Данная заявка имеет приоритет предварительной заявки на патент США серийный №61/899,903, поданной 5 ноября 2013 года, полное содержание которой включено в данное описание посредством ссылки.
Данное изобретение относится к пролекарствам антагониста NMDA (N-метил-D-аспартата), (S)-1-фенил-2-(пиридин-2-ил)этанамина, и к их применению в лечении депрессии и депрессивных расстройств, в частности большого депрессивного расстройства (MDD), а также для лечения боли (такой как невропатическая боль). Пролекарства (S)-1-фенил-2-(пиридин-2-ил)этанамина можно применять также для лечения синдрома Ретта, суицидального мышления, биполярного расстройства (включая биполярную депрессию), обсессивно-компульсивного расстройства, отравления газом зарином и эпилептического состояния. Изобретение также относится к фармацевтическим композициям, содержащим эти пролекарства, и к способам их получения.
Боль в той или иной форме является широко распространенной частью жизни людей. Боль в результате травмы и боль после хирургического вмешательства часто является временной, но может быть тяжелой и может быть продолжительной. Невропатическая боль, такая как боль при диабетической невропатии и постгерпетической невралгии сильно воздействует на страдающих от нее людей. Каждый год десять миллионов людей по всему миру, включая пациентов в конце их жизни, страдают от боли без адекватного лечения.
Депрессия поражает примерно 120 миллионов людей по всему миру. Симптомы депрессии включают, без ограничения, подавленное настроение, потерю интереса или удовольствия, ощущение вины или низкой самооценки, нарушение сна или аппетита, низкую энергичность и плохую концентрацию или любую их комбинацию. Эти проблемы могут быть хроническими или рекуррентными и могут приводить к существенному ухудшению способности у индивидуума заботиться о себе или выполнять повседневные обязанности. Депрессия является лидирующей причиной нетрудоспособности, измеряемой годами жизни, прожитыми в состоянии инвалидности (YLD), и четвертым лидирующим фактором, вносящим вклад в глобальное бремя болезней, измеряемое годами жизни, скорректированными по нетрудоспособности (DALY; т.е. суммой лет потери потенциальной жизни вследствие преждевременной смерти и лет продуктивной жизни, потерянных вследствие инвалидности), в 2000 году. По прогнозам к 2020 году депрессия достигнет второго места в рейтинге DALY, рассчитанном для всех возрастов, как для мужчин, так и для женщин. В настоящее время депрессия уже является второй причиной DALY в возрастной категории 15-44 лет для обоих полов.
Дигидрохлорид (S)-1-фенил-2-(пиридин-2-ил)этанамина был открыт для лечения MDD (большое депрессивное расстройство) путем внутривенной инфузии (Gerard Sanacora et al, постер, представленный 6 декабря 2012 года на 51-й ежегодной конференции Американского колледжа нейропсихофармакологии в Голливуде, шт. Флорида, США). Другие родственные источники информации включают WO 1993/020052, WO 2000/056324 и WO 2000/63175. Для удобства было бы полезно иметь возможность вводить это лекарственное средство в пероральной лекарственной форме. Однако проблема, связанная с такой пероральной лекарственной формой дигидрохлорида (S)-1-фенил-2-(пиридин-2-ил)этанамина, заключается в том, что он может стать общедоступным для злоупотребления путем внутривенного введения, например путем измельчения таблеточной пероральной лекарственной формы дигидрохлорида (S)-1-фенил-2-(пиридин-2-ил)этанамина с последующей немедленной инъекцией полученной измельченной пероральной лекарственной формы дигидрохлорида (S)-1-фенил-2-(пиридин-2-ил)этанамина. Предполагается, что пролекарства (S)-1-фенил-2-(пиридин-2-ил)этанамина разлагаются в организме человека с образованием (S)-1-фенил-2-(пиридин-2-ил)этанамина, так что при пероральном введении пролекарства (S)-1-фенил-2-(пиридин-2-ил)этанамина будут разлагаться с высвобождением терапевтически эффективной дозы (S)-1-фенил-2-(пиридин-2-ил)этанамина. Но если пролекарство по настоящему изобретению вводить внутривенно, то по предварительной оценке пролекарство будет высвобождать (S)-1-фенил-2-(пиридин-2-ил)этанамин в концентрации ниже Cmax (максимальная концентрация), с меньшей скоростью, чем если бы соответствующую дозу (S)-1-фенил-2-(пиридин-2-ил)этанамина вводили внутривенно. Применение пролекарств (S)-1-фенил-2-(пиридин-2-ил)этанамина может улучшить клинический профиль безопасности (S)-1-фенил-2-(пиридин-2-ил)этанамина (например, в условиях передозировки или злоупотребления лекарственным средством (например, измельчения таблетки)). Таким образом, в итоге, введение (S)-1-фенил-2-(пиридин-2-ил)этанамина непосредственно в пероральную композицию может приводить к злоупотреблениям. Соединения по настоящему изобретению метаболизируются in vivo с образованием (S)-1-фенил-2-(пиридин-2-ил)этанамина, но медленнее, чем при введении (S)-1-фенил-2-(пиридин-2-ил)этанамина внутривенно, и, следовательно, не будут поощрять потенциальное злоупотребление (S)-1-фенил-2-(пиридин-2-ил)этанамином.
Описание графических материалов
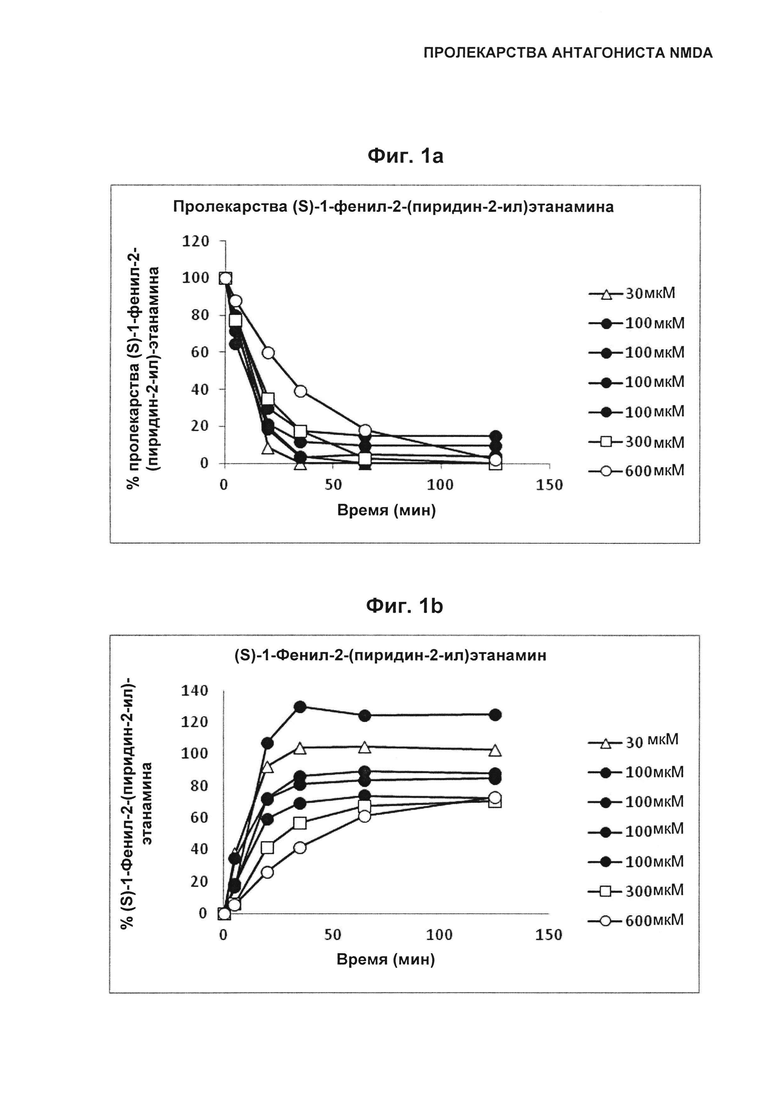
Фиг. 1а и 1b иллюстрируют превращение пролекарства (S)-1-фенил-2-(пиридин-2-ил)этанамина (Пример 5) в (S)-1-фенил-2-(пиридин-2-ил)этанамин во время инкубирования в кишечной среде человека. В частности, Фиг. 1а иллюстрирует концентрацию пролекарства (S)-1-фенил-2-(пиридин-2-ил)этанамина (Пример 5) (начальные концентрации 30, 100, 300 и 600 мкМ) по мере его превращения в (S)-1-фенил-2-(пиридин-2-ил)этанамин во время инкубирования в кишечной среде человека. Фиг. 1b иллюстрирует концентрацию (S)-1-фенил-2-(пиридин-2-ил)этанамина по мере его превращения из пролекарства (S)-1-фенил-2-(пиридин-2-ил)этанамина (Пример 5, начальные концентрации 30, 100, 300 и 600 мкМ) во время инкубирования в кишечной среде человека.
Краткое изложение сущности изобретения
Согласно настоящему изобретению предложено соединение формулы (I):
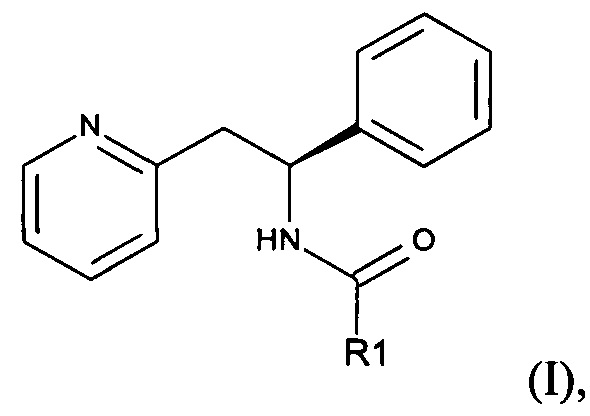
где R1 представляет собой С1-6алкилС(O)O(С1-6алкокси) или
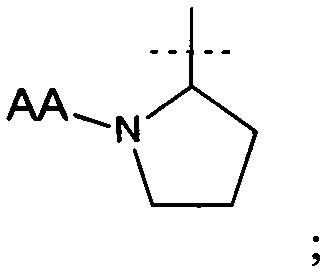
АА представляет собой связанную пептидной связью природную аминокислоту; или его фармацевтически приемлемая соль.
Алкил имеет прямую или разветвленную цепь и содержит 1-6, например 1-4, атомов углерода. Алкил представляет собой, например, метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изобутил или трет-бутил.
Алкокси имеет прямую или разветвленную цепь и содержит 1-6, например 1-4, атомов углерода. Алкокси представляет собой, например, метокси, этокси, н-пропокси, изопропокси, н-бутокси, втор-бутокси, изобутокси или трет-бутокси.
Подробное описание изобретения
В одном аспекте настоящего изобретения предложено соединение формулы (I), где R1 представляет собой С1-6алкилС(O)O(С1-6алкокси), например С1-4алкилС(O)O(С1-4алкокси). Примеры включают: (CH3)2CHC(O)OCH2O, (СН3)2СНС(O)ОСН(СН(СН3)2)O, СН3С(O)ОСН(СН3)O или (СН3)2СНС(O)ОСН(СН3)O.
В другом аспекте настоящего изобретения предложено соединение формулы (I), где R1 представляет собой
и АА представляет собой связанную пептидной связью природную аминокислоту.
В еще одном аспекте настоящего изобретения предложено соединение формулы (I), где R1 представляет собой
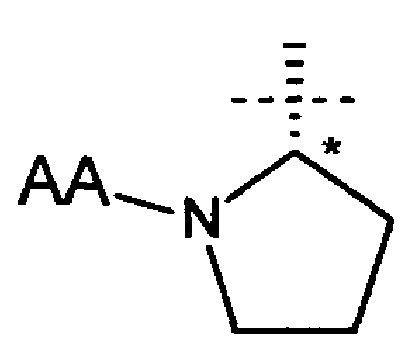
(хиральный центр * имеет S абсолютную конфигурацию).
В дополнительном аспекте настоящего изобретения предложено соединение формулы (I), где АА представляет собой, например, глицин, аланин, валин, лейцин, изолейцин, серин, треонин, цистеин, цистин, метионин, аспарагиновую кислоту, глутаминовую кислоту, аспарагин, глутамин, лизин, гидроксилизин, аргинин, гистидин, фенилаланин, тирозин, триптофан, пролин или гидроксипролин. В еще одном аспекте АА выбран из группы, состоящей из тирозина, триптофана, фенилаланина, лейцина, аргинина, гистидина, лизина и валина. В другом аспекте АА выбран из группы, состоящей из тирозина, аргинина, гистидина, лизина и валина. В еще одном дополнительном аспекте АА представляет собой валин.
Подходящей фармацевтически приемлемой солью является, например, соль присоединения кислоты, такая как гидрохлорид, гидробромид, сульфат, фосфат, ацетат, фумарат, малеат, тартрат, лактат, цитрат, пируват, сукцинат, оксалат, метансульфонат, пара-толуолсульфонат, бисульфат, бензолсульфонат, этансульфонат, малонат, ксинафоат, аскорбат, олеат, никотинат, сахаринат, адипат, формиат, гликолят, L-лактат, D-лактат, аспартат, малат, L-тартрат, D-тартрат, стеарат, 2-фуроат, 3-фуроат, нападисилат (нафталин-1,5-дисульфонат или нафталин-1-(сульфоновая кислота)-5-сульфонат), эдисилат (этан-1,2-дисульфонат или этан-1-(сульфоновая кислота)-2-сульфонат), изетионат (2-гидроксиэтилсульфонат), 2-мезитиленсульфонат, 2-нафталинсульфонат, D-манделат, L-манделат, 2,5-дихлорбензолсульфонат, циннамат или бензоат.
Соединение формулы (I), которое представляет собой:
(S)-(1-фенил-2-(пиридин-2-ил)этилкарбамоилокси)метилизобутират;
2-метил-1-((S)-1-фенил-2-(пиридин-2-ил)этилкарбамоилокси)пропил-изобутират;
2-метил-1-((S)-1-фенил-2-(пиридин-2-ил)этилкарбамоилокси)пропил-изобутират, диастереомер 1;
2-метил-1-((S)-1-фенил-2-(пиридин-2-ил)этилкарбамоилокси)пропил-изобутират, диастереомер 2;
1-((S)-1-фенил-2-(пиридин-2-ил)этилкарбамоилокси)этилацетат;
1-((S)-1-фенил-2-(пиридин-2-ил)этилкарбамоилокси)этилацетат, диастереомер 1;
1-((S)-1-фенил-2-(пиридин-2-ил)этилкарбамоилокси)этилацетат, диастереомер 2;
1-((S)-1-фенил-2-(пиридин-2-ил)этилкарбамоилокси)этилизобутират;
1-((S)-1-Фенил-2-(пиридин-2-ил)этилкарбамоилокси)этилизобутират, диастереомер 1;
1-((S)-1-фенил-2-(пиридин-2-ил)этилкарбамоилокси)этилизобутират, диастереомер 2;
(S)-1-((S)-2-амино-3-метилбутаноил)-N-((S)-1-фенил-2-(пиридин-2-ил)этил)пирролидин-2-карбоксамид;
(S)-1-((S)-2,6-диаминогексаноил)-N-((S)-1-фенил-2-(пиридин-2-ил)этил)пирролидин-2-карбоксамид;
(S)-1-((S)-2-амино-3-(1Н-имидазол-4-ил)пропаноил)-N-((S)-1-фенил-2-(пиридин-2-ил)этил)пирролидин-2-карбоксамид;
2-((S)-2-((S)-1-((5)-2-амино-3-(4-гидроксифенил)пропаноил)пирролидин-2-карбоксамидо)-2-фенилэтил)пиридин;
2-((S)-2-((S)-1-((S)-2-амино-3-(1Н-индол-3-ил)пропаноил)пирролидин-2-карбоксамидо)-2-фенилэтил)пиридин;
(S)-1-((S)-2-амино-3-фенилпропаноил)-N-((S)-1-фенил-2-(пиридин-2-ил)этил)пирролидин-2-карбоксамид; или
(S)-1-((S)-2-амино-4-метилпентаноил)-N-((S)-1-фенил-2-(пиридин-2-ил)этил)пирролидин-2-карбоксамид;
или фармацевтически приемлемую соль любого из вышеуказанных соединений.
Соединения по изобретению могут быть получены, применяя способы, описанные в данной области, или применяя способы, описанные в Примерах. Соединение (S)-1-фенил-2-(пиридин-2-ил)этанамин может быть получено, например, по методологии, описанной в ЕР-0633879, и содержание этого документа включено в данное описание посредством ссылки.
Соединения по настоящему изобретению, где R1 представляет собой
могут быть синтезированы в результате химического процесса, который протекает через промежуточный 2-((S)-2-фенил-2-((S)-пирролид-2-инкарбоксамидо)этил)пиридин.
Таким образом, в другом аспекте настоящего изобретения предложено соединение 2-((S)-2-фенил-2-((S)-пирролид-2-инкарбоксамидо)этил)пиридин:
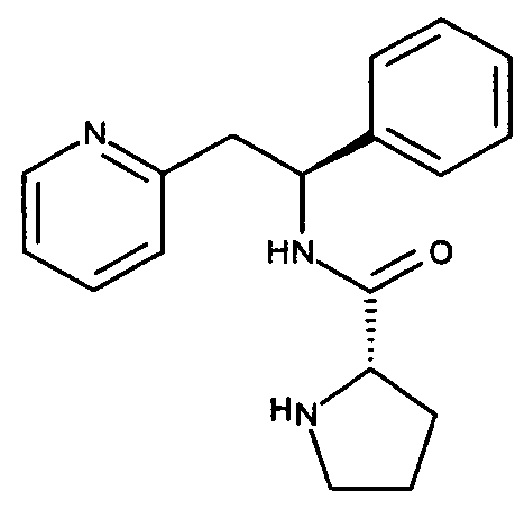
или его соль, где указанная соль представляет собой, например, соль гидрохлорид, гидробромид, сульфат, фосфат, ацетат, метансульфонат, пара-толуолсульфонат, формиат или бензоат.
В дополнительном аспекте настоящего изобретения предложено промежуточное соединение хлорид 2-((S)-2-фенил-2-((S)-пирролидин-2-ийкарбоксамидо)этил)пиридиния
Соединение формулы (I) или его фармацевтически приемлемую соль можно применять в лечении депрессии (такой как большое депрессивное расстройство, например в лечении резистентного глубокого депрессивного расстройства).
Соединение формулы (I) или его фармацевтически приемлемую соль можно применять в лечении боли (такой как невропатическая боль, хроническая боль, фантомная боль, ноцицептивная боль, психогенная боль, инцидентная боль или боль при переломах).
Соединение формулы (I) или его фармацевтически приемлемую соль можно применять в лечении синдрома Ретта, суицидального мышления, биполярного расстройства, обсессивно-компульсивного расстройства, отравления газом зарином или эпилептического состояния.
Таким образом, согласно настоящему изобретению предложено соединение формулы (I) или его фармацевтически приемлемая соль, как определено в данном документе, для применения в терапии. Следовательно, термин "пролекарство" в данном документе может относиться к соединению формулы (I) в форме соли или в форме свободного основания.
В дополнительном аспекте настоящего изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, как определено в данном документе, в изготовлении лекарственного средства для применения в терапии.
В дополнительном аспекте настоящего изобретения предложен способ введения (S)-1-фенил-2-(пиридин-2-ил)этанамина пациенту, включающий введение соединения формулы (I) или его фармацевтически приемлемой соли пациенту, где указанное соединение формулы (I) метаболизируется в организме указанного пациента с образованием (S)-1-фенил-2-(пиридин-2-ил)этанамина.
В контексте настоящего описания изобретения термин "терапия" также охватывает "профилактику", если нет конкретных указаний обратного. Термины "терапевтический" и "терапевтически" должны толковаться соответствующим образом.
Профилактика, как ожидается, будет особенно актуальна для лечения лиц, которые ранее уже имели эпизод или по иным причинам считаются имеющими повышенный риск заболевания или состояния, о котором идет речь. Лица, имеющие риск развития заболевания или состояния, обычно включают лиц, имеющих семейную историю данного заболевания или состояния, или лиц, идентифицированных генетическим тестированием или скринингом как особенно восприимчивых к развитию данного заболевания или состояния.
Согласно изобретению также предложен способ лечения депрессии, включающий введение пациенту, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, как определено в данном документе.
Согласно изобретению также предложен способ лечения MDD, включающий введение пациенту, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, как определено в данном документе.
Согласно изобретению также предложен способ лечения боли, включающий введение пациенту, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, как определено в данном документе.
Согласно изобретению также предложен способ лечения невропатической боли, хронической боли, фантомной боли, ноцицептивной боли, психогенной боли, инцидентной боли или боли при переломах, включающий введение пациенту, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, как определено в данном документе.
Применение соединения формулы (I) или его фармацевтически приемлемой соли для лечения депрессии.
Применение соединения формулы (I) или его фармацевтически приемлемой соли для лечения боли.
Для вышеупомянутых терапевтических применений вводимая дозировка, разумеется, будет варьировать в зависимости от используемого соединения, способа введения, требуемого лечения и показанного расстройства. Например, суточная дозировка соединения по изобретению при введении ингаляцией может находиться в диапазоне от 0,05 микрограммов на килограмм массы тела (мкг/кг) до 100 микрограммов на килограмм массы тела (мкг/кг). Альтернативно, если соединение вводят перорально, то ежесуточная дозировка соединения по изобретению может находиться в диапазоне от 0,01 микрограммов на килограмм массы тела (мкг/кг) до 100 миллиграммов на килограмм массы тела (мг/кг).
Соединения формулы (I) и их фармацевтически приемлемые соли можно применять сами по себе, но обычно их будут вводить в форме фармацевтической композиции, в которой соединение формулы (I)/соль (активный ингредиент) присутствует совместно с фармацевтически приемлемым вспомогательным веществом, разбавителем или носителем. Стандартные методики для выбора и получения подходящих фармацевтических композиций описаны, например, в "Pharmaceuticals-The Science of Dosage Form Designs", M.E. Aulton, Churchill Livingstone, 1988.
В зависимости от способа введения фармацевтическая композиция будет содержать предпочтительно от 0,05 до 99% масс. (массовые проценты), более предпочтительно от 0,05 до 80% масс., еще более предпочтительно от 0,10 до 70% масс. и еще более предпочтительно от 0,10 до 50% масс. активного ингредиента, где все массовые проценты рассчитаны от общей массы композиции. В некоторых воплощениях фармацевтическая композиция содержит 0,5% масс. активного ингредиента. В некоторых воплощениях фармацевтическая композиция содержит 20% масс. активного ингредиента.
Согласно настоящему изобретению также предложена фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль, как определено в данном документе, совместно с фармацевтически приемлемым вспомогательным веществом, разбавителем или носителем.
Согласно настоящему изобретению предложен также способ получения фармацевтической композиции по изобретению, включающий смешивание соединения формулы (I) или его фармацевтически приемлемой соли, как определено а данном документе, с фармацевтически приемлемым вспомогательным веществом, разбавителем или носителем.
Для перорального введения соединение по изобретению может быть смешано с вспомогательным веществом или носителем, например лактозой, сахарозой, сорбитом, маннитом; крахмалом, например картофельным крахмалом, кукурузным крахмалом или амилопектином; производным целлюлозы; связующим, например желатином или поливинилпирролидоном; и/или смазывающим веществом, например стеаратом магния, стеаратом кальция, полиэтиленгликолем, воском, парафином и т.п., и затем спрессовано в таблетки. Если требуются таблетки, покрытые оболочкой, то ядра, полученные так, как описано выше, могут быть покрыты концентрированным раствором сахара, который может содержать, например, аравийскую камедь, желатин, тальк и диоксид титана. Альтернативно, таблетка может быть покрыта подходящим полимером, растворенным в легколетучем органическом растворителе.
Для получения мягких желатиновых капсул соединение по изобретению может быть смешано, например, с растительным маслом или полиэтиленгликолем. Твердые желатиновые капсулы могут содержать гранулы соединения с использованием любых вышеупомянутых эксципиентов для таблеток. Твердые желатиновые капсулы могут быть заполнены также жидкими или полутвердыми композициями соединения по изобретению.
Соединения по изобретению можно также вводить в сочетании с другими соединениями, используемыми для лечения вышеуказанных состояний.
Приведенные ниже Примеры иллюстрируют настоящее изобретение. В Примерах использованы некоторые методы, и эти методы описаны далее.
Жидкостную хроматографию высокого давления (ЖХВД) выполняли на обращенно-фазной (ОФ) колонке. Применяли линейный градиент, используя, например, подвижную фазу А (0,1% муравьиной кислоты в H2O MilliQ, или 0,1% NH3 в H2O MilliQ, или 10 мМ NH4OAc и 5% CH3CN в H2O MilliQ, или 0,05% трифторуксусной кислоты в H2O MilliQ, или NH4HCO3 в H2O MilliQ (10 мМ)) и В (СН3ОН или CH3CN). Масс-спектрометрические (МС) анализы выполняли с электрораспылительной ионизацией в режиме регистрации положительных и/или отрицательных ионов (ЭРИ+/-), с фотоионизацией при атмосферном давлении (ФИАД+/-) и/или с химической ионизацией при атмосферном давлении (ХИАД+/-).
Газовую хроматографию (ГХ) выполняли на газовом хроматографе (ГХ), оснащенном масс-спектрометром (МС) или пламенно-ионизационным детектором (ПИД). МС источником ионов был либо электронный удар (ЭУ), либо химическая ионизация (ХИ, газ-реактант: метан). Для разделения использовали капиллярную колонку, например DB-5MS (J&W Scientific). Применяли линейный температурный градиент.
Суперкритическую флюидную хроматографию (СФХ) выполняли на прямо-фазной колонке. Применяли изократический поток с использованием подвижной фазы А (CO2) и, например, подвижной фазы В (МеОН, EtOH или IPA).
Альтернативно, жидкостную хроматографию высокого давления (ЖХВД) выполняли на прямо-фазной колонке. Применяли линейный градиент или изократический поток с использованием, например, подвижной фазы А (гептан) и В (EtOH или IPA).
ЯМР-спектры регистрировали на ЯМР-спектрометре 300 МГц (или более высокое поле), снабженном детектором подходящей конфигурации. Спектры регистрировали при температуре окружающей среды, если не указано иное. Химические сдвиги приведены в миллионных долях (м.д.) в направлении снижения и увеличения от уровня сигнала TMS (триметилсилан) (0,00 м.д.). Использовали следующие эталонные сигналы: TMS δ 0.00, или остаточный сигнал растворителя DMSO-d6 δ 2.49, CD3OD δ 3.30, ацетон-d6 2.04, CDCl3 δ 7.25, или D2O δ 4.79 (если не указано иное). Мультиплетности резонанса обозначены s, d, t, q, m, br и app, означающие синглет, дублет, триплет, квартет, мультиплет, уширенный и кажущийся соответственно.

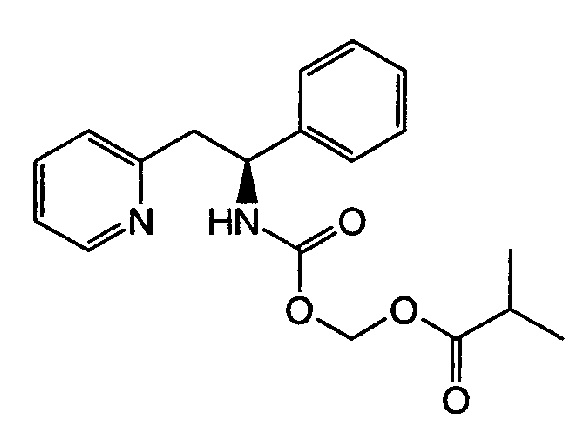
Пример 1
(S)-(1-Фенил-2-(пиридин-2-ил)этилкарбамоилокси)метилизобутират
Стадия А
Хлорметилизобутират
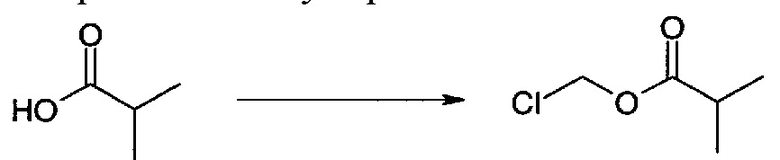
К изомасляной кислоте (0,600 мл, 6,47 ммоль) в DCM (6 мл) добавляли бикарбонат натрия (2092 мг, 24,91 ммоль), гидросульфат тетрабутиламмония (220 мг, 0,65 ммоль) и воду (6 мл). При быстром перемешивании добавляли хлорметилсульфохлоридат (0,767 мл, 7,44 ммоль) при к.т., и реакционную смесь затем перемешивали при к.т.в течение ночи. Реакционную смесь затем разбавляли DCM (10 мл), промывали водой (2×10 мл), сушили над Na2SO4, фильтровали и концентрировали с получением хлорметилизобутирата (746 мг, 84%), который использовали на следующей стадии без дополнительной очистки.
1Н ЯМР (500 МГц, CDCl3) δ м.д. 1.17 (m, 6 Н), 2.62 (m, 1 Н), 5.72 (s, 2 Н).
Стадия В
(S)-(1-Фенил-2-(пиридин-2-ил)этилкарбамоилокси)метилизобутират
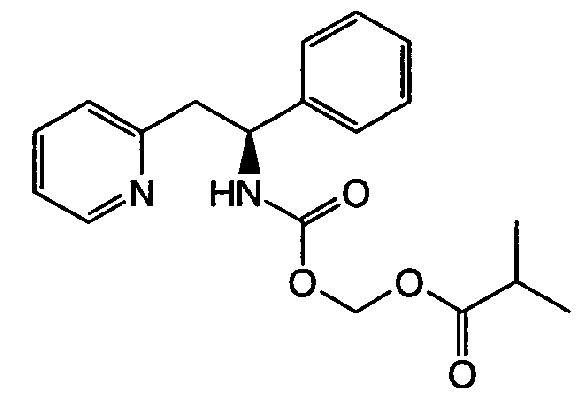
Карбонат цезия (1543 мг, 4,74 ммоль) и йодид тетрабутиламмония (1749 мг, 4,74 ммоль) добавляли к (S)-1-фенил-2-(пиридин-2-ил)этанамину (313 мг, 1,58 ммоль) в безводном DMF (8 мл) при к.т. Газ диоксид углерода барботировали в реакционную смесь в течение 30 мин, после чего добавляли хлорметилизобутират (647 мг, 4,74 ммоль) в DMF (2 мл). Реакционную смесь перемешивали при к.т., продолжали барботирование газа CO2 в течение ночи и продолжали перемешивать на протяжении выходных дней без добавления дополнительного газа СО2. Реакционную смесь разбавляли водой и экстрагировали EtOAc (3×), объединенные органические слои промывали водой (2×), рассолом, сушили над Na2SO4 и концентрировали. Очистку выполняли колоночной хроматографией, используя градиент EtOAc в гептане (0-60%), с получением указанного в заголовке соединения (239 мг, 44,2%).
1Н ЯМР (500 МГц, DMSO-d6) δ м.д. 1.00 (m, 6 Н), 2.46 (m, 1 Н, частично скрыт в DMSO-d6), 3.09 (m, 2 Н), 5.04 (m, 1 Н), 5.52 (m, 2 Н), 7.17-7.24 (m, 3 Н), 7.27-7.34 (m, 4 Н), 7.65 (td, 1 Н), 8.24 (d, 1 Н), 8.49 (m, 1 Н).
Пример 2
2-Метил-1-((S)-1-фенил-2-(пиридин-2-ил)этилкарбамоилокси)-пропилизобутират
Существует два разных диастереомера соединения Примера 2 благодаря двум возможным конфигурациям по атому углерода, показанному звездочкой *. Они упоминаются как соединение Примера 2, диастереомер 1, и соединение Примера 2, диастереомер 2. Их абсолютная конфигурация не определена.
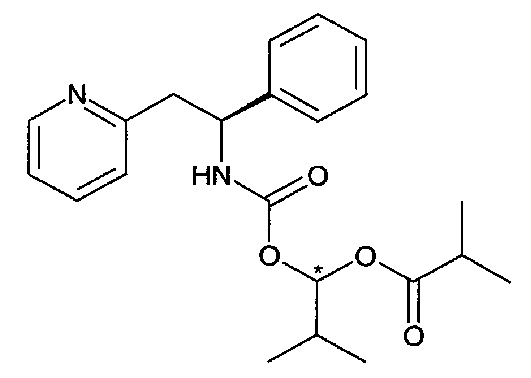
Стадия А
2-Метил-1-((4-(метилсульфонил)фенокси)карбонилокси)-пропилизобутират
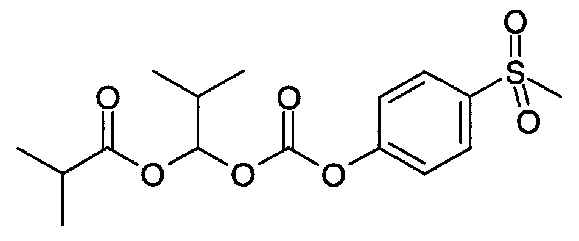
(1) 4-(Метилмеркапто)фенол (8,46 г, 57,30 ммоль) переносили в DCM (60 мл), и затем реакционную колбу охлаждали до 0°C, после чего добавляли 1-хлор-2-метилпропилкарбонохлоридат (4,27 мл, 28,65 ммоль). По каплям добавляли раствор 4-метилморфолина (7,87 мл, 71,63 ммоль) в DCM (40 мл) в течение 50 минут при 0°C, и полученную смесь перемешивали при этой температуре в течение 5 минут и в конце перемешивали при к.т. в течение 150 минут. Реакционную смесь промывали водой (2×), сушили над Na2SO4, фильтровали и упаривали с получением 1-хлор-2-метилпропил-4-(метилтио)фенилкарбоната (13,84 г), который использовали на следующей стадии без дополнительной очистки.
(2) Смесь 1-хлор-2-метилпропил-4-(метилтио)фенилкарбоната (3,50 г, 12,74 ммоль), оксида серебра(I) (2,95 г, 12,74 ммоль) и изомасляной кислоты (13,00 мл, 140,12 ммоль) в атмосфере аргона нагревали до 95°C в течение 2 часов. Реакционную смесь охлаждали до к.т. и перемешивали при к.т. в течение ночи, затем разбавляли МТВЕ, фильтровали через диатомитовую землю и снова промывали МТВЕ. Объединенные фильтраты промывали водой (4×25 мл), насыщенным водным бикарбонатом натрия (2×25 мл), сушили (Na2SO4) и упаривали с получением 3,56 г 2-метил-1-((4-(метилтио)фенокси)-карбонилокси)пропилизобутирата, который использовали на следующей стадии без дополнительной очистки.
(3) 2-Метил-1-((4-(метилтио)фенокси)карбонилокси)пропилизобутират (3,56 г, 10,91 ммоль) в смесь ацетона (30 мл) и воды (7,50 мл), после чего порциями добавляли оксон (13,41 г, 21,81 ммоль) в течение 5 минут, затем перемешивали при к.т. в течение 2 часов. Реакционную смесь фильтровали, и фильтрат промывали МТВЕ (2×50 мл), объем сокращали до 50 мл (отгоняя ацетон), и затем полученную смесь разделяли между МТВЕ и водой. Водный слой экстрагировали МТВЕ, и объединенные органические фазы сушили над Na2SO4, фильтровали и упаривали с получением 1,89 г указанного в заголовке соединения, которое использовали на следующей стадии без дополнительной очистки.
Стадия В:
2-Метил-1-((S)-1-фенил-2-(пиридин-2-ил)этилкарбамоилокси)-пропилизобутират
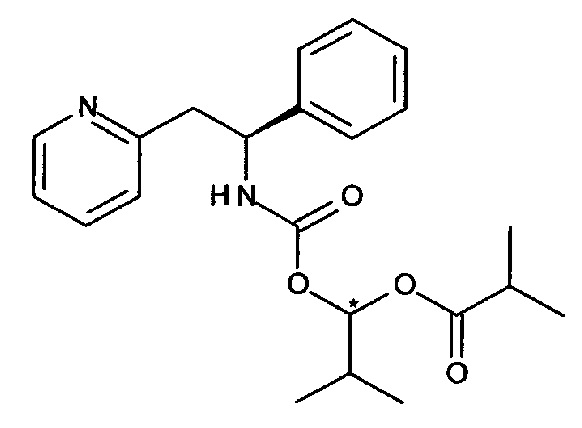
В перемешиваемую смесь (S)-1-фенил-2-(пиридин-2-ил)этанамина (0,215 г, 1,08 ммоль) и бикарбоната натрия (0,182 г, 2,17 ммоль) в ацетонитриле (3 мл) добавляли 2-метил-1-((4-(метилсульфонил)фенокси)карбонилокси)пропил-изобутират (0,389 г, 1,08 ммоль) в ацетонитриле (2 мл), и реакционную смесь перемешивали при к.т. в течение 2 часов. Смесь разделяли между EtOAc и насыщенным водным раствором NaHCO3, органический слой промывали насыщенным водным раствором NaHCO3, сушили (Na2SO4) и упаривали с получением 413 мг вещества, которое очищали колоночной хроматографией, используя градиент EtOAc в гептане (0-50%), с получением 235 мг продукта в виде смеси двух диастереомеров 2-метил-1-(S)-1-фенил-2-(пиридин-2-ил)этилкарбамоилокси)пропилизобутирата.
Анализ и разделение диастереомеров выполняли на Chiralpak AD-H, 4,6×250 мм; 5 мкм, используя 10% МеОН/90% CO2 при скорости потока 3 мл/мин, и Chiralpak AD-H, 20×250 мм; 5 мкм, используя 10% МеОН/90% CO2 при скорости потока 50 мл/мин, соответственно.
Пример 2, диастереомер 1
105 мг диастереомера 1 было получено в результате хирального разделения как элюирующийся первым диастереомер с оптической чистотой 99%. Смесь ротамеров:
1Н ЯМР (500 МГц, DMSO-d6) δ м.д. 0.66-1.07 (m), 1.86 (m), 2.39 (m), 2.98-3.21 (m), 5.01 (m), 6.31 (m), 7.10-7.26 (m), 7.26-7.37 (m), 7.56-7.73 (m), 8.10 (d), 8.43-8.55 (m). Общее число протонов в спектре: 28. Соотношение ротамеров основной/минорный: 1/0,15.
МС (ЭРИ+ХИАД+) m/z 385 (М+Н)+
Пример 2, диастереомер 2
104 мг диастереомера 2 было получено как элюирующийся вторым диастереомер. Смесь ротамеров:
1Н ЯМР (500 МГц, DMSO-d6) δ м.д. 0.48-0.69 (m), 0.75-0.87 (m), 0.87-1.07 (m), 1.73 (m), 1.85 (m), 2.40 (m), 2.98 (m), 3.03-3.18 (m), 4.97 (m), 6.22-6.38 (m), 7.13-7.26 (m), 7.26-7.37 (m), 7.65 (m), 7.78 (d), 8.06 (d), 8.48 (m). Общее число протонов в спектре: 28. Соотношение ротамеров основной/минорный: 1/0,17.
МС (ЭРИ+ХИАД+) m/z 385 (М+Н)+.
Оптическая чистота =99%
Пример 3
1-((S)-1-Фенил-2-(пиридин-2-ил)этилкарбамоилокси)этилацетат
Существует два разных диастереомера соединения Примера 3, поскольку возможны две конфигурации по атому углерода, обозначенному звездочкой *. Эти диастереомеры названы соединение Примера 3, диастереомер 1, и соединение Примера 3, диастереомер 2. Их абсолютные конфигурации не определены.
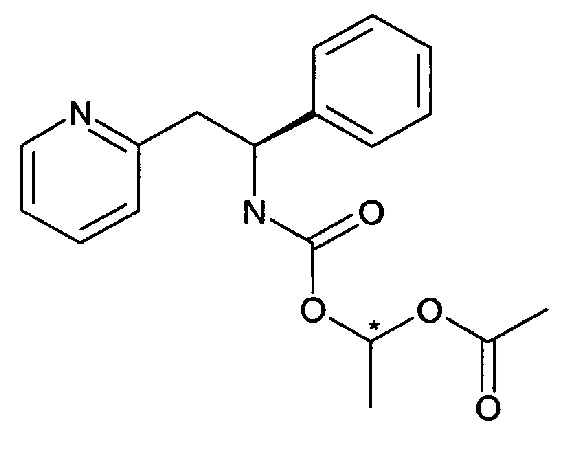
Стадия А:
1-((4-(Метилсульфонил)фенокси)карбонилокси)этилацетат
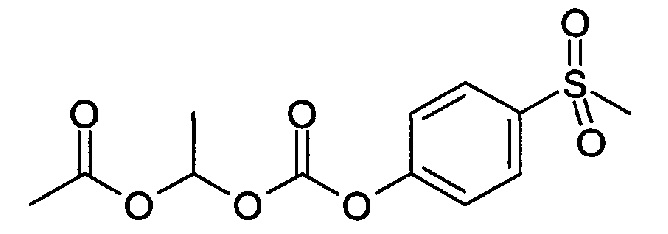
(1) Смесь 1-хлорэтил 4-(метилтио)фенилкарбоната (4,6 г, 18,65 ммоль), оксида серебра(I) (4,32 г, 18,65 ммоль) и уксусной кислоты (11,75 мл, 205,1 ммоль) в атмосфере аргона нагревали до 95°C в течение 2 ч. Реакционную смесь охлаждали до к.т. и разбавляли МТВЕ, фильтровали через диатомитовую землю и промывали дополнительным МТВЕ. Объединенные фильтраты промывали водой (4×25 мл), насыщенным водным раствором бикарбоната натрия (2×25 мл), сушили (Na2SO4) и упаривали с получением 1,79 г 1-((4-(метилтио)фенокси)карбонилокси)этилацетата, который использовали на следующей стадии без дополнительной очистки.
(2) 1-((4-(Метилтио)фенокси)карбонилокси)этилацетат (1,79 г, 6,62 ммоль) переносили в смесь ацетона (16 мл) и воды (4,00 мл). Порциями добавляли Оксон (8,14 г, 13,24 ммоль) в течение 5 минут, затем перемешивали при к.т. в течение ночи. Реакционную смесь фильтровали, и фильтрат промывали МТВЕ (2×50 мл), объем сокращали до примерно 50 мл (отгоняя ацетон). Продукт разделяли между МТВЕ и водой. Водный слой экстрагировали МТВЕ, и объединенные органические фазы сушили (Na2SO4), фильтровали и упаривали с получением 832 мг указанного в заголовке соединения, которое использовали на следующей стадии без дополнительной очистки.
Стадия В:
1-((S)-1-Фенил-2-(пиридин-2-ил)этилкарбамоилокси)этилацетат
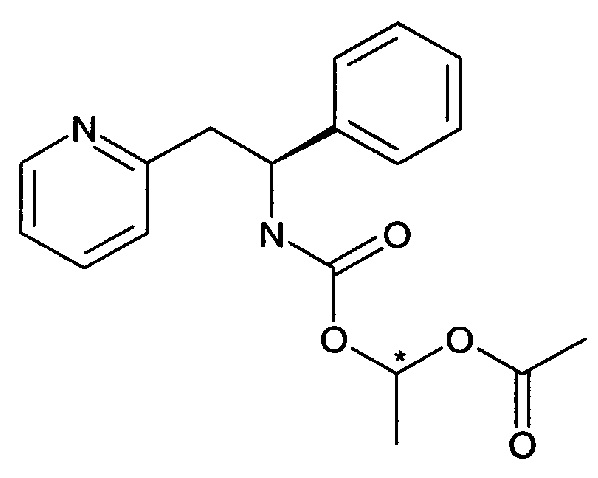
В перемешиваемую смесь (S)-1-фенил-2-(пиридин-2-ил)этанамина (198 мг, 1 ммоль) и бикарбоната натрия (168 мг, 2,00 ммоль) в ацетонитриле (4 мл) добавляли 1-((4-(метилсульфонил)фенокси)карбонилокси)этилацетат (302 мг, 1,00 ммоль) в ацетонитриле (1 мл), и эту реакционную смесь перемешивали при к.т. в течение 4 часов. Реакционную смесь затем разделяли между EtOAc и насыщенным водным раствором NaHCO3, органический слой промывали насыщенным водным раствором NaHCO3, рассолом, сушили над Na2SO4 и упаривали. Эту смесь предварительно очищали колоночной хроматографией, используя градиент EtOAc в гептане (0-50%), с получением 160 мг указанного в заголовке соединения в виде смеси двух его диастереомеров.
Анализ и разделение диастереомеров выполняли на Phenomenex LuxC4, 4,6×250 мм; 5 мкм, используя 30% MeOH+DEA/70% CO2 при скорости потока 3 мл/мин, и Phenomenex LuxC4, 20×250 мм; 5 мкм, используя 25% MeOH+DEA/75% CO2 при скорости потока 50 мл/мин, соответственно.
Пример 3, диастереомер 1
29 мг диастереомера 1 было получено путем хирального разделения как элюирующийся первым диастереомер с оптической чистотой 99%.
1Н ЯМР (500 МГц, DMSO-d6) δ м.д. 1.31 (d, 3 Н), 1.92 (s, 3 Н), 3.00-3.16 (m, 2 Н), 5.00 (td, 1 Н), 6.52 (q, 1 Н), 7.14-7.25 (m, 3 Н), 7.30 (d, 4 Н), 7.65 (t, 1 Н), 8.13 (d, 1 Н), 8.50 (d, 1 Н). Сигналы в спектре были уширены, и расщепление DMSO-сигнала нельзя было видеть.
13С ЯМР (126 МГц, DMSO-d6) δ 19.6, 20.7, 44.6, 54.9, 88.7, 121.6, 123.8, 126.4, 126.9, 128.3, 136.2, 143.0, 149.0, 153.2, 158.0, 168.6 м.д. МС (ЭРИ+) m/z 328 (М+Н)+.
Пример 3, диастереомер 2
39 мг диастереомера 2 было получено как элюирующийся вторым диастереомер.
1Н ЯМР (500 МГц, DMSO-d6) δ м.д. 1.32 (d, 3 Н), 1.95 (s, 3 Н), 3.01-3.18 (m, 2 Н), 5.00 (td, 1 Н), 6.53 (q, 1 Н), 7.13-7.24 (m, 3 Н), 7.29 (d, 4 Н), 7.65 (t, 1 Н), 8.17 (d, 1 Н), 8.49 (d, 1 Н). Сигналы в спектре были уширены, и расщепление DMSO-сигнала нельзя было видеть.
13С ЯМР (126 МГц, DMSO-d6) δ 19.6, 20.7, 44.4, 54.9, 88.6, 121.6, 123.7, 126.4, 126.9, 128.3, 136.2, 143.0, 149.0, 153.1, 158.0, 168.5. МС (ЭРИ+) m/z 329 (М+Н)+.
Оптическая чистота 98%.
Пример 4
1-((S)-1-Фенил-2-(пиридин-2-ил)этилкарбамоилокси)этилизобутират
Существует два разных диастереомера соединения Примера 4, поскольку возможны две конфигурации по атому углерода, обозначенному звездочкой *. Эти диастереомеры названы соединением Примера 4, диастереомер 1, и соединение Примера 4, диастереомер 2. Их абсолютные конфигурации не определены.
Стадия А:
1-((4-(Метилсульфонил)фенокси)карбонилокси)этилизобутират
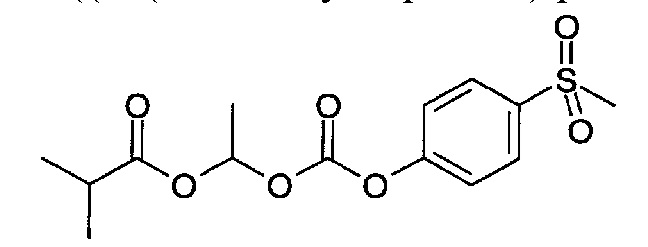
(1) 4-(Метилмеркапто)фенол (4,91 г, 35,00 ммоль) переносили в DCM (100 мл) и охлаждали до 0°C. Добавляли 1-хлорэтилхлорформиат (1,888 мл, 17,50 ммоль). По каплям добавляли раствор 4-метилморфолина (4,81 мл, 43,74 ммоль) в DCM (20 мл) в течение 10 минут при 0°C, и полученную смесь перемешивали при этой температуре в течение 5 минут. Реакционную смесь перемешивали при к.т. в течение 180 минут, затем разбавляли DCM, промывали водой (2×), сушили над Na2SO4, фильтровали и упаривали с получением 1-хлорэтил-4-(метилтио)фенилкарбоната (6,94 г), который использовали на следующей стадии без дополнительной очистки.
(2) Смесь 1-хлорэтил-4-(метилтио)фенилкарбоната (2,3 г, 9,32 ммоль), оксида серебра(I) (2,160 г, 9,32 ммоль) и изомасляной кислоты (9,51 мл, 102,55 ммоль) в атмосфере аргона нагревали до 95°C в течение 2 ч. Реакционную смесь охлаждали до к.т. и разбавляли МТВЕ, фильтровали через диатомитовую землю, промывали дополнительным МТВЕ. Объединенные фильтраты промывали водой (4×25 мл), насыщенным водным раствором бикарбоната натрия (2×25 мл), сушили (Na2SO4) и упаривали с получением 1,16 г продукта, который использовали на следующей стадии без дополнительной очистки.
(3) 1-((4-(Метилтио)фенокси)карбонилокси)этилизобутират (1,16 г, 3,89 ммоль) переносили в смесь ацетона (12 мл) и воды (3,00 мл). Порциями добавляли Оксон (4,78 г, 7,78 ммоль) в течение 5 минут, затем перемешивали при к.т. в течение ночи. Реакционную смесь фильтровали, и фильтрат промывали МТВЕ (2×50 мл), объем сокращали до приблизительно 50 мл, отгоняя ацетон, затем разделяли между МТВЕ и водой. Водный слой экстрагировали МТВЕ, и объединенные органические фазы сушили над Na2SO4, фильтровали и упаривали с получением 531 мг указанного в заголовке соединения, который использовали на следующей стадии без дополнительной очистки.
Стадия В
1-((S)-1-Фенил-2-(пиридин-2-ил)этилкарбамоилокси)этилизобутират
В перемешиваемую смесь (S)-1-фенил-2-(пиридин-2-ил)этанамина (0,300 г, 1,51 ммоль) и бикарбоната натрия (0,254 г, 3,03 ммоль) в ацетонитриле (6 мл) добавляли 1-((4-(метилсульфонил)фенокси)карбонилокси)этилизобутират (0,500 г, 1,51 ммоль) в ацетонитриле (2 мл), и эту реакционную смесь перемешивали в течение ночи. Смесь разделяли между EtOAc и насыщенным водным раствором NaHCO3, органический слой промывали насыщенным водным раствором NaHCO3, рассолом, сушили над Na2SO4 и упаривали с получением 0,70 г вещества, которое очищали колоночной хроматографией, используя градиент EtOAc в гептане (0-50%), с получением указанного в заголовке соединения (0,309 г, 57,2%) в виде смеси двух диастереомеров.
Анализ и разделение диастереомеров выполняли на Phenomenex LuxC4, 4,6×250 мм; 5 мкм, используя 15% MeOH+DEA/85% CO2 при скорости потока 3 мл/мин, и Phenomenex LuxC4, 20×250 мм; 5 мкм, используя 15% MeOH+DEA/85% СО2 при скорости потока 50 мл/мин, соответственно.
Пример 4, диастереомер 1
25 мг диастереомера 1 было получено путем хирального разделения как элюирующийся первым диастереомер с оптической чистотой 99%. Смесь ротамеров:
1Н ЯМР (500 МГц, DMSO-d6) δ м.д. 0.86-1.05 (m), 1.19 (d), 1.32 (d), 2.31-2.44 (m), 2.98-3.20 (m), 4.94-5.07 (m), 6.48 (d), 6.52 (q), 7.15-7.25 (m), 7.25-7.34 (m), 7.60-7.70 (m), 7.77 (d), 8.06-8.18 (m), 8.44-8.55 (m). Общее число протонов в спектре: 24. Соотношение основной/минорный: 1:0,07.
МС (ЭРИ+ХИАД+) m/z 357 (М+Н)+.
УФ чистота =100%.
Пример 4, диастереомер 2
25 мг диастереомера 2 было получено как элюирующийся вторым изомер. Смесь ротамеров:
1Н ЯМР (500 МГц, DMSO-d6) δ м.д. 0.82-0.94 (m), 0.94-1.04 (m), 1.25 (d), 1.32 (d), 2.24-2.32 (m), 2.34-2.44 (m), 3.00-3.17 (m), 5.02 (td), 6.48 (d), 6.52 (q), 7.15-7.25 (m), 7.25-7.38 (m), 7.55-7.77 (m), 8.16 (d), 8.44-8.57 (m). Общее число протонов в спектре: 24. Соотношение основной/минорный: 1:0,08.
МС (ЭРИ+ХИАД+) m/z 357 (М+Н)+.
УФ чистота =100%.
Оптическая чистота =99%.
В Примерах 5-12 использовали общее промежуточное соединение, получение которого описано ниже.
Получение общего промежуточного соединения для Примеров 5-12
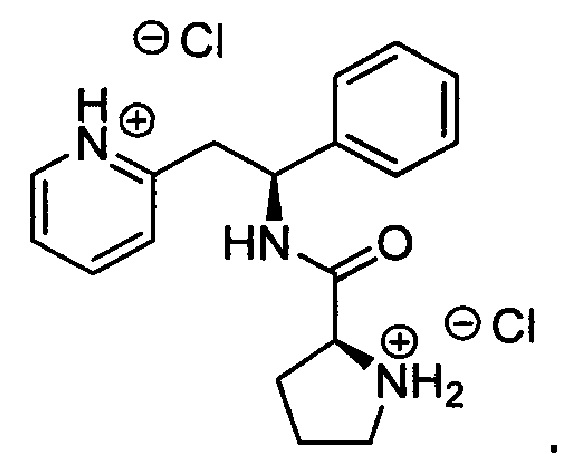
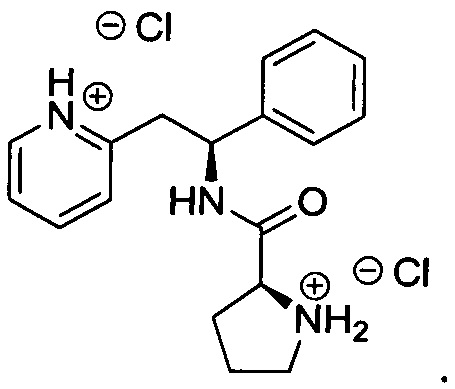
Дихлорид 2-((S)-2-фенил-2-((S)-пирролидин-2-ийкарбоксамидо)этил)-пиридиния (который также имеет название дигидрохлорид (S)-N-((S)-1-фенил-2-(пиридин-2-ил)этил)пирролидин-2-карбоксамида)
СТАДИЯ А
(S)-трет-Бутил-2-((S)-1-фенил-2-(пиридин-2-ил)этилкарбамоил)-пирролидин-1-карбоксилат
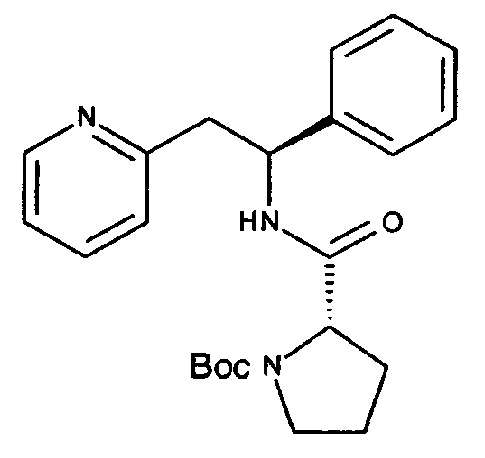
ТЗР (50% масс. в DMF, 0,86 мл, 1,47 ммоль) по каплям добавляли в раствор (1S)-1-фенил-2-(пиридин-2-ил)этан-1-амина (200 мг, 0,737 ммоль), Вос-L-пролина (159 мг, 0,737 ммоль) и DIPEA (0,64 мл, 3,69 ммоль) в CH2Cl2 (7,4 мл) при 0°C. Реакционную смесь постепенно нагревали до к.т. при перемешивании в течение ночи. Органический слой промывали дважды 5% водным раствором NaHCO3, один раз рассолом, сушили над Na2SO4, фильтровали и концентрировали. Продукт очищали с использованием 50 г Snap колонки, элюируя с градиентом МеОН и CH2Cl2 (0% МеОН/100% CH2Cl2→10% МеОН/90% CH2Cl2), с получением указанного в заголовке соединения, 305 мг (>100%).
1Н ЯМР (300 МГц, CDCl3): м.д. 1.35 (s, 9 Н), 1.70-1.90 (m, 2 Н), 2.05-2.19 (m, 2 Н), 3.13 (dd, 1 Н), 3.29-3.39 (m, 1 Н), 3.46-3.62 (m, 2 Н), 4.20-4.29 (m, 2 Н), 5.39 (q, 1 Н), 6.91 (d, 1 Н), 7.09-7.14 (m, 1 Н), 7.16-7.23 (m, 5 Н), 7.49 (td, 1 Н), 8.45-8.59 (m, 1 Н).
Альтернативный способ:
Boc-L-Pro-OH (2,0 г, 9,29 ммоль) растворяли в безводном DMF (15 мл). Добавляли HATU (3,7 г, 9,76 ммоль) и основание Хюнига (5,3 мл, 30,66 ммоль), и смесь перемешивали при к.т. в течение 30 минут. Затем в раствор добавляли дихлорид 2-[(2S)-2-азаниумил-2-фенилэтил]пиридин-1-ия (2,5 г, 9,29 ммоль), и смесь перемешивали в течение 3 часов 30 минут при к.т. Добавляли воду, и смесь экстрагировали 3×EtOAc. Органическую фазу промывали рассолом, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Продукт очищали на SiO2 колонке, элюируя с градиентом (0% МеОН/50% EtOAc/50% гептан →0% МеОН/100% EtOAc/0% гептан →10% МеОН/90% EtOAc/0% гептан), и затем на С-18 колонке, элюируя с градиентом МеОН и воды (от 0 до 100% МеОН), с получением 2,9 г (78%) указанного в заголовке соединения.
1Н ЯМР (300 МГц, CDCl3): м.д. 1.35 (s, 9 Н), 1.70-1.90 (m, 2 Н), 2.05-2.19 (m, 2 Н), 3.13 (dd, J=13.8, 7.1 Гц, 1 Н), 3.29-3.39 (m, 1 Н), 3.46-3.62 (m, 2 Н), 4.20-4.29 (m, 1 Н), 5.39 (q, J=6.8 Гц, 1 Н), 6.91 (d, J=7.3 Гц, 1 Н), 7.09-7.14 (m, 1 Н), 7.16-7.23 (m, 5 Н), 7.49 (td, J=7.63, 1.8 Гц, 1 Н), 8.45-8.59 (m, 1 Н).
СТАДИЯ В
Дигидрохлорид (S)-N-((S)-1-фенил-2-(пиридин-2-ил)этил)пирролидин-2-карбоксамида
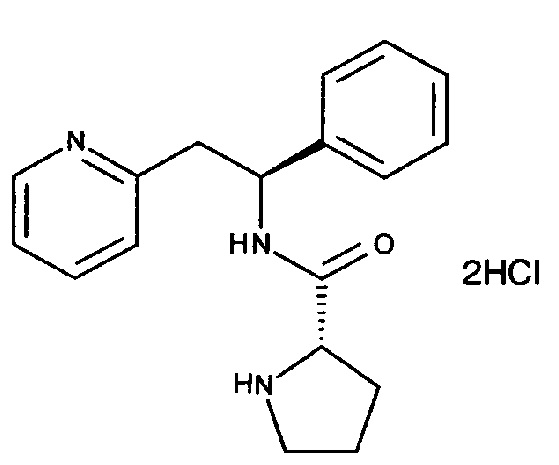
При 0°C (S)-трет-бутил-2-((S)-1-фенил-2-(пиридин-2-ил)этилкарбамоил)-пирролидин-1-карбоксилат (305 мг, 0,771 ммоль) растворяли в растворе 4М HCl в 1,4-диоксане (7,7 мл, 30,8 ммоль). Реакционную смесь постепенно подогревали до к.т. при перемешивании в течение ночи. Летучие вещества удаляли под вакуумом, и продукт растирали в МТВЕ. Твердое вещество выделяли фильтрованием на воронке Бюхнера, промывали МТВЕ и сушили под вакуумом с получением 263 мг (93%) указанного в заголовке соединения в виде дигидрохлоридной соли.
1Н ЯМР (300 МГц, DMSO-d6): м.д. 1.61-1.84 (m, 3 Н), 2.30-2.36 (m, 1 Н), 3.05-3.11 (m, 2 Н), 3.39-3.47 (m, 2 Н), 4.05-4.20 (m, 2 Н), 5.34 (q, 1 Н), 7.17-7.41 (m, 5 Н), 8.20-8.42 (m, 1 Н), 8.67-8.75 (m, 1 Н), 9.45-9.62 (m, 2 Н).
Альтернативный способ:
трет-Бутил-(2S)-2-{[(1S)-1-фенил-2-(пиридин-2-ил)этил]карбамоил}пирролидин-1-карбоксилат (2,9 г, 7,33 ммоль) растворяли в растворе 4М HCl в 1,4-диоксане (73 мл, 293,31 ммоль). Реакционную смесь перемешивали при к.т. в течение 3 ч. Летучие вещества удаляли под вакуумом, и продукт растирали в МТВЕ. Твердое вещество выделяли фильтрованием на воронке Бюхнера, промывали МТВЕ и сушили под вакуумом с получением 2,7 г (100%) (2S)-2-{[(1S)-1-фенил-2-(пиридин-1-иум-2-ил)этил]карбамоил}-пирролидин-1-ия.
1Н ЯМР (300 МГц, D2O): м.д. 1.66-1.83 (m, 3 Н), 2.20-2.26 (m, 1 Н), 3.14 (t, J=6.9 Гц, 2 Н), 3.34 (dd, J=14.1, 8.2 Гц, 1 Н), 3.51 (dd, J=14.4, 7.3 Гц, 1 Н), 4.18 (dd, J=8.5, 6.2 Гц, 1 Н), 5.11 (t, J=7.9 Гц, 1 Н), 7.06-7.10 (m, 2 Н), 7.15-7.21 (m, 3 Н), 7.61-7.70 (m, 2 Н), 8.23 (dt, J=7.9, 1.5 Гц, 1 Н), 8.37 (d, J=5.8 Гц, 1 Н).
Пример 5
(S)-1-((S)-2-Амино-3-метилбутаноил)-N-((S)-1-фенил-2-(пиридин-2-ил)-этил)пирролидин-2-карбоксамид
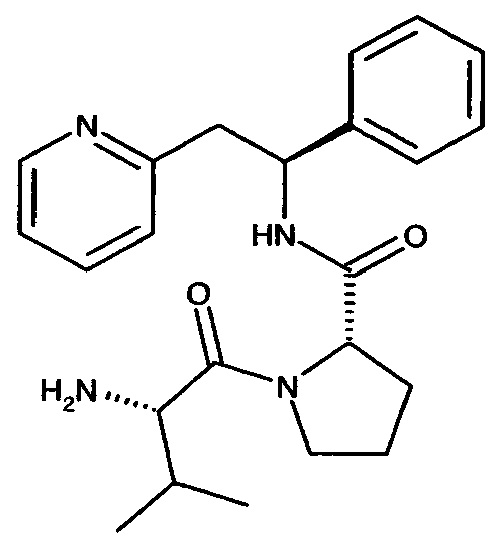
Указанное в заголовке соединение было получено в виде дигидрохлорида (S)-1-((S)-2-амино-3-метилбутаноил)-N-((S)-1-фенил-2-(пиридин-2-ил)этил)пирролидин-2-карбоксамида.
СТАДИЯ А
трет-Бутил-(S)-3-метил-1-оксо-1-((S)-2-((S)-1-фенил-2-(пиридин-2-ил)этилкарбамоил)пирролидин-1-ил)бутан-2-илкарбамат
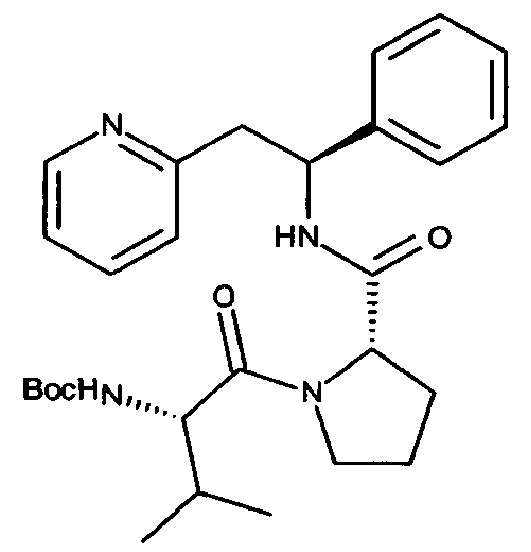
ТЗР (50% масс. в DMF, 0,83 мл, 1,42 ммоль) по каплям добавляли в раствор дигидрохлоридной соли (S)-N-((S)-1-фенил-2-(пиридин-2-ил)этил)пирролидин-2-карбоксамида (263 мг, 0,714 ммоль), Boc-L-валина (155 мг, 0,714 ммоль) и DIPEA (0,62 мл, 3,57 ммоль) в DCM (7 мл) при 0°C. Реакционную смесь постепенно нагревали до к.т. при перемешивании в течение ночи. Органический слой промывали дважды 5% водным раствором NaHCO3, один раз рассолом, сушили над Na2SO4, фильтровали и концентрировали. Продукт очищали с использование 50 г Snap колонки, элюируя с градиентом МеОН и CH2Cl2 (5% МеОН/95% CH2Cl2→10% МеОН/90% CH2Cl2), с получением указанного в заголовке соединения, 258 мг (73%).
1Н ЯМР (300 МГц, CDCl3): м.д. 0.91 (d, 3 Н), 1.00 (d, 3 Н), 1.43 (s, 9 Н), 1.85-1.99 (m, 2 Н), 2.00-2.16 (m, 1 Н), 2.17-2.21 (m, 1 Н), 3.12-3.27 (m, 2 Н), 3.47-3.62 (m, 1 Н), 3.63-3.78 (m, 1 Н), 4.32 (dd, 1 Н), 4.58 (d, 1 Н), 5.23-5.35 (m, 2 Н), 6.97 (d, 1 Н), 7.10 (dd, 1 Н), 7.14-7.30 (m, 5 Н), 7.50 (td, 1 Н), 7.84 (d, 1 Н), 8.48 (dd, 1 Н).
СТАДИЯ В
Дигидрохлорид (S)-1-((S)-2-амино-3-метилбутаноил)-N-((S)-1-фенил-2-(пиридин-2-ил)этил)пирролидин-2-карбоксамида
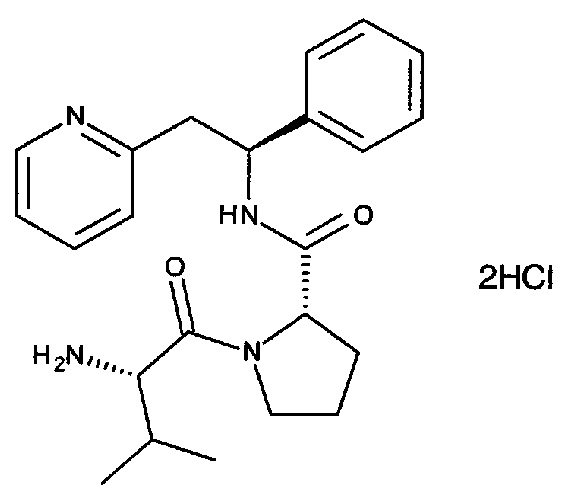
трет-Бутил-(S)-3-метил-1-оксо-1-((S)-2-((S)-1-фенил-2-(пиридин-2-ил)-этилкарбамоил)пирролидин-1-ил)бутан-2-илкарбамат (258 мг, 0,522 ммоль) растворяли в растворе 4М HCl в 1,4-диоксане (5,2 мл, 30,8 ммоль). Реакционную смесь перемешивали в течение ночи. Летучие вещества удаляли под вакуумом, и продукт растирали в EtOAc. Твердое вещество выделяли фильтрованием на воронке Бюхнера, затем растирали в Et2O. После фильтрования на воронке Бюхнера твердое вещество сушили под вакуумом с получением 200 мг (82%) указанного в заголовке соединения в виде дигидрохлоридной соли. Указанное в заголовке соединение в виде дигидрохлоридной соли затем превращали в свободное основание способами, известными специалисту в данной области техники. Альтернативно, указанное в заголовке соединение может быть получено в виде фумаратной соли способами, известными специалисту в данной области техники.
1Н ЯМР (300 МГц, CD3OD): м.д. 0.97 (d, 3 Н), 1.04 (d, 3 Н), 1.68-1.79 (m, 1 Н), 1.86-2.05 (m, 2 Н), 2.13-2.27 (m, 2 Н), 3.50-3.61 (m, 3 Н), 3.68-3.75 (m, 1 Н), 4.02 (d, 1 Н), 4.45 (dd, 1 Н), 5.40 (dd, 1 Н), 7.31-7.43 (m, 5 Н), 7.90 (t, 1 Н), 8.02 (d, 1 Н), 8.51 (td, 1 Н), 8.75 (d, 1 Н).
[М+Н]+=395,27
Пример 6
(S)-1-((S)-2,6-Диаминогексаноил)-N-((S)-1-фенил-2-(пиридин-2-ил)этил)пирролидин-2-карбоксамид
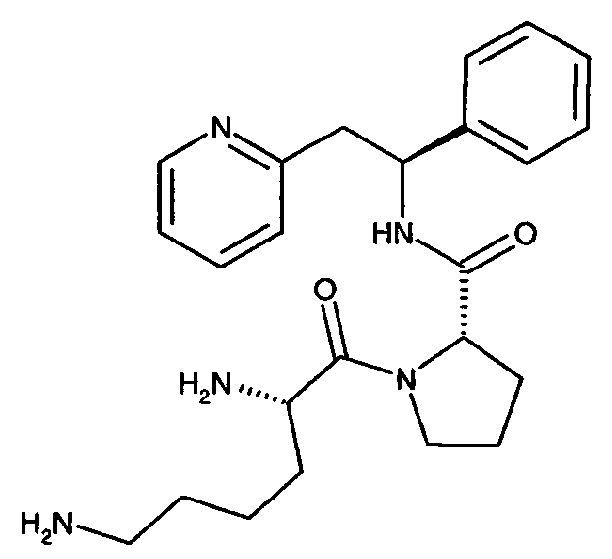
Указанное в заголовке соединение было получено в виде трихлорида 2-[(2S)-2-{[(2S)-1-[(2S)-2,5-диазаниумилгексаноил]пирролидин-2-ил]формамидо}-2-фенилэтил]пиридин-1-ия
СТАДИЯ А
трет-Бутил-(S)-6-оксо-6-((S)-2-((S)-1-фенил-2-(пиридин-2-ил)-этилкарбамоил)пирролидин-1-ил)гексан-1,5-диилдикарбамат
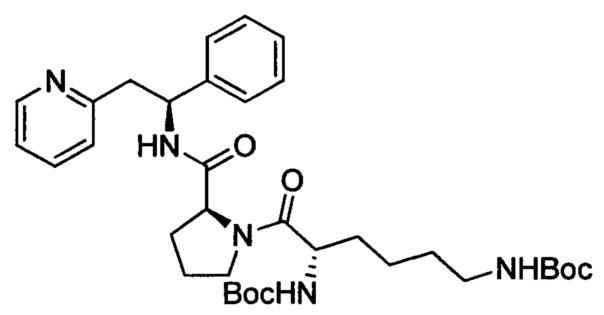
Дициклогексиламмониевую соль N-альфа, N-эпсилон-бис(трет-бутоксикарбонил)-L-лизина (287 мг, 0,54 ммоль) растворяли в безводном DMF (4 мл). Добавляли HATU (217 мг, 0,57 ммоль) и DIPEA (0,21 мл, 1,19 ммоль), и эту смесь перемешивали при к.т. в течение 30 минут. Затем в раствор добавляли дихлорид (2S)-2-{[(1S)-1-фенил-2-(пиридин-1-иум-2-ил)этил]-карбамоил}пирролидин-1-ия (200 мг, 0,54 ммоль), и смесь перемешивали в течение 18 ч при к.т. Добавляли воду, и смесь экстрагировали 3×EtOAc. Органическую фазу промывали рассолом, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Продукт очищали на SiO2 колонке, элюируя с градиентом (0% МеОН/50% EtOAc/50% гептана →0% МеОН/100% EtOAc/0% гептана →10% МеОН/90% EtOAc/0% гептана), затем на С-18 колонке, элюируя с градиентом МеОН и воды (от 0 до 100% МеОН), с получением 80 мг (24%) указанного в заголовке соединения.
1Н ЯМР (300 МГц, CDCl3): м.д. 1.34-1.39 (m, 2 Н), 1.41 (s, 9 Н), 1.43 (s, 9 Н), 1.52-1.64 (m, 3 Н), 1.68-2.02 (m, 4 Н), 2.08-2.22 (m, 2 Н), 3.02-3.16 (m, 3 Н), 3.20-3.28 (m, 1 Н), 3.48-3.57 (m, 1 Н), 3.60-3.70 (m, 1 Н), 4.40-4.51 (m, 1 Н), 4.54-4.57 (m, 1 Н), 4.97-5.08 (m, 1 Н), 5,29-5.39 (m, 2 Н), 7.00 (d, 1 Н), 7.10-7.32 (m, 6 Н), 7.52 (td, 1 Н), 7.86 (d, 1 Н), 8.49 (d, 1 Н).
СТАДИЯ В
Синтез трихлорида 2-[(2S)-2-{[(2S)-1-[(2S)-2,5-диазаниумилгексаноил]-пирролидин-2-ил]формамидо}-2-фенилэтил]пиридин-1-ия
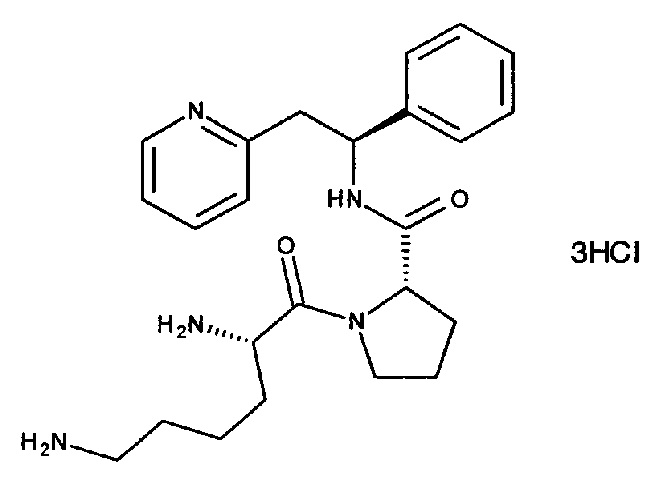
трет-Бутил-(S)-6-оксо-6-((S)-2-((S)-1-фенил-2-(пиридин-2-ил)-этилкарбамоил)пирролидин-1-ил)гексан-1,5-диилдикарбамат (80 мг, 0,13 ммоль) растворяли в растворе 4М HCl в 1,4-диоксане (1,6 мл, 6,4 ммоль). Реакционную смесь перемешивали при к.т. в течение 18 часов. Летучие вещества удаляли под вакуумом, и продукт растирали в МТВЕ. Твердое вещество выделяли фильтрованием на воронке Бюхнера, промывали МТВЕ и сушили под вакуумом. Твердое вещество растворяли в воде и подвергали сублимационной сушке с получением 60 мг (88%) указанного в заголовке соединения.
1Н ЯМР (300 МГц, D2O): м.д. 1.23-1.35 (m, 2 Н), 1.46-1.59 (m, 3 Н), 1.65-1.80 (m, 4 Н), 2.04-2.13 (m, 1 Н), 2.80 (t, 2 Н), 3.31-3.43 (m, 2 Н), 3.47-3.57 (m, 2 Н), 4.16 (t, 1 Н), 4.28 (t, 1 Н), 5.11 (t, 1 Н), 7.10-7.13 (m, 2 Н), 7.16-7.26 (m, 3 Н), 7.68-7.73 (m, 2 H), 8.31 (td, 1 H), 8.42 (dd, 1 H);
[M+H]+=424,2.
Пример 7
(S)-1-((S)-2-Амино-3-(1Н-имидазол-4-ил)пропаноил)-N-((S)-1-фенил-2-(пиридин-2-ил)этил)пирролидин-2-карбоксамид
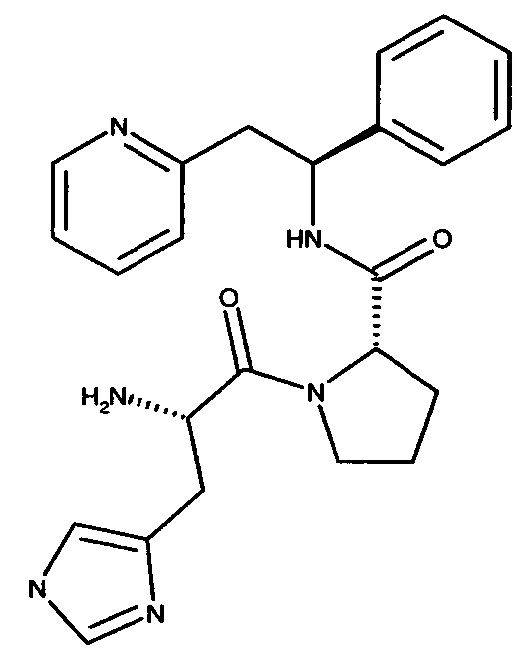
Указанное в заголовке соединение было получено в виде трихлорида 2-[(2S)-2-{[(2S)-1-[(2S)-2-азаниумил-3-(1Н-имидазол-1-иум-4-ил)пропаноил]пирролидин-2-ил]формамидо}-2-фенилэтил]пиридин-1-ия
СТАДИЯ А
трет-Бутил-(S)-3-(1Н-имидазол-4-ил)-1-оксо-1-((S)-2-((S)-1-фенил-2-(пиридин-2-ил)этилкарбамоил)пирролидин-1-ил)пропан-2-илкарбамат
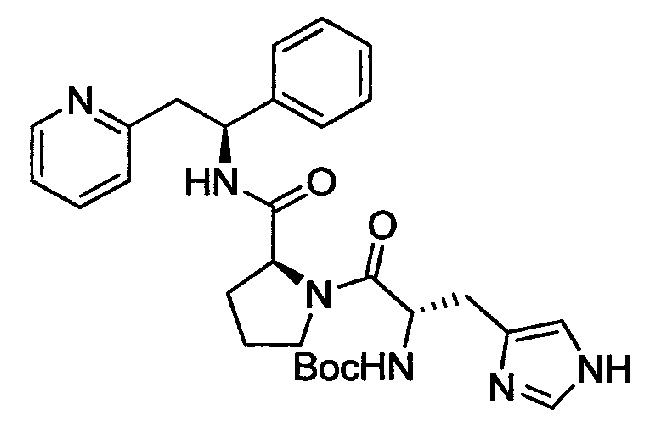
Boc-His-OH (166 мг, 0,65 ммоль) растворяли в безводном DMF (4 мл). Добавляли HATU (260 мг, 0,68 ммоль) и DIPEA (0,38 мл, 2,85 ммоль), и эту смесь перемешивали при к.т. в течение 30 минут. Затем добавляли хлорид 2-((S)-2-фенил-2-((S)-пирролидин-2-ийкарбоксамидо)этил)пиридиния (240 мг, 0,65 ммоль), и смесь перемешивали в течение 66 часов при к.т. Добавляли воду, и смесь экстрагировали 3× EtOAc. Органическую фазу промывали рассолом, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Продукт очищали на SiO2 колонке, элюируя с градиентом МеОН и CH2Cl2 (от 0% до 13% МеОН), затем на С-18 колонке, элюируя с градиентом МеОН и воды (от 0 до 100% МеОН), с получением 100 мг (29%) указанного в заголовке соединения.
1Н ЯМР (300 МГц, CDCl3): м.д. 1.44 (s, 9 Н), 1.77-2.00 (m, 3 Н), 2.05-2.19 (m, 1 Н), 3.03-3.14 (m, 2 Н), 3.23 (dd, 1 Н), 3.40 (dd, 1 Н), 3.52-3.58 (m, 1 Н), 4.50-4.62 (m, 2 Н), 5,43-5.50 (m, 2 Н), 6.91-6.97 (m, 2 Н), 7.13-7.27 (m, 5 Н), 7.53 (t, 1 Н), 7.65 (s, 1 Н), 8.53 (d, 1 Н), 8.67 (d, 1 Н).
СТАДИЯ В
Трихлорид 2-((S)-2-((S)-1-((S)-2-аммонио-3-(1Н-имидазол-1-иум-4-ил)пропаноил)пирролидин-2-карбоксамидо)-2-фенилэтил)пиридиния
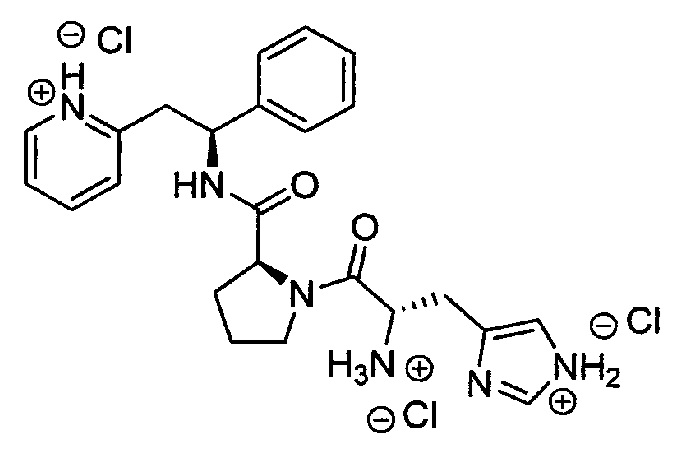
трет-Бутил-(S)-3-(1Н-имидазол-4-ил)-1-оксо-1-((S)-2-((S)-1-фенил-2-(пиридин-2-ил)этилкарбамоил)пирролидин-1-ил)пропан-2-илкарбамат (100 мг, 0,19 ммоль) растворяли в растворе 4М HCl в 1,4-диоксане (2,3 мл, 9,39 ммоль). Реакционную смесь перемешивали при к.т. в течение 18 ч. Летучие вещества удаляли под вакуумом, и продукт растирали в МТВЕ. Твердое вещество выделяли фильтрованием на воронке Бюхнера, промывали МТВЕ и сушили под вакуумом. Твердое вещество растворяли в воде и подвергали сублимационной сушке с получением 85 мг (69%) указанного в заголовке соединения.
1Н ЯМР (300 МГц, D2O): м.д. 1.50-1.58 (m, 1 Н), 1.69-1.76 (m, 2 Н), 2.07-2.14 (m, 1 Н), 3.12-3.21 (m, 3 Н), 3.38 (dd, 7.3 Гц, 1 Н), 3.48-3.56 (m, 2 Н), 4.32 (t, 1 Н), 4.43 (t, 1 Н), 5.14 (t, 1 Н), 7.08-7.12 (m, 2 Н), 7.15-7.23 (m, 3 Н), 7.26 (s, 1 Н), 7.65-7.70 (m, 2 Н), 8.26 (td, 1 Н), 8.39 (d, 1 Н), 8.49 (d, 1 Н);
[М+Н]+=433,2;
[M+Na]+=455,1.
Пример 8
Дигидрохлорид 2-((S)-2-((S)-1-((S)-2-амино-3-(4-гидроксифенил)-пропаноил)пирролидин-2-карбоксамидо)-2-фенилэтил)пиридина
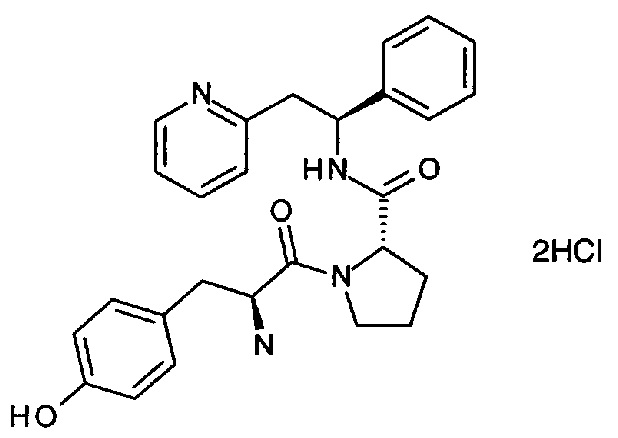
Указанное в заголовке соединение может иметь название дихлорид 2-[(2S)-2-{[(2S)-1-[(2S)-2-азаниумил-3-(4-гидроксифенил)пропаноил]пирролидин-2-ил]формамидо}-2-фенилэтил]пиридин-1-ия
СТАДИЯ А
трет-Бутил-(S)-3-(4-гидроксифенил)-1-оксо-1-((S)-2-((S)-1-фенил-2-(пиридин-2-ил)этилкарбамоил)пирролидин-1-ил)пропан-2-илкарбамат
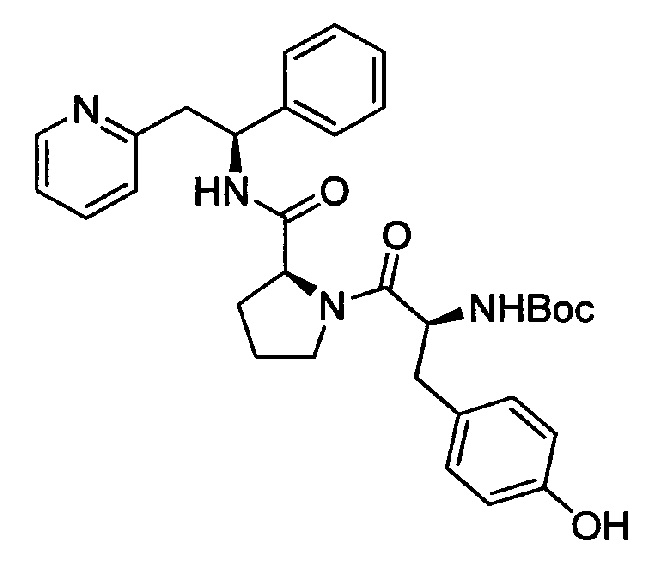
Вос-Tyr-ОН (229 мг, 0,81 ммоль) растворяли в безводном DMF (5 мл). Добавляли HATU (325 мг, 0,86 ммоль) и DIPEA (0,47 мл, 2,69 ммоль), и эту смесь перемешивали при к.т. в течение 30 минут. Затем в раствор добавляли хлорид 2-((S)-2-фенил-2-((S)-пирролидин-2-иякарбоксамидо)этил)пиридиния (300 мг, 0,81 ммоль), и смесь перемешивали в течение 66 ч при к.т. Добавляли воду, и смесь экстрагировали 3× EtOAc. Органическую фазу промывали рассолом, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Продукт очищали на SiO2 колонке, элюируя с градиентом (0% МеОН/50% EtOAc/50% гептана →0% МеОН/100% EtOAc/0% гептана →15% МеОН/85% EtOAc/0% гептана), затем на С-18 колонке, элюируя с градиентом МеОН и воды (от 0 до 100% МеОН), с получением 180 мг (40%) указанного в заголовке соединения.
1Н ЯМР (300 МГц, CDCl3): м.д. 1.44 (s, 9 Н), 1.73-2.02 (m, 3 Н), 2.16-2.22 (m, 1 H), 2.92-3.09 (m, 4 H), 3.35-3.42 (m, 1 H), 3.48-3.51 (m, 1 H), 3.53-3.68 (m, 1 H), 4.48-4.51 (m, 1 H), 4.65-4.73 (m, 1 H), 5.06-5.14 (m, 1 H), 5.43 (d, 1 H), 6.61 (d, 1 H), 6.86 (d,, 2 H), 6.94-6.99 (m, 2 H), 7.08-7.26 (m, 6 H), 7.42 (dd, 1 H), 8.49 (d, 1 H), 8.63 (s, 1 H).
СТАДИЯ В:
Дихлорид 2-((S)-2-((S)-1-((S)-2-аммонио-3-(4-гидроксифенил)пропаноил)-пирролидин-2-карбоксамидо)-2-фенилэтил)пиридиния
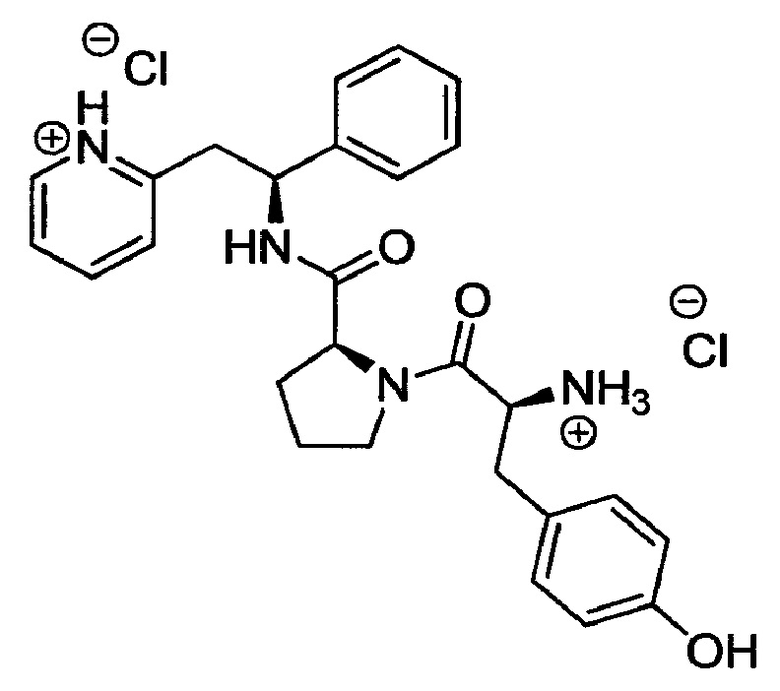
трет-Бутил-(S)-3-(4-гидроксифенил)-1-оксо-1-((S)-2-((S)-1-фенил-2-(пиридин-2-ил)этилкарбамоил)пирролидин-1-ил)пропан-2-илкарбамат (180 мг, 0,32 ммоль) растворяли в растворе 4М HCl в 1,4-диоксане (3,2 мл, 12,89 ммоль). Реакционную смесь перемешивали при к.т. в течение 18 часов. Летучие вещества удаляли под вакуумом, и продукт растирали в МТВЕ. Твердое вещество выделяли фильтрованием на воронке Бюхнера, промывали МТВЕ и сушили под вакуумом. Твердое вещество растворяли в воде и подвергали сублимационной сушке с получением 150 мг (88%) указанного в заголовке соединения.
1Н ЯМР (300 МГц, D2O): м.д. 1.48-1.57 (m, 1 Н), 1.68-1.73 (m, 2 Н), 1.98-2.05 (m, 1 Н), 2.68-2.76 (m, 1 Н), 2.93-3.00 (m, 1 Н), 3.10-3.19 (m, 1 Н), 3.40-3.63 (m, 3 Н), 4.20-4.28 (m, 2 Н), 5.14 (t, 1 Н), 6.65 (d, 2 Н), 6.96 (d, 2 Н), 7.13-7.22 (m, 5 Н), 7.63-7.66 (m, 2 Н), 8.19-8.22 (m, 1 Н), 8.40 (d, 1 Н);
[М+Н]+=459,2.
Пример 9
Дигидрохлорид 2-((S)-2-((S)-1-((S)-2-амино-3-(1Н-индол-3-ил)пропаноил)-пирролидин-2-карбоксамидо)-2-фенилэтил)пиридина
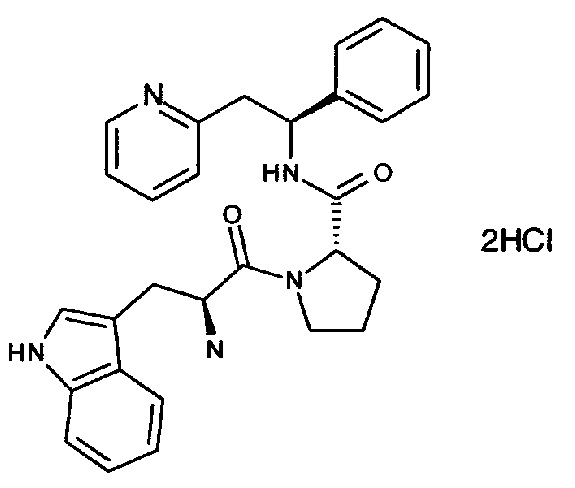
Указанное в заголовке соединение также может иметь название дихлорид 2-[(2S)-2-{[(2S)-1-[(2S)-2-азаниумил-3-(1Н-индол-3-ил)пропаноил]-пирролидин-2-ил]формамидо}-2-фенилэтил]пиридин-1-ия.
СТАДИЯ А
трет-Бутил-(S)-3-(1Н-индол-3-ил)-1-оксо-1-((S)-2-((S)-1-фенил-2-(пиридин-2-ил)этилкарбамоил)пирролидин-1-ил)пропан-2-илкарбамат
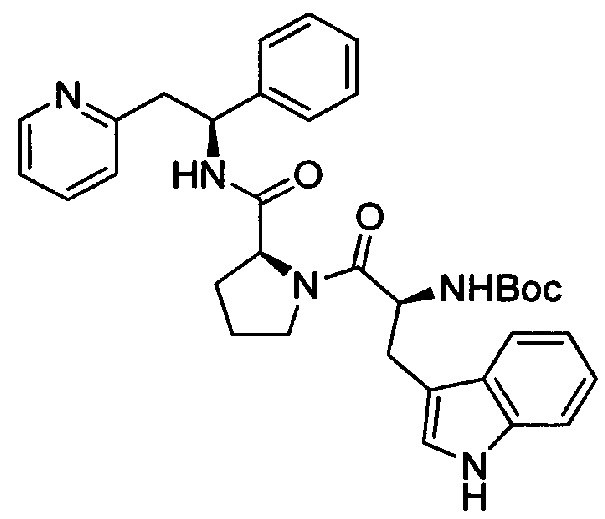
Вос-Trp-ОН (207 мг, 0,68 ммоль) растворяли в безводном DMF (4 мл). Добавляли HATU (271 мг, 0,71 ммоль) и DIPEA (0,39 мл, 2,24 ммоль), и эту смесь перемешивали при к.т. в течение 30 минут. Затем добавляли дихлорид (2S)-2-{[(1S)-1-фенил-2-(пиридин-1-иум-2-ил)этил]карбамоил}пирролидин-1-ия (250 мг, 0,68 ммоль), и смесь перемешивали в течение 18 часов при к.т. Добавляли воду, и смесь экстрагировали 3×EtOAc. Органическую фазу промывали рассолом, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Продукт очищали на SiO2 колонке, элюируя с градиентом (0% МеОН/50% EtOAc/50% гептана →0% МеОН/100% EtOAc/0% гептана →15% МеОН/85% EtOAc/0% гептана), с получением 410 мг (количественный выход) указанного в заголовке соединения.
1Н ЯМР (300 МГц, CDCl3): м.д. 1.44 (s, 9 Н), 1.77-1.89 (m, 3 Н), 2.14-2.24 (m, 1 Н), 3.03-3.24 (m, 4 Н), 3.28-3.36 (m, 2 Н), 3.54-3.65 (m, 1 Н), 4.52-4.58 (m, 1 Н), 4.79-4.88 (m, 1 Н), 5,12-5.27 (m, 1 Н), 5.34-5.41 (m, 1 Н), 6.63 (d, 1 Н), 6.94-7.31 (m, 8 Н), 7.38-7.53 (m, 3 Н), 7.62-7.67 (m, 1 Н), 8.59 (d, 1 Н), 9.87 (s, 1 Н).
СТАДИЯ В
дихлорид 2-((S)-2-((S)-1-((S)-2-аммонио-3-(1Н-индол-3-ил)пропаноил)-пирролидин-2-карбоксамидо)-2-фенилэтил)пиридиния
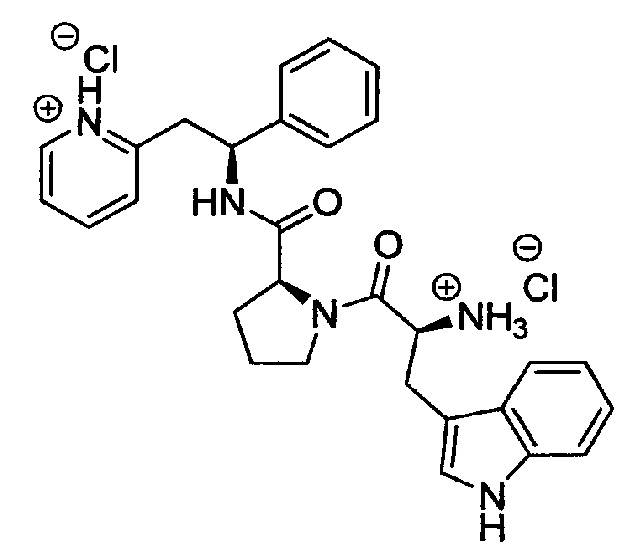
трет-Бутил-(S)-3-(1Н-индол-3-ил)-1-оксо-1-((S)-2-((S)-1-фенил-2-(пиридин-2-ил)этилкарбамоил)пирролидин-1-ил)пропан-2-илкарбамат (395 мг, 0,68 ммоль) растворяли в растворе 1М HCl в Et2O (27,0 мл, 27,20 ммоль). Реакционную смесь перемешивали при к.т. в течение 4 ч. Продукт растирали в Et2O, и твердое вещество выделяли фильтрованием на воронке Бюхнера, промывали Et2O и сушили под вакуумом. Твердое вещество растворяли в воде и подвергали сублимационной сушке с получением 175 мг (44%) указанного в заголовке соединения.
1Н ЯМР (300 МГц, D2O): м.д. 1.41-1.53 (m, 1 Н), 1.63-1.72 (m, 2 Н), 1.93-2.04 (m, 1 Н), 2.85 (dd, 1 Н), 2.94-3.55 (m, 5 Н), 4.19-4.31 (m, 2 Н), 5.08 (t, 1 Н), 6.90-7.31 (m, 9 Н), 7.38 (d, 1 Н), 7.55-7.58 (m, 2 Н), 8.15 (t, 1 Н), 8.31 (d, 1 Н);
[М+Н]+=482,2,
[M+Na]+=504,1.
Пример 10
(S)-1-((S)-2-Амино-3-фенилпропаноил)-N-((S)-1-фенил-2-(пиридин-2-ил)этил)пирролидин-2-карбоксамид
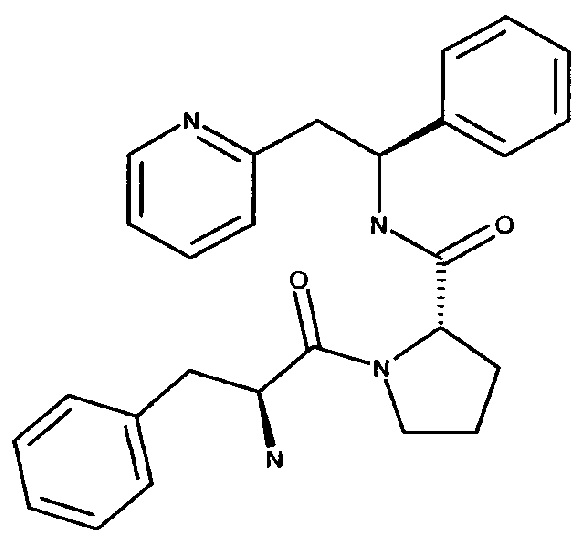
Указанное в заголовке соединение было получено в виде дихлорида 2-[(2S)-2-{[(2S)-1-[(2S)-2-азаниумил-3-фенилпропаноил]пирролидин-2-ил]формамидо}-2-фенилэтил]пиридин-1-ия
СТАДИЯ А
трет-Бутил-(S)-1-оксо-3-фенил-1-((S)-2-((S)-1-фенил-2-(пиридин-2-ил)этилкарбамоил)пирролидин-1-ил)пропан-2-илкарбамат
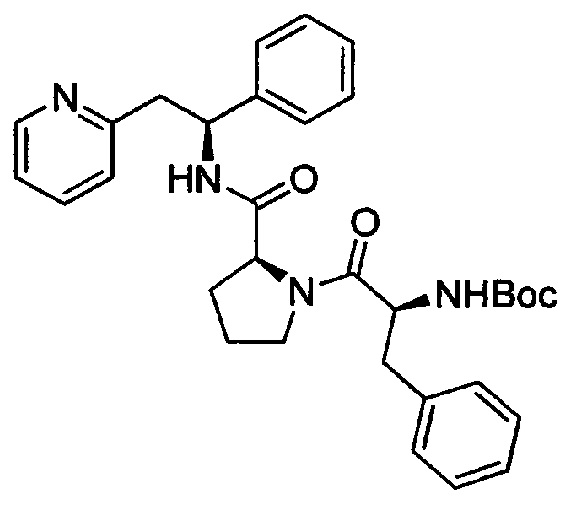
Boc-Phe-OH (180 мг, 0,68 ммоль) растворяли в безводном DMF (4 мл). Добавляли HATU (271 мг, 0,71 ммоль) и DIPEA (0,39 мл, 2,24 ммоль), и эту смесь перемешивали при к.т. в течение 30 минут. Затем в раствор добавляли дихлорид (2S)-2-{[(1S)-1-фенил-2-(пиридин-1-иум-2-ил)этил]карбамоил}-пирролидин-1-ия (250 мг, 0,68 ммоль), и смесь перемешивали в течение 18 ч при к.т. Добавляли воду, и смесь экстрагировали 3×EtOAc. Органическую фазу промывали рассолом, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Продукт очищали на SiO2 колонке, элюируя с градиентом (0% МеОН/50% EtOAc/50% гептана →0% МеОН/100% EtOAc/0% гептана →15% МеОН/85% EtOAc/0% гептана), затем на С-18 колонке, элюируя с градиентом МеОН и воды (от 0 до 100% МеОН), с получением 170 мг (46%) указанного в заголовке соединения.
1Н ЯМР (300 МГц, CDCl3): м.д. 1.44 (s, 9 Н), 1.73-1.91 (m, 3 Н), 2.10-2.19 (m, 1 H), 3.10-3.40 (m, 3 H), 3.45-3.61 (m, 2 H), 4.27-4.35 (m, 1 H), 4.50-4.56 (m, 1 H), 4.60-4.69 (m, 1 H), 5,17-5.41 (m, 3 H), 6.93-7.03 (m, 2 H), 7.06-7.32 (m, 9 H), 7.47-7.56 (m, 1 H), 7.82 (d, 1 H), 8.48 (d, 1 H).
СТАДИЯ В
Дихлорид 2-((S)-2-((S)-1-((S)-2-аммонио-3-фенилпропаноил)пирролидин-2-карбоксамидо)-2-фенилэтил)пиридиния
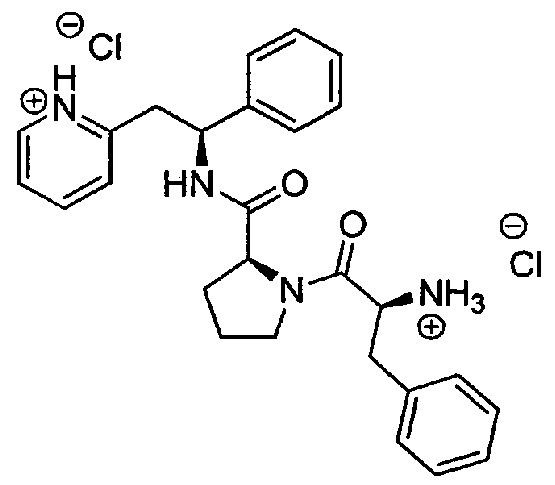
трет-Бутил-(S)-1-оксо-3-фенил-1-((S)-2-((S)-1-фенил-2-(пиридин-2-ил)-этилкарбамоил)пирролидин-1-ил)пропан-2-илкарбамат (170 мг, 0,31 ммоль) растворяли в растворе 1М HCl в Et2O (12,5 мл, 12,53 ммоль). Реакционную смесь перемешивали при к.т. в течение 18 часов. Продукт растирали в Et2O. Твердое вещество выделяли фильтрованием на воронке Бюхнера, промывали Et2O и сушили под вакуумом. Твердое вещество растворяли в воде и подвергали сублимационной сушке с получением 150 мг (93%) указанного в заголовке соединения.
1Н ЯМР (300 МГц, D2O): м.д. 1.39-1.48 (m, 1 Н), 1.58-1.65 (m, 2 Н), 1.92-1.99 (m, 1 Н), 2.66-2.74 (m, 1 Н), 2.95-3.10 (m, 2 Н), 3.29-3.45 (m, 3 Н), 4.15-4.27 (m, 2 Н), 5.09 (t, 1 Н), 6.97-7.17 (m, 10 Н), 7.59-7.64 (m, 2 Н), 8.18 (t, 1 Н), 8.35 (d, 1 Н);
[М+Н]+=443,3,
[M+Na]+=465,2.
Пример 11
(S)-1-((S)-2-Амино-4-метилпентаноил)-N-((S)-1-фенил-2-(пиридин-2-ил)этил)пирролидин-2-карбоксамид
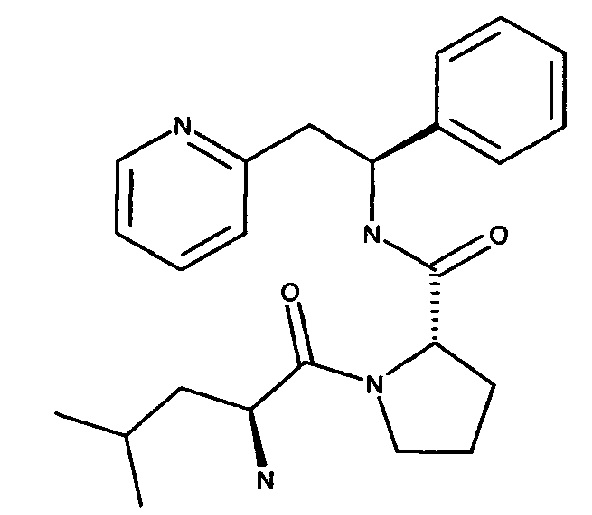
Указанное в заголовке соединение было получено в виде дихлорида 2-[(2S)-2-{[(2S)-1-[(2S)-2-азаниумил-4-метилпентаноил]пирролидин-2-ил]-формамидо}-2-фенилэтил]пиридин-1-ия
СТАДИЯ А
трет-Бутил-(S)-4-метил-1-оксо-1-((S)-2-((S)-1-фенил-2-(пиридин-2-ил)этилкарбамоил)пирролидин-1-ил)пентан-2-илкарбамат
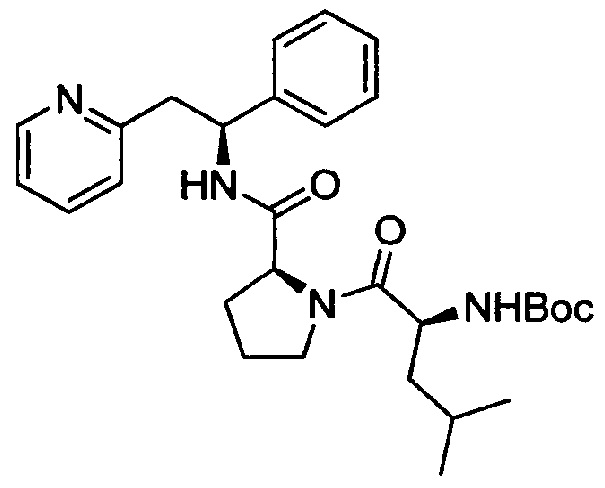
N-(трет-Бутоксикарбонил)-L-лейцин (157 мг, 0,68 ммоль) растворяли в безводном DMF (4 мл). Добавляли HATU (271 мг, 0,71 ммоль) и DIPEA (0,39 мл, 2,24 ммоль), и эту смесь перемешивали при к.т. в течение 30 минут. Затем в раствор добавляли дихлорид (2S)-2-{[(1S)-1-фенил-2-(пиридин-1-иум-2-ил)этил]карбамоил}пирролидин-1-ия (250 мг, 0,68 ммоль), и смесь перемешивали в течение 18 часов при к.т. Добавляли воду, и смесь экстрагировали 3×EtOAc. Органическую фазу промывали рассолом, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Продукт очищали на SiO2 колонке, элюируя с градиентом (0% МеОН/50% EtOAc/50% гептана →0% МеОН/100% EtOAc/0% гептана →15% МеОН/85% EtOAc/0% гептана), затем на С-18 колонке, элюируя с градиентом МеОН и воды (от 0 до 100% МеОН), с получением 200 мг (58%) указанного в заголовке соединения.
1Н ЯМР (300 МГц, CDCl3): м.д. 0.91 (d, 3 Н), 0.98 (d, 3 Н), 1.34-1.52 (m, 1 Н), 1.41 (s, 9 Н), 1.70-1.84 (m, 2 Н), 1.85-1.96 (m, 3 Н), 2.11-2.18 (m, 1 Н), 3.07-3.27 (m, 2 Н), 3.47-3.56 (m, 1 Н), 3.61-3.71 (m, 1 Н), 4.43-4.56 (m, 2 Н), 5.14-5.18 (m, 1 Н), 5,26-5.34 (m, 1 Н), 6.98 (d, J=7.7 Гц, 1 H), 7.08-7.25 (m, 5 Н), 7.52 (t, 1 Н), 7.83 (d, 1 Н), 8.48 (d, 1 Н).
СТАДИЯ В
Дихлорид 2-((S)-2-((S)-1-((S)-2-аммонио-4-метилпентаноил)пирролидин-2-карбоксамидо)-2-фенилэтил)пиридиния
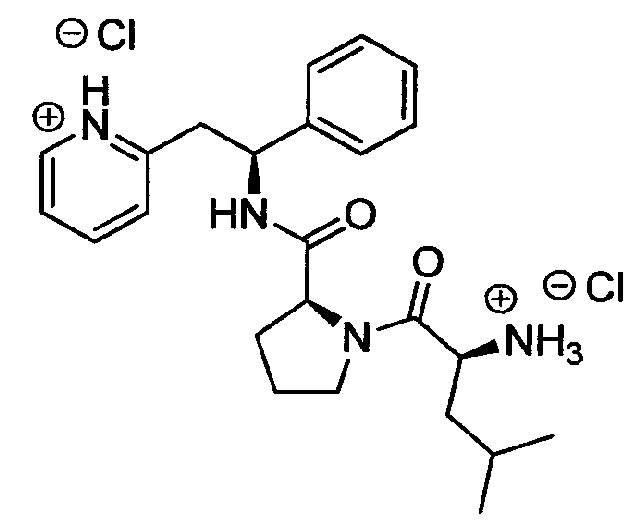
трет-Бутил-(S)-4-метил-1-оксо-1-((S)-2-((S)-1-фенил-2-(пиридин-2-ил)-этилкарбамоил)пирролидин-1-ил)пентан-2-илкарбамат (200 мг, 0,39 ммоль) растворяли в растворе 1М HCl в Et2O (15,7 мл, 15,7 ммоль). Реакционную смесь перемешивали при к.т. в течение 18 ч. Продукт растирали в Et2O. Твердое вещество выделяли фильтрованием на воронке Бюхнера, промывали Et2O и сушили под вакуумом. Твердое вещество растворяли в воде и подвергали сублимационной сушке с получением 130 мг (69%) указанного в заголовке соединения.
1Н ЯМР (300 МГц, D2O): м.д. 0.72 (d, 6 Н), 1.31-1.49 (m, 4 Н), 1.65-1.74 (m, 2 Н), 1.93-2.02 (m, 1 Н), 3.23-3.49 (m, 4 Н), 3.99-4.04 (m, 1 Н), 4.16-4.21 (m, 1 Н), 5.07 (t, 1 Н), 7.08-7.20 (m, 5 Н), 7.63-7.67 (m, 2 Н), 8.22 (td, 1 Н), 8.37 (dd, 1 Н);
[М+Н]+=409,2,
[M+Na]+=431,2.
Пример 12
Тетрахлорид 2-[(2S)-2-{[(2S)-1-[(2S)-2-азаниумил-5-{[азаниумил(иминиумил)метил]амино}пентаноил]-пирролидин-2-ил]-формамидо}-2-фенилэтил]пиридин-1-ия
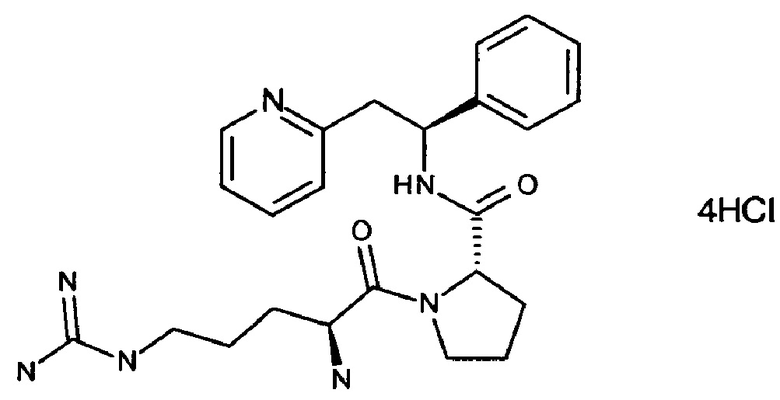
СТАДИЯ А
трет-Бутил-N-[(1Z)-{[(4S)-4-{[(трет-бутокси)карбонил]амино}-5-оксо-5-[(2S)-2-{[(1S)-1-фенил-2-(пиридин-2-ил)этил]карбамоил}пирролидин-1-ил]пентил]амино}({[(трет-бутокси)карбонил]имино})метил]карбамат
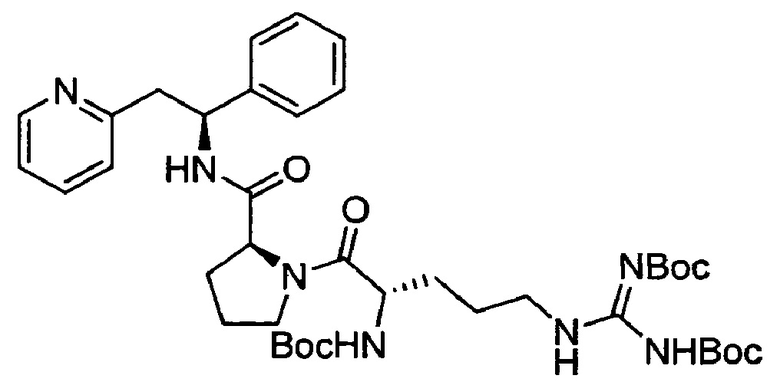
Boc-Arg(Boc)2-OH (644 мг, 1,36 ммоль) растворяли в безводном DMF (7 мл). Добавляли HATU (542 мг, 1,43 ммоль) и DIPEA (0,78 мл, 4,48 ммоль), и эту смесь перемешивали при к.т. в течение 30 минут. Затем в раствор добавляли дихлорид (2S)-2-{[(1S)-1-фенил-2-(пиридин-1-иум-2-ил)этил]карбамоил}-пирролидин-1-ия (500 мг, 1,36 ммоль), и смесь перемешивали в течение 18 ч при к.т. Добавляли воду, и смесь экстрагировали 3×EtOAc. Органическую фазу промывали рассолом, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Продукт очищали на SiO2 колонке, элюируя с градиентом (0% МеОН/50% EtOAc/50% гептана →0% МеОН/100% EtOAc/0% гептана →10% МеОН/90% EtOAc/0% гептана), затем на С-18 колонке, элюируя с градиентом МеОН и воды (от 0 до 100% МеОН), с получением 200 мг (20%) указанного в заголовке соединения.
1Н ЯМР (300 МГц, CDCl3): м.д. 1.43 (s, 9 Н), 1.46 (s, 9 Н), 1.51 (s, 9 Н), 1.57-1.76 (m, 4 Н), 1.86-1.99 (m, 3 Н), 2.12-2.25 (m, 1 Н), 3.11 (dd, 1 Н), 3.23-3.30 (m, 1 Н), 3.58-3.95 (m, 4 Н), 4.42 (t, 1 Н), 4.56 (d, 1 Н), 5,31-5.38 (m, 2 Н), 6.98 (d, 1 Н), 7.17-7.33 (m, 5 Н), 7.54 (t, 1 Н), 7.93 (d, 1 Н), 8.56 (d, 1 Н).
СТАДИЯ В
Тетрахлорид 2-[(2S)-2-{[(2S)-1-[(2S)-2-азаниумил-5-{[азаниумил(иминиумил)метил]амино}пентаноил]пирролидин-2-ил]формамидо}-2-фенилэтил]пиридин-1-ия
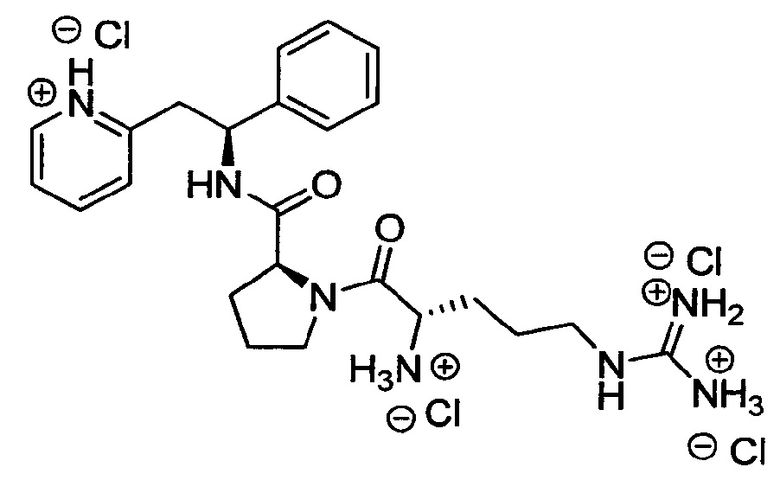
трет-Бутил-N-[(1Z)-{[(4S)-4-{[(трет-бутокси)карбонил]амино}-5-оксо-5-[(2S)-2-{[(1S)-1-фенил-2-(пиридин-2-ил)этил]карбамоил}пирролидин-1-ил]-пентил]амино}({[(трет-бутокси)карбонил]имино})метил]карбамат (200 мг, 0,27 ммоль) растворяли в растворе 1М HCl в Et2O (5,3 мл, 10,64 ммоль). Реакционную смесь перемешивали при к.т. в течение 18 часов. Продукт растирали в Et2O. Твердое вещество выделяли фильтрованием на воронке Бюхнера, промывали Et2O и сушили под вакуумом. Твердое вещество растворяли в воде и подвергали сублимационной сушке с получением 140 мг (88%) указанного в заголовке соединения.
1Н ЯМР (300 МГц, D2O): м.д. 1.31-1.57 (m, 3 Н), 1.66-1.78 (m, 4 Н), 2.00-2.10 (m, 1 Н), 2.99 (t, 2 Н), 3.28-3.40 (m, 4 Н), 4.16 (t, 1 Н), 4.26 (t, 1 Н), 5.09 (t, 1 Н), 7.10-7.13 (m, 2 Н), 7.16-7.21 (m, 3 Н), 7.67-7.71 (m, 2 Н), 8.27 (t, 1 Н), 8.40 (d, 1 Н);
[М+Н]+=452,2.
БИОЛОГИЧЕСКАЯ АКТИВНОСТЬ
Пролекарства, описанные в данном документе, предусмотрены для перорального введения субъектам, страдающим от депрессии или боли.
Пример 13
Этот Пример иллюстрирует, что разные пролекарства (S)-1-фенил-2-(пиридин-2-ил)этанамина, где R1 представляет собой C1-6алкилС(О)О(С1-6алкокси), можно использовать для достижения различных скоростей превращения (т.е. медленного или быстрого превращения) в (S)-1-фенил-2-(пиридин-2-ил)этанамин с целью выявления подходящего PK (фармакокинетического) профиля. Следовательно, варьирование R1 для получения разных пролекарств приводит в результате к разным фармакокинетическим профилям (S)-1-фенил-2-(пиридин-2-ил)этанамина. Этот Пример также иллюстрирует, что ожидаемая потеря отсутствует при пероральном воздействии, когда пролекарство превращается в свободное основание. Предполагается, что превращение пролекарства, где R1 представляет собой С1-6алкилС(O)O(С1-6алкокси), в (S)-1-фенил-2-(пиридин-2-ил)этанамин происходит посредством начального ферментативный гидролиз функциональной группы, такой как сложноэфирная группа, с последующим спонтанным превращением в (S)-1-фенил-2-(пиридин-2-ил)этанамин. Предполагается, что вовлеченными в гидролиз ферментами являются некоторые неспецифические, мощные эстеразы, что может быть продемонстрировано при ингибировании превращения с использованием неселективного ингибитора эстераз, но не при ингибировании превращения с использованием селективных ингибиторов. Предполагается, что такие эстеразы распределены по всему организму человека. Кишечная среда человека (HIF), печеночная фракция S9 человека и цельная кровь человека были использованы для исследования фармакокинетических свойств пролекарств (S)-1-фенил-2-(пиридин-2-ил)этанамина, а также скорости образования и степени образования (S)-1-фенил-2-(пиридин-2-ил)этанамина в разных отделах организма. Скорость образования (S)-1-фенил-2-(пиридин-2-ил)этанамина из разных пролекарств (S)-1-фенил-2-(пиридин-2-ил)этанамина значительно различается во всех протестированных анализируемых образцах, как показано в Таблице 1.
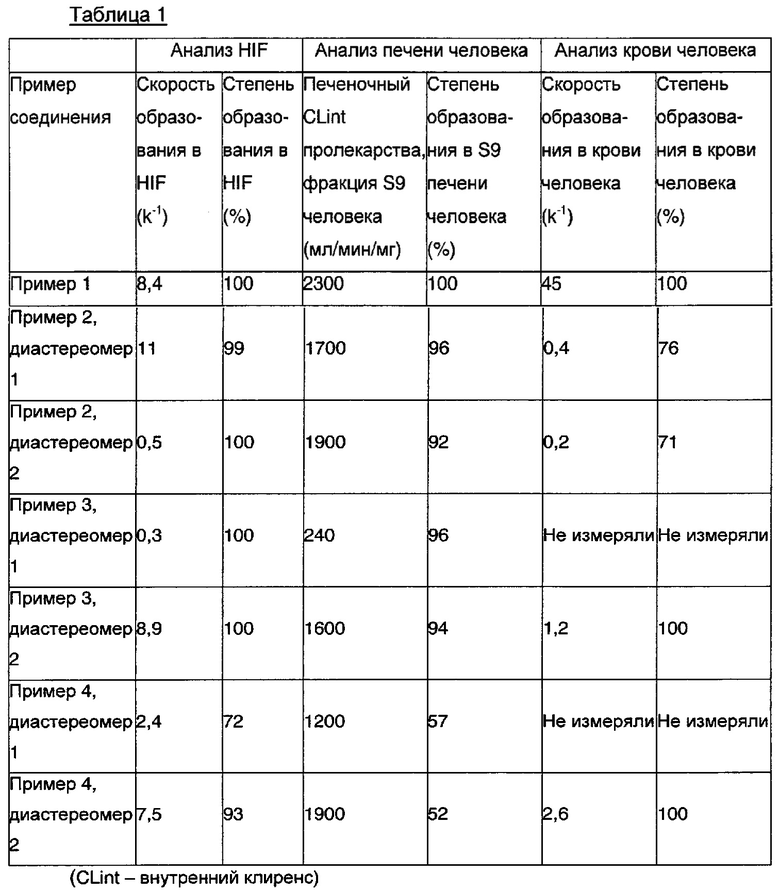
Результаты in vitro, представленные в Таблице 1, показывают, что разные фармакокинетические профили в плазме крови (S)-1-фенил-2-(пиридин-2-ил)этанамина могут быть достигнуты при использовании разных пролекарств (S)-1-фенил-2-(пиридин-2-ил)этанамина. Следовательно, выбор конкретного пролекарства будет обеспечивать разные достигаемые фармакокинетические профили (S)-1-фенил-2-(пиридин-2-ил)этанамина, то есть разные пролекарства могут быть использованы для осуществления медленного или быстрого превращения в (S)-1-фенил-2-(пиридин-2-ил)этанамин. Анализы кишечных сред человека (HIF), печени человека и крови человека, использованные для получения результатов, представленных в Таблице 1, были описаны в Malmborg J & Ploeger ВА, J Pharmacol Toxicol Methods (2013) May-Jun 67(3) 203-13, который включен в данное описание посредством ссылки.
Пример 14
Этот Пример иллюстрирует, что разные пролекарства (S)-1-фенил-2-(пиридин-2-ил)этанамина, где R1 представляет собой 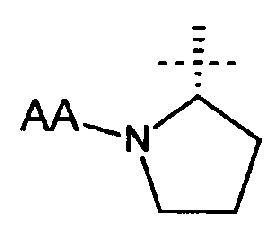 , можно использовать для достижения различных скоростей превращения (т.е. медленных или быстрых) в (S)-1-фенил-2-(пиридин-2-ил)этанамин с целью выявления подходящего PK профиля. Следовательно, варьирование R1 для получения разных пролекарств приводит в результате к разным фармакокинетическим профилям (S)-1-фенил-2-(пиридин-2-ил)этанамина. Этот Пример также иллюстрирует, что ожидаемая потеря отсутствует при пероральном воздействии, когда пролекарство превращается в свободное основание.
, можно использовать для достижения различных скоростей превращения (т.е. медленных или быстрых) в (S)-1-фенил-2-(пиридин-2-ил)этанамин с целью выявления подходящего PK профиля. Следовательно, варьирование R1 для получения разных пролекарств приводит в результате к разным фармакокинетическим профилям (S)-1-фенил-2-(пиридин-2-ил)этанамина. Этот Пример также иллюстрирует, что ожидаемая потеря отсутствует при пероральном воздействии, когда пролекарство превращается в свободное основание.
Предполагается, что превращение пролекарства, соединения формулы (I), где R1 представляет собой:
, в (S)-1-фенил-2-(пиридин-2-ил)этанамин происходит посредством ферментативного гидролиза по С-концу пролина с высвобождением дипептида и (S)-1-фенил-2-(пиридин-2-ил)этанамина. Предполагается, что вовлеченным в гидролиз ферментом является дипептидилпептидаза IV (DPPIV), что может быть продемонстрировано ингибированием превращения селективным ингибитором этого фермента. Предполагается, что DPPIV распределена по всему организму человека. Кишечная среда человека (HIF), печеночная фракция S9 человека и цельная кровь человека были использованы для исследования фармакокинетических свойств пролекарств (S)-1-фенил-2-(пиридин-2-ил)этанамина, а также скорости образования и степени образования (S)-1-фенил-2-(пиридин-2-ил)этанамина в разных отделах организма. Клетки Сасо-2 были использованы для оценки потенциала проницаемости и всасывания пролекарств (S)-1-фенил-2-(пиридин-2-ил)этанамина. Скорость образования (S)-1-фенил-2-(пиридин-2-ил)этанамина из разных пролекарств (S)-1-фенил-2-(пиридин-2-ил)этанамина различалась в протестированных аналитических образцах, особенно в крови человека, как показано в Таблице 2. Различающаяся проницаемость, показанная разными пролекарствами (S)-1-фенил-2-(пиридин-2-ил)этанамина, свидетельствует о том, что системное воздействие пролекарства (S)-1-фенил-2-(пиридин-2-ил)этанамина будет варьировать в зависимости от выбранного пролекарства.
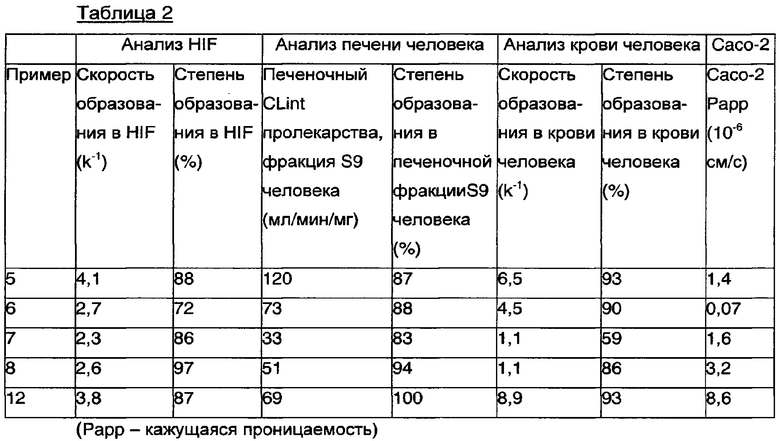
Результаты in vitro, представленные в Таблице 1, показывают, что разные фармакокинетические профили в плазме крови (S)-1-фенил-2-(пиридин-2-ил)этанамина могут быть достигнуты при использовании разных пролекарств (S)-1-фенил-2-(пиридин-2-ил)этанамина. Следовательно, разные фармакокинетические профили (S)-1-фенил-2-(пиридин-2-ил)этанамина могут быть достигнуты при использовании разных пролекарств.
Пример 15
Этот Пример иллюстрирует, что пероральные дозы пролекарства (S)-1-фенил-2-(пиридин-2-ил)этанамина имеют сходный фармакокинетический профиль с профилем при внутривенной 1-часовой инфузии (S)-1-фенил-2-(пиридин-2-ил)этанамина.
На основе оценочных данных in vitro (S)-1-фенил-2-(пиридин-2-ил)этанамин (ланицемин) будет иметь хорошую пероральную биодоступность (>75%) у человека, но выше, чем допустимая Cmax при введении в виде внутривенного болюса (относительно его профиля безопасности в виде 1-часовой инфузии). Одним критерием выбора, использованным для оценки применимости и пригодности пролекарства (S)-1-фенил-2-(пиридин-2-ил)этанамина, был потенциал пролекарства сохранять характеристики перорального введения/1-часовой внутривенной инфузии (S)-1-фенил-2-(пиридин-2-ил)этанамина (например, Tmax, Cmax, AUC) с одновременным снижением Cmax активной группировки, когда пролекарство вводят непосредственно в кровоток, тем самым ослабляя потенциальное злоупотребление (например склонность к злоупотреблению) и/или связанные с Cmax проблемы безопасности. В качестве средств идентификации пролекарств с подходящими фармакокинетическими профилями у человека были созданы мультикомпартментальные, физиологически обоснованные фармакокинетические модели (РВРK) как для (S)-1-фенил-2-(пиридин-2-ил)этанамина, так и для каждого рассматриваемого пролекарства. РВРK модели были проверены с использованием группы известных пролекарств, для которых PK данные для людей были приемлемыми (смотри Malmborg J & Ploeger ВА, J Pharmacol Toxicol Methods (2013) May-Jun 67(3) 203-13, который включен в данное описание посредством ссылки). Константы скорости, использованные в этой модели, были основаны на исследованиях стабильности in vitro (т.е. в кишечной среде, крови человека и т.д.), проведенных как с пролекарствами, так и с (S)-1-фенил-2-(пиридин-2-ил)этанамином (значения для указанных примеров представлены в Таблицах 1 и 2).
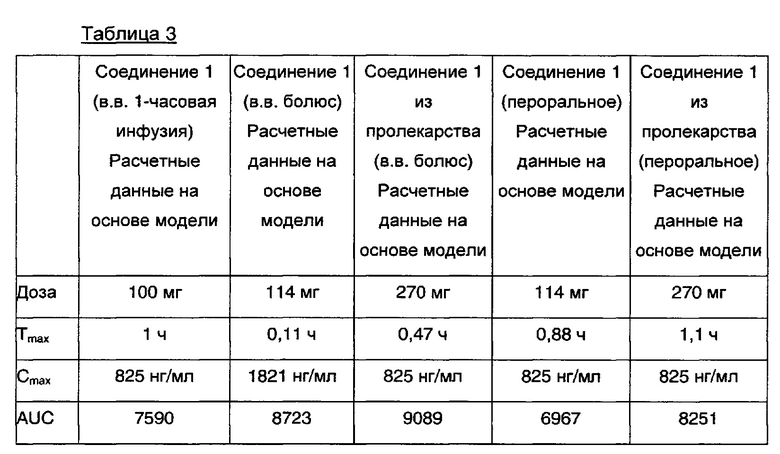
В Таблице 3 представлены ключевые фармакокинетические прогностические данные для соединения Примера 5, одного из расщепляемых ферментом DPPIV пролекарств. Как показано в Таблице 3, экспозиция Cmax (S)-1-фенил-2-(пиридин-2-ил)этанамина увеличивается более чем в 2 раза, когда пероральную форму дозы измельчают и незаконно внутривенно (в.в.) вводят в виде болюса. Напротив, (S)-1-фенил-2-(пиридин-2-ил)этанамин, высвобождаемый из перорально введенного пролекарства, имеет PK профиль, сходный с профилем 1-часовой инфузии (S)-1-фенил-2-(пиридин-2-ил)этанамина без удваивания Cmax, как при введении в виде болюса внутривенно. Кроме того, поскольку превращение в (S)-1-фенил-2-(пиридин-2-ил)этанамин осуществляется селективно ферментом DPPIV, ингибитор DPPIV можно использовать для остановки превращения в случае целенаправленного или непреднамеренного злоупотребления, что является дополнительной функцией безопасности, заложенной в разработку DPPIV пролекарства.
В Таблице 3 Соединение 1 представляет собой (S)-1-фенил-2-(пиридин-2-ил)этанамин. В Таблице 3 отобраны все дозы, которые содержат эквивалентные количества Соединения 1 [(S)-1-фенил-2-(пиридин-2-ил)этанамин в форме свободного основания], т.е. каждая доза содержит одинаковое количество Соединения 1 и потенциально доступное для высвобождения (S)-1-фенил-2-(пиридин-2-ил)этанамина в форме свободного основания. Таким образом, смоделированные концентрации Cmax Соединения 1, свободного основания, можно сравнивать непосредственно с различными введениями (S)-1-фенил-2-(пиридин-2-ил)этанамина и пролекарством (S)-1-фенил-2-(пиридин-2-ил)этанамина, как показано в Таблице 3.
Пример 16
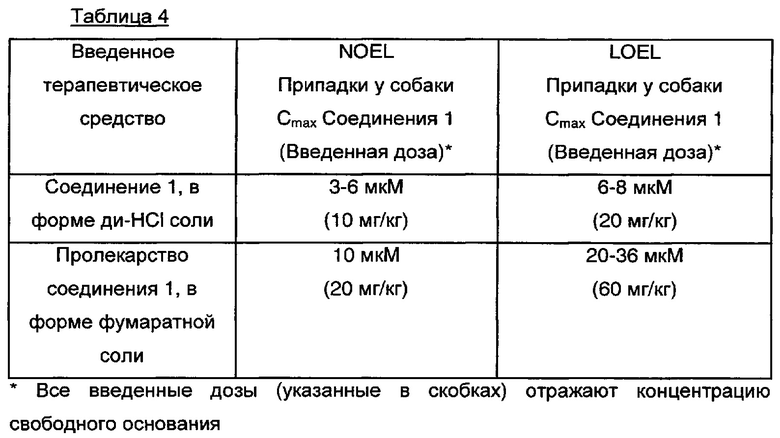
Этот пример иллюстрирует, что когда собакам вводили дозы пролекарства дигидрохлорида (S)-1-фенил-2-(пиридин-2-ил)этанамина (Пример 5) и дигидрохлорида (S)-1-фенил-2-(пиридин-2-ил)этанамина, риск припадков, выраженный в NOEL и LOEL, был меньше при использовании пролекарства.
Дигидрохлорид (S)-1-фенил-2-(пиридин-2-ил)этанамина и носитель вводили один раз в сутки через пероральный зонд (вода). Композиции готовили дважды, один раз в неделю. Дозировочные композиции хранили и охлаждали (2-8°C) в контейнерах из стекла янтарного цвета до тех пор, пока они не потребуются для введения. Подготавливали серию концентраций дигидрохлорида (S)-1-фенил-2-(пиридин-2-ил)этанамина в носителе с сохранением объема дозы для всех доз (1 мл/кг). Индивидуальные дозы рассчитывали на самую последнюю массу тела собак, использованных в исследовании.
Пролекарство (S)-1-фенил-2-(пиридин-2-ил)этанамина в виде фумаратной соли (Пример 5) и носитель вводили один раз в сутки в течение 14 суток через пероральный зонд приблизительно в одно и то же время ежедневно (±1 час) (носителем является 0,3М глюконовая кислота рН 3,0). Подготавливали серию концентраций пролекарства (S)-1-фенил-2-(пиридин-2-ил)этанамина в виде фумаратной соли в носителе (например 3, 10 и 30 мг/мл пролекарства (S)-1-фенил-2-(пиридин-2-ил)этанамина) с сохранением объема дозы для всех доз (2 мл/кг). Дозировочные композиции были нагреты до комнатной температуры при перемешивании в течение по меньшей мере 30 минут перед введением дозы и непрерывно на протяжении процедуры введения. Индивидуальные дозы рассчитывали на самую последнюю массу тела собак, использованных в исследовании.
В Таблице 4 Соединение 1 представляет собой (S)-1-фенил-2-(пиридин-2-ил)этанамин. Таблица 4 иллюстрирует наивысшие уровни в крови, при которых припадки отсутствуют (уровень, не вызывающий видимых отрицательных эффектов, NOEL), и наименьшие уровни в крови, при которых припадки имеют место (наименьший уровень, вызывающий видимый отрицательный эффект, LOEL), после введения дигидрохлорида Соединения 1 и после введения пролекарства соединения 1 в форме фумаратной соли. В каждом случае измеряют наивысшую концентрацию в крови (Cmax) (S)-1-фенил-2-(пиридин-2-ил)этанамина в форме свободного основания. В первом случае (S)-1-фенил-2-(пиридин-2-ил)этанамин в форме свободного основания образуется непосредственно из Соединения 1, введенного в форме дигидрохлоридной соли. Во втором случае (S)-1-фенил-2-(пиридин-2-ил)этанамин в форме свободного основания образуется в результате превращения in vivo из пролекарства Соединения 1, введенного в форме фумаратной соли. NOEL для припадков был выше, когда вводили пролекарство (S)-1-фенил-2-(пиридин-2-ил)этанамина в форме фумаратной соли, по сравнению с образованием (S)-1-фенил-2-(пиридин-2-ил)этанамина, когда (S)-1-фенил-2-(пиридин-2-ил)этанамин вводили в форме дигидрохлоридной соли.
Пример 17
Этот пример иллюстрирует, что увеличение доз пролекарства (S)-1-фенил-2-(пиридин-2-ил)этанамина, соединения Примера 5, вызывает менее чем пропорциональное увеличение концентрации (S)-1-фенил-2-(пиридин-2-ил)этанамина в кишечной среде человека, как проиллюстрировано на Фиг. 1а и Фиг. 1b. Как следствие, это затрудняет получение высоких концентраций (S)-1-фенил-2-(пиридин-2-ил)этанамина при введении пролекарства (S)-1-фенил-2-(пиридин-2-ил)этанамина по сравнению с прямым введением (S)-1-фенил-2-(пиридин-2-ил)этанамина, которые требуются в целях злоупотребления лекарственным средством. Без связи с теорией, пролекарства (S)-1-фенил-2-(пиридин-2-ил)этанамина, в которых присутствует R1, расщепляются в первую очередь ферментом DPPIV. Существует потенциал для эндогенного и/или экзогенного модулирования DPPIV с целью воздействия на превращение пролекарства в (S)-1-фенил-2-(пиридин-2-ил)этанамин.
Исходные растворы пролекарства (S)-1-фенил-2-(пиридин-2-ил)этанамина (0,33, 1, 3 и 6 ммоль/л) подготавливали путем добавления объема (8,25, 25, 75 и 150 мкл) 20 ммоль/л раствора пролекарства (S)-1-фенил-2-(пиридин-2-ил)этанамина, растворенного в DMSO, к FaSSIF-v2 (491,75, 475, 425 и 350 мкл). Затем 10 мкл исходного раствора пролекарства (S)-1-фенил-2-(пиридин-2-ил)этанамина добавляли в надосадочную жидкость (90 мкл HIF) в стеклянном флаконе, смешивали в течение 1 минуты и впрыскивали в ЖХ-УФ систему. Начальная концентрация при инкубированиях составляла 33, 100, 300 и 600 мкмоль/л, и образцы отбирали через 5, 20, 35, 65 и 125 минут после начала инкубирования. Эксперимент по инкубированию с (S)-1-фенил-2-(пиридин-2-ил)этанамином (мкмоль/л) использовали в качестве стандартного образца (одноточечная калибровка) для определения скорости образования и концентрации (S)-1-фенил-2-(пиридин-2-ил)этанамина. Анализ осуществляли с использованием детектора на фотодиодной матрице (использовали аналитическую длину волны 261 нм), соединенного с Waters Acquity UPLC. Использовали колонку ВЕН С-18, 1,7 мкм, 2,1×50 мм внутренний диаметр, при 40°C и использовали 0,2 мкм встроенный фильтр предварительной очистки. Подвижные фазы состояли из А: 0,03% TFA в H2O (об./об.) и В: 0,03% TFA в ацетонитриле (об./об.). Пример градиента: начальный 1% В линейно изменялся до 95% В за 7 минут, после чего следовала выдержка при 95% В в течение 0,4 минут при скорости потока 0,6 мл/мин. Скорость превращения из пролекарства в (S)-1-фенил-2-(пиридин-2-ил)этанамин определяли путем аппроксимации измеренных концентраций пролекарства и (S)-1-фенил-2-(пиридин-2-ил)этанамина в зависимости от времени.
Пример 18
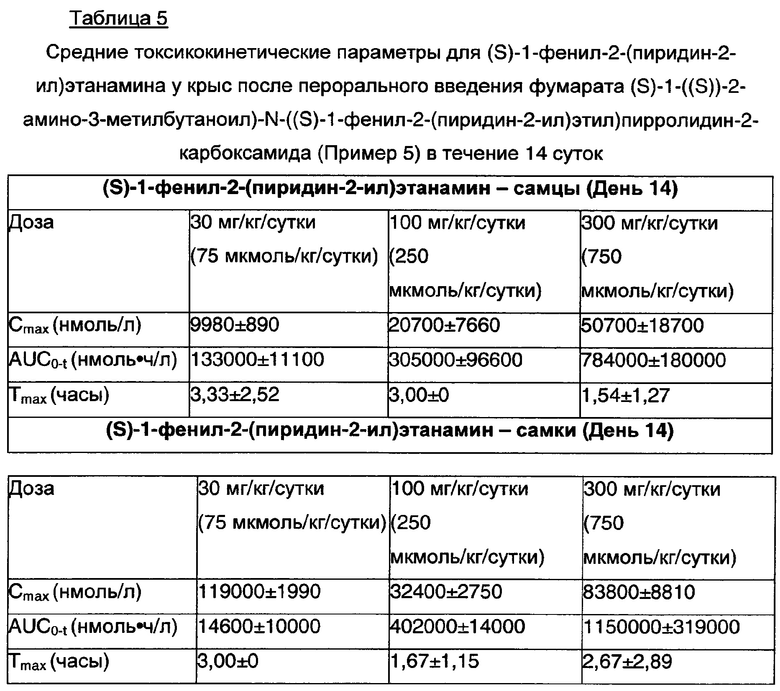
В соответствии с данными in vitro из Примера 17, исследования in vivo (Пример 18, приведенный ниже) на крысах иллюстрируют, что экспозиция Cmax (S)-1-фенил-2-(пиридин-2-ил)этанамина, как описано выше, увеличивается менее чем пропорционально после перехода от терапевтических до сверхтерапевтических доз пролекарства (S)-1-фенил-2-(пиридин-2-ил)этанамина. Результаты этих исследований in vivo представлены в Таблице 5.
52 самцам и 52 самкам крыс вводили либо (а) пролекарство (S)-1-фенил-2-(пиридин-2-ил)этанамина в виде фумаратной соли в носителе (0,3М глюконовая кислота, рН 3,0), либо (б) носитель (0,3М глюконовая кислота, рН 3,0) один раз в сутки в течение 14 суток приблизительно в одно и то же время каждый день (09:26+/-124 минуты). Дозы (30, 100 и 300 мг/кг пролекарства (S)-1-фенил-2-(пиридин-2-ил)этанамина вводили в дозовом объеме 10 мл/кг. Через 14 суток собирали образцы крови через 24 часа после введения дозы из латеральной хвостовой вены у зафиксированных животных. Образцы крови собирали с использованием капиллярных пробирок Sarstedt Minivette РОСТ, нейтральная система, 50 мкл. Кровь затем переносили в предварительно охлажденные пробирки, содержащие охлажденный во льду цитрат натрия, встряхивали 5-10 раз рукой и замораживали в сухом льду в пределах 10 секунд от сбора. Образцы цельной крови перевозили замороженными на сухом льду в лабораторию Covance Laboratories Inc. для анализа. Образцы анализировали в отношении концентраций пролекарства (S)-1-фенил-2-(пиридин-2-ил)этанамина и (S)-1-фенил-2-(пиридин-2-ил)этанамина. Вся аналитическая работа была проведена в лаборатории Covance Laboratories, Inc., Madison, Wisconsin, с использованием ЖХ/МС/МС аналитического метода, разработанного и утвержденного этой лабораторией.
Таблица 5 иллюстрирует токсикокинетические параметры после введения крысам терапевтических и сверхтерапевтических доз пролекарства (S)-1-фенил-2-(пиридин-2-ил)этанамина.
| название | год | авторы | номер документа |
|---|---|---|---|
| Спироконденсированные пирролидиновые производные в качестве ингибиторов деубиквитилирующих ферментов (DUB) | 2017 |
|
RU2730552C2 |
| Новые соединения | 2016 |
|
RU2734256C2 |
| НОВЫЕ СОЕДИНЕНИЯ | 2015 |
|
RU2691389C2 |
| Бициклические азотсодержащие соединения как агонисты М1 мускариновых рецепторов | 2015 |
|
RU2685230C2 |
| ФАРМАЦЕВТИЧЕСКИЕ СОЕДИНЕНИЯ | 2020 |
|
RU2840815C2 |
| Бициклические конденсированные гетероарильные или арильные соединения в качестве модуляторов IRAK4 | 2016 |
|
RU2684324C1 |
| Гетероциклические ингибиторы МСТ4 | 2017 |
|
RU2771875C2 |
| ЗАМЕЩЕННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СУЛЬФОНАМИДНЫЕ СОЕДИНЕНИЯ, ПОЛЕЗНЫЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ TRPA 1 | 2014 |
|
RU2675792C2 |
| СОЕДИНЕНИЯ, ЦЕЛЕНАПРАВЛЕННО ВОЗДЕЙСТВУЮЩИЕ НА BRM, И СВЯЗАННЫЕ С НИМИ СПОСОБЫ ПРИМЕНЕНИЯ | 2019 |
|
RU2797832C2 |
| ХИНОЛИНКАРБОКСАМИДНЫЕ И ХИНОЛИНКАРБОНИТРИЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ mGLuR2-НЕГАТИВНЫХ АЛЛОСТЕРИЧЕСКИХ МОДУЛЯТОРОВ, КОМПОЗИЦИИ И ИХ ПРИМЕНЕНИЕ | 2012 |
|
RU2610262C2 |
Предлагаются пролекарства антагониста NMDA, (S)-1-фенил-2-(пиридин-2-ил)этанамина формулы (I):
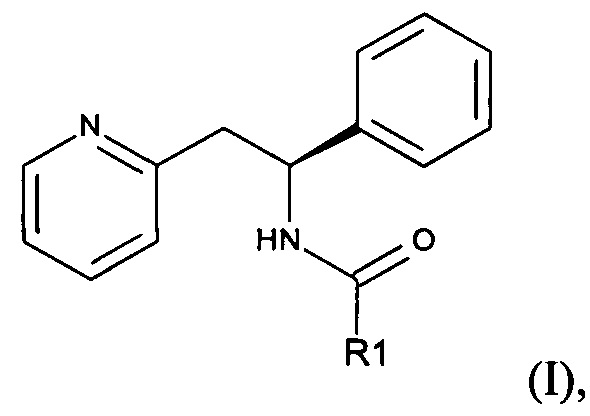
где R1 представляет собой
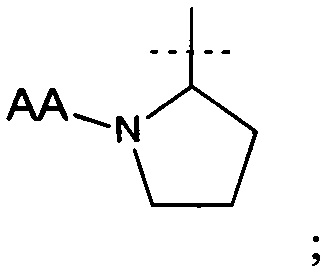
АА представляет собой связанную пептидной связью природную аминокислоту; или его фармацевтически приемлемая соль, полезные для лечения депрессии (в частности, большого депрессивного расстройства) или боли. 12 н. и 3 з.п. ф-лы, 1 ил., 5 табл., 18 пр.
1. Соединение формулы (I):
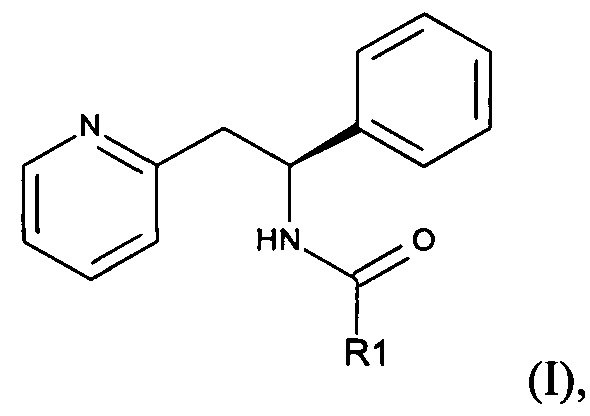
где R1 представляет собой
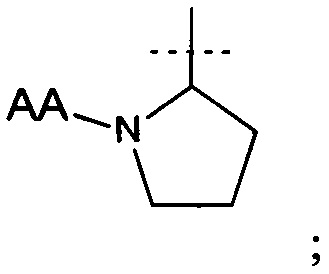
АА представляет собой связанную пептидной связью природную аминокислоту; или его фармацевтически приемлемая соль.
2. Соединение по п. 1, выбранное из группы, состоящей из
(S)-1-((S)-2-амино-3-метилбутаноил)-N-((S)-1-фенил-2-(пиридин-2-ил)этил)пирролидин-2-карбоксамида;
(S)-1-((S)-2,6-диаминогексаноил)-N-((S)-1-фенил-2-(пиридин-2-ил)этил)пирролидин-2-карбоксамида;
(S)-1-((S)-2-амино-3-(1Н-имидазол-4-ил)пропаноил)-N-((S)-1-фенил-2-(пиридин-2-ил)этил)пирролидин-2-карбоксамида;
2-((S)-2-((S)-1-((S)-2-амино-3-(4-гидроксифенил)пропаноил)пирролидин-2-карбоксамидо)-2-фенилэтил)пиридина;
2-((S)-2-((S)-1-((S)-2-амино-3-(1Н-индол-3-ил)пропаноил)пирролидин-2-карбоксамидо)-2-фенилэтил)пиридина;
(S)-1-((S)-2-амино-3-фенилпропаноил)-N-((S)-1-фенил-2-(пиридин-2-ил)этил)пирролидин-2-карбоксамида; или
(S)-1-((S)-2-амино-4-метилпентаноил)-N-((S)-1-фенил-2-(пиридин-2-ил)этил)пирролидин-2-карбоксамида;
или фармацевтически приемлемая соль любого из вышеуказанных соединений.
3. Соединение (S)-1-((S)-2-амино-3-метилбутаноил)-N-((S)-1-фенил-2-(пиридин-2-ил)этил)пирролидин-2-карбоксамид или его фармацевтически приемлемая соль.
4. Фармацевтическая композиция, обладающая активностью антагониста NMDA (N-метил-D-аспартат), содержащая эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли по любому из пп. 1-3 совместно с фармацевтически приемлемым вспомогательным веществом, разбавителем или носителем.
5. Применение соединения формулы (I) или его фармацевтически приемлемой соли по любому из пп. 1-3 в изготовлении лекарственного средства для лечения депрессии.
6. Применение соединения формулы (I) или его фармацевтически приемлемой соли по любому из пп. 1-3 для лечения депрессии.
7. Применение по п. 5 или 6, где расстройство депрессия представляет собой большое депрессивное расстройство.
8. Способ лечения депрессии, включающий введение пациенту, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли по любому из пп. 1-3.
9. Способ по п. 8, где расстройство депрессия представляет собой большое депрессивное расстройство.
10. Способ лечения боли, включающий введение пациенту, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли по любому из пп. 1-3.
11. Применение соединения формулы (I) или его фармацевтически приемлемой соли по любому из пп. 1-3 в изготовлении лекарственного средства для лечения синдрома Ретта, суицидального мышления, биполярного расстройства, обсессивно-компульсивного расстройства, отравления газом зарином или эпилептического состояния.
12. Применение соединения формулы (I) или его фармацевтически приемлемой соли по любому из пп. 1-3 для лечения синдрома Ретта, суицидального мышления, биполярного расстройства, обсессивно-компульсивного расстройства, отравления газом зарином или эпилептического состояния.
13. Способ лечения синдрома Ретта, суицидального мышления, биполярного расстройства, обсессивно-компульсивного расстройства, отравления газом зарином или эпилептического состояния, включающий введение пациенту, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли по любому из пп. 1-3.
14. Соединение 2-((S)-2-фенил-2-((S)-пирролид-2-инкарбоксамидо)-этил)-пиридин или его соль:
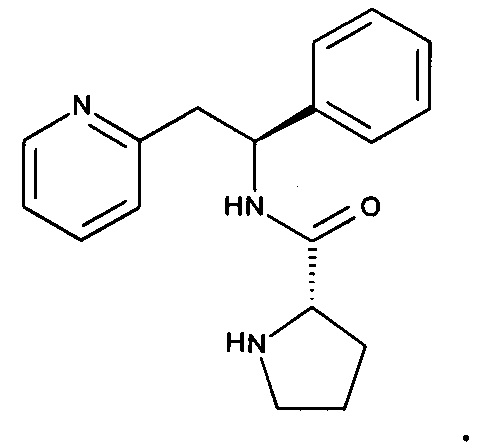
15. Соединение 2-((S)-2-фенил-2-((S)-пирролидин-2-ийкарбоксамидо)-этил)-пиридиния дихлорид:
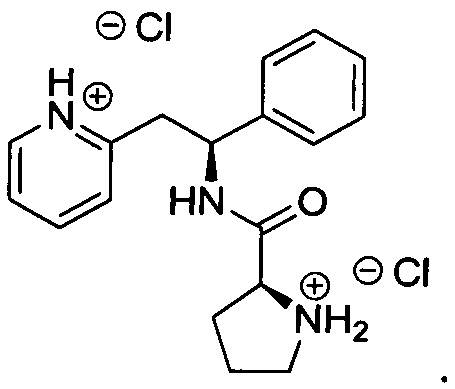
| (S)-α-ФЕНИЛ-2-ПИРИДИНЭТАНАМИН И ЕГО СОЛЬ С НЕОРГАНИЧЕСКИМИ КИСЛОТАМИ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ЛЕКАРСТВЕННЫЙ ПРЕПАРАТ НА ИХ ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ СУДОРОГ | 1993 |
|
RU2128650C1 |
| Способ получения производных пиридилаллиламина или их солей, или их смеси цис- и транс-изомеров, или индивидуальных изомеров | 1981 |
|
SU1103794A3 |
| ГНДРОВИНТОВОЙ ПРЕСС-МОЛОТ | 0 |
|
SU356035A1 |

