[Область техники]
Настоящее изобретение относится к производному 1,3,4-оксадиазола, обладающему ингибирующей активностью в отношении гистондеацетилазы 6 (HDAC6), его оптическому изомеру, его фармацевтически приемлемой соли, применению для получения терапевтического лекарственного средства, способу лечения с применением указанного соединения, фармацевтической композиции, содержащей указанное соединение, и способу его получения.
[Уровень техники]
Посттрансляционные модификации, такие как ацетилирование в клетках, являются очень важными регуляторными модулями в центре биологических процессов и строго контролируются рядом ферментов. Гистоны представляют собой коровые белки, из которых состоит хроматин, выступающие в роли катушек, на которые наматывается ДНК, чтобы помочь конденсации ДНК. Кроме того, баланс между ацетилированием и деацетилированием гистонов играет очень важную роль в экспрессии генов.
Гистондеацетилазы (HDAC) представляют собой ферменты, которые удаляют ацетильную группу в лизиновых остатках гистоновых белков, из которых состоит хроматин, которые, как известно, связаны с сайленсингом генов и индуцируют остановку клеточного цикла, ингибирование ангиогенеза, иммунную регуляцию, гибель клеток и тому подобное (Hassig et al., Curr. Opin. Chem. Biol. 1997, 1, 300-308). Кроме того, сообщалось, что ингибирование функции фермента HDAC индуцирует гибель раковых клеток путем снижения активности факторов, связанных с выживаемостью раковых клеток, и активации факторов, связанных с гибелью раковых клеток, in vivo (Warrell et al, J. Natl. Cancer Inst. 1998, 90, 1621-1625).
У человека известно 18 HDAC, которые подразделяются на 4 группы в зависимости от их гомологии с дрожжевыми HDAC. Так, 11 HDAC, использующих цинк в качестве кофактора, можно подразделить на три группы: класс I (HDAC 1, 2, 3 и 8), класс II (IIa: HDAC 4, 5, 7 и 9; IIb: HDAC 6 и 10) и класс IV (HDAC11). Кроме того, 7 HDAC класса III (SIRT 1-7) используют NAD+ в качестве кофактора вместо цинка (Bolden et al., Nat. Rev. Drug. Discov. 2006, 5(9), 769-784).
Различные ингибиторы HDAC находятся на стадии доклинической или клинической разработки. Однако до сих пор в качестве противораковых агентов известны только неселективные ингибиторы HDAC, при этом вориностат (SAHA) и ромидепсин (FK228) были одобрены для лечения кожной Т-клеточной лимфомы, а панобиностат (LBH-589) был одобрен для лечения множественной миеломы. Однако известно, что неселективные ингибиторы HDAC в общем случае в высоких дозах вызывают побочные эффекты, такие как усталость и тошнота, и тому подобное (Piekarz et al., Pharmaceuticals 2010, 3, 2751-2767). Сообщается, что эти побочные эффекты вызваны ингибированием HDAC класса I, и в связи с этими побочными эффектами неселективные ингибиторы HDAC были ограничены для разработки лекарственных средств в областях, отличных от противоопухолевых агентов (Witt et al., Cancer Letters 277 (2009) 8.21).
Между тем сообщалось, что селективное ингибирование HDAC класса II может не проявлять токсичности, наблюдаемой при ингибировании HDAC класса I, и если будет разработан селективный ингибитор HDAC класса II, то побочные эффекты, такие как токсичность, вызванная неселективным ингибированием HDAC, могут быть устранены, и, таким образом, селективный ингибитор HDAC может быть разработан в качестве эффективного терапевтического агента для различных заболеваний (Matthias et al., Mol. Cell. Biol. 2008, 28, 1688-1701). HDAC6, одна из HDAC класса IIb, в основном присутствует в цитоплазме и, как известно, участвует в деацетилировании ряда негистоновых субстратов (HSP90, кортактин и тому подобное), включая тубулиновые белки (Yao et al., Mol. Cell 2005, 18, 601-607). HDAC6 имеет два каталитических домена, и С-конец цинк-пальцевого домена может связываться с убиквитинированными белками. Поскольку субстратами HDAC6 может быть множество негистоновых белков, она, как известно, играет важную роль в различных заболеваниях, таких как рак, воспалительные заболевания, аутоиммунные заболевания, неврологические заболевания и нейродегенеративные расстройства, и тому подобное (Santo et al., Blood 2012 119: 2579-258; Vishwakarma et al., International Immunopharmacology 2013, 16, 72-78; Hu et al., J. Neurol. Sci. 2011, 304, 1-8).
Общей структурной особенностью различных ингибиторов HDAC является то, что они состоят из кэп-группы, линкерной группы и цинк-связывающей группы (ZBG), как показано на структуре вориностата ниже. Многие исследователи изучали ингибирующую активность и селективность ферментов посредством структурных модификаций кэп-группы и линкерной группы. Известно, что среди указанных групп цинк-связывающая группа играет более важную роль в ингибировании ферментативной активности и селективности (Wiest et al., J. Org. Chem. 2013 78: 5051-5065; Methot et al., Bioorg. Med. Chem. Lett. 2008, 18, 973-978).
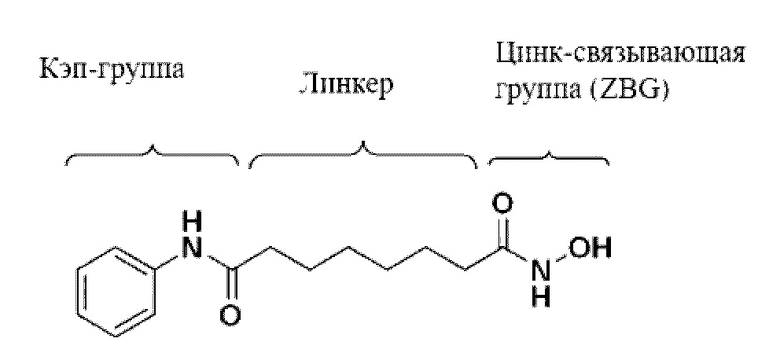
Большинство цинк-связывающих групп представляют собой гидроксамовую кислоту или бензамид, и среди них производные гидроксамовой кислоты проявляют сильное ингибирующее действие в отношении HDAC, но им присущи такие проблемы, как низкая биодоступность и сильная нецелевая активность. Поскольку бензамид содержит анилин, существует проблема, заключающаяся в образовании токсичных метаболитов in vivo (Woster et al., Med. Chem. Commun. 2015, публикация в Интернете).
Таким образом, для лечения рака, воспалительных заболеваний, аутоиммунных заболеваний, неврологических заболеваний и нейродегенеративных расстройств и тому подобного требуется разработка селективного ингибитора HDAC6, содержащего цинк-связывающую группу с улучшенной биодоступностью без побочных эффектов, в отличие от неселективных ингибиторов с побочными эффектами.
[Краткое описание изобретения]
[Техническая задача]
Задачей настоящего изобретения является обеспечение производного 1,3,4-оксадиазола, обладающего селективной ингибирующей активностью в отношении гистондеацетилазы 6 (HDAC6), его оптического изомера или его фармацевтически приемлемой соли.
Другой задачей настоящего изобретения является обеспечение фармацевтической композиции, содержащей производное 1,3,4-оксадиазола, обладающее селективной ингибирующей активностью в отношении HDAC6, его оптического изомера или его фармацевтически приемлемой соли.
Еще одна задача настоящего изобретения состоит в обеспечении способа их получения.
Еще одна задача настоящего изобретения заключается в обеспечении фармацевтической композиции, содержащей указанные соединения, для предотвращения или лечения заболеваний, опосредованных гистондеацетилазой 6 (HDAC6), включая инфекционные заболевания; новообразования; эндокринные, пищевые и метаболические заболевания; психические и поведенческие расстройства; неврологические заболевания; заболевания глаза и его придаточного аппарата; заболевания системы кровообращения; респираторные заболевания; заболевания пищеварительной системы; заболевания кожи и подкожных тканей; заболевания опорно-двигательного аппарата и соединительной ткани; или врожденные пороки развития, изменения или хромосомные аномалии.
Другой задачей настоящего изобретения является обеспечение применения указанных соединений для получения лекарственного средства для предотвращения или лечения заболеваний, опосредованных HDAC6.
Другой задачей настоящего изобретения является обеспечение способа предотвращения или лечения заболеваний, опосредованных HDAC6, включая введение терапевтически эффективного количества композиции, содержащей соединения, как описано выше.
[Техническое решение]
Авторы настоящего изобретения обнаружили производное 1,3,4-оксадиазола, обладающее ингибирующей активностью в отношении гистондеацетилазы 6 (HDAC6), для ингибирования или лечения заболеваний, опосредованных HDAC6, и осуществили настоящее изобретение.
Производное 1,3,4-оксадиазола
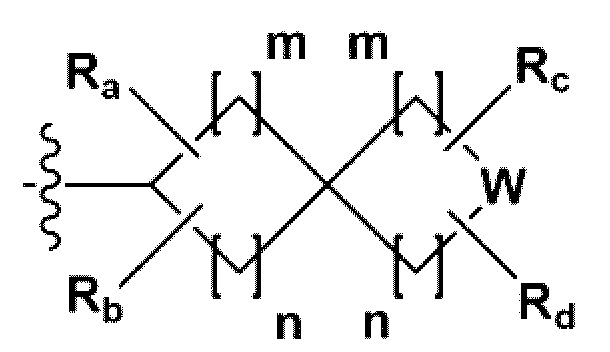
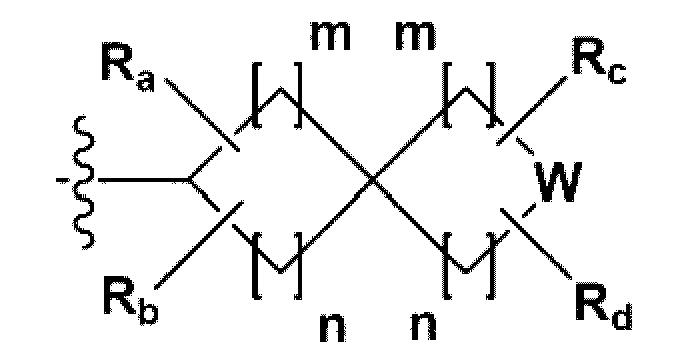
В одном общем аспекте в настоящем изобретении предложено производное 1,3,4-оксадиазола, представленное химической формулой I ниже, его оптический изомер, или его фармацевтически приемлемая соль:
[Химическая формула I]
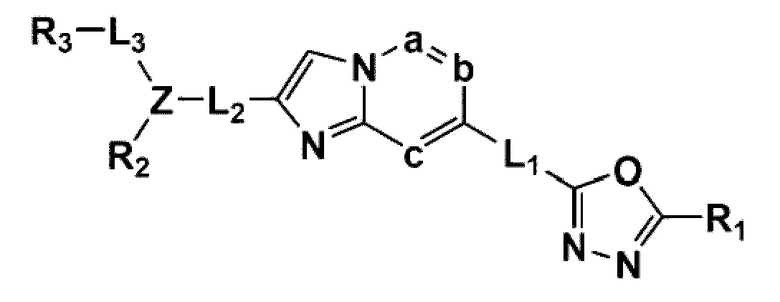
в химической формуле I,
L1, L2 и L3 каждый независимо представляет собой -(С0-С2алкил)-;
a, b и с каждый независимо представляет собой N или CR4 {где a, b и с не могут представлять собой N одновременно, и R4 представляет собой -Н, -X или -О(С1-С4алкил)};
Z представляет собой N, О, S или отсутствует {где в случае, когда Z отсутствует, R2 также отсутствует, и L2 и L3 связаны напрямую};
R1 представляет собой -CH2X или -СХ3;
R2 представляет собой -Н, - (С1-С4алкил), -С (=O)-RA, -С(=O)-ORB или -С(=O)-NRCRD {где, когда Z представляет собой 0 или S, R2 отсутствует};
RA представляет собой -(С1-С4алкил), -(С1-С4алкил)-O-(C1-С4алкил), -(С1-С4алкил)-С(=O)-O(С1-С4алкил), -арил, -гетероарил, -NRA1RA2,
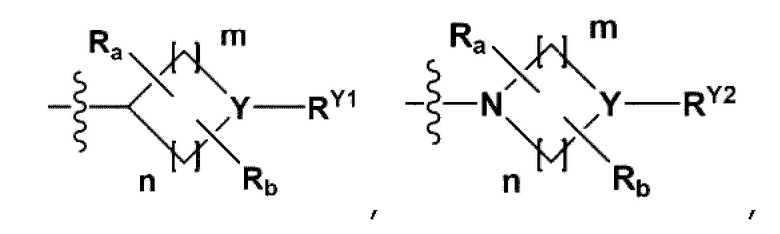
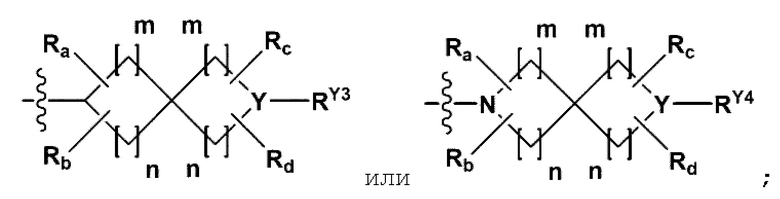
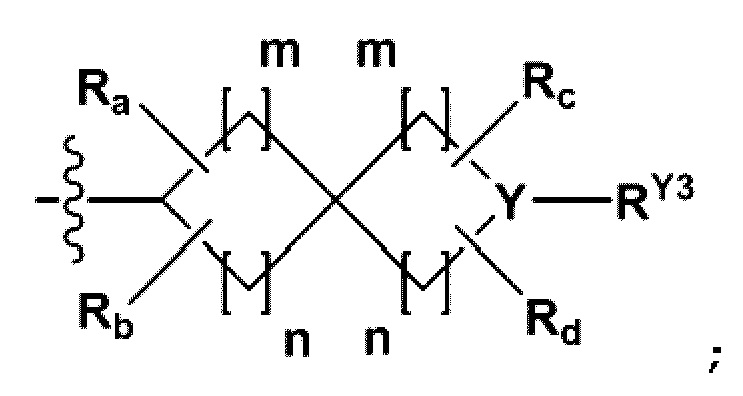
RB - RD каждый независимо представляет собой -Н, -(C1-С4алкил), - (С1-С4алкил) -О- (С1-С4алкил), - (С1-С4алкил) -С (=O)-О(C1-С4алкил), -(С3-С7циклоалкил), -арил или -гетероарил; Y представляет собой N, СН, О или S(=O)2, когда Y представляет собой N или СН, каждый из RY1 - RY4 независимо представляет собой -Н, -X, -ОН, -(С1-С4алкил), -(С3-С7циклоалкил), -(С2-С6 гетероциклоалкил), -(С1-С4алкил)-О-(C1-С4алкил), -(С1-С4алкил) -С(=O)-О(С1-С4алкил), -С(=O) - (С1-С4алкил), -С(=O)-О(С1-С4алкил), -С(=O)-NRA3RA4, -С(=O)-(С3-С7циклоалкил), -С(=O)-(С2-С6гетероциклоалкил), -S(=O)2-(С1-С4алкил), -арил, -(C1-С4алкил)-арил, -гетероарил, -(С1-С4алкил)-гетероарил, аминозащитную группу, или
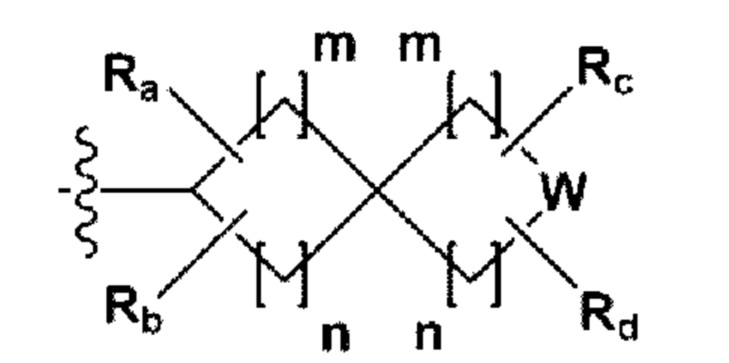
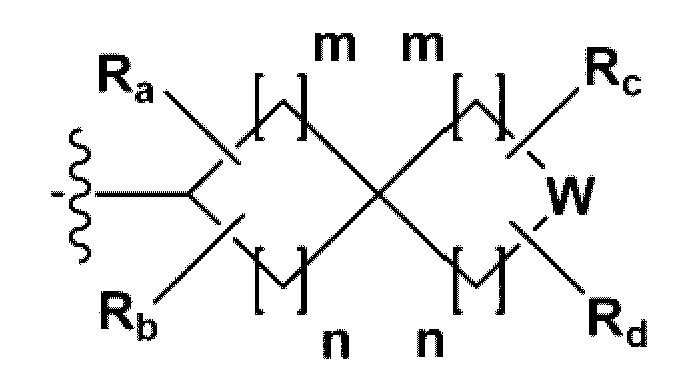
{где по меньшей мере один Н -(С1-С4алкил), -(С3-С7циклоалкил), -(С1-С4алкил)-О-(С1-С4алкил), -(С1-С4алкил) -С(=O)-О(С1-С4алкил), -С(=O)-(С1-С4алкил), -С(=O)-(С3-С7циклоалкил), -С(=O)-(С2-С6 гетероциклоалкил) и -S(=O)2-(С1-С4алкил) может быть замещен -X или -ОН; по меньшей мере один Н арила, -(С1-С4алкил)-арила, гетероарила и -(С1-С4алкил)-гетероарила может быть замещен группой - (С1-С4 алкил), -О-(С1-С4алкил), -X, -ОН или -CF3; -(С2-С6гетероциклоалкил), -гетероарил или -(С1-С4алкил)-гетероарил может содержать в кольце атомы N, О или S; и W представляет собой NH, СН2 или O};
когда Y представляет собой О или S(=O)2, RY1 - RY4 отсутствуют;
m и n каждый независимо представляет собой целое число 1, 2 или 3;
Ra - Rd каждый независимо представляет собой -Н или -(C1-С4алкил);
R3 представляет собой -Н, -(С1-С4алкил), -(С1-С4алкил)-О(C1-С4алкил), -(С1-С4алкил)-С(=O)-О(С1-С4алкил), -С(=O)-О(С1-С4алкил), -(С3-С7циклоалкил), -(С2-С6 гетероциклоалкил), -адамантил, -арил или -гетероарил {где по меньшей мере один Н -(С1-С4алкила) может быть замещен -X или -ОН; по меньшей мере один -Н -(С3-С7циклоалкила), -(С2-С6 гетероциклоалкила), -адамантила, -арила и -гетероарила может быть каждый независимо замещен группой -X, -ОН, -(C1-С4алкил), -O(С1-С4алкил), -(С=O)-(С1-С4алкил), -С(=O)-O(С1-С4алкил), -CF3, -CF2H, -OCF3, -NRA5RA6, -S(=O)2-(С1-С4алкил), -арил или -гетероарил};
RA1 - RA6 каждый независимо представляет собой -Н или -(C1-С4алкил); и
X представляет собой F, Cl, Br или I.
Согласно одному из вариантов реализации настоящего изобретения, в химической формуле I выше,
L1, L2 и L3 каждый независимо представляет собой -(С0-С1алкил)-;
a, b и с каждый независимо представляет собой N или CR4 {где a, b и с не могут представлять собой N одновременно, и R4 представляет собой -Н или -X};
Z представляет собой N, О или отсутствует {где в случае, когда Z отсутствует, R2 также отсутствует), и L2 и L3 связаны напрямую};
R1 представляет собой -СН2Х или -СХ3;
R2 представляет собой -Н, -(С1-С4алкил) или -С(=O)-RA { где, когда Z представляет собой O, R2 отсутствует};
RA представляет собой -NRA1RA2,
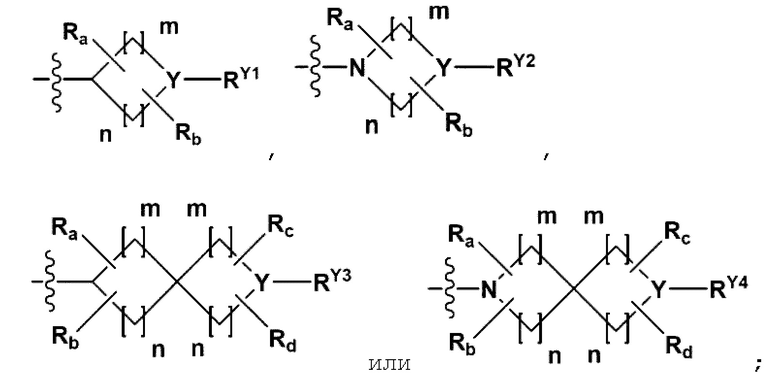
Y представляет собой N, СН, O или S(=O)2;
когда Y представляет собой N или СН, каждый из RY1-RY4 независимо представляет собой -Н, -(С1-С4алкил), -(С3-С7циклоалкил), -(С2-С6 гетероциклоалкил), -С(=O)-(С1-С4алкил), -С(=O)-O(С1-С4алкил), -С(=O)-NRA3RA4, -С(=O)-(С3-С7циклоалкил), -С(=O)-(С2-С6 гетероциклоалкил), -S(=O)2-(С1-С4алкил), -арил, -гетероарил или
{где по меньшей мере один Н - (С1-С4алкила), -(С3-С7циклоалкила), -С(=O)-(С1-С4алкила), -С(=O)-(С3-С7циклоалкила), -С(=O)-(С2-С6 гетероциклоалкила) и -S(=O)2-(С1-С4алкила) может быть замещен -X или -ОН; по меньшей мере один Н арила и гетероарила может быть замещен группой - (С1-С4алкил), -О-(С1-С4алкил), -X, -ОН или -CF3; -(С2-С6 гетероциклоалкил) или -гетероарил может содержать в кольце атомы N, О или S; и W представляет собой NH, СН2 или О};
когда Y представляет собой О или S (=O)2, RY1 - RY4 отсутствуют;
каждый из m и n независимо представляет собой целое число 1 или 2;
Ra - Rd каждый независимо представляет собой -Н или -(C1-С4алкил);
R3 представляет собой - (С3-С7циклоалкил), - (С2-С6 гетероциклоалкил), -адамантил, -арил или -гетероарил {где по меньшей мере один -Н -(С3-С7циклоалкила), -(С2-С6 гетероциклоалкила), -адамантила, -арила и -гетероарила каждый может быть независимо замещен группой -X, -ОН, -(С1-С4алкил), -О(С1-С4алкил), -(С=O)-(С1-С4алкил), -С(=O)-О(С1-С4алкил), -CF3, -CF2H, -OCF3, -NRA5RA6, -S(=O)2-(С1-С4алкил), -арил или -гетероарил;}
RA1 - RA6 каждый независимо представляет собой -Н или -(C1-С4алкил); и
X может представлять собой F, Cl или Br.
Кроме того, согласно другому варианту реализации настоящего изобретения в химической формуле I выше,
L1 и L3 каждый независимо представляет собой -(С0алкил)-;
L2 представляет собой -(С1алкил)-;
a, b и с каждый независимо представляет собой CR4 {где R4 представляет собой -Н или -X};
Z представляет собой N, О или отсутствует {где в случае, когда Z отсутствует, R2 также отсутствует, и L2 и L3 связаны напрямую};
R1 представляет собой -CF2H или CF3;
R2 представляет собой -Н или -С(=O)-RA {где, когда Z представляет собой O, R2 отсутствует};
Ra представляет собой 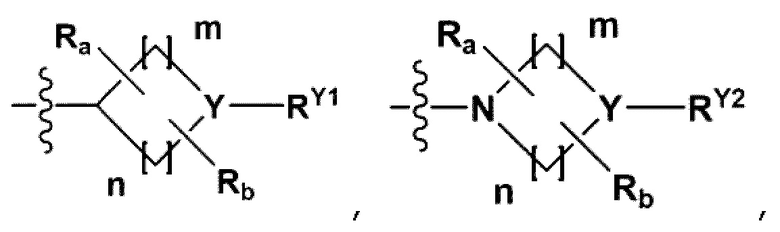
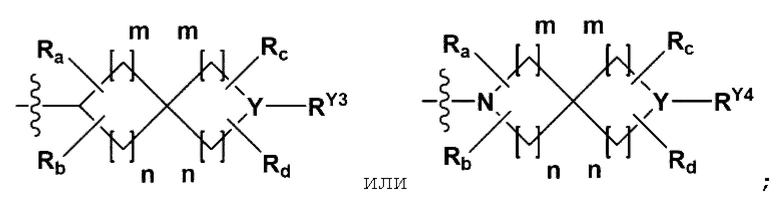
Y представляет собой N;
RY1 - RY4 каждый независимо представляет собой -(С1-С4алкил),
-(С3-С7циклоалкил), -(С2-С6 гетероциклоалкил), -С(=O)-(С1-С4алкил), -С(=O)-О(С1-С4алкил), -С(=O)-NRA3RA4, -С(=O)-(С3-С7циклоалкил), -С(=O)-(С2-С6 гетероциклоалкил), -S(=O)2-(С1-С4алкил), -гетероарил или
{где по меньшей мере один Н -(С1-С4алкила), -(С3-С7циклоалкила), -С(=O)-(С1-С4алкила), -С(=O)-(С3-С7циклоалкила), -С(=O)-(С2-С6гетероциклоалкила) и -S(=O)2-(С1-С4алкила) может быть замещен -X или -ОН; -(С2-С6гетероциклоалкил) может содержать в кольце атомы N, О или S; и W представляет собой СН2 или О};
каждый из m и n независимо представляет собой целое число 1 или 2;
Ra - Rd каждый независимо представляет собой -Н или -(C1-С4алкил);
R3 представляет собой -(С3-С7циклоалкил), -адамантил, -арил или -гетероарил {где по меньшей мере один -Н -(С3-С7циклоалкила), -адамантила, -арила и -гетероарила каждый может быть независимо замещен группой -X, -(С1-С4алкил), -O(С1-С4алкил), -(C=O)-(C1-С4алкил), -CF3 или -S (=O)2-(С1-С4алкил)};
RA1 - RA6 каждый независимо представляет собой -Н или -(C1-С4алкил); и
X может представлять собой F или Cl.
Кроме того, согласно еще одному варианту реализации настоящего изобретения в химической формуле I выше,
L1, L2 или L3 каждый независимо представляет собой -(С0-С1алкил)-;
a, b и с каждый независимо представляет собой N или CR4 {где a, b и с не могут представлять собой N одновременно, и R4 представляет собой -Н или -X};
Z представляет собой N;
R1 представляет собой -СН2Х или -СХ3;
R2 представляет собой -С (=O)-RA;
RA представляет собой 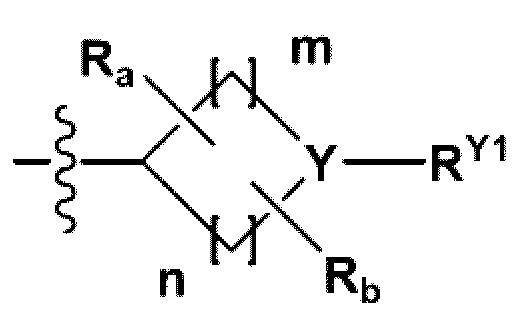 или
или
Y представляет собой N, СН, O или S(=O)2;
когда Y представляет собой N или СН, RY1 и RY3 каждый независимо представляет собой -Н, -(С1-С4алкил), -(С3-С7циклоалкил), - (С2-С6гетероциклоалкил), -С(=O)-(С1-С4алкил), -С(=O)-O(С1-С4алкил), -С(=O)-NRA3RA4, -С(=O)-(С3-С7циклоалкил), -С(=O)-(С2-С6гетероциклоалкил), -S(=O)2-(С1-С4алкил), -арил, -гетероарил или
{где по меньшей мере один Н -(С1-С4алкила), -(С3-С7циклоалкила), -С(=O)-(С1-С4алкила), -С(=O)-(С3-С7циклоалкила), -С(=O)-(С2-С6гетероциклоалкила) и -S(=O)2-(С1-С4алкила) может быть замещен -X или -ОН; по меньшей мере один Н арила и гетероарила может быть замещен группой - (С1-С4алкил), -О-(С1-С4алкил), -X, -ОН или -CF3; -(С2-С6 гетероциклоалкил) или -гетероарил может содержать в кольце атомы N, О или S; и W представляет собой NH, СН2 или О};
когда Y представляет собой О или S (=O)2, RY1 и RY3 отсутствуют;
каждый из m и n независимо представляет собой целое число 1 или 2;
Ra - Rd каждый независимо представляет собой -Н или -(С1-С4алкил);
R3 представляет собой -С(=O)-О(С1-С4алкил), -(С3-С7циклоалкил), -(С2-С6 гетероциклоалкил), -адамантил, -арил или -гетероарил {где по меньшей мере один -Н -(С3-С7циклоалкила), -(С2-С6гетероциклоалкила), -адамантила, -арила и -гетероарила каждый независимо может быть замещен группой -X, -ОН, -(C1-С4алкил), -О(С1-С4алкил), -(С=O)-(С1-С4алкил), -С (=O)-О(C1-С4алкил), -CF3, -CF2H, -OCF3, -NRA5RA6, -S(=O)2-(С1-С4алкил), -арил или -гетероарил;}
RA3 - RA6 каждый независимо представляет собой -Н или -(C1-С4алкил); и
X может представлять собой F, Cl или Br.
Кроме того, согласно еще одному варианту реализации настоящего изобретения в химической формуле I выше,
L1, L2 и L3 каждый независимо представляет собой -(С0-С1алкил)-;
a, b и с каждый независимо представляет собой N или CR4 {где a, b и с не могут представлять собой N одновременно, и R4 представляет собой -Н или -X};
Z представляет собой N;
R1 представляет собой -СН2Х или -СХ3;
R2 представляет собой -С(=O)-RA;
RA представляет собой -NRA1RA2,
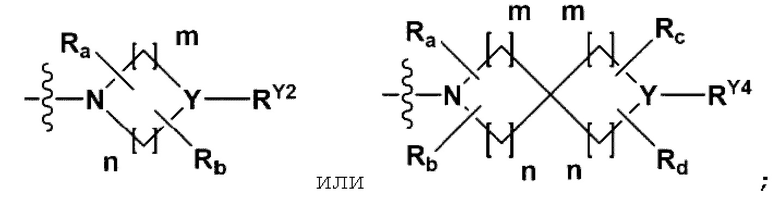
каждый из m и n независимо представляет собой целое число 1 или 2;
Ra - Rd каждый независимо представляет собой -Н или -(C1-С4алкил);
Y представляет собой N, СН, O или S(=O)2; когда Y представляет собой N или СН, RY2 и RY4 каждый независимо представляет собой -Н, -(С1-С4алкил), -(С3-С7циклоалкил), -(С2-С6 гетероциклоалкил), -С(=O)-(С1-С4алкил), -С(=O)-O(С1-С4алкил), -С(=O)-NRA3RA4, -С(=O)-(С3-С7циклоалкил), -С (=O)-(С2-С6 гетероциклоалкил), -S(=O)2-(С1-С4алкил), -арил или -гетероарил {где по меньшей мере один Н -(С1-С4алкила), -(С3-С7циклоалкила), -С(=O)-(С1-С4алкила), -С(=O)-(С3-С7циклоалкила), -С(=O)-(С2-С6 гетероциклоалкила) и -S(=O)2-(С1-С4алкила) может быть замещен -X или -ОН; по меньшей мере один Н арила и гетероарила может быть замещен группой -(С1-С4алкил), -О-(С1-С4 алкил), -X, -ОН или -CF3; и -(С2-С6гетероциклоалкил) или -гетероарил может содержать в кольце атомы N, О или S};
когда Y представляет собой О или S(=O)2, RY2 и RY4 отсутствуют;
R3 представляет собой -С(=O)-О(С1-С4алкил), -(С3-С7циклоалкил), -(С2-С6 гетероциклоалкил), -адамантил, -арил или -гетероарил {где по меньшей мере один -Н-(С3-С7циклоалкила), -(С2-С6 гетероциклоалкила), -адамантила, -арила и -гетероарила каждый независимо может быть замещен группой -X, -ОН, -(C1-С4алкил), -О(C1-С4алкил), -(С=O)-(С1-С4аалкил), -C(=O)-O(C1-С4алкил), -CF3, -CF2H, -OCF3, -NRA5RA6, -S(=O)2-(С1-С4алкил), -арил или -гетероарил;}
RA1 - RA6 каждый независимо представляет собой -Н или -(C1-С4алкил); и
X может представлять собой F, Cl или Br.
Кроме того, согласно еще одному варианту реализации настоящего изобретения в химической формуле I выше,
L1, L2 и L3 каждый независимо представляет собой -(С0-С1алкил) -;
a, b и с каждый независимо представляет собой N или CR4 {где a, b и с не могут представлять собой N одновременно, и R4 представляет собой -Н или -X};
Z представляет собой N, О или отсутствует {где в случае, когда Z отсутствует, R2 также представляет собой отсутствует, и L2 и L3 связаны напрямую};
R1 представляет собой -СН2Х или -СХ3;
R2 представляет собой -Н, -(С1-С4алкил) {где, когда Z представляет собой О, R2 представляет собой отсутствует};
R3 представляет собой -С(=O)-О(С1-С4алкил), -(С3-С7циклоалкил), -(С2-С6 гетероциклоалкил), -адамантил, -арил или -гетероарил {где по меньшей мере один -Н -(С3-С7циклоалкила), -(С2-С6гетероциклоалкила), -адамантила, -арила и -гетероарила каждый независимо может быть замещен группой -X, -ОН, -(C1-С4алкил), -О(С1-С4алкил), -(С=O)-(C1-С4алкил), -C(=O)-O(C1-С4алкил), -CF3, -CF2H, -OCF3, -NRA5RA6, -S(=O)2-(С1-С4алкил), -арил или -гетероарил;}
RA5 и RA6 каждый независимо представляет собой -Н или -(C1-С4алкил); и
X может представлять собой F, Cl или Br.
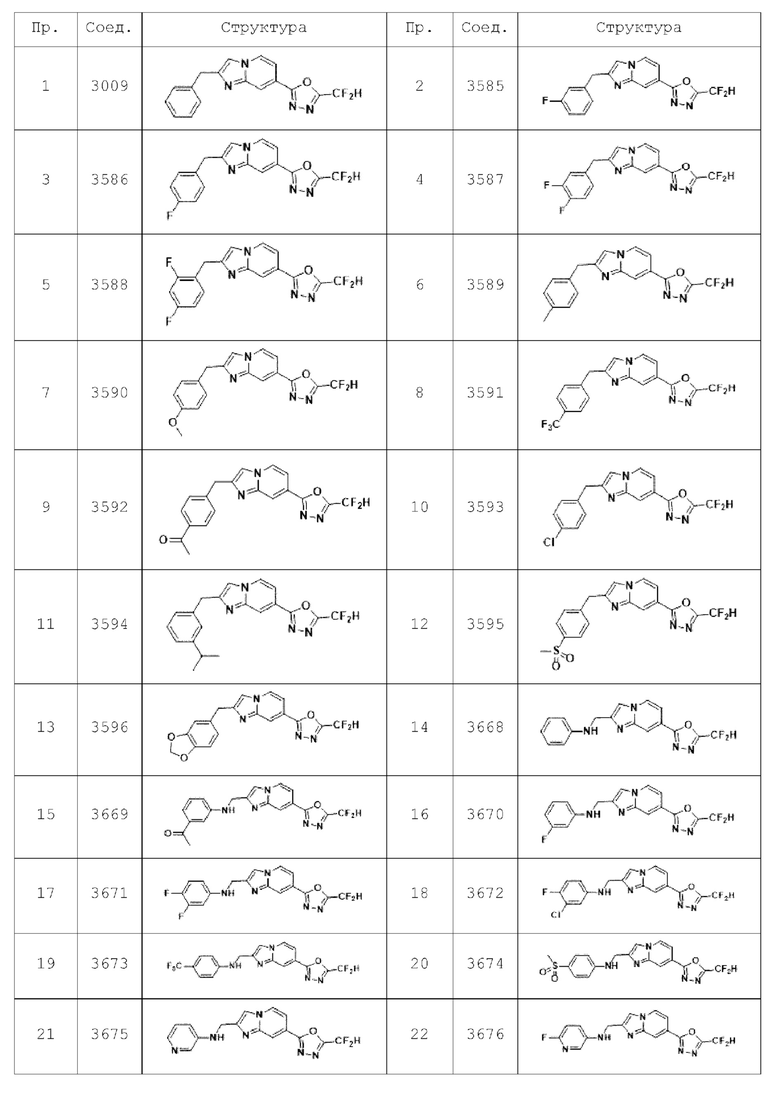
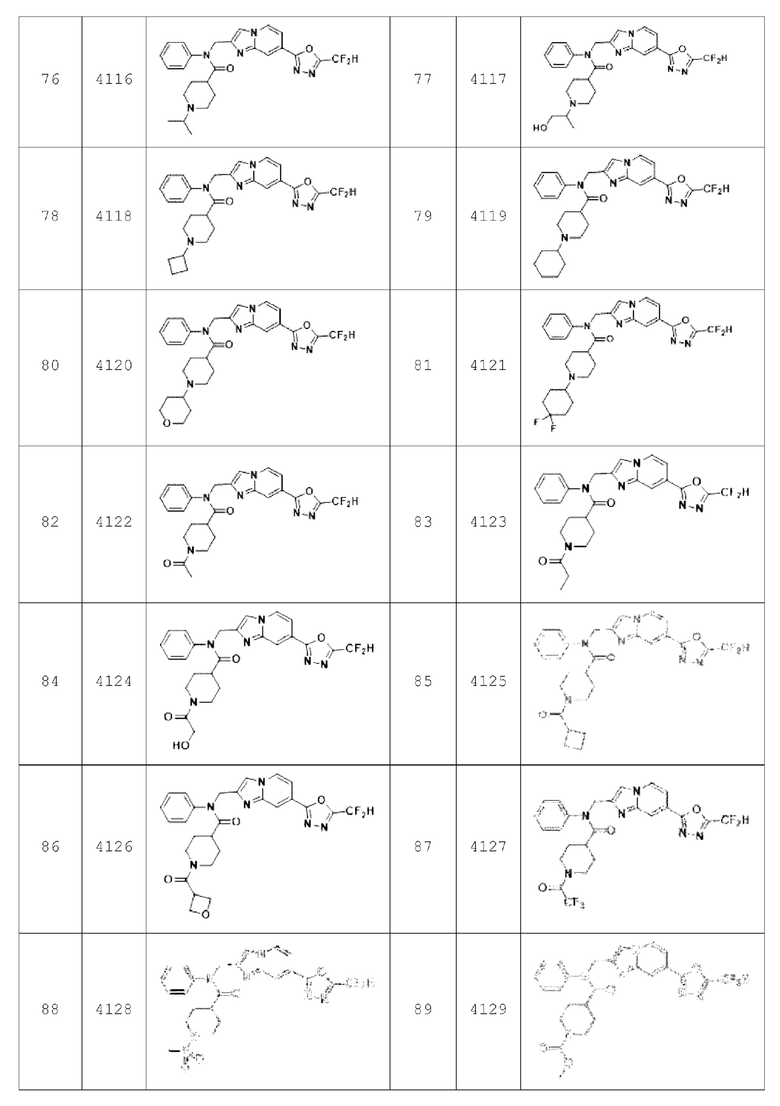
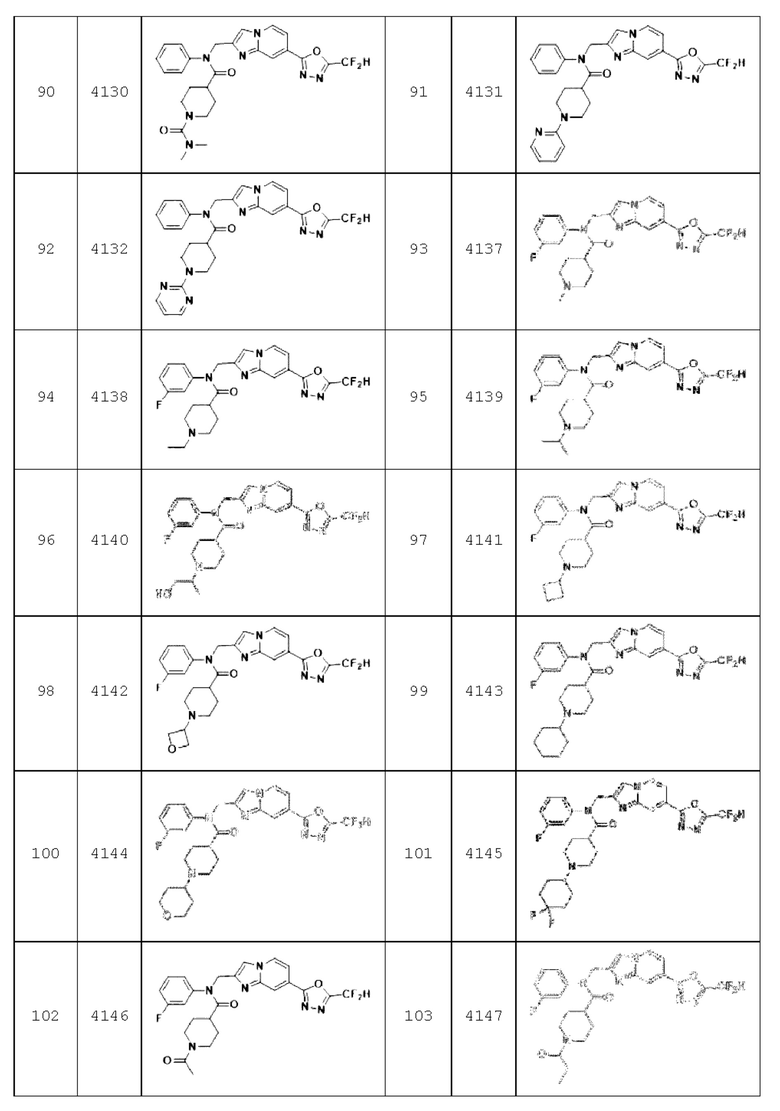
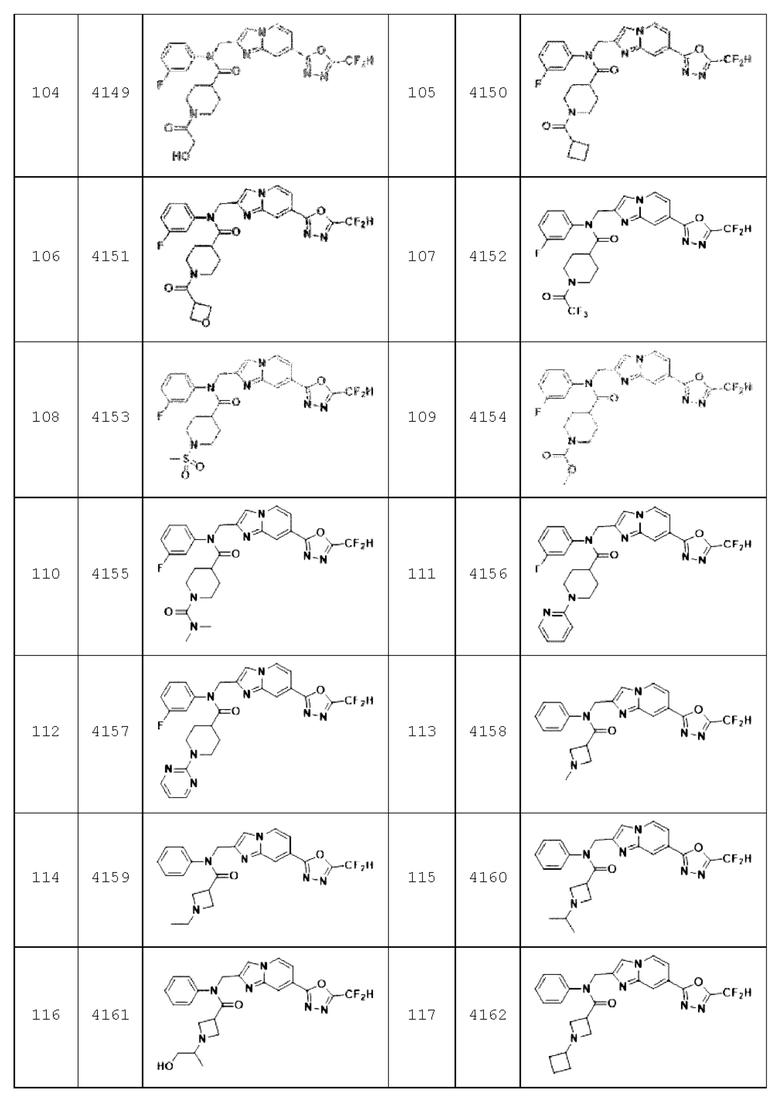
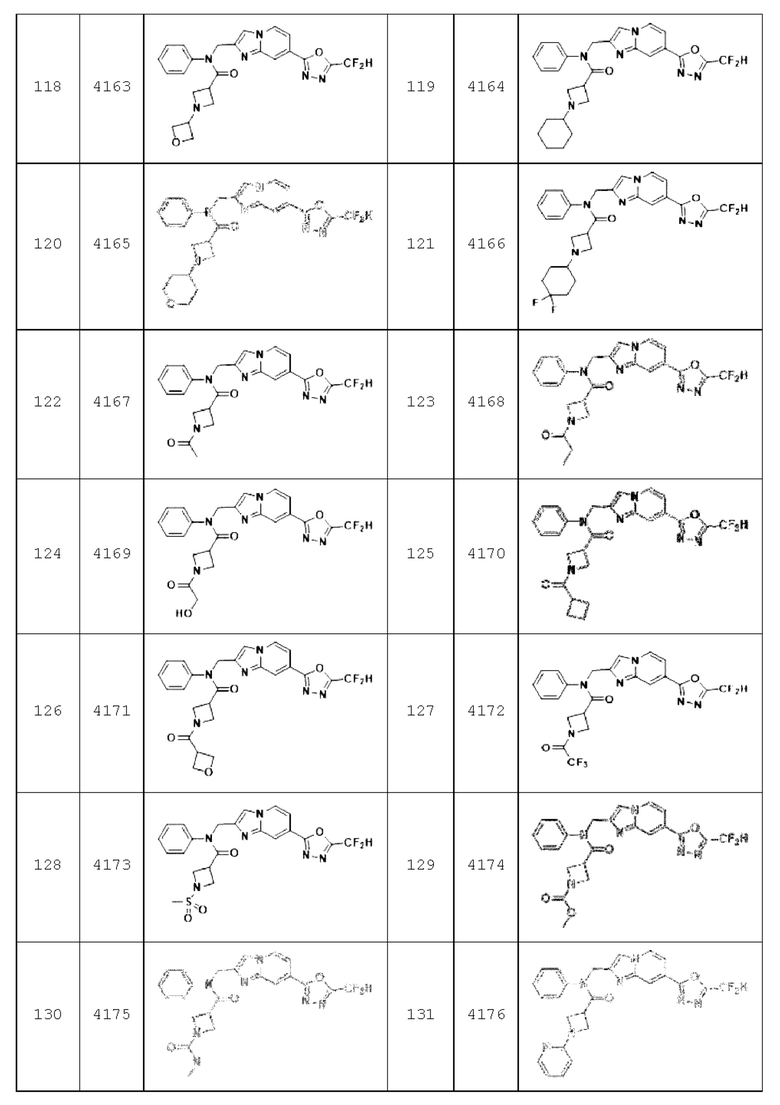
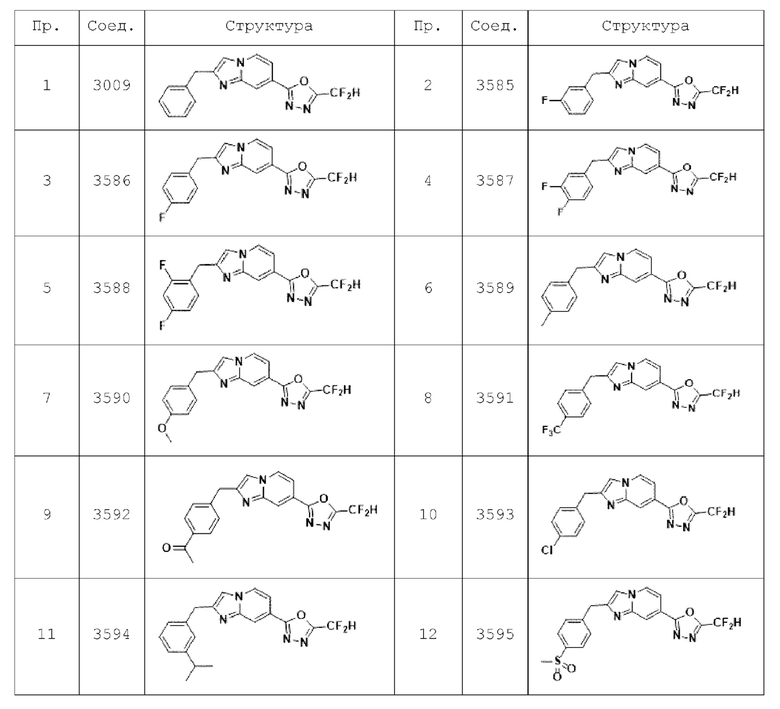
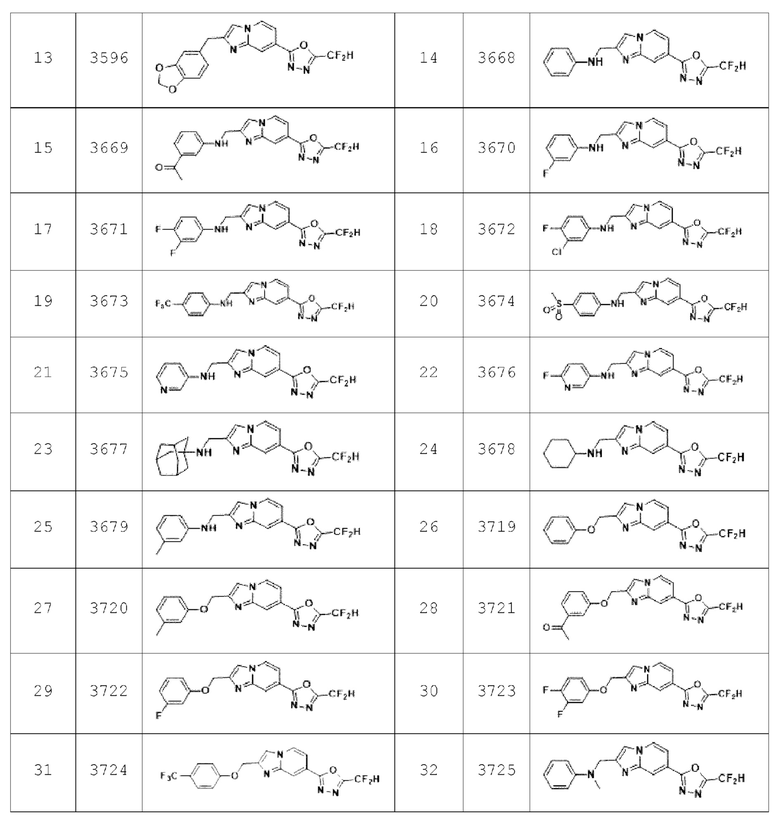
Конкретные соединения, представленные химической формулой I согласно настоящему изобретению, показаны в Таблице 1 ниже.
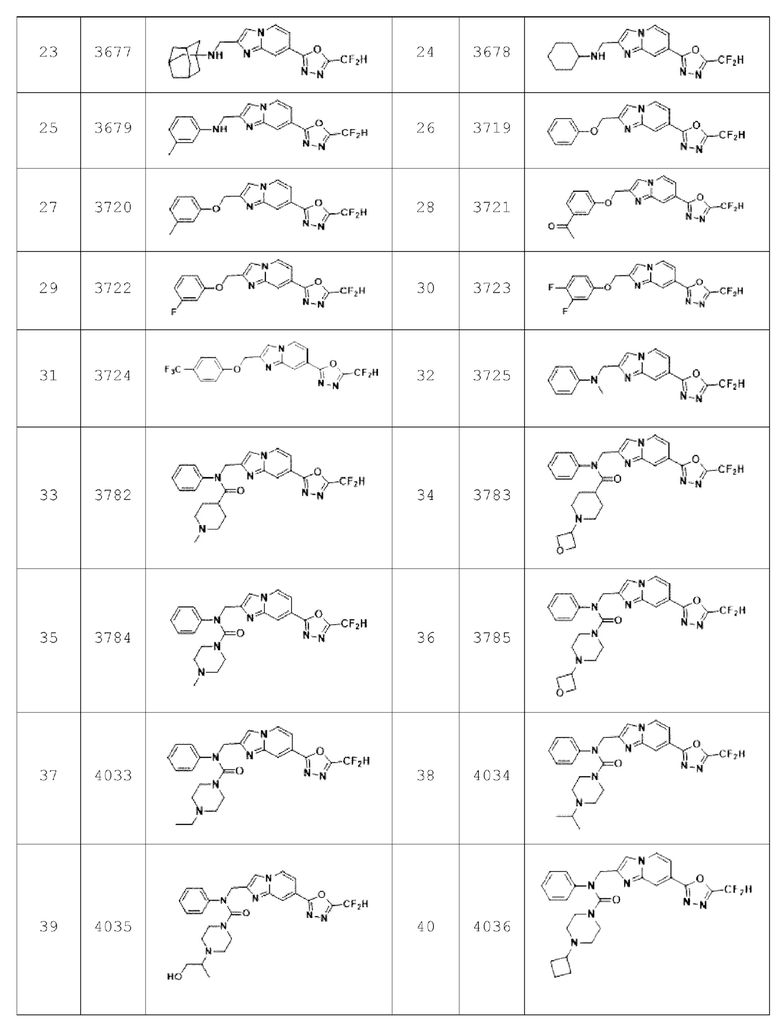
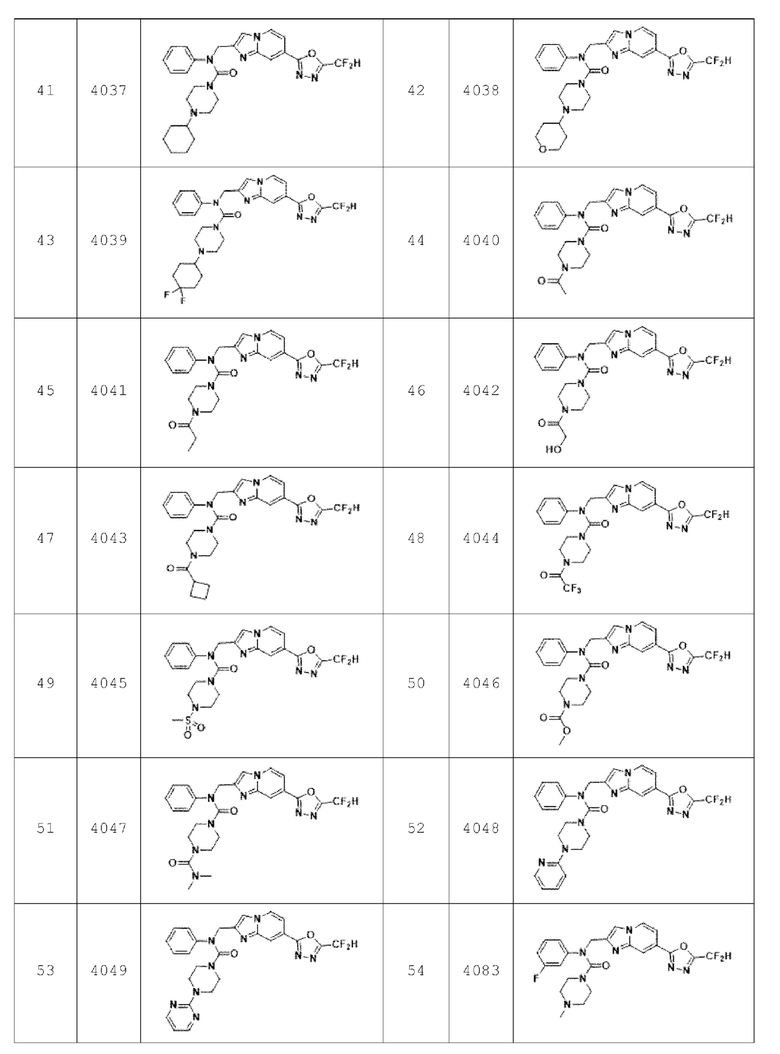
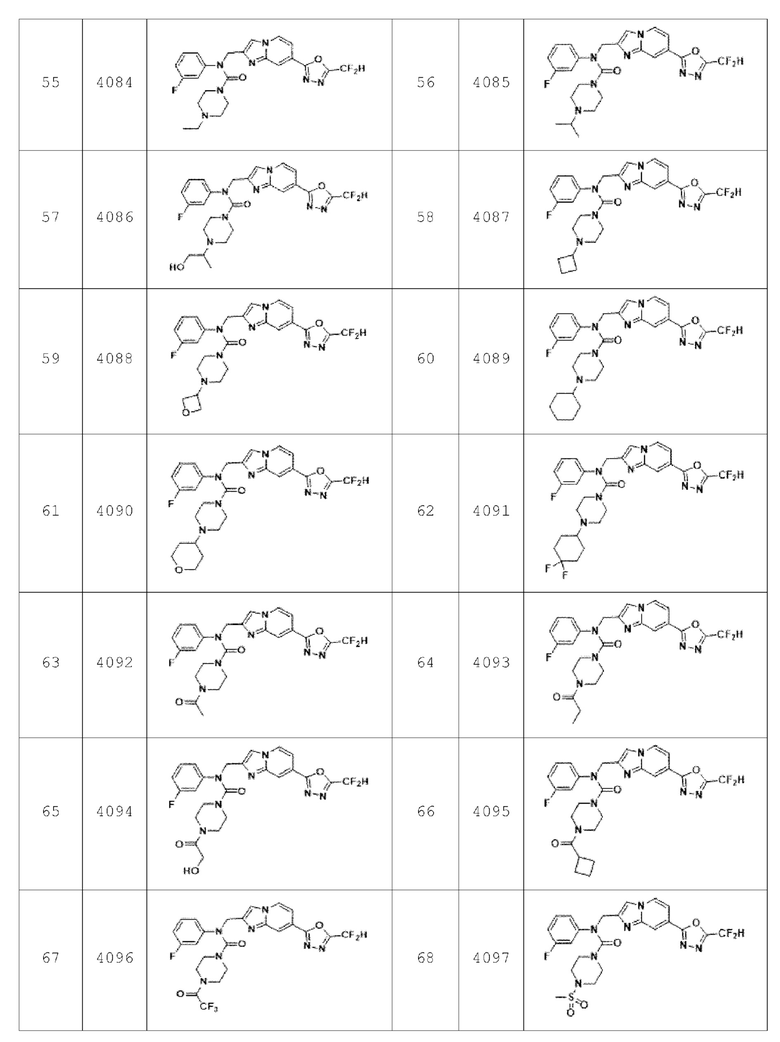
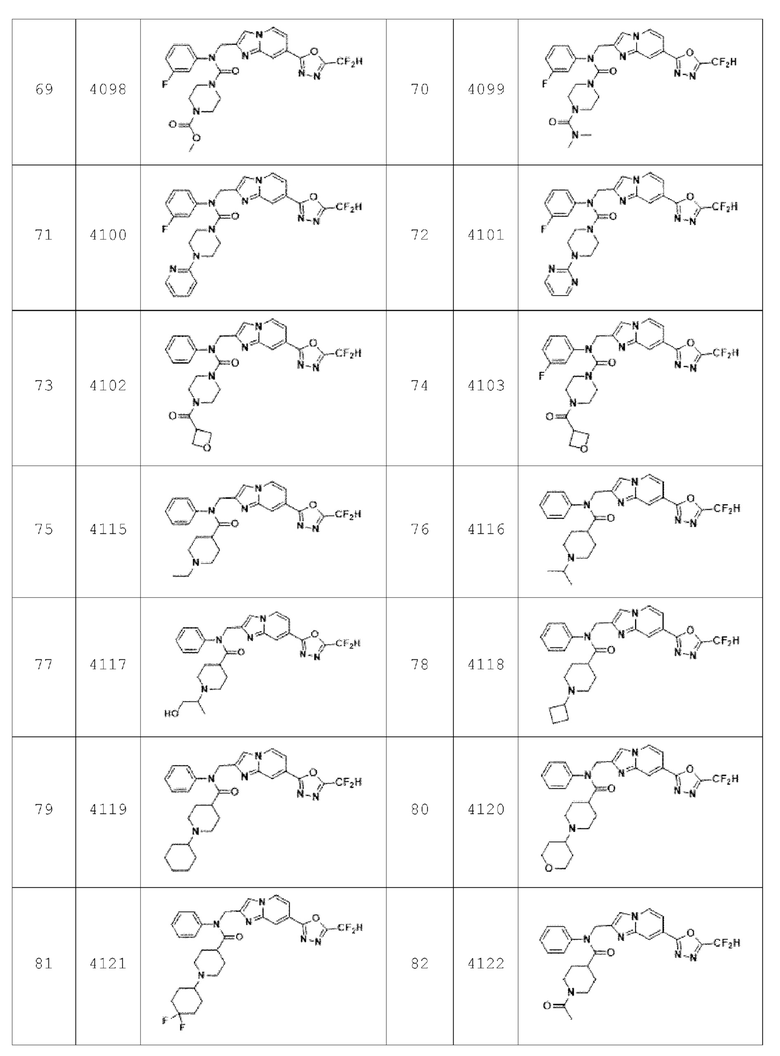
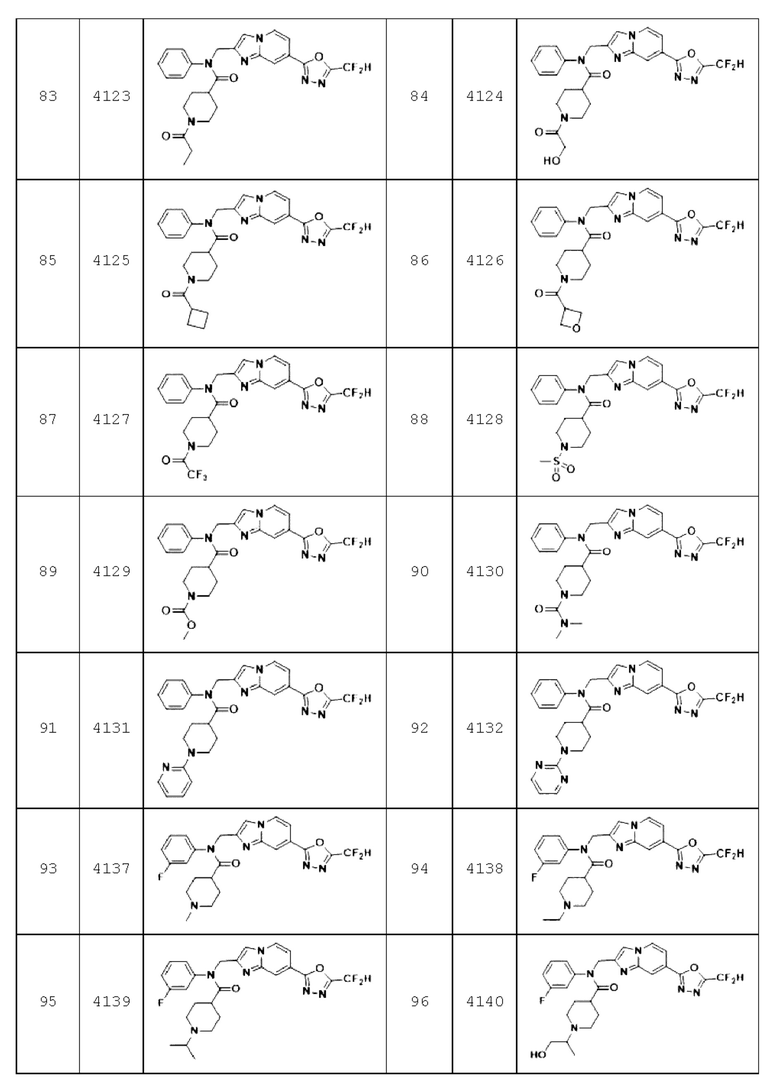
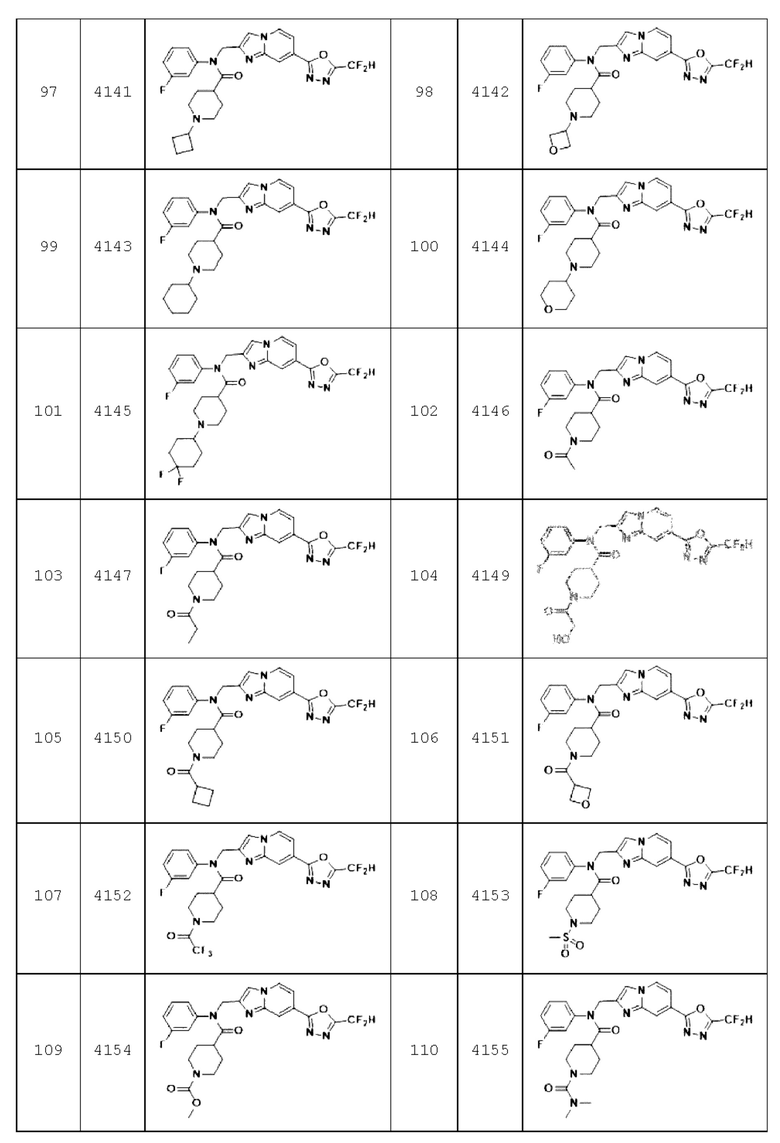
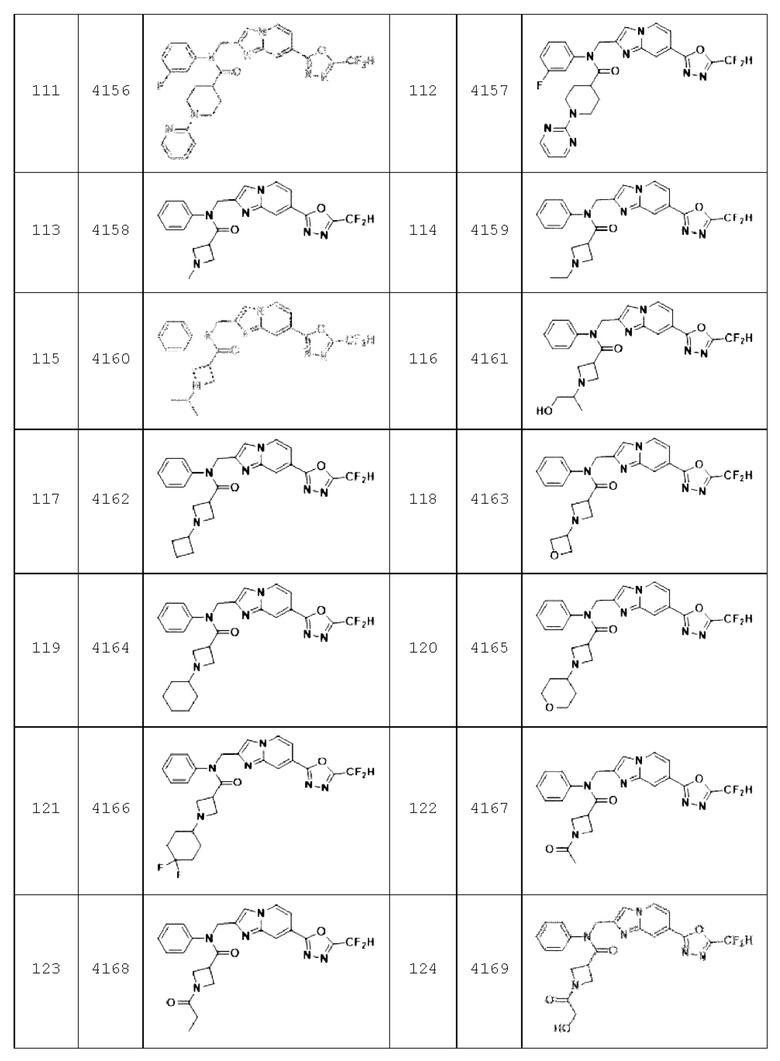
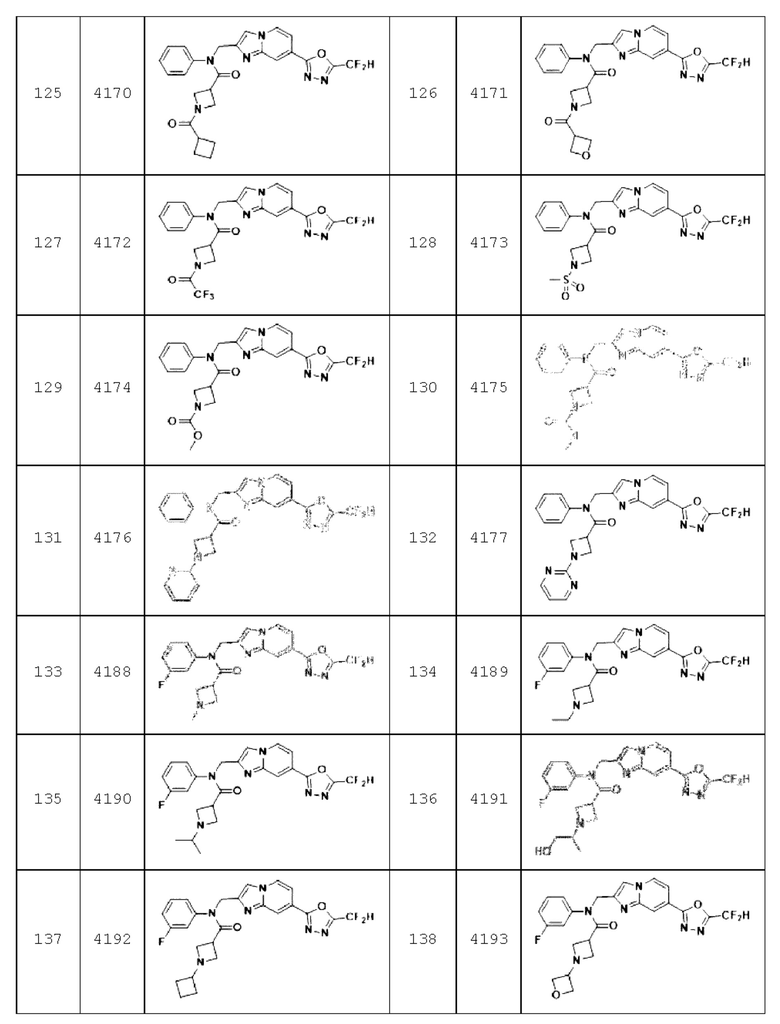
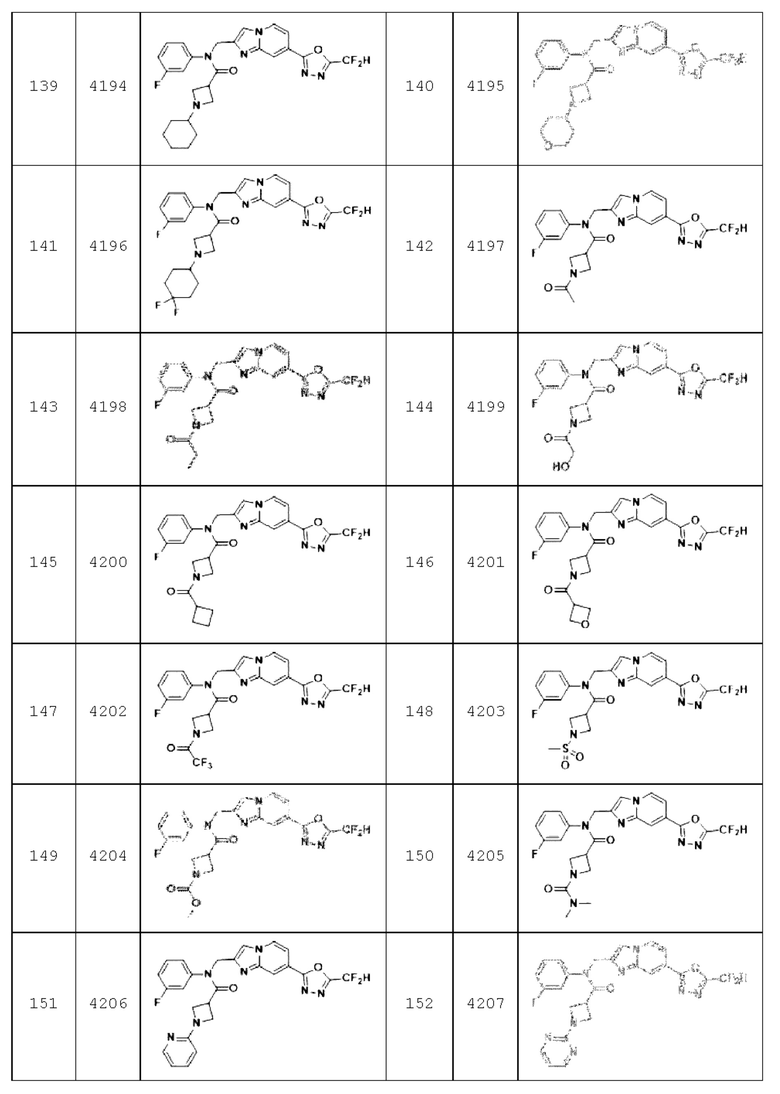
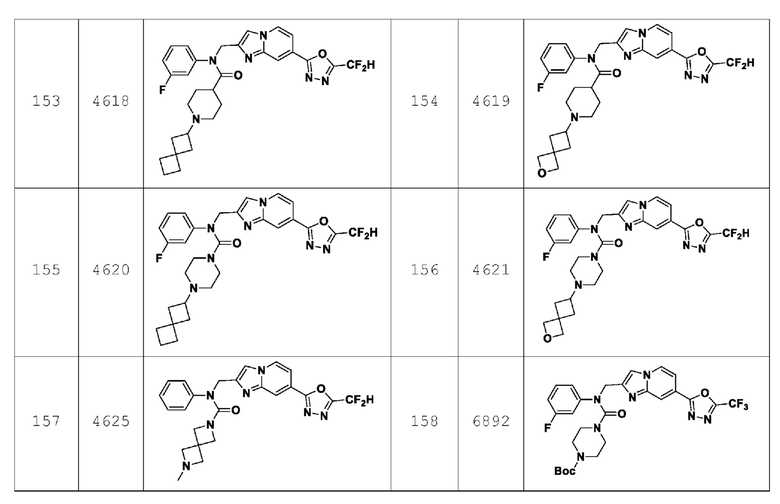
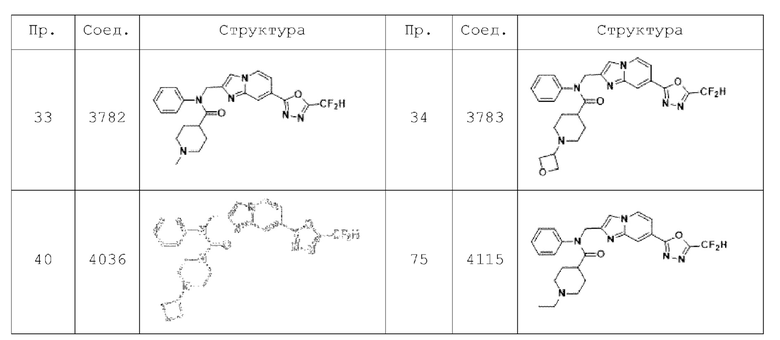
В соответствии с вариантом реализации настоящего изобретения конкретные соединения, представленные химической формулой I согласно настоящему изобретению, могут быть показаны в Таблице 2 ниже:
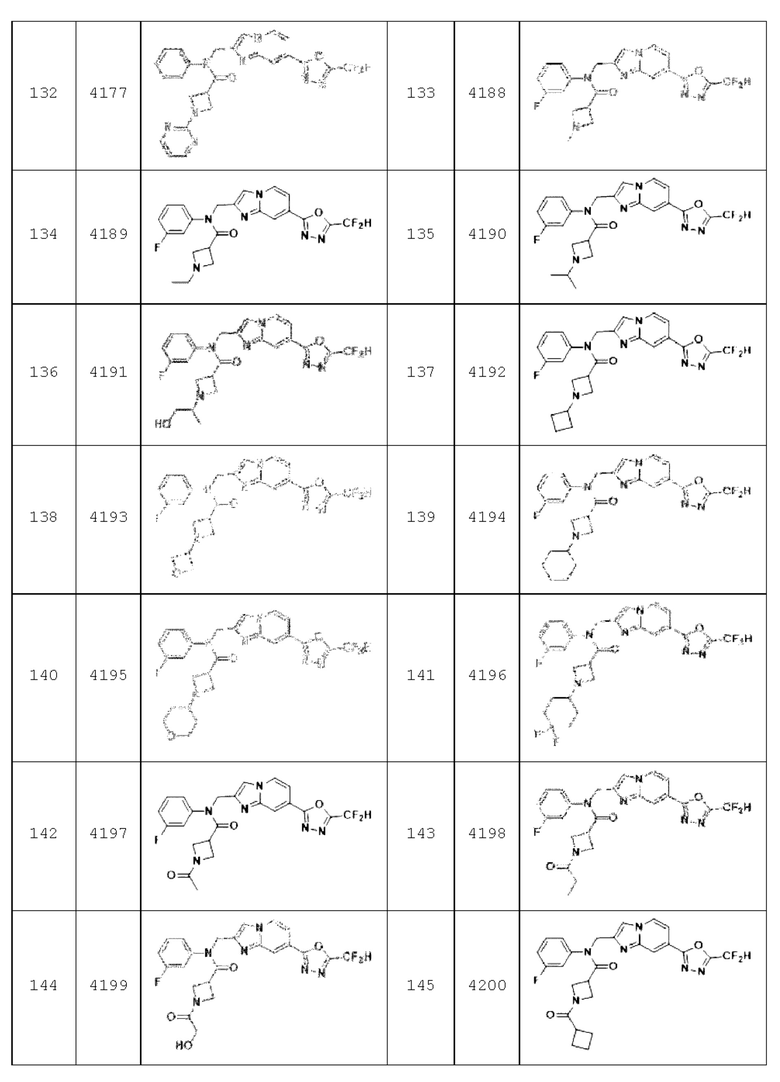
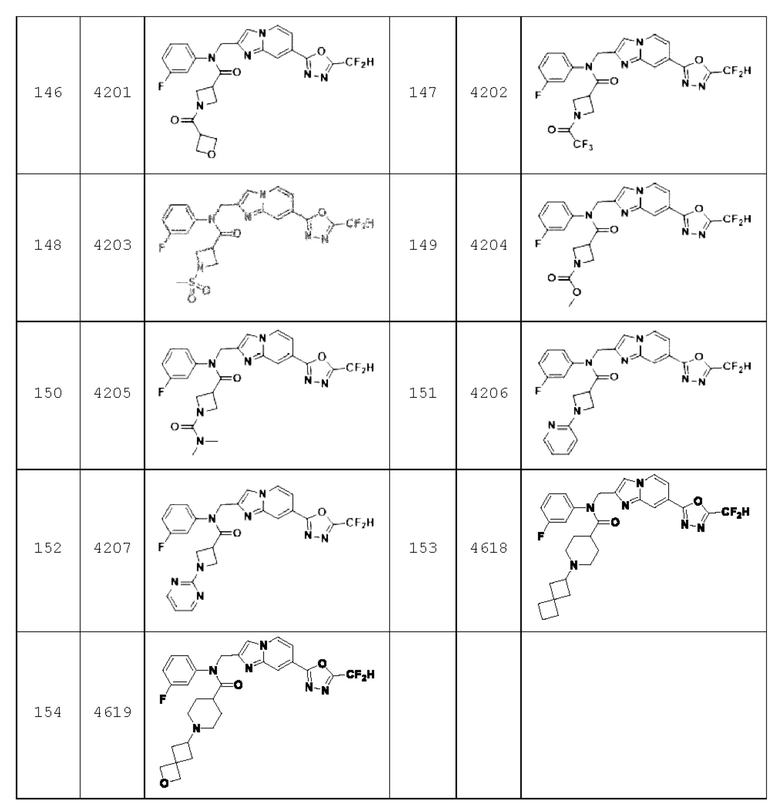 В соответствии с другим вариантом реализации настоящего изобретения конкретные соединения, представленные химической формулой I согласно настоящему изобретению, могут быть показаны в Таблице 3 ниже:
В соответствии с другим вариантом реализации настоящего изобретения конкретные соединения, представленные химической формулой I согласно настоящему изобретению, могут быть показаны в Таблице 3 ниже:
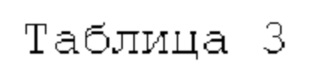

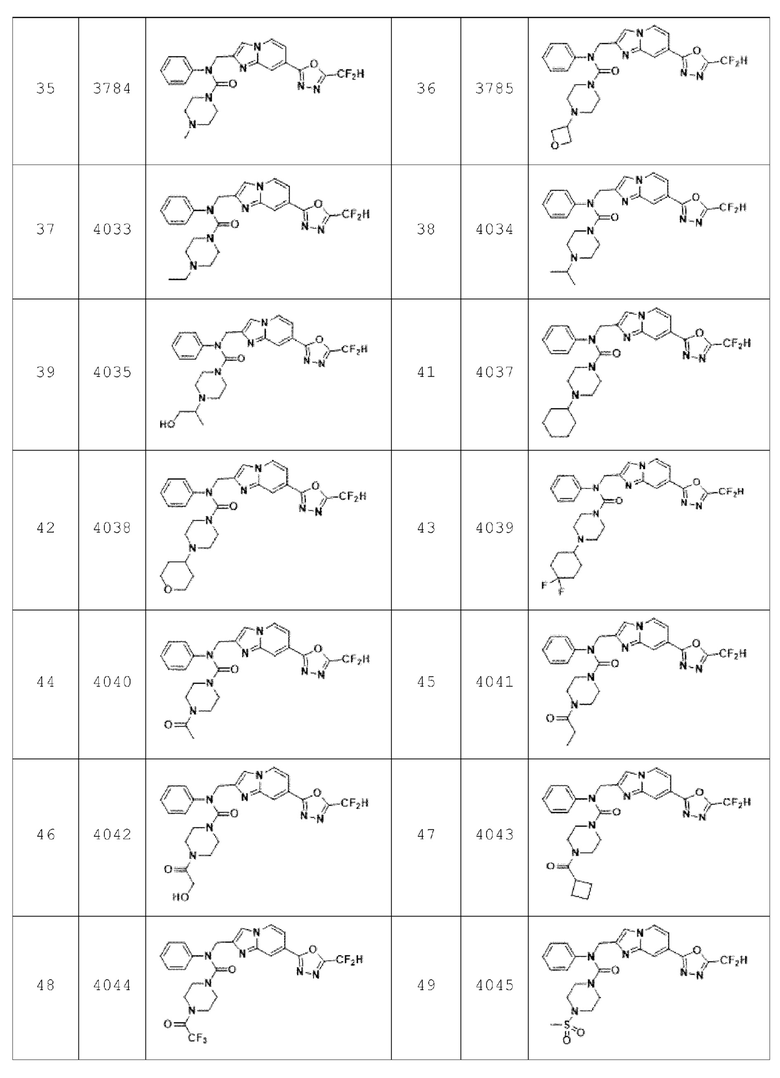
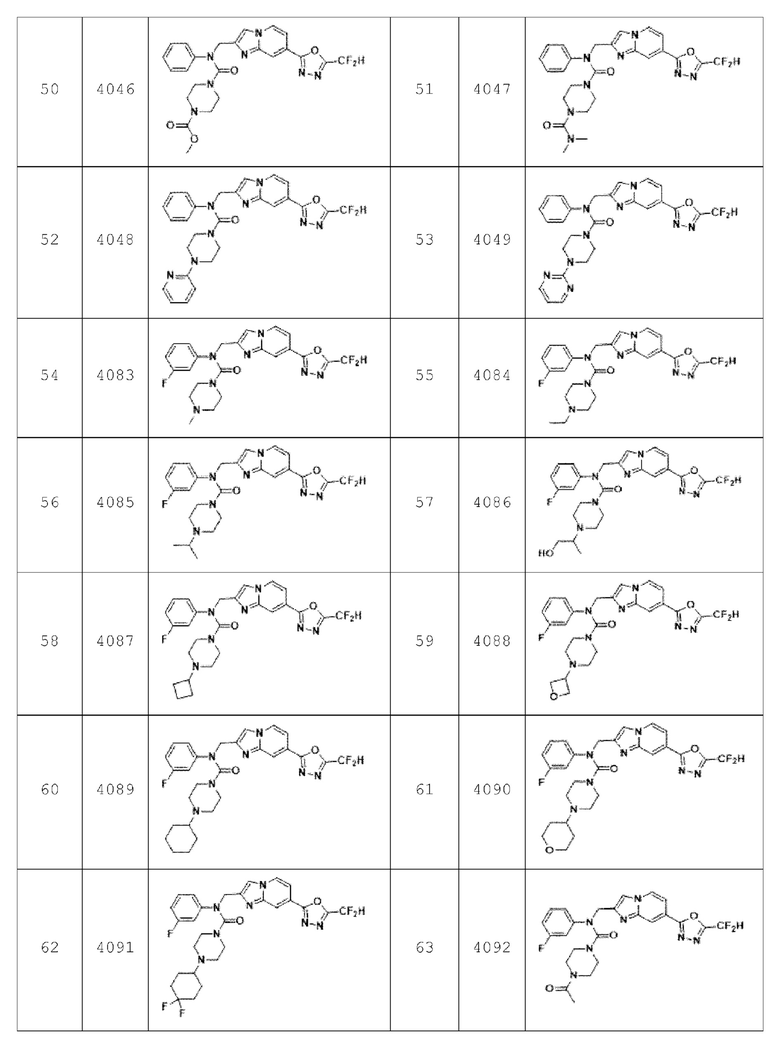
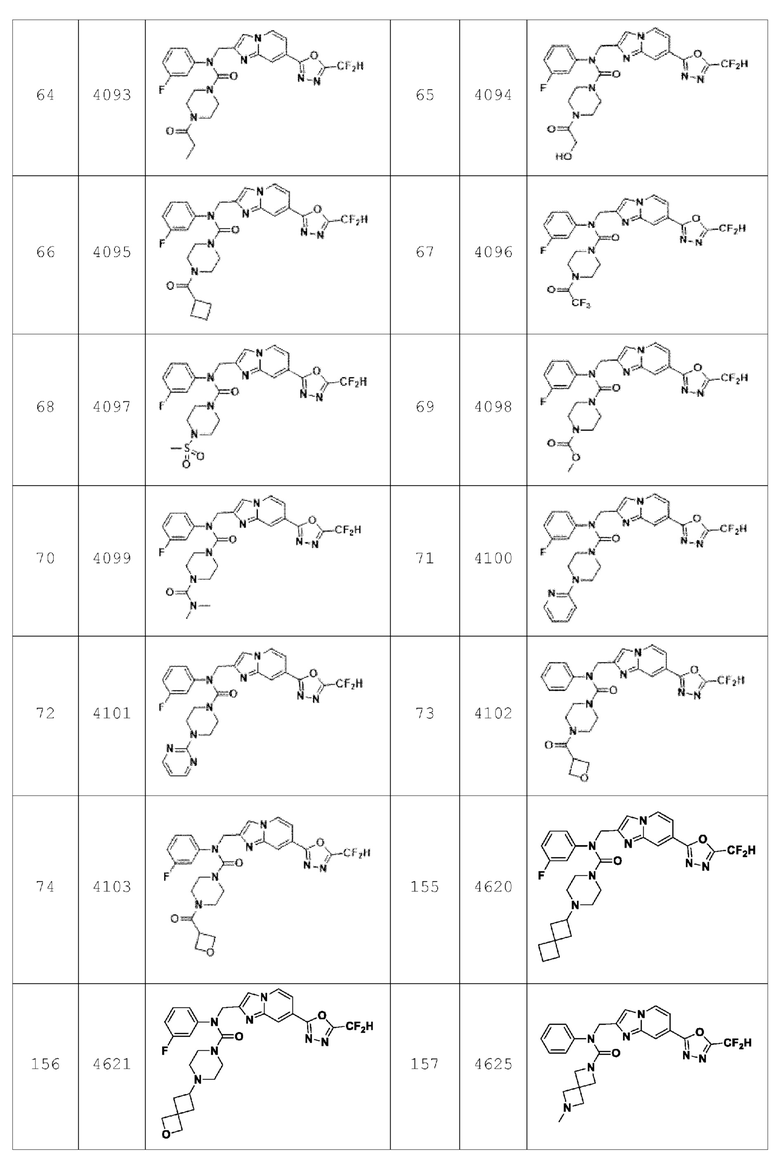
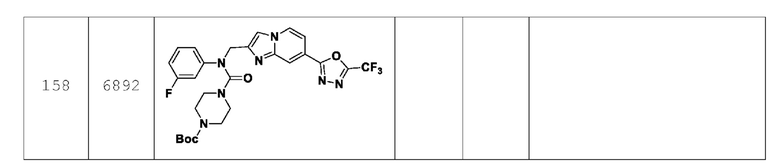
Согласно еще одному варианту реализации настоящего изобретения конкретные соединения, представленные химической формулой I согласно настоящему изобретению, могут быть показаны в Таблице 4 ниже:
В настоящем изобретении фармацевтически приемлемая соль относится к соли, обычно используемой в фармацевтической промышленности, например, может включать неорганические ионные соли, полученные из кальция, калия, натрия и магния, и тому подобные, соли неорганических кислот, полученные из хлористоводородной кислоты, азотной кислоты, фосфорной кислоты, бромноватой кислоты, йодноватой кислоты, перхлорной кислоты и серной кислоты, и тому подобные; соли органических кислот, полученные из уксусной кислоты, трифторуксусной кислоты, лимонной кислоты, малеиновой кислоты, янтарной кислоты, щавелевой кислоты, бензойной кислоты, винной кислоты, фумаровой кислоты, миндальной кислоты, пропионовой кислоты, молочной кислоты, гликолевой кислоты, глюконовой кислоты, галактуроновой кислоты, глутаминовой кислоты, глутаровой кислоты, глюкуроновой кислоты, аспарагиновой кислоты, аскорбиновой кислоты, угольной кислоты, ванильной кислоты, йодистоводородной кислоты, и тому подобные; соли сульфоновой кислоты, полученные из метансульфоновой кислоты, этансульфоновой кислоты, бензолсульфоновой кислоты, п-толуолсульфоновой кислоты и нафталинсульфоновой кислоты, и тому подобные; соли аминокислот, полученные из глицина, аргинина, лизина, и тому подобные; и соли аминов, полученные из триметиламина, триэтиламина, аммиака, пиридина, пиколина, и тому подобные, но типы солей, указанные в настоящем изобретении, не ограничены этими солями, перечисленными выше.
Предпочтительные соли в настоящем изобретении включают гидрохлорид, фосфат, сульфат, трифторацетат, цитрат, бромат, малеат или тартрат.
Соединение, представленное химической формулой I согласно настоящему изобретению, может содержать один или более асимметричных атомов углерода, благодаря чему способно существовать в виде рацемата, рацемической смеси, одного энантиомера, диастереомерной смеси и каждого диастереомера. Эти изомеры могут быть разделены с помощью обычных методов, например, путем разделения, например, с помощью колоночной хроматографии, ВЭЖХ или тому подобного, соединения, представленного химической формулой I. Альтернативно, стереоизомеры каждого из соединений, представленных химической формулой I, могут быть стереоспецифически синтезированы с использованием оптически чистых исходных материалов и/или реагентов с известным пространственным расположением. Способ получения производного 1,3,4-оксадиазола В настоящем изобретении предложен способ получения производного 1,3,4-оксадиазола, представленного химической формулой I, его оптического изомера или его фармацевтически приемлемой соли.
В настоящем изобретении предпочтительный способ получения производного 1,3,4-оксадиазола, представленного химической формулой I, его оптического изомера или фармацевтически приемлемой соли, является таким же, как и в схемах реакции 1-4 ниже, который также включает способы получения, модифицированные в объеме, очевидном для специалистов в данной области техники.
[Схема реакции 1]
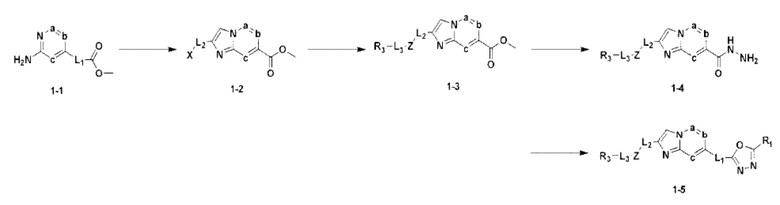
На схеме реакции 1 соединение 1-1 вступает во взаимодействие с 1,3-дихлорпропан-2-оном для синтеза бициклического соединения 1-2 с последующим замещением различными функциональными группами для синтеза соединения 1-3, с последующим взаимодействием с гидразином для синтеза гидразидного соединения 1-4. Наконец, проводят реакцию циклизации с дифторуксусным ангидридом для синтеза целевого соединения 1-5.
Соединение, полученное с помощью приведенной выше схемы реакции, представляет собой соединение 3009.
Схема реакции 2
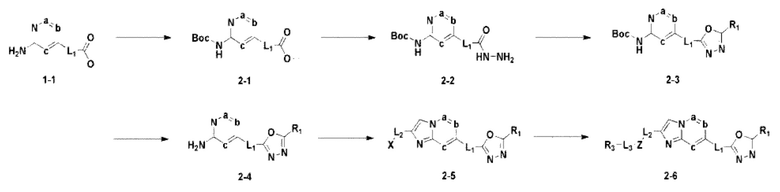
На схеме реакции 2 синтезируют соединение 2-1, в котором в соединение 1-1 введена защитная группа, которое затем вступает в реакцию с гидразином для синтеза гидразидного соединения 2-2. Для синтеза соединения 2-3 проводят реакцию циклизации с фторзамещенным уксусным ангидридом, и затем удаляют защитную группу в кислотных условиях для синтеза соединения 2-4. При взаимодействии с 1,3-дихлорпропан-2-оном синтезируют бициклическое соединение 2-5 и проводят взаимодействие с различными функциональными группами для синтеза целевого соединения 2-6.
Соединения, полученные в соответствии с указанной выше схемой реакции, представляют собой соединения 3585, 3586, 3587, 3588, 3589, 3590, 3591, 3592, 3593, 3594, 3595, 3596, 3668, 3669, 3670, 3671, 3672, 3673, 3674, 3675, 3676, 3677, 3678, 3679, 3719, 3720, 3721, 3722, 3723, 3724 и 3725.
Схема реакции 3
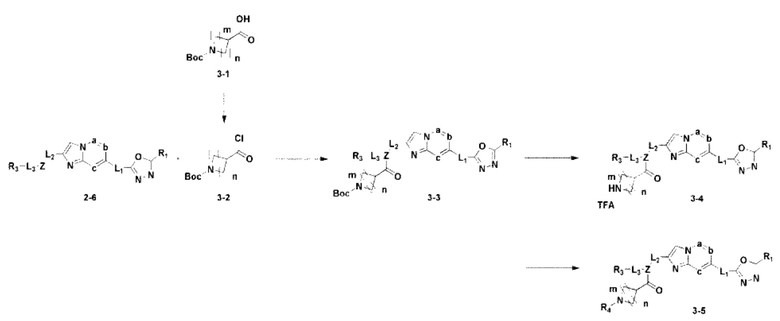
Схема реакции 3 иллюстрирует способ синтеза соединения, имеющего амидную структуру, где соединение 2-6, синтезированное в схеме реакции 2, вступает во взаимодействие с соединением 3-2, содержащим ацетилхлоридную функциональную группу, в основных условиях, для синтеза соединения 3-3. Синтезируют соединение 3-4, из которого удалена защитная группа в кислотных условиях, и проводят взаимодействие с различными функциональными группами для синтеза целевого соединения 3-5.
Соединения, полученные в соответствии с указанной выше схемой реакции, представляют собой соединения 3782, 3783, 4115, 4116, 4117, 4118, 4119, 4120, 4121, 4122, 4123, 4124, 4125, 4126, 4127, 4128, 4129, 4130, 4131, 4132, 4137, 4138, 4139, 4140, 4141, 4142, 4143, 4144, 4145, 4146, 4147, 4148, 4150, 4151, 4152, 4153, 4154, 4155, 4156, 4157, 4158, 4159, 4160, 4161, 4162, 4163, 4164, 4165, 4166, 4167, 4168, 4169, 4170, 4171, 4172, 4173, 4174, 4175, 4176, 4177, 4188, 4189, 4190, 4191, 4192, 4193, 4194, 4195, 4196, 4197, 4198, 4199, 4200, 4201, 4202, 4203, 4204, 4204, 4205, 4206, 4207, 4618 и 4619.
Схема реакции 4
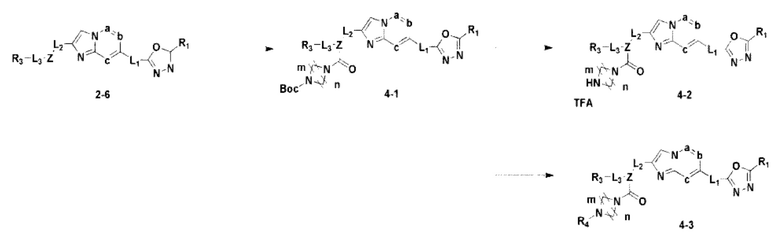
Схема реакции 4 иллюстрирует способ синтеза соединения, имеющего структуру мочевины, где соединение 2-6, синтезированное в схеме реакции 2, вступает во взаимодействие с трифосгеном и аминным соединением в основных условиях, для синтеза соединения 4-1. Синтезируют соединение 4-2, из которого удалена защитная группа в кислотных условиях, и проводят взаимодействие с различными функциональными группами для синтеза целевого соединения 4-3.
Соединения, полученные в соответствии с указанной выше схемой реакции, представляют собой соединения 3784, 3785, 4033, 4034, 4035, 4036, 4037, 4038, 4039, 4040, 4041, 4042, 4043, 4044, 4045, 4046, 4047, 4048, 4049, 4083, 4084, 4085, 4086, 4087, 4088, 4089, 4090, 4091, 4092, 4093, 4094, 4095, 4096, 4097, 4098, 4099, 4100, 4101, 4102, 4103, 4620, 4621, 4625 и 6892.
Композиция, содержащая производные 1,3,4-оксадиазола, ее применение, и способ лечения с ее применением
В настоящем изобретении предложена фармацевтическая композиция для предотвращения или лечения заболеваний, опосредованных гистондеацетилазой 6, содержащая в качестве активного ингредиента соединение, представленное химической формулой I ниже, его оптический изомер или его фармацевтически приемлемую соль:
[Химическая формула I]
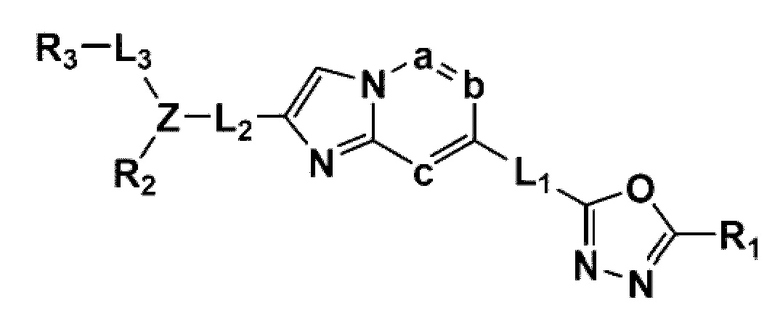
Химическая формула I является такой же, как определено выше.
Фармацевтическая композиция согласно настоящему изобретению проявляет замечательный эффект в предотвращении или лечении заболеваний, опосредованных гистондеацетилазой 6, путем селективного ингибирования гистондеацетилазы 6.
Заболевания, опосредованные гистондеацетилазой 6, включают инфекционные заболевания, такие как прионное заболевание; новообразования, такие как доброкачественные опухоли (например, миелодиспластический синдром) или злокачественные опухоли (например, множественная миелома, лимфома, лейкоз, рак легкого, колоректальный рак, рак толстой кишки, рак предстательной железы, эпителиальная карцинома мочевыводящих путей, рак молочной железы, меланома, рак кожи, рак печени, рак головного мозга, рак желудка, рак яичника, рак поджелудочной железы, рак головы и шеи, рак полости рта или глиома); эндокринные, пищевые и метаболические заболевания, такие как болезнь Вильсона, амилоидоз или диабет; психические и поведенческие расстройства, такие как депрессия или синдром Ретта; неврологические заболевания, такие как атрофия центральной нервной системы (например, болезнь Хантингтона, спинальная мышечная атрофия (СМА), спинальная мозжечковая атаксия (SCA)), нейродегенеративные заболевания (например, болезнь Альцгеймера), двигательные расстройства (например, болезнь Паркинсона), нейропатия (например, наследственная нейропатия (болезнь Шарко-Мари-Тута), спорадическая нейропатия, воспалительная нейропатия, медикаментозная нейропатия), двигательная нейропатия (например, амиотрофический боковой склероз (ALS)) или демиелинизация центральной нервной системы (например, рассеянный склероз (MS)); заболевания глаз и прилежаащих органов, такие как увеит; заболевания системы кровообращения, такие как фибрилляция предсердий, инсульт и тому подобное; респираторные заболевания, такие как астма; заболевания пищеварительного тракта, такие как алкогольное заболевание печени, воспалительное заболевание кишечника, болезнь Крона, язвенное заболевание кишечника и тому подобное; заболевания кожи и подкожных тканей, такие как псориаз; заболевания опорно-двигательного аппарата и соединительной ткани, такие как ревматоидный артрит, остеоартрит, системная красная волчанка (СКВ) и тому подобное; или врожденные пороки развития, изменения и хромосомные аномалии, такие как аутосомно-доминантная поликистозная болезнь почки, а также включают симптомы или заболевания, связанные с аномальными функциями гистондеацетилазы.
Фармацевтически приемлемая соль представляет собой такую же, как описано выше для фармацевтически приемлемой соли соединения, представленного химической формулой I согласно настоящему изобретению.
Фармацевтическая композиция согласно настоящему изобретению может дополнительно содержать один или более фармацевтически приемлемых носителей для введения, в дополнение к соединению, представленному химической формулой I, его оптическому изомеру или его фармацевтически приемлемой соли. Фармацевтически приемлемый носитель можно применять путем смешивания физиологического раствора, стерильной воды, раствора Рингера, забуференного физиологического раствора, раствора декстрозы, раствора мальтодекстрина, глицерина, этанола и одного или более из этих ингредиентов, и, при необходимости, могут быть добавлены другие обычные добавки, такие как антиоксиданты, буферы, бактериостатические агенты и тому подобное. Кроме того, инъекционные составы, такие как водные растворы, суспензии, эмульсии и тому подобное, пилюли, капсулы, гранулы или таблетки могут быть приготовлены путем дополнительного добавления разбавителей, диспергирующих агентов, поверхностно-активных веществ, связующих веществ и смазывающих веществ. Соответственно, композиция согласно настоящему изобретению может представлять собой пластырь, жидкость, пилюлю, капсулу, гранулу, таблетку, суппозиторий или тому подобное. Эти составы могут быть получены обычным способом, используемым в данной области техники, или способом, описанным в Remington's Pharmaceutical Science (последняя редакция). Mack Publishing Company, Easton PA, и приготовлены в виде различных составов в зависимости от соответствующих заболеваний или ингредиентов.
Композицию согласно настоящему изобретению можно вводить перорально или парентерально (например, внутривенно, подкожно, внутрибрюшинно или местно) в зависимости от желаемого способа, и диапазон доз варьируется в зависимости от массы тела пациента, возраста, пола, состояния здоровья, питания, времени введения, способа введения, скорости выведения и тяжести заболевания и тому подобного. Суточная доза соединения, представленного химической формулой I согласно настоящему изобретению, может составлять примерно от 1 до 1000 мг/кг, предпочтительно от 5 до 100 мг/кг, и ее можно вводить один раз в сутки или разделять на несколько раз в сутки.
Фармацевтическая композиция согласно настоящему изобретению может дополнительно содержать один или более активных ингредиентов, проявляющих одинаковые или аналогичные лекарственные эффекты в дополнение к соединению, представленному химической формулой I выше, его оптическому изомеру или его фармацевтически приемлемой соли.
В настоящем изобретении предложен способ предотвращения или лечения заболеваний, опосредованных гистондеацетилазой 6, включая введение терапевтически эффективного количества соединения, представленного химической формулой I, его оптического изомера или его фармацевтически приемлемой соли.
Термин "терапевтически эффективное количество", используемый в настоящем изобретении, относится к количеству соединения, представленного химической формулой I, которое эффективно для предотвращения или лечения заболеваний, опосредованных гистондеацетилазой 6.
Кроме того, в настоящем изобретении предложен способ селективного ингибирования HDAC6 путем введения соединения, представленного химической формулой I, его оптического изомера или его фармацевтически приемлемой соли, млекопитающему, включая человека.
Способ предотвращения или лечения заболеваний, опосредованных гистондеацетилазой 6 согласно настоящему изобретению, также включает введение соединения, представленного химической формулой I, для лечения самого заболевания до начала симптома, а также для ингибирования или предотвращения его симптома. При лечении заболевания профилактическая или терапевтическая доза конкретного активного ингредиента может варьироваться в зависимости от природы и тяжести заболевания или состояния, а также от способа введения активного ингредиента. Доза и частота приема будут варьироваться в зависимости от возраста, массы тела и ответа отдельных пациентов. Подходящий режим дозирования может быть легко выбран человеком, обладающим обычными знаниями в данной области техники, учитывая эти факторы как должное. Кроме того, способ предотвращения или лечения заболеваний, опосредованных гистондеацетилазой 6 согласно настоящему изобретению, может дополнительно включать введение терапевтически эффективного количества дополнительного активного агента, подходящего для лечения заболевания, совместно с соединением, представленным химической формулой I, при этом дополнительный активный агент может проявлять синергетический или вспомогательный эффект совместно с соединением, представленным химической формулой I.
Настоящее изобретение также направлено на обеспечение применения соединения, представленного химической формулой I выше, его оптического изомера или его фармацевтически приемлемой соли для получения лекарственного средства для лечения заболеваний, опосредованных гистондеацетилазой 6. Соединение, представленное химической формулой I выше для получения лекарственного средства, может быть смешано с приемлемыми адъювантами, разбавителями, носителями и тому подобным, и может быть получено в виде сложного состава с другими активными агентами для обеспечения синергетического эффекта активных ингредиентов.
Сущности, упомянутые в применениях, композициях и способах лечения согласно настоящему изобретению, применяются одинаково, если они несовместимы друг с другом.
[Преимущества изобретения]
Соединение, представленное химической формулой I выше согласно настоящему изобретению, его оптический изомер или его фармацевтически приемлемая соль, способно селективно ингибировать гистондеацетилазу 6 (HDAC6), тем самым оказывая удивительно превосходное профилактическое или терапевтическое действие на заболевания, опосредованные HDAC6.
[Наилучший вариант реализации]
Далее настоящее изобретение будет более подробно описано с помощью примеров и экспериментальных примеров. Однако они являются только примерами настоящего изобретения, и объем настоящего изобретения ими не ограничивается.
Получение производных 1,3,4-оксадиазола согласно настоящему изобретению
Конкретный способ получения соединения, представленного химической формулой I, является следующим.
Пример 1: Синтез Соединения 3009, 2-(2-бензилимидазо[1,2-а]пиридин-7-ил)-5-(дифторметил)-1,3,4-оксадиазола
[Стадия 1] Синтез метил-2-(хлорметил)-имидазо[1,2-а]пиридин-7-карбоксилата
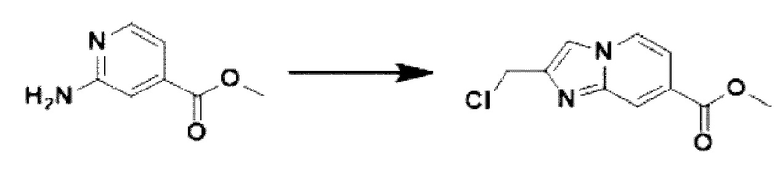
Метил-2-аминоизоникотинат (1,000 г, 6,572 ммоль) и 1,3-дихлорпропан-2-он (1,085 г, 8,544 ммоль) растворяли в этаноле (5 мл)/1,2-диметоксиэтане (6 мл), и полученный раствор перемешивали при комнатной температуре в течение 1 часа, а затем перемешивали при 90°С в течение 16 часов. Затем температуру понижали до комнатной температуры для завершения реакции. В реакционную смесь выливали воду, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 24 г; этилацетат/гексан = от 5% до 70%) и концентрировали с получением целевого соединения (0,200 г, 13,5%) в виде твердого вещества бежевого цвета.
[Стадия 2] Синтез метил-2-бензилимидазо[1,2-а]пиридин-7-карбоксилата
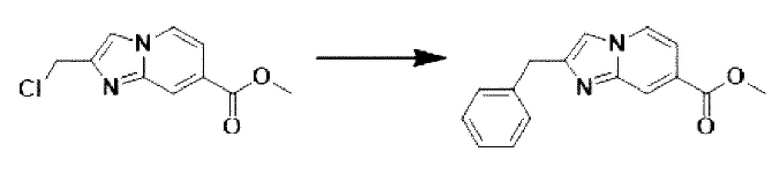
Метил-2-(хлорметил)-имидазо[1,2-а] пиридин-7-карбоксилат (0,200 г, 0,890 ммоль), полученный на стадии 1, фенилбороновую кислоту (0,217 г, 1,781 ммоль), дихлорид бис-((трифенил)-фосфин)-палладия (II) (Pd(PPh3)2Cl2, 0,062 г, 0,089 ммоль) и карбонат калия (0,369 г, 2,671 ммоль) растворяли в 1,4-диоксане (8 мл)/воде (2 мл) при комнатной температуре и полученный раствор перемешивали при 105°С в течение 16 часов. Затем температуру понижали до комнатной температуры для завершения реакции. В реакционную смесь выливали воду, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 24 г; этилацетат/гексан = от 5% до 70%) и концентрировали с получением целевого соединения (0,076 г, 32,1%) в виде твердого вещества белого цвета.
[Стадия 3] Синтез 2-бензилимидазо[1,2-а]пиридин-7-карбогидразида
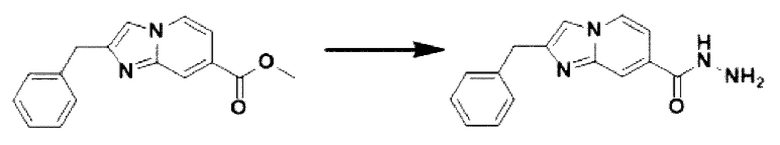
Метил-2-бензилимидазо[1,2-а]пиридин-7-карбоксилат (0,075 г, 0,282 ммоль), полученный на стадии 2, и моногидрат гидразина (0,068 мл, 1,408 ммоль) растворяли в этаноле (5 мл) при комнатной температуре и полученный раствор нагревали с обратным холодильником в течение 16 часов. Затем температуру понижали до комнатной температуры для завершения реакции. В реакционную смесь вливали воду, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении, в результате чего получали указанное в целевое соединение (0,074 г, 98,7%) в виде твердого вещества белого цвета.
[Стадия 4] Синтез соединения 3009
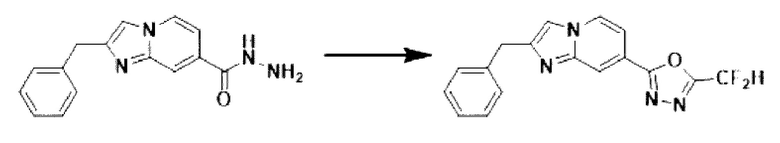
2-бензилимидазо[1,2-а]пиридин-7-карбогидразид (0,065 г, 0,244 ммоль), полученный на стадии 3, и имидазол (0,050 г, 0,732 ммоль) растворяли в дихлорметане (5 мл), и добавляли при комнатной температуре 2,2-дифторуксусный ангидрид (0,152 мл, 1,220 ммоль), и нагревали с обратным холодильником в течение 16 часов. Затем температуру понижали до комнатной температуры для завершения реакции. В реакционную смесь вливали воду, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (картридж SiO2, 4 г; этилацетат/гексан = от 5% до 60%) и концентрировали с получением целевого соединения (0,030 г, 37,7%) в виде твердого вещества ярко-красного цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,29 (м, 1Н), 8,15-8,13 (м, 1Н), 7,49-7,46 (м, 1Н), 7,36-7,32 (м, 5Н), 7,29-7,26 (м, 1Н), 6,95 (т, J=51,7 Гц, 1Н), 4,22 (с, 2Н);
МСНР (ЭР) m/z 327,2 (М++1).
Пример 2: Синтез Соединения 3585, 2-(дифторметил)-5-(2-(3-фторбензил)-имидазо[1,2-а]пиридин-7-ил)-1,3,4-оксадиазола
[Стадия 1] Синтез метил-2-((трет-бутоксикарбонил)-амино)-изоникотината
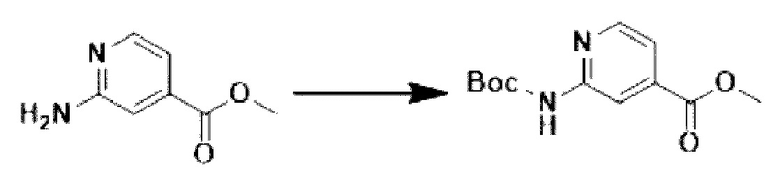
Метил-2-аминоизоникотинат (20,000 г, 131,449 ммоль) и ди-трет-бутилдикарбонат (37,295 г, 170,884 ммоль) растворяли в трет-бутаноле (800 мл) при комнатной температуре. Полученный раствор перемешивали при 60°С в течение 16 часов, и затем температуру понижали до комнатной температуры для прекращения реакции. Выпавшее в осадок твердое вещество фильтровали, промывали этанолом и сушили с получением целевого соединения (26,000 г, 78,4%) в виде твердого вещества белого цвета.
[Стадия 2] Синтез трет-бутил-(4-(гидразинкарбонил-)пиридин-2-ил)-карбамата
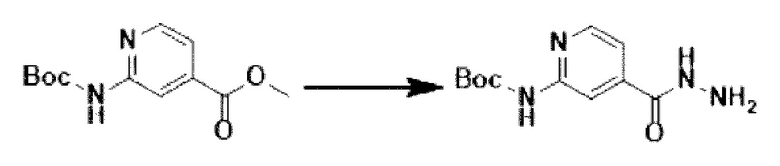
Метил-2-((трет-бутоксикарбонил-)амино)-изоникотинат (26,000 г, 103,064 ммоль), полученный на стадии 1, и моногидрат гидразина (100,182 мл, 2,061 моль) растворяли в метаноле (800 мл) при комнатной температуре. Полученный раствор перемешивали при той же температуре в течение 16 часов. К полученному продукту добавляли метанол (500 мл), затем фильтровали через пластиковый фильтр с получением органического слоя, и полученный органический слой концентрировали с получением целевого соединения (25,000 г, 96,2%) в виде твердого вещества белого цвета.
[Стадия 3] Синтез трет-бутил-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-пиридин-2-ил)-карбамата
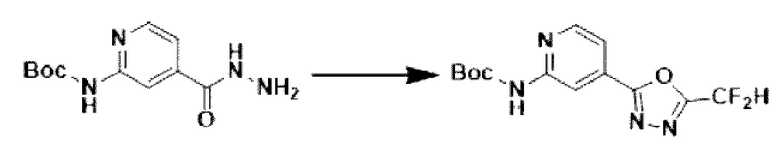
Трет-бутил-(4-(гидразинкарбонил)-пиридин-2-ил)-карбамат (20,000 г, 79,280 ммоль), полученный на стадии 2, и триэтиламин (55,250 мл, 396,401 ммоль) растворяли в тетрагидрофуране (600 мл), и добавляли при комнатной температуре 2,2-дифторуксусный ангидрид (49,281 мл, 396,401 ммоль), и нагревали с обратным холодильником в течение 16 часов. Затем температуру понижали до комнатной температуры для завершения реакции. В реакционную смесь вливали насыщенный водный раствор хлорида аммония с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. В концентрат вливали этилацетат (40 мл) и гексан (200 мл), суспендировали и фильтровали с получением твердого вещества, и полученное твердое вещество промывали гексаном и сушили с получением целевого соединения (11,500 г, 46,5%) в виде твердого вещества белого цвета.
[Стадия 4] Синтез 4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-пиридин-2-амина
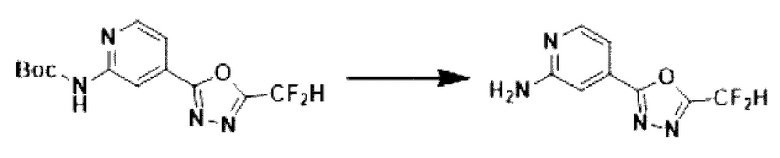
Трет-бутил-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-пиридин-2-ил)-карбамат (11,500 г, 36,826 ммоль), полученный на стадии 3, растворяли в дихлорметане (300 мл) и добавляли трифторуксусную кислоту (28,199 мл, 368,259 ммоль) при 0°С, и перемешивали при комнатной температуре в течение 4 часов. После удаления растворителя из реакционной смеси при пониженном давлении в концентрат вливали насыщенный водный раствор гидрокарбоната натрия (150 мл) и суспендировали с последующей фильтрацией с получением твердого вещества. Полученное твердое вещество промывали водой и сушили с получением целевого соединения (7,500 г, 96,0%) в виде твердого вещества белого цвета.
[Стадия 5] Синтез 2-(2-(хлорметил)-имидазо[1,2-а]пиридин-7-ил)-5-(дифторметил)-1,3,4-оксадиазола
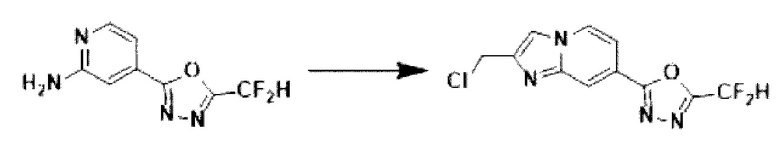
4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-пиридин-2-амин (7,500 г, 35,351 ммоль), полученный на стадии 4, 1,3-дихлорпропан-2-он (6,732 г, 53,026 ммоль) и гидрокарбонат натрия (14,848 г, 176,753 ммоль) растворяли в 1,4-диоксане (250 мл) при комнатной температуре. Реакционную смесь нагревали с обратным холодильником в течение 16 часов, а затем понижали температуру до комнатной температуры для прекращения реакции. Реакционную смесь фильтровали через пластиковый фильтр для удаления твердых веществ, и фильтрат очищали с помощью колоночной хроматографии (картридж SiO2, 80 г; этилацетат/гексан = от 10 до 90%) и концентрировали с получением целевого соединения (7,000 г, 69,6%) в виде твердого вещества бежевого цвета.
[Стадия 6] Синтез соединения 3585
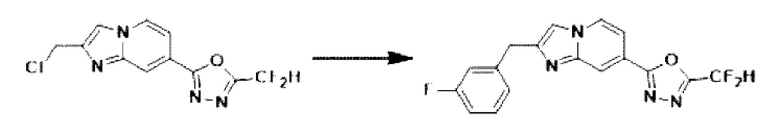
2-(2-(хлорметил)-имидазо[1,2-а]пиридин-7-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,050 г, 0,176 ммоль), полученный на стадии 5, (3-фторфенил)-бороновую кислоту (0,049 г, 0,351 ммоль), дихлорид бис-(трифенилфосфин)-палладия (II) (Pd(PPh3)2Cl2, 0,012 г, 0,018 ммоль) и карбонат калия (0,073 г, 0,527 ммоль) растворяли в 1,4-диоксане (4 мл)/воде (1 мл) при комнатной температуре, и полученный раствор перемешивали при 100°С в течение 1 часа. Затем температуру понижали до комнатной температуры для завершения реакции. В реакционную смесь вливали воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором, сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (картридж SiO2, 4 г; этилацетат/гексан = от 0% до 70%) и концентрировали с получением целевого соединения (0,023 г, 38,0%) в виде твердого вещества розового цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,27-8,19 (м, 1Н), 8,10 (дд, J=7,1, 1,0 Гц, 1Н), 7,44 (дд, J=7,1, 1,7 Гц, 1Н), 7,34 (с, 1Н), 7,23 (тд, J=8,0, 6,0 Гц, 1Н), 7,05 (дт, J=7,6, 1,2 Гц, 1Н), 7,00-6,70 (м, 3Н), 4,13 (с, 2Н);
МСНР (ЭР) m/z 345,9 (M++1).
Пример 3: Синтез Соединения 3586, 2-(дифторметил)-5-(2-(4-фторбензил)-имидазо[1,2-а]пиридин-7-ил)-1,3,4-оксадиазола
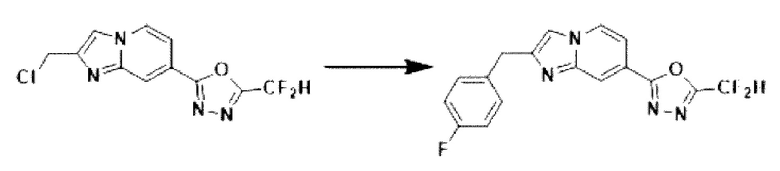
2-(2-(хлорметил)-имидазо[1,2-а]пиридин-7-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,050 г, 0,176 ммоль), полученный на стадии 5 примера 2, (4-фторфенил)-бороновую кислоту (0,049 г, 0,351 ммоль), дихлорид бис-(трифенилфосфин)-палладия (II) (Pd(PPh3)2Cl2, 0,012 г, 0,018 ммоль) и карбонат калия (0,073 г, 0,527 ммоль) растворяли в 1,4-диоксане (4 мл)/воде (1 мл) при комнатной температуре, и полученный раствор перемешивали при 100°С в течение 1 часа. Затем температуру понижали до комнатной температуры для завершения реакции. В реакционную смесь вливали воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором, сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (картридж SiO2, 4 г; этилацетат/гексан = от 0% до 70%) и концентрировали с получением целевого соединения (0,030 г, 49,6%) в виде твердого вещества розового цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,37-8,29 (м, 1Н), 8,18 (дд, J=7,1, 1,0 Гц, 1Н), 7,53 (дд, J=7,1, 1,7 Гц, 1Н), 7,39 (д, J=0,9 Гц, 1Н), 7,36-7,30 (м, 2Н), 7,10-6,81 (м, 3Н), 4,20 (с, 2Н);
МСНР (ЭР) m/z 346,0 (М++1).
Пример 4: Синтез Соединения 3587, 2-(2-(2-(3,4-дифторбензил)-имидазо[1,2-а]пиридин-7-ил)-5-(дифторметил)-1,3,4-оксадиазола
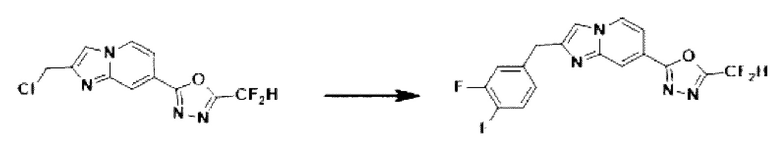
2-(2-(хлорметил)-имидазо[1,2-а]пиридин-7-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,050 г, 0,176 ммоль), полученный на стадии 5 примера 2, (3,4-дифторфенил)-бороновую кислоту (0,055 г, 0,351 ммоль), дихлорид бис-(трифенилфосфин)-палладия (II) (Pd(PPh3)2Cl2, 0,012 г, 0,018 ммоль) и карбонат калия (0,073 г, 0,527 ммоль) растворяли в 1,4-диоксане (4 мл)/воде (1 мл) при комнатной температуре, и полученный раствор перемешивали при 100°С в течение 1 часа. Затем температуру понижали до комнатной температуры для завершения реакции. В реакционную смесь вливали воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором, сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (картридж SiO2, 4 г; этилацетат/гексан = от 0% до 70%) и концентрировали с получением целевого соединения (0,033 г, 51,9%) в виде твердого вещества розового цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,38-8,29 (м, 1Н), 8,21 (дд, J=7,1, 1,0 Гц, 1Н), 7,55 (дд, J=7,1, 1,7 Гц, 1Н), 7,45 (с, 1Н), 7,21-6,81 (м, 4Н), 4,19 (с, 2Н);
МСНР (ЭР) m/z 364,0 (М++1).
Пример 5: Синтез Соединения 3588, 2-(2-(2-(2,4-дифторбензил)-имидазо[1,2-а]пиридин-7-ил)-5-(дифторметил)-1,3,4-оксадиазола

2-(2-(хлорметил)-имидазо[1,2-а]пиридин-7-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,050 г, 0,176 ммоль), полученный на стадии 5 примера 2, (2,4-дифторфенил)-бороновую кислоту (0,055 г, 0,351 ммоль), дихлорид бис-(трифенилфосфин)-палладия (II) (Pd(PPh3)2Cl2, 0,012 г, 0,018 ммоль) и карбонат калия (0,073 г, 0,527 ммоль) растворяли в 1,4-диоксане (4 мл)/воде (1 мл) при комнатной температуре, и полученный раствор перемешивали при 100°С в течение 1 часа. Затем температуру понижали до комнатной температуры для завершения реакции. В реакционную смесь вливали воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором, сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (картридж SiO2, 4 г; этилацетат/гексан = от 0% до 70%) и концентрировали с получением целевого соединения (0,031 г, 48,7%) в виде твердого вещества розового цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,37-8,29 (м, 1Н), 8,20 (дд, J=7,1, 1,0 Гц, 1Н), 7,54 (дд, J=7,1, 1,7 Гц, 1Н), 7,47 (с, 1Н), 7,37 (тд, J=8,7, 8,3, 6,4 Гц, 1Н), 7,11-6,80 (м, 3Н), 4,22 (с, 2Н);
МСНР (ЭР) m/z 364,0 (M++1).
Пример 6: Синтез Соединения 3589, 2-(дифторметил)-5-(2-(4-метилбензил)-имидазо[1,2-а]пиридин-7-ил)-1,3,4-оксадиазола
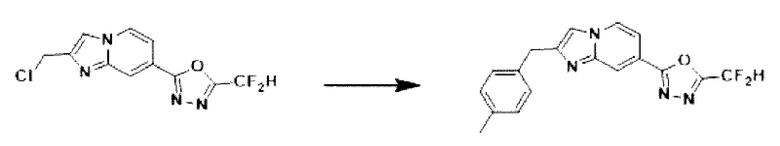
2-(2-(хлорметил)-имидазо[1,2-а]пиридин-7-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,050 г, 0,176 ммоль), полученный на стадии 5 примера 2, п-толилбороновую кислоту (0,048 г, 0,351 ммоль), дихлорид бис-(трифенилфосфин)-палладия (II) (Pd(PPh3)2Cl2, 0,012 г, 0,018 ммоль) и карбонат калия (0,073 г, 0,527 ммоль) растворяли в 1,4-диоксане (4 мл)/воде (1 мл) при комнатной температуре, и полученный раствор перемешивали при 100°С в течение 1 часа. Затем температуру понижали до комнатной температуры для завершения реакции. В реакционную смесь вливали воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором, сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (картридж SiO2, 4 г; этилацетат/гексан = от 0% до 70%) и концентрировали с получением целевого соединения (0,028 г, 46,8%) в виде твердого вещества розового цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,39-8,28 (м, 1Н), 8,15 (дд, J=7,1, 1,0 Гц, 1Н), 7,51 (дд, J=7,1, 1,7 Гц, 1Н), 7,37 (т, J=0,8 Гц, 1Н), 7,26 (д, J=8,0 Гц, 2Н), 7,19-7,15 (м, 2Н), 6,95 (т, J=51,7 Гц, 1Н), 4,20 (с, 2Н), 2,37 (с, 3Н);
МСНР (ЭР) m/z 341,3 (М++1).
Пример 7: Синтез Соединения 3590, 2-(дифторметил)-5-(2-(4-метилбензил)-имидазо[1,2-а]пиридин-7-ил)-1,3,4-оксадиазола
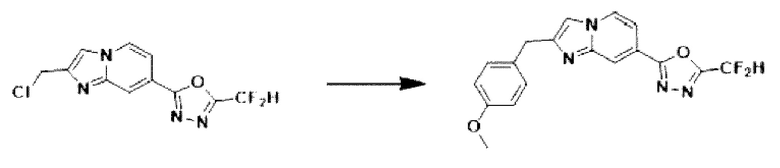
2-(2-(хлорметил)-имидазо[1,2-а]пиридин-7-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,050 г, 0,176 ммоль), полученный на стадии 5 примера 2, (4-метоксифенил)-бороновую кислоту (0,053 г, 0,351 ммоль), дихлорид бис-(трифенилфосфин)-палладия (II) (Pd(PPh3)2Cl2, 0,012 г, 0,018 ммоль) и карбонат калия (0,073 г, 0,527 ммоль) растворяли в 1,4-диоксане (4 мл)/воде (1 мл) при комнатной температуре, и полученный раствор перемешивали при 100°С в течение 1 часа. Затем температуру понижали до комнатной температуры для завершения реакции. После удаления растворителя из реакционной смеси при пониженном давлении, концентрат очищали с помощью хроматографии (SiO2 пластина, 20×20×1 мм; этилацетат/гексан = от 0% до 70%) и концентрировали с получением целевого соединения (0,015 г, 24,0%) в виде твердого вещества розового цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,29-8,16 (м, 1Н), 8,06 (дд, J=7,1, 1,0 Гц, 1Н), 7,41 (дд, J=7,1, 1,7 Гц, 1Н), 7,19 (т, J=4,3 Гц, 3Н), 7,00-6,71 (м, 3Н), 4,08 (с, 2Н), 3,73 (с, 3Н);
МСНР (ЭР) m/z 356,9 (М++1).
Пример 8: Синтез Соединения 3591, 2-(дифторметил)-5-(2-(4-трифторметил)-бензил)-имидазо[1,2-а]пиридин-7-ил)-1,3,4-оксадиазола
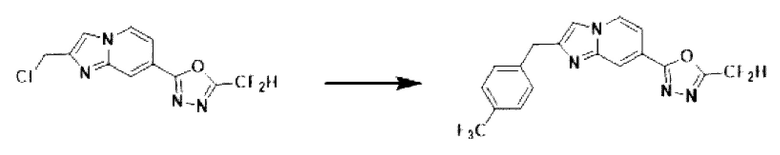
2-(2-(хлорметил)-имидазо[1,2-а]пиридин-7-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,050 г, 0,176 ммоль), полученный на стадии 5 примера 2, (4-(трифторметил)-фенил)-бороновую кислоту (0,067 г, 0,351 ммоль), дихлорид бис-(трифенилфосфин)-палладия (II) (Pd(PPh3)2Cl2, 0,012 г, 0,018 ммоль) и карбонат калия (0,073 г, 0,527 ммоль) растворяли в 1,4-диоксане (4 мл)/воде (1 мл) при комнатной температуре, и полученный раствор перемешивали при 100°С в течение 1 часа.
Затем температуру понижали до комнатной температуры для завершения реакции. После удаления растворителя из реакционной смеси при пониженном давлении, концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; этилацетат/гексан = от 0% до 70%) и концентрировали с получением целевого соединения (0,029 г, 41,9%) в виде твердого вещества розового цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,40-8,27 (м, 1Н), 8,20 (дд, J=7,1, 0,9 Гц, 1Н), 7,61 (д, J=8,0 Гц, 2Н), 7,54 (дд, J=7,1, 1,7 Гц, 1Н), 7,48 (д, J=8,0 Гц, 2Н), 7,44 (с, 1Н), 6,96 (т, J=51,7 Гц, 1Н), 4,28 (с, 2Н);
МСНР (ЭР) m/z 394,7 (М++1).
Пример 9: Синтез Соединения 3592, 1-(4-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-фенил)-этан-1-она
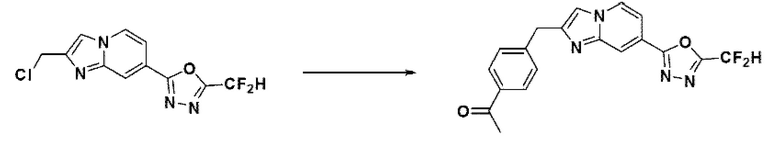
2-(2-(хлорметил)-имидазо[1,2-а]пиридин-7-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,050 г, 0,176 ммоль), полученный на стадии 5 примера 2, (4-ацетилфенил)-бороновую кислоту (0,058 г, 0,351 ммоль), дихлорид бис-(трифенилфосфин)-палладия (II) (Pd(PPh3)2Cl2, 0,012 г, 0,018 ммоль) и карбонат калия (0,073 г, 0,527 ммоль) растворяли в 1,4-диоксане (4 мл)/воде (1 мл) при комнатной температуре, и полученный раствор перемешивали при 100°С в течение 1 часа. Затем температуру понижали до комнатной температуры для завершения реакции. После удаления растворителя из реакционной смеси при пониженном давлении, концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; этилацетат/гексан = от 0% до 70%) и концентрировали с получением целевого соединения (0,021 г, 32,5%) в виде твердого вещества розового цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,35-8,30 (м, 1Н), 8,19 (дд, J=7,1, 1,0 Гц, 1Н), 7,99-7,92 (м, 2Н), 7,70 (ддд, J=12,0, 8,3, 1,4 Гц, 1Н), 7,58-7,50 (м, 1Н), 7,49-7,43 (м, 3Н), 6,96 (т, J=51,7 Гц, 1Н), 4,29 (с, 2Н), 2,62 (с, 3Н);
МСНР (ЭР) m/z 369,3 (M++1).
Пример 10: Синтез Соединения 3593, 2-(2-(2-(4-хлорбензил)-имидазо[1,2-а]пиридин-7-ил)-5-(дифторметил)-1,3,4-оксадиазола
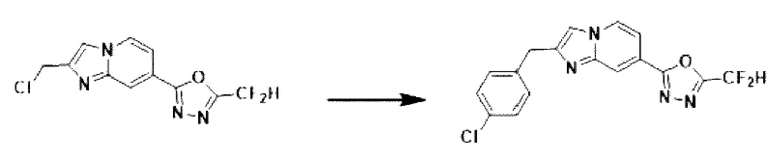
2-(2-(хлорметил)-имидазо[1,2-а]пиридин-7-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,050 г, 0,176 ммоль), полученный на стадии 5 примера 2, (4-фторфенил)-бороновую кислоту (0,055 г, 0,351 ммоль), дихлорид бис-(трифенилфосфин)-палладия (II) (Pd(PPh3)2Cl2, 0,012 г, 0,018 ммоль) и карбонат калия (0,073 г, 0,527 ммоль) растворяли в 1,4-диоксане (4 мл)/воде (1 мл) при комнатной температуре, и полученный раствор перемешивали при 100°С в течение 1 часа. Затем температуру понижали до комнатной температуры для завершения реакции. После удаления растворителя из реакционной смеси при пониженном давлении, концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; этилацетат/гексан = от 0% до 70%) и концентрировали с получением целевого соединения (0,012 г, 18,9%) в виде твердого вещества розового цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,32 (дт, J=1,7, 0,8 Гц, 1Н), 8,18 (дд, J=7,1, 0,9 Гц, 1Н), 7,52 (дд, J=7,1, 1,7 Гц, 1Н), 7,39 (kb, J=0,8 Гц, 1Н), 7,35-7,27 (м, 4Н), 6,95 (т, J=51,7 Гц, 1Н), 4,19 (с, 2Н);
МСНР (ЭР) m/z 362,9 (М++1).
Пример 11: Синтез Соединения 3594, 2-(дифторметил)-5-(2-(3-изопропилбензил)-имидазо[1,2-а]пиридин-7-ил)-1,3,4-оксадиазола
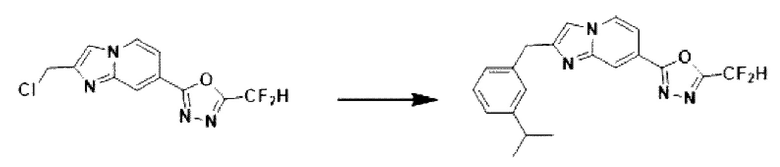
2-(2-(хлорметил)-имидазо[1,2-а]пиридин-7-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,050 г, 0,176 ммоль), полученный на стадии 5 примера 2, (3-изопропилфенил)-бороновую кислоту (0,058 г, 0,351 ммоль), дихлорид бис-(трифенилфосфин)-палладия (II) (Pd(PPh3)2Cl2, 0,012 г, 0,018 ммоль) и карбонат калия (0,073 г, 0,527 ммоль) растворяли в 1,4-диоксане (4 мл)/воде (1 мл) при комнатной температуре, и полученный раствор перемешивали при 100°С в течение 1 часа. Затем температуру понижали до комнатной температуры для завершения реакции. После удаления растворителя из реакционной смеси при пониженном давлении, концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; этилацетат/гексан = от 0% до 70%) и концентрировали с получением целевого соединения (0,032 г, 49,5%) в виде твердого вещества розового цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,33 (с, 1Н), 8,17 (дд, J=7,1, 0,9 Гц, 1Н), 7,52 (дд, J=7,1, 1,7 Гц, 1Н), 7,37 (д, J=0,8 Гц, 1Н), 7,32-7,26 (м, 1Н), 7,23 (д, J=1,8 Гц, 1Н), 7,20-7,13 (м, 2Н), 6,95 (т, J=51,7 Гц, 1Н), 4,22 (с, 2Н), 2,92 (п, J=6,9 Гц, 1Н), 1,28 (д, J=6,9 Гц, 6Н);
МСНР (ЭР) m/z 370,0 (М++1).
Пример 12: Синтез Соединения 3595, 2-(дифторметил)-5-(2-(4-(метилсульфонил)-бензил)-имидазо[1,2-а]пиридин-7-ил)-1,3,4-оксадиазола
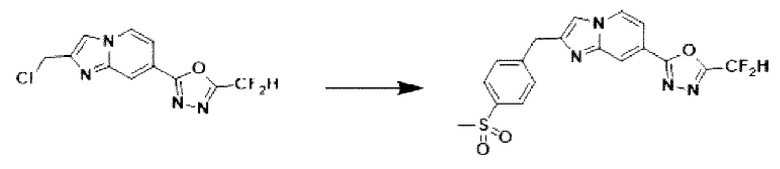
2-(2-(хлорметил)-имидазо[1,2-а]пиридин-7-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,050 г, 0,176 ммоль), полученный на стадии 5 примера 2, (4-(метилсульфонил)-фенил)-бороновую кислоту (0,070 г, 0,351 ммоль), дихлорид бис-(трифенилфосфин)-палладия (II) (Pd(PPh3)2Cl2, 0,012 г, 0,018 ммоль) и карбонат калия (0,073 г, 0,527 ммоль) растворяли в 1,4-диоксане (4 мл)/воде (1 мл) при комнатной температуре, и полученный раствор перемешивали при 100°С в течение 1 часа.
Затем температуру понижали до комнатной температуры для завершения реакции. После удаления растворителя из реакционной смеси при пониженном давлении, концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,033 г, 46,5%) в виде твердого вещества розового цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,33 (дт, J=1,7, 0,8 Гц, 1Н), 8,22 (дд, J=7,1, 0,9 Гц, 1Н), 7,95-7,87 (м, 2Н), 7,56 (тд, J=6,6, 1,8 Гц, 3Н), 7,49 (д, J=0,8 Гц, 1Н), 6,96 (т, J=51,7 Гц, 1Н), 4,31 (с, 2Н), 3,06 (с, 3Н);
МСНР (ЭР) m/z 405,1 (М++1).
Пример 13: Синтез Соединения 3596, 2-(2-(бензо[d][1,3]диоксол-5-илметил)-имидазо[1,2-а]пиридин-7-ил)-5-(дифторметил)-1,3,4-оксадиазола
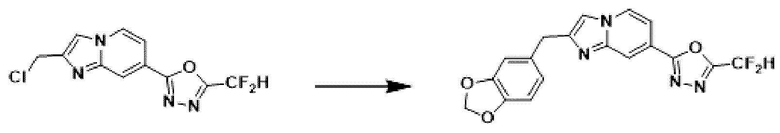
2-(2-(хлорметил)-имидазо[1,2-а]пиридин-7-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,050 г, 0,176 ммоль), полученный на стадии 5 примера 2, бензо[d][1,3]диоксол-5-илбороновую кислоту (0,058 г, 0,351 ммоль), дихлорид бис-(трифенилфосфин)-палладия (II) (Pd(PPh3)2Cl2, 0,012 г, 0,018 ммоль) и карбонат калия (0,073 г, 0,527 ммоль) растворяли в 1,4-диоксане (4 мл)/воде (1 мл) при комнатной температуре, и полученный раствор перемешивали при 100°С в течение 1 часа.
Затем температуру понижали до комнатной температуры для завершения реакции. После удаления растворителя из реакционной смеси при пониженном давлении, концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,041 г, 63,0%) в виде твердого вещества розового цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,32 (д, J=1,6 Гц, 1Н), 8,18 (дд, J=7,1, 1,0 Гц, 1Н), 7,52 (дд, J=7,1, 1,7 Гц, 1Н), 7,41 (с, 1Н), 7,10-6,77 (м, 4Н), 5,96 (с, 2Н), 4,15 (с, 2Н);
МСНР (ЭР) m/z 372,0 (М++1).
Пример 14: Синтез Соединения 3668, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-анилина
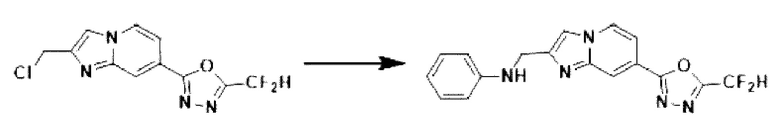
2-(2-(хлорметил)-имидазо[1,2-а]пиридин-7-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,050 г, 0,176 ммоль), полученный на стадии 5 примера 2, анилин (0,024 мл, 0,263 ммоль), карбонат калия (0,036 г, 0,263 ммоль) и иодид калия (0,015 г, 0,088 ммоль) растворяли в N,N-диметилформамиде (3 мл) при комнатной температуре и полученный раствор перемешивали при той же температуре в течение 18 часов. В концентрат, полученный путем удаления растворителя из реакционной смеси при пониженном давлении, вливали насыщенный водный раствор хлорида аммония с последующей экстракцией дихлорметаном. Затем полученный продукт фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (картридж SiO2, 4 г; этилацетат/гексан = от 0% до 70%) и концентрировали, получая целевое соединение (0,050 г, 83,4%) в виде твердого вещества желтого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,33 (дт, J=1,7, 0,8 Гц, 1Н), 8,21 (дд, J=7,2, 1,0 Гц, 1Н), 7,68 (д, J=0,9 Гц, 1Н), 7,54 (дд, J=7,1, 1,7 Гц, 1Н), 7,25-7,18 (м, 2Н), 6,96 (т, J=51,7 Гц, 1Н), 6,80-6,70 (м, 3Н), 4,62 (д, J=0,8 Гц, 2Н);
МСНР (ЭР) m/z 342,9 (М++1).
Пример 15: Синтез Соединения 3669, 1-(3-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-амино)-фенил)-этан-1-она
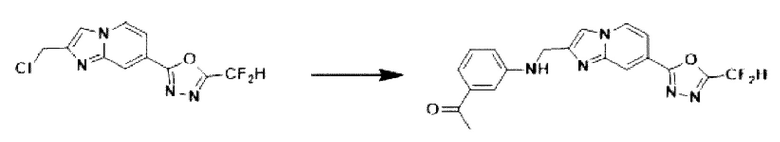
2-(2-(хлорметил)-имидазо[1,2-а]пиридин-7-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,050 г, 0,176 ммоль), полученный на стадии 5 примера 2, м-толуидин (0,036 г, 0,263 ммоль), карбонат калия (0,036 г, 0,263 ммоль) и иодид калия (0,015 г, 0,088 ммоль) растворяли в N,N-диметилформамиде (3 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В концентрат, полученный путем удаления растворителя из реакционной смеси при пониженном давлении, вливали насыщенный водный раствор хлорида аммония с последующей экстракцией дихлорметаном. Затем полученный продукт фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (картридж SiO2, 4 г; этилацетат/гексан = от 0% до 70%) и концентрировали, получая целевое соединение (0,040 г, 59,4%) в виде твердого вещества желтого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,38-8,29 (м, 1Н), 8,24 (дд, J=7,1, 1,0 Гц, 1Н), 7,74-7,67 (м, 1Н), 7,57 (дд, J=7,1, 1,7 Гц, 1Н), 7,36-7,32 (м, 2Н), 7,31-7,28 (м, 1Н), 7,11-6,82 (м, 2Н), 4,68-4,64 (м, 2Н), 2,59 (с, 3Н);
МСНР (ЭР) m/z 385,0 (M++1).
Пример 16: Синтез Соединения 3670, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-3-фторанилина
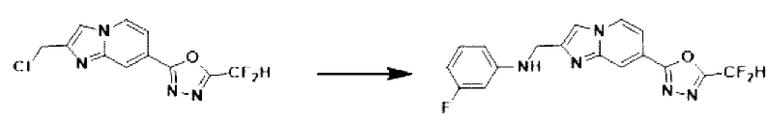
2-(2-(хлорметил)-имидазо[1,2-а]пиридин-7-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,050 г, 0,176 ммоль), полученный на стадии 5 примера 2, 3-фторанилин (0,036 г, 0,263 ммоль), карбонат калия (0,036 г, 0,263 ммоль) и иодид калия (0,015 г, 0,088 ммоль) растворяли в N,N-диметилформамиде (3 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В концентрат, полученный путем удаления растворителя из реакционной смеси при пониженном давлении, вливали насыщенный водный раствор хлорида аммония с последующей экстракцией дихлорметаном. Затем полученный продукт фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (картридж SiO2, 4 г; этилацетат/гексан = от 0% до 70%) и концентрировали, получая целевое соединение (0,040 г, 59,4%) в виде твердого вещества желтого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,34 (дт, J=1,7, 0,8 Гц, 1Н), 8,23 (дд, J=7,1, 1,0 Гц, 1Н), 7,68 (д, J=0,8 Гц, 1Н), 7,57 (дд, J=7,1, 1,7 Гц, 1Н), 7,18-6,76 (м, 2Н), 6,54-6,32 (м, 3Н), 4,59 (д, J=0,8 Гц, 2Н);
МСНР (ЭР) m/z 360,9 (М++1).
Пример 17: Синтез Соединения 3671, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-3,4-дифторанилина
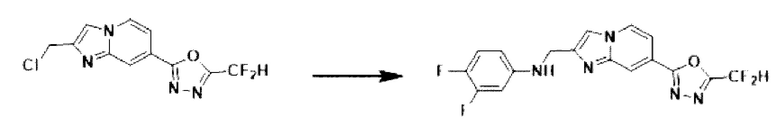
2-(2-(хлорметил)-имидазо[1,2-а]пиридин-7-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,050 г, 0,176 ммоль), полученный на стадии 5 примера 2, 3,4-дифторанилин (0,029 г, 0,263 ммоль), карбонат калия (0,036 г, 0,263 ммоль) и иодид калия (0,015 г, 0,088 ммоль) растворяли в N,N-диметилформамиде (3 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В концентрат, полученный путем удаления растворителя из реакционной смеси при пониженном давлении, вливали насыщенный водный раствор хлорида аммония с последующей экстракцией дихлорметаном. Затем полученный продукт фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (картридж SiO2, 4 г; этилацетат/гексан = от 0% до 70%) и концентрировали, получая целевое соединение (0,024 г, 38,0%) в виде твердого вещества желтого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,38 (с, 1Н), 8,27 (дд, J=7,1, 1,0 Гц, 1Н), 7,69 (д, J=0,8 Гц, 1Н), 7,63 (дд, J=7,1, 1,7 Гц, 1Н), 7,11-6,80 (м, 2Н), 6,52 (ддд, J=12,5, 6,6, 2,9 Гц, 1Н), 6,42 (ддд, J=8,9, 3,2, 1,6 Гц, 1Н), 4,57 (д, J=0,8 Гц, 2Н);
МСНР (ЭР) m/z 378,1 (M++1).
Пример 18: Синтез Соединения 3672, 3-хлор-N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-4-фторанилина
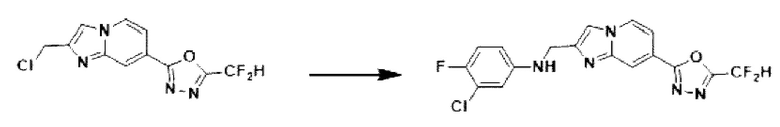
2-(2-(хлорметил)-имидазо[1,2-а]пиридин-7-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,050 г, 0,176 ммоль), полученный на стадии 5 примера 2, 3-хлор-4-фторанилин (0,038 г, 0,263 ммоль), карбонат калия (0,036 г, 0,263 ммоль) и иодид калия (0,015 г, 0,088 ммоль) растворяли в N,N-диметилформамиде (3 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В концентрат, полученный путем удаления растворителя из реакционной смеси при пониженном давлении, вливали насыщенный водный раствор хлорида аммония с последующей экстракцией дихлорметаном. Затем полученный продукт фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (картридж SiO2, 4 г; этилацетат/гексан = от 0% до 70%) и концентрировали, получая целевое соединение (0,041 г, 59,3%) в виде твердого вещества желтого цвета.
1Н ЯМР (400 МГц, CDCl3) δ 8,37 (д, J=1,5 Гц, 1Н), 8,26 (дд, J=7,1, 1,0 Гц, 1Н), 7,69 (д, J=0,8 Гц, 1Н), 7,62 (дд, J=7,1, 1,7 Гц, 1Н), 7,10-6,80 (м, 2Н), 6,73 (дд, J=6,0, 2,9 Гц, 1Н), 6,57 (ддд, J=8,9, 3,8, 2,9 Гц, 1Н), 4,57 (д, J=0,8 Гц, 2Н);
МСНР (ЭР) m/z 396,0 (M++1).
Пример 19: Синтез Соединения 3673, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-4-(трифторметил)-анилина
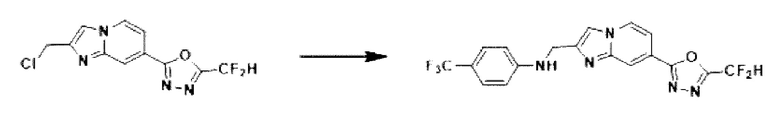
2-(2-(хлорметил)-имидазо[1,2-а]пиридин-7-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,050 г, 0,176 ммоль), полученный на стадии 5 примера 2, 4-(трифторметил)-анилин (0,042 г, 0,263 ммоль), карбонат калия (0,036 г, 0,263 ммоль) и иодид калия (0,015 г, 0,088 ммоль) растворяли в N,N-диметилформамиде (3 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В концентрат, полученный путем удаления растворителя из реакционной смеси при пониженном давлении, вливали насыщенный водный раствор хлорида аммония с последующей экстракцией дихлорметаном. Затем полученный продукт фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (картридж SiO2, 4 г; этилацетат/гексан = от 0% до 70%) и концентрировали, получая указанное в заголовке соединение (0,029 г, 40,3%) в виде твердого вещества желтого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,36 (дт, J=1,6, 0,7 Гц, 1Н), 8,25 (дд, J=7,1, 1,0 Гц, 1Н), 7,68 (д, J=0,7 Гц, 1Н), 7,60 (дд, J=7,1, 1,7 Гц, 1Н), 7,47-7,39 (м, 2Н), 6,97 (т, J=51,7 Гц, 1Н), 6,74 (д, J=8,6 Гц, 2Н), 4,66 (с, 2Н);
МСНР (ЭР) m/z 411,0 (М++1).
Пример 20: Синтез Соединения 3674, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-4-(метилсульфонил)-анилина
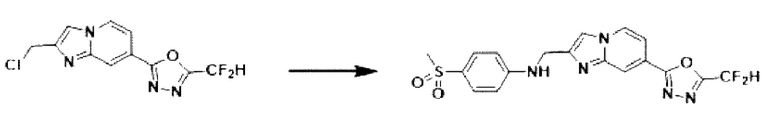
2-(2-(хлорметил)-имидазо[1,2-а]пиридин-7-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,050 г, 0,176 ммоль), полученный на стадии 5 примера 2, 4-(метилсульфонил)-анилин (0,045 г, 0,263 ммоль), карбонат калия (0,036 г, 0,263 ммоль) и иодид калия (0,015 г, 0,088 ммоль) растворяли в N,N-диметилформамиде (3 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В концентрат, полученный путем удаления растворителя из реакционной смеси при пониженном давлении, вливали насыщенный водный раствор хлорида аммония с последующей экстракцией дихлорметаном. Затем полученный продукт фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (картридж SiO2, 4 г; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,034 г, 46,2%) в виде твердого вещества желтого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,33 (дт, J=1,7, 0,8 Гц, 1Н), 8,26 (дд, J=7,1, 1,0 Гц, 1Н), 7,76-7,71 (м, 2Н), 7,69 (д, J=0,8 Гц, 1Н), 7,58 (дд, J=7,2, 1,7 Гц, 1Н), 6,97 (т, J=51,7 Гц, 1Н), 6,79-6,71 (м, 2Н), 4,66 (д, J=4,3 Гц, 2Н), 3,02 (с, 3Н);
МСНР (ЭР) m/z 420,3 (М++1).
Пример 21: Синтез Соединения 3675, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-пиридин-3-амина
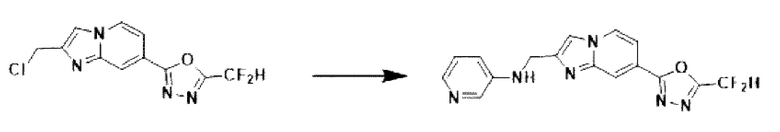
2-(2-(хлорметил)-имидазо[1,2-а]пиридин-7-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,050 г, 0,176 ммоль), полученный на стадии 5 примера 2, пиридин-3-амин (0,025 г, 0,263 ммоль), карбонат калия (0,036 г, 0,263 ммоль) и иодид калия (0,015 г, 0,088 ммоль) растворяли в N,N-диметилформамиде (3 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В концентрат, полученный путем удаления растворителя из реакционной смеси при пониженном давлении, вливали насыщенный водный раствор хлорида аммония с последующей экстракцией дихлорметаном. Затем полученный продукт фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (картридж SiO2, 4 г; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,037 г, 61,5%) в виде твердого вещества желтого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,33 (дт, J=1,7, 0,8 Гц, 1Н), 8,24 (дд, J=7,1, 1,0 Гц, 1Н), 8,19 (д, J=2,8 Гц, 1Н), 8,05-7,99 (м, 1Н), 7,70 (с, 1Н), 7,56 (дд, J=7,1, 1,7 Гц, 1Н), 7,17-6,82 (м, ЗН), 4,62 (с, 2Н);
МСНР (ЭР) m/z 343,1 (M++1).
Пример 22: Синтез Соединения 3676, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-6-фторпиридин-3-амина
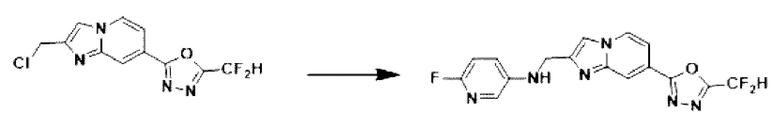
2-(2-(хлорметил)-имидазо[1,2-а]пиридин-7-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,050 г, 0,176 ммоль), полученный на стадии 5 примера 2, 6-фторпиридин-3-амин (0,030 г, 0,263 ммоль), карбонат калия (0,036 г, 0,263 ммоль) и иодид калия (0,015 г, 0,088 ммоль) растворяли в N,N-диметилформамиде (3 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В концентрат, полученный путем удаления растворителя из реакционной смеси при пониженном давлении, вливали насыщенный водный раствор хлорида аммония с последующей экстракцией дихлорметаном. Затем полученный продукт фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (картридж SiO2, 4 г; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,033 г, 52,1%) в виде твердого вещества желтого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,35 (дт, J=1,7, 0,9 Гц, 1Н), 8,26 (дд, J=7,1, 1,0 Гц, 1Н), 7,73-7,68 (м, 1Н), 7,66 (дд, J=3,1, 1,9 Гц, 1Н), 7,60 (дд, J=7,1, 1,7 Гц, 1Н), 7,16 (ддд, J=8,8, 6,7, 3,1 Гц, 1Н), 6,97 (т, J=51,7 Гц, 1Н), 6,79 (ддд, J=8,7, 3,4, 0,6 Гц, 1Н), 4,59 (с, 2Н);
МСНР (ЭР) m/z 362,1 (М++1).
Пример 23: Синтез Соединения 3677, (3s,5s,7s)-N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-адамантан-1-амина
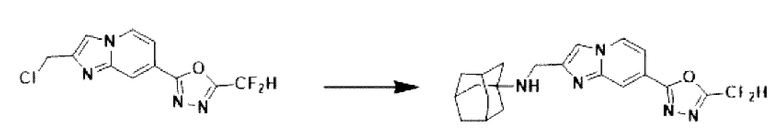
2-(2-(хлорметил)-имидазо[1,2-а]пиридин-7-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,050 г, 0,176 ммоль), полученный на стадии 5 примера 2, (3s,5s,7s)-адамантан-1-амин (0,040 г, 0,263 ммоль), карбонат калия (0,036 г, 0,263 ммоль) и иодид калия (0,015 г, 0,088 ммоль) добавляли к N,N-диметилформамиду (3 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В концентрат, полученный путем удаления растворителя из реакционной смеси при пониженном давлении, вливали насыщенный водный раствор хлорида аммония с последующей экстракцией дихлорметаном. Затем полученный продукт фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 25%) и концентрировали с получением целевого соединения (0,023 г, 32,8%) в виде твердого вещества желтого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,29 (п, J=0,8 Гц, 1Н), 8,23 (дд, J=7,1, 0,9 Гц, 1Н), 7,84 (с, 1Н), 7,50 (дд, J=7,1, 1,7 Гц, 1Н), 6,95 (т, J=51,7 Гц, 1Н), 4,10 (с, 2Н), 2,18-2,09 (м, 3Н), 1,84 (д, J=2,9 Гц, 6Н), 1,70 (кв, J=12,8 Гц, 6Н);
МСНР (ЭР) m/z 399,8 (М++1).
Пример 24: Синтез Соединения 3678, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-циклогексанамина
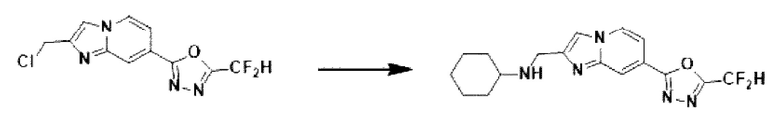
2-(2-(хлорметил)-имидазо[1,2-а]пиридин-7-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,050 г, 0,176 ммоль), полученный на стадии 5 примера 2, циклогексанамин (0,026 г, 0,263 ммоль), карбонат калия (0,036 г, 0,263 ммоль) и иодид калия (0,015 г, 0,088 ммоль) растворяли в N,N-диметилформамиде (3 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В концентрат, полученный путем удаления растворителя из реакционной смеси при пониженном давлении, вливали насыщенный водный раствор хлорида аммония с последующей экстракцией дихлорметаном. Затем полученный продукт фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 25%) и концентрировали с получением целевого соединения (0,024 г, 39,3%) в виде твердого вещества желтого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,30 (дт, J=1,7, 0,8 Гц, 1Н), 8,24 (дд, J=7,1, 0,9 Гц, 1Н), 7,74 (с, 1Н), 7,53 (дд, J=7,1, 1,7 Гц, 1Н), 6,96 (т, J=51,7 Гц, 1Н), 4,09 (с, 2Н), 2,61 (тт, J=10,0, 3,7 Гц, 1Н), 2,39 (с, 1Н), 2,01 (д, J=12,0 Гц, 2Н), 1,85-1,71 (м, 2Н), 1,70-1,59 (м, 1Н), 1,39-1,13 (м, 4Н);
МСНР (ЭР) m/z 347,7 (М++1).
Пример 25: Синтез Соединения 3679, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-3-метиланилина
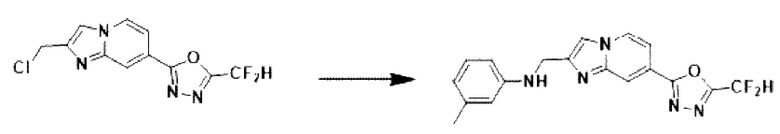
2-(2-(хлорметил)-имидазо[1,2-а]пиридин-7-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,050 г, 0,176 ммоль), полученный на стадии 5 примера 2, м-толуидин (0,028 г, 0,263 ммоль), карбонат калия (0,036 г, 0,263 ммоль) и иодид калия (0,015 г, 0,088 ммоль) растворяли в N,N-диметилформамиде (3 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В концентрат, полученный путем удаления растворителя из реакционной смеси при пониженном давлении, вливали насыщенный водный раствор хлорида аммония с последующей экстракцией дихлорметаном. Затем полученный продукт фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (картридж SiO2, 4 г; этилацетат/гексан = от 0% до 70%) и концентрировали, получая целевое соединение (0,032 г, 51,3%) в виде твердого вещества желтого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,31 (п, J=0,8 Гц, 1Н), 8,19 (дд, J=7,1, 1,0 Гц, 1Н), 7,67 (д, J=0,9 Гц, 1Н), 7,52 (дд, J=7,1, 1,7 Гц, 1Н), 7,14-6,77 (м, 2Н), 6,63-6,50 (м, 3Н), 4,60 (д, J=0,9 Гц, 2Н), 2,29 (с, 3Н);
МСНР (ЭР) m/z 356,2 (М++1).
Пример 26. Синтез Соединения 3719, 2-(дифторметил)-5-(2-(феноксиметил)-имидазо[1,2-а]пиридин-7-ил)-1,3,4-оксадиазола
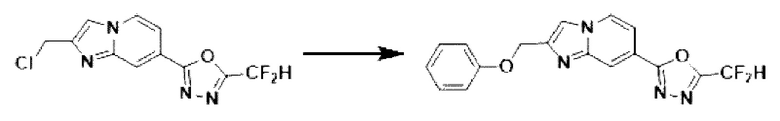
2-(2-(хлорметил)-имидазо[1,2-а]пиридин-7-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,040 г, 0,141 ммоль), полученный на стадии 5 примера 2, фенол (0,019 мл, 0,211 ммоль) и карбонат калия (0,039 г, 0,281 ммоль) растворяли в N,N-диметилформамиде (2 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 2 часов. В концентрат, полученный путем удаления растворителя из реакционной смеси при пониженном давлении, вливали насыщенный водный раствор хлорида аммония с последующей экстракцией дихлорметаном. Затем полученный продукт фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; этилацетат/гексан = от 0% до 50%) и концентрировали с получением целевого соединения (0,032 г, 66,5%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,38 (д, J=1,6 Гц, 1Н), 8,28 (дд, J=7,1, 1,0 Гц, 1Н), 7,86-7,79 (м, 1Н), 7,60 (дд, J=7,1,1,7 Гц, 1Н), 7,38-7,30 (м, 2Н), 7,12-6,81 (м, 4Н), 5,39-5,35 (м, 2Н);
МСНР (ЭР) m/z 342,7 (М++1).
Пример 27: Синтез Соединения 3720, 2-(дифторметил)-5-(2-((м-толилокси)-метил)-имидазо[1,2-а]пиридин-7-ил)-1,3,4-оксадиазола
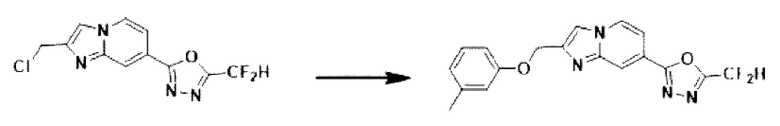
2-(2-(хлорметил)-имидазо[1,2-а]пиридин-7-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,040 г, 0,141 ммоль), полученный на стадии 5 примера 2, м-крезол (0,023 г, 0,211 ммоль) и карбонат калия (0,039 г, 0,281 ммоль) растворяли в N,N-диметилформамиде (2 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 2 часов. В концентрат, полученный путем удаления растворителя из реакционной смеси при пониженном давлении, вливали насыщенный водный раствор хлорида аммония с последующей экстракцией дихлорметаном. Затем полученный продукт фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; этилацетат/гексан = от 0% до 50%) и концентрировали с получением целевого соединения (0,037 г, 73,9%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,38 (дд, J=1,8, 0,9 Гц, 1Н), 8,27 (дд, J=7,2, 1,0 Гц, 1Н), 7,82 (д, J=0,9 Гц, 1Н), 7,59 (дд, J=7,1, 1,7 Гц, 1Н), 7,22 (т, J=7,8 Гц, 1Н), 7,12-6,78 (м, 4Н), 5,36 (д, J=0,8 Гц, 2Н), 2,37 (с, 3Н);
МСНР (ЭР) m/z 356,7 (М++1).
Пример 28: Синтез Соединения 3721, 1-(3-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метокси)-фенил)-этан-1-она
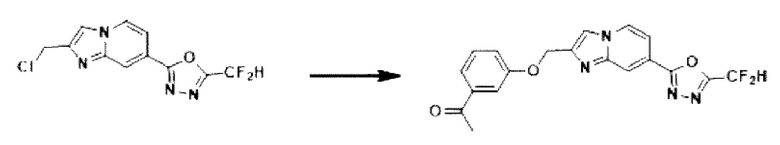
2-(2-(хлорметил)-имидазо[1,2-а]пиридин-7-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,040 г, 0,141 ммоль), полученный на стадии 5 примера 2, 1-(3-гидроксифенил)-этан-1-он (0,029 г, 0,211 ммоль) и карбонат калия (0,039 г, 0,281 ммоль) растворяли в N,N-диметилформамиде (2 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 2 часов. В концентрат, полученный путем удаления растворителя из реакционной смеси при пониженном давлении, вливали насыщенный водный раствор хлорида аммония с последующей экстракцией дихлорметаном. Затем полученный продукт фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; этилацетат/гексан = от 0% до 50%) и концентрировали с получением целевого соединения (0,010 г, 18,5%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,28 (дт, J=1,7, 0,8 Гц, 1Н), 8,19 (дд, J=7,1, 1,0 Гц, 1Н), 7,91-7,86 (м, 2Н), 7,74 (д, J=0,8 Гц, 1Н), 7,50 (дд, J=7,1, 1,7 Гц, 1Н), 7,05-6,74 (м, 3Н), 5,33 (д, J=0,7 Гц, 2Н), 2,50 (с, 3Н);
МСНР (ЭР) m/z 384,8 (М++1).
Пример 29: Синтез Соединения 3722, 2-(дифторметил)-5-(2-((3-фторфенокси)-метил)-имидазо[1,2-а]пиридин-7-ил)-1,3,4-оксадиазола
2-(2-(хлорметил)-имидазо[1,2-а]пиридин-7-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,040 г, 0,141 ммоль), полученный на стадии 5 примера 2, 3-фторфенол (0,024 г, 0,211 ммоль) и карбонат калия (0,039 г, 0,281 ммоль) растворяли в N,N-диметилформамиде (2 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 2 часов. В концентрат, полученный путем удаления растворителя из реакционной смеси при пониженном давлении, вливали насыщенный водный раствор хлорида аммония с последующей экстракцией дихлорметаном. Затем полученный продукт фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; этилацетат/гексан = от 0% до 50%) и концентрировали с получением целевого соединения (0,036 г, 71,1%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,38 (дт, J=1,7, 0,8 Гц, 1Н), 8,29 (дд, J=7,1, 0,9 Гц, 1Н), 7,83 (т, J=0,8 Гц, 1Н), 7,61 (дд, J=7,1, 1,7 Гц, 1Н), 7,28 (д, J=6,9 Гц, 1Н), 7,11-6,68 (м, 4Н), 5,35 (д, J=0,8 Гц, 2Н);
МСНР (ЭР) m/z 360,7 (М++1).
Пример 30: Синтез Соединения 3723, 2-(дифторметил)-5-(2-((3,4-дифторфенокси)-метил)-имидазо[1,2-а]пиридин-7-ил)-1,3,4-оксадиазола
2-(2-(хлорметил)-имидазо[1,2-а]пиридин-7-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,040 г, 0,141 ммоль), полученный на стадии 5 примера 2, 3,4-дифторфенол (0,027 г, 0,211 ммоль) и карбонат калия (0,039 г, 0,281 ммоль) растворяли в N,N-диметилформамиде (2 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 2 часов. В концентрат, полученный путем удаления растворителя из реакционной смеси при пониженном давлении, вливали насыщенный водный раствор хлорида аммония с последующей экстракцией дихлорметаном. Затем полученный продукт фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; этилацетат/гексан = от 0% до 50%) и концентрировали с получением целевого соединения (0,032 г, 60,2%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,37 (дт, J=1,7, 0,8 Гц, 1Н), 8,29 (дд, J=7,1, 1,0 Гц, 1Н), 7,82 (д, J=0,8 Гц, 1Н), 7,60 (дд, J=7,1, 1,7 Гц, 1Н), 7,15-6,73 (м, 4Н), 5,30 (д, J=0,8 Гц, 2Н);
МСНР (ЭР) m/z 378,7 (М++1).
Пример 31: Синтез Соединения 3724, 2-(дифторметил)-5-(2-((4-(трифторметил)-фенокси)-метил)-имидазо[1,2-а]пиридин-7-ил)-1,3,4-оксадиазола
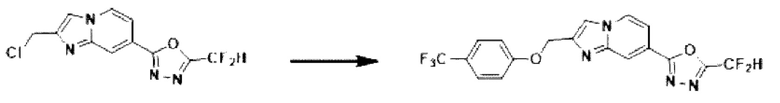
2-(2-(хлорметил)-имидазо[1,2-а]пиридин-7-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,040 г, 0,141 ммоль), полученный на стадии 5 примера 2, 4-(трифторметил)-фенол (0,034 г, 0,211 ммоль) и карбонат калия (0,039 г, 0,281 ммоль) растворяли в N,N-диметилформамиде (2 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 2 часов. В концентрат, полученный путем удаления растворителя из реакционной смеси при пониженном давлении, вливали насыщенный водный раствор хлорида аммония с последующей экстракцией дихлорметаном. Затем полученный продукт фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; этилацетат/гексан = от 0% до 50%) и концентрировали с получением целевого соединения (0,051 г, 88,5%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,33-8,28 (м, 1Н), 8,20 (дд, J=7,1, 1,0 Гц, 1Н), 7,74 (д, J=0,8 Гц, 1Н), 7,52 (ддд, J=10,9, 8,1, 1,3 Гц, 3Н), 7,08-6,71 (м, 3Н), 5,33 (д, J=0,8 Гц, 2Н);
МСНР (ЭР) m/z 410,7 (М++1).
Пример 32: Синтез Соединения 3725, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-метиланилина
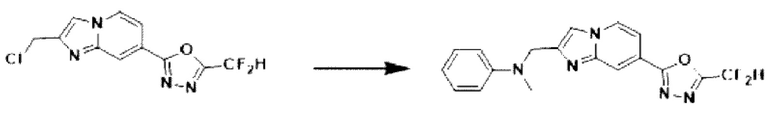
2-(2-(хлорметил)-имидазо[1,2-а]пиридин-7-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,050 г, 0,176 ммоль), полученный на стадии 5 примера 2, N-метиланилин (0,028 г, 0,263 ммоль), карбонат калия (0,036 г, 0,263 ммоль) и иодид калия (0,015 г, 0,088 ммоль) растворяли в N,N-диметилформамиде (3 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В концентрат, полученный путем удаления растворителя из реакционной смеси при пониженном давлении, вливали насыщенный водный раствор хлорида аммония с последующей экстракцией дихлорметаном. Затем полученный продукт фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; гексан/этилацетат = от 0% до 50%) и концентрировали с получением целевого соединения (0,051 г, 81,7%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,33 (дт, J=1,7, 0,9 Гц, 1Н), 8,15 (дд, J=7,1, 1,0 Гц, 1Н), 7,53-7,48 (м, 2Н), 7,30-7,23 (м, 2Н), 7,10-6,75 (м, 4Н), 4,80 (д, J=0,9 Гц, 2Н), 3,16 (с, 3Н);
МСНР (ЭР) m/z 355,7 (М++1).
Пример 33: Синтез Соединения 3782, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-1-метил-N-фенилпиперидин-4-карбоксамида
[Стадия 1] Синтез трет-бутил-4-(хлоркарбонил)-пиперидин-1-карбоксилата

1-(трет-бутоксикарбонил)-пиперидин-4-карбоновую кислоту (0,200 г, 0,872 ммоль) растворяли в дихлорметане (10 мл) и добавляли оксалилхлорид (2,00 M раствор, 0,567 мл, 1,134 ммоль) и N,N-диметилформамид (0,007 мл, 0,087 ммоль) при 0°С и перемешивали при комнатной температуре в течение 2 часов. После удаления растворителя из реакционной смеси при пониженном давлении, получали целевое соединение (0,216 г, 100,0%) в виде твердого вещества желтого цвета без дополнительной очистки.
[Стадия 2] Синтез трет-бутил-4-(((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-(фенил)-карбамоил)-пиперидин-1-карбоксилата
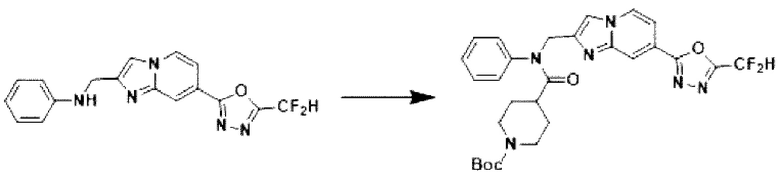
К раствору, в котором N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-анилин (0,200 г, 0,586 ммоль), полученный в Примере 14, был растворен в дихлорметане (20 мл) при 0°С, добавляли трет-бутил-4-(хлоркарбонил)-пиперидин-1-карбоксилат (0,189 г, 0,762 ммоль), полученный на стадии 1, и триэтиламин (0,245 мл, 1,758 ммоль), и перемешивали при комнатной температуре в течение 16 часов. В концентрат, полученный путем удаления растворителя из реакционной смеси при пониженном давлении, вливали насыщенный водный раствор хлорида аммония с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором, сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (картридж SiO2, 12 г; этилацетат/гексан = от 0% до 30%) и концентрировали, получая целевое соединение (0,200 г, 61,8%) в виде твердого вещества желтого цвета.
[Стадия 3] Синтез N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилпиперидин-4-карбоксамида
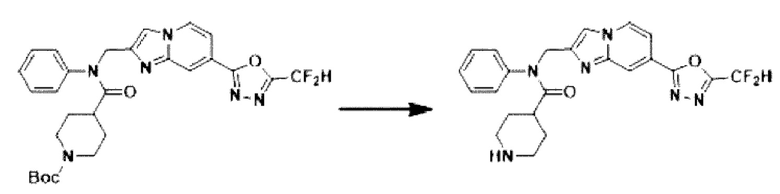
Трет-бутил-4-(((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-(фенил)-карбамоил)-пиперидин-1-карбоксилат (0,120 г, 0,217 ммоль), полученный на стадии 2, и трифторуксусную кислоту (0,333 мл, 4,343 ммоль) растворяли в дихлорметане (5 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 3 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором, сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении, и получали целевое соединение (0,098 г, 99,7%) в виде коричневого геля без дополнительной очистки.
[Стадия 4] Синтез соединения 3782
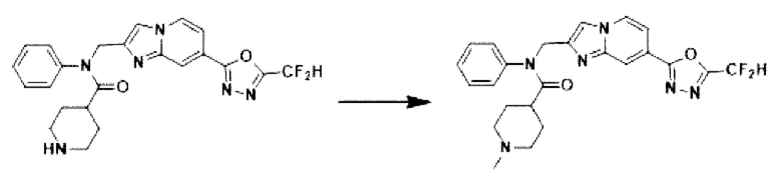
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилпиперидин-4-карбоксамид (0,050 г, 0,111 ммоль), полученный на стадии 3, формальдегид (0,007 г, 0,221 ммоль), уксусную кислоту (0,006 мл, 0,111 ммоль) и триацетоксиборгидрид натрия (0,070 г, 0,332 ммоль) растворяли в дихлорметане (3 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором, сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,038 г, 73,7%) в виде твердого вещества белого цвета.
1Н ЯМР (400 МГц, CDCl3) δ 8,29 (дт, J=1,7, 0,8 Гц, 1Н), 8,23 (дд, J=7,1, 1,0 Гц, 1Н), 7,78 (с, 1Н), 7,53 (дд, J=7,1, 1,7 Гц, 1Н), 7,46-7,36 (м, 3Н), 7,27-7,21 (м, 2Н), 6,95 (т, J=51,7 Гц, 1Н), 5,04 (с, 2Н), 2,89 (с, 2Н), 2,25 (с, 4Н), 1,93 (кв, J=11,6, 11,0 Гц, 3Н), 1,71 (с, 3Н);
МСНР (ЭР) m/z 468,1 (М++1).
Пример 34: Синтез Соединения 3783, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-1-(оксетан-3-ил)-N-фенилпиперидин-4-карбоксамида
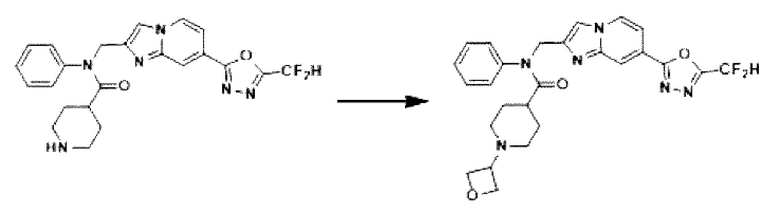
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилпиперидин-4-карбоксамид (0,050 г, 0,111 ммоль), полученный на стадии 3 Примера 33, оксетан-3-он (0,016 г, 0,221 ммоль), уксусную кислоту (0,006 мл, 0,111 ммоль) и триацетоксиборгидрид натрия (0,070 г, 0,332 ммоль) растворяли в дихлорметане (3 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором, сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,042 г, 74,7%) в виде твердого вещества белого цвета.
1Н ЯМР (400 МГц, CDCl3) δ 8,29 (дт, J=1,6, 0,8 Гц, 1Н), 8,23 (дд, J=7,1, 0,9 Гц, 1Н), 7,79 (с, 1Н), 7,53 (дд, J=7,1, 1,7 Гц, 1Н), 7,46-7,33 (м, 3Н), 7,27-7,22 (м, 2Н), 6,95 (т, J=51,7 Гц, 1Н), 5,04 (с, 2Н), 4,59 (д, J=6,6 Гц, 4Н), 3,38 (с, 1Н), 2,70 (с, 2Н), 2,25 (д, J=12,0 Гц, 1Н), 1,92 (кв, J=11,8, 11,2 Гц, 2Н), 1,67 (с, 4Н);
МСНР (ЭР) m/z 508,9 (М++1).
Пример 35: Синтез Соединения 3784, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-4-метил-N-фенилпиперазин-1-карбоксамида
[Стадия 1] Синтез трет-бутил-4-(((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-(фенил)-карбамоил)-пиперазин-1-карбоксилата
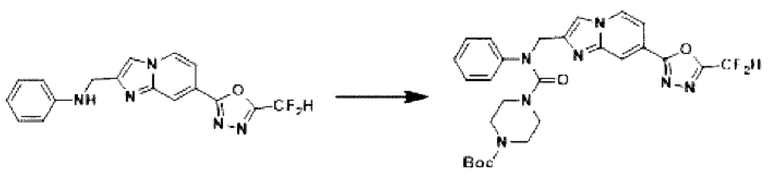
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-анилин (0,200 г, 0,586 ммоль), полученный в Примере 14, трифосген (0,174 г, 0,586 ммоль) и N,N-диизопропилэтиламин (0,510 мл, 2,930 ммоль) растворяли в дихлорметане (15 мл), и полученный раствор перемешивали при комнатной температуре в течение 10 минут. Затем добавляли трет-бутилпиперазин-1-карбоксилат (0,142 г, 0,762 ммоль) и дополнительно перемешивали при той же температуре в течение 16 часов. В реакционную смесь вливали воду, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (картридж SiO2, 12 г; этилацетат/гексан = от 5% до 50%) и концентрировали с получением целевого соединения (0,160 г, 49,3%) в виде твердого вещества белого цвета.
[Стадия 2] Синтез N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилпиперазин-1-карбоксамида 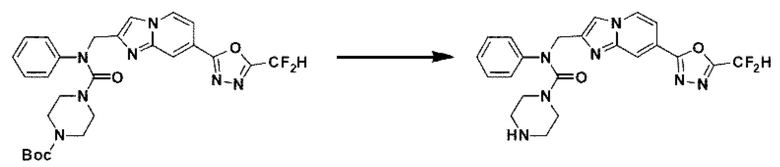
Трет-бутил-4-(((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)имидазо[1,2-а]пиридин-2-ил)-метил)-(фенил)-карбамоил)-пиперазин-1-карбоксилат (0,100 г, 0,181 ммоль), полученный на стадии 1, растворяли в дихлорметане (15 мл) и добавляли трифторуксусную кислоту (0,277 мл, 3,613 ммоль) при 0°С, и перемешивали при комнатной температуре в течение 16 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном, а затем фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя. После концентрирования при пониженном давлении получали целевое соединение (0,099 г, 96,6%) в виде твердого вещества типа пены без дополнительной очистки.
[Стадия 3] Синтез соединения 3784

N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилпиперазин-1-карбоксамид (0,050 г, 0,110 ммоль), полученный на стадии 2, параформальдегид (0,007 г, 0,221 ммоль) и уксусную кислоту (0,006 мл, 0,110 ммоль) растворяли в дихлорметане (5 мл), и полученный раствор перемешивали при комнатной температуре в течение 1 часа. Затем добавляли триацетоксиборгидрид натрия (0,070 г, 0,331 ммоль) и дополнительно перемешивали при той же температуре в течение 16 часов. В реакционную смесь вливали воду, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; метанол/дихлорметан = 10%) и концентрировали с получением целевого соединения (0,030 г, 58,2%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, MeOD) δ 8,59 (м, 1Н), 8,20 (м, 1Н), 7,97 (м, 1Н), 7,56-7,54 (м, 1Н), 7,39-7,35 (м, 2Н), 7,26-7,13 (м, 4Н), 5,03 (с, 2Н), 3,32 (м, 4Н), 2,36 (м, 4Н), 2,29 (с, 3Н);
МСНР (ЭР) m/z 468,3 (М++1).
Пример 36: Синтез Соединения 3785, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-4-(оксетан-3-ил)-N-фенилпиперазин-1-карбоксамида
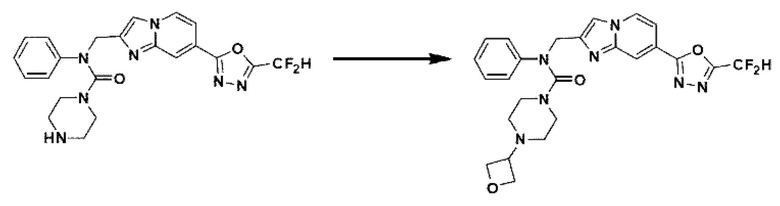
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилпиперазин-1-карбоксамид (0,050 г, 0,110 ммоль), полученный на стадии 2 примера 35, оксетан-3-он (0,014 мл, 0,221 ммоль) и уксусную кислоту (0,006 мл, 0,110 ммоль) растворяли в дихлорметане (5 мл), и полученный раствор перемешивали при комнатной температуре в течение 1 часа. Затем добавляли триацетоксиборгидрид натрия (0,070 г, 0,331 ммоль) и дополнительно перемешивали при той же температуре в течение 16 часов. В реакционную смесь вливали воду, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; метанол/дихлорметан = 10%) и концентрировали с получением целевого соединения (0,028 г, 49,8%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, MeOD) δ 8,58 (м, 1Н), 8,19 (м, 1Н), 7,96 (с, 1Н), 7,55-7,52 (м, 1Н), 7,39-7,34 (м, 2Н), 7,26-7,13 (м, 4Н), 5,02 (с, 2Н), 4,63 (м, 2Н), 4,52 (м, 2Н), 3,41 (м, 1Н), 3,32 (м, 4Н), 2,17 (м, 4Н);
МСНР (ЭР) m/z 510,1 (М++1).
Пример 37: Синтез Соединения 4033, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-4-этил-N-фенилпиперазин-1-карбоксамида
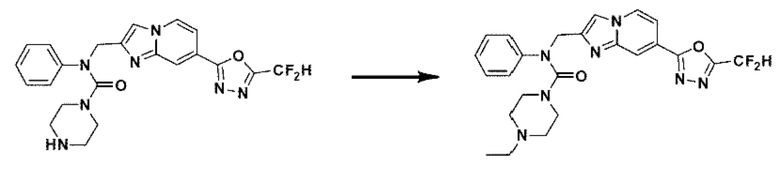
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилпиперазин-1-карбоксамид (0,100 г, 0,221 ммоль), полученный на стадии 2 примера 35, ацетальдегид (0,019 г, 0,441 ммоль), уксусную кислоту (0,013 мл, 0,221 ммоль) и триацетоксиборгидрид натрия (0,140 г, 0,662 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и перемешивали полученный раствор при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,027 г, 25,4%) в виде твердого вещества желтого цвета.
1H ЯМР (700 МГц, MeOD) δ 8,59 (дд, J=7,2, 1,0 Гц, 1Н), 8,21 (дт, J=1,7, 0,8 Гц, 1Н), 7,98 (д, J=0,8 Гц, 1Н), 7,55 (дд, J=7,1, 1,7 Гц, 1Н), 7,38-7,13 (м, 6Н), 5,03 (д, J=0,8 Гц, 2Н), 3,32-3,27 (м, 4Н), 2,38 (кв, J=7,3 Гц, 2Н), 2,31 (т, J=5,1 Гц, 4Н), 1,06 (т, J=7,2 Гц, 3Н);
МСНР (ЭР) m/z 482,2 (М++1).
Пример 38: Синтез Соединения 4034, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-4-изолролил-N-фенилпиперазин-1-карбоксамида
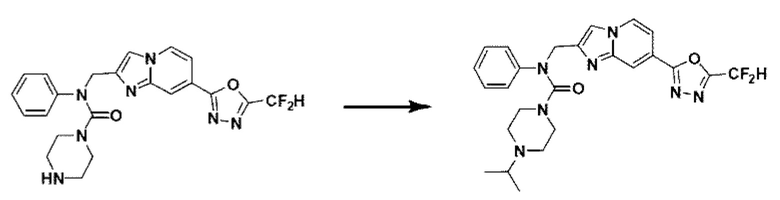
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилпиперазин-1-карбоксамид (0,050 г, 0,110 ммоль), полученный на стадии 2 примера 35, пропан-2-он (0,013 г, 0,221 ммоль), уксусную кислоту (0,006 мл, 0,110 ммоль) и триацетоксиборгидрид натрия (0,070 г, 0,331 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,030 г, 54,9%) в виде твердого вещества желтого цвета.
1H ЯМР (400 МГц, MeOD) δ 8,53 (дд, J=7,1, 1,0 Гц, 1Н), 8,22 (дт, J=1,8, 0,8 Гц, 1Н), 7,92 (с, 1Н), 7,55 (дд, J=7,1, 1,7 Гц, 1Н), 7,39-7,02 (м, 6Н), 5,02 (с, 2Н), 3,30 (т, J=5,1 Гц, 4Н), 2,65 (п, J=6,5 Гц, 1Н), 2,40 (т, J=5,1 Гц, 4Н), 1,02 (д, J=6,5 Гц, 6Н);
МСНР (ЭР) m/z 496,5 (М++1).
Пример 39: Синтез Соединения 4035, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-4-(1-гидроксипропан-2-ил)-N-фенилпиперазин-1-карбоксамида
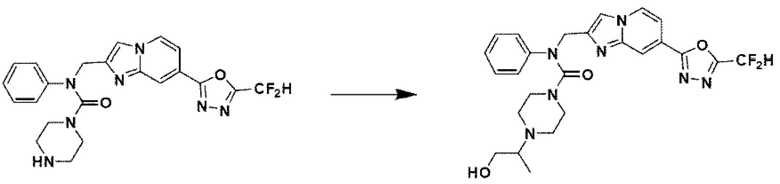
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилпиперазин-1-карбоксамид (0,100 г, 0,221 ммоль), полученный на стадии 2 примера 35, 1-гидроксипропан-2-он (0,033 г, 0,441 ммоль), уксусную кислоту (0,013 мл, 0,221 ммоль) и триацетоксиборгидрид натрия (0,140 г, 0,662 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и перемешивали полученный раствор при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,011 г, 9,8%) в виде твердого вещества желтого цвета.
1Н ЯМР (400 МГц, CDCl3) δ 8,31-8,27 (м, 1Н), 8,20 (дт, J=7,2, 1,4 Гц, 1Н), 7,79-7,74 (м, 1Н), 7,51 (тт, J=5,4, 1,7 Гц, 1Н), 7,38-7,30 (м, 2Н), 7,27-7,21 (м, 2Н), 7,20-7,09 (м, 1Н), 7,09-6,80 (м, 1Н), 5,07 (с, 2Н), 3,33 (д, J=23,0 Гц, 4Н), 3,24-3,15 (м, 2Н), 2,40 (с, 2Н), 2,12 (с, 2Н), 2,04 (с, 1Н), 1,28 (с, 3Н);
МСНР (ЭР) m/z 512,3 (М++1).
Пример 40: Синтез Соединения 4036, 4-циклобутил-N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилпиперазин-1-карбоксамида
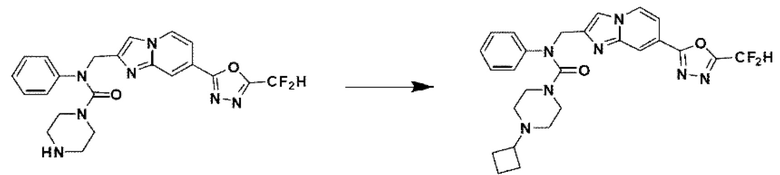
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилпиперазин-1-карбоксамид (0,100 г, 0,221 ммоль), полученный на стадии 2 примера 35, циклобутанон (0,031 г, 0,441 ммоль), уксусную кислоту (0,013 мл, 0,221 ммоль) и триацетоксиборгидрид натрия (0,140 г, 0,662 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и перемешивали полученный раствор при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,028 г, 25,0%) в виде твердого вещества желтого цвета.
1H ЯМР (700 МГц, MeOD) δ 8,60 (дд, J=1,1, 1,0 Гц, 1Н), 8,21 (п, J=0,8 Гц, 1Н), 7,98 (д, J=0,8 Гц, 1Н), 7,56 (дд, J=7,1, 1,7 Гц, 1Н), 7,40-7,14 (м, 6Н), 5,03 (д, J=0,8 Гц, 2Н), 3,29 (т, J=5,1 Гц, 4Н), 2,73-2,66 (м, 1Н), 2,18 (т, J=5,1 Гц, 4Н), 2,06-1,98 (м, 2Н), 1,89-1,79 (м, 2Н), 1,70 (ддт, J=12,8, 10,0, 5,7 Гц, 2Н);
МСНР (ЭР) m/z 508,3 (М++1).
Пример 41: Синтез Соединения 4037, 4-циклогексил-N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилпиперазин-1-карбоксамида
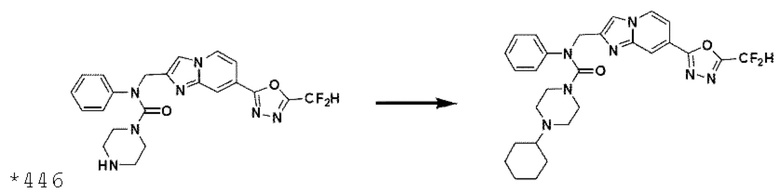
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилпиперазин-1-карбоксамид (0,100 г, 0,221 ммоль), полученный на стадии 2 примера 35, циклогексанон (0,043 г, 0,441 ммоль), уксусную кислоту (0,013 мл, 0,221 ммоль) и триацетоксиборгидрид натрия (0,140 г, 0,662 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и перемешивали полученный раствор при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,031 г, 26,2%) в виде твердого вещества желтого цвета.
1H ЯМР (700 МГц, MeOD) δ 8,57 (д, J=7,1 Гц, 1Н), 8,22-8,20 (м, 1Н), 7,96 (с, 1Н), 7,55 (дд, J=7,1, 1,7 Гц, 1Н), 7,41-7,09 (м, 6Н), 5,04-5,01 (м, 2Н), 3,29 (т, J=5,1 Гц, 4Н), 2,43 (т, J=5,0 Гц, 4Н), 2,23 (тт, J=11,9, 3,6 Гц, 1Н), 1,86-1,74 (м, 4Н), 1,63 (дт, J=12,6, 3,3 Гц, 1Н), 1,24 (квт, J=12,9, 3,2 Гц, 2Н), 1,19-1,07 (м, 3Н); МСНР (ЭР) m/z 536,2 (М++1).
Пример 42: Синтез Соединения 4038, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенил-4-(тетрагидро-2Н-пиран-4-ил)-пиперазин-1-карбоксамида
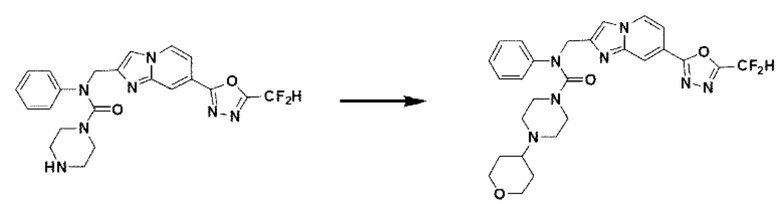
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилпиперазин-1-карбоксамид (0,100 г, 0,221 ммоль), полученный на стадии 2 примера 35, тетрагидро-4Н-пиран-4-он (0,044 г, 0,441 ммоль), уксусную кислоту (0,013 мл, 0,221 ммоль) и триацетоксиборгидрид натрия (0,140 г, 0,662 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,016 г, 13,5%) в виде твердого вещества желтого цвета.
1Н ЯМР (700 МГц, MeOD) δ 8,53 (д, J=7,1 Гц, 1Н), 8,23-8,20 (м, 1Н), 7,92 (с, 1Н), 7,55 (дд, J=7,1, 1,7 Гц, 1Н), 7,36 (дд, J=8,5, 7,4 Гц, 2Н), 7,29-7,05 (м, 4Н), 5,02 (с, 2Н), 3,97 (дд, J=11,3, 4,4 Гц, 2Н), 3,36 (тд, J=12,0, 1,9 Гц, 2Н), 3,31-3,28 (м, 4Н), 2,43 (т, J=5,1 Гц, 5Н), 1,75 (ддд, J=12,4, 4,3, 2,1 Гц, 2Н), 1,47 (квд, J=12,2, 4,5 Гц, 2Н);
МСНР (ЭР) m/z 538,1 (М++1).
Пример 43: Синтез Соединения 4039, 4-(4,4-дифторциклогексил)-N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилпиперазин-1-карбоксамида
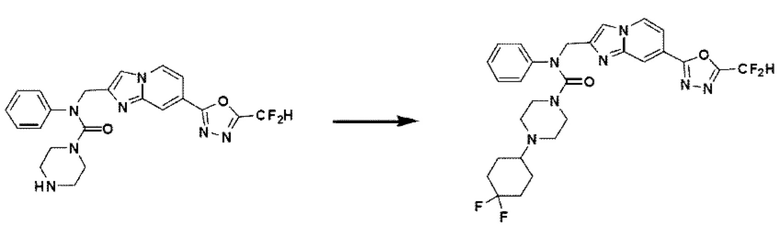
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилпиперазин-1-карбоксамид (0,100 г, 0,221 ммоль), полученный на стадии 2 примера 35, 4,4-дифторциклогексан-1-он (0,059 г, 0,441 ммоль), уксусную кислоту (0,013 мл, 0,221 ммоль) и триацетоксиборгидрид натрия (0,140 г, 0,662 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и перемешивали полученный раствор при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,034 г, 27,0%) в виде твердого вещества желтого цвета.
1H ЯМР (700 МГц, MeOD) δ 8,47 (дд, J=7,1, 1,0 Гц, 1Н), 8,21 (дт, J=1,7, 0,8 Гц, 1Н), 7,88 (д, J=0,8 Гц, 1Н), 7,54 (дд, J=7,1, 1,7 Гц, 1Н), 7,37-7,32 (м, 2Н), 7,25-7,00 (м, 4Н), 5,01 (с, 2Н), 3,30-3,26 (м, 4Н), 2,47-2,27 (м, 5Н), 2,08-2,04 (м, 2Н), 1,81 (д, J=13,1 Гц, 2Н), 1,72 (дддд, J=30,7, 17,4, 13,1, 4,2 Гц, 2Н), 1,57-1,49 (м, 2Н);
МСНР (ЭР) m/z 572,3 (М++1).
Пример 44: Синтез Соединения 4040, 4-ацетил-N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилпиперазин-1-карбоксамида
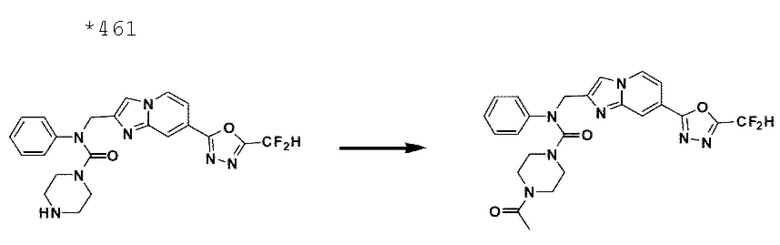
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилпиперазин-1-карбоксамид (0,100 г, 0,221 ммоль), полученный на стадии 2 примера 35, ацетилхлорид (0,031 мл, 0,441 ммоль) и триэтиламин (0,092 мл, 0,662 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор хлорида аммония, экстрагировали дихлорметаном, фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,018 г, 16,5%) в виде твердого вещества желтого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,37 (с, 1Н), 8,24 (д, J=7,1 Гц, 1Н), 7,79 (с, 1Н), 7,59 (д, J=7,1 Гц, 1Н), 7,37 (дд, J=8,4, 7,2 Гц, 2Н), 7,33-7,28 (м, 2Н), 7,21-7,13 (м, 1Н), 6,95 (т, J=51,7 Гц, 1Н), 5,07 (с, 2Н), 3,43 (т, J=5,3 Гц, 2Н), 3,30 (с, 4Н), 3,23 (т, J=5,4 Гц, 2Н), 2,04 (с, 3Н);
МСНР (ЭР) m/z 496,0 (М++1).
Пример 45: Синтез Соединения 4041, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенил-4-пропионилпиперазин-1-карбоксамида
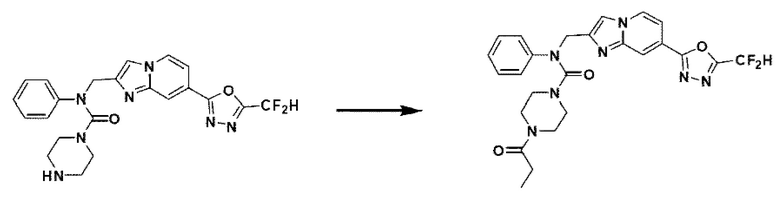
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилпиперазин-1-карбоксамид (0,100 г, 0,221 ммоль), полученный на стадии 2 Примера 35, пропионилхлорид (0,041 г, 0,441 ммоль) и триэтиламин (0,092 мл, 0,662 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор хлорида аммония, экстрагировали дихлорметаном, фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,014 г, 12,5%) в виде твердого вещества желтого цвета.
1Н ЯМР (700 МГц, MeOD) δ 8,58 (дд, J=7,1, 1,0 Гц, 1Н), 8,20 (дт, J=1,7, 0,8 Гц, 1Н), 8,00-7,98 (м, 1Н), 7,54 (дд, J=7,1, 1,7 Гц, 1Н), 7,40-7,14 (м, 6Н), 5,06-5,03 (м, 2Н), 3,44-3,38 (м, 4Н), 3,32-3,24 (м, 4Н), 2,35 (кв, J=7,5 Гц, 2Н), 1,07 (т, J=7,5 Гц, 3Н);
МСНР (ЭР) m/z 510,3 (М++1).
Пример 46: Синтез Соединения 4042, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-4-(2-гидроксиацетил)-N-фенилпиперазин-1-карбоксамида
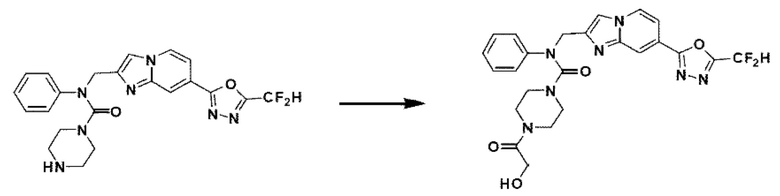
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилпиперазин-1-карбоксамид (0,100 г, 0,221 ммоль), полученный на стадии 2 примера 35, 2-гидроксиацетилхлорид (0,042 г, 0,441 ммоль) и триэтиламин (0,092 мл, 0,662 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор хлорида аммония, экстрагировали дихлорметаном, фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,029 г, 25,7%) в виде твердого вещества желтого цвета.
1Н ЯМР (400 МГц, CDCl3) δ 8,42 (с, 1Н), 8,26 (д, J=7,1 Гц, 1Н), 7,80 (с, 1Н), 7,64 (с, 1Н), 7,43-7,30 (м, 4Н), 7,24-7,16 (м, 1Н), 6,95 (т, J=51,7 Гц, 1Н), 5,07 (д, J=0,7 Гц, 2Н), 4,09 (с, 2Н), 3,50 (т, J=5,3 Гц, 2Н), 3,29 (д, J=9,9 Гц, 4Н), 3,10 (т, J=5,2 Гц, 2Н);
МСНР (ЭР) m/z 513,2 (М++1).
Пример 47: Синтез Соединения 4043, 4-(циклобутанкарбонил)-N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилпиперазин-1-карбоксамида
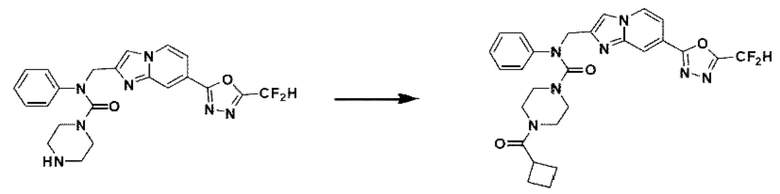
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилпиперазин-1-карбоксамид (0,100 г, 0,221 ммоль), полученный на стадии 2 Примера 35, циклобутанкарбонилхлорид (0,052 г, 0,441 ммоль) и триэтиламин (0,092 мл, 0,662 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор хлорида аммония, экстрагировали дихлорметаном, фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,030 г, 25,4%) в виде твердого вещества желтого цвета.
1H ЯМР (700 МГц, MeOD) δ 8,59 (дд, J=7,1, 1,0 Гц, 1Н), 8,21 (дт, J=1,7, 0,8 Гц, 1Н), 8,02-7,98 (м, 1Н), 7,56 (дд, J=7,1, 1,7 Гц, 1Н), 7,44-7,14 (м, 6Н), 5,06-5,03 (м, 2Н), 3,42-3,36 (м, 2Н), 3,29-3,22 (м, 6Н), 2,28-2,17 (м, 3Н), 2,14 (ддддд, J=12,2, 11,0, 8,8, 3,7, 1,8 Гц, 2Н), 2,02-1,94 (м, 1Н), 1,84-1,78 (м, 1Н);
МСНР (ЭР) m/z 537,1 (М++1).
Пример 48: Синтез Соединения 4044, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенил-4-(2,2,2-трифторацетил)-пиперазин-1-карбоксамида
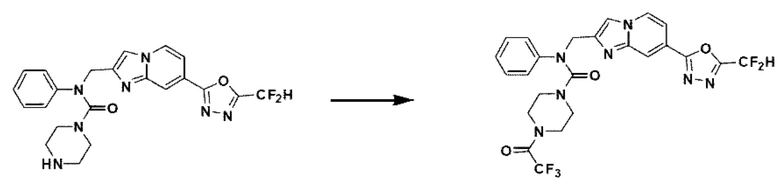
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилпиперазин-1-карбоксамид (0,050 г, 0,110 ммоль), полученный на стадии 2 примера 35, 1,1,1,5,5,5-гексафторпентан-2,4-дион (0,046 г, 0,221 ммоль) и триэтиламин (0,046 мл, 0,331 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор хлорида аммония, экстрагировали дихлорметаном, фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,041 г, 67,7%) в виде твердого вещества желтого цвета.
1H ЯМР (400 МГц, MeOD) δ 8,59 (дд, J=7,2, 1,0 Гц, 1Н), 8,22 (дт, J=1,7, 0,8 Гц, 1Н), 8,00 (д, J=0,7 Гц, 1Н), 7,56 (дд, J=7,2, 1,7 Гц, 1Н), 7,44-7,09 (м, 6Н), 5,06 (д, J=0,8 Гц, 2Н), 3,51 (кв, J=5,3 Гц, 4H), 3,36 (дд, J=6,7, 4,4 Гц, 4Н);
МСНР (ЭР) m/z 550,4 (М++1).
Пример 49: Синтез Соединения 4045, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-4-(метилсульфонил)-N-фенилпиперазин-1-карбоксамида
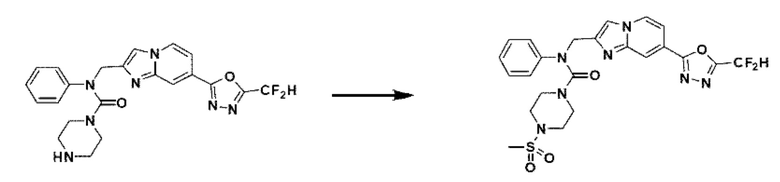
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилпиперазин-1-карбоксамид (0,050 г, 0,110 ммоль), полученный на стадии 2 примера 35, метансульфонилхлорид (0,017 мл, 0,221 ммоль) и триэтиламин (0,046 мл, 0,331 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор хлорида аммония, экстрагировали дихлорметаном, фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,012 г, 20,5%) в виде твердого вещества желтого цвета.
1H ЯМР (400 МГц, MeOD) δ 8,59 (дд, J=7,1, 1,0 Гц, 1Н), 8,22 (дт, J=1,7, 0,8 Гц, 1Н), 7,99 (д, J=0,8 Гц, 1Н), 7,56 (дд, J=7,1, 1,7 Гц, 1Н), 7,45-7,06 (м, 6Н), 5,04 (д, J=0,7 Гц, 2Н), 3,39-3,35 (м, 4Н), 3,07-3,02 (м, 4Н), 2,80 (с, 3Н); МСНР (ЭР) m/z 532,4 (М++1).
Пример 50: Синтез Соединения 4046, метил-4-(((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-(фенил)-карбамоил)-пиперазин-1-карбоксилата

N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилпиперазин-1-карбоксамид (0,100 г, 0,221 ммоль), полученный на стадии 2 Примера 35, метилхлорформиат (0,042 г, 0,441 ммоль) и триэтиламин (0,092 мл, 0,662 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор хлорида аммония, экстрагировали дихлорметаном, фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,034 г, 30,1%) в виде твердого вещества желтого цвета.
1Н ЯМР (700 МГц, MeOD) δ 8,59 (дд, J=7,1, 1,0 Гц, 1Н), 8,21 (дт, J=1,7, 0,8 Гц, 1Н), 7,99 (д, J=0,8 Гц, 1Н), 7,55 (дд, J=7,1, 1,7 Гц, 1Н), 7,44-7,11 (м, 6Н), 5,04 (д, J=0,8 Гц, 2Н), 3,66 (с, 3Н), 3,29 (д, J=5,9 Гц, 4Н), 3,26 (дд, J=7,0, 3,5 Гц, 4Н);
МСНР (ЭР) m/z 511,8 (М++1).
Пример 51: Синтез Соединения 4047, N1-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N4,N4-диметил-N1-фенилпиперазин-1,4-дикарбоксамида
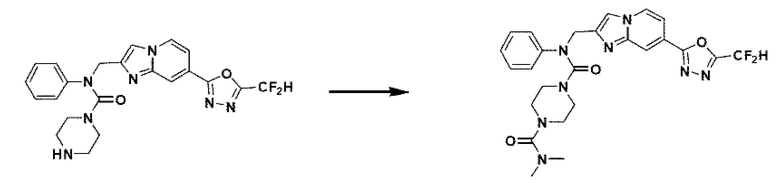
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилпиперазин-1-карбоксамид (0,100 г, 0,221 ммоль), полученный на стадии 2 Примера 35, хлориангидрид диметилкарбаминовой кислоты (0,047 г, 0,441 ммоль) и триэтиламин (0,092 мл, 0,662 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор хлорида аммония, экстрагировали дихлорметаном, фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,028 г, 24,2%) в виде твердого вещества желтого цвета.
1H ЯМР (700 МГц, MeOD) δ 8,59 (дд, J=7,1, 1,0 Гц, 1Н), 8,21 (дт, J=1,7, 0,8 Гц, 1Н), 7,99 (д, J=0,8 Гц, 1H), 7,55 (дд, J=7,1, 1,7 Гц, 1Н), 7,43-7,07 (м, 6Н), 5,04 (д, J=0,8 Гц, 2Н), 3,32-3,28 (м, 4Н), 3,09-3,05 (м, 4Н), 2,81 (с, 6Н);
МСНР (ЭР) m/z 525,0 (М++1).
Пример 52: Синтез Соединения 4048, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенил-4-(пиридин-2-ил)-пиперазин-1-карбоксамида
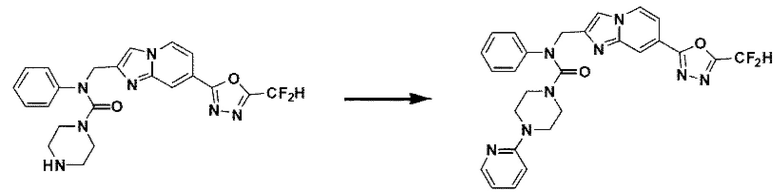
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилпиперазин-1-карбоксамид (0,065 г, 0,143 ммоль), полученный на стадии 2 примера 35, 2-хлорпиридин (0,033 г, 0,287 ммоль), карбонат цезия (0,093 г, 0,287 ммоль) и палладий RuPhos G2 (0,006 г, 0,007 ммоль) растворяли в 1,4-диоксане (2 мл) при комнатной температуре, и полученный раствор перемешивали при 100°С в течение 18 часов. Затем температуру понижали до комнатной температуры для завершения реакции. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном, фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 30%) и концентрировали с получением целевого соединения (0,011 г, 14,5%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,32 (с, 1Н), 8,24 (д, J=7,1 Гц, 1Н), 8,20-8,16 (м, 1Н), 7,79 (с, 1Н), 7,53 (д, J=7,1 Гц, 1Н), 7,39-7,29 (м, 5Н), 7,16 (т, J=7,1 Гц, 1Н), 6,95 (т, J=51,7 Гц, 1Н), 6,74 (с, 2Н), 5,10 (д, J=2,3 Гц, 2Н), 3,45 (с, 6Н), 1,63 (с, 2Н);
МСНР (ЭР) m/z 531,4 (М++1).
Пример 53: Синтез Соединения 4049, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенил-4-(пиримидин-2-ил)-пиперазин-1-карбоксамида
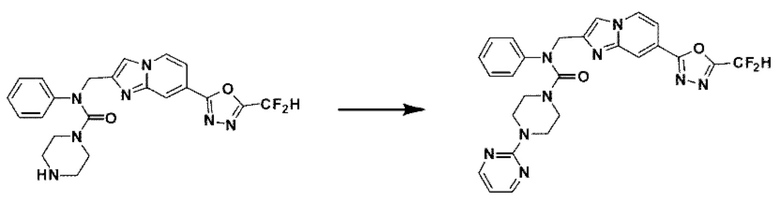
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилпиперазин-1-карбоксамид (0,100 г, 0,221 ммоль), полученный на стадии 2 примера 35, 2-хлорпиримидин (0,051 г, 0,441 ммоль) и карбонат калия (0,061 г, 0,441 ммоль) растворяли в ацетонитриле (2 мл)/N, N-диметилформамиде (2 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В концентрат, полученный путем удаления растворителя из реакционной смеси при пониженном давлении, вливали насыщенный водный раствор хлорида аммония с последующей экстракцией дихлорметаном. Затем полученный продукт фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,015 г, 12,8%) в виде твердого вещества желтого цвета.
1H ЯМР (700 МГц, MeOD) δ 8,56 (д, J=7,0 Гц, 1Н), 8,31-8,23 (м, 2Н), 7,97 (д, J=9,4 Гц, 1Н), 7,76-7,73 (м, 1Н), 7,59 (д, J=7,1 Гц, 1Н), 7,40-7,08 (м, 6Н), 6,58 (т, J=4,8 Гц, 1Н), 5,06 (с, 2Н), 3,66-3,62 (м, 4Н), 3,36-3,34 (м, 4Н);
МСНР (ЭР) m/z 532,0 (М++1).
Пример 54: Синтез Соединения 4083, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-4-метилпиперазин-1-карбоксамида
[Стадия 1] Синтез трет-бутил-4-(((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-(3-фторфенил)-карбамоил)-пиперазин-1-карбоксилата
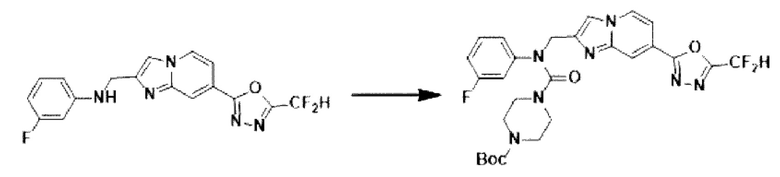
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-3-фторанилин (0,200 г, 0,557 ммоль), полученный в Примере 16, трет-бутилпиперазин-1-карбоксилат (0,135 г, 0,724 ммоль), трифосген (0,165 г, 0,557 ммоль) и N,N-диизопропилэтиламин (0,485 мл, 2,783 ммоль) растворяли в дихлорметане (15 мл), и полученный раствор перемешивали при 0°С в течение 1 часа и дополнительно перемешивали при комнатной температуре в течение 18 часов. В реакционную смесь вливали воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором, сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 40 г; этилацетат/гексан = от 0% до 60%) и концентрировали с получением целевого соединения (0,240 г, 75,4%) в виде твердого вещества белого цвета.
[Стадия 2] Синтез N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперазин-1-карбоксамида
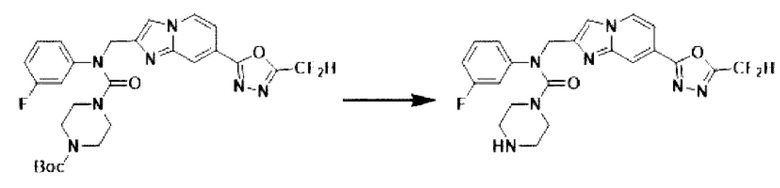
Трет-бутил-4-(((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-(3-фторфенил)-карбамоил)-пиперазин-1-карбоксилат (2,000 г, 3,499 ммоль), полученный на стадии 1, и трифторуксусную кислоту (5,359 мл, 69,984 ммоль) растворяли в дихлорметане (30 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 3 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором, сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении, и получали целевое соединение (1,650 г, 100,0%) в виде коричневого геля без дополнительной очистки.
[Стадия 3] Синтез соединения 4083
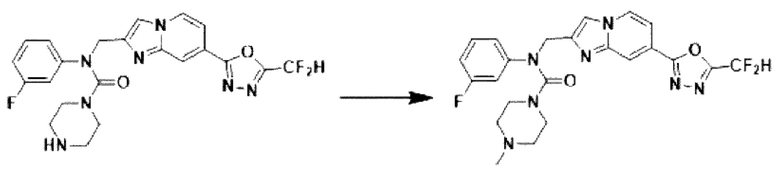
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперазин-1-карбоксамид (0,080 г, 0,170 ммоль), полученный на стадии 2, формальдегид (0,010 г, 0,339 ммоль), уксусную кислоту (0,010 мл, 0,170 ммоль) и триацетоксиборгидрид натрия (0,108 г, 0,509 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и перемешивали полученный раствор при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,018 г, 21,8%) в виде твердого вещества желтого цвета.
1H ЯМР (700 МГц, MeOD) δ 8,59 (дд, J=7,2, 1,0 Гц, 1Н), 8,22 (дт, J=1,8, 0,9 Гц, 1Н), 7,99 (д, J=0,7 Гц, 1Н), 7,56 (дд, J=7,1, 1,7 Гц, 1Н), 7,37 (тд, J=8,4, 6,6 Гц, 1Н), 7,26 (т, J=51,6 Гц, 1Н), 7,09-7,03 (м, 2Н), 6,90 (тдд, J=8,3, 2,5, 0,9 Гц, 1Н), 5,04 (д, J=0,8 Гц, 2H), 3,32 (с, 2Н), 3,28-3,25 (м, 2Н), 2,31 (т, J=5,0 Гц, 4Н), 2,25 (с, 3Н);
МСНР (ЭР) m/z 486,1 (М++1).
Пример 55: Синтез Соединения 4084, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-4-этил-N-(3-фторфенил)-пиперазин-1-карбоксамида
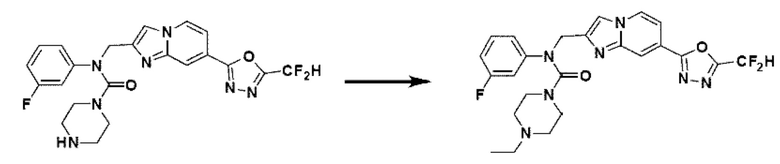
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперазин-1-карбоксамид (0,080 г, 0,170 ммоль), полученный на стадии 2 примера 54, ацетальдегид (0,015 г, 0,339 ммоль), уксусную кислоту (0,010 мл, 0,170 ммоль) и триацетоксиборгидрид натрия (0,108 г, 0,509 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и перемешивали полученный раствор при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,022 г, 26,0%) в виде твердого вещества желтого цвета.
1H ЯМР (700 МГц, MeOD) δ 8,59 (дд, J=7,1, 1,0 Гц, 1Н), 8,21 (дт, J=1,7, 0,8 Гц, 1Н), 7,99 (д, J=0,8 Гц, 1Н), 7,56 (дд, J=7,1, 1,7 Гц, 1Н), 7,37 (тд, J=8,3, 6,5 Гц, 1Н), 7,27 (т, J=51,6 Гц, 1Н), 7,09-7,04 (м, 2Н), 6,90 (тдд, J=8,4, 2,4, 0,9 Гц, 1Н), 5,04 (д, J=0,8 Гц, 2Н), 3,33 (дд, J=3,6, 2,0 Гц, 4Н), 2,40 (кв, J=7,2 Гц, 2Н), 2,35 (т, J=5,1 Гц, 4Н), 1,07 (т, J=7,2 Гц, 3Н);
МСНР (ЭР) m/z 500,1 (М++1).
Пример 56: Синтез Соединения 4085, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-4-изопропилпиперазин-1-карбоксамида
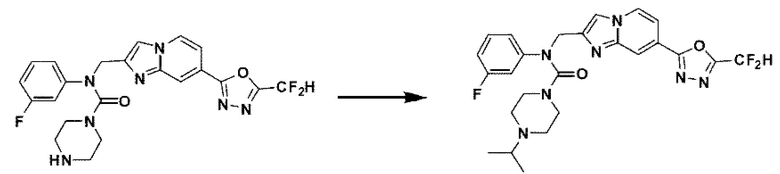
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперазин-1-карбоксамид (0,080 г, 0,170 ммоль), полученный на стадии 2 примера 54, пропан-2-он (0,020 г, 0,339 ммоль), уксусную кислоту (0,010 мл, 0,170 ммоль) и триацетоксиборгидрид натрия (0,108 г, 0,509 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и перемешивали полученный раствор при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,024 г, 27,5%) в виде твердого вещества желтого цвета.
1H ЯМР (700 МГц, MeOD) δ 8,59 (дд, J=7,1, 1,0 Гц, 1Н), 8,22 (дт, J=1,7, 0,8 Гц, 1Н), 7,99 (д, J=0,8 Гц, 1Н), 7,56 (дд, J=7,2, 1,7 Гц, 1Н), 7,37 (тд, J=8,3, 6,6 Гц, 1Н), 7,27 (т, J=51,6 Гц, 1Н), 7,08-7,04 (м, 2Н), 6,90 (тдд, J=8,4, 2,4, 0,9 Гц, 1Н), 5,04 (д, J=0,8 Гц, 2Н), 3,33-3,31 (м, 4Н), 2,65 (п, J=6,5 Гц, 1Н), 2,42 (т, J=5,1 Гц, 4Н), 1,03 (д, J=6,5 Гц, 6Н);
МСНР (ЭР) m/z 514,4 (М++1).
Пример 57: Синтез Соединения 4086, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-4-(1-гидроксипропан-2-ил)-пиперазин-1-карбоксамида
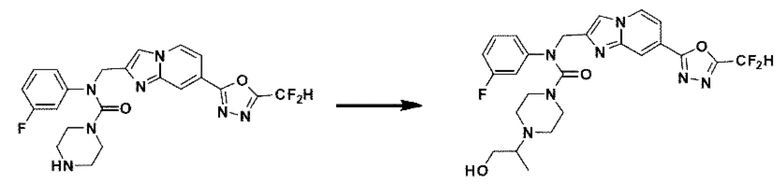
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперазин-1-карбоксамид (0,080 г, 0,170 ммоль), полученный на стадии 2 примера 54, 1-гидроксипропан-2-он (0,025 г, 0,339 ммоль), уксусную кислоту (0,010 мл, 0,170 ммоль) и триацетоксиборгидрид натрия (0,108 г, 0,509 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и перемешивали полученный раствор при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,022 г, 24,5%) в виде твердого вещества желтого цвета.
1Н ЯМР (700 МГц, MeOD) δ 8,60 (дд, J=1,1, 1,0 Гц, 1Н), 8,23 (дт, J=1,8, 0,9 Гц, 1Н), 8,00 (д, J=0,8 Гц, 1Н), 7,56 (дд, J=1,1, 1,1 Гц, 1Н), 7,36 (тд, J=8,3, 6,6 Гц, 1Н), 7,26 (т, J=51,7 Гц, 1Н), 7,08-7,03 (м, 2Н), 6,92-6,86 (м, 1Н), 5,04 (д, J=0,8 Гц, 2Н), 3,53 (дд, J=11,1, 6,8 Гц, 1Н), 3,41 (дд, J=11,2, 5,6 Гц, 1Н), 3,33-3,30 (м, 4Н), 2,65 (тд, J=6,8, 5,7 Гц, 1Н), 2,50 (дд, J=10,6, 6,3, 3,8 Гц, 2Н), 2,43 (дд, J=10,9, 6,2, 2Н), 0,96 (д, 3Н);
МСНР (ЭР) m/z 530,1 (М++1).
Пример 58: Синтез Соединения 4087, 4-циклобутил-N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперазин-1-карбоксамида
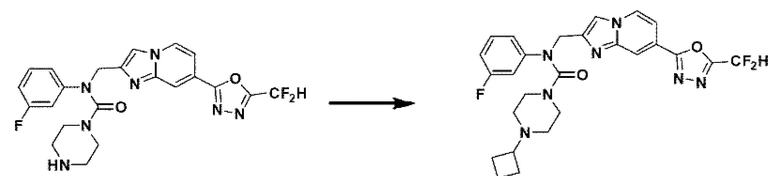
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперазин-1-карбоксамид (0,080 г, 0,170 ммоль), полученный на стадии 2 примера 54, циклобутанон (0,024 г, 0,339 ммоль), уксусную кислоту (0,010 мл, 0,170 ммоль) и триацетоксиборгидрид натрия (0,108 г, 0,509 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и перемешивали полученный раствор при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,025 г, 28,0%) в виде твердого вещества желтого цвета.
1H ЯМР (700 МГц, MeOD) δ 8,55 (дд, J=7,1, 1,0 Гц, 1Н), 8,24-8,21 (м, 1Н), 7,94 (с, 1Н), 7,56 (дд, J=7,1, 1,7 Гц, 1Н), 7,34 (тд, J=8,4, 6,5 Гц, 1Н), 7,20 (т, J=51,7 Гц, 1Н), 7,05-6,99 (м, 2Н), 6,90-6,85 (м, 1Н), 5,03 (с, 2Н), 3,31 (д, J=7,4 Гц, 4Н), 2,74 (с, 1Н), 2,24 (с, 4Н), 2,06-2,00 (м, 2Н), 1,90-1,79 (м, 2Н), 1,71 (дтд, J=15,6, 10,8, 10,3, 8,2 Гц, 2Н);
МСНР (ЭР) m/z 526,1 (М++1).
Пример 59: Синтез Соединения 4088, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-4-(оксетан-3-ил)-пиперазин-1-карбоксамида
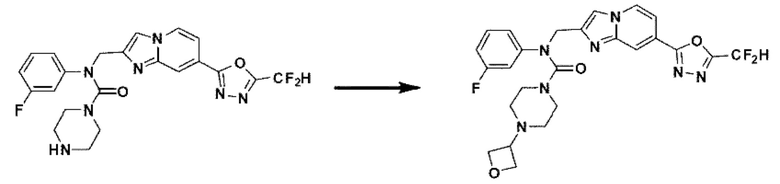
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперазин-1-карбоксамид (0,080 г, 0,170 ммоль), полученный на стадии 2 примера 54, оксетан-3-он (0,024 г, 0,339 ммоль), уксусную кислоту (0,010 мл, 0,170 ммоль) и триацетоксиборгидрид натрия (0,108 г, 0,509 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и перемешивали полученный раствор при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,019 г, 21,2%) в виде твердого вещества желтого цвета.
1H ЯМР (700 МГц, MeOD) δ 8,54 (дд, J=7,1, 1,0 Гц, 1Н), 8,23 (дт, J=1,8, 0,8 Гц, 1Н), 7,94 (д, J=0,8 Гц, 1Н), 7,56 (дд, J=7,1, 1,7 Гц, 1Н), 7,34 (тд, J=8,4, 6,4 Гц, 1Н), 7,20 (т, J=51,6 Гц, 1Н), 7,05-6,99 (м, H), 6,88 (тдд, J=8,3, 2,5, 0,9 Гц, 1Н), 5,05-4,98 (м, 2Н), 4,66 (т, J=6,7 Гц, 2Н), 4,55 (т, J=6,2 Гц, 2Н), 3,47 (с, 1Н), 3,36-3,34 (м, 4Н), 2,24 (с, 4Н);
МСНР (ЭР) m/z 529,2 (М++1).
Пример 60: Синтез Соединения 4089, 4-циклогексил-N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперазин-1-карбоксамида
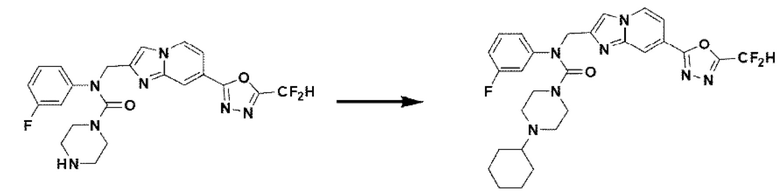
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперазин-1-карбоксамид (0,080 г, 0,170 ммоль), полученный на стадии 2 примера 54, циклогексанон (0,033 г, 0,339 ммоль), уксусную кислоту (0,010 мл, 0,170 ммоль) и триацетоксиборгидрид натрия (0,108 г, 0,509 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и перемешивали полученный раствор при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,018 г, 19,2%) в виде твердого вещества желтого цвета.
1H ЯМР (700 МГц, MeOD) δ 8,54 (дд, J=7,1, 1,0 Гц, 1Н), 8.23 (т, J=1,1 Гц, 1Н), 7,94 (с, 1Н), 7,56 (дд, J=7,1, 1,7 Гц, 1Н), 7,34 (тд, J=8,5, 6,7 Гц, 1Н), 7,20 (т, J=51,6 Гц, 1Н), 7,07-6,95 (м, 2Н), 6,87 (ддд, J=9,5, 8,0, 2,5 Гц, 1Н), 5,03 (с, 2Н), 3,32 (с, 4Н), 2,50 (с, 4Н), 2,28 (с, 1Н), 1,84 (д, J=11,2 Гц, 2Н), 1,82-1,76 (м, 2Н), 1,64 (д, J=12,6 Гц, 1Н), 1.24 (ддд, J=16,0, 11,2, 3,2 Гц, 2Н), 1,21-1,13 (м, 2Н), 1Н (д, J=12,9, 1Н);
МСНР (ЭР) m/z 554,4 (М++1).
Пример 61: Синтез Соединения 4090, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-4-(тетрагидро-2Н-пиран-4-ил)-пиперазин-1-карбоксамида
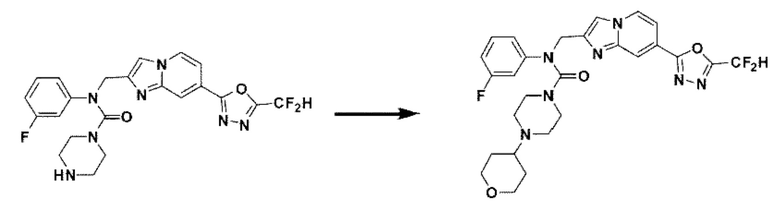
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперазин-1-карбоксамид (0,080 г, 0,170 ммоль), полученный на стадии 2 примера 54, тетрагидро-4Н-пиран-4-он (0,034 г, 0,339 ммоль), уксусную кислоту (0,010 мл, 0,170 ммоль) и триацетоксиборгидрид натрия (0,108 г, 0,509 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и перемешивали полученный раствор при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,028 г, 29,7%) в виде твердого вещества желтого цвета.
1H ЯМР (700 МГц, MeOD) δ 8,53 (дт, J=6,8, 3,2 Гц, 1Н), 8,23 (с, 1Н), 7,93 (д, J=2,5 Гц, 1Н), 7,56 (д, J=7,0 Гц, 1Н), 7,34 (кв, J=7,8 Гц, 1Н), 7,18 (тт, J=51,7, 2,9 Гц, 1Н), 7,02 (т, J=10,6 Гц, 2Н), 6,87 (тд, J=8,3, 2,6 Гц, 1Н), 5,03 (с, 2Н), 4,00-3,96 (м, 2Н), 3,41-3,32 (м, 7Н), 2,48 (с, 4Н), 1,77 (д, J=12,5 Гц, 2Н), 1,50 (дд, J=12,2, 4,3 Гц, 2Н);
МСНР (ЭР) m/z 556,1 (М++1).
Пример 62: Синтез Соединения 4091, 4-(4,4-дифторциклогексил)-N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперазин-1-карбоксамида
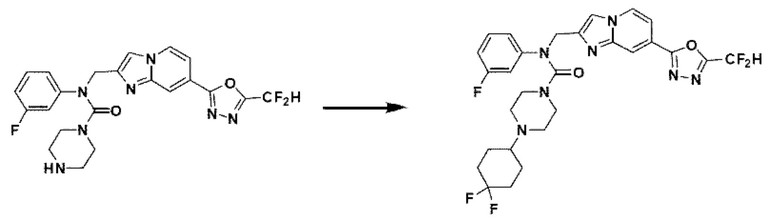
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперазин-1-карбоксамид (0,080 г, 0,170 ммоль), полученный на стадии 2 примера 54, 4,4-дифторциклогексан-1-он (0,046 г, 0,339 ммоль), уксусную кислоту (0,010 мл, 0,170 ммоль) и триацетоксиборгидрид натрия (0,108 г, 0,509 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и перемешивали полученный раствор при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,030 г, 30,0%) в виде твердого вещества желтого цвета.
1Н ЯМР (400 МГц, CDCl3) δ 8,28 (с, 1Н), 8,21 (д, J=7,0 Гц, 1Н), 7,75 (д, J=0,8 Гц, 1Н), 7,53 (д, J=7,2 Гц, 1Н), 7,32 (д, J=7,8 Гц, 1Н), 7,09-6,78 (м, 4Н), 5,07 (с, 2Н), 4,01-3,87 (м, 1Н), 3,30 (с, 2Н), 2,40 (с, 3Н), 2,23-2,05 (м, 3Н), 1,98-1,83 (м, 4Н), 1,76 (дт, J=12,3, 6,3 Гц, 4Н);
МСНР (ЭР) m/z 590,3 (М++1).
Пример 63: Синтез Соединения 4092, 4-ацетил-N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперазин-1-карбоксамида
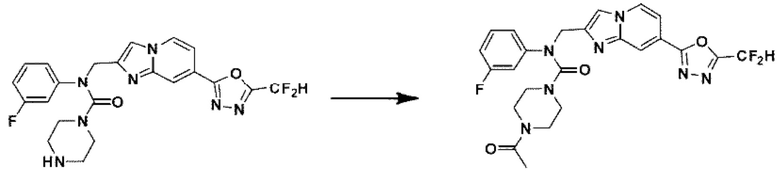
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперазин-1-карбоксамид (0,080 г, 0,170 ммоль), полученный на стадии 2 примера 54, ацетилхлорид (0,024 мл, 0,339 ммоль) и триэтиламин (0,071 мл, 0,509 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор хлорида аммония, экстрагировали дихлорметаном, фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,037 г, 42,5%) в виде твердого вещества желтого цвета.
1Н ЯМР (400 МГц, CDCl3) δ 8,32 (с, 1Н), 8,23 (дд, J=7,1, 1,0 Гц, 1Н), 7,77 (с, 1Н), 7,56 (дд, J=7,1, 1,7 Гц, 1Н), 7,32 (тд, J=8,2, 6,5 Гц, 1Н), 7,13-6,79 (м, 4Н), 5,07 (с, 2Н), 3,47 (дд, J=6,6, 4,0 Гц, 2Н), 3,33 (с, 4Н), 3,26-3,22 (м, 2Н), 2,05 (с, 3Н);
МСНР (ЭР) m/z 514,3 (М++1).
Пример 64: Синтез Соединения 4093, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-4-пропионилпиперазин-1-карбоксамида
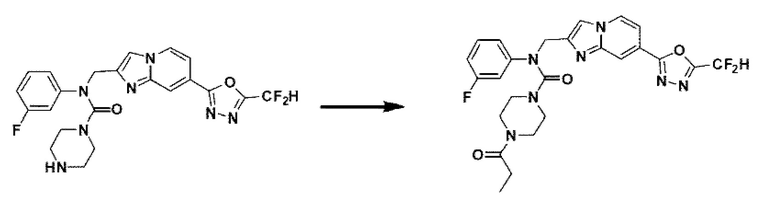
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперазин-1-карбоксамид (0,080 г, 0,170 ммоль), полученный на стадии 2 Примера 54, пропионилхлорид (0,031 г, 0,339 ммоль) и триэтиламин (0,071 мл, 0,509 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор хлорида аммония, экстрагировали дихлорметаном, фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,031 г, 34,6%) в виде твердого вещества желтого цвета.
1H ЯМР (700 МГц, MeOD) δ 8,59 (дд, J=7,1, 1,0 Гц, 1Н), 8,21 (дт, J=1,7, 0,8 Гц, 1Н), 8,02-7,99 (м, 1Н), 7,55 (дд, J=7,1, 1,7 Гц, 1Н), 7,37 (тд, J=8,2, 6,4 Гц, 1Н), 7,27 (т, J=51,7 Гц, 1Н), 7,13-7,06 (м, 2Н), 6,91 (тдд, J=8,4, 2,5, 0,9 Гц, 1Н), 5,06 (д, J=0,8 Гц, 2Н), 3, 49-3, 42 (м, 4Н), 3,35-3,34 (м, 2Н), 3,30-3,27 (м, 2Н), 2,37 (кв, J=7,5 Гц, 2Н), 1,08 (т, J=7,5 Гц, 3H);
МСНР (ЭР) m/z 528,1 (М+ +1).
Пример 65: Синтез Соединения 4094, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-4-(2-гидроксиацетил)-пиперазин-1-карбоксамида
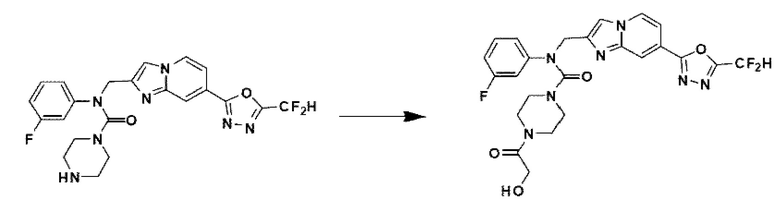
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперазин-1-карбоксамид (0,080 г, 0,170 ммоль), полученный на стадии 2 примера 54, 2-гидроксиацетилхлорид (0,032 г, 0,339 ммоль) и триэтиламин (0,071 мл, 0,509 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор хлорида аммония, экстрагировали дихлорметаном, фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,014 г, 15,6%) в виде твердого вещества желтого цвета.
1H ЯМР (700 МГц, MeOD) δ 8,60 (дд, J=1,1, 1,0 Гц, 1Н), 8,23 (дт, J=1,7, 0,8 Гц, 1Н), 8,01 (д, J=0,8 Гц, 1Н), 7,57 (дд, J=7,1, 1,7 Гц, 1Н), 7,38 (тд, J=8,2, 6,5 Гц, 1Н), 7,26 (т, J=51,7 Гц, 1Н), 7,13-7,06 (м, 2Н), 6,92 (тдд, J=8,3, 2,5, 0,9 Гц, 1Н), 5,06 (д, J=0,8 Гц, 2Н), 4,19 (с, 2Н), 3,74 (п, J=6,6 Гц, 1Н), 3,48 (т, J=5,3 Гц, 2Н), 3,26-3,19 (м, 1Н), 1,39 (д, J=6,7 Гц, 4Н);
МСНР (ЭР) m/z 530,0 (М+ +1).
Пример 66: Синтез Соединения 4095, 4-(циклобутанкарбонил)-N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперазин-1-карбоксамида
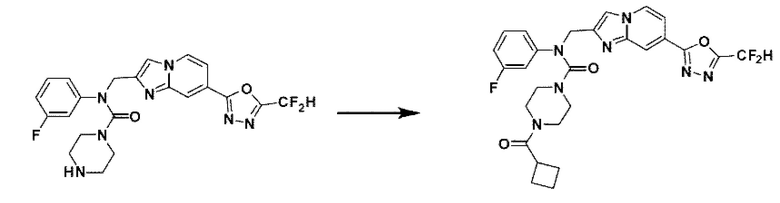
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперазин-1-карбоксамид (0,080 г, 0,170 ммоль), полученный на стадии 2 Примера 54, циклобутанкарбонилхлорид (0,040 г, 0,339 ммоль) и триэтиламин (0,071 мл, 0,509 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор хлорида аммония, экстрагировали дихлорметаном, фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 2 0×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,027 г, 28,7%) в виде твердого вещества желтого цвета.
1H ЯМР (700 МГц, MeOD) δ 8,60 (дд, J=1,1, 1,0 Гц, 1Н), 8,24-8,21 (м, 1Н), 8,01 (с, 1Н), 7,57 (дд, J=7,1, 1,7 Гц, 1Н), 7,37 (тд, J=8,2, 6,5 Гц, 1Н), 7,26 (т, J=51,7 Гц, 1Н), 7,13-7,06 (м, 2Н), 6,92 (тдд, J=8,4, 2,5, 0,9 Гц, 1Н), 5,06 (с, 2Н), 3, 46-3, 43 (м, 2Н), 3,37 (тд, J=8,7, 1,1 Гц, 1Н), 3,32 (с, 1Н), 3,3 0-3,2 6 (м, 4Н), 2,2 8-2,19 (м, 3H), 2,17-2,10 (м, 2Н), 2,04-1,95 (м, 1Н), 1,84-1,78 (м, 1Н);
МСНР (ЭР) m/z 553,9 (М+ +1).
Пример 67: Синтез Соединения 4096, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-4-(2,2,2-трифторацетил)-пиперазин-1-карбоксамида
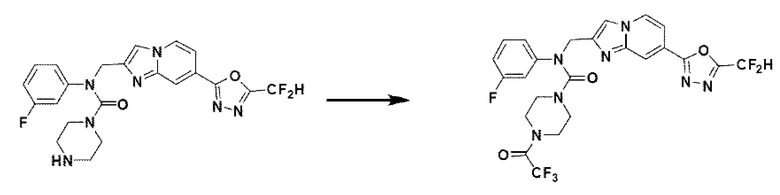
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперазин-1-карбоксамид (0,050 г, 0,106 ммоль), полученный на стадии 2 примера 54, 1,1,1,5,5,5-гексафторпентан-2,4-дион (0,044 г, 0,212 ммоль) и триэтиламин (0,044 мл, 0,318 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор хлорида аммония, экстрагировали дихлорметаном, фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,038 г, 63,1%) в виде твердого вещества желтого цвета.
1Н ЯМР (400 МГц, MeOD) δ 8,59 (дд, J=7,2, 1,0 Гц, 1Н), 8,23 (дт, J=1,6, 0,8 Гц, 1Н), 8,01 (д, J=0,7 Гц, 1Н), 7,57 (дд, J=7,2, 1,7 Гц, 1Н), 7,42-6,86 (м, 5Н), 5,07 (д, J=0,8 Гц, 2Н), 3,56 (кв, J=5,0 Гц, 4Н), 3,38 (тд, J=7,2, 6,2, 4,0 Гц, 4Н);
МСНР (ЭР) m/z 568,4 (М+ +1).
Пример 68: Синтез Соединения 4097, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-4-(метилсульфонил)-пиперазин-1-карбоксамида

N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперазин-1-карбоксамид (0,050 г, 0,106 ммоль), полученный на стадии 2 примера 54, метансульфонилхлорид (0,016 мл, 0,212 ммоль) и триэтиламин (0,044 мл, 0,318 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор хлорида аммония, экстрагировали дихлорметаном, фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,015 г, 25,7%) в виде твердого вещества желтого цвета.
1H ЯМР (400 МГц, MeOD) δ 8,60 (дд, J=7,2, 1,0 Гц, 1Н), 8,23 (дт, J=1,7, 0,8 Гц, 1Н), 8,01 (д, J=0,8 Гц, 1Н), 7,57 (дд, J=7,1, 1,7 Гц, 1Н), 7,46-6,87 (м, 5Н), 5,06 (д, J=0,7 Гц, 2Н), 3,43-3,36 (м, 4Н), 3,13-3,0 6 (м, 4Н), 2,81 (с, 3H);
МСНР (ЭР) m/z 550,4 (М+ +1).
Пример 69: Синтез Соединения 4098, метил-4-(((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-(3-фторфенил)-карбамоил)-пиперазин-1-карбоксилата
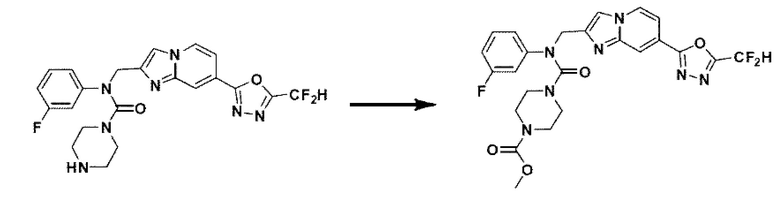
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперазин-1-карбоксамид (0,080 г, 0,170 ммоль), полученный на стадии 2 Примера 54, метилхлорформиат (0,032 г, 0,339 ммоль) и триэтиламин (0,071 мл, 0,509 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор хлорида аммония, экстрагировали дихлорметаном, фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,032 г, 35,6%) в виде твердого вещества желтого цвета.
1H ЯМР (700 МГц, MeOD) δ 8,55 (дд, J=7,1, 1,0 Гц, 1Н), 8,24 (дт, J=1,8, 0,9 Гц, 1Н), 7,96 (с, 1Н), 7,57 (дд, J=7,1, 1,7 Гц, 1Н), 7,35 (тд, J=8,4, 6,5 Гц, 1Н), 7,20 (т, J=51,6 Гц, 1Н), 7,07-7,02 (м, 2Н), 6,92-6,8 6 (м, 1Н), 5,05-5,02 (м, 2Н), 3,67 (с, 3H), 3,36-3,34 (м, 4Н), 3,28 (дд, J=6,6, 3,7 Гц, 4Н);
МСНР (ЭР) m/z 531,3 (М+ +1).
Пример 70: Синтез Соединения 4099, N1-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N1-(3-фторфенил)-N4,N4-диметилпиперазин-1,4-дикарбоксамида
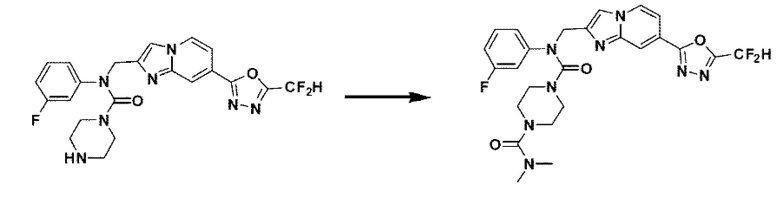
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперазин-1-карбоксамид (0,080 г, 0,170 ммоль), полученный на стадии 2 Примера 54, хлорангидрид диметилкарбаминовой кислоты (0,036 г, 0,339 ммоль) и триэтиламин (0,071 мл, 0,509 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор хлорида аммония, экстрагировали дихлорметаном, фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,018 г, 19,6%) в виде твердого вещества желтого цвета.
1Н ЯМР (700 МГц, MeOD) δ 8,58-8,55 (м, 1Н), 8,26-8,24 (м, 1Н), 7,97 (с, 1Н), 7,59 (дд, J=7,1, 1,7 Гц, 1Н), 7,36 (тд, J=8,4, 6,4 Гц, 1Н), 7,20 (t, J=51,7 Гц, 1Н), 7,06-7,02 (m, 2Н), 6,89 (тдд, J=8,3, 2,4, 1,0 Гц, 1Н), 5,05 (с, 2Н), 3,32-3,30 (м, 4Н), 3, 12-3, 10 (м, 4Н), 2,82 (с, 6Н);
МСНР (ЭР) m/z 542,9 (М+ +1).
Пример 71: Синтез Соединения 4100, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-4-(пиридин-2-ил)-пиперазин-1-карбоксамида
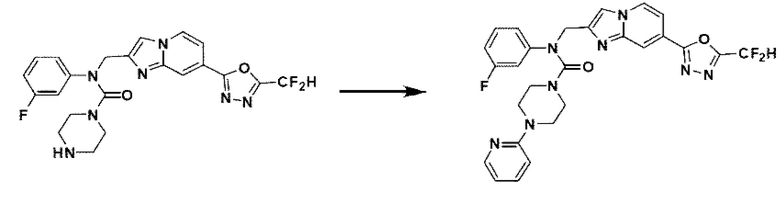
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперазин-1-карбоксамид (0,066 г, 0,140 ммоль), полученный на стадии 2 примера 54, 2-хлорпиридин (0,032 г, 0,280 ммоль), карбонат цезия (0,0 91 г, 0,280 ммоль) и палладий RuPhos G2 (0,005 г, 0,007 ммоль) растворяли в 1,4-диоксане (2 мл) при комнатной температуре, и полученный раствор перемешивали при 100°С в течение 18 часов.
Затем температуру понижали до комнатной температуры для завершения реакции. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 30%) и концентрировали с получением целевого соединения (0,014 г, 18,2%) в виде твердого вещества белого цвета.
1Н ЯМР (400 МГц, CDCl3) δ 8,32 (с, 1Н), 8,25 (д, J=7,0 Гц, 1Н), 8,21-8,17 (м, 1Н), 7,78 (с, 1Н), 7,59-7,52 (м, 1Н), 7,42 (ддт, J=10,9, 7,4, 1,6 Гц, 1Н), 7,36-7,29 (м, 1Н), 7,14-6,79 (м, 5Н), 6,61 (д, J=8,3 Гц, 1Н), 5,10 (с, 2Н), 3,55 (д, J=57,6 Гц, 6Н), 1,80-1,56 (м, 2Н);
МСНР (ЭР) m/z 54 9,4 (М+ +1).
Пример 72: Синтез Соединения 4101, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-4-(пиримидин-2-ил)-пиперазин-1-карбоксамида
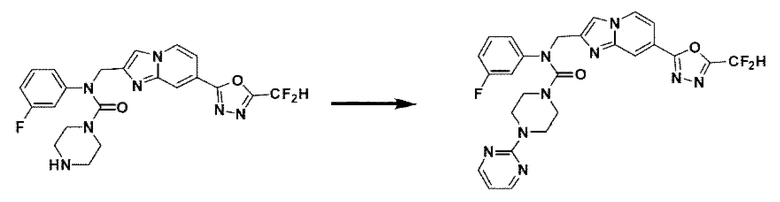
N-((7-(5-(дифторметил)-1,3,4-океадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперазин-1-карбоксамид (0,080 г, 0,170 ммоль), полученный на стадии 2 примера 54, 2-хлорпиримидин (0,039 г, 0,339 ммоль) и карбонат калия (0, 047 г, 0,339 ммоль) растворяли в ацетонитриле (2 мл)/N,N-диметилформамиде (2 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В концентрат, полученный путем удаления растворителя из реакционной смеси при пониженном давлении, вливали насыщенный водный раствор хлорида аммония с последующей экстракцией дихлорметаном. Затем полученный продукт фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,015 г, 16,1%) в виде твердого вещества желтого цвета.
1H ЯМР (700 МГц, MeOD) δ 8,50 (дд, J=7,1, 1,8 Гц, 1Н), 8,28 (д, J=4,8 Гц, 1Н), 8,24 (д, J=1,6 Гц, 1Н), 7,93 (с, 1Н), 7,56 (дд, J=7,1, 1,7 Гц, 1Н), 7,34 (тд, J=8,1, 6,4 Гц, 1Н), 7,15 (тд, J=51,7, 2,0 Гц, 1Н), 7,07-7,02 (м, 2Н), 6,87 (тд, J=8,3, 2,4 Гц, 1Н), 6,57 (т, J=4,7 Гц, 1Н), 5,06 (с, 2Н), 3,70-3,66 (м, 4Н), 3,4 0-3,35 (м, 4Н);
МСНР (ЭР) m/z 550,2 (М+ +1).
Пример 73: Синтез Соединения 4102, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-4-(оксетан-3-карбонил)-N-фенилпиперазин-1-карбоксамида
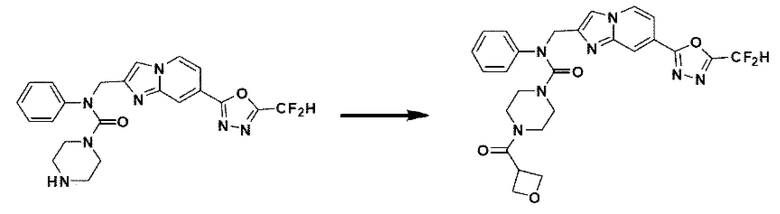
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилпиперазин-1-карбоксамид (0,050 г, 0,110 ммоль), полученный на стадии 2 примера 35, оксетан-3-карбоновую кислоту (0,023 г, 0,221 ммоль), 1-[бис-(диметиламино)-метилен]-1Н-1,2,3-триазоло[4,5-b]пиридиния3-оксид гексафторфосфат (HATU, 0,050 г, 0,132 ммоль) и триэтиламин (0,043 мл, 0,331 ммоль) растворяли в дихлорметане (5 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (картридж SiO2, 12 г; этилацетат/гексан = от 0% до 30%) и концентрировали с получением целевого соединения (0,015 г, 25,3%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,34-8,29 (м, 1Н), 8,24 (дт, J=7.1, 1,0 Гц, 1Н), 7,77 (дд, J=5,3, 0,7 Гц, 1Н), 7,55 (дд, J=7.2, 1,7 Гц, 1Н), 7,36 (тт, J=7,4, 1,9 Гц, 2Н), 7,28-7,25 (м, 2Н), 7,21-7,13 (м, 1Н), 6,95 (т, J=51,7 Гц, 1Н), 5,07 (д, J=3,3 Гц, 2Н), 4,85 (дд, J=7,1, 5,9 Гц, 1Н), 4,76 (дд, J=8,7, 5,9 Гц, 1Н), 3,44 (дт, J=10,2, 5,4 Гц, 2Н), 3,31 (с, 2Н), 3,24 (ддт, J=10,3, 7,4, 3,5 Гц, 4Н), 3,01 (т, J=5,3 Гц, 1Н), 2,04 (с, 2Н);
МСНР (ЭР) m/z 538,5 (М+ +1).
Пример 74: Синтез Соединения 4103, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-4-(оксетан-3-карбонил)-пиперазин-1-карбоксамида
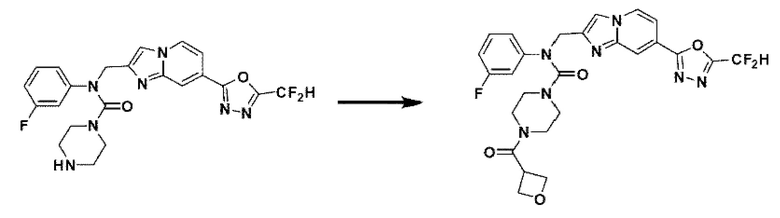
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперазин-1-карбоксамид (0,050 г, 0,106 ммоль), полученный на стадии 2 примера 54, оксетан-3-карбоновую кислоту (0,022 г, 0,212 ммоль), 1-[бис-(диметиламино)-метилен]-1Н-1,2,3-триазоло[4,5-b]пиридиния 3-оксид гексафторфосфат (HATU, 0,048 г, 0,127 ммоль) и триэтиламин (0,044 мл, 0,318 ммоль) растворяли в дихлорметане (5 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (картридж SiO2, 12 г; этилацетат/гексан = от 0% до 3 0%) и концентрировали с получением целевого соединения (0,024 г, 4 0,7%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,31 (дд, J=1,7, 0,9 Гц, 1Н), 8,23 (дд, J=7,1, 1,0 Гц, 1Н), 7,76 (дд, J=4,5, 0,7 Гц, 1Н), 7,55 (дд, J=7,1, 1,8 Гц, 1Н), 7,32 (тдд, J=8,5, 6,6, 2,0 Гц, 1Н), 7,11-6,79 (м, 4Н), 5,07 (д, J=3,0 Гц, 2Н), 4,87 (дд, J=7,1, 5,9 Гц, 1Н), 4,77 (дд, J=8,7, 6,0 Гц, 1Н), 3,48 (дт, J=10,5, 5,4 Гц, 2Н), 3,34 (с, 2Н), 3,31-3,16 (м, 4Н), 3,05 (т, J=5,3 Гц, 1Н);
МСНР (ЭР) m/z 556,5 (М+ +1).
Пример 75: Синтез Соединения 4115, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-1-этил-N-фенилпиперидин-4-карбоксамида
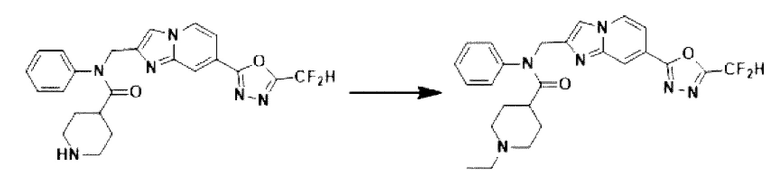
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилпиперидин-4-карбоксамид (0,040 г, 0,088 ммоль), полученный на стадии 3 Примера 33, ацетальдегид (0,010 мл, 0,177 ммоль) и уксусную кислоту (0,005 мл, 0,088 ммоль) растворяли в дихлорметане (0,5 мл), и полученный раствор перемешивали при комнатной температуре в течение 1 часа. Затем добавляли триацетоксиборгидрид натрия (0,056 г, 0,265 ммоль) и дополнительно перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном, фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; метанол/дихлорметан = от 0% до 2 0%) и концентрировали с получением целевого соединения (0,003 г, 7,8%) в виде желтого геля.
1H ЯМР (400 МГц, CDCl3) δ 8,30 (д, J=1,6 Гц, 1Н), 8,24 (дд, J=0,9, 7,1 Гц, 1Н), 7,77 (с, 1Н), 7,54 (дд, J=1,8, 7,1 Гц, 1Н), 7,4 6-7,3 6 (м, 3H), 7,27-7,21 (м, 2Н), 6,95 (т, J=51,7 Гц, 1Н), 5,04 (с, 2Н), 3,09 (с, 2Н), 2,55 (с, 2Н), 2,41 (с, 1Н), 2,02 (с, 1Н), 1,97 (д, J=10,3 Гц, 3H), 1,80 (с, 2Н), 1,28 (с, 1Н), 1,15 (с, 3H);
МСНР (ЭР) m/z 481,3 (М+ +1).
Пример 76: Синтез Соединения 4116, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-1-изопропил-N-фенилпиперидин-4-карбоксамида
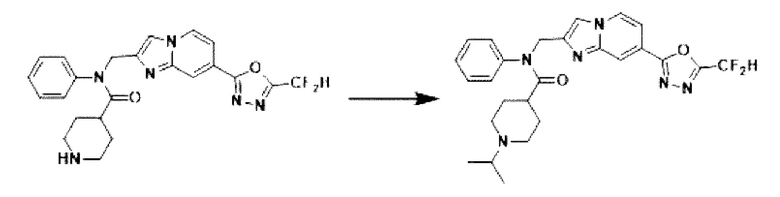
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилпиперидин-4-карбоксамид (0,024 г, 0,053 ммоль), полученный на стадии 3 примера 33, пропан-2-он (0,006 г, 0,106 ммоль) и уксусную кислоту (0,003 мл, 0,053 ммоль) растворяли в дихлорметане (1 мл), и полученный раствор перемешивали при комнатной температуре в течение 1 часа. Затем добавляли триацетоксиборгидрид натрия (0,034 г, 0,159 ммоль) и дополнительно перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном, фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; метанол/дихлорметан = от 0% до 20%) и концентрировали с получением целевого соединения (0,006 г, 22,5%) в виде твердого вещества желтого цвета.
1H ЯМР (700 МГц, MeOD) δ 8,58 (дд, J=1,0, 7,1 Гц, 1Н), 8,23 (дт, J = 0,8, 1,8 Гц, 1Н), 7,94-7,89 (м, 1Н), 7,58 (дд, J = 1,7, 7,1 Гц, 1Н), 7,47-7,38 (м, 3H), 7,32-7,27 (м, 2Н), 5,11 (с, 2Н), 3,40 (д, J=12,2 Гц, 2Н), 2,78 (с, 2Н), 2,65 (д, J=11,8 Гц, 1Н), 2,12-2,05 (м, 2Н), 2,04-1,99 (м, 2Н), 1,96 (с, 1Н), 1,27 (д, J=6,7 Гц, 6Н);
МСНР (ЭР) m/z 4 95,4 (М+ +1).
Пример 77: Синтез Соединения 4117, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-1-(1-гидроксипропан-2-ил)-N-фенилпиперидин-4-карбоксамида
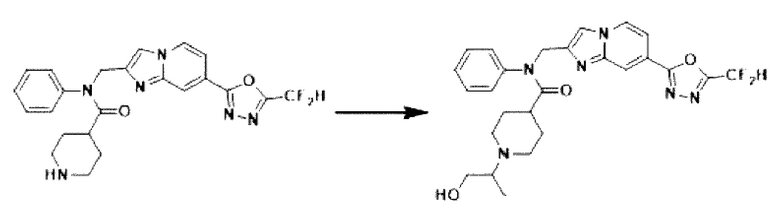
N-((7-(5-(дифторметил)-1,3,4-океадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилпиперидин-4-карбоксамид (0, 040 г, 0,088 ммоль), полученный на стадии 3 примера 33, 1-гидроксипропан-2-он (0,012 м, 0,177 ммоль), уксусную кислоту (0,005 мл, 0,088 ммоль) и триацетоксиборгидрид натрия (0,056 г, 0,265 ммоль) растворяли в дихлорметане (1 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном, фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; метанол/ дихлорметан = от 0% до 2 0%) и концентрировали с получением целевого соединения (0,009 г, 19,1%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,31 (с, 1Н), 8,24 (д, J=7,1 Гц, 1Н), 7,74 (с, 1Н), 7,55 (дд, J=1,7, 7,1 Гц, 1Н), 7,41 (тд, J=5,8, 8,3 Гц, 3H), 7,26-7,18 (м, 2Н), 6,96 (т, J=51,7 Гц, 1Н), 5,04 (с, 2Н), 3,71 (дд, J=4,2, 12,2 Гц, 1Н), 3,48 (дд, J=8,6, 12,0 Гц, 1Н), 3,30 (с, 1Н), 3,14 (с, 2Н), 2,80 (с, 1Н), 2,50 (д, J=26,3 Гц, 2Н), 2,05 (с, 2Н), 1,97-1,82 (м, 2Н), 1,06 (д, J=6,7 Гц, 3H);
МСНР (ЭР) m/z 510,55 (М+ +1).
Пример 78: Синтез Соединения 4118, 1-циклобутил-N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилпиперидин-4-карбоксамида
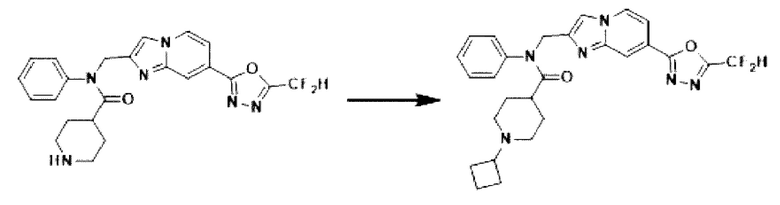
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилпиперидин-4-карбоксамид (0,040 г, 0,088 ммоль), полученный на стадии 3 Примера 33, циклобутанон (0,013 мл, 0,177 ммоль) и уксусную кислоту (0,005 мл, 0,088 ммоль) растворяли в дихлорметане (0,5 мл), и полученный раствор перемешивали при комнатной температуре в течение 1 часа. Затем добавляли триацетоксиборгидрид натрия (0,056 г, 0,265 ммоль) и дополнительно перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном, фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; метанол/дихлорметан = от 0% до 2 0%) и концентрировали с получением целевого соединения (0,012 г, 27,2%) в виде твердого вещества бледно-красного цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,31 (с, 1Н), 8,24 (д, J=7,1 Гц, 1Н), 7,74 (с, 1H), 7,55 (дд, J=1,7, 7,1 Гц, 1H), 7,41 (тд, J=5,8, 8,3 Гц, 3H), 7,26-7,18 (м, 2Н), 6,96 (т, J=51,7 Гц, 1Н), 5,04 (с, 2Н), 3,71 (дд, J=4,2, 12,2 Гц, 1Н), 3,48 (дд, J=8,6, 12,0 Гц, 1Н), 3,30 (с, 1Н), 3,14 (с, 2Н), 2,80 (с, 1Н), 2,50 (д, J=26,3 Гц, 2Н), 2,05 (с, 2Н), 1,97-1,82 (м, 2Н), 1,06 (д, J=6,7 Гц, 3H);
МСНР (ЭР) m/z 507,3 (М+ +1).
Пример 79: Синтез Соединения 4119, 1-циклогексил-N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилпиперидин-4-карбоксамида
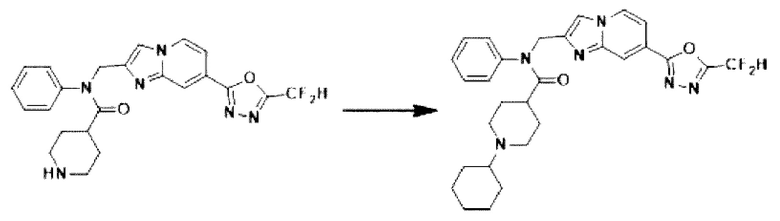
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилпиперидин-4-карбоксамид (0,040 г, 0,088 ммоль), полученный на стадии 3 Примера 33, циклогексанон (0,018 мл, 0,177 ммоль) и уксусную кислоту (0,005 мл, 0,088 ммоль) растворяли в дихлорметане (0,5 мл), и полученный раствор перемешивали при комнатной температуре в течение 1 часа. Затем добавляли триацетоксиборгидрид натрия (0,056 г, 0,265 ммоль) и дополнительно перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном, фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; метанол/дихлорметан = от 0% до 20%) и концентрировали с получением целевого соединения (0,004 г, 7,6%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, MeOD) δ 8,58 (дд, J=1,0, 7,1 Гц, 1Н), 8,25-8,22 (м, 1Н), 7,92 (с, 1Н), 7,57 (дд, J=1,7, 7,1 Гц, 1Н), 7,47-7,36 (м, 3H), 7,31-7,15 (м, 3H), 5,10 (с, 2Н), 2,84 (с, 1Н), 2,58 (дд, J=12,7, 22,7 Гц, 3H), 2,11-1,81 (м, 8Н), 1,68 (д, J=13,0 Гц, 1Н), 1,41-1,31 (м, 5Н), 1,20 (дд, J=11,7, 23,7 Гц, 2Н);
МСНР (ЭР) m/z 535,3 (М+ +1).
Пример 80: Синтез Соединения 4120, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенил-1-(тетрагидро-2Н-пиран-4-ил)-пиперидин-4-карбоксамида
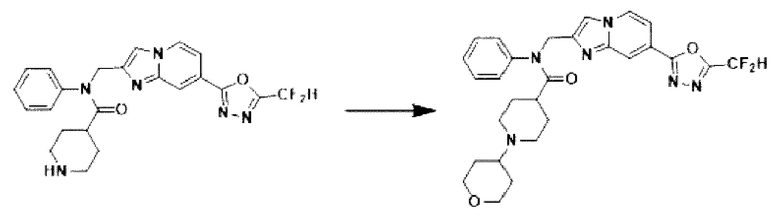
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилпиперидин-4-карбоксамид (0,050 г, 0,111 ммоль), полученный на стадии 3 примера 33, тетрагидро-4Н-пиран-4-он (0,020 мл, 0,221 ммоль), уксусную кислоту (0,006 мл, 0,111 ммоль) и триацетоксиборгидрид натрия (0,070 г, 0,332 ммоль) растворяли в дихлорметане (1 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном, фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; метанол/дихлорметан = от 0% до 20%) и концентрировали с получением целевого соединения (0,012 г, 2 0,7%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CDCl3) 5 8,31-8,28 (м, 1Н), 8,23 (дд, J=1,0, 7,1 Гц, 1Н), 7,78 (с, 1Н), 7,53 (дд, J=1,7, 7,1 Гц, 1Н), 7,46-7,32 (м, 3H), 7,27-7,20 (м, 2Н), 6,95 (т, J=51,7 Гц, 1Н), 5,04 (с, 2Н), 3,99 (дд, J=4,2, 11,4 Гц, 2Н), 3,34 (тд, J=2,0, 11,8 Гц, 2Н), 2,92 (с, 2Н), 2,34 (д, J=64,4 Гц, 2Н), 1,88 (д, J=14,6 Гц, 4Н), 1,76-1,48 (м, 6Н);
МСНР (ЭР) m/z 537,3 (М+ +1).
Пример 81: Синтез Соединения 4121, 1-(4,4-дифторциклогексил)-N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилпиперидин-4-карбоксамида
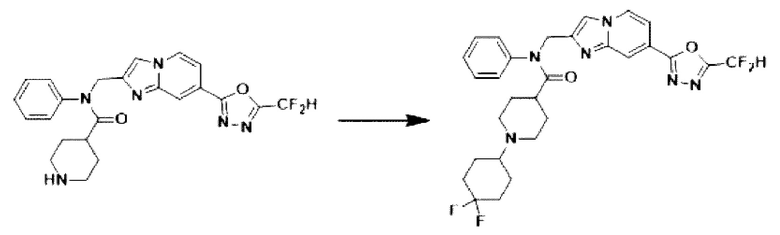
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а] пиридин-2-ил)-метил)-N-фенилпиперидин-4-карбоксамид (0,040 г, 0,088 ммоль), полученный на стадии 3 примера 33, 4,4-дифторциклогексан-1-он (0,024 г, 0,177 ммоль), уксусную кислоту (0,005 мл, 0,088 ммоль) и триацетоксиборгидрид натрия (0,056 г, 0,265 ммоль) растворяли в дихлорметане (1 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном, фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; метанол/дихлорметан = от 0% до 20%) и концентрировали с получением целевого соединения (0,014 г, 2 8,2%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8, 29 (с, 1Н), 8,23 (д, J=7,1 Гц, 1Н), 7,78 (с, 1Н), 7,53 (дд, J=1,8, 7,1 Гц, 1Н), 7,46-7,32 (м, 3H), 7,27-7,19 (м, 2Н), 5,04 (с, 2Н), 3,93 (дт, J=3,5, 7,0 Гц, 1Н), 2,85 (с, 2Н), 2,41-1,51 (м, 15Н);
МСНР (ЭР) m/z 571,4 (М+ +1).
Пример 82: Синтез Соединения 4122, 1-ацетил-N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилпиперидин-4-карбоксамида
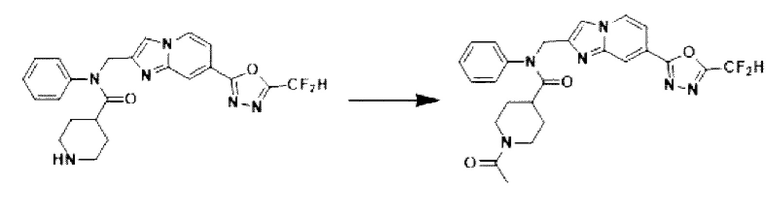
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилпиперидин-4-карбоксамид (0,041 г, 0,091 ммоль), полученный на стадии 3 примера 33, ацетилхлорид (0,031 мл, 0,181 ммоль) и триэтиламин (0,038 мл, 0,272 ммоль) растворяли в дихлорметане (1 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном, фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; метанол/дихлорметан = от 0% до 7%) и концентрировали с получением целевого соединения (0,012 г, 26,6%) в виде твердого вещества белого цвета.
1Н ЯМР (400 МГц, CDCl3) δ 8,30 (с, 1Н), 8,23 (д, J=7,2 Гц, 1Н), 7,77 (с, 1Н), 7,54 (д, J=7,2 Гц, 1Н), 7,41 (дкв, J=7,0, 13,7 Гц, 3H), 7,31-7,22 (м, 3H), 6,95 (т, J=51,7 Гц, 1Н), 5,14-4,95 (м, 2Н), 4,53 (д, J - 13,3 Гц, 1Н), 3,77 (д, J=13,5 Гц, 1Н), 2,84 (т, J - 13,0 Гц, 1Н), 2,49 (д, J=11,1 Гц, 1Н), 2,34 (т, J=12,4 Гц, 1Н), 2,06 (с, 3H), 1,75 (дд, J=14,2, 4 9,2 Гц, 4Н);
МСНР (ЭР) m/z 4 94,9 (М+ +1).
Пример 83: Синтез Соединения 4123, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенил-1-пропионилпиперидин-4-карбоксамида
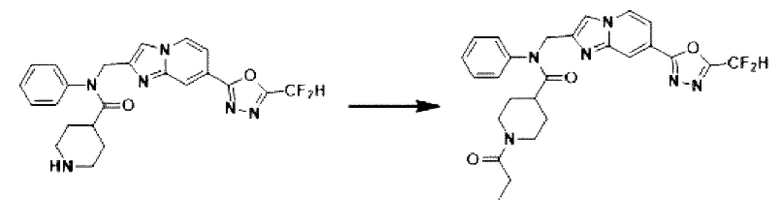
N-((7-(5-(дифторметил)-1,3,4-океадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилпиперидин-4-карбоксамид (0,041 г, 0,091 ммоль), полученный на стадии 3 Примера 33, пропионилхлорид (0,017 г, 0,181 ммоль) и триэтиламин (0,038 мл, 0,272 ммоль) растворяли в дихлорметане (1 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном, фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; метанол/дихлорметан = от 0% до 7%) и концентрировали с получением целевого соединения (0,023 г, 49,0%) в виде твердого вещества оранжевого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,32-8,26 (м, 1Н), 8,23 (дд, J=0,9, 7,1 Гц, 1Н), 7,76 (с, 1Н), 7,53 (дд, J=1,7, 7,1 Гц, 1Н), 7,47-7,33 (м, 3H), 7,29-7,21 (м, 2Н), 6,95 (т, J=51,7 Гц, 1Н), 5,10-4,95 (м, 2Н), 4,55 (д, J=13,4 Гц, 1Н), 3,81 (д, J=13,6 Гц, 1Н), 2, 84-2, 74 (м, 1Н), 2,50 (ддт, J=4,3, 10,3, 15,0 Гц, 1Н), 2,31 (пд, J=4,11, 7,5, 8,6 Гц, 3H), 1,85-1,59 (м, 4Н), 1,12 (т, J=7,5 Гц, 3H);
МСНР (ЭР) m/z 509,2 (М+ +1).
Пример 84: Синтез Соединения 4124, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-1-(2-гидроксиацетил)-N-фенилпиперидин-4-карбоксамида
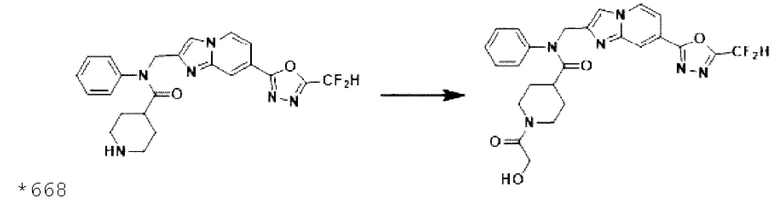
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилпиперидин-4-карбоксамид (0,041 г, 0,091 ммоль), полученный на стадии 3 примера 33,2-гидроксиуксусную кислоту (0,014 г, 0,181 ммоль), триэтиламин (0,038 мл, 0,272 ммоль) и 1-[бис-(диметиламино)-метилен]-1Н-1,2,3-триазоло[4,5-b]пиридиния 3-оксид гексафторфосфат (HATU, 0,052 г, 0,136 ммоль) растворяли в дихлорметане (1 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали воду, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; метанол/дихлорметан = от 0% до 7%) и концентрировали с получением целевого соединения (0,012 г, 25,5%) в виде твердого вещества оранжевого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,33-8,29 (м, 1Н), 8,23 (дд, J=0,9, 7,1 Гц, 1Н), 7,76 (с, 1Н), 7,54 (дд, J=1,7, 7,1 Гц, 1Н), 7,47-7,38 (м, 3H), 7,27 (д, J=1,1 Гц, 3H), 6,95 (т, J=51,7 Гц, 1Н), 5,03 (д, J=2,9 Гц, 2Н), 4,48 (д, J=13,3 Гц, 1Н), 4,18-4,03 (м, 2Н), 3,62 (с, 1Н), 3,47 (д, J=13,7 Гц, 1Н), 2,83-2,72 (м, 1Н), 2,54 (тд, J=4,1, 10,1, 10,5 Гц, 2Н), 1,85-1,64 (м, 4Н);
МСНР (ЭР) m/z 511,3 (М+ +1).
Пример 85: Синтез Соединения 4125, 1-(циклобутанкарбонил)-N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилпиперидин-4-карбоксамида
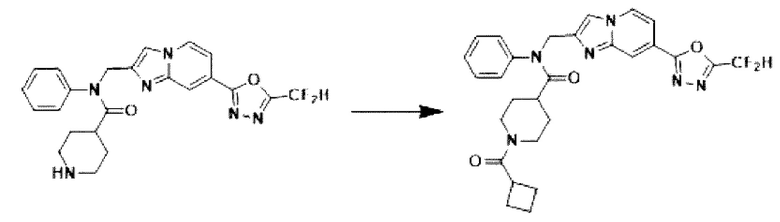
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилпиперидин-4-карбоксамид (0,041 г, 0,091 ммоль), полученный на стадии 3 Примера 33, циклобутанкарбонилхлорид (0,021 г, 0,181 ммоль) и триэтиламин (0,038 мл, 0,272 ммоль) растворяли в дихлорметане (1 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном, фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина, 20×20×1 мм; метанол/дихлорметан = от 0% до 7%) и концентрировали с получением целевого соединения (0,020 г, 4 0,9%) в виде оранжевого геля.
1Н ЯМР (400 МГц, CDCl3) δ 8,30 (дт, J=0,8, 1,7 Гц, 1Н), 8,23 (дд, J=1,0, 7,1 Гц, 1Н), 7,77 (с, 1Н), 7,53 (дд, J=1,7, 7,1 Гц, 1Н), 7,4 6-7,35 (м, 3H), 7,2 8-7,24 (м, 2Н), 6,95 (т, J=51,7 Гц, 1Н), 5,10-4,95 (м, 2Н), 4,52 (д, J=13,6 Гц, 1Н), 3,66 (д, J=13,4 Гц, 1Н), 3,25-3,15 (м, 1Н), 2,71 (т, J=11,7 Гц, 1Н), 2,48 (ддт, J=4,4, 10,4, 15,0 Гц, 1Н), 2,39-2,25 (м, 4Н), 2,19-2,03 (м, 1Н), 1,99-1,79 (м, 2Н), 1,77-1,61 (м, 4Н);
МСНР (ЭР) m/z 535,1 (М+ +1).
Пример 86: Синтез Соединения 4126, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-1-(оксетан-3-карбонил)-N-фенилпиперидин-4-карбоксамида
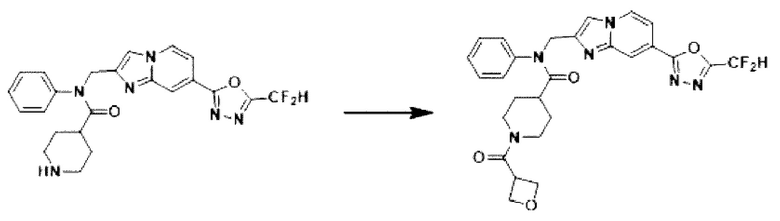
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилпиперидин-4-карбоксамид (0,050 г, 0,111 ммоль), полученный на стадии 3 примера 33, оксетан-3-карбоновую кислоту (0,023 г, 0,221 ммоль), 1-[бис-(диметиламино)-метилен]-1Н-1,2,3-триазоло[4,5-b]пиридиния 3-оксид гексафторфосфат (HATU, 0,063 г, 0,166 ммоль) и триэтиламин (0,043 мл, 0,332 ммоль) растворяли в дихлорметане (1 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном, фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; метанол/дихлорметан = от 0% до 7%) и концентрировали с получением целевого соединения (0,007 г, 12,0%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,31 (с, 1Н), 8,23 (дд, J=1,0, 7,1 Гц, 1Н), 7,76 (с, 1Н), 7,55 (дд, J=1,7, 7,1 Гц, 1Н), 7,48-7,35 (м, 3H), 7,30-7,23 (м, 2Н), 7,02 (д, J=51,7 Гц, 1Н), 5,03 (Д, J=4,3 Гц, 2Н), 4,93 (дд, J - 5,9, 7,2 Гц, 1Н), 4,85 (дд, J=5,9, 7,2 Гц, 1Н), 4,76 (ддд, J - 5,8, 8,7, 10,1 Гц, 2Н), 4,52 (д, J=13,4 Гц, 1Н), 3,96 (тт, J=7,2, 8,7 Гц, 1Н), 3,30 (д, J=13,6, 1Н), 2,78-2,70 (м, 1Н), 2,50 (д, J=4,8, 1Н), 2,46-2,38 (м, 1Н), 1Н);
МСНР (ЭР) m/z 537,2 (М+ +1).
Пример 87: Синтез Соединения 4127, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенил-1-(2,2,2-трифторацетил)-пиперидин-4-карбоксамида
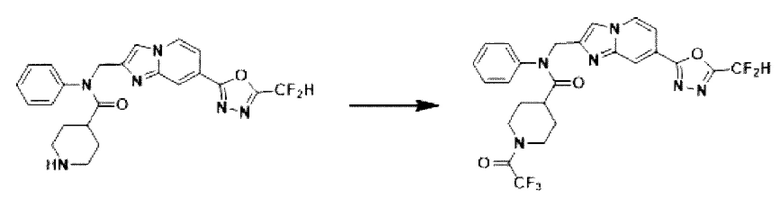
N-((7-(5-(дифторметил)-1,3,4-океадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилпиперидин-4-карбоксамид (0,041 г, 0,091 ммоль), полученный на стадии 3 примера 33, 2,2,2-трифторуксусный ангидрид (0,025 мл, 0,181 ммоль) и триэтиламин (0,038 мл, 0,272 ммоль) растворяли в дихлорметане (1 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном, фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; метанол/дихлорметан = от 0% до 7%) и концентрировали с получением целевого соединения (0,020 г, 4 0,0%) в виде твердого вещества бледно-красного цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,33-8,26 (м, 1Н), 8,23 (дд, J=0,9, 7,1 Гц, 1Н), 7,77 (с, 1Н), 7,54 (дд, J=1,7, 7,1 Гц, 1Н), 7,43 (ддд, J=5,9, 7,9, 10,5 Гц, 3H), 7,28 (д, J=6,2 Гц, 2Н), 6,95 (т, J=51,7 Гц, 1Н), 5,03 (с, 2Н), 4,45-4,33 (м, 1Н), 3,97 (д, J=14,0 Гц, 1Н), 3,01-2,88 (м, 1Н), 2,72-2,53 (м, 2Н), 1,94-1,65 (м, 4Н);
МСНР (ЭР) m/z 549,3 (М+ +1).
Пример 88: Синтез Соединения 4128, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-1-(метилсульфонил)-N-фенилпиперидин-4-карбоксамида
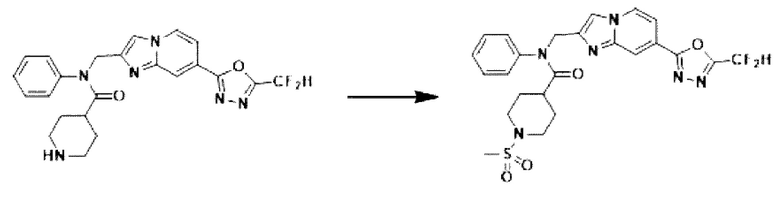
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилпиперидин-4-карбоксамид (0,041 г, 0,091 ммоль), полученный на стадии 3 примера 33, метансульфонилхлорид (0,014 мл, 0,181 ммоль) и триэтиламин
(0,038 мл, 0,272 ммоль) растворяли в дихлорметане (1 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном, фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; метанол/дихлорметан = от 0% до 7%) и концентрировали с получением целевого соединения (0,008 г, 15,6%) в виде твердого вещества белого цвета.
1Н ЯМР (400 МГц, CDCl3) δ 8,32 (с, 1Н), 8,25 (дд, J=1,0, 7,1 Гц, 1Н), 7,78 (с, 1Н), 7,56 (дд, J=1,7, 7,1 Гц, 1Н), 7,48-7,36 (м, 3H), 7,28-7,25 (м, 2Н), 6,95 (т, J=51,7 Гц, 1Н), 5,05 (с, 2Н), 3,74 (дд, J=6,0, 10,1 Гц, 2Н), 2,74 (с, 3H), 2,54 (тд, J=2,9, 11,9 Гц, 2Н), 2,43-2,34 (м, 1Н), 2,00-1,86 (м, 2Н), 1,75 (дд, J=3,6, 13,6 Гц, 2Н);
МСНР (ЭР) m/z 531,1 (М+ +1).
Пример 89: Синтез Соединения 4129, метил-4-(((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-(фенил)-карбамоил)-пиперидин-1-карбоксилата
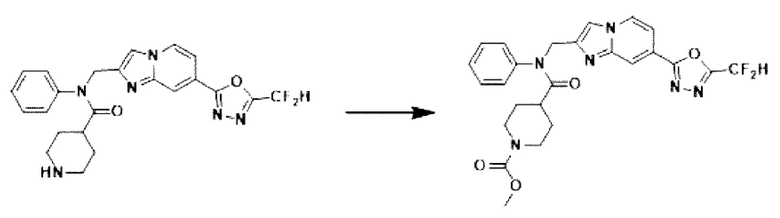
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилпиперидин-4-карбоксамид (0,041 г, 0,091 ммоль), полученный на стадии 3 примера 33, метилхлорформиат (0,017 г, 0,181 ммоль) и триэтиламин (0,025 мл, 0,181 ммоль) растворяли в дихлорметане (1 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали воду, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; метанол/дихлорметан = от 0% до 7%) и концентрировали с получением целевого соединения (0,020 г, 43,7%) в виде твердого вещества бледно-оранжевого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,29 (дт, J=0,9, 1,8 Гц, 1Н), 8,23 (дд, J=0,9, 7,1 Гц, 1Н), 7,77 (с, 1Н), 7,53 (дд, J=1,7, 7,1 Гц, 1Н), 7,4 6-7,35 (м, 3H), 7,2 6-7,23 (м, 2Н), 6,95 (т, J=51,7 Гц, 1Н), 5,03 (с, 2Н), 4,09 (с, 2Н), 3,67 (с, 3H), 2,53 (с, 2Н), 2,43 (тт, J=3,8, 11,2 Гц, 1Н), 1,76 (квд, J=4,4, 11,9, 12,4 Гц, 2Н), 1,62 (д, J=12,8 Гц, 2Н);
МСНР (ЭР) m/z 511,1 (М+ +1).
Пример 90: Синтез Соединения 4130, N4-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N1,N1-диметил-N-фенилпиперидин-1,4-дикарбоксамида
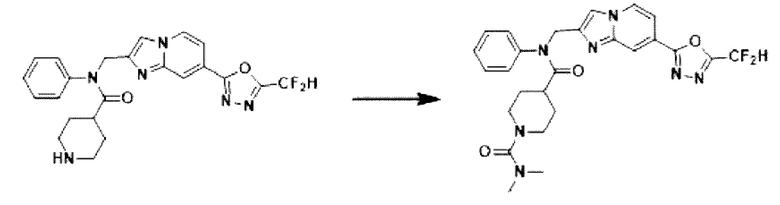
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилпиперидин-4-карбоксамид (0,041 г, 0,091 ммоль), полученный на стадии 3 примера 33, хлорангидрид диметилкарбаминовой кислоты (0,019 г, 0,181 ммоль) и триэтиламин (0, 025 мл, 0,181 ммоль) растворяли в дихлорметане (1 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном, фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; метанол/дихлорметан = от 0% до 7%) и концентрировали с получением целевого соединения (0,024 г, 51,4%) в виде красного геля.
1H ЯМР (400 МГц, CDCl3) δ 8,29 (дт, J=0,8, 1,7 Гц, 1Н), 8,23 (дд, J=1,0, 7,1 Гц, 1Н), 7,77 (с, 1Н), 7,53 (дд, J=1,7, 7,1 Гц, 1Н), 7,40 (дддд, J=2,3, 4,6, 6,8, 11,7 Гц, 3H), 7,27-7,23 (м, 2Н), 6,95 (т, J=51,7 Гц, 1Н), 5,03 (с, 2Н), 3,59 (дд, J=3,8, 13,3 Гц, 2Н), 2,79 (с, 6Н), 2,44 (дтд, J=5,1, 11,5, 12,2, 16,5 Гц, 3H), 1,82 (квд, J=4,0, 12,6 Гц, 2Н), 1,64 (дд, J=3,5, 13,7 Гц, 2Н);
МСНР (ЭР) m/z 524,4 (М+ +1).
Пример 91: Синтез Соединения 4131, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенил-1-(пиридин-2-ил)-пиперидин-4-карбоксамида
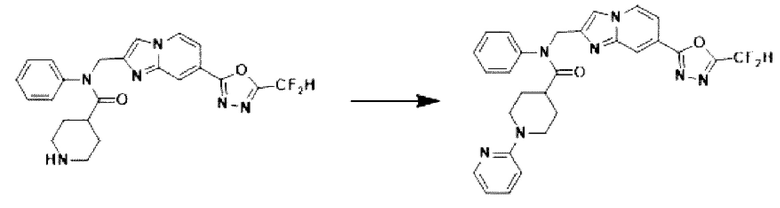
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилпиперидин-4-карбоксамид (0,025 г, 0,055 ммоль), полученный на стадии 3 примера 33, 2-бромпиридин (0,017 г, 0,111 ммоль), карбонат цезия (0,036 г, 0,111 ммоль) и палладий RuPhos G2 (0,002 г, 0,003 ммоль) растворяли в 1,4-диоксане (0,5 мл) при комнатной температуре, и полученный
раствор перемешивали при 120°С в течение 18 часов. Затем температуру понижали до комнатной температуры для завершения реакции. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; метанол/дихлорметан = от 0% до 7%) и концентрировали с получением целевого соединения (0,006 г, 19,1%) в виде твердого вещества желтого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8, 30 (п, J=0,8 Гц, 1Н), 8,23 (дд, J=1,0, 7,1 Гц, 1Н), 8,15 (ддд, J=0,9, 2,0, 4,9 Гц, 1Н), 7,79 (с, 1Н), 7,54 (дд, J=1,7, 7,1 Гц, 1Н), 7,48-7,37 (м, 4Н), 7,31-7,26 (м, 2Н), 6,95 (т, J=51,7 Гц, 1Н), 6, 63-6,55 (м, 2Н), 5,05 (с, 2Н), 4,25 (д, J=13,1 Гц, 2Н), 2,57 (дт, J=11,9, 34,6 Гц, 3H), 1,90 (квд, J=4,2, 12,7 Гц, 2Н), 1,73 (д, J=13,1 Гц, 2Н);
МСНР (ЭР) m/z 530,3 (М+ +1).
Пример 92: Синтез Соединения 4132, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенил-1-(пиримидин-2-ил)-пиперидин-4-карбоксамида
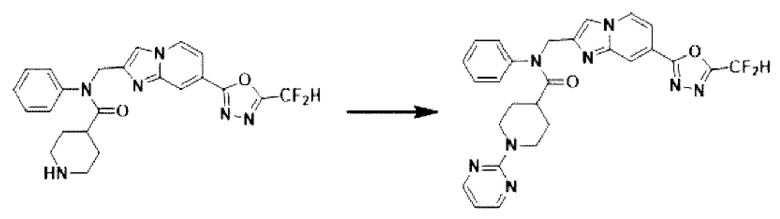
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилпиперидин-4-карбоксамид (0,041 г, 0,091 ммоль), полученный на стадии 3 примера 33, 2-хлорпиримидин (0,021 г, 0,181 ммоль) и карбонат калия (0,038 г, 0,272 ммоль) растворяли в N,N-диметилформамиде (0,5 мл) / ацетонитриле (0,5 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия вливали в концентрат, полученный путем удаления растворителя из реакционной смеси при пониженном давлении, с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; метанол/дихлорметан = от 0% до 7%) и концентрировали с получением целевого соединения (0,022 г, 45,6%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,29-8,25 (м, 3H), 8,22 (дд, J=1,0, 7,1 Гц, 1Н), 7,78 (с, 1Н), 7,52 (дд, J=1,7, 7,1 Гц, 1Н), 7,47-7,36 (м, 3H), 7,29-7,26 (м, 2Н), 6,95 (т, J=51,7 Гц, 1Н), 6,44 (т, J=4,7 Гц, 1Н), 5,04 (с, 2Н), 4,71 (дт, J=3,4, 13,5 Гц, 2Н), 2,70-2,52 (м, 3H), 1,83 (дкв, J=5,8, 7,6, 19,8 Гц, 2Н), 1,70 (дд, J=3,6, 13,9 Гц, 2Н);
МСНР (ЭР) m/z 531,3 (М+ +1).
Пример 93: Синтез Соединения 4137, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-1-метилпиперидин-4-карбоксамида
[Стадия 1] Синтез трет-бутил-4-(хлоркарбонил)-пиперидин-1-карбоксилата

1-(трет-бутоксикарбонил)-пиперидин-4-карбоновую кислоту (1,000 г, 4,361 ммоль), оксалилхлорид (2,00 М сухой раствор в ДХМ, 2,835 мл, 5,670 ммоль) и N,N-диметилформамид (0,034 мл, 0,436 ммоль) растворяли в дихлорметане (25 мл) при 0°С, и полученный раствор перемешивали при комнатной температуре в течение 2 часов. После удаления растворителя из реакционной смеси при пониженном давлении, получали целевое соединение (1,080 г, 100,0%) в виде твердого вещества желтого цвета без дополнительной очистки.
[Стадия 2] Синтез трет-бутил-4-(((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-(3-фторфенил)-карбамоил)-пиперидин-1-карбоксилата
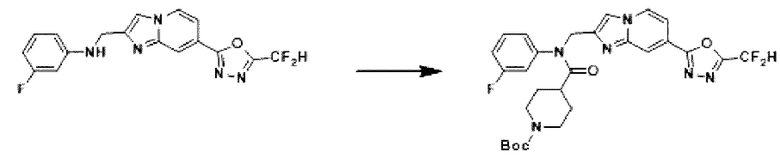
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-3-фторанилин (1,000 г, 2,783 ммоль), полученный в Примере 16, трет-бутил-4-(хлоркарбонил)-пиперидин-1-карбоксилат (1,034 г, 4,175 ммоль) и триэтиламин (1,164 мл, 8,349 ммоль) растворяли в дихлорметане (15 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали водный раствор хлорида аммония с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 24 г; этилацетат/гексан = от 0% до 90%) и концентрировали с получением целевого соединения (0,680 г, 4 2,8%) в виде твердого вещества бледно-оранжевого цвета.
[Стадия 3] Синтез N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперидин-4-карбоксамида
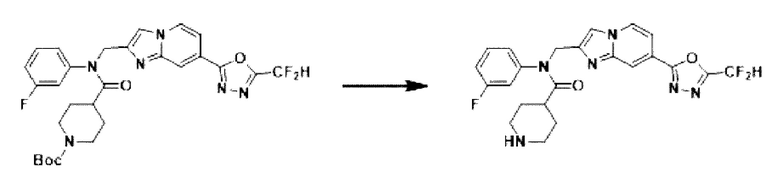
Трет-бутил-4-(((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-(3-фторфенил)-карбамоил)-пиперидин-1-карбоксилат (1000 г, 1,753 ммоль), полученный на стадии 2, и трифторуксусную кислоту (2,684 мл, 35,053 ммоль) растворяли в дихлорметане (30 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 3 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором, сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении, и получали целевое соединение (0,824 г, 99,9%) в виде коричневого геля без дополнительной очистки.
[Стадия 4] Синтез соединения 4137
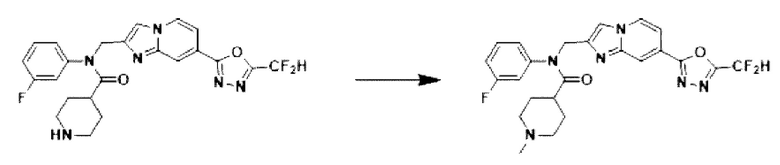
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперидин-4-карбоксамид (0,060 г, 0,128 ммоль), полученный на стадии 3, формальдегид (35,00%, 0,022 г, 0,255 ммоль), уксусную кислоту (0,007 мл, 0,128 ммоль) и триацетоксиборгидрид натрия (0,081 г, 0,383 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и перемешивали полученный раствор при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,024 г, 38,8%) в виде твердого вещества желтого цвета.
1Н ЯМР (700 МГц, CDCl3) δ 8, 29 (д, J=1,7 Гц, 1Н), 8,24 (дд, J=7,1, 1,0 Гц, 1Н), 7,79 (с, 1Н), 7,54 (дд, J=7,1, 1,7 Гц, 1Н), 7,39 (тд, J=8,1, 6,2 Гц, 1Н), 7,12-6,86 (м, 4Н), 5,01 (с, 2Н), 2,92-2,80 (м, 2Н), 2,29-2,16 (м, 5Н), 1,94-1,86 (м, 2Н), 1,82-1,72 (м, 3H);
МСНР (ЭР) m/z 485,3 (М+ +1).
Пример 94: Синтез Соединения 4138, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-1-этил-N-(3-фторфенил)-пиперидин-4-карбоксамида
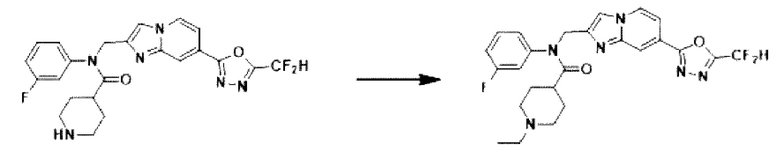
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперидин-4-карбоксамид (0,060 г, 0,128 ммоль), полученный на стадии 3 примера 93, ацетальдегид (0,011 г, 0,255 ммоль), уксусную кислоту (0,007 мл, 0,128 ммоль) и триацетоксиборгидрид натрия (0,081 г, 0,383 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и перемешивали полученный раствор при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,031 г, 48,8%) в виде твердого вещества желтого цвета.
1Н ЯМР (400 МГц, CDCl3) δ 8,30 (дт, J=1,7, 0,8 Гц, 1Н), 8,24 (ддд, J=7,1, 2,9, 1,0 Гц, 1Н), 7,78 (д, J=8,3 Гц, 1Н), 7,55 (дд, J=7,2, 1,7 Гц, 1Н), 7,40 (тдд, J=8,2, 6,3, 1,8 Гц, 1Н), 7,13-6,80 (м, 4Н), 5,02 (с, 2Н), 3,00 (с, 1Н), 2,50-2,21 (м, 3H), 1, 9 9-1, 62 (м, 7Н), 1, 12 (с, 3H); МСНР (ЭР) m/z 499,3 (М+ +1).
Пример 95: Синтез Соединения 4139, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-1-изопропилпиперидин-4-карбоксамида
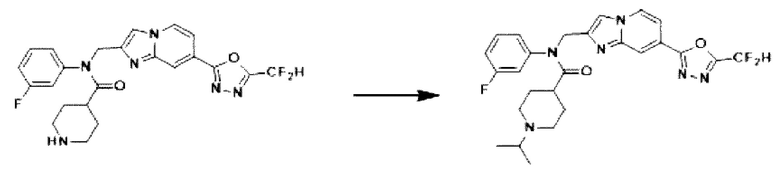
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперидин-4-карбоксамид (0,060 г, 0,128 ммоль), полученный на стадии 3 примера 93, пропан-2-он (0,015 г, 0,255 ммоль), уксусную кислоту (0,007 мл, 0,128 ммоль) и триацетоксиборгидрид натрия (0,081 г, 0,383 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и перемешивали полученный раствор при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,018 г, 27,5%) в виде твердого вещества желтого цвета.
1Н ЯМР (700 МГц, CDCl3) δ 8, 30 (д, J=1,6 Гц, 1Н), 8,24 (дд, J=7,1, 1,0 Гц, 1Н), 7,78 (с, 1Н), 7,55 (дд, J=7,1, 1,7 Гц, 1Н), 7,39 (тд, J=8,1, 6,3 Гц, 1Н), 7,13-6,85 (м, 4Н), 5,02 (с, 2Н), 2,86 (с, 2Н), 2,68 (с, 1Н), 2,29-2,19 (м, 1Н), 1,91 (с, 4Н), 1,66 (с, 2Н), 1,00 (с, 6Н);
МСНР (ЭР) m/z 512,9 (М+ +1).
Пример 96: Синтез Соединения 4140, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-1-(1-гидроксипропан-2-ил)-пиперидин-4-карбоксамида
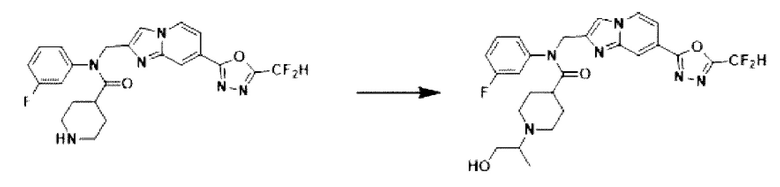
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперидин-4-карбоксамид (0,060 г, 0,128 ммоль), полученный на стадии 3 примера 93, 1-гидроксипропан-2-он (0,019 г, 0,255 ммоль), уксусную кислоту (0,007 мл, 0,128 ммоль) и триацетоксиборгидрид натрия (0,081 г, 0,383 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и перемешивали полученный раствор при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,019 г, 28,2%) в виде твердого вещества желтого цвета.
1H ЯМР (700 МГц, CDCl3) δ 8,31 (дт, J=1,7, 0,8 Гц, 1Н), 8,25 (дд, J=7,1, 1,0 Гц, 1Н), 7,78 (с, 1Н), 7,56 (дд, J=7,1, 1,7 Гц, 1Н), 7,41 (тд, J=8,1, 6,3 Гц, 1Н), 7,12 (дт, J=8,3, 4,6 Гц, 1Н), 7,0 9-7,04 (м, 2Н), 7,04-6,8 8 (м, 1Н), 5,02 (с, 2Н), 3,40 (д, J=64,6 Гц, 3H), 2,98-2,69 (м, 3H), 1,96 (д, J=11,2 Гц, 2Н), 1,84 (с, 2Н), 0, 93-0, 8 0 (м, 5Н);
МСНР (ЭР) m/z 529,3 (М+ +1).
Пример 97: Синтез Соединения 4141, 1-циклобутил-N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперидин-4-карбоксамида
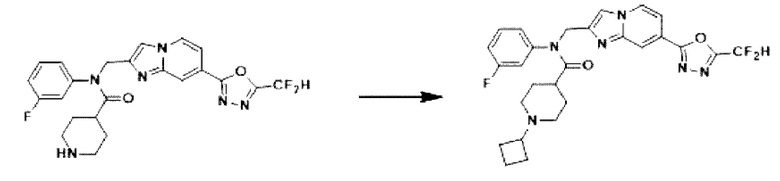
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперидин-4-карбоксамид (0,060 г, 0,128 ммоль), полученный на стадии 3 примера 93, циклобутанон (0,018 г, 0,255 ммоль), уксусную кислоту (0,007 мл, 0,128 ммоль) и триацетоксиборгидрид натрия (0,081 г, 0,383 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и перемешивали полученный раствор при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,022 г, 32,9%) в виде твердого вещества желтого цвета.
1H ЯМР (700 МГц, CDCl3) δ 8,29 (д, J=1,6 Гц, 1Н), 8,24 (дд, J=7,1, 1,0 Гц, 1Н), 7,78 (с, 1Н), 7,55 (дд, J=7,1, 1,7 Гц, 1Н), 7,39 (тд, J=8,1, 6,3 Гц, 1Н), 7,10 (дт, J=8,7, 4,7 Гц, 1Н), 7,07-6,87 (м, 3H), 5,02 (с, 2Н), 2,87 (с, 2Н), 2,61 (с, 1Н), 2,30-2,19 (м, 1Н), 1,98 (с, 2Н), 1,90 (д, J=10,9 Гц, 4Н), 1,65 (с, 4Н), 1,56-1,44 (м, 2Н);
МСНР (ЭР) m/z 525,2 (М+ +1).
Пример 98: Синтез Соединения 4142, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-1-(оксетан-3-ил)-пиперидин-4-карбоксамида
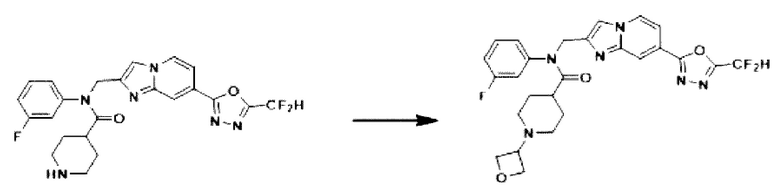
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперидин-4-карбоксамид (0,060 г, 0,128 ммоль), полученный на стадии 3 примера 93, оксетан-3-он (0,018 г, 0,255 ммоль), уксусную кислоту (0,007 мл, 0,128 ммоль) и триацетоксиборгидрид натрия (0,081 г, 0,383 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и перемешивали полученный раствор при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,032 г, 47,7%) в виде твердого вещества желтого цвета.
1H ЯМР (700 МГц, CDCl3) δ 8,30 (дд, J=1,8, 0,9 Гц, 1Н), 8,24 (дд, J=7,1, 1,0 Гц, 1Н), 7,78 (с, 1Н), 7,55 (дд, J=7,1, 1,7 Гц, 1Н), 7,40 (тд, J=8,1, 6,3 Гц, 1Н), 7,15-7,09 (м, 1Н), 7,09-6,84 (м, 3H), 5,02 (с, 2Н), 4,61 (с, 4Н), 3,40 (с, 1Н), 2,73 (с, 2Н), 2,34-2,21 (м, 1Н), 1,93 (д, J=12,2 Гц, 2Н), 1,66 (с, 4Н);
МСНР (ЭР) m/z 527,0 (М+ +1).
Пример 99: Синтез Соединения 4143, 1-циклогексил-N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперидин-4-карбоксамида
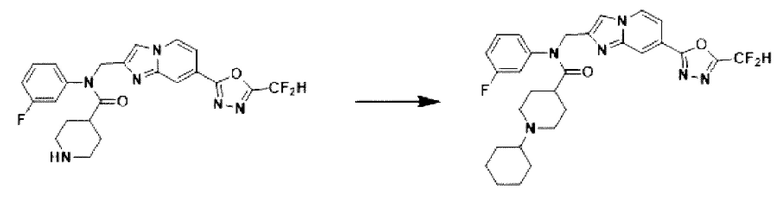
N-((7-(5-(дифторметил)-1,3,4-океадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперидин-4-карбоксамид (0,060 г, 0,128 ммоль), полученный на стадии 3 примера 93, циклогексанон (0,025 г, 0,255 ммоль), уксусную кислоту (0,007 мл, 0,128 ммоль) и триацетоксиборгидрид натрия (0,081 г, 0,383 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и перемешивали полученный раствор при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,014 г, 19,9%) в виде твердого вещества желтого цвета.
1H ЯМР (700 МГц, CDCl3) δ 8,32-8,26 (м, 1Н), 8,23 (дд, J=7,1, 1,0 Гц, 1Н), 7,78 (с, 1Н), 7,53 (дд, J=7,1, 1,7 Гц, 1Н), 7,38 (тд, J=8,1, 6,3 Гц, 1Н), 7,13-6,85 (м, 4Н), 5,01 (с, 2Н), 2,92 (с, 2Н), 2,35-2,19 (м, 2Н), 2,10-1,95 (м, 2Н), 1,92-1,73 (м, 8Н), 1,61 (д, J=13,1 Гц, 2Н), 1,21 (д, J=14,6 Гц, 4Н); МСНР (ЭР) m/z 553,1 (М++1).
Пример 100: Синтез Соединения 4144, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-1-(тетрагидро-2Н-пиран-4-ил)-пиперидин-4-карбоксамида
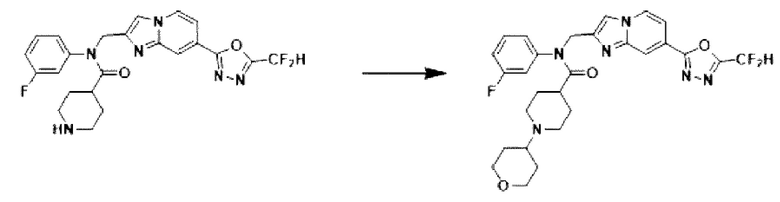
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперидин-4-карбоксамид (0,060 г, 0,128 ммоль), полученный на стадии 3 примера 93, тетрагидро-4Н-пиран-4-он (0,026 г, 0,255 ммоль), уксусную кислоту (0,007 мл, 0,128 ммоль) и триацетоксиборгидрид натрия (0,081 г, 0,383 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и перемешивали полученный раствор при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,016 г, 22,6%) в виде твердого вещества желтого цвета.
1H ЯМР (700 МГц, CDCl3) δ 8,33-8,27 (м, 1Н), 8,24 (дд, J=7,1, 1,0 Гц, 1Н), 7,83-7,74 (м, 1Н), 7,55 (дд, J=7,1, 1,7 Гц, 1Н), 7,40 (тд, J=8,1, 6,3 Гц, 1Н), 7,10 (ддд, J=8,4, 5,5, 2,2 Гц, 1Н), 7,09-6,86 (м, 3Н), 5,02 (с, 2Н), 4,04-3,95 (м, 2Н), 3,35 (т, J=11,7 Гц, 2Н), 2,93 (с, 2Н), 2,45 (д, J=22,4 Гц, 1Н), 2,31-2,20 (м, 1Н), 1,89 (д, J=10,6 Гц, 4Н), 1,67 (с, 4Н), 1,56 (д, J=11,5 Гц, 2Н);
МСНР (ЭР) m/z 555,3 (М++1).
Пример 101: Синтез Соединения 4145, 1-(4,4-дифторциклогексил)-N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперидин-4-карбоксамида
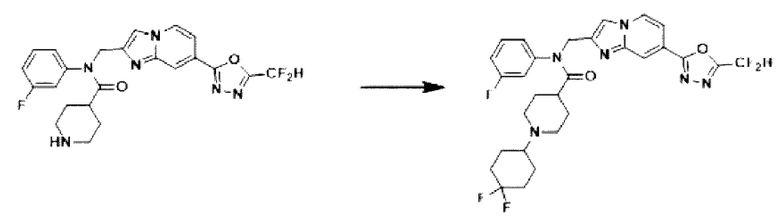
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперидин-4-карбоксамид (0,060 г, 0,128 ммоль), полученный на стадии 3 примера 93, 4,4-дифторциклогексан-1-он (0,034 г, 0,255 ммоль), уксусную кислоту (0,007 мл, 0,128 ммоль) и триацетоксиборгидрид натрия (0,081 г, 0,383 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и перемешивали полученный раствор при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,026 г, 34,6%) в виде твердого вещества желтого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,31-8,27 (м, 1Н), 8,23 (дд, J=7,1, 1,0 Гц, 1Н), 7,78 (с, 1Н), 7,53 (дд, J=7,1, 1,7 Гц, 1Н), 7,39 (тд, J=8,1, 6,3 Гц, 1Н), 7,15-6,77 (м, 4Н), 5,01 (с, 2Н), 2,87 (с, 2Н), 2,36 (с, 1Н), 2,30-2,21 (м, 1Н), 2,10 (д, J=11,6 Гц, 2Н), 1,9 6 (с, 2Н), 1, 92-1, 54 (м, 1Н);
МСНР (ЭР) m/z 589,2 (М++1).
Пример 102: Синтез Соединения 4146, 1-ацетил-N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперидин-4-карбоксамида
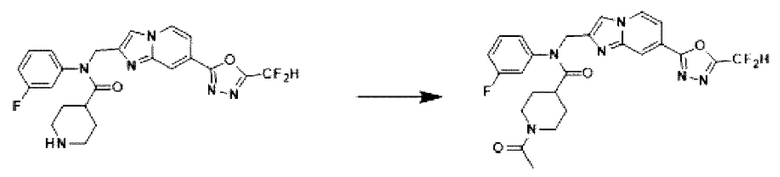
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-a]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперидин-4-карбоксамид (0,060 г, 0,128 ммоль), полученный на стадии 3 примера 93, ацетилхлорид (0,018 мл, 0,255 ммоль) и триэтиламин (0,053 мл, 0,383 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор хлорида аммония, экстрагировали дихлорметаном, фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,021 г, 32,1%) в виде твердого вещества желтого цвета.
1H ЯМР (700 МГц, CDCl3) δ 8, 46 (с, 1Н), 8, 34-8, 26 (м, 1Н), 7,82 (с, 1Н), 7,69 (дд, J=7,1, 1,7 Гц, 1Н), 7,45 (тд, J=8,2, 6,3 Гц, 1Н), 7,17-7,13 (м, 2Н), 7,12-7,09 (м, 1Н), 6,97 (т, J=51,7 Гц, 1Н), 5,06 (с, 2Н), 4,55 (д, J=13,6 Гц, 1Н), 3,80 (д, J=13,6 Гц, 1Н), 2,88 (т, J=13,8 Гц, 1Н), 2,53 (д, J=11,2 Гц, 1Н), 2, 4 4-2, 34 (м, 1Н), 2,07 (с, 3Н), 1, 84-1, 63 (м, 4Н);
МСНР (ЭР) m/z 513,0 (М++1).
Пример 103: Синтез Соединения 4147, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-1-пропионилпиперидин-4-карбоксамида
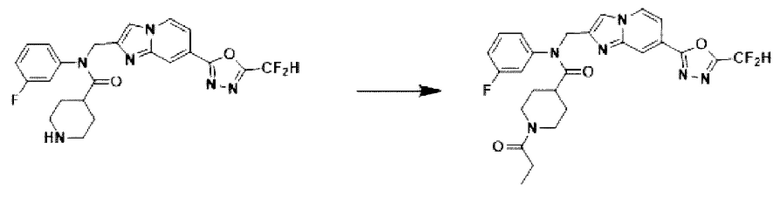
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперидин-4-карбоксамид (0,060 г, 0,128 ммоль), полученный на стадии 3 Примера 93, пропионилхлорид (0,024 г, 0,255 ммоль) и триэтиламин (0, 053 мл, 0,383 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор хлорида аммония, экстрагировали дихлорметаном, фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,010 г, 14,9%) в виде твердого вещества желтого цвета.
1H ЯМР (700 МГц, CDCl3) δ 8,33 (с, 1Н), 8,25 (дд, J=7,1, 1,0 Гц, 1Н), 7,78 (с, 1Н), 7,57 (дд, J=7,2, 1,6 Гц, 1Н), 7,42 (тд, J=8,1, 6,2 Гц, 1Н), 7,16-7,05 (м, 3Н), 6,96 (т, J=51,7 Гц, 1Н), 5,08-4,94 (м, 2Н), 4,57 (дд, J=11,2, 6,8 Гц, 1Н), 3,83 (д, J=13,7 Гц, 1Н), 2,88-2,74 (м, 1Н), 2,52 (тд, J=11,8, 11,0, 5,7 Гц, 1Н), 2,33 (дддд, J=23,0, 17,7, 15,3, 10,0 Гц, 4Н), 1,83-1,74 (м, 1Н), 1,74-1,61 (м, 2Н), 1,13 (т, J=7,5, 3Н);
МСНР (ЭР) m/z 527,0 (М++1).
Пример 104: Синтез Соединения 4149, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-1-(2-гидроксиацетил)-пиперидин-4-карбоксамида
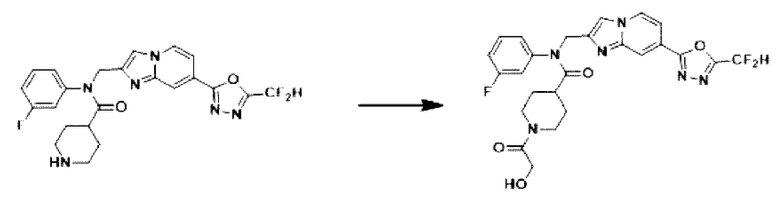
N-((7-(5-(дифторметил)-1,3,4-океадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперидин-4-карбоксамид (0,060 г, 0,128 ммоль), полученный на стадии 3 Примера 93, 2-гидроксиацетилхлорид (0,024 г, 0,255 ммоль), 1-[бис-(диметиламино)-метилен]-1Н-1,2,3-триазоло[4,5-b]пиридиния 3-оксид гексафторфосфат (HATU, 0,097 г, 0,255 ммоль) и N,N-диизопропилэтиламин (0,044 мл, 0,255 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор хлорида аммония, экстрагировали дихлорметаном, фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,017 г, 25,2%) в виде твердого вещества желтого цвета.
1Н ЯМР (400 МГц, CDCl3) δ 8,31 (дт, J=1,7, 0,9 Гц, 1Н), 8,24 (дд, J=7,1, 1,0 Гц, 1Н), 7,76 (с, 1Н), 7,55 (дд, J=7,1, 1,7 Гц, 1Н), 7,42 (тд, J=8,3, 6,3 Гц, 1Н), 7,16-6,78 (м, 4Н), 5,01 (с, 2Н), 4,48 (д, J=13,7 Гц, 1Н), 4,18-4,01 (м, 2Н), 3,48 (д, J=13,7 Гц, 1Н), 2, 86-2,73 (м, 1Н), 2, 63-2, 44 (м, 2Н), 1,84-1,62 (м, 4Н);
МСНР (ЭР) m/z 529,0 (М++1).
Пример 105: Синтез Соединения 4150, 1-(циклобутанкарбонил)-N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперидин-4-карбоксамида
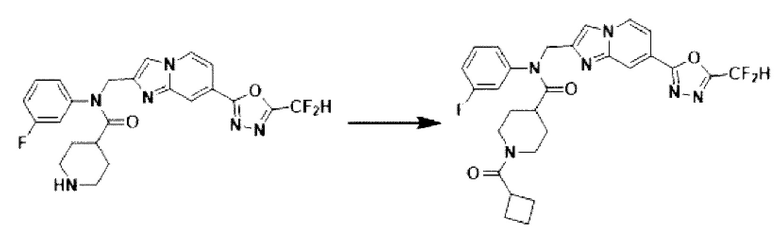
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-a]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперидин-4-сульфонамид (0,050 г, 0,099 ммоль), полученный на стадии 3 примера 93, циклобутанкарбонилхлорид (0,023 г, 0,197 ммоль) и триэтиламин (0,041 мл, 0,296 ммоль) растворяли в дихлорметане (1 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном, фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; метанол/дихлорметан = 0% до 7%) и концентрировали, а затем полученный продукт снова очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; метанол/1% водный раствор дихлорметана = 0% до 7%) и концентрировали с получением целевого соединения (0,011 г, 20,0%) в виде желтого геля.
1H ЯМР (400 МГц, CDCl3) δ 8,31 (д, J=1,6 Гц, 1Н), 8,24 (дд, J=1,0, 7,1 Гц, 1Н), 7,77 (с, 1Н), 7,55 (дт, J=1,2, 7,0 Гц, 1Н), 7,41 (тд, J=6,3, 8,3 Гц, 1Н), 7,16-6,80 (м, 4Н), 5,01 (д, J=3,4 Гц, 2Н), 4,53 (д, J=13,3 Гц, 1Н), 3,68 (д, J=13,6 Гц, 1Н), 3,21 (п, J=8,5 Гц, 1Н), 2,74 (т, J=12,7 Гц, 1Н), 2,48 (д, J=12,2 Гц, 1Н), 2,34 (ддд, J=9,3, 11,8, 20,7 Гц, 3Н), 2,12 (дд, J=3,6, 8,2, 11,8 Гц, 2Н), 2,00-1,59 (м, 6Н);
МСНР (ЭР) m/z 553,4 (М++1).
Пример 106: Синтез Соединения 4151, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-1-(оксетан-3-карбонил)-пиперидин-4-карбоксамида
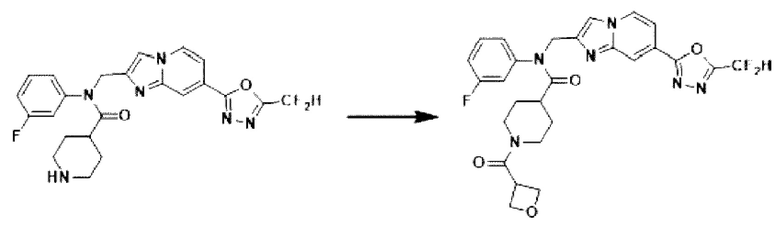
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперидин-4-карбоксамид (0,050 г, 0,106 ммоль), полученный на стадии 3 примера 93, оксетан-3-карбоновую кислоту (0,022 г, 0,213 ммоль), 1-[бис-(диметиламино)-метилен]-1Н-1,2,3-триазоло[4,5-b]пиридиния 3-оксида гексафторфосфат (HATU, 0,061 г, 0,159 ммоль) и триэтиламин (0,044 мл, 0,319 ммоль) растворяли в дихлорметане (1 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном, фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; метанол/дихлорметан = 0% до 7%) и концентрировали, а затем полученный продукт снова очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; метанол/1% водный раствор дихлорметана = 0% до 7%) и концентрировали с получением целевого соединения (0,016 г, 27,3%) в виде твердого вещества желтого цвета.
1Н ЯМР (400 МГц, CDCl3) δ 8,34-8,28 (м, 1Н), 8,24 (дд, J=1,0, 7,1 Гц, 1Н), 7,76 (с, 1Н), 7,56 (дд, J=1,7, 7,2 Гц, 1Н), 7,42 (тд, J=6,2, 8,2 Гц, 1Н), 7,16-6,82 (м, 4Н), 5,01 (д, J=1,6 Гц, 2Н), 4,93 (дд, J=5,9, 7,2 Гц, 1Н), 4,86 (дд, J=5,9, 7,2 Гц, 1Н), 4,77 (тд, J=5,9, 8,6 Гц, 2Н), 4,53 (д, J=13,4 Гц, 1Н), 4,01-3,91 (м, 1Н), 3,31 (д, J=13,5 Гц, 1Н), 2,77 (т, J=11,9, 1Н), 2,56-2,38 (м, 2Н), 1,71 (д, J=18,8 Гц, 4Н);
МСНР (ЭР) m/z 555,4 (М++1).
Пример 107: Синтез Соединения 4152, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-1-(2,2,2-трифторацетил)-пиперидин-4-карбоксамида
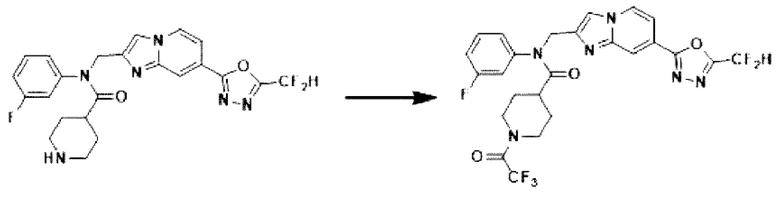
N-((7-(5-(дифторметил)-1,3,4-океадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперидин-4-карбоксамид (0,050 г, 0,106 ммоль), полученный на стадии 3 примера 93, 2,2,2-трифторуксусный ангидрид (0,045 г, 0,213 ммоль) и триэтиламин (0,044 мл, 0,319 ммоль) растворяли в дихлорметане (1 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном, фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; метанол/дихлорметан = от 0% до 7%) и концентрировали с получением целевого соединения (0,020 г, 33,6%) в виде твердого вещества коричневого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,33-8,28 (м, 1Н), 8,24 (дд, J=1,0, 7,1 Гц, 1Н), 7,77 (с, 1Н), 7,56 (дд, J=1,7, 7,2 Гц, 1Н), 7,43 (тд, J=6,3, 8,3 Гц, 1Н), 7,17-6,77 (м, 4Н), 5,01 (с, 2Н), 4,46-4,34 (м, 1Н), 3,98 (д, J=14,0 Гц, 1Н), 3,06-2,94 (м, 1Н), 2,74-2,53 (м, 1Н), 1,94-1,68 (м, 4Н);
МСНР (ЭР) m/z 566,7 (М++1).
Пример 108: Синтез Соединения 4153, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-1-(метилсульфонил)-пиперидин-4-карбоксамида
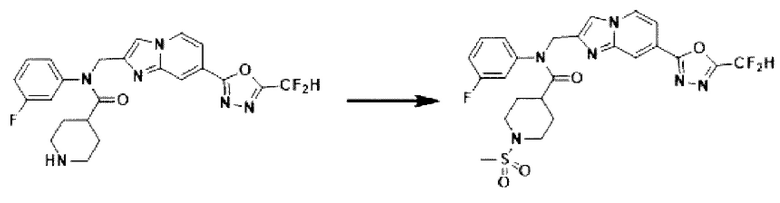
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперидин-4-карбоксамид (0,050 г, 0,106 ммоль), полученный на стадии 3 примера 93, метансульфонилхлорид (0,016 мл, 0,213 ммоль) и триэтиламин (0,044 мл, 0,319 ммоль) растворяли в дихлорметане (1 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном, фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; метанол/дихлорметан = от 0% до 7%) и концентрировали с получением целевого соединения (0,005 г, 8,7%) в виде твердого вещества коричневого цвета.
1Н ЯМР (400 МГц, CDCl3) δ 8,32 (д, J=1,5 Гц, 1Н), 8,25 (дд, J=1,0, 7,2 Гц, 1Н), 7,78 (с, 1Н), 7,57 (дд, J=1,7, 7,1 Гц, 1Н), 7,46-7,39 (м, 1Н), 7,16-6,77 (м, 4Н), 5,02 (с, 2Н), 3,76 (д, J=12,3 Гц, 2Н), 2,75 (с, 3Н), 2,57 (т, J=11,6 Гц, 2Н), 2,40 (д, J=11,4 Гц, 1Н), 2,01-1,84 (м, 2Н), 1,75 (д, J=13,5 Гц, 2Н);
МСНР (ЭР) m/z 549,4 (М++1).
Пример 109: Синтез Соединения 4154, метил-4-(((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-(3-фторфенил)-карбамоил)-пиперидин-1-карбоксилата
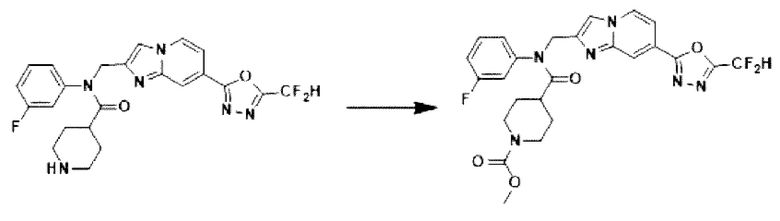
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперидин-4-карбоксамид (0,050 г, 0,106 ммоль), полученный на стадии 3 примера 93, метилхлорформиат (0,020 г, 0,213 ммоль) и триэтиламин (0,044 мл, 0,319 ммоль) растворяли в дихлорметане (1 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном, фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; метанол/дихлорметан = от 0% до 7%) и концентрировали с получением целевого соединения (0,021 г, 35,2%) в виде твердого вещества красного цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,32-8,27 (м, 1Н), 8,23 (дд, J=1,0, 7,2 Гц, 1Н), 7,77 (с, 1Н), 7,54 (дд, J=1,7, 7,1 Гц, 1Н), 7,40 (тд, J=6,4, 8,3 Гц, 1Н), 7,16-6,80 (м, 4Н), 5,01 (с, 2Н), 4,10 (с, 2Н), 3,67 (с, 3Н), 2,65-2,37 (м, 3Н), 1,75 (квд, J=4,4, 11,9, 12,4 Гц, 2Н), 1,62 (д, J=13,1 Гц, 2Н);
МСНР (ЭР) m/z 529,1 (М++1).
Пример 110: Синтез Соединения 4155, N4-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N4-(3-фторфенил)-N1,N1-диметилпиперидин-1,4-дикарбоксамида
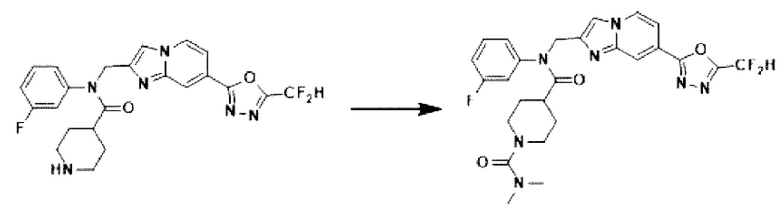
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперидин-4-карбоксамид (0,050 г, 0,106 ммоль), полученный на стадии 3 примера 93, хлорангидрид диметилкарбаминовой кислоты (0,023 г, 0,213 ммоль) и триэтиламин (0,044 мл, 0,319 ммоль) растворяли в дихлорметане (1 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном, фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; метанол/дихлорметан = от 0% до 7%) и концентрировали с получением целевого соединения (0,019 г, 33,5%) в виде твердого вещества бледно-красного цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,32-8,28 (м, 1Н), 8,24 (дд, J=1,0, 7,1 Гц, 1Н), 7,78 (с, 1Н), 7,55 (дд, J=1,7, 7,1 Гц, 1Н), 7,41 (тд, J=6,4, 8,3 Гц, 1Н), 7,15-6,81 (м, 4Н), 5,02 (с, 2Н), 3,61 (д, J=13,2 Гц, 2Н), 2,80 (с, 6Н), 2,57-2,37 (м, 3Н), 1,82 (квд, J=4,0, 12,6 Гц, 2Н), 1,64 (д, J=12,8 Гц, 2Н);
МСНР (ЭР) m/z 542,0 (М++1).
Пример 111: Синтез Соединения 4156, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-1-(пиридин-2-ил)-пиперидин-4-карбоксамида
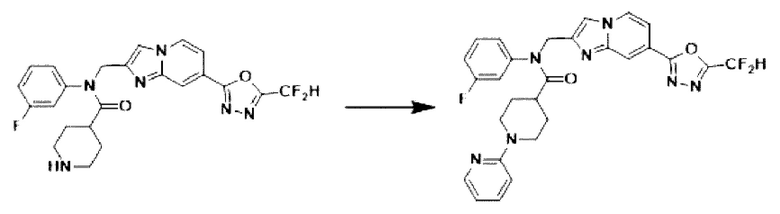
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперидин-4-карбоксамид (0,050 г, 0,106 ммоль), полученный на стадии 3 примера 93, 2-бромпиридин (0,034 г, 0,213 ммоль), RuPhos палладий G2 (0,004 г, 0,005 ммоль) и карбонат цезия (0,069 г, 0,213 ммоль) растворяли в 1,4-диоксане (1 мл) при комнатной температуре, и полученный раствор перемешивали при 120°С в течение 18 часов. Затем температуру понижали до комнатной температуры для завершения реакции. Насыщенный водный раствор гидрокарбоната натрия вливали в концентрат, полученный путем удаления растворителя из реакционной смеси при пониженном давлении, с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; метанол/дихлорметан = от 0% до 7%) и концентрировали с получением целевого соединения (0,013 г, 22,2%) в виде твердого вещества коричневого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8, 33-8, 29 (м, 1Н), 8,23 (дд, J=1,0, 7,1 Гц, 1Н), 8,16 (ддд, J=0,8, 2,0, 5,0 Гц, 1Н), 7,79 (с, 1Н), 7,55 (дд, J=1,7, 7,1 Гц, 1Н), 7,48-7,36 (м, 2Н), 7,16-6,80 (м, 4Н), 6,65-6,55 (м, 2Н), 5,03 (с, 2Н), 4,26 (д, J=13,5 Гц, 2Н), 2,65 (т, J=12,6 Гц, 2Н), 2,53 (с, 1Н), 1,89 (квд, J=4,1, 12,5 Гц, 2Н), 1,72 (д, J=13,1 Гц, 2Н);
МСНР (ЭР) m/z 548,2 (М++1).
Пример 112: Синтез Соединения 4157, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-1-(пиримидин-2-ил)-пиперидин-4-карбоксамида
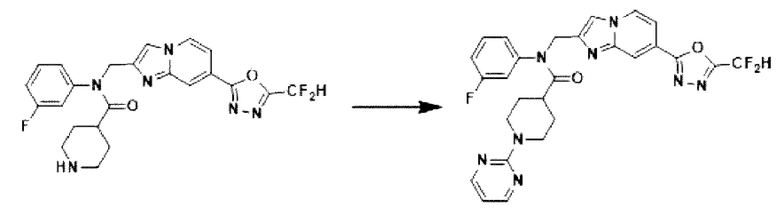
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперидин-4-карбоксамид (0,050 г, 0,106 ммоль), полученный на стадии 3 примера 93, 2-хлорпиримидин (0,02 4 г, 0,213 ммоль) и карбонат калия (0,04 4 г, 0,319 ммоль) растворяли в N,N-диметилформамиде (0,5 мл)/ацетонитриле (0,5 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В концентрат, полученный путем удаления растворителя из реакционной смеси при пониженном давлении, вливали насыщенный водный раствор гидрокарбоната натрия с последующей экстракцией дихлорметаном. Затем полученный продукт фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. К концентрату добавляли дихлорметан (3 мл), затем перемешивали, осажденное твердое вещество фильтровали, промывали дихлорметаном и сушили с получением целевого соединения (0,018 г, 30,5%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,31-8,26 (м, 3Н), 8,23 (дд, J=0,9, 7,1 Гц, 1Н), 7,78 (с, 1Н), 7,54 (дд, J=1,7, 7,1 Гц, 1Н), 7,43 (тд, J=6,5, 8,3 Гц, 1Н), 7,15-6,81 (м, 4Н), 6,46 (т, J=4,7 Гц, 1Н), 5,02 (с, 2Н), 4,73 (дт, J=3,5, 13,4 Гц, 2Н), 2,69 (т, J=13,0 Гц, 2Н), 2,56 (д, J=11,5 Гц, 1Н), 1,83 (квд, J=4,2, 12,0, 12,5 Гц, 2Н), 1,71 (д, J=13,0 Гц, 2Н);
МСНР (ЭР) m/z 549,4 (М++1).
Пример 113: Синтез Соединения 4158, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-1-метил-N-фенилазетидин-3-карбоксамида
[Стадия 1] Синтез трет-бутил-3-(хлоркарбонил)-азетидин-1-карбоксилата
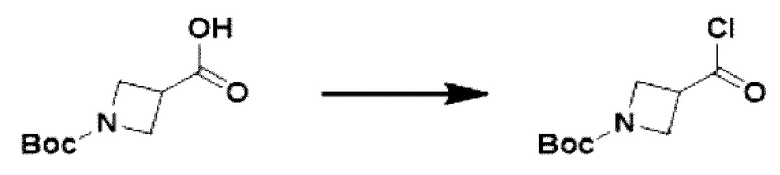
1-(трет-бутоксикарбонил)-азетидин-3-карбоновую кислоту (1,200 г, 5,964 ммоль) растворяли в дихлорметане (15 0 мл) и добавляли оксалилхлорид (2,00 М раствор в ДХМ, 3,578 мл, 7,156 ммоль) и N,N-диметилформамид (0, 046 мл, 0, 596 ммоль) при 0°C, и перемешивали при комнатной температуре в течение 2 часов. После удаления растворителя из реакционной смеси при пониженном давлении, получали целевое соединение (1,250 г, 95,4%) в виде бесцветного масла без дополнительной очистки.
[Стадия 2] Синтез трет-бутил-3-(((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-(фенил)-карбамоил)-азетидин-1-карбоксилата
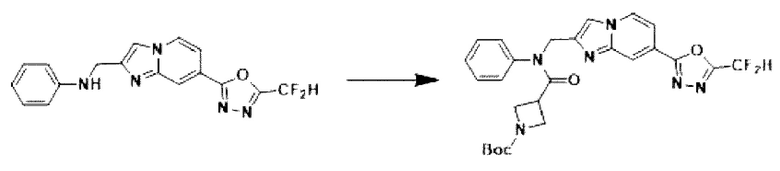
К раствору, в котором растворяли N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-анилин (1,500 г, 4,395 ммоль), полученный в Примере 14, и триэтиламин (1,838 мл, 13,184 ммоль) в дихлорметане (150 мл) при комнатной температуре, добавляли трет-бутил-3-(хлоркарбонил)-азетидин-1-карбоксилат (1,255 г, 5,713 ммоль) и перемешивали при той же температуре в течение 16 часов. В реакционную смесь вливали насыщенный водный раствор хлорида аммония, экстрагировали дихлорметаном, фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 4 0 г; этил ацетат/гексан = от 5% до 60%) и концентрировали с получением целевого соединения (1,600 г, 69,4%) в виде твердого вещества бежевого цвета.
[Стадия 3] Синтез N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилазетидин-3-карбоксамида
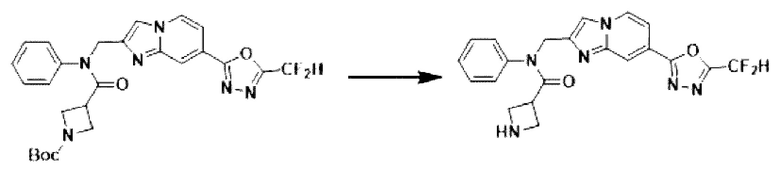
Трет-бутил-3-(((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-(фенил)-карбамоил)-азетидин-1-карбоксилат (0.600 г, 1.144 ммоль), полученный на стадии 2, и трифторуксусную кислоту (1.752 мл, 22.87 8 ммоль) растворяли в дихлорметане (7 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 2 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором, сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении, и получали целевое соединение (0,485 г, 99,9%) в виде коричневого геля без дополнительной очистки.
[Стадия 4] Синтез соединения 4158
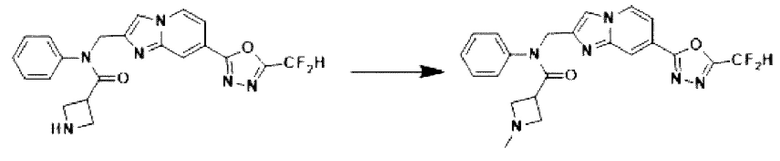
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилазетидин-3-карбоксамид (0,050 г, 0,118 ммоль), полученный на стадии 3, формальдегид (0,007 г, 0,236 ммоль), уксусную кислоту (0,007 мл, 0,118 ммоль) и триацетоксиборгидрид натрия (0,075 г, 0,353 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,014 г, 27,1%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,31 (дт, J=1,8, 0,8 Гц, 1Н), 8,23 (дд, J=7,1, 1,0 Гц, 1Н), 7,79 (с, 1Н), 7,54 (дд, J=7,1, 1,7 Гц, 1Н), 7,44-7,34 (м, 3Н), 7,19-7,13 (м, 2Н), 6,95 (т, J=51,7 Гц, 1Н), 5,05 (с, 2Н), 3,3 9-3,2 8 (м, 5Н), 2,35 (с, 3Н);
МСНР (ЭР) m/z 439,3 (М++1).
Пример 114: Синтез Соединения 4159, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-1-этил-N-фенилазетидин-3-карбоксамида
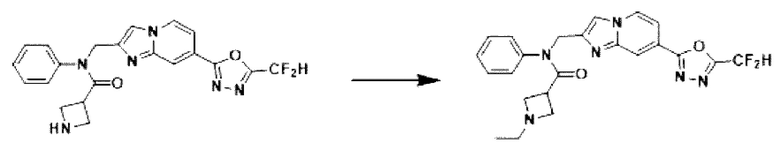
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилазетидин-3-карбоксамид (0,050 г, 0,118 ммоль), полученный на стадии 3 примера 113, ацетальдегид (0,010 г, 0,236 ммоль), уксусную кислоту (0,007 мл, 0,118 ммоль) и триацетоксиборгидрид натрия (0,075 г, 0,353 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,022 г, 41,3%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,31 (дт, J=1,7, 0,8 Гц, 1Н), 8,23 (дд, J=7,1, 1,0 Гц, 1Н), 7,78 (с, 1Н), 7,54 (дд, J=7,1, 1,7 Гц, 1Н), 7,4 5-7,34 (м, 3Н), 7,21-7,13 (м, 2Н), 6,95 (т, J=51,7 Гц, 1Н), 5,06 (с, 2Н), 3,39 (с, 5Н), 2,60 (с, 2Н), 0,99 (т, J=7,1 Гц, 3Н);
МСНР (ЭР) m/z 453,4 (М++1).
Пример 115: Синтез Соединения 4160, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-1-изопропил-N-фенилазетидин-3-карбоксамида
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-a]пиридин-2-ил)-метил)-N-фенилазетидин-3-карбоксамид (0,050 г, 0,118 ммоль), полученный на стадии 3 примера 113, пропан-2-он (0,014 г, 0,236 ммоль), уксусную кислоту (0,007 мл, 0,118 ммоль) и триацетоксиборгидрид натрия (0,075 г, 0,353 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,024 г, 4 3,7%) в виде твердого вещества белого цвета.
1Н ЯМР (400 МГц, CDCl3) δ 8,31 (дт, J=1,8, 0,8 Гц, 1Н), 8,23 (дд, J=7,2, 1,0 Гц, 1Н), 7,76 (с, 1Н), 7,54 (дд, J=7,1, 1,7 Гц, 1Н), 7,4 4-7,35 (м, 3Н), 7,2 0-7,13 (м, 2Н), 6,95 (т, J=51,7 Гц, 1Н), 5,06 (с, 2Н), 3,40 (с, 5Н), 2,63 (с, 1Н), 1,01 (д, J=5,4 Гц, 6Н);
МСНР (ЭР) m/z 467,4 (М++1).
Пример 116: Синтез Соединения 4161, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-1-(1-гидроксипропан-2-ил)-N-фенилазетидин-3-карбоксамида
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилазетидин-3-карбоксамид (0,050 г, 0,118 ммоль), полученный на стадии 3 примера 113, 1-гидроксипропан-2-он (0,017 г, 0,236 ммоль), уксусную кислоту (0,007 мл, 0,118 ммоль) и триацетоксиборгидрид натрия (0,075 г, 0,353 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,029 г, 51,0%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,32 (дт, J=1,7, 0,8 Гц, 1Н), 8,24 (дд, J=7,1, 1,0 Гц, 1Н), 7,80-7,73 (м, 1Н), 7,55 (дд, J=7,1, 1,7 Гц, 1Н), 7,44-7,36 (м, 3Н), 7,20-7,14 (м, 2Н), 6,95 (т, J=51,7 Гц, 1Н), 5,06 (с, 2Н), 3,64-3,28 (м, 7Н), 2,64 (с, 1Н), 1,02 (д, J=6,5 Гц, 3Н);
МСНР (ЭР) m/z 483,4 (М++1).
Пример 117: Синтез Соединения 4162, 1-циклобутил-17- ((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилазетидин-3-карбоксамида
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилазетидин-3-карбоксамид (0,050 г, 0,118 ммоль), полученный на стадии 3 примера 113, циклобутанон (0,017 г, 0,236 ммоль), уксусную кислоту (0,007 мл, 0,118 ммоль) и триацетоксиборгидрид натрия (0,075 г, 0,353 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,018 г, 31,9%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,32 (дт, J=1,7, 0,8 Гц, 1Н), 8,23 (дд, J=7,2, 1,0 Гц, 1Н), 7,76 (с, 1Н), 7,55 (дд, J=7,1, 1,7 Гц, 1Н), 7,44-7,35 (м, 3Н), 7,16 (дд, J=7,8, 1,9 Гц, 2Н), 6,95 (т, J=51,7 Гц, 1Н), 5,06 (с, 2Н), 3,60-3,28 (м, 5Н), 2,04 (д, J=11,2 Гц, 4Н), 1,84 (с, 1Н), 1,71 (кв, J=9,2 Гц, 2Н);
МСНР (ЭР) m/z 479,0 (М++1).
Пример 118: Синтез Соединения 4163, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-1-(оксетан-3-ил)-N-фенилазетидин-3-карбоксамида
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилазетидин-3-карбоксамид (0,050 г, 0,118 ммоль), полученный на стадии 3 примера 113, оксетан-3-он (0,017 г, 0,236 ммоль), уксусную кислоту (0,007 мл, 0,118 ммоль) и триацетоксиборгидрид натрия (0,075 г, 0,353 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,019 г, 33,6%) в виде твердого вещества белого цвета.
1Н ЯМР (400 МГц, CDCl3) δ 8,32 (дт, J=1,6, 0,8 Гц, 1Н), 8,23 (дд, J=7,1, 1,0 Гц, 1Н), 7,78 (с, 1Н), 7,54 (дд, J=7,1, 1,7 Гц, 1Н), 7,4 5-7,3 6 (м, 3Н), 7,21-7,15 (м, 2Н), 6,95 (т, J=51,7 Гц, 1Н), 5,06 (с, 2Н), 4,69 (т, J=6,8 Гц, 2Н), 4,53 (т, J=6,1 Гц, 2Н), 3,81 (с, 1Н), 3,54-3,29 (м, 5Н);
МСНР (ЭР) m/z 481,4 (М++1).
Пример 119: Синтез Соединения 4164, 1-циклогексил-N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилазетидин-3-карбоксамида
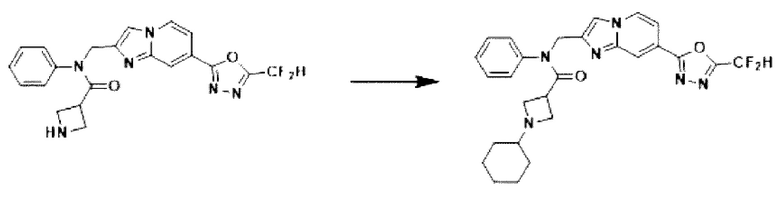
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-a]пиридин-2-ил)-метил)-N-фенилазетидин-3-карбоксамид (0,050 г, 0,118 ммоль), полученный на стадии 3 примера 113, циклогексанон (0,023 г, 0,236 ммоль), уксусную кислоту (0,007 мл, 0,118 ммоль) и триацетоксиборгидрид натрия (0,075 г, 0,353 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,028 г, 4 6,9%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,34-8,29 (м, 1Н), 8,23 (дд, J=7,2, 1,0 Гц, 1Н), 7,77 (с, 1Н), 7,54 (дд, J=7,1, 1,7 Гц, 1Н), 7,44-7,34 (м, 3Н), 7,17 (дд, J=8,0, 1,7 Гц, 2Н), 6,95 (т, J=51,7 Гц, 1Н), 5,06 (с, 2Н), 3,40 (с, 4Н), 1,73 (д, J=10,2 Гц, 5Н), 1,61 (с, 2Н), 1,17 (д, J=9,2 Гц, 5Н);
МСНР (ЭР) m/z 507,4 (М++1).
Пример 120: Синтез Соединения 4165, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенил-1-(тетрагидро-2Н-пиран-4-ил)-азетидин-3-карбоксамида
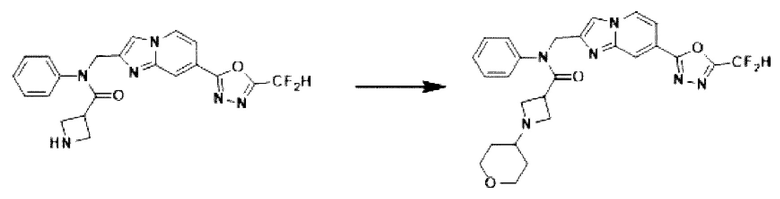
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилазетидин-3-карбоксамид (0,050 г, 0,118 ммоль), полученный на стадии 3 примера 113, тетрагидро-4Н-пиран-4-он (0,024 г, 0,236 ммоль), уксусную кислоту (0,007 мл, 0,118 ммоль) и триацетоксиборгидрид натрия (0,075 г, 0,353 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,023 г, 38,4%) в виде твердого вещества белого цвета.
1Н ЯМР (400 МГц, CDCl3) δ 8,32-8,30 (м, 1Н), 8,23 (дд, J=7,1, 1,0 Гц, 1Н), 7,77 (с, 1Н), 7,54 (дд, J=7,1, 1,7 Гц, 1Н), 7, 44-7, 34 (м, 3Н), 7,20-7,14 (м, 2Н), 6,95 (т, J=51,7 Гц, 1Н), 5,06 (с, 2Н), 3,94 (д, J=11,8 Гц, 2Н), 3,35 (тд, J=11,4, 2,3 Гц, 6Н), 2,35 (с, 1Н), 1,84-1,69 (м, 1Н), 1,64 (д, J=12,8 Гц, 2Н), 1,28 (с, 2Н);
МСНР (ЭР) m/z 509,0 (М++1).
Пример 121: Синтез Соединения 4166, 1-(4,4-дифторциклогексил)-N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилазетидин-3-карбоксамида
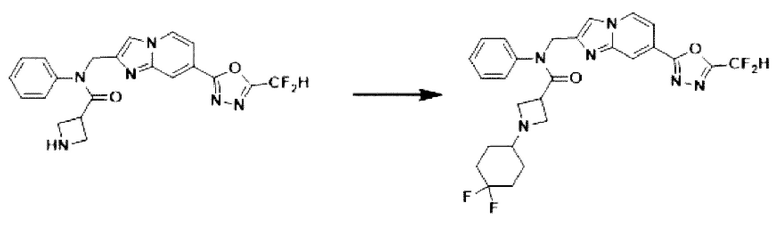
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилазетидин-3-карбоксамид (0,050 г, 0,118 ммоль), полученный на стадии 3 примера 113, 4,4-дифторциклогексан-1-он (0,032 г, 0,236 ммоль), уксусную кислоту (0,007 мл, 0,118 ммоль) и триацетоксиборгидрид натрия (0,075 г, 0,353 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,031 г, 48,5%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,31 (дт, J=1,7, 0,8 Гц, 1Н), 8,23 (дд, J=7,1, 1,0 Гц, 1Н), 7,78 (с, 1Н), 7,54 (дд, J=7,1, 1,7 Гц, 1Н), 7,44-7,34 (м, 3Н), 7,22-7,16 (м, 2Н), 6,95 (т, J=51,7 Гц, 1Н), 5,05 (с, 2Н), 3,28 (кв, J=7,5, 6,9 Гц, 1Н), 3,19 (с, 4Н), 2,21 (с, 1Н), 2,00 (с, 2Н), 1,68 (кв, J=14,1 Гц, 4Н), 1,39 (с, 2Н);
МСНР (ЭР) m/z 543,5 (М++1).
Пример 122: Синтез Соединения 4167, 1-ацетил-N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилазетидин-3-карбоксамида
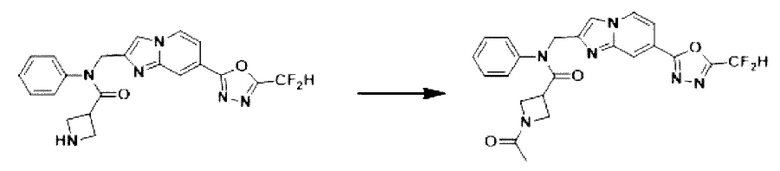
N-((7-(5-(дифторметил)-1,3,4-океадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилазетидин-3-карбоксамид (0,050 г, 0,118 ммоль), полученный на стадии 3 примера 113, ацетилхлорид (0,017 мл, 0,236 ммоль) и триэтиламин (0,049 мл, 0,353 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор хлорида аммония, экстрагировали дихлорметаном, фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,020 г, 36,4%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8, 39 (с, 1Н), 8,29 (д, J=7,0 Гц, 1Н), 7,82 (с, 1Н), 7,62 (д, J=7,1 Гц, 1Н), 7,42 (дт, J=9,3, 6,4 Гц, 3Н), 7,25-7,19 (м, 2Н), 6,96 (т, J=51,7 Гц, 1Н), 5,13-5,06 (м, 2Н), 4,54-4,40 (м, 1Н), 4,06 (дд, J=9,6, 6,5 Гц, 1Н), 3,93 (т, J=8,5 Гц, 1Н), 3,71 (т, J=9,4 Гц, 1Н), 3,37 (ддд, J=15,3, 9,0, 6,3 Гц, 1Н), 1,83 (с, 3Н);
МСНР (ЭР) m/z 466,9 (М++1).
Пример 123: Синтез Соединения 4168, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенил-1-пропионилазетидин-3-карбоксамида
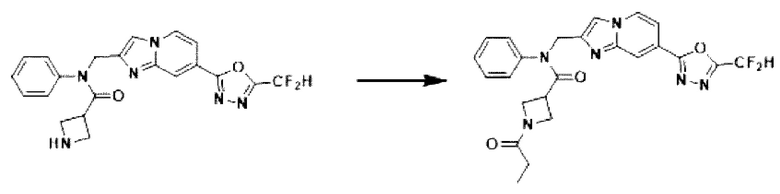
N-((7-(5-(дифторметил)-1,3,4-океадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилазетидин-3-карбоксамид (0,050 г, 0,118 ммоль), полученный на стадии 3 примера 113, пропионилхлорид (0,022 г, 0,236 ммоль) и триэтиламин (0,049 мл, 0,353 ммоль) растворяли в дихлорметане (5 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (картридж SiO2, 12 г; этилацетат/гексан = от 0% до 3 0%) и концентрировали с получением целевого соединения (0,032 г, 5 6,5%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,36-8,31 (м, 1Н), 8,25 (дд, J=7,1, 1,0 Гц, 1Н), 7,80 (д, J=0,7 Гц, 1Н), 7,57 (дд, J=7,1, 1,7 Гц, 1Н), 7,46-7,36 (м, 3Н), 7,20 (дд, J=8,1, 1,6 Гц, 2Н), 6,96 (т, J=51,7 Гц, 1Н), 5,18-4,96 (м, 2Н), 4,45 (дд, J=8,1, 6,2 Гц, 1Н), 4,05 (дд, J=9,5, 6,5 Гц, 1Н), 3,91 (т, J=8,4 Гц, 1Н), 3,71 (т, J=9,3 Гц, 1Н), 3,36 (тт, J=9,0, 6,3 Гц, 1Н), 2,07 (n, J=7,5 Гц, 2H), 1,09 (т, J=7,5 Гц, 3Н);
МСНР (ЭР) m/z 480,9 (М++1).
Пример 124: Синтез Соединения 4169, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-1-(2-гидроксиацетил)-N-фенилазетидин-3-карбоксамида
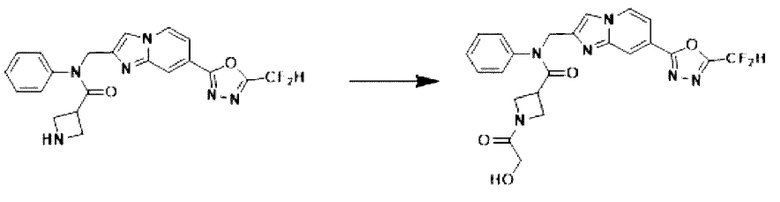
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилазетидин-3-карбоксамид (0,050 г, 0,118 ммоль), полученный на стадии 3 примера 113, 2-гидроксиацетилхлорид (0,022 г, 0,236 ммоль) и триэтиламин (0,049 мл, 0,353 ммоль) растворяли в дихлорметане (5 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (картридж SiO2, 12 г; этилацетат/гексан = от 0% до 30%) и концентрировали с получением целевого соединения (0,030 г, 52,8%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, MeOD) δ 8,58 (дд, J=7,1, 1,0 Гц, 1Н), 8,23 (дт, J=1,7, 0,8 Гц, 1Н), 7,98 (д, J=0,7 Гц, 1Н), 7,57 (дд, J=7,2, 1,7 Гц, 1Н), 7,48-7,11 (м, 6Н), 5,14 (дд, J=2,2, 0,7 Гц, 2Н), 4,48 (дд, J=9,0, 6,2 Гц, 1Н), 4,22-4,17 (м, 1Н), 4,14-4,07 (м, 1Н), 3,82 (т, J=9,4 Гц, 1Н), 3,57-3,48 (м, 1Н), 3,22 (кв, J=7,3 Гц, 2Н);
МСНР (ЭР) m/z 483,0 (М++1).
Пример 125: Синтез Соединения 4170, 1-(циклобутанкарбонил)-N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилазетидин-3-карбоксамида
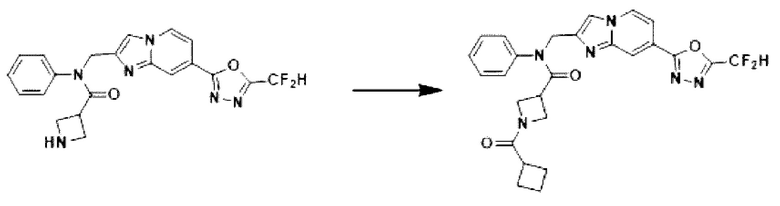
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-a]пиридин-2-ил)-метил)-N-фенилазетидин-3-карбоксамид (0,050 г, 0,118 ммоль), полученный на стадии 3 примера 113, циклобутанкарбонилхлорид (0,028 г, 0,236 ммоль) и триэтиламин (0,049 мл, 0,353 ммоль) растворяли в дихлорметане (5 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (картридж SiO2, 12 г; этил ацетат/гексан = от 0% до 30%) и концентрировали с получением целевого соединения (0,029 г, 48,6%) в виде твердого вещества белого цвета.
1Н ЯМР (400 МГц, CDCl3) δ 8,35 (с, 1Н), 8,25 (дд, J=7,1, 1,0 Гц, 1Н), 7,80 (д, J=0,7 Гц, 1Н), 7,58 (дд, J=7,2, 1,7 Гц, 1Н), 7,4 7-7,3 6 (м, 3Н), 7,22-7,15 (м, 2Н), 6,9 6 (т, J=51,7 Гц, 1Н), 5,15-4,9 6 (м, 2Н), 4, 44-4, 30 (м, 1Н), 4,04 (дд, J=9,5, 6,5 Гц, 1Н), 3,85 (т, J=8,4 Гц, 1Н), 3,70 (т, J=9,4 Гц, 1Н), 3,35 (тт, J=8,9, 6,4 Гц, 1Н), 3,09-2,92 (м, 1Н), 2,43-2,21 (м, 3Н), 2,14-1,8 9 (м, 3Н);
МСНР (ЭР) m/z 507,4 (М++1).
Пример 126: Синтез Соединения 4171, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-1-(оксетан-3-карбонил)-Ы-фенилазетидин-3-карбоксамида
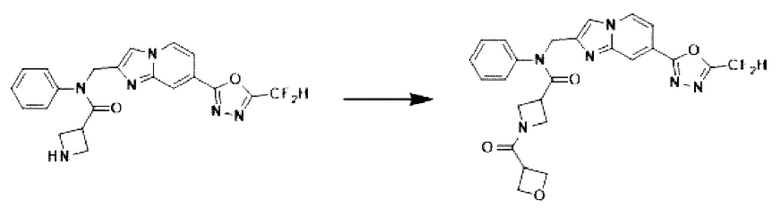
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилазетидин-3-карбоксамид (0,050 г, 0,118 ммоль), полученный на стадии 3 примера 113, оксетан-3-карбонилхлорид (0,028 г, 0,236 ммоль) и триэтиламин (0,049 мл, 0,353 ммоль) растворяли в дихлорметане (5 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (картридж SiO2, 12 г; этил ацетат / гексан = от 0% до 3 0%) и концентрировали с получением целевого соединения (0,039 г, 65,1%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8, 38 (с, 1Н), 8,26 (дд, J=7,0, 1,0 Гц, 1Н), 7,78 (с, 1Н), 7,61 (д, J=7,1 Гц, 1Н), 7,47-7,38 (м, 3Н), 7,24-7,18 (м, 2Н), 6,96 (т, J=51,7 Гц, 1Н), 5,09 (д, J=2,9 Гц, 2Н), 4,90 (дд, J=7,0, 5,8 Гц, 1Н), 4,83 (дд, J=7,0, 5,7 Гц, 1Н), 4,72 (ддд, J=14,5, 8,6, 5,8 Гц, 2Н), 4,40-4,32 (м, 1Н), 4,11-4,04 (м, 1Н), 3,83 (т, J=8,4 Гц, 1Н), 3,79-3,71 (м, 2Н), 3,39 (ддд, J=15,1, 8,9, 8,3 Гц, 1Н);
МСНР (ЭР) m/z 509,4 (М++1).
Пример 127: Синтез Соединения 4172, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенил-1-(2,2,2-трифторацетил)-азетидин-3-карбоксамида
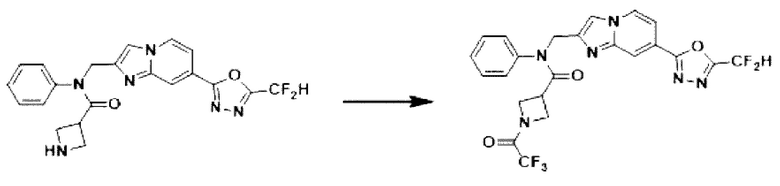
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-a]пиридин-2-ил)-метил)-N-фенилазетидин-3-карбоксамид (0,050 г, 0,118 ммоль), полученный на стадии 3 примера 113, 1,1,1,5,5,5-гексафторпентан-2,4-дион (0,049 г, 0,236 ммоль) и триэтиламин (0,049 мл, 0,353 ммоль) растворяли в дихлорметане (5 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (картридж SiO2, 12 г; этилацетат/гексан = от 0% до 30%) и концентрировали с получением целевого соединения (0,034 г, 55,5%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,36 (с, 1Н), 8,26 (дд, J=7,1, 1,0 Гц, 1Н), 7,79 (с, 1Н), 7,59 (дд, J=7,1, 1,7 Гц, 1Н), 7,51-7,37 (м, 3Н), 7,25-7,19 (м, 2Н), 6,96 (т, J=51,7 Гц, 1Н), 5,09 (д, J=2,9 Гц, 2Н), 4,76-4,60 (м, 1Н), 4,27 (дд, J=10,4, 6,6 Гц, 1Н), 4,22-4,10 (м, 1Н), 3,87 (т, J=9,9 Гц, 1Н), 3,51 (тт, J=9,1, 6,5 Гц, 1Н);
МСНР (ЭР) m/z 520,9 (М++1).
Пример 128: Синтез Соединения 4173, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-1-(метилсульфонил)-N-фенилазетидин-3-карбоксамида
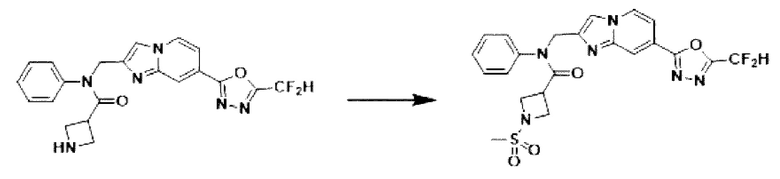
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-a]пиридин-2-ил)-метил)-N-фенилазетидин-3-карбоксамид (0,050 г, 0,118 ммоль), полученный на стадии 3 примера 113, метансульфонилхлорид (0,018 мл, 0,236 ммоль) и триэтиламин (0,049 мл, 0,353 ммоль) растворяли в дихлорметане (5 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (картридж SiO2, 12 г; этилацетат/гексан = от 0% до 3 0%) и концентрировали с получением целевого соединения (0,032 г, 54,1%) в виде твердого вещества белого цвета.
1Н ЯМР (400 МГц, CDCl3) δ 8,37 (с, 1Н), 8,26 (дд, J=7,2, 1,0 Гц, 1Н), 7,80-7,75 (м, 1Н), 7,60 (дд, J=7,1, 1,7 Гц, 1Н), 7, 47-7, 38 (м, 3Н), 7,24-7,15 (м, 2Н), 6,96 (т, J=51,7 Гц, 1Н), 5,09 (с, 2Н), 4,16 (дд, J=8,1, 7,1 Гц, 2Н), 3,71 (т, J=8,3 Гц, 2Н), 3,4 7-3,32 (м, 1Н), 2,91 (с, 3Н);
МСНР (ЭР) m/z 503,4 (М++1).
Пример 129: Синтез Соединения 4174, метил-3-(((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-(фенил)-карбамоил)-азетидин-1-карбоксилата
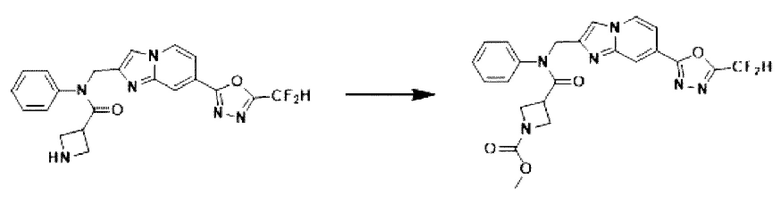
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилазетидин-3-карбоксамид (0,050 г, 0,118 ммоль), полученный на стадии 3 примера 113, метилхлорформиат (0,022 г, 0,236 ммоль) и триэтиламин (0,049 мл, 0,353 ммоль) растворяли в дихлорметане (5 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (картридж SiO2, 12 г; этилацетат/гексан = от 0% до 30%) и концентрировали с получением целевого соединения (0,023 г, 40,5%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,36 (с, 1Н), 8,26 (дд, J=7,1, 1,0 Гц, 1Н), 7,82 (с, 1Н), 7,59 (дд, J=7,2, 1,7 Гц, 1Н), 7,46-7,36 (м, 3Н), 7,22-7,16 (м, 2Н), 6,96 (т, J=51,7 Гц, 1Н), 5,08 (с, 2Н), 4,19 (с, 2Н), 3,74 (т, J=8,6 Гц, 2Н), 3,64 (с, 3Н), 3,36 (тт, J=8,8, 6,4 Гц, 1Н);
МСНР (ЭР) m/z 482,8 (М++1).
Пример 130: Синтез Соединения 4175, N3-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N1,N1-диметил-N3-фенилазетидин-1,3-дикарбоксамида
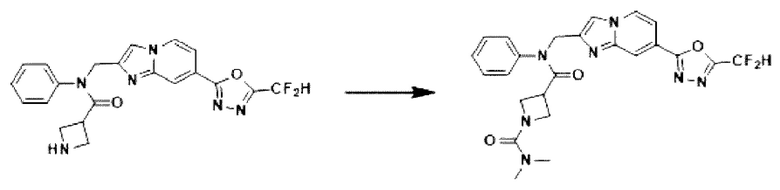
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилазетидин-3-карбоксамид (0,050 г, 0,118 ммоль), полученный на стадии 3 примера 113, хлорангидрид диметилкарбаминовой кислоты (0,025 г, 0,236 ммоль) и триэтиламин (0,04 9 мл, 0,353 ммоль) растворяли в дихлорметане (5 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (картридж SiO2, 12 г; этилацетат/гексан = от 0% до 30%) и концентрировали с получением целевого соединения (0,030 г, 51,4%) в виде твердого вещества белого цвета.
1Н ЯМР (400 МГц, CDCl3) δ 8, 34 (с, 1Н), 8,26 (дд, 77=7,1, 1,0 Гц, 1Н), 7,84-7,80 (м, 1Н), 7,58 (дд, J=7,1, 1,7 Гц, 1Н), 7,45-7,34 (м, 3Н), 7,22-7,15 (м, 2Н), 6,95 (т, J=51,7 Гц, 1Н), 5,08 (с, 2Н), 4,18 (дд, 77=8,0, 6,8 Гц, 2Н), 3,70 (дд, 77=9,0, 8,0 Гц, 2Н), 3,35 (тт, J=9,0, 6,8 Гц, 1Н), 2,82 (с, 6Н);
МСНР (ЭР) m/z 4 95,9 (М++1).
Пример 131: Синтез Соединения 4176, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенил-1-(пиридин-2-ил)-азетидин-3-карбоксамида
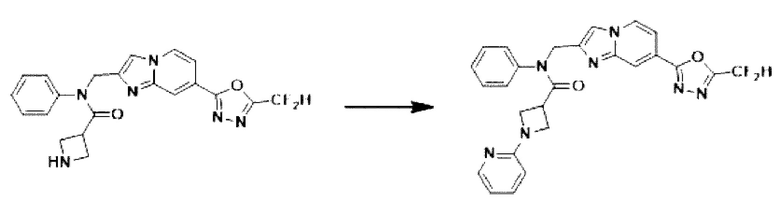
N-((7-(5-(дифторметил)-1,3,4-океадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенилазетидин-3-карбоксамид (0,050 г, 0,118 ммоль), полученный на стадии 2 примера 113, 2-хлорпиридин (0,027 г, 0,236 ммоль), карбонат цезия (0,077 г, 0,236 ммоль) и палладий RuPhos G2 (0,005 г, 0,006 ммоль) растворяли в 1,4-диоксане (2 мл) при комнатной температуре, и полученный раствор перемешивали при 10 0°С в течение 18 часов. Затем температуру понижали до комнатной температуры для завершения реакции. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 30%) и концентрировали с получением целевого соединения (0,028 г, 47,4%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,31 (дт, J=1,7, 0,8 Гц, 1Н), 8,23 (дд, J=7,1, 1,0 Гц, 1Н), 8,11 (ддд, J=5,1, 1,8, 0,9 Гц, 1Н), 7,82 (д, J=0,7 Гц, 1Н), 7,54 (дд, J=7,1, 1,7 Гц, 1Н), 7, 49-7, 35 (м, 4Н), 7,25-7,21 (м, 2Н), 6,95 (т, J=51,7 Гц, 1Н), 6,61 (ддд, J=7,1, 5,1, 1,0 Гц, 1Н), 6,35-6,27 (м, 1Н), 5,08 (с, 2Н), 4,23 (т, J=7,2 Гц, 2Н), 3,86 (т, J=8,1 Гц, 2Н), 3,61-3,48 (м, 1Н);
МСНР (ЭР) m/z 502,4 (М++1).
Пример 132: Синтез Соединения 4177, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенил-1-(пиримидин-2-ил)-азетидин-3-карбоксамида
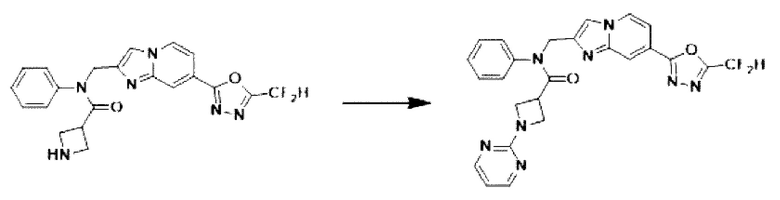
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-a]пиридин-2-ил)-метил)-N-фенилазетидин-3-карбоксамид (0,050 г, 0,118 ммоль), полученный на стадии 3 примера 113, 2-хлорпиримидин (0,027 г, 0,236 ммоль) и карбонат калия (0,033 г, 0,236 ммоль) растворяли в ацетонитриле (2 мл)/N,N-диметилформамиде (2 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В концентрат, полученный путем удаления растворителя из реакционной смеси при пониженном давлении, вливали насыщенный водный раствор хлорида аммония с последующей экстракцией дихлорметаном. Затем полученный продукт фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,018 г, 30,4%) в виде твердого вещества белого цвета.
1Н ЯМР (400 МГц, CDCl3) δ 8, 33-8, 28 (м, 3Н), 8,23 (дд, J=7,1, 1,0 Гц, 1Н), 7,82 (д, J=0,7 Гц, 1Н), 7,54 (дд, J=7,2, 1,7 Гц, 1Н), 7,47-7,36 (м, 3Н), 7,26-7,18 (м, 2Н), 6,95 (т, J=51,7 Гц, 1Н), 6,54 (т, J=4,8 Гц, 1Н), 5,09 (с, 2Н), 4,36 (дд, J=8,5, 6,5 Гц, 2Н), 3,95 (т, J=8,6 Гц, 2Н), 3,51 (тт, J=8,7, 6,5 Гц, 1Н);
МСНР (ЭР) m/z 503,4 (М++1).
Пример 133: Синтез Соединения 4188, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-1-метилазетидин-3-карбоксамида
[Стадия 1] Синтез трет-бутил-3-(((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-(3-фторфенил)-карбамоил)-азетидин-1-карбоксилата
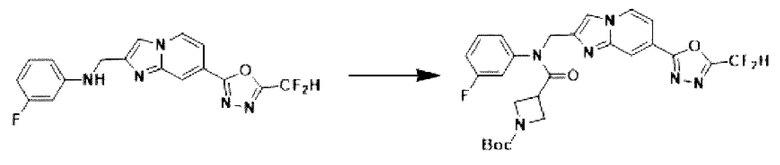
К раствору, в котором растворяли N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-3-фторанилин (1,500 г, 4,175 ммоль), полученный в Примере 16, и триэтиламин (1,746 мл, 12,524 ммоль) в дихлорметане (150 мл) при комнатной температуре, добавляли трет-бутил-3-(хлоркарбонил)-азетидин-1-карбоксилат (1,192 г, 5,427 ммоль), полученный на стадии 1 Примера 113, и перемешивали при той же температуре в течение 1 6 часов. В реакционную смесь вливали насыщенный водный раствор хлорида аммония, экстрагировали дихлорметаном, фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 40 г; этил ацетат/гексан = от 5% до 60%) и концентрировали, получая целевое соединение (1200 г, 53,0%) в виде твердого вещества желтого цвета.
[Стадия 2] Синтез N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-азетидин-3-карбоксамида
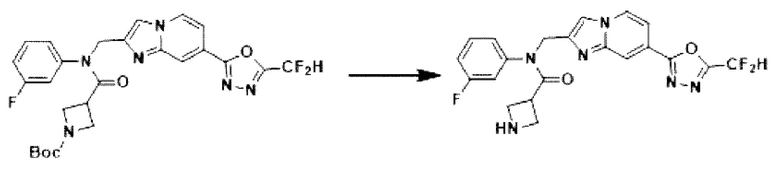
Трет-бутил-4-(((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-(3-фторфенил)-карбамоил)-азетидин-1-карбоксилат (0,700 г, 1,290 ммоль), полученный на стадии 1, и трифторуксусную кислоту (1,976 мл, 25,806 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 2 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором, сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении, и получали целевое соединение (0,570 г, 9 9,9%) в виде коричневого геля без дополнительной очистки.
[Стадия 3] Синтез соединения 4188
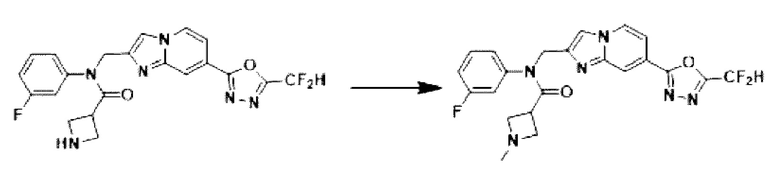
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-азетидин-3-карбоксамид (0,050 г, 0,113 ммоль), полученный на стадии 2, формальдегид (0,007 г, 0,226 ммоль), уксусную кислоту (0,006 мл, 0,113 ммоль) и триацетоксиборгидрид натрия (0,072 г, 0,339 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном, фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,016 г, 31,0%) в виде твердого вещества белого цвета.
1Н ЯМР (400 МГц, CDCl3) δ 8,33-8,30 (м, 1Н), 8,24 (дд, J=7,1, 1,0 Гц, 1Н), 7,79 (с, 1Н), 7,55 (дд, J=7,1, 1,7 Гц, 1Н), 7,38 (тд, J=8,3, 6,3 Гц, 1Н), 7,14-6,80 (м, 4Н), 5,03 (с, 2Н), 3,45 (с, 2Н), 3,36 (с, 3Н), 2,39 (с, 3Н);
МСНР (ЭР) m/z 457,4 (М++1).
Пример 134: Синтез Соединения 4189, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-1-этил-N-(3-фторфенил)-азетидин-3-карбоксамида
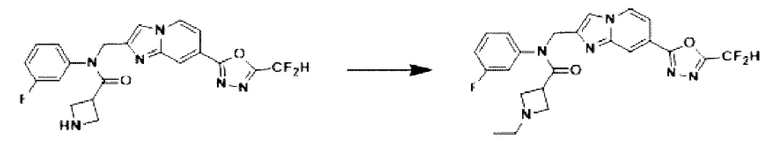
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-азетидин-3-карбоксамид (0,060 г, 0,136 ммоль), полученный на стадии 2 примера 133, ацетальдегид (0,012 г, 0,271 ммоль), уксусную кислоту (0,008 мл, 0,136 ммоль) и триацетоксиборгидрид натрия (0,086 г, 0,407 ммоль) растворяли в дихлорметане (5 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (картридж SiO2, 12 г; этилацетат/гексан = от 0% до 3 0%) и концентрировали с получением целевого соединения (0,021 г, 32,9%) в виде твердого вещества белого цвета.
1Н ЯМР (400 МГц, CDCl3) δ 8,33-8,30 (м, 1Н), 8,24 (дд, J=7,1, 1,0 Гц, 1Н), 7,78 (с, 1Н), 7,56 (дд, J=7,1, 1,7 Гц, 1Н), 7,43-7,34 (м, 1Н), 7,14-6,78 (м, 4Н), 5,04 (с, 2Н), 3,38 (с, 4Н), 2,59 (с, 1Н), 1,68 (с, 5Н);
МСНР (ЭР) m/z 471,5 (М++1).
Пример 135: Синтез Соединения 4190, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-1-изопропилазетидин-3-карбоксамида
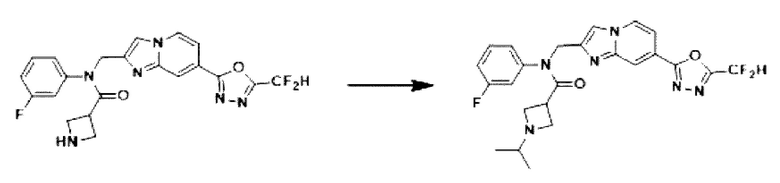
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-азетидин-3-карбоксамид (0,060 г, 0,136 ммоль), полученный на стадии 2 примера 133, пропан-2-он (0,016 г, 0,271 ммоль), уксусную кислоту (0,008 мл, 0,136 ммоль) и триацетоксиборгидрид натрия (0,086 г, 0,407 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и перемешивали полученный раствор при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,025 г, 38,0%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,32 (дд, J=1,7, 0,9 Гц, 1Н), 8,24 (дд, J=7,1, 1,0 Гц, 1Н), 7,78 (с, 1Н), 7,55 (дд, J=7,1, 1,7 Гц, 1Н), 7,38 (тд, J=8,3, 6,3 Гц, 1Н), 7,14-6,80 (м, 4Н), 5,03 (с, 2Н), 3,43 (с, 2Н), 3,35 (с, 3Н), 2,54 (с, 1Н), 0,97 (д, J=6, 3 Гц, 6Н);
МСНР (ЭР) m/z 485,5 (М++1).
Пример 136: Синтез Соединения 4191, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-1-(1-гидроксипропан-2-ил)-азетидин-3-карбоксамида
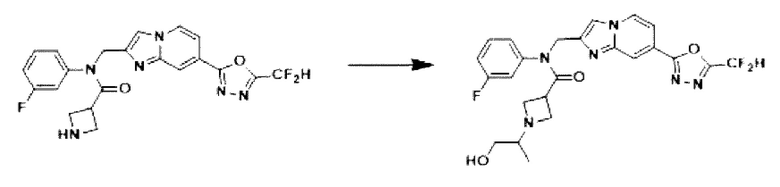
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-азетидин-3-карбоксамид (0,050 г, 0,113 ммоль), полученный на стадии 2 примера 133, 1-гидроксипропан-2-он (0,017 г, 0,226 ммоль), уксусную кислоту (0,006 мл, 0,113 ммоль) и триацетоксиборгидрид натрия (0,072 г, 0,339 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и перемешивали полученный раствор при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,020 г, 35,4%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,32 (дд, J=1,7, 0,9 Гц, 1Н), 8,24 (дд, J=7,1, 1,0 Гц, 1Н), 7,78 (с, 1Н), 7,55 (дд, J=7,1, 1,7 Гц, 1Н), 7, 4 3-7, 34 (м, 1Н), 7,13-6,81 (м, 4Н), 5,03 (с, 2Н), 3,60-3,45 (м, 4Н), 3,43-3,30 (м, 3Н), 2,62 (д, J=9,4 Гц, 1Н), 1,00 (д, J=6,5 Гц, 3Н);
МСНР (ЭР) m/z 501,4 (М++1).
Пример 137: Синтез Соединения 4192, 1-циклобутил-N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-азетидин-3-карбоксамида
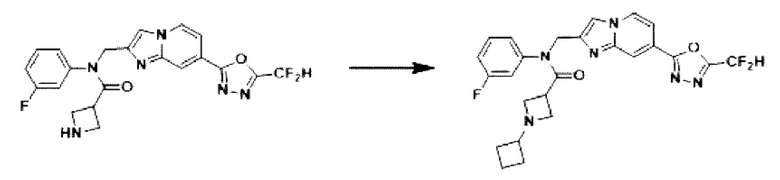
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-азетидин-3-карбоксамид (0,050 г, 0,113 ммоль), полученный на стадии 2 примера 133, циклобутанон (0,016 г, 0,226 ммоль), уксусную кислоту (0,006 мл, 0,113 ммоль) и триацетоксиборгидрид натрия (0,072 г, 0,339 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и перемешивали полученный раствор при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,024 г, 42,8%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,32 (дт, J=1,7, 0,8 Гц, 1Н), 8,24 (дд, J=7,1, 1,0 Гц, 1Н), 7,79 (с, 1Н), 7,55 (дд, J=7,1, 1,7 Гц, 1Н), 7,38 (тд, J=8,2, 6,2 Гц, 1Н), 7,12-6,81 (м, 4Н), 5,03 (с, 2Н), 3,23 (д, J=61,3 Гц, 5Н), 2,01-1,94 (м, 2Н), 1,86-1,59 (м, 5Н);
МСНР (ЭР) m/z 497,4 (М++1).
Пример 138: Синтез Соединения 4193, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-1-(оксетан-3-ил)-азетидин-3-карбоксамида
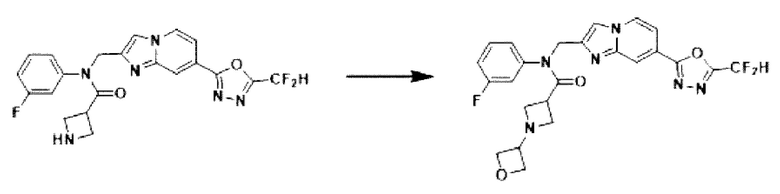
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-азетидин-3-карбоксамид (0,050 г, 0,113 ммоль), полученный на стадии 2 примера 133, оксетан-3-он (0,016 г, 0,226 ммоль), уксусную кислоту (0,006 мл, 0,113 ммоль) и триацетоксиборгидрид натрия (0,072 г, 0,339 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и перемешивали полученный раствор при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,032 г, 56,8%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,32 (дт, J=1,7, 0,8 Гц, 1Н), 8,24 (дд, J=7,1, 1,0 Гц, 1Н), 7,79 (с, 1Н), 7,55 (дд, J=7,1, 1,7 Гц, 1Н), 7,39 (тд, J=8,2, 6,2 Гц, 1Н), 7,14-6,79 (м, 4Н), 5,03 (с, 2Н), 4,67 (т, J=6,7 Гц, 2Н), 4,49 (дд, J=6,7, 5,2 Гц, 2Н), 3,74 (ддд, J=11,9, 6,7, 5,2 Гц, 1Н), 3,44-3,32 (м, 3Н), 3,28 (д, J=5,6 Гц, 2Н);
МСНР (ЭР) m/z 499,4 (М++1).
Пример 139: Синтез Соединения 4194, 1-циклогексил-N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-азетидин-3-карбоксамида
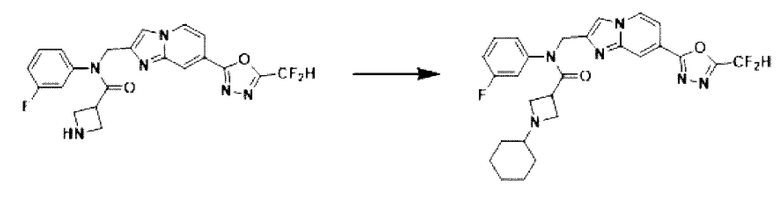
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-азетидин-3-карбоксамид (0,050 г, 0,113 ммоль), полученный на стадии 2 примера 133, циклогексанон (0,022 г, 0,226 ммоль), уксусную кислоту (0,006 мл, 0,113 ммоль) и триацетоксиборгидрид натрия (0,072 г, 0,339 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и перемешивали полученный раствор при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,034 г, 57,4%) в виде твердого вещества белого цвета.
1Н ЯМР (400 МГц, CDCl3) δ 8,32 (дт, J=1,6, 0,8 Гц, 1Н), 8,24 (дд, J=7,1, 1,0 Гц, 1Н), 7,77 (с, 1Н), 7,55 (дд, J=7,1, 1,7 Гц, 1Н), 7,38 (тд, J=8,3, 6,3 Гц, 1Н), 7,14-6,78 (м, 4Н), 5,03 (с, 2Н), 3,45 (д, J=29,4 Гц, 5Н), 2,28 (с, 1Н), 1,74 (д, J=10,1 Гц, 4Н), 1,61 (с, 1Н), 1,23-1,05 (м, 5Н);
МСНР (ЭР) m/z 525,1 (М++1).
Пример 140: Синтез Соединения 4195, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-1-(тетрагидро-2Н-пиран-4-ил)-азетидин-3-карбоксамида
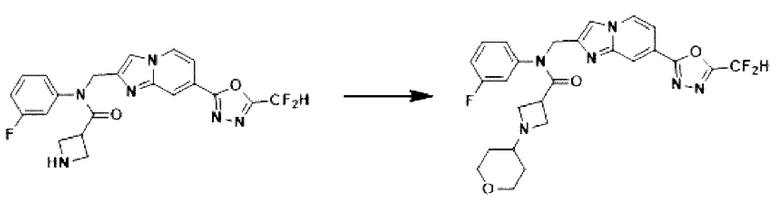
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-азетидин-3-карбоксамид (0,050 г, 0,113 ммоль), полученный на стадии 2 примера 133, тетрагидро-4Н-пиран-4-он (0,023 г, 0,226 ммоль), уксусную кислоту (0,006 мл, 0,113 ммоль) и триацетоксиборгидрид натрия (0,072 г, 0,339 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и перемешивали полученный раствор при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,015 г, 25,2%) в виде твердого вещества белого цвета.
1Н ЯМР (400 МГц, CDCl3) δ 8,32 (дт, J=1,6, 0,8 Гц, 1Н), 8,23 (дд, J=7,1, 1,0 Гц, 1Н), 7,78 (с, 1Н), 7,55 (дд, J=7,1, 1,7 Гц, 1Н), 7,38 (тд, J=8,3, 6,3 Гц, 1Н), 7,13-6,79 (м, 4Н), 5,03 (с, 2Н), 3,92 (дт, J=11,6, 3,7 Гц, 2Н), 3,41-3,15 (м, 7Н), 2,36-2,22 (м, 1Н), 1,61 (д, J=13,0 Гц, 2Н), 1,31 (д, J=4,3 Гц, 2Н);
МСНР (ЭР) m/z 527,5 (М++1).
Пример 141: Синтез Соединения 4196, 1-(4,4-дифторциклогексил)-N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-азетидин-3-карбоксамида
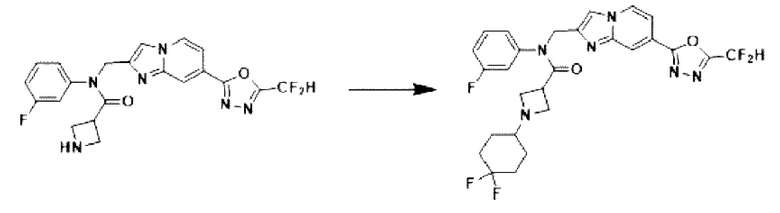
N-((7-(5-(дифторметил)-1,3,4-океадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-азетидин-3-карбоксамид (0,050 г, 0,113 ммоль), полученный на стадии 2 примера 133, 4,4-дифторциклогексан-1-он (0,030 г, 0,226 ммоль), уксусную кислоту (0,006 мл, 0,113 ммоль) и триацетоксиборгидрид натрия (0,072 г, 0,339 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и перемешивали полученный раствор при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,021 г, 33,1%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,32 (дт, J=1,7, 0,8 Гц, 1Н), 8,23 (дд, J=7,1, 1,0 Гц, 1Н), 7,78 (с, 1Н), 7,55 (дд, J=7,1, 1,7 Гц, 1Н), 7,38 (тд, J=8,4, 6,5 Гц, 1Н), 7,15-6,77 (м, 4Н), 5,03 (с, 2Н), 3,34-3,12 (м, 5Н), 2,22 (с, 1Н), 2,00 (дд, J=12,7, 5,4 Гц, 2Н), 1,69 (кв, J=17,0, 15,4 Гц, 4Н), 1,40 (д, J=11,4 Гц, 2Н);
МСНР (ЭР) m/z 561,3 (М++1).
Пример 142: Синтез Соединения 4197, 1-ацетил-N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-азетидин-3-карбоксамида
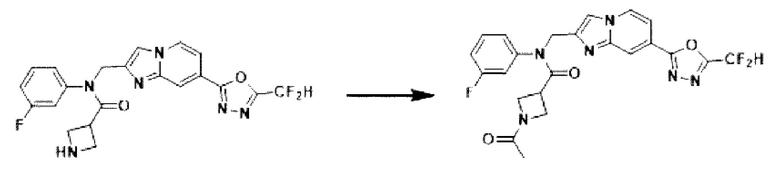
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-азетидин-3-карбоксамид (0,050 г, 0,113 ммоль), полученный на стадии 2 примера 133, ацетилхлорид (0,016 мл, 0,226 ммоль) и триэтиламин (0,047 мл, 0,339 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,024 г, 43,8%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,36 (д, J=1,6 Гц, 1Н), 8,26 (дд, J=7,2, 1,0 Гц, 1Н), 7,81 (с, 1Н), 7,60 (дд, J=7,2, 1,7 Гц, 1Н), 7,41 (тд, J=8,0, 6,4 Гц, 1Н), 7,18-6,77 (м, 4Н), 5,15-4,97 (м, 2Н), 4,50-4,42 (м, 1Н), 4,06 (дд, J=9,5, 6,4 Гц, 1Н), 3,97 (т, J=8,5 Гц, 1Н), 3,81-3,69 (м, 1Н), 3,38 (тт, J=8,9, 6,3 Гц, 1Н), 1,84 (с, 3Н);
МСНР (ЭР) m/z 485,0 (М++1).
Пример 143: Синтез Соединения 4198, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-1-изопропилазетидин-3-карбоксамида
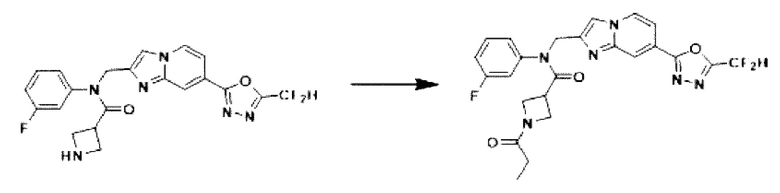
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-азетидин-3-карбоксамид (0,050 г, 0,113 ммоль), полученный на стадии 2 примера 133, пропионилхлорид (0,021 г, 0,226 ммоль) и триэтиламин (0,047 мл, 0,339 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,027 г, 47,9%) в виде твердого вещества белого цвета.
1Н ЯМР (400 МГц, CDCl3) δ 8,35 (с, 1Н), 8,26 (д, J=7,1 Гц, 1Н), 7,80 (с, 1Н), 7,58 (дд, J=7,1, 1,7 Гц, 1Н), 7,41 (кв, J=7,6 Гц, 1Н), 7,17-6,81 (м, 4Н), 5,14-4,97 (м, 2Н), 4,51-4,38 (м, 1Н), 4,06 (дд, J=9,5, 6,5 Гц, 1Н), 3,95 (т, J=8,4 Гц, 1Н), 3,76 (т, J=9,3 Гц, 1Н), 3,38 (тт, J=8,8, 6,2 Гц, 1Н), 2,14-2,0 0 (м, 2Н), 1,10 (т, J=7,5 Гц, 3Н);
МСНР (ЭР) m/z 499,5 (М++1).
Пример 144: Синтез Соединения 4199, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-1-(2-гидроксиацетил)-азетидин-3-карбоксамида
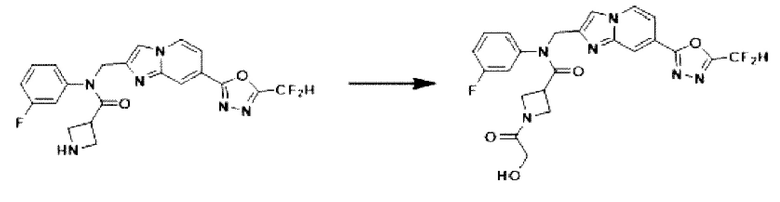
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-азетидин-3-карбоксамид (0,050 г, 0,113 ммоль), полученный на стадии 2 примера 133, 2-гидроксиацетилхлорид (0,021 г, 0,226 ммоль) и триэтиламин (0,047 мл, 0,339 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,012 г, 21,2%) в виде твердого вещества белого цвета.
1Н ЯМР (400 МГц, CDCl3) δ 8,33 (д, J=2,0 Гц, 1Н), 8,25 (дд, J=7,2, 1,1 Гц, 1Н), 7,79 (с, 1Н), 7,57 (дт, J=7,1, 1,6 Гц, 1Н), 7,42 (тд, J=8,1, 6,2 Гц, 1Н), 7,18-6,79 (м, 4Н), 5,05 (д, J=4,0 Гц, 2Н), 4,38 (т, J=7,3 Гц, 1Н), 4,21-4,14 (м, 1Н), 4,03-3,81 (м, 4Н), 3,49 (ддд, J=15,2, 8,9, 6,3 Гц, 1Н); МСНР (ЭР) m/z 501,0 (М++1).
Пример 145: Синтез Соединения 4200, 1-(циклобутанкарбонил)-N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-азетидин-3-карбоксамида
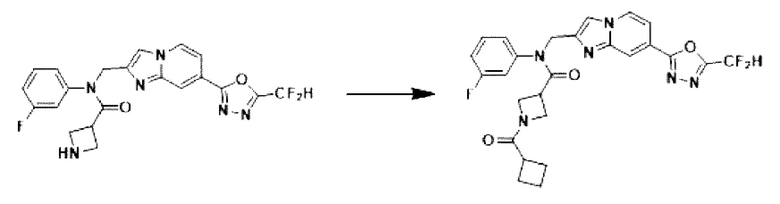
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-азетидин-3-карбоксамид (0,050 г, 0,113 ммоль), полученный на стадии 2 примера 133, циклобутанкарбонилхлорид (0,027 г, 0,226 ммоль) и триэтиламин (0,047 мл, 0,339 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,038 г, 64,1%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,38 (с, 1Н), 8,27 (д, J=7,1 Гц, 1Н), 7,81 (с, 1Н), 7,62 (д, J=7,1 Гц, 1Н), 7,41 (тд, J=8,3, 6,2 Гц, 1Н), 7,17-6,77 (м, 4Н), 5,15-4,96 (м, 2Н), 4,43-4,31 (м, 1Н), 4,04 (дд, J=9,4, 6,5 Гц, 1Н), 3,89 (т, J=8,4 Гц, 1Н), 3,75 (т, J=9,3 Гц, 1Н), 3,38 (ддд, J=15,1, 8,9, 6,3 Гц, 1Н), 3,00 (дд, J=9,4, 7,6 Гц, 1Н), 2,28 (дп, J=27,9, 9,2, 8,8 Гц, 3Н), 2,13-1,90 (м, 3Н);
МСНР (ЭР) m/z 525,1 (М++1).
Пример 146: Синтез Соединения 4201, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-1-(оксетан-3-карбонил)-азетидин-3-карбоксамида
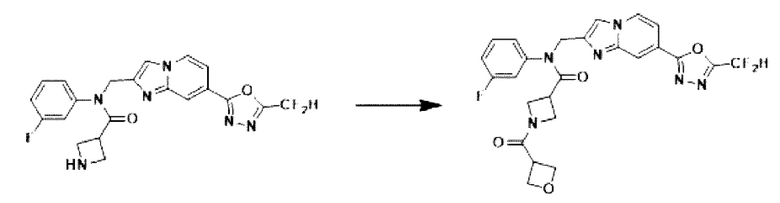
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-азетидин-3-карбоксамид (0,050 г, 0,113 ммоль), полученный на стадии 2 примера 133, оксетан-3-карбонилхлорид (0,027 г, 0,226 ммоль) и триэтиламин (0,047 мл, 0,339 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,033 г, 55,5%) в виде твердого вещества белого цвета.
1Н ЯМР (400 МГц, CDCl3) δ 8,35 (д, J=1,7 Гц, 1Н), 8,25 (дд, J = 7,1, 1,0 Гц, 1Н), 7,78 (с, 1Н), 7,58 (дд, J = 7,1, 1,7 Гц, 1Н), 7,42 (тд, J = 8,0, 6,1 Гц, 1Н), 7,17-6,79 (м, 4Н), 5,14-4,98 (м, 2Н), 4,90 (дд, J = 7,0, 5,8 Гц, 1Н), 4,83 (дд, J=7,0, 5,8 Гц, 1Н), 4,72 (ддд, J=12,0, 8,6, 5,8 Гц, 2Н), 4,36 (дд, J = 8,1, 6,1 Гц, 1Н), 4,09 (дд, J = 9,7, 6,4 Гц, 1Н), 3,89-3,71 (м, 3Н), 3,39 (тт, J = 8,8, 6,3 Гц, 1Н);
МСНР (ЭР) m/z 527,1 (М++1).
Пример 147: Синтез Соединения 4202, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-1-(2,2,2-трифторацетил)-азетидин-3-карбоксамида
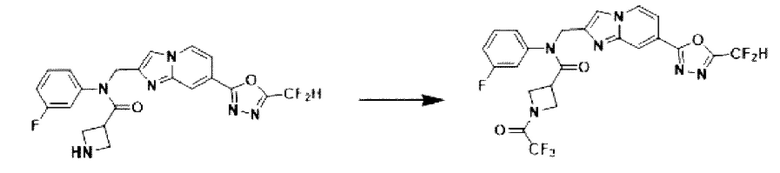
N-((7-(5-(дифторметил)-1,3,4-океадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-азетидин-3-карбоксамид (0,060 г, 0,136 ммоль), полученный на стадии 2 примера 133, 1,1,1,5,5,5-гексафторпентан-2,4-дион (0,056 г, 0,271 ммоль) и триэтиламин (0,057 мл, 0,407 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,031 г, 42,5%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,37 (с, 1Н), 8,27 (дд, J=7,1, 1,0 Гц, 1Н), 7,80 (с, 1Н), 7,60 (дд, J=7,2, 1,7 Гц, 1Н), 7,44 (тд, J=8,3, 6,3 Гц, 1Н), 7,15 (тдд, J=8,3, 2,5, 1,1 Гц, 1Н), 7,11-6,81 (м, 3Н), 5,07 (д, J=2,3 Гц, 2Н), 4, 74-4, 62 (м, 1Н), 4,33-4,12 (м, 2Н), 3,92 (т, J=9,8 Гц, 1Н), 3,53 (тт, J=9,1, 6,5 Гц, 1Н);
МСНР (ЭР) m/z 539,0 (М++1).
Пример 148: Синтез Соединения 4203, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-1-(метилсульфонил)-азетидин-3-карбоксамида
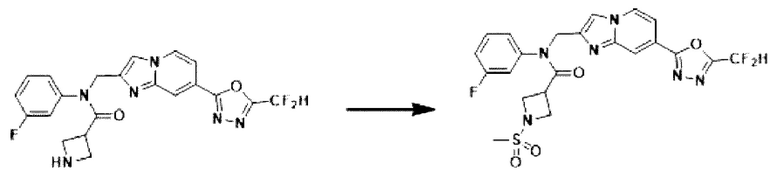
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-азетидин-3-карбоксамид (0,060 г, 0,136 ммоль), полученный на стадии 2 примера 133, метансульфонилхлорид (0,021 мл, 0,271 ммоль) и триэтиламин (0,057 мл, 0,407 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,030 г, 42,5%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,37 (с, 1Н), 8,27 (дд, J=7,2, 1,0 Гц, 1Н), 7,79 (д, J=0,7 Гц, 1Н), 7,61 (дд, J=7,1, 1,7 Гц, 1Н), 7,5 0-7,3 6 (м, 1Н), 7,19-7,10 (м, 1Н), 7,10-6,7 9 (м, 3Н), 5,07 (с, 2Н), 4,17 (дд, J=8,1, 7,1 Гц, 2Н), 3,75 (т, J=8,4 Гц, 2Н), 3,49-3,35 (м, 1Н), 2,91 (с, 3Н); МСНР (ЭР) m/z 521,1 (М++1).
Пример 149: Синтез соединения 4204, метил-3-(((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-(3-фторфенил)-карбамоил)-азетидин-1-карбоксилата
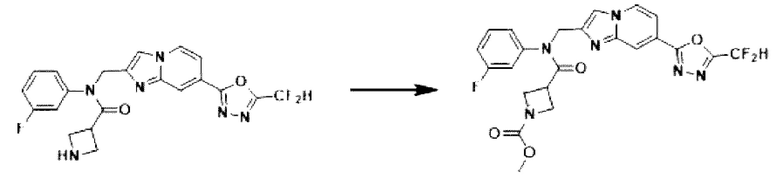
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-азетидин-3-карбоксамид (0,050 г, 0,113 ммоль), полученный на стадии 2 примера 133, метилхлорформиат (0,021 г, 0,226 ммоль) и триэтиламин (0,047 мл, 0,339 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,031 г, 54,8%) в виде твердого вещества белого цвета.
1Н ЯМР (400 МГц, CDCl3) δ 8,37-8,33 (м, 1Н), 8,26 (дд, J=7,1, 1,0 Гц, 1Н), 7,82 (с, 1Н), 7,59 (дд, J=7,2, 1,8 Гц, 1Н), 7,41 (тд, J=8,3, 6,2 Гц, 1Н), 7,17-6,78 (м, 4Н), 5,06 (с, 2Н), 4,19 (с, 2Н), 3,78 (т, J=8,6 Гц, 2Н), 3,65 (с, 3Н), 3,37 (тт, J=8,9, 6,4 Гц, 1Н);
МСНР (ЭР) m/z 501,0 (М++1).
Пример 150: Синтез Соединения 4205, N3-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N3-(3-фторфенил)-N1,N1-диметилазетидин-1,3-дикарбоксамида
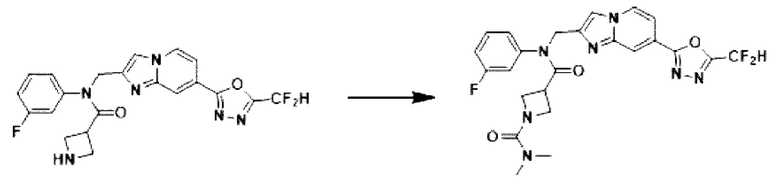
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-азетидин-3-карбоксамид (0,050 г, 0,113 ммоль), полученный на стадии 2 примера 133, хлорангидрид диметилкарбаминовой кислоты (0,024 г, 0,226 ммоль) и триэтиламин (0,047 мл, 0,339 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,024 г, 41,4%) в виде твердого вещества белого цвета.
1Н ЯМР (400 МГц, CDCl3) δ 8,34 (дд, J=1,8, 0,9 Гц, 1Н), 8,26 (дд, J=7,1, 1,0 Гц, 1Н), 7,82 (с, 1Н), 7,58 (дд, J=7,2, 1,7 Гц, 1Н), 7,40 (тд, J=8,3, 6,3 Гц, 1Н), 7,15-6,77 (м, 4Н), 5,05 (с, 2Н), 4,17 (дд, J=8,0, 6,7 Гц, 2Н), 3,74 (т, J=8,5 Гц, 2Н), 3,36 (тт, J=8,9, 6,7 Гц, 1Н), 2,82 (с, 6Н);
МСНР (ЭР) m/z 514,0 (М++1).
Пример 151: Синтез Соединения 4206, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-1-(пиридин-2-ил)-азетидин-3-карбоксамида
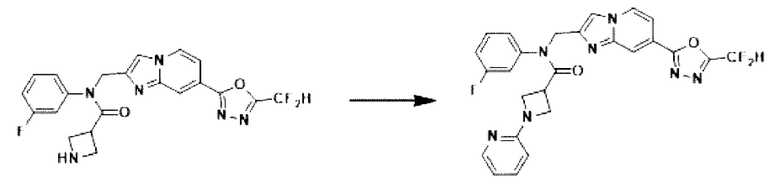
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-азетидин-3-карбоксамид (0,050 г, 0,113 ммоль), полученный на стадии 2 примера 133, 2-хлорпиридин (0,026 г, 0,226 ммоль), карбонат цезия (0,074 г, 0,226 ммоль) и палладий RuPhos G2 (0,004 г, 0,006 ммоль) растворяли в 1,4-диоксане (2 мл) при комнатной температуре, и полученный раствор перемешивали при 100°С в течение 18 часов.
Затем температуру понижали до комнатной температуры для завершения реакции. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 3 0%) и концентрировали с получением целевого соединения (0,012 г, 20,4%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,36-8,30 (м, 1Н), 8,24 (ддд, J=7,1, 6,0, 1,0 Гц, 1Н), 8,12 (ддд, J=5,1, 1,9, 0,9 Гц, 1Н), 7,80 (д, J=8,0 Гц, 1Н), 7,59-7,51 (м, 1Н), 7,50-7,37 (м, 2Н), 7,15-6,81 (м, 4Н), 6,62 (ддд, J=7,2, 5,0, 1,0 Гц, 1Н), 6,29 (д, J=8,4 Гц, 1Н), 5,06 (с, 2Н), 4,21 (т, J=7,1 Гц, 2Н), 3,88 (т, J=8,2 Гц, 2Н), 3,64 - 3,54 (м, 1Н); МСНР (ЭР) m/z 520,5 (М++1).
Пример 152: Синтез Соединения 4207, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-1-(пиримидин-2-ил)-азетидин-3-карбоксамида
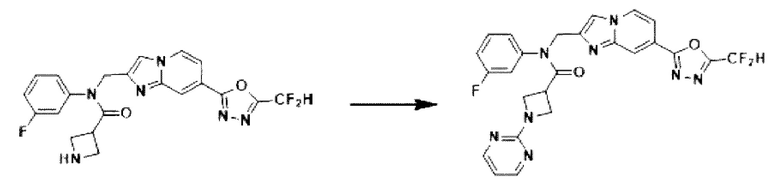
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-азетидин-3-карбоксамид (0,050 г, 0,113 ммоль), полученный на стадии 2 примера 133, 2-хлорпиримидин (0,026 г, 0,226 ммоль) и карбонат калия (0,031 г, 0,226 ммоль) растворяли в ацетонитриле (2 мл)/N,N-диметилформамиде (2 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В концентрат, полученный путем удаления растворителя из реакционной смеси при пониженном давлении, вливали насыщенный водный раствор хлорида аммония с последующей экстракцией дихлорметаном. Затем полученный продукт фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,041 г, 69,7%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,35-8,29 (м, 3Н), 8,24 (дд, J=7,1, 1,0 Гц, 1Н), 7,82 (с, 1Н), 7,56 (дд, J=7,1, 1,7 Гц, 1Н), 7,41 (тд, J=8,2, 6,3 Гц, 1Н), 7,16-6,80 (м, 4Н), 6,56 (т, J=4,8 Гц, 1Н), 5,07 (с, 2Н), 4,36 (дд, J=8,6, 6,5 Гц, 2Н), 4,00 (т, J=8,6 Гц, 2Н), 3,54 (тт, J=8,7, 6,4 Гц, 1Н);
МСНР (ЭР) m/z 521,4 (М++1).
Пример 153: Синтез Соединения 4618, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-1-(спиро[3,3]гептан-2-ил)-пиперидин-4-карбоксамида
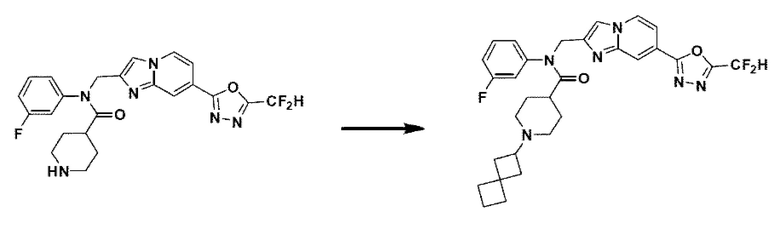
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперидин-4-карбоксамид (0,04 0 г, 0,085 ммоль), полученный на стадии 3 примера 93, спиро[3,3]гептан-2-он (0,019 г, 0,170 ммоль), уксусную кислоту (0,011 мл, 0,085 ммоль) и триацетоксиборгидрид натрия (0,054 г, 0,255 ммоль) растворяли в дихлорметане (5 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 3 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,020 г, 41,7%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,30 (с, 1Н), 8,23 (д, J=7,1 Гц, 1Н), 7,55 (д, J=6,9 Гц, 1Н), 7,39 (дд, J=14,6, 8,2 Гц, 1Н), 7,14-6,79 (м, 5Н), 5,01 (с, 2Н), 3,13 (д, J=41,6 Гц, 1Н), 2,84 (с, 2Н), 2,24 (с,2Н), 1,89 (дт, J=53,6, 40,7 Гц, 12Н), 1,57-1,37 (м, 2Н);
МСНР (ЭР) m/z 565,5 (М++1).
Пример 154: Синтез Соединения 4619, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-1-(2-оксаспиро[3.3]гептан-6-ил)-пиперидин-4-карбоксамида
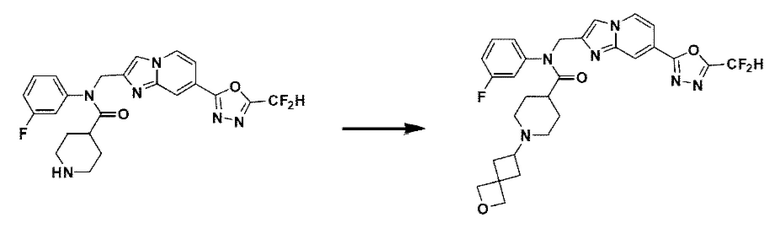
N-((7-(5-(дифторметил)-1,3,4-океадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперидин-4-карбоксамид (0,040 г, 0,085 ммоль), полученный на стадии 3 примера 93, 2-оксаспиро[3,3]гептан-6-он (0,019 г, 0,170 ммоль), уксусную кислоту (0,005 мл, 0,085 ммоль) и триацетоксиборгидрид натрия (0,054 г, 0,255 ммоль) растворяли в дихлорметане (5 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 3 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,020 г, 41,5%) в виде твердого вещества белого цвета.
1Н ЯМР (400 МГц, CDCl3) δ 8,30 (с, 1Н), 8,23 (д, J=7,1 Гц, 1Н), 7,55 (д, J=7,1 Гц, 1Н), 7,39 (дд, J=14,5, 8,1 Гц, 1Н), 7,14-6,79 (м, 5Н), 5,01 (с, 2Н), 4,69 (с, 2Н), 4,59 (с, 2Н), 3,23 (с, 1Н), 2,83 (с, 2Н), 2,30 (д, J=47,7 Гц, 4Н), 2,04 (с, 2Н), 1,96-1,77 (м, 2Н), 1,50 (с, 2Н);
МСНР (ЭР) m/z 568,2 (М++1).
Пример 155: Синтез Соединения 4620, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-1-(спиро[3,3]гептан-2-ил)-пиперидин-4-карбоксамида
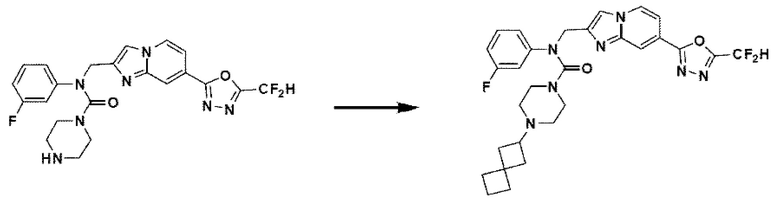
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперазин-1-карбоксамид (0,040 г, 0,085 ммоль), полученный на стадии 2 примера 54, спиро[3,3] гептан-2-он (0,019 г, 0,170 ммоль), уксусную кислоту (0,005 мл, 0,085 ммоль) и триацетоксиборгидрид натрия (0,054 г, 0,255 ммоль) растворяли в дихлорметане (5 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 3 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,020 г, 41,7%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,28 (с, 1Н), 8,20 (д, J=7,1 Гц, 1Н), 7,74 (с, 1H), 7,51 (дд, J=7,1, 1,6 Гц, 1H), 7,32-7,23 (м, 1H), 7,09-6,76 (м, 4H), 5,06 (с, 2Н), 3,33 (с, 4Н), 2,14 (д, J=27,7 Гц, 6Н), 2, 03-1, 94 (м, 3Н), 1, 94-1, 8 6 (м, 2Н), 1,82 (дт, J=8,1, 6,0 Гц, 3Н);
МСНР (ЭР) m/z 566,4 (М++1).
Пример 156: Синтез Соединения 4621, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-1-(2-оксаспиро[3.3]гептан-6-ил)-пиперидин-4-карбоксамида
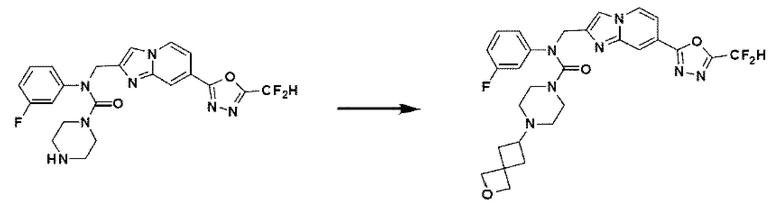
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-(3-фторфенил)-пиперазин-1-карбоксамид (0,040 г, 0,085 ммоль), полученный на стадии 2 примера 54, 2-оксаспиро [3,3] гептан-6-он (0,019 г, 0,170 ммоль), уксусную кислоту (0,005 мл, 0,085 ммоль) и триацетоксиборгидрид натрия (0,054 г, 0,255 ммоль) растворяли в дихлорметане (5 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 3 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 10%) и концентрировали с получением целевого соединения (0,020 г, 41,5%) в виде твердого вещества белого цвета.
1Н ЯМР (400 МГц, CDCl3) δ 8,28 (с, 1Н), 8,20 (д, J=7,1 Гц, 1Н), 7,74 (с, 1Н), 7,51 (дд, J=7,1, 1,6 Гц, 1Н), 7,33-7,23 (м, 1Н), 7,09-6,75 (м, 4Н), 5,05 (с, 2Н), 4,69 (с, 2Н), 4,58 (с, 2Н), 3,30 (с, 4Н), 2,46 (с, 1Н), 2,37 (с, 2Н), 2,25-2,04 (м, 3Н), 1,95 (д, J=17,0 Гц, 2Н);
МСНР (ЭР) m/z 568,0 (М++1).
Пример 157: Синтез Соединения 4625, N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-6-метил-N-фенил-2,6-диазаспиро[3.3]гептан-2-карбоксамида
[Стадия 1] Синтез трет-бутил-6-(((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-(фенил)-карбамоил)-2,6-диазаспиро[3,3]гептан-2-карбоксилата
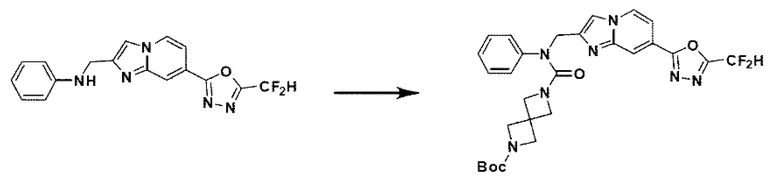
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-анилин (0,200 г, 0,586 ммоль), полученный в Примере 14, трет-бутил-2,6-диазаспиро[3.3] гептан-2-карбоксилат (0,081 г, 0,410 ммоль), трифосген (0,174 г, 0,586 ммоль) и N,N-диизопропилэтиламин (0,510 мл, 2,930 ммоль) растворяли в дихлорметане (10 мл), и полученный раствор перемешивали при 0°С в течение 1 часа и дополнительно перемешивали при комнатной температуре в течение 18 часов. В реакционную смесь вливали воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором, сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (картридж SiO2, 12 г; этилацетат/гексан = от 0% до 60%) и концентрировали с получением целевого соединения (0,210 г, 63,4%) в виде твердого вещества коричневого цвета.
[Стадия 2] Синтез N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил-N-фенил-2,6-диазаспиро[3.3]гептан-2-карбоксамида
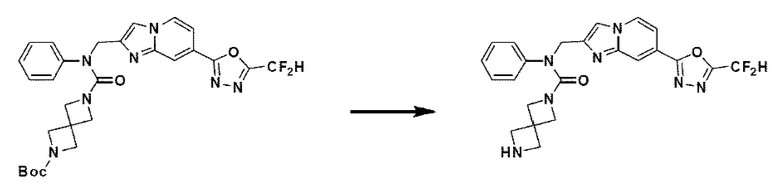
Трет-бутил-6-(((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-(фенил)-карбамоил)-2,6-диазаспиро[3.3]гептан-2-карбоксилат (0,210 г, 0,371 ммоль), полученный на стадии 1, и трифторуксусную кислоту (0,569 мл, 7,426 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 2 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным водным раствором, сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении, и получали целевое соединение (0,172 г, 99,5%) в виде желтого геля без дополнительной очистки.
[Стадия 3] Синтез соединения 4625
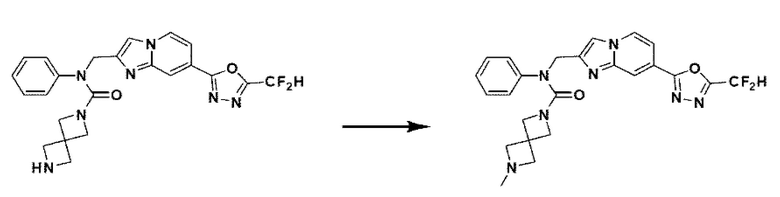
N-((7-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-N-фенил-2,6-диазаспиро[3.3]гептан-2-карбоксамид (0,172 г, 0,370 ммоль), полученный на стадии 2, формальдегид (0,022 г, 0,739 ммоль), уксусную кислоту (0,021 мл, 0,370 ммоль) и триацетоксиборгидрид натрия (0,235 г, 1,109 ммоль) растворяли в дихлорметане (5 мл) при комнатной температуре, и полученный раствор перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали насыщенный водный раствор гидрокарбоната натрия, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; дихлорметан/метанол = от 0% до 2 0%) и концентрировали с получением целевого соединения (0,100 г, 56,4%) в виде твердого вещества желтого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,27 (с, 1Н), 8,19 (дт, J=7,8, 3,9 Гц, 1Н), 7,81 (с, 1Н), 7,50 (дд, J=7,1, 1,7 Гц, 1Н), 7,38-7,29 (м, 4Н), 7,25-7,19 (м, 1Н), 6,94 (дд, J=54,3, 49,1 Гц, 1Н), 5,02 (с, 2Н), 3,64 (с, 4Н), 3,18 (с, 4Н), 2,21 (с, 3Н);
МСНР (ЭР) m/z 480,3 (М++1).
Пример 158: Синтез Соединения 6892, трет-бутил-4-((3-фторфенил)-((7-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-карбамоил)-пиперазин-1-карбоксилата
[Стадия 1] Синтез трет-бутил-(4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)-пиридин-2-ил)-карбамата
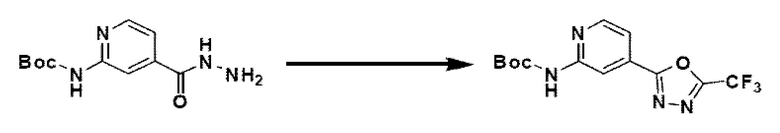
Трет-бутил-(4-(гидразинкарбонил)-пиридин-2-ил)-карбамат (2,600 г, 10,306 ммоль), полученный на стадии 2 примера 2, и триэтиламин (14,365 мл, 103,064 ммоль) растворяли в тетрагидрофуране (150 мл), добавляли трифторуксусный ангидрид (7,279 мл, 51,532 ммоль) при комнатной температуре и нагревали с обратным холодильником в течение 16 часов. Затем температуру понижали до комнатной температуры для завершения реакции. К реакционной смеси добавляли насыщенный водный раствор хлорида аммония с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Этилацетат (30 мл) и гексан (10 0 мл) вливали в концентрат, суспендировали и фильтровали с получением твердого вещества, полученное твердое вещество промывали гексаном и сушили с получением целевого соединения (1,500 г, 44,1%) в виде твердого вещества белого цвета.
[Стадия 2] Синтез 4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)-пиридин-2-амина
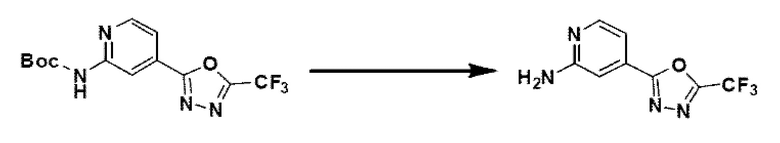
Трет-бутил-(4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)-пиридин-2-ил)-карбамат (1,500 г, 4,542 ммоль), полученный на стадии 1, растворяли в дихлорметане (70 мл). Затем добавляли трифторуксусную кислоту (6,956 мл, 90,835 ммоль) при 0°С, и полученный раствор перемешивали при комнатной температуре в течение 4 часов. После удаления растворителя из реакционной смеси при пониженном давлении в концентрат вливали насыщенный водный раствор гидрокарбоната натрия (50 мл) и суспендировали с последующей фильтрацией с получением твердого вещества. Полученное твердое вещество промывали водой и сушили, в результате чего получали целевое соединение (1,030 г, 98,5%) в виде твердого вещества желтого цвета.
[Стадия 3] Синтез 2-(2-(хлорметил)-имидазо[1,2-а]пиридин-7-ил)-5-(трифторметил)-1,3,4-оксадиазола
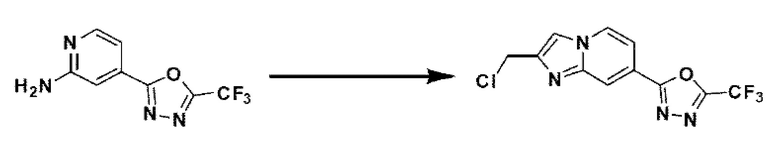
4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)-пиридин-2-амин (1,100 г, 4,779 ммоль), полученный на стадии 2, 1,3-дихлорпропан-2-он (1,214 г, 9,559 ммоль) и гидрокарбонат натрия (2,008 г, 23,897 ммоль) растворяли в 1,4-диоксане (60 мл) при комнатной температуре. Реакционную смесь нагревали с обратным холодильником в течение 1 6 часов, а затем понижали температуру до комнатной температуры для прекращения реакции. Реакционную смесь фильтровали через пластиковый фильтр для удаления твердых веществ, фильтрат очищали с помощью колоночной хроматографии (картридж SiO2, 4 0 г; этилацетат/гексан = от 5% до 7 0%) и концентрировали с получением целевого соединения (0,850 г, 5 8,8%) в виде твердого вещества бежевого цвета.
[Стадия 4] Синтез 3-фтор-N-((7-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-анилина
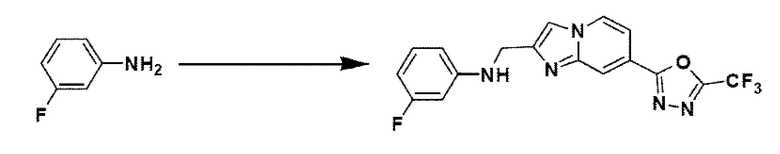
3-(2-(хлорметил)-имидазо[1,2-а]пиридин-7-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,311 г, 1,028 ммоль), полученный на стадии 3, 3-фторанилин (0,228 г, 2,055 ммоль), карбонат калия (0,213 г, 1,541 ммоль) и иодид калия (0,085 г, 0,514 ммоль) растворяли в N,N-диметилформамиде (6 мл) при комнатной температуре, и полученный раствор перемешивали при 60°С в течение 18 часов. Затем температуру понижали до комнатной температуры для завершения реакции. В реакционную смесь вливали водный раствор хлорида аммония с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором, сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; этилацетат/гексан = от 0% до 5 0%) и концентрировали с получением целевого соединения (0,050 г, 12,9%) в виде твердого вещества желтого цвета.
[Стадия 5] Синтез соединения 6892
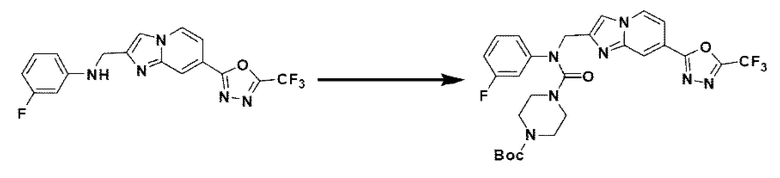
3-фтор-N-((7-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)-имидазо[1,2-а]пиридин-2-ил)-метил)-анилин (0,050 г, 0,133 ммоль), полученный на стадии 4, бис-(трихлорметил)-карбонат (0,039 г, 0,133 ммоль) и N,N-диизопропилэтиламин (0,115 мл, 0,663 ммоль) растворяли в дихлорметане (4 мл), и полученный раствор перемешивали при комнатной температуре в течение 10 минут. Затем добавляли трет-бутилпиперазин-1-карбоксилат (0,032 г, 0,172 ммоль) и дополнительно перемешивали при той же температуре в течение 18 часов. В реакционную смесь вливали воду, экстрагировали дихлорметаном и фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии (SiO2 пластина 20×20×1 мм; этилацетат/гексан = от 0% до 100%) и концентрировали с получением целевого соединения (0,040 г, 51,3%) в виде твердого вещества коричневого цвета.
1H ЯМР (400 МГц, ацетон-d6) δ 8,69 (д, J=7,1 Гц, 1Н), 8,25 (д, J=1,7 Гц, 1Н), 8,08 (с, 1Н), 7,53 (дд, J=7,1, 1,8 Гц, 1Н), 7,36 (тд, J=8,3, 6,7 Гц, 1Н), 7,14 (ддт, J=8,0, 5,3, 2,3 Гц, 2Н), 6,86 (тд, J=8,4, 2,4 Гц, 1Н), 5,09 (с, 2Н), 3,28 (квд, J=6,2, 5,0, 2,5 Гц, 8Н), 1,42 (с, 10Н);
МСНР (ЭР) m/z 591,1 (М++1).
Протокол измерения и анализа активности соединений согласно настоящему изобретению
<Экспериментальный пример 1> Анализ ингибирования активности фермента HDAC in vitro
Чтобы подтвердить селективность соединений, представленных химической формулой I согласно настоящему изобретению, к HDAC6 в экспериментах по ингибированию активности ферментов HDAC1 и HDAC6, был проведен эксперимент сравнения с использованием материала, который уже был разработан в качестве контрольной группы.
Ферментную активность HDAC измеряли с использованием набора HDAC Fluorimetric Drug Discovery Kit (Enzo Life Sciences, Inc., BML-AK511, 516). Для теста активности фермента HDAC1 использовали рекомбинантный HDAC1 человека (BML-SE456) в качестве источника фермента и Fluor de Lys®-SIRT1 (BNL-KI177) в качестве субстрата. После внесения 5-кратного разведения соединений в 9 6-луночный планшет в каждую лунку планшета добавляли 0,3 мкг фермента и 10 мкМ субстрата и оставляли для осуществления реакции при 30°С в течение 60 минут. Затем добавляли Fluor de Lys® Developer II (BML-KI176) и осуществляли взаимодействие в течение 30 минут для завершения реакции, а затем измеряли значения флуоресценции (Ех 360, Em 460) с использованием многопланшетного ридера (Flexstation 3, Molecular Device). Ферменты HDAC6 тестировали с использованием рекомбинантного HDAC6 (382180) человека от Calbiochem Inc., в соответствии с тем же протоколом, что и метод испытания активности фермента HDAC1. Что касается конечных значений результатов, соответствующие значения IC50 рассчитывали с использованием программы GraphPad Prism 4.0, и их результаты приведены ниже в таблице 5.

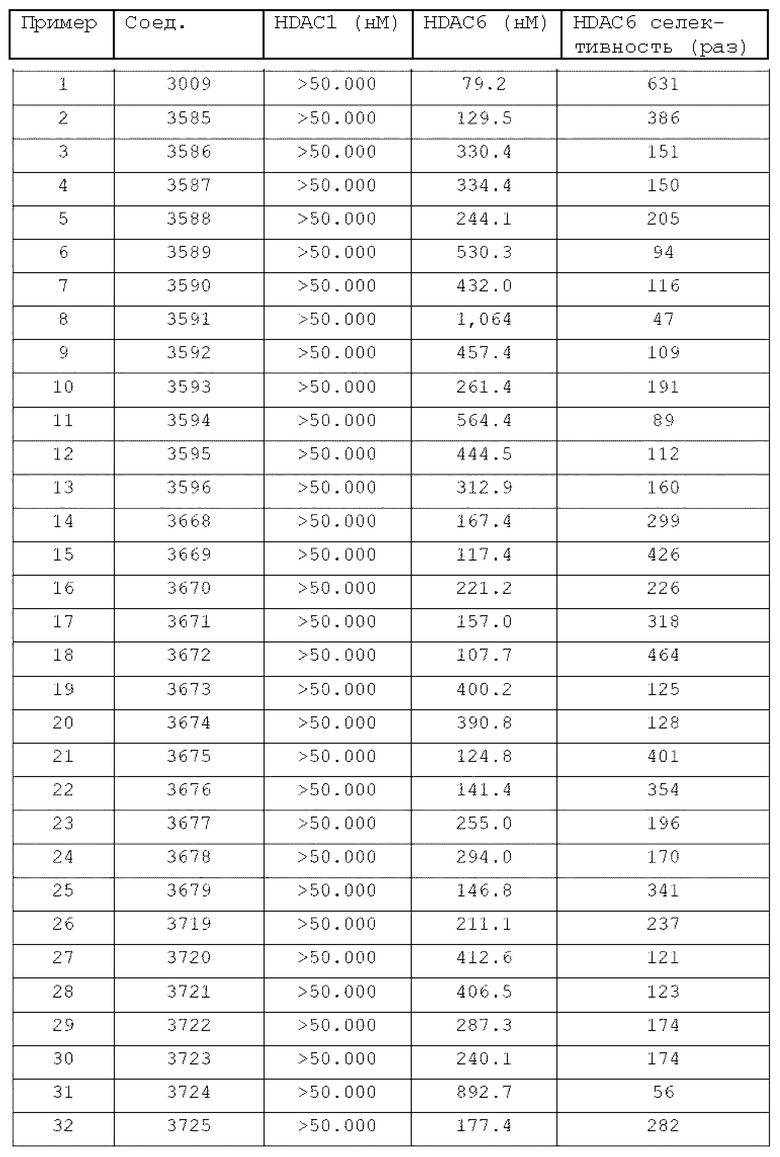
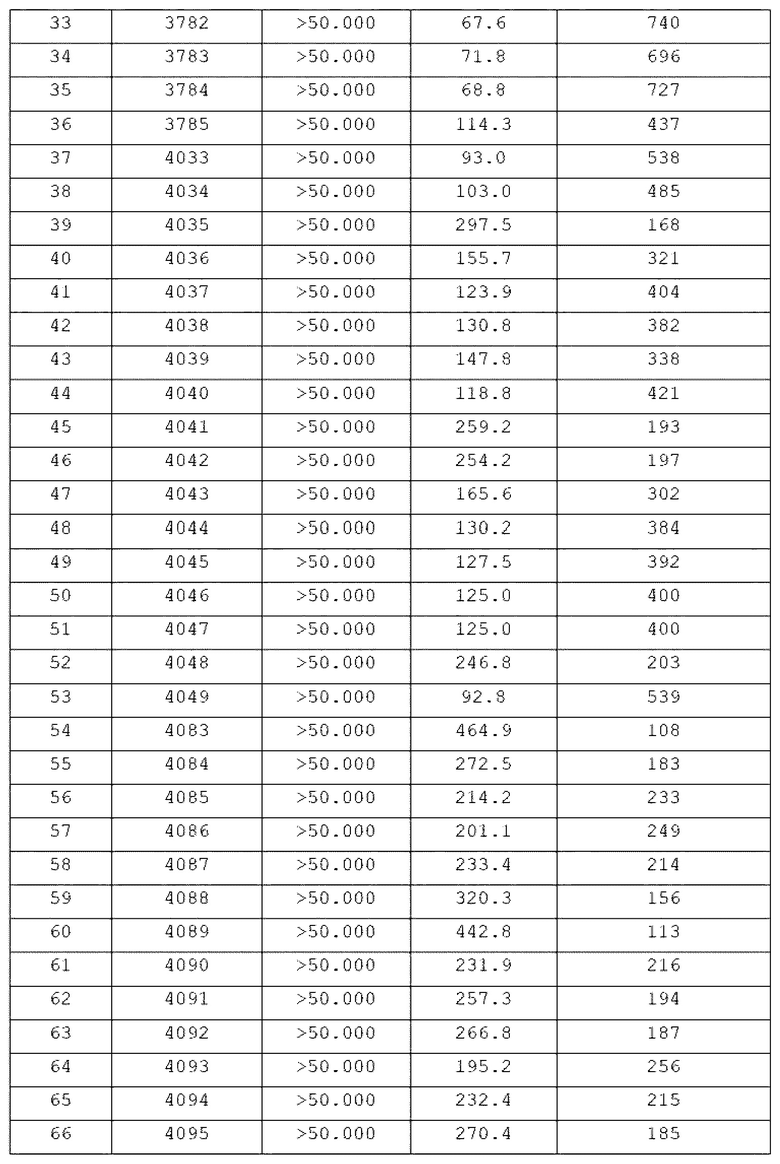
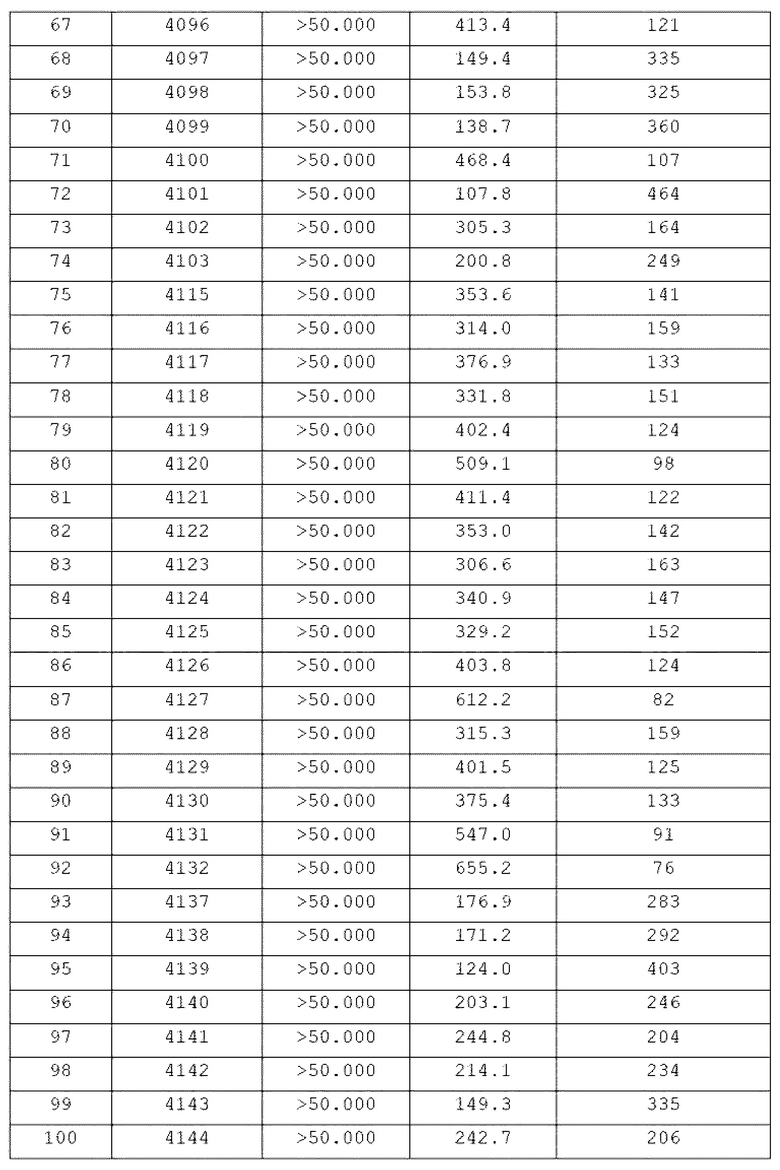
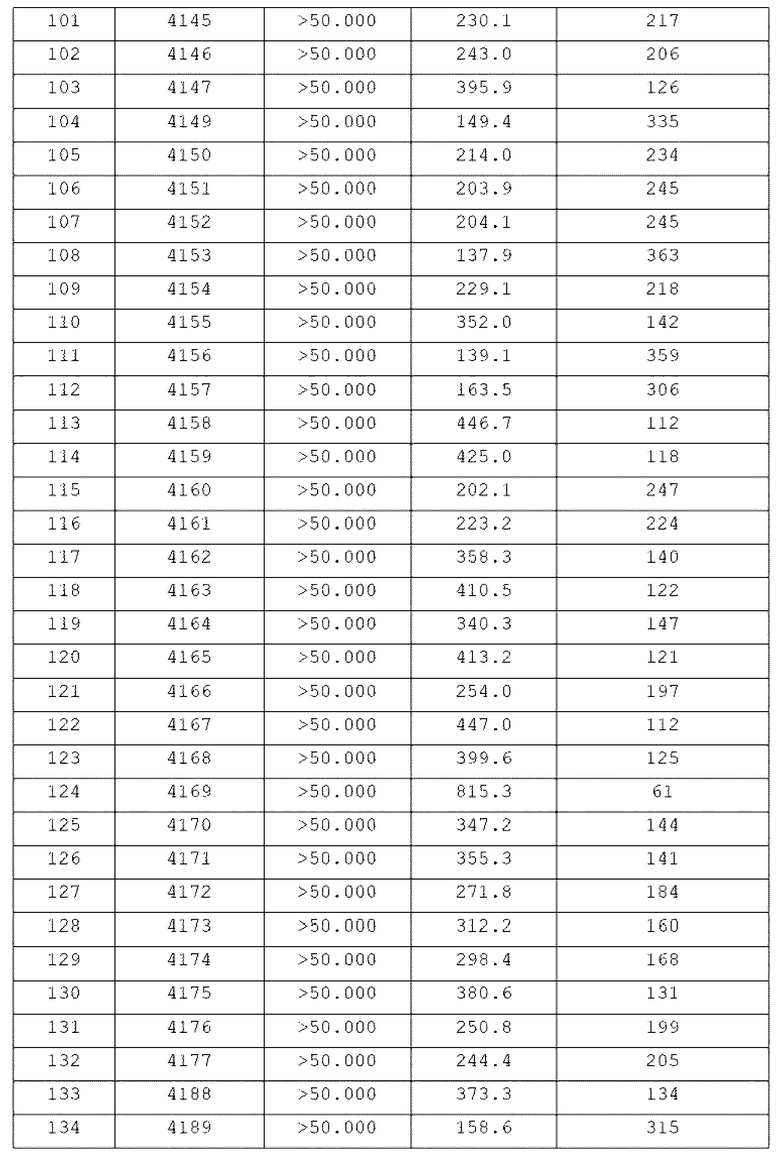
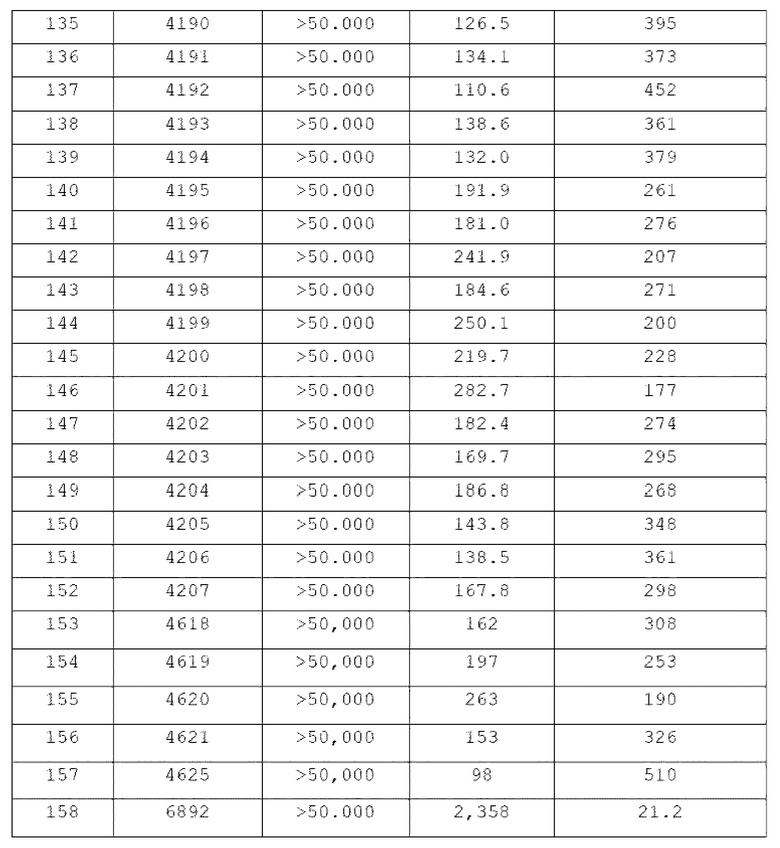
Как показано в таблице 5 выше, из результатов анализа ингибирования активности для HDAC1 и HDAC6 было обнаружено, что производные 1,3,4-оксадиазола согласно настоящему изобретению, их оптический изомер или их фармацевтически приемлемая соль проявляли селективную ингибирующую активность HDAC6, которая от примерно 21 до примерно 740 раз выше, чем у HDAC1.
<Экспериментальный пример 2> Анализ эффекта in vitro HDAC6-специфических ингибиторов на митохондриальный аксональный транспорт
Было проанализировано влияние специфических ингибиторов HDAC6 на митохондриальный аксональный транспорт. В частности, чтобы подтвердить, действительно ли соединения, представленные химической формулой I согласно настоящему изобретению, селективно ингибируют активность HDAC6 и увеличивают ацетилирование тубулина, который является основным субстратом HDAC6, тем самым улучшая скорость митохондриального аксонального транспорта, сниженную при обработке амилоидом-бета в нейронных аксонах, был проведен эксперимент сравнения с использованием материала, который уже был разработан в качестве контрольной группы.
Гиппокампальные нейроны из эмбрионов крыс линии Спраг-Доули (SD) на 17-18 день эмбриона (Е17-18) культивировали в течение 7 дней во внеклеточной культуральной чашке, покрытой матриксом, для визуализации, а затем обрабатывали 1М пептидными фрагментами амилоида-бета. Через 24 часа обрабатывали соединением на 8 день культивирования in vitro, а через 3 часа обрабатывали MitoTracker Red CMXRos (Life Technologies, Нью-Йорк, США) в течение последних 5 минут для окрашивания митохондрий. Что касается аксонального транспорта окрашенных митохондрий нейронов, скорости транспорта каждой митохондрии определяли, используя программное обеспечение для анализа IMARIS (BITPLANE, Цюрих, Швейцария), путем получения изображений, используя конфокальный микроскоп (Leica 5Р8; Leica Microsystems, Великобритания) с интервалами в 1 секунду в течение 1 минуты.
В результате было подтверждено, что производное 1,3,4-оксадиазола согласно настоящему изобретению, его оптический изомер или его фармацевтически приемлемые соли продемонстрировали улучшающий эффект на скорости митохондриального аксонального транспорта.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ 1,3,4-ОКСАДИАЗОЛА В КАЧЕСТВЕ ИНГИБИТОРА ГИСТОНДЕАЦЕТИЛАЗЫ 6 И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2021 |
|
RU2822180C1 |
| ПРОИЗВОДНЫЕ 1,3,4-ОКСАДИАЗОЛА В КАЧЕСТВЕ ИНГИБИТОРА ГИСТОНДЕАЦЕТИЛАЗЫ 6 И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2021 |
|
RU2810081C1 |
| НОВЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРА ГИСТОНДЕАЦЕТИЛАЗЫ 6 И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2021 |
|
RU2814288C1 |
| НОВЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРА ГИСТОНДЕАЦЕТИЛАЗЫ 6 И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ИХ | 2021 |
|
RU2817736C1 |
| 1,3,4,-ОКСАДИЗОЛАМИДНОЕ ПРОИЗВОДНОЕ СОЕДИНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРА ГИСТОНДЕАЦЕТИЛАЗЫ 6 И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2016 |
|
RU2700696C2 |
| СПИРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ | 2016 |
|
RU2745069C2 |
| ЗАМЕЩЕННЫЕ ИМИДАЗОПИРИДИНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ HDM2 | 2013 |
|
RU2690663C2 |
| НОВОЕ ПРОИЗВОДНОЕ ПИПЕРИДИНА И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ИНГИБИРОВАНИЯ АУТОТАКСИНА | 2022 |
|
RU2834850C2 |
| ИМИДАЗОПИРИДИНИЛЬНЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ НЕЙРОДЕГЕНЕРАТИВНЫХ РАССТРОЙСТВ | 2020 |
|
RU2833052C2 |
| ПРОИЗВОДНЫЕ 1,3,4-ОКСАДИАЗОЛСУЛЬФАМИДА В КАЧЕСТВЕ ИНГИБИТОРА ГИСТОНДЕАЦЕТИЛАЗЫ 6 И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2016 |
|
RU2695227C1 |
Настоящее изобретение относится к производным 1,3,4-оксадиазола, обладающим ингибирующей активностью в отношении гистондеацетилазы 6 (HDAC6), а именно к соединению формулы I или его фармацевтически приемлемой соли, в котором L1, L2 и L3 каждый независимо представляет собой -(C0-C1алкил)-; a, b и c каждый независимо представляет собой CR4 и R4 представляет собой -H; Z представляет собой N, O или отсутствует, при этом, когда Z отсутствует, R2 также отсутствует, и L2 и L3 связаны напрямую; R1 представляет собой -CF2Н или -CX3; R2 представляет собой -H, -(C1-C4алкил) или -C(=O)-RA, при этом, когда Z представляет собой O, R2 отсутствует; RA представляет собой
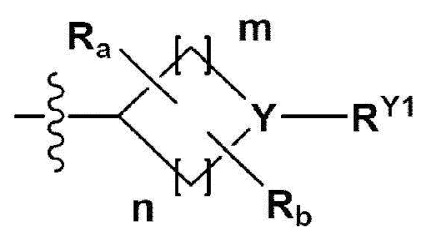 ,
, 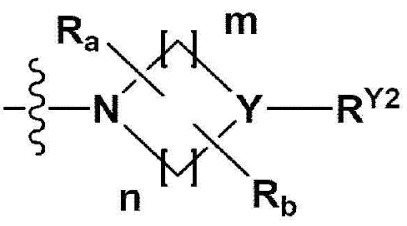 или
или  ;
;
Y представляет собой N, когда Y представляет собой N, каждый из RY1, RY2 и RY4 независимо представляет собой -(C1-C4алкил), -(C3-C7циклоалкил), -(C2-C6гетероциклоалкил), содержащий атом N или O в кольце, -C(=O)-(C1-C4алкил), -C(=O)-O(C1-C4алкил), -C(=O)-NRA3RA4, -C(=O)-(C3-C7циклоалкил), -C(=O)-(C2-C6гетероциклоалкил), содержащий один атом O в кольце, -S(=O)2-(C1-C4алкил), -С4-С5гетероарил, содержащий один или два атома N в кольце, или
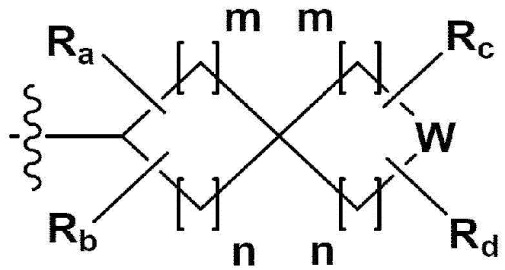 ,
,
где по меньшей мере один H -(C1-C4алкила), -(C3-C7циклоалкила) и -C(=O)-(C1-C4алкила) может быть замещен -X или -OH; и W представляет собой CH2 или O; m и n каждый независимо представляет собой целое число 1 или 2; Ra - Rd каждый независимо представляет собой -H; R3 представляет собой -(C3-C7циклоалкил), -адамантил, -С6арил, -С5гетероарил, содержащий один атом N в кольце, или С7гетероарил, содержащий два атома O в кольце, где по меньшей мере один -H -С6арила или -С5гетероарила, содержащего один атом N в кольце, может быть каждый независимо замещен группами -X, -(C1-C4алкил), -O(C1-C4алкил), -(C=O)-(C1-C4алкил), -CF3, или -S(=O)2-(C1-C4алкил); RA3 и RA4 каждый независимо представляет собой -(C1-C4алкил); и X представляет собой F, Cl или Br. Изобретение также относится к фармацевтической композиции, способу ингибирования HDAC6 и применению для получения лекарственного средства для ингибирования HDAC6 на основе указанных соединений. Технический результат заключается в получении альтернативных соединений, обладающих ингибирующей активностью к HDAC6, которые могут найти применение в медицине для лечения HDAC6-опосредованных заболеваний. 4 н. и 9 з.п. ф-лы, 5 табл., 158 пр.
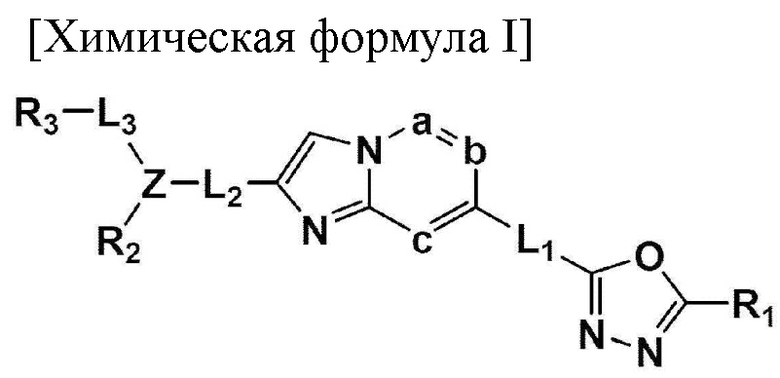
1. Производное 1,3,4-оксадиазола, обладающее селективной ингибирующей активностью в отношении гистондеацетилазы 6 (HDAC6), представленное химической формулой I, или его фармацевтически приемлемая соль:
[Химическая формула I]
 ,
,
где в вышеуказанной химической формуле I:
L1, L2 и L3 каждый независимо представляет собой -(C0-C1алкил)-;
a, b и c каждый независимо представляет собой CR4 и R4 представляет собой -H;
Z представляет собой N, O или отсутствует, при этом, когда Z отсутствует, R2 также отсутствует и L2 и L3 связаны напрямую;
R1 представляет собой -CF2Н или -CX3;
R2 представляет собой -H, -(C1-C4алкил) или -C(=O)-RA, при этом, когда Z представляет собой O, R2 отсутствует;
RA представляет собой
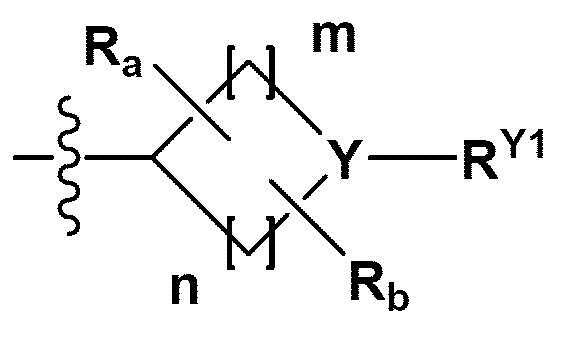 ,
, 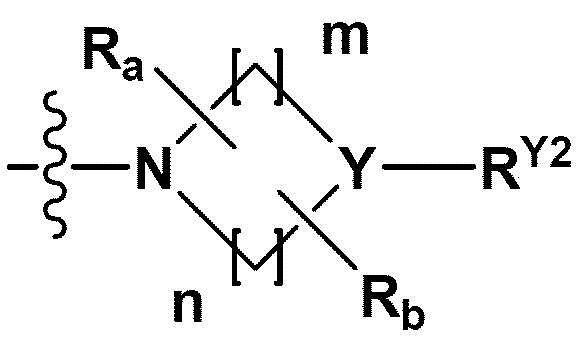
или 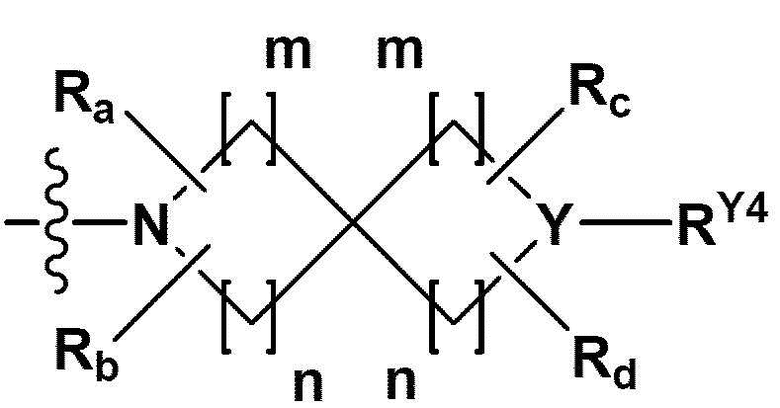 ;
;
Y представляет собой N,
когда Y представляет собой N, каждый из RY1, RY2 и RY4 независимо представляет собой -(C1-C4алкил), -(C3-C7циклоалкил), -(C2-C6гетероциклоалкил), содержащий атом N или O в кольце, -C(=O)-(C1-C4алкил), -C(=O)-O(C1-C4алкил), -C(=O)-NRA3RA4, -C(=O)-(C3-C7циклоалкил), -C(=O)-(C2-C6гетероциклоалкил), содержащий один атом O в кольце, -S(=O)2-(C1-C4алкил), -С4-С5гетероарил, содержащий один или два атома N в кольце, или
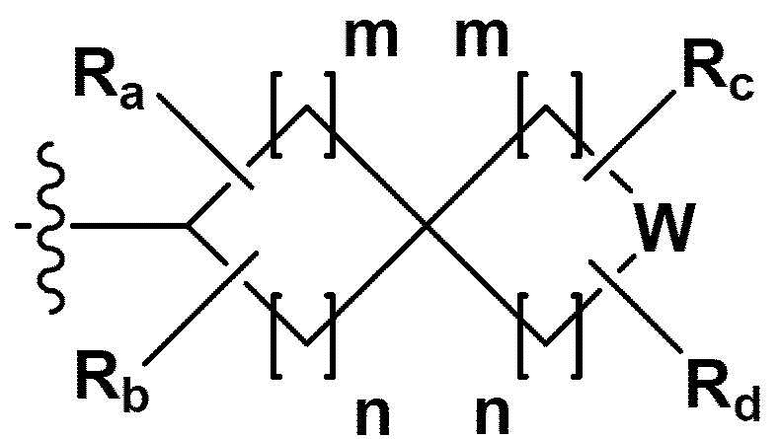 ,
,
где по меньшей мере один H -(C1-C4алкила), -(C3-C7циклоалкила) и -C(=O)-(C1-C4алкила) может быть замещен -X или -OH; и W представляет собой CH2 или O;
m и n каждый независимо представляет собой целое число 1 или 2;
Ra - Rd каждый независимо представляет собой -H;
R3 представляет собой -(C3-C7циклоалкил), -адамантил, -С6арил, -С5гетероарил, содержащий один атом N в кольце, или -С7гетероарил, содержащий два атома O в кольце, где по меньшей мере один -H -С6арила или -С5гетероарила, содержащего один атом N в кольце, может быть каждый независимо замещен группами -X, -(C1-C4алкил), -O(C1-C4алкил), -(C=O)-(C1-C4алкил), -CF3 или -S(=O)2-(C1-C4алкил);
RA3 и RA4 каждый независимо представляет собой -(C1-C4алкил); и
X представляет собой F, Cl или Br.
2. Производное 1,3,4-оксадиазола, обладающее селективной ингибирующей активностью в отношении гистондеацетилазы 6 (HDAC6), представленное химической формулой I, или его фармацевтически приемлемая соль по п. 1, отличающиеся тем, что в химической формуле I выше:
L1 и L3 каждый независимо представляет собой -(C0алкил)-;
L2 представляет собой -(C1алкил)-;
a, b и c представляют собой CR4, где R4 представляет собой -H;
Z представляет собой N, O или отсутствует, при этом, когда Z отсутствует, R2 также отсутствует и L2 и L3 связаны напрямую;
R1 представляет собой -CF2H или CF3;
R2 представляет собой -H или -C(=O)-RA, где Z представляет собой O, R2 отсутствует;
RA представляет собой 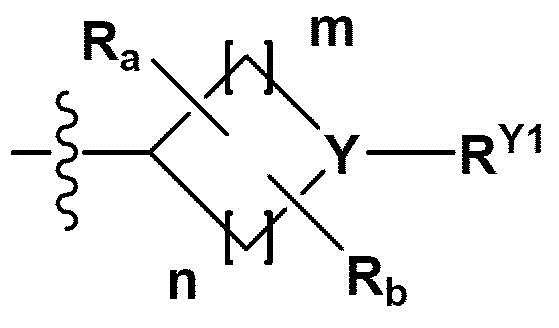 ,
, 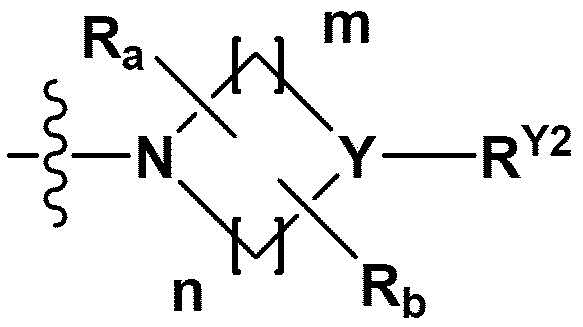
или 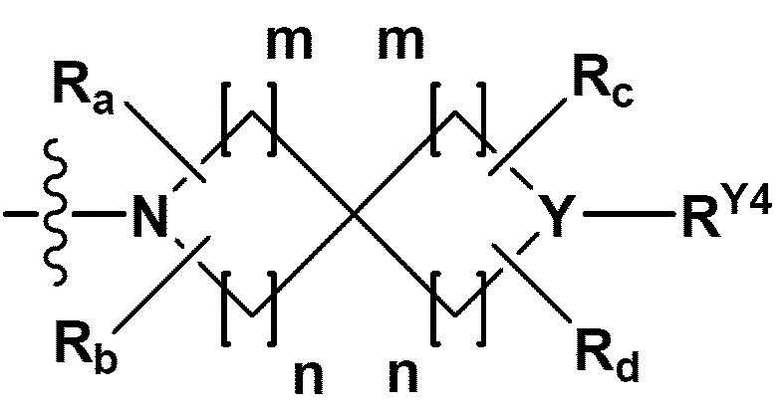 ;
;
Y представляет собой N;
RY1, RY2 и RY4 каждый независимо представляет собой -(C1-C4алкил), -(C3-C7циклоалкил), -(C2-C6гетероциклоалкил), содержащий атом N или O в кольце, -C(=O)-(C1-C4алкил), -C(=O)-O(C1-C4алкил), -C(=O)-NRA3RA4, -C(=O)-(C3-C7циклоалкил), -C(=O)-(C2-C6гетероциклоалкил), содержащий один атом O в кольце, -S(=O)2-(C1-C4алкил), -С4-С5гетероарил, содержащий один или два атома N в кольце, или
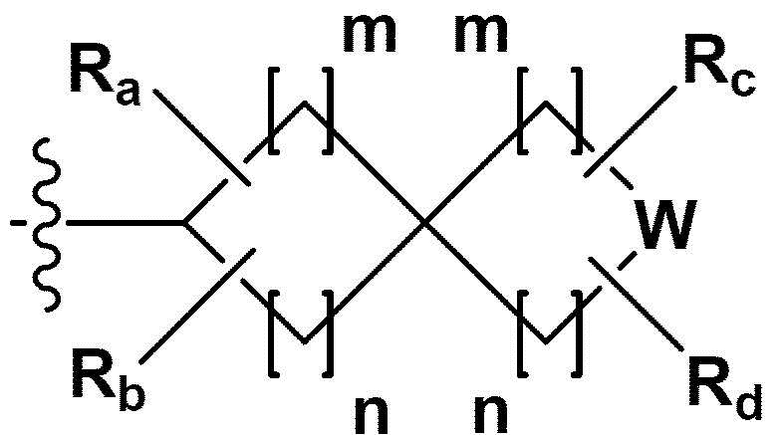 ,
,
где по меньшей мере один H -(C1-C4алкила), -(C3-C7циклоалкила) или -C(=O)-(C1-C4алкила) может быть замещен -X или -OH; и W представляет собой CH2 или O;
каждый из m и n независимо представляет собой целое число 1 или 2;
Ra - Rd каждый независимо представляет собой -H;
R3 представляет собой -(C3-C7циклоалкил), -адамантил, -С6арил, -С5гетероарил, содержащий один атом N в кольце, или -С7гетероарил, содержащий два атома O в кольце, где каждый из по меньшей мере одного -H в -С6ариле или -С5гетероариле, содержащем один атом N в кольце, может быть независимо замещен группами -X, -(C1-C4алкил), -O(C1-C4алкил), -(C=O)-(C1-C4алкил), -CF3 или -S(=O)2-(C1-C4алкил);
RA3 и RA4 каждый независимо представляет собой -(C1-C4алкил); и
X представляет собой F или Cl.
3. Производное 1,3,4-оксадиазола, обладающее селективной ингибирующей активностью в отношении гистондеацетилазы 6 (HDAC6), представленное химической формулой I, или его фармацевтически приемлемая соль по п. 1, отличающиеся тем, что в химической формуле I выше:
L1, L2 или L3 каждый независимо представляет собой -(C0-C1алкил)-;
a, b и c каждый независимо представляет собой CR4 и R4 представляет собой -H;
Z представляет собой N;
R1 представляет собой -CF2Н или -CX3;
R2 представляет собой -C(=O)-RA;
RA представляет собой 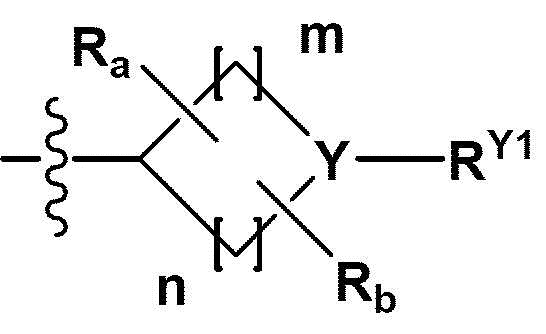 ;
;
Y представляет собой N;
когда Y представляет собой N, RY1 представляет собой -(C1-C4алкил), -(C3-C7циклоалкил), -(C2-C6гетероциклоалкил), содержащий атом N или O в кольце, -C(=O)-(C1-C4алкил), -C(=O)-O(C1-C4алкил), -C(=O)-NRA3RA4, -C(=O)-(C3-C7циклоалкил), -C(=O)-(C2-C6гетероциклоалкил), содержащий один атом O в кольце, -S(=O)2-(C1-C4алкил), -С4-С5гетероарил, содержащий один или два атома N в кольце, или
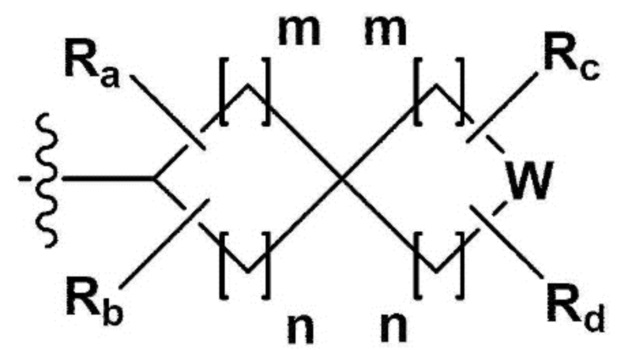 ,
,
где по меньшей мере один H -(C1-C4алкила), -(C3-C7циклоалкила) или -C(=O)-(C1-C4алкила) может быть замещен -X или -OH; и W представляет собой CH2 или O;
каждый из m и n независимо представляет собой целое число 1 или 2;
Ra - Rd каждый независимо представляет собой -H;
R3 представляет собой -(C3-C7циклоалкил), -адамантил, -С6арил, -С5гетероарил, содержащий один атом N в кольце, или -С7гетероарил, содержащий два атома O в кольце, где по меньшей мере один -H -С6арила или -С5гетероарила, содержащего один атом N в кольце, каждый независимо может быть замещен группами -X, -(C1-C4алкил), -O(C1-C4алкил), -(C=O)-(C1-C4алкил), -CF3 или -S(=O)2-(C1-C4алкил);
RA3 и RA4 каждый независимо представляет собой -(C1-C4алкил); и
X представляет собой F, Cl или Br.
4. Производное 1,3,4-оксадиазола, обладающее селективной ингибирующей активностью в отношении гистондеацетилазы 6 (HDAC6), представленное химической формулой I, или его фармацевтически приемлемая соль по п. 1, отличающиеся тем, что в вышеуказанной химической формуле I:
L1, L2 и L3 каждый независимо представляет собой -(C0-C1алкил)-;
a, b и c каждый независимо представляет собой CR4 и R4 представляет собой -H;
Z представляет собой N;
R1 представляет собой -CF2Н или -CX3;
R2 представляет собой -C(=O)-RA;
RA представляет собой
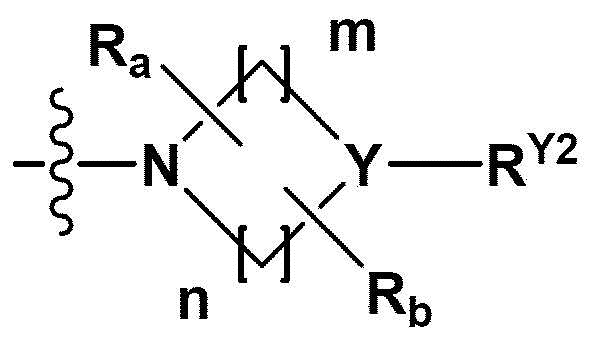 или
или 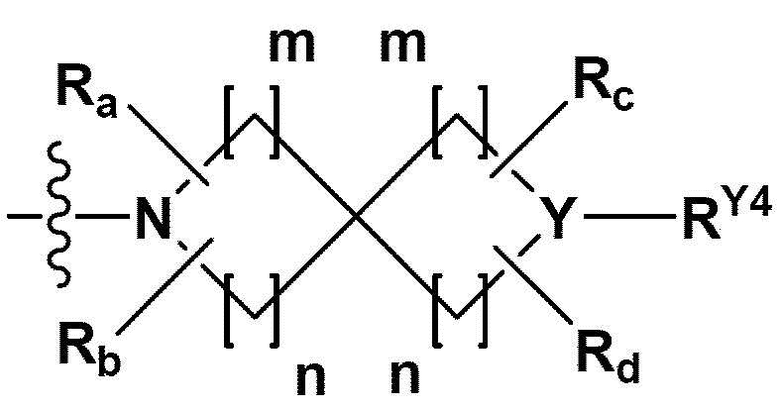 ;
;
Y представляет собой N;
когда Y представляет собой N, каждый из RY2 и RY4 независимо представляет собой -(C1-C4алкил), -(C3-C7циклоалкил), -(C2-C6гетероциклоалкил), содержащий атом N или O в кольце, -C(=O)-(C1-C4алкил), -C(=O)-O(C1-C4алкил), -C(=O)-NRA3RA4, -C(=O)-(C3-C7циклоалкил), -C(=O)-(C2-C6гетероциклоалкил), содержащий один атом O в кольце, -S(=O)2-(C1-C4алкил), -С4-С5гетероарил, содержащий один или два атома N в кольце, или
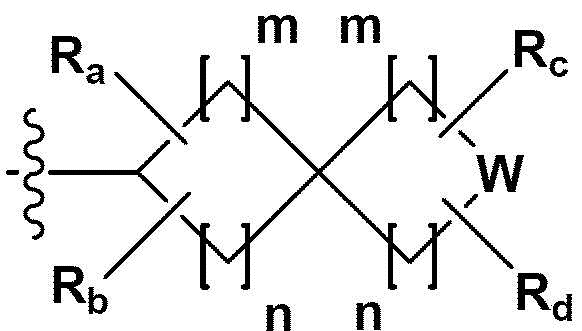 ,
,
где по меньшей мере один H -(C1-C4алкила), -(C3-C7циклоалкила) или -C(=O)-(C1-C4алкила) может быть замещен -X или -OH; и W представляет собой CH2 или O;
каждый из m и n независимо представляет собой целое число 1 или 2;
Ra - Rd каждый независимо представляет собой -H;
R3 представляет собой -(C3-C7циклоалкил), -адамантил, -С6арил, -С5гетероарил, содержащий один атом N в кольце, или -С7гетероарил, содержащий два атома О в кольце, где по меньшей мере один -H -С6арила или -С5гетероарила, содержащего один атом N в кольце, каждый независимо может быть замещен группами -X, -(C1-C4алкил), -O(C1-C4алкил), -(C=O)-(C1-C4алкил), -CF3 или -S(=O)2-(C1-C4алкил);
RA3 и RA4 каждый независимо представляет собой -(C1-C4алкил); и
X представляет собой F, Cl или Br.
5. Производное 1,3,4-оксадиазола, обладающее селективной ингибирующей активностью в отношении гистондеацетилазы 6 (HDAC6), представленное химической формулой I, или его фармацевтически приемлемая соль по п. 1, отличающиеся тем, что в вышеуказанной химической формуле I:
L1, L2 и L3 каждый независимо представляет собой -(C0-C1алкил)-;
a, b и c каждый независимо представляет собой CR4 и R4 представляет собой -H;
Z представляет собой N, O или отсутствует, при этом, когда Z отсутствует, R2 также отсутствует и L2 и L3 связаны напрямую;
R1 представляет собой -CF2Н или -CX3;
R2 представляет собой -H или -(C1-C4алкил), при этом, когда Z представляет собой O, R2 отсутствует;
R3 представляет собой -(C3-C7циклоалкил), -адамантил, -С6арил, -С5гетероарил, содержащий один атом N в кольце, или –С7гетероарил, содержащий два атома O в кольце, где по меньшей мере один -H в -С6ариле или -С5гетероариле, содержащем один атом N в кольце, каждый независимо может быть замещен группами -X, -(C1-C4алкил), -O(C1-C4алкил), -(C=O)-(C1-C4алкил), -CF3 или -S(=O)2-(C1-C4алкил); и
X представляет собой F, Cl или Br.
6. Производное 1,3,4-оксадиазола, обладающее селективной ингибирующей активностью в отношении гистондеацетилазы 6 (HDAC6), или его фармацевтически приемлемая соль по п. 1, отличающиеся тем, что они представляют собой любое из соединений, перечисленных в следующей таблице:
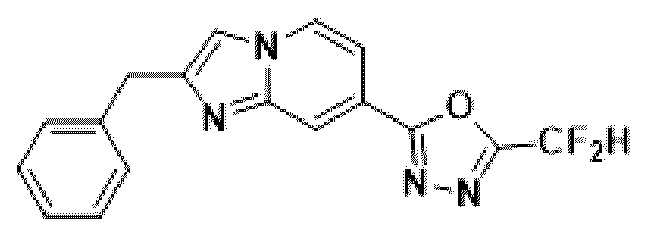
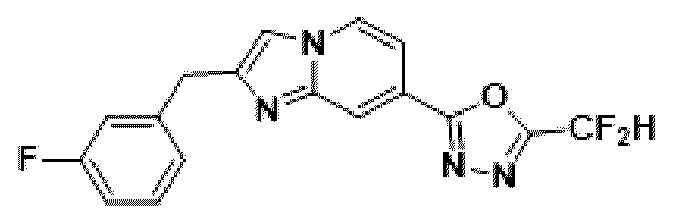
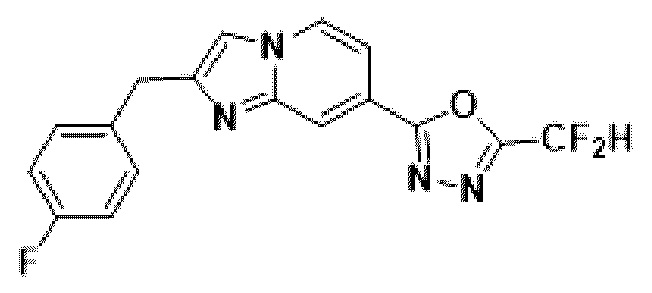
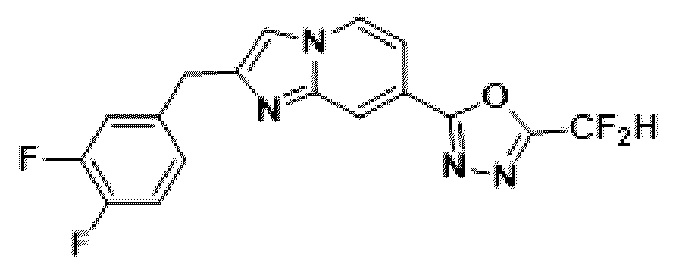
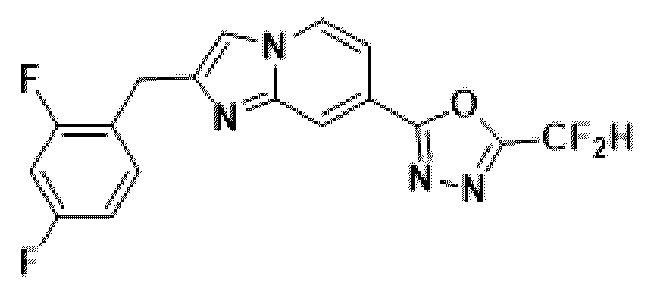
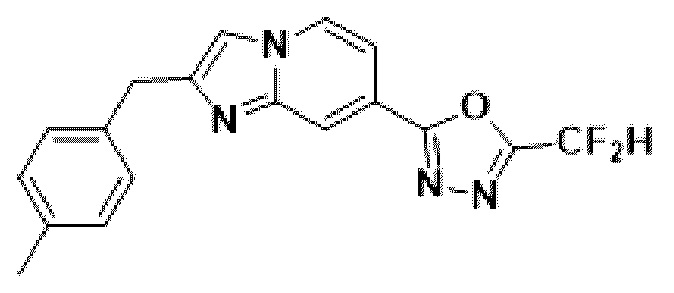
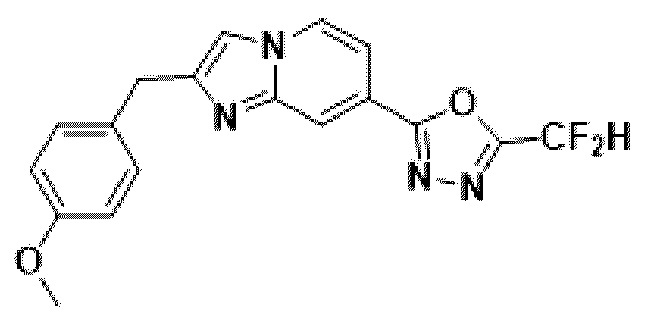
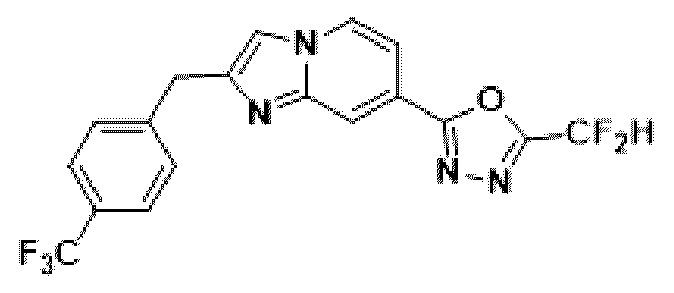
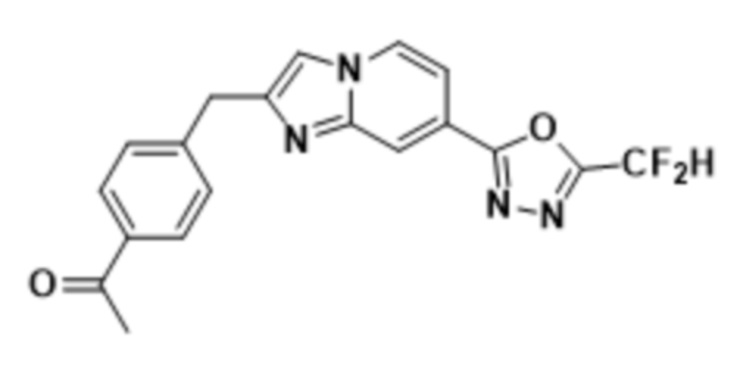
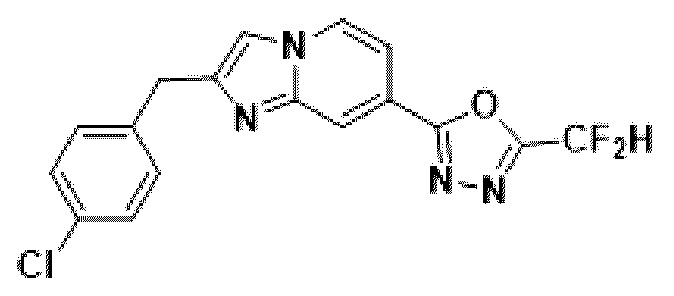
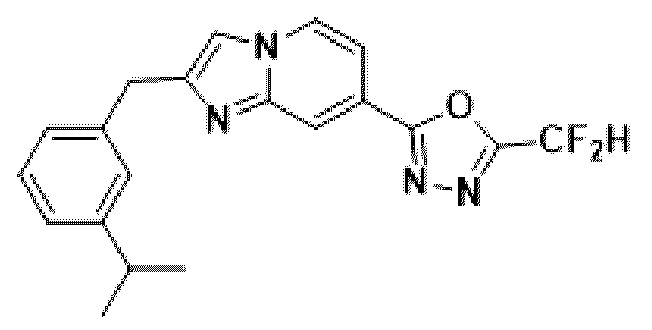
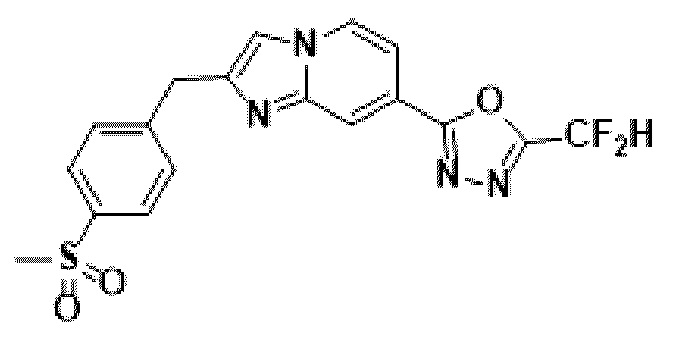
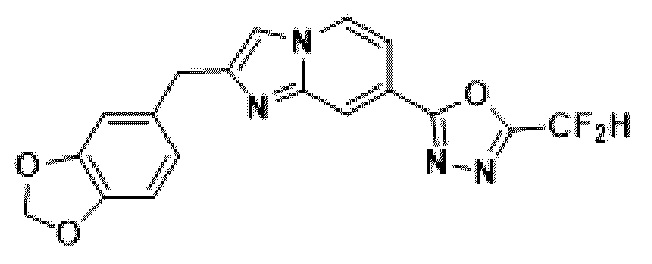
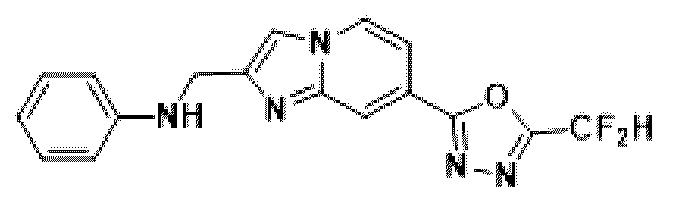
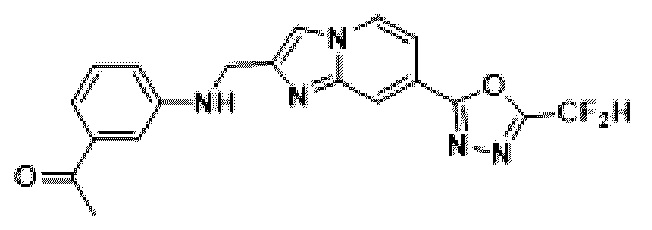
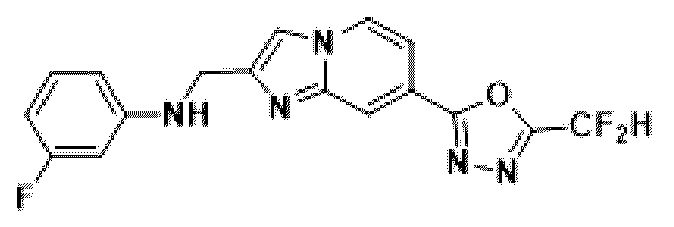
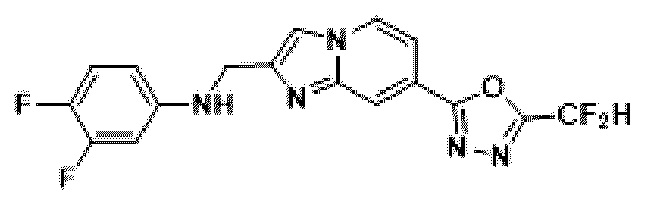
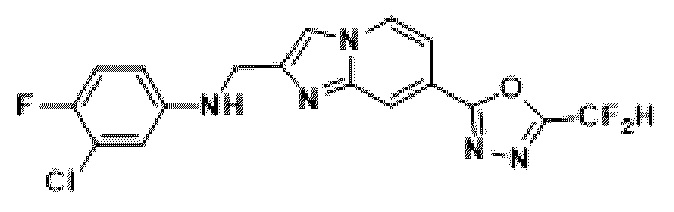
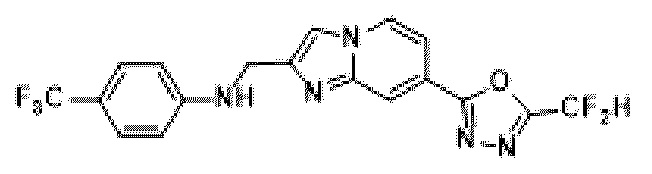
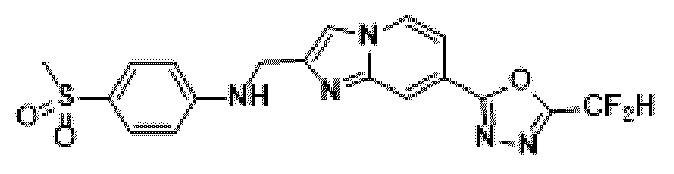
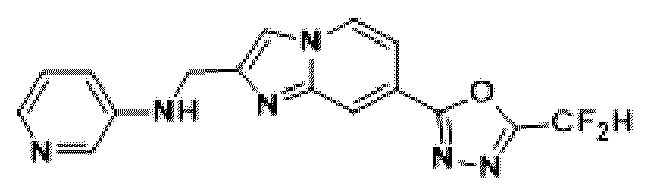
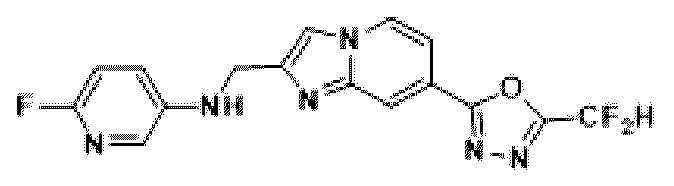
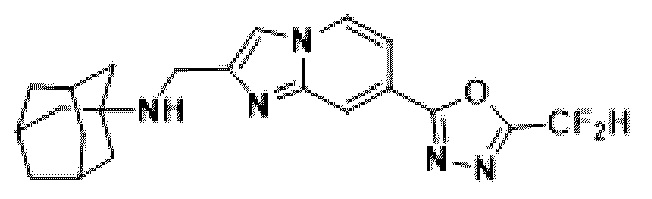
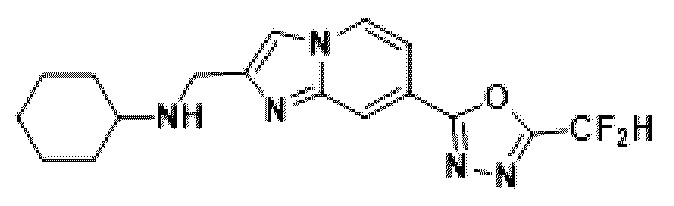
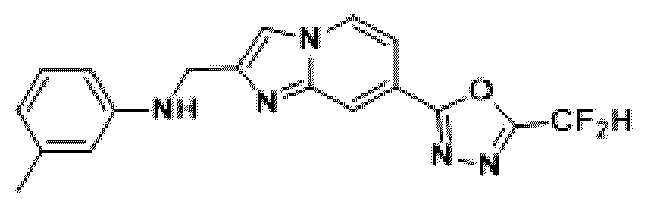
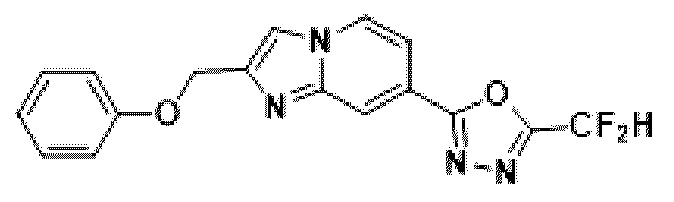
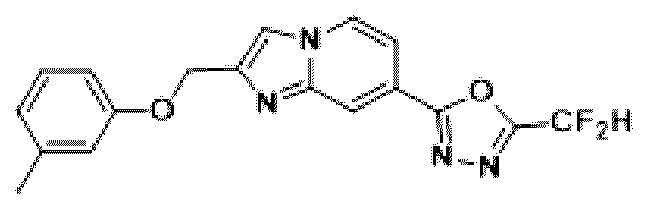
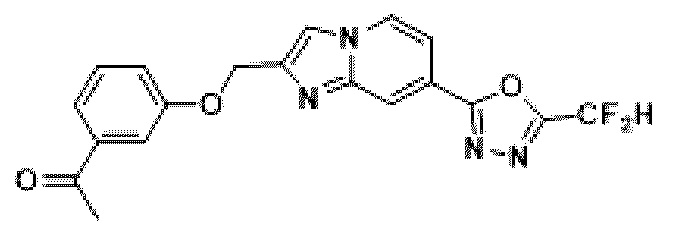
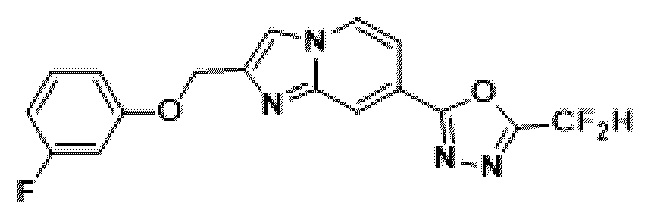
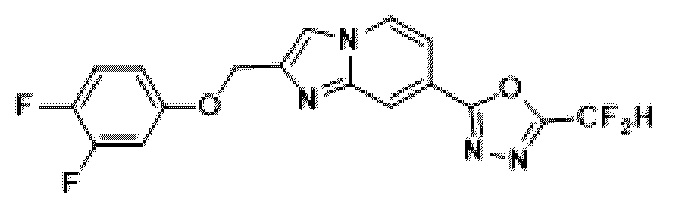
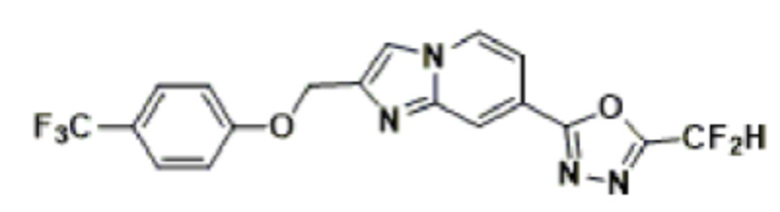
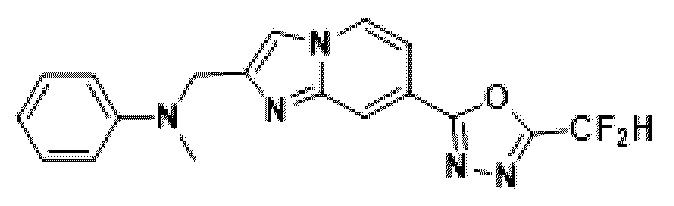
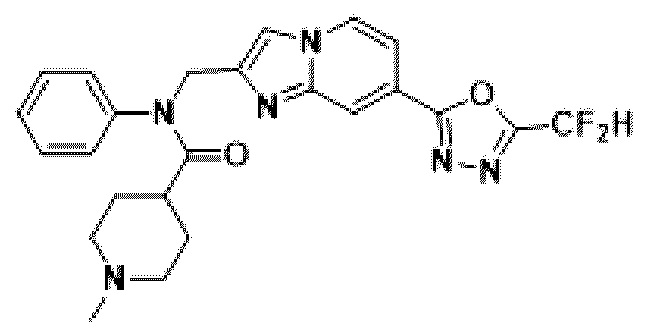
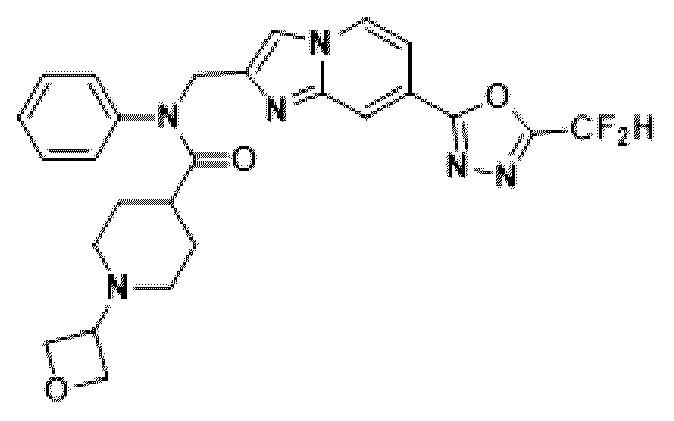
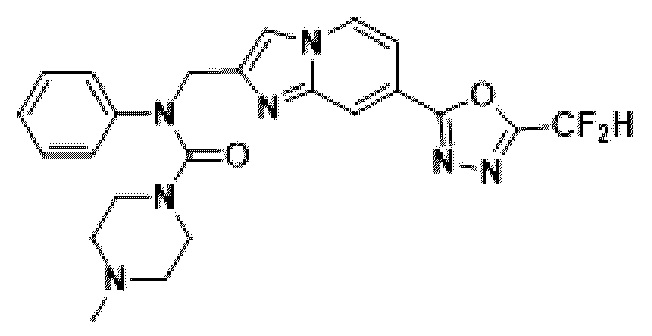
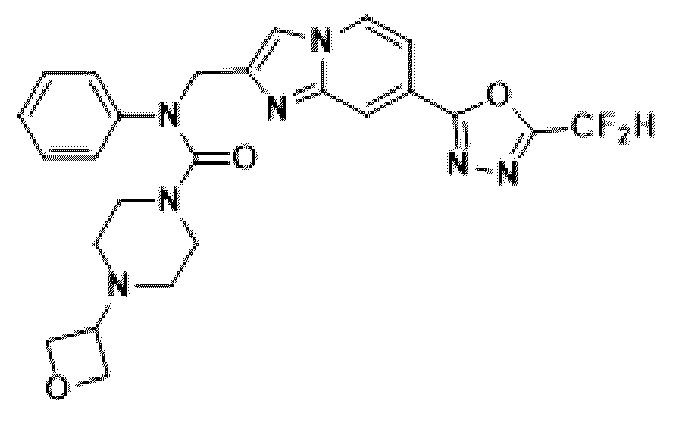
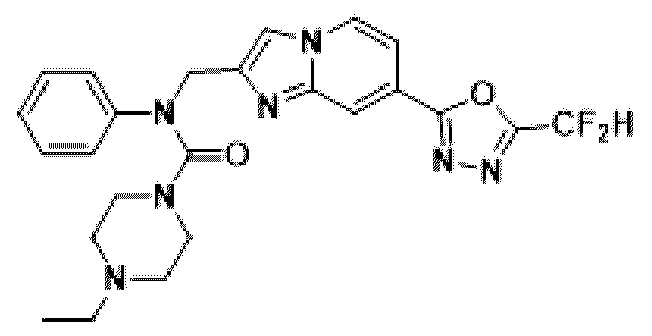
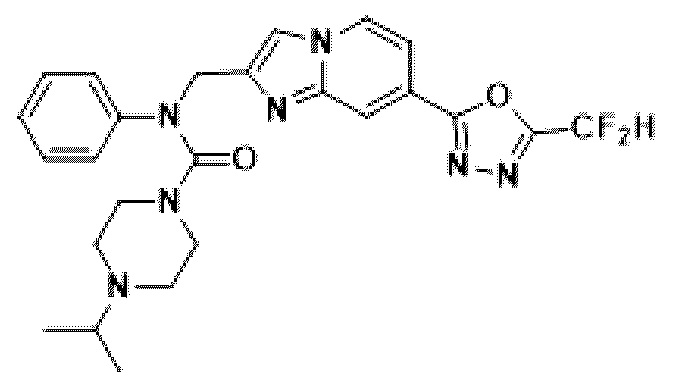
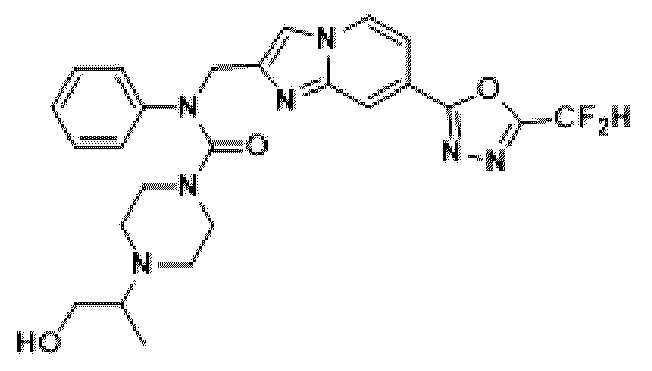
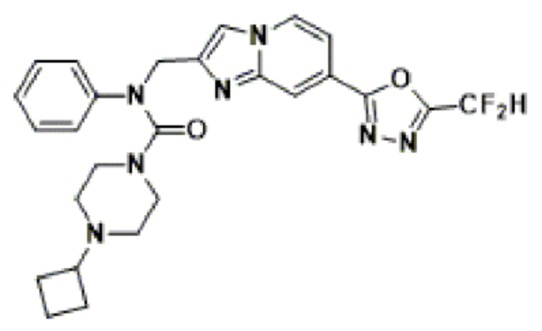
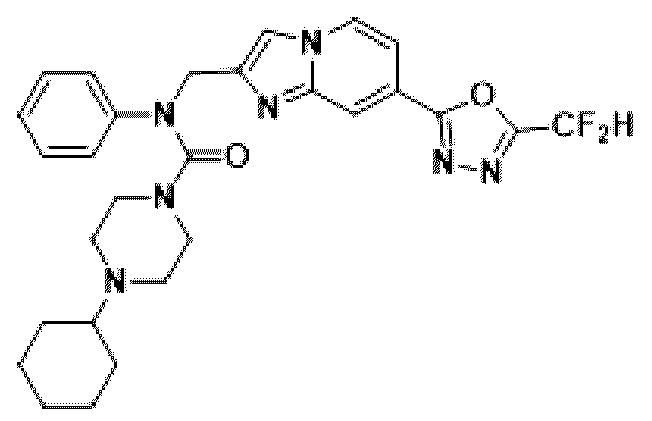
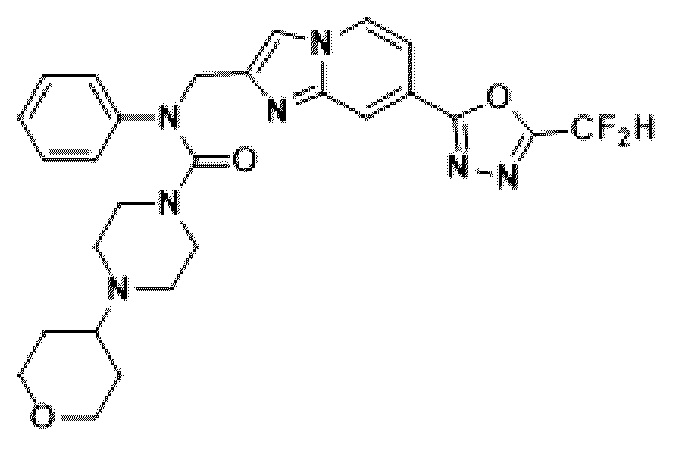
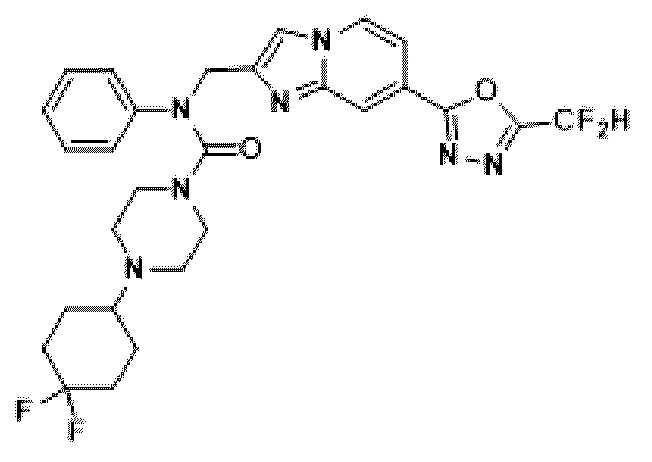
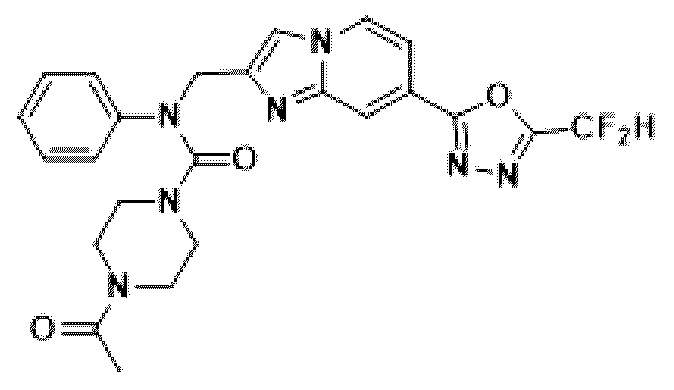
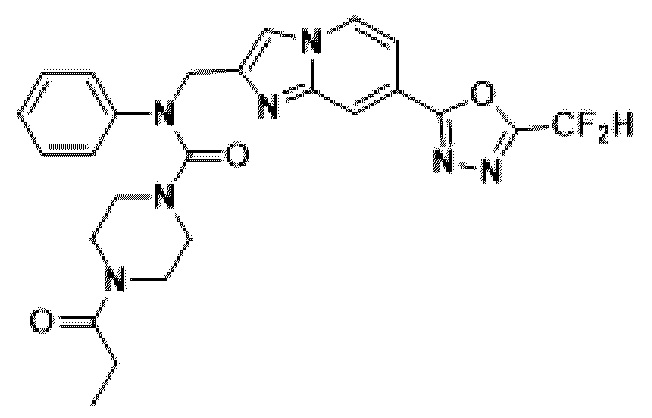
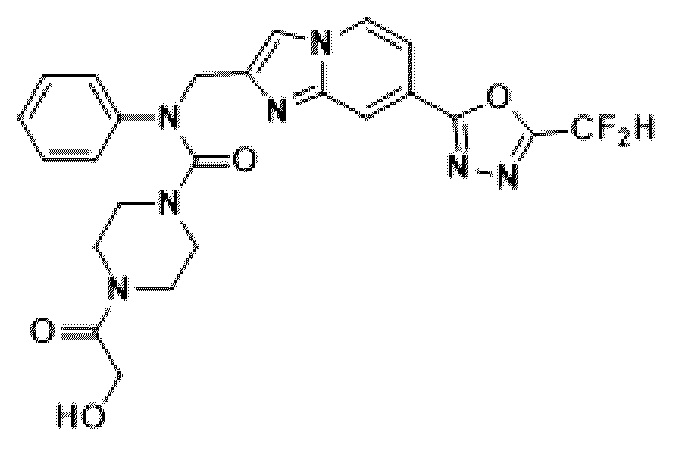
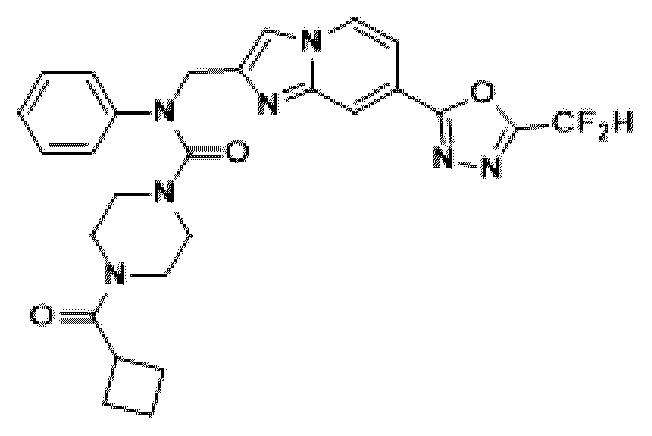
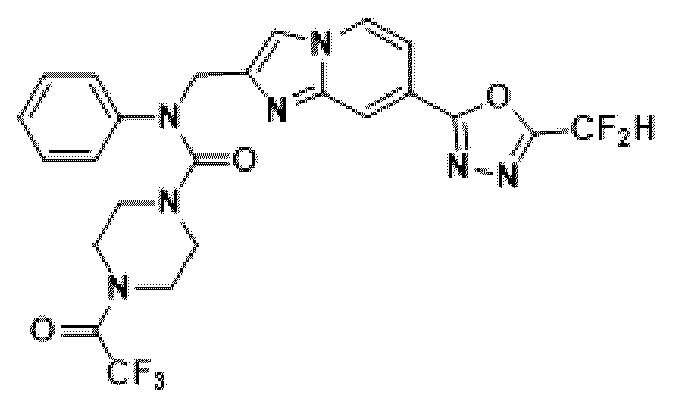
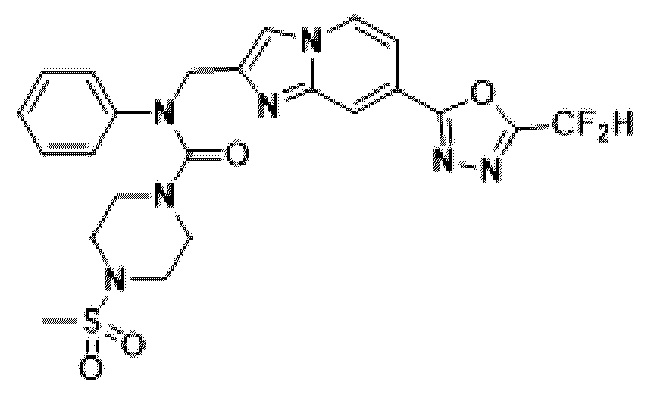
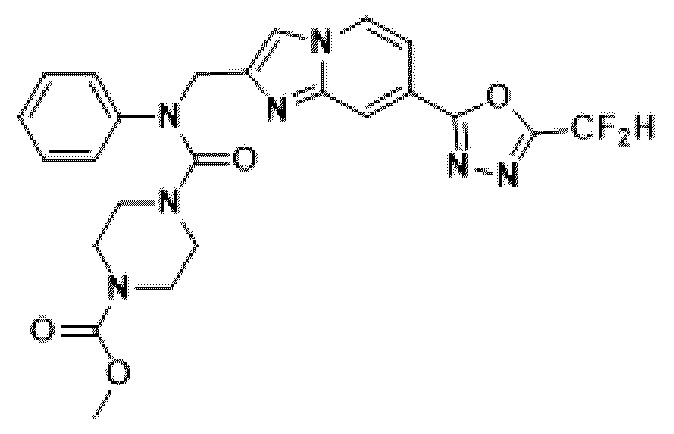
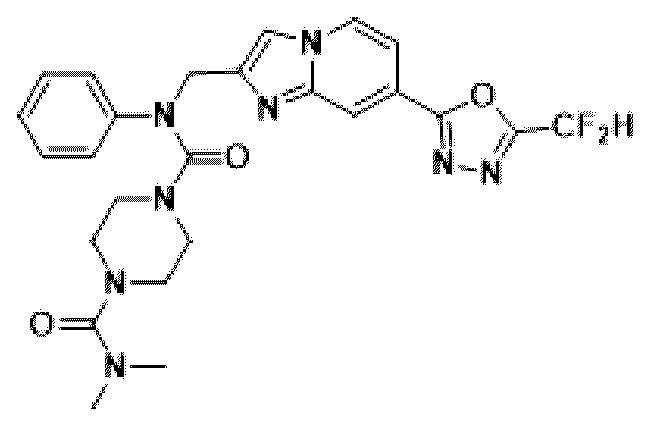
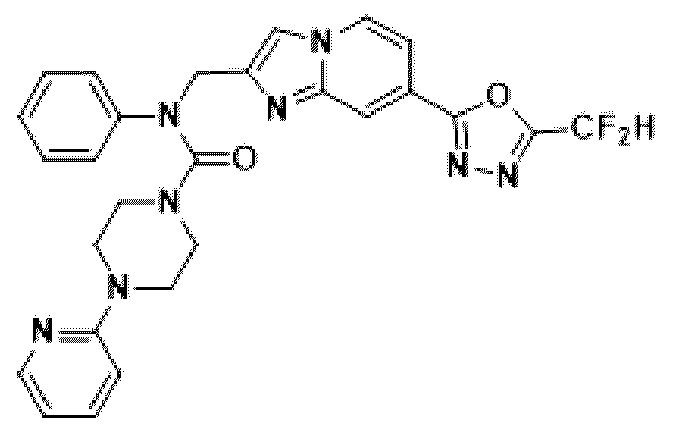
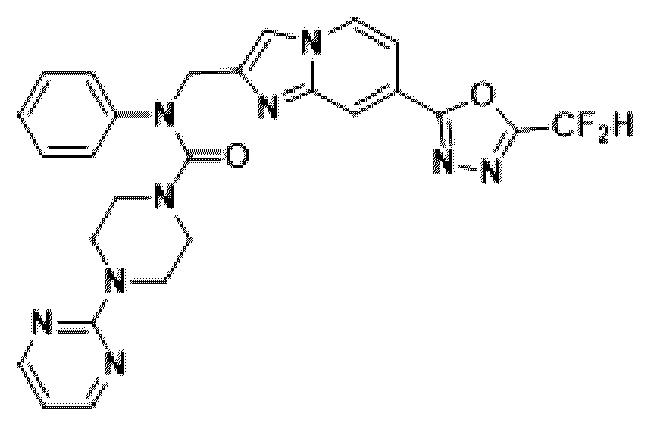
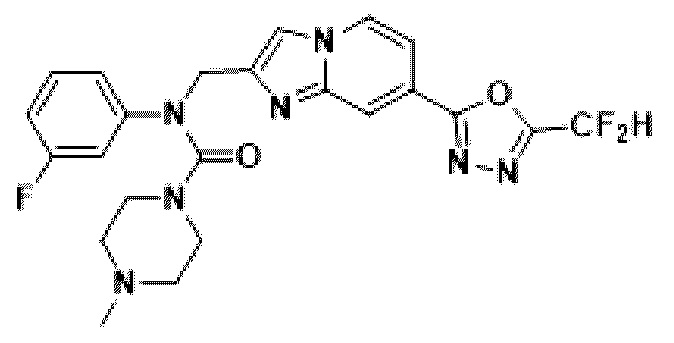
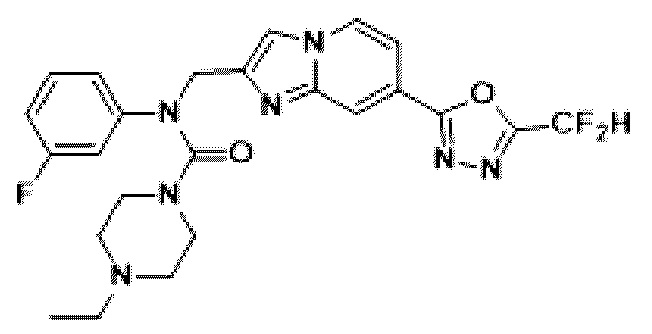
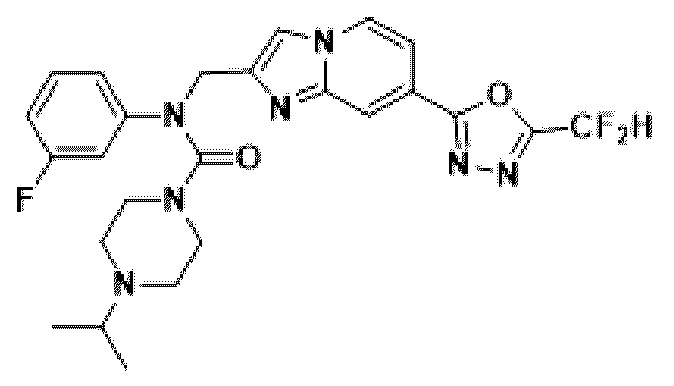
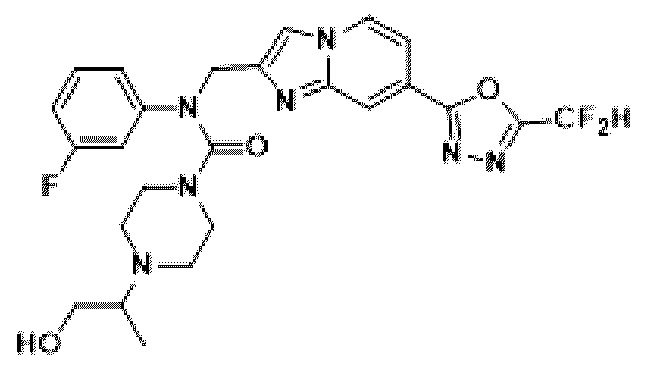
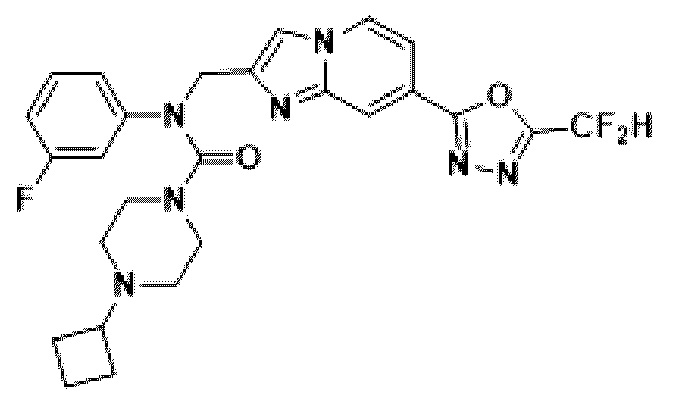
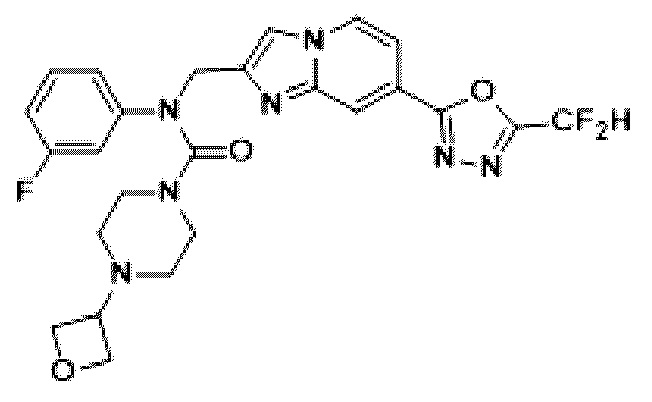
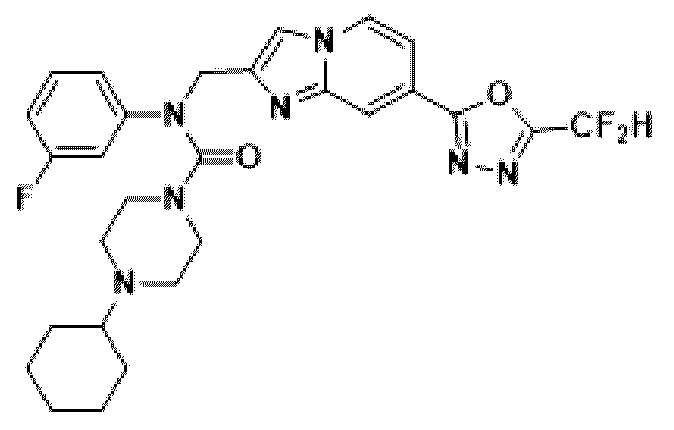
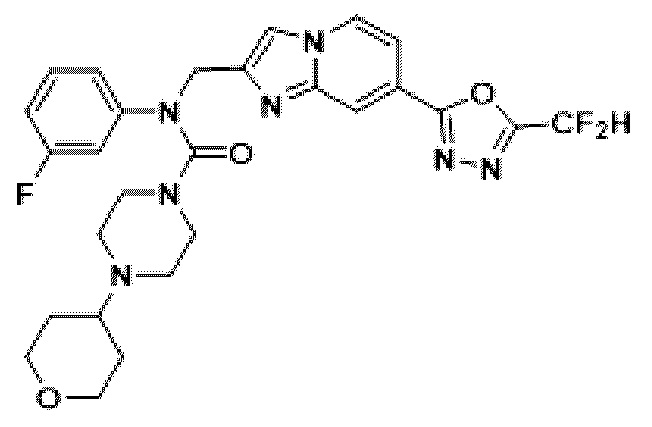
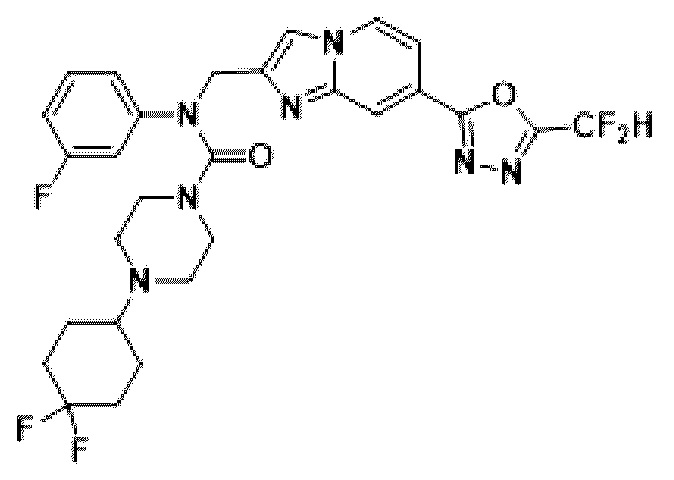
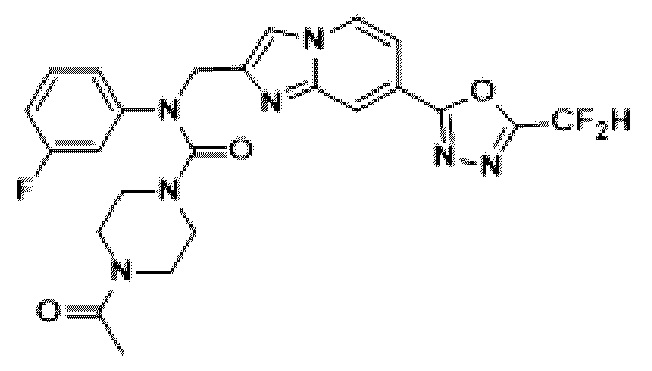
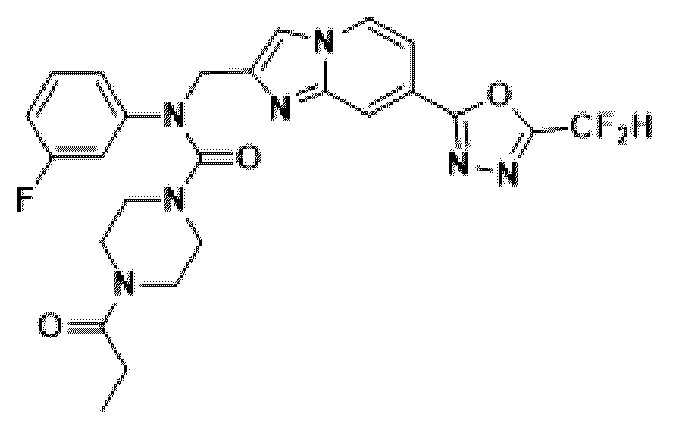
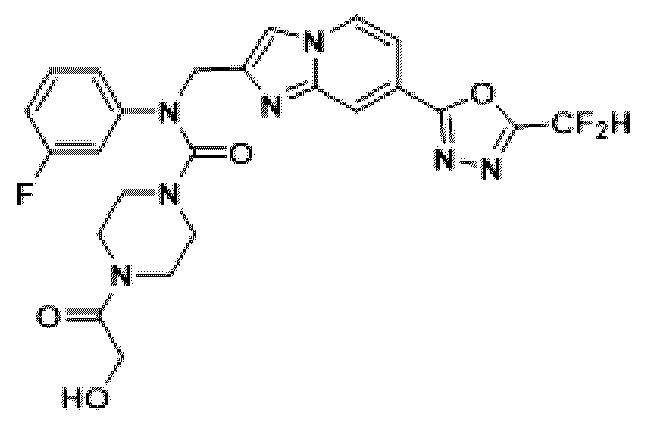
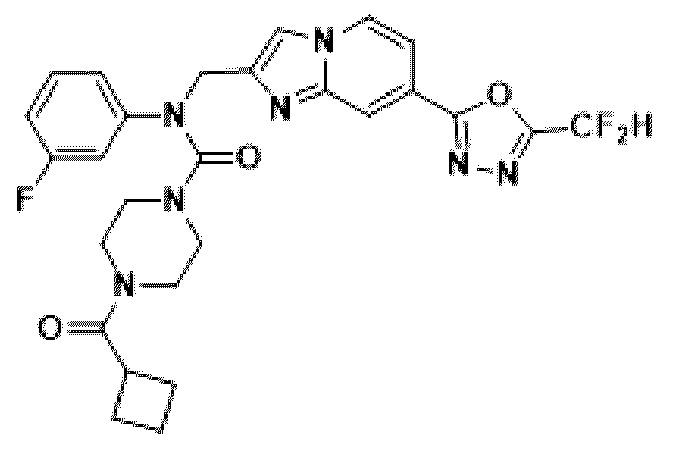
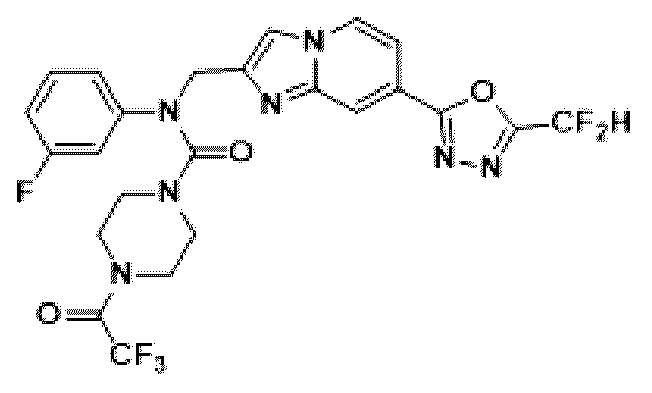
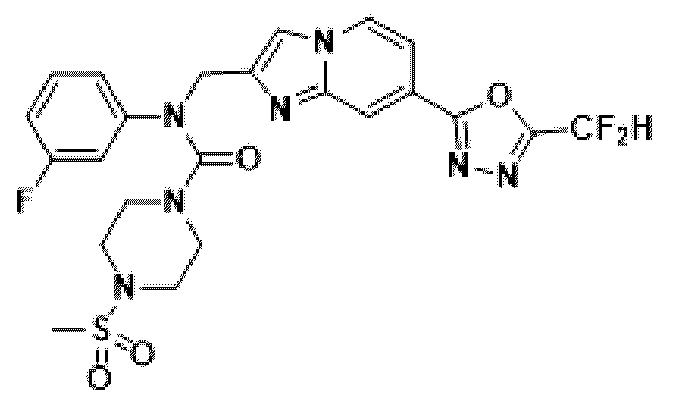
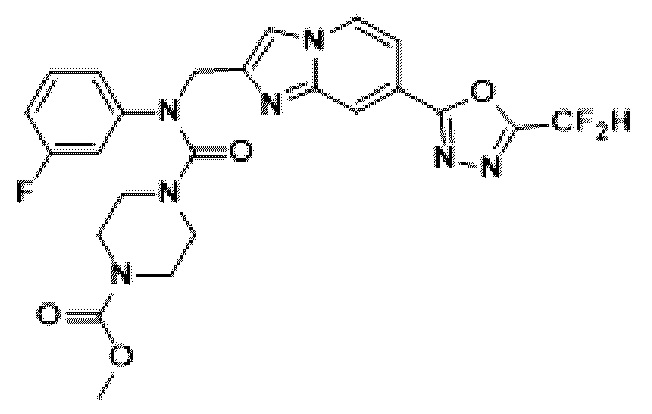
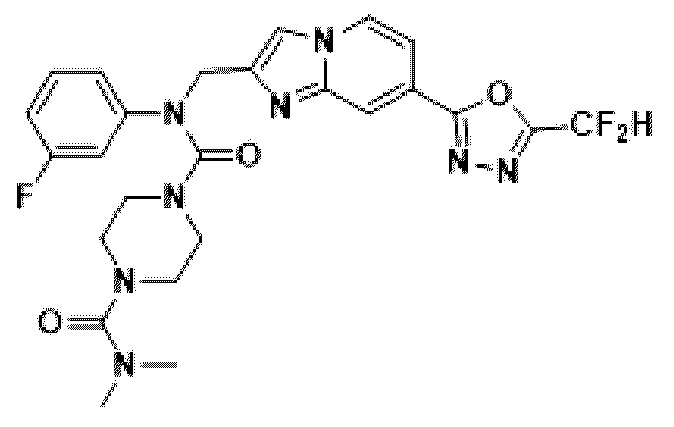
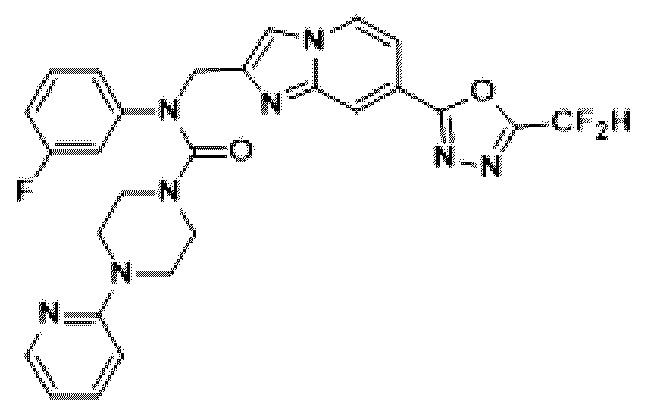
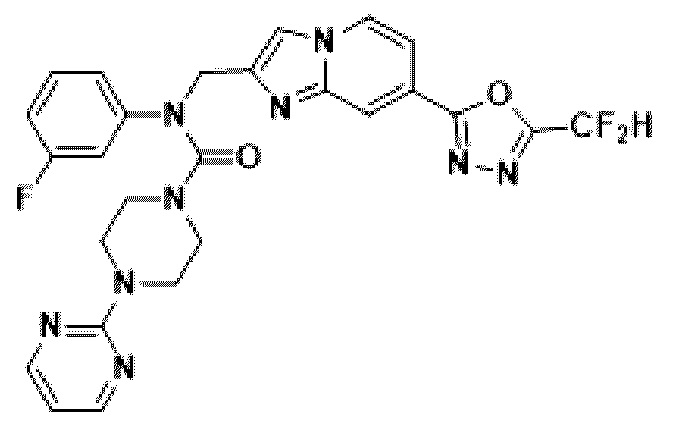
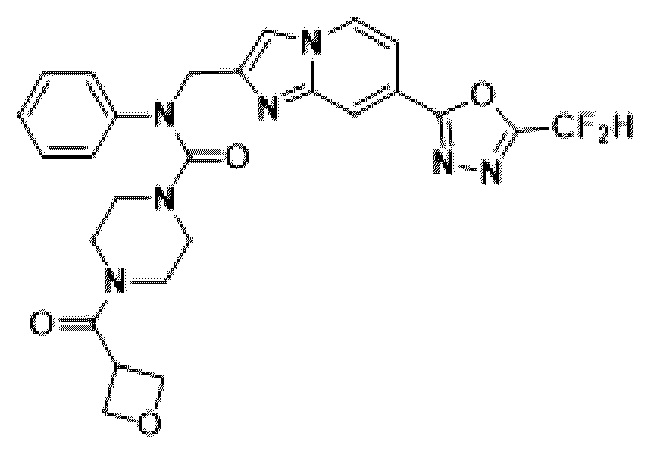
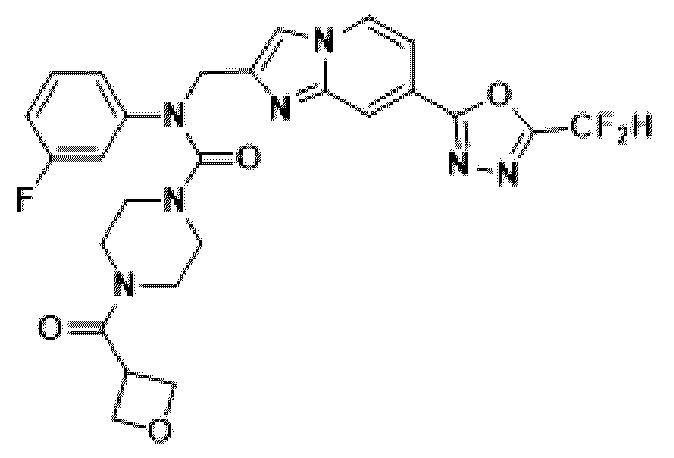
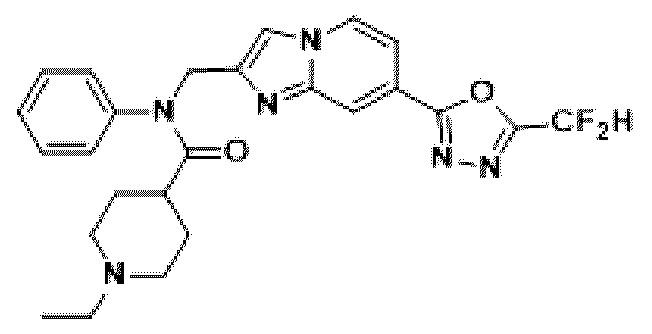
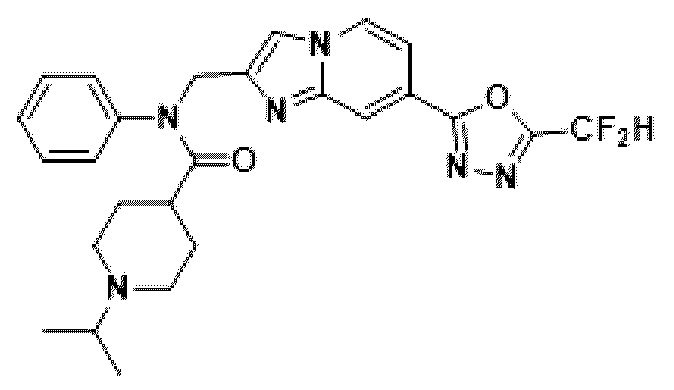
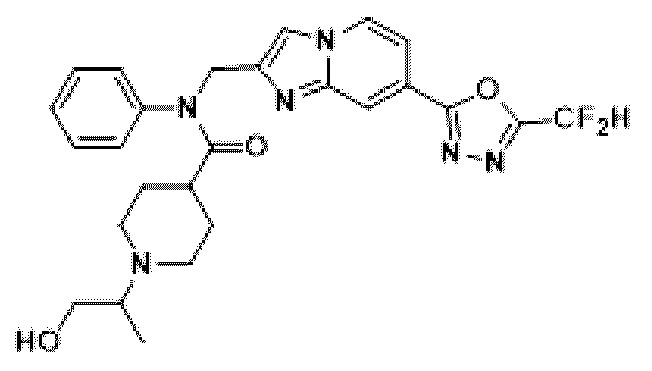
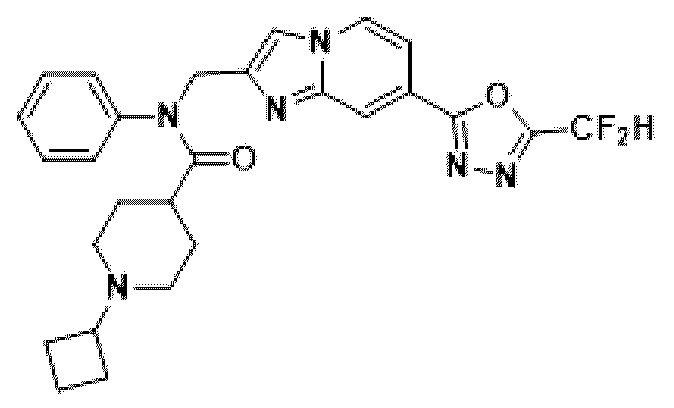
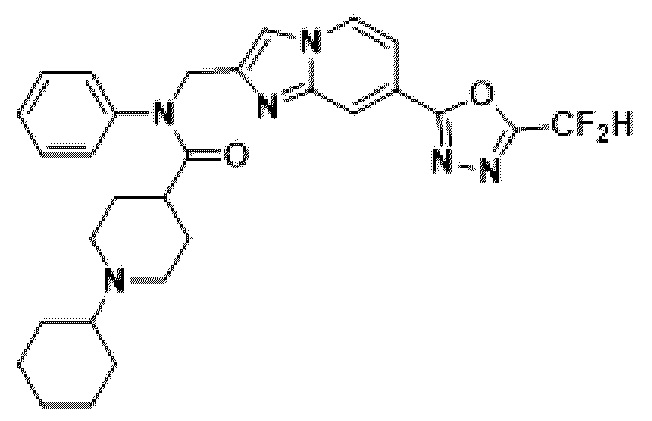
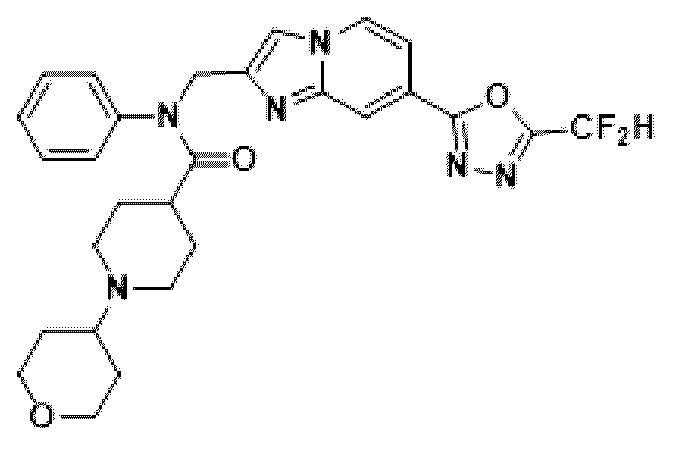
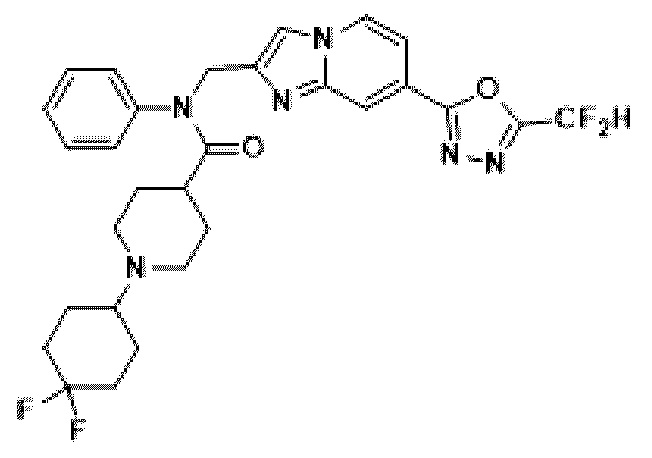
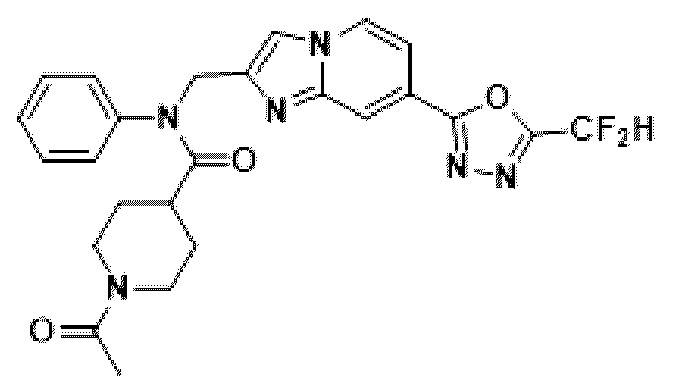
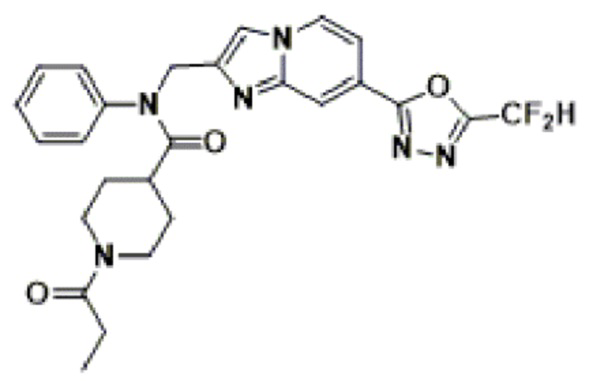
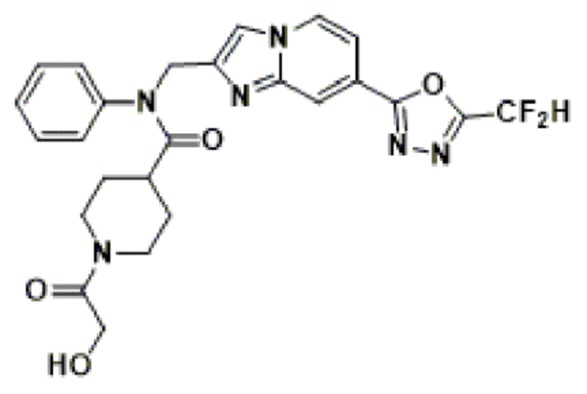
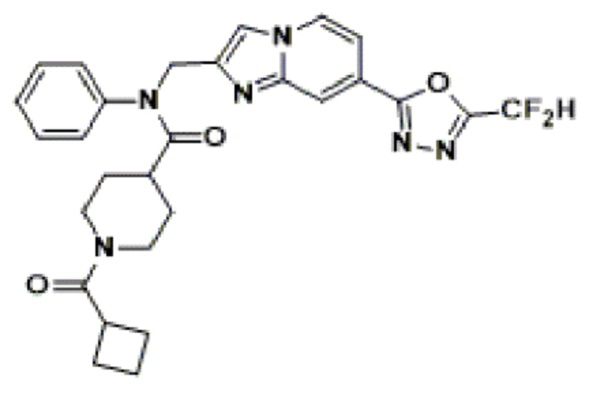
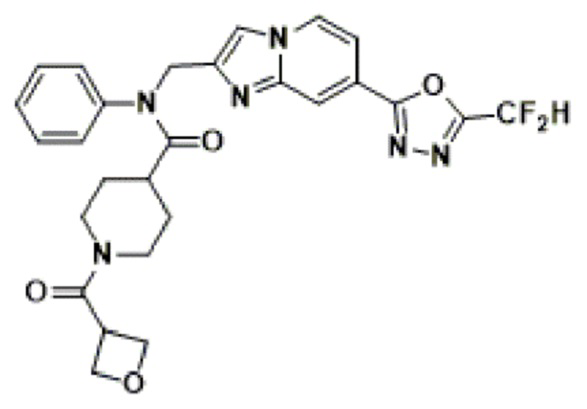
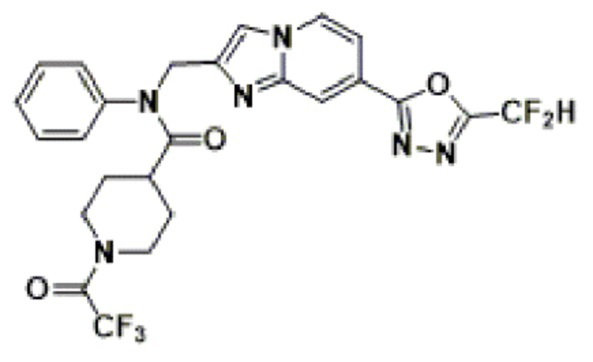
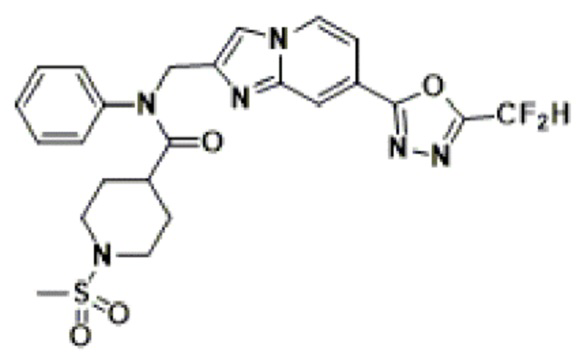
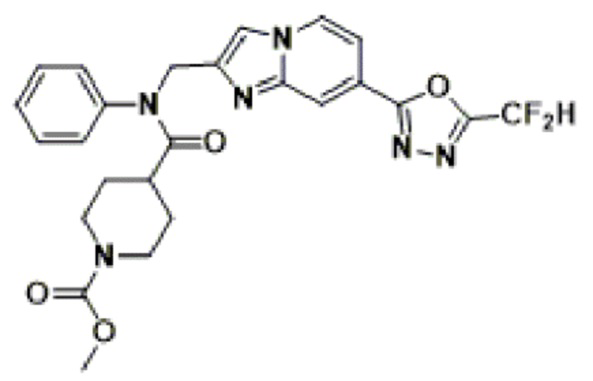
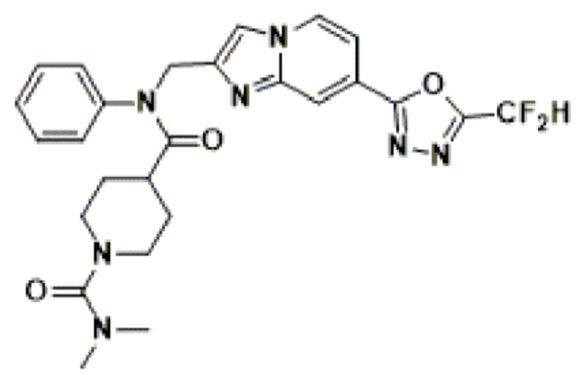
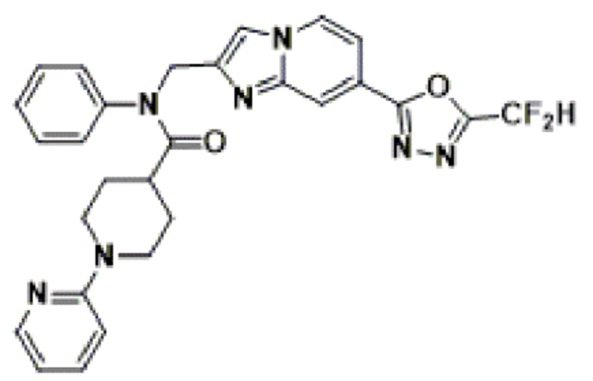
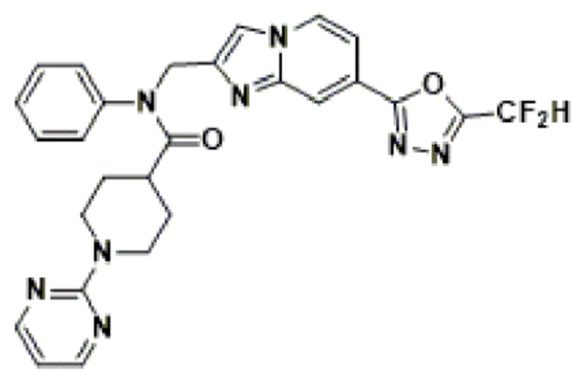
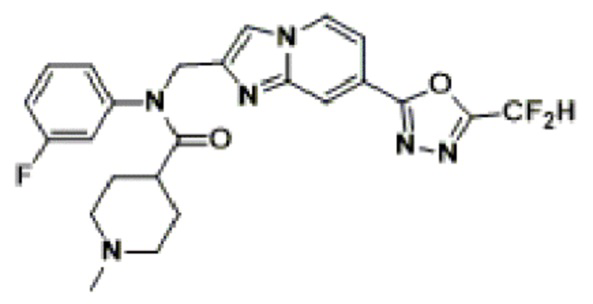
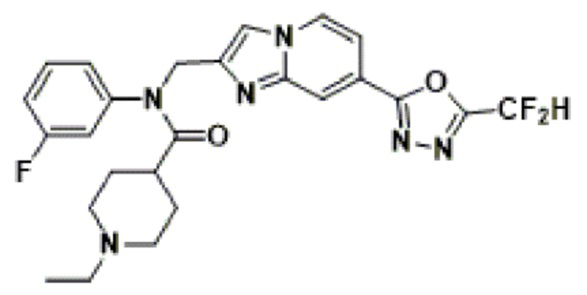
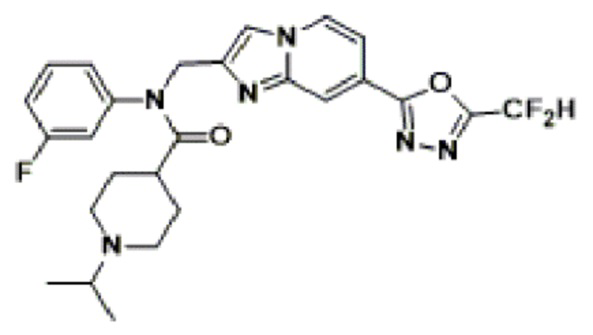
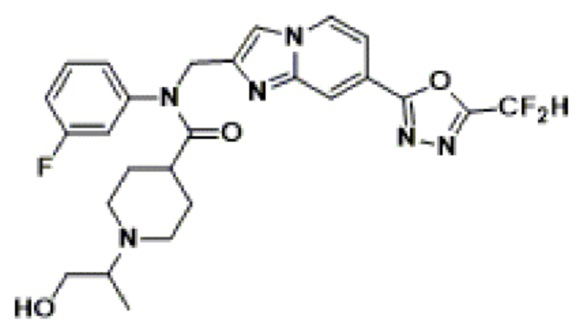
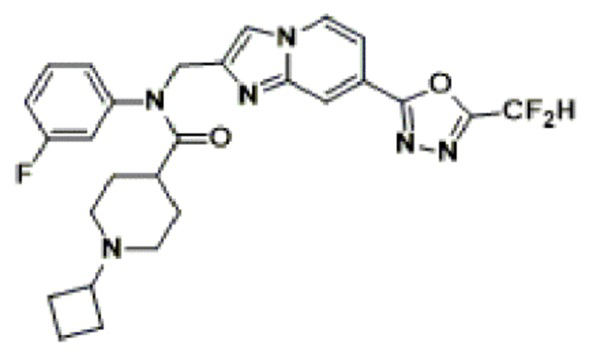
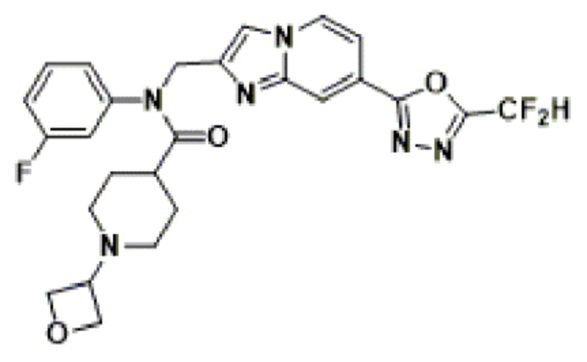
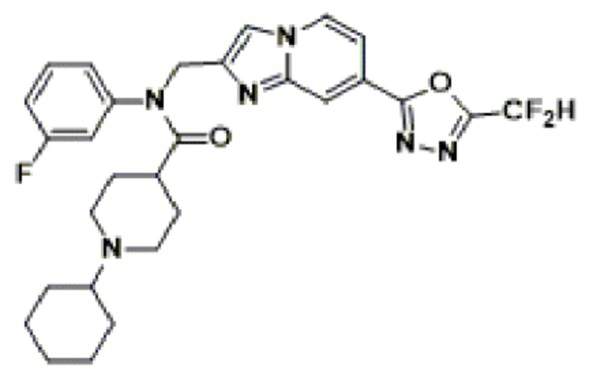
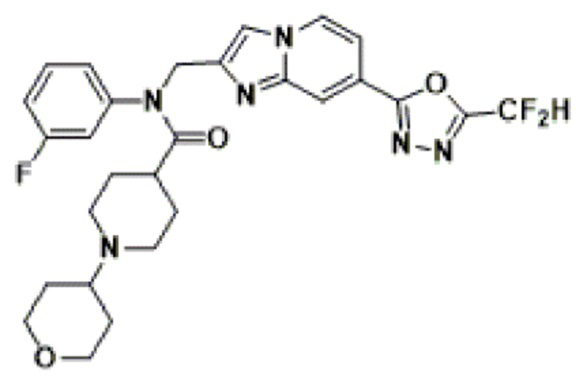
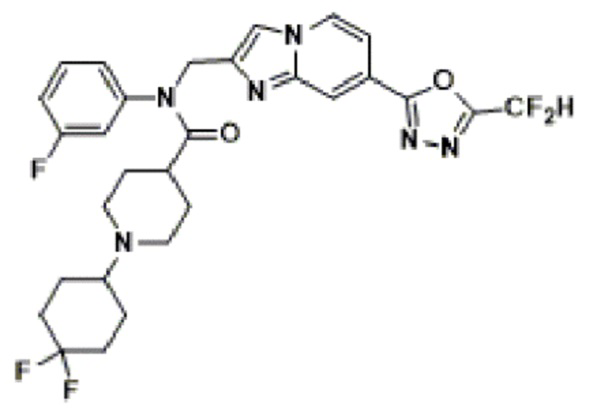
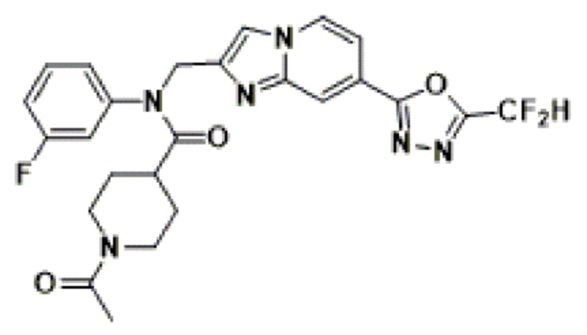
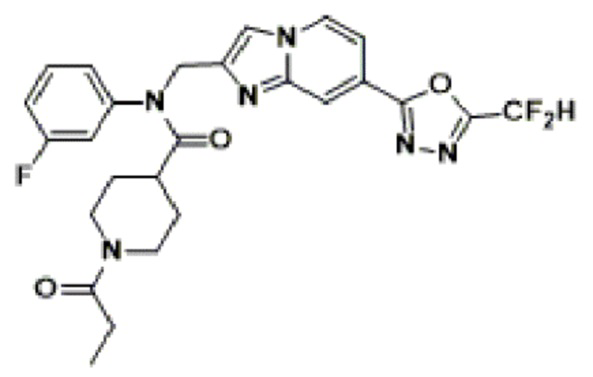
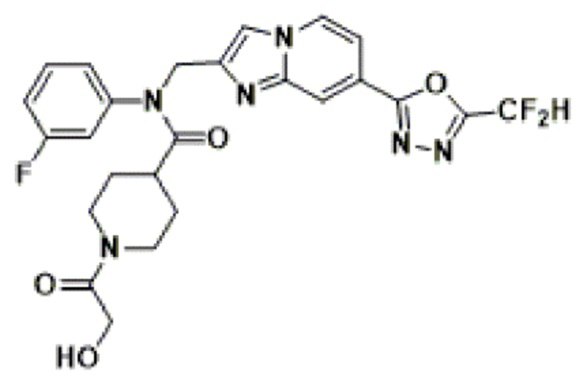
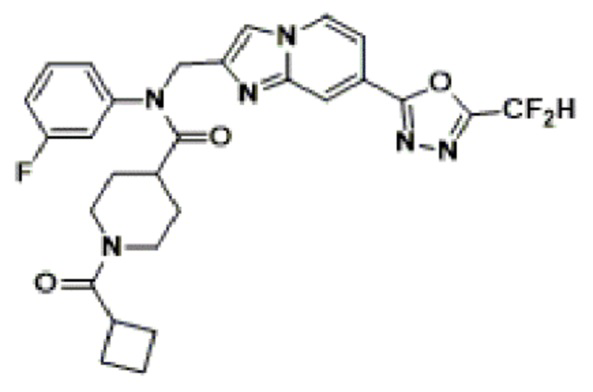
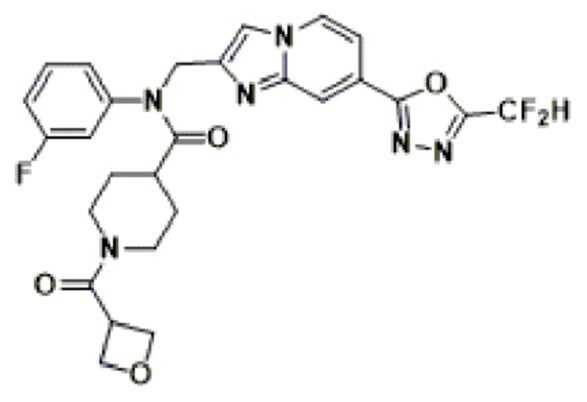
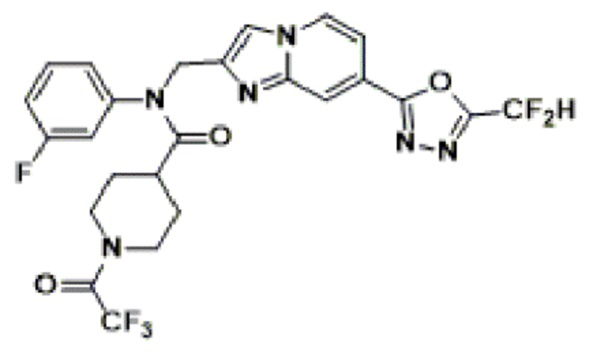
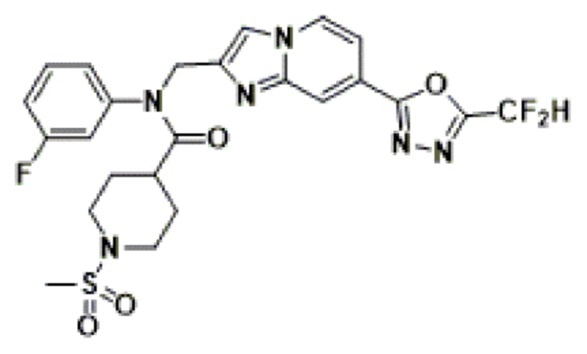
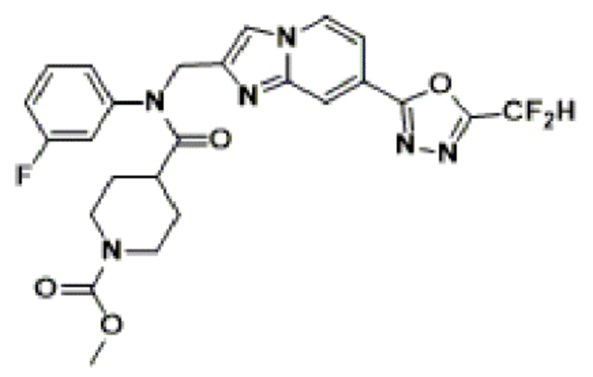
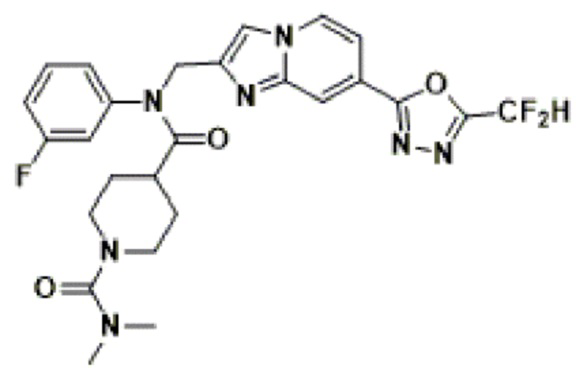
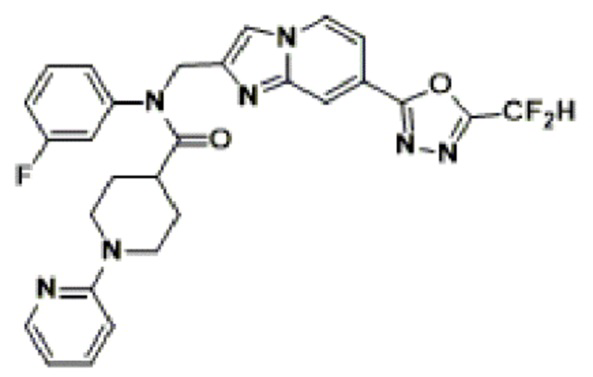
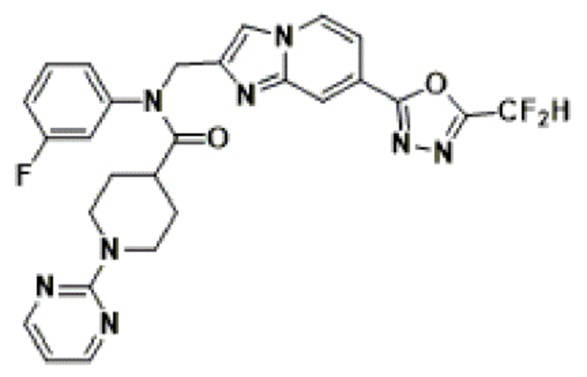
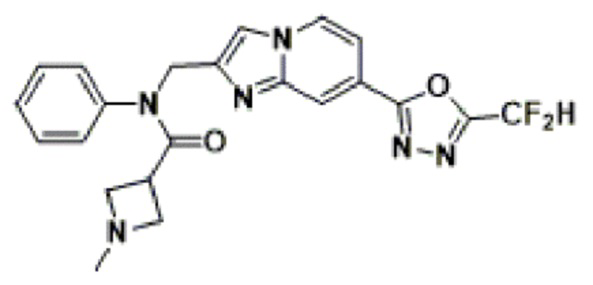
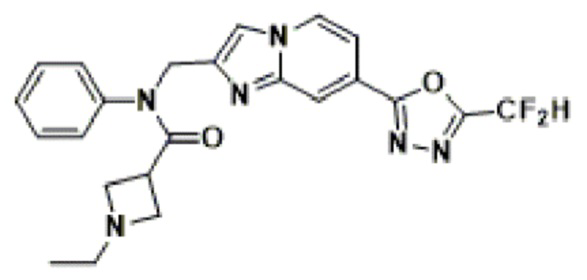
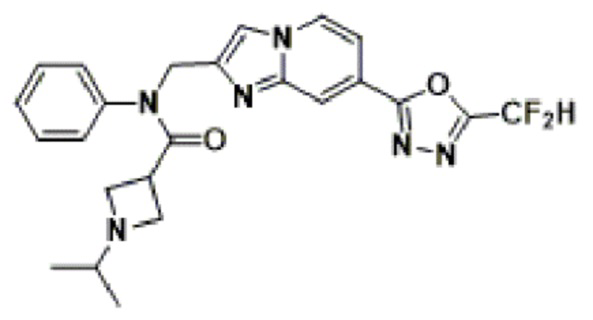
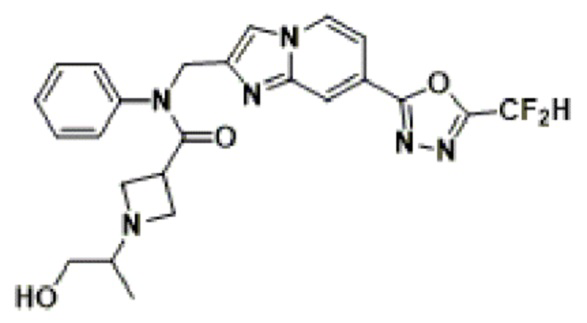
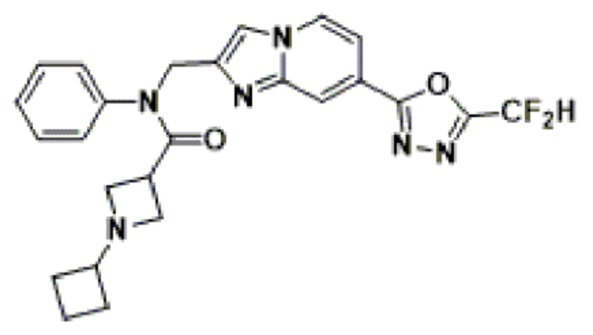
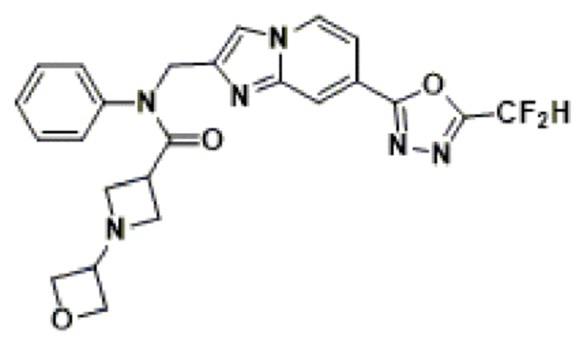
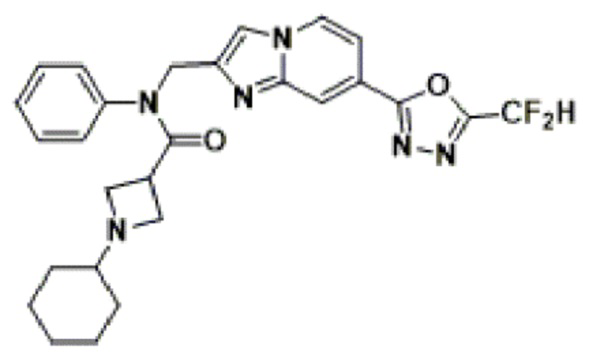
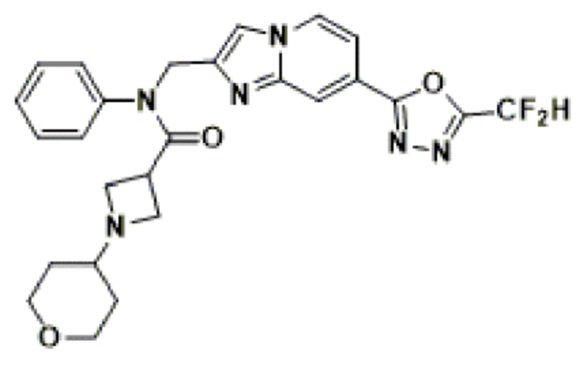
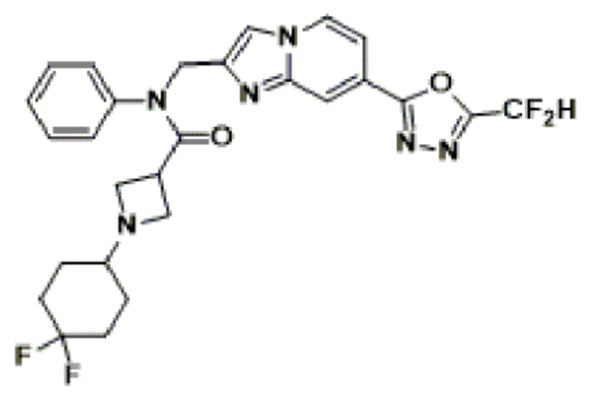
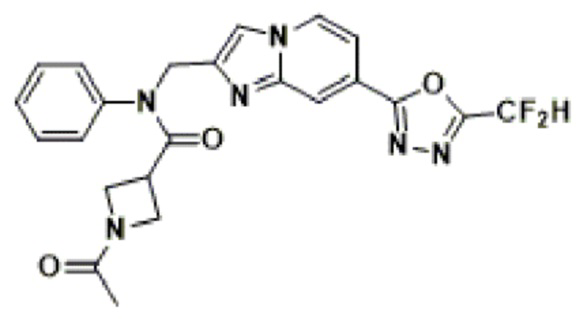
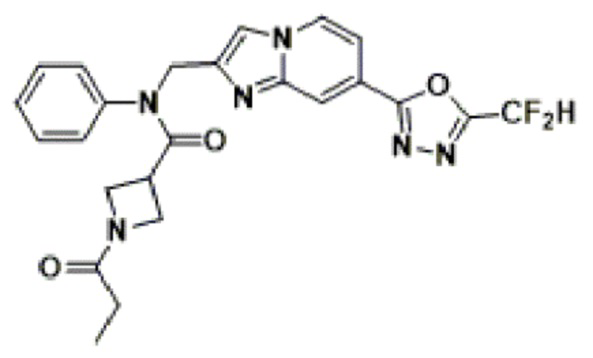
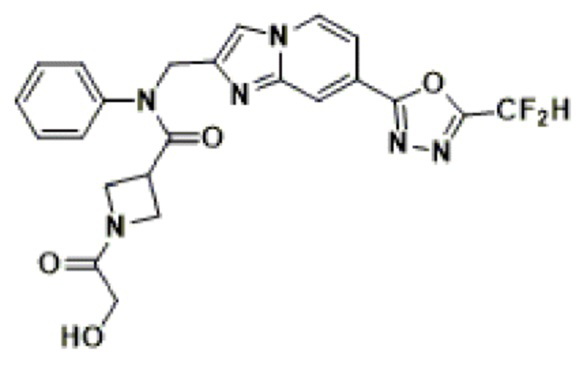
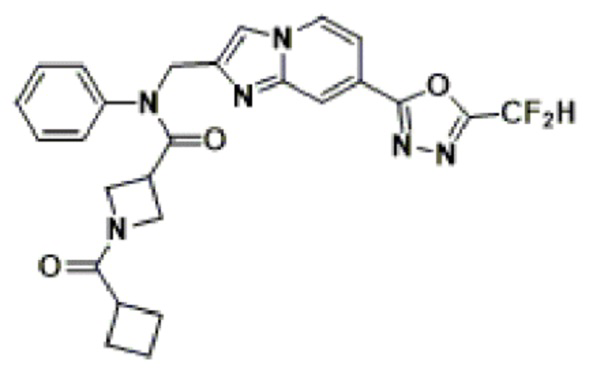
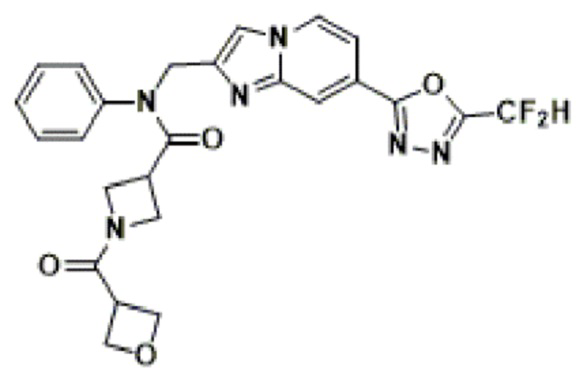
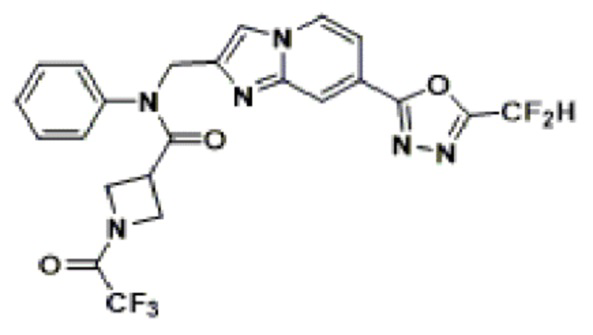
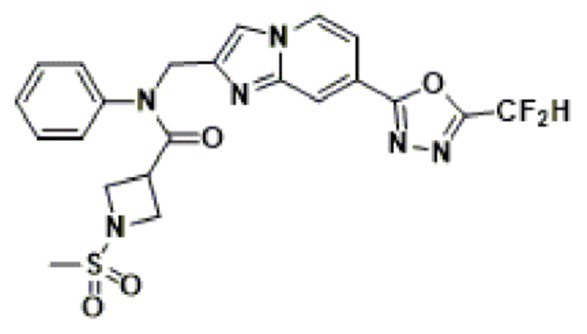
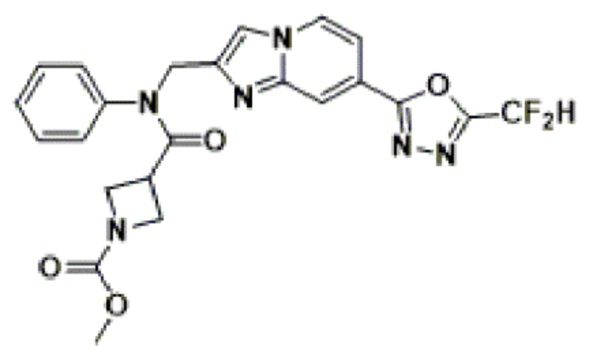
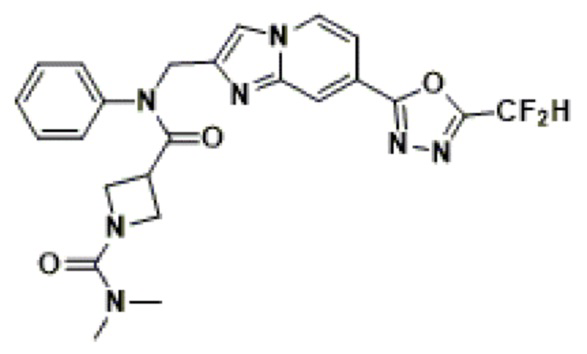
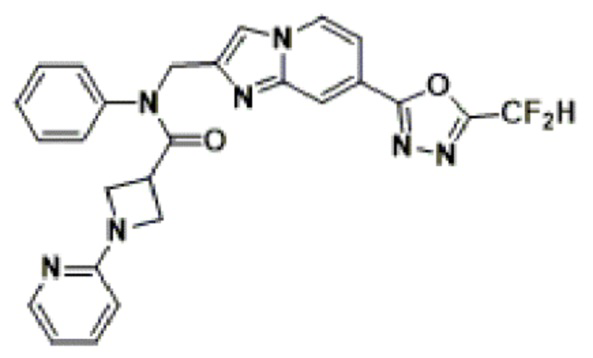
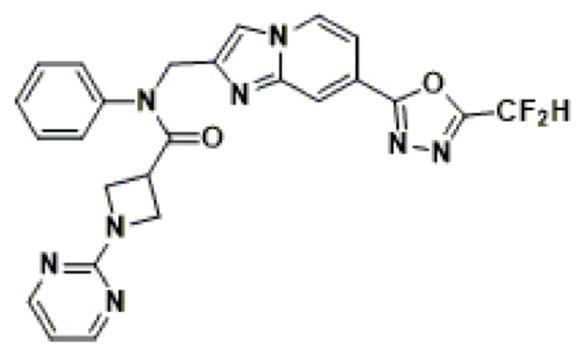
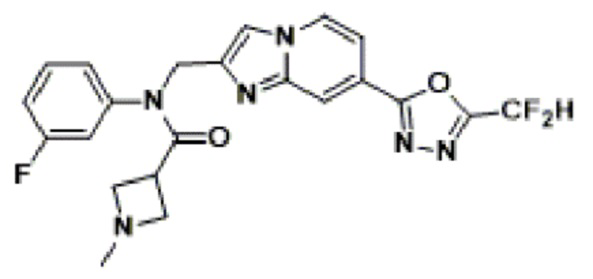
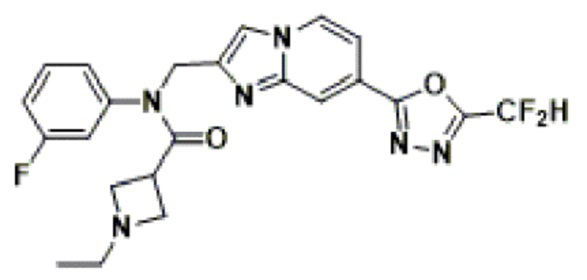
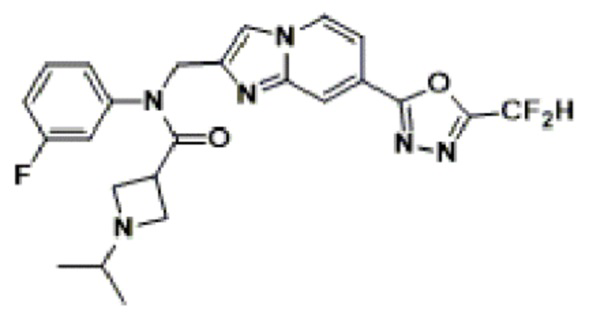
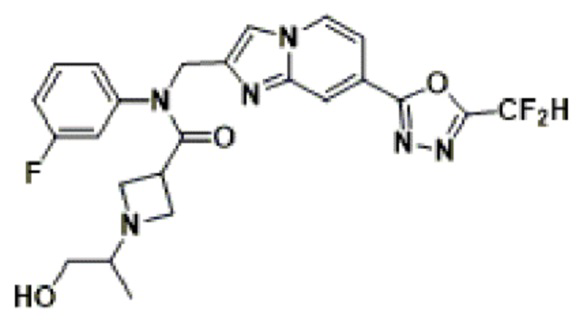
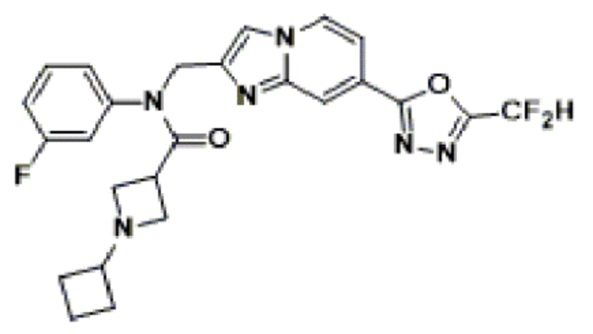
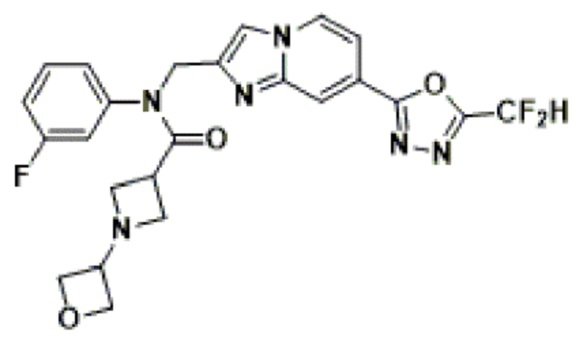
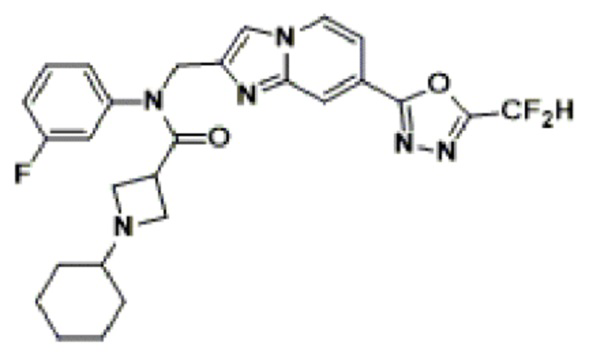
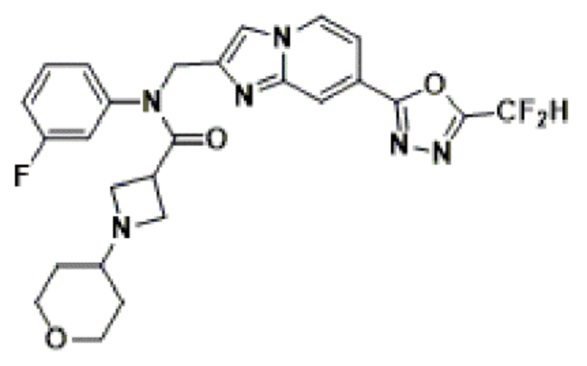
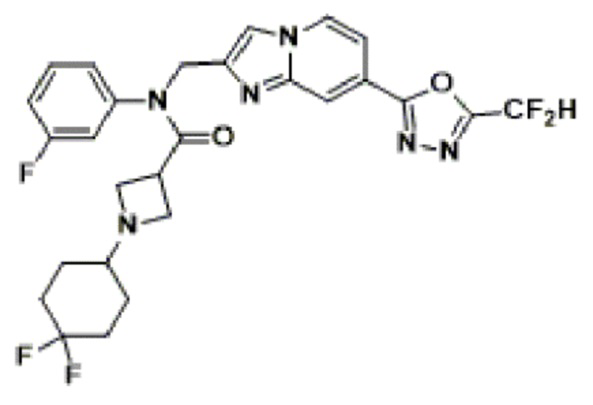
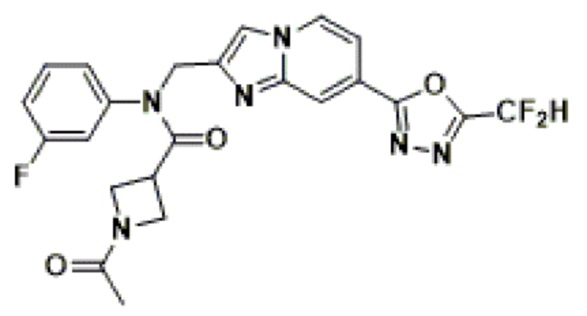
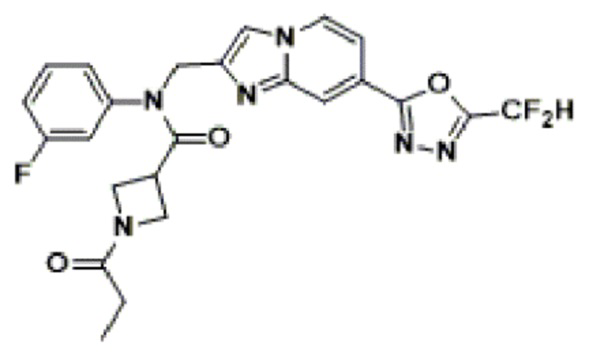
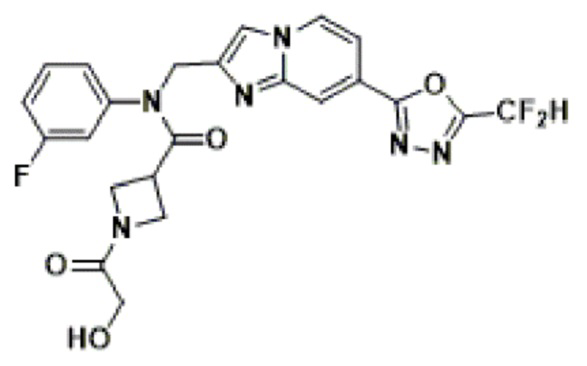
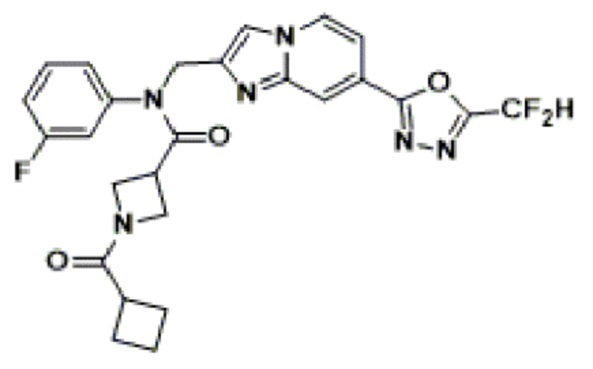
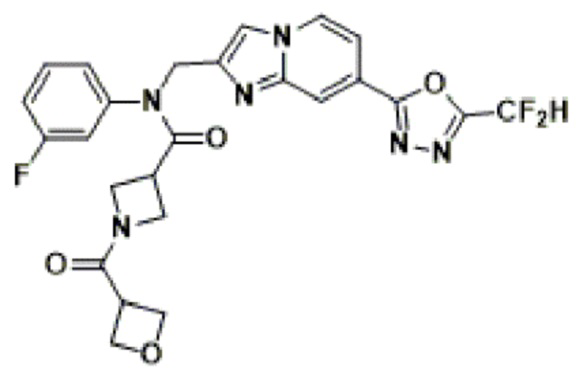
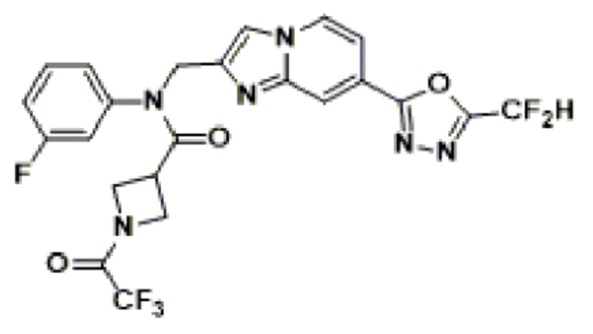
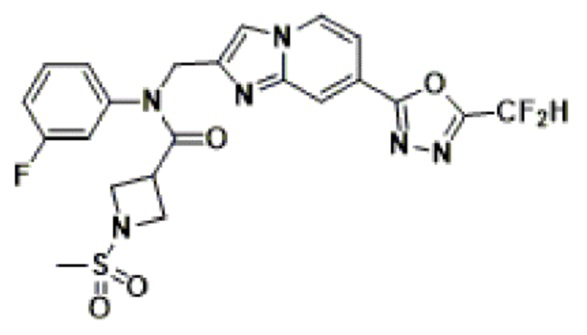
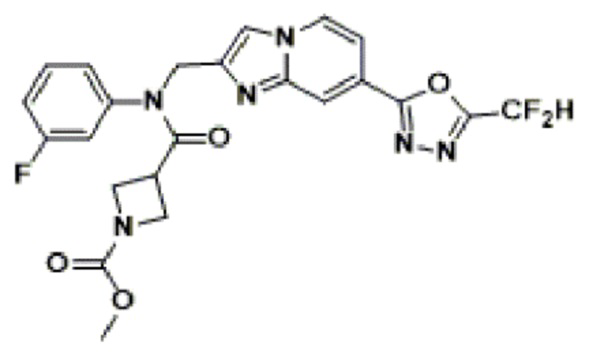
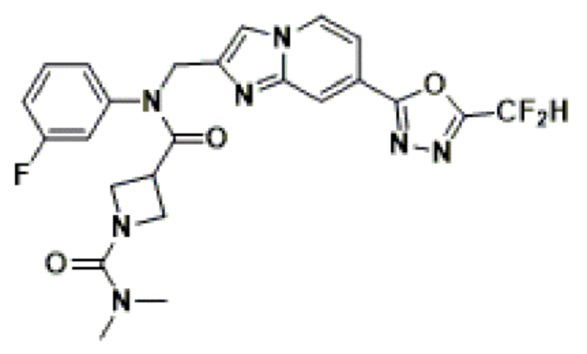
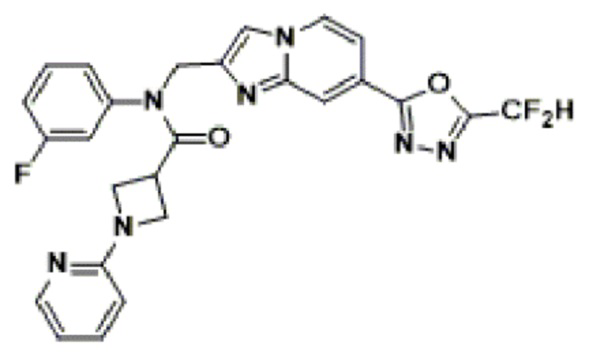
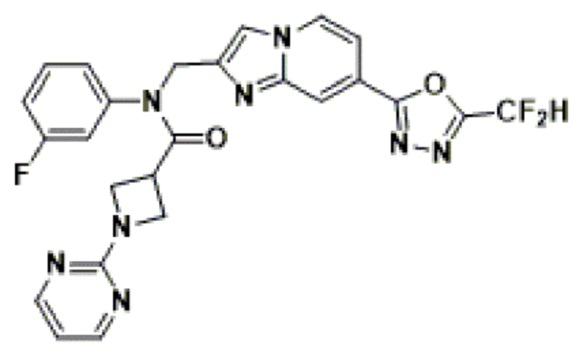
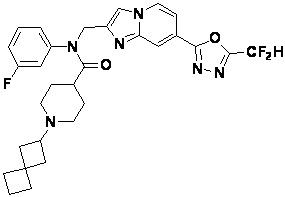
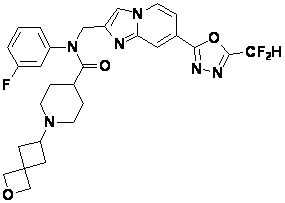
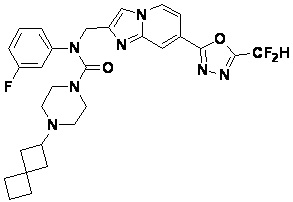
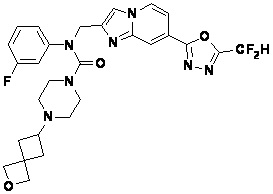
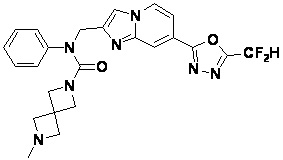
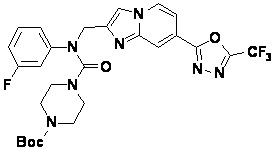
7. Производное 1,3,4-оксадиазола, обладающее селективной ингибирующей активностью в отношении гистондеацетилазы 6 (HDAC6), или его фармацевтически приемлемая соль по п. 6, отличающиеся тем, что они представляют собой любое из соединений, перечисленных в следующей таблице:
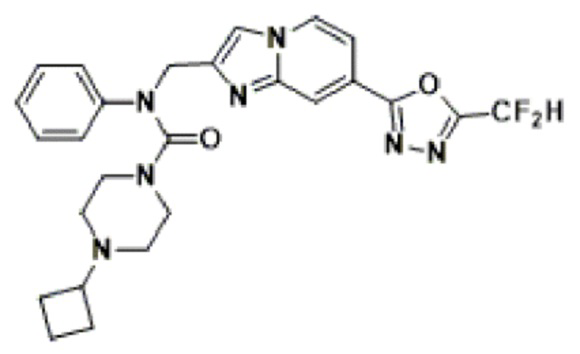
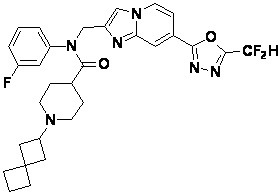
8. Производное 1,3,4-оксадиазола, обладающее селективной ингибирующей активностью в отношении гистондеацетилазы 6 (HDAC6), или его фармацевтически приемлемая соль по п. 6, отличающиеся тем, что они представляют собой любое из соединений, перечисленных в следующей таблице:
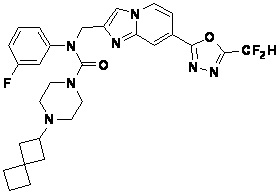
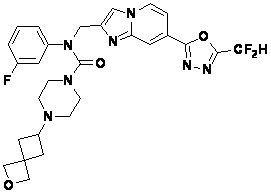
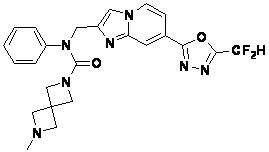
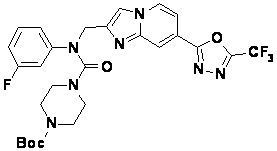
9. Производное 1,3,4-оксадиазола, обладающее селективной ингибирующей активностью в отношении гистондеацетилазы 6 (HDAC6), или его фармацевтически приемлемая соль по п. 6, отличающиеся тем, что они представляют собой любое из соединений, перечисленных в следующей таблице:
10. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении гистондеацетилазы-6, содержащая в качестве активного ингредиента терапевтически эффективное количество соединения, представленного химической формулой I, или его фармацевтически приемлемой соли по любому из пп. 1-9 и фармацевтически приемлемый носитель.
11. Фармацевтическая композиция по п. 10, отличающаяся тем, что указанная композиция применяется для предотвращения или лечения заболеваний, опосредованных гистондеацетилазой-6, и
где указанные заболевания, опосредованные гистондеацетилазой 6, представляют собой инфекционные заболевания; новообразования; эндокринные, пищевые и метаболические заболевания; психические и поведенческие расстройства; неврологические заболевания; заболевания глаза и его придаточного аппарата; заболевания системы кровообращения; респираторные заболевания; заболевания пищеварительной системы; заболевания кожи и подкожных тканей; заболевания опорно-двигательного аппарата и соединительной ткани; или врожденные пороки развития, изменения или хромосомные аномалии.
12. Способ ингибирования гистондеацетилазы 6, включающий введение в качестве активного ингредиента терапевтически эффективного количества соединения, представленного химической формулой I, или его фармацевтически приемлемой соли по любому из пп. 1-9.
13. Применение соединения, представленного химической формулой I, или его фармацевтически приемлемой соли по любому из пп. 1-9 для получения лекарственного средства для ингибирования гистондеацетилазы-6.
| WO 2013066835 A2, 10.05.2013 | |||
| WO 2017222951 A1, 28.12.2017 | |||
| RU 2709207 C2, 17.12.2019 | |||
| 1,3,4,-ОКСАДИЗОЛАМИДНОЕ ПРОИЗВОДНОЕ СОЕДИНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРА ГИСТОНДЕАЦЕТИЛАЗЫ 6 И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2016 |
|
RU2700696C2 |
| ПРОИЗВОДНЫЕ 1,3,4-ОКСАДИАЗОЛСУЛЬФАМИДА В КАЧЕСТВЕ ИНГИБИТОРА ГИСТОНДЕАЦЕТИЛАЗЫ 6 И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2016 |
|
RU2695227C1 |
| БИЦИКЛИЧЕСКИЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ FGFR | 2007 |
|
RU2466130C2 |

