ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
По настоящей заявке испрашивается приоритет согласно заявке на патент США U.S.S.N №61/762087, поданной 7 февраля 2013 г., и U.S.S.N. 61/900706, поданной 6 ноября 2013 г., содержание каждой из которых полностью включено в настоящую заявку посредством ссылки.
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Возрастная макулярная дегенерация (ВМД) является основной причиной слепоты у людей старше 55 лет; у людей моложе 55 лет основной причиной является диабетическая ретинопатия (ДР) (Klein, 1994; Williams, 2004). Оба заболевания характеризуются ростом новых кровеносных сосудов - хороидальной неоваскуляризации при ВМД и ретинальной неоваскуляризации при ДР (Freund, 1993; Speicher, 2003; Zarbin, 2004). Макулярный отек развивается, когда слезная жидкость и белковые отложения скапливаются на поверхности или под глазной макулой (желтой центральной областью сетчатки) и вызывают ее расширение и вздутие (отек). Диабетический макулярный отек (ДМО) имеет похожую причину - подтекание макулярных капилляров. ДМО является наиболее частой причиной потери зрения как при пролиферативной, так и при непролиферативной ДР. Тромбоз центральной вены сетчатки и ее ответвлений является второй по распространенности причиной сосудистой патологии после ДР и приводит к резкому снижению остроты зрения и сопровождается макулярным отеком. Таким образом, антиангиогенные терапии применимы в борьбе со всеми этими состояниями.
Было показано, что αv интегрины участвуют в глазном ангиогенезе. Экспрессия αv интегринов повышена при разных заболеваниях или состояниях, таких как ВМД и ДР и в мышиной модели индуцированной кислородом ретинопатии или в модели ретинопатии недоношенных (Takagi, 2002). Также αvβ3 экспрессируется в новых сосудах после фотокоагуляции, но не в нормальных хороидальных сосудах, в модели индуцированной лазером хороидальной неоваскуляризации ВМД (Kamizuru, 2001). Было показано, что введение антагонистов αv интегрина, таких как циклического пептида RGD, ингибирует ретинальную и хороидальную неоваскуляризацию (Friedlander, 1996; Chavakis, 2002; Luna, 1996; Riecke, 2001; Yasukawa, 2004).
Ингибиторы ангиогенеза, действующие на фактор роста сосудистого эндотелия (VEGF, vascular endothelial growth factor), другие факторы (например, фактор роста фибробластов (FGF, fibroblast growth factor), тромбоцитарный фактор роста (PDGF, platelet-derived growth factor), хемокины (например, IL8, SDF1, G-CSF), рецепторы (например, CXCR1, FGF-R, PlGFR, PDGFR, рецепторы Tie), внутриклеточные медиаторы (например, киназа c-kit, фосфоинозитид-3-киназа, протеинкиназа C) и внеклеточные медиаторы (например, интегрины, кадгерины), а также ингибиторы проангиогенных мишеней (например, фосфоинозитид-3-киназы) исследовали на предмет лечения ВМД и ДР. Однако применение этих лекарственных средств ограничено по разным причинам, таким как токсичность и сложность введения. Более того, разработка исследованных или проходящих в настоящее время клинические исследования для лечения ВМД и ДР ингибиторов αv интегринов не является успешной из-за плохой окулярной фармакокинетики и/или высокого содержания эксципиента/носителя (например, хлорида бензалкония и маннита), которое, как известно, оказывает повреждающее действие на глаз.
Таким образом, остается необходимость улучшения соединений, композиций и способов лечения ВМД, ДР, ДМО и макулярного отека после окклюзии вены сетчатки, являющихся безопасными, эффективными и удобными для введения. Настоящее изобретение направлено на удовлетворение этой потребности.
КРАТКОЕ СОДЕРЖАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение представляет новые фторсодержащие соединения, являющиеся антагонистами αv интегрина, имеющие формулу I:
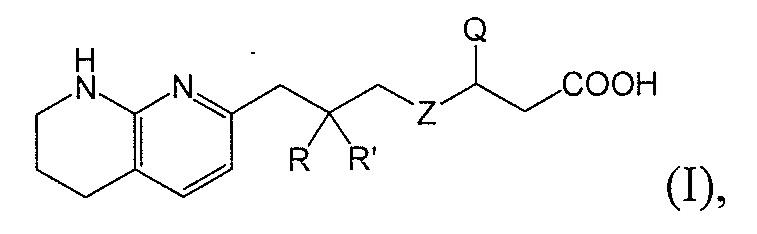
или их фармацевтически приемлемую соль или сольват.
Настоящее изобретение представляет фармацевтическую композицию, содержащую соединение изобретения, или его фармацевтически приемлемую соль или сольват, и фармацевтически приемлемый носитель или эксципиент.
Настоящее изобретение также представляет фармацевтическую композицию, содержащую соединение изобретения, или его фармацевтически приемлемую соль или сольват, и фармацевтически приемлемый носитель или эксципиент, и, кроме того, активный ингредиент, выбираемый из группы, состоящей из а) антагониста интегрина α5β1, b) цитотоксического/антипролиферативного агента, c) ингибитора эпидермального, фибробластного или тромбоцитарного фактора роста, d) ингибитора VEGF, e) ингибитора Flk-1/KDR, Flt-1, Tck/Tie-2 или Tic-1 и f) ингибитора фосфоинозитид-3-киназы и их смеси.
Кроме того, настоящее изобретение представляет фармацевтическую композицию, содержащую соединение изобретения, или его фармацевтически приемлемую соль или сольват, и фармацевтически приемлемый носитель или эксципиент, и, кроме того, активный ингредиент, выбираемый из группы, состоящей из а) антагониста интегрина α5β1, b) цитотоксического/антипролиферативного агента, c) ингибитора эпидермального, фибробластного или тромбоцитарного фактора роста, d) ингибитора VEGF и f) ингибитора фосфоинозитид-3-киназы и их смеси.
Настоящее изобретение представляет способ лечения или предотвращения заболевания или состояния у субъекта, включающий введение нуждающемуся в них субъекту терапевтически эффективного количества соединения настоящего изобретения, или его фармацевтически приемлемой соли или сольвата или терапевтически эффективного количества фармацевтической композиции настоящего изобретения. В одном аспекте изобретение представляет лечение заболевания или состояния. В одном аспекте изобретение представляет предотвращение заболевания или состояния. В одном аспекте соединение или фармацевтическую композицию настоящего изобретения, вводят местно.
Настоящее изобретение представляет способ лечения или предотвращения заболевания или состояния, опосредованного αv интегрином, у субъекта, включающий введение нуждающемуся в них субъекту терапевтически эффективного количества соединения настоящего изобретения, или его фармацевтически приемлемой соли или сольвата или терапевтически эффективного количества фармацевтической композиции настоящего изобретения. В одном аспекте заболевание или состояние является заболеванием или состоянием, в котором задействован ангиогенез. В другом аспекте заболевание или состояние является заболеванием или состоянием, в котором задействован глазной ангиогенез.
Настоящее изобретение представляет способ лечения или предотвращения заболевания или состояния, опосредованного αvβ3 и/или αvβ5 интегрином, у субъекта, включающий введение нуждающемуся в них субъекту терапевтически эффективного количества соединения настоящего изобретения, или его фармацевтически приемлемой соли или сольвата или терапевтически эффективного количества фармацевтической композиции настоящего изобретения. В одном аспекте заболевание или состояние является заболеванием или состоянием, в котором задействован глазной ангиогенез. В одном аспекте заболевание или состояние является макулярной дегенерацией. В одном аспекте заболевание или состояние является возрастной макулярной дегенерацией (ВМД). В одном аспекте заболевание или состояние является диабетической ретинопатией (ДР). В одном аспекте заболевание или состояние является диабетическим макулярным отеком (ДМО). В одном аспекте заболевание или состояние является макулярным отеком после окклюзии вены сетчатки.
Кроме того, настоящее изобретение представляет способ лечения или предотвращения ВМД, ДР, ДМО или макулярного отека после окклюзии вены сетчатки, включающий введение нуждающемуся в них субъекту терапевтически эффективного количества соединения настоящего изобретения, или его фармацевтически приемлемой соли или сольвата или терапевтически эффективного количества фармацевтической композиции настоящего изобретения. В одном аспекте изобретение представляет лечение ВМД, ДР, ДМО или макулярного отека после окклюзии вены сетчатки. В одном аспекте изобретение представляет предотвращение ВМД, ДР, ДМО или макулярного отека после окклюзии вены сетчатки.
Кроме того, настоящее изобретение представляет способ лечения или предотвращения заболевания или состояния у субъекта, включающий введение нуждающемуся в них субъекту терапевтически эффективного количества соединения настоящего изобретения, или его фармацевтически приемлемой соли или сольвата или терапевтически эффективного количества фармацевтической композиции настоящего изобретения, в сочетании с одной или несколькими терапиями для лечения или предотвращения заболевания или состояния. В одном аспекте заболевание или состояние опосредовано αv интегрином. В другом аспекте заболевание или состояние опосредовано αvβ3 и/или αvβ5 интегрином. В одном аспекте заболевание или состояние является заболеванием или состоянием, в котором задействован ангиогенез. В другом аспекте заболевание или состояние является заболеванием или состоянием, в котором задействован глазной ангиогенез. В одном аспекте терапия является анти-VEGF терапией. В другом аспекте анти-VEGF терапию вводят интравитреально.
Настоящее изобретение представляет применение соединения настоящего изобретения, или его фармацевтически приемлемой соли или сольвата для лечения или предотвращения заболевания или состояния у субъекта. В одном аспекте применение имеет целью лечение заболевания или состояния. В одном аспекте применение имеет целью предотвращение заболевания или состояния. В одном аспекте соединение или фармацевтическую композицию настоящего изобретения составляют в форме для местного введения.
Настоящее изобретение представляет применение соединения настоящего изобретения, или его фармацевтически приемлемой соли или сольвата в производстве лекарственного средства для лечения или предотвращения заболевания или состояния у субъекта. В одном аспекте изобретение предусматривает лечение заболевания или состояния. В одном аспекте изобретение предусматривает предотвращение заболевания или состояния. В одном аспекте лекарственное средство составляют в форме для местного применения.
Если иное не определено, все технические и научные термины, применяемые в данной заявке, имеют то же значение, что и общепринятое у обычных специалистов в области техники, к которой относится изобретение. В случае конфликта настоящее описание, включая определения, будет иметь преимущественное значение. В данном описании единственное число также включает множественное число, если иное четко не определено контекстом. Хотя на практике или при тестировании настоящего изобретения можно применять способы и материалы, похожие или эквивалентные описанным в данной заявке, подходящие способы и материалы описаны ниже. Все публикации, патентные заявки, патенты и другие ссылки, упомянутые в данной заявке, включены посредством ссылки. Ссылки, указанные в данной заявке, не признаются известными из уровня техники заявленного изобретения. Кроме того, материалы, способы и примеры приводятся только в иллюстративных целях и не являются ограничивающими изобретение.
Прочие отличительные признаки и преимущества изобретения станут очевидными на основе последующего подробного описания и формулы изобретения.
КРАТКОЕ ОПИСАНИЕ РИСУНКОВ
Фигура 1. Столбчатая диаграмма, показывающая число кровеносных сосудов (уровень) в исследовании куриной хориоаллантоисной мембраны.
Фигура 2. Столбчатая диаграмма, показывающая распределение соединения А1 в плазме и в глазу кролика.
Фигура 3. Столбчатая диаграмма, показывающая распределение соединения А2 в плазме и в глазу кролика.
Фигура 4. Столбчатая диаграмма, показывающая распределение соединения А3 в плазме и в глазу кролика.
Фигура 5. Типичные изображения флуоресцеиновой ангиографии глаза животных в день 35 после двукратного местного введения один раз в день (А) 50 мкл соединения А1, (B) 50 мкл соединения А2, (C) 50 мкл инертного носителя.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Считается, что многие заболевания и состояния можно лечить или предотвращать, ингибируя процессы, опосредованные αv интегринами. Таким образом, антагонисты αv интегрина представляют собой полезный класс лекарственных средств для лечения или предотвращения этих заболеваний и состояний. Интегрины являются гетеродимерными трансмембранными белками, с помощью которых клетки прикрепляются и взаимодействуют с внеклеточным матриксом и другими клетками. Интегрины αv являются ключевыми рецепторами, опосредующими клеточную миграцию и ангиогенез. Антагонисты интегринов αvβ3 и αvβ5 применимы в лечении и предотвращении резорбции кости, остеопороза, сосудистого рестеноза, диабетической ретинопатии, макулярной дегенерации, ангиогенеза, атеросклероза, воспаления, вирусного заболевания, опухолевого роста и метастазирования.
Также было обнаружено, что интегрины αv принимают участие в глазном ангиогенезе, процессе, который приводит к развитию разных глазных болезней, таких как возрастная макулярная дегенерация (ВМД), диабетическая ретинопатия (ДР), диабетический макулярный отек (ДМО) и макулярный отек после окклюзии вены сетчатки (Freund, 1993; Speicher, 2003; Zarbin, 2004). Проангиогенные факторы роста, включая VEGF и FGF, активированы при ВМД и ДР, что в свою очередь стимулирует экспрессию αv интегрина. В хорошо разработанной мышиной модели индуцированной кислородом ретинопатии или в модели ретинопатии недоношенных αv интегрины и лиганд остеопонтин экспрессируются на повышенном уровне в неоваскулярных эндотелиальных клетках во время пика роста сосудов сетчатки (Takagi, 2002). Было показано, что циклические пептиды, имитирующие мотив связывания аргинин-глицин-аспарагин (RGD, arginine-glycine-asparagine), через который αv интегрины связываются с лигандами внеклеточного матрикса, ингибируют ретинальную неоваскуляризацию в мышиной модели индуцированной кислородом ретинопатии при разных способах введения (например, подкожном, внутрибрюшинном, периокулярном или местном введении) (Friedlander, 1996; Chavakis, 2002; Luna, 1996; Riecke, 2001). Также в модели (крысиной) индуцированной лазером хороидальной неоваскуляризации, широко распространенной модели ВМД, интегрины αvβ3 и фактор Виллебранда экспрессируются на эндотелиальных клетках новых сосудов после фотокоагуляции, но не в нормальных хороидальных сосудах (Kamizuru, 2001). В данной модели интравитреальная инъекция циклического пептида RGD существенно снижает развитие хороидальной неоваскуляризации (Yasukawa, 2004). У человека экспрессию αvβ3 и αvβ5, которые не экспрессируются в нормальной ретинальной ткани, наблюдают в сосудистых клетках в глазах пациентов с ДР (Friedlander, 1996; Luna, 1996), а высокие уровни экспрессии αvβ5 в основном наблюдают в тканях глаза у пациентов с ВМД (Friedlander, 1996).
Заболевания и состояния сетчатки (расположенной на задней поверхности глаза), включая макулярную дегенерацию, ДР, ВМД и макулярный отек после окклюзии вены сетчатки, с трудом поддаются лечению посредством системного введения (например, перорального, внутривенного, интраназального введения или ингаляции), поскольку доступ к сетчатке из системного кровотока затруднен из-за гематоретинального барьера. Таким образом, одобренные в настоящее время средства лечения (например, анти-VEGF белки или химически модифицированный анти-VEGF аптамер) макулярной дегенерации, ДМО и макулярного отека после окклюзии вены сетчатки необходимо вводить повторно посредством интраокулярной инъекции (интравитреального введения).
Многие ингибиторы ангиогенеза, действующие на фактор роста сосудистого эндотелия (VEGF) (например, аптамер VEGF, пегаптаниб, и моноклональные антитела к VEGF или к рецептору VEGF (VEGFR), бевацизумаб, ранибизумаб, афлиберцепт) исследуют на предмет лечения ВМД и ДР. Однако лишь пегаптаниб, ранибизумаб и афлиберцепт одобрены Управлением по контролю за качеством пищевых продуктов и медикаментов (FDA, Food and Drug Administration). Более того, все действующие на VEGF лекарственные средства необходимо вводить посредством интравитреальной инъекции для лечения ВМД и ДР. Интравитреальная инъекция требует введения адекватной анестезии и микробицида широкого спектра действия, и введения иглы шприца внутрь глаза в асептических условиях, обуславливая таким образом необходимость проведения процедуры введения в медицинском кабинете. Например, раздел «Дозировка и способ введения» в инструкции по медицинскому применению препарата ранибизумаб описывает сложные требования введения лекарственного средства безопасным и эффективным способом: все содержимое флакона ранибизумаба отбирают 5-микронной 19 g иглой с фильтром, соединенной с 1 см3 туберкулиновым шприцем в асептических условиях; иглу с фильтром следует утилизировать после отбора содержимого флакона и заменить стерильной 30 g 0,5 дюймовой иглой для интравитреальной инъекции; содержимое следует слить, пока конец поршня не достигнет отметки 0,05 мл на шприце; процедуру интравитреальной инъекции следует проводить в контролируемых асептических условиях (например, с применением стерильных перчаток, стерильной салфетки и стерильного зеркала для века).
Кроме практических недостатков и ограничений, связанных с необходимостью делать интравитреальную инъекцию для лечения глазных болезней, действующие только на VEGF лекарственные средства действуют на ангиогенез, стимулированный VEGF, но не на ангиогенез, стимулированный другими факторами роста, включая фибробластный фактор роста (FGF) и тромбоцитарный фактор роста (PDGF). Действие на ангиогенные молекулы, отличные от VEGF или дополняющие VEGF, может идентифицировать более эффективные и безопасные ингибиторы интраокулярной неоваскуляризации. Потенциальные целевые молекулы включают факторы роста (например, ангиопоетин, FGF, HGF, IGF-1, PDGF-B, PlGF), хемокины (например, IL8, SDF1, G-CSF), рецепторы (например, CXCR1, FGF-R, PlGFR, PDGFR, рецепторы Tie), внутриклеточные медиаторы (например, киназа c-kit, фосфоинозитид-3-киназа, протеинкиназа C) и внеклеточные медиаторы (например, интегрины, кадгерины). Действительно, было показано, что несколько лекарственных средств, которые действуют не специфично на VEGF, обладают антиангиогенным действием в глазах: пазопаниб (блокирующий рецепторы PDGF, c-Kit, рецептор FGF и c-fms) подавляет хороидальную неоваскуляризацию в мышиных моделях; и PKC412 (блокирующий PKC, рецептор VEGF, рецептор PDGF и изоформы SCF-R) снижает макулярный отек при диабете (Doukas, 2008; Takahashi, 2009; Campochiara, 2004). Также изучают средства лечения, комбинирующие действие на VEGF и ингибирование одного из указанных прочих факторов роста. Например, клиническое исследование фазы 3 проводится для изучения комбинации ранибизумаба и антитела к PDGF, называемого E10030 (ClinTrials.gov, NCT01944839). Однако эти средства лечения ограничиваются угрозами безопасности и сложностью введения, такими как гепатотоксичность, наблюдаемая после перорального введения PKC412 и интравитреальное введение ранибизумаба и E10030.
Другой подход лечения или предотвращения глазных болезней заключается в селективном действии на определенную проангиогенную цель, такую как PI3K. Ингибитор PI3K широкого спектра действия LY294002 подавляет ретинальную или хороидальную неоваскуляризацию после интраокулярной инъекции у грызунов (Yang, 2009). Дополнительные альтернативные средства лечения ВМД и ДР, включая интравитреальное введение нескольких низкомолекулярных ингибиторов, препятствующих ангиогенезу (например, антагонистов рецептора фибронектина, JSM6427 (Clin Trials.gov, NCT00536016) и ATN-161 (Wang, 2011), ингибиторы тирозинфосфатазы белка сосудистого эндотелия (ClinTrials.gov, NCT01702441) и ингибиторы mTOR, сиролимус и Palomar 529 (Jacot, 2011)) проходят исследование на животных или в рамках клинических исследований. Более того, было показано, что комбинированные терапии, включая множество участников проангиогенного пути передачи сигнала и несколько селективных ингибиторов (например, комбинирование ангиостатической терапии с аптамером VEGF, антагонистом интегрина и протеолитическим фрагментом триптофановой тРНК-синтазы) ингибируют глазной ангиогенез (Dorrell, 2007). Несмотря на эти исследования и клинические исследования не было сообщений о терапии с подходящим профилем эффективности и безопасности.
Недавние достижения технологии доставки лекарственных средств, включая технологию приготовления лекарственных средств, полимерную химию, нанотехнологию, микроприспособления для доставки лекарственных средств и хирургические технологии, предложили новые варианты и возможности для местного введения лекарственного средства в глаз. Эти технологии включают применение гидрогелей, мукоадгезивных полимеров, циклодекстринов, нанокомпозитных составов, мицеллярных и липидных наночастиц, ниосом, микроэмульсии, микросфер и получение производных неактивной формы лекарства. Например, нанокомпозиты применяют для доставки диклофенака (Cao, 2011), и было показано, что местное введение непафенака снижает распространенность микроангиопатии в животных моделях ДР (Kern, 2007) и индуцированной кислородом ретинопатии (Yanni, 2010). Также технологию наночастиц применяли для усиления проникновения через поверхность гидрофобных соединений, таких как глюкокортикоиды, в расположенные сзади структуры глаза (Diebold, 2010), и было показано, что инъекция наночастиц в стекловидное тело обеспечивает интраретинальную локализацию на протяжении нескольких месяцев после введения исходной дозы и, таким образом, может применяться в качестве локализованного депо высвобождения лекарственного средства (Bourges, 2003). Изучалось или изучается в настоящее время местное введение с помощью глазных капель (например, состава глазных капель, включающего TG100572, который ингибирует FGF, PDGF и VEGF (Doukas, 2008), или ингибиторы тирозинкиназы (TKI) (например, сорафениб (WO2013/000909), антагонисты рецептора брадикинина (ClinTrials.gov, NCT01319487) или антимикробный агент, скваламин (ClinTrials.gov, NCT01678963)). Однако не было показано, что какой-либо из этих подходов подходит для замещения принятых в настоящее время стандартных анти-VEGF средств лечения, требующих интравитреальной инъекции.
Пероральное или местное введение лекарственных средств, ингибирующих αv интегрины (например, циклического пентапептидного ингибитора αvβ3 и αvβ5, цикло-RGDfV, циленгетида и непептидного антагониста αvβ3 и αvβ5, JNJ-26076713 и EMD478761), например, посредством резервуарного импланта на основе поливинилового спирта, изучали ранее или продолжают изучать в рамках клинических исследований для лечения ВМД и ДР (Friedlander, 1996; Santulli, 2008; Fu, 2007). Цикло-RGDfV исследовали на мышиной модели ретинопатии недоношенных, применяя его местное введение (Riecke, 2000); однако соединение необходимо было вводить шесть раз в день из-за плохой фармакокинетики соединения (т.е. количества соединения, которое распределяется в сетчатке после введения в виде глазных капель и затем поддерживает необходимую концентрацию в сетчатке между введениями средства). Кроме того, пептид составляли в форме, содержащей высокие уровни хлорида бензалкония и маннита, о которых известно, что они вызывают повреждение глаза. В результате разработка местного введения не была успешной. На сегодняшний день последним подходом к лечению глазного ангиогенеза является интравитреальная инъекция ALG-1001 (синтетического олигопептидного ингибитора αvβ3, αvβ5 и α5β1), который нельзя вводить местно.
Настоящее изобретение относится к новым фторсодержащим соединениям, являющимся антагонистами αv интегринов, в частности интегринов αvβ3 и/или αvβ5. Соединения настоящего изобретения или их фармацевтически приемлемые соли и сольваты применимы в лечении или предотвращении резорбции кости, остеопороза, сосудистого рестеноза, атеросклероза, воспаления, вирусного заболевания, роста опухоли и метастазирования. В частности, соединения настоящего изобретения, или их фармацевтически приемлемые соли или сольваты и фармацевтические композиции, содержащие эти соединения, являются эффективными в отношении лечения макулярной дегенерации, ДР, ДМО и макулярного отека после окклюзии вены сетчатки при местном применении.
Предпринятые ранее попытки применения низкомолекулярных антагонистов интегринов посредством местного введения не были успешными, поскольку эти соединения не обладают соответствующими физико-химическими свойствами (например, липофильностью, молекулярным размером и полярной поверхностью), обеспечивающими доставку терапевтически эффективных количеств этих соединений посредством удобной формы и режима дозирования. Неожиданно было обнаружено, что соединения настоящего изобретения, распределяются в сетчатке после местного введения в терапевтически эффективных количествах для ингибирования функции интегринов αvβ3 и αvβ5 и таким образом лечат или предотвращают ретинальный ангиогенез. Соединения настоящего изобретения обладают преимуществами, такими как улучшенная активность, селективность, проникновение в ткани, период полужизни и/или метаболическая стабильность и эффективное распределение в сетчатку в терапевтически эффективных количествах посредством удобного местного введения в глаза.
Соединения настоящего изобретения
Настоящее изобретение относится к новым фторсодержащим соединениям формулы I:
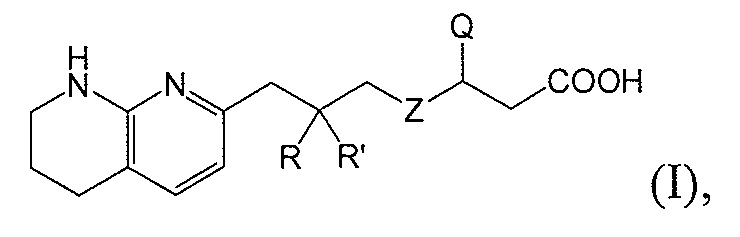
или к их фармацевтически приемлемой соли или сольвату, где:
Z является 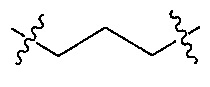 или
или 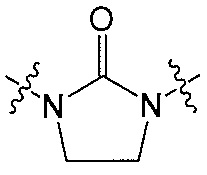 ;
;
R и R’ независимо друг от друга обозначают H или F, или R и R’ вместе с атомом углерода, к которому они присоединены, образуют 3- или 4-членное карбоциклическое или гетероциклическое кольцо;
Q является 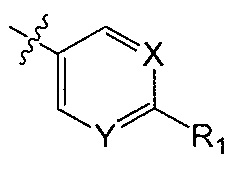 или
или 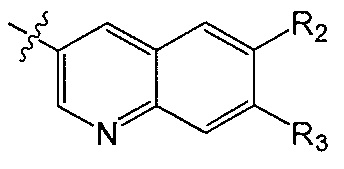 ;
;
X является CH или N;
Y является CH или N;
R1 является C1-C4 алкилом, замещенным 1, 2, 3, 4, 5, 6, 7, 8 или 9 атомами фтора, или C1-C6 алкоксигруппой, замещенным 0, 1, 2, 3, 4, 5, 6 или 7 атомами фтора; и
R2 и R3 независимо друг от друга обозначают H, F, CH2F, CHF2 или CF3, при условии, что один из R2 и R3 не является H,
при условии, что соединение формулы (I) включает по меньшей мере один атом фтора.
Соединения настоящего изобретения включают по меньшей мере один атом фтора. В одном аспекте соединения настоящего изобретения включают по меньшей мере один атом фтора в заместителе R или R’. В другом аспекте соединения настоящего изобретения включают по меньшей мере один атом фтора в заместителе R1. В другом аспекте соединения настоящего изобретения включают по меньшей мере один атом фтора в заместителе R2 или R3. Фторирование по любому определенному положению, такое как в соединениях настоящего изобретения, не изучали или не предполагали.
В одном аспекте Z является  . В другом аспекте Z является
. В другом аспекте Z является  .
.
В одном аспекте и R, и R’ являются H. В другом аспекте и R, и R’ являются F. В другом аспекте R является H и R’ является F.
В другом аспекте R и R’ вместе с атомом углерода, к которому они присоединены, образуют 3- или 4-членное карбоциклическое или гетероциклическое кольцо. В другом аспекте R и R’ вместе с атомом углерода, к которому они присоединены, образуют 4-членное гетероциклическое кольцо. В другом аспекте 4-членное гетероциклическое кольцо является окситановым кольцом. Например, окситановое кольцо является окситан-3-ил кольцом или окситан-2-ил кольцом.
В одном аспекте Q является 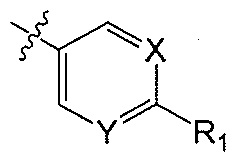 . В одном аспекте X является N и Y является CH. В другом аспекте и X, и Y являются CH. В другом аспекте и X, и Y являются N.
. В одном аспекте X является N и Y является CH. В другом аспекте и X, и Y являются CH. В другом аспекте и X, и Y являются N.
В одном аспекте R1 является алкилом с прямой цепью C1-C4 или разветвленным C3-C4 алкилом и замещен 1, 2, 3, 4, 5, 6, 7, 8 или 9 атомами фтора. В другом аспекте R1 является метилом, этилом, пропилом или бутилом и замещен 1, 2, 3, 4, 5, 6, 7, 8 или 9 атомами фтора. В другом аспекте R1 является метилом, замещенным 1, 2 или 3 атомами фтора. В другом аспекте R1 является CF3.
В одном аспекте R1 является алкоксигруппой с прямой цепью C1-C6 или разветвленной C3-C4 алкоксигруппой и замещен 1, 2, 3, 4, 5, 6 или 7 атомами фтора. В другом аспекте R1 является метокси-, этокси-, пропокси- или бутокси-группой и замещен 1, 2, 3, 4, 5, 6 или 7 атомами фтора. В другом аспекте R1 является метокси-группой, замещенной 0, 1, 2 или 3 атомами фтора. В другом аспекте R1 является OCH3, OCH2F, OCHF2 или OCF3. В другом аспекте R1 является OCHF2.
В другом аспекте Q является 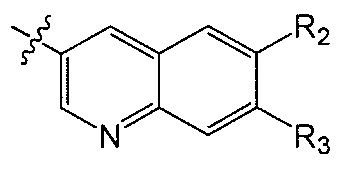 .
.
В одном аспекте R2 является F. В другом аспекте R2 является F и R3 является H. В другом аспекте R2 является CH2F, CHF2 или CF3.
В одном аспекте R3 является F. В другом аспекте R3 является F и R2 является H. В другом аспекте R3 является CH2F, CHF2 или CF3. В другом аспекте R3 является CF3. В другом аспекте R3 является CF3 и R2 является H.
В одном аспекте и R2 и R3 являются F.
В одном аспекте Z является 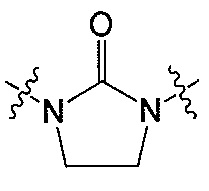 и Q является
и Q является 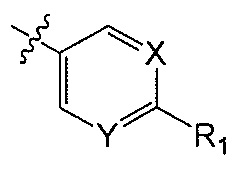 .
.
В другом аспекте Z является 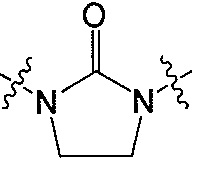 ; Q является
; Q является 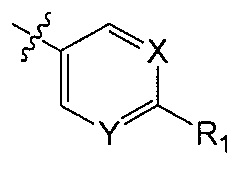 ; и R, и R’ являются H.
; и R, и R’ являются H.
В другом аспекте Z является 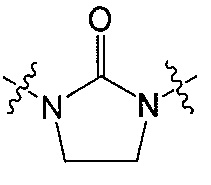 ; Q является
; Q является 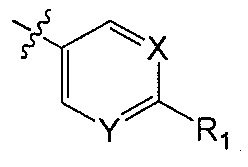 ; и R, и R’ являются H; и R1 является OCH3, OCH2F, OCHF2 или OCF3. В другом аспекте X является N и Y является CH; и R1 является OCHF2.
; и R, и R’ являются H; и R1 является OCH3, OCH2F, OCHF2 или OCF3. В другом аспекте X является N и Y является CH; и R1 является OCHF2.
В одном аспекте Z является 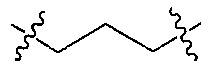 и Q является
и Q является 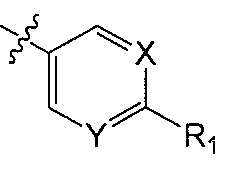 .
.
В другом аспекте Z является 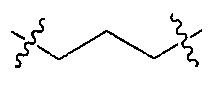 ; Q является
; Q является 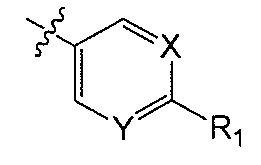 ; и X, и Y являются N. В другом аспекте R1 является метилом, замещенным 1, 2 или 3 атомами фтора. В другом аспекте R1 является CF3.
; и X, и Y являются N. В другом аспекте R1 является метилом, замещенным 1, 2 или 3 атомами фтора. В другом аспекте R1 является CF3.
В другом аспекте Z является 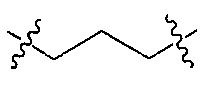 ; Q является
; Q является 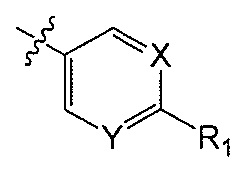 ; и X является N и Y является CH. В другом аспекте R1 является OCH3, OCH2F, OCHF2 или OCF3. В другом аспекте R1 является OCHF2.
; и X является N и Y является CH. В другом аспекте R1 является OCH3, OCH2F, OCHF2 или OCF3. В другом аспекте R1 является OCHF2.
В одном аспекте Z является 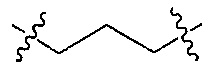 и Q является
и Q является 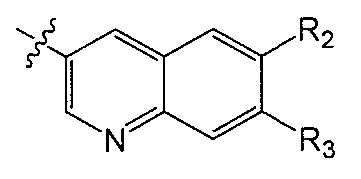 .
.
В одном аспекте соединение настоящего изобретения имеет формулу II:
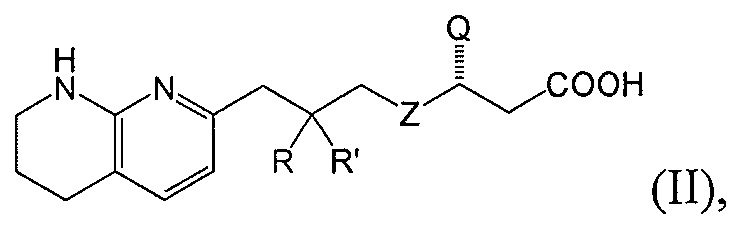
или является фармацевтически приемлемой солью или сольватом указанного соединения, где каждая из вариабельных групп соответствует описанию выше. Соединения настоящего изобретения включают соединения формулы II, где вариабельные группы проиллюстрированы в разных аспектах формулы I выше.
Типичные соединения настоящего изобретения включают соединения, перечисленные в таблице 1.
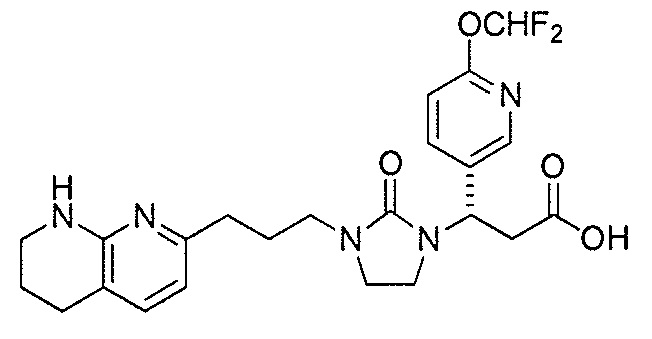
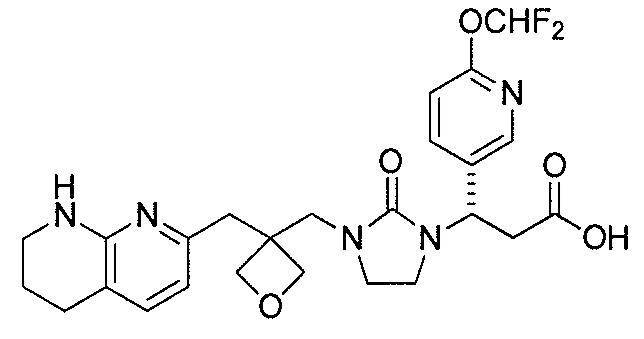
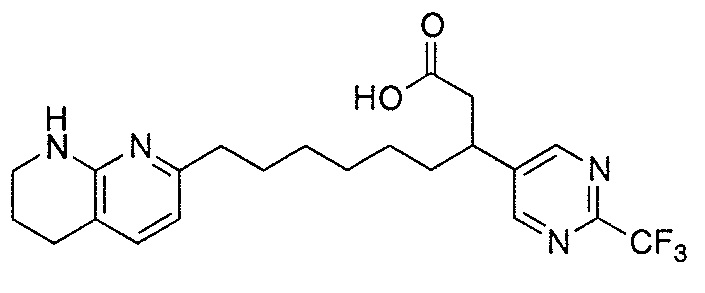
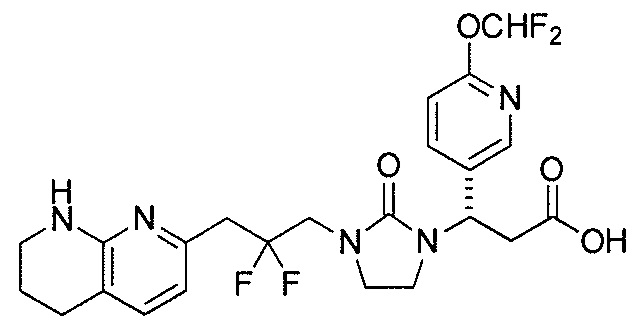
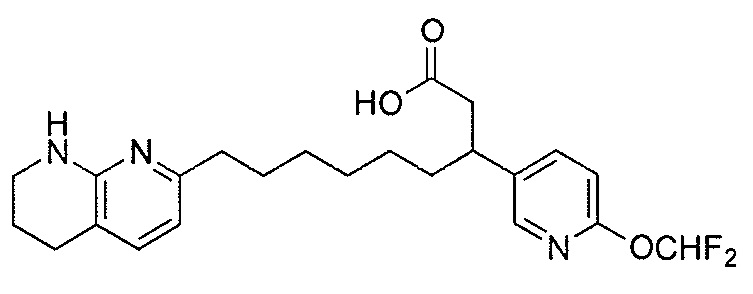
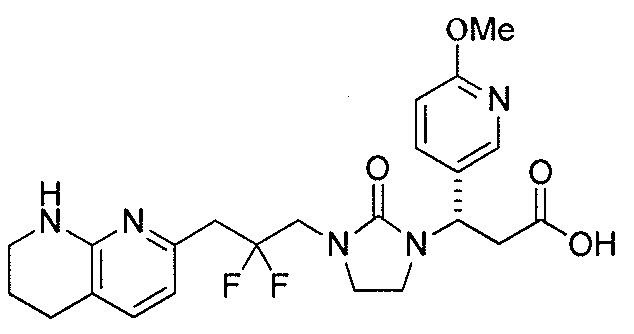
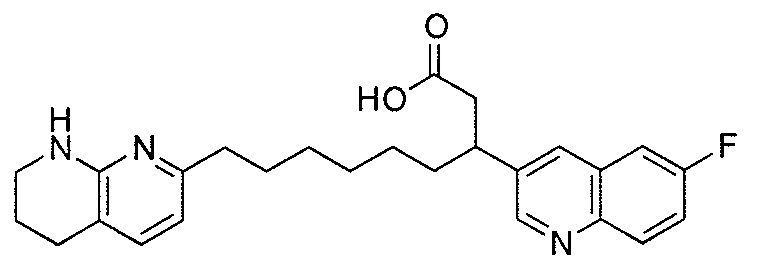
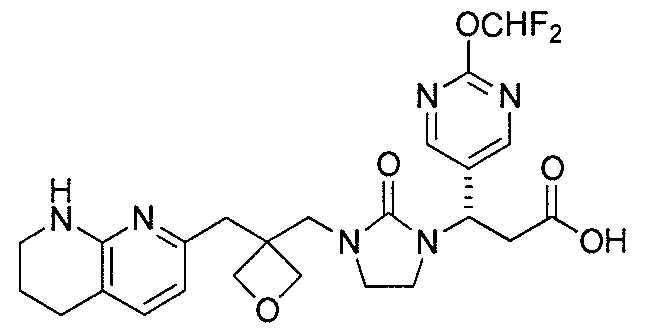
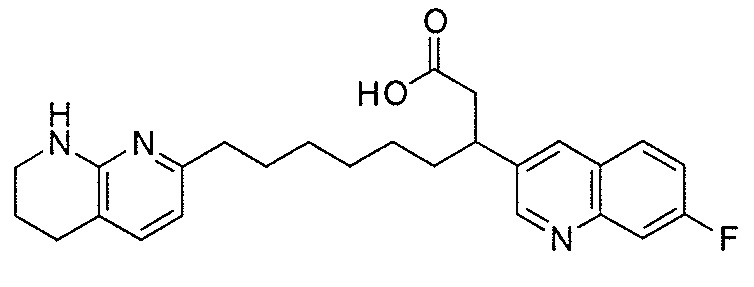
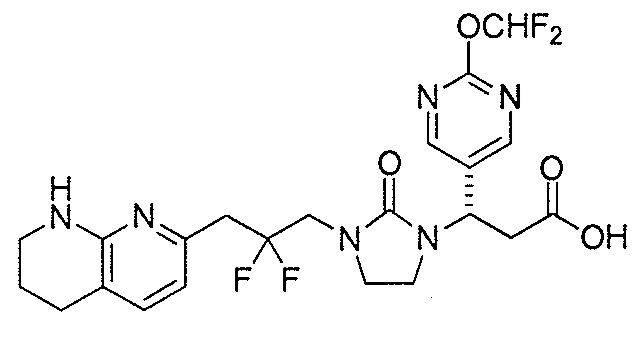
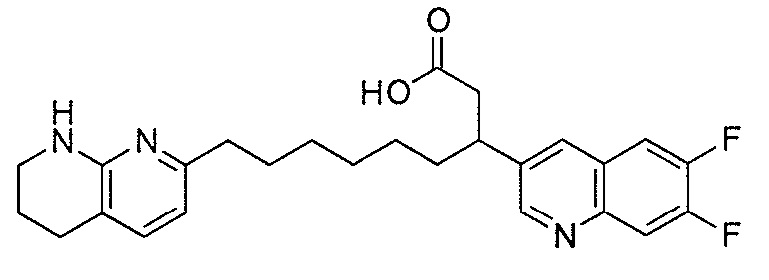

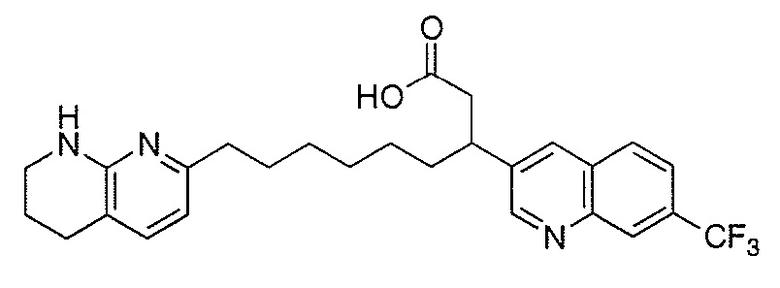
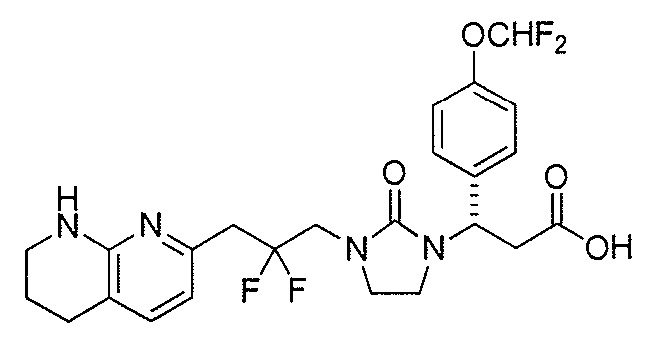
В одном аспекте соединение настоящего изобретения выбирают из соединений A1, A2 и A3, или их фармацевтически приемлемой соли или сольвата. В другом аспекте соединение настоящего изобретения выбирают из соединений A1 и A2, или их фармацевтически приемлемой соли или сольвата. В другом аспекте соединение настоящего изобретения является соединением A1, или его фармацевтически приемлемой солью или сольватом.
В одном аспекте соединение настоящего изобретения является фармацевтически приемлемой солью. В одном аспекте соединение настоящего изобретения является сольватом. В другом аспекте соединение настоящего изобретения является гидратом.
В одном аспекте соединение настоящего изобретения ингибирует активность αv интегринов (например, αvβ3 и αvβ5) в субмикромолярной концентрации, например, ниже 1 мкМ, 0,8 мкМ, 0,6 мкМ, 0,5 мкМ, 0,2 мкМ или 0,1 мкМ.
В одном аспекте соединение настоящего изобретения ингибирует клеточную адгезию к витронектину через αv интегрин (например, αvβ3 и αvβ5) при половинной ингибирующей концентрации (IC50), равной 2,0E-07 M или менее, по данным исследования человеческих эндотелиальных клеток микрососудов дермы (HMVEC). В другом аспекте соединение настоящего изобретения ингибирует клеточную адгезию к витронектину через αv интегрин (например, αvβ3 и αvβ5) при IC50, равной 2,5E-08 M или менее, по данным исследования HMVEC. В другом аспекте соединение настоящего изобретения ингибирует клеточную адгезию к витронектину через αv интегрин (например, αvβ3 и αvβ5) при IC50, равной 1,0E-08 M или менее, по данным исследования HMVEC. В одном аспекте соединение настоящего изобретения ингибирует клеточную адгезию к витронектину через αv интегрин (например, αvβ3 и αvβ5) при IC50, равной 2,5E-07 M или менее, по данным исследования крысиных эндотелиальных клеток микрососудов легких (RLMVEC). В другом аспекте соединение настоящего изобретения ингибирует клеточную адгезию к витронектину через αv интегрин (например, αvβ3 и αvβ5) при IC50, равной 3,5E-08 M или менее, по данным исследования RLMVEC. В одном аспекте соединение настоящего изобретения ингибирует клеточную адгезию к витронектину через αv интегрин (например, αvβ3 и αvβ5) при IC50, равной или менее 2,0E-08 M или менее, по данным исследования кроличьих эндотелиальных клеток аорты (RAEC). В другом аспекте соединение настоящего изобретения ингибирует клеточную адгезию к витронектину через αv интегрин (например, αvβ3 и αvβ5) при IC50, равной 1,0E-08 M или менее, по данным исследования RAEC.
В одном аспекте соединение настоящего изобретения ингибирует или уменьшает образование кровеносных сосудов в ткани или органе, in vivo или in vitro. В одном аспекте соединение настоящего изобретения снижает образование кровеносных сосудов ниже 90%, 80%, 70%, 60%, 50%, 40%, 30%, 20%, 10% или 5% по сравнению с необработанным контролем. В другом аспекте соединение настоящего изобретения снижает образование кровеносных сосудов ниже 60%, 50%, 40%, 30%, 20%, 10% или 5% по сравнению с необработанным контролем. В другом аспекте соединение настоящего изобретения снижает образование кровеносных сосудов ниже 40%, 30%, 20%, 10% или 5% по сравнению с необработанным контролем. В одном аспекте ткань является тканью глаза, такой как ретинальная ткань. В одном аспекте орган является глазом.
В одном аспекте соединение настоящего изобретения эффективно распределяется на задней поверхности глаза, например, в сетчатке, после местного введения. В одном аспекте соединение настоящего изобретения эффективно распределяется в сетчатке в течение 12 часов, 10 часов, 8 часов, 6 часов, 4 часов, 2 часов или 1 часа после местного введения в глаз. В другом аспекте соединение настоящего изобретения эффективно распределяется в сетчатке в течение 8 часов, 6 часов, 4 часов, 2 часов или 1 часа после местного введения в глаз.
Соединения настоящего изобретения можно удобно получить с помощью ряда способов, известных специалистам в данной области техники. Соединения, соответствующие каждой из описанных в данной заявке формул, можно получить в соответствии со следующими процедурами из коммерчески доступных исходных материалов или из исходных материалов, которые можно получить с помощью описанных в литературе способов. Эти способы показывают получение типичных соединений настоящего изобретения. Подразумевается, что соединения настоящего изобретения, отличные от показанных на следующих схемах, можно получить с помощью этих схем с модификациями, широко известными в данной области техники (например, с помощью другого исходного материала, изменения растворителей в реакции или корректировки продолжительности реакции или температуры).
Схема 1
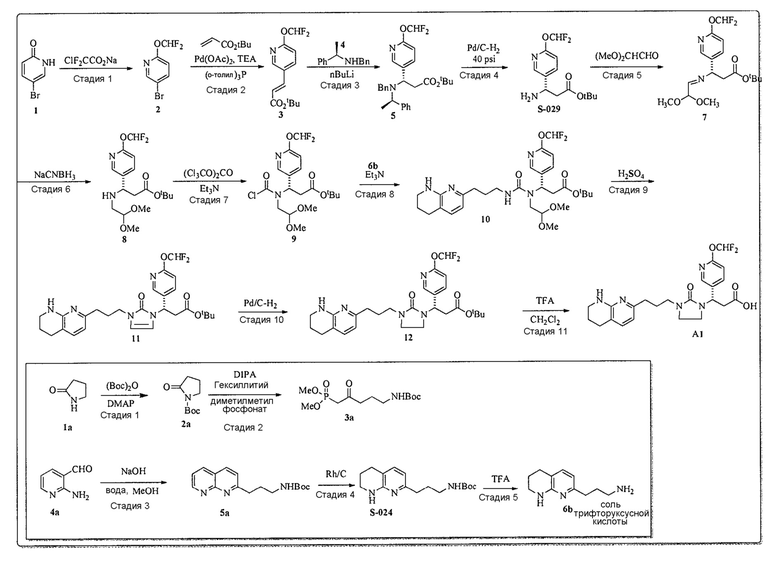
Схема 2
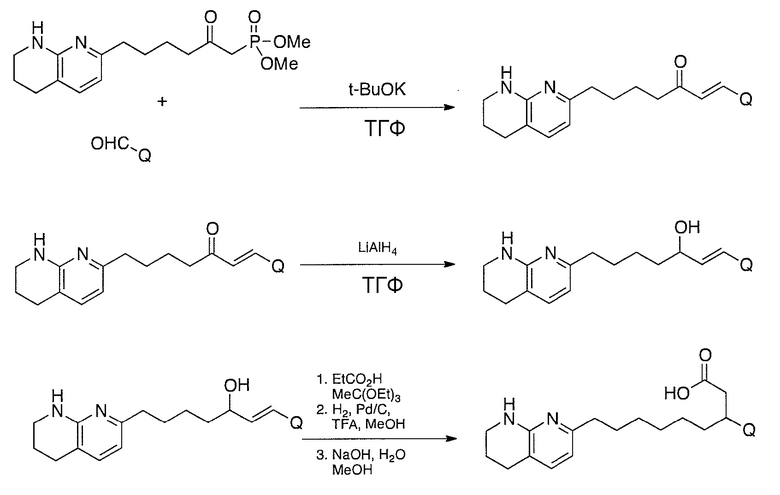
Схема 3
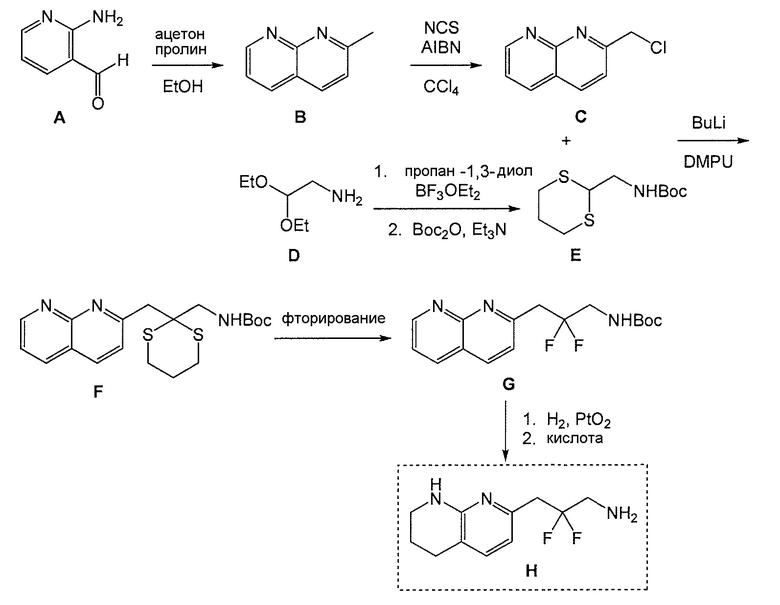
Схема 4
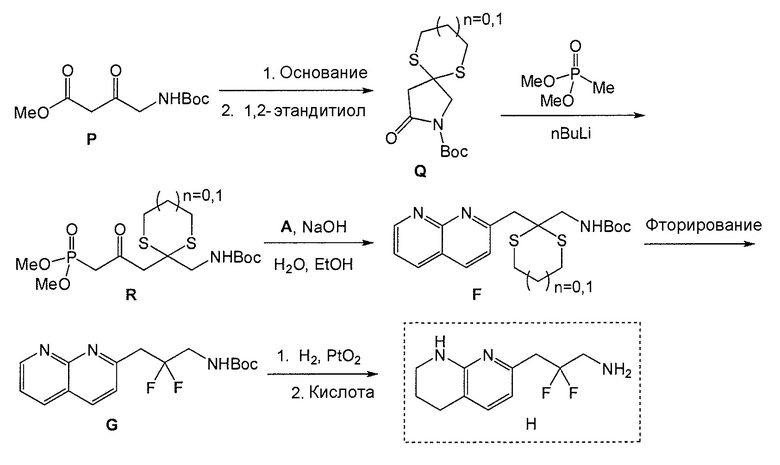
Схема 5
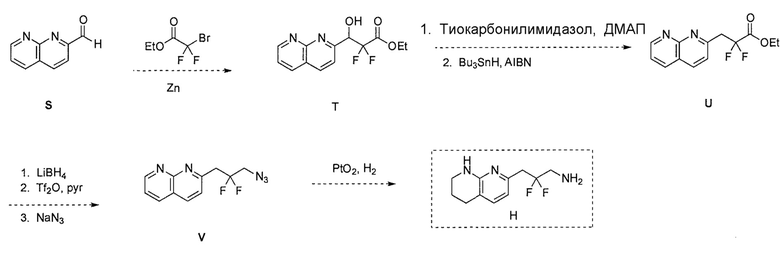
Схема 6
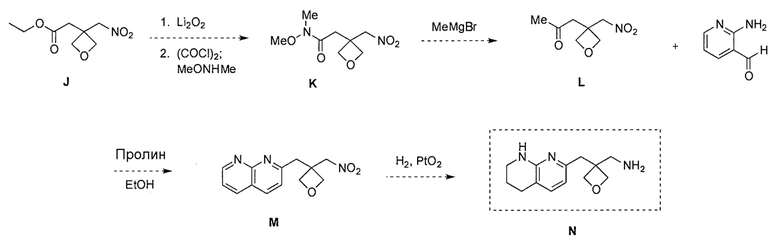
Схема 7
Схема 8
Соединения настоящего изобретения могут содержать один или несколько асимметричных центров и, таким образом, существовать в виде рацематов и рацемических смесей, единых энантиомеров, диастереомерных смесей и отдельных диастереомеров. Дополнительные центры асимметрии могут присутствовать в зависимости от природы разных заместителей в молекуле. Каждый центр асимметрии будет независимо образовывать два оптических изомера. Предполагается, что все возможные оптические изомеры и диастереомеры в смесях, и соединения в чистом или частично очищенном виде включены в объем изобретения. Подразумевается, что изобретение включает все подобные изомерные формы этих соединений.
Независимый синтез данных диастереомеров или их хроматографическое разделение можно провести, с помощью соответствующих известных в данной области техники модификаций методологии, раскрытой в данной заявке. Их абсолютную стереохимию можно определить с помощью рентгеновской кристаллографии кристаллических продуктов или кристаллических промежуточных продуктов, которые получают, при необходимости, с помощью реагента, содержащего асимметричный центр с известной абсолютной конфигурацией.
При желании, рацемические смеси соединений можно разделить так, чтобы выделить отдельные энантиомеры. Разделение можно провести с помощью способов, хорошо известных в данной области техники, таких как контактирование рацемической смеси соединений с энантиомерно чистым соединением для образования диастереомерной смеси, а затем разделение отдельных диастереомеров стандартными способами, такими как фракционная кристаллизация или хроматография. Диастереомерная смесь часто является смесью диастереомерных солей, образованной при контактировании рацемической смеси соединений с энантиомерно чистой кислотой или основанием. Диастереомерные производные можно затем превратить в чистые энантиомеры с помощью расщепления добавленного хирального остатка. Рацемическую смесь соединений также можно разделить напрямую с помощью хроматографических способов, применяющих хиральные стационарные фазы, хорошо известные в данной области техники.
В качестве альтернативы любой энантиомер соединения можно получить с помощью стереоселективного синтеза, применяя оптически чистые исходные материалы или реагенты известной конфигурации с помощью способов, хорошо известные в данной области техники.
Некоторые из соединений настоящего изобретения могут существовать как в несольватированных, так и в сольватированных формах, таких как, например, гидраты.
“Сольват” означает форму добавления растворителя, содержащую либо стехиометрическое, либо нестехиометрическое количество молекул растворителя. Некоторые соединения имеют тенденцию захватывать фиксированное молярное соотношение молекул растворителя в кристаллическом твердом состоянии, образуя таким образом сольват. Если растворителем является вода, образующийся сольват является гидратом. Когда растворителем является спирт, образующийся сольват является алколатом. Гидраты образуются комбинацией одной или нескольких молекул воды и других веществ (например, соединения изобретения), в которой вода сохраняет молекулярное состояния в виде H2O, такая комбинация может образовывать один или несколько гидратов. В гидратах молекулы воды соединены побочными валентностями за счет сил межмолекулярного взаимодействия, в частности водородных мостиков. Твердые гидраты содержат воду в виде так называемой кристаллической воды в стехиометрических соотношениях, где молекулы воды не обязательно являются одинаковыми в отношении состояния связывания. Примеры гидратов включают полуторагидраты, моногидраты, дигидраты и тригидраты. Гидраты солей соединения настоящего изобретения являются одинаково подходящими.
Для применения в медицине соли соединений настоящего изобретения относятся к нетоксичным «фармацевтически приемлемым солям». Однако другие соли могут быть применимы при получении соединений настоящего изобретения или их фармацевтически приемлемых солей. Соли, которые включены в термин «фармацевтически приемлемые соли», относятся к нетоксичным солям соединений настоящего изобретения, которые можно получить с помощью реакции свободного основания и подходящей органической или неорганической кислоты. Типичные соли включают следующие: ацетат, бензолсульфонат, бензоат, бикарбонат, бисульфат, битартрат, борат, юромид, камсилат, карбонат, хлорид, клавуланат, цитрат, дигидрохлорид, эдетат, эдизилат, эстолт, эсилат, фумарат, глюцептат, глюконат, глутамат, гликолилласанилат, гексилрезорцинат, гидрабамин, гидробромид, гидрохлорид, гидроксинафтоат, йодид, изотионат, лактат, лактобионат, лаурат, малат, малеат, манделат, месилат, метилбромид, метилнитрат, метилсульфат, мукат, напсилат, нитрат, аммониевые соль N-метилгюкамина, олеат, оксалат, памоат (эмбонат), пальмитат, пантотенат, фосфат/дифосфат, полигалактуронат, салицилат, стеарат, сульфат, основную уксуснокислую соль, сукцинат, таннат, тартрат, теоклат, тозилат, триэтиодид и валерат. Более того, в тех случаях, когда соединения изобретения, содержат остаток кислоты, их фармацевтически приемлемые соли могут включать соли щелочных металлов, например, соли натрия или калия; соли щелочноземельных металлов, например, соли кальция или магния; и соли, образованные подходящими органическими лигандами, например, четвертичные аммониевые соли, которые можно получить из аммиака или органических аминов, таких как, например, диэтиламин, триэтиламин, этилдиизопропиламин, прокаин, дибензиламин, N-метилморфолин, дигидроабиэтиламин или метилпиперидин.
Объем изобретения включает неактивные формы соединений настоящего изобретения. Как правило, такие неактивные формы лекарственных средств являются функциональными производными соединений настоящего изобретения, которые легко могут превратиться in vivo в необходимое соединение. Таким образом, в способах лечения по изобретению, термин «введение» должен включать лечение различных описанных заболеваний и состояний, с помощью специально раскрытого соединения или с помощью соединения, которое может не быть раскрыто специально, но превращается в описанное соединение in vivo после введения пациенту. Принятые процедуры для отбора и приготовления подходящих производных неактивного лекарственного соединения описаны, например, в “Design of Prodrugs,” ed. H. Bundgaard, Elsevier, 1985. Metabolites of these compounds include active species produced upon introduction of compounds of the invention into the biological milieu.
Изобретение также включает один или несколько метаболитов соединения настоящего изобретения.
Настоящее изобретение также включает меченные дейтерием соединения формулы I или II или соединения, перечисленные в таблице 1, где атом водорода заменен атомом дейтерия. Меченные дейтерием соединения включают атом дейтерия с распространенностью дейтерия, существенно превышающей встречающуюся в природе распространенность дейтерия, например, 0,015%.
Термин «фактор обогащения дейтерием», как применяют в данной заявке, обозначает соотношения распространенности дейтерия и встречающейся природе распространенности дейтерия. В одном аспекте соединение настоящего изобретения имеет фактор обогащения дейтерием, равный по меньшей мере 3500 (52,5% включение дейтерия в каждом атоме дейтерия), по меньшей мере 4000 (60% включение дейтерия), по меньшей мере 4500 (67,5% включение дейтерия), по меньшей мере 5000 (75% дейтерия), по меньшей мере 5500 (82,5% включение дейтерия), по меньшей мере 6000 (90% включение дейтерия), по меньшей мере 6333,3 (95% включение дейтерия), по меньшей мере 6466,7 (97% включение дейтерия), по меньшей мере 6600 (99% включение дейтерия) или по меньшей мере 6633,3 (99,5% включение дейтерия).
Меченные дейтерием соединения можно получить с помощью любой из ряда техник, известных в данной области. Например, меченные дейтерием соединения формулы I или II или соединения, перечисленные в таблице 1, можно обычно получить, проводя процедуры, раскрытые на схемах и/или в примерах, описанных в данной заявке, посредством замены легко доступного меченного дейтерием реагента немеченным дейтерием реагентом.
Соединение настоящего изобретения или его фармацевтическая соль или сольват, которые содержат упомянутые выше атом(ы) дейтерия, включены в объем изобретения. Более того, замещение дейтерия, т.е. 2H, может предоставить определенные терапевтические преимущества, являющиеся результатом повышенной метаболической стабильности, например, повышенного времени полужизни in vivo и/или пониженной дозировки.
В одном аспекте настоящее изобретение относится к способу синтеза соединения настоящего изобретения или его фармацевтически приемлемой соли или сольвата.
Фармацевтические композиции настоящего изобретения
Настоящее изобретение относится к фармацевтическим композициям, содержащим соединение настоящего изобретения в качестве активного ингредиента. В одном аспекте изобретение представляет фармацевтическую композицию, содержащую по меньшей мере одно соединение формулы I или II или его фармацевтически приемлемую соль или сольват и одно или несколько фармацевтически приемлемых носителей или эксципиентов. В одном аспекте изобретение представляет фармацевтическую композицию, содержащую по меньшей мере одно соединение, выбранное из таблицы 1. В другом аспекте изобретение представляет фармацевтическую композицию, содержащую по меньшей мере одно соединение, выбранное из соединений A1, A2 и A3. В другом аспекте изобретение представляет фармацевтическую композицию, содержащую по меньшей мере одно соединение, выбранное из соединений A1 и A2. В другом аспекте изобретение представляет фармацевтическую композицию, содержащую соединение A1.
Как применяют в данной заявке термин «композиция» охватывает продукт, содержащий определенные ингредиенты в определенном количестве, а также любой продукт, который получается, прямо или косвенно, из комбинации ингредиентов в определенных количествах.
Соединения настоящего изобретения можно составлять в формах для перорального введения, в таких формах как таблетки, капсулы (каждая из которых включает составы для пролонгированного высвобождения или отложенного высвобождения), пилюли, порошки, гранулы, настойки, суспензии, сиропы и эмульсии. Соединения изобретения также можно составлять в форме для внутривенного (разовая доза или инфузия), внутрибрюшинного, местного (например, глазных капель), подкожного, внутримышечного или трансдермального (например, повязки) введения, все применяемые формы хорошо известны обычным специалистам в фармацевтической области. Предпочтительно, чтобы соединения изобретения для лечения макулярной дегенерации, ДР, ДМО или макулярного отека после окклюзии вены сетчатки, составляли для местного введения, например, в форме глазных капель.
Для местного окулярного введения композиции представляют в виде офтальмологического состава, включающего соединение настоящего изобретения, в концентрации, равной приблизительно между 0,01 и приблизительно 5 процентов по весу, предпочтительно, между приблизительно 0,1 и приблизительно 5,0 процентов по весу, более предпочтительно, между приблизительно 0,5 и приблизительно 5,0 процентов по весу, наиболее предпочтительно, между приблизительно 0,8 и приблизительно 3,0 процентов по весу.
Офтальмологический состав изобретения, может быть в форме водного раствора, включающего водный инертный носитель.
Водный инертный компонент офтальмологического состава может включать воду и по меньшей мере один офтальмологически приемлемый эксципиент. Предпочтительно, чтобы водный инертный носитель включал раствор одного или нескольких офтальмологически приемлемых эксципиентов в воде.
Подходящие офтальмологически приемлемые эксципиенты включают выбираемые из группы, состоящей из усиливающего растворимость агента, хелатирующего агента, консерванта, тонического агента, повышающего вязкость/суспендирующего агента, буфера и изменяющего рН агента и их смеси. Предпочтительно, чтобы офтальмологически приемлемый эксципиент выбирали из группы, состоящей из усиливающего растворимость агента, хелатирующего агента, консерванта, тонического агента, повышающего вязкость/суспендирующего агента и изменяющего рН агента и их смеси.
Можно применять любой подходящий офтальмологически приемлемый усиливающий растворимость агент. Примеры усиливающего растворимость агента включают циклодекстрин, такой как выбираемый из группы, состоящей из гидроксипропил-β-циклодекстрина, метил-β-циклодекстрина, случайно метилированного β-циклодекстрина, этилированного β-циклодекстрина, триацетил-β-циклодекстрина, перацетилированного β-циклодекстрина, карбоксиметил-β-циклодекстрина, гидроксиэтил-β-циклодекстрина, 2-гидрокси-3-(триметиламмоний)пропил-β-циклодекстрина, глюкозил-β- циклодекстрина, сульфатированного β-циклодекстрина (S-β-CD), мальтозил-β-циклодекстрина, β-циклодекстринсульфобутилового эфира, разветвленного β-циклодекстрина, гидроксипропил-γ-циклодекстрина, случайно метилированного γ-циклодекстрина и триметил-γ-циклодекстрина и их смесей. Предпочтительно, чтобы усиливающие растворимость агенты включали β-циклодекстринсульфобутиловый эфир, гидроксипропил-β-циклодекстрин, сульфатированный β-циклодекстрин (S-β-CD) и мальтозил-β-циклодекстрин и их смеси. β-циклодекстринсульфобутиловый эфир является особенно предпочтительным усиливающим растворимость агентом. Усиливающий(ие) растворимость агент(ы) можно добавлять в количестве, равном приблизительно от 1 до приблизительно 20% по весу, предпочтительно, приблизительно от 1 до приблизительно 10% по весу, и более предпочтительно, приблизительно от 5 до приблизительно 10% по весу.
Можно применять любой подходящий офтальмологически приемлемый хелатирующий агент. Примеры подходящего офтальмологически приемлемого хелатирующего агента включают выбираемые из группы, состоящей из этилендиаминтетрауксусной кислоты и ее солей металлов, динатрия эдетата, тринатрия эдетата и тетранатрия эдетата и их смесей. Динатрия эдетат является особенно предпочтительным хелатирующим агентом. Хелатирующий агент(ы) можно добавлять в количестве, равном приблизительно от 0,001 приблизительно до 0,05% по весу, предпочтительно, приблизительно от 0,001 до приблизительно 0,02% по весу, и более предпочтительно, приблизительно от 0,002 до приблизительно 0,01% по весу, и наиболее предпочтительно, приблизительно от 0,002 до приблизительно 0,005% по весу.
Предпочтительно, чтобы водный инертный носитель включал консервант. Предпочтительные консерванты включают выбираемые из группы, состоящей из четвертичных солей аммония, таких как галоиды бензалкония (предпочтительно, хлорида бензалкония), хлоргексидина глюконата, бензетония хлорида, цетилпиридина хлорида, бензилбромида, нитрата фенилртути, ацетата фенилртути, неодеканоата фенилртути, мертиолата, метилпарабена, пропилпарабена, сорбиновой кислоты, сорбата калия, бензоата натрия, пропионата натрия, этил-п-гидроксибензоата, пропиламинопропилбигуанида и бутил-п-гидроксибензоата, сорбиновой кислоты и их смесей. Более предпочтительно, чтобы консервант был четвертичной солью аммония, такой как галиды бензалкония (предпочтительно, хлорида бензалкония), хлоргексидина глюконатом, бензетония хлоридом, цетилпиридина хлоридом, сорбатом калия, бензоатом натрия, этил-п-гидроксибензоатом, бутил-п-гидроксибензоатом или пропиламинопропилбигуанидом и их смесями. Пропиламинопропилбигуанид является особенно предпочтительным консервантом. Консервант(ы) можно применять в количестве, равном приблизительно от 0,00001 приблизительно до 0,0001% по весу, предпочтительно, приблизительно от 0,00001 приблизительно до 0,00008% по весу, более предпочтительно, приблизительно от 0,00002 приблизительно до 0,00005% по весу.
Водный инертный носитель может также включать тонический агент для регуляции тоничности (осмотического давления) для обеспечения офтальмологически совместимого состава. Тонический агент можно выбирать из группы, состоящей из гликоля (такого как пропиленгликоль, диэтиленгликоль, триэтиленгликоль), глицерола, декстрозы, глицерина, маннита, хлорида калия и хлорида натрия и их смеси. Предпочтительно выбирать тонический агент из группы, состоящей из глицерина, маннита, хлорида калия и хлорида натрия. Более предпочтительно применять маннит и/или хлорид натрия (и наиболее предпочтительно, их смесь). Тонический агент(ы) можно применять в количестве, равном приблизительно от 0,05 приблизительно до 8% по весу, предпочтительно, приблизительно от 0,1 приблизительно до 6% по весу, более предпочтительно, приблизительно от 0,1 приблизительно до 4% по весу, наиболее предпочтительно, приблизительно от 0,2 приблизительно до 4% по весу.
При применении смеси маннита и хлорида натрия в качестве тонических агентов предпочтительно, чтобы отношение веса маннита: хлорида натрия составляло приблизительно от 4:1 приблизительно до 15:1, более предпочтительно, приблизительно от 6:1 приблизительно до 14:1, или 8:1 приблизительно до 14:1 и, в частности, particularly приблизительно от 10:1 приблизительно до 12:1. При применении маннита в качестве единственного тонического агента предпочтительно применять его в концентрации, равной приблизительно от 4,5 приблизительно до 6,5% по весу и, более предпочтительно, в концентрации, равной приблизительно от 5,0 приблизительно до 5,5% по весу. При применении хлорида натрия в качестве единственного тонического агента его применяют в концентрации, равной приблизительно от 0,05 приблизительно до 8% по весу, предпочтительно приблизительно от 0,1 приблизительно до 6% по весу, более предпочтительно приблизительно от 0,1 приблизительно до 4% по весу и наиболее предпочтительно приблизительно от 0,2 приблизительно до 4% по весу.
Предпочтительно, чтобы водный инертный носитель также содержал повышающий вязкость/суспендирующий агент. Подходящие повышающие вязкость/суспендирующие агенты включают выбираемые из группы, состоящей из производных целлюлозы, таких как метилцеллюлоза, этилцеллюлоза, гидроксиэтилцеллюлоза, полиэтиленгликоли (такие как полиэтиленгликоль 300, полиэтиленгликоль 400), карбоксиметицеллюлоза, гидроксипропилметилцеллюлоза и сшитые полимеры акриловой кислоты (карбомеры), такие как полимеры акриловой кислоты, сшитые с полиалкениловыми эфирами или дивинилгликолем (карбополы - такие как карбопол 934, карбопол 934P, карбопол 971, карбопол 974 и карбопол 974P), и их смеси. В предпочтительных вариантах осуществления настоящего изобретения повышающий вязкость/суспендирующий агент является карбомером, более предпочтительно, карбополом 974P. Повышающий вязкость/суспендирующий агент(ы) могут присутствовать в количестве, равном приблизительно от 0,05 приблизительно до 2% по весу, предпочтительно приблизительно от 0,1 приблизительно до 1% по весу, более предпочтительно приблизительно от 0,2 приблизительно до 0,8% по весу и наиболее предпочтительно приблизительно от 0,3 приблизительно до 0,5% по весу.
Чтобы приблизить состав к офтальмологически приемлемому значению рН (обычно диапазон рН составляет приблизительно от 5,0 до приблизительно 9,0, более предпочтительно приблизительно от 5,5 приблизительно до 8,5, в частности, приблизительно от 6,0 приблизительно до 8,5, приблизительно от 7,0 приблизительно до 8,5, приблизительно от 7,2 приблизительно до 7,7, приблизительно от 7,1 приблизительно до 7,9 или приблизительно от 7,5 приблизительно до 8,0), в состав может входить изменяющий рН агент. Изменяющий рН агент является типичной неорганической кислотой или гидроксидом металла, выбираемым из группы гидроксида калия, гидроксида натрия и соляной кислоты и их смесей, и, предпочтительно, гидроксида натрия и/или соляной кислоты. Это кислотные и/или основные изменяющие рН агенты добавляют для изменения состава для достижения офтальмологически приемлемого диапазона рН. Следовательно, не обязательно существует необходимость применять и кислоту, и основание в зависимости от состава, и может быть достаточно добавлять или кислоту, или основание, чтобы подвести рН смеси до желаемого диапазона.
Водный инертный носитель может также содержать буферный агент для стабилизации рН. При применении буфер выбирают из группы, состоящей из фосфатного буфера (такого как дигидрофосфат натрия и гидрофосфат натрия), боратного буфера (такого как борная кислота или ее соли, включая динатрийтетраборат), цитратного буфера (такого как лимонная кислота или ее соли, включая цитрат натрия) и ε-аминокапроновой кислоты и их смесей. Буферный агент(ы) могут присутствовать в количестве, равном приблизительно от 0,05 приблизительно до 5% по весу, предпочтительно, приблизительно от 0,1 приблизительно до 5% по весу, более предпочтительно, приблизительно от 0,2 приблизительно до 5% по весу и наиболее предпочтительно, приблизительно от 0,5 приблизительно до 5% по весу.
Офтальмологический состав для местного введения в глаз может далее включать увлажняющий агент. В любом варианте осуществления настоящего изобретения увлажняющий агент предпочтительно является неионным увлажняющим агентом. Более предпочтительно, чтобы увлажняющий агент был водорастворимым или набухающим. Наиболее предпочтительно, чтобы увлажняющий агент был водорастворимым. “Водорастворимый” следует понимать так, как применяют в стандартных текстах, таких как “Handbook of Pharmaceutical Excipients” (Raymond C Rowe, Paul J Sheskey and Sian C Owen, Fifth Edition, Pharmaceutical Press and American Pharmacists Association 2006). Подходящие классы увлажняющих агентов включают выбираемые из группы, состоящие из полиоксипропилен-полиоксиэтиленовых блочных сополимеров (полоксамеров), полиэтоксилированных эфиров касторовых масел, полиоксиэтиленовых сорбитановых эфиров (полисорбатов), полимеров оксиэтилированного октилфенола (Tyloxapol), полиоксил-40-стеарата, эфиров жирных кислот и гликоля, эфиров жирных кислот и глицерина, жирных эфиров сахарозы и жирных эфиров полиоксиэтилена и их смесей.
Специальные примеры подходящих увлажняющих агентов включают выбираемые из группы, состоящей из: полиоксиэтилен-полиоксипропиленовых блочных сополимеров (полоксамеров), таких как: полиоксиэтилен (160) полиоксипропилен (30) гликоль [Pluronic F68], полиоксиэтилен (42) полиоксипропилен (67) гликоль [Pluronic P123], полиоксиэтилен (54) полиоксипропилен (39) гликоль [Pluronic P85], полиоксиэтилен (196) полиоксипропилен (67) гликоль [Poloxamer 407, Pluronic F127], полиоксиэтилен (20) полиоксипропилен (20) гликоль [Pluronic L44], полиоксиэтилированных сорбитановых эфиров (полисорбатов), таких как поли(оксиэтилен)сорбитана монопальмиат (полисорбат 40), поли(оксиэтилен)сорбитана моностеарат (полисорбат 60), поли(оксиэтилен)сорбитана тристеарат (полисорбат 65), поли(оксиэтилен)сорбитана моноолеат (полисорбат 80), поли(оксиэтилен)сорбитана монолаурат, поли(оксиэтилен)сорбитана триолеат, полиэтоксилированные эфиры касторовых масел, такие как полиоксиэтилен гидрогенированное касторовое масло 10, полиоксиэтилен гидрогенированное касторовое масло 40, полиоксиэтилен гидрогенированное касторовое масло 50 и полиоксиэтилен гидрогенированное касторовое масло 60, полиоксил-40-стеарат, жирные эфиры сахарозы и полиоксиэтиленовые жирные эфиры и их смеси.
Предпочтительно, чтобы увлажняющий агент выбирали из группы, состоящей из полиоксиэтилен-полиоксипропиленовых блочных сополимеров (полоксамеров), таких как: полиоксиэтилен (160) полиоксипропилен (30) гликоль [Pluronic F68], полиоксиэтилен (42) полиоксипропилен (67) гликоль [Pluronic P123], полиоксиэтилен (54) полиоксипропилен (39) гликоль [Pluronic P85], полиоксиэтилен (196) полиоксипропилен (67) гликоль [Poloxamer 407, Pluronic F127] и полиоксиэтилен (20) полиоксипропилен (20) гликоль [Pluronic L44], полиоксиэтиленовых сорбитановых эфиров (полисорбатов), таких как поли(оксиэтилен)сорбитана монопальмиат (полисорбат 40), поли(оксиэтилен)сорбитана моностеарат (полисорбат 60), поли(оксиэтилен)сорбитана тристеарат (полисорбат 65), поли(оксиэтилен)сорбитана моноолеат (полисорбат 80), поли(оксиэтилен)сорбитана монолаурат и поли(оксиэтилен)сорбитана триолеат и их смесей.
Более предпочтительно, чтобы увлажняющий агент был полиоксиэтилен-полиоксипропиленовым блочным сополимером (полоксамером). Примеры подходящих полоксамеров включают полиоксиэтилен (160) полиоксипропилен (30) гликоль [Pluronic F68], полиоксиэтилен (42) полиоксипропилен (67) гликоль [Pluronic P123], полиоксиэтилен (54) полиоксипропилен (39) гликоль [Pluronic P85], полиоксиэтилен (196) полиоксипропилен (67) гликоль [Poloxamer 407, Pluronic F127] и полиоксиэтилен (20) полиоксипропилен (20) гликоль [Pluronic L44] и их смеси.
Другие предпочтительные увлажняющие агенты выбирают из группы, состоящей из полиоксиэтилен (42) полиоксипропилен (67) гликоль [Pluronic P123], полиоксиэтилен (54) полиоксипропилен (39) гликоль [Pluronic P85], полиоксиэтилен (196) полиоксипропилен (67) гликоль [Poloxamer 407, Pluronic F127] и их смесей.
Особенно предпочтительным увлажняющим агентом является полиоксиэтилен (196) полиоксипропилен (67) гликоль [Poloxamer 407, Pluronic F127].
Особенно предпочтительные составы для местного введения в глаз по настоящему изобретению включают соединение настоящего изобретения, усиливающий растворимость агент, хелатирующий агент, консервант, тонический агент, повышающий вязкость/суспендирующий агент, буфер и изменяющий рН агент. Более предпочтительные составы содержат водный раствор β-циклодекстрина, боратную соль, борную кислоту, хлорид натрия, динатрия эдетат и пропиламинопропилбигуанид.
В одном аспекте офтальмологический состав настоящего изобретения, находится в форме раствора, такого как один из следующих:
Офтальмологический состав настоящего изобретения, также может быть в форме геля или полугеля или в обеих формах; в форме желе; суспензии; эмульсии; масла; мази; крема или спрея.
Офтальмологический гель, полугель, желе, суспензия, эмульсия, масло, мазь, крем или спрей могут содержать различные добавки, включенные в установленном порядке, такие как буферные агенты (например, фосфатные буферы, боратные буферы, цитратные буферы, тартратные буферы, ацетатные буферы, аминокислоты, ацетат натрия, цитрат натрия и подобные), тонические агенты (например, сахариды, такие как сорбит, глюкоза и маннит, многоатомные спирты, такие как глицерин, концентрированный глицерин, ПЭГ и пропиленгликоль, соли, такие как хлорид натрия), консерванты или антисептики (например, хлорид бензалкония, хлорид бензалкония, п-оксибензоаты, такие как метил-п-оксибензоат или этил-п-оксибензоат, бензиловый спирт, фенилэтиловый спирт, сорбиновая кислота или ее соли, тимеросал, хлорбутанол и подобные), усиливающие растворимость агенты (например, циклодекстрины и их производные, водорастворимые полимеры, такие как поливинилпирролидон, сурфактанты, такие как тилоксапол, полисорбаты), изменяющие рН вещества (например, соляная кислота, уксусная кислота, фосфорная кислота, гидроксид натрия, гидроксид калия, гидроксид аммония и подобные), загустители (например, гидроксиэтилцеллюлоза, гидроксипропилцеллюлоза, метилцеллюлоза, гидроксипропилметилцеллюлоза, карбоксиметилцеллюлоза и их соли), хелатирующие агенты (например, эдетат натрия, цитрат натрия, конденсированный фосфат натрия) и подобные. Каждая из этих добавок может быть в количестве или в концентрации, схожей с описанными выше для офтальмологического состава в форме раствора.
Более того, соединения изобретения можно составлять в форме для местного введения посредством включения в новые офтальмологические составы, включая, но не ограничиваясь микроэмульсиями, липосомами, ниосомами, гелями, гидрогелем, наночастицами и наносуспензией.
1. Микроэмульсии
Микроэмульсии являются дисперсией воды и масла, усиленной комбинацией сурфактанта и сосурфактанта таким образом, чтобы уменьшить поверхностное натяжение на границе раздела. Эти системы обычно характеризуются повышенной термодинамической стабильностью, малым размером капель (приблизительно 100 нм) и прозрачным внешним видом. Прозрачный внешний вид обусловлен высоким содержанием дисперсии внутренней фазы, размер которой варьирует в пределах 100-1000 ангстрем. Процессы образования микроэмульсий, подходящих для применения в офтальмологических составах, описаны в Vandamne TF. Prog Retinal Eye Res 2002; 21:15-34, включенной посредством ссылки.
2. Липосомы
Липосомы являются липидными везикулами, содержащими водяное ядро и активно изучаемыми в отношении доставки в глаз разных активных ингредиентов. В зависимости от природы выбранной липидной композиции липосомы могут обеспечить пролонгированное высвобождение лекарственного средства.
3. Ниосомы
Ниосомы являются двухслойными структурными везикулами, составленными из неионного сурфактанта и способными инкапсулировать как липофильные, так и гидрофильные соединения. Они могут высвобождать лекарственное средство вне зависимости от рН и усиливают окулярную биодоступность. Ниосомы являются микроскопическими ламеллярными структурами, которые образуются на смеси неионного сурфактанта алкилового или диалкилового полиглицеринового класса эфира и холестерина с последующим гидратированием в водной среде. Структурно ниосомы похожи на липосомы тем, что они сформированы бислоем. Однако бислой в случае ниосом состоит из неионных поверхностно-активных агентов, а не фосфолипидов в случае липосом. Ниосомы могут быть однослойными или многослойными в зависимости от способа их приготовления. Они способны захватывать гидрофильные и гидрофобные растворы. Они обладают высокой стабильностью и лишены многих недостатков, ассоциированных с липосомами, таким как высокая стоимость и разная степень чистоты фосфолипидов. Свойства ниосом и процесс их приготовления хорошо известны специалистам в данной области техники, см., например, Wagh VD et al., J Pharm Res 2010; 3(7):1558-1563; Kaur H et al., Int J Pharm Sci Rev Res 2012; 15(1):113-120, каждая из которых включена посредством ссылки.
4. Гели
Офтальмологические гели составлены из мукоадгезивных полимеров, которые обеспечивают локализованную доставку активного ингредиента в глаз. Такие полимеры обладают свойством, называемым биоадгезивностью, обозначающей прикрепление носителя лекарственного средства к определенной биологической ткани. Данные полимеры могут увеличивать время контактирования лекарственного средства с биологическими тканями и, таким образом, улучшать окулярную биодоступность. Выбор полимера имеет ключевое значение для кинетики высвобождения лекарственного средства из лекарственной формы. Некоторые биоадгезивные полимеры доступны с разной степенью мукоадгезивной активности. Некоторые примеры являются карбоксиметилцеллюлозой, карбополом, поликарбофилом и альгинатом натрия. Обзор применения форм составления средства в виде геля для доставки лекарственного средства в глаз представлен в Ali Yet al., Adv Drug Deliv Rev 2006; 58: 1258-1268, которая включена посредством ссылки.
5. Гидрогели
Гидрогели являются трехмерными, гидрофильными, полимерными сетями, способными захватывать большие количества воды или биологических жидкостей. Время удержания можно значительно увеличить, составляя средство в форме гидрогеля. Образование геля можно вызвать изменением температуры и рН. Полоксамеры, наиболее распространенные полимеры, содержат гидрофобную часть в центре, окруженную гидрофильной частью. При этом их широко применяют для увеличения времени удержания. Недавние достижения в применении гидрогелей для доставки лекарственных средств в глаз описаны в Gaudana R, Jwala J, Boddu SHS, Mitra AK. Pharm Res. 2009; 26(5):1197-1216, которая включена посредством ссылки.
6. Наночастицы
Наночастицы определяют как частицы с диаметром менее 1 мкм, включающие разные биодеградируемые или небиодеградируемые полимеры, липид, фосфолипиды или металлы. Их можно классифицировать как наносферы или нанокапсулы в зависимости от того, распределено ли лекарственное средство однородно или покрыто полимерным материалом. Захват и распределение наночастиц зависит от их размера. Недавно был составлен обзор применения наночастиц в доставке лекарственного средства в глаз Hing et al., Int. J. Ophthalmol 2013; 6:390-396, который включен посредством ссылки.
7. Наносуспензии
Наносуспензии определяют как субмикронные коллоидные системы, которые состоят из обладающих плохой водорастворимостью лекарственных средств, суспендированных в соответствующей среде для диспергирования, стабилизированных сурфактантами. Как правило, наносуспензии состоят из коллоидных носителей, таких как полимерные смолы, инертные в естественных условиях. Наносуспензии усиливают растворимость лекарственного средства, а, значит, и его биодоступность. В отличие от микроэмульсий наносуспензии не являются раздражающими. Заряд поверхности наночастиц усиливает их адгезию к роговице. Недавно был составлен обзор применения наносуспензий в доставке лекарственного средства в глаз Rabinow, Nature Rev Drug Disc 2004; 785-796, который включен посредством ссылки.
Соединения настоящего изобретения можно также вводить в форме состава, подходящего для местной доставки в глаз. Подробные описания состава, подходящего для местной доставки в глаз, приведены в J.D. Bartlett and S. D. Jaanus, “Clinical Ocular Pharmacology”, 2008, Elsevier Health Sciences, которая включена посредством ссылки.
Соединения изобретения можно также соединять с растворимыми полимерами в качестве направляемых носителей лекарственного средства. Такие полимеры могут включать Поливинилпирролидон, сополимер пирана, полигидроксипропилметакриламид-фенол, полигидроксиэтиласпертамид-фенол и полиэтиленоксид-полилизин, замещенный пальмитоиловыми остатками. Более того, соединения изобретения можно соединять с классом биодеградируемых полимеров, применимых для обеспечения контролируемого высвобождения лекарственного средства, например, с полилактидной кислотой, полигликолевой кислотой, сополимерами полилактидной и полигликолевой кислот, полиэпсилонкапролактоном, полигидроксимасляной кислотой, полиортоэфирами, полиацеталями, полидигидропиранами, полицианоакрилатами и сшитыми или амфипатичными блочными сополимерами гидрогелей.
Настоящее изобретение также представляет фармацевтическую композицию, содержащую соединение изобретения или его фармацевтически приемлемую соль или сольват и фармацевтически приемлемый носитель или эксципиент и, кроме того, активный ингредиент, выбираемый из группы, состоящей из а) антагониста интегрина α5β1, b) цитотоксического/антипролиферативного агента, c) ингибитора эпидермального, фибробластного или тромбоцитарного факторов роста, d) ингибитора VEGF, e) ингибитора Flk-1/KDR, Flt-1, Tck/Tie-2 или Tic-1 и f) ингибитора фосфоинозитид-3-киназы, и их смесей.
Настоящее изобретение далее представляет фармацевтическую композицию, содержащую соединение настоящего изобретения или его фармацевтически приемлемую соль или сольват и фармацевтически приемлемый носитель или эксцципиент и, кроме того, активный ингредиент, выбираемый из группы, состоящей из а) антагониста интегрина α5β1, b) цитотоксического/антипролиферативного агента, c) ингибитора эпидермального, фибробластного или тромбоцитарного факторов роста, d) ингибитора VEGF, e) ингибитора фосфоинозитид-3-киназы, и их смесей.
Неограничивающими примерами антагонистов интегрина α5β1 являются (S)-2-((R)-2-((S)-2-((S)-2-((S)-1-ацетилпирролидин-2-карбоксамидо)-3-(1H-имидазол-5-ил)пропанамидо)-3-гидроксипропанамидо)-3-меркаптопропанамидо)сукцинамид и JSM6427, описанные в Stragies, R. et al., J. Med. Chem. 2007, 50:3786-3794, включенные в данную заявку посредством ссылки.
Неограничивающими примерами цитотоксических/антипролиферативных агентов являются таксол, винкристин, винбластин и доксорубицин.
Неограничивающими примерами ингибиторов эпидермального, фибробластного или тромбоцитарного факторов роста являются пазопаниб и сунитиниб.
Неограничивающими примерами ингибиторов фактора роста сосудистого эндотелия (VEGF) являются бевацизумаб и ранибизумаб.
Неограничивающими примерами ингибиторов фосфоинозитид-3-киназы являются инделасиб и 2-морфодин-4-ил-8-фенилхроман-4-он.
Способы применения
Соединения изобретения, как правило, обладают субмикромолярной ингибирующей активностью в отношении интегринов αv, таких как αvβ3 и αvβ5. Ингибирование функции αvβ3 и αvβ5 интегринов препятствует пролиферации эндотелиальных клеток. Пролиферация эндотелиальных клеток может привести к нежелательной неоваскуляризации или ангиогенезу, в частности к хороидальной неоваскуляризации хориокапилляров через оболочку Бруха, приводя в конечном счете к просачиванию крови и белков под макулу. Кровотечение, просачивание и рубцевание из этих кровеносных сосудов впоследствии вызывает необратимое повреждение фоторецепторов и резкой потери зрения в отсутствие лечения.
Диабетическая ретинопатия, тесно связанное состояние, является результатом изменений микрососудов сетчатки. Вызванная гипергликемией гибель интрамуральных перицитов и утолщение базальной мембраны приводят к функциональной недостаточности стенок сосудов в сетчатке, которая нарушает гематоретинальный барьер и делает кровеносные сосуды сетчатки более проницаемыми. Через поврежденные кровеносные сосуды подтекает жидкость и липиды в макулу, часть сетчатки, которая обеспечивает подробное зрение, вызывая отек макулы. Впоследствии это может прогрессировать до развития состояния, называемого макулярным отеком.
Соответственно, ВМД, ДР и ДМО и макулярный отек после окклюзии центральной вены сетчатки (тромбоза) можно лечить или предотвращать посредством введения (например, местного введения) соединений или фармацевтических композиций настоящего изобретения.
Настоящее изобретение представляет способ лечения или предотвращения заболевания или состояния у субъекта, включающий введение нуждающемуся в нем субъекту терапевтически эффективного количества соединения изобретения или его фармацевтически приемлемой соли или сольвата или терапевтически эффективного количества фармацевтической композиции изобретения. В одном аспекте изобретение представляет лечение заболевания или состояния. В одном аспекте изобретение представляет предотвращение заболевания или состояния.
В одном аспекте соединение или фармацевтическую композицию изобретения вводят местно. В другом аспекте соединение или фармацевтическую композицию изобретения вводят в виде офтальмологического раствора. В другом аспекте соединение или фармацевтическую композицию изобретения вводят в виде офтальмологической эмульсии, суспензии, геля или полугеля. В другом аспекте соединение или фармацевтическую композицию изобретения вводят в виде офтальмологического желе, масла, мази, крема или спрея.
Соединения или фармацевтические композиции изобретения вводят в дозировках, эффективных для ингибирования функции αvβ3 и/или αvβ5 интегринов, а, значит, и для лечения или предотвращения заболевания или состояния, опосредованного αvβ3 и/или αvβ5 интегрином.
Настоящее изобретение представляет способ лечения или предотвращения заболевания или состояния, опосредованного αv интегрином, у субъекта, включающий введение нуждающегося в нем субъекту терапевтически эффективного количества соединения изобретения или его фармацевтически приемлемой соли или сольвата или терапевтически эффективного количества фармацевтической композиции изобретения. В одном аспекте заболевание или состояние является заболеванием или состоянием, в котором задействован ангиогенез. В другом аспекте заболевание или состояние является заболеванием или состоянием, в котором задействован глазной ангиогенез.
Настоящее изобретение также представляет способ лечения или предотвращения опосредованного αvβ3 и/или αvβ5 интегрином заболевания или состояния у субъекта, включающий введение нуждающегося в нем субъекту терапевтически эффективного количества соединения изобретения или его фармацевтически приемлемой соли или сольвата или терапевтически эффективного количества фармацевтической композиции изобретения. В одном аспекте заболевание или состояние является заболеванием или состоянием, в котором задействован глазной ангиогенез. В одном аспекте заболевание или состояние является макулярной дегенерацией. В одном аспекте заболевание или состояние является возрастной макулярной дегенерацией (ВМД). В одном аспекте заболевание или состояние является диабетической ретинопатией (ДР). В одном аспекте заболевание или состояние является диабетическим макулярным отеком (ДМО). В одном аспекте заболевание или состояние является макулярным отеком после окклюзии вены сетчатки.
Настоящее изобретение далее представляет способ лечения или предотвращения ВМД, ДР, ДМО или макулярным отеком после окклюзии вены сетчатки, включающий введение нуждающегося в нем субъекту терапевтически эффективного количества соединения изобретения или его фармацевтически приемлемой соли или сольвата или терапевтически эффективного количества фармацевтической композиции изобретения. В одном аспекте изобретение представляет лечение ВМД, ДР, ДМО или макулярного отека после окклюзии вены сетчатки. В одном аспекте изобретение представляет предотвращение ВМД, ДР, ДМО или макулярного отека после окклюзии вены сетчатки.
Настоящее изобретение далее представляет способ лечения или предотвращения заболевания или состояния субъекта, включающий введение нуждающегося в нем субъекту терапевтически эффективного количества соединения изобретения или его фармацевтически приемлемой соли или сольвата или терапевтически эффективного количества фармацевтической композиции изобретения в сочетании со второй терапией для лечения или предотвращения заболевания или состояния. В одном аспекте заболевание или состояние опосредовано αv интегрином. В другом аспекте заболевание или состояние опосредовано αvβ3 и/или αvβ5 интегрином. В одном аспекте заболевание или состояние является заболеванием или состоянием, в котором задействован ангиогенез. В другом аспекте заболевание или состояние является заболеванием или состоянием, в котором задействован глазной ангиогенез. В одном аспекте вторая терапия включает введение одного или нескольких из следующего: а) антагонист интегрина α5β1, b) цитотоксический/антипролиферативный агент, c) ингибитор эпидермального, фибробластного или тромбоцитарного фактора роста, d) ингибитор VEGF, e) ингибитор Flk-1/KDR, Flt-1, Tck/Tie-2 или Tic-1 и f) ингибитор фосфоинозитид-3-киназы и их смесь. В другом аспекте вторая терапия включает введение одного или нескольких из следующего: а) антагонист интегрина α5β1, b) цитотоксический/антипролиферативный агент, c) ингибитор эпидермального, фибробластного или тромбоцитарного фактора роста, d) ингибитор VEGF и e) ингибитор фосфоинозитид-3-киназы и их смесь. В другом аспекте вторая терапия включает введение ингибитора VEGF. В другом аспекте ингибитор VEGF является бевацизумабом или ранибизумабом.
Вторую терапию можно вводить посредством любого способа введения, включая пероральное введение в таких формах, как таблетки, капсулы (каждая из которых включает составы для пролонгированного высвобождения или отложенного высвобождения), пилюли, порошки, гранулы, эликсиры, настойки, суспензии, сиропы, эмульсии, внутривенное введение (разовая доза или инфузия), внутрибрюшинное введение, местное введение (например, глазных капель), подкожное введение, внутримышечное введение, трансдермальное (например, повязки) введение и интравитреальное введение. В одном аспекте вторую терапию вводят посредством интравитреальной инъекции.
Введение второй терапии в комбинации, как правило, проводят на протяжении определенного периода времени (обычно минут, часов, дней или недель в зависимости от выбранной комбинации). Предполагается, что «комбинированная терапия» может включать, но, как правило, не включает введение двух или более из этих терапевтических агентов как части отдельных режимов монотерапии, которые случайно и произвольно приводят к комбинациям настоящего изобретения. Предполагается, что «комбинированная терапия» может включать введение данных терапевтических агентов последовательным способом, в котором каждый терапевтический агент вводят в разное время, а также введение данных терапевтических агентов или по меньшей мере двух терапевтических агентов практически одновременно.
В соответствии со способом по изобретению, отдельные компоненты комбинации можно вводить по отдельности в разные времена на протяжении курса терапии или параллельно в виде раздельных или единых форм комбинации. Таким образом, следует понимать настоящее изобретение как включающее все такие режимы одновременного или поочередного лечения, и соответствующим образом следует интерпретировать термин «введение». Следует понимать, что объем комбинаций соединений изобретения с другими агентами, применимыми для лечения опосредованных αv интегрином состояний, включает в принципе любую комбинацию с фармацевтической композицией для лечения макулярной дегенерации, ДР, ДМО или макулярного отека после окклюзии вены сетчатки. Когда способ по изобретению представляет комбинированное лечение составом настоящего изобретения, вводимым местно в глаза, и анти-VEGF белком или аптамером, процедуры, дозировка и количество анти-VEGF белка или аптамера являются такими, как описанные в Информации для пациента для данных агентов.
Режим дозирования с применением соединений изобретения выбирают в соответствии с рядом факторов, включающих тип, виды, возраст, вес, пол и медицинское состояние пациента; степень тяжести состояния, подлежащего лечению; и конкретное применяемое соединение или его соль. Обычный терапевт, ветеринар или клиницист может легко определить и прописать эффективное количество лекарственного средства, которое требуется для предотвращения, противодействия или остановки прогресса состояния.
В способах по изобретению соединения, подробно описанные в данной заявке, могут образовывать активный ингредиент, и, как правило, их вводят в виде смеси с подходящими фармацевтическими растворителями, эксципиентами или носителями (вместе называемыми в данной заявке как «носитель»), выбранными соответствующим образом для местного введения в глаз и соответствующие принятым фармацевтическим практикам.
В целях изобретения будут применять следующие определения (если иное прямо не указано):
«Соединение изобретения», «соединения изобретения», «соединение настоящего изобретения» или «соединения настоящего изобретения» относятся к соединению (соединениям), раскрытым в данной заявке, например, соединение (соединения) изобретения включает соединение (соединения) по любой из формул, описанных в данной заявке, включая формулу I и II и/или соединение (соединения), точно раскрытые в данной заявке. При любом применении термина в контексте изобретения следует понимать, что ссылка сделана на свободное основание и соответствующие его фармацевтически приемлемые соли или сольваты, принимая во внимание то, что это допустимо и/или соответствует обстоятельствам.
«Фармацевтический» или «фармацевтически приемлемый», как применяют в данной заявке в виде прилагательного, обозначает в основном нетоксичный и в основном не вредный для реципиента.
Под «фармацевтической композицией» далее понимают, что носитель, растворитель, сольвент, эксципиент и соль должны быть совместимыми с активным ингредиентом состава (например, с соединением настоящего изобретения). Специалисты в данной области техники понимают, что термины «фармацевтический состав» и «фармацевтическая композиция» являются в основном взаимозаменяемыми и таким образом применяются в целях данной заявки.
«Раствор» относится к прозрачной гомогенной жидкой лекарственной форме, которая содержит одно или несколько химических веществ, растворенных в растворителе или в смеси взаиморастворимых растворителей. Из-за того, что молекулы вещества терапевтического агента в растворе распределены равномерно, применение растворов в качестве готовой лекарственной формы, как правило, обеспечивает единообразие доз при введении и высокую точность при разведении раствора или другом смешивании. «Раствор», как раскрыто в данной заявке, предусматривает любые изменения на основании текущего состояния данной области техники или изменения, вносимые специалистами в данной области техники.
«Суспензия» относится к жидкой лекарственной форме, которая содержит твердые частицы, диспергированные в жидком носителе. «Суспензия», как раскрыто в данной заявке, предусматривает любые изменения на основании текущего состояния данной области техники или изменения, вносимые специалистами в данной области техники.
«Эксципиент» применяют в данной заявке для включения любого другого соединения, которое не является терапевтически или биологически активным соединением и может содержаться в или в сочетании с одним или несколькими соединениями настоящего изобретения. Сам по себе эксципиент должен быть фармацевтически или биологически приемлемым или подходящим (например, эксципиент должен быть в целом нетоксичным для субъекта). «Эксципиент» включает одно такое соединение и также может включать множество эксципиентов. В целях настоящего раскрытия термины «эксципиент» и «носитель» применяют как взаимозаменяемые на протяжении всего описания настоящей заявки.
«Терапевтически эффективное количество» относится к такому количеству лекарственного средства, которое вызовет биологический или медицинский ответ в ткани, системе, животном или человеке, который ищет исследователь или клиницист.
«Лечить», «лечение» или «обработка» относится к уменьшению симптомов, маркеров и/или любых негативных эффектов заболевания или состояния любой приемлемой степени у субъекта, который в настоящее время имеет заболевание или состояние. В некоторых вариантах осуществления изобретения лечение можно вводить субъекту, который проявляет лишь ранние признаки заболевания или состояния с целью снижения риска развития заболевания или состояния. В некоторых вариантах осуществления изобретения «лечить», «лечение» или «обработка» относится к уменьшению интенсивности одного или нескольких симптомов заболевания или состояния. Например, уменьшение интенсивности одного или нескольких симптомов заболевания или состояния включает снижение степени тяжести, частоты и/или продолжительности одного или нескольких симптомов заболевания или состояния.
«Предотвращать», «предотвращение» или «предотвращая» относится к любому способу частичного или полного предотвращения или отсрочки начала одного или нескольких симптомов или свойств заболевания или состояния. Предотвращение можно провести у субъекта, который не проявляет каких-либо признаков заболевания или состояния.
«Субъект» означает человека или животное (в случае животного, как правило, млекопитающего). В одном аспекте субъект является человеком.
Термин «симптом» определяют как признак заболевания, болезни, повреждения или отклонений в организме. Симптомы чувствует или отмечает индивид, имеющий симптом, но они легко могут остаться незамеченными для других. Других определяют как не являющихся медицинскими работниками.
«Антагонист αv интегрина» относится к соединению, которое связывается с и ингибирует или нарушает функцию либо αvβ3, либо αvβ5, или к соединению, которое связывается с и ингибирует или нарушает функцию как αvβ3, так и αvβ5 (т.е. двойной αvβ3/αvβ5 антагонист). Соединения связываются с рецепторами как антагонисты, блокируя или препятствуя связыванию природного агониста, такого как витронектин, не вызывая сами по себе биологический ответ.
«Резорбция кости» относится к процессу, с помощью которого остеокласты разрушают кость.
«Алкил» относится к алкилу с прямой цепью или к разветвленному алкилу с определенным числом атомов углерода (например, C1-C4 алкил) или с любым числом атомов углерода в данном интервале (метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил и т.п.).
«Алкоксигруппа» относится к алкоксидам с прямой цепью или к разветвленным алкоксидам с определенным числом атомов углерода (например, C1-C6 алкоксигруппа) или с любым числом атомов углерода в данном интервале (метоксигруппа, этоксигруппа, пропоксигруппа, изопропоксигруппа, бутоксигруппа, изобутоксигруппа, трет-бутоксигруппа и т.п.).
«Карбоциклическое кольцо» относится к насыщенному циклоалкилу с определенным числом атомов углерода (т.е. C3 или C4), такому как циклопропил и циклобутил.
«Гетероциклическое кольцо» относится к насыщенному гетероциклическому кольцу с определенным числом атомов углерода (т.е. C3 или C4), далее включающему один дополнительный гетероатом, выбираемый из N, O и S.
Термин «около» относится к интервалам значений, которые могут быть на 15%, 10%, 8%, 5%, 3%, 2%, 1% или 0,5% больше или меньше определенного значения. Например, «около 10%» может составлять от 8,5% или 11,5%. В одном варианте осуществления изобретения термин «около» относится к интервалу значений, которые на 5% больше или меньше определенного значения. В другом варианте осуществления изобретения термин «около» относится к интервалу значений, которые на 2% больше или меньше определенного значения. В другом варианте осуществления изобретения термин «около» относится к интервалу значений, которые на 1% больше или меньше определенного значения.
ПРИМЕРЫ
Пример 1. Синтез (S)-3-(6-(дифторметокси)-пиридин-3-ил)-3-(2-оксо-3-(3-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)пропил)имидазолидин-1-ил)пропионовой кислоты (Соединение A1)
Соединение A1 образуют, применяя схему конвергентного синтеза, как показанная на схеме 1: фрагмент 6b реагирует с фрагментом 9 для образования соединения 10, которое далее реагирует в три стадии для образования Соединения A1.
Синтез фрагмента 6b
трет-бутил 2-оксопирролидин-1-карбоксилат (2a): К перемешанному раствору соединения 1a (10,0 г, 117 ммоль, 1,0 эквив.) в ДХМ (дихлорметане) добавляли (Boc)2O (25,5 г, 117 ммоль, 1,00 эквив.) и ДМАП (0,022 г, 0,180 ммоль, 0,001 эквив.) при комнатной температуре и перемешивали в течение 12 ч. После расхода исходного материала (по данным наблюдений с помощью тонкослойной хроматографии) летучие вещества удаляли под сниженным давлением для образования соединения 2a (19,6 г, 90,3%) в виде коричневого сиропа.
ТСХ: 50% EtOAc/гексан (Rf: 0,40)
1H ЯМР(400 МГц, CDCl3): β 3,74 (т, J=6,8 Гц, 2H), 2,50 (т, J=8,0 Гц, 2H), 2,01 (т, J=7,6 Гц, 2H), 1,52(с, 9H)
трет-бутил (5-(диметоксифосфорил)-4-оксопентил)карбамат (3a): К перемешанному раствору iPr2NH (2,99 мл, 21,8 ммоль, 1,35 эквив.) в ТГФ, охлажденному до -10°C, медленно добавляли гексиллитий (8,79 мл, 20,0 ммоль, 1,24 эквив.). Реакционную смесь охлаждали до -60°C, добавляли диметилметилфосфонат (2,20 мл, 20,9 ммоль, 1,29 эквив.) и перемешивали в течение 1 ч. Затем температуру повышали до -40°C и соединение 2a (3,0 г, 16,2 ммоль, 1,0 эквив.) вводили в реакционную смесь и продолжали перемешивать в течение еще 1 ч. После расхода исходного материала раствор 2н H2SO4 (20 мл) медленно добавляли к реакции и перемешивали при 0°C в течение 15 минут. Водный слой экстрагировали EtOAc (2×25 мл). Объединенные органические экстракты промывали водой (25 мл), солевым раствором (25 мл), высушивали над Na2SO4, фильтровали и выпаривали под сниженным давлением для образования соединения 3a в виде коричневой жидкости (5,0 г, сырой).
ТСХ: 80% EtOAc/гексан (Rf: 0,30)
1H ЯМР (400 МГц, CDCl3): δ 4,85 (уш.с, 1H, Exc), 3,80-3,72 (м, 8H), 3,13-3,07 (м, 2H), 2,67 (т, J=6,8 Гц, 2H), 1,87-1,76 (м, 2H), 1,43 (с, 9H)
ЖХ-МС (m/z) = 308,3 [M+H]+ при комнатной температуре 2,67 (99,1% чистота)
трет-бутил (3-(1,8-нафтиридин-2-ил)пропил)карбамат (5a): К перемешанному раствору соединения 4a (0,500 г, 4,09 ммоль, 1,0 эквив.) и соединения 3a (1,26 г, сырой, 1,0 эквив.) в MeOH (9,17 мл) добавляли 50% раствор NaOH (0,314 мл) и перемешивали реакционную смесь при 50°C в течение 10 ч. После расхода исходного материала (по данным наблюдений с помощью тонкослойной хроматографии) летучие вещества удаляли, сырой остаток растворяли в EtOAc (15 мл) и промывали органический слой водой (2×15 мл). Отделенный органический слой высушивали над Na2SO4, фильтровали и концентрировали под сниженным давлением для образования коричневого сиропа, который очищали с помощью колоночной хроматографии на нейтральной окиси алюминия (80% EtOAc: гексан) для образования соединения 5a (0,980 г, 83,3%) в виде беловатого твердого вещества.
ТСХ: EtOAc
1H ЯМР (500 МГц, CDCl3): δ 9,09 (с, 1H), 8,17-8,15 (м, 1H), 8,10 (д, J=8,0 Гц, 1H), 7,45 (т, J=8,0 Гц, 1H), 7,41 (т, J=15,0, 1H), 4,76 (уш.с, 1H, Exc), 3,25-3,21 (м, 2H), 3,09 (т, J=10,0 Гц, 2H), 2,14-2,08 (м, 2H), 1,42 (с, 9H)
ЖХ-МС (m/z) = 288 [M-H]- при комнатной температуре 2,86 (94,7%)
трет-бутил (3-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)пропил)карбамат (S-024): К перемешанному раствору соединения 5a (0,25 г, 0,87 ммоль, 1,00 эквив.) в метаноле (5 мл) добавляли Rh/C (каталитический, 5% по весу) в атмосфере N2 и перемешивали при комнатной температуре в течение 8 ч в атмосфере водорода (под давлением в баллоне). После расхода исходного материала реакционную смесь фильтровали через прокладку CELITE®, промывали MeOH (5 мл). Фильтрат выпаривали под сниженным давлением для образования соединения S-024 (0,18 г, 71,1%) в виде белого твердого вещества.
ТСХ: EtOAc
1H ЯМР (400 МГц, CDCl3): δ 7,05 (д, J=7,6 Гц, 1H), 6,34 (д, J=7,2 Гц, 1H), 5,44 (с, 1H), 4,78 (уш.с, 1H, Exc), 3,41-3,38 (м, 2H), 3,16 (д, J=6,0 Гц, 2H), 2,68(т, J=6,0 Гц, 2H), 2,59 (т, J=7,6 Гц, 2H), 1,93-1,81(м, 4H), 1,44 (с, 9H)
ЖХ-МС (m/z) = 292,3 [M+H]+ при комнатной температуре 3,41 (97,9% чистота)
3-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)пропан-1-амин (6b): К перемешанному раствору S-024 (0,25 г, 0,85 ммоль, 1,00 эквив.) в ДХМ (5 мл), охлажденному до 0°C, добавляли трифторуксусную кислоту (0,13 мл, 1,69 ммоль, 2,00 эквив.). Реакцию нагревали до комнатной температуры и перемешивали в течение 4 ч. После расхода исходного материала (по данным наблюдений с помощью тонкослойной хроматографии) реакционную смесь концентрировали под сниженным давлением для образования сырого соединения 6b (0,30 г) в виде густого сиропа, который применяли без очистки на следующей стадии.
Синтез Фрагмента 9 и завершение синтеза
5-бром-2-(дифторметокси)пиридин (2): К перемешанному раствору соединения 1 (4,50 г, 25,8 ммоль, 1,0 эквив.) в безводном MeCN (80 мл), добавляли 2-хлор-2,2-дифторацетат натрия (4,89 г, 31,0 ммоль, 1,20 эквив.) при комнатной температуре и перемешивали при 70°C в течение 48 ч. После расхода исходного материала (по данным наблюдений с помощью тонкослойной хроматографии) реакционную смесь доводили до комнатной температуры и разводили раствором NH4Cl (30 мл). Водный слой экстрагировали EtOAc (2×40 мл). Объединенные органические слои промывали солевым раствором (2×50 мл), высушивали над безводным Na2SO4, фильтровали и концентрировали под сниженным давлением для образования сырого соединения, которое очищали с помощью колоночной хроматографии (20% EtOAc/гексан) для образования соединения 2 (3,2 г, 57%) в виде бледно-желтого сиропа.
ТСХ: 5% EtOAc/гексан (Rf: 0,5) z
1H ЯМР (400 МГц, CDCl3): δ 8,25 (д, J=2,8 Гц, 1H), 7,82 (дд, J=2,4, 6,4 Гц, 1H), 7,40 (т, J=72,8 Гц, 1H), 6,83 (д, J=8,8 Гц, 1H)
ЖХ-МС (m/z) = 224,7 [M+H]+ при комнатной температуре 4,22 (98,2% чистота)
(E)-трет-бутил 3-(6-(дифторметокси)пиридин-3-ил)акрилат (3): К перемешанному раствору трет-бутилакрилата (9,99 г, 78,1 ммоль, 3,50 эквив.) добавляли Et3N (8,5 мл, 60,2 ммоль, 2,70 эквив.), N-метилпирролидин (20 мл), тритолилфосфин (1,17 г, 3,52 ммоль, 0,16 эквив.), а затем Pd(OAc)2 (0,50 г, 2,22 ммоль, 0,09 эквив.). Температуру постепенно повышали до 90°C и по каплям добавляли соединение 2 (5,00 г, 22,3 ммоль, 1,0 эквив.) в н-метил-2-пирролидоне (10 мл) и перемешивали при 90°C в течение 12 ч. После расхода исходного материала (по данным наблюдений с помощью тонкослойной хроматографии) реакционную смесь фильтровали через прокладку CELITE®, промывали EtOAc (50 мл). Объединенный фильтрат промывали холодной водой (2×50 мл), а затем NaOCl (50 мл), солевым раствором (50 мл). Органический слой высушивали над безводным Na2SO4, фильтровали и концентрировали под сниженным давлением для образования сырого остатка, который очищали с помощью колоночной хроматографии (3% EtOAc/гексан) для образования соединения 3 (4,0 г, 66%) в виде желтого твердого вещества.
ТСХ: 5% EtOAc/гексан (Rf: 0,5)
1H ЯМР (400 МГц, CDCl3): δ 8,28 (д, J=2,4 Гц, 1H), 7,88 (дд, J=2,0, 6,4 Гц, 1H), 7,56 (д, J=16,0 Гц, 1H), 7,55 (т, J=45,6 Гц, 1H), 6,91 (д, J=8,4 Гц, 1H), 6,34 (д, J=16,0 Гц, 1H), 1,53 (с, 9H)
LC-MS: m/z = 272 [M+H]+ при комнатной температуре 4,16 (99,5% чистота)
(S)-трет-бутил 3-(бензил((R)-1-фенилэтил)амино)-3-(6-метоксипиридин-3-ил)пропионат (5): К перемешанному раствору соединения 4 (0,39 г, 1,85 ммоль, 2,0 эквив.) в ТГФ (5 мл), охлажденном до -30°C, добавляли n-BuLi (0,66 мл, 1,65 ммоль, 1,79 эквив.) и затем охлаждали до -78°C. Соединение 3 (0,25 г, 0,92 ммоль, 1,0 эквив.), растворенное в ТГФ (3 мл), добавляли к реакционной смеси, перемешивали в течение 30 мин и останавливали реакцию насыщенным хлоридом аммония. Реакционную смесь экстрагировали EtOAc (2×20 мл). Объединенные органические экстракты промывали 10% AcOH, солевым раствором, который высушивали над безводным Na2SO4, фильтровали и концентрировали под сниженным давлением для получения сырого соединения (смеси 3 и 5, 0,17 г) в виде густого сиропа, который напрямую применяли на следующей стадии.
ТСХ: 5% EtOAc/гексан (Rf: 0,5)
ЖХ-МС (m/z) = 483 [M+H]+ при комнатной температуре 4,66 (75,1% чистота)
Синтез (S)-трет-бутил 3-амино-3-(6-(дифторметокси)пиридин-3-ил)пропионата (S-029): К перемешанному раствору соединения 5 (0,80 г, сырая смесь) в EtOAc (5 мл) и AcOH (0,5 мл) добавляли 20% Pd(OH)2 (50 мг) в атмосфере N2. Реакционную смесь перемешивали в атмосфере H2 (40 фунт/кв.дюйм) при комнатной температуре в течение 2 ч. После расхода исходного материала (по данным наблюдений с помощью тонкослойной хроматографии) реакционную смесь фильтровали через прокладку CELITE®. Фильтрат концентрировали под сниженным давлением для образования сырого соединения, которое очищали с помощью колоночной хроматографии (2% MeOH/ДХМ) для образования S-029 (0,3 г, 63%) в виде желтого сиропа.
ТСХ: 5% MeOH/ДХМ (Rf: 0,3)
1H ЯМР (400 МГц, CDCl3): δ 8,17 (д, J=2,8 Гц, 1H), 7,78 (дд, J=2,4, 6,4 Гц, 1H), 7,44 (т, 73,2 Гц, 1H), 6,88 (д, J=8,4 Гц, 1H), 4,43-4,40 (м, 1H), 2,65-2,56 (м, 2H), 1,42 (с, 9H)
ЖХ-МС (m/z)= 274 [M+H]+ при комнатной температуре 2,76 (99,8% чистота)
(S,E)-трет-бутил 3-(6-(трет-бутокси)пиридин-3-ил)-3-((2,2-диметоксиэтилиден)амино)пропионат (7): К перемешанному раствору диметоксиацетальдегида (0,44 мл, 2,50 ммоль, 1,20 эквив., 60% в воде) в ДХМ (10 мл), охлажденном до 0°C, добавляли безводный MgSO4 (10 г), а затем S-029 (600 мг, 2,08 ммоль, 1,0 эквив.) в ДХМ (5 мл). Реакцию продолжали проводить при комнатной температуре в течение 2 ч и фильтровали через прокладку CELITE®, фильтрат концентрировали под сниженным давлением для образования соединения 7 (650 мг, сырое) в виде желтой жидкости, которую применяли на следующей стадии без какой-либо очистки.
ТСХ: 5% MeOH/ДХМ (Rf: 0,5)
(S)-трет-бутил 3-(6-(дифторметокси)пиридин-3-ил)-3-((2,2-диметоксиэтитл)амино)пропионат (8): К перемешанному раствору соединения 7 (0,65 г, сырое, 1,0 эквив.) в MeOH (7 мл), охлажденному до 0°C, добавляли NaBH(CN)3 (0,13 г, 2,09 ммоль, 1,20 эквив.) и перешивали реакционную смесь при комнатной температуре в течение 2 ч. После расхода исходного материала (по данным наблюдений с помощью тонкослойной хроматографии) MeOH удаляли под сниженным давлением для получения сырого остатка, который разводили водой (10 мл) и экстрагировали EtOAc (2×10 мл). Объединенные органические экстракты высушивали над безводным Na2SO4, фильтровали и концентрировали под сниженным давлением для образования сырого материала, который очищали с помощью колоночной хроматографии (2% MeOH/ДХМ) для образования соединения 8 (0,52 г, 79%) в виде густого сиропа.
ТСХ: 5% MeOH/ДХМ (Rf: 0,7)
1H ЯМР (400 МГц, CDCl3): δ 8,13 (д, J=2,0 Гц, 1H), 7,75 (дд, J=2,4, 6,0 Гц, 1H), 7,44 (т, J=73,2 Гц, 1H), 6,87 (д, J=8,4 Гц, 1H), 4,43-4,37 (м, 2H), 4,06-4,02 (м, 1H), 3,60-3,54 (м, 2H), 3,35 (с, 3H) 3,31 (с, 3H), 2,66-2,57 (м, 2H), 1,39 (с, 9H)
ЖХ-МС (m/z) = 377 [M+H]+ при комнатной температуре 2,96 (92,3% чистота)
(S)-трет-бутил 3-(6-(дифторметокси)пиридин-3-ил)-3-(1-(2,2-диметоксиэтил)-3-(3-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)пропил)уреидо)пропионат (10): К перемешанному раствору соединения 8 (375 мг, 0,99 ммоль, 1,0 эквив.) в безводном ДХМ (5 мл), охлажденному до 0°C, добавляли трифосген (1,50 мл, 2,99 ммоль, 3,00 эквив., 20% в PhMe), а затем Et3N (0,30 мл, 2,09 ммоль, 2,10 эквив.). Реакционную смесь медленно доводили до комнатной температуры и перемешивали в течение 2 ч. После расхода исходного материала летучие вещества выпаривали для получения сырого соединения 9, которое напрямую без очистки применяли на следующей стадии. Раствор соединения 9 в дихлорэтане (2 мл) добавляли к раствору соединения 6b (400 мг, 1,32 ммоль, 1,32 эквив.) в ДХМ (5 мл), Et3N (0,55 мл, 3,98 ммоль, 4,00 эквив.) при 0°C и перемешивали при комнатной температуре в течение 4 ч. После расхода исходного материала (по данным наблюдений с помощью тонкослойной хроматографии) реакционную смесь концентрировали под сниженным давлением для образования сырого остатка, который очищали с помощью колоночной хроматографии (2% MeOH/ДХМ) для образования соединения 10 (0,40 г, 67%) в виде густого сиропа.
ТСХ: 5% MeOH/ДХМ (Rf: 0,2)
1H ЯМР (400 МГц, CDCl3): δ 8,13 (д, J=2,8 Гц, 1H), 7,79 (дд, J=2,4, 6,4 Гц, 1H), 7,62 (тт, J=72,8 Гц, 1H), 7,12 (д, J=6,4 Гц, 1H), 6,86 (д, J=8,4 Гц, 1H), 6,36 (д, J=3,6 Гц, 1H), 6,22 (т, J=4,8 Гц, 1H), 5,75 (т, J=7,6 Гц, 1H), 4,26 (т, J=5,2 Гц, 1H), 3,45-3,38 (м, 8H), 3,27-3,13 (м, 3H), 2,99-2,93 (м, 2H), 2,71-2,59 (м, 5H), 1,93-1,83 (м, 5H), 1,39 (с, 9H)
ЖХ-МС (m/z) = 594 [M+H]+ при комнатной температуре 3,42 (88,1% чистота)
(S)-трет-бутил 3-(6-(дифторметокси)пиридин-3-ил)-3-(2-оксо-3-(3-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)пропил)-2,3-дигидро-1H-имидазол-1-ил)пропионат (11): К перемешанному раствору соединения 10 (0,20 г, 0,34 ммоль, 1,0 эквив.) в ТГФ (4 мл) при -10°C добавляли 1 M серную кислоту (0,8 мл). Реакционную смесь медленно нагревали до комнатной температуры и перемешивали в течение 10 ч. После расхода исходного материала (по данным наблюдений с помощью жидкостной хроматографии с масс-спектрометрией) удаляли тетрагидрофуран и нейтрализовали сырой остаток гидроксидом натрия (50% по весу) до pH~7. Водный слой экстрагировали 5% MeOH/ДХМ (3×20 мл) и высушивали объединенные органические экстракты над безводным Na2SO4, фильтровали и концентрировали под сниженным давлением для образования соединения 11 (0,22 г, сырое) в виде сиропа.
ТСХ: 10% MeOH/ДХМ (Rf: 0,5)
ЖХ-МС (m/z) = 530 [M+H]+ при комнатной температуре 4,06 (72,8% чистота)
(S)-трет-бутил 3-(6-(дифторметокси)пиридин-3-ил)-3-(2-оксо-3-(3-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)пропил)имидазолидин-1-ил)пропионат (12): К перемешанному раствору соединения 11 (0,45 г, сырой, 1,0 эквив.) в EtOH (8 мл) добавляли 20% Pd/C (200 мг) в атмосфере N2. Реакционную смесь перемешивали в атмосфере H2 (40 фунт/кв.дюйм) при комнатной температуре в течение 36 ч. После расхода исходного материала реакционную смесь фильтровали через прокладку CELITE® и концентрировали фильтрат под сниженным давлением для образования сырого соединения 12, который очищали с помощью хиральной препаративной ВЭЖХ для образования соединения 12 (45,0 мг, сырое) в виде беловатого твердого вещества.
ТСХ: 10% MeOH/ДХМ (Rf: 0,5)
ЖХ-МС (m/z) = 532,6 [M+H]+ при комнатной температуре 3,99 (80,1% чистота)
(S)-3-(6-(дифторметокси)пиридин-3-ил)-3-(2-оксо-3-(3-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)пропил)имидазолидин-1-ил)пропионовая кислота (Соединение A1): К перемешанному раствору соединения 12 (0,40 г, сырое, 1,0 эквив.) в ДХМ (2 мл), охлажденному до -10°C, добавляли трифторуксусную кислоту (0,5 мл) в атмосфере N2. Реакционную смесь медленно доводили до комнатной температуры и перемешивали в течение 2 ч; после расхода исходного материала летучие вещества выпаривали для получения сырого (400 мг) соединения, которое очищали с помощью хиральной препаративной ВЭЖ для образования соединения А1 в виде беловатого твердого вещества.
ТСХ: 10% MeOH/ДХМ (Rf: 0,3)
1H ЯМР (400 МГц, CD3OD): δ 8,20 (д, J=2,4 Гц, 1H), 7,85 (дд, J=2,4, 6,4 Гц, 1H), 7,53 (т,, J=2,4 Гц, 1H), 7,50 (д, J=7,2 Гц, 1H), 6,98 (д, J=8,4 Гц, 1H), 6,57 (д, J=7,2 Гц, 1H), 5,51 (дд, J=3,6, 8,0 Гц, 1H), 3,68-3,61 (м, 1H), 3,52-3,46 (м, 3H), 3,38 (м, 1H), 3,24-3,17 (м, 1H), 3,07-2,98 (м, 2H), 2,90-2,62 (м, 6H), 2,09-1,81 (м, 4H).
ЖХ-МС (m/z) = 476 [M+H]+ при комнатной температуре 2,78 (97,9% чистота)
Чистота ВЭЖХ: 96,4%; хиральная чистота: 99%
Соединения настоящего изобретения, описанные в примерах 2-7, в которых Z является -CH2CH2CH2-, синтезировали, применяя общую схему реакции, показанную на схеме 2. Диметил(2-оксо-6-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)гексил)фосфонат добавляли к фторированному азотистому гетероциклическому (Q) альдегиду для образования гепт-1-ен-3-она. Гепт-1-ен-3-он восстанавливали до соответствующего гепт-1-ен-3-ола с помощью алюмогидрида лития или боргидрида натрия. Гепт-1-ен-3-ол затем реагировал с пропионовой кислотой в 1,1,1-триэтоксиэтане и образующийся в результате сырой продукт перегруппировки восстанавливали водородом и палладием на углеродном катализаторе до соответствующего продукта восстановления олефина, который затем реагировал с водным основанием для образования конечных соединений нонановой кислоты.
Пример 2. Синтез 9-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)-3-(2-(трифторметил)пиримидин-5-ил)нонановой кислоты (Соединения A2)
(E)-7-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)-1-(2-(трифторметил)пиримидин-5-ил)гепт-1-ен-3-он
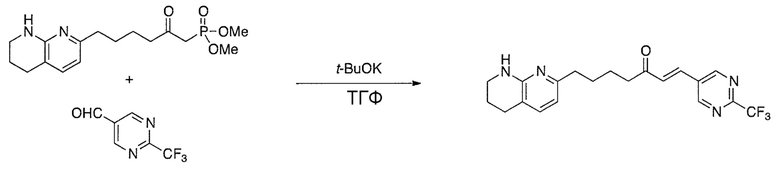
В атмосфере азота к диметил(2-оксо-6-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)гексил)фосфонату (3,40 г, 10,0 ммоль, 1,00 эквив.; Coleman, P. J. et al., J. Med. Chem. 2004, 47:4829-4837) в ТГФ (10 мл) при 23°C добавляли 2-(трифторметил)пиримидин-5-карбальдегид (1,76 г, 10,0 ммоль, 1,00 эквив.) и t-BuOK (1,01 г, 9,00 ммоль, 0,900 эквив.). После перемешивания в течение 10 мин при 23°C реакционную смесь сразу наносили на силикагель и очищали с помощью колоночной хроматографии на силикагеле, элюируя CH2Cl2/MeOH для образования 2,10 г заявленного соединения (54% выход).
1H ЯМР (300 МГц, CDCl3, 23°C, δ): 9,03 (с, 2H), 7,50 (д, J=16,2 Гц, 1H), 7,07 (д, J=7,2 Гц, 1H), 6,93 (д, J=16,2 Гц, 1H), 6,35 (д, J=7,2 Гц, 1H), 5,17 (уш.с, 1H), 3,42-3,37 (м, 2H), 2,79-2,64 (м, 4H), 2,62-2,55 (м, 2H), 1,95-1,85 (м, 2H), 1,77-1,66 (м, 4H), 19F ЯМР (282 МГц, CDCl3, 23°C, δ): -70,3 (с, 3F).
(E)-7-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)-1-(2-(трифторметил)пиримидин-5-ил)гепт-1-ен-3-ол
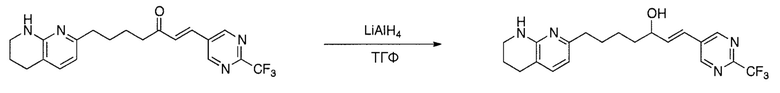
В атмосфере азота к (E)-7-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)-1-(2-(трифторметил)пиримидин-5-ил)гепт-1-ен-3-ону (1,20 г, 3,07 ммоль, 1,00 эквив.) в ТГФ (15 мл) при -78°C добавляли LiAlH4 (1,0 M в ТГФ, 3,07 мл, 3,07 ммоль, 1,00 эквив.). После перемешивания в течение 10 мин при -78°C к реакционной смеси последовательно добавляли H2O (116 мкл), 15% NaOH водн. (116 мкл) и H2O (348 мкл). Реакционную смесь нагревали до 23°C и фильтровали через прокладку CELITE®. Фильтрат концентрировали в вакууме и остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя CH2Cl2/MeOH для образования 560 мг заявленного соединения (46% выход).
1H ЯМР (300 МГц, CDCl3, 23°C, δ): 8,86 (с, 2H), 7,06 (д, J=7,2 Гц, 1H), 6,66 (д, J=16,2 Гц, 1H), 6,53 (дд, J=16,2 Гц, 4,5 Гц, 1H), 6,34 (д, J=7,2 Гц, 1H), 4,81 (уш.с, 1H), 4,50-4,40 (м, 1H), 3,42-3,37 (м, 2H), 2,70-2,50 (м, 4H), 1,96-1,40 (м, 8H), 19F ЯМР (282 МГц, CDCl3, 23°C, δ): -70,1 (с, 3F).
9-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)-3-(2-(трифторметил)пиримидин-5-ил)нонановая кислота (Соединение A2)
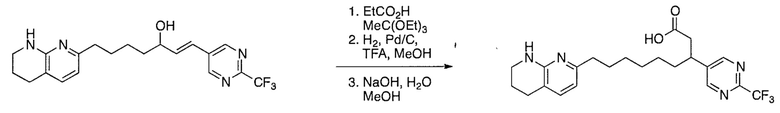
В атмосфере азота к (E)-7-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)-1-(2-(трифторметил)пиримидин-5-ил)гепт-1-ен-3-олу (560 мг, 1,43 ммоль, 1,00 эквив.) в MeC(OEt)3 (14 мл) при 23°C добавляли EtCO2H (107 мкл, 1,43 ммоль, 1,00 эквив.). После перемешивания в течение 2 ч при 140°C реакционную смесь сразу наносили на силикагель и очищали с помощью колоночной хроматографии на силикагеле, элюируя гексанами/EtOAc для образования сырого продукта перегруппировки, который применяли на следующей стадии без дальнейшей очистки.
В воздухе к полученному выше остатку в MeOH-TFA (10 мл - 1 мл) при 23°C добавляли 10% Pd/C (103 мг, 0,0969 ммоль, 6,78% молярный) и H2 с помощью баллона. После перемешивания в течение 1 ч при 23°C реакционную смесь фильтровали через прокладку CELITE®. Фильтрат концентрировали в вакууме для образования сырого продукта восстановления олефина, который применяли на следующей стадии без дальнейшей очистки.
В воздухе к полученному выше остатку в MeOH (10 мл) при 23°C добавляли 15% NaOH водн. (2,7 мл). После перемешивания в течение 20 мин при 60°C реакционную смесь нейтрализовали 3н HCl и концентрировали в вакууме для удаления MeOH. Оставшийся водный раствор экстрагировали EtOAc (3×10 мл) и промывали объединенные органические фазы NaHCO3 водн. (2×5 мл) и высушивали (MgSO4). Фильтрат концентрировали в вакууме и очищали остаток с помощью колоночной хроматографии на силикагеле, элюируя CH2Cl2/MeOH для образования 280 мг заявленного соединения (45% выход в 3 стадии).
1H ЯМР (300 МГц, CDCl3, 23°C, δ): 8,79 (с, 2H),7,24 (д, J=7,2 Гц, 1H), 6,25 (д, J=7,2 Гц, 1H), 3,48-3,40 (м, 2H), 3,38-3,32 (м, 1H), 2,75-2,52 (м, 4H), 1,95-1,80 (м, 4H), 1,75-1,58 (м, 4H), 1,40-1,18 (м, 6H), 19F ЯМР (282 МГц, CDCl3, 23°C, δ): -70,1 (с, 3F).
Пример 3. Синтез 3-(6-(дифторметокси)пиридин-3-ил)-9-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)нонановой кислоты (Соединение A3)
6-(дифторметокси)никотинальдегид
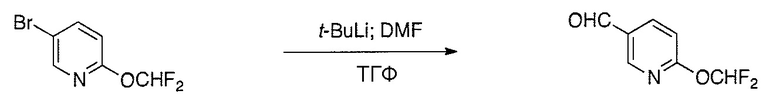
В атмосфере азота к 5-бром-2-(дифторметокси)пиридину (448 мг, 2,00 ммоль, 1,00 эквив.; Ando, M. et al., Org. Lett. 2006, 8:3805-3808) в тетрагидрофурану (10 мл) при -78°C по каплям добавляли t-BuLi (1,7 М в пентане, 2,35 мл, 4,00 ммоль, 2,00 эквив.) в течение 5 мин. После перемешивания в течение 20 мин при -78°C в реакционную смесь добавляли ДМФ (0,54 мл, 7,0 ммоль, 3,5 эквив.). После перемешивания в течение 20 мин при -78°C к реакционной смеси добавляли 1н HCl водн. (10 мл) и нагревали реакционную смесь до 23°C. Фазы разделяли и водную фазу экстрагировали EtOAc (3×5 мл). Объединенные органические фазы промывали солевым раствором (10 мл) и высушивали (MgSO4). Фильтрат концентрировали в вакууме и очищали остаток с помощью колоночной хроматографии на силикагеле, элюируя гексанами/EtOAc для образования 105 мг заявленного соединения (30% выход).
1H ЯМР (300 МГц, CDCl3, 23°C, δ): 10,05 (с, 1H), 8,69 (д, J=2,1 Гц, 1H), 8,24 (дд, J=8,4 Гц, 2,4 Гц, 1H), 7,56 (т, J=72,3 Гц, 1H), 7,04 (д, J=8,4 Гц, 1H), 19F ЯМР (282 МГц, CDCl3, 23°C, δ): -89,8 (д, J=72,3 Гц, 2F).
(E)-1-(6-(дифторметокси)пиридин-3-ил)-7-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)гепт-1-ен-3-он
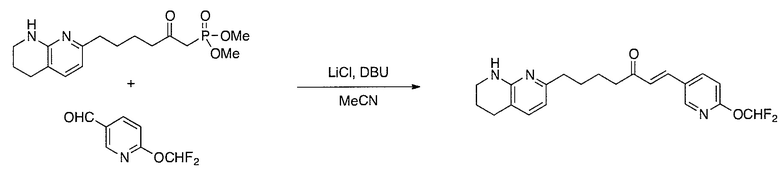
В атмосфере азота к диметил(2-оксо-6-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)гексил)фосфонату (1,57 г, 4,62 ммоль, 1,00 эквив.) в MeCN (11 мл) при 23°C добавляли 6-(дифторметокси)никотинальдегид (800 мг, 4,62 ммоль, 1,00 эквив.), LiCl (196 мг, 4,62 ммоль, 1,00 эквив.) и DBU (0,725 мл, 4,85 ммоль, 1,05 эквив.). После перемешивания в течение 1 ч при 50°C реакционную смесь охлаждали до 23°C и фильтровали через прокладку CELITE®. Фильтрат концентрировали в вакууме и остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя CH2Cl2/MeOH для образования 1,27 г заявленного соединения (71% выход).
1H ЯМР (300 МГц, CDCl3, 23°C, δ): 8,32 (д, J=2,4 Гц, 1H),7,92 (дд, J=8,4 Гц, 2,4 Гц, 1H), 7,49 (т, J=72,3 Гц, 1H), 7,47 (д, J=16,2 Гц, 1H), 7,06 (д, J=7,2 Гц, 1H), 6,93 (д, J=8,7 Гц, 1H), 6,70 (д, J=16,2 Гц, 1H), 6,35 (д, J=7,2 Гц, 1H), 4,89 (уш.с, 1H), 3,42-3,36 (м, 2H), 2,76-2,64 (м, 4H), 2,62-2,56 (м, 2H), 1,94-1,85 (м, 2H), 1,80-1,66 (м, 4H). 19F ЯМР (282 МГц, CDCl3, 23°C, δ): -89,2 (д, J=72,3 Гц, 2F).
(E)-1-(6-(дифторметокси)пиридин-3-ил)-7-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)гепт-1-ен-3-ол
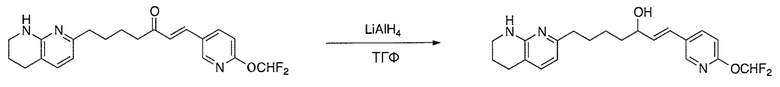
В атмосфере азота к (E)-1-(6-(дифторметокси)пиридин-3-ил)-7-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)гепт-1-ен-3-ону (1,27 г, 3,28 ммоль, 1,00 эквив.) в ТГФ (33 мл) при 0°C добавляли LiAlH4 (1,0 M в ТГФ, 3,28 мл, 3,28 ммоль, 1,00 эквив.). После перемешивания в течение 10 мин при 0°C к реакционной смеси последовательно добавляли H2O (124 мкл), 15% NaOH водн. (124 мкл) и H2O (372 мкл). Реакционную смесь нагревали до 23°C и фильтровали через прокладку CELITE®. Фильтрат концентрировали в вакууме и остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя CH2Cl2/MeOH для образования 1,05 г заявленного соединения (82% выход).
1H ЯМР (300 МГц, CDCl3, 23°C, δ): 8,22 (д, J=2,4 Гц, 1H),7,84 (дд, J=8,4 Гц, 2,4 Гц, 1H), 7,49 (т, J=72,3 Гц, 1H), 7,05 (д, J=7,2 Гц, 1H), 6,88 (д, J=8,7 Гц, 1H), 6,66 (д, J=16,2 Гц, 1H), 6,55 (дд, J=16,2 Гц, 4,5 Гц, 1H), 6,33 (д, J=7,2 Гц, 1H), 4,84 (уш.с, 1H), 4,52-4,43 (м, 1H), 3,40-3,37 (м, 2H), 2,72-2,51 (м, 4H), 1,95-1,40 (м, 8H). 19F ЯМР (282 МГц, CDCl3, 23°C, δ): -89,0 (д, J=72,5 Гц, 2F).
3-(6-(дифторметокси)пиридин-3-ил)-9-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)нонановая кислота (Соединение A3)
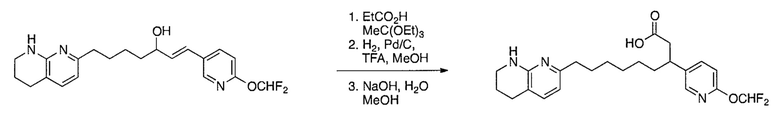
В атмосфере азота к (E)-1-(6-(дифторметокси)пиридин-3-ил)-7-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)гепт-1-ен-3-олу (1,05 г, 2,70 ммоль, 1,00 эквив.) в MeC(OEt)3 (27 мл) при 23°C добавляли EtCO2H (201 мкл, 2,70 ммоль, 1,00 эквив.). После перемешивания в течение 2 ч при 140°C в реакционную смесь добавляли ДМФ (0,54 мл, 7,0 ммоль, 3,5 эквив.). После перемешивания в течение 20 мин при -78°C реакционную смесь сразу наносили на силикагель и очищали с помощью колоночной хроматографии на силикагеле, элюируя гексанами/EtOAc для образования сырого продукта перегруппировки, который применяли на следующей стадии без дальнейшей очистки.
В воздухе к полученному выше остатку в MeOH-TFA (10 мл - 1 мл) при 23°C добавляли 10% Pd/C (176 мг, 0,165 ммоль, 6,11% молярн.) и H2 с помощью баллона. После перемешивания в течение 1 ч при 23°C реакционную смесь фильтровали через прокладку CELITE®. Фильтрат концентрировали в вакууме для образования сырого продукта восстановления олефина, который применяли на следующей стадии без дальнейшей очистки.
В воздухе к полученному выше остатку в MeOH (10 мл) при 23°C добавляли 15% NaOH водн. (4,4 мл). После перемешивания в течение 20 мин при 60°C реакционную смесь нейтрализовали 3н HCl и концентрировали в вакууме для удаления MeOH. Оставшийся водный раствор экстрагировали EtOAc (3×10 мл) и объединенные органические фазы промывали NaHCO3 водн. (2×5 мл) и высушивали (MgSO4). Фильтрат концентрировали в вакууме и остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя CH2Cl2/MeOH для образования 400 мг заявленного соединения (34% выход в 3 стадии).
1H ЯМР (300 МГц, CDCl3, 23°C, δ): 8,06 (д, J=2,4 Гц, 1H),7,66 (дд, J=8,4 Гц, 2,4 Гц, 1H), 7,43 (т, J=72,3 Гц, 1H), 7,20 (д, J=8,7 Гц, 1H), 6,84 (д, J=7,2 Гц, 1H), 6,25 (д, J=7,2 Гц, 1H), 3,46-3,40 (м, 2H), 3,38-3,28 (м, 1H), 2,79-2,40 (м, 4H), 1,95-1,80 (м, 4H), 1,75-1,62 (м, 4H), 1,40-1,20 (м, 6H). 19F ЯМР (282 МГц, CDCl3, 23°C, δ): -88,3 (д, J=72,5 Гц, 2F).
Пример 4. Синтез 3-(6-фторхинолин-3-ил)-9-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)нонановой кислоты (Соединения A4)
6-фторхинолин-3-карбальдегид
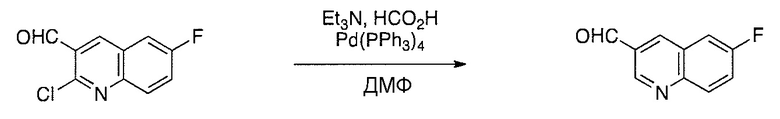
В атмосфере азота к 2-хлор-6-фторхинолин-3-карбальдегиду (2,03 г, 9,68 ммоль, 1,00 эквив.) в ДМФ (10 мл) при 23°C добавляли триэтиламин (16,2 мл, 116 ммоль, 12,0 эквив.), Pd(PPh3)4 (559 мг, 0,484 ммоль, 5,00% молярн.) и муравьиную кислоту (1,29 мл, 34,2 ммоль, 5,40 эквив.). После перемешивания в течение 1 ч при 100°C реакционную смесь охлаждали до 23°C и добавляли воду (40 мл) и EtOAc (30 мл). Фазы разделяли и водную фазу экстрагировали EtOAc (3×30 мл). Объединенные органические фазы промывали солевым раствором (50 мл) и высушивали (MgSO4). Фильтрат концентрировали в вакууме и очищали остаток с помощью колоночной хроматографии на силикагеле, элюируя гексанами/EtOAc для образования 734 мг заявленного соединения (43% выход).
1H ЯМР (300 МГц, CDCl3, 23°C, δ): 10,27 (с, 1H), 9,34 (д, J=2,1 Гц, 1H), 8,60 (д, J=1,8 Гц, 1H), 8,21 (дд, J=9,0 Гц, 4,8 Гц, 1H), 7,70-7,60 (м, 2H). 19F ЯМР (282 МГц, CDCl3, 23°C, δ): -110,8 (м, 1F).
(E)-1-(6-фторхинолин-3-ил)-7-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)гепт-1-ен-3-он
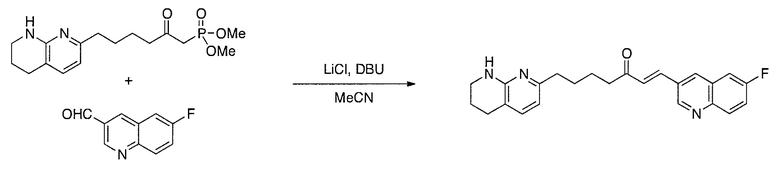
В атмосфере азота к диметил(2-оксо-6-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)гексил)фосфонату (900 мг, 2,64 ммоль, 1,10 эквив.) в MeCN (22 мл) при 23°C добавляли 6-фторхинолин-3-карбальдегид (420 мг, 2,40 ммоль, 1,00 эквив.), LiCl (101 мг, 2,.40 ммоль, 1,00 эквив.) и DBU (0,377 мл, 2,52 ммоль, 1,05 эквив.). После перемешивания в течение 1 ч при 75°C реакционную смесь охлаждали до 23°C и фильтровали через прокладку CELITE®. Фильтрат концентрировали в вакууме и остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя CH2Cl2/MeOH для образования 900 мг заявленного соединения (96% выход).
1H ЯМР (300 МГц, CDCl3, 23°C, δ): 9,06 (д, J=2,4 Гц, 1H), 8,21 (д, J=2,1 Гц, 1H),8,11 (дд, J=10,6 Гц, 5,7 Гц, 1H), 7,66 (д, J=16,2 Гц, 1H), 7,58-7,43 (м, 2H), 7,06 (д, J=7,2 Гц, 1H), 6,96 (д, J=16,2 Гц, 1H), 6,37 (д, J=7,2 Гц, 1H), 4,76 (уш.с, 1H), 3,43-3,35 (м, 2H), 2,78-2,65 (м, 4H), 2,63-2,56 (м, 2H), 1,94-1,85 (м, 2H), 1,82-1,66 (м, 4H). 19F ЯМР (282 МГц, CDCl3, 23°C, δ): -111,9 (м, 1F).
(E)-1-(6-фторхинолин-3-ил)-7-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)гепт-1-ен-3-ол
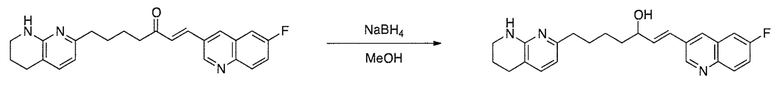
В воздухе к (E)-1-(6-фторхинолин-3-ил)-7-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)гепт-1-ен-3-ону (490 мг, 1,26 ммоль, 1,00 эквив.) в MeOH (29 мл) при 0°C добавляли NaBH4 (71,5 мг, 1,89 ммоль, 1,5 эквив.). После перемешивания в течение 1 ч при 0°C в реакционную смесь добавляли 1н HCl водн. (10 мл) и концентрировали в вакууме для удаления MeOH. Остаток нейтрализовали NaHCO3 водн. и добавляли EtOAc (10 мл). Фазы разделяли и водную фазу экстрагировали EtOAc (3×20 мл). Объединенные органические фазы промывали солевым раствором (30 мл) и высушивали (MgSO4). Фильтрат концентрировали в вакууме и очищали остаток с помощью колоночной хроматографии на силикагеле, элюируя CH2Cl2/MeOH для образования 490 мг заявленного соединения (99% выход).
1H ЯМР (300 МГц, CDCl3, 23°C, δ): 8,95 (с, 1H),8,06 (дд, J=10,6 Гц, 5,7 Гц, 1H), 7,99 (с, 1H),7,50-7,40 (м, 2H), 7,06 (д, J=7,2 Гц, 1H), 6,75 (д, J=16,2 Гц, 1H), 6,49 (дд, J=16,2 Гц, 4,5 Гц, 1H), 6,34 (д, J=7,2 Гц, 1H), 4,94 (уш.с, 1H), 4,47-4,39 (м, 1H), 3,42-3,38 (м, 2H), 2,70-2,47 (м, 4H), 1,96-1,45 (м, 8H). 19F ЯМР (282 МГц, CDCl3, 23°C, δ): -111,8 (м, 1F).
3-(6-фторхинолин-3-ил)-9-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)нонановая кислота (Соединение A4)
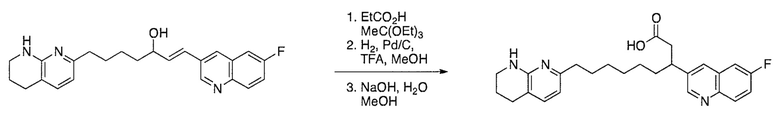
В атмосфере азота к (E)-1-(6-(фторхинолин-3-ил)-7-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)гепт-1-ен-3-олу (489 мг, 1,25 ммоль, 1,00 эквив.) в MeC(OEt)3 (12 мл) при 23°C добавляли EtCO2H (93,3 мкл, 1,25 ммоль, 1,00 эквив.).После перемешивания в течение 2 ч при 140°C реакционную смесь сразу наносили на силикагель и очищали с помощью колоночной хроматографии на силикагеле, элюируя гексанами/EtOAc для образования сырого продукта перегруппировки, который применяли на следующей стадии без дальнейшей очистки.
В воздухе к полученному выше остатку в MeOH-TFA (10 мл - 1 мл) при 23°C добавляли 10% Pd/C (128 мг, 0,121 ммоль, 9,68% молярн.) и H2 с помощью баллона. После перемешивания в течение 1 ч при 23°C реакционную смесь фильтровали через прокладку CELITE®. Фильтрат концентрировали в вакууме для образования сырого продукта восстановления олефина, который применяли на следующей стадии без дальнейшей очистки.
В воздухе к полученному выше остатку в MeOH (10 мл) при 23°C добавляли 15% NaOH водн. (3,0 мл). После перемешивания в течение 20 мин при 60°C реакционную смесь нейтрализовали 3н HCl и концентрировали в вакууме для удаления MeOH. Оставшийся водный раствор экстрагировали EtOAc (3×10 мл) и объединенные органические фазы промывали NaHCO3 водн. (2×5 мл) и высушивали (MgSO4). Фильтрат концентрировали в вакууме и остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя CH2Cl2/MeOH для образования 500 мг заявленного соединения (92% выход в 3 стадии).
1H ЯМР (300 МГц, CD3OD, 23°C, δ): 8,78 (с, 1H), 8,11 (с, 1H),8,00-7,93 (м, 1H), 7,52-7,42 (м, 2H), 7,31 (д, J=7,2 Гц, 1H), 6,35 (д, J=7,2 Гц, 1H), 3,38-3,20 (м, 3H), 2,77-2,42 (м, 4H), 1,90-1,20 (м, 14H). 19F ЯМР (282 МГц, CD3OD, 23°C, δ): -110,9 (м, 1F).
Пример 5. Синтез 3-(7-фторхинолин-3-ил)-9-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)нонановой кислоты (Соединения A5)
(E)-1-(7-фторхинолин-3-ил)-7-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)гепт-1-ен-3-он
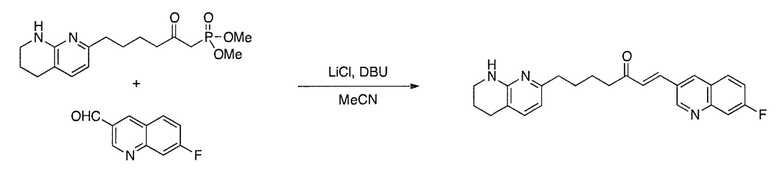
В атмосфере азота к диметил(2-оксо-6-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)гексил)фосфонату (749 мг, 2,20 ммоль, 1,10 эквив.) в MeCN (22 мл) при 23°C добавляли 7-фторхинолин-3-карбальдегид (350 мг, 2,00 ммоль, 1,00 эквив.; Sato, I. et al., Synthesis 2004, 9:1419-1428), LiCl (84,8 мг, 2,00 ммоль, 1,00 эквив.) и DBU (0,314 мл, 2,10 ммоль, 1,05 эквив.). После перемешивания в течение 1 ч при 75°C реакционную смесь охлаждали до 23°C и фильтровали через прокладку CELITE®. Фильтрат концентрировали в вакууме и остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя CH2Cl2/MeOH для образования 570 мг заявленного соединения (73% выход).
1H ЯМР (300 МГц, CDCl3, 23°C, δ): 9,10 (д, J=2,4 Гц, 1H), 8,28 (д, J=2,1 Гц, 1H),7,87 (дд, J=9,0 Гц, 6,0 Гц, 1H), 7,74 (дд, J=9,9 Гц, 2,4 Гц, 1H), 7,69 (д, J=16,2 Гц, 1H), 7,42-7,33 (м, 1H), 7,11 (д, J=7,2 Гц, 1H), 6,94 (д, J=16,2 Гц, 1H), 6,37 (д, J=7,2 Гц, 1H), 5,41 (уш.с, 1H), 3,43-3,37 (м, 2H), 2,78-2,58 (м, 6H), 1,93-1,85 (м, 2H), 1,81-1,69 (м, 4H). 19F ЯМР (282 МГц, CDCl3, 23°C, δ): -107,0 (м, 1F).
(E)-1-(7-фторхинолин-3-ил)-7-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)гепт-1-ен-3-ол
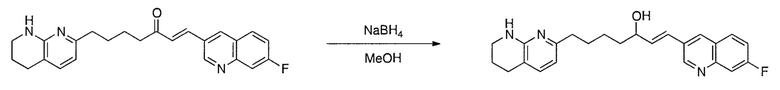
В воздухе к (E)-1-(7-фторхинолин-3-ил)-7-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)гепт-1-ен-3-ону (300 мг, 0,770 ммоль, 1,00 эквив.) в MeOH (8 мл) при 0°C добавляли NaBH4 (87,4 мг, 2,31 ммоль, 3,00 эквив.). После перемешивания в течение 30 мин при 0°C в реакционную смесь добавляли 1н HCl водн. (10 мл) и концентрировали в вакууме для удаления MeOH. Остаток нейтрализовали NaHCO3 водн. и добавляли EtOAc (10 мл). Фазы разделяли и водную фазу экстрагировали EtOAc (3×20 мл). Объединенные органические фазы промывали солевым раствором (30 мл) и высушивали (MgSO4). Фильтрат концентрировали в вакууме и очищали остаток с помощью колоночной хроматографии на силикагеле, элюируя CH2Cl2/MeOH для образования 210 мг заявленного соединения (70% выход).
1H ЯМР (300 МГц, CDCl3, 23°C, δ): 8,98 (с, 1H), 8,07 (с, 1H),7,81 (дд, J=9,0 Гц, 6,0 Гц, 1H), 7,78 (дд, J=9,9 Гц, 2,4 Гц, 1H), 7,63 (уш.с, 1H), 7,39-7,28 (м, 1H), 6,78 (д, J=16,2 Гц, 1H), 6,47 (дд, J=16,2 Гц, 4,5 Гц, 1H), 6,36 (д, J=7,2 Гц, 1H), 4,48-4,41 (м, 1H), 3,48-3,41 (м, 2H), 2,79-2,67 (м, 4H), 1,97-1,48 (м, 8H). 19F ЯМР (282 МГц, CDCl3, 23°C, δ): -109,9 (м, 1F).
3-(7-фторхинолин-3-ил)-9-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)нонановая кислота (Соединение A5)
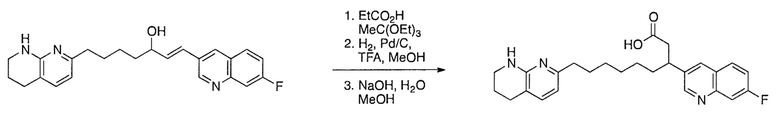
В атмосфере азота к (E)-1-(7-фторхинолин-3-ил)-7-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)гепт-1-ен-3-олу (730 мг, 1,71 ммоль, 1,00 эквив.) в MeC(OEt)3 (17 мл) при 23°C добавляли EtCO2H (128 мкл, 1,71 ммоль, 1,00 эквив.). После перемешивания в течение 2 ч при 140°C реакционную смесь сразу наносили на силикагель и очищали с помощью колоночной хроматографии на силикагеле, элюируя гексанами/EtOAc для образования сырого продукта перегруппировки, который применяли на следующей стадии без дальнейшей очистки.
В воздухе к полученному выше остатку в MeOH-TFA (10 мл-1 мл) при 23°C добавляли 10% Pd/C (125 мг, 0,117 ммоль, 6,84% молярн.) и H2 с помощью баллона. После перемешивания в течение 1 ч при 23°C реакционную смесь фильтровали через прокладку CELITE®. Фильтрат концентрировали в вакууме для образования сырого продукта восстановления олефина, который применяли на следующей стадии без дальнейшей очистки.
В воздухе к полученному выше остатку в MeOH (10 мл) при 23°C добавляли 15% NaOH водн. (3,0 мл). После перемешивания в течение 20 мин при 60°C реакционную смесь нейтрализовали 3н HCl и концентрировали в вакууме для удаления MeOH. Оставшийся водный раствор экстрагировали EtOAc (3×10 мл) и объединенные органические фазы промывали NaHCO3 водн. (2×5 мл) и высушивали (MgSO4). Фильтрат концентрировали в вакууме и остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя CH2Cl2/MeOH для образования 480 мг заявленного соединения (64% выход в 3 стадии).
1H ЯМР (300 МГц, CD3OD, 23°C, δ): 8,79 (с, 1H), 8,21 (с, 1H),8,00-7,91 (м, 1H), 7,62-7,57 (м, 1H), 7,48-7,38 (м, 2H), 6,47 (д, J=7,2 Гц, 1H), 3,48-3,30 (м, 3H), 2,80-2,52 (м, 4H), 1,90-1,20 (м, 14H). 19F ЯМР (282 МГц, CD3OD, 23°C, δ): -111,9 (м, 1F).
Пример 6. Синтез 3-(6,7-дифторхинолин-3-ил)-9-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)нонановой кислоты (Соединения A6)
6,7-дифторхинолин-3-карбальдегид
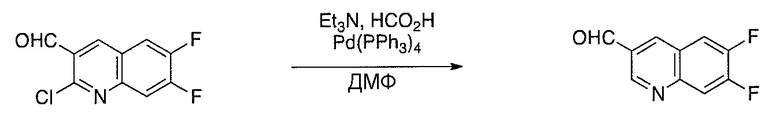
В атмосфере азота к 2-хлор-6,7-дифторхинолин-3-карбальдегиду (1,44 г, 6,33 ммоль, 1,00 эквив.) в ДМФ (6,3 мл) при 23°C добавляли триэтиленамин (10,6 мг, 76,0 ммоль, 12,00 эквив.), Pd(PPh3)4 (366 мг, 0,317 ммоль, 5,00% молярн.) и муравьиную кислоту (1,29 мл, 34,2 ммоль, 5,40 эквив.). После перемешивания в течение 1 ч при 100°C реакционную смесь охлаждали до 23°C и добавляли воду (30 мл) и EtOAc (20 мл). Фазы разделяли и водную фазу экстрагировали EtOAc (3×20 мл). Объединенные органические фазы промывали солевым раствором (50 мл) и высушивали (MgSO4). Фильтрат концентрировали в вакууме и очищали остаток с помощью колоночной хроматографии на силикагеле, элюируя гексанами/EtOAc для образования 500 мг заявленного соединения (41% выход).
1H ЯМР (300 МГц, CDCl3, 23°C, δ): 10,26 (с, 1H), 9,35 (д, J=1,2 Гц, 1H), 8,60 (д, J=1,5 Гц, 1H), 7,97 (дд, J=10,8 Гц, 7,5 Гц, 1H), 7,97 (дд, J=9,0 Гц, 8,7 Гц, 1H). 19F ЯМР (282 МГц, CDCl3, 23°C, δ): -125,3 (м, 1F), -132,3 (м, 1F).
(E)-1-(6,7-дифторхинолин-3-ил)-7-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)гепт-1-ен-3-он
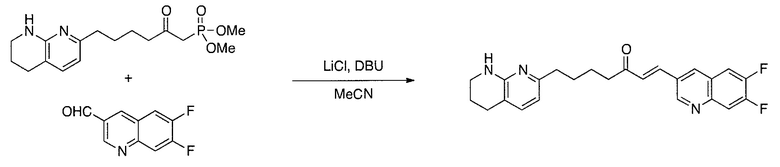
В атмосфере азота к диметил(2-оксо-6-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)гексил)фосфонату (599 мг, 1,76 ммоль, 1,10 эквив.) в MeCN (5 мл) при 23°C добавляли 6,7-дифторхинолин-3-карбальдегид (310 мг, 1,60 ммоль, 1,00 эквив.), LiCl (67,8 мг, 1,60 ммоль, 1,00 эквив.) и DBU (0,251 мл, 1,68 ммоль, 1,05 эквив.). После перемешивания в течение 1 ч при 75°C реакционную смесь охлаждали до 23°C и фильтровали через прокладку CELITE®. Фильтрат концентрировали в вакууме и остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя CH2Cl2/MeOH для образования 570 мг заявленного соединения (84% выход).
1H ЯМР (300 МГц, CDCl3, 23°C, δ): 9,07 (д, J=2,4 Гц, 1H), 8,20 (д, J=2,1 Гц, 1H),7,87 (дд, J=10,8 Гц, 7,5 Гц, 1H), 7,66 (д, J=16,2 Гц, 1H), 7,62-7,53 (м, 1H), 7,06 (д, J=7,2 Гц, 1H), 6,93 (д, J=16,2 Гц, 1H), 6,36 (д, J=7,2 Гц, 1H), 4,77 (уш.с, 1H), 3,43-3,38 (м, 2H), 2,79-2,58 (м, 6H), 1,96-1,85 (м, 2H), 1,81-1,69 (м, 4H). 19F ЯМР (282 МГц, CDCl3, 23°C, δ): -129,1 (м, 1F), -133,6 (м, 1F).
(E)-1-(6,7-дифторхинолин-3-ил)-7-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)гепт-1-ен-3-ол
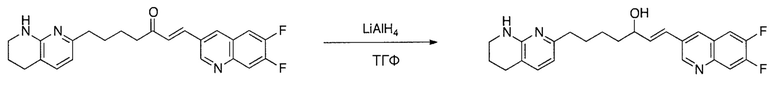
В атмосфере азота к (E)-1-(6,7-дифторхинолин-3-ил)-7-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)гепт-1-ен-3-ону (1,03 г, 2,53 ммоль, 1,00 эквив.) в ТГФ (25 мл) при 0°C добавляли LiAlH4 (1,0 M в ТГФ, 2,53 мл, 2,53 ммоль, 1,00 эквив.). После перемешивания в течение 10 мин при 0°C к реакционной смеси последовательно добавляли H2O (96 мкл), 15% NaOH водн. (96 мкл) и H2O (288 мкл). Реакционную смесь нагревали до 23°C и фильтровали через прокладку CELITE®. Фильтрат концентрировали в вакууме и остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя CH2Cl2/MeOH для образования 780 мг заявленного соединения (75% выход).
1H ЯМР (300 МГц, CDCl3, 23°C, δ): 8,95 (д, J=2,4 Гц, 1H), 8,00 (д, J=2,1 Гц, 1H),7,81 (дд, J=10,8 Гц, 7,5 Гц, 1H), 7,52 (д, J=16,2 Гц, 1H), 7,21 (д, J=7,2 Гц, 1H), 6,76 (д, J=16,2 Гц, 1H), 6,48 (дд, J=16,2 Гц, 4,5 Гц, 1H), 6,34 (д, J=7,2 Гц, 1H), 4,48-4,42 (м, 1H), 3,47-3,41 (м, 2H), 2,79-2,67 (м, 4H), 1,97-1,47 (м, 8H). 19F ЯМР (282 МГц, CDCl3, 23°C, δ): -132,1 (м, 1F), -135,1 (м, 1F).
3-(6,7-дифторхинолин-3-ил)-9-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)нонановая кислота (Соединение A6)
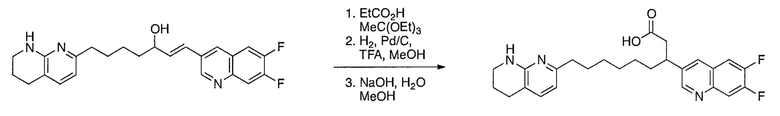
В атмосфере азота к (E)-1-(6,7-дифторхинолин-3-ил)-7-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)гепт-1-ен-3-олу (780 мг, 1,90 ммоль, 1,00 эквив.) в MeC(OEt)3 (19 мл) при 23°C добавляли EtCO2H (142 мкл, 1,90 ммоль, 1,00 эквив.). После перемешивания в течение 2 ч при 140°C реакционную смесь сразу наносили на силикагель и очищали с помощью колоночной хроматографии на силикагеле, элюируя гексанами/EtOAc для образования сырого продукта перегруппировки, который применяли на следующей стадии без дальнейшей очистки.
В воздухе к полученному выше остатку в MeOH-TFA (10 мл - 1 мл) при 23°C добавляли 10% Pd/C (127 мг, 0,119 ммоль, 6,26% молярн.) и H2 с помощью баллона. После перемешивания в течение 1 ч при 23°C реакционную смесь фильтровали через прокладку CELITE®. Фильтрат концентрировали в вакууме для образования сырого продукта восстановления олефина, который применяли на следующей стадии без дальнейшей очистки.
В воздухе к полученному выше остатку в MeOH (10 мл) при 23°C добавляли 15% NaOH водн. (3,2 мл). После перемешивания в течение 20 мин при 60°C реакционную смесь нейтрализовали 3н HCl и концентрировали в вакууме для удаления MeOH. Оставшийся водный раствор экстрагировали EtOAc (3×10 мл) и объединенные органические фазы промывали NaHCO3 водн. (2×5 мл) и высушивали (MgSO4). Фильтрат концентрировали в вакууме и остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя CH2Cl2/MeOH для образования 500 мг заявленного соединения (58% выход в 3 стадии).
1H ЯМР (300 МГц, CDCl3, 23°C, δ): 8,79 (с, 1H), 7,97 (с, 1H),7,90-7,81 (м, 1H), 7,58-7,47 (м, 1H), 7,24 (д, J=7,2 Гц, 1H), 6,23 (д, J=7,2 Гц, 1H), 3,48-3,32 (м, 3H), 2,80-2,57 (м, 4H), 1,95-1,20 (м, 14H). 19F ЯМР (282 МГц, CDCl3, 23°C, δ): -132,3 (м, 1F), -135,5 (м, 1F).
Пример 7. Синтез 9-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)-3-(7-(трифторметил)хинолин-3-ил)нонановой кислоты (Соединения A7)
2-хлор-7-йодхинолин-3-карбальдегид
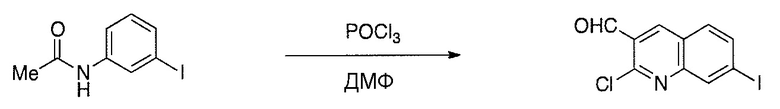
В атмосфере азота к POCl3 (14,9 мл, 160 ммоль, 7,00 эквив.) при 0°C добавляли ДМФ (4,40 мл, 57,1 ммоль, 2,50 эквив.). После перемешивания в течение 10 мин при 0°C к реакционной смеси добавляли N-(3-йодфенил)ацетамид (5,96 г, 22,8 ммоль, 1,00 эквив.; Pialat, A. et al., Org. Lett. 2013, 15:1764-1767). После перемешивания в течение 17 ч при 75°C реакционную смесь выливали на лед. Фазы разделяли и водную фазу экстрагировали CH2Cl2 (3×50 мл). Объединенные органические фазы промывали солевым раствором (100 мл) и высушивали (MgSO4). Фильтрат концентрировали в вакууме и очищали остаток с помощью колоночной хроматографии на силикагеле, элюируя CH2Cl2/MeOH для образования 2,9 г заявленного соединения (40% выход).
1H ЯМР (300 МГц, CDCl3, 23°C, δ): 10,55 (с, 1H), 8,72 (с, 1H), 8,52 (с, 1H), 7,93 (дд, J=8,4 Гц, 1,5 Гц, 1H), 7,69 (д, J=8,4 Гц, 1H).
2-хлор-7-(трифторметил)хинолин-3-карбальдегид
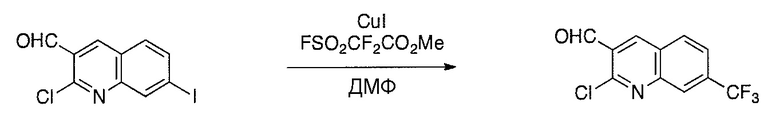
В атмосфере азота к 2-хлор-7-йодхинолин-3-карбальдегиду (2,90 г, 9,13 ммоль, 1,00 эквив.) в ДМФ (18 мл) при 23°C добавляли CuI (4,35 г, 22,8 ммоль, 2,50 эквив.) и FSO2CF2CO2Me (11,6 мл, 91,3 ммоль, 10,0 эквив.). После перемешивания в течение 2 ч при 95°C, реакционную смесь охлаждали до 23°C и фильтровали через прокладку CELITE®. Фильтрат концентрировали в вакууме и остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя гексанами/EtOAc для образования 1,5 г заявленного соединения (63% выход).
1H ЯМР (300 МГц, CDCl3, 23°C, δ): 10,60 (с, 1H), 8,82 (с, 1H), 8,39 (с, 1H), 8,14 (д, J=8,4 Гц, 1H), 7,84 (д, J=8,4 Гц, 1H). 19F ЯМР (282 МГц, CDCl3, 23°C, δ): -63,2 (с, 3F).
7-(трифторметил)хинолин-3-карбальдегид
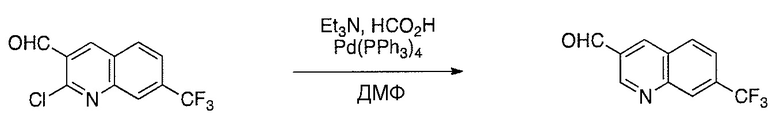
В атмосфере азота к 2-хлор-7-(трифторметил)хинолин-3-карбальдегиду (1,50 г, 5,78 ммоль, 1,00 эквив.) в ДМФ (5,8 мл) при 23°C добавляли триэтиленамин (9,67 мг, 69,4 ммоль, 12,0 эквив.), Pd(PPh3)4 (334 мг, 0,289 ммоль, 5,00% молярн.) и муравьиную кислоту (1,18 мл, 31,2 ммоль, 5,40 эквив.). После перемешивания в течение 1 ч при 100°C реакционную смесь охлаждали до 23°C и добавляли воду (30 мл) и EtOAc (20 мл). Фазы разделяли и водную фазу экстрагировали EtOAc (3×20 мл). Объединенные органические фазы промывали солевым раствором (50 мл) и высушивали (MgSO4). Фильтрат концентрировали в вакууме и очищали остаток с помощью колоночной хроматографии на силикагеле, элюируя гексанами/EtOAc для образования 412 мг заявленного соединения (32% выход).
1H ЯМР (300 МГц, CDCl3, 23°C, δ): 10,32 (с, 1H), 9,48 (д, J=1,5 Гц, 1H), 8,71 (д, J=1,5 Гц, 1H), 8,51 (с, 1H), 8,15 (д, J=8,4 Гц, 1H), 7,86 (д, J=8,4 Гц, 1H). 19F ЯМР (282 МГц, CDCl3, 23°C, δ): -63,1 (с, 3F).
(E)-7-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)-1-(7-(трифторметил)хинолин-3-ил)гепт-1-ен-3-он
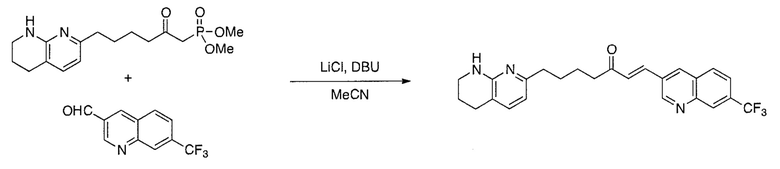
В атмосфере азота к диметил(2-оксо-6-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)гексил)фосфонату (685 мг, 2,01 ммоль, 1,10 эквив.) в MeCN (9 мл) при 23°C добавляли 7-(трифторметил)хинолин-3-карбальдегид (412 мг, 1,83 ммоль, 1,00 эквив.), LiCl (77,6 мг, 1,83 ммоль, 1,00 эквив.) и DBU (0,287 мл, 1,92 ммоль, 1,05 эквив.). После перемешивания в течение 1 ч при 75°C реакционную смесь охлаждали до 23°C и фильтровали через прокладку CELITE®. Фильтрат концентрировали в вакууме и остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя CH2Cl2/MeOH для образования 706 мг заявленного соединения (88% выход).
1H ЯМР (300 МГц, CDCl3, 23°C, δ): 9,19 (д, J=2,4 Гц, 1H), 8,42 (д, J=2,1 Гц, 1H), 8,31 (д, J=2,1 Гц, 1H),8,00 (д, J=9,0 Гц, 1H), 7,79 (д, J=9,0 Гц, 1H), 7,69 (д, J=16,2 Гц, 1H), 7,04 (д, J=7,2 Гц, 1H), 6,99 (д, J=16,2 Гц, 1H), 6,37 (д, J=7,2 Гц, 1H), 4,78 (уш.с, 1H), 3,41-3,37 (м, 2H), 2,80-2,58 (м, 6H), 1,93-1,85 (м, 2H), 1,81-1,69 (м, 4H). 19F ЯМР (282 МГц, CDCl3, 23°C, δ): -62,8 (с, 3F).
(E)-7-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)-1-(7-(трифторметил)хинолин-3-ил)гепт-1-ен-3-ол
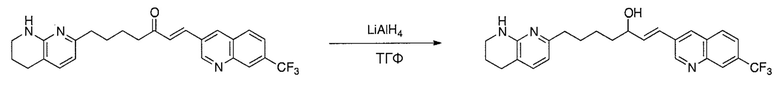
В атмосфере азота к (E)-7-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)-1-(7-(трифторметил)хинолин-3-ил)гепт-1-ен-3-ону (705 мг, 1,60 ммоль, 1,00 эквив.) в ТГФ (16 мл) при 0°C добавляли LiAlH4 (1,0 M в ТГФ, 1,60 мл, 1,60 ммоль, 1,00 эквив.). После перемешивания в течение 10 мин при 0°C к реакционной смеси последовательно добавляли H2O (54 мкл), 15% NaOH водн. (54 мкл) и H2O (162 мкл). Реакционную смесь нагревали до 23°C и фильтровали через прокладку CELITE®. Фильтрат концентрировали в вакууме и остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя CH2Cl2/MeOH для образования 515 мг заявленного соединения (73% выход).
1H ЯМР (300 МГц, CDCl3, 23°C, δ): 9,08 (д, J=2,4 Гц, 1H), 8,37 (д, J=2,1 Гц, 1H), 8,08 (д, J=2,1 Гц, 1H),7,91 (д, J=9,0 Гц, 1H), 7,71 (д, J=9,0 Гц, 1H), 7,06 (д, J=7,2 Гц, 1H), 6,79 (д, J=16,2 Гц, 1H), 6,53 (дд, J=16,2 Гц, 4,5 Гц, 1H), 6,34 (д, J=7,2 Гц, 1H), 4,89 (уш.с, 1H), 4,48-4,40 (м, 1H), 3,43-3,37 (м, 2H), 2,75-2,57 (м, 4H), 1,97-1,42 (м, 8H). 19F ЯМР (282 МГц, CDCl3, 23°C, δ): -62,6 (с, 3F).
9-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)-3-(7-(трифторметил)хинолин-3-ил)нонановая кислота (Соединение A7)
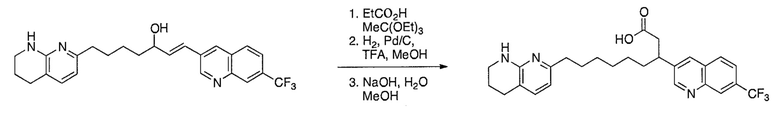
В атмосфере азота к (E)-7-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)-1-(7-(трифторметил)хинолин-3-ил)гепт-1-ен-3-олу (515 мг, 1,17 ммоль, 1,00 эквив.) в MeC(OEt)3 (12 мл) при 23°C добавляли EtCO2H (87,3 мкл, 1,17 ммоль, 1,00 эквив.). После перемешивания в течение 2 ч при 140°C реакционную смесь сразу наносили на силикагель и очищали с помощью колоночной хроматографии на силикагеле, элюируя гексанами/EtOAc для образования сырого продукта перегруппировки, который применяли на следующей стадии без дальнейшей очистки.
В воздухе к полученному выше остатку в MeOH-TFA (10 мл - 1 мл) при 23°C добавляли 10% Pd/C (66,6 мг, 0,0626 ммоль, 5,35% молярн.) и H2 с помощью баллона. После перемешивания в течение 1 ч при 23°C реакционную смесь фильтровали через прокладку CELITE®. Фильтрат концентрировали в вакууме для образования сырого продукта восстановления олефина, который применяли на следующей стадии без дальнейшей очистки.
В воздухе к полученному выше остатку в MeOH (10 мл) при 23°C добавляли 15% NaOH водн. (4,4 мл). После перемешивания в течение 20 мин при 60°C реакционную смесь нейтрализовали 3н HCl и концентрировали в вакууме для удаления MeOH. Оставшийся водный раствор экстрагировали EtOAc (3×10 мл) и объединенные органические фазы промывали NaHCO3 водн. (2×5 мл) и высушивали (MgSO4). Фильтрат концентрировали в вакууме и остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя CH2Cl2/MeOH для образования 300 мг заявленного соединения (53% выход в 3 стадии).
1H ЯМР (300 МГц, CDCl3, 23°C, δ): 8,93 (с, 1H), 8,40 (с, 1H), 8,03 (с, 1H),7,91 (д, J=9,0 Гц, 1H), 7,70 (д, J=9,0 Гц, 1H), 7,24 (д, J=7,2 Гц, 1H), 6,23 (д, J=7,2 Гц, 1H), 3,48-3,40 (м, 3H), 2,80-2,59 (м, 4H), 1,95-1,20 (м, 14H). 19F ЯМР (282 МГц, CDCl3, 23°C, δ): -62,7 (с, 3F).
Пример 8. Синтез (S)-3-(6-(дифторметокси)пиридин-3-ил)-3-(2-оксо-3-((3-((5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)метил)оксетан-3-ил)метил)имидазолидин-1-ил)пропионовой кислоты
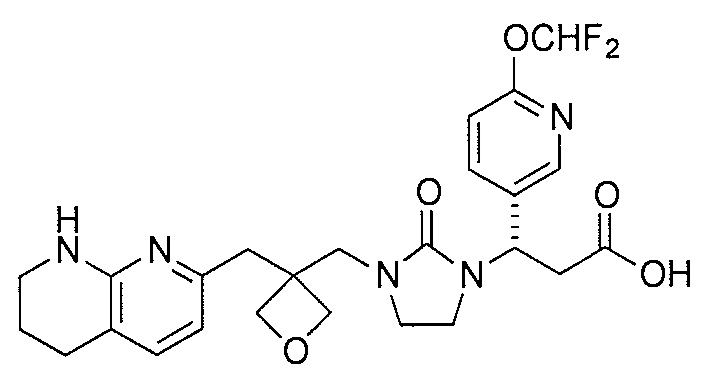
Способ синтеза является таким же, как в примере 1, за исключением замещения на стадии 8 (3-((5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)метил)оксетан-3-ил)метанамина соединением 6b, и схема синтеза продолжается с применением тех же условий реакции.
Пример 9. Синтез (S)-3-(3-(2,2-дифтор-3-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)пропил)-2-оксоимидазолидин-1-ил)-3-(6-(дифторметокси)пиридин-3-ил)пропионовой кислоты
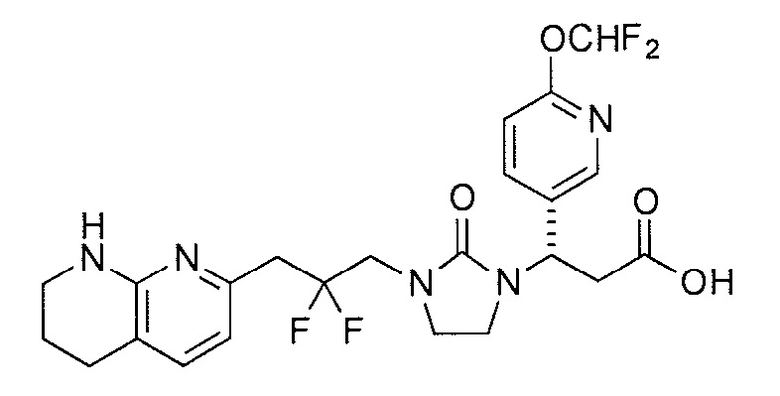
Способ синтеза является таким же, как в примере 1, за исключением замещения на стадии 8 2,2-дифтор-3-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)пропан-1-амина соединением 6b, и схема синтеза продолжается с применением тех же условий реакции.
Синтез 2,2-дифтор-3-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)пропан-1-амина проводят, как показано на схеме 3.
Пример 10. Синтез (S)-3-(3-(2,2-дифтор-3-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)пропил)-2-оксоимидазолидин-1-ил)-3-(6-метоксипиридин-3-ил)пропионовой кислоты
Схема синтеза является такой же, как в примере 9, за исключением того, что во время синтеза на стадии 1 применяют 2-хлорацетат натрия вместо 2-хлор-2,2-дифторацетата натрия. Синтез проводят в тех же условиях, что и в примере 3.
Пример 10-1. Схема 3
Схема 3
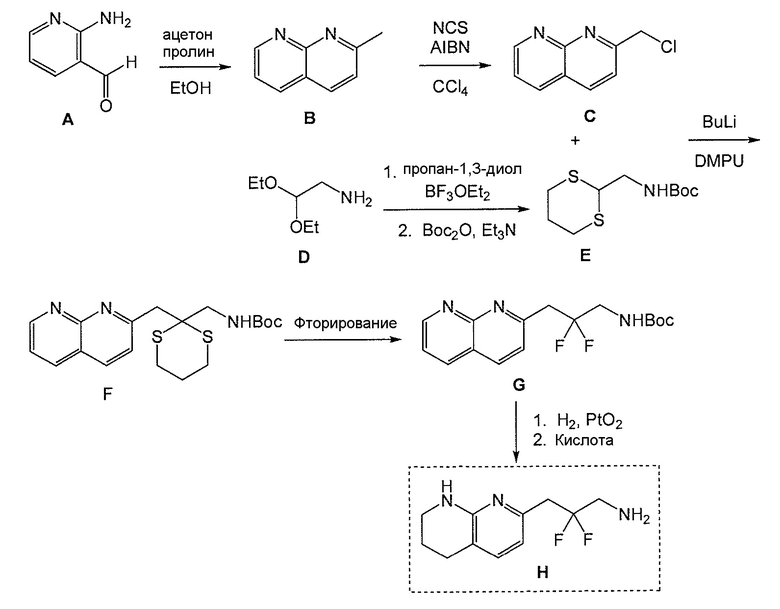
Приготовление промежуточных соединений C и E подробно описано в литературе и показано выше (для C; WO2011150156 и для E; Seebach, D. et al., Liebigs Ann. Chem., 1994, 701-717). Образование дианиона E было описано и применено для вытеснения замещенных бензиловых хлоридов (Bradshaw, B. et al., Org. Biomol. Chem., 2008, 6:2138-2157.). Это событие схожим образом приводит к образованию комплексного дитиана F. Фтородесульфурирование тиокеталей было описано для нескольких реагентов (Sondej, S. C. et al., J. Org. Chem., 1986, 51:3508-13.); промежуточное соединение G восстанавливается и становится незащищенным, как описано в литературе (US20040038963). Фрагмент H применяют в известном способе для образования целевых соединений.
Пример 11. Тестирование соединений настоящего изобретения в исследованиях клеточной адгезии
Способность соединений блокировать адгезию трех первичных клеточных культур - человеческих эндотелиальных клеток микрососудов дермы (HMVEC), крысиных эндотелиальных клеток микрососудов легких (RLMVEC) и кроличьих эндотелиальных клеток аорты (RAEC) - к покрытым витронектином планшетам определяли, применяя следующую процедуру. Этот тест показывает ингибирование взаимодействия αv интегрина на клеточной поверхности с лигандом витронектином.
Приготовление адгезивных планшетов. 96-луночные планшеты покрывали витронектином в фосфатно-солевом буферном растворе, pH7,4, инкубируя 50 мкл раствора (10 мкг/мл) в течение 1,5 ч при комнатной температуре или в течение ночи при 4°C. Планшеты блокировали 1% БСА в фосфатно-солевом буферном растворе (30 мин при комнатной температуре) и промывали фосфатно-солевым буферным раствором.
Культивирование клеток и обработка. Клетки HMVEC (пассажи (p)9-14) (из Lonza, Allendale, штат Нью-Джерси), клетки RLMVEC (p4-14) (из Vec Technology, Rensselaer, штат Нью-Йорк) и клетки RAEC (p4-14) (из CellBiologics, Chicago, штат Иллинойс) применяли для тестирования соединения. Клетки выращивали во флаконах для культивирования ткани T175 и снимали мягкой обработкой Accutase (Life Technologies) в течение 3 минут. После промывки клетки в суспензии в RPMI-1640 (Life Technologies) обрабатывали кальцеином-AM (5 мкМ) (Life Technologies) в течение 30 мин при 37°C и ресуспендировали в среде RPMI без фенолового красного, содержащей 10% фетальной бычьей сыворотки.
Исследование адгезии. Аликвоты клеточной суспензии вносили в лунки при плотности 1,0×105 клеток/лунка (RLMVEC) и 5,0×104 (HMVEC и RAEC). Тестируемые соединения добавляли одновременно с клетками. Планшеты инкубировали в течение 1,5 ч при 37°C. Клетки, которые не прикрепились во время этой инкубации, удаляли мягким промыванием. Промывание проводили в 2 цикла аспирации супернатанта и добавляли 100 мкл предварительно нагретого свежего фосфатно-солевого буфера Дульбекко (Life Technologies). Флуоресценцию оставшихся клеток измеряли, применяя многорежимный планшетный спектрофотометр (Victor 2V, PerkinElmer) при длинах волн возбуждения/испускания, равных 485/535 нм. Соединения тестировали, начиная с максимальной концентрации, равной 1 мкМ по полулогарифмической схеме разведения. Значения IC50 рассчитывали с помощью Prism 5 (GraphPad, штат Калифорния), фиксируя нижнюю часть кривых на контрольном значении для флуоресценции пустых лунок.
Как показано в таблице 2, все фторсодержащие антагонисты αv активны в отношении ингибирования клеточной адгезии к витронектину через αv интегрин. Нефлуоресцирующий контрольный антагонист, L-845704, показан для сравнения.
адгезию разных клеточных культур к витронектину
Пример 12. Исследование антиангиогенной активности с помощью куриной хориоаллантоисной мембраны (CAM)
Поверхности CAM пересаживали внутри желатиновых губок, заполненных концентрациями тестируемых веществ и 50 нг VEGF, растворенного в фосфатно-солевом буферном растворе. Необработанные CAM получали только VEGF и фосфатно-солевой буферный раствор. Погрешности представляют собой стандартную ошибку среднего, N=5, значения P для обработанных групп рассчитывали посредством сравнения с необработанной группой (*p<0,05, **p<0,01, ***p<0,001).
Приготовление тестируемого вещества: Тестируемые образцы и стандарты растворяли в фосфатно-солевом буферном растворе и стерилизовали, пропуская через шприцевой фильтр (0,22 мкм). hVEGF (SIGMA) 50 нг/мкл готовили в стерильном фосфатно-солевом буферном растворе.
Пересадка: Желатиновую губку (Abogel) нарезали на кусочки приблизительно 2 мм3 и заполняли необходимым тестируемым веществом или фосфатно-солевым буферным раствором и VEGF. Трансплантат помещали на CAM.
Яйца: Оплодотворенные куриные яйца получали из инкубатора, мыли и дезинфицировали спиртом. Отбирали 1 мл альбумина с помощью шприца и инкубировали в течение 8 дней. Трансплантаты помешали на развивающиеся САМ и далее инкубировали до дня 12. В день 12 CAM фиксировали 4% формальдегидом в фосфатно-солевом буферном растворе, готовили срезы и снимали изображения.
Получение изображений: Фиксированные CAM снимали под постоянным освещением и под увеличением с помощью стереомикроскопа, снабженного цифровой камерой (CANON).
Анализ изображений: Изображения анализировали с помощью MS PowerPoint, оставляя размер изображений постоянным. Вокруг трансплантата рисовали кольцо и сохраняли размер постоянным. Считали число кровеносных сосудов, пересекающих кольцо, в каждой исследуемой группе.
Статистический анализ: данные анализировали в MS Excel 2007.
Как показано на фигуре 1, каждое из соединений A1 и A2 обладает антиангиогенной активностью в исследовании куриных CAM и значительно уменьшает число кровеносных сосудов по сравнению с необработанным контролем.
Пример 13. Распределение в плазме, внутриглазной жидкости, стекловидном теле и в сетчатке после местного введения в глаза голландских кроликов.
Концентрации в плазме и окулярное распределение (во внутриглазной жидкости, стекловидном теле и в сетчатке) соединений A1, A2 и A3 определяли после местного введения в глаза голландских кроликов. Тестируемые соединения вводили в каждый глаз в объеме 50 мкл/глаз в концентрации, равной 1,0-2,5 мг/мл (соединение A2, 1,0 мг/мл; соединения A1 и A3 в концентрации 2,5 мг/мл). Образцы плазмы и разных окулярных тканей собирали в заранее определенные временные точки (1,0 и 8,0 часов для соединения A1; 0,5 и 8 часов для соединений A2 и A3). Внутриглазную жидкость, стекловидное тело и сетчатку отбирали из каждого глаза в каждой временной точке после приема дозы. Также регистрировали вес. Концентрации соединений в плазме и в глазу определяли с помощью жидкостной хроматографии/ тандемной масс-спектрометрии.
Введение доз животным: Действие соединений A1, A2 и A3 оценивали в голландских кроликах. Исследование не было слепым. Каждое соединение дозировали как n=3/временная точка всего для девяти кроликов. Кроликов содержали по одному в клетке. Животных не ограничивали в еде, поставляли пищу и воду ad libitum.
Животным вводили анестезию в соответствии с протоколом дозирования 13IA5 IACUC. Каждый кролик получал разовую дозу тестируемого состава посредством местного окулярного введения в оба глаза во время 0 в день введения дозы. Образцы плазмы и окулярные образцы собрали в заранее определенных временных точках. Во временных точках 30 минут и 1 час животным вводили анестезию на весь период исследования. Во временной точке 8 часов животные восстанавливались после введения дозы и их умерщвляли для сбора образцов.
В каждой временной точке отбирали приблизительно 0,5 мл крови и помещали в охлажденные Na-гепариновые пробирки, содержащие лимонную кислоту. Образцы крови центрифугировали со скоростью 3000 g в течение 5 минут для скорейшего получения плазмы. До анализа образцы хранили в замороженном виде при -80°C. Животных умерщвляли в соответствии с протоколом 13IA5 IACUC и немедленно энуклеировали оба глаза. После энуклеации каждый глаз промывали фосфатно-солевым буферным раствором. Отбирали окулярные образцы из обоих глаз каждого животного и регистрировали вес. Все образцы сразу замораживали на сухом льду и хранили при температуре от -60 до -80°C для анализа.
Анализ образцов плазмы и окулярных образцов: Для определения соединений А1, А2 и А3 в образцах плазмы и в окулярных образцах кролика разработали способ жидкостной хроматографии/тандемной масс-спектрометрии. Стандартную кривую, полученную до исследования, анализировали, чтобы определить специфичность, интервал и нижний предел количественного определения способа.
Как показано на фигурах 2, 3 и 4, каждое из соединений A1, A2 и A3 эффективно распределяются в сетчатку.
Следующие примеры офтальмологических составов представлены в иллюстративных целях:
Пример 14. Офтальмологический состав соединения A1
Активные соединения добавляли к раствору боратного буфера, содержащему β- циклодекстринсульфобутиловый эфир, динатрия эдетат и пропиламинопропилбигуанид, растворенные в стерильной воде для инъекций в тарированном стерильном сосуде. pH раствора доводили до 7,5 добавлением соляной кислоты. Композицию стерилизуют фильтрованием через 0,45-микронный фильтр.
Пример 15. Офтальмологический состав соединения A1
Активные соединения добавляли к раствору боратного буфера, содержащему гидроксипропил β-циклодекстрин, динатрия эдетат и пропиламинопропилбигуанид, растворенные в стерильной воде для инъекций в тарированном стерильном сосуде. pH раствора доводили до 7,5 добавлением соляной кислоты. Композицию стерилизуют фильтрованием через 0,45-микронный фильтр.
Пример 16. Офтальмологический состав соединения A2
Активные соединения добавляли к раствору боратного буфера, содержащему β-циклодекстринсульфобутиловый эфир, динатрия эдетат и пропиламинопропилбигуанид, растворенные в стерильной воде для инъекций в тарированном стерильном сосуде. pH раствора доводили до 7,5 добавлением соляной кислоты. Композицию стерилизуют фильтрованием через 0,45-микронный фильтр.
Пример 17. Офтальмологический состав соединения A3
Активные соединения добавляли к раствору боратного буфера, содержащему β-циклодекстринсульфобутиловый эфир, динатрия эдетат и пропиламинопропилбигуанид, растворенные в стерильной воде для инъекций в тарированном стерильном сосуде. pH раствора доводили до 7,5 добавлением соляной кислоты. Композицию стерилизуют фильтрованием через 0,45-микронный фильтр.
Пример 18. Оценка безопасности и эффективности наносимых местно тестовых соединений в модели индуцированной лазером хороидальной неоваскуляризации в голландских кроликах.
В этих исследованиях применяли здоровых самцов кроликов весом от 1,5 до 2,0 кг. Животных взвешивали перед введением дозы и во время эвтаназии и чаще, если это требовалось. Съемку глазного дна в исходный период и флуоресцеиновую ангиографию проводили для каждого животного перед индукцией хороидальной неоваскуляризации.
Животным вводили анестезию посредством внутримышечной инъекции гидрохлорида кетамина (20 мг/кг) и ксилазина (2 мг/кг) для индукции хороидальной неоваскуляризации, фотографии глазного дна, флуоресцеиновой ангиографии и интравитреальных инъекций. Кроликов держали на изофлуране (приблизительно до 3%) с кислородом (приблизительно от 1 до 2 л/мин), если это было необходимо. Одну каплю местного пропаракаингидрохлоридного анестетика (0,5%) наносили в каждый глаз перед проведением процедур. Дополнительную местную окулярную анестезию применяли во время процедуры, если в этом была необходимость.
Хороидальную неоваскуляризацию индуцировали лазерной фотокоагуляцией. Внешний диодный лазер наводили на сетчатку с помощью лазерных контактных линз и биомикроскопа со щелевой лампой. В День 1 оба глаза каждого животного обрабатывали лазерной коагуляцией со следующими настройками лазера:
Количество пятен: 12-15 пятен на глаз
Интервал мощности: 50-200 мВт
Размер пятна: 20-100 мкм
Время: 0,05-0,1 секунд
После обработки лазером 50 мкл раствора 25 мкг/мл VEGF (доза 1,25 мкг) инъецировали в стекловидное тело каждого глаза. Ежедневные макроскопические исследования глаз проводили на протяжении всего периода исследования.
Клинические офтальмологические исследования (биомикроскопию со щелевой лампой и непрямую офтальмоскопию), фотографирование глазного дна и флуоресцеиновую ангиографию проводили в исходный период и затем каждую неделю на протяжении 6 недель после индукции. Исследования оценивали с помощью системы баллов Макдоналда-Шеддака (McDonald-Shadduck). Получение изображений с помощью оптической когерентной томографии проводили каждую неделю для получения диагностических изображений во время исследований.
В последний день исследования отбор проб проводили сразу перед введением дозы AM и через 2 часа после введения дозы. Образцы крови центрифугировали со скоростью 3000 g в течение 5 минут для скорейшего получения плазмы. До анализа образцы хранили в замороженном виде при -80°C. При завершении исследования животных умерщвляли в соответствии с протоколом 13IA5 IACUC и немедленно энуклеировали оба глаза. После энуклеации каждый глаз промывали фосфатно-солевым буферным раствором. Отбирали окулярные образцы (внутриглазной жидкости, стекловидного тела и сосудистой оболочки) из обоих глаз каждого животного и регистрировали вес. Все образцы сразу замораживали на сухом льду и хранили при температуре от -60 до -80°C для анализа.
Как показано на фигуре 5, соединение A1 или соединение A2 эффективно снижали индуцированную лазером хороидальную неоваскуляризацию по сравнению с контрольным инертным носителем.
ЭКВИВАЛЕНТЫ
Специалисты в данной области техники распознают или будут иметь возможность применять не более чем рутинные экспериментальные исследования, многие эквиваленты специальных вариантов осуществления изобретения и способы, описанные в данной заявке. Предполагается, что такие эквиваленты должны быть включены в объем настоящего изобретения.
Все патенты, патентные заявки и ссылки на литературу, приведенные в данной заявке, явным образом включены в данную заявку посредством ссылки.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ НАФТИРИДИНА В КАЧЕСТВЕ АНТАГОНИСТОВ АЛЬФА V БЕТА 6 ИНТЕГРИНА ДЛЯ ЛЕЧЕНИЯ, В ЧАСТНОСТИ, ФИБРОЗНЫХ ЗАБОЛЕВАНИЙ | 2015 |
|
RU2692775C2 |
| СОЕДИНЕНИЯ ПИПЕРИДИНИЛА, СЕЛЕКТИВНО СВЯЗЫВАЮЩИЕ ИНТЕГРИНЫ | 2003 |
|
RU2333210C2 |
| ИНГИБИТОРЫ ИНТЕГРИНА AVB6 | 2018 |
|
RU2769702C2 |
| ИНГИБИТОРЫ TrkA КИНАЗЫ, ОСНОВАННЫЕ НА НИХ КОМПОЗИЦИИ И СПОСОБЫ | 2015 |
|
RU2672583C2 |
| ПРОИЗВОДНЫЕ 3-КАРБОКСИПРОПИЛ-АМИНОТЕТРАЛИНА И РОДСТВЕННЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ АНТАГОНИСТОВ MU-ОПИОИДНОГО РЕЦЕПТОРА | 2008 |
|
RU2482107C2 |
| СОЕДИНЕНИЯ, ОБЛАДАЮЩИЕ ИЗБИРАТЕЛЬНОСТЬЮ В ОТНОШЕНИИ РЕЦЕПТОРОВ РЕТИНОИДА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ (ВАРИАНТЫ), СПОСОБ МОДУЛИРОВАНИЯ ПРОЦЕССОВ (ВАРИАНТЫ), СПОСОБ ПОДАВЛЕНИЯ ПАТОЛОГИЧЕСКИХ СОСТОЯНИЙ | 1993 |
|
RU2144913C1 |
| НОВЫЕ ПРОИЗВОДНЫЕ ПИРИДИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2004 |
|
RU2366659C2 |
| ПРОИЗВОДНОЕ НИТРОИМИДАЗОЛА ПРОТИВ ТУБЕРКУЛЕЗА ЛЕГКИХ | 2016 |
|
RU2675622C1 |
| ИНГИБИТОРЫ СЕРИН/ТРЕОНИН КИНАЗЫ ДЛЯ ЛЕЧЕНИЯ ГИПЕРПРОЛИФЕРАТИВНЫХ ЗАБОЛЕВАНИЙ | 2013 |
|
RU2644947C2 |
| ТИОФЕНИЛЬНЫЕ И ПИРРОЛИЛЬНЫЕ АЗЕПИНЫ В КАЧЕСТВЕ ЛИГАНДОВ СЕРОТОНИНОВОГО 5-HT РЕЦЕПТОРА И ИХ ПРИМЕНЕНИЕ | 2007 |
|
RU2434872C2 |
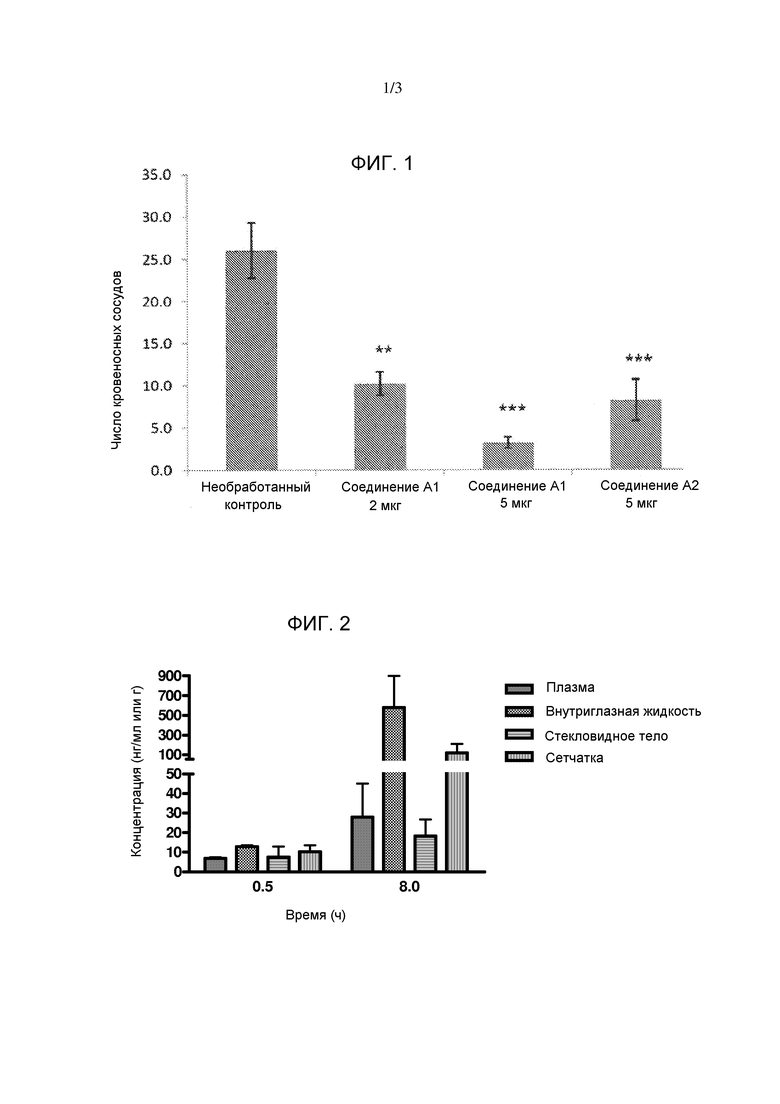
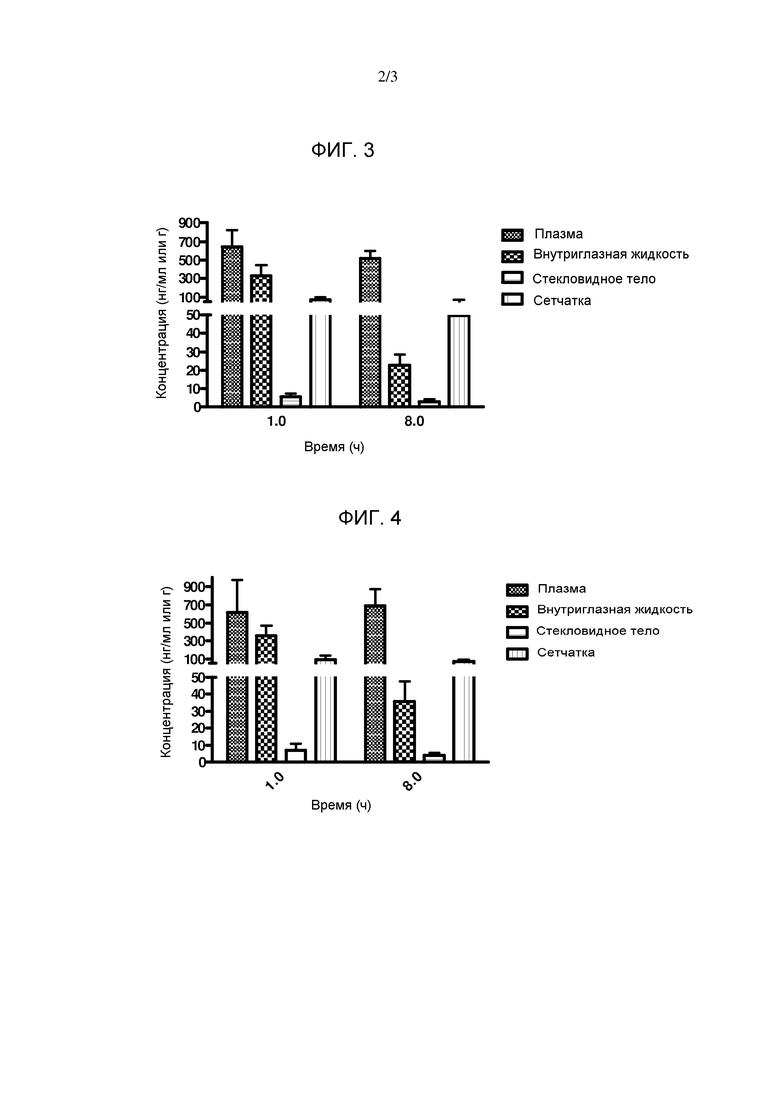
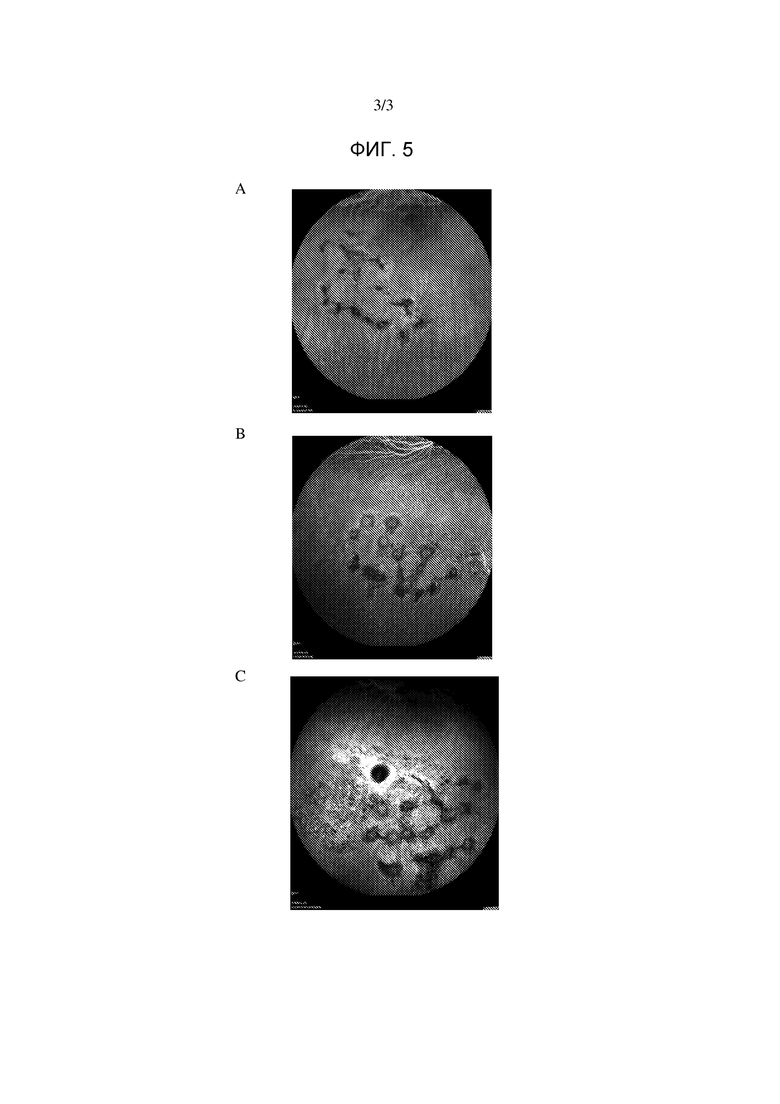
Изобретение относится к области органической химии, а именно к гетероциклическому соединению формулы I или к его фармацевтически приемлемой соли, где Z является 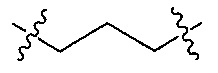 или
или 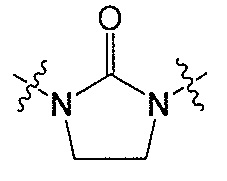 ; R и R’ обозначают H; Q является
; R и R’ обозначают H; Q является 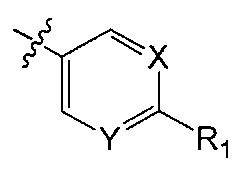 или
или 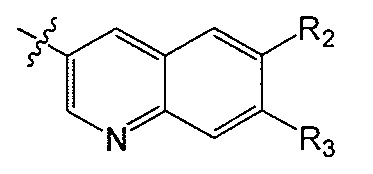 ; X является CH или N; Y является CH или N; R1 является C1-C4 алкилом, замещенным 1, 2, 3, 4, 5, 6, 7, 8 или 9 атомами фтора, или C1-C6 алкоксигруппой, замещенной 1, 2, 3, 4, 5, 6 или 7 атомами фтора; и R2 и R3 независимо друг от друга обозначают H, F, CH2F, CHF2 или CF3, при условии, что один из R2 и R3 не является H, при условии, что соединение формулы I содержит по меньшей мере один атом фтора. Также изобретение относится к фармацевтической композиции на основе соединения формулы I и применению соединения формулы I. Технический результат: получены новые гетероциклические соединения, полезные для лечения или предотвращения заболевания, опосредованного αv интегрином. 4 н. и 33 з.п. ф-лы, 5 ил., 9 табл., 18 пр.
; X является CH или N; Y является CH или N; R1 является C1-C4 алкилом, замещенным 1, 2, 3, 4, 5, 6, 7, 8 или 9 атомами фтора, или C1-C6 алкоксигруппой, замещенной 1, 2, 3, 4, 5, 6 или 7 атомами фтора; и R2 и R3 независимо друг от друга обозначают H, F, CH2F, CHF2 или CF3, при условии, что один из R2 и R3 не является H, при условии, что соединение формулы I содержит по меньшей мере один атом фтора. Также изобретение относится к фармацевтической композиции на основе соединения формулы I и применению соединения формулы I. Технический результат: получены новые гетероциклические соединения, полезные для лечения или предотвращения заболевания, опосредованного αv интегрином. 4 н. и 33 з.п. ф-лы, 5 ил., 9 табл., 18 пр.
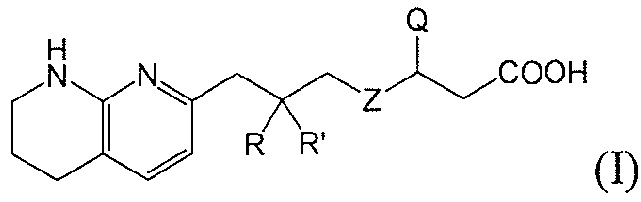
1. Соединение формулы I:
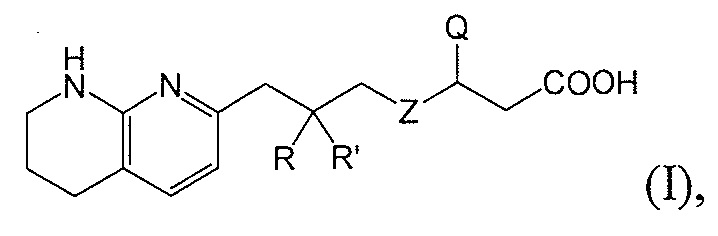
или его фармацевтически приемлемая соль, где:
Z является  или
или 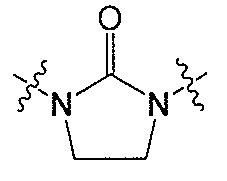 ;
;
R и R’ обозначают H;
Q является 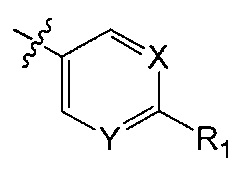 или
или 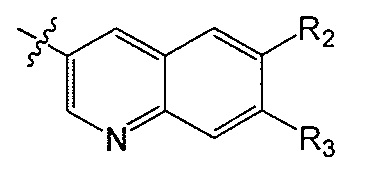 ;
;
X является CH или N;
Y является CH или N;
R1 является C1-C4 алкилом, замещенным 1, 2, 3, 4, 5, 6, 7, 8 или 9 атомами фтора, или C1-C6 алкоксигруппой, замещенной 1, 2, 3, 4, 5, 6 или 7 атомами фтора; и
R2 и R3 независимо друг от друга обозначают H, F, CH2F, CHF2 или CF3, при условии, что один из R2 и R3 не является H,
при условии, что соединение формулы I содержит по меньшей мере один атом фтора.
2. Соединение по п.1, где Q является 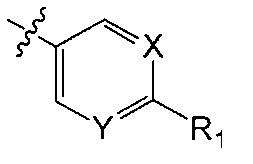 .
.
3. Соединение по п.2, где X является N и Y является CH.
4. Соединение по п.2, где X и Y каждый является CH.
5. Соединение по п.2, где X и Y каждый является N.
6. Соединение по п.2, где R1 является C1-C4 алкилом с прямой цепью или разветвленным C3-C4 алкилом и является замещенным 1, 2, 3, 4, 5, 6, 7, 8 или 9 атомами фтора.
7. Соединение по п.2, где R1 является метилом, замещенным 1, 2 или 3 атомами фтора.
8. Соединение по п.7, где R1 является CF3.
9. Соединение по п.2, где R1 является C1-C6 алкоксигруппой с прямой цепью или разветвленной C3-C6 алкоксигруппой и является замещенным 1, 2, 3, 4, 5, 6 или 7 атомами фтора.
10. Соединение по п.9, где R1 является метокси, замещенным 1, 2 или 3 атомами фтора.
11. Соединение по п.10, где R1 является OCHF2.
12. Соединение по п.2, где Z является  и R1 является метилом, замещенным 1, 2 или 3 атомами фтора, или метокси, замещенным 1, 2 или 3 атомами фтора.
и R1 является метилом, замещенным 1, 2 или 3 атомами фтора, или метокси, замещенным 1, 2 или 3 атомами фтора.
13. Соединение по п.12, где X является N и Y является CH; и R1 является OCHF2.
14. Соединение по п.12, где X и Y каждый является N; и R1 является CF3.
15. Соединение по п.2, где Z является  ; X является N и Y является CH; и R1 является метокси, замещенным 1, 2 или 3 атомами фтора.
; X является N и Y является CH; и R1 является метокси, замещенным 1, 2 или 3 атомами фтора.
16. Соединение по п.15, где R1 является OCHF2.
17. Соединение по п.1, имеющее формулу II:
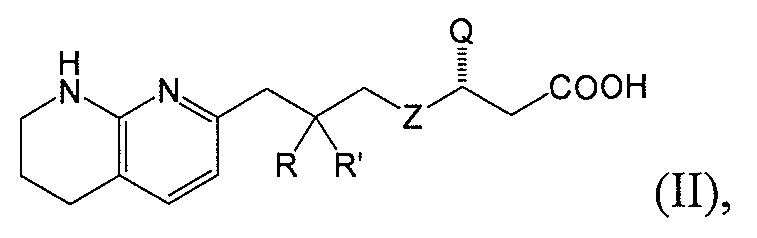
или его фармацевтически приемлемая соль.
18. Соединение по п.1, выбираемое из группы, состоящей из:
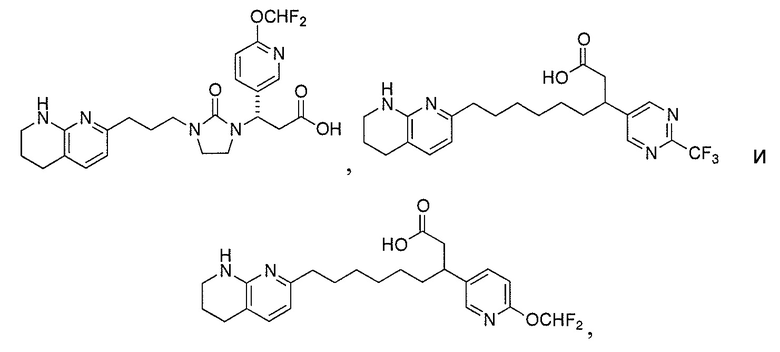
или его фармацевтически приемлемая соль.
19. Соединение по п.18, где соединение является 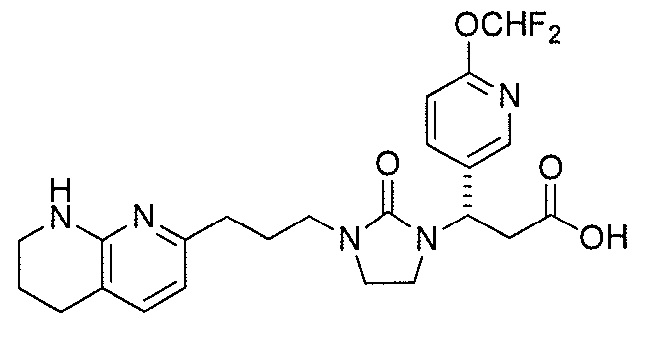 , или его фармацевтически приемлемая соль.
, или его фармацевтически приемлемая соль.
20. Соединение по п.1, выбранное из группы, состоящей из:
или его фармацевтически приемлемая соль.
21. Фармацевтическая композиция для лечения или предотвращения заболевания или состояния, опосредованного αv интегрином, содержащая эффективное количество соединения по п.1 или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель или эксципиент.
22. Фармацевтическая композиция по п.21, содержащая водный инертный носитель.
23. Фармацевтическая композиция по п.22, где водный инертный носитель содержит офтальмологически приемлемый эксципиент, выбираемый из группы, состоящей из усиливающего растворимость агента, хелатирующего агента, консерванта, тонического агента и буфера и их смеси.
24. Фармацевтическая композиция по п.23, где усиливающий растворимость агент является циклодекстрином.
25. Фармацевтическая композиция по п.24, где циклодекстрин выбирают из группы, состоящей из гидроксипропил-β-циклодекстрина, метил-β-циклодекстрина, случайно метилированного β-циклодекстрина, этилированного β-циклодекстрина, триацетил-β-циклодекстрина, перацетилированного β-циклодекстрина, карбоксиметил-β-циклодекстрина, гидроксиэтил-β-циклодекстрина, 2-гидрокси-3-(триметиламмоний)пропил-β-циклодекстрина, глюкозил-β- циклодекстрина, сульфатированного β-циклодекстрина (S-β-CD), мальтозил-β-циклодекстрина, β-циклодекстринсульфобутилового эфира, разветвленного β-циклодекстрина, гидроксипропил-γ-циклодекстрина, случайно метилированного γ-циклодекстрина и триметил-γ-циклодекстрина и их смеси.
26. Фармацевтическая композиция по п.23, где хелатирующий агент выбирают из группы, состоящей из этилендиаминтетрауксусной кислоты и ее солей металлов и их смеси.
27. Фармацевтическая композиция по п.23, где консервант выбирают из группы, состоящей из галоида бензалкония, хлоргексидина глюконата, бензетония хлорида, цетилпиридина хлорида, бензилбромида, нитрата фенилртути, ацетата фенилртути, неодеканоата фенилртути, мертиолата, метилпарабена, пропилпарабена, сорбиновой кислоты, сорбата калия, бензоата натрия, пропионата натрия, этил-п-гидроксибензоата, пропиламинопропилбигуанида и бутил-п-гидроксибензоата, сорбиновой кислоты и их смеси.
28. Фармацевтическая композиция по п.23, где тонический агент выбирают из группы, состоящей из гликоля, глицерола, декстрозы, глицерина, маннита, хлорида калия и хлорида натрия и их смеси.
29. Фармацевтическая композиция по п.23, где фармацевтическую композицию составляют в форме для местного глазного введения.
30. Применение соединения по п.1 в лечении или предотвращении заболевания или состояния, опосредованного αv интегрином, у субъекта.
31. Применение по п.30, где αv интегрин представляет собой αvβ3 или αvβ5 интегрин.
32. Применение по п.30, где заболевание или состояние выбирают из группы, состоящей из макулярной дегенерации, диабетической ретинопатии (ДР), макулярного отека, диабетического макулярного отека (ДМО) и макулярного отека после окклюзии вены сетчатки.
33. Применение по п.30, где соединение вводят местно.
34. Применение соединения по п.1 в производстве лекарственного средства для лечения или предотвращения заболевания или состояния, опосредованного αv интегрином, у субъекта.
35. Применение по п.34, где αv интегрин представляет собой αvβ3 или αvβ5 интегрин.
36. Применение по п.34, где заболевание или состояние выбирают из группы, состоящей из макулярной дегенерации, диабетической ретинопатии (ДР), макулярного отека, диабетического макулярного отека (ДМО) и макулярного отека после окклюзии вены сетчатки.
37. Применение по п.34, где лекарственное средство вводят местно.
| Хранилище сочной сельскохозяйственной продукции | 1980 |
|
SU1040098A1 |
| Аппарат для автоматического тартания нефти из буровых | 1923 |
|
SU3095A1 |
| WO 2002007730 A1, 31.01.2002. | |||

