ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к областям как органической химии, так и фармацевтики, в частности, к соединению общей формулы (I), фармацевтически приемлемым солям присоединения кислоты соединения общей формулы (I), способу их получения, фармацевтической композиции, содержащей их, и применению соединения общей формулы (I) для лечения и/или предупреждения психических болезней депрессивного типа.
ПРЕДПОСЫЛКИ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
Депрессия, как полагают, представляет собой аффективное расстройство, симптом психической болезни, который главным образом характеризуется депрессивным состоянием. Клинически это выражается сериями симптомов, в том числе депрессивным состоянием, брадифренией, немногословностью, сниженной активностью и потерей интереса к работе и т. д. Как сообщает ВОЗ, депрессия стала четвертым по распространенности заболеванием во всем мире. К 2020 году она, вероятно, станет вторым по распространенности заболеванием, сразу после заболеваний сердечно-сосудистой системы. Сейчас в Китае насчитывается около 26 миллионов пациентов с депрессией, только 10% из которых при этом имеют возможность получить нормальное медикаментозное лечение. Таким образом антидепрессивные лекарственные средства, безусловно, будут иметь огромный потенциальный рынок.
Как предполагалось многочисленными исследованиями, изменение моноаминергических нейромедиаторов нервных центров, допамина и холинэргика, изменчивость их соответствующей рецепторной функции и нейроэндокринной дисфункции, вероятно, играют важную роль в возникновении и развитии депрессии. На данный момент принцип лечения депрессии должен быть направлен на регулирование содержания моноаминов-нейромедиаторов в гипоталамусе, их рецепторных функций и восстановление нормальной нейроэндокринной функции.
В настоящее время медикаментозное лечение все еще представляет собой основной способ лечения депрессии. То, что патогенез депрессии является сложным, связанным со многими факторами, например, социальной психологией, наследственностью, биохимическими изменениями человеческого тела и нейроэндокринологией, подтверждается литературными источниками. Антидепрессивное лекарственное средство может иметь разнообразные виды мишеней, например, рецепторы, концентрацию моноаминов-нейромедиаторов и цитокин. Различные антидепрессивные лекарственные средства воздействуют через различные мишени. Первое поколение антидепрессивных лекарственных средств относится к ингибиторам моноаминоксидазы. Тем не менее, их селективность и необратимый ингибирующий эффект на фермент приводят к токсическому повреждению печени, что имеет определенные токсические и побочные эффекты; поэтому их постепенно заменяют на трициклические антидепрессивные лекарственные препараты. Такие широкоприменяемые медикаменты включают доксепин, амитриптилин и кломипрамин и т.д. Несмотря на то что они оказывают лучший терапевтический эффект на эндогенную депрессию, в частности, обладают более чем 80% эффективностью при эмоциональной депрессии, потере интереса и пессимизме, считают, что эти лекарственные средства обладают более высокой кардиотоксичностью и большим количеством побочных реакций. Избирательный ингибитор обратного захвата 5-HT (SSRI) появился в конце 1980-х, как вид нового антидепрессивного лекарственного средства. Их до сих пор применяют в Европе и США в качестве общепринятого антидепрессивного лекарственного средства первой линии, потому что оно сохраняет классический эффект антидепрессанта и значительно снижает побочные реакции, вызванные другими рецепторами. Такие широкоприменяемые медикаменты включают флуоксетин, пароксетин, сертралин, циталопрам и флувоксамин и т.д. Они всасываются через желудок и кишечный тракт и метаболизируются в организме, таким образом вызывают желудочно-кишечную дисфункцию, а некоторые из них вызывают также половую дисфункцию. Помимо этого, клинические исследования показали, что для таких синтезированных лекарственных средств, которые были предназначены для нацеливания на отдельную мишень, было тяжело достичь удовлетворительного эффекта. До сих пор не было разработано идеального антидепрессивного лекарственного средства, обладающего лучшей эффективностью и меньшими токсическими и побочными эффектами.
Заявка на патент Китая (заявка № 201010169679.9) раскрывает соединение N-изобутил-5'-метокси-3',4'-метилендиоксициннамамида, который представлял собой алкалоид, выделенный из Piper laetispicum C. DC, растения семейства Piperaceae. Его структура приведена ниже. Как было показано в экспериментах на животных, соединение N-изобутил-5'-метокси-3',4'-метилендиоксициннамамида обладает значительным антидепрессивным эффектом.
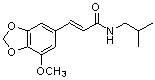
N-изобутил-5'-метокси-3',4'-метилендиоксициннамамид.
В настоящее время существует ограниченный ресурс растения Piper laetispicum C. DC, с низким содержанием в нем соединения N-изобутил-5'-метокси-3',4'-метилендиоксициннамамида. Экстракция и выделение этого соединения только из растения плохо удовлетворяет требованиям фундаментального исследования и клинического исследования. Таким образом, настоящее изобретение сфокусировано на способе химического синтеза N-изобутил-5'-метокси-3',4'-метилендиоксициннамамида и его производных с тем, чтобы получать молекулы лекарственного средства с более высокой антидепрессивной активностью.
В настоящем изобретении были синтезированы N-изобутил-5'-метокси-3',4'-метилендиоксициннамамид (I-1) и его производные, и вещества, обладающие антидепрессивными активностями, были подвергнуты скринингу с помощью разнообразных типов мышиных моделей депрессии. Впоследствии были обнаружены серии молекул лекарственного средства, которые обладают значительными антидепрессивными эффектами.
ПОДРОБНОЕ ОПИСАНИЕ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
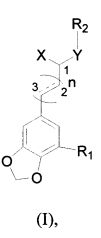
Целью настоящего изобретения является предоставление соединения общей формулы (I) и его фармацевтически приемлемых солей присоединения кислоты,
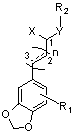
(I),
где
R1 представляет собой H, OH, F, Cl, Br, I, OCH3, OCF3, OCHF2, OCH2F, CF3, CHF2, CH2F, CH3, CH3CH2, CF3CH2, CN, NO2, NH2 или COOR3; где указанный R3 представляет собой H, C1~гидрокарбил с неразветвленной цепью, C3~C10 гидрокарбил с разветвленной цепью, C3~C10циклический гидрокарбил или C6~C10ароматический гидрокарбил;
n равняется 0, 1, 2 или 3, и элементарное звено 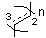 содержит по меньшей мере одну одинарную или двойную углерод-углеродную связь;
содержит по меньшей мере одну одинарную или двойную углерод-углеродную связь;
X представляет собой =O, =S, H, SH или SR3;
Y представляет собой N или NR3, O или S, где указанный R3 представляет собой H, C1~C10 гидрокарбил с неразветвленной цепью; C3~C10 гидрокарбил с разветвленной цепью; C3~C10циклический гидрокарбил или C6~C10ароматический гидрокарбил;
R2 представляет собой H, C1~C10 гидрокарбил с неразветвленной цепью, C3~C10 гидрокарбил с разветвленной цепью, C3~C10циклический гидрокарбил, C6~C10ароматический гидрокарбил, C1~C10-гидроксиалкил или группу производных N-замещенного пиперазина; или R2 представляет собой группу, которая образует тетрагидропирролильную, пиперидильную или гексаметилениминовую группу с соседним Y.
Предпочтительно R1 представляет собой -CF3;
n равняется 0, 1, 2 или 3, и элементарное звено содержит по меньшей мере одну одинарную или двойную углерод-углеродную связь;
X представляет собой =O;
Y представляет собой N или NH;
R2 представляет собой H, C1~C10 гидрокарбил с неразветвленной цепью, C3~C10 гидрокарбил с разветвленной цепью, C3~C10циклический гидрокарбил, C6~C10ароматический гидрокарбил, C1~C10-гидроксиалкил или группу производных N-замещенного пиперазина; или R2 представляет собой группу, которая образует тетрагидропирролильную, пиперидильную или гексаметилениминовую группу с соседним Y.
Другое предпочтительное производное замещенного циннамамида представлено структурой следующей общей формулы (II).
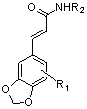
(II),
где
R1 представляет собой H, OH, F, Cl, Br, I, OCH3, OCF3, OCHF2, OCH2F, CF3, CHF2, CH2F, CH3, CH3CH2, CF3CH2, CN, NO2, NH2 или COOR3; где указанный R3 представляет собой C1~C10 гидрокарбил с неразветвленной цепью, C3~C10 гидрокарбил с разветвленной цепью, C3~C10циклический гидрокарбил или C6~C10ароматический гидрокарбил;
R2 представляет собой H, C1~C10 гидрокарбил с неразветвленной цепью, C3~C10 гидрокарбил с разветвленной цепью, C3~C10циклический гидрокарбил, C6~C10ароматический гидрокарбил, C1~C10-гидроксиалкил или группу производных N-замещенного пиперазина.
Наиболее предпочтительно, структура настоящего соединения и его фармацевтически приемлемые соли присоединения кислоты представлены в виде следующих соединений.
N-Изобутил-5'-метокси-3',4'-метилендиоксициннамамид (I-1)
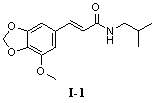
N-Изобутил-5'-нитро-3',4'-метилендиоксициннамамид (I-2)
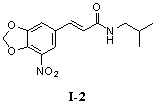
N-Изобутил-5'-йод-3',4'-метилендиоксициннамамид (I-3)
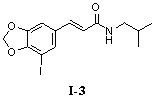
N-Изобутил-5'-хлор-3',4'-метилендиоксициннамамид (I-4)
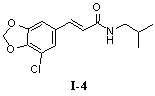
N-Изобутил-5'-трифторметил-3',4'-метилендиоксициннамамид (I-5)
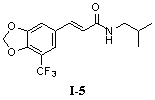
N-Изобутил-5-(5'-метокси-3',4'-метилендиоксифенил)-пентадиенамид (I-6)
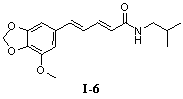
N-Изобутил-3',4'-метилендиоксициннамамид (I-7)
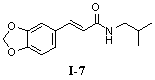
N,N-Диметил-5'-трифторметил-3',4'-метилендиоксициннамамид (I-8)
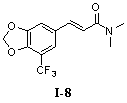
N,N-Диэтил-5'-трифторметил-3',4'-метилендиоксициннамамид (I-9)

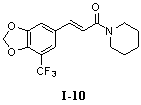
1-(5'-Трифторметил-3',4'-метилендиоксициннамил)-пиперидин (I-10)
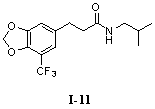
N-Изобутил-3-(5'-трифторметил-3',4'-метилендиоксифенил)-пропионамид (I-11)
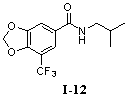
N-Изобутил-5-трифторметил-3,4-метилендиоксибензамид (I-12)
1-(5-Трифторметил-3,4-метилендиоксибензоил)-пиперидин (I-13)

В соответствии с настоящим изобретением указанные фармацевтически приемлемые соли присоединения кислоты настоящего соединения получают путем осуществления реакции со следующими кислотами: серной кислотой, соляной кислотой, бромистоводородной кислотой, фосфорной кислотой, винной кислотой, фумаровой кислотой, малеиновой кислотой, лимонной кислотой, уксусной кислотой, муравьиной кислотой, метансульфоновой кислотой, п-толуолсульфоновой кислотой, щавелевой кислотой или янтарной кислотой; предпочтительно, указанные фармацевтически приемлемые соли присоединения кислоты настоящего соединения представляют собой хлористоводородные соли.
В другом аспекте настоящее изобретение обеспечивает способ получения соединений общей формулы (I).
Предпочтительно соединение общей формулы (I) представляет собой производное замещенного N-изобутилциннамамида. Соединение общей формулы (I) получают следующими путями синтеза.
Производное замещенной коричной кислоты получают с помощью реакции Виттига или реакции Виттига-Хорнера между производным замещенного пипероналя и этоксиформилметилентрифенилфосфином или триэтилфосфоноацетатом.
Далее полученное производное замещенной коричной кислоты ацилируют с получением его ацилированного производного (в том числе ацилгалогенида, азида, ангидрида, активного сложного эфира), а затем осуществляют реакцию ацилированного производного с органическим амином с получением амидного производного. Альтернативно, производное замещенной коричной кислоты реагирует с органическим амином и конденсирующим средством (HATU, HBTU, EDCI, DCC и т.д.) с получением амидного производного.
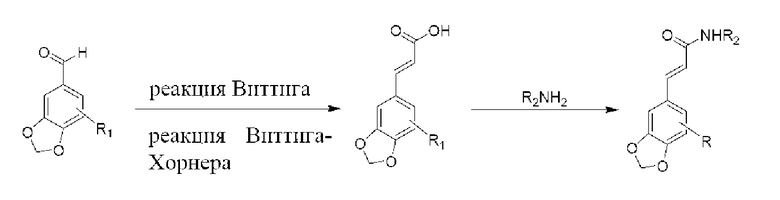
В соответствии с настоящим изобретением, наиболее удобным способом синтеза является получение амидного соединения с помощью реакции амидирования кислоты, соответствующей конечному продукту.
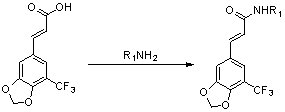
Предпочтительную структуру производного 5'-трифторметил-3',4'-метилендиоксициннамамида получают с помощью следующих путей синтеза.
Используют 5'-трифторметил-3',4'-метилендиоксикоричную кислоту в качестве исходного материала для получения ее ацилированного производного (в том числе ацилгалогенида, азида, ангидрида, активного сложного эфира), а затем осуществляют реакцию ацилированного производного с органическим амином с получением амидного производного. Альтернативно осуществляют реакцию 5'-трифторметил-3',4'-метилендиоксикоричной кислоты с органическим амином и конденсирующим средством (HATU, HBTU, EDCI, DCC и т.д.) с получением амидного производного;
 .
.
Предпочтительно, ацилгалогенид используют для прямого ацилирования.
В соответствии с настоящим изобретением указанные фармацевтически приемлемые соли присоединения кислоты по настоящему соединению получают с помощью обычной реакции кислотно-основной нейтрализации. Например, соответствующие соли присоединения кислоты по настоящему изобретению получают путем осуществления реакции настоящего соединения со следующими кислотами: серной кислотой, соляной кислотой, бромистоводородной кислотой, фосфорной кислотой, винной кислотой, фумаровой кислотой, малеиновой кислотой, лимонной кислотой, уксусной кислотой, муравьиной кислотой, метансульфоновой кислотой, п-толуолсульфоновой кислотой, щавелевой кислотой или янтарной кислотой, предпочтительно указанные фармацевтически приемлемые соли присоединения кислоты по настоящему соединению представляют собой хлористоводородные соли.
В другом аспекте настоящего изобретения обеспечивают фармацевтические композиции, содержащие указанное соединение по настоящему изобретению или его фармацевтически приемлемые соли присоединения кислоты.
В соответствии с настоящим изобретением фармацевтические композиции могут быть получены в виде любых лекарственных форм. Лекарственные формы включают таблетки, например, таблетки в сахарной оболочке, таблетки, покрытые пленочной оболочкой, таблетки с энтеросолюбильным покрытием или таблетки с пролонгированным действием; капсулы, например, твердые капсулы, мягкие капсулы или капсулы с пролонгированным действием; пероральные растворы; защечные таблетки; гранулы; гранулы, которые принимают после растворения в кипятке; пилюли; порошкообразные лекарственные формы; пасты, например, мази, пластыри; пеллеты; суспензии; порошки; водные растворы, например, инъекции; суппозитории; крема; спреи; капли и пластыри небольшого размера.
В соответствии с настоящим изобретением, соединение можно предпочтительно получать в составе стандартной лекарственной формы.
В соответствии с настоящим изобретением композиция содержит 0,1~1000 мг указанного соединения в качестве действующего ингредиента на стандартную лекарственную форму, и остатком является фармацевтически приемлемое(ые) вспомогательное(ые) вещество(а). Указанное(ые) фармацевтически приемлемое(ые) вспомогательное(ые) вещество(а) составляет(ют) 0,01~99,99 вес. % от общего веса состава.
В соответствии с настоящим изобретением, медицинское применение и дозировку указанной композиции определяют на основании состояний пациентов, например, 1~3 раза в день и 1~10 таблеток за один прием.
В соответствии с настоящим изобретением указанная композиция может быть получена в виде перорально вводимой лекарственной формы или инъекции.
Композиция, где указанную перорально вводимую лекарственную форму выбирают из одного из следующего: капсулы, таблетки, капельные пилюли, гранулы, концентрированные пилюли и пероральные растворы.
Композиция, где указанные инъекции выбирают из одного из следующего: инъекционные растворы, лиофилизированные порошки для инъекций и водные инъекции.
В соответствии с настоящим изобретением указанная перорально вводимая лекарственная форма настоящей фармацевтической композиции как правило содержит обычное вспомогательное(ые) вещество(а), например, связующие средства, объемообразующие средства, разбавители, средства для таблетирования, смазывающие вещества, распадающиеся вещества, окрашивающие вещества, вкусовые вещества, увлажняющие средства, и, при необходимости, на таблетки можно нанести покрытие.
Подходящие объемообразующие средства включают целлюлозу, маннит, лактозу и другие аналогичные объемообразующие средства. Подходящие распадающиеся вещества включают крахмал, поливинилпирролидон (PVP) и производные крахмала (предпочтительно натрия крахмалгликолят). Подходящие смазывающие вещества включают, например, стеарат магния. Подходящие увлажняющие средства включают додецилсульфат натрия.
Обычно, твердые препараты для перорального введения могут быть получены обычными способами, такими как перемешивание, заполнение и таблетирование и т.д. Постоянное перемешивание обеспечивает равномерное распределение действующего вещества в композициях, которые имеют большое количество объемообразующего средства.
В соответствии с настоящим изобретением пероральные жидкие препараты могут быть, например, водорастворимыми или жирорастворимыми суспензиями, растворами, эмульсиями, сиропами или настойками, или высушенными продуктами, которые перед применением могут быть ресуспендированы водой или другими подходящими носителями. Жидкие препараты могут содержать обычные добавки, например суспендирующие средства, например сорбит, сироп, метилцеллюлозу, желатин, гидроксиэтилцеллюлозу, карбоксиметилцеллюлозу, гель стеарата алюминия или гидрогенизированный пищевой жир; эмульгирующие средства, например, лецитин, сорбитан моноолеат или гуммиарабик; неводные носители, которыми могут быть пищевое масло, например, миндальное масло, фракционированное кокосовое масло, сложные эфиры глицерина, пропиленгликоль или этанол; и консерванты, например метилпарабен, нипазол или сорбиновая кислота. И, при необходимости, могут быть включены традиционные отдушки или окрашивающие вещества.
Также как и в случае инъекций, полученная жидкая стандартная лекарственная форма содержит активный(е) компонент(ы) по настоящему изобретению и стерильный(е) носитель(и). В соответствии с типом носителя(ей) и концентрацией активного(ых) компонента(ов), указанный активный(е) компонент(ы) может(могут) быть растворен(ы) или суспендирован(ы). Как правило, растворы получают с помощью растворения активного(ых) компонента(ов) в носителях, стерилизации с помощью фильтрования, наполнения подходящего флакона или ампулы и запаивания. В носители также могут добавлять некоторые фармацевтически приемлемые наполнители, например, анестезирующие средства местного действия, консерванты и буферные вещества. Для того чтобы улучшить стабильность, композицию по настоящему изобретению после того, как наполнили флакон, можно заморозить, а затем обработать в вакууме для удаления воды.
В соответствии с настоящим изобретением указанную фармацевтическую композицию получают в составе, в который необязательно могут быть добавлены фармацевтически приемлемые носители. Указанные носители выбирают из сахарного спирта, например, маннита, сорбита, ксилита; аминокислот, например, цистеина-гидрохлорида, метионина, глицина; двунатриевого EDTA, кальциево-нитриевой соли EDTA; неорганических солей, например, карбонатов, фосфатов одновалентных щелочных металлов или их водных растворов, хлорида натрия, хлорида калия, пиросульфита натрия, бисульфита натрия, тиосульфата натрия, карбоната кальция, бикарбоната кальция; стеаратов, например, стеарата кальция, стеарата магния; неорганических кислот, например, соляной кислоты, серной кислоты, фосфорной кислоты; органических кислот, например, уксусной кислоты, витамина C; солей органических кислот, например, ацетатов, лактата натрия; олигосахаридов, полисахаридов, целлюлоз и их производных, например, мальтозы, глюкозы, фруктозы, декстрана, сахарозы, лактозы, циклодекстрина (такого как β-циклодекстрин), крахмала; меркаптоуксусной кислоты; кремнеорганических производных; альгината; желатина; PVP, глицерина; Tween-80; агара; поверхностно-активных веществ; полиэтиленгликоля; фосфолипидных материалов; каолина; тальковой пудры и т.д.
В соответствии с настоящим изобретением указанную фармацевтическую композицию можно применять в комбинации с другими антидепрессивными лекарственными препаратами. Другими словами, за исключением соединения по настоящему изобретению, есть один или несколько видов антидепрессивных лекарственных средств, которые применяют клинически для профилактики и лечения психических болезней, например, нефазодон, сульпирид, алпразолам, сереназ, буспирон, тандоспирон, метилфенидат, флуоксетин, пароксетин, сертралин, циталопрам, лексапро, флувоксамин, ребоксетин, венлафаксин, флюанксол, мелитрацен и невростан и т.д.
В соответствии с настоящим изобретением, как показано в экспериментах на животных, замещенный циннамамид и его производные могут значительно сокращать длительность неподвижности в тесте принудительного плавания и в тесте подвешивания мышей за хвост, которые представляют собой две модели депрессии на животных при приобретенном поведенческом отчаянии. Они оказывают антагонистический эффект на активность резерпина в расходовании моноамина. Таким образом, замещенный циннамамид и его производные могут быть применены в качестве лекарственного средства для лечения и профилактики психических болезней депрессивного типа.
В другом аспекте настоящего изобретения обеспечивают применение соединения общей формулы (I) в получении медикамента для предупреждения и лечения психических болезней депрессивного типа.
В соответствии с настоящим изобретением, благоприятные эффекты соединения и композиции для предупреждения и лечения психических болезней подтверждают посредством следующих экспериментальных данных.
Тест 1 Модель депрессии “приобретенное отчаяние” в тесте подвешивания мышей за хвост
1 Материал
1.1 Реагенты
Соединения I-1, I-2, I-3, I-4, I-5, I-6, I-7, I-8, I-9, I-10, I-11, I-12 и I-13 синтезировали в соответствии со способом по настоящему изобретению, с чистотой более 95%. Их добавляли в 2% водный раствор Tween-80 с получением раствора, содержащего перед применением 1 мг/мл соединения лекарственного средства.
Гидрохлорид флуоксетина был изготовлен Patheon Inc. (Франция) и отдельно упакован Eli Lilly (Сучжоу) Pharmaceutical Inc. со спецификацией 20 мг/таблетка и серийным номером 81958. Перед экспериментом его растворяли в 2% водном растворе Tween-80 с получением раствора, содержащего 1 мг/мл соединения лекарственного средства.
1.2 Животные
Мышей C57BL/6 приобретали у Beijing Vital River Experimental Animal Co. Ltd. Номер сертификации животных SCXK (Beijing) 2006-0009.
1.3 Оборудование
Многофункциональное регистрирующее автономную деятельность у мышей устройство YLS-1A было предоставлено Shandong Institute of Medical Instruments.
2. Способ
240 самцов мышей C57BL/6 с весом 18~22 г в возрасте 6~8 недель кормили для адаптации в течение 2~3 дня.
Эксперимент 1
110 мышей выбирали случайным образом для определения величины их автономной деятельности. Мышей помещали в многофункциональное регистрирующее автономную деятельность у мышей устройство YLS-1A, а после 1 мин адаптации подсчитывали величину деятельности мышей в период времени с конца 1ой мин до 4ой мин. Отбирали 96 мышей с величиной автономной деятельности от 70 до 140, случайным образом разделяли на 8 групп и внутрижелудочно вводили соединения лекарственного средства в дозе, приведенной в таблице 1, один раз в день последовательно в течение 7 дней. Контрольной группе растворителя вводили 2% водный раствор Tween-80 в таком же объеме. Всех мышей помещали в регистрирующее автономную деятельность устройство спустя 30 мин после введения на 6ой день. После 1 мин адаптации величину деятельности мышей подсчитывали за период времени с конца 1ой мин до 4ой мин.
Спустя 30 мин после введения на 7ой день мышей фиксировали на опоре с помощью прорезиненной ткани на расстоянии 1 см от конца хвоста мыши, подвешивая мышей вниз головой. Голова мыши находилась на высоте около 30 см над столом, и ее поле зрения закрывали от соседней мыши с помощью пластинки. Как правило, мышь пыталась бороться в надежде преодолеть ненормальное положение тела. Тем не менее, после некоторого периода времени мыши эпизодически показывали неподвижность, отображающую безысходность. Для каждой мыши определяли суммарную длительность неподвижности в пределах 6 мин, которую рассматривали как “время отчаяния”. Причем под неподвижностью понимали то, что конечности мыши не двигаются, за исключением дыхания.
Эксперимент 2
130 мышей выбирали случайным образом для определения величины их автономной деятельность. Способ был аналогичен таковому из эксперимента 1. Отбирали 108 мышей с величиной автономной деятельности от 70 до 140, случайным образом разделяли на 9 групп и внутрижелудочно вводили соединения лекарственного средства в дозе, приведенной в таблице 2, один раз в день последовательно в течение 7 дней. Контрольной группе растворителя вводили 2% водный раствор Tween-80 в таком же объеме. Спустя 30 мин после введения на 6ой день определяли величину автономной деятельности каждой мыши. Спустя 30 мин после введения на 7ой день, определяли длительность неподвижности в тесте подвешивания за хвост. Способ был аналогичен таковому из эксперимента 1.
Статистические данные
Использовали аналитическое программное обеспечение SPSS10.0 и результаты анализировали с помощью способа однофакторного дисперсионного анализа для того, чтобы сравнить межгрупповую значимость.
3. Результаты
Эксперимент 1 использовали для оценки эффекта соединений I-1, I-2, I-3, I-4, I-5, I-6 на величину автономной деятельности и время неподвижности мыши в тесте подвешивания за хвост. Как показано в таблице 1, при сравнении с контрольной группой растворителя, одна неделя внутрижелудочного введения флуоксетина гидрохлорида в дозе 10 мг/кг не влияет на автономную деятельность мыши и может значительно уменьшать длительность неподвижности в тесте подвешивания за хвост (p<0,01). Внутрижелудочное введение соединения I-4 или I-5 в дозе 10 мг/кг может также существенно уменьшать длительность неподвижности мышей в тесте подвешивания за хвост (p<0,05, p<0,01), но не влияет на автономную деятельность мышей. В случае других обработанных групп, длительность неподвижности мыши в тесте подвешивания за хвост была снижена до различных степеней, но не имела статистической разницы. По сравнению с I-1, I-4 или I-5 оказывает лучший эффект на продолжительность борьбы мышей в тесте подвешивания за хвост (p<0,05).
Таблица 1. Эффекты различных соединений на величину автономной деятельности и длительность неподвижности мышей в тесте подвешивания за хвост
Примечание: при сравнении с контрольной группой растворителя, *p<0,05, **p<0,01; при сравнении с I-1, ∆ p<0,05.
Эксперимент 2 использовали для оценки эффекта соединений I-5, I-8, I-9, I-10, I-11, I-12 и I-13 на величину автономной деятельности мышей и длительность неподвижности в тесте подвешивания за хвост. Как показано в таблице 2, при сравнении с контрольной группой растворителя, одна неделя внутрижелудочного введения I-5, I-9, I-10, I-11, I-12, I-13 в дозе 10 мг/кг может значительно уменьшить длительность неподвижности мыши в тесте подвешивания за хвост (p<0,01), но не влияет на автономную деятельность мышей. Было проиллюстрировано, что данные соединения имеют определенную антидепрессивную активность без возбуждения центральной нервной системы.
Таблица 2. Эффект различных соединений на величину автономной деятельности и длительность неподвижности мыши в тесте подвешивания за хвост
Примечание: при сравнении с контрольной группой растворителя, **p<0,01.
Тест 2 Тест антиблефароптоза, вызванного резерпином, на модели депрессии
1 Материал
1.1 Реагенты
Соединения I-1, I-2, I-3, I-4, I-5, I-6, I-7, I-8, I-9, I-10, I-11, I-12 и I-13 синтезировали в соответствии со способом настоящего изобретения (чистота>95%). 2% водный раствор Tween-80 добавляли для получения раствора, содержащего 1 мг/мл соединения лекарственного средства.
Флуоксетина гидрохлорид был изготовлен Patheon Inc. (Франция) и отдельно упакован Eli Lilly (Сучжоу) Pharmaceutical Inc. со спецификацией 20 мг/таблетка и номером партии 81958. Перед экспериментом его растворяли в 2% водном растворе Tween-80 для получения раствора, содержащего 1 мг/мл соединения лекарственного средства.
Инъекцию резерпина приобретали у Shanghai Fudan Fuhua Pharmaceutical Co., Ltd. со спецификацией 1 мг/мл и серийным номером x070302.
1.2 Животные
Мышей C57BL/6 приобретали у Beijing Vital River Experimental Animal Co. Ltd. Номер сертификации животных SCXK (Beijing) 2006-0009.
1.3 Оборудование
Многофункциональное регистрирующее автономную деятельность у мышей устройство YLS-1A было предоставлено Shandong Institute of Medical Instruments.
2. Способ
240 самцов мышей C57BL/6 с весом 18~22 г в возрасте 6~8 недель кормили для адаптации в течение 2~3 дней.
Эксперимент 1
120 самцов мышей C57BL/6 выбирали случайным образом для определения величины их автономной деятельности. Мышей помещали в многофункциональное регистрирующее автономную деятельность у мышей устройство YLS-1A и после 1 мин адаптации подсчитывали величину деятельности мышей за период времени с конца 1ой мин до 4ой мин. Отбирали 96 мышей с величиной автономной деятельности от 70 до 140, случайным образом разделяли на 8 групп и внутрижелудочно вводили им соединения лекарственного средства в дозе, приведенной в таблице 3, один раз в день последовательно в течение 7 дней. Контрольной группе растворителя вводили 2% водный раствор Tween-80 в таком же объеме. За исключением нормальной контрольной группы, спустя 30 мин после последнего введения, каждой группе внутрибрюшинно инъецировали резерпин в дозе 4 мг/кг. После этого наблюдали за проявлением акинезии, блефароптоза и наблюдали за внешней температурой тела.
I Акинезия: спустя один час после внутрибрюшинной инъекции резерпина мышей помещали в центр круга диаметром 7,5 см для наблюдения в течение 15 с, чтобы рассчитать соотношение “выхода из круга”.
II Блефароптоз: спустя один час после внутрибрюшинной инъекции резерпина наблюдали смыкание глаз мышей и оценивали с помощью следующих нормативов: веки открыты, 0; веки закрыты на 1/4, 1; веки закрыты на 2/4, 2; веки закрыты на 3/4, 3; веки закрыты полностью, 4.
III Температура поверхности тела: через два часа после внутрибрюшинной инъекции резерпина измеряли внешнюю температуру живота мышей.
Эксперимент 2
120 мышей C57BL/6 выбирали случайным образом для определения величины их автономной деятельности. Способ был аналогичен таковому из эксперимента 1. Отбирали 100 мышей с величиной автономной деятельности от 70 до 140, случайным образом разделяли на 10 групп и внутрижелудочно вводили соединения лекарственного средства в дозе, приведенной в таблице 4, один раз в день последовательно в течение 7 дней. Мышам в нормальной группе и контрольной группе растворителя вводили 2% водный раствор Tween-80 в таком же объеме. Исключая нормальную контрольную группу, спустя 30 мин после последнего введения каждой группе внутрибрюшинно инъецировали резерпин в дозе 4 мг/кг. После этого наблюдали за проявлением акинезии, блефароптоза и наблюдали за внешней температурой тела. Способ наблюдения был аналогичен таковому из эксперимента 1.
Статистические данные
Использовали аналитическое программное обеспечение SPSS10.0 и анализировали результаты с помощью способа однофакторного дисперсионного анализа для того, чтобы сравнить межгрупповую значимость.
3 Результаты
Полное исчезновение резерпина, как полагают, представляет собой везикулярный ингибитор обратного захвата, который производит медиаторы вне везикулы, и кроме того приводит к легкой деградации медиаторов под действием моноаминоксидазы. Таким образом, NE, E, DA, 5-HT и им подобные истощаются, что приводит к физиологическим или поведенческим изменениям, следовательно, наблюдают симптомы депрессии.
Как показано в эксперименте 1, после введения резерпина наблюдали акинезию, блефароптоз и пониженную внешнюю температуру тела. При сравнении с контрольной группой растворителя лекарственное средство положительного контроля, флуоксетина гидрохлорид (10 мг/кг), может заметно улучшить соотношение “выхода из круга”, и заметно повысить внешнюю температуру тела у мышей, и значительно снизить степень смыкания глаз у мышей (p<0,01). При сравнении с контрольной группой растворителя значительно снижается блефароптоз, значительно улучшается соотношение “выхода из круга” и значительно повышается внешняя температура тела (p<0,05, p<0,01) в группах I-1, I-2, I-3, I-4, I-5, I-6. По сравнению с I-1, степень смыкания глаз у мышей в группах I-3, I-4, I-5 была снижена (p<0,01); соотношение “выхода из круга” в группах I-2, I-3, I-4, I-5, I-6 заметно улучшалось (p<0,05). Соединения I-4 и I-5 оказывают лучший эффект на снижение акинезии, блефароптоза и повышение внешней температуры тела.
Таблица 3. Эффект каждого соединения на смыкание глаз и акинезию, вызванную резерпином (±SD)
Примечание: при сравнении с контрольной группой растворителя, *p<0,05, **p<0,01; при сравнении с I-1, ∆p<0,05, ∆∆p<0,01.
В эксперименте 2 оценивали антагонистический эффект соединений I-1, I-8, I-9, I-10, I-11, I-12 и I-13 с помощью внутрибрюшинной инъекции резерпина. Как показано в таблице 4, при сравнении с нормальной группой, степень смыкания глаз у мышей в контрольной группе растворителя значительно усиливалась (p<0,01), но внешняя температура тела значительно понижались и соотношение “выхода из круга” значительно ухудшалось (p<0,01). Было проиллюстрировано то, что мышиная модель депрессии, вызванная внутрибрюшинной инъекцией резерпина, является успешной. При сравнении с контрольной группой растворителя лекарственное средство положительного контроля, гидрохлорид флуоксетина, в дозе 10 мг/кг может заметно улучшить соотношение “выхода из круга”, и значительно снизить степень смыкания глаз и повысить внешнюю температуру тела (p<0,01). Соединения I-1, I-8, I-9, I-11, I-12 и I-13 в дозе 10 мг/кг могут значительно снизить блефароптоз, повысить внешнюю температуру тела и улучшить соотношение “выхода из круга”, и улучшение было статистически значимым (p<0,05, p<0,01). Соединение I-10 в дозе 10 мг/кг может значительно снизить блефароптоз мышей (p<0,01), повысить внешнюю температуру тела и улучшить соотношение “выхода из круга” до определенной степени, но без статистически значимой разницы. По сравнению с соединением I-1, соединения I-11, I-12 и I-13 оказывают значительно лучший эффект на блефароптоз, температуру поверхности тела и соотношение “выхода из круга” у мышей (p<0,05, p<0,01), и I-9 и I-10 оказывают более значительное снижение блефароптоза (p<0,05). В отношении снижения степени смыкания глаз и повышения внешней температуры тела мышей, I-13, до определенной степени, оказывает лучшее эффект, чем флуоксетин (p<0,05). В отношении снижения блефароптоза, повышения внешней температуры тела и улучшения соотношения “выхода из круга” у мышей, I-11, I-12 и I-13 оказывают лучшие эффекты.
Таблица 4. Эффект каждого соединения на смыкание глаз, акинезию и внешнюю температуру тела, вызванные резерпином (±SD)
гидрохлорид
Примечание: при сравнении с нормальной группой, ∆∆ p<0,01; при сравнении с контрольной группой растворителя, *p<0,05, **p<0,01; при сравнении с группой, получающей флуоксетина гидрохлорид, #p<0,05, ##p<0,01; при сравнении с I-1, †p<0,05, †† p<0,01.
Тест 3 Эксперимент принудительного плавания на мышах
1 Материал
1.1 Реагенты
Соединения I-5, I-10, I-13 синтезировали в соответствии со способом по настоящему изобретению (чистота>95%). 2% водный раствор Tween-80 добавляли для получения раствора, содержащего 1 мг/мл соединения лекарственного средства.
Флуоксетина гидрохлорид был изготовлен Patheon Inc. (Франция) и отдельно упакован Eli Lilly (Сучжоу) Pharmaceutical Inc. со спецификацией 20 мг/таблетка и серийным номером 81958. Перед экспериментом его растворяли в 2% водном растворе Tween-80 для получения раствора, содержащего 1 мг/мл соединения лекарственного средства.
1.2 Животные
Мышей C57BL/6 приобретали у Beijing Vital River Experimental Animal Co. Ltd. Номер сертификации животных SCXK (Beijing) 2006-0009.
1.3 Оборудование
Многофункциональное регистрирующее автономную деятельность у мышей устройство YLS-1A было предоставлено Shandong Institute of Medical Instruments.
2. Способ
После 1~2 дней адаптирующего питания 80 самцов мышей C57BL/6 в возрасте 6~8 недель и весом 18~22 г помещали в многофункциональное регистрирующее автономную деятельность у мышей устройство YLS-1A. После 1 мин адаптации, величину деятельности у мышей подсчитывали за период времени с конца 1ой мин до 4ой мин. Отбирали 60 мышей с величиной автономной деятельности от 70 до 140, случайным образом разделяли на 6 групп и внутрижелудочно вводили соединения лекарственного средства в дозе, приведенной в таблице 5, один раз в день последовательно в течение 7 дней. Контрольной группе растворителя вводили 2% водный раствор Tween-80 в таком же объеме. После введения на 6ой день мышей помещали в цилиндрический аквариум с глубиной воды 10 см и 25°С, чтобы заставить мышей плавать. Спустя 15 мин мышей доставали, сушили и возвращали в клетку. Через 24 часа, спустя 30 мин после последнего внутрижелудочного введения, мышей помещали в стеклянную бутылку с диаметром 10 см, высотой 30 см и глубиной воды 10 см, и температура воды в бутылке составляла 25°С. Мышей разделяла непрозрачная перегородка, так чтобы они не влияли одна на другую. Спустя 2 мин адаптации, с конца 2ой мин до 6ой мин фиксировали суммарную длительность неподвижности. Под указанным состоянием неподвижности подразумевают, что мыши прекращают борьбу или плавают на поверхности воды, и все тело демонстрирует лишь незначительное изгибание со слабыми движениями конечности для того, чтобы держать свою голову на плаву над водой с ноздрями, выставленными на воздух.
Статистические данные
Использовали аналитическое программное обеспечение SPSS10.0 и анализировали результаты с помощью способа однофакторного дисперсионного анализа для того, чтобы сравнить межгрупповую значимость.
3 Результаты
Как показано в результатах, при сравнении с контрольной группой растворителя, соединения I-5, I-10, I-13 оказывают эффект на сокращение длительности неподвижности в пределах дозировки в тесте принудительного плавания на мышах. Оно было статистически значимым (p<0,05, p<0,01). В тесте принудительного плавания на мышах I-5 показало дозозависимый эффект на длительность неподвижности.
Таблица 5. Эффект производных на длительность неподвижности в тесте принудительного плавания на мышах
Примечание: при сравнении с контрольной группой растворителя, *p<0,05,**p<0,01.
На основании вышеуказанных экспериментов можно сделать следующий вывод:
1. В модели депрессии “приобретенное отчаяние” в тесте подвешивания мышей за хвост, введение соединений I-4, I-5, I-9, I-10, I-11, I-12 и I-13 в дозе 10 мг/кг в течение 7 дней может значительно сокращать длительность неподвижности в тесте подвешивания мышей за хвост.
2. В тесте антиблефароптоза, вызванного резерпином, на модели депрессии, введение соединений I-4, I-5, I-8, I-9, I-10, I-11, I-12 и I-13 в дозе 10 мг/кг в течение 7 дней оказывает противоположный эффект на снижение температуры поверхности тела, на проявление акинезии и снижает степень смыкания глаз мышей, вызванные резерпином, что таким образом указывает, что соединения по настоящему изобретению обладают модулирующим эффектом на обратный захват 5-HT, NE и DA.
3. В тесте принудительного плавания на мышах I-5, I-10 и I-13 настоящего соединения могут сокращать длительность неподвижности в тесте принудительного плаванья, и I-5 показал дозозависимый эффект на длительность неподвижности в тесте принудительного плавания на мышах.
4. При сравнении с фармакологическим эффектом I-1, антидепрессивный эффект I-4, I-5, I-9, I-10, I-11, I-12 и I-13 в тестируемой модели при тестируемой дозе до некоторой степени повышается.
Таким образом, было подтверждено, что указанные производные замещенного циннамамида по настоящему изобретению обладают лучшей антидепрессивной активностью, чем любое из уровня техники.
ПОДРОБНОЕ ОПИСАНИЕ ПРЕДПОЧТИТЕЛЬНЫХ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ
Следующие примеры даны только с целью иллюстрации настоящего изобретения. Синтез и соответствующие данные, которые касаются идентификации структуры обычных соединений, представлены в следующих примерах. Следующие примеры даны только с целью иллюстрация и не предназначены ограничивать объем настоящего изобретения никаким образом. Любое простое улучшение в соответствии с сущностью настоящего изобретения следует оценивать как входящее в объем защиты настоящего изобретения.
Пример 1. N-изобутил-5'-метокси-3',4'-метилендиоксициннамамид (I-1)
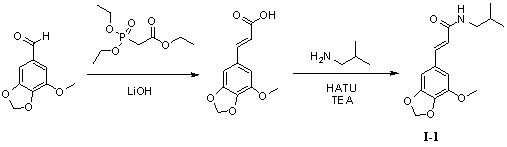
Триэтилфосфоноацетат (300 мг, 1,3 ммоль), безводный тетрагидрофуран (10 мл) и гидроксид лития (163 мг, 3,9 ммоль) добавляли в 50 мл трехгорлую колбу и нагревали до 70°С для осуществления реакции в течение 1 часа под защитой азота. 3,4-Метилендиокси-5-метоксибензальдегид (200 мг, 1,1 ммоль) растворяли в 5 мл безводного тетрагидрофурана, и получаемый раствор по каплям стекал в колбу в течение 0,5 часа. Реакционный раствор оставляли для осуществления реакции при 70°С в течение 10 часов. Для того чтобы контролировать реакцию использовали тонкослойную хроматографию (TLC). Нагревание не прекращали до тех пор, пока реакция не была завершена. Получаемый в результате реакционный раствор концентрировали до сухого вещества путем ротационного выпаривания. Добавляли 20 мл дистиллированной воды, чтобы растворить твердое вещество для образования раствора. В вышеуказанный раствор медленно по каплям добавляли 2N гидрохлорид, чтобы скорректировать pH до 2,0 и постоянно перемешивали в течение 1 часа с предоставлением светло-желтому твердому веществу возможности осадиться. Твердое вещество собирали посредством фильтрации при пониженном давлении, а затем применяли способ вакуумной сушки для получения промежуточного соединения 5'-метокси-3',4'-метилендиоксикоричной кислоты (180 мг, 74%).
5'-Метокси-3',4'-метилендиоксикоричную кислоту (210 мг, 0,81 ммоль), изобутенамин (71 мг, 0,97 ммоль) и триэтиламин (122 мг, 1,2 ммоль) растворяли в 10 мл безводного дихлорметана, перемешивали в течение 15 минут в условиях ледяной бани и медленно добавляли HATU (368 мг, 0,97 ммоль). Получаемый в результате раствор продолжали перемешивать в условиях ледяной бани в течение 2 часов. После того как прекращали взбалтывание, добавляли 20 мл воды и встряхивали, чтобы отделить органическую фазу. Водную фазу экстрагировали дихлорметаном (2×20 мл). После объединения органических фаз для высушивания использовали безводный сульфат натрия и получаемый в результате раствор концентрировали до сухого состояния при пониженном давлении. Исходный продукт очищали с помощью колоночной хроматографии с силикагелем (подвижная фаза: петролейный эфир/этилацетат=3/1) для получения N-изобутил-5'-метокси-3',4'-метилендиоксициннамамида (160 мг, 71%).
1H ЯМР(CDCl3, 400 МГц): δ 7,50 (1H, d, J=15,6 Гц), 6,72 (1H, d, J=1,2 Гц), 6,67 (1H, d, J=1,6 Гц), 6,25 (1H, d, J=15,6 Гц), 6,00 (2H, s), 5,69 (1H, br), 3,91 (3H, s), 3,22 (2H, t, J=6,8 Гц), 1,84 (1H, m), 0,96 (3H, s), 0,95 (3H, s);
13C ЯМР (CDCl3, 100 МГц) δ 166,19, 149,49, 143,85, 140,90, 136,93, 129,97, 119,64, 109,18, 102,05, 101,04, 56,83, 47,33, 28,87, 20,38;
ESI-MS: 278,1 [M+H]+
Пример 2. N-Изобутил-5'-нитро-3',4'-метилендиоксициннамамид (I-2)

Триэтилфосфоноацетат (300 мг, 1,3 ммоль), безводный тетрагидрофуран (10 мл) и гидроксид лития (163 мг, 3,9 ммоль) добавляли в 50 мл трехгорлую колбу и нагревали до 70°С для осуществления реакции в течение 1 часа под защитой азота. 3,4-Метилендиокси-5-нитробензальдегид (215 мг, 1,1 ммоль) растворяли в 5 мл безводного тетрагидрофурана, и получаемый раствор по каплям стекал в колбу в течение 0,5 часа. Реакционный раствор оставляли для осуществления реакции при 70°С в течение 10 часов. Для того чтобы контролировать реакцию использовали тонкослойную хроматографию (TLC). Нагревание не прекращали до тех пор, пока реакция не была завершена. Получаемый в результате реакционный раствор концентрировали до сухого вещества путем ротационного выпаривания. Добавляли 20 мл дистиллированной воды, чтобы растворить твердое вещество для образования раствора. В вышеуказанный раствор медленно по каплям добавляли 2N гидрохлорид, чтобы скорректировать pH до 2,0 и постоянно перемешивали в течение 1 часа с предоставлением желтому твердому веществу возможности осадиться. Твердое вещество собирали посредством фильтрации при пониженном давлении, а затем применяли способ вакуумной сушки для получения промежуточного соединения 5'-нитро-3',4'-метилендиоксикоричной кислоты (190 мг, 73%).
5'-Нитро-3',4'-метилендиоксикоричную кислоту (190 мг, 0,80 ммоль), изобутенамин (71 мг, 0,97 ммоль) и триэтиламин (122 мг, 1,2 ммоль) растворяли в 10 мл безводного дихлорметана, перемешивали в течение 15 минут в условиях ледяной бани и медленно добавляли HATU (368 мг, 0,97 ммоль). Получаемый в результате раствор продолжали перемешивать в условиях ледяной бани в течение 2 часов. После того как прекращали взбалтывание, добавляли 20 мл воды и встряхивали, чтобы отделить органическую фазу. Водную фазу экстрагировали дихлорметаном (2×20 мл). После объединения органических фаз для высушивания использовали безводный сульфат натрия и получаемый в результате раствор концентрировали до сухого состояния при пониженном давлении. Исходный продукт очищали с помощью колоночной хроматографии с силикагелем (подвижная фаза: петролейный эфир/этилацетат=3/1) для получения N-изобутил-5'-нитро-3',4'-метилендиоксициннамамида (140 мг, 60%).
1H ЯМР(CDCl3, 400 МГц): δ 7,75 (1H, d, J=1,2 Гц), 7,53 (1H, d, J=15,2 Гц), 7,19 (1H, d, J=1,6 Гц), 6,36 (1H, d, J=15,6 Гц), 6,26 (2H, s), 5,72 (1H, br), 3,23 (2H, t, J=6,8 Гц), 1,85 (1H, m), 0,97 (3H, s), 0,96 (3H, s);
13C ЯМР (CDCl3, 100 МГц): δ 165,32, 151,37, 144,62, 138,53, 132,27, 129,83, 122,31, 117,12, 111,64, 104,20, 47,42, 28,23, 20,36;
ESI-MS: 293,1 [M+H]+
Пример 3. N-изобутил-5'-йод-3',4'-метилендиоксициннамамид (I-3)
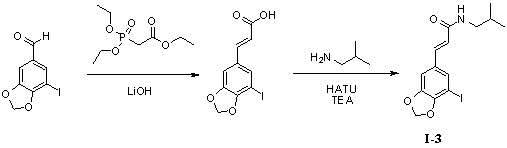
Триэтилфосфоноацетат (300 мг, 1,3 ммоль), безводный тетрагидрофуран (10 мл) и гидроксид лития (163 мг, 3,9 ммоль) добавляли в 50 мл трехгорлую колбу и нагревали до 70°С для осуществления реакции в течение 1 часа под защитой азота. 3,4-Метилендиокси-5-йодбензальдегид (300 мг, 1,1 ммоль) растворяли в 5 мл безводного тетрагидрофурана, и получаемый раствор по каплям стекал в колбу в течение 0,5 часа. Реакционный раствор оставляли для осуществления реакции при 70°С в течение 10 часов. Для того чтобы контролировать реакцию использовали тонкослойную хроматографию (TLC). Нагревание не прекращали до тех пор, пока реакция не была завершена. Получаемый в результате реакционный раствор концентрировали до сухого вещества путем ротационного выпаривания. Добавляли 20 мл дистиллированной воды, чтобы растворить твердое вещество для образования раствора. В вышеуказанный раствор медленно по каплям добавляли 2N гидрохлорид, чтобы скорректировать pH до 2,0 и постоянно перемешивали в течение 1 часа с предоставлением желтому твердому веществу возможности осадиться. Твердое вещество собирали посредством фильтрации при пониженном давлении, а затем применяли способ вакуумной сушки для получения промежуточного соединения 5'-йод-3',4'-метилендиоксикоричной кислоты (245 мг, 70%).
5'-Йод-3',4'-метилендиоксикоричную кислоту (245 мг, 0,77 ммоль), изобутенамин (71 мг, 0,97 ммоль) и триэтиламин (122 мг, 1,2 ммоль) растворяли в 10 мл безводного дихлорметана, перемешивали в течение 15 минут в условиях ледяной бани и медленно добавляли HATU (368 мг, 0,97 ммоль). Получаемый в результате раствор продолжали перемешивать в условиях ледяной бани в течение 2 часов. После того как прекращали взбалтывание, добавляли 20 мл воды и встряхивали, чтобы отделить органическую фазу. Водную фазу экстрагировали дихлорметаном (2×20 мл). После объединения органических фаз для высушивания использовали безводный сульфат натрия и получаемый в результате раствор концентрировали до сухого состояния при пониженном давлении. Исходный продукт очищали с помощью колоночной хроматографии с силикагелем (подвижная фаза: петролейный эфир/этилацетат=5/1) для получения N-изобутил-5'-йод-3',4'-метилендиоксициннамамида (200 мг, 69%).
1H ЯМР (CDCl3, 400 МГц): δ 7,45 (1H, d, J=15,6 Гц), 7,29 (1H, d, J=1,2 Гц), 6,92 (1H, d, J=1,2 Гц), 6,23 (1H, d, J=15,2 Гц), 6,05 (2H, s), 5,63 (1H, br), 3,21 (2H, t, J=6,8 Гц), 1,84 (1H, m), 0,96 (3H, s), 0,95 (3H, s);
13C ЯМР (CDCl3, 100 МГц): δ 165,93, 150,75, 147,20, 139,35, 131,53, 131,49, 120,47, 106,46, 106,85, 101,29, 70,86, 47,35, 28,86, 20,39;
ESI-MS: 374,0 [M+H]+
Пример 4. N-изобутил-5'-хлор-3',4'-метилендиоксициннамамид (I-4)
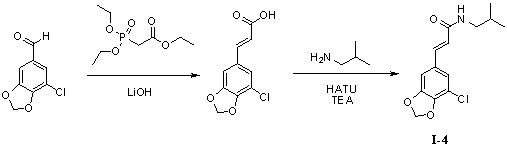
Триэтилфосфоноацетат (300 мг, 1,3 ммоль), безводный тетрагидрофуран (10 мл) и гидроксид лития (163 мг, 3,9 ммоль) добавляли в 50 мл трехгорлую колбу, и нагревали до 70°С для осуществления реакции в течение 1 часа под защитой азота. 3,4-Метилендиокси-5-хлорбензальдегид (200 мг, 1,1 ммоль) растворяли в 5 мл безводного тетрагидрофурана, и получаемый раствор по каплям стекал в колбу в течение 0,5 часа. Реакционный раствор оставляли для осуществления реакции при 70°С в течение 10 часов. Для того чтобы контролировать реакцию использовали тонкослойную хроматографию (TLC). Нагревание не прекращали до тех пор, пока реакция не была завершена. Получаемый в результате реакционный раствор концентрировали до сухого вещества путем ротационного выпаривания. Добавляли 20 мл дистиллированной воды, чтобы растворить твердое вещество для образования раствора. В вышеуказанный раствор медленно по каплям добавляли 2N гидрохлорид, чтобы скорректировать pH до 2,0 и постоянно перемешивали в течение 1 часа с предоставлением светло-желтому твердому веществу возможности осадиться. Твердое вещество собирали посредством фильтрации при пониженном давлении, а затем применяли способ вакуумной сушки для получения промежуточного соединения 5'-хлор-3',4'-метилендиоксикоричной кислоты (175 мг, 70%).
5'-Хлор-3',4'-метилендиоксикоричную кислоту (175 мг, 0,77 ммоль), изобутенамин (71 мг, 0,97 ммоль) и триэтиламин (122 мг, 1,2 ммоль) растворяли в 10 мл безводного дихлорметана, перемешивали в течение 15 минут в условиях ледяной бани и медленно добавляли HATU (368 мг, 0,97 ммоль). Получаемый в результате раствор продолжали перемешивать в условиях ледяной бани в течение 2 часов. После того как прекращали взбалтывание, добавляли 20 мл воды и встряхивали, чтобы отделить органическую фазу. Водную фазу экстрагировали дихлорметаном (2×20 мл). После объединения органических фаз для высушивания использовали безводный сульфат натрия и получаемый в результате раствор концентрировали до сухого состояния при пониженном давлении. Исходный продукт очищали с помощью колоночной хроматографии с силикагелем (подвижная фаза: петролейный эфир/этилацетат=5/1) для получения N-изобутил-5'-хлор-3',4'-метилендиоксициннамамида (160 мг, 74%).
1H ЯМР(CDCl3, 400 МГц): δ 7,40 (1H, d, J=15,6 Гц), 6,92 (1H, d, J=1,2 Гц), 6,81 (1H, d, J=1,2 Гц), 6,18 (1H, d, J=15,6 Гц), 6,00 (2H, s), 5,62 (1H, br), 3,15 (2H, t, J=6,8 Гц), 1,77 (1H, m), 0,89 (3H, s), 0,88 (3H, s);
13C ЯМР (CDCl3, 100 МГц): δ 165,87, 149,23, 145,59, 139,62, 130,52, 123,68, 120,60, 114,43, 105,57, 102,41, 47,34, 28,85, 20,37;
ESI-MS: 282,1 [M+H]+
Пример 5. N-изобутил-5'-трифторметил-3',4'-метилендиоксициннамамида (I-5)
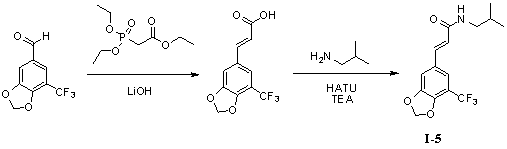
Триэтилфосфоноацетат (300 мг, 1,3 ммоль), безводный тетрагидрофуран (10 мл) и гидроксид лития (163 мг, 3,9 ммоль) добавляли в 50 мл трехгорлую колбу и нагревали до 70°С для осуществления реакции в течение 1 часа под защитой азота. 3,4-Метилендиокси-5-трифторметилбензальдегид (240 мг, 1,1 ммоль) растворяли в 5 мл безводного тетрагидрофурана, и получаемый раствор по каплям стекал в колбу в течение 0,5 часа. Реакционный раствор оставляли для осуществления реакции при 70°С в течение 10 часов. Для того, чтобы контролировать реакцию использовали тонкослойную хроматографию (TLC). Нагревание не прекращали до тех пор, пока реакция не была завершена. Получаемый в результате реакционный раствор концентрировали до сухого вещества путем ротационного выпаривания. Добавляли 20 мл дистиллированной воды, чтобы растворить твердое вещество для образования раствора. В вышеуказанный раствор медленно по каплям добавляли 2N гидрохлорид, чтобы скорректировать pH до 2,0 и постоянно перемешивали в течение 1 часа для получения осадка светло-желтого твердого вещества. Твердое вещество собирали посредством фильтрации при пониженном давлении, а затем применяли способ вакуумной сушки для получения промежуточного соединения 5'-трифторметил-3',4'-метилендиоксикоричной кислоты (170 мг, 59%).
5'-Трифторметил-3',4'-метилендиоксикоричную кислоту (170 мг, 0,65 ммоль), изобутенамин (57 мг, 0,78 ммоль) и триэтиламин (100 мг, 0,97 ммоль) растворяли в 10 мл безводного дихлорметана, перемешивали в течение 15 минут в условиях ледяной бани и медленно добавляли HATU (300 мг, 0,78 ммоль). Получаемый в результате раствор продолжали перемешивать в условиях ледяной бани в течение 2 часов. После того как прекращали взбалтывание, добавляли 20 мл воды и встряхивали, чтобы отделить органическую фазу. Водную фазу экстрагировали дихлорметаном (2×20 мл). После объединения органических фаз для высушивания использовали безводный сульфат натрия и получаемый в результате раствор концентрировали до сухого состояния при пониженном давлении. Исходный продукт очищали с помощью колоночной хроматографии с силикагелем (подвижная фаза: петролейный эфир/этилацетат=5/1) для получения N-изобутил-5'-трифторметил-3',4'-метилендиоксициннамамида (140 мг, 70%).
1H ЯМР(CDCl3, 400 МГц): δ 7,53 (1H, d, J=15,6 Гц), 7,17 (1H, d, J=1,2 Гц), 7,11 (1H, d, J=1,2 Гц), 6,29 (1H, d, J=15,6 Гц), 6,13 (2H, s), 5,65 (1H, br), 3,22 (2H, t, J=6,8 Гц), 1,84 (1H, m), 0,97 (3H, s), 0,95 (3H, s);
13C ЯМР (CDCl3, 100 МГц): δ 165,97, 149,60, 146,31, 139,08, 129,81, 123,80, 121,63, 121,29, 119,36, 109,49, 103,09, 47,42, 28,84, 20,35;
ESI-MS: 316,1 [M+H]+
Пример 6. N-изобутил-5-(5'-метокси-3',4'-метилендиоксифенил)-пентадиенамид (I-6)
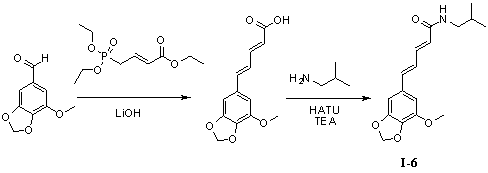
Триэтил-4-фосфонокротонат (325 мг, 1,3 ммоль), безводный тетрагидрофуран (10 мл) и гидроксид лития (163 мг, 3,9 ммоль) добавляли в 50 мл трехгорлую колбу и нагревали до 70°С для осуществления реакции в течение 1 часа под защитой азота. 3,4-Метилендиокси-5-метоксибензальдегид (200 мг, 1,1 ммоль) растворяли в 5 мл безводного тетрагидрофурана, и получаемый раствор по каплям стекал в колбу в течение 0,5 часа. Реакционный раствор оставляли для осуществления реакции при 70°С в течение 10 часов. Для того чтобы контролировать реакцию использовали тонкослойную хроматографию (TLC). Нагревание не прекращали до тех пор, пока реакция не была завершена. Получаемый в результате реакционный раствор концентрировали до сухого вещества путем ротационного выпаривания. Добавляли 20 мл дистиллированной воды, чтобы растворить твердое вещество для образования раствора. В вышеуказанный раствор медленно по каплям добавляли 2N гидрохлорид, чтобы скорректировать pH до 2,0 и постоянно перемешивали в течение 1 часа для получения осадка светло-желтого твердого вещества. Твердое вещество собирали посредством фильтрации при пониженном давлении, а затем применяли способ вакуумной сушки для получения промежуточного соединения 5-(5'-метокси-3',4'-метилендиоксифенил)-пентадиеновой кислоты (125 мг, 65%).
5-(5'-Метокси-3',4'-метилендиоксифенил)-пентадиеновую кислоту (125 мг, 0,65 ммоль), изобутенамин (57 мг, 0,78 ммоль) и триэтиламин (100 мг, 0,97 ммоль) растворяли в 10 мл безводного дихлорметана, перемешивали в течение 15 минут в условиях ледяной бани и медленно добавляли HATU (300 мг, 0,78 ммоль). Получаемый в результате раствор продолжали перемешивать в условиях ледяной бани в течение 2 часов. После того как прекращали взбалтывание, добавляли 20 мл воды и встряхивали, чтобы отделить органическую фазу. Водную фазу экстрагировали дихлорметаном (2×20 мл). После объединения органических фаз для высушивания использовали безводный сульфат натрия и получаемый в результате раствор концентрировали до сухого состояния при пониженном давлении. Исходный продукт очищали с помощью колоночной хроматографии с силикагелем (подвижная фаза: петролейный эфир/этилацетат=3/1) для получения N-изобутил-5-(5'-метокси-3',4'-метилендиоксифенил)-пентадиенамида (140 мг, 71%).
1H ЯМР(CDCl3, 400 МГц): δ 7,29 (1H, dd, J1=10,0 Гц, J2=12,8 Гц), 6,69-6,58 (3H, m), 6,51 (1H, s), 5,91 (2H, s), 5,87 (1H, d, J=14,8 Гц), 5,55 (1H, br), 3,85 (3H, S), 3,12 (2H, t, J=6,4 Гц), 1,76 (1H, m), 0,88 (3H, s), 0,86 (3H, s);
13C ЯМР (CDCl3, 100 МГц): δ 166,45, 149,46, 143,82, 140,94, 138,97, 136,15, 131,52, 125,42, 123,83, 108,08, 101,94, 100,28, 56,78, 47,26, 28,87, 20,40;
ESI-MS: 304,2 [M+H]+
Пример 7. N-изобутил-3',4'-метилендиоксициннамамид (I-7)
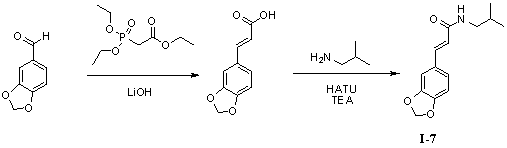
Триэтилфосфоноацетат (300 мг, 1,3 ммоль), безводный тетрагидрофуран (10 мл) и гидроксид лития (163 мг, 3,9 ммоль) добавляли в 50 мл трехгорлую колбу и нагревали до 70°С для осуществления реакции в течение 1 часа под защитой азота. 3,4-(Метилендиокси)бензальдегид (165 мг, 1,1 ммоль) растворяли в 5 мл безводного тетрагидрофурана, и получаемый раствор по каплям стекал в колбу в течение 0,5 часа. Реакционный раствор оставляли для осуществления реакции при 70°С в течение 10 часов. Для того чтобы контролировать реакцию использовали тонкослойную хроматографию (TLC). Нагревание не прекращали до тех пор, пока реакция не была завершена. Получаемый в результате реакционный раствор концентрировали до сухого вещества путем ротационного выпаривания. Добавляли 20 мл дистиллированной воды, чтобы растворить твердое вещество для образования раствора. В вышеуказанный раствор медленно по каплям добавляли 2N гидрохлорид, чтобы скорректировать pH до 2,0 и постоянно перемешивали в течение 1 часа для получения осадка светло-желтого твердого вещества. Твердое вещество собирали посредством фильтрации при пониженном давлении, а затем применяли способ вакуумной сушки для получения промежуточного соединения 3',4'-метилендиоксикоричной кислоты (180 мг, 80%).
3',4'-Метилендиоксикоричную кислоту (180 мг, 0,94 ммоль), изобутенамин (83 мг, 1,12 ммоль) и триэтиламин (142 мг, 1,4 ммоль) растворяли в 10 мл безводного дихлорметана, перемешивали в течение 15 минут в условиях ледяной бани и медленно добавляли HATU (425 мг, 1,12 ммоль). Получаемый в результате раствор продолжали перемешивать в условиях ледяной бани в течение 2 часов. После того как прекращали взбалтывание, добавляли 20 мл воды и встряхивали, чтобы отделить органическую фазу. Водную фазу экстрагировали дихлорметаном (2×20 мл). После объединения органических фаз для высушивания использовали безводный сульфат натрия и получаемый в результате раствор концентрировали до сухого состояния при пониженном давлении. Исходный продукт очищали с помощью колоночной хроматографии с силикагелем (подвижная фаза: петролейный эфир/этилацетат=3/1) для получения N-изобутил-3',4'-метилендиоксициннамамида (188 мг, 81%).
1H ЯМР(CDCl3, 400 МГц): δ 7,45 (1H, d, J=20,4 Гц), 6,90 (1H, s), 6,88 (1H, d, J=10,8 Гц), 6,68 (1H, d, J=10,8 Гц), 6,22 (1H, d, J=20,8 Гц), 5,96 (1H, br), 5,89 (2H, s), 3,13 (2H, t, J=8,8 Гц), 1,77 (1H, m), 0,88 (3H, s), 0,86 (3H, s);
13C ЯМР (CDCl3, 100 МГц): δ 166,53, 149,10, 148,36, 140,58, 129,57, 123,92, 119,36, 108,65, 106,54, 101,59, 47,35, 28,87, 20,35;
ESI-MS: 248,1 [M+H]+
Пример 8. N,N-диметил-5'-трифторметил-3',4'-метилендиоксициннамамид (I-8)
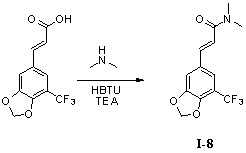
5'-Трифторметил-3',4'-метилендиоксикоричную кислоту (200 мг, 0,77 ммоль), диметиламин (1,54 ммоль) в безводном тетрагидрофуране и триэтиламин (233 мг, 2,3 ммоль) растворяли в 20 мл безводного дихлорметана и перемешивали в условиях ледяной бани в течение 15 мин. В получаемый раствор медленно добавляли HBTU (352 мг, 0,92 ммоль) и перемешивали в течение 2 часов в условиях ледяной бани. После того как прекращали взбалтывание, добавляли 20 мл воды и встряхивали, чтобы отделить органическую фазу. Водную фазу экстрагировали дихлорметаном (2×20 мл). После объединения органических фаз для высушивания использовали безводный сульфат натрия и получаемый в результате раствор концентрировали до сухого состояния при пониженном давлении. Исходный продукт очищали с помощью колоночной хроматографии с силикагелем (подвижная фаза: петролейный эфир/этилацетат=3/1) для получения N,N-диметил-5'-трифторметил-3',4'-метилендиоксициннамамида (200 мг, 90%).
1H ЯМР (CDCl3, 400 МГц): δ 7,57 (1H, d, J=15,6 Гц), 7,18 (1H, s), 7,15 (1H, s), 6,78 (1H, d, J=15,6 Гц), 6,14 (2H, s), 3,19 (3H, s), 3,08 (3H, s);
13C ЯМР (CDCl3, 100 МГц): δ 166,24, 149,44, 146,15, 140,62, 130,01, 123,89, 121,19, 119,26, 117,28, 109,40, 102,91, 37,43, 35,98;
19F ЯМР (CDCl3, 400 МГц): δ -61,48
ESI-MS: 310,1 [M+Na]+
Пример 9. N,N-диэтил-5'-трифторметил-3',4'-метилендиоксициннамамид (I-9)

5'-Трифторметил-3',4'-метилендиоксикоричную кислоту (300 мг, 1,15 ммоль), диэтиламин (170 мг, 2,3 ммоль) и триэтиламин (350 мг, 3,45 ммоль) растворяли в 10 мл безводного дихлорметана и перемешивали в условиях ледяной бани в течение 15 мин. В получаемый раствор медленно добавляли HBTU (530 мг, 1,38 ммоль) и перемешивали в течение 2 часов в условиях ледяной бани. После того как прекращали взбалтывание, добавляли 20 мл воды и встряхивали, чтобы отделить органическую фазу. Водную фазу экстрагировали дихлорметаном (2×20 мл). После объединения органических фаз для высушивания использовали безводный сульфат натрия и получаемый в результате раствор концентрировали до сухого состояния при пониженном давлении. Исходный продукт очищали с помощью колоночной хроматографии с силикагелем (подвижная фаза: петролейный эфир/этилацетат=5/1) для получения N,N-диэтил-5'-трифторметил-3',4'-метилендиоксициннамамида (350 мг, 96%).
1H ЯМР (CDCl3, 400 МГц): δ 7,61 (1H, d, J=15,2 Гц), 7,17 (1H, s), 7,16 (1H, s), 6,71 (1H, d, J=15,2 Гц), 6,14 (2H, s), 3,53-3,46 (4H, m), 1,28 (3H, t, J=7,2 Гц), 1,20 (3H, t, J=7,2 Гц);
13C ЯМР (CDCl3, 100 МГц): δ 165,23, 149,42, 146,08, 140,59, 130,16, 123,91, 121,20, 119,22, 117,66, 109,36, 102,89, 42,30, 41,13, 15,13, 13,19;
19F NMR(CDCl3, 400 МГц): δ -61,48
ESI-MS: 338,1 [M+Na]+
Пример 10. 1-(5'-трифторметил-3',4'-метилендиоксициннамил)-пиперидин (I-10)
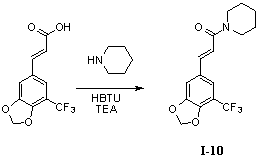
5'-Трифторметил-3',4'-метилендиоксикоричную кислоту (300 мг, 1,15 ммоль), пиперидин (195 мг, 2,3 ммоль) и триэтиламин (350 мг, 3,45 ммоль) растворяли в 10 мл безводного дихлорметана и перемешивали в условиях ледяной бани в течение 15 мин. В получаемый раствор медленно добавляли HBTU (530 мг, 1,38 ммоль) и перемешивали в течение 2 часов в условиях ледяной бани. После того как прекращали взбалтывание, добавляли 20 мл воды и встряхивали чтобы отделить органическую фазу. Водную фазу экстрагировали дихлорметаном (2×20 мл). После объединения органических фаз для высушивания использовали безводный сульфат натрия и получаемый в результате раствор концентрировали до сухого состояния при пониженном давлении. Исходный продукт очищали с помощью колоночной хроматографии с силикагелем (подвижная фаза: петролейный эфир/этилацетат=5/1) для получения 1-(5'-трифторметил-3',4'-метилендиоксициннамил)-пиперидина (350 мг, 92%).
1H ЯМР (CDCl3, 400 МГц): δ 7,55 (1H, d, J=15,6 Гц), 7,18 (1H, s), 7,15 (1H, s), 6,80 (1H, d, J=15,6 Гц), 6,14 (2H, s), 3,63 (4H, br), 1,72-1,68 (2H, m), 1,64-1,60 (4H, m);
13C ЯМР (CDCl3, 100 МГц): δ 164,86, 149,43, 146,06, 140,47, 130,19, 123,93, 121,23, 119,06, 117,64, 109,43, 102,89, 47,07, 43,44, 26,77, 25,68, 24,64;
19F NMR(CDCl3, 400 МГц): δ -61,46
ESI-MS: 350,1 [M+Na]+
Пример 11. N-изобутил-3-(5'-трифторметил-3',4'-метилендиоксифенил)-пропионамид (I-11)
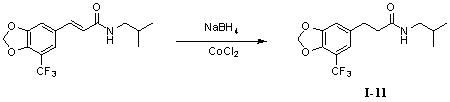
N-Изобутил-5'-трифторметил-3',4'-метилендиоксициннамамид (200 мг, 0,63 ммоль) растворяли в 30 мл метанола и добавляли CoCl2·6H2O (600 мг, 2,54 ммоль) в условиях ледяной бани. После 0,5 часа взбалтывания, в полученный раствор порционно добавляли NaBH4 (195 мг, 5,1 ммоль), нагревали до комнатной температуры спустя 1 час и продолжали перемешивать в течение 2 часов. После того как прекращали взбалтывание, раствор выпаривали до сухого состояния. Неочищенный продукт экстрагировали дихлорметаном (2×20 мл). После объединения органических фаз для высушивания использовали безводный сульфат натрия и получаемый в результате раствор концентрировали до сухого состояния при пониженном давлении. Исходный продукт очищали с помощью колоночной хроматографии с силикагелем (подвижная фаза: петролейный эфир/этилацетат=5/1) для получения N-изобутил-3-(5'-трифторметил-3',4'-метилендиоксифенил)-пропионамида (140 мг, 70%).
1H ЯМР (CDCl3, 400 МГц): δ 6,86 (1H, s), 6,85 (1H, s), 6,06 (2H, s), 5,55 (1H, br), 3,06 (2H, t, J=6,4 Гц), 2,93 (2H, t, J=7,2 Гц), 2,45 (2H, t, J=7,2 Гц), 1,75-1,68 (1H, m), 0,87 (3H, s), 0,85 (3H, s);
13C ЯМР (CDCl3, 100 МГц): δ 171,44, 148,91, 143,54, 135,21, 124,21, 121,51, 117,42, 112,01, 102,29, 46,89, 38,39, 31,27, 28,44, 19,98;
19F ЯМР (CDCl3, 400 МГц): δ -119,72;
ESI-MS: 340,1 [M+Na]+
Пример 12. N-изобутил-5-трифторметил-3,4-метилендиоксибензамид (I-12)
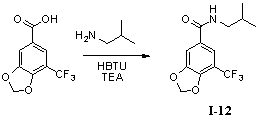
5-Трифторметил-3,4-метилендиоксибензойную кислоту (260 мг, 1,11 ммоль), изобутиламин (162 мг, 2,22 ммоль) и триэтиламин (330 мг, 3,33 ммоль) растворяли в 10 мл безводного дихлорметана и перемешивали в условиях ледяной бани в течение 15 мин. В получаемый раствор медленно добавляли HBTU (500 мг, 1,33 ммоль) и перемешивали в течение 2 часов в условиях ледяной бани. После того как прекращали взбалтывание, добавляли 20 мл воды и встряхивали, чтобы отделить органическую фазу. Водную фазу экстрагировали дихлорметаном (2×20 мл). После объединения органических фаз для высушивания использовали безводный сульфат натрия и получаемый в результате раствор концентрировали до сухого состояния при пониженном давлении. Исходный продукт очищали с помощью колоночной хроматографии с силикагелем (подвижная фаза: петролейный эфир/этилацетат=3/1) для получения N-изобутил-5-трифторметил-3,4-метилендиоксибензамида (250 мг, 78%).
1H ЯМР (CDCl3, 400 МГц): δ 7,46 (1H, s), 7,41 (1H, s), 6,37 (1H, br), 6,16 (2H, s), 3,26 (2H, t, J=6,4 Гц), 1,95-1,85 (1H, m), 0,98 (3H, s), 0,96 (3H, s);
13C ЯМР (CDCl3, 100 МГц): δ 165,73, 149,27, 147,56, 129,47, 123,79, 121,08, 117,73, 110,40, 103,19, 47,57, 28,59, 20,16;
19F ЯМР (CDCl3, 400 МГц): δ -61,45;
ESI-MS: 312,1 [M+Na]+
Пример 13. 1-(5-трифторметил-3,4-метилендиоксибензоил)-пиперидин (I-13)
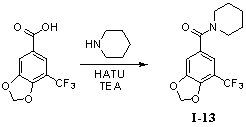
5-Трифторметил-3,4-метилендиоксибензойную кислоту (260 мг, 1,11 ммоль), пиперидин (190 мг, 2,22 ммоль) и триэтиламин (330 мг, 3,33 ммоль) растворяли в 10 мл безводного дихлорметана и перемешивали в условиях ледяной бани в течение 15 мин. В получаемый раствор медленно добавляли HBTU (500 мг, 1,33 ммоль) и перемешивали в течение 2 часов в условиях ледяной бани. После того как прекращали взбалтывание, добавляли 20 мл воды и встряхивали, чтобы отделить органическую фазу. Водную фазу экстрагировали дихлорметаном (2×20 мл). После объединения органических фаз для высушивания использовали безводный сульфат натрия и получаемый в результате раствор концентрировали до сухого состояния при пониженном давлении. Исходный продукт очищали с помощью колоночной хроматографии с силикагелем (подвижная фаза: петролейный эфир/этилацетат=3/1) для получения 1-(5-трифторметил-3,4-метилендиоксибензоил)-пиперидина (280 мг, 87%).
1H ЯМР (CDCl3, 400 МГц): δ 7,11 (1H, s), 7,03 (1H, s), 6,14 (2H, s), 3,64-3,41 (4H, m), 1,69-1,62 (6H, m);
13C ЯМР (CDCl3, 100 МГц): δ 168,37, 148,91, 146,11, 130,45, 123,82, 121,12, 117,72, 110,69, 102,91, 102,91, 29,73, 24,53;
19F ЯМР (CDCl3, 400 МГц): δ -61,48;
ESI-MS: 324,1 [M+Na]+
Пример 14. Получение таблеток N-изобутил-5'-трифторметил-3',4'-метилендиоксициннамамида
Брали N-изобутил-5'-трифторметил-3',4'-метилендиоксициннамамид (I-5), смешивали с крахмалом, декстрином, микрокристаллической целлюлозой и стеаратом магния в соответствии с обычным способом получения во влажные гранулы. Таблетки получали с помощью устройства для штамповки таблеток и осуществляли этап нанесения покрытия для получения покрытых оболочкой таблеток. Каждая таблетка содержала 20 мг N-изобутил-5'-трифторметил-3',4'-метилендиоксициннамамида. Применение: дважды в день и 1~2 таблетки за один прием.
Пример 15. Получение капсул N-изобутил-5'-трифторметил-3',4'-метилендиоксициннамамида
К хорошо перемешанным N-изобутил-5'-трифторметил-3',4'-метилендиоксициннамамиду (I-5), лактозе и гидроксипропилцеллюлозе (HPC), которые были просеянные с использованием сита 60, добавляли соответствующее количество Tween-80, и затем добавляли 3% водный раствор гидроксипропилметилцеллюлозы (HMPC), и пропускали через сито 20. Получаемые гранулы подвергали воздушной сушке в сушильной камере. В высушенный материал добавляли тальковую пудру, хорошо перемешивали и загружали в оболочку капсулы. Каждая капсула содержала 20 мг N-изобутил-5'-трифторметил-3',4'-метилендиоксициннамамида. Применение: дважды в день и 1~2 капсулы за один прием.
Указанные применения по настоящему изобретению в области фармацевтики ни коем образом полностью не ограничены тем, что приведено в данном документе. Указанный исходный медикамент представляет собой любые соединения или их фармацевтически приемлемые соли присоединения кислоты, описанные в настоящем изобретении.
Указанные лекарственные формы ни коем образом полностью не ограничены тем, что приведено в данном документе. Указанные соединения могут быть получены в еще других фармацевтически приемлемых лекарственных формах, например, капельные пилюли, составы с продолжительным действием и т.д.
| название | год | авторы | номер документа |
|---|---|---|---|
| БИЦИКЛИЧЕСКИЕ ГЕТЕРОАРИЛЬНЫЕ ПРОИЗВОДНЫЕ И ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2017 |
|
RU2725886C1 |
| ПРОИЗВОДНОЕ С КОНДЕНСИРОВАННЫМ КОЛЬЦОМ В КАЧЕСТВЕ ИНГИБИТОРА РЕЦЕПТОРА A | 2018 |
|
RU2748993C1 |
| ПРИМЕНЕНИЕ СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ ХРОНИЧЕСКОЙ ТРЕВОГИ ИЛИ ОСТРОЙ ТРЕВОГИ | 2017 |
|
RU2738648C2 |
| ПРОИЗВОДНОЕ ТРИАЗОЛА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2022 |
|
RU2832307C2 |
| ТИЕНОДИАЗЕПИНОВЫЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2795005C2 |
| МАКРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ, ВЫПОЛНЯЮЩЕЕ ФУНКЦИИ ИНГИБИТОРА WEE1, И ВАРИАНТЫ ЕГО ПРИМЕНЕНИЯ | 2018 |
|
RU2783243C2 |
| ФЕНИЛАТНОЕ ПРОИЗВОДНОЕ, ПОЛУЧЕНИЕ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРИМЕНЕНИЕ | 2017 |
|
RU2744975C2 |
| Ингибитор CDK4/6 | 2017 |
|
RU2747311C2 |
| СОДЕРЖАЩЕЕ ЗАМЕСТИТЕЛЬ, ПРЕДСТАВЛЯЮЩИЙ СОБОЙ БУТАН, В ГЕТЕРОЦИКЛИЧЕСКОМ КОЛЬЦЕ ПРОИЗВОДНОЕ ПИРИДОНА ДЛЯ ЛЕЧЕНИЯ ФИБРОЗА И ВОСПАЛИТЕЛЬНЫХ ЗАБОЛЕВАНИЙ | 2017 |
|
RU2738844C2 |
| ПИРИМИДОИМИДАЗОЛЬНЫЕ СОЕДИНЕНИЯ, ПРИМЕНЯЕМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ДНК-PK | 2020 |
|
RU2796163C1 |
Настоящее изобретение относится к производным замещенного циннамамида, а именно соединениям общей формулы (I) или их фармацевтически приемлемым солям присоединения кислоты, где R1 представляет собой ОН, F, Cl, Br, I, ОСН3, OCF3, OCHF2, OCH2F, CF3, CHF2, CH2F, CH3, CH3CH2, CF3CH2, NO2; n равняется 0, 1, 2 или 3, и элементарное звено  содержит по меньшей мере одну одинарную или двойную углерод-углеродную связь; X представляет собой =O, =S; Y представляет собой N или NR3, где указанный R3 представляет собой Н, C1~С10гидрокарбил с неразветвленной цепью; С3~С10гидрокарбил с разветвленной цепью; R2 представляет собой Н, C1~С10гидрокарбил с неразветвленной цепью, группу С3~С10гидрокарбила с разветвленной цепью; или R2 представляет собой группу, которая образует тетрагидропирролильную, пиперидильную или гексаметилениминовую группу с соседним Y; при этом когда n равняется 1, R1 не является ОН и ОСН3, а также к способам их получения, согласно которому производные замещенного пипероналя выбирают в качестве исходных материалов для получения производных замещенного циннамамида путем реакции Виттига и реакции конденсации кислоты с амином. Изобретение также относится к фармацевтической композиции на основе соединений формулы I и их применению для предупреждения и лечения психических болезней депрессивного типа.4 н. и 10 з.п. ф-лы, 5 табл.,15 пр.
содержит по меньшей мере одну одинарную или двойную углерод-углеродную связь; X представляет собой =O, =S; Y представляет собой N или NR3, где указанный R3 представляет собой Н, C1~С10гидрокарбил с неразветвленной цепью; С3~С10гидрокарбил с разветвленной цепью; R2 представляет собой Н, C1~С10гидрокарбил с неразветвленной цепью, группу С3~С10гидрокарбила с разветвленной цепью; или R2 представляет собой группу, которая образует тетрагидропирролильную, пиперидильную или гексаметилениминовую группу с соседним Y; при этом когда n равняется 1, R1 не является ОН и ОСН3, а также к способам их получения, согласно которому производные замещенного пипероналя выбирают в качестве исходных материалов для получения производных замещенного циннамамида путем реакции Виттига и реакции конденсации кислоты с амином. Изобретение также относится к фармацевтической композиции на основе соединений формулы I и их применению для предупреждения и лечения психических болезней депрессивного типа.4 н. и 10 з.п. ф-лы, 5 табл.,15 пр.
(I).
1. Соединение общей формулы (I) или его фармацевтически приемлемые соли присоединения кислоты,
где R1 представляет собой ОН, F, Cl, Br, I, ОСН3, OCF3, OCHF2, OCH2F, CF3, CHF2, CH2F, CH3, CH3CH2, CF3CH2, NO2;
n равняется 0, 1, 2 или 3, и элементарное звено содержит по меньшей мере одну одинарную или двойную углерод-углеродную связь;
X представляет собой =O, =S;
Y представляет собой N или NR3, где указанный R3 представляет собой Н, C1~С10гидрокарбил с неразветвленной цепью; С3~С10гидрокарбил с разветвленной цепью;
R2 представляет собой Н, C1~С10гидрокарбил с неразветвленной цепью, группу С3~С10гидрокарбила с разветвленной цепью; или R2 представляет собой группу, которая образует тетрагидропирролильную, пиперидильную или гексаметилениминовую группу с соседним Y; при этом когда n равняется 1, R1 не является ОН и ОСН3.
2. Соединение или его фармацевтически приемлемые соли присоединения кислоты по п. 1, где
R1 представляет собой -CF3;
n равняется 0, 1, 2 или 3, и содержит по меньшей мере одну одинарную или двойную углерод-углеродную связь;
X представляет собой =O;
Y представляет собой N или NH;
R2 представляет собой Н, C1~С10гидрокарбил с неразветвленной цепью, группу С3~С10гидрокарбила с разветвленной цепью; или R2 представляет собой группу, которая образует тетрагидропирролильную, пиперидильную или гексаметилениминовую группу с соседним Y.
3. Соединение или его фармацевтически приемлемые соли присоединения кислоты по п. 1, где производные замещенного циннамамида общей формулы (II),
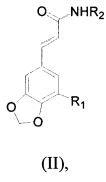
где R1 представляет собой F, Cl, Br, I, OCF3, OCHF2, OCH2F, CF3, CHF2, CH2F, CH3, CH3CH2, CF3CH2, NO2,;
R2 представляет собой H, С1~С10гидрокарбил с неразветвленной цепью, группу С3~С10гидрокарбила с разветвленной цепью.
4. Соединение или его фармацевтически приемлемые соли присоединения кислоты, причем соединение выбрано из группы, включающей
N-изобутил-5′-нитро-3′,4′-метилендиоксициннамамид;
N-изобутил-5′-йод-3′,4′-метилендиоксициннамамид;
N-изобутил-5′-хлор-3′,4′-метилендиоксициннамамид;
N-изобутил-5′-трифторметил-3′,4′-метилендиоксициннамамид;
N-изобутил-5-(5′-метокси-3′,4′-метилендиоксифенил)-пентадиенамид;
N,N-диметил-5′-трифторметил-3′,4′-метилендиоксициннамамид;
N,N-диэтил-5′-трифторметил-3′,4′-метилендиоксициннамамид;
1-(5′-трифторметил-3′,4′-метилендиоксициннамил)-пиперидин;
N-изобутил-3-(5′-трифторметил-3′,4′-метилендиоксифенил)-пропионамид;
N-изобутил-5-трифторметил-3,4-метилендиоксибензамид и
1-(5-трифторметил-3,4-метилендиоксибензоил)-пиперидин.
5. Соединение или его фармацевтически приемлемые соли присоединения кислоты по любому из пп. 1-4, где указанные подходящие соли присоединения кислоты получены путем осуществления реакции соединения общей формулы (I) со следующими кислотами: серной кислотой, соляной кислотой, бромистоводородной кислотой, фосфорной кислотой, винной кислотой, фумаровой кислотой, малеиновой кислотой, лимонной кислотой, уксусной кислотой, муравьиной кислотой, метансульфоновой кислотой, п-толуолсульфоновой кислотой, щавелевой кислотой или янтарной кислотой.
6. Фармацевтическая композиция для предупреждения и лечения психических болезней депрессивного типа, содержащая соединение по любому из пп. 1-5 или его фармацевтически приемлемые соли присоединения кислоты.
7. Фармацевтическая композиция по п. 6, где указанная композиция дополнительно содержит фармацевтически приемлемый(приемлемые) носитель(носители).
8. Способ получения соединения или его фармацевтически приемлемых солей присоединения кислоты по любому из пп. 1-5, включающий следующие этапы:
a. осуществления реакции Виттига или реакции Виттига-Хорнера производного замещенного пипероналя с этоксиформилметилентрифенилфосфином или триэтилфосфоноацетатом с получением производного замещенной коричной кислоты;
b. получения ацилированного производного, в том числе ацилгалогенида, азида, ангидрида, активного сложного эфира, производного замещенной коричной кислоты из производного замещенной коричной кислоты, и осуществление реакции ацилированного производного с органическим амином с получением амидного производного.
9. Способ получения соединения или его фармацевтически приемлемых солей присоединения кислоты по любому из пп. 1-5, включающий следующие этапы:
a. осуществления реакции Виттига или реакции Виттига-Хорнера производного замещенного пипероналя с этоксиформилметилентрифенилфосфином или триэтилфосфоноацетатом с получением производного замещенной коричной кислоты;
b. осуществление реакции производного замещенной коричной кислоты с органическим амином и конденсирующим средством HATU (гексафторфосфат О-(7-аза-бензотриазол-1-ил)-N,N,N′,N′-тетраметилурония), HBTU (гексафторфосфат 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурония), EDCI (1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорид)) или DCC (N,N′-дициклогексилкарбодиимид) и т.д. с получением амидного производного.
10. Способ получения соединения или его фармацевтически приемлемых солей присоединения кислоты по любому из пп. 1-5, включающий следующие этапы:
применение 5′-трифторметил-3′,4′-метилендиоксикоричной кислоты в качестве исходного материала для получения его ацилированного производного, в том числе ацилгалогенида, азида, ангидрида, активного сложного эфира, и осуществление реакции ацилированного производного с органическим амином с получением амидного производного.
11. Способ получения соединения или его фармацевтически приемлемых солей присоединения кислоты по любому из пп. 1-5, включающий следующие этапы:
осуществление реакции 5′-трифторметил-3′,4′-метилендиоксикоричной кислоты с органическим амином и конденсирующим средством HATU, HBTU, EDCI, DCC и т.д. с получением амидного производного.
12. Способ получения соединения или его фармацевтически приемлемых солей присоединения кислоты по любому из пп. 1-5, включающий следующие этапы:
получение производного, содержащего одинарную углерод-углеродную связь в его боковой цепи, путем восстановления производного, содержащего двойную углерод-углеродную связь в его боковой цепи, каталитической гидрогенизацией или борогидридом натрия.
13. Применение любого соединения или его фармацевтически приемлемых солей присоединения кислоты по любому из пп. 1-3 в получении медикамента для предупреждения и лечения психических болезней депрессивного типа.
14. Применение соединения в получении медикамента для предупреждения и лечения психических болезней депрессивного типа, где указанное соединение выбрано из группы, состоящей из:
N-изобутил-5′-нитро-3′,4′ -метилендиоксициннамамид;
N-изобутил-5′-йод-3′,4′-метилендиоксициннамамид;
N-изобутил-5′-хлор-3′,4′-метилендиоксициннамамид;
N-изобутил-5′-трифторметил-3′,4′-метилендиоксициннамамид;
N-изобутил-5-(5′-метокси-3′,4′-метилендиоксифенил)-пентадиенамид;
N,N-диметил-5′-трифторметил-3′,4′-метилендиоксициннамамид;
N,N-диэтил-5′-трифторметил-3′,4′-метилендиоксициннамамид;
1-(5′-трифторметил-3′,4′-метилендиоксициннамил)-пиперидин;
N-изобутил-3-(5′-трифторметил-3′,4′-метилендиоксифенил)-пропионамид;
N-изобутил-5-трифторметил-3,4-метилендиоксибензамид и
1-(5-трифторметил-3,4-метилендиоксибензоил)-пиперидин.
| Смесь для изготовления литейных форм и стержней в нагреваемой оснастке | 1988 |
|
SU1532182A1 |
| WO 2008028314 A1 13.03.2008 | |||
| JOCOBS et al, Amidts of Piper analogo var.nigrinodum, J.Indian Chem | |||
| Soc.,1999,v.76,p.713-717 LIU et al, Chemical constituents of Piper austrosinens, Natural Product Research and Development, 1995, v.7, no.2, p | |||
| Прибор для промывания газов | 1922 |
|
SU20A1 |
| База данных СА (онлайн) WANG et al, Beijing Yixueyuan Xuebao, 1982,v.14, | |||

