в течение 1 часа. Реакционную смесь охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 12 г; метанол/дихлорметан=0%-15%) с получением N-(3-хлор-4-фторфенил)-4-циклобутил-N-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)пиперазин-1-сульфонамида в виде белого твердого вещества (0,038 г, 33,9%).
1H ЯМР (400 МГц, CDCl3) δ 9,24 (с, 1 H), 8,37 (дд, 1 H, J=8,2, 2,2 Гц), 7,60 ~ 7,58 (м, 1 H), 7,54 ~ 7,52 (м, 1 H), 7,35 ~ 7,31 (м, 1 H), 7,26 ~ 7,07 (м, 1 H), 7,05 (с, 0,25 H), 6,92 (с, 0,5 H), 6,80 (с, 0,25 H), 5,00 (с, 2 H), 3,50 ~ 3,15 (м, 4 H), 2,78 ~ 2,75 (м, 1 H), 2,53 ~ 2,04 (м, 6 H), 1,93 ~ 1,59 (м, 4 H); LRMS (ES) m/z 557,3 (M++1).
Пример 56: Соединение 11672, N-(3-хлор-4-фторфенил)-N-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4-(оксетан-3-ил)пиперазин-1-сульфонамид
[Стадия 1] метил 6-(((N-(3-хлор-4-фторфенил)-4-(оксетан-3-ил)пиперазин)-1-сульфонамидо)метил)никотинат
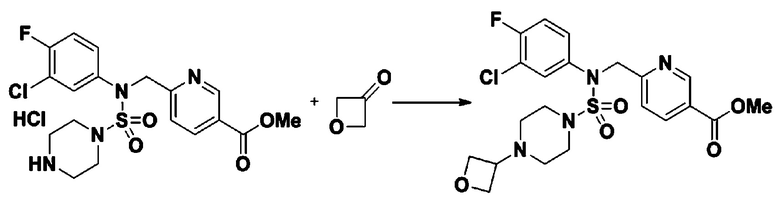
Раствор метил 6-((N-(3-хлор-4-фторфенил)пиперазин-1-сульфонамидо)метил)никотинат гидрохлорида (0,150 г, 0,313 ммоль) и оксетан-3-она (0,030 мл, 0,469 ммоль) в дихлорметане (10 мл) смешивали при комнатной температуре с триацетоксиборгидридом натрия (0,133 г, 0,626 ммоль). Реакционную смесь перемешивали при той же температуре в течение 18 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 12 г; метанол/дихлорметан=0%-15%) с получением метил 6-(((N-(3-хлор-4-фторфенил)-4-(оксетан-3-ил)пиперазин)-1-сульфонамидо)метил)никотината в виде желтого твердого вещества (0,154 г, 98,6%).
[Стадия 2] N-(3-хлор-4-фторфенил)-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-4-(оксетан-3-ил)пиперазин-1-сульфонамид
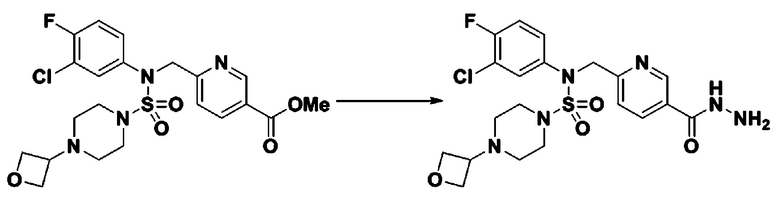
метил 6-(((N-(3-хлор-4-фторфенил)-4-(оксетан-3-ил)пиперазин)-1-сульфонамидо)метил)никотинат (0,154 г, 0,309 ммоль) и гидразин моногидрат (0,450 мл, 9,259 ммоль) смешивали при комнатной температуре в этаноле (8 мл)/воде (2 мл), и затем смесь перемешивали при 80°C в течение 18 часов. Реакционную смесь охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Указанное в заголовке соединение использовали без дополнительной очистки (N-(3-хлор-4-фторфенил)-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-4-(оксетан-3-ил)пиперазин-1-сульфонамид, 0,053 г, 34,4%, белое твердое вещество).
[Стадия 3] Соединение 11672
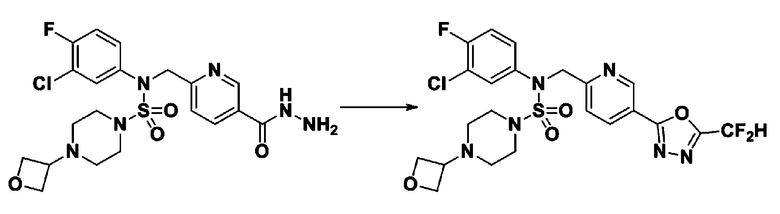
Раствор N-(3-хлор-4-фторфенил)-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-4-(оксетан-3-ил)пиперазин-1-сульфонамида (0,053 г, 0,106 ммоль) и триэтиламина (0,030 мл, 0,212 ммоль) в тетрагидрофуране (15 мл) смешивали при комнатной температуре с 2,2-дифторуксусным ангидридом (0,040 мл, 0,319 ммоль), и перемешивали при 80°С в течение 1 часа. Реакционную смесь охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 12 г; метанол/дихлорметан=0%-15%) с получением N-(3-хлор-4-фторфенил)-N-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4-(оксетан-3-ил)пиперазин-1-сульфонамида в виде белого твердого вещества (0,032 г, 53,9%).
1H ЯМР (400 МГц, CDCl3) δ 9,23~9,22 (м, 1 H), 8,36 (дд, 1 H, J=8,2, 2,2 Гц), 7,61~7,59 (м, 1 H), 7,54~7,52 (м, 1 H), 7,34~7,30 (м, 1 H), 7,13~7,06 (м, 1 H), 7,05 (с, 0,25 H), 6,92 (с, 0,5 H), 6,80 (с, 0,25 H), 5,01 (с, 2 H), 4,65~4,62 (м, 2 H), 4,56~4,53 (м, 2 H), 3,50~3,42 (м, 1 H), 3,40~3,20 (м, 4 H), 2,40~2,20 (м, 4 H); LRMS (ES) m/z 559,2 (M++1).
Пример 57: Соединение 11673, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-4-изопропил-N-фенилпиперазин-1-сульфонамид
[Стадия 1] метил 4-(((4-изопропил-N-фенилпиперазин)-1-сульфонамидо)метил)бензоат
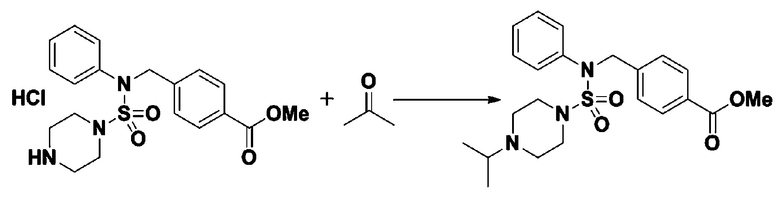
Раствор метил 4-((N-фенилпиперазин-1-сульфонамидо)метил)бензоат гидрохлорида (0,150 г, 0,352 ммоль) и пропан-2-она (0,039 мл, 0,528 ммоль) в дихлорметане (10 мл) смешивали при комнатной температуре с триацетоксиборгидридом натрия (0,149 г, 0,704 ммоль). Реакционную смесь перемешивали при той же температуре в течение 18 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 12 г; метанол/дихлорметан=0%-15%) с получением метил 4-(((4-изопропил-N-фенилпиперазин)-1-сульфонамидо)метил)бензоата в виде белого твердого вещества (0,142 г, 93,4%).
[Стадия 2] N-(4-(гидразинкарбонил)бензил)-4-изопропил-N-фенилпиперазин-1-сульфонамид
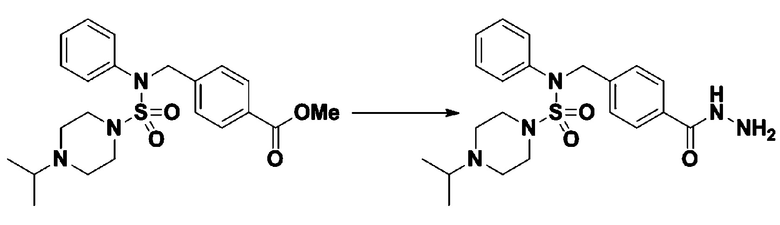
метил 4-(((4-изопропил-N-фенилпиперазин)-1-сульфонамидо)метил)бензоата (0,149 г, 0,345 ммоль) и гидразин моногидрат (0,503 мл, 10,358 ммоль) смешивали при комнатной температуре в этаноле (8 мл)/воде (2 мл), и затем смесь перемешивали при 80°C в течение 18 часов. Реакционную смесь охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Указанное в заголовке соединение использовали без дополнительной очистки (N-(4-(гидразинкарбонил)бензил)-4-изопропил-N-фенилпиперазин-1-сульфонамид, 0,135 г, 90,6%, белое твердое вещество).
[Стадия 3] Соединение 11673
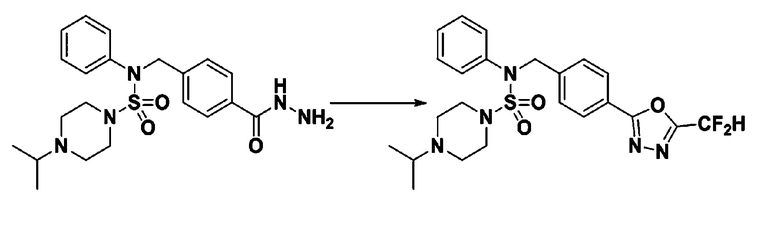
Раствор N-(4-(гидразинкарбонил)бензил)-4-изопропил-N-фенилпиперазин-1-сульфонамида (0,135 г, 0,313 ммоль) и триэтиламина (0,087 мл, 0,626 ммоль) в тетрагидрофуране (15 мл) смешивали при комнатной температуре с 2,2-дифторуксусным ангидридом (0,117 мл, 0,938 ммоль), и перемешивали при 80°С в течение 1 часа. Реакционную смесь охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 12 г; метанол/дихлорметан=0%-15%) с получением N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-4-изопропил-N-фенилпиперазин-1-сульфонамида в виде белого твердого вещества (0,108 г, 70,2%).
1H ЯМР (400 МГц, CDCl3) δ 8,01 ~ 7,99 (м, 2 H), 7,42 ~ 7,40 (м, 2 H), 7,30 ~ 7,22 (м, 5 H), 7,02 (с, 0,25 H), 6,89 (с, 0,5 H), 6,76 (с, 0,25 H), 4,89 (с, 2 H), 3,50 ~ 3,10 (м, 4 H), 3,00 ~ 2,20 (м, 5 H), 1,80 ~ 1,40 (м, 1 H),1,20 ~ 0,82 (м, 5 H); LRMS (ES) m/z 492,3 (M++1).
Пример 58: Соединение 11674, 4-циклобутил-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенилпиперазин-1-сульфонамид
[Стадия 1] метил 4-(((4-циклобутил-N-фенилпиперазин)-1-сульфонамидо)метил)бензоат
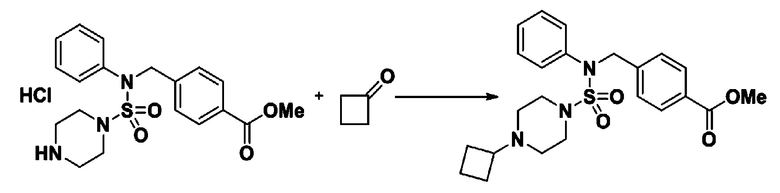
Раствор метил 4-((N-фенилпиперазин-1-сульфонамидо)метил)бензоат гидрохлорида (0,150 г, 0,352 ммоль) и циклобутанона (0,039 мл, 0,528 ммоль) в дихлорметане (15 мл) смешивали при комнатной температуре с триацетоксиборгидридом натрия (0,149 г, 0,704 ммоль). Реакционную смесь перемешивали при той же температуре в течение 18 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 12 г; метанол/дихлорметан=0%-15%) с получением метил 4-(((4-циклобутил-N-фенилпиперазин)-1-сульфонамидо)метил)бензоата в виде белого твердого вещества (0,154 г, 98,6%).
[Стадия 2] 4-циклобутил-N-(4-(гидразинкарбонил)бензил)-N-фенилпиперазин-1-сульфонамид
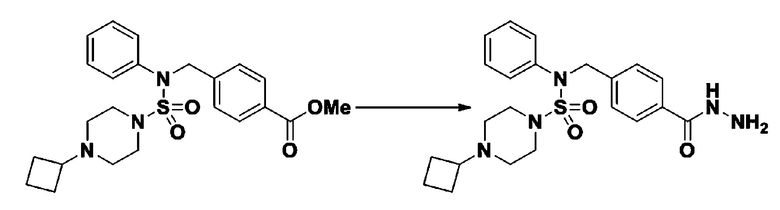
метил 4-(((4-циклобутил-N-фенилпиперазин)-1-сульфонамидо)метил)бензоата (0,154 г, 0,347 ммоль) и гидразин моногидрат (0,506 мл, 10,416 ммоль) смешивали при комнатной температуре в этаноле (8 мл)/воде (2 мл), и затем смесь перемешивали при 80°C в течение 18 часов. Реакционную смесь охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Указанное в заголовке соединение использовали без дополнительной очистки (4-циклобутил-N-(4-(гидразинкарбонил)бензил)-N-фенилпиперазин-1-сульфонамид, 0,140 г, 90,9%, белое твердое вещество).
[Стадия 3] Соединение 11674
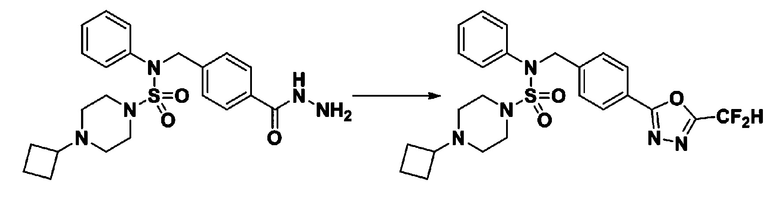
Раствор 4-циклобутил-N-(4-(гидразинкарбонил)бензил)-N-фенилпиперазин-1-сульфонамида (0,140 г, 0,316 ммоль) и триэтиламина (0,088 мл, 0,631 ммоль) в тетрагидрофуране (15 мл) смешивали при комнатной температуре с 2,2-дифторуксусным ангидридом (0,118 мл, 0,947 ммоль), и перемешивали при 80°С в течение 1 часа. Реакционную смесь охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 12 г; метанол/дихлорметан=0%-15%) с получением 4-циклобутил-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенилпиперазин-1-сульфонамида в виде белого твердого вещества (0,085 г, 53,5%).
1H ЯМР (400 МГц, CDCl3) δ 8,01 ~ 7,99 (м, 2 H), 7,41 ~ 7,39 (м, 2 H), 7,32 ~ 7,23 (м, 5 H), 7,02 (с, 0,25 H), 6,89 (с, 0,5 H), 6,76 (с, 0,25 H), 4,88 (с, 2 H), 3,71 ~ 3,08 (м, 4 H), 2,40 ~ 2,20 (м, 3 H), 2,20 ~ 2,00 (м, 3 H), 2,00 ~ 1,40 (м, 5 H); LRMS (ES) m/z 503,9 (M++1).
Пример 59: Соединение 11702, 2-((4-(((N-метил-N-(2-(4-метилпиперазин-1-ил)этил)сульфамоил)(фенил)амино)метил))фенил)-5-(дифторметил)-1,3,4-оксадиазол
[Стадия 1] трет-бутил 4-(2-гидроксиэтил)пиперазин-1-карбоксилат
Раствор 2-(пиперазин-1-ил)этан-1-ола (0,943 мл, 7,681 ммоль) и ди-трет-бутилдикарбоната (1,760 г, 8,065 ммоль) в тетрагидрофуране (5 мл) перемешивали при комнатной температуре в течение 18 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 24 г; метанол/дихлорметан=0%-10%) с получением трет-бутил 4-(2-гидроксиэтил)пиперазин-1-карбоксилата в виде бесцветного масла (1,750 г, 98,9%).
[Стадия 2] трет-бутил 4-(2-((метилсульфонил)окси)этил)пиперазин-1-карбоксилат
Раствор трет-бутил 4-(2-гидроксиэтил)пиперазин-1-карбоксилата (1,750 г, 7,598 ммоль), метансульфонилхлорида (0,706 мл, 9,118 ммоль) и триэтиламина (1,589 мл, 11,398 ммоль) в дихлорметане (20 мл) перемешивали при комнатной температуре в течение 18 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 24 г; метанол/дихлорметан=0%-10%) с получением трет-бутил 4-(2-((метилсульфонил)окси)этил)пиперазин-1-карбоксилата в виде белого твердого вещества (1,850 г, 78,9%).
[Стадия 3] метил 4-((фениламино)метил)бензоат
Раствор анилина (4,902 мл, 53,688 ммоль), метил 4-(бромметил)бензоата (13,528 г, 59,057 ммоль), йодида калия (4,456 г, 26,844 ммоль) и карбоната калия (11,130 г, 80,533 ммоль) в N,N-диметилформамиде (50 мл) перемешивали при комнатной температуре в течение 18 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 80 г; этилацетат/гексан=0%-25%) с получением метил 4-((фениламино)метил)бензоата в виде желтой жидкости (7,800 г, 60,2%).
[Стадия 4] метил 4-(((N-(трет-бутоксикарбонил)сульфамоил)(фенил)амино)метил)бензоат
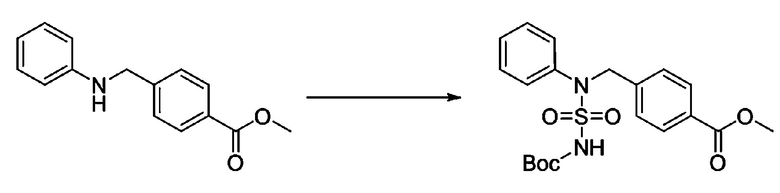
Раствор хлорсульфонилизоцианата (0,704 г, 4,973 ммоль) и трет-бутанола (0,456 мл, 4,766 ммоль) в дихлорметане (20 мл) перемешивали при 0°C в течение 1 часа, и смешивали с метил 4-((фениламино)метил)бензоата (1,000 г, 4,144 ммоль) и триэтиламина (0,866 мл, 6,217 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение дополнительных 18 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 40 г; этилацетат/гексан=0%-30%) с получением метил 4-(((N-(трет-бутоксикарбонил)сульфамоил)(фенил)амино)метил)бензоата в виде белого твердого вещества (1,190 г, 68,3%).
[Стадия 5] метил 4-(((N-(трет-бутоксикарбонил)-N-метилсульфамоил)(фенил)амино)метил)бензоат
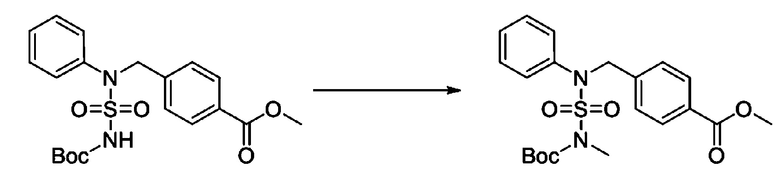
К раствору метил 4-(((N-(трет-бутоксикарбонил)сульфамоил)(фенил)амино)метил)бензоата (1,400 г, 3,330 ммоль) в N,N-диметилформамиде (5 мл) медленно добавляли гидрид натрия (60,00%, 0,200 г, 4,994 ммоль) при 0°C, и смесь перемешивали в течение 0,5 часа. Реакционную смесь обрабатывали иодметаном (0,311 мл, 4,994 ммоль), и перемешивали при комнатной температуре в течение дополнительных 3 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 40 г; этилацетат/гексан=0%-30%) с получением метил 4-(((N-(трет-бутоксикарбонил)-N-метилсульфамоил)(фенил)амино)метил)бензоата в виде белого твердого вещества (1,095 г, 75,7%).
[Стадия 6] метил 4-(((N-метилсульфамоил)(фенил)амино)метил)бензоат
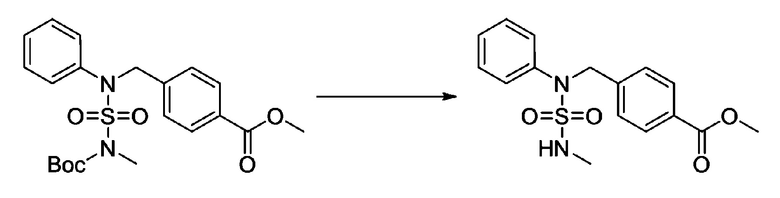
Раствор метил 4-(((N-(трет-бутоксикарбонил)-N-метилсульфамоил)(фенил)амино)метил)бензоата (1,095 г, 2,520 ммоль) и хлористоводородной кислоты (4,00 M раствор 1,4-диоксан, 6,300 мл, 25,201 ммоль) в 1,4-диоксане (3 мл) перемешивали при комнатной температуре в течение 18 часов, и концентрировали при пониженном давлении для удаления растворителя. Остаток разбавляли диэтиловым эфиром (20 мл), и перемешивали. Полученные осадки собирали фильтрованием и сушили с получением метил 4-(((N-метилсульфамоил)(фенил)амино)метил)бензоата в виде белого твердого вещества (0,711 г, 84,4%).
[Стадия 7] трет-бутил 4-(2-((N-(4-(метоксикарбонил)бензил)-N-фенилсульфамоил)(метил)амино)этил)пиперазин-1-карбоксилат
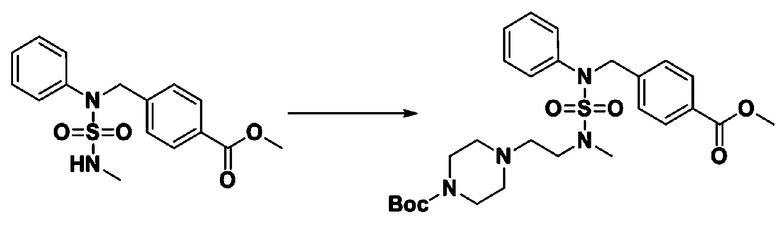
Раствор метил 4-(((N-метилсульфамоил)(фенил)амино)метил)бензоата (0,711 г, 2,126 ммоль), трет-бутил 4-(2-((метилсульфонил)окси)этил)пиперазин-1-карбоксилаты (0,787 г, 2,552 ммоль) и гидрида натрия (60,00%, 0,102 г, 2,552 ммоль) в N,N-диметилформамиде (5 мл) перемешивали при комнатной температуре в течение 18 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 40 г; этилацетат/гексан=0%-80%) с получением трет-бутил 4-(2-((N-(4-(метоксикарбонил)бензил)-N-фенилсульфамоил)(метил)амино)этил)пиперазин-1-карбоксилата в виде бесцветной жидкости (0,732 г, 62,9%).
[Стадия 8] метил 4-(((N-метил-N-(2-(пиперазин-1-ил)этил)сульфамоил)(фенил)амино)метил)бензоат гидрохлорид
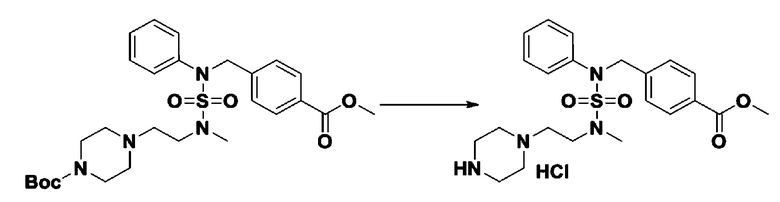
Раствор трет-бутил 4-(2-((N-(4-(метоксикарбонил)бензил)-N-фенилсульфамоил)(метил)амино)этил)пиперазин-1-карбоксилата (0,732 г, 1,338 ммоль) и хлористоводородной кислоты (4,00 M раствор 1,4-диоксан, 3,345 мл, 13,381 ммоль) в 1,4-диоксане (10 мл) перемешивали при комнатной температуре в течение 18 часов, и концентрировали при пониженном давлении для удаления растворителя. Остаток разбавляли диэтиловым эфиром (20 мл) и перемешивали. Полученные осадки собирали фильтрованием и сушили с получением метил 4-(((N-метил-N-(2-(пиперазин-1-ил)этил)сульфамоил)(фенил)амино)метил)бензоат гидрохлорида в виде твердого вещества цвета слоновой кости (0,480 г, 74,3%).
[Стадия 9] метил 4-(((N-метил-N-(2-(4-метилпиперазин-1-ил)этил)сульфамоил)(фенил)амино)метил)бензоат
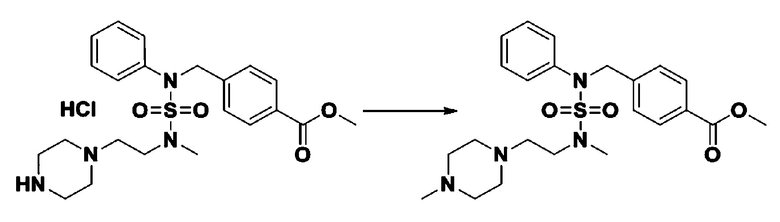
Раствор метил 4-(((N-метил-N-(2-(пиперазин-1-ил)этил)сульфамоил)(фенил)амино)метил)бензоат гидрохлорида (0,080 г, 0,166 ммоль) и параформальдегида (0,010 г, 0,331 ммоль) в дихлорметане (5 мл) перемешивали при комнатной температуре в течение 0,5 часа, и смешивали с триацетоксиборгидридом натрия (0,070 г, 0,331 ммоль). Реакционную смесь перемешивали при той же температуре в течение дополнительных 18 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 12 г; метанол/дихлорметан=0%-10%) с получением метил 4-(((N-метил-N-(2-(4-метилпиперазин-1-ил)этил)сульфамоил)(фенил)амино)метил)бензоата в виде бесцветного масла (0,054 г, 70,8%).
[Стадия 10] 4-(((N-метил-N-(2-(4-метилпиперазин-1-ил)этил)сульфамоил)(фенил)амино)метил)бензогидразид
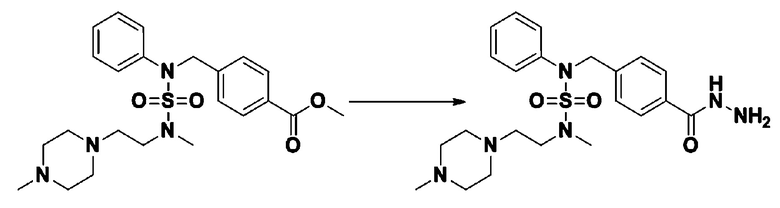
Раствор метил 4-(((N-метил-N-(2-(4-метилпиперазин-1-ил)этил)сульфамоил)(фенил)амино)метил)бензоата (0,054 г, 0,117 ммоль) и гидразина моногидрата (0,057 мл, 1,172 ммоль) в этаноле (3 мл) перемешивали при 80°С в течение 18 часов, и охлаждали до комнатной температуры для прекращения реакции. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Остаток разбавляли бикарбонатом натрия (5 мл) и перемешивали. Полученные осадки собирали фильтрованием и сушили с получением 4-(((N-метил-N-(2-(4-метилпиперазин-1-ил)этил)сульфамоил)(фенил)амино)метил)бензогидразида в виде белого твердого вещества (0,025 г, 46,3%).
[Стадия 11] Соединение 11702
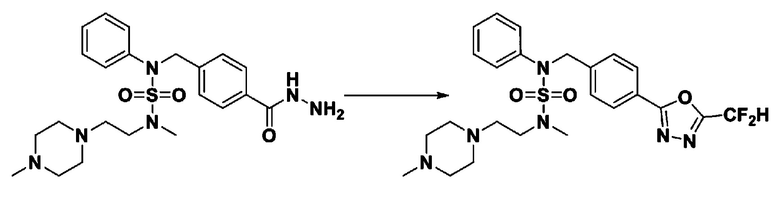
Раствор 4-(((N-метил-N-(2-(4-метилпиперазин-1-ил)этил)сульфамоил)(фенил)амино)метил)бензогидразида (0,025 г, 0,054 ммоль), 2,2-дифторуксусного ангидрида (0,067 мл, 0,543 ммоль) и триэтиламина (0,038 мл, 0,271 ммоль) в тетрагидрофуране (5 мл) перемешивали при 80°С в течение 1 часа, и охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией дихлорметаном. Двухфазную смесь пропускали через пластиковую фритту, для удаления твердого остатка и водного слоя, и собранный органический слой концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; метанол/дихлорметан=0%-10%) с получением 2-((4-(((N-метил-N-(2-(4-метилпиперазин-1-ил)этил)сульфамоил)(фенил)амино)метил))фенил)-5-(дифторметил)-1,3,4-оксадиазола в виде бесцветного масла (0,023 г, 81,4%).
1H ЯМР (400 МГц, CD3OD) δ8,01 (д, 2H, J=8,4 Гц), 7,53 (д, 2H,J=8,4 Гц), 7,54 ~ 7,25 (м, 6H), 7,22 (т, 1H, J=51,5 Гц), 5,00 (с, 2H), 3,23 (т, 2H, J=6,9 Гц), 2,88 (с, 3H), 2,47 ~ 2,43 (м, 10H), 2,27 (с, 3H); LRMS (ES) m/z 521,4 (M++1)
Пример 60: Соединение 11704, 2-((4-(((N-метил-N-(2-(4-(оксетан-3-ил)пиперазин-1-ил)этил)сульфамоил)(фенил)амино)метил))фенил)-5-(дифторметил)-1,3,4-оксадиазол
[Стадия 1] метил 4-(((N-метил-N-(2-(4-(оксетан-3-ил)пиперазин-1-ил)этил)сульфамоил)(фенил)амино)метил)бензоат
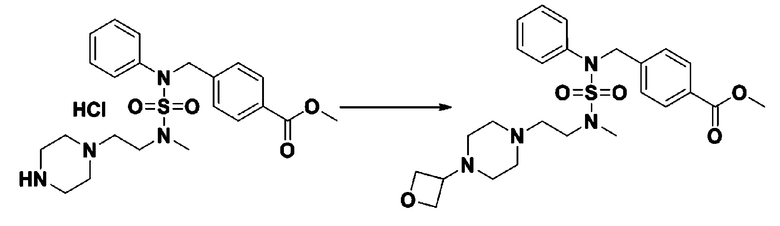
Раствор метил 4-(((N-метил-N-(2-(пиперазин-1-ил)этил)сульфамоил)(фенил)амино)метил)бензоат гидрохлорида (0,080 г, 0,166 ммоль) и оксетан-3-она (0,024 г, 0,331 ммоль) в дихлорметане (5 мл) перемешивали при комнатной температуре в течение 0,5 часа, и смешивали с триацетоксиборгидридом натрия (0,070 г, 0,331 ммоль). Реакционную смесь перемешивали при той же температуре в течение дополнительных 18 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 12 г; метанол/дихлорметан=0%-10%) с получением метил 4-(((N-метил-N-(2-(4-(оксетан-3-ил)пиперазин-1-ил)этил)сульфамоил)(фенил)амино)метил)бензоата в виде бесцветной жидкости (0,075 г, 90,6%).
[Стадия 2] 4-(((N-метил-N-(2-(4-(оксетан-3-ил)пиперазин-1-ил)этил)сульфамоил)(фенил)амино)метил)бензогидразид
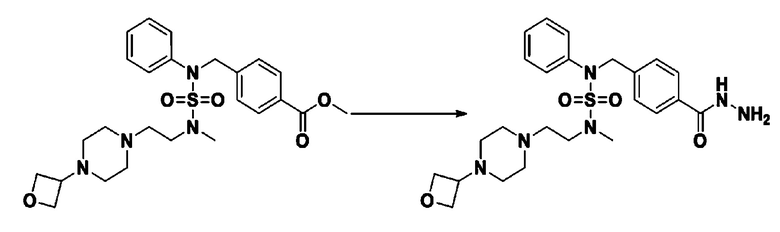
Раствор метил 4-(((N-метил-N-(2-(4-(оксетан-3-ил)пиперазин-1-ил)этил)сульфамоил)(фенил)амино)метил)бензоата (0,075 г, 0,150 ммоль) и гидразина моногидрата (0,073 мл, 1,500 ммоль) в этаноле (3 мл) перемешивали при 80°С в течение 18 часов, и охлаждали до комнатной температуры для прекращения реакции. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Неочищенный продукт использовали без дополнительной очистки (4-(((N-метил-N-(2-(4-(оксетан-3-ил)пиперазин-1-ил)этил)сульфамоил)(фенил)амино)метил)бензогидразид, 0,071 г, 94,2%, бесцветная жидкость).
[Стадия 3] Соединение 11704
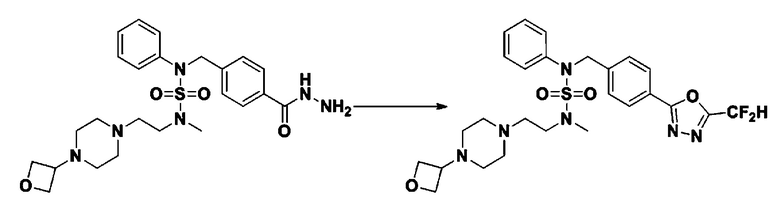
Раствор 4-(((N-метил-N-(2-(4-(оксетан-3-ил)пиперазин-1-ил)этил)сульфамоил)(фенил)амино)метил)бензогидразида (0,071 г, 0,141 ммоль), 2,2-дифторуксусного ангидрида (0,176 мл, 1,413 ммоль) и триэтиламина (0,098 мл, 0,706 ммоль) в тетрагидрофуране (5 мл) перемешивали при 80°С в течение 1 часа, и охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией дихлорметаном. Двухфазную смесь пропускали через пластиковую фритту, для удаления твердого остатка и водного слоя, и собранный органический слой концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; метанол/дихлорметан=0%-10%) с получением 2-((4-(((N-метил-N-(2-(4-(оксетан-3-ил)пиперазин-1-ил)этил)сульфамоил)(фенил)амино)метил))фенил)-5-(дифторметил)-1,3,4-оксадиазола в виде желтой жидкости (0,031 г, 39,0%).
1H ЯМР (400 МГц, CD3OD) δ 8,01 (дт, 1H, J=8,6, 1,8 Гц), 7,53 (дт, 1H, J=2,5, 1,8 Гц), 7,41 ~ 7,32 (м, 4H), 7,29 ~ 7,25 (м, 1H), 7,22 (т, 1H, J=51,6 Гц), 4,99 (с, 2H), 4,67 (т, 2H, J=6,8 Гц), 4,57 (т, 2H, J=6,3 Гц), 3,50 ~ 3,44 (м, 1H), 3,23 (т, 2H, J=6,9 Гц), 2,87 (с, 3H), 2,52 ~ 2,45 (м, 4H), 2,47 (т, 2H, J=6,9 Гц), 2,36 ~ 2,33 (м, 4H) ; LRMS (ES) m/z 563,4 (M++1)
Пример 61: Соединение 11713, 2-((4-(((N-метил-N-(2-(4-ацетилпиперазин-1-ил)этил)сульфамоил)(фенил)амино)метил))фенил)-5-(дифторметил)-1,3,4-оксадиазол
[Стадия 1] метил 4-(((N-(2-(4-ацетилпиперазин-1-ил)этил)-N-метилсульфамоил)(фенил)амино)метил)бензоат
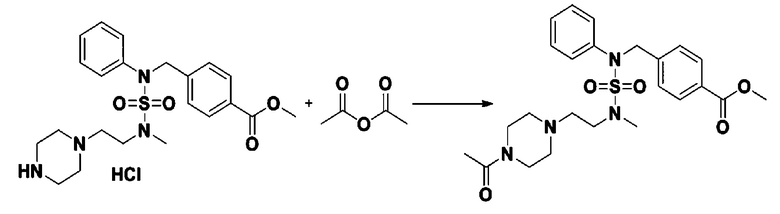
Раствор метил 4-(((N-метил-N-(2-(пиперазин-1-ил)этил)сульфамоил)(фенил)амино)метил)бензоат гидрохлорида (0,040 г, 0,083 ммоль), триэтиламина (0,023 мл, 0,166 ммоль) и уксусного ангидрида (0,017 г, 0,166 ммоль) в дихлорметане (2 мл) перемешивали при комнатной температуре в течение 2 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией дихлорметаном. Двухфазную смесь пропускали через пластиковую фритту, для удаления твердого остатка и водного слоя, и собранный органический слой концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; этилацетат/гексан=0%-30%) с получением метил 4-(((N-(2-(4-ацетилпиперазин-1-ил)этил)-N-метилсульфамоил)(фенил)амино)метил)бензоата в виде бесцветной жидкости (0,039 г, 96,4%).
[Стадия 2] 4-(((N-метил-N-(2-(4-ацетилпиперазин-1-ил)этил)сульфамоил)(фенил)амино)метил)бензогидразид
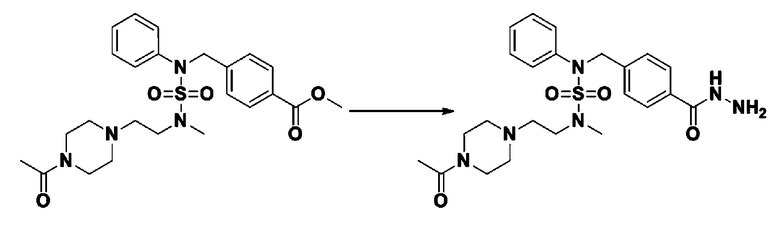
Раствор метил 4-(((N-(2-(4-ацетилпиперазин-1-ил)этил)-N-метилсульфамоил)(фенил)амино)метил)бензоата (0,039 г, 0,080 ммоль) и гидразина моногидрата (0,039 мл, 0,798 ммоль) в этаноле (2 мл) перемешивали при 80°С в течение 18 часов, и охлаждали до комнатной температуры для прекращения реакции. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Остаток разбавляли насыщенным водным раствором бикарбоната натрия (5 мл) и перемешивали. Полученные осадки собирали фильтрованием, промывали гексаном и сушили с получением 4-(((N-метил-N-(2-(4-ацетилпиперазин-1-ил)этил)сульфамоил)(фенил)амино)метил)бензогидразида в виде желтого масла (0,037 г, 95,1%).
[Стадия 3] Соединение 11713
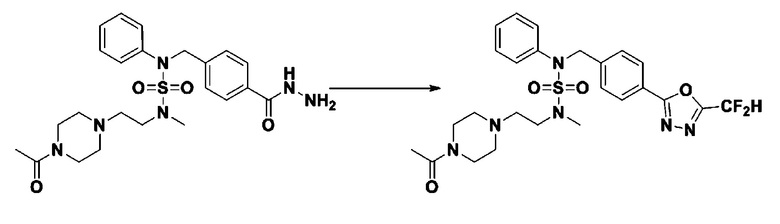
Раствор 4-(((N-метил-N-(2-(4-ацетилпиперазин-1-ил)этил)сульфамоил)(фенил)амино)метил)бензогидразида (0,037 г, 0,076 ммоль), 2,2-дифторуксусного ангидрида (0,019 мл, 0,152 ммоль) и триэтиламина (0,021 мл, 0,152 ммоль) в тетрагидрофуране (2 мл) перемешивали при 80°С в течение 0,5 часоа и охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией дихлорметаном. Двухфазную смесь пропускали через пластиковую фритту, для удаления твердого остатка и водного слоя, и собранный органический слой концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; метанол/дихлорметан=0%-5%) с получением 2-((4-(((N-метил-N-(2-(4-ацетилпиперазин-1-ил)этил)сульфамоил)(фенил)амино)метил))фенил)-5-(дифторметил)-1,3,4-оксадиазола в виде желтой жидкости (0,009 г, 21,6%).
1H ЯМР (400 МГц, CD3OD) δ 8,02 (д, 2H, J=8,4 Гц), 7,53 (д, 2H, J=8,4 Гц), 7,42 ~ 7,26 (м, 5H), 7,22 (т, 1H, J=51,6 Гц), 5,01 (с, 2H), 3,55 (т, 2H, J=5,0 Гц), 3,51 (т, 2H, J=5,0 Гц), 3,26 (т, 2H, J=6,6 Гц), 2,89 (с, 3H), 2,52 ~ 2,49 (м, 4H), 2,44 (т, 2H, J=5,0 Гц), 2,09 (с, 3H); LRMS (ES) m/z 549,4 (M++1)
Пример 62: Соединение 11714, 2-((4-(((N-метил-N-(2-(4-(метилсульфонил)пиперазин-1-ил)этил)сульфамоил)(фенил)амино)метил))фенил)-5-(дифторметил)-1,3,4-оксадиазол
[Стадия 1] метил 4-(((N-метил-N-(2-(4-(метилсульфонил)пиперазин-1-ил)этил)сульфамоил)(фенил)амино)метил)бензоат
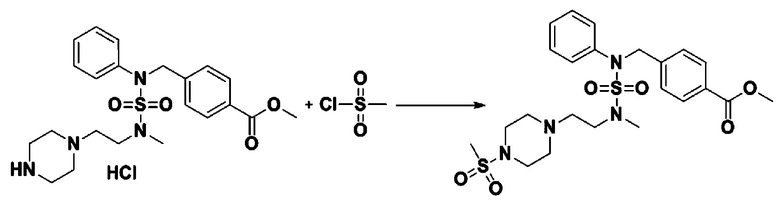
Раствор метил 4-(((N-метил-N-(2-(пиперазин-1-ил)этил)сульфамоил)(фенил)амино)метил)бензоат гидрохлорида (0,040 г, 0,083 ммоль), триэтиламина (0,023 мл, 0,166 ммоль) и метансульфонилхлорида (0,013 мл, 0,166 ммоль) в дихлорметане (2 мл) перемешивали при комнатной температуре в течение 2 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией дихлорметаном. Двухфазную смесь пропускали через пластиковую фритту, для удаления твердого остатка и водного слоя, и собранный органический слой концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; этилацетат/гексан=0%-30%) с получением метил 4-(((N-метил-N-(2-(4-(метилсульфонил)пиперазин-1-ил)этил)сульфамоил)(фенил)амино)метил)бензоата в виде желтого твердого вещества (0,043 г, 99,0%).
[Стадия 2] 4-(((N-метил-N-(2-(4-(метилсульфонил)пиперазин-1-ил)этил)сульфамоил)(фенил)амино)метил)бензогидразид
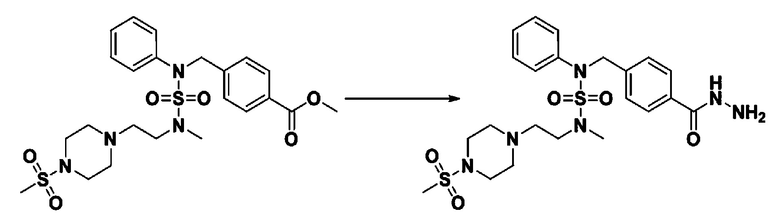
Раствор метил 4-(((N-метил-N-(2-(4-(метилсульфонил)пиперазин-1-ил)этил)сульфамоил)(фенил)амино)метил)бензоата (0,043 г, 0,082 ммоль) и гидразина моногидрата (0,040 мл, 0,820 ммоль) в этаноле (2 мл) перемешивали при 80°С в течение 18 часов, и охлаждали до комнатной температуры для прекращения реакции. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Остаток разбавляли насыщенным водным раствором бикарбоната натрия (5 мл) и перемешивали. Полученные осадки собирали фильтрованием и сушили с получением 4-(((N-метил-N-(2-(4-(метилсульфонил)пиперазин-1-ил)этил)сульфамоил)(фенил)амино)метил)бензогидразида в виде бесцветной жидкости (0,039 г, 90,7%).
[Стадия 3] Соединение 11714
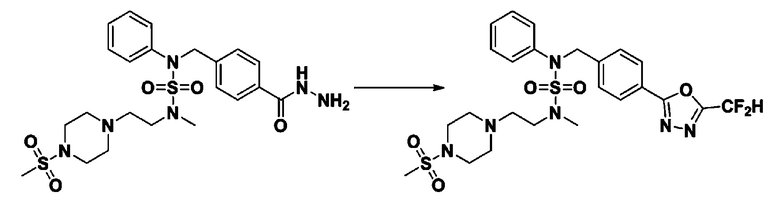
Раствор 4-(((N-метил-N-(2-(4-(метилсульфонил)пиперазин-1-ил)этил)сульфамоил)(фенил)амино)метил)бензогидразида (0,039 г, 0,074 ммоль), 2,2-дифторуксусного ангидрида (0,018 мл, 0,149 ммоль) и триэтиламина (0,021 мл, 0,149 ммоль) в тетрагидрофуране (2 мл) перемешивали при 80°С в течение 0,5 часа, и охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией дихлорметаном. Двухфазную смесь пропускали через пластиковую фритту, для удаления твердого остатка и водного слоя, и собранный органический слой концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; метанол/дихлорметан=0%-5%) с получением 2-((4-(((N-метил-N-(2-(4-(метилсульфонил)пиперазин-1-ил)этил)сульфамоил)(фенил)амино)метил))фенил)-5-(дифторметил)-1,3,4-оксадиазола в виде желтого масла (0,011 г, 25,3%).
1H ЯМР (400 МГц, CD3OD) δ 8,02 (д, 2H, J=8,5 Гц), 7,54 (д, 2H, J=8,6 Гц), 7,42 ~ 7,26 (м, 5H), 7,22 (т, 1H, J=51,7 Гц), 5,00 (с, 2H), 3,24 (т, 2H, J=6,6 Гц), 3,19 ~ 3,17 (м, 4H), 2,89 (с, 3H), 2,81 (с, 3H), 2,56 ~ 2,54 (м, 4H), 2,50 (т, 2H, J=6,6 Гц); LRMS (ES) m/z 585,3 (M++1)
Пример 63: Соединение 11787, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенил-2,7-диазаспиро[3.5]нонан-7-сульфонамид гидрохлорид
[Стадия 1] трет-бутил 7-((1H-имидазол-1-ил)сульфонил)-2,7-диазаспиро[3.5]нонан-2-карбоксилат
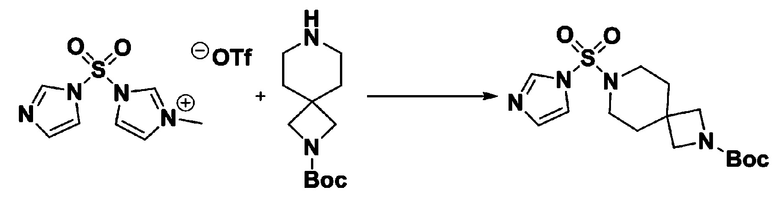
Раствор 1-((1H-имидазол-1-ил)сульфонил)-3-метил-1H-имидазол-3-ий трифторметансульфоната (2,000 г, 5,520 ммоль) и трет-бутил 2,7-диазаспиро[3.5]нонан-2-карбоксилата (1,187 г, 5,244 ммоль) в ацетонитриле (20 мл) перемешивали при комнатной температуре в течение 18 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 40 г; метанол/дихлорметан=0%-10%) с получением трет-бутил 7-((1H-имидазол-1-ил)сульфонил)-2,7-диазаспиро[3.5]нонан-2-карбоксилата в виде белого твердого вещества (0,830 г, 42,2%).
[Стадия 2] 1-((2-(трет-бутоксикарбонил)-2,7- диазаспиро[3.5]нонан-7-ил)сульфонил)-3-метил-1H-имидазол-3-ий трифторметансульфонат
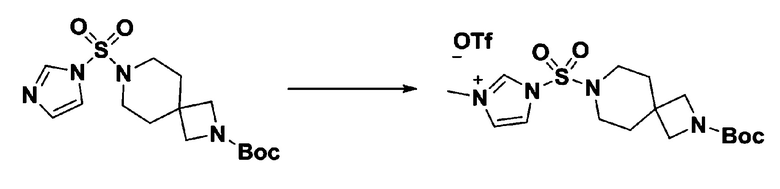
Раствор трет-бутил 7-((1H-имидазол-1-ил)сульфонил)-2,7-диазаспиро[3.5]нонан-2-карбоксилата (0,900 г, 2,525 ммоль) и метил трифторметансульфоната (0,305 мл, 2,777 ммоль) в дихлорметане (10 мл) перемешивали при 0°C в течение 3 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Осадки собирали фильтрованием, промывали дихлорметаном и сушили с получением 1-((2-(трет-бутоксикарбонил)-2,7-диазаспиро[3.5]нонан-7-ил)сульфонил)-3-метил-1H-имидазол-3-ий трифторметансульфоната в виде белого твердого вещества (1,282 г, 97,5%).
[Стадия 3] трет-бутил 7-(N-фенилсульфамоил)-2,7-диазаспиро[3.5]нонан-2-карбоксилат
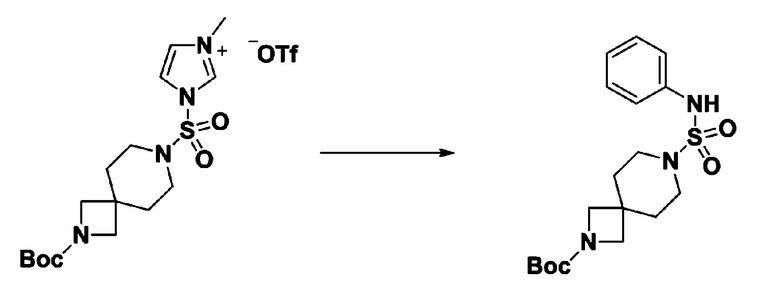
Раствор 1-((2-(трет-бутоксикарбонил)-2,7- диазаспиро[3.5]нонан-7-ил)сульфонил)-3-метил-1H-имидазол-3-ий трифторметансульфоната (1,282 г, 2,463 ммоль) и анилина (0,270 мл, 2,955 ммоль) в ацетонитриле (10 мл) перемешивали при комнатной температуре в течение 18 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 24 г; этилацетат/гексан=0%-50%) с получением трет-бутил 7-(N-фенилсульфамоил)-2,7-диазаспиро[3.5]нонан-2-карбоксилата в виде бесцветной жидкости (0,192 г, 20,4%).
[Стадия 4] трет-бутил 7-(N-(4-(метоксикарбонил)бензил)-N-фенилсульфамоил)-2,7-диазаспиро[3.5]нонан-2-карбоксилат
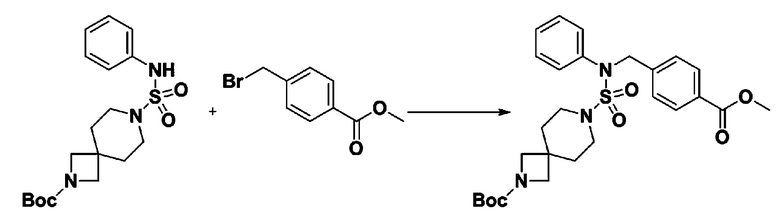
Раствор трет-бутил 7-(N-фенилсульфамоил)-2,7-диазаспиро[3.5]нонан-2-карбоксилата (0,192 г, 0,503 ммоль), метил 4-(бромметил)бензоата (0,138 г, 0,604 ммоль) и гидрида натрия (60,00%, 0,030 г, 0,755 ммоль) в N,N-диметилформамиде (5 мл) перемешивали при комнатной температуре в течение 18 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; этилацетат/гексан=0%-30%) с получением трет-бутил 7-(N-(4-(метоксикарбонил)бензил)-N-фенилсульфамоил)-2,7-диазаспиро[3.5]нонан-2-карбоксилата в виде белого твердого вещества (0,151 г, 56,5%).
[Стадия 5] трет-бутил 7-(N-(4-(гидразинкарбонил)бензил)-N-фенилсульфамоил)-2,7-диазаспиро[3.5]нонан-2-карбоксилат
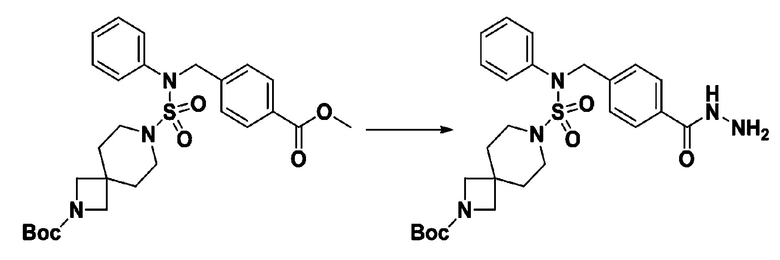
Раствор трет-бутил 7-(N-(4-(метоксикарбонил)бензил)-N-фенилсульфамоил)-2,7-диазаспиро[3.5]нонан-2-карбоксилата (0,075 г, 0,142 ммоль) и гидразина моногидрата (0,069 мл, 1,416 ммоль) в этаноле (5 мл) перемешивали при 80°С в течение 18 часов, и охлаждали до комнатной температуры для прекращения реакции. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Остаток разбавляли насыщенным водным раствором бикарбоната натрия (5 мл) и перемешивали. Полученные осадки собирали фильтрованием и сушили с получением трет-бутил 7-(N-(4-(гидразинкарбонил)бензил)-N-фенилсульфамоил)-2,7-диазаспиро[3.5]нонан-2-карбоксилата в виде белого твердого вещества (0,073 г, 97,3%).
[Стадия 6] трет-бутил 7-(N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенилсульфамоил)-2,7-диазаспиро[3.5]нонан-2-карбоксилат
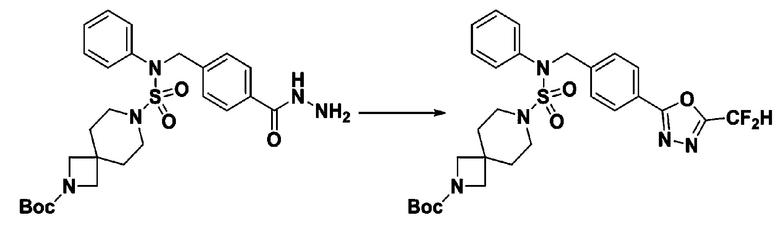
Раствор трет-бутил 7-(N-(4-(гидразинкарбонил)бензил)-N-фенилсульфамоил)-2,7-диазаспиро[3.5]нонан-2-карбоксилата (0,073 г, 0,138 ммоль), 2,2-дифторуксусного ангидрида (0,051 мл, 0,413 ммоль) и триэтиламина (0,058 мл, 0,413 ммоль) в тетрагидрофуране (3 мл) перемешивали при 80°С в течение 0,5 часа, и охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; этилацетат/гексан=0%-50%) с получением трет-бутил 7-(N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенилсульфамоил)-2,7-диазаспиро[3.5]нонан-2-карбоксилата в виде белого твердого вещества (0,081 г, 99,7%).
[Стадия 7] Соединение 11787
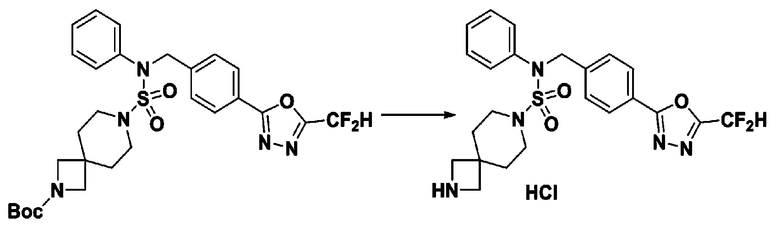
Раствор трет-бутил 7-(N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенилсульфамоил)-2,7-диазаспиро[3.5]нонан-2-карбоксилата (0,081 г, 0,137 ммоль) и хлористоводородной кислоты (4,00 M раствор, 0,343 мл, 1,374 ммоль) в 1,4-диоксане (5 мл) перемешивали при комнатной температуре в течение 2 часов, и концентрировали при пониженном давлении для удаления растворителя. Остаток разбавляли диэтиловым эфиром (10 мл), и перемешивали. Полученные осадки собирали фильтрованием и сушили с получением N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенил-2,7-диазаспиро[3.5]нонан-7-сульфонамид гидрохлорида в виде желтого твердого вещества (0,067 г, 92,9%).
1H ЯМР (400 МГц, CD3OD) δ 8,02 (д, 2H, J=8,1 Гц), 7,52 (д, 2H, J=8,2 Гц), 7,41~7,24 (м, 5H), 7,21 (т, 1H, J=51,6 Гц), 4,98 (с, 2H), 3,85 (с, 4H), 3,24~3,22 (м, 4H), 1,89~1,86 (м, 4H); LRMS (ES) m/z 490,3 (M++1)
Пример 64: Соединение 11788, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-2-(оксетан-3-ил)-N-фенил-2,7-диазаспиро[3.5]нонан-7-сульфонамид
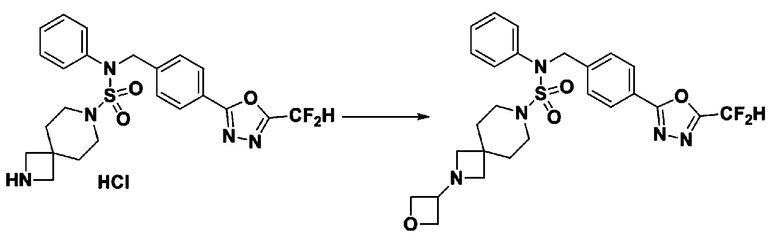
Раствор N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенил-2,7-диазаспиро[3.5]нонан-7-сульфонамид гидрохлорида (0,030 г, 0,057 ммоль) и оксетан-3-она (0,008 г, 0,114 ммоль) в дихлорметане (3 мл) перемешивали при комнатной температуре в течение 1 часа, и смешивали с триацетоксиборгидридом натрия (0,024 г, 0,114 ммоль). Реакционную смесь перемешивали при той же температуре в течение дополнительных 3 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; метанол/дихлорметан=0%-10%) с получением N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-2-(оксетан-3-ил)-N-фенил-2,7-диазаспиро[3.5]нонан-7-сульфонамида в виде белого твердого вещества (0,021 г, 67,5%).
1H ЯМР (400 МГц, CD3OD) δ 8,01 (д, 2H, J=8,3 Гц), 7,52 (д, 2H, J=8,2 Гц), 7,41~7,39 (м, 2H), 7,35~7,31 (м, 2H), 7,27~7,23 (м, 1H), 7,21 (т, 1H, J=51,7 Гц), 4,99 (с, 2H), 4,74 (т, 2H, J=6,8 Гц), 4,48 (дд, 1H, J=6,8, 5,0 Гц), 3,20~3,16 (м, 8H), 1,79~1,76 (м, 4H); LRMS (ES) m/z 546,3 (M++1)
Пример 65: Соединение 11789, 2-циклобутил-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенил-2,7-диазаспиро[3.5]нонан-7-сульфонамид
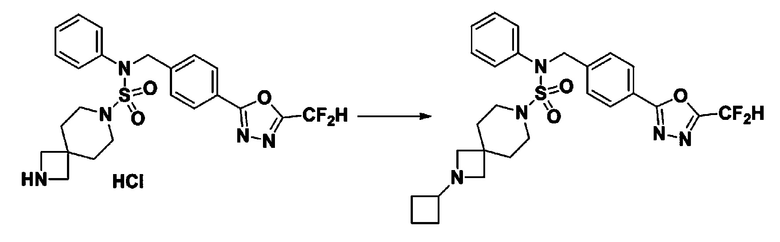
Раствор N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенил-2,7-диазаспиро[3.5]нонан-7-сульфонамид гидрохлорида (0,030 г, 0,057 ммоль) и циклобутанона (0,008 г, 0,114 ммоль) в дихлорметане (3 мл) перемешивали при комнатной температуре в течение 1 часа, и смешивали с триацетоксиборгидридом натрия (0,024 г, 0,114 ммоль). Реакционную смесь перемешивали при той же температуре в течение дополнительных 3 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; метанол/дихлорметан=0%-10%) с получением 2-циклобутил-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенил-2,7-диазаспиро[3.5]нонан-7-сульфонамида в виде бесцветной жидкости (0,023 г, 74,2%).
1H ЯМР (400 МГц, CD3OD) δ 8,01 (д, 2H, J=8,4 Гц), 7,51 (д, 2H, J=8,5 Гц), 7,40~7,37 (м, 2H), 7,35~7,31 (м, 2H), 7,28~ 7,24 (м, 1H), 7,22 (т, 1H, J=51,6 Гц), 3,61 (с, 4H), 3,22~ 3,19 (м, 4H), 2,24~2,16 (м, 2H), 2,10~2,01 (м, 2H), 1,91~1,83 (м, 2H), 1,82~1,79 (м, 4H); LRMS (ES) m/z 544,3 (M++1)
Пример 66: Соединение 11823, (R)-N-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3,4-диметил-N-(м-толил)пиперазин-1-сульфонамид
[Стадия 1] трет-бутил (R)-4-((1H-имидазол-1-ил)сульфонил)-2-метилпиперазин-1-карбоксилат
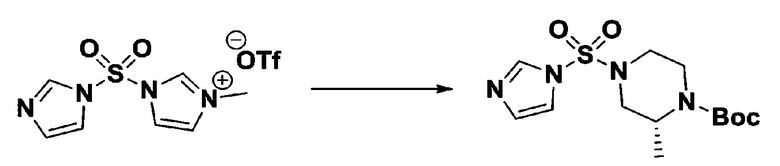
Раствор 1-((1H-имидазол-1-ил)сульфонил)-3-метил-1H-имидазол-3-ий трифторметансульфоната (9,600 г, 26,497 ммоль) и трет-бутил (R)-2-метилпиперазин-1-карбоксилата (5,838 г, 29,147 ммоль) в ацетонитриле (100 мл) перемешивали при комнатной температуре в течение 16 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида аммония, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 40 г; этилацетат/гексан=0%-40%) с получением трет-бутил (R)-4-((1H-имидазол-1-ил)сульфонил)-2-метилпиперазин-1-карбоксилата в виде желтого твердого вещества (4,600 г, 52,5%).
[Стадия 2] (R)-1-((4-(трет-бутоксикарбонил)-3-метилпиперазин-1-ил)сульфонил)-3-метил-1H-имидазол-3-ий трифторметансульфонат
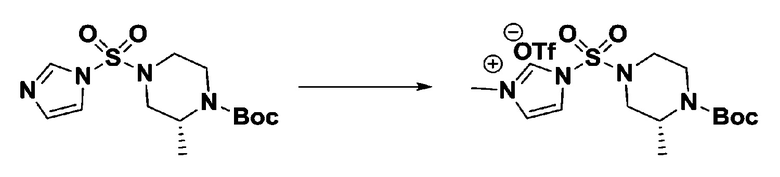
Раствор трет-бутил (R)-4-((1H-имидазол-1-ил)сульфонил)-2-метилпиперазин-1-карбоксилата (4,600 г, 13,923 ммоль) и метил трифторметансульфоната (1,527 мл, 13,923 ммоль) в дихлорметане (150 мл) перемешивали при 0°C в течение 3 часов, и концентрировали при пониженном давлении для удаления растворителя. Неочищенный продукт использовали без дополнительной очистки ((R)-1-((4-(трет-бутоксикарбонил)-3-метилпиперазин-1-ил)сульфонил)-3-метил-1H-имидазол-3-ий трифторметансульфонат, 6,700 г, 97,3%, белое твердое вещество).
[Стадия 3] трет-бутил (R)-2-метил-4-(N-(м-толил)сульфамоил)пиперазин-1-карбоксилат
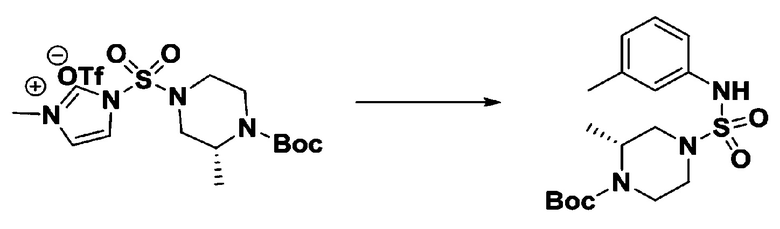
Смесь (R)-1-((4-(трет-бутоксикарбонил)-3-метилпиперазин-1-ил)сульфонил)-3-метил-1H-имидазол-3-ий трифторметансульфоната (3,350 г, 6,775 ммоль) и м-толуидин гидрохлорида (1,070 г, 7,452 ммоль) в ацетонитриле (100 мл), полученную при температуре окружающей среды, нагревали с обратным холодильником в течение 16 часов, и охлаждали до температуры окружающей среды. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида аммония, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 24 г; этилацетат/гексан=0%-30%) с получением трет-бутил (R)-2-метил-4-(N-(м-толил)сульфамоил)пиперазин-1-карбоксилата в виде желтого твердого вещества (1,100 г, 43,9%).
[Стадия 4] трет-бутил (R)-4-(N-((5-(метоксикарбонил)пиридин-2-ил)метил)-N-(м-толил)сульфамоил)-2-метилпиперазин-1-карбоксилат
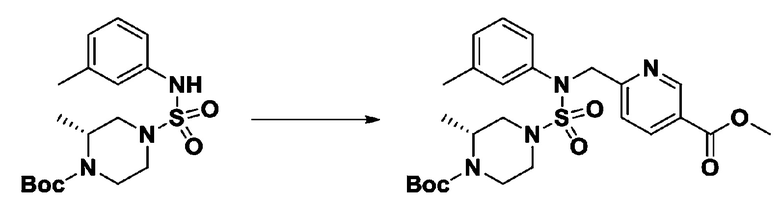
Раствор трет-бутил (R)-2-метил-4-(N-(м-толил)сульфамоил)пиперазин-1-карбоксилата (1,100 г, 4,082 ммоль) и карбоната калия (0,846 г, 6,123 ммоль) в N,N-диметилформамиде (50 мл) перемешивали при комнатной температуре в течение 30 мин и смешивали с метил-6-(бромметил)никотинатом (1,033 г, 4,490 ммоль) и иодидом калия (0,339 г, 2,041 ммоль). Реакционную смесь перемешивали при той же температуре в течение дополнительных 12 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида аммония, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 12 г; этилацетат/гексан=0%-60%) с получением трет-бутил (R)-4-(N-((5-(метоксикарбонил)пиридин-2-ил)метил)-N-(м-толил)сульфамоил)-2-метилпиперазин-1-карбоксилата в виде желтого твердого вещества (1,180 г, 55,7%).
[Стадия 5] метил (R)-6-(((3-метил-N-(м-толил)пиперазин)-1-сульфонамидо)метил)никотинат гидрохлорид
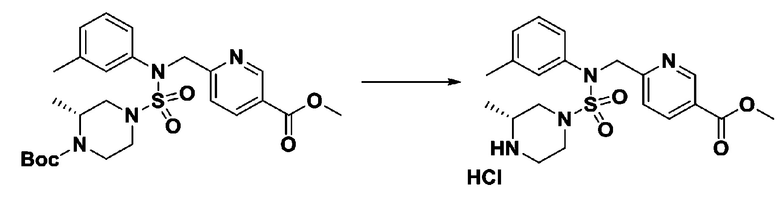
Раствор трет-бутил (R)-4-(N-((5-(метоксикарбонил)пиридин-2-ил)метил)-N-(м-толил)сульфамоил)-2-метилпиперазин-1-карбоксилата (1,100 г, 2,121 ммоль) и хлорида водорода (4,00 M раствор в 1,4-диоксане, 1,060 мл, 4,242 ммоль) в дихлорметане (50 мл) перемешивали при комнатной температуре в течение 5 часов, и концентрировали при пониженном давлении для удаления растворителя. Неочищенный продукт использовали без дополнительной очистки (метил (R)-6-(((3-метил-N-(м-толил)пиперазин)-1-сульфонамидо)метил)никотинат гидрохлорид, 0,850 г, 88,1%, желтое твердое вещество).
[Стадия 6] метил (R)-6-(((3,4-диметил-N-(м-толил)пиперазин)-1-сульфонамидо)метил)никотинат
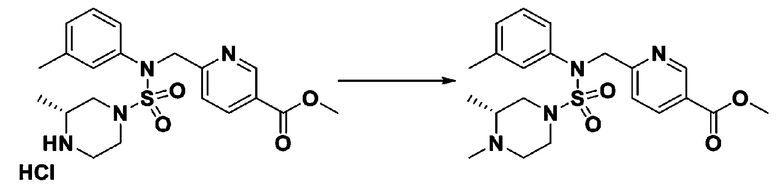
Раствор метил (R)-6-(((3-метил-N-(м-толил)пиперазин)-1-сульфонамидо)метил)никотинат гидрохлорида (0,300 г, 0,659 ммоль), параформальдегида (0,040 г, 1,319 ммоль), уксусной кислоты (0,045 мл, 0,791 ммоль) и триацетоксиборгидрида натрия (0,279 г, 1,319 ммоль) в дихлорметане (10 мл) перемешивали при комнатной температуре в течение 12 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором бикарбоната натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; метанол/дихлорметан=0%-5%) с получением метил (R)-6-(((3,4-диметил-N-(м-толил)пиперазин)-1-сульфонамидо)метил)никотината в виде желтого твердого вещества (0,120 г, 42,1%).
[Стадия 7] (R)-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-3,4-диметил-N-(м-толил)пиперазин-1-сульфонамид
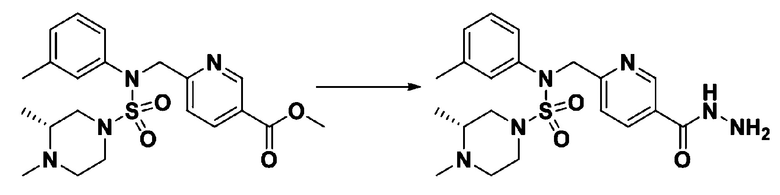
Раствор метил (R)-6-(((3,4-диметил-N-(м-толил)пиперазин)-1-сульфонамидо)метил)никотината (0,120 г, 0,253 ммоль) и гидразина моногидрата (0,123 мл, 2,529 ммоль) в этаноле (10 мл) перемешивали при 90°C в течение 12 часов, и охлаждали до комнатной температуры для прекращения реакции. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Затем к концентрату добавляли воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором бикарбоната натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Указанное в заголовке соединение использовали без дополнительной очистки ((R)-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-3,4-диметил-N-(м-толил)пиперазин-1-сульфонамид, 0,100 г, 83,3%, белое твердое вещество).
[Стадия 8] Соединение 11823
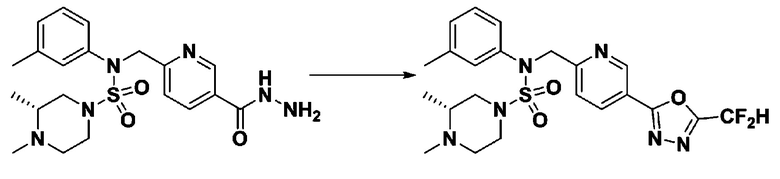
Раствор (R)-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-3,4-диметил-N-(м-толил)пиперазин-1-сульфонамида (0,100 г, 0,231 ммоль), триэтиламина (0,161 мл, 1,156 ммоль) и 2,2-дифторуксусного ангидрида (0,086 мл, 0,694 ммоль) в тетрагидрофуране (10 мл) перемешивали при 70°С в течение 12 часов, и охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида аммония, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; метанол/дихлорметан=0%-5%) с получением (R)-N-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3,4-диметил-N-(м-толил)пиперазин-1-сульфонамида в виде желтого твердого вещества (0,082 г, 72,0%).
1H ЯМР (400 МГц, CDCl3) δ 9,21 (д, 1 H, J=2,1 Гц), 8,36 (д, 1 H, J=8,3 Гц), 7,73 (д, 1 H, J=8,2 Гц), 7,26-7,21 (м, 3 H), 7,19-7,17 (м, 1,3 H), 6,94 (с, 0,5 H), 6,81 (с, 0,2 H), 5,12 (с, 2 H), 3,58-3,48 (м, 2 H), 3,00-2,93 (м, 1 H), 2,75-2,71 (м, 1 H), 2,62-2,57 (м, 1 H), 2,32 (с, 3 H), 2,26 (с, 3 H), 2,25-2,20 (м, 1 H), 2,19-2,09 (м, 1 H), 1,04 (д, 3 H, J=6,3 Гц); LRMS (ES) m/z 493,3 (M++1).
Пример 67: Соединение 11824, (R)-N-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-метил-4-(оксетан-3-ил)-N-(м-толил)пиперазин-1-сульфонамид
[Стадия 1] метил (R)-6-(((3-метил-4-(оксетан-3-ил)-N-(м-толил)пиперазин)-1-сульфонамидо)метил)никотинат
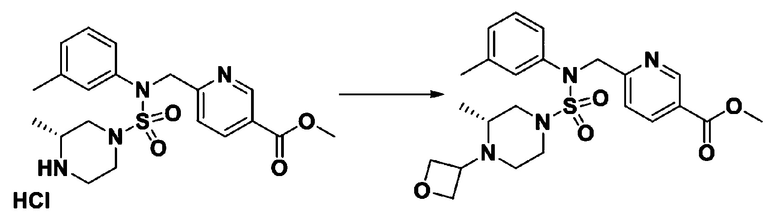
Раствор метил (R)-6-(((3-метил-N-(м-толил)пиперазин)-1-сульфонамидо)метил)никотинат гидрохлорида (0,300 г, 0,659 ммоль) и оксетан-3-она (0,095 г, 1,319 ммоль), уксусной кислоты (0,045 мл, 0,791 ммоль) и триацетоксиборгидрида натрия (0,279 г, 1,319 ммоль) в дихлорметане (10 мл) перемешивали при комнатной температуре в течение 12 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором бикарбоната натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; метанол/дихлорметан=0%-5%) с получением метил (R)-6-(((3-метил-4-(оксетан-3-ил)-N-(м-толил)пиперазин)-1-сульфонамидо)метил)никотината в виде желтого твердого вещества (0,150 г, 47,9%).
[Стадия 2] (R)-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-3-метил-4-(оксетан-3-ил)-N-(м-толил)пиперазин-1-сульфонамид
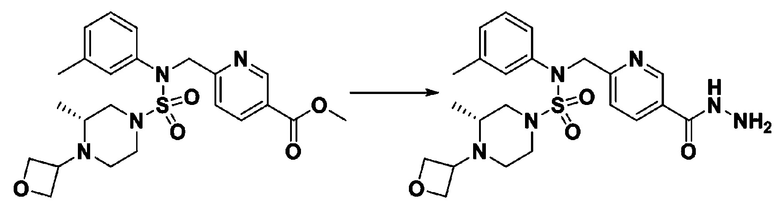
Раствор метил (R)-6-(((3-метил-4-(оксетан-3-ил)-N-(м-толил)пиперазин)-1-сульфонамидо)метил)никотината (0,150 г, 0,316 ммоль) и гидразина моногидрата (0,154 мл, 3,161 ммоль) в этаноле (10 мл) перемешивали при 90°C в течение 12 часов, и охлаждали до комнатной температуры для прекращения реакции. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Затем к концентрату добавляли воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором бикарбоната натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Указанное в заголовке соединение использовали без дополнительной очистки ((R)-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-3-метил-4-(оксетан-3-ил)-N-(м-толил)пиперазин-1-сульфонамид, 0,100 г, 66,7%, белое твердое вещество).
[Стадия 3] Соединение 11824
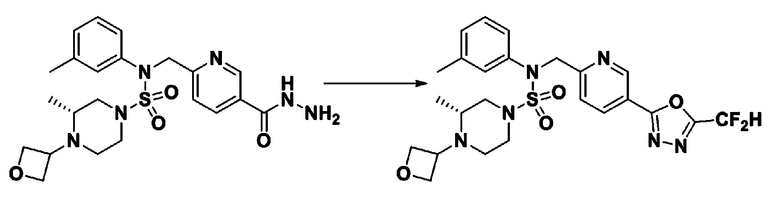
Раствор (R)-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-3-метил-4-(оксетан-3-ил)-N-(м-толил)пиперазин-1-сульфонамида (0,100 г, 0,211 ммоль), триэтиламина (0,147 мл, 1,054 ммоль) и 2,2-дифторуксусного ангидрида (0,079 мл, 0,632 ммоль) в тетрагидрофуране (10 мл) перемешивали при 70°С в течение 12 часов, и охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида аммония, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; этилацетат/гексан=0%-80%) с получением (R)-N-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-метил-4-(оксетан-3-ил)-N-(м-толил)пиперазин-1-сульфонамида в виде желтого масла (0,073 г, 64,8%).
1H ЯМР (400 МГц, CDCl3) δ 9,23-9,21 (м, 1 H), 8,37 (д, 1 H, J=8,2 Гц), 7,72 (д, 1 H, J=7,2 Гц), 7,28-7,26 (м, 1 H), 7,22-7,21 (м, 2 H), 7,08-7,06 (м, 1 H), 7,05 (с, 0,2 H), 6,94 (с, 0,5 H), 6,81 (с, 0,3 H), 5,11 (с, 2 H), 4,63-4,55 (м, 4 H), 3,75-3,71 (м, 1 H), 3,42-3,33 (м, 2 H), 3,15-3,13 (м, 1 H), 2,87-2,82 (м, 1 H), 2,61-2,57 (м, 1 H), 2,41-2,35 (м, 1 H), 2,33 (с, 3 H), 2,16-2,10 (м, 1 H), 0,88 (д, 3 H, J=6,5 Гц); LRMS (ES) m/z 535,1 (M++1).
Пример 68: Соединение 11825, (R)-N-(3-хлорфенил)-N-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3,4-диметилпиперазин-1-сульфонамид
[Стадия 1] трет-бутил (R)-4-(N-(3-хлорфенил)сульфамоил)-2-метилпиперазин-1-карбоксилат
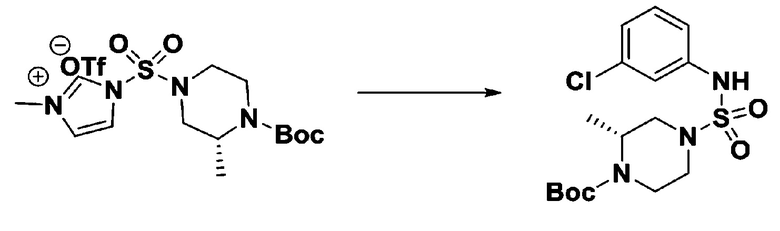
Смесь (R)-1-((4-(трет-бутоксикарбонил)-3-метилпиперазин-1-ил)сульфонил)-3-метил-1H-имидазол-3-ий трифторметансульфоната (3,700 г, 7,482 ммоль) и 3-хлоранилина (1,050 г, 8,231 ммоль) в ацетонитриле (100 мл), полученную при температуре окружающей среды, нагревали с обратным холодильником в течение 16 часов, и охлаждали до температуры окружающей среды. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида аммония, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 24 г; этилацетат/гексан=0%-30%) с получением трет-бутил (R)-4-(N-(3-хлорфенил)сульфамоил)-2-метилпиперазин-1-карбоксилата в виде белого твердого вещества (0,610 г, 20,9%).
[Стадия 2] трет-бутил (R)-4-(N-(3-хлорфенил)-N-((5-(метоксикарбонил)пиридин-2-ил)метил)сульфамоил)-2-метилпиперазин-1-карбоксилат
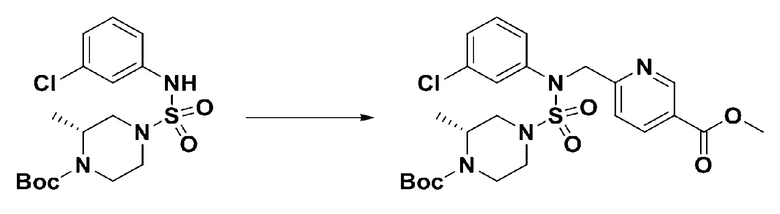
Раствор трет-бутил (R)-4-(N-(3-хлорфенил)сульфамоил)-2-метилпиперазин-1-карбоксилата (0,610 г, 1,565 ммоль) и карбоната калия (0,324 г, 2,347 ммоль) в N,N-диметилформамиде (50 мл) перемешивали при комнатной температуре в течение 30 мин и смешивали с метил-6-(бромметил)никотинатом (0,396 г, 1,721 ммоль) и иодидом калия (0,130 г, 0,782 ммоль). Реакционную смесь перемешивали при той же температуре в течение дополнительных 12 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида аммония, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 12 г; этилацетат/гексан=0%-40%) с получением трет-бутил (R)-4-(N-(3-хлорфенил)-N-((5-(метоксикарбонил)пиридин-2-ил)метил)сульфамоил)-2-метилпиперазин-1-карбоксилата в виде желтого твердого вещества (0,660 г, 78,3%).
[Стадия 3] метил (R)-6-(((N-(3-хлорфенил)-3-метилпиперазин)-1-сульфонамидо)метил)никотинат гидрохлорид
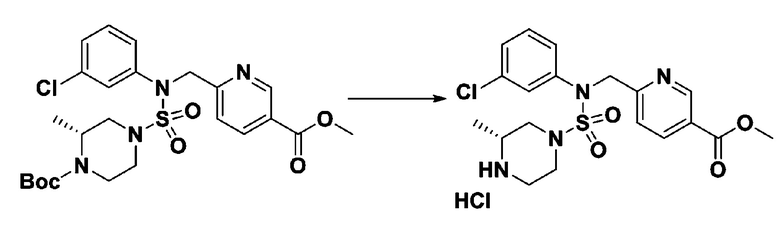
Раствор трет-бутил (R)-4-(N-(3-хлорфенил)-N-((5-(метоксикарбонил)пиридин-2-ил)метил)сульфамоил)-2-метилпиперазин-1-карбоксилата (0,660 г, 1,224 ммоль) и хлорида водорода (4,00 M раствор в 1,4-диоксане, 0,612 мл, 2,449 ммоль) в дихлорметане (30 мл) перемешивали при комнатной температуре в течение 5 часов, и концентрировали при пониженном давлении для удаления растворителя. Неочищенный продукт использовали без дополнительной очистки (метил (R)-6-(((N-(3-хлорфенил)-3-метилпиперазин)-1-сульфонамидо)метил)никотинат гидрохлорид, 0,450 г, 77,3%, желтое твердое вещество).
[Стадия 4] метил (R)-6-(((N-(3-хлорфенил)-3,4-диметилпиперазин)-1-сульфонамидо)метил)никотинат
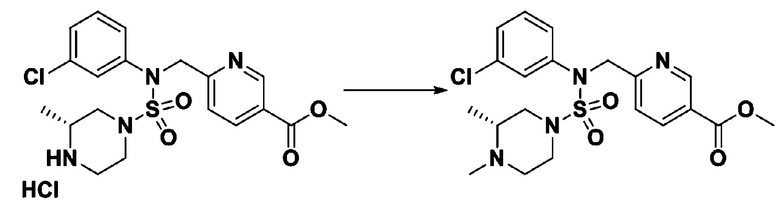
Раствор метил (R)-6-(((N-(3-хлорфенил)-3-метилпиперазин)-1-сульфонамидо)метил)никотинат гидрохлорида (0,210 г, 0,442 ммоль), параформальдегида (0,027 г, 0,883 ммоль), уксусной кислоты (0,030 мл, 0,530 ммоль) и триацетоксиборгидрида натрия (0,187 г, 0,883 ммоль) в дихлорметане (10 мл) перемешивали при комнатной температуре в течение 12 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором бикарбоната натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; метанол/дихлорметан=0%-5%) с получением метил (R)-6-(((N-(3-хлорфенил)-3,4-диметилпиперазин)-1-сульфонамидо)метил)никотината в виде желтого твердого вещества (0,115 г, 57,5%).
[Стадия 5] (R)-N-(3-хлорфенил)-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-3,4-диметилпиперазин-1-сульфонамид
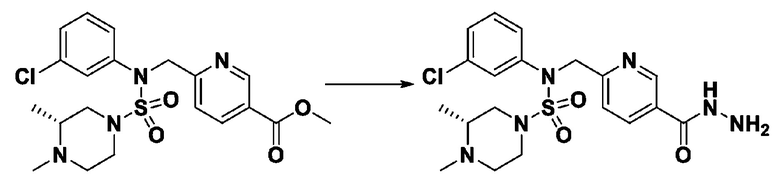
Раствор метил (R)-6-(((N-(3-хлорфенил)-3,4-диметилпиперазин)-1-сульфонамидо)метил)никотината (0,115 г, 0,254 ммоль) и гидразина моногидрата (0,123 мл, 2,539 ммоль) в этаноле (10 мл) перемешивали при комнатной температуре в течение 12 часов, и концентрировали при пониженном давлении для удаления растворителя. Затем к концентрату добавляли воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором бикарбоната натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Указанное в заголовке соединение использовали без дополнительной очистки ((R)-N-(3-хлорфенил)-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-3,4-диметилпиперазин-1-сульфонамид, 0,090 г, 78,3%, белое твердое вещество).
[Стадия 6] Соединение 11825
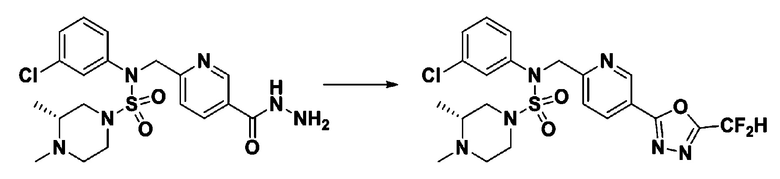
Раствор (R)-N-(3-хлорфенил)-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-3,4-диметилпиперазин-1-сульфонамида (0,090 г, 0,199 ммоль), триэтиламина (0,138 мл, 0,993 ммоль) и 2,2-дифторуксусного ангидрида (0,074 мл, 0,596 ммоль) в тетрагидрофуране (10 мл) перемешивали при 70°С в течение 12 часов, и охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида аммония, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; метанол/дихлорметан=0%-5%) с получением (R)-N-(3-хлорфенил)-N-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3,4-диметилпиперазин-1-сульфонамида в виде желтого твердого вещества (0,061 г, 59,8%).
1H ЯМР (400 МГц, CDCl3) δ 9,25-9,23 (м, 1 H), 8,39-8,36 (м, 1 H), 7,68-7,66 (м, 1 H), 7,50-7,49 (м, 1 H), 7,36-7,34 (м, 1 H), 7,34-7,21 (м, 2 H), 7,07 (с, 0,3 H), 6,94 (с, 0,5 H), 6,81 (с, 0,2 H), 5,10 (с, 2 H), 3,58-3,47 (м, 2 H), 3,00-2,95 (м, 1 H), 2,76-2,72 (м, 1 H), 2,63-2,58 (м, 1 H), 2,27 (с, 3 H), 2,24-2,19 (м, 1 H), 2,10-2,09 (м, 1 H), 1,04 (д, 3 H, J=6,3 Гц); LRMS (ES) m/z 513,3 (M++1).
Пример 69: Соединение 11826, (R)-N-(3-хлорфенил)-N-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-метил-4-(оксетан-3-ил)пиперазин-1-сульфонамид
[Стадия 1] метил (R)-6-(((N-(3-хлорфенил)-3-метил-4-(оксетан-3-ил)пиперазин)-1-сульфонамидо)метил)никотинат
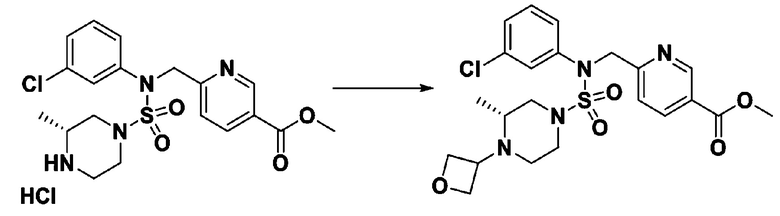
Раствор метил (R)-6-(((N-(3-хлорфенил)-3-метилпиперазин)-1-сульфонамидо)метил)никотинат гидрохлорида (0,200 г, 0,421 ммоль), и оксетан-3-она (0,061 г, 0,841 ммоль) и уксусной кислоты (0,029 мл, 0,505 ммоль) в дихлорметане (10 мл) перемешивали при комнатной температуре в течение 30 мин и смешивали с триацетоксиборгидридом натрия (0,178 г, 0,841 ммоль). Реакционную смесь перемешивали при той же температуре в течение дополнительных 12 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором бикарбоната натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; метанол/дихлорметан=0%-5%) с получением метил (R)-6-(((N-(3-хлорфенил)-3-метил-4-(оксетан-3-ил)пиперазин)-1-сульфонамидо)метил)никотината в виде желтого твердого вещества (0,100 г, 48,0%).
[Стадия 2] (R)-N-(3-хлорфенил)-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-3-метил-4-(оксетан-3-ил)пиперазин-1-сульфонамид
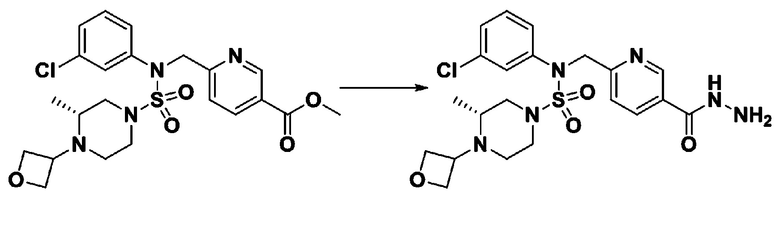
Раствор метил (R)-6-(((N-(3-хлорфенил)-3-метил-4-(оксетан-3-ил)пиперазин)-1-сульфонамидо)метил)никотината (0,100 г, 0,202 ммоль) и гидразин моногидрата (0,098 мл, 2,020 ммоль) в этаноле (10 мл) перемешивали при 90°C в течение 12 часов, и охлаждали до комнатной температуры для прекращения реакции. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Затем к концентрату добавляли воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором бикарбоната натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Неочищенный продукт использовали без дополнительной очистки ((R)-N-(3-хлорфенил)-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-3-метил-4-(оксетан-3-ил)пиперазин-1-сульфонамид, 0,085 г, 85,0%, белое твердое вещество).
[Стадия 3] Соединение 11826
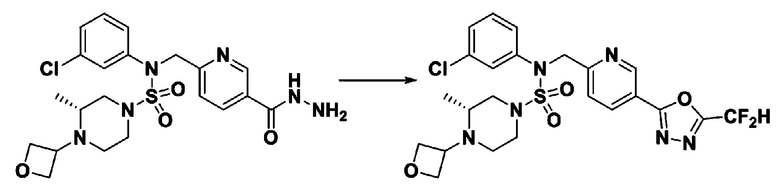
Раствор (R)-N-(3-хлорфенил)-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-3-метил-4-(оксетан-3-ил)пиперазин-1-сульфонамида (0,085 г, 0,172 ммоль), триэтиламина (0,120 мл, 0,859 ммоль) и 2,2-дифторуксусного ангидрида (0,064 мл, 0,515 ммоль) в тетрагидрофуране (10 мл) перемешивали при 70°С в течение 12 часов, и охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида аммония, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; этилацетат/гексан=0%-80%) с получением (R)-N-(3-хлорфенил)-N-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-метил-4-(оксетан-3-ил)пиперазин-1-сульфонамида в виде желтого масла (0,053 г, 55,6%).
1H ЯМР (400 МГц, CDCl3) δ 9,26-9,23 (м, 1 H), 8,39-8,36 (м, 1 H), 7,65 (д, 1 H, J=8,3 Гц), 7,50-7,49 (м, 1 H), 7,36-7,33 (м, 1 H), 7,29-7,23 (м, 2 H), 7,07 (с, 0,3 H), 6,94 (с, 0,5 H), 6,81 (с, 0,2 H), 5,09 (с, 2 H), 4,64-4,55 (м, 4 H), 3,75-3,71 (м, 1 H), 3,40-3,33 (м, 2 H), 3,17-3,15 (м, 1 H), 2,88-2,83 (м, 1 H), 2,62-2,58 (м, 1 H), 2,39-2,38 (м, 1 H), 2,15-2,10 (м, 1 H), 0,87 (д, 3 H, J=6,5 Гц); LRMS (ES) m/z 555,3 (M++1).
Пример 70: Соединение 11827, (S)-N-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3,4-диметил-N-(м-толил)пиперазин-1-сульфонамид
[Стадия 1] трет-бутил (S)-4-((1H-имидазол-1-ил)сульфонил)-2-метилпиперазин-1-карбоксилат
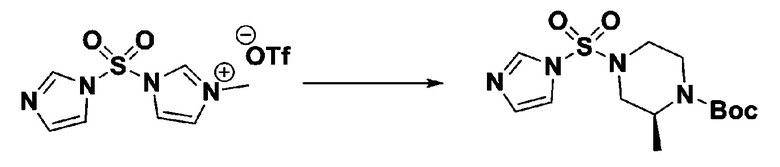
Раствор 1-((1H-имидазол-1-ил)сульфонил)-3-метил-1H-имидазол-3-ий трифторметансульфоната (8,900 г, 24,565 ммоль) и трет-бутил (S)-2-метилпиперазин-1-карбоксилата (5,412 г, 27,022 ммоль) в ацетонитриле (5 мл) перемешивали при комнатной температуре в течение 16 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида аммония, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 40 г; этилацетат/гексан=0%-50%) с получением трет-бутил (S)-4-((1H-имидазол-1-ил)сульфонил)-2-метилпиперазин-1-карбоксилата в виде желтого твердого вещества (5,050 г, 62,2%).
[Стадия 2] (S)-1-((4-(трет-бутоксикарбонил)-3-метилпиперазин-1-ил)сульфонил)-3-метил-1H-имидазол-3-ий трифторметансульфонат
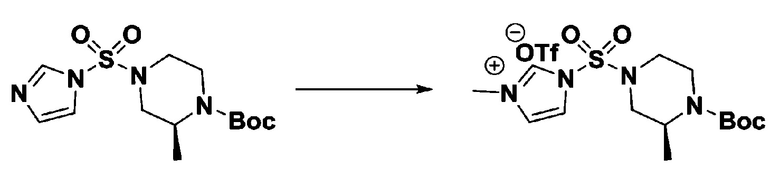
Раствор трет-бутил (S)-4-((1H-имидазол-1-ил)сульфонил)-2-метилпиперазин-1-карбоксилата (5,000 г, 15,133 ммоль) и метил трифторметансульфоната (1,660 мл, 15,133 ммоль) в дихлорметане (150 мл) перемешивали при 0°C в течение 3 часов, и концентрировали при пониженном давлении для удаления растворителя. Неочищенный продукт использовали без дополнительной очистки ((S)-1-((4-(трет-бутоксикарбонил)-3- метилпиперазин-1-ил)сульфонил)-3-метил-1H-имидазол-3-ий трифторметансульфонат, 5,800 г, 77,5%, белое твердое вещество).
[Стадия 3] трет-бутил (S)-2-метил-4-(N-(м-толил)сульфамоил)пиперазин-1-карбоксилат
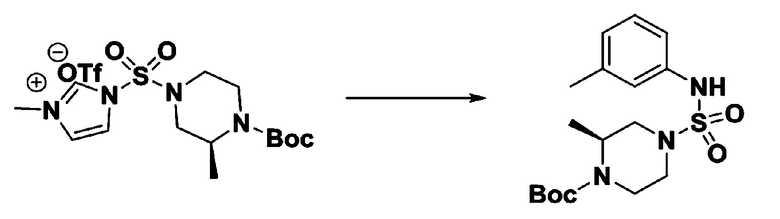
Смесь (S)-1-((4-(трет-бутоксикарбонил)-3-метилпиперазин-1-ил)сульфонил)-3-метил-1H-имидазол-3-ий трифторметансульфоната (4,000 г, 8,089 ммоль) и м-толуидин гидрохлорида (1,278 г, 8,898 ммоль) в ацетонитриле (100 мл), полученную при температуре окружающей среды, нагревали с обратным холодильником в течение 16 часов, и охлаждали до температуры окружающей среды. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида аммония, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 24 г; этилацетат/гексан=0%-30%) с получением трет-бутил (S)-2-метил-4-(N-(м-толил)сульфамоил)пиперазин-1-карбоксилата в виде желтого твердого вещества (1,160 г, 38,8%).
[Стадия 4] трет-бутил (S)-4-(N-((5-(метоксикарбонил)пиридин-2-ил)метил)-N-(м-толил)сульфамоил)-2-метилпиперазин-1-карбоксилат
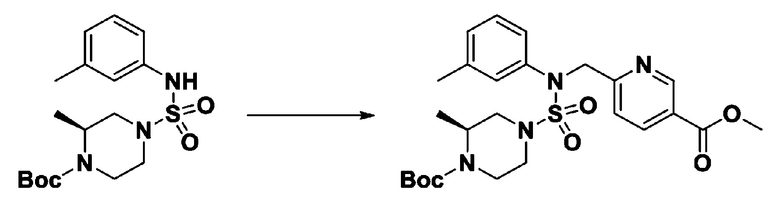
Раствор трет-бутил (S)-2-метил-4-(N-(м-толил)сульфамоил)пиперазин-1-карбоксилата (1,100 г, 2,977 ммоль) и карбоната калия (0,617 г, 4,466 ммоль) в N,N-диметилформамиде (50 мл) перемешивали при комнатной температуре в течение 30 мин и смешивали с метил-6-(бромметил)никотинатом (0,753 г, 3,275 ммоль) и иодидом калия (0,247 г, 1,489 ммоль). Реакционную смесь перемешивали при той же температуре в течение дополнительных 12 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида аммония, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 24 г; этилацетат/гексан=0%-40%) с получением трет-бутил (S)-4-(N-((5-(метоксикарбонил)пиридин-2-ил)метил)-N-(м-толил)сульфамоил)-2-метилпиперазин-1-карбоксилата в виде желтого твердого вещества (1,150 г, 74,5%).
[Стадия 5] метил (S)-6-(((3-метил-N-(м-толил)пиперазин)-1-сульфонамидо)метил)никотинат гидрохлорид
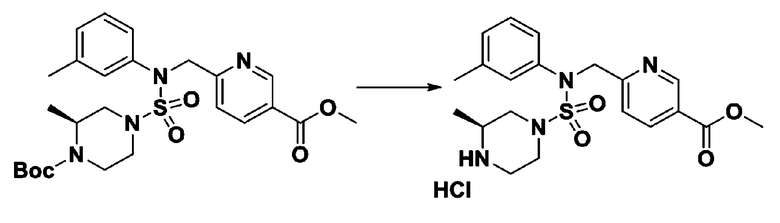
Раствор трет-бутил (S)-4-(N-((5-(метоксикарбонил)пиридин-2-ил)метил)-N-(м-толил)сульфамоил)-2-метилпиперазин-1-карбоксилата (1,100 г, 2,121 ммоль) и хлорида водорода (4,00 M раствор в 1,4-диоксане, 1,060 мл, 4,242 ммоль) в дихлорметане (30 мл) перемешивали при комнатной температуре в течение 5 часов, и концентрировали при пониженном давлении для удаления растворителя. Неочищенный продукт использовали без дополнительной очистки (метил (S)-6-(((3-метил-N-(м-толил)пиперазин)-1-сульфонамидо)метил)никотинат гидрохлорид, 0,900 г, 93,3%, желтое твердое вещество).
[Стадия 6] метил (S)-6-(((3,4-диметил-N-(м-толил)пиперазин)-1-сульфонамидо)метил)никотинат
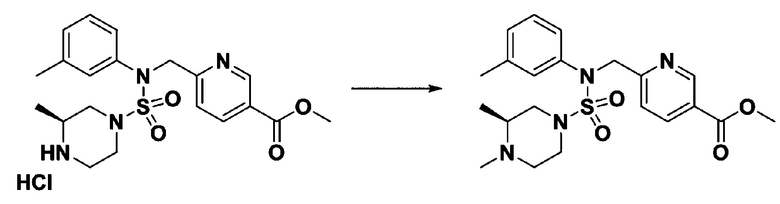
Раствор метил (S)-6-(((3-метил-N-(м-толил)пиперазин)-1-сульфонамидо)метил)никотинат гидрохлорида (0,200 г, 0,440 ммоль), параформальдегида (0,026 г, 0,879 ммоль) и уксусной кислоты (0,030 мл, 0,528 ммоль) в дихлорметане (10 мл) перемешивали при комнатной температуре в течение 30 мин и смешивали с триацетоксиборгидридом натрия (0,186 г, 0,879 ммоль). Реакционную смесь перемешивали при той же температуре в течение дополнительных 12 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором бикарбоната натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; метанол/дихлорметан=0%-5%) с получением метил (S)-6-(((3,4-диметил-N-(м-толил)пиперазин)-1-сульфонамидо)метил)никотината в виде желтого твердого вещества (0,120 г, 63,1%).
[Стадия 7] (S)-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-3,4-диметил-N-(м-толил)пиперазин-1-сульфонамид
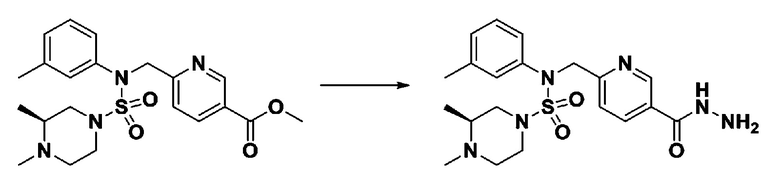
Раствор метил (S)-6-(((3,4-диметил-N-(м-толил)пиперазин)-1-сульфонамидо)метил)никотината (0,120 г, 0,277 ммоль) и гидразина моногидрата (0,135 мл, 2,774 ммоль) в этаноле (10 мл) перемешивали при 90°C в течение 12 часов, и охлаждали до комнатной температуры для прекращения реакции. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Затем к концентрату добавляли воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором бикарбоната натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Неочищенный продукт использовали без дополнительной очистки ((S)-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-3,4-диметил-N-(м-толил)пиперазин-1-сульфонамид, 0,092 г, 76,7%, белое твердое вещество).
[Стадия 8] Соединение 11827
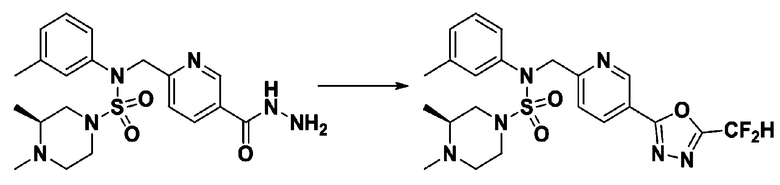
Раствор (S)-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-3,4-диметил-N-(м-толил)пиперазин-1-сульфонамида (0,092 г, 0,213 ммоль), триэтиламина (0,148 мл, 1,063 ммоль) и 2,2-дифторуксусного ангидрида (0,079 мл, 0,638 ммоль) в тетрагидрофуране (10 мл) перемешивали при комнатной температуре в течение 12 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида аммония, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; метанол/дихлорметан=0%-5%) с получением (S)-N-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3,4-диметил-N-(м-толил)пиперазин-1-сульфонамида в виде белого твердого вещества (0,067 г, 64,0%).
1H ЯМР (400 МГц, CDCl3) δ 9,21-9,20 (м, 1 H), 8,37-8,34 (м, 1 H), 7,73 (д, 1 H, J=8,2 Гц), 7,28-7,17 (м, 3 H), 7,07 (с, 0,3 H), 7,06-7,03 (м, 1 H), 6,93 (с, 0,5 H), 6,81 (с, 0,2 H), 5,12 (с, 2 H), 3,59-3,48 (м, 2 H), 3,00-2,93 (м, 1 H), 2,76-2,71 (м, 1 H), 2,63-2,57 (м, 1 H), 2,32 (с, 3 H), 2,27 (с, 3 H), 2,21-2,19 (м, 1 H), 2,12-2,08 (м, 1 H), 1,04 (д, 3 H, J=6,3 Гц); LRMS (ES) m/z 493,3 (M++1).
Пример 71: Соединение 11828, (S)-N-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-метил-4-(оксетан-3-ил)-N-(м-толил)пиперазин-1-сульфонамид
[Стадия 1] метил (S)-6-(((3-метил-4-(оксетан-3-ил)-N-(м-толил)пиперазин)-1-сульфонамидо)метил)никотинат
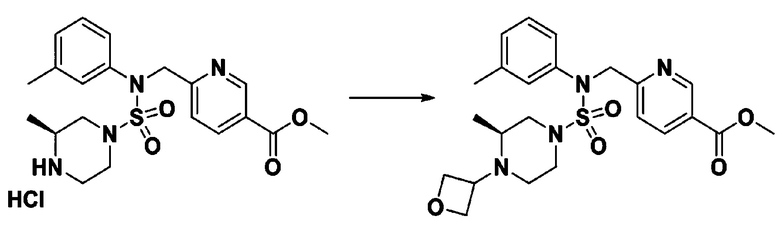
Раствор метил (S)-6-(((3-метил-N-(м-толил)пиперазин)-1-сульфонамидо)метил)никотинат гидрохлорида (0,200 г, 0,440 ммоль), оксетан-3-она (0,063 г, 0,879 ммоль) и уксусной кислоты (0,030 мл, 0,528 ммоль) в дихлорметане (10 мл) перемешивали при комнатной температуре в течение 30 мин и смешивали с триацетоксиборгидридом натрия (0,186 г, 0,879 ммоль). Реакционную смесь перемешивали при той же температуре в течение дополнительных 12 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором бикарбоната натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; метанол/дихлорметан=0%-5%) с получением метил (S)-6-(((3-метил-4-(оксетан-3-ил)-N-(м-толил)пиперазин)-1-сульфонамидо)метил)никотината в виде желтого твердого вещества (0,130 г, 62,3%).
[Стадия 2] (S)-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-3-метил-4-(оксетан-3-ил)-N-(м-толил)пиперазин-1-сульфонамид
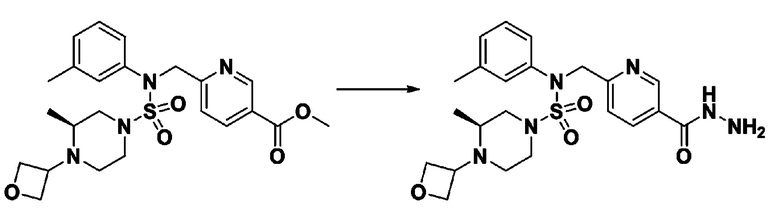
Раствор метил (S)-6-(((3-метил-4-(оксетан-3-ил)-N-(м-толил)пиперазин)-1-сульфонамидо)метил)никотината (0,130 г, 0,274 ммоль) и гидразина моногидрата (0,133 мл, 2,739 ммоль) в этаноле (10 мл) перемешивали при 90°C в течение 12 часов, и охлаждали до комнатной температуры для прекращения реакции. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Затем к концентрату добавляли воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором бикарбоната натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Неочищенный продукт использовали без дополнительной очистки ((S)-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-3-метил-4-(оксетан-3-ил)-N-(м-толил)пиперазин-1-сульфонамид, 0,100 г, 76,9%, белое твердое вещество).
[Стадия 3] Соединение 11828
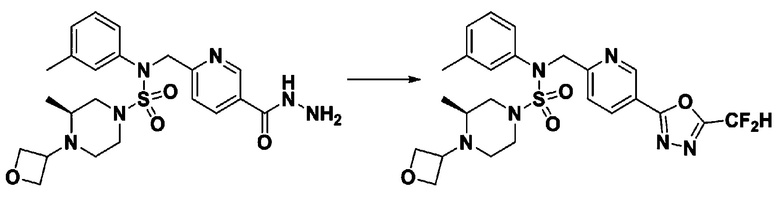
Раствор (S)-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-3-метил-4-(оксетан-3-ил)-N-(м-толил)пиперазин-1-сульфонамида (0,100 г, 0,211 ммоль), триэтиламина (0,147 мл, 1,054 ммоль) и 2,2-дифторуксусного ангидрида (0,079 мл, 0,632 ммоль) в тетрагидрофуране (10 мл) перемешивали при 70°С в течение 12 часов, и охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида аммония, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; этилацетат/гексан=0%-80%) с получением (S)-N-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-метил-4-(оксетан-3-ил)-N-(м-толил)пиперазин-1-сульфонамида в виде желтого масла (0,079 г, 70,1%).
1H ЯМР (400 МГц, CDCl3) δ 9,22-9,20 (м, 1 H), 8,37-8,35 (м, 1 H), 7,71 (д, 1 H, J=8,2 Гц), 7,28-7,20 (м, 3 H), 7,08 (с, 0,3 H), 7,07-7,05 (м, 1 H), 6,94 (с, 0,5 H), 6,81 (с, 0,2 H), 5,11 (с, 2 H), 4,65-4,56 (м, 4 H), 3,74-3,71 (м, 1 H), 3,39-3,33 (м, 2 H), 3,15-3,13 (м, 1 H), 2,87-2,82 (м, 1 H), 2,61-2,58 (м, 1 H), 2,49-2,35 (м, 1 H), 2,33 (с, 3 H), 2,15-2,10 (м, 1 H), 0,87 (д, 3 H, J=6,5 Гц); LRMS (ES) m/z 535,1 (M++1).
Пример 72: Соединение 11829, (S)-N-(3-хлорфенил)-N-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3,4-диметилпиперазин-1-сульфонамид
[Стадия 1] трет-бутил (S)-4-(N-(3-хлорфенил)сульфамоил)-2-метилпиперазин-1-карбоксилат
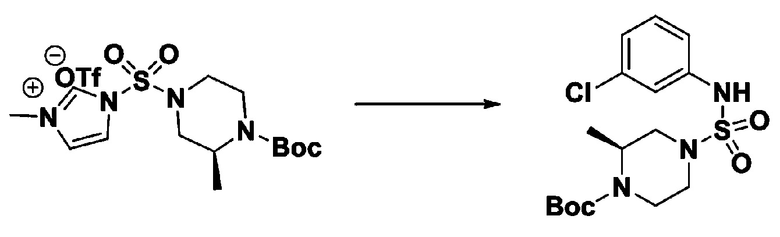
Смесь (S)-1-((4-(трет-бутоксикарбонил)-3-метилпиперазин-1-ил)сульфонил)-3-метил-1H-имидазол-3-ий трифторметансульфоната (3,000 г, 6,067 ммоль) и 3-хлоранилина (0,851 г, 6,673 ммоль) в ацетонитриле (100 мл), полученную при температуре окружающей среды, нагревали с обратным холодильником в течение 16 часов, и охлаждали до температуры окружающей среды. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида аммония, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 24 г; этилацетат/гексан=0%-30%) с получением трет-бутил (S)-4-(N-(3-хлорфенил)сульфамоил)-2-метилпиперазин-1-карбоксилата в виде желтого твердого вещества (0,660 г, 27,9%).
[Стадия 2] трет-бутил (S)-4-(N-(3-хлорфенил)-N-((5-(метоксикарбонил)пиридин-2-ил)метил)сульфамоил)-2-метилпиперазин-1-карбоксилат
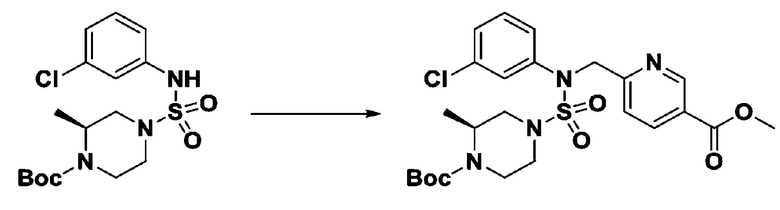
Раствор трет-бутил (S)-4-(N-(3-хлорфенил)сульфамоил)-2-метилпиперазин-1-карбоксилата (0,660 г, 1,693 ммоль) и карбоната калия (0,351 г, 2,539 ммоль) в N,N-диметилформамиде (30 мл) перемешивали при комнатной температуре в течение 30 мин и смешивали с метил-6-(бромметил)никотинатом (0,428 г, 1,862 ммоль) и иодидом калия (0,140 г, 0,846 ммоль). Реакционную смесь перемешивали при той же температуре в течение дополнительных 12 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида аммония, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 12 г; этилацетат/гексан=0%-50%) с получением трет-бутил (S)-4-(N-(3-хлорфенил)-N-((5-(метоксикарбонил)пиридин-2-ил)метил)сульфамоил)-2-метилпиперазин-1-карбоксилата в виде желтого твердого вещества (0,690 г, 75,6%).
[Стадия 3] метил (S)-6-(((N-(3-хлорфенил)-3-метилпиперазин)-1-сульфонамидо)метил)никотинат гидрохлорид
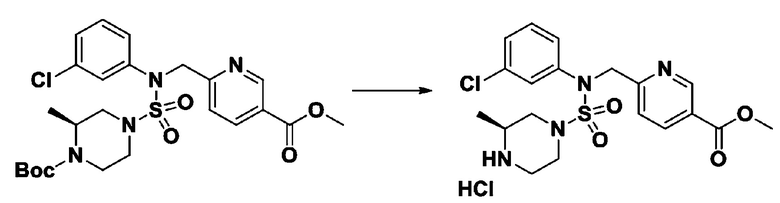
Раствор трет-бутил (S)-4-(N-(3-хлорфенил)-N-((5-(метоксикарбонил)пиридин-2-ил)метил)сульфамоил)-2-метилпиперазин-1-карбоксилата (0,690 г, 1,280 ммоль) и хлорида водорода (4,00 M раствор в 1,4-диоксане, 0,640 мл, 2,560 ммоль) в дихлорметане (30 мл) перемешивали при комнатной температуре в течение 5 часов, и концентрировали при пониженном давлении для удаления растворителя. Неочищенный продукт использовали без дополнительной очистки (метил (S)-6-(((N-(3-хлорфенил)-3-метилпиперазин)-1-сульфонамидо)метил)никотинат гидрохлорид, 0,460 г, 75,6%, желтое твердое вещество).
[Стадия 4] метил (S)-6-(((N-(3-хлорфенил)-3,4-диметилпиперазин)-1-сульфонамидо)метил)никотинат
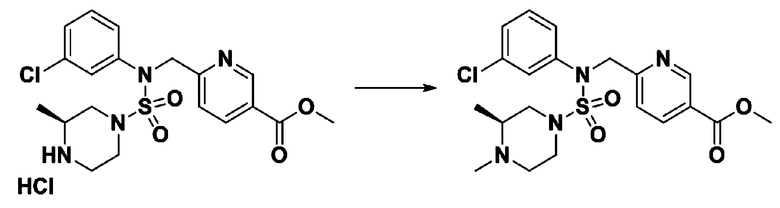
Раствор метил (S)-6-(((N-(3-хлорфенил)-3-метилпиперазин)-1-сульфонамидо)метил)никотинат гидрохлорида (0,200 г, 0,421 ммоль), параформальдегида (0,025 г, 0,841 ммоль) и уксусной кислоты (0,029 мл, 0,505 ммоль) в дихлорметане (10 мл) перемешивали при комнатной температуре в течение 30 мин и смешивали с триацетоксиборгидридом натрия (0,178 г, 0,841 ммоль). Реакционную смесь перемешивали при той же температуре в течение дополнительных 12 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором бикарбоната натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; метанол/дихлорметан=0%-5%) с получением метил (S)-6-(((N-(3-хлорфенил)-3,4-диметилпиперазин)-1-сульфонамидо)метил)никотината в виде желтого твердого вещества (0,140 г, 73,5%).
[Стадия 5] (S)-N-(3-хлорфенил)-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-3,4-диметилпиперазин-1-сульфонамид
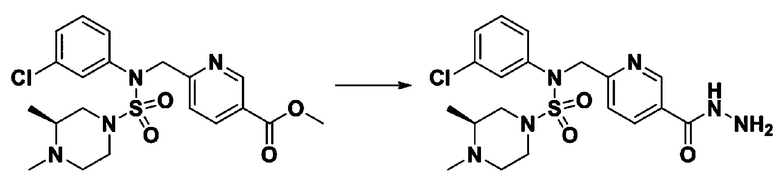
Раствор метил (S)-6-(((N-(3-хлорфенил)-3,4-диметилпиперазин)-1-сульфонамидо)метил)никотината (0,140 г, 0,309 ммоль) и гидразина моногидрата (0,150 мл, 3,091 ммоль) в этаноле (10 мл) перемешивали при 90°C в течение 12 часов, и охлаждали до комнатной температуры для прекращения реакции. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Затем к концентрату добавляли воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором бикарбоната натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Указанное в заголовке соединение использовали без дополнительной очистки ((S)-N-(3-хлорфенил)-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-3,4-диметилпиперазин-1-сульфонамид, 0,110 г, 78,6%, белое твердое вещество).
[Стадия 6] Соединение 11829
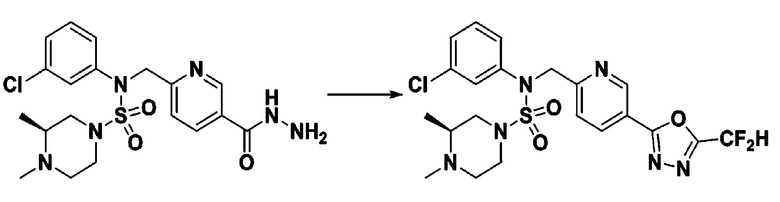
Раствор (S)-N-(3-хлорфенил)-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-3,4-диметилпиперазин-1-сульфонамида (0,110 г, 0,243 ммоль), триэтиламина (0,169 мл, 1,214 ммоль) и 2,2-дифторуксусного ангидрида (0,091 мл, 0,729 ммоль) в тетрагидрофуране (10 мл) перемешивали при 70°С в течение 12 часов, охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида аммония, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; метанол/дихлорметан=0%-5%) с получением (S)-N-(3-хлорфенил)-N-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3,4-диметилпиперазин-1-сульфонамида в виде желтого твердого вещества (0,085 г, 68,2%).
1H ЯМР (400 МГц, CDCl3) δ 9,25-9,23 (м, 1 H), 8,39-8,36 (м, 1 H), 7,67 (д, 1 H, J=8,2 Гц), 7,50-7,49 (м, 1 H), 7,36-7,33 (м, 1 H), 7,27-7,21 (м, 2 H), 7,07 (с, 0,3 H), 6,94 (с, 0,5 H), 6,81 (с, 0,2 H), 5,10 (с, 2 H), 3,58-3,48 (м, 2 H), 3,02-2,95 (м, 1 H), 2,77-2,73 (м, 1 H), 2,64-2,58 (м, 1 H), 2,27 (с, 3 H), 2,24-2,19 (м, 1 H), 2,18-2,09 (м, 1 H), 1,04 (д, 3 H, J=6,3 Гц); LRMS (ES) m/z 513,3 (M++1).
Пример 73: Соединение 11830, (S)-N-(3-хлорфенил)-N-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-метил-4-(оксетан-3-ил)пиперазин-1-сульфонамид
[Стадия 1] метил (S)-6-(((N-(3-хлорфенил)-3-метил-4-(оксетан-3-ил)пиперазин)-1-сульфонамидо)метил)никотинат
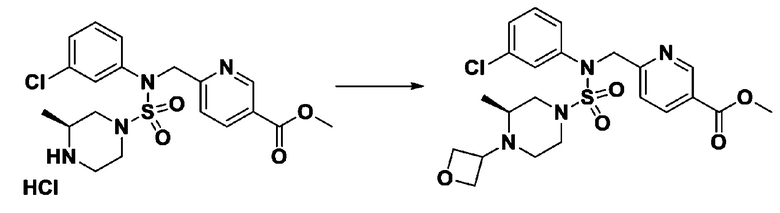
Раствор метил (S)-6-(((N-(3-хлорфенил)-3-метилпиперазин)-1-сульфонамидо)метил)никотинат гидрохлорида (0,200 г, 0,421 ммоль), оксетан-3-она (0,059 г, 0,841 ммоль) и уксусной кислоты (0,029 мл, 0,505 ммоль) в дихлорметане (10 мл) перемешивали при комнатной температуре в течение 30 мин и смешивали с триацетоксиборгидридом натрия (0,178 г, 0,841 ммоль). Реакционную смесь перемешивали при той же температуре в течение дополнительных 12 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором бикарбоната натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; метанол/дихлорметан=0%-5%) с получением метил (S)-6-(((N-(3-хлорфенил)-3-метил-4-(оксетан-3-ил)пиперазин)-1-сульфонамидо)метил)никотината в виде желтого твердого вещества (0,110 г, 52,8%).
[Стадия 2] (S)-N-(3-хлорфенил)-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-3-метил-4-(оксетан-3-ил)пиперазин-1-сульфонамид
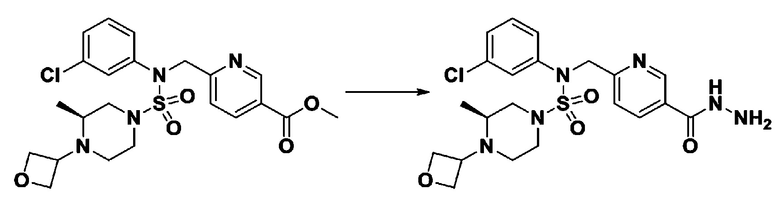
Раствор метил (S)-6-(((N-(3-хлорфенил)-3-метил-4-(оксетан-3-ил)пиперазин)-1-сульфонамидо)метил)никотината (0,110 г, 0,222 ммоль) и гидразин моногидрата (0,108 мл, 2,222 ммоль) в этаноле (10 мл) перемешивали при 90°C в течение 12 часов, и охлаждали до комнатной температуры для прекращения реакции. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Затем к концентрату добавляли воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором бикарбоната натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Указанное в заголовке соединение использовали без дополнительной очистки ((S)-N-(3-хлорфенил)-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-3-метил-4-(оксетан-3-ил)пиперазин-1-сульфонамид, 0,083 г, 75,5%, белое твердое вещество).
[Стадия 3] Соединение 11830
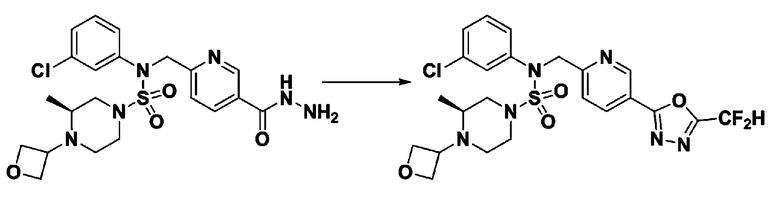
Раствор (S)-N-(3-хлорфенил)-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-3-метил-4-(оксетан-3-ил)пиперазин-1-сульфонамида (0,083 г, 0,168 ммоль), триэтиламина (0,117 мл, 0,838 ммоль) и 2,2-дифторуксусного ангидрида (0,063 мл, 0,503 ммоль) в тетрагидрофуране (10 мл) перемешивали при 70°С в течение 12 часов, и охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида аммония, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; этилацетат/гексан=0%-80%) с получением (S)-N-(3-хлорфенил)-N-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-метил-4-(оксетан-3-ил)пиперазин-1-сульфонамида в виде белого твердого вещества (0,059 г, 63,4%).
1H ЯМР (400 МГц, CDCl3) δ 9,26-9,24 (м, 1 H), 8,39-8,36 (м, 1 H), 7,66 (д, 1 H, J=8,2 Гц), 7,50-7,49 (м, 1 H), 7,36-7,33 (м, 1 H), 7,30-7,23 (м, 2 H), 7,07 (с, 0,2 H), 6,95 (с, 0,5 H), 6,82 (с, 0,3 H), 5,09 (с, 2 H), 4,65-4,56 (м, 4 H), 3,75-3,71 (м, 1 H), 3,40-3,34 (м, 2 H), 3,15-3,14 (м, 1 H), 2,88-2,83 (м, 1 H), 2,62-2,59 (м, 1 H), 2,40-2,38 (м, 1 H), 2,15-2,10 (м, 1 H), 0,87 (д, 3 H, J=6,5 Гц); LRMS (ES) m/z 555,3 (M++1).
Пример 74: Соединение 11831, N-(3-хлорфенил)-4-циклобутил-N-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)пиперазин-1-сульфонамид
[Стадия 1] трет-бутил 4-(N-(3-хлорфенил)сульфамоил)пиперазин-1-карбоксилат
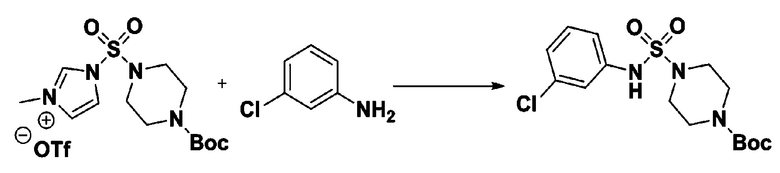
Раствор 1-((4-(трет-бутоксикарбонил)пиперазин-1-ил)сульфонил)-3-метил-1H-имидазол-3-ий трифторметансульфоната (4,750 г, 9,886 ммоль) и 3-хлоранилина (1,241 мл, 11,863 ммоль) в ацетонитриле (50 мл) перемешивали при комнатной температуре в течение 18 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 40 г; этилацетат/гексан=0%-80%) с получением трет-бутил 4-(N-(3-хлорфенил)сульфамоил)пиперазин-1-карбоксилата в виде белого твердого вещества (2,770 г, 74,5%).
[Стадия 2] трет-бутил 4-(N-(3-хлорфенил)-N-((5-(метоксикарбонил)пиридин-2-ил)метил)сульфамоил)пиперазин-1-карбоксилат
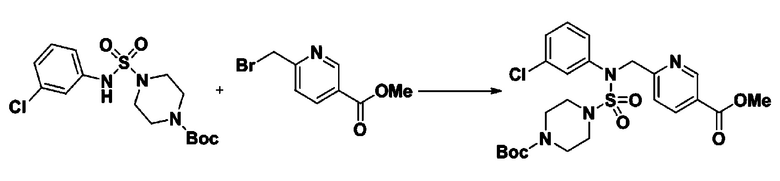
Раствор трет-бутил 4-(N-(3-хлорфенил)сульфамоил)пиперазин-1-карбоксилата (1,000 г, 2,660 ммоль) и гидрида натрия (60,00%, 0,213 г, 5,321 ммоль) в N,N-диметилформамиде (30 мл) перемешивали при 0°C в течение 10 мин, и смешивали с метил 6-(бромметил)никотинатом (0,734 г, 3,193 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение дополнительных 18 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 12 г; этилацетат/гексан=0%-80%) с получением трет-бутил 4-(N-(3-хлорфенил)-N-((5-(метоксикарбонил)пиридин-2-ил)метил)сульфамоил)пиперазин-1-карбоксилата в виде желтого твердого вещества (1,200 г, 85,9%).
[Стадия 3] метил 6-((N-(3-хлорфенил)пиперазин-1-сульфонамидо)метил)никотинат гидрохлорид
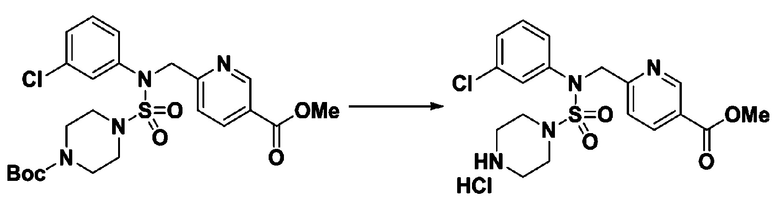
Раствор трет-бутил 4-(N-(3-хлорфенил)-N-((5-(метоксикарбонил)пиридин-2-ил)метил)сульфамоил)пиперазин-1-карбоксилата (1,900 г, 3,619 ммоль) в дихлорметане (50 мл) смешивали при комнатной температуре с хлористоводородной кислотой (4,00 M раствор в диоксане, 3,619 мл, 14,476 ммоль), и перемешивали при той же температуре в течение 3 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Указанное в заголовке соединение использовали без дополнительной очистки (метил 6-((N-(3-хлорфенил)пиперазин-1-сульфонамидо)метил)никотинат гидрохлорид, 1,200 г, 71,9%, желтое твердое вещество).
[Стадия 4] метил 6-(((N-(3-хлорфенил)-4-циклобутилпиперазин)-1-сульфонамидо)метил)никотинат
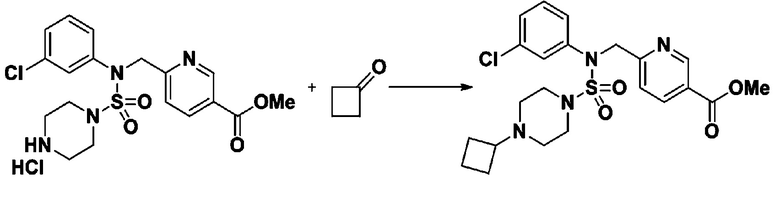
Раствор метил 6-((N-(3-хлорфенил)пиперазин-1-сульфонамидо)метил)никотинат гидрохлорида (0,300 г, 0,650 ммоль) и циклобутанона (0,058 мл, 0,780 ммоль) в дихлорметане (12 мл) перемешивали при комнатной температуре в течение 10 минут, и смешивали с триацетоксиборгидридом натрия (0,413 г, 1,951 ммоль). Реакционную смесь перемешивали при той же температуре в течение дополнительных 18 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; метанол/дихлорметан=0%-15%) с получением метил 6-(((N-(3-хлорфенил)-4-циклобутилпиперазин)-1-сульфонамидо)метил)никотината в виде желтого твердого вещества (0,174 г, 55,9%).
[Стадия 5] N-(3-хлорфенил)-4-циклобутил-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)пиперазин-1-сульфонамид
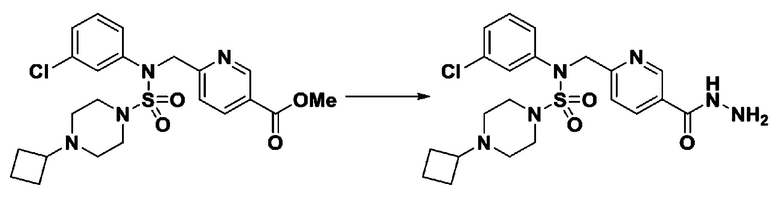
метил 6-(((N-(3-хлорфенил)-4-циклобутилпиперазин)-1-сульфонамидо)метил)никотинат (0,174 г, 0,363 ммоль) и гидразин моногидрат (0,530 мл, 10,898 ммоль) смешивали при комнатной температуре в этаноле (10 мл), и затем смесь перемешивали при 80°C в течение 18 часов. Реакционную смесь охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Указанное в заголовке соединение использовали без дополнительной очистки (N-(3-хлорфенил)-4-циклобутил-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)пиперазин-1-сульфонамид, 0,168 г, 96,5%, желтое твердое вещество).
[Стадия 6] Соединение 11831
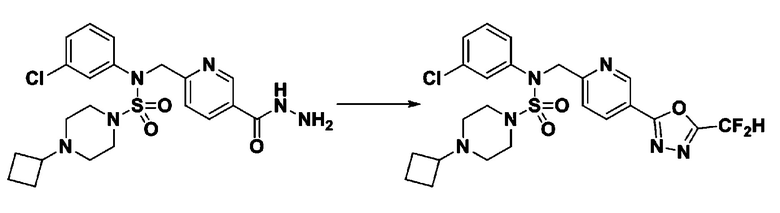
Раствор N-(3-хлорфенил)-4-циклобутил-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)пиперазин-1-сульфонамида (0,168 г, 0,351 ммоль) и триэтиламина (0,244 мл, 1,754 ммоль) в тетрагидрофуране (15 мл) смешивали при комнатной температуре с 2,2-дифторуксусным ангидридом (0,131 мл, 1,052 ммоль), и перемешивали при 80°С в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 12 г; метанол/дихлорметан=0%-15%) с получением N-(3-хлорфенил)-4-циклобутил-N-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)пиперазин-1-сульфонамида в виде желтого твердого вещества (0,082 г, 43,4%).
1H ЯМР (400 МГц, CD3OD) δ 9,23 (д, 1H, J=1,7 Гц,), 8,36 (дд, 1H, J=8,2, 2,2 Гц), 7,66 (д, 1H, J=8,2 Гц), 7,49-7,48 (м, 1H), 7,36-7,33 (м, 1H), 7,28-7,20 (м, 2H), 7,07 (с, 0,25H), 6,94 (с, 0,5H), 6,81 (с, 0,25H), 5,09 (с, 2H), 3,32-3,28 (м, 4H), 2,83-2,70 (м, 1H), 2,34-2,30 (м, 4H), 2,05-1,99 (м, 2H), 1,88-1,84 (м, 2H), 1,73-1,63 (м, 2H); LRMS (ES) m/z 539,2 (M++1)
Пример 75: Соединение 11832, N-(3-хлорфенил)-N-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4-(оксетан-3-ил)пиперазин-1-сульфонамид
[Стадия 1] метил 6-(((N-(3-хлорфенил)-4-(оксетан-3-ил)пиперазин)-1-сульфонамидо)метил)никотинат
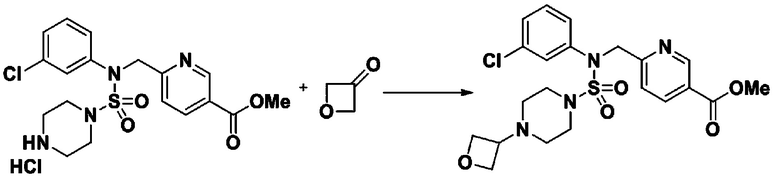
Раствор метил 6-((N-(3-хлорфенил)пиперазин-1-сульфонамидо)метил)никотинат гидрохлорида (0,300 г, 0,650 ммоль) и оксетан-3-она (0,050 мл, 0,780 ммоль) в дихлорметане (15 мл) перемешивали при комнатной температуре в течение 10 минут, и смешивали с триацетоксиборгидридом натрия (0,413 г, 1,951 ммоль). Реакционную смесь перемешивали при той же температуре в течение дополнительных 18 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; метанол/дихлорметан=0%-15%) с получением метил 6-(((N-(3-хлорфенил)-4-(оксетан-3-ил)пиперазин)-1-сульфонамидо)метил)никотината в виде желтого твердого вещества (0,211 г, 67,5%).
[Стадия 2] N-(3-хлорфенил)-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-4-(оксетан-3-ил)пиперазин-1-сульфонамид
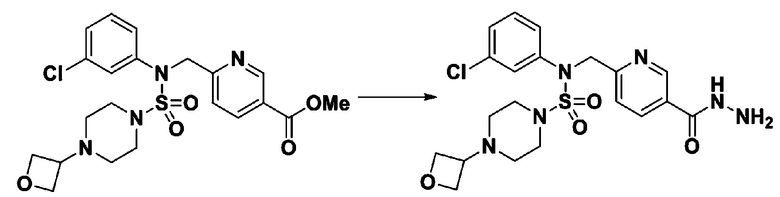
метил 6-(((N-(3-хлорфенил)-4-(оксетан-3-ил)пиперазин)-1-сульфонамидо)метил)никотинат (0,211 г, 0,439 ммоль) и гидразин моногидрат (0,640 мл, 13,161 ммоль) смешивали при комнатной температуре в этаноле (10 мл), и затем смесь перемешивали при 80°C в течение 18 часов. Реакционную смесь охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Указанное в заголовке соединение использовали без дополнительной очистки (N-(3-хлорфенил)-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-4-(оксетан-3-ил)пиперазин-1-сульфонамид, 0,188 г, 89,1%, желтое твердое вещество).
[Стадия 3] Соединение 11832
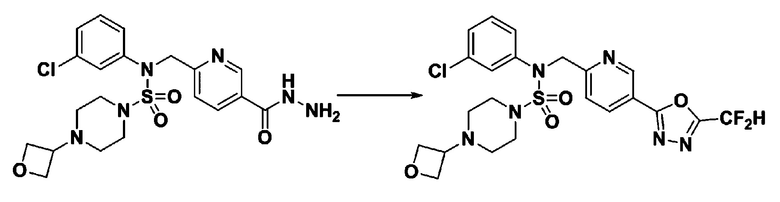
Раствор N-(3-хлорфенил)-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-4-(оксетан-3-ил)пиперазин-1-сульфонамида (0,188 г, 0,391 ммоль) и триэтиламина (0,272 мл, 1,954 ммоль) в тетрагидрофуране (15 мл) смешивали при комнатной температуре с 2,2-дифторуксусным ангидридом (0,146 мл, 1,173 ммоль), и перемешивали при 80°С в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; метанол/дихлорметан=0%-15%) с получением N-(3-хлорфенил)-N-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4-(оксетан-3-ил)пиперазин-1-сульфонамида в виде желтого твердого вещества (0,094 г, 44,5%).
1H ЯМР (400 МГц, CD3OD) δ 9,23 (д, 1H, J=1,7 Гц,), 8,37 (дд, 1H, J=8,2, 2,2 Гц), 7,65 (д, 1H, J=8,2 Гц), 7,50-7,47 (м, 1H), 7,36-7,33 (м, 1H), 7,30-7,22 (м, 2H), 7,07 (с, 0,25H), 6,94 (с, 0,5H), 6,81 (с, 0,25H), 5,09 (с, 2H), 4,66-4,63 (м, 2H), 4,56-4,53 (м, 2H), 3,50-3,44 (м, 1H), 3,31-3,29 (м, 4H), 2,31-2,29 (м, 4H); LRMS (ES) m/z 541,3 (M++1).
Пример 76: Соединение 11833, N-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4-(оксетан-3-ил)-N-(м-толил)пиперазин-1-сульфонамид
[Стадия 1] трет-бутил 4-(N-(м-толил)сульфамоил)пиперазин-1-карбоксилат
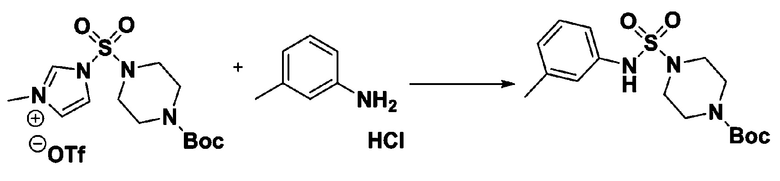
Раствор 1-((4-(трет-бутоксикарбонил)пиперазин-1-ил)сульфонил)-3-метил-1H-имидазол-3-ий трифторметансульфоната (4,750 г, 9,886 ммоль) и м-толуидин гидрохлорида (1,704 г, 11,863 ммоль) в ацетонитриле (50 мл) перемешивали при комнатной температуре в течение 18 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 12 г; этилацетат/гексан=0%-30%) с получением трет-бутил 4-(N-(м-толил)сульфамоил)пиперазин-1-карбоксилата в виде белого твердого вещества (2,110 г, 60,0%).
[Стадия 2] трет-бутил 4-(N-((5-(метоксикарбонил)пиридин-2-ил)метил)-N-(м-толил)сульфамоил)пиперазин-1-карбоксилат
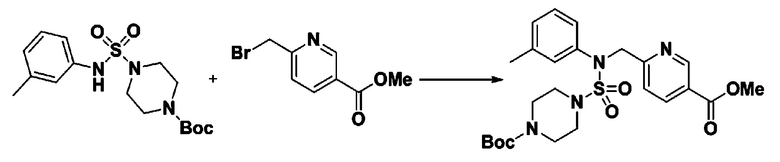
Раствор трет-бутил 4-(N-(м-толил)сульфамоил)пиперазин-1-карбоксилата (1,000 г, 2,813 ммоль) и гидрида натрия (60,00%, 0,225 г, 5,627 ммоль) в N,N-диметилформамиде (30 мл) перемешивали при 0°C в течение 10 минут, и смешивали с метил 6-(бромметил)никотинатом (0,777 г, 3,376 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение дополнительных 18 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 40 г; этилацетат/гексан=0%-80%) с получением трет-бутил 4-(N-((5-(метоксикарбонил)пиридин-2-ил)метил)-N-(м-толил)сульфамоил)пиперазин-1-карбоксилата в виде желтого твердого вещества (1,170 г, 82,4%).
[Стадия 3] метил 6-((N-(м-толил)пиперазин-1-сульфонамидо)метил)никотинат гидрохлорид
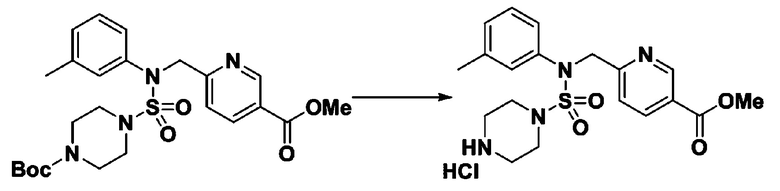
Раствор трет-бутил 4-(N-((5-(метоксикарбонил)пиридин-2-ил)метил)-N-(м-толил)сульфамоил)пиперазин-1-карбоксилата (1,300 г, 2,576 ммоль) в дихлорметане (50 мл) смешивали при комнатной температуре с хлористоводородной кислотой (4,00 M раствор в диоксане, 2,576 мл, 10,305 ммоль), и перемешивали при той же температуре в течение 3 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Указанное в заголовке соединение использовали без дополнительной очистки (метил 6-((N-(м-толил)пиперазин-1-сульфонамидо)метил)никотинат гидрохлорид, 0,980 г, 86,3%, красное твердое вещество).
[Стадия 4] метил 6-(((4-(оксетан-3-ил)-N-(м-толил)пиперазин)-1-сульфонамидо)метил)никотинат
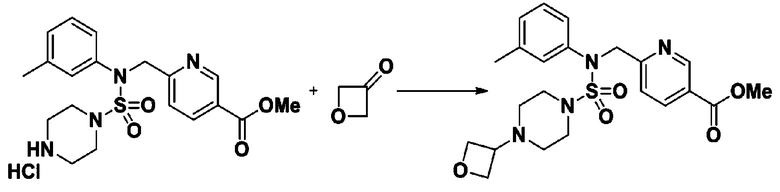
Раствор метил 6-((N-(м-толил)пиперазин-1-сульфонамидо)метил)никотинат гидрохлорида (0,300 г, 0,680 ммоль) и оксетан-3-она (0,053 мл, 0,816 ммоль) в дихлорметане (15 мл) перемешивали при комнатной температуре в течение 10 минут, и смешивали с триацетоксиборгидридом натрия (0,433 г, 2,041 ммоль). Реакционную смесь перемешивали при той же температуре в течение дополнительных 18 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; метанол/дихлорметан=0%-15%) с получением метил 6-(((4-(оксетан-3-ил)-N-(м-толил)пиперазин)-1-сульфонамидо)метил)никотината в виде желтого твердого вещества (0,248 г, 79,1%).
[Стадия 5] N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-4-(оксетан-3-ил)-N-(м-толил)пиперазин-1-сульфонамид
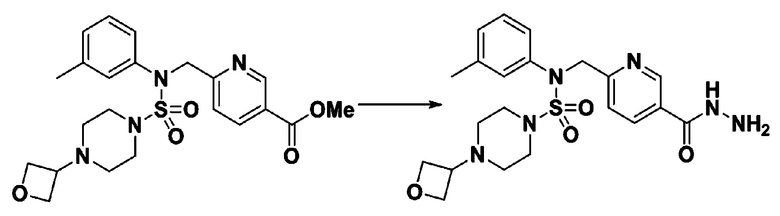
Метил 6-(((4-(оксетан-3-ил)-N-(м-толил)пиперазин)-1-сульфонамидо)метил)никотинат (0,248 г, 0,538 ммоль) и гидразин моногидрат (0,785 мл, 16,155 ммоль) смешивали при комнатной температуре в этаноле (10 мл), и затем смесь перемешивали при 80°C в течение 18 часов. Реакционную смесь охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Указанное в заголовке соединение использовали без дополнительной очистки (N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-4-(оксетан-3-ил)-N-(м-толил)пиперазин-1-сульфонамид, 0,177 г, 71,4%, желтое твердое вещество).
[Стадия 6] Соединение 11833
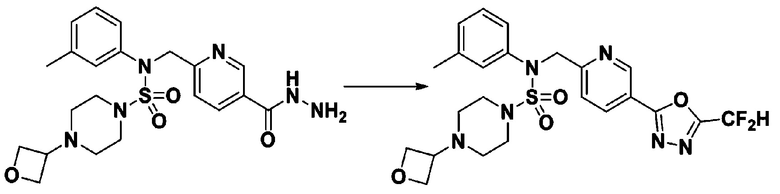
Раствор N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-4-(оксетан-3-ил)-N-(м-толил)пиперазин-1-сульфонамида (0,177 г, 0,384 ммоль) и триэтиламина (0,268 мл, 1,922 ммоль) в тетрагидрофуране (15 мл) смешивали при комнатной температуре с 2,2-дифторуксусным ангидридом (0,143 мл, 1,153 ммоль), и перемешивали при 80°С в течение 1 часа. Реакционную смесь охлаждали до комнатной температуры для прекращения реакции. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток хроматографировали (SiO2, картридж 4 г; метанол/дихлорметан=0%-15%) с получением N-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4-(оксетан-3-ил)-N-(м-толил)пиперазин-1-сульфонамида в виде желтого твердого вещества (0,130 г, 65,0%).
1H ЯМР (400 МГц, CD3OD) δ 9,20-9,19 (м, 1H), 8,35 (дд, 1H, J=8,2, 2,2 Гц), 7,70 (д, 1H, J=8,2 Гц,), 7,26 (с, 1H), 7,24-7,18 (м, 2H), 7,06-7,05 (м, 1H), 7,03 (с, 0,25H), 6,93 (с, 0,5H), 6,81 (с, 0,25H), 5,10 (с, 2H), 4,66-4,62 (м, 2H), 4,56-4,53 (м, 2H), 3,50-3,43 (м, 1H), 3,30-3,28 (м, 4H), 2,37-2,29 (м, 7H); LRMS (ES) m/z 521,4 (M++1).
Измерение активности соединений настоящего изобретения и протокол анализа
Экспериментальный пример 1: анализ ингибирования ферментативной активности HDAC ( in vitro )
Для исследования селективности HDAC6 соединений формулы I по настоящему изобретению с помощью анализа ингибирования ферментативной активности HDAC1 и HDAC6, эксперимент проводили с использованием обычного вещества в качестве контроля.
Ферментативную активность HDAC измеряли с использованием набора HDAC Fluorimetric Drug Discovery (BML-AK511, 516, Enzo Life Science). Для теста ферментативной активности HDAC1 в качестве источника фермента использовали рекомбинантный HDAC1 человека (BML-SE456), а в качестве субстрата использовали Fluor de Lysⓡ-ʺSIRT1 (BNL-KI177). 5-кратное разбавление соединения высевали в 96-луночный планшет, и затем в каждую лунку планшета добавляли 0,3 мкг фермента и 10 мкМ субстрата и давали возможность взаимодействовать при 30°С в течение 60 минут. Затем к этому добавляли Fluor de Lysⓡ-Developer II (BML-KI176) и давали возможность взаимодействовать в течение 30 минут, после чего измеряли значение флуоресценции (Ex 360, Em 460) с использованием многофункционального ридера для планшетов (Flexstation 3, Molecular Device). Фермент HDAC6 тестировали с использованием рекомбинантного HDAC6 человека (382180) (Calbiochem) в соответствии с тем же протоколом, что и метод определения активности фермента HDAC1. На основе полученных значений, каждое значение IC50 рассчитывали с использованием программы GraphPad Prism4.0.
[Таблица 2] Результаты анализа ингибирования ферментативной активности HDAC
Как можно видеть в таблице 2 выше, производные 1,3,4-оксадиазолсульфамида, их стереоизомеры или их фармацевтически приемлемые соли по настоящему изобретению показали приблизительно 142 до приблизительно 7142 раз более высокую селективную ингибирующую активность HDAC6 в анализах ингибирования активности HDAC1 и HDAC6.
Экспериментальный пример 2: Анализ влияния HDAC6-специфических ингибиторов на аксональный транспорт митохондрий (in vitro)
Был проведен анализ влияния HDAC6-специфических ингибиторов на аксональный транспорт митохондрий. В частности, чтобы исследовать, селективно ли соединения, представленные формулой I, по настоящему изобретению, ингибируют активность HDAC6 для увеличения ацетилирования тубулина, который является основным субстратом HDAC6, тем самым улучшая скорость аксонального переноса митохондрий, снижаемую при обработке бета-амилоида в аксонах нейрона, проводили сравнительный эксперимент с использованием соединения, которое уже было разработано в качестве контроля.
Нейроны гиппокампа из эмбрионов крыс Sprague-Dawley (SD) в 17-18 день эмбрионального развития (E17-18) культивировали в планшете с покрытием внеклеточным матриксом для визуализации в течение 7 дней и затем обрабатывали 1 мкМ бета-амилоидных пептидов. Через 24 часа нейроны обрабатывали соединениями в течение 3 часов на 8-й день in vitro и обрабатывали MitoTracker Red CMXRos (Life Technologies, NY, USA) в течение последних 5 минут для окрашивания митохондрий. Аксональный транспорт окрашенных митохондрий был отображен с использованием конфокального микроскопа (Leica SP8; Leica Microsystems, UK) с интервалом в 1 секунду в течение 1 минуты, а скорость переноса в секунду каждого митохондрия определяли с использованием программного обеспечения для анализа IMARIS (BITPLANE, Zurich, Швейцария).
В результате было обнаружено, что производные 1,3,4-оксадиазолсульфамида, их стереоизомеры или фармацевтически приемлемые соли по настоящему изобретению улучшают скорость аксонального переноса митохондрий.
Изобретение относится к новым соединениям формулы I, их стереоизомерам или их фармацевтически приемлемым солям, а также к фармацевтическим композициям на их основе и способу лечения опосредованных заболеваний. Технический результат: получены новые соединения, обладающие ингибирующей активностью в отношении гистондеацетилазы. 4 н. и 6 з.п. ф-лы, 2 табл., 78 пр.
[Формула I]
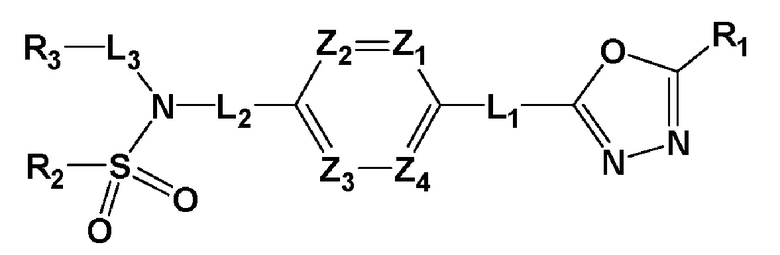
1. Производное 1,3,4-оксадиазолсульфамида, представленное следующей формулой I, его стереоизомер или его фармацевтически приемлемая соль:
[Формула I]
где L1, L2 или L3 каждый независимо представляет собой связь или -(C1-C2 алкилен)-;
Z1-Z4 каждый независимо представляет собой N или CRZ, где RZ представляет собой -H или -X;
R1 представляет собой -CX2H или -CX3;
R2 представляет собой
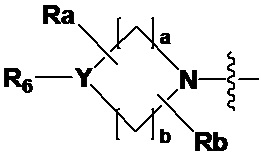 ,
, 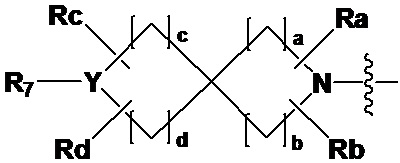
или -NR8R9,
где Y представляет собой -N-, -O- или -S(=O)2-,
a-d каждый независимо представляет собой целое число 1, 2 или 3,
Ra-Rd каждый независимо представляет собой -H или -(C1-C4 алкил),
когда Y представляет собой -N-, R6 и R7 каждый независимо представляет собой -H, -(C1-C4 алкил), -C(=O)-(C1-C4 алкил), -C(=O)-CF2H, -S(=O)2-(C1-C4 алкил), -(C3-C7 циклоалкил) или -(C2-C4 гетероциклоалкил), содержащий один атом О в кольце,
и, когда Y представляет собой -O- или -S(=O)2-, R6 и R7 отсутствуют,
R8 и R9 каждый независимо представляет собой -H, -(C1-C4 алкил) или -(C1-C4 алкил)-(6-членный гетероциклоалкил), где 6-членный гетероциклоалкил содержит два атома N в кольце, где по меньшей мере один H из -(C1-C4 алкил)-(6-членного гетероциклоалкила) может быть замещен -(C1-C4 алкилом), -C(=O)-(C1-C4 алкилом), -S(=O)2-(C1-C4 алкилом) или оксетанилом; и
R3 представляет собой -C6-арил, -пиридинил, где по меньшей мере один H из –С6-арила или -пиридинила может быть замещен -X или -(C1-C4 алкил); и
X представляет собой F или Cl.
2. Производное 1,3,4-оксадиазолсульфамида, представленное формулой I, его стереоизомер или его фармацевтически приемлемая соль по п.1,
где L1 или L3 каждый независимо представляет собой связь;
L2 представляет собой -(C1 алкилен)-;
Z1-Z4 каждый независимо представляет собой N или CRZ, где RZ представляет собой -H или -X;
R1 представляет собой -CX2H или -CX3;
R2 представляет собой
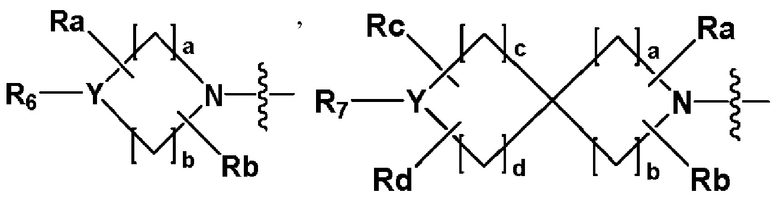 или -NR8R9,
или -NR8R9,
где Y представляет собой -N-, -O- или -S(=O)2-,
a-d каждый независимо представляет собой целое число 1 или 2,
Ra-Rd каждый независимо представляет собой -H или -(C1-C4 алкил),
когда Y представляет собой -N-, R6 и R7 каждый независимо представляет собой -H, -(C1-C4 алкил), -C(=O)-(C1-C4 алкил), -S(=O)2-(C1-C4 алкил), -(C3-C7 циклоалкил) или -(C2-C4 гетероциклоалкил), содержащий один атом О в кольце,
и, когда Y представляет собой -O- или -S(=O)2-, R6 и R7 отсутствуют,
каждый R8 и R9 независимо представляет собой -Н, (С1-С4 алкил) или -(С1-С4 алкил)-(6-членный гетероциклоалкил), где 6-членный гетероциклоалкил содержит два атома N в кольце, где по меньшей мере один H из -(C1-C4 алкил)-(6-членного гетероциклоалкила) может быть замещен -(C1-C4 алкилом), -C(=O)-(C1-C4 алкилом), -S(=O)2-(C1-C4 алкилом) или оксетанилом;
R3 представляет собой –С6-арил или -пиридинил, где по меньшей мере один Н из –С6-арила или -пиридинила может быть замещен -X; и
X представляет собой F или Cl.
3. Производное 1,3,4-оксадиазолсульфамида, представленное формулой I, его стереоизомер или его фармацевтически приемлемая соль по п. 2,
где L1 или L3 каждый независимо представляет собой связь;
L2 представляет собой -(C1 алкилен)-;
Z1-Z4 каждый независимо представляет собой N или CRZ, где RZ представляет собой -H или -X;
R1 представляет собой -CF2H или -CF3;
R2 представляет собой
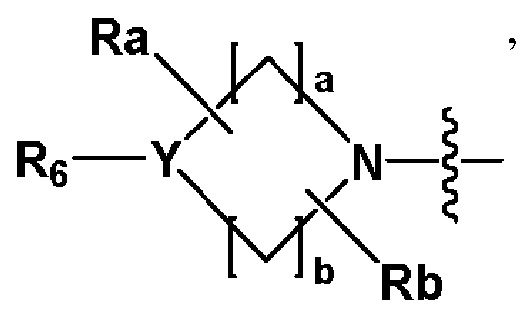
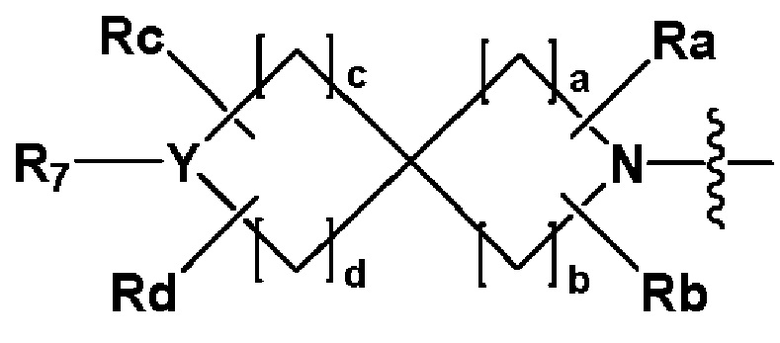 ,
,
где Y представляет собой -N- или -S(=O)2-,
a-d каждый независимо представляет собой целое число 1 или 2,
Ra-Rd каждый независимо представляет собой -H или -(C1-C4 алкил),
когда Y представляет собой -N-, R6 и R7, каждый независимо представляет собой -H, -(C1-C4 алкил), -C(=O)-(C1-C4 алкил), -S(=O)2-(C1-C4 алкил), -(C3-C7 циклоалкил) или -(C2-C4 гетероциклоалкил), содержащий один атом О в кольце,
и, когда Y представляет собой -S(=O)2-, R6 и R7 отсутствуют;
R3 представляет собой –С6-арил или -пиридинил, где по меньшей мере один Н из –С6-арила или -пиридинила может быть замещен -X; и
X представляет собой F или Cl.
4. Производное 1,3,4-оксадиазолсульфамида, представленное формулой I, его стереоизомер или его фармацевтически приемлемая соль по п. 3,
где L1 или L3 каждый независимо представляет собой связь;
L2 представляет собой -(C1 алкилен)-;
Z1-Z4 каждый независимо представляет собой N или CRZ, где RZ представляет собой -H или -X;
R1 представляет собой -CF2H или -CF3;
R2 представляет собой
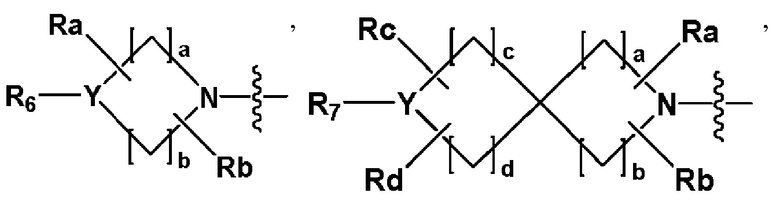
где Y представляет собой -N- или -S(=O)2-,
a и b равны 2, c и d равны 1,
Ra-Rd каждый независимо представляет собой -H или -(C1-C4 алкил),
когда Y представляет собой -N-, R6 и R7 каждый независимо представляет собой -H, -(C1-C4 алкил), -C(=O)-(C1-C4 алкил), -S(=O)2-(C1-C4 алкил), -(C3-C7 циклоалкил) или -(C2-C4 гетероциклоалкил), содержащий один атом О в кольце,
и, когда Y представляет собой -S(=O)2-, R6 и R7 отсутствуют;
R3 представляет собой –С6-арил или -пиридинил, где по меньшей мере один Н из –С6-арила или -пиридинила может быть замещен -F; и
X представляет собой F или Cl.
1. Производное 1,3,4-оксадиазолсульфамида, представленное формулой I, его стереоизомер или его фармацевтически приемлемая соль по п. 1, где соединение, представленное формулой I, выбрано из группы, состоящей из соединений, описанных в следующей таблице:
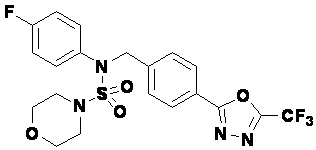
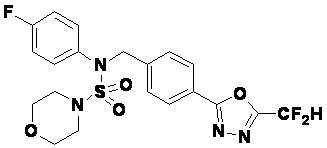
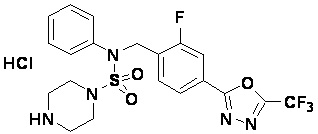
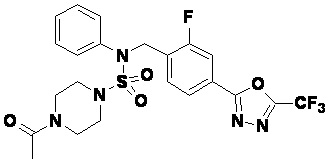
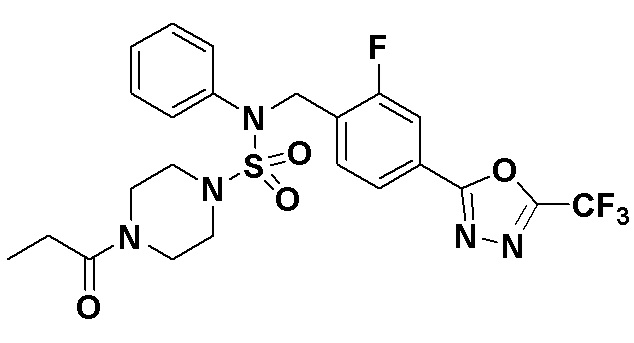
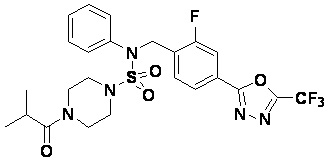
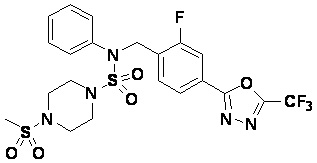
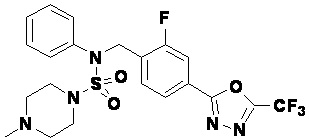
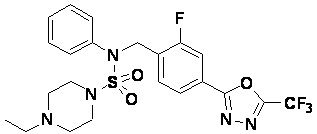
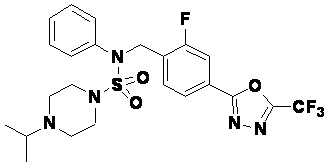
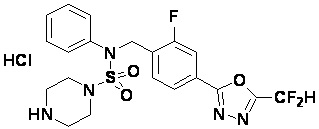
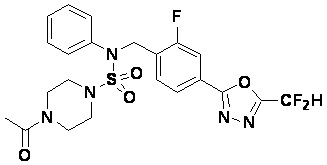
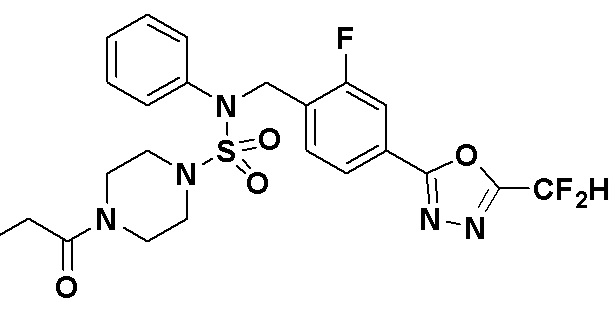
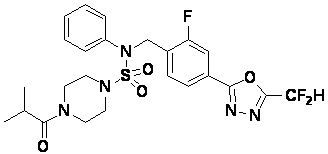
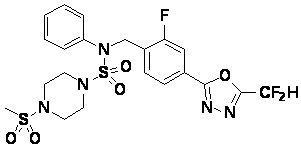
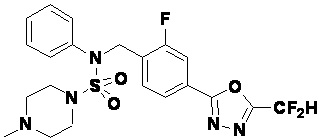
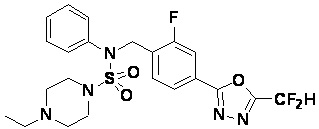
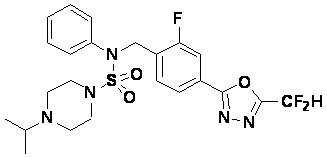
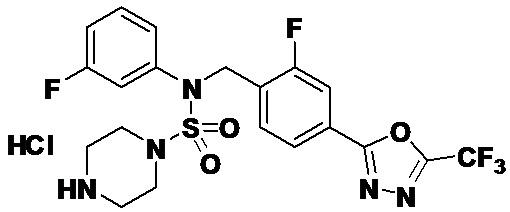
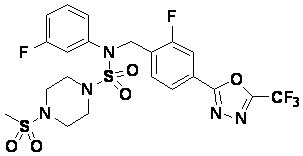
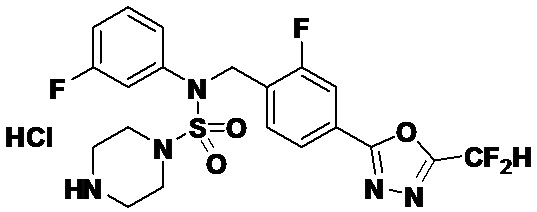
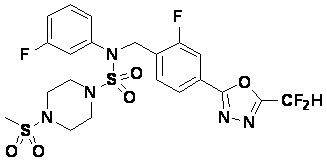
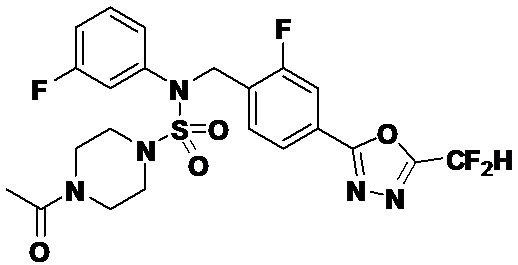
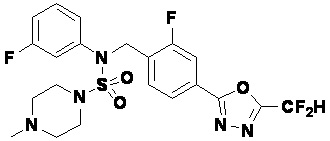
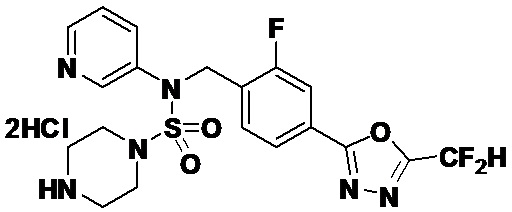
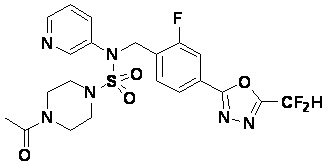
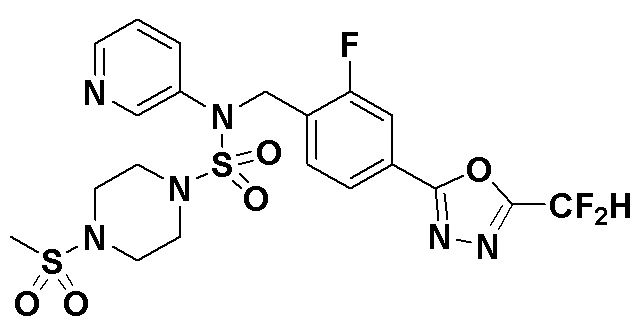
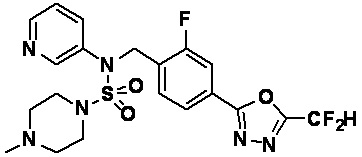
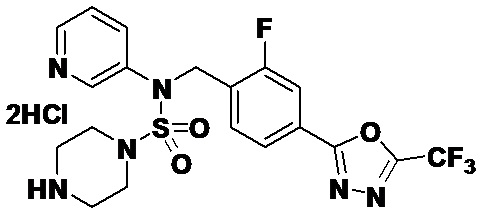
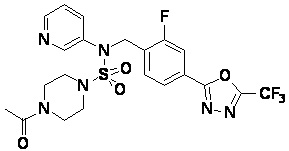
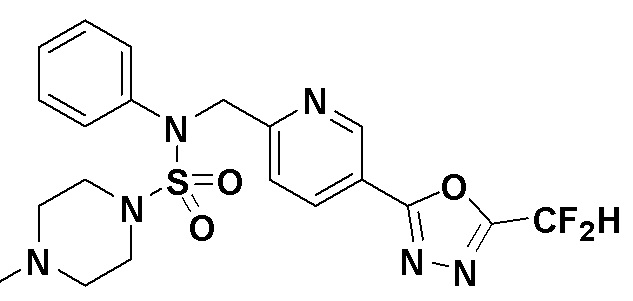
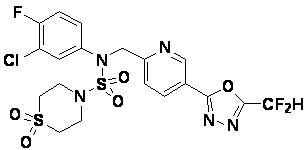
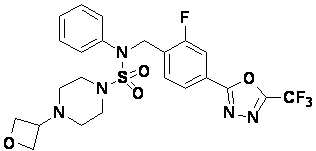
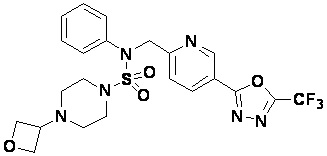
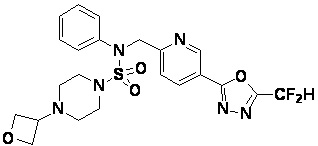
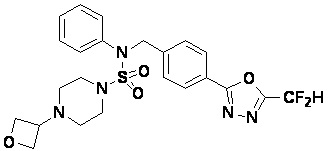
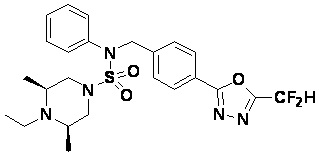
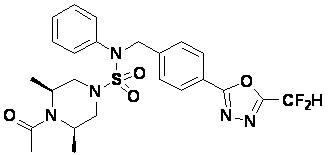
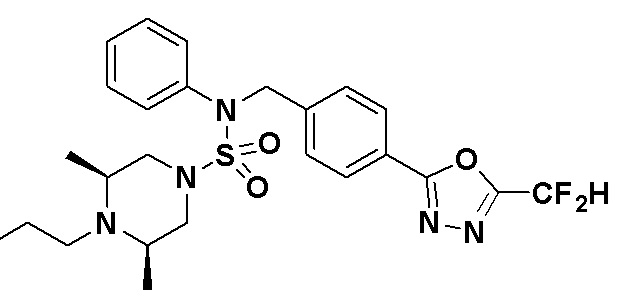
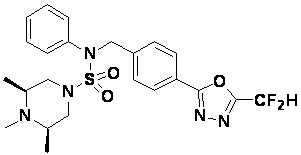
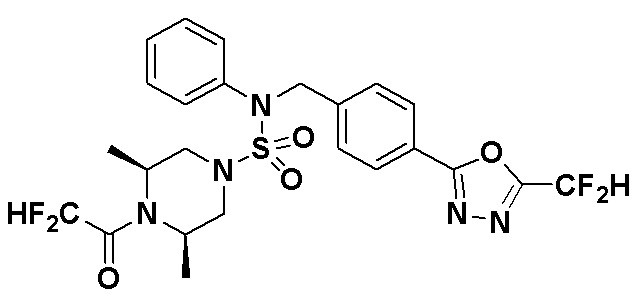
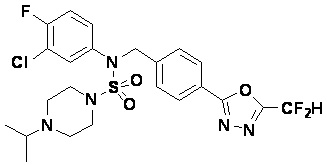
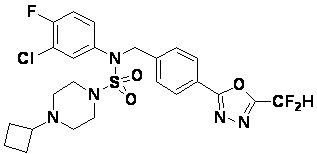
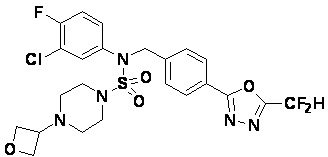
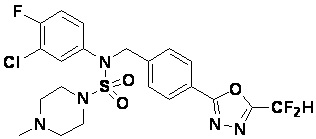
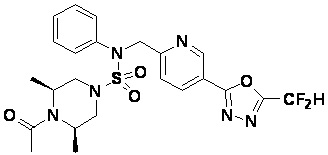
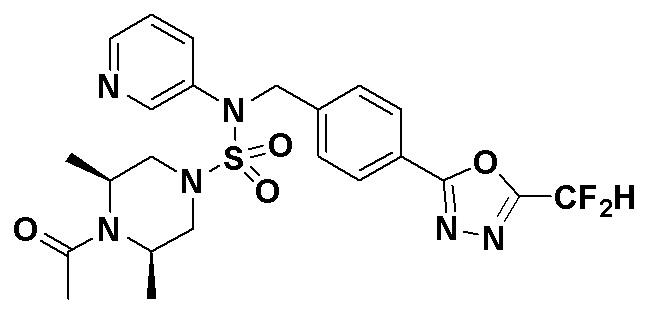
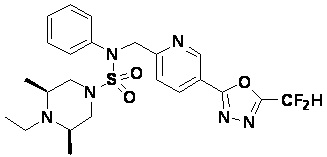
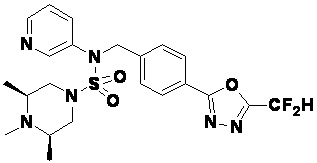
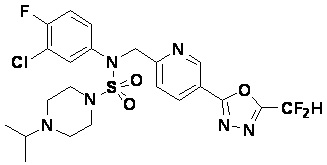
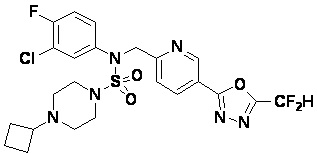
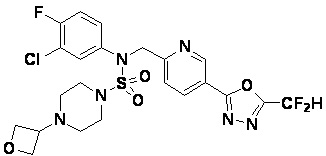
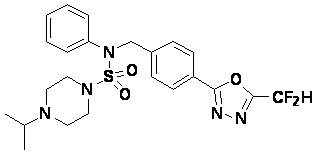
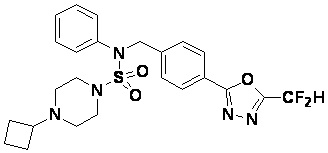
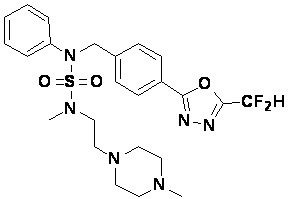
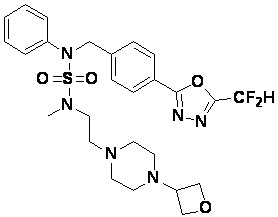
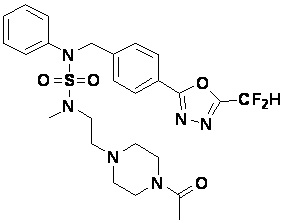
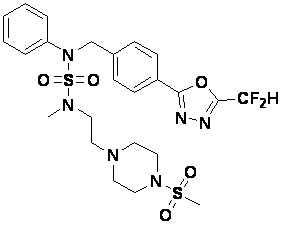
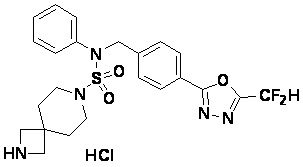
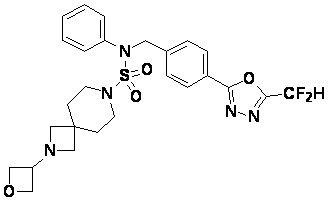
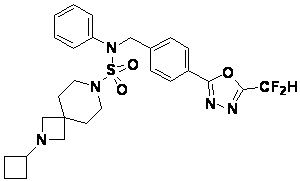
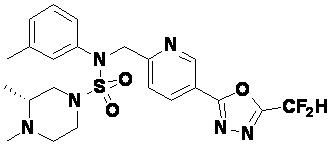
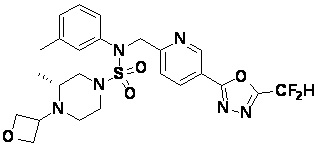
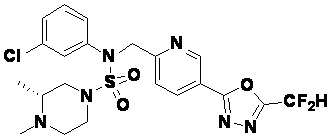
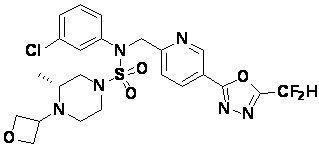
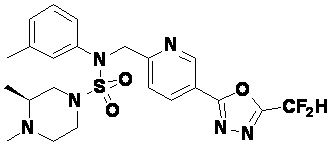
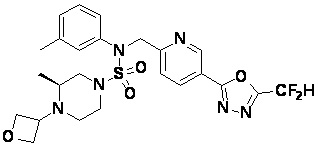
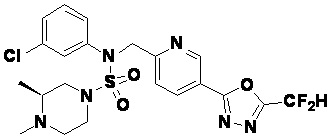
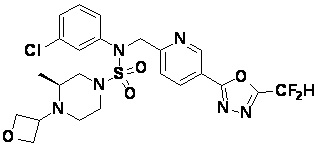
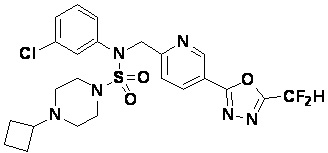
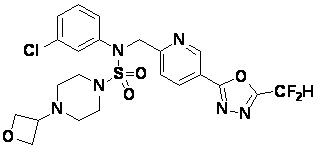
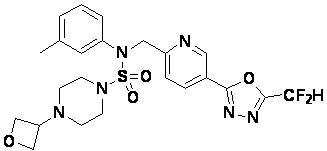
6. Производное 1,3,4-оксадиазолсульфамида, представленное формулой I, его стереоизомер или его фармацевтически приемлемая соль по п.5, где соединение, представленное формулой I, выбрано из группы, состоящей из соединений, описанных в следующей таблице:
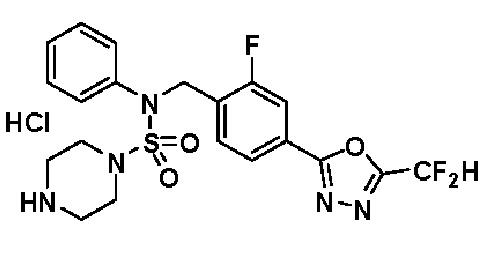
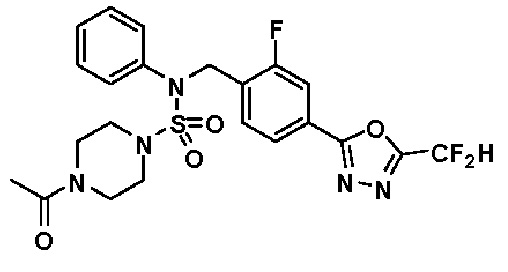
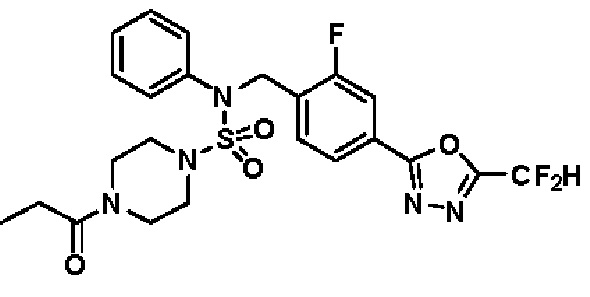
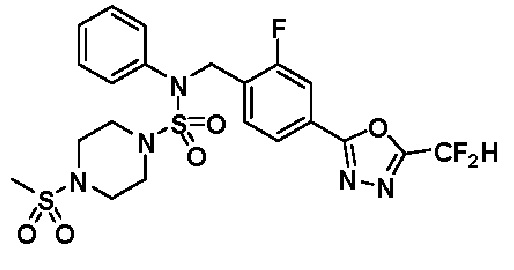
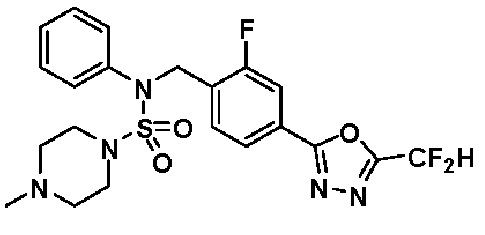
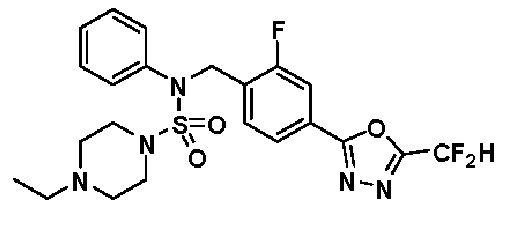
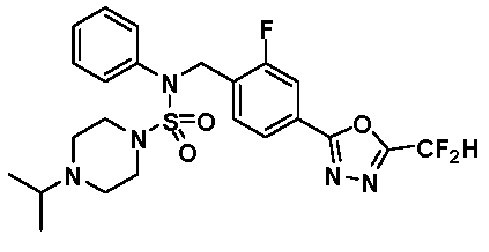
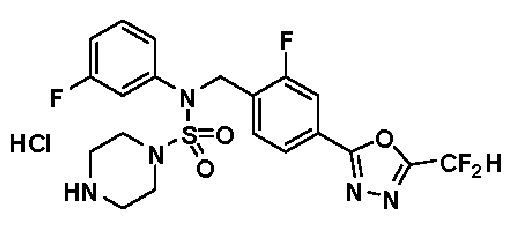
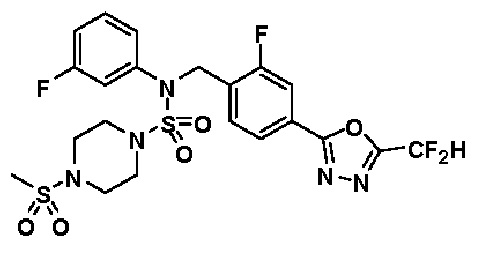
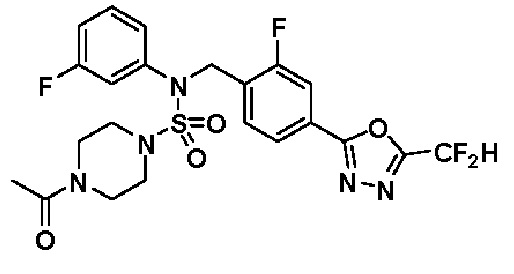
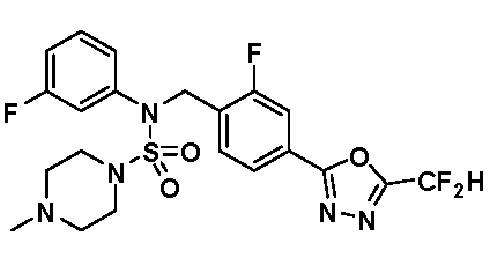
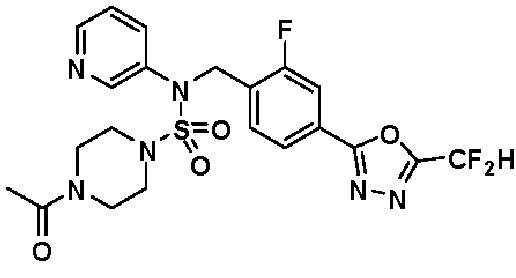
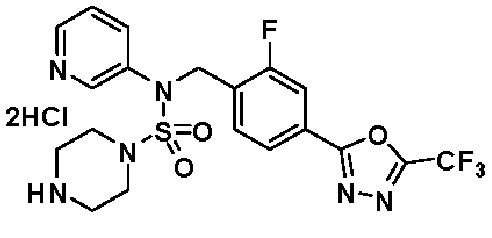
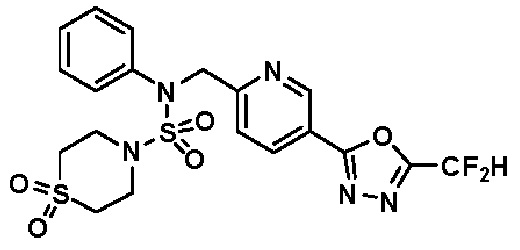
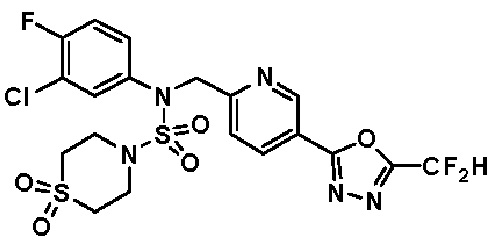
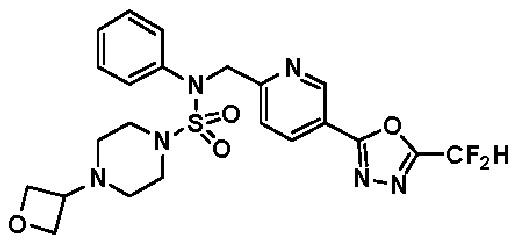
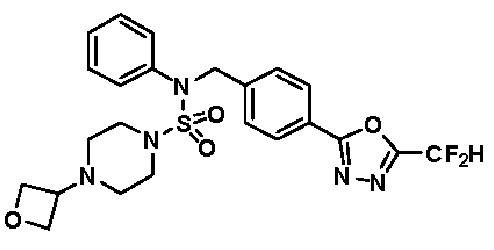
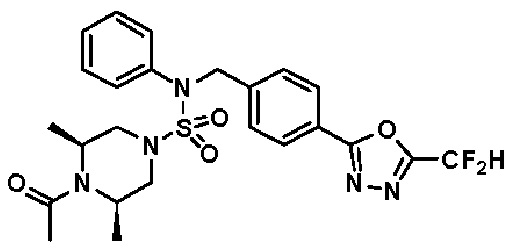
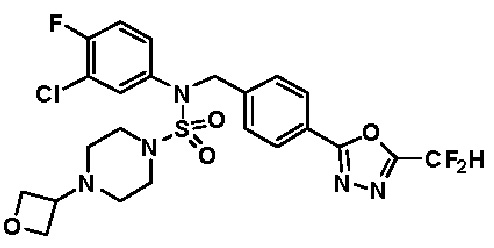
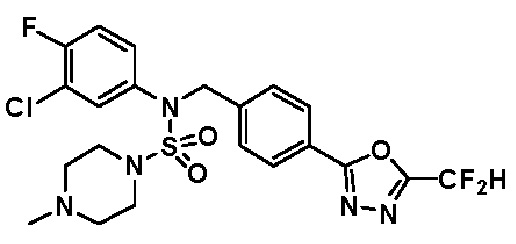
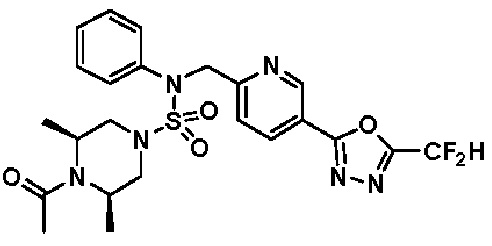
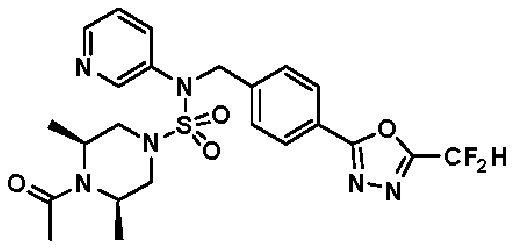
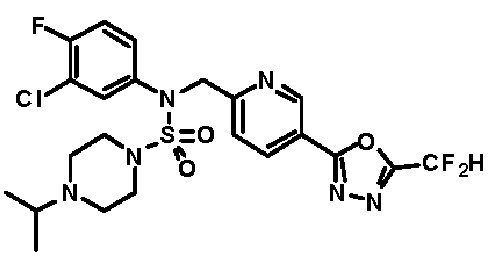
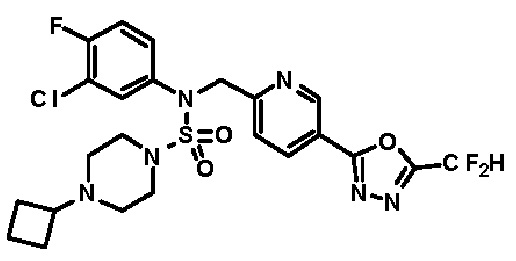
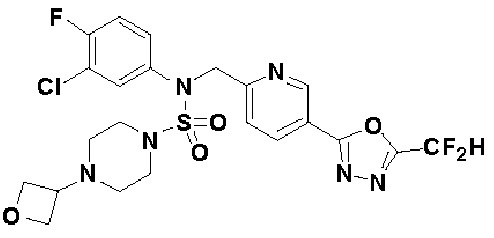
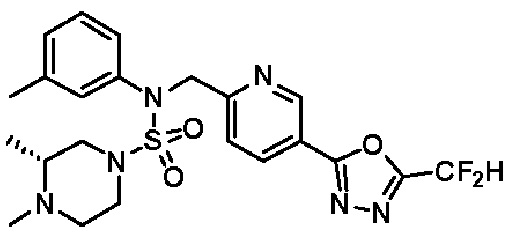
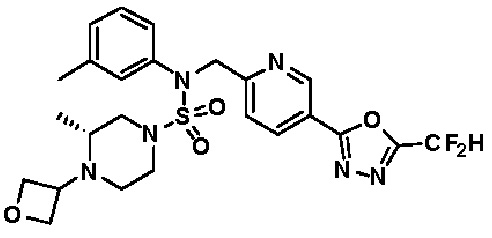
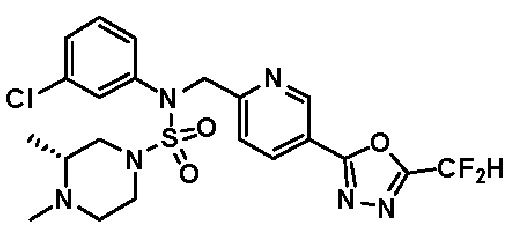
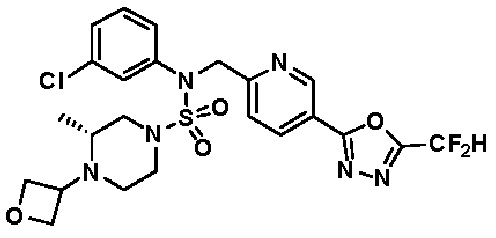
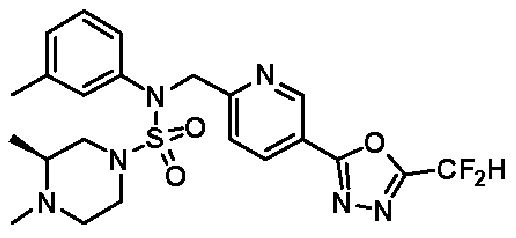
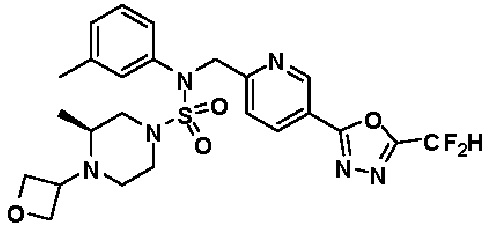
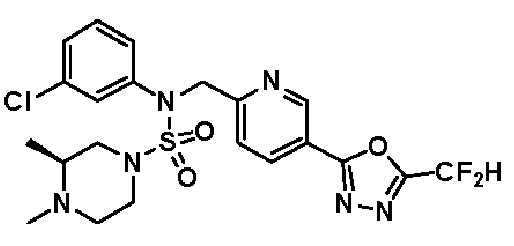
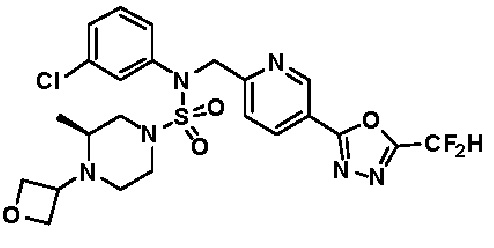
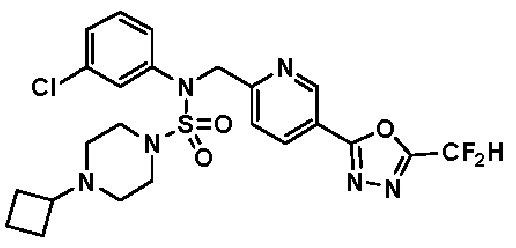
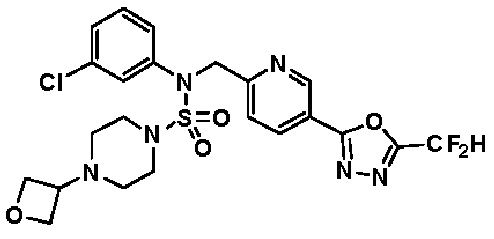
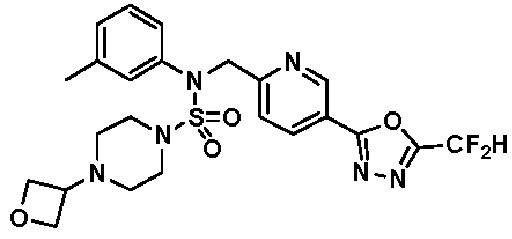
7. Производное 1,3,4-оксадиазолсульфамида, представленное формулой I, его стереоизомер или его фармацевтически приемлемая соль по п. 6, где соединение, представленное формулой I, выбрано из группы, состоящей из соединений, описанных в следующей таблице:
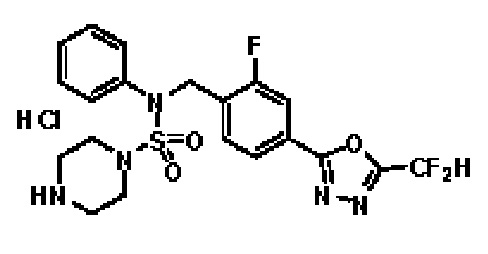
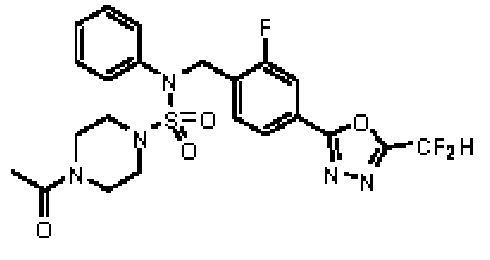
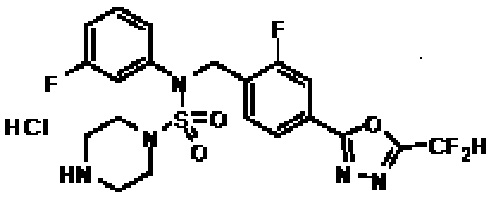
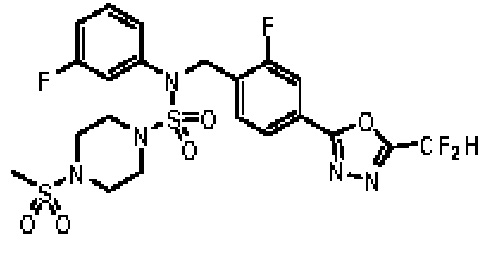
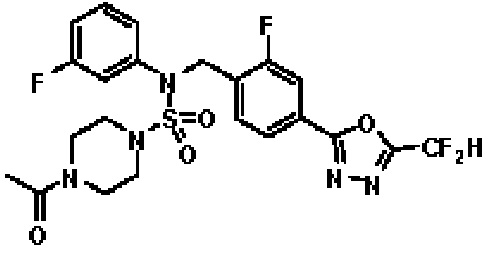
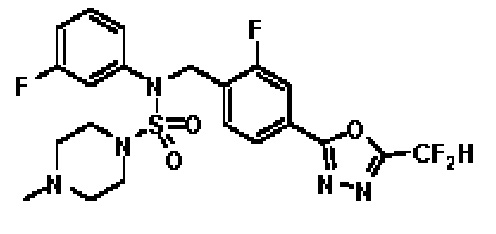
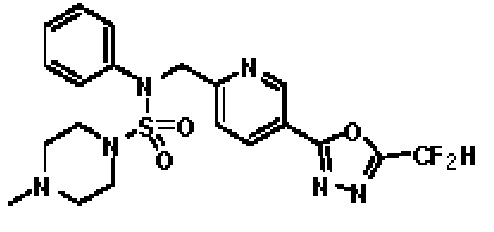
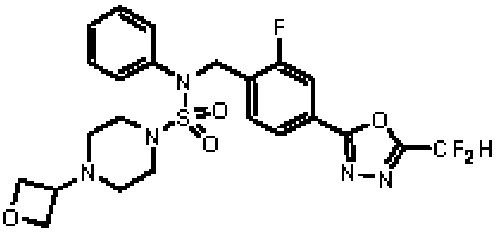
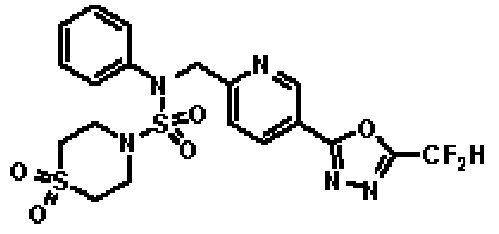
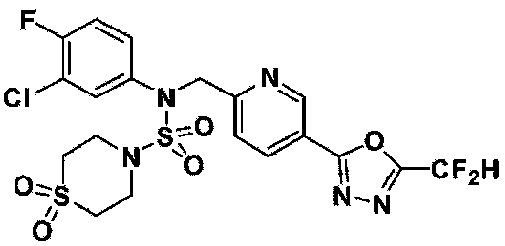
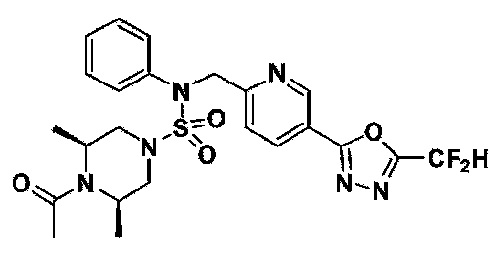
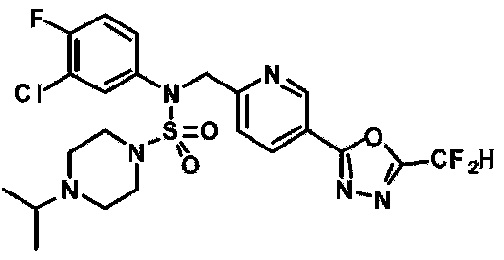
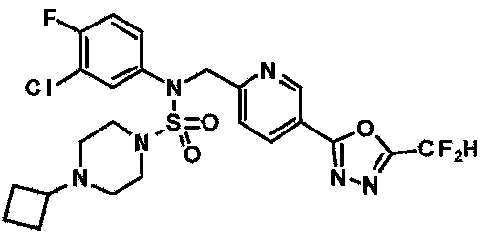
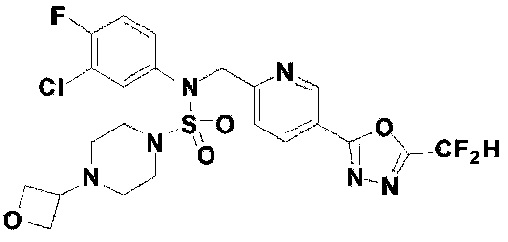
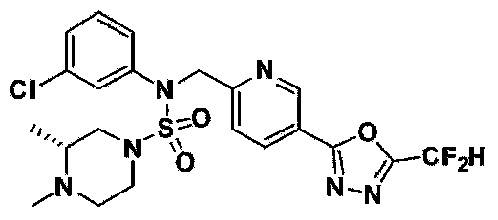
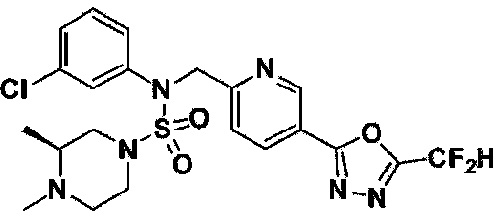
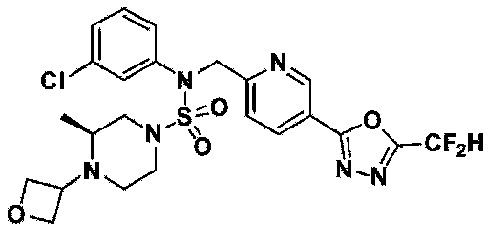
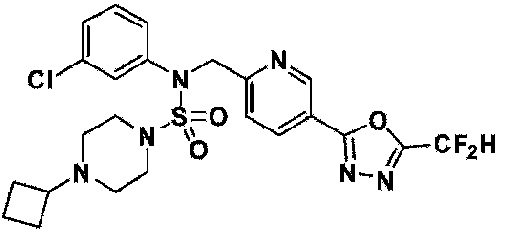
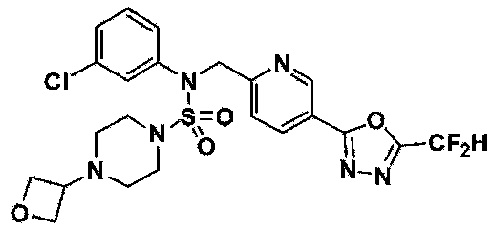
8. Фармацевтическая композиция для профилактики или лечения заболевания, опосредованного гистондеацетилазой, содержащая в качестве активного ингредиента терапевтически эффективное количество соединения, представленного формулой I, его стереоизомера или его фармацевтически приемлемой соли по любому из пп.1-7 и фармацевтически приемлемый носитель.
9. Способ лечения заболевания, опосредованного гистондеацетилазой, включающий введение терапевтически эффективного количества соединения, представленного формулой I, его стереоизомера или его фармацевтически приемлемой соли по любому из пп.1-7.
10. Применение соединения, представленного формулой I, его стереоизомера или его фармацевтически приемлемой соли по любому из пп.1-7 при получении лекарственного средства для лечения заболевания, опосредованного гистондеацетилазой.
| US2012289495A1, 15.11.2012 | |||
| US2007293530A1, 20.12.2007 | |||
| Manku S | |||
| et al, Synthesis and evaluation of lysine derived sulfamides as histone deacetylase inhibitors, Bioorganic & Medicinal Chemistry Letters, 19, 7, стр.:1866 - 1870, 2009 | |||
| Pal; Dilipkumar, Hydroxamic acid - A novel molecule for anticancer therapy, Journal of Advanced Pharmaceutical Technology & Research, 3, 2, стр.:92 - 99, 2012 | |||
| ИНГИБИТОРЫ ДЕАЦЕТИЛАЗЫ И ИХ ПРИМЕНЕНИЕ | 2009 |
|
RU2515611C2 |

