Изобретение относится к органической и фармацевтической химии, а именно к фторсодержащим арилнитроциклопропанам, конкретно к E-2-арил-2-трифторметил-1-нитроциклопропанам общей формулы I, обладающим противоопухолевой активностью, и к способу их получения.
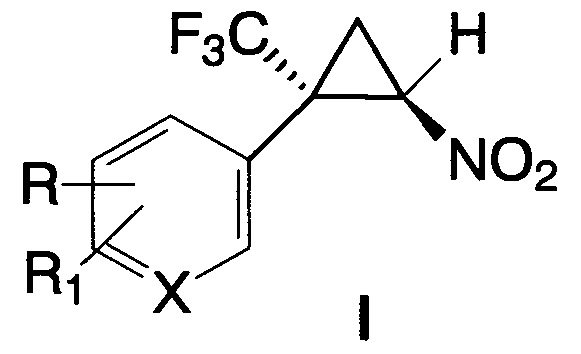
где X=С, R=R1=Н; X=С, R=Cl, R1=Н; X=С, R=R1=Cl; X=С, R=Br, R1=Н;
X=С, R=R1=Br; X=С, R=C1-С3-алкил, R1=Н; X=С, R=NO2, R1=Н;
X=С, R=CO2Alk, R1=Н; X=С, R=CF3, R1=Н; X=С, R=ОМе, R1=Н;
X=С, R=R1=ОМе; X=С, R=F, R1=Н; X=С, R-R1=-OCH2O-; X=N, R=R1=Н.
Заявляемые соединения, их свойства и способы получения в литературе не описаны.
Известно, что соединения, содержащие циклопропановое ядро, проявляют широкий спектр биологической активности [ , Baird M.S. Current Med. Chem., 1995, 2 (1), 511-542; Top. Curr. Chem., 2000, 207, 1-67; Wessjohann L.A., Brandt W. Chem. Rev., 2003, 103 (4), 1625-1647]. Ярким примером противоопухолевых агентов, имеющих в своей структуре циклопропильный фрагмент, являются антибиотики класса дуокармицина, в частности (+)-ятакемицин (1) [Mullins Е. A., Shi R., Eichman В. F. Nature Chemical Biology, 2017, 13, 1003-1005].
, Baird M.S. Current Med. Chem., 1995, 2 (1), 511-542; Top. Curr. Chem., 2000, 207, 1-67; Wessjohann L.A., Brandt W. Chem. Rev., 2003, 103 (4), 1625-1647]. Ярким примером противоопухолевых агентов, имеющих в своей структуре циклопропильный фрагмент, являются антибиотики класса дуокармицина, в частности (+)-ятакемицин (1) [Mullins Е. A., Shi R., Eichman В. F. Nature Chemical Biology, 2017, 13, 1003-1005].
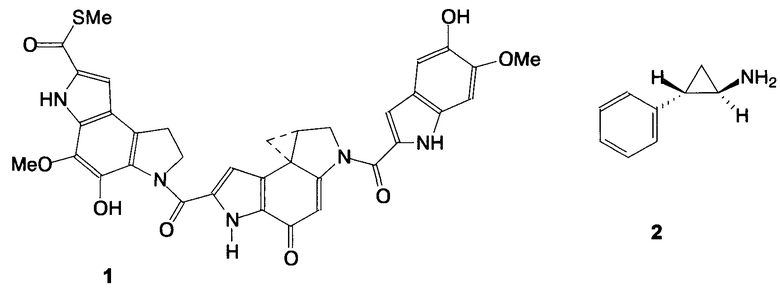
К одному из перспективных соединений с точки зрения создания новых противоопухолевых препаратов относится транилципромин (2). Это обусловлено тем, что он является ингибитором лизинспецифической деметилазы 1 (LSD1) - энзима, вовлеченного в механизм регулирования роста в онкогенезе. В настоящее время соединение 2 рассматривается как начальный член ряда для молекулярного дизайна противоопухолевых препаратов [Lee М. G., Wynder С, Schmidt D. М., McCafferty D. G., Shiekhattar R. Chem. Biol., 2006, 13 (6), 563-567]. Показано, что введение гидрофобных заместителей в циклопропильное ядро транилципромина приводит к усилению его ингибирующей активности в отношении LSD1 вследствие улучшения взаимодействия с гидрофобными карманами белка-мишени [Vianello P., Botrugno О.А., Сарра A. et al. Europ. J. Med. Chem., 2014, 86, 352-363]. В качестве гидрофобного заместителя может выступать трифторметильная группа, введение которой в биоактивные молекулы в целях улучшения их биологических свойств представляет собой перспективное направление в дизайне новых лекарственных средств [Gillis Е.Р., Eastman K.J., Hill M.D. et al., J. Med. Chem., 2015, 58 (21), 8315-8359; Wang J.,  Acenall J.L. et al, Chem. Rev., 2014, 114 (4), 2432-2506]. Предшественниками трифторметилсодержащих 1-амино-2-арилциклопропанов, потенциально обладающих противоопухолевым действием, служат соответствующие нитропроизводные, которые и являются предметом настоящего изобретения.
Acenall J.L. et al, Chem. Rev., 2014, 114 (4), 2432-2506]. Предшественниками трифторметилсодержащих 1-амино-2-арилциклопропанов, потенциально обладающих противоопухолевым действием, служат соответствующие нитропроизводные, которые и являются предметом настоящего изобретения.
В литературе известны структурные аналоги заявляемых соединений.
Описан 2-фенил-1-трифторметил-1-нитроциклопропан 3, который был получен по реакции стирола с трифторметилнитродиазометаном в присутствии тетраацетата диродия [О'Bannon P. Е., Dailey W. P. Tetrahedron, 1990, 46 (21), 7341-7358]. Однако, во-первых, соединение 3 было выделено в виде смеси геометрических изомеров, во-вторых, используемый для его получения трифторметилнитродиазометан взрывоопасен, к тому же быстро разлагается при комнатной температуре до ядовитого трифторнитрозометана, а в-третьих, родиевый катализатор дорог.
Описаны 2-трифторметил-3-арилнитроциклопропаны 4, получаемые взаимодействием солей трифторэтилдифенилсульфония с нитростиролами в присутствии основания (реакция Кори-Чайковски) [Hock K.J., Hommelsheim R., Mertens L., Ho J., Nguyen T.V., Koenigs R. M. J. Org. Chem., 2017, 82 (15), 8220-8227]. Однако способ получения исходных циклопропанирующих реагентов весьма трудоемок и требует использования высокого давления.
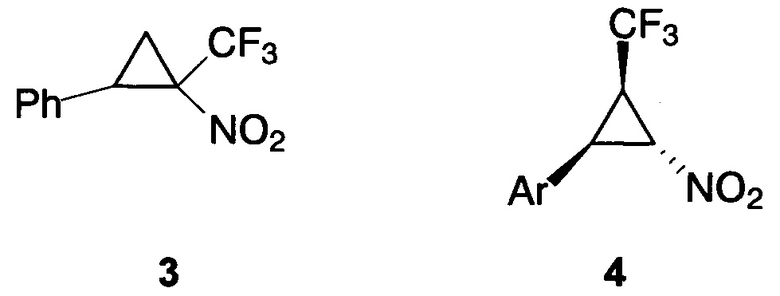
Данные о биологической активности, в том числе противоопухолевой, приведенных структурных аналогов заявляемых соединений в литературе отсутствуют.
Задачей настоящего изобретения является создание ранее неизвестных трифторметилсодержащих 2-арил-1-нитроциклопропанов, включающих комбинацию фармакофорных элементов транилципромина (циклопропановое и арильное ядра) и функциональной нитрогруппы, способной к восстановлению до аминогруппы.
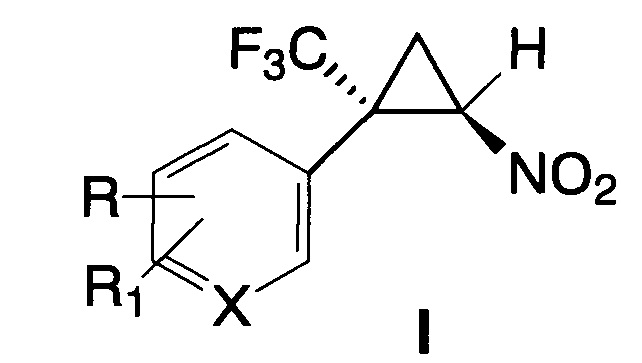
Поставленная задача решается новыми E-2-арил-2-трифторметил-1-нитроциклопропанами общей формулы I

где X=С, R=R1=Н; X=С, R=Cl, R1=Н; X=С, R=R1=Cl; X=С, R=Br, R1=Н;
X=С, R=R1=Br; X=С, R=C1-С3-алкил, R1=Н; X=С, R=NO2, R1=Н;
X=С, R=CO2Alk, R1=Н; X=С, R=CF3, R1=Н; X=С, R=ОМе, R1=Н;
X=С, R=R1=ОМе; X=С, R=F, R1=Н; X=С, R-R1=-OCH2O-; X=N, R=R1=Н,
обладающими противоопухолевой активностью в отношении клеток рака прямой кишки и острого миелогенного лейкоза человека, а также способом получения соединений общей формулы I, включающим взаимодействие соответствующего α-(трифторметил)стирола с бромнитрометаном в присутствии основания в полярном растворителе при комнатной температуре, причем бромнитрометан и основание используют в эквимолярных количествах в 4-15-кратном мольном избытке по отношению к α-(трифторметил)стиролу и вводят порциями.
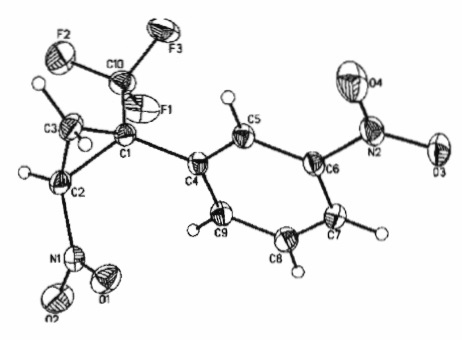
Способ получения заявляемых соединений представляет собой тандемную реакцию нуклеофильного присоединения депротонированного под действием основания бромнитрометана к двойной связи α-(трифторметил)стирола и последующего внутримолекулярного нуклеофильного замещения брома образующимся С-анионным центром, что приводит к формированию циклопропильного ядра (схема). Способ по изобретению характеризуется высокой стереоселективностью: заявляемые соединения имеют E-конфигурацию. Их строение установлено на основе данных 1Н-, 13С- и 19F-ЯМР-спектроскопии, масс-спектрометрии и элементного анализа, а пространственное транс-расположение нитро- и трифторметильной групп в циклопропановом ядре доказано с помощью рентгеноструктурного анализа и двумерной ЯМР-спектроскопии (1H-19F HOESY).
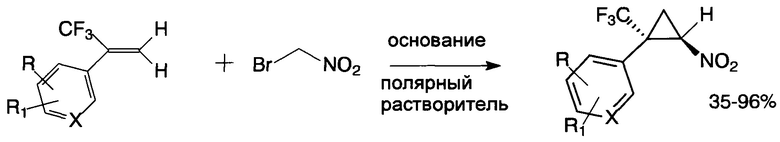
Циклопропанирующий реагент представляет собой доступный бромнитрометан [Fishwick B.R., Rowles D.K., Stirling C.J.M. Bromonitromethane - a versatile electrophile. J. Chem. Soc. Perkin Trans. 1, 1986, (7), 1171-1179] в тандеме с эквимолярным количеством основания. Процесс идет под действием как неорганического основания (например, карбонат калия, карбонат натрия, карбонат цезия, гидроксид лития в форме моногидрата, фосфат калия в форме тригидрата, оксид кальция), так и органического (например, 1,2-диметил-1,4,5,6-тетрагидропиримидин, 1,8-диазабицикло [5.4.0]ундец-7-ен, 1,5-диазабицикдо[4.3.0]нон-5-ен, триэтиламин, N-метилморфолин), в полярных растворителях (например, N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидон, диметилсульфоксид, 1,1,3,3-тетраметилмочевина, ацетонитрил) при комнатной температуре. Для неорганических оснований предпочтительным растворителем является диметилсульфоксид, для органических - 1,1,3,3-тетраметилмочевина или N,N-диметилформамид.
Важным условием достижения высоких выходов заявляемых соединений является использование избытков бромнитрометана и основания (4-15-кратных в мольном выражении) по отношению к α-(трифторметил)стиролу и введение их в реакцию порциями (в течение 12-48 ч). Количество циклопропанирующего реагента, продолжительность реакции и в конечном итоге выход соединений I зависят от строения исходного α-(трифторметил)стирола и от природы используемого основания.
Так, для получения E-2-арил-2-трифторметил-1-нитроциклопропанов формулы I, содержащих электроноакцепторную группу в бензольном ядре (X=С, R=Cl, Br, NO2, CO2Alk или CF3, R1=Н; X=С, R=R1=Cl или Br) либо электроноакцепторное ароматическое ядро (X=N, R=R1=Н), предпочтительно использовать карбонат калия в качестве основания и диметилсульфоксид в качестве растворителя: при этом для получения 64-96%-ных выходов заявляемых соединений необходимы 4-6-кратные мольные избытки бромнитрометана и К2СО3 при их двух-трехпорционном прибавлении и 18-24-часовая продолжительность реакции. Однако в случае α-(трифторметил)стиролов, несущих алкильную группу в бензольном ядре (X=С, R=C1-С3-алкил, R1=Н) или не имеющих заместителей в нем (X=С, R=R1=Н), выходы соответствующих 2-арил-2-трифторметил-1-нитроциклопропанов не превышали 15% при порционном добавлении (5-10 порций в течение 48 ч) 10-15-кратного мольного избытка бромнитрометана и К2СО3. А в случае α-(трифторметил)стиролов, несущих высоко электронодонорную группу в бензольном ядре (X=С, R=ОМе или F, R1=Н; X=С, R=R1=ОМе; X=С, R-R1=-OCH2O-), соответствующие 2-арил-2-трифторметил-1-нитроциклопропаны не были выделены вовсе вследствие крайне низкой конверсии указанных α-(трифторметил)стиролов на фоне порционного добавления (10 порций в течение 48 ч) 15-кратных мольных избытков бромнитрометана и К2СО3. Получить E-2-арил-2-трифторметил-1-нитроциклопропаны из малореакционноспособных α-(трифторметил)стиролов удалось, правда с низкими (от 10 до 40%) выходами, при использовании в качестве основания фосфата калия в виде K3PO4×3H2O или оксида кальция СаО, при этом 12-15-кратные избытки бромнитрометана и неорганического основания прибавляли порциями в течение 48 ч.
Для получения с выходами выше 50% E-2-арил-2-трифторметил-1-нитроциклопропанов (I), не имеющих заместителей в бензольном ядре или содержащих электронодонорные группы в нем, было предложено осуществлять циклопропанирование соответствующего α-(трифторметил)стирола в присутствии органического основания, предпочтительно 1,2-диметил-1,4,5,6- тетрагидропиримидина. Использование 1,2-диметил-1,4,5,6-тетрагидропиримидина позволяет значительно ускорить реакцию и существенно сократить количество бромнитрометана, видимо, за счет протекания циклопропанирования в гомогенных условиях. Процесс получения соответствующих E-2-арил-2-трифторметил-1-нитроциклопропанов включает двухпорционное прибавление 6-кратных мольных избытков бромнитрометана и 1,2-диметил-1,4,5,6-тетрагидропиримидина к раствору α-(трифторметил)стирола в N,N-диметилформамиде или 1,1,3,3-тетраметилмочевине и продолжается 22 ч. В наиболее неблагоприятных случаях высоко электронодонорных заместителей в α-(трифторметил)стиролах (X=С, R=ОМе, R1= =Н; X=С, R=R1=ОМе; X=С, R-R1=-ОСН2О-) выходы заявляемых соединений составляют 35-55% (35-45%).
Безусловно, можно использовать 1,2-диметил-1,4,5,6-тетрагидропиримидин в качестве основания и для получения 2-арил-2-трифторметил-1-нитроциклопропанов формулы I, содержащих электроноакцепторную группу в бензольном ядре (X=С, R=Cl, Br, NO2, CO2Alk или CF3, R1=Н; X=С, R=R1=Cl или Br) либо электроноакцепторное ароматическое ядро (X=N, R=R1=Н), однако для таких заявляемых соединений использование карбоната калия предпочтительнее с точки зрения удешевления процесса и более простой процедуры обработки реакции.
В итоге констатируем, что заявляемый способ получения E-2-арил-2-трифторметил-1-нитроциклопропанов общей формулы I характеризуется доступностью исходных реагентов, простотой осуществления, высокими выходами и стереоселективностью.
Неожиданно оказалось, что большинство полученных по изобретению соединений I, рассматриваемых в первую очередь как прекурсоры для потенциально противоопухолевых трифторметилсодержащих 1-амино-2-арилциклопропанов, сами обладают противоопухолевой активностью. Цитотоксическое действие заявляемых Е-2-арил-2-трифторметил-1-нитроциклопропанов определяли в отношении опухолевых клеток человека линий НСТ116 (рак толстой кишки) и HL-60 (острый миелогенный лейкоз) с помощью МТТ-теста - по способности восстанавливать желтый тетразолиевый краситель - 3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолийбромид - в нерастворимый формазан, который имеет пурпурное окрашивание дегидрогеназами митохондрий живых клеток.
Результаты исследования цитотоксичности, выраженные в величинах концентрации, необходимой для гибели 50% клеток (IC50), приведены в таблице.
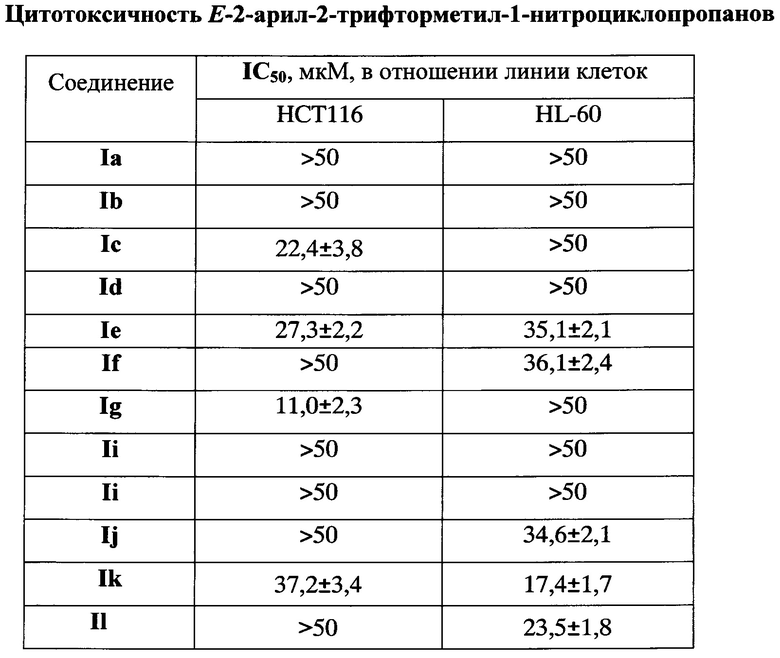
Технический результат изобретения - создание новых E-2-трифторметил-2-арил-1-нитроциклопропанов, обладающих противоопухолевой активностью, в частности в отношении клеток рака толстой кишки и острого миелогенного лейкоза человека и разработка простого и эффективного способа их получения.
Для получения исходных α-(трифторметил)стиролов использовали две синтетические схемы. Низкокипящие α-(трифторметил)стиролы (с температурой кипения ниже 200°С) получали высокотемпературной дегидратацией 2-арил-1,1,1-трифторпропан-2-олов пятиокисью фосфора по методу Тарранта [Tarrant P., Taylor R.E. J. Org. Chem., 1959, 24 (2), 238-239; Казенникова Г.В., Талалаева Т.В., Зимин А.В., Кочешков К.А. Изв. АН СССР. Отд. хим. наук, 1961, 1066]. Высококипящие α-(трифторметил)стиролы синтезировали по реакции Виттига кипячением смеси соответствующего α,α,α-трифторацетофенона, метилтрифенилфосфонийбромида или - йодида (1.25 экв.) и карбоната калия (1.7 экв.) в сухом 1,2-диметоксиэтане или диоксане в течение 5 ч [Сиган А.Л., Голубев А.С., Беляева Е.В. и др. Изв. АН. Сер. хим., 2019, (1), 99-103]. Бромнитрометан получали согласно методике, приведенной в работе [Fishwick B.R., Rowles D.K., Stirling C.J.M. J. Chem. Soc. Perkin Trans. 1, 1986, (7), 1171-1179].
Спектры ЯМР 1Н, 13С{1Н} и 19F{1Н} регистрировали на спектрометре Bruker Avance™ 400. Рабочая частота по протонам составляла 400.93 МГц, по ядрам 13С - 100.925 МГц, по ядрам 19F - 376.38 МГц. В качестве растворителя использовали дейтерохлороформ. Химические сдвиги протонов определяли относительно тетраметилсилана как внутреннего стандарта, ядер 13С определяли относительно сигнала CDCl3 (77.0 м.д.) и пересчитывали к сигналу SiMe4, ядер 19F определяли относительно трифторуксусной кислоты как внешнего стандарта и пересчитывали к сигналу CFCl3. Масс-спектры получали на приборе Finnigan Polaris Q (ионная ловушка, энергия ионизирующих электронов 70 эВ) методом прямого ввода образца. Элементный анализ выполняли в лаборатории элементного анализа ИНЭОС РАН. Рентгеноструктурный анализ проводили на дифрактометре Bruker SMART АРЕХ2 CCD, двумерную 1H-19F HOESY-ЯМР-спектроскопию осуществляли на спектрометре Bruker Avance™ 400.
ТСХ-контроль проводили на пластинах фирмы Merck (60 F-254, 0.25 мм). Для очистки веществ колоночной хроматографией использовали силикагель фирмы Merck (Kieselgel 60, 0.063-0.200 мм). В качестве элюента применяли смеси петролейного эфира (ПЭ) с этилацетатом (ЭА). Все используемые растворители очищали по стандартным методикам.
Для биологических экспериментов использовали линии клеток человека НСТ116 (рак толстой кишки) и HL-60 (острый миелогенный лейкоз), которые были протестированы в American Type Culture Collection, США. Клетки НСТ116 культивировали в среде DMEM, клетки HL-60 - в среде RPMI-1640. В культуральные среды добавляли следующие компоненты до приведенных ниже конечных концентраций: эмбриональную телячью сыворотку (HyClone, US) - до 5%, L-глутамин (ПанЭко, Россия) - до 2 мМ, пенициллин (ПанЭко, Россия) - до 100 мкг/мл, стрептомицин (ПанЭко, Россия) - до 100 ед/мл. В экспериментах использовали культуры в логарифмической фазе роста. Для профилактики микоплазменного заражения использовали препарат Mycokill (ПанЭко, Россия). Перед началом экспериментов проводили не менее трех пассажей на свободной от антимикоплазменного препарата среде.
E-2-Арил-2-трифторметил-1-нитроциклопропаны (I) хранили в виде 10 мМ стоковых растворов в ДМСО (ПанЭко, Россия) в замороженном виде при -20°С и размораживали непосредственно перед использованием.
Изобретение иллюстрируется конкретными примерами осуществления, приведенными ниже.
Пример 1. Получение E-1-хлор-4-[2-нитро-1-(трифторметил)циклопропил]-бензола (Ia).
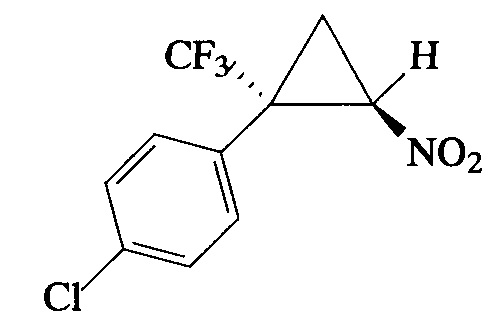
К суспензии карбоната калия (1.93 г, 14 ммоль) в ДМСО (50 мл) прибавляют сначала 1-хлор-4-(3,3,3-трифторпроп-1-ен-2-ил)бензол (1.5 г, 7 ммоль), а затем при перемешивании в течение 20 мин бромнитрометан (1.96 г, 14 ммоль). Через 2 ч перемешивания прибавляют в течение 2 мин еще порцию карбоната калия (1.93 г, 14 ммоль) и бромнитрометана (1.96 г, 14 ммоль). После 16-часового перемешивания бордово-красную реакционную массу обрабатывают ледяной водой (300 мл) и экстрагируют диэтиловым эфиром (3×50 мл). Объединенные органические фазы промывают насыщенным раствором NaCl (30 мл), сушат над сульфатом магния, фильтруют и упаривают на роторном испарителе. Остаток очищают колоночной хроматографией на силикагеле (элюент ПЭ:ЭА=7:1). Получают 1.78 г (96%) соединения Ia в виде бесцветных кристаллов с т. пл. 35-36°С. Спектр 1Н-ЯМР, δ 7.40 (д, 2Н, J=8.3 Гц), 7.33 (д, 2Н, J=8.4 Гц), 4.90 (дд, 1Н, J=7.4, 4.9 Гц), 2.51 (дд, 1Н, J=8.5, 3.7 Гц), 2.08 (дд, 1Н, J=9.3, 5.6 Гц); спектр 13С-ЯМР, δ: 136.34, 131.65, 129.45, 126.78, 123.44 (кв, J=275.8 Гц), 60.42, 37.22 (кв, J=34.4 Гц), 16.42; спектр 19F-ЯМР, δ: - 70.11 (с, CF3). Масс-спектр, m/z (I/Imax,%): 262 [M-HNO2]+(30), 199 [M-HNO2-F]+(15), 183 [M-HNO2-Cl]+(20), 164 [M-HNO2-F-Cl]+(64), 149 [M-CF3-HNO2]+(15), 139 [M-CF3-CH2=CHNO]+(100). Вычислено для C10H7ClF3NO2,%: С 45.22; H 2.66; N 5.27. Найдено, % С 45.19; Н 2.62; N 5.22.
Пример 2. Получение E-1-бром-4-[2-нитро-1-(трифторметил)циклопропил]-бензола (Ib).
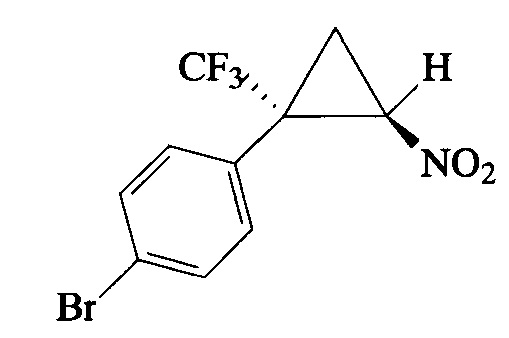
Соединение Ib получают по методике, аналогичной описанной в примере 1, из 1-бром-4-(3,3,3-трифторпроп-1-ен-2-ил)бензола, используя 6-кратный избыток бромнитрометана и карбоната калия и осуществляя добавление этих реагентов тремя порциями в течение 24 ч. Выход 82%. Бесцветные кристаллы с т. пл. 58-59°С. Спектр 1Н-ЯМР, δ: 7.56 (д, 1H, J=8.5 Гц), 7.26 (д, 1Н, J=8.3 Гц), 4.90 (дд, 1Н, J=7.5, 4.9 Гц), 2.52 (д, 1Н, J=5.3 Гц), 2.08 (т, 1H, J=7.5 Гц); спектр 13С-ЯМР, δ: 132.43, 131.89, 127.28, 124.62, 123.35 (кв, J=275.9 Гц), 60.35, 37.31 (кв, J=34.6 Гц), 16.40 (кв, J=2.2 Гц); спектр 19F-ЯМР, δ: - 70.10 (с, CF3). Масс-спектр, m/z (I/Imax, %): 262 [M-HNO2]+(30), 243 [M-HNO2-F]+(6), 224 [M-HNO2-F2]+(16), 183 [M-CF3-CH2=CHNO]+(100). Вычислено для C10H7BrF3NO2, %: С 38.74; H 2.28; N 4.52. Найдено, %: С 38.79; Н 2.34; N 4.41.
Пример 3. Получение E-3-[2-нитро-1-(трифторметил)цикло-пропил]пиридина (Ic).
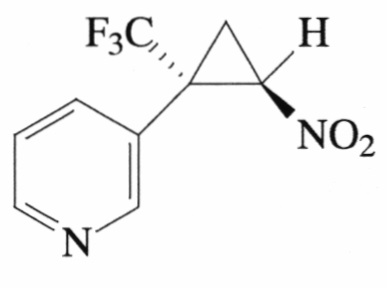
Соединение Ic получают по методике, описанной в примере 1, из 3-(3,3,3-трифторпроп-1-ен-2-ил)пиридина. Выход 70%. Бесцветные кристаллы с т. пл. 49°С. Спектр 1Н-ЯМР, δ: 8.70 (с, 2Н), 7.69 (д, 1Н, J=7.9 Гц), 7.36 (дд, 1Н J=7.9, 4.8 Гц), 4.95 (дд, 1Н, J=7.6, 4.9 Гц), 2.58 (дд, 1H, J=4.5, 2.9 Гц), 2.15 (т, 1Н, J=7.6 Гц); спектр 13С-ЯМР, δ: 151.44, 151.15, 137.67, 124.64, 123.72, 123.29 (кв, J=275.9 Гц), 59.80, 35.71 (кв, J=35.0 Гц), 16.07 (кв, J=2.1 Гц); спектр 19F-ЯМР, δ: - 69.99 (с, CF3). Вычислено для C9H7F3N2O2, %: С 46.56; Н 3.04; N 12.07. Найдено, %: С 46.52; Н 3.01; N 12.02.
Пример 4. Получение E-1-нитро-3-[2-нитро-1-(трифторметил)циклопропил]-бензола (Id).
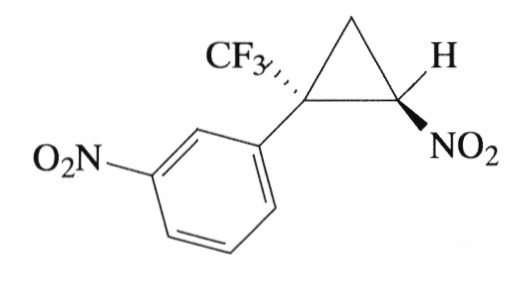
Соединение Id получают по методике, описанной в примере 1, из 1-нитро-3-(3,3,3-трифторпроп-1-ен-2-ил)бензола. Выход 64%. Бесцветные кристаллы с т. пл. 77°С. Спектр 1H- ЯМР, δ: 8.45-8.32 (м, 2Н), 7.71 (д, 1Н, J=7.5 Гц), 7.63 (т, 1Н, J=7.9 Гц), 4.98 (дд, 1Н, J=7.5, 4.9 Гц), 2.62 (дд, 1Н, J=8.6, 3.7 Гц), 2.19 (с, 1Н); спектр 13С-ЯМР, δ: 148.54, 136.05, 130.39, 130.37, 125.76, 125.09, 123.19 (кв, J=276.1 Гц), 60.35, 37.26 (кв, J=35.0 Гц), 16.65 (д, J=1.7 Гц); спектр 19F ЯМР, δ: - 69.75 (с, CF3). Масс-спектр, m/z (I/Imax) %): 277 [M+H]+(1), 229 [M-HNO2]+(6), 210 [M-HNO2-F]+(2), 182 [M-HNO2-F-C2H4]+(25), 150 [M-CF3-CH2=CHNO]+(100). Вычислено для C10H7F3N2O4, %: С 43.49; H 2.55; N 10.14. Найдено, %: С 43.31; Н 2.49; N 10.14.
Пример 5. Получение E-1,2-дихлор-4-[2-нитро-1-(трифторметил)цикло-пропил]-бензола (Ie).
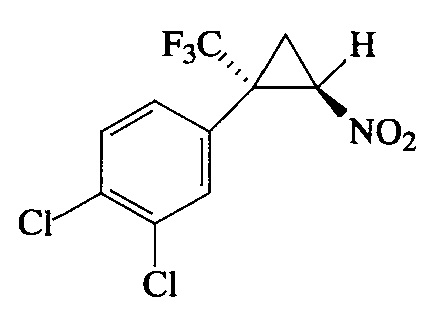
Соединение Ie получают по методике, описанной в примере 1, из 1,2-дихлор-4-(3,3,3-трифторпроп-1-ен-2-ил)бензола. Выход 87%. Бесцветные кристаллы с т. пл. 50°С. Спектр 1Н-ЯМР, δ: 7.51 (с, 1H), 7.50 (д, 1H, J=8.3 Гц), 7.22 (дд, 1Н, J=8.3, 2.0 Гц), 4.91 (дд, 1Н, J=7.5, 4.9 Гц), 2.52 (ддд, 1Н, J=4.9, 4.1, 1.6 Гц), 2.10 (т, 1H, J=7.5 Гц); спектр 13С-ЯМР, δ: 134.81, 133.39, 132.42, 131.15, 129.45, 128.36, 123.23 (кв, J=275.9 Гц), 60.34, 36.86 (кв, J=34.8 Гц); спектр 19F-ЯМР, δ: - 69.91 (с, CF3). Масс-спектр, m/z (I/Imax, %): 299 [М]+(27), 252 [M-HNO2]+(71), 233 [M-HNO2-F]+(21), 217 [M-HNO2-Cl]+(28), 198 [M-HNO2-F-Cl]+(40), 183 [M-2Cl-HNO2]+(45), 173 [M-CF3-CH2=CHNO]+(100). Вычислено для C10H7Cl2F3NO2, %: С 40.03; H 2.0.2; N 4.67. Найдено, %: С 40.00; Н 2.05; N 4.65.
Пример 6. Получение метилового эфира E-4-[2-нитро-1-(трифтор-метил)циклопропил] бензойной кислоты (If).
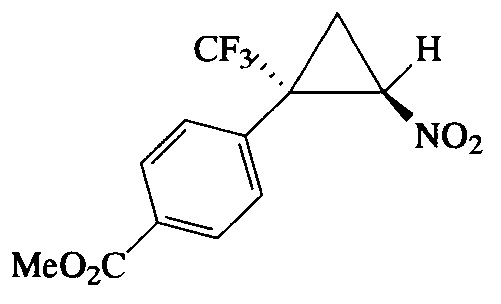
Соединение If получалют по методике, описанной в примере 1, из метилового эфира 4-(3,3,3-трифторпроп-1-ен-2-ил)бензойной кислоты. Выход 94%. Бесцветные кристаллы с т. пл. 62-63°С. Спектр 1H-ЯМР, δ: 8.07 (д, 2Н, J=8.3 Гц,), 7.47 (д, 2Н, J=8.3 Гц), 4.92 (дд, 1H, J=7.5, 4.9 Гц), 3.93 (с, 3Н), 2.55 (дд, 1H, J=8.8, 3.4 Гц), 2.10 (т, 1H, J=7.5 Гц); спектр 13С-ЯМР, δ: 166.16, 132.96, 131.75, 130.46, 130.22, 123.37 (кв, J=276.0 Гц), 60.39, 52.33, 37.62 (кв, J=34.6 Гц), 16.44 (кв, J=1.6 Гц); спектр 19F-ЯМР, δ: - 69.79 (с, CF3). Масс-спектр, m/z (I/Imax, %): 277 [M+H]+(2), 229 [M-HNO2]+(11), 210 [M-HNO2-F]+(11), 182 [M-HNO2-F-C2H4]+(21), 150 [M-CF3-CH2=CHNO]+(100). Вычислено для C12H10F3NO4, %: С 49.84; H 3.49; N 4.84. Найдено, %: С 49.82; Н 3.33; N 4.71.
Пример 7. Получение E-1-[2-нитро-1-(трифторметил)циклопропил]-3-(трифторметил)бензола (Ig).
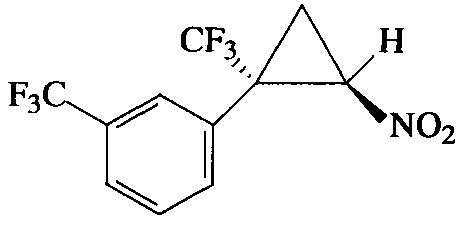
Соединение Ig получают по методике, описанной в примере 1, из 1-(трифторметил)-3-(3,3,3-трифторпроп-1-ен-2-ил)бензола. Выход 70%. Бесцветные кристаллы с т. пл. 55°С. Спектр 1H-ЯМР, δ: 7.75-7.70 (м, 1Н), 7.68 (с, 1Н), 7.53-7.59 (м, 2Н), 4.95 (дд, 1Н, J=7.5, 4.9 Гц), 2.65-2.48 (м, 1Н), 2.14 (т, 1H, J=7.5 Гц); спектр 13С-ЯМР, δ: 133.52, 131.68 (кв, J=33.0 Гц), 129.74, 129.43, 127.37 (кв, J=3.7 Гц), 126.97 (кв, J=3.6 Гц), 123.52 (кв, J=272.4 Гц), 123.33 (кв, J=275.9 Гц), 60.30, 37.46 (кв, J=34.7 Гц), 16.44 (кв, J=1.9 Гц); спектр 19F-ЯМР, δ: - 62.79 (с, CF3), - 69.98 (с, CF3). Масс-спектр, m/z (I/Imax, %): 299 [М]+(1), 280 [M-F]+(13), 252 [M-HNO2]+(25), 233 [M-HNO2-F]+(20), 213 [M-HNO2-HF2]+(42), 183 [M-CF3-HNO2]+(21), 173 [M-CF3-CH2=CHNO]+(100). Вычислено для C11H7F6NO2, %: С 44.16; H 2.36; N 4.68. Найдено, %: С 43.94; Н 2.48; N 4.51.
Пример 8. Получение E-[2-нитро-1-(трифторметил)циклопропил]бензола (In).
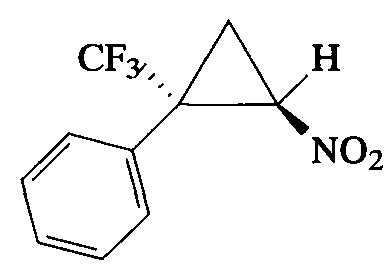
К раствору 1-(3,3,3-трифторпроп-1-ен-2-ил)бензола (0.34 г, 2 ммоль) и 1,2-диметил-1,4,5,6-тетрагидропиримидина (0.672 г, 6 ммоль) в 10 мл N,N-диметилформамида при перемешивании прибавляют по каплям бромнитрометан (0.84 г, 6 ммоль) при 5°С. Реакционную смесь перемешивают 6 ч при 20°С, после чего повторно прибавляют 1,2-диметил-1,4,5,6-тетрагидропиримидин (0.672 г, 6 ммоль) и бромнитрометан (0.84 г, 6 ммоль) и продолжают перемешивание в течение 16 ч. Контроль протекания реакции (спектр ЯМР 19F) свидетельствует о полной конверсии исходного α-(трифторметил)стирола к этому моменту. Основное количество N,N-диметилформамида удаляют в вакууме масляного насоса (2 мм рт.ст.), остаток обрабатывают водой (100 мл) и экстрагируют диэтиловым эфиром (3×30 мл). Объединенные органические фазы промывают насыщенным раствором NaCl, сушат над MgSO4, фильтруют и упаривают на роторном испарителе. Остаток очищают колоночной хроматографией на силикагеле (элюент ПЭ:ЭА=6:1). Получают 0.28 г (62%) соединения Ih в виде бесцветных кристаллов с т. пл. 47-48°С. Спектр 1Н-ЯМР, δ: 7.52-7.32 (м, 5Н), 4.91 (дд, 1Н, J=7.4, 4.9 Гц), 2.54 (т, 1H J=5.2 Гц), 2.07 (т, 1H, J=7.4 Гц); спектр 13С-ЯМР, δ: 130.31, 130.00, 129.09, 128.22, 123.65 (кв, J=275.7 Гц), 60.45, 37.90 (кв, J=34.4 Гц), 16.40 (кв, J=2.2 Гц); спектр 19F-ЯМР, δ: - 70.12 (с, CF3). Масс-спектр, m/z (I/Imax, %): 232 [М+Н]+(5), 184 [M-HNO2]+(100), 164 [M-HNO2-HF]+(56), 115 [M-CF3-HNO2]+(87), 105 [M-CF3-CH2=CHNO]+(37). Вычислено для C10H8F3NO2, %: С 51.96; Н 3.49; N 6.06. Найдено, %: С 51.91; Н 3.35; N 5.94.
Пример 9. Получение E-1-метокси-4-[2-нитро-1-(трифторметил)циклопропил]-бензола (Ii).

К раствору 1-метокси-4-(3,3,3-трифторпроп-1-ен-2-ил)бензола (0.40 г, 2 ммоль) в 15 мл тетраметилмочевины при перемешивании прибавляют 1,2-диметил-1,4,5,6-тетрагидропиримидин (0.672 г, 6 ммоль), а затем бромнитрометан (0.84 г, 6 ммоль) при 20°С. Реакционную смесь перемешивают 6 ч при 20°С, после чего повторно прибавляют 1,2-диметил-1,4,5,6-тетрагидропиримидин (0.672 г, 6 ммоль) и бромнитрометан (0.84 г, 6 ммоль) и продолжают перемешивание еще 16 ч. Контроль протекания реакции (спектр ЯМР 19F) свидетельствует о 77%-ной конверсии исходного α-(трифторметил)стирола к этому моменту. Реакционную массу выливают в 200 мл воды и экстрагируют диэтиловым эфиром (2×30 мл). Объединенные органические фазы промывают водой (2×30 мл), сушат над MgSO4, упаривают на роторном испарителе. Остаток очищают колоночной хроматографией на силикагеле (элюент ПЭ:ЭА=6:1). Получают 0.23 г (45%) соединения Ii в виде бесцветного масла. Спектр 1Н-ЯМР, δ: 7.34-7.26 (м, 2Н), 6.91 (д, 2Н, J=8.7 Гц), 4.87 (дд, 1H, J=7.4, 4.9 Гц), 3.82 (с, 3Н), 2.55-2.44 (м, 1Н), 2.03 (т, 1H, J=7.4 Гц); спектр 13С-ЯМР, δ: 160.73, 131.54, 123.75 (кв, J=275.6 Гц), 119.93, 114.51, 60.59, 55.27, 37.29 (кв, J=34.4 Гц), 16.45 (кв, J=2.1 Гц); спектр 19F-ЯМР, δ: - 70.40 (с, CF3). Вычислено для C11H10F3NO3, %: С 50.58; Н 3.86; N 5.36. Найдено, %: С 50.84; Н 4.08; N 5.18.
Пример 10. Получение E-5-[2-нитро-1-(трифторметил)циклопропил]бензо-[d][1,3]диоксола (Ij).
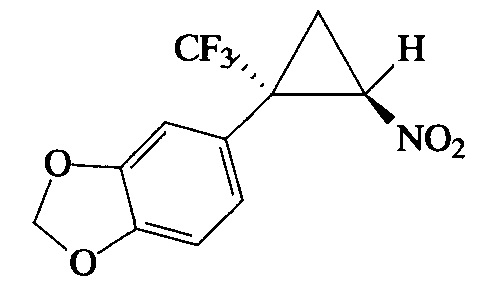
Соединение Ij получают по методике, описанной в примере 8, из 5-(3,3,3-трифторпроп-1-ен-2-ил)бензо[d][1,3]диоксола. Выход 55%. Бесцветные кристаллы с т. пл. 88-89°С. Спектр 1Н-ЯМР, δ: 6.88 (дд, 1Н, J=8.0, 1.3 Гц), 6.83 (с, 1Н), 6.82 (д, 1Н, J=8.1 Гц), 6.02 (дд, 1Н, J=2.3, 1.2 Гц), 4.85 (дд, 1Н, J=7.5, 4.9 Гц), 2.56-2.40 (м, 1H), 2.02 (т, 1Н, J=7.4 Гц); спектр 13С-ЯМР, δ: 149.07, 148.10, 124.45, 123.61 (кв, J=277.2 Гц), 121.37, 110.34, 108.76, 101.65, 60.66, 37.55 (кв, J=34.5 Гц), 16.65 (кв, J=2.0 Гц); спектр 19F-ЯМР, δ: - 70.31 (с, CF3). Масс-спектр, m/z (I/Imax, %): 275 [М]+(22), 228 [M-HNO2]+(51), 199 [M-HNO2-CHO]+(35), 171 [M-HNO2-CHO-CO]+(64), 151 [M-HNO2-CHO-CO-HF]+(100), 149 [M-CF3-CH2=CHNO]+(66). Вычислено для C11H8F3NO4, %: С 48.01; H 2.93; N 5.09. Найдено, %: С 48.61; Н 2.22; N 5.14.
Пример 11. Получение E-1,2-диметокси-4-[2-нитро-1-(трифторметил)циклопропил]бензола (Ik).
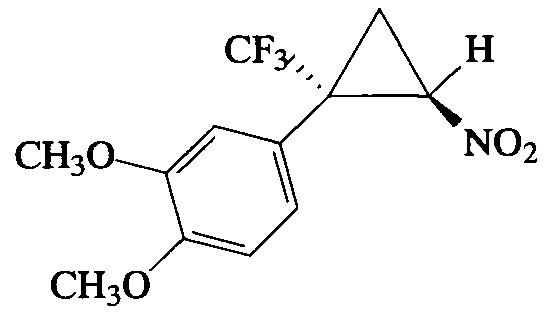
Соединение Ik получают по методике, описанной в примере 8, из 1,2-диметокси-4-(3,3,3-трифторпроп-1-ен-2-ил)бензола. Выход 35%. Бесцветные кристаллы с т. пл. 54-55°С. Спектр 1Н-ЯМР, δ: 6.97 (дд, 1Н, J=8.3, 1.9 Гц), 6.87 (д, 1Н, J=8.3 Гц), 6.84 (кв, 1Н, J=1.5 Гц), 4.88 (дд, 1H, J=7.4, 4.9 Гц), 3.90 (с, 3Н), 3.89 (с, 3Н), 2.57-2.47 (м, 1Н), 2.03 (т, 1Н, J=7.4 Гц); спектр 13С-ЯМР, δ: 150.32, 149.13, 123.72 (кв, J=275.7 Гц), 123.18, 120.20, 112.71, 111.22, 60.67 (кв, J=2.6 Гц), 55.96, 55.84, 37.54 (кв, J=34.5 Гц), 16.50 (кв, J=2.1 Гц); спектр 19F-ЯМР, δ: - 70.26 (с, CF3). Масс-спектр, m/z (I/Imax, %): 291 [М]+(84), 245 [M-NO2]+(100), 214 [M-NO2-2CH3]+(43), 165 [M-CF3-CH2=CHNO]+(57), 145 [M-NO2-2CH3-CF3]+(62). Вычислено для C12H12F3NO4, %: С 49.49; H 4.15; N 4.81. Найдено, %: С 49.34; Н, 4.27; N, 4.74.
Пример 12. Получение E-1-нитро-4-[2-нитро-1-(трифторметил)циклопропил]-бензола (II).
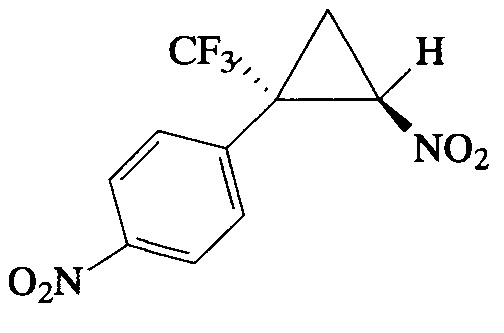
Соединение II получают по методике, описанной в примере 1, из 1-нитро-4-(3,3,3-трифторпроп-1-ен-2-ил)бензола. Выход 87%. Бесцветные кристаллы с т. пл. 77°С. Спектр 1Н-ЯМР, δ: 8.28 (д, 2Н, J=8.8 Гц), 7.60 (д, 2Н, J=8.7 Гц), 4.97 (дд, 1Н, J=7.6, 4.9 Гц), 2.58 (м, 1Н), 2.19 (т, 1H, J=7.6 Гц); спектр 13С-ЯМР, δ: 148.91, 135.16, 131.59, 124.22, 123.11 (кв, J=276.1 Гц), 60.35, 37.30 (кв, J=34.8 Гц), 16.64 (кв, J=2.0 Гц); спектр 19F-ЯМР, δ: - 69.57 (с, CF3). Вычислено для C10H7F3N2O4, %: С 43.49; Н 2.55; N 10.14. Найдено, %: С 43.47; Н 2.49; N 10.09.
Пример 13. Получение E-1-фтор-4-[2-нитро-1-(трифторметил)циклопропил]-бензола (Im).
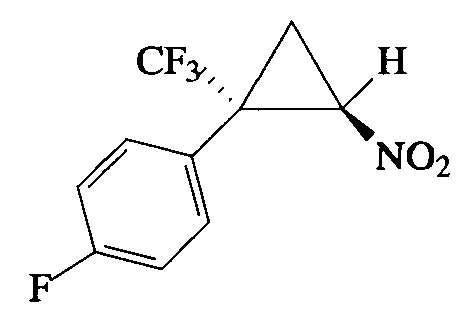
Соединение Im получают по методике, описанной в примере 8, из 1-фтор-4-(3,3,3-трифторпроп-1-ен-2-ил)бензола. Выход 68%. Бесцветное масло. Спектр 1Н-ЯМР, δ: 7.38 (дд, 2Н, J=8.4, 5.2 Гц), 7.11 (т, 2Н, J=8.6 Гц), 4.90 (дд, 1Н, J=7.5, 4.9 Гц), 2.62-2.40 (м, 1H), 2.08 (т, 1Н, J=7.5 Гц); спектр 13С-ЯМР, δ: 163.58 (д, J=250.1 Гц), 132.26 (д, J=8.7 Гц), 124.14 (д, J=3.3 Гц), 123.53 (кв, J=275.8 Гц), 116.29 (д, J=22.1 Гц), 60.46, 37.10 (кв, J=34.6 Гц), 16.46 (кв, J=2.0 Гц); спектр 19F-ЯМР, δ: - 70.33 (с, 3F), - 110.48 (с, 1F). Вычислено для C10H7F4NO2, %: С 48.21; Н 2.83; N 5.62. Найдено, %: С 48.49; Н 2.99; N 5.29.
Пример 14. Получение E-1-метил-4-[2-нитро-1-(трифторметил)циклопропил]-бензола (In).
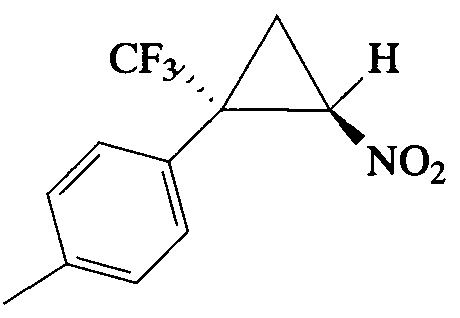
Соединение In получают по методике, описанной в примере 8, из 1-метил-4-(3,3,3-трифторпроп-1-ен-2-ил)бензола. Выход 58%. Бесцветные кристаллы с т. пл. 47-48°С (пентан). Спектр 1H-ЯМР, δ: 7.27 (д, 2Н, J=8.2 Гц), 7.21 (д, 2Н, J=8.1 Гц), 4.88 (дд, 1Н, J=7.5, 4.9 Гц), 2.57-2.47 (м, 1H), 2.38 (с, 3Н), 2.04 (т, 1Н, J=7.4 Гц); спектр 13С-ЯМР, δ: 140.11, 130.13, 129.82, 125.17, 123.73 (кв, J=275.9 Гц), 60.52, 37.63 (кв, J=34.4 Гц), 21.27, 16.38 (кв, J=2.1 Гц); спектр 19F-ЯМР, δ: - 70.23 (с, CF3). Масс-спектр, m/z (I/Imax, %): 246 [М+Н]+(7), 198 [M-HNO2]+(52), 164 [M-HNO2-CH3F]+(78), 129 [M-CF3-HNO2]+(51), 119 [M-CF3-CH2=CHNO]+(100). Вычислено для C11H10F3NO2, %: С 53.88; H 4.11; N 5.71. Найдено, %: С 53.97; Н 4.46; N 5.49.
МТТ-тест для исследования цитотоксичности
Клетки рассевают в лунки 96-луночного планшета (NUNC, США) (5000 клеток в 190 мкл культуральной среды) и инкубируют 24 ч при 37°С в увлажненной атмосфере с 5%-ным содержанием CO2. В лунки вносят по 10 мкл растворов исследуемых веществ в культуральной среде, приготовленых серийными разведениями из исходных растворов в ДМСО (10 мМ) до конечных концентраций 0,1; 0,2; 0,4; 0,8; 1,6; 3,2; 6; 12; 25 и 50 мкМ. Контролем служат интактные клетки. Клетки инкубируют 72 ч при 37°С в увлажненной атмосфере с 5%-ным содержанием CO2. За 1 ч до окончания инкубации в лунки вносят по 20 мкл водного раствора МТТ (5 мг/мл, ПанЭко, Россия). После окончания инкубации культуральную среду отбирают, клетки ресуспендируют в 100 мкл ДМСО и измеряют оптическую плотность раствора на планшетном спектрофотометре Multiscan FC (ThermoScientific, США) при длине волны 571 нм. Процент клеток, выживших при действии каждой дозы соединения, подсчитывают как частное от деления средней оптической плотности в лунках после инкубации с данной дозой к средней оптической плотности контрольных лунок (значения последних приняты за 100%). Каждую концентрацию соединения изучают с трехкратной статистикой, приводят усредненные данные по результатам трех экспериментов, погрешности не превышают 10%.
| название | год | авторы | номер документа |
|---|---|---|---|
| 2-АМИНОЗАМЕЩЕННЫЕ 6-МЕТОКСИ-4-ТРИФТОРМЕТИЛ-9Н-ПИРИМИДО[4,5b]ИНДОЛЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ПРИМЕНЕНИЕ И ПРЕДШЕСТВЕННИКИ | 2016 |
|
RU2625316C1 |
| Способ совместного получения 1-(2-амино-4-R-фенил)- и 2-(2-амино-4-R-фенил)бензотриазолов | 2023 |
|
RU2825731C1 |
| ФТОРСОДЕРЖАЩИЕ ПРОИЗВОДНЫЕ ТЕВИНОЛА И ОРВИНОЛА И СПОСОБЫ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ) | 2012 |
|
RU2506265C1 |
| Способ получения полиядерных тетрааминов, содержащих мостиковые атомы | 2018 |
|
RU2691737C1 |
| ФОСФОИНДОЛЫ КАК ИНГИБИТОРЫ ВИЧ | 2005 |
|
RU2393163C2 |
| Спироконденсированные производные 2,3-дигидроиндола, их применение в офтальмологии | 2017 |
|
RU2712039C2 |
| БЕЗДИАФРАГМЕННЫЙ ЭЛЕКТРОСИНТЕЗ ЗАМЕЩЕННЫХ ПИРИДО[1,2-а]БЕНЗИМИДАЗОЛОВ | 2014 |
|
RU2556001C1 |
| Замещенные 4-арил-гексагидро-7Н-имидазоло[1,5-b][1,2]оксазин-7-оны и способ их получения | 2018 |
|
RU2670097C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПЕРФТОРАЛКИЛЦИАНО- ИЛИ ПЕРФТОРАЛКИЛЦИАНОФТОРБОРАТОВ | 2011 |
|
RU2575352C2 |
| 3-Арил-4-(трифторметил)-4H-спиро[хромено[3,4-c]пирролидин-1,3'-оксиндолы], обладающие высокой цитотоксической активностью в отношении к клеткам линии карциномы шейки матки человека HeLa, и способ их получения | 2021 |
|
RU2774705C1 |
Изобретение относится к E-2-арил-2-трифторметил-1-нитроциклопропанам общей формулы I, где X=С, R=R1=Н; X=С, R=Cl, R1=Н; X=С, R=R1=Cl; X=С, R=Br, R1=Н; X=С, R=R1=Br; X=С, R=C1-С3-алкил, R1=Н; X=С, R=NO2, R1=Н; X=С, R=CO2Alk, R1=Н; X=С, R=CF3, R1=Н; X=С, R=ОМе, R1=Н; X=С, R=R1=ОМе; X=С, R=F, R1=Н; X=С, R-R1=-ОСН2O-; X=N, R=R1=Н. Также предложен способ получения соединений I, заключающийся в порционном прибавлении 4-15-кратных мольных избытков бромнитрометана и неорганического (предпочтительно карбонат калия) или органического (предпочтительно 1,1-диметил-1,4,5,6-тетрагидропиримидин) основания к раствору соответствующего α-(трифторметил)стирола в полярном растворителе, предпочтительно диметилсульфоксиде (в случае неорганического основания) и 1,1,3,3-тетраметилмочевине или N,N-диметилформамиде (в случае органического основания) при комнатной температуре. Технический результат – получены новые соединения, которые могут найти применение в медицине в качестве противоопухолевых агентов в отношении клеток рака прямой кишки и миелогенного лейкоза человека. 2 н. и 5 з.п. ф-лы, 1 табл., 14 пр.
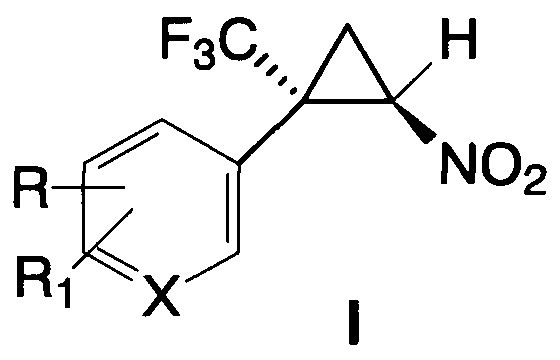
1. E-2-Арил-2-трифторметил-1-нитроциклопропаны общей формулы I
 ,
,
где X=С, R=R1=Н; X=С, R=Cl, R1=Н; X=С, R=R1=Cl; X=С, R=Br, R1=Н;
X=С, R=R1=Br; X=С, R=C1-С3-алкил, R1=Н; X=С, R=NO2, R1=Н;
X=С, R=CO2Alk, R1=Н; X=С, R=CF3, R1=Н; X=С, R=ОМе, R1=Н;
X=С, R=R1=ОМе; X=С, R=F, R1=Н; X=С, R-R1=-ОСН2O-; X=N, R=R1=Н.
2. Соединения по п. 1, обладающие противоопухолевой активностью, в частности, в отношении клеток рака толстой кишки и миелогенного лейкоза человека.
3. Способ получения соединений по п. 1, включающий взаимодействие соответствующего α-(трифторметил)стирола с бромнитрометаном в присутствии неорганического или органического основания в полярном растворителе, таком как диметилсульфоксид, N,N-диметилформамид, N,N-диметилацетамид, 1,1,3,3-тетраметилмочевина, N-метилпирролидон, ацетонитрил, при комнатной температуре, причем бромнитрометан и основание используют в эквимолярных количествах в 4-15-кратном мольном избытке по отношению к α-(трифторметил)стиролу и вводят порциями.
4. Способ по п. 3, отличающийся тем, что в качестве неорганического основания используют карбонат натрия, карбонат калия, карбонат цезия, фосфат калия, гидроксид лития, оксид кальция, предпочтительно карбонат калия.
5. Способ по п. 3 или 4, отличающийся тем, что в случае неорганического основания в качестве полярного растворителя предпочтительно используют диметилсульфоксид.
6. Способ по п. 3, отличающийся тем, что в качестве органического основания используют 1,2-диметил-1,4,5,6-тетрагидропиримидин, 1,8-диазабициклоундец-7-ен, 1,5-диазабицикло[4.3.0]нон-5-ен, N-метилпиперидин, триэтиламин, предпочтительно 1,2-диметил-1,4,5,6-тетрагидропиримидин.
7. Способ по п. 3 или 6, отличающийся тем, что в случае органического основания в качестве полярного растворителя предпочтительно используют 1,1,3,3-тетраметилмочевину или N,N-диметилформамид.
| KATHARINA J | |||
| HOCK et al | |||
| The Journal of Organic Chemistry, 82 (15), 2017, pp | |||
| Застежка для чулочных подвязок | 1928 |
|
SU8220A1 |
| LEE М | |||
| G | |||
| et al | |||
| Chemistry & Biology, 13 (6), 2006, pp | |||
| РАДИОПРИЕМНИК - ВОЛНОМЕР | 1923 |
|
SU563A1 |
| СПОСОБ ПОЛУЧЕНИЯ 1-АЛКИЛ-1,2-ДИЭТИЛЦИКЛОПРОПАНОВ | 2000 |
|
RU2183614C1 |

