ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к новым фармацевтическим соединениям, фармацевтическим композициям, содержащим эти соединения, и их применению для ингибирования активности диацилглицерол-ацилтрансферазы 2 (DGAT2).
УРОВЕНЬ ТЕХНИКИ ИЗОБРЕТЕНИЯ
Триглицериды или триацилглицеролы (TAG) представляют собой главную форму накопления энергии у млекопитающих. TAG образуются путем последовательной этерификации глицерина тремя жирными кислотами с различной длиной цепи и степенью насыщения (1). TAG, синтезированные в кишечнике или печени, упакованы в хиломикроны или липопротеины очень низкой плотности (VLDL), соответственно, и экспортируются в периферические ткани, где они гидролизуются до составляющих их жирных кислот и глицерина липопротеинлипазой (LPL). Получающиеся в результате неэтерифицированные жирные кислоты (NEFA) могут либо подвергаться дальнейшему метаболизму для получения энергии, либо переэтерификации и хранению.
При нормальных физиологических условиях энергоплотный TAG остается секвестрированным в различных жировых запасах до тех пор, пока не возникнет потребность в его высвобождении, после чего он гидролизуется до глицерина и свободных жирных кислот, которые затем высвобождаются в кровоток. Этот процесс строго регулируется противоположными действиями инсулина и гормонов, таких как катехоламины, которые способствуют отложению и мобилизации запасов TAG в различных физиологических условиях. В послеобеденных условиях инсулин действует, чтобы ингибировать липолиз, тем самым, ограничивая высвобождение энергии в форме NEFA и обеспечивая надлежащее хранение пищевых липидов в жировых запасах. Однако у пациентов с сахарным диабетом типа 2 способность инсулина подавлять липолиз улучшается, и поток NEFA из адипоцитов несообразно повышается. Это, в свою очередь, приводит к увеличению доставки липидов в ткани, такие как мышцы и печень. В отсутствие энергетической потребности TAG и другие липидные метаболиты, такие как диацилглицерол (DAG), могут накапливаться и вызывать потерю чувствительности к инсулину (2). Резистентность к инсулину в мышцах характеризуется сниженным поглощением глюкозы и накоплением гликогена, в то время как в печени потеря передачи сигналов инсулина приводит к нарушению регуляции выработки глюкозы и избыточной выработке TAG-богатого VLDL, что является признаком диабета 2 типа (3). Считается, что повышенная секреция TAG-обогащенных VLDL, так называемых частиц VLDL1, стимулирует выработку малого плотного липопротеина низкой плотности (sdLDL), проатерогенной субфракции LDL, которая связана с повышенным риском ишемической болезни сердца (4).
Диацилглицерол-ацилтрансферазы (DGAT) катализируют терминальную стадию синтеза TAG, в частности, этерификацию жирной кислоты диацилглицеролом, что приводит к образованию TAG. У млекопитающих были охарактеризованы два фермента DGAT (DGAT1 и DGAT2). Хотя эти ферменты катализируют одну и ту же ферментативную реакцию, их соответствующие аминокислотные последовательности не связаны, и они состоят в разных семействах генов. Мыши, несущие нарушение в гене, кодирующем DGAT1, устойчивы к ожирению, вызванному питанием, и имеют повышенные энергетические затраты и активность (5). У мышей Dgat1-/- наблюдается нарушение регуляции постабсорбтивного высвобождения хиломикронов и накопление липидов в энтероцитах (6). Предполагается, что метаболически благоприятный фенотип, наблюдаемый у этих мышей, обусловлен потерей экспрессии DGAT1 в кишечнике (7). Важно отметить, что, несмотря на нарушение лактации у самок мышей Dgat1-/-, эти животные сохраняют способность синтезировать TAG, что свидетельствует о существовании дополнительных ферментов DGAT. Это наблюдение и выделение второго DGAT из гриба Mortierella rammaniana привело к идентификации и характеристике DGAT2 (8).
DGAT2 высоко экспрессируется в печени и жировой ткани и, в отличие от DGAT1, обладает превосходной субстратной специфичностью для DAG (8). Удаление гена DGAT2 у грызунов приводит к нарушению внутриутробного роста, тяжелой липемии, нарушению барьерной функции кожи и ранней послеродовой смерти (9). Из-за летальности, вызванной потерей DGAT2, большая часть нашего понимания физиологической роли DGAT2 получена из исследований, проведенных с антисмысловыми олигонуклеотидами (ASO) на моделях метаболических заболеваний у грызунов. В этих условиях ингибирование печеночной DGAT2 приводило к улучшению профиля липопротеинов в плазме (снижению общего холестерина и TAG) и снижению липидной нагрузки в печени, что сопровождалось улучшенной чувствительностью к инсулину и контролем уровня глюкозы во всем организме (10-12). Хотя молекулярные механизмы, лежащие в основе этих наблюдений, полностью не выяснены, ясно, что подавление DGAT2 приводит к подавлению экспрессии множества генов, кодирующих белки, участвующие в липогенезе, включая белки-1c, связывающие регуляторные элементы стерола (SREBP1c), и стеароил-КоА-десатуразу 1 (SCD1) (11, 12). Параллельно индуцируются окислительные пути, о чем свидетельствует повышенная экспрессия генов, таких как карнитин-пальмитоил трансфераза 1 (СРТ1) (11). Конечным результатом этих изменений является снижение уровней печеночных липидов DAG и TAG, что, в свою очередь, приводит к улучшению чувствительности к инсулину в печени. Кроме того, ингибирование DGAT2 подавляет секрецию TAG VLDL в печени и снижает уровень холестерина в циркулирующей крови. Наконец, уровни аполипопротеина B в плазме (APOB) были супрессированы, возможно, из-за сниженной поставки TAG для липидизации вновь синтезированного белка APOB (10, 12). Благоприятное влияние ингибирования DGAT2 как на гликемический контроль, так и на профиль холестерина в плазме позволяет предположить, что эта мишень может быть полезной при лечении метаболических заболеваний (11). Кроме того, наблюдение, что подавление активности DGAT2 приводит к уменьшению накопления липидов в печени, позволяет предположить, что ингибиторы этого фермента могут быть полезны в лечении неалкогольного стеатогепатита (NASH), широко распространенного заболевания печени, характеризующегося отложением избыточного жира в печени.
В последние годы в литературе (13-19) и патентных заявках (WO2013150416, WO2013137628, US20150259323, WO2015077299, WO2016036633, WO2016036638, WO2016036636) сообщалось о нескольких низкомолекулярных ингибиторах DGAT2.
1. Coleman, R. A. и D. G. Mashek. 2011. Chem Rev 111: 6359-6386.
2. Erion, D. M. и G. I. Shulman. 2010. Nat Med 16: 400-402.
3. Choi, S. H. и H. N. Ginsberg. 2011. Trends Endocrinol Metab 22: 353-363.
4. St-Pierre, A. C. с соавт. 2005. Arterioscler Thromb Vasc Biol 25: 553-559.
5. Smith, S. J. с соавт. 2000. Nat Genet 25: 87-90.
6. Buhman, K. K. с соавт. 2002. J Biol Chem 277: 25474-25479.
7. Lee, B., A. M. с соавт. 2010. J Lipid Res 51: 1770-1780.
8. Yen, C. L. с соавт. 2008. J Lipid Res 49: 2283-2301.
9. Stone, S. J. с соавт. 2004. J Biol Chem 279: 11767-11776.
10. Liu, Y. с соавт. 2008. Biochim Biophys Acta 1781: 97-104.
11. Choi, C. S. с соавт. 2007. J Biol Chem 282: 22678-22688.
12. Yu, X. X. с соавт. 2005. Hepatology 42: 362-371.
13. Qi, J. с соавт. J. Lipid. Res. 2012, 53 (6), 1106-16.
14. Wurie, H. R. с соавт. FEBS. J. 2012, 279 (17), 3033-47;
15. Kim, M. O. с соавт. Biol. Pharm. Bull. 2013, 36 (7), 1167-73
16. Lee, K. с соавт. Org. Biomol. Chem. 2013, 11 (5), 849-58
17. Kim, M. O. с соавт. Biol. Pharm. Bull. 2014, 37 (10), 1655-1660.
18. Futatsugi, K. с соавт. J Med Chem 2015, 58 (18), 7173-85.
19. Imbriglio, J. E. с соавт. J. Med. Chem. 2015, 58 (23), 9345-9353.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящая заявка направлена на соединения формулы (I) и (Ia)

(I)

(Ia)
где
D1 и D2 каждый независимо представляет собой N или СН;
R1 представляет собой Н или (С1-С2)алкил, необязательно замещенный одним или двумя заместителями, каждый из которых независимо выбран из фтора и (С3-С6)циклоалкила;
R2 представляет собой Н или фтор;
R3 представляет собой  ,
,  или
или 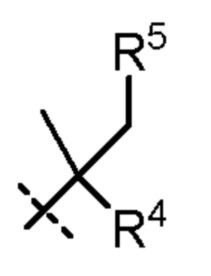 ;
;
R4 представляет собой H, циано или (C1-C4)алкил, необязательно замещенный одним или двумя заместителями, каждый из которых независимо выбран из -OH и -S(O)2R6;
R5 представляет собой Н или -OH; и
R6 представляет собой (C1-C4)алкил;
или его фармацевтически приемлемая соль.
Настоящее изобретение также направлено на кристалл, содержащий соединение, имеющее структуру:
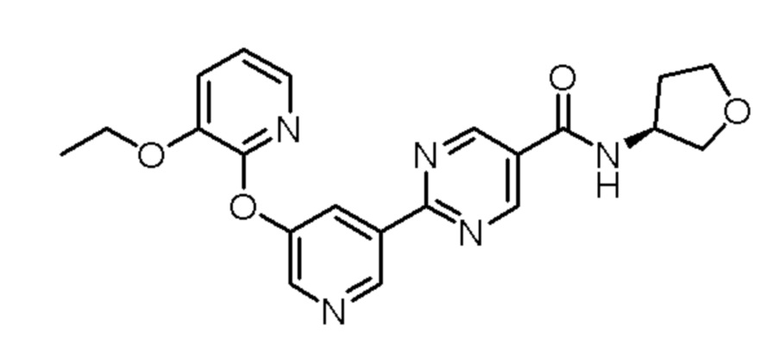
или его фармацевтически приемлемую соль.
Настоящее изобретение также направлено на фармацевтические композиции, которые содержат соединение формулы (I) или (Ia) или фармацевтически приемлемую соль указанного соединения, присутствующего в терапевтически эффективном количестве, в смеси по меньшей мере с одним фармацевтически приемлемым эксципиентом.
Кроме того, настоящее изобретение направлено на фармацевтические композиции, которые содержат соединение формулы (I) или (Ia) или фармацевтически приемлемую соль указанного соединения, присутствующего в терапевтически эффективном количестве, в смеси по меньшей мере с одним фармацевтически приемлемым эксципиентом и дополнительно содержащие по меньшей мере один дополнительный фармацевтический агент, выбранный из группы, состоящей из противовоспалительного средства, антидиабетического средства и средства, регулирующего холестерин/липиды.
В другом варианте осуществления способ по настоящему изобретению предназначен для лечения гиперлипидемии, диабета I типа, сахарного диабета II типа, идиопатического диабета I типа (типа Ib), латентного аутоиммунного сахарного диабета взрослых (LADA), диабета типа 2 с ранним началом (EOD), атипичного диабета молодых (YOAD), диабета зрелого возраста у молодых (MODY), диабета, связанного с недоеданием, гестационного диабета, ишемической болезни сердца, ишемического инсульта, рестеноза после ангиопластики, заболевания периферических сосудов, перемежающейся хромоты, инфаркта миокарда, дислипидемии, послеобеденной липемии, состояний нарушенной толерантности к глюкозе (IGT), состояний нарушенной концентрации глюкозы в плазме натощак, метаболического ацидоза, кетоза, артрита, ожирения, остеопороза, гипертонии, застойной сердечной недостаточности, гипертрофии левого желудочка, заболевания периферических артерий, диабетической ретинопатии, дегенерации желтого пятна, катаракты, диабетической нефропатии, гломерулосклероза, хронической почечной недостаточности, диабетической невропатии, метаболического синдрома, синдрома X, предменструального синдрома, стенокардии, тромбоза, атеросклероза, преходящих ишемических атак, инсульта, сосудистого рестеноза, гипергликемии, гиперинсулинемии, гипертриглицеридемии, резистентности к инсулину, нарушения метаболизма глюкозы, эректильной дисфункции, заболеваний кожи и соединительных тканей, изъязвления стопы и язвенного колита, эндотелиальной дисфункции и нарушенной податливости сосудов, гипер-апо-B-липопротеинемии, болезни Альцгеймера, шизофрении, ухудшения когнитивной деятельности, воспалительного заболевания кишечника, язвенного колита, болезни Крона и синдрома раздраженного кишечника, неалкогольного стеатогепатита (NASH) или неалкогольной жировой дистрофии печени (NAFLD) у людей.
В другом варианте осуществления способ снижает портальную гипертензию, синтетическую способность печеночного белка, гипербилирубинемию или энцефалопатию.
Настоящее изобретение также направлено на способ снижения по меньшей мере на одну степень тяжести по классифицирующим системам оценки неалкогольной жировой дистрофии печени или неалкогольного стеатогепатита, снижения уровня сывороточных маркеров активности неалкогольного стеатогепатита, снижения активности болезни неалкогольного стеатогепатита или снижения медицинских последствий неалкогольного стеатогепатита у людей, включающий стадию введения человеку, нуждающемуся в таком лечении диабета, включающий введение пациенту, нуждающемуся в этом, эффективного количества соединения формулы (I) или (Ia) или фармацевтически приемлемой соли указанного соединения.
Настоящее изобретение также направлено на способ лечения жирового гепатоза, неалкогольной жировой дистрофии печени, неалкогольного стеатогепатита, неалкогольного стеатогепатита с фиброзом печени, неалкогольного стеатогепотита с циррозом печени или неалкогольного стеатогепатита с циррозом и гепатоцеллюлярной карциномой, метаболического заболевания, состояния или расстройства или заболевания, состояния или расстройства, связанного с метаболизмом, у людей, включающий стадию введения человеку, нуждающемуся в таком лечении, включающий стадию введения пациенту терапевтически эффективного количества соединения формулы (I) или (Ia) или фармацевтически приемлемой соли указанного соединения.
Настоящее изобретение также направлено на способ лечения жирового гепатоза, неалкогольной жировой дистрофии печени, неалкогольного стеатогепатита, неалкогольного стеатогепатита с фиброзом печени, неалкогольного стеатогепотита с циррозом печени или неалкогольного стеатогепатита с циррозом и гепатоцеллюлярной карциномой, метаболического заболевания, состояния или расстройства или заболевания, состояния или расстройства, связанного с метаболизмом, у людей, включающий стадию введения человеку, нуждающемуся в таком лечении, включающий стадию введения пациенту, нуждающемуся в таком лечении, терапевтически эффективного количества двух отдельных фармацевтических композиций, включающих
(i) первую композицию, которая содержит соединение формулы (I) или (Ia) или фармацевтически приемлемую соль указанного соединения, присутствующие в терапевтически эффективном количестве, в смеси по меньшей мере с одним фармацевтически приемлемым эксципиентом; и
(ii) вторую композицию, содержащую по меньшей мере один дополнительный фармацевтический агент, выбранный из группы, состоящей из противовоспалительного средства, антидиабетического средства и средства, регулирующего холестерин/липиды, и антидиабетического средства, и по меньшей мере один фармацевтически приемлемый эксципиент.
Следует понимать, что как предшествующее общее описание, так и последующее подробное описание являются только примерными и пояснительными и не ограничивают изобретение, как оно заявлено.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Фигура 1 представляет собой характеристическую рентгеновскую порошковую дифрактограмму, демонстрирующую кристаллическую форму 1 примера 1 (вертикальная ось: интенсивность (CPS); горизонтальная ось: два тета (градусы)).
Фигура 2 представляет собой характеристическую рентгеновскую порошковую дифрактограмму, демонстрирующую кристаллическую форму 2 примера 1 (вертикальная ось: интенсивность (CPS); горизонтальная ось: два тета (градусы)).
На фигурах 3 и 4 обобщены эффекты перорального введения примера 1 на уровни триглицеридов в плазме и печени у крыс Sprague Dawley, получавших западную диету, соответственно.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение может быть более легко понято при обращении к следующему подробному описанию примерных вариантов осуществления изобретения и примеров, включенных в настоящий документ.
Следует понимать, что настоящее изобретение не ограничено конкретными синтетическими способами получения, которые, конечно, могут изменяться. Также следует понимать, что используемая в настоящем описании терминология предназначена только для описания конкретных вариантов осуществления и не предназначена для ограничения. В настоящем описании и в последующей формуле изобретения будут упомянуты ряд терминов, которые должны иметь следующие значения:
В контексте настоящего описания единственное число может означать единственное число и множественное число. В контексте пункта(ов) настоящей формулы изобретения при использовании в сочетании со словом "содержащий",единственное число может означать единственное число и множественное число. В контексте настоящего описания "другой" может означать по меньшей мере второй или более.
Термин "приблизительно" относится к относительному термину, обозначающему приближение плюс или минус 10% от номинального значения, в одном варианте осуществления он относится к плюс или минус 5%, в другом варианте - к плюс или минус 2%. Для области настоящего раскрытия этот уровень приближения является подходящим, если специально не указано значение, требующее более узкого диапазона.
"Соединения" при использовании в настоящем документе включают любое фармацевтически приемлемое производное или изменение, включая конформационные изомеры (например, цис- и транс-изомеры) и все оптические изомеры (например, энантиомеры и диастереомеры), рацемические, диастереомерные и другие смеси таких изомеров, а также сольваты, гидраты, изоморфы, полиморфы, таутомеры, сложные эфиры, солевые формы и пролекарства. Выражение "пролекарство" относится к соединениям, которые являются предшественниками лекарственного средства, которые после введения высвобождают лекарственное средство in vivo посредством какого-либо химического или физиологического процесса (например, пролекарство при доведении до физиологического pH или посредством действия фермента превращается в желаемую форму лекарственного средства). Типичные пролекарства после расщепления высвобождают соответствующую свободную кислоту, и такие гидролизуемые образующие сложные эфиры остатки соединений по настоящему изобретению включают, но не ограничиваются ими, те, которые имеют карбоксильный остаток, где свободный водород замещен (C1-C4)алкилом, (C2-C7)алканоилоксиметилом, 1-(алканоилокси)этилом, имеющим от 4 до 9 атомов углерода, 1-метил-1-(алканоилокси)этилом, имеющим от 5 до 10 атомов углерода, алкоксикарбонилоксиметилом, имеющим от 3 до 6 атомов углерода, 1-(алкоксикарбонилокси)этилом, имеющим от 4 до 7 атомов углерода, 1-метил-1-(алкоксикарбонилокси)этилом, имеющим от 5 до 8 атомов углерода, N-(алкоксикарбонил)аминометилом, имеющим от 3 до 9 атомов углерода, 1-(N-(алкоксикарбонил)амино)этилом, имеющим от 4 до 10 атомов углерода, 3-фталидилом, 4-кротонолактонилом, гамма-бутиролактон-4-илом, ди-N,N-(C1-C2)алкиламино(C2-C3)алкилом (таким как β-диметиламиноэтил), карбамоил-(C1-C2)алкилом, N,N-ди(C1-C2)алкилкарбамоил-(C1-C2)алкилом и пиперидино-, пирролидино- или морфолино(C2-C3)алкилом.
В контексте настоящего описания стрелка " " или волнистая линия "
" или волнистая линия " " обозначают точку присоединения заместителя к другой группе.
" обозначают точку присоединения заместителя к другой группе.
Под "алкилом" подразумевается насыщенный углеводород с прямой цепью или насыщенный углеводород с разветвленной цепью. Примерами таких алкильных групп (при условии, что указанная длина охватывает конкретный пример) являются метил, этил, пропил, изопропил, бутил, втор-бутил, третичный бутил, изобутил, пентил, изопентил, неопентил, третичный пентил, 1-метилбутил, 2-метилбутил, 3-метилбутил, гексил, изогексил, гептил и октил.
Термин "арил" означает карбоциклическую ароматическую систему, содержащую одно, два или три кольца, в которых такие кольца могут быть конденсированы. Если кольца конденсированы, одно из колец должно быть полностью ненасыщенным, и конденсированное кольцо(а) может быть полностью насыщенным, частично ненасыщенным или полностью ненасыщенным. Термин "конденсированный" означает, что присутствует второе кольцо (то есть присоединено или образовано) за счет наличия двух смежных атомов, являющихся общими (т.е. совместно используемыми) с первым кольцом. Термин "конденсированный" эквивалентен термину "соединенный". Термин "арил" охватывает ароматические радикалы, такие как фенил, нафтил, тетрагидронафтил, инданил, бифенил, бензо[b][1,4]оксазин-3(4H)-онил, 2,3-дигидро-1H-инденил и 1,2,3,4-тетрагидронафталинил.
"Циклоалкил" относится к неароматическому кольцу, которое полностью гидрогенизировано, имеющему одно, два или три кольца, где такие кольца могут быть конденсированы, где "конденсированный" определено выше. Циклоалкил также включает бициклические структуры, которые могут быть мостиковыми или спироциклическими по природе, причем каждое отдельное кольцо в бицикле варьируется от 3 до 8 атомов. Примеры таких карбоциклических колец включают циклопропил, циклобутил, циклопентил и циклогексил.
Термин "гетероарил" означает ароматическую карбоциклическую систему, содержащую один, два, три или четыре гетероатома, выбранных независимо из кислорода, азота и серы, и имеющую одно, два или три кольца, где такие кольца могут быть конденсированы, где "конденсированный" определено выше. Термин "гетероарил" включает, но не ограничивается ими, фурил, тиенил, оксазолил, тиазолил, имидазолил, пиразолил, триазолил, тетразолил, изоксазолил, изотиазолил, оксадиазолил, тиадиазолил, пиридинил, пиридиазинил, пиримидинил, пиразинил, пиридин-2(1H)-онил, пиридазин-2(1H)-онил, пиримидин-2(1H)-онил, пиразин-2(1H)- онил, имидазо[1,2-a]пиридинил, пиразоло[1,5-a]пиридинил, 5,6,7,8-тетрагидроизохинолинил, 5,6,7,8-тетрагидрохинолинил, 6,7-дигидро-5H-циклопента[b]пиридинил, 6,7-дигидро-5H-циклопента[c]пиридинил, 1,4,5,6-тетрагидроциклопента[c]пиразолил, 2,4,5,6-тетрагидроциклопента[c]пиразолил, 5,6-дигидро-4H-пирроло[1,2-b]пиразолил, 6,7-дигидро-5H-пирроло[1,2-b][1,2,4]триазолил, 5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиридинил, 4,5,6,7-тетрагидропиразоло[1,5-a]пиридинил, 4,5,6,7-тетрагидро-1H-индазолил и 4,5,6,7-тетрагидро-2H-индазолил.
Следует понимать, что если карбоциклический или гетероциклический остаток может быть связан или иным образом присоединен к обозначенному субстрату через различные атомы кольца без обозначения конкретной точки присоединения, то предполагаются все возможные точки, будь то через атом углерода или, например, трехвалентный атом азота. Например, термин "пиридил" означает 2-, 3- или 4-пиридил, термин "тиенил" означает 2- или 3-тиенил и так далее.
"Пациент" относится к теплокровным животным, таким как, например, морские свинки, мыши, крысы, песчанки, кошки, кролики, собаки, крупный рогатый скот, козы, овцы, лошади, обезьяны, шимпанзе и люди.
Под "фармацевтически приемлемым" подразумевается, что вещество или композиция должны быть химически и/или токсикологически совместимыми с другими ингредиентами, содержащимися в составе, и/или млекопитающим, которого ими лечат.
В контексте настоящего описания выражения "реакционно-инертный растворитель" и "инертный растворитель" относятся к растворителю или их смеси, которые не взаимодействуют с исходными веществами, реагентами, промежуточными соединениями или продуктами таким образом, который отрицательно влияет на выход желаемого продукта.
В контексте настоящего описания термин "селективность" или "селективный" относится к большему действию соединения в первом анализе по сравнению с действием того же соединения во втором анализе. Например, в "селективных для ЖКТ" соединениях первый анализ относится к периоду полураспада соединения в кишечнике, а второй анализ относится к периоду полураспада соединения в печени.
"Терапевтически эффективное количество" означает количество соединения по настоящему изобретению, которое (i) лечит или предотвращает конкретное заболевание, состояние или расстройство, (ii) ослабляет, облегчает или устраняет один или несколько симптомов конкретного заболевания, состояния или расстройства, или (iii) предотвращает или задерживает появление одного или нескольких симптомов конкретного заболевания, состояния или расстройства, описанных в настоящем документе.
В контексте настоящего описания термин "лечащий", "лечить" или "лечение" охватывает как превентивное, то есть профилактическое, так и паллиативное лечение, то есть облегчение, ослабление или замедление прогрессирования заболевания (или состояния) пациента или любого повреждения ткани, связанного с заболеванием.
Соединения по настоящему изобретению могут содержать асимметричные или хиральные центры и, следовательно, существовать в разных стереоизомерных формах. Если не указано иное, предполагается, что все стереоизомерные формы соединений по настоящему изобретению, а также их смеси, включая рацемические смеси, составляют часть настоящего изобретения. Кроме того, настоящее изобретение охватывает все геометрические и позиционные изомеры. Например, если соединение по настоящему изобретению включает в себя двойную связь или конденсированное кольцо, как цис-, так и транс-формы, а также смеси включены в объем изобретения.
Хиральные соединения по изобретению (и их хиральные предшественники) могут быть получены в энантиомерно-обогащенной форме с использованием хроматографии, обычно жидкостной хроматографии высокого давления (ЖХВД) или сверхкритической флюидной хроматографии (СФХ), на смоле с асимметричной стационарной фазой и с подвижной фазой, состоящей из углеводорода, обычно гептана или гексана, содержащего от 0 до 50% изопропанола, обычно от 2 до 20%, и от 0 до 5% алкиламина, обычно 0,1% диэтиламина (ДЭА) или изопропиламина. Концентрация элюента дает обогащенную смесь.
Диастереомерные смеси могут быть разделены на их индивидуальные диастереоизомеры на основе их физико-химических различий способами, хорошо известными специалистам в данной области, такими как хроматография и/или фракционная кристаллизация. Энантиомеры могут быть разделены путем превращения энантиомерной смеси в диастереомерную смесь путем взаимодействия с соответствующим оптически активным соединением (например, хиральным вспомогательным веществом, таким как хиральный спирт или хлорангидрид кислоты Мошера), разделения диастереоизомеров и превращения (например, гидролизом) отдельных диастереоизомеров в соответствующие чистые энантиомеры. Энантиомеры также могут быть разделены с использованием хиральной колонки ВЭЖХ. Альтернативно, конкретные стереоизомеры могут быть синтезированы с использованием оптически активного исходного вещества, путем асимметричного синтеза с использованием оптически активных реагентов, субстратов, катализаторов или растворителей или путем превращения одного стереоизомера в другой путем асимметричного превращения.
Когда соединения по настоящему изобретению обладают двумя или более стереогенными центрами и абсолютная или относительная стереохимия указана в названии, обозначения R и S относятся соответственно к каждому стереогенному центру в порядке возрастания цифровой последовательности (1, 2, 3 и т.д.) в соответствии с традиционными последовательностями номеров, используемыми в ИЮПАК (международный союз теоретической и прикладной химии), для каждой молекулы. Когда соединения по настоящему изобретению обладают одним или несколькими стереогенными центрами, и в названии или структуре не указана стереохимия, подразумевается, что название или структура предназначены для охвата всех форм соединения, включая рацемическую форму.
Соединения по настоящему изобретению могут содержать олефиноподобные двойные связи. Когда такие связи присутствуют, соединения по изобретению существуют в виде цис- и транс-конфигураций и в виде их смесей. Термин "цис" относится к ориентации двух заместителей относительно друг друга и плоскости кольца (либо оба "вверх", либо оба "вниз"). Аналогично, термин "транс" относится к ориентации двух заместителей относительно друг друга и плоскости кольца (заместители находятся на противоположных сторонах кольца).
Также возможно, что промежуточные соединения и соединения по настоящему изобретению могут существовать в разных таутомерных формах, и все такие формы включены в объем изобретения. Термин "таутомер" или "таутомерная форма" относится к структурным изомерам с различными энергиями, которые являются взаимопревращаемыми через барьер с низкой энергией. Например, протонные таутомеры (также известные как прототропные таутомеры) включают взаимопревращения посредством миграции протона, такие как изомеризации кето-енола и имин-енамина.
Валентные таутомеры включают взаимопревращения путем реорганизации некоторых связывающих электронов.
В объем заявленных соединений по настоящему изобретению включены все стереоизомеры, геометрические изомеры и таутомерные формы соединений формулы (I), включая соединения, проявляющие более одного типа изомерии, и смеси одного или нескольких из них. Также включены кислотно-аддитивные или основные соли, в которых противоион является оптически активным, например, D-лактат или L-лизин, или рацемическим, например, DL-тартрат или DL-аргинин.
Настоящее изобретение включает все фармацевтически приемлемые изотопно-меченые соединения формулы (I), где один или несколько атомов заменены атомами, имеющими одинаковый атомный номер, но атомную массу или массовое число, отличные от атомной массы или массового числа, обычно встречающихся в природе.
Примеры изотопов, подходящих для включения в соединения по изобретению, включают изотопы водорода, такие как 2H и 3H, углерода, такие как 11C, 13C и 14C, хлора, такие как 36Cl, фтора, такие как 18F, йода, такие как 123I, 124I и 125I, азота, такие как 13N и 15N, кислорода, такие как 15O, 17O и 18O, фосфора, такие как 32P, и серы, такие как 35S.
Некоторые изотопно-меченые соединения формулы (I), например, те, которые включают радиоактивный изотоп, полезны в исследованиях распределения лекарственного средства и/или субстрата в ткани. Радиоактивные изотопы тритий, то есть 3H, и углерод-14, то есть 14C, особенно полезны для этой цели ввиду их легкого включения и готовых средств обнаружения.
Замена более тяжелыми изотопами, такими как дейтерий, то есть 2H, может дать определенные терапевтические преимущества в результате большей метаболической стабильности, например, увеличенный период полужизни in vivo или сниженные требования к дозировке, и, следовательно, может быть предпочтительной в некоторых обстоятельствах.
Замена позитрон-излучающими изотопами, такими как 11C, 18F, 15O и 13N, может быть полезна в исследованиях позитронно-эмиссионной томографией (ПЭТ) для изучения степени занятости рецептора субстратом.
Изотопно-меченые соединения формулы (I), как правило, могут быть получены обычными методами, известными специалистам в данной области техники, или способами, аналогичными тем, которые описаны в прилагаемых примерах и получениях, с использованием подходящих изотопно-меченых реагентов вместо ранее используемых немеченых реагентов.
Соединения по настоящему изобретению могут быть выделены и использованы сами по себе или, когда это возможно, в форме их фармацевтически приемлемой соли. Термин "соли" относится к неорганическим и органическим солям соединения по настоящему изобретению. Эти соли могут быть получены in situ во время окончательного выделения и очистки соединения или путем отдельной обработки соединения подходящей органической или неорганической кислотой или основанием и выделения образованной таким образом соли. Кислоты, которые используются для получения фармацевтически приемлемых кислотно-аддитивных солей вышеупомянутых основных соединений по настоящему изобретению, представляют собой кислоты, которые образуют нетоксичные кислотно-аддитивные соли (т.е. соли, содержащие фармакологически приемлемые анионы, такие как гидрохлоридные, гидробромидные, гидроиодидные, нитратные, сульфатные, бисульфатные, фосфатные, кислые фосфатные, ацетатные, лактатные, цитратные, кислые цитратные, тартратные, битартратные, сукцинатные, малеатные, фумаратные, глюконатные, сахаратные, бензоатные, метансульфонатные, этансульфонатные, бензолсульфонатные, нафтилатные, мезилатные, глюкогептонатные, лактобионатные, лаурилсульфонатные, гексафторфосфатные, бензолсульфонатные, тозилатные, формиатные, трифторацетатные, оксалатные, безилатные, пальмитатные, памоатные, малонатные, стеаратные, лауратные, малатные, боратные, п-толуолсульфатные и памоатные (то есть 1,1'-метилен-бис(2-гидрокси-3-нафтоат)) соли).
Изобретение также относится к основно-аддитивным солям соединений по настоящему изобретению. Химические основания, которые могут быть использованы в качестве реагентов для получения фармацевтически приемлемых основных солей тех соединений по настоящему изобретению, которые имеют кислотную природу, представляют собой те, что образуют нетоксичные основные соли с такими соединениями. Такие нетоксичные основные соли включают, но не ограничиваются этим, соли, полученные из таких фармакологически приемлемых катионов, таких как катионы щелочных металлов (например, литий, калий и натрий) и катионы щелочноземельных металлов (например, кальций и магний), аммоний или водорастворимые соли присоединения амина, такие как N-метилглюкамин-(меглумин), тетраметиламмоний, тетраэтиламмоний, метиламин, диметиламин, триметиламин, триэтиламин, этиламин и низший алканоламмоний, и другие основные соли фармацевтически приемлемых органических аминов. См., например, Berge с соавт. J. Pharm. Sci. 66, 1-19 (1977).
Некоторые соединения по настоящему изобретению могут существовать в более чем одной кристаллической форме (обычно именуемые как "полиморфы"). Полиморфы могут быть получены кристаллизацией в различных условиях, например, с использованием разных растворителей или разных смесей растворителей для перекристаллизации; кристаллизации при разных температурах; и/или различных режимов охлаждения, варьирующихся от очень быстрого до очень медленного охлаждения во время кристаллизации. Полиморфы также могут быть получены путем нагревания или плавления соединения по настоящему изобретению с последующим постепенным или быстрым охлаждением. Присутствие полиморфов может быть определено с помощью твердотельной ЯМР-спектроскопии, ИК-спектроскопии, дифференциальной сканирующей калориметрии, порошковой рентгеновской дифракции или иными подобными методами.
В одном варианте осуществления R3 представляет собой  .
.
В другом варианте осуществления R3 представляет собой 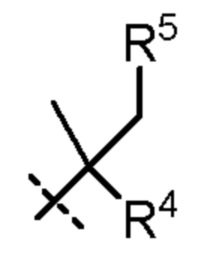 .
.
В дополнительном варианте осуществления R1 представляет собой метил.
В еще одном варианте осуществления R4 представляет собой H, -CH2OH или циано.
В другом варианте осуществления соединение представляет собой
(S)-2-(5-((3-этокси-5-фторпиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамид;
N-(2-цианопропан-2-ил)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)пиримидин-5-карбоксамид;
2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(3-метил-1,1-диоксидотетрагидротиофен-3-ил)пиримидин-5-карбоксамид;
2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(1-гидрокси-2-метилпропан-2-ил)пиримидин-5-карбоксамид;
(S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамид;
(S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(3-(гидроксиметил)тетрагидрофуран-3-ил)пиримидин-5-карбоксамид;
(R)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(3-(гидроксиметил)тетрагидрофуран-3-ил)пиримидин-5-карбоксамид;
2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(2-метил-1-(метилсульфонил)пропан-2-ил)пиримидин-5-карбоксамид;
(S)-2-(5-((3-(2-фторэтокси)пиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамид;
3-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(1-гидрокси-2-метилпропан-2-ил)-1,2,4-триазин-6-карбоксамид;
N-(1,3-дигидрокси-2-метилпропан-2-ил)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)пиримидин-5-карбоксамид;
(S)-3-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)-1,2,4-триазин-6-карбоксамид;
N-(1,1-диоксидотетрагидротиофен-3-ил)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)пиримидин-5-карбоксамид;
(R)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамид; или
(S)-2-(5-((3-этоксипиразин-2-ил)окси)пиридин-3-ил)-N-(1-гидрокси-2-метилпропан-2-ил)пиримидин-5-карбоксамид;
или его фармацевтически приемлемую соль.
В другом варианте осуществления соединение представляет собой:
(R)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(3-(гидроксиметил)тетрагидрофуран-3-ил)пиримидин-5-карбоксамид;
(S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамид; или
(S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(3-(гидроксиметил)тетрагидрофуран-3-ил)пиримидин-5-карбоксамид, или его фармацевтически приемлемую соль.
В дополнительном варианте осуществления соединение имеет структуру:

или его фармацевтически приемлемая соль.
В дополнительном варианте осуществления соединение формулы (I) или (Ia) или соль соединения присутствует в фармацевтической композиции в терапевтически эффективном количестве в смеси по меньшей мере с одним фармацевтически приемлемым эксципиентом.
В дополнительном варианте осуществления композиция дополнительно содержит по меньшей мере один дополнительный фармацевтический агент, выбранный из группы, состоящей из противовоспалительного средства, антидиабетического средства и средства, регулирующего холестерин/липиды.
В варианте осуществления способ лечения диабета включает введение эффективного количества соединения по настоящему изобретению или фармацевтически приемлемой соли указанного соединения пациенту, нуждающемуся в этом.
В другом варианте осуществления способ лечения метаболического заболевания, состояния или расстройства или заболевания, состояния или расстройства, связанного с метаболизмом, включает стадию введения пациенту терапевтически эффективного количества соединения по настоящему изобретению или фармацевтически приемлемой соли указанного соединения.
В другом варианте осуществления способ лечения состояния, выбранного из группы, состоящей из гиперлипидемии, диабета I типа, сахарного диабета II типа, идиопатического диабета I типа (типа Ib), латентного аутоиммунного сахарного диабета взрослых (LADA), диабета типа 2 с ранним началом (EOD), атипичного диабета молодых (YOAD), диабета зрелого возраста у молодых (MODY), диабета, связанного с недоеданием, гестационного диабета, ишемической болезни сердца, ишемического инсульта, рестеноза после ангиопластики, заболевания периферических сосудов, перемежающейся хромоты, инфаркта миокарда, дислипидемии, послеобеденной липемии, состояний нарушенной толерантности к глюкозе (IGT), состояний нарушенной концентрации глюкозы в плазме натощак, метаболического ацидоза, кетоза, артрита, ожирения, остеопороза, гипертонии, застойной сердечной недостаточности, гипертрофии левого желудочка, заболевания периферических артерий, диабетической ретинопатии, дегенерации желтого пятна, катаракты, диабетической нефропатии, гломерулосклероза, хронической почечной недостаточности, диабетической невропатии, метаболического синдрома, синдрома X, предменструального синдрома, ишемической болезни сердца, стенокардии, тромбоза, атеросклероза, инфаркта миокарда, преходящих ишемических атак, инсульта, сосудистого рестеноза, гипергликемии, гиперинсулинемии, гипертриглицеридемии, резистентности к инсулину, нарушения метаболизма глюкозы, состояний нарушенной толерантности к глюкозе, состояний нарушенной концентрации глюкозы в плазме натощак, ожирения, эректильной дисфункции, заболеваний кожи и соединительных тканей, изъязвления стопы и язвенного колита, эндотелиальной дисфункции и нарушенной податливости сосудов, гипер-апо-B-липопротеинемии, болезни Альцгеймера, шизофрении, ухудшения когнитивной деятельности, воспалительного заболевания кишечника, язвенного колита, болезни Крона и синдрома раздраженного кишечника, неалкогольного стеатогепатита (NASH) или неалкогольной жировой дистрофии печени (NAFLD), включает введение эффективного количества соединения по настоящему изобретению или фармацевтически приемлемой соли указанного соединения.
В дополнительном варианте осуществления способ лечения метаболического заболевания, состояния или расстройства или заболевания, состояния или расстройства, связанного с метаболизмом, включает стадию введения пациенту, нуждающемуся в таком лечении, двух отдельных фармацевтических композиций, включающих
(i) первую композицию в соответствии с настоящим изобретением; и
(ii) вторую композицию, содержащую по меньшей мере один дополнительный фармацевтический агент, выбранный из группы, состоящей из средства против ожирения и антидиабетического средства и по меньшей мере одного фармацевтически приемлемого эксципиента.
В еще одном дополнительном варианте осуществления способ по настоящему изобретению осуществляют, когда указанную первую композицию и указанную вторую композицию вводят одновременно.
В еще одном варианте осуществления способ по настоящему изобретению осуществляют, когда первую композицию и указанную вторую композицию вводят последовательно и в любом порядке.
В одном варианте осуществления, когда вводят две композиции, первую композицию и вторую композицию вводят одновременно. В другом варианте осуществления первую композицию и вторую композицию вводят последовательно и в любом порядке.
Соединения по настоящему изобретению могут быть синтезированы синтетическими путями, которые включают способы, аналогичные тем, которые хорошо известны в области химии, особенно в свете описания, содержащегося в настоящем документе. Исходные вещества обычно доступны из коммерческих источников, таких как Aldrich Chemicals (Милуоки, Висконсин), или могут быть легко получены с использованием способов, хорошо известных специалистам в данной области техники (например, получены способами, в основном описанными в Louis F. Fieser и Mary Fieser, Reagents for Organic Synthesis, v. 1-19, Wiley, Нью-Йорк (изд. 1967-1999) или Beilsteins Handbuch der organischen Chemie, 4, Aufl. ed. Springer-Verlag, Берлин, включая приложения (также доступны через онлайн базу данных Beilstein)). Многие из соединений, использованных в настоящем описании, относятся к соединениям, которые представляют большой научный интерес и коммерческую необходимость, или получены из них, и, соответственно, многие такие соединения являются коммерчески доступными или описаны в литературе, или их легко получить из других общедоступных веществ способами, о которых сообщается в литературе.
В иллюстративных целях схемы реакций, изображенные ниже, предоставляют потенциальные пути для синтеза соединений по настоящему изобретению, а также ключевых промежуточных соединений. Более подробное описание отдельных стадий реакции см. в разделе "Примеры" ниже. Специалистам в данной области будет понятно, что другие пути синтеза могут быть использованы для синтеза соединений по изобретению. Хотя конкретные исходные вещества и реагенты обсуждаются ниже, другие исходные вещества и реагенты могут быть легко использованы вместо для обеспечения разнообразных производных и/или условий реакции. Кроме того, многие из соединений, полученных способами, описанными ниже, могут быть дополнительно модифицированы в свете настоящего раскрытия с использованием обычной химии, хорошо известной специалистам в данной области техники.
При получении соединений формулы I следует отметить, что некоторые из способов получения, полезных для получения соединений, описанных в настоящем документе, могут требовать защиты отдаленной функциональной группы (например, первичный амин, вторичный амин, карбоксил в предшественниках формулы I). Необходимость такой защиты будет варьироваться в зависимости от характера отдаленной функциональной группы и условий способов получения. Необходимость такой защиты легко определяется специалистом в данной области техники. Использование таких способов защиты/удаления защиты также известно специалистам в данной области. Общее описание защитных групп и их использования см. в T.W. Greene, Protective Groups in Organic Synthesis, John Wiley & Sons, Нью-Йорк, 1991.
Например, некоторые соединения содержат функциональные группы первичных аминов или карбоновых кислот, которые могут мешать реакциям на других участках молекулы, если оставить их незащищенными. Соответственно, такие функциональные группы могут быть защищены соответствующей защитной группой, которая может быть удалена на последующей стадии. Подходящие защитные группы для защиты амина и карбоновой кислоты включают защитные группы, обычно используемые в синтезе пептидов (такие как N-трет-бутоксикарбонил (Boc), бензилоксикарбонил (Cbz) и 9-флуоренилметиленоксикарбонил (Fmoc) для аминов и низшие алкиловые или бензиловые эфиры для карбоновых кислот), которые, как правило, не являются химически активными в описанных условиях реакции и обычно могут быть удалены без химического изменения других функциональных групп в соединениях формулы I и Ia.
Схемы реакций, описанные ниже, предназначены для обеспечения общего описания методологии, используемой в получении соединений по настоящему изобретению. Некоторые из соединений по настоящему изобретению содержат один хиральный центр. На следующих схемах общие способы получения соединений показаны либо в рацемической, либо в энантиомернообогащенной форме. Специалисту в данной области техники будет очевидно, что все синтетические превращения могут быть проведены точно одинаковым образом, независимо от того, являются ли вещества энантиомернообогащенными или рацемическими. Кроме того, разделение на желаемое оптически активное вещество может происходить в любой желаемой точке последовательности с использованием хорошо известных способов, таких как описанные в настоящем документе и в литературе по химии.
На схемах реакций I и II переменные D1, D2, R1, R2 и R3 являются такими, как описано в кратком описании, если не указано иное. Переменная R представляет собой метил или этил. Схема реакции I описывает общие процедуры, которые могут быть использованы для получения соединений по настоящему изобретению, имеющих формулу (I).
Схема реакции I
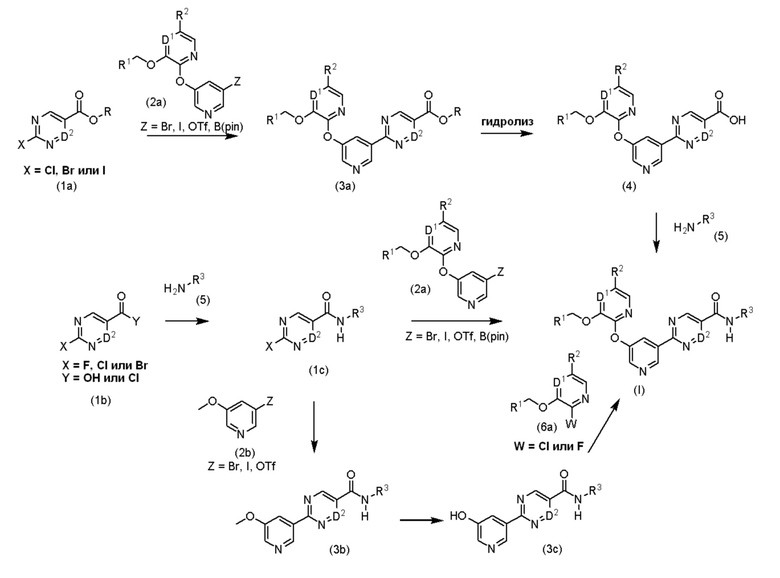
Соединения формулы (I) могут быть синтезированы, исходя из соответствующих промежуточных соединений, посредством способов, описанных в литературе, такой как J. Med. Chem., 2007, 50, 2990-3003; Monatsh Chem, 2012, 143, 1575-1592; J. Med. Chem., 2011, 54, 6342-6363; Org. Proc. Res. Dev. 2014, 18, 1145-1152; Angew. Chem. Int. Ed. 2011, 50, 9943; J. Am. Chem. Soc. 2005, 127, 8146; J. Org. Chem. 2008, 73, 284; Org. Lett. 2002, 4, 973; Org. Lett., 2011, 13, 1840-1843; Metal Catalyzed Cross-Coupling Reactions and More, Wiley-VCH, Weinheim, Germany, 2014, 3, 995; Applications of Transition Metal Catalysis in Drug Discovery and Development, John Wiley & Sons, Inc., Hoboken, New Jersey, USA, 2012, 3, 97. Промежуточные соединения (1a) и (1b) являются коммерчески доступными и/или могут быть получены посредством способов, известных специалистам в данной области техники. Например, промежуточные соединения (1a) и (1b) могут быть синтезированы посредством способов, описанных в литературе, такой как: J. Med. Chem. 2000, 43, 3995; Org. Proc. Res. Dev. 2010, 14, 936. Промежуточные соединения (2a) и (2b) являются коммерчески доступными или описаны в литературе и могут быть получены посредством способов, известных специалистам в данной области техники, включая те, что описаны ниже (схема реакции II).
Промежуточное соединение (3а) может быть получено из промежуточных соединений (1а) и (2а) в реакции сочетания, опосредованной переходным металлом. Один из галогенангидридов (1a) или (2a) может быть превращен в металлоорганический реагент, такой как бороновая кислота, цинкат, станнан или производное Гриньяра, с использованием способов, хорошо известных специалистам в данной области техники. Полученный металлоорганический реагент может затем взаимодействовать с другим галоидным промежуточным соединением в реакции кросс-сочетания, катализируемой переходным металлом. Предпочтительно промежуточное соединение (2а) превращают в цинкат и сочетают с промежуточным соединением (1а) с использованием палладиевого или никелевого катализатора в инертном по отношению к реакции растворителе, таком как толуол, 1,2-диметоксиэтан, диоксан, ДМСО, ДМФА или ТГФ, в присутствии подходящего лиганда и основания, такого как трет-бутоксид натрия, калия или лития или карбонат цезия, при температуре от 10°C до 130°C посредством способов, описанных в литературе, такой как: J. Med. Chem., 2007, 50, 2990-3003; Monatsh Chem, 2012, 143, 1575-1592; J. Med. Chem., 2011, 54, 6342-6363; Org. Proc. Res. Dev. 2014, 18, 1145-1152, или других способов, известных специалистам в данной области техники.
Промежуточное соединение (4) может быть получено из сложного эфира (3а) посредством реакции гидролиза в условиях, хорошо известных специалистам в данной области техники. Предпочтительно промежуточное соединение (3a, R=метил или этил) обрабатывают водным основанием, таким как гидроксид натрия, гидроксид лития или гидроксид калия, в подходящем растворителе или смеси растворителей, состоящей из воды, метанола и/или ТГФ, при температуре от 20°C до 60°C.
Соединения формулы (I) могут быть получены из кислоты (4) и амина (5) в условиях образования амидов, хорошо известных специалистам в данной области техники, с использованием реагентов сочетания, таких как ангидрид пропанфосфоновой кислоты (T3P), 1,1'-карбонилдиимидазол (CDI), бензотриазол-1-илокситрис(диметиламино)фосфонийгексафторфосфат (BOP), 2-(1H-7-азабензотриазол-1-ил)-1,1,3,3-тетраметилуроний гексафторфосфат метанаминий (HATU), оксалилхлорид, O-бензотриазол-1-ил-N,N,N',N'-тетраметилурония гексафторфосфат (HBTU), 2-хлор-1,3-диметилимидазолиния хлорид (DMC), N-(3-диметиламинопропил)-N'-этилкарбодиимид (EDCI) или 1-гидроксибензотриазол (HOBT), в инертном по отношению к реакции растворителе, таком как ацетонитрил, дихлорметан (ДХМ), ДМФА, ДМСО или ТГФ, в присутствии основания, такого как триэтиламин, N-метил-морфолин или N,N-диизопропилэтиламин, при температуре от 10°C до 90°C, предпочтительно от 20°C до 65°C.
Альтернативно, соединения формулы (I) могут быть получены путем двухстадийной последовательности из промежуточного соединения (1b) и амина (5) посредством реакции амидного сочетания с получением промежуточного соединения (1c) с последующей металл-опосредованной реакцией сочетания с арилгалогенидом (2а). Предпочтительно промежуточное соединение (1с) получают из хлорангидрида кислоты (1b, Y=Cl) и амина (5) в присутствии основания, такого как триэтиламин или N,N-диизопропилэтиламин, в инертном по отношению к реакции растворителе, таком как дихлорметан, при температуре от -20°C до 30°C, предпочтительно от -20°C до 0°C. Альтернативно, промежуточное соединение (1c) может быть получено из кислоты (1b, Y=OH) и амина (5) в присутствии реагента амидного сочетания, такого как ангидрид пропанфосфоновой кислоты (T3P), 1,1'-карбонилдиимидазол (CDI), бензотриазол-1-илокситрис(диметиламино)фосфонийгексафторфосфат (BOP), 2-(1H-7-азабензотриазол-1-ил)-1,1,3,3-тетраметилуроний гексафторфосфат метанаминий (HATU), O-бензотриазол-1-ил-N,N,N',N'-тетраметилурония гексафторфосфат (HBTU), 2-хлор-1,3-диметилимидазолиния хлорид (DMC), N-(3-диметиламинопропил)-N'-этилкарбодиимид (EDCI) или 1-гидроксибензотриазол (HOBT), в инертном по отношению к реакции растворителе, таком как ацетонитрил, дихлорметан, ДМФА, ДМСО или ТГФ, в присутствии основания, такого как триэтиламин, N-метил-морфолин или N,N-диизопропилэтиламин, при температуре от 10°C до 90°C. Соединения формулы (I) могут затем быть получены из галогенидов (1c) и (2a) в реакции сочетания, опосредованной переходным металлом. Один из галогенидов (1c) или (2a) может быть превращен в металлоорганический реагент, такой как бороновая кислота, цинкат, станнан или производное Гриньяра, с использованием способов, хорошо известных специалистам в данной области техники. Полученный металлоорганический реагент может затем взаимодействовать с другим галоидным промежуточным соединением в реакции кросс-сочетания, катализируемой переходным металлом. Предпочтительно промежуточное соединение (2a) превращают в цинкат и сочетают с промежуточным соединением (1c) с использованием палладиевого или никелевого катализатора в инертном по отношению к реакции растворителе, таком как толуол, 1,2-диметоксиэтан, диоксан, ДМСО, ДМФА или ТГФ, в присутствии подходящего лиганда и основания, такого как трет-бутоксид натрия, калия или лития или карбонат цезия, при температуре от 10°C до 130°C посредством способов, описанных в литературе, такой как: J. Med. Chem., 2007, 50, 2990-3003; Monatsh Chem, 2012, 143, 1575-1592; J. Med. Chem., 2011, 54, 6342-6363; Org. Proc. Res. Dev. 2014, 18, 1145-1152, или других способов, известных специалистам в данной области техники.
Альтернативно, соединения формулы (I) могут быть получены из промежуточного соединения (1c) путем трехступенчатой последовательности, включающей добавление гетероарилгалогенида (2b) с последующим деметилированием и добавлением арилгалогенида (6a). Промежуточное соединение (3b) может быть получено посредством реакции сочетания, опосредованной переходным металлом, начиная с галогенидов (1c) и (2b). Один из галогенидов (1c) или (2b) может быть превращен в металлоорганический реагент, такой как бороновая кислота, цинкат, станнан или производные Гриньяра, с использованием способов, хорошо известных специалистам в данной области техники. Полученный металлоорганический реагент может затем взаимодействовать с другим галоидным промежуточным соединением в реакции кросс-сочетания, катализируемой переходным металлом. Предпочтительно промежуточное соединение (2b) превращают в цинкат и сочетают с промежуточным соединением (1c) с использованием палладиевого или никелевого катализатора в инертном по отношению к реакции растворителе, таком как толуол, 1,2-диметоксиэтан, диоксан, ДМСО, ДМФА или ТГФ, в присутствии подходящего лиганда и основания, такого как трет-бутоксид натрия, калия или лития или карбонат цезия, при температуре от 10°C до 130°C посредством способов, описанных в литературе, такой как: J. Med. Chem., 2007, 50, 2990-3003; Monatsh Chem, 2012, 143, 1575-1592; J. Med. Chem., 2011, 54, 6342-6363; Org. Proc. Res. Dev. 2014, 18, 1145-1152, или других способов, известных специалистам в данной области техники. Промежуточное соединение (3c) может быть получено из промежуточного соединения (3b) посредством деметилирования с использованием галогенводородных кислот, таких как бромистоводородная, оснований, таких как гидроксид натрия или алкоксид натрия, трибромида бора, тиола или других способов, известных специалистам в данной области техники. Например, деметилирование может быть осуществлено посредством способов, описанных в литературе, такой как: Arch Pharm Res 2008, 31, 305-309; Tetrahedron, 2005, 61, 7833-7863; Protecting Groups in Organic Synthesis, John Wiley & Sons, Inc., Hoboken, New Jersey, USA, 2007, 370-382. Соединения формулы (I) затем могут быть получены из гетероарилгалогенида (6a) в реакции нуклеофильного ароматического замещения спиртом (3c) в инертном по отношению к реакции растворителе, таком как диметилсульфоксид (ДМСО), N,N-диметилформамид (ДМФА), ацетонитрил или тетрагидрофуран (ТГФ), в присутствии подходящего основания, такого как карбонат цезия, триэтиламин (ТЭА) или N,N-диизопропилэтиламин (ДИПЭА), при температуре от 20°C до 160°C. Предпочтительно промежуточные соединения (6a) и (3c) взаимодействуют в ДМСО, ТГФ или ацетонитриле в присутствии триэтиламина или N,N-диизопропилэтиламина при температуре от 100°C до 160°C с получением соединений формулы (I) посредством способов, описанных в литературе, такой как: Tetrahedron 2005, 62 6000-6005, Journal of Medicinal Chemistry, 2015, 58(7), 3036-3059.
Схема реакции II в общих чертах описывает синтез промежуточных соединений (2а).
Схема реакции II
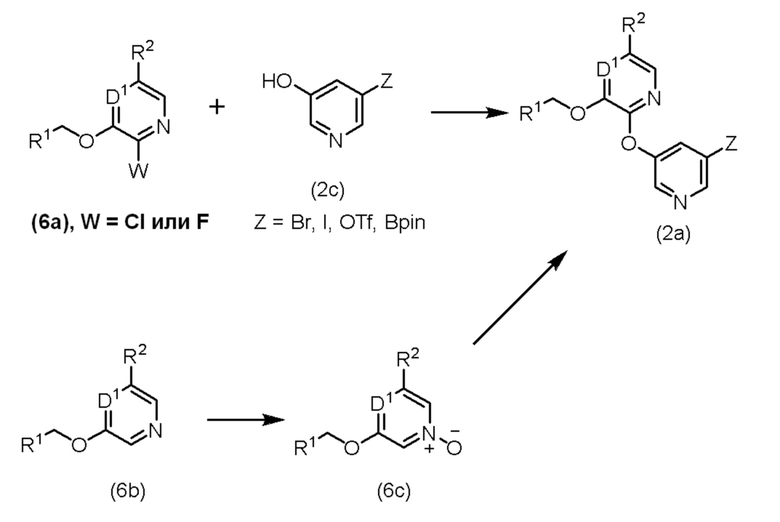
Промежуточные соединения (6a), (6b) и (2c) являются коммерчески доступными или описаны в литературе и могут быть получены посредством способов, известных специалистам в данной области техники. Промежуточное соединение (2a) может быть синтезировано посредством реакции нуклеофильного ароматического замещения гетероарилгалогенида (6a) гидроксипиридином (2c) в инертном по отношению к реакции растворителе, таком как диметилсульфоксид (ДМСО), N,N-диметилформамид (ДМФА), ацетонитрил, N-метил-2-пирролидинон (NMP) или тетрагидрофуран (ТГФ), в присутствии подходящего основания, такого как карбонат цезия, карбонат калия, триэтиламин (ТЭА) или N,N-диизопропилэтиламин (ДИПЭА), при температуре от 20°C до 160°C. Предпочтительно промежуточные соединения (6a) и (2c) взаимодействуют в ДМСО, NMP или ацетонитриле в присутствии триэтиламина или N,N-диизопропилэтиламина при температуре от 100°C до 160°C с получением промежуточного соединения (2a) с использованием способов, описанных в литературе, такой как: Tetrahedron 2005, 62 6000-6005, Journal of Medicinal Chemistry, 2015, 58(7), 3036-3059. Альтернативно, промежуточное соединение (2a) может быть синтезировано путем образования эфира, промотированного переходным металлом, между гидроксиароматическим участником сочетания (2c) и ароматическим галогенидом (6a) с использованием способов, таких как те, что описаны в: Advanced Synthesis & Catalysis, 2011, 353, 3403-3414; Chemistry - A European Journal, 2015, 21, 8727-8732; Synlett 2012, 23, 101; J. Org. Chem. 2009, 74, 7187; Org. Lett. 2007, 9, 643; Angew. Chem. Int. Ed. 2011, 50, 9943; J. Am. Chem. Soc. 2005, 127, 8146; J. Org. Chem. 2008, 73, 284; Org. Lett. 2002, 4, 973. Соответствующие исходные вещества (6a) и (2c) могут быть обработаны солью металла, такой как хлорид меди (I), бромид меди (I) или йодид меди (I), и лигандом, таким как 2,2,6,6-тетраметилгептан-3,5-дион, 1,10-фенантролин или другим подходящим лигандом, в инертном по отношению к реакции растворителе, таком как толуол, ДМСО или ДМФА, в присутствии основания, такого как карбонат калия, карбонат цезия или фосфат калия, при температуре от 80°C до 120°C. Предпочтительно соответствующие исходные вещества (6a) и (2c) обрабатывают хлоридом меди (I) и 2,2,6,6-тетраметилгептан-3,5-дионом в толуоле в присутствии карбоната цезия при температуре от 100°C до 120°C.
Альтернативно, промежуточное соединение (2a) может быть получено в результате двухстадийной последовательности, включающей образование N-оксида (6c) с последующим добавлением гидроксилпиридина (2c). N-оксид (6c) может быть получен с помощью окислителей, таких как м-хлорпероксибензойная кислота, пероксид водорода, перманганат калия или других окислителей, известных специалистам в данной области техники, в инертном по отношению к реакции растворителе, таком как дихлорметан, 1,2-дихлорэтан или ацетонитрил, при температуре от 0°C до 25°C. Предпочтительно промежуточное соединение (6b) подвергают взаимодействию в дихлорметане с м-хлорпероксибензойной кислотой при температуре от 10°C до 25°C с получением промежуточного соединения (6c). Промежуточное соединение (2a) может быть получено из промежуточного соединения (6c) и промежуточного соединения (2c) в присутствии бромтрипирролидинофосфония гексафторфосфата (PyBroP) в инертном по отношению к реакции растворителе, таком как тетрагидрофуран, дихлорметан или диоксан, при температуре от 10°C до 25°C. Предпочтительно промежуточное соединение (6с) подвергают взаимодействию с промежуточным соединением (2с) в присутствии бромтрипирролидинофосфония гексафторфосфата в тетрагидрофуране при температуре от 10°C до 25°C, как описано в Org. Lett., 2011, 13, 1840-1843.
АГЕНТЫ ДЛЯ КОМБИНИРОВАНИЯ
Соединения по настоящему изобретению могут быть введены в виде монотерапии или в комбинации с одним или несколькими дополнительными терапевтическими агентами. Под "введением в комбинации" или "комбинированной терапией" подразумевается, что соединение по настоящему изобретению и один или несколько дополнительных терапевтических агентов вводят параллельно млекопитающему, подвергающемуся лечению. При введении в комбинации каждый компонент может быть введен одновременно или последовательно в любом порядке в разные моменты времени. Таким образом, каждый компонент может быть введен отдельно, но достаточно близко по времени, чтобы обеспечить желаемый терапевтический эффект. Таким образом, способы профилактики и лечения, описанные в настоящем документе, включают применение агентов для комбинирования.
Агенты для комбинирования вводят млекопитающему в терапевтически эффективном количестве. Под "терапевтически эффективным количеством" подразумевается количество соединения по настоящему изобретению, которое при введении млекопитающему в виде монотерапии или в комбинации с дополнительным терапевтическим агентом, является эффективным для лечения желаемого заболевания/состояния, например, ожирения, диабета и сердечно-сосудистых заболеваний, например, антигипертензивные вещества, и ишемической болезни сердца.
Примеры подходящих антидиабетических средств включают, например, инсулины, метфомин, ингибиторы ДППIV, агонисты GLP-1, аналоги и миметики, ингибиторы SGLT1 и SGLT2. Подходящие антидиабетические агенты включают ингибитор ацетил-КоА-карбоксилазы (ACC), такой как те, которые описаны в WO2009144554, WO2003072197, WO2009144555 и WO2008065508, ингибитор диацилглицерол-O-ацилтрансферазы 1 (DGAT-1), такой как те, которые описаны в WO09016462 или WO2010086820, AZD7687 или LCQ908, ингибиторы моноацилглицерол-O-ацилтрансферазы, ингибитор фосфодиэстеразы (PDE)-10, активатор AMPK, сульфонилмочевину (например, ацетогексамид, хлорпропамид, диабинез, глибенкламид, глипизид, глибурид, глимепирид, гликлазид, глипентид, гликвидон, глизоламид, толазамид и толбутамид), меглитинид, ингибитор α-амилазы (например, тендамистат, трестатин и AL-3688), ингибитор α-глюкозидгидролазы (например, акарбозу), ингибитор α-глюкозидазы (например, адипозин, камиглибоз, эмиглитат, миглитол, воглибоз, прадимицин-Q и сальбостатин), агонист PPARγ (например, балаглитазон, циглитазон, дарглитазон, энглитазон, изаглитазон, пиоглитазон и росиглитазон), агонист PPAR α/γ (например, CLX-0940, GW-1536, GW-1929, GW-2433, KRP-297, L-796449, LR-90, MK-0767 и SB-219994), бигуанид (например, метформин), модулятор глюкагоноподобного пептида 1 (GLP-1), такой как агонист (например, эксендин-3 и эксендин-4), лираглутид, альбиглютид, эксенатид (Баета®), альбиглютид, ликсисенатид, дулаглутид, семаглутид, NN-9924, TTP-054, ингибитор протеин-тирозинфосфатазы-1B (PTP-1B) (например, тродусквемин, экстракт гиртиозала и соединения, описанные Zhang S. с соавт., Drug Discovery Today, 12(9/10), 373-381 (2007)), активатор SIRT-1 (например, ресвератрол, GSK2245840 или GSK184072), ингибитор дипептидилпептидазы IV (ДПП-IV) (например, те, которые в WO2005116014, ситаглиптин, вилдаглиптин, алоглиптин, дутоглиптин, линаглиптин и саксаглиптин), стимулятор секреции инсулина, ингибитор окисления жирных кислот, антагонист А2, ингибитор амино-концевой киназы c-jun (JNK), активаторы глюкокиназы (GKa), такие как те, которые описаны в WO2010103437, WO2010103438, WO2010013161, WO2007122482, TTP-399, TTP-355, TTP-547, AZD1656, ARRY403, MK-0599, TAK-329, AZD5658 или GKM-001, инсулин, миметик инсулина, ингибитор гликогенфосфорилазы (например, GSK1362885), агонист рецептора VPAC2, ингибиторы SGLT2, такие как описанные E.C. Chao с соавт. Nature Reviews Drug Discovery 9, 551-559 (июль 2010), включая дапаглифлозин, канаглифлозин, эмпаглифлозин, тофоглифлозин (CSG452), эртуглифлозин, ASP-1941, THR1474, TS-071, ISIS388626 и LX4211, а также те, которые в WO2010023594, модулятор рецептора глюкагона, такой как описанный в Demong, D.E. с соавт. Annual Reports in Medicinal Chemistry 2008, 43, 119-137, модуляторы GPR119, в частности агонисты, такие как те, которые описаны в WO2010140092, WO2010128425, WO2010128414, WO2010106457, Jones, R.M. с соавт. в Medicinal Chemistry 2009, 44, 149-170 (например, MBX-2982, GSK1292263, APD597 и PSN821), производные или аналоги FGF21, такие как те, что описаны в Kharitonenkov A. с соавт., Current Opinion in Investigation Drugs 2009, 10(4)359-364, модуляторы рецепторов TGR5 (также называемых GPBAR1), в частности агонисты, такие как те, которые описаны в Zhong, M., Current Topics in Medicinal Chemistry, 2010, 10(4), 386-396 и INT777, агонисты GPR40, такие как те, которые описаны в Medina, J.C., Annual Reports in Medicinal Chemistry, 2008, 43, 75-85, включая, но не ограничиваясь ими, модуляторы TAK-875, GPR120, в частности агонисты, активаторы рецептора с высоким сродством к никотиновой кислоте (HM74A) и ингибиторы SGLT1, такие как GSK1614235. Дополнительный репрезентативный список антидиабетических средств, которые можно комбинировать с соединениями по настоящему изобретению, можно найти, например, на стр. 28, строка 35 по стр. 30, строка 19 WO2011005611. Предпочтительными антидиабетическими средствами являются метформин и ингибиторы ДПП-IV (например, ситаглиптин, вильдаглиптин, алоглиптин, дутоглиптин, линаглиптин и саксаглиптин). Другие противодиабетические средства могут включать ингибиторы или модуляторы ферментов карнитин-пальмитоилтрансферазы, ингибиторы фруктоза-1,6-дифосфатазы, ингибиторы альдозаредуктазы, ингибиторы минералокортикоидных рецепторов, ингибиторы TORC2, ингибиторы CCR2 и/или CCR5, ингибиторы изоформ PKC (например, PKCα, PKCβ, PKCγ), ингибиторы синтетазы жирных кислот, ингибиторы серин-пальмитоилтрансферазы, модуляторы GPR81, GPR39, GPR43, GPR41, GPR105, Kv1.3, ретинол-связывающий белок 4, глюкокортикоидный рецептор, соматостатиновые рецепторы (например, SSTR1, SSTR2, SSTR3 и SSTR5), ингибиторы или модуляторы PDHK2 или PDHK4, ингибиторы MAP4K4, модуляторы семейства IL1, включая IL1бета, модуляторы RXRальфа. Кроме того, подходящие антидиабетические средства включают механизмы, перечисленные Carpino, P.A., Goodwin, B. Expert Opin. Ther. Pat, 2010, 20(12), 1627-51.
Подходящие средства против ожирения включают ингибиторы 11β-гидроксистероиддегидрогеназы-1 (11β-HSD типа 1), ингибитор стеароил-КоА-десатуразы-1 (SCD-1), агонисты MCR-4, агонисты холецистокинина-A (CCK-A), ингибиторы обратного захвата моноаминов (такие как сибутрамин), симпатомиметические агенты, адренергические агонисты β3, агонисты допамина (такие как бромокриптин), аналоги меланоцит-стимулирующего гормона, агонисты 5HT2c, антагонисты гормона, концентрирующего меланин, лептин (белок OB), аналоги лептина, агонисты лептина, антагонисты галанина, ингибиторы липазы (такие как тетрагидролипстатин, т.е. орлистат), аноректические средства (такие как агонист бомбезина), антагонисты нейропептида-Y (например, антагонисты NPY Y5), PYY3-36 (включая их аналоги), тиромиметические агенты, дегидроэпиандростерон или его аналог, агонисты или антагонисты глюкокортикоидов, антагонисты орексина, агонисты глюкагоноподобного пептида-1, цилиарные нейротрофические факторы (такие как Аксокин™, доступный от Regeneron Pharmaceuticals, Inc., Тарритаун, Нью-Йорк, и Procter & Gamble Company, Цинциннати, Огайо), ингибиторы человеческого агути-связанного белка (AGRP), антагонисты грелина, антагонисты гистамина 3 или обратные агонисты, агонисты нейромедина U, ингибиторы MTP/ApoB (например, кишечные селективные ингибиторы MTP, такие как дирлотапид), опиоидный антагонист, антагонист орексина, комбинацию налтрексона с бупроприоном и тому подобное.
Предпочтительные агенты против ожирения для применения в аспектах комбинирования по настоящему изобретению включают кишечно-селективные ингибиторы MTP (например, дирлотапид, митратапид и имплитапид, R56918 (CAS № 403987) и CAS № 913541-47-6), агонисты CCKa (например, N-бензил-2-[4-(1H-индол-3-илметил)-5-оксо-1-фенил-4,5-дигидро-2,3,6,10b-тетрааза-бензо[e]азулен-6-ил]-N-изопропилацетамид, описанный в публикации PCT № WO 2005/116034 или публикации США № 2005-0267100 A1), агонисты 5HT2c (например, лоркасерин), агонист MCR4 (например, соединения, описанные в US 6,818,658), ингибитор липазы (например, Цетилистат), PYY3-36 (в контексте настоящего документа "PYY3-36" включает аналоги, такие как пегилированный PYY3-36, например, те, которые описаны в публикации США 2006/0178501), опиоидные антагонисты (например, налтрексон), комбинацию налтрексона с бупропионом, олеоил-эстрон (CAS № 180003-17-2), обинепитид (TM30338), прамлинтид (Симлин®), тезофензин (NS2330), лептин, лираглутид, бромокриптин, орлистат, эксенатид (Баета®), AOD-9604 (CAS № 221231-10-3), фентермин и топирамат (торговое наименование: Qsymia) и сибутрамин. Предпочтительно соединения по настоящему изобретению и комбинированные терапевтические средства вводят в сочетании с физической нагрузкой и разумной диетой.
Соединения по настоящему изобретению могут быть применены в комбинации со средствами, регулирующими холестерин (включая средства, снижающие уровень холестерина), такими как ингибитор липазы, ингибитор ГМГ-КоА-редуктазы, ингибитор ГМГ-КоА-синтазы, ингибитор экспрессии гена ГМГ-КоА-редуктазы, ингибитор экспрессии гена ГМГ-КоА-синтазы, ингибитор секреции MTP/Apo B, ингибитор CETP, ингибитор абсорбции желчной кислоты, ингибитор абсорбции холестерина, ингибитор синтеза холестерина, ингибитор сквален-синтетазы, ингибитор сквален-эпоксидазы, ингибитор сквален-циклазы, комбинированный ингибитор сквален-эпоксидазы/сквален-циклазы, фибрат, ниацин, ионообменная смола, антиоксидант, ингибитор ACAT или секвестрант желчных кислот, или агент, такой как мипомерсен.
Примеры подходящих средств для снижения уровня холестерина/липидов и терапевтических средств, влияющих на липидный профиль, включают в себя: ингибиторы ГМГ-КоА-редуктазы (например, правастатин, ловастатин, аторвастатин, симвастатин, флувастатин, NK-104 (также известный как итавастатин, или нисвастатин или нисбастатин) и ZD-22 (также известный как розувастатин, или атавастатин, или визастатин)); ингибиторы сквален-синтетазы; фибраты; секвестранты желчных кислот (такие как квестран); ингибиторы ACAT; ингибиторы MTP; ингибиторы липооксигеназы; ингибиторы всасывания холестерина; и ингибиторы белка, переносящего эфиры холестерина. Другие атеросклеротические средства включают модуляторы PCSK9.
В другом варианте осуществления соединение формулы I может быть введено совместно со средствами для лечения неалкогольного стеатогепатита (NASH) и/или неалкогольной жировой дистрофии печени (NAFLD), такими как Орлистат, TZD и другие инсулин-сенсибилизирующие агенты, аналоги FGF21, метформин, этиловые эфиры омега-3-кислоты (например, Ловаза), фибраты, ингибиторы ГМГ-КоА-редуктазы, Эзитимбе, Пробукол, Урсодезоксихолевая кислота, агонисты TGR5, агонисты FXR, витамин E, Бетаин, Пентоксифиллин, антагонисты CB1, Карнитин, N-ацетилцистеин, восстановленный глутатион, лоркасерин, комбинация налтрексона с бупроприоном, ингибиторы SGLT2, фентермин, топирамат, аналоги инкретина (GLP и GIP) и блокаторы рецепторов ангиотензина.
В другом варианте осуществления дополнительный фармацевтический агент выбран из группы, состоящей из цистеамина или его фармацевтически приемлемой соли, цистамина или его фармацевтически приемлемой соли, антиоксидантного соединения, лецитина, комплекса витаминов B, препаратов соли желчной кислоты, антагонистов каннабиноидного рецептора-1 (CB1), обратных агонистов каннабиноидного рецептора-1 (CB1), регуляторов активности рецептора, активируемого пролифератором пероксисом, соединения бензотиазепина или бензотиепина, антисмысловой конструкции РНК для ингибирования протеин-тирозинфосфатазы PTPRU, связанного с гетероатомом замещенного пиперидина и его производных, производного азациклопентана, способного ингибировать стеароил-кофермент-альфа-дельта-9-дезатуразу, ациламидного соединения, обладающего повышающей секрецию или индуцирующей активностью адипонектина, соединения четвертичного аммония, глатирамера ацетата, белков пентраксина, ингибитора ГМГ-КоА-редуктазы, N-ацетилцистеина, изофлавонового соединения, макролидного антибиотика, ингибитора галектина, антитела или любой их комбинации.
Дополнительные терапевтические агенты включают антикоагулянт или ингибиторы коагуляции, анти-тромбоцитарные средства или ингибиторы тромбоцитов, ингибиторы тромбина, тромболитические или фибринолитические средства, антиаритмические средства, антигипертензивные средства, блокаторы кальциевых каналов (L-типа и Т-типа), сердечные гликозиды, диуретики, антагонисты минералокортикоидных рецепторов, NO-донорные агенты, такие как органонитраты, NO-промотирующие агенты, такие как ингибиторы фосфодиэстеразы, средства, снижающие уровень холестерина/липидов и терапевтические средства, влияющие на липидный профиль, антидиабетические средства, антидепрессанты, противовоспалительные средства (стероидные и нестероидные), средства против остеопороза, заместительную гормональную терапию, пероральные контрацептивы, средства против ожирения, средства против тревоги, антипролиферативные средства, противоопухолевые средства, средства против язвенной болезни и гастроэзофагеальной рефлюксной болезни, гормон роста и/или стимуляторы секреции гормона роста, миметики щитовидной железы (включая антагонист рецептора гормонов щитовидной железы), антиинфекционные средства, антивирусные средства, антибактериальные средства и противогрибковые средства.
Агенты, используемые в условиях отделения интенсивной терапии, включают, например, добутамин, допамин, эпинефрин, нитроглицерин, нитропруссид и т.д.
Агенты для комбинирования, полезные для лечения васкулита, включают, например, азатиоприн, циклофосфамид, микофенолат, мофетил, ритуксимаб и т.д.
В другом варианте осуществления настоящее изобретение предоставляет комбинацию, в которой второй агент представляет собой по меньшей мере один агент, выбранный из ингибитора фактора Xa, антикоагулянтного средства, анти-тромбоцитарного средства, ингибитора тромбина, тромболитического средства и фибринолитического средства. Типичные ингибиторы фактора Ха включают апиксабан и ривароксабан. Примеры подходящих антикоагулянтов для применения в комбинации с соединениями по настоящему изобретению включают гепарины (например, нефракционированные и низкомолекулярные гепарины, такие как эноксапарин и дальтепарин).
В другом предпочтительном варианте осуществления второй агент представляет собой по меньшей мере один агент, выбранный из варфарина, дабигатрана, нефракционированного гепарина, низкомолекулярного гепарина, синтетического пентасахарида, гирудина, аргатробанов, аспирина, ибупрофена, напроксена, сулиндака, индометацина, мефенамата, дроксикама, диклофенака, сульфинпиразона, пироксикама, тиклопидина, клопидогреля, тирофибана, эптифибатида, абциксимаба, мелагатрана, дисульфатогирудина, активатора тканевого плазминогена, модифицированного активатора тканевого плазминогена, анистреплазы, урокиназы и стрептокиназы.
Предпочтительным вторым агентом является по меньшей мере одно анти-тромбоцитарное средство. Особенно предпочтительными анти-тромбоцитарными средствами являются аспирин и клопидогрель.
Термин "анти-тромбоцитарные средства" (или агенты, ингибирующие тромбоциты) в контексте настоящего описания обозначает агенты, которые ингибируют функцию тромбоцитов, например, путем ингибирования агрегации, адгезии или гранулярной секреции тромбоцитов. Агенты включают, но не ограничиваются ими, различные известные нестероидные противовоспалительные лекарственные средства (NSAIDS), такие как аспирин, ибупрофен, напроксен, сулиндак, индометацин, мефенамат, дроксикам, диклофенак, сульфинпиразон, пироксикам и их фармацевтически приемлемые соли или пролекарства. Из NSAIDS предпочтительными являются аспирин (ацетилсалициловая кислота или ASA) и ингибиторы COX-2, такие как ЦЕЛЕБРЕКС или пироксикам. Другие подходящие агенты, ингибирующие тромбоциты, включают антагонисты IIb/IIIa (например, тирофибан, эптифибатид и абциксимаб), антагонисты рецептора тромбоксана-A2 (например, ифетробан), ингибиторы синтетазы тромбоксана-A2, ингибиторы PDE-III (например, Плетал, дипиридамол) и их фармацевтически приемлемые соли или пролекарства.
Термин "анти-тромбоцитарные средства" (или агенты, ингибирующие тромбоциты) в контексте настоящего описания также предназначен для включения антагонистов рецептора ADP (аденозиндифосфата), предпочтительно антагонистов пуринергических рецепторов P2Y1 и P2Y12, причем P2Y12 являются еще более предпочтительными. Предпочтительные антагонисты рецептора P2Y12 включают тикагрелор, прасугрель, тиклопидин и клопидогрель, включая их фармацевтически приемлемые соли или пролекарства. Клопидогрель является еще более предпочтительным агентом. Тиклопидин и клопидогрель также являются предпочтительными соединениями, поскольку известно, что они мягко воздействуют на желудочно-кишечный тракт при применении.
Термин "ингибиторы тромбина" (или антитромбиновые агенты) в контексте настоящего описания обозначает ингибиторы сериновой протеазы тромбина. При ингибировании тромбина различные процессы, опосредованные тромбином, такие как тромбин-опосредованная активация тромбоцитов (например, агрегация тромбоцитов и/или гранулярная секреция ингибитора-1 активатора плазминогена и/или серотонина) и/или образование фибрина, нарушены. Специалистам в данной области техники известен ряд ингибиторов тромбина, и предполагается, что эти ингибиторы будут использоваться в комбинации с соединениями по настоящему изобретению. Такие ингибиторы включают, но не ограничиваются ими, производные бораргинина, борпептиды, дабигатран, гепарины, гирудин, аргатробан и мелагатран, включая их фармацевтически приемлемые соли и пролекарства. Производные бораргинина и борпептиды включают N-ацетильные и пептидные производные бороновой кислоты, такие как производные альфа-аминобороновой кислоты по С-концу лизина, орнитина, аргинина, гомоаргинина и их соответствующих аналогов изотиоурония. В контексте настоящего описания термин "гирудин" включает подходящие производные или аналоги гирудина, именуемые в настоящем документе гирулогами, такие как дисульфатогирудин. Термин "тромболитики" или "фибринолитические средства" (или "тромболитики" или "фибринолитики") в контексте настоящего описания обозначает средства, которые лизируют кровяные сгустки (тромбы). Такие агенты включают активатор тканевого плазминогена (природный или рекомбинантный) и его модифицированные формы, анистреплазу, урокиназу, стрептокиназу, тенектеплазу (TNK), ланотеплазу (nPA), ингибиторы фактора VIIa, ингибиторы PAI-1 (т.е. инактиваторы ингибиторов активатора тканевого плазминогена), ингибиторы альфа2-антиплазмина и анизоилированный комплекс активатора стрептокиназы плазминогена, включая их фармацевтически приемлемые соли или пролекарства. В контексте настоящего описания термин "анистреплаза" относится к анизоилированному комплексу активатора стрептокиназы плазминогена, как описано, например, в ЕР 028,489, описание которого тем самым включено в настоящий документ посредством ссылки. В контексте настоящего описания термин "урокиназа" предназначен для обозначения как урокиназы с двойной цепью, так и одноцепочечной урокиназы, причем последняя также именуется в настоящем документе как проурокиназа.
Примеры подходящих антиаритмических средств включают в себя: агенты класса I (такие как пропафенон); агенты класса II (такие как метопролол, атенолол, карвадиол и пропранолол); агенты класса III (такие как соталол, дофетилид, амиодарон, азимилид и ибутилид); агенты класса IV (такие как дитиазем и верапамил); вещества, открывающие K+-каналы, такие как ингибиторы IAch и ингибиторы IKur (например, соединения, такие как те, которые раскрыты в WO01/40231).
Соединения по настоящему изобретению могут быть применены в комбинации с антигипертензивными средствами, и такая антигипертензивная активность легко определяется специалистами в данной области техники в соответствии со стандартными анализами (например, измерение артериального давления). Примеры подходящих антигипертензивных средства включают: альфа-адренергические блокаторы; бета-адренергические блокаторы; блокаторы кальциевых каналов (например, дилтиазем, верапамил, нифедипин и амлодипин); вазодилататоры (например, гидралазин), диуретики (например, хлортиазид, гидрохлортиазид, флуметиазид, гидрофлуметиазид, бендрофлуметиазид, метилхлортиазид, трихлорметиазид, политиазид, бензтиазид, трикринафен этакриновую кислоту, хлорталидон, торсемид, фуросемид, музолимин, буметанид, триамтренен, амилорид, спиронолактон); ингибиторы ренина; ингибиторы АПФ (например, каптоприл, зофеноприл, фозиноприл, эналаприл, кераноприл, цилазоприл, делаприл, пентоприл, квинаприл, рамиприл, лизиноприл); антагонисты рецептора AT-1 (например, лозартан, ирбесартан, валсартан); антагонисты рецептора ЕТ (например, ситаксентан, атрсентан и соединения, раскрытые в патентах США №№ 5,612,359 и 6,043,265); двойной антагонист ET/AII (например, соединения, раскрытые в WO 00/01389); ингибиторы нейтральной эндопептидазы (НЭП); ингибиторы вазопепсидазы (двойные ингибиторы НЭП-АПФ) (например, гемопатрилат и нитраты). Типичным антиангинальным средством является ивабрадин.
Примеры подходящих блокаторов кальциевых каналов (L-типа или Т-типа) включают дилтиазем, верапамил, нифедипин и амлодипин и мибефрадил.
Примеры подходящих сердечных гликозидов включают наперстянку и уабаин.
В одном варианте осуществления соединение формулы I может быть введено совместно с одним или несколькими диуретиками. Примеры подходящих диуретиков включают (a) петлевые диуретики, такие как фуросемид (такой как ЛАЗИКС™), торсемид (такой как ДЕМАДЕКС™), беметанид (такой как БУМЕКС™) и этакриновая кислота (такая как ЭДЕКРИН™); (b) диуретики тиазидного типа, такие как хлортиазид (такие как ДИУРИЛ™, ЭЗИДРЕКС™ или ГИДРОДИУРИЛ™), гидрохлортиазид (такие как МИКРОЗИД™ или ОРЕТИК™), бензтиазид, гидрофлуметиазид (такие как САЛУРОН™), бендрофлуметиазид, метилхлортиазид, политиазид, трихлорметиазид и индапамид (такой как ЛОЗОЛ™); (c) диуретики фталимидинового типа, такие как хлорталидон (такой как ГИГРОТОН™) и метолазон (такой как ЗАРОКСОЛИН™); (d) диуретики хиназолинового типа, такие как хинетазон; и (e) калийсберегающие диуретики, такие как триамтерен (такой как ДИРЕНИУМ™) и амилорид (такой как МИДАМОР™ или МОДУРЕТИК™).
В другом варианте осуществления соединение формулы I может быть введено совместно с петлевым диуретиком. В еще одном варианте осуществления петлевой диуретик выбран из фуросемида и торсемида. В еще одном варианте осуществления одно или несколько соединений формулы I или Ia могут быть введены совместно с фуросемидом. В еще одном варианте осуществления одно или несколько соединений формулы I или Ia могут быть введены совместно с торсемидом, который необязательно может представлять собой форму торсемида с контролируемым или модифицированным высвобождением.
В другом варианте осуществления соединение формулы I может быть введено совместно с диуретиком тиазидного типа. В еще одном варианте осуществления диуретик тиазидного типа выбран из группы, состоящей из хлортиазида и гидрохлортиазида. В еще одном варианте осуществления одно или несколько соединений формулы I или Ia могут быть введены совместно с хлортиазидом. В еще одном варианте осуществления одно или несколько соединений формулы I или Ia могут быть введены совместно с гидрохлортиазидом.
В другом варианте осуществления одно или несколько соединений формулы I или Ia могут быть введены совместно с диуретиком фталимидинового типа. В еще одном варианте осуществления диуретик фталимидинового типа представляет собой хлорталидон. Примеры подходящих антагонистов минералокортикоидных рецепторов включают спиронолактон и эплеренон. Примеры подходящих ингибиторов фосфодиэстеразы включают: ингибиторы ФДЭ III (такие как цилостазол); и ингибиторы ФДЭ V (такие как силденафил).
Специалистам в данной области техники будет понятно, что соединения по настоящему изобретению могут также применяться в сочетании с другими сердечно-сосудистыми или цереброваскулярными методами лечения, включая ЧКВ, стентирование, стенты с лекарственным покрытием, терапию стволовыми клетками и медицинские устройства, такие как имплантированные кардиостимуляторы, дефибрилляторы или сердечную ресинхронизирующую терапию.
Дозировка дополнительного фармацевтического средства обычно зависит от ряда факторов, включая здоровье субъекта, которого лечат, степень желаемого лечения, природу и тип параллельной терапии, если таковая имеется, и частоту лечения и природу желаемого эффекта. Как правило, диапазон доз дополнительного фармацевтического агента находится в диапазоне от приблизительно 0,001 мг до приблизительно 100 мг на килограмм веса тела человека в день, предпочтительно от приблизительно 0,1 мг до приблизительно 10 мг на килограмм веса тела человека в день. Однако может также потребоваться некоторая вариабельность общего диапазона доз в зависимости от возраста и веса субъекта, которого лечат, предполагаемого пути введения, конкретного средства против ожирения, которое вводят, и тому подобного. Определение диапазонов доз и оптимальных дозировок для конкретного пациента также находится в пределах компетенции специалиста в данной области техники, имея преимущество настоящего раскрытия.
Согласно способам лечения по изобретению соединение по настоящему изобретению или комбинацию соединения по настоящему изобретению и по меньшей мере одного дополнительного фармацевтического агента (именуемого в настоящем документе как "комбинация") вводят субъекту, нуждающемуся в таком лечении, предпочтительно в форме фармацевтической композиции. В комбинированном аспекте изобретения соединение по настоящему изобретению и по меньшей мере один другой фармацевтический агент (например, другое средство против ожирения) могут быть введены либо раздельно, либо в виде фармацевтической композиции, содержащей и то, и другое. Обычно предпочтительно, чтобы такое введение было пероральным.
Когда комбинацию соединения по настоящему изобретению и по меньшей мере одного другого фармацевтического агента вводят вместе, такое введение может быть последовательным во времени или одновременным. Одновременное введение лекарственных комбинаций обычно является предпочтительным. Для последовательного введения соединение по настоящему изобретению и дополнительный фармацевтический агент могут быть введены в любом порядке. Обычно предпочтительно, чтобы такое введение было пероральным. Особенно предпочтительно, чтобы такое введение было пероральным и одновременным. Когда соединение по настоящему изобретению и дополнительный фармацевтический агент вводят последовательно, введение каждого из них может быть одним и тем же или разными способами.
Согласно способам по изобретению соединение по настоящему изобретению или комбинацию предпочтительно вводят в форме фармацевтической композиции. Соответственно, соединение по настоящему изобретению или комбинация могут быть введены пациенту раздельно или вместе в виде любой обычной пероральной, ректальной, трансдермальной, парентеральной (например, внутривенной, внутримышечной или подкожной), интрацистернальной, интравагинальной, интраперитонеальной, для местного применения (например, порошок, мазь, крем, спрей или лосьон), буккальной или назальной дозированной формы (например, спрей, капли или ингалятор).
Соединения по изобретению или комбинации могут быть введены в чистом виде, но обычно их вводят в смеси с одним или несколькими подходящими фармацевтическими эксципиентами, адъювантами, разбавителями или носителями, известными в данной области техники и выбранными с учетом предполагаемого пути введения и стандартной фармацевтической практики. Соединение по изобретению или комбинация могут быть составлены в смесь для предоставления дозированных форм с немедленным, отсроченным, модифицированным, непрерывным, прерывистым или контролируемым высвобождением в зависимости от желаемого пути введения и специфичности профиля высвобождения, соответствующего терапевтическим потребностям.
Фармацевтическая композиция содержит соединение по изобретению или комбинацию в количестве, обычно находящемся в диапазоне от приблизительно 1% до приблизительно 75%, 80%, 85%, 90% или даже 95% (по массе) композиции, обычно в диапазоне от приблизительно 1%, 2% или 3% до приблизительно 50%, 60% или 70%, чаще в диапазоне от приблизительно 1%, 2% или 3% до менее 50%, таком как приблизительно 25%, 30% или 35%.
Способы получения различных фармацевтических композиций с определенным количеством активного соединения известны специалистам в данной области. Например, см. Remington: The Practice of Pharmacy, Lippincott Williams and Wilkins, Балтимор, штат Мэриленд, 20-е дополн. изд. 2000.
Композиции, подходящие для парентерального введения, обычно содержат фармацевтически приемлемые стерильные водные или неводные растворы, дисперсии, суспензии или эмульсии и стерильные порошки для разведения в стерильные инъекционные растворы или дисперсии. Примеры подходящих водных и неводных носителей или разбавителей (включая растворители и носители) включают воду, этанол, полиолы (пропиленгликоль, полиэтиленгликоль, глицерин и тому подобное), их подходящие смеси, триглицериды, включая растительные масла, такие как оливковое масло, и инъецируемые органические сложные эфиры, такие как этилолеат. Предпочтительным носителем является сложный эфир каприловой/каприновой кислоты с глицерином или пропиленгликолем торговой марки Миглиол® (например, Миглиол® 812, Миглиол® 829, Миглиол® 840), доступный от Condea Vista Co., Кранфорд, Нью-Джерси. Можно поддерживать, например, надлежащую текучесть путем использования покрытия, такого как лецитин, путем поддержания требуемого размера частиц в случае дисперсий и путем использования поверхностно-активных веществ.
Эти композиции для парентерального введения могут также содержать эксципиенты, такие как консервирующие, смачивающие, эмульгирующие и диспергирующие агенты. Предотвращение загрязнения композициями микроорганизмами может быть достигнуто с помощью различных антибактериальных и противогрибковых агентов, например, парабенов, хлорбутанола, фенола, сорбиновой кислоты и тому подобного. Также может быть желательным включение изотонических агентов, например, сахаров, хлорида натрия и тому подобного. Пролонгированная абсорбция инъецируемых фармацевтических композиций может быть достигнута за счет использования агентов, способных замедлять абсорбцию, например, алюминия моностеарата и желатина.
Твердые дозированные формы для перорального введения включают капсулы, таблетки, жевательные таблетки, пастилки, пилюли, порошки и препараты, состоящие из множества частиц (гранулы). В таких твердых дозированных формах соединение по настоящему изобретению или комбинацию смешивают по меньшей мере с одним инертным эксципиентом, разбавителем или носителем. Подходящие эксципиенты, разбавители или носители включают такие вещества, как цитрат или дикальцийфосфат натрия, и/или (a) один или несколько наполнителей или сухих разбавителей (например, микрокристаллическая целлюлоза (доступная как Авицел™ от FMC Corp.), крахмалы, лактоза, сахароза, маннит, кремниевая кислота, ксилит, сорбит, декстроза, гидрофосфат кальция, декстрин, альфа-циклодекстрин, бета-циклодекстрин, полиэтиленгликоль, жирные кислоты со средней длиной цепи, оксид титана, оксид магния, оксид алюминия и тому подобное); (b) одно или несколько связующих веществ (например, карбоксиметилцеллюлоза, метилцеллюлоза, гидроксипропилцеллюлоза, гидроксипропилметилцеллюлоза, желатин, гуммиарабик, этилцеллюлоза, поливиниловый спирт, пуллулан, прежелатинизированный крахмал, агар, трагакант, альгинаты, желатин, поливинилпирролидон, сахароза, аравийская камедь и тому подобное); (c) один или несколько увлажнителей (например, глицерин и тому подобное); (d) один или несколько разрыхлителей (например, агар-агар, карбонат кальция, крахмал картофеля или тапиоки, альгиновая кислота, некоторые комплексные силикаты, карбонат натрия, лаурилсульфат натрия, натрия крахмал гликолят (доступный как Эксплотаб™ от Edward Mendell Co.), сшитый поливинилпирролидон, кроскармеллоза натрия A-типа (доступная как Ак-ди-сол™), полиакрилин калия (ионообменная смола) и тому подобное); (e) один или несколько замедлителей растворения (например, парафин и тому подобное); (f) один или несколько ускорителей абсорбции (например, четвертичные аммониевые соединения и тому подобное); (g) один или несколько смачивающих агентов (например, цетиловый спирт, глицерина моностеарат и тому подобное); (h) один или несколько адсорбентов (например, каолин, бентонит и тому подобное); и/или (i) один или несколько смазывающих веществ (например, тальк, стеарат кальция, стеарат магния, стеариновая кислота, полиоксилстеарат, цетанол, тальк, гидрогенизированное касторовое масло, сложные эфиры сахарозы и жирных кислот, диметилполисилоксан, микрокристаллический воск, желтый пчелиный воск, белый пчелиный воск, твердые полиэтиленгликоли, лаурилсульфат натрия и тому подобное). В случае капсул и таблеток дозированные формы также могут содержать буферные агенты.
Твердые композиции подобного типа также могут быть использованы в качестве наполнителей в желатиновых капсулах с мягким или твердым наполнением, используя такие наполнители, как лактоза или молочный сахар, а также высокомолекулярные полиэтиленгликоли и тому подобное.
Твердые дозированные формы, такие как таблетки, драже, капсулы и гранулы, могут быть получены с покрытиями и оболочками, такими как энтеросолюбильные покрытия и другие, хорошо известные в данной области техники. Они также могут содержать опалесцирующие компоненты и могут также быть такого состава, что они высвобождают соединение по настоящему изобретению и/или дополнительный фармацевтический агент с задержкой. Примерами композиций для заливки, которые могут быть использованы, являются полимерные вещества и воски. Лекарственное средство также может быть в микроинкапсулированной форме, если необходимо, с одним или несколькими из вышеупомянутых эксципиентов.
Для таблеток активный агент обычно составляет менее 50% (по массе) состава, например, менее чем приблизительно 10%, как например, 5% или 2,5% по массе. Преобладающая часть состава содержит наполнители, разбавители, дезинтегрирующие агенты, смазывающие вещества и, необязательно, ароматизаторы. Состав этих эксципиентов хорошо известен в данной области техники. Часто наполнители/разбавители содержат смеси двух или более из следующих компонентов: микрокристаллическая целлюлоза, маннит, лактоза (все типы), крахмал и дикальцийфосфат. Смеси наполнитель/разбавитель обычно составляют менее 98% состава и предпочтительно менее 95%, например, 93,5%. Предпочтительные дезинтегрирующие агенты включают Ак-ди-сол™, Эксплотаб™, крахмал и лаурилсульфат натрия. При наличии дезинтегрирующий агент обычно составляет менее 10% состава или менее 5%, например, приблизительно 3%. Предпочтительным смазывающим веществом является стеарат магния. При наличии смазывающеее вещество обычно составляет менее 5% состава или менее 3%, например, приблизительно 1%.
Таблетки могут быть изготовлены стандартными способами таблетирования, например прямым прессованием или влажной, сухой грануляцией или грануляцией в расплаве, процессом застывания расплава и экструзией. Ядра таблеток могут быть однослойными или многослойными и могут быть покрыты соответствующими покрытиями, известными в данной области техники.
Жидкие дозированные формы для перорального введения включают фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы и эликсиры. В дополнение к соединению по настоящему изобретению или комбинации жидкая дозированная форма может содержать инертные разбавители, обычно используемые в данной области техники, такие как вода или другие растворители, солюбилизирующие агенты и эмульгаторы, такие как, например, этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, диметилформамид, масла (например, хлопковое масло, арахисовое масло, масло зародышей кукурузы, оливковое масло, касторовое масло, масло кунжутных семян и тому подобное), Миглиол® (доступный от CONDEA Vista Co., Кранфорд, Нью-Джерси), глицерин, тетрагидрофурфуриловый спирт, полиэтиленгликоли и сложные эфиры жирных кислот и сорбитана или смеси этих веществ и тому подобное.
Помимо таких инертных разбавителей, композиция также может содержать наполнители, такие как смачивающие агенты, эмульгирующие и суспендирующие агенты, подсластители, ароматизаторы и отдушки.
Пероральные жидкие формы соединений по изобретению или комбинаций включают растворы, в которых активное соединение полностью растворено. Примеры растворителей включают все фармацевтически применимые растворители, подходящие для перорального введения, особенно те, в которых соединения по изобретению проявляют хорошую растворимость, например, полиэтиленгликоль, полипропиленгликоль, пищевые масла и системы на основе глицерилов и глицеридов. Системы на основе глицерилов и глицеридов могут включать, например, продукты следующих торговых марок (и соответствующие непатентованные продукты): Каптекс™ 355 EP (глицерил-трикаприлат/капрат, от Abitec, Колумбус, Огайо), Кродамол GTC/C (триглицерид со средней длиной цепи, от Croda, Cowick Hall, Великобритания) или Лабрафак™ CC (триглициды со средней длиной цепи, от Gattefosse), Каптекс™ 500P (глицерил-триацетат, то есть триацетин от Abitec), Капмул™ MCM (моно- и диглицериды со средней длиной цепи, от Abitec), Миглиол™ 812 (каприловый/каприновый триглицерид, от Condea, Кранфорд, Нью-Джерси), Миглиол™ 829 (каприловый/каприновый/янтарный триглицерид, от Condea), Миглиол™ 840 (пропиленгликоль дикаприлат/дикапрат, от Condea), Лабрафил M1944CS (олеоил макрогол-6 глицериды, от Gattefosse), Пецеол™ (глицерилмоноолеат, от Gattefosse) и Майсин™ 35-1 (глицерилмоноолеат, от Gattefosse). Особый интерес представляют триглицеридные масла со средней длиной цепи (приблизительно от C8 до C10). Эти растворители часто составляют преобладающую часть композиции, то есть более чем приблизительно 50%, обычно более чем приблизительно 80%, например, приблизительно 95% или 99%. Адъюванты и добавки также могут быть включены с растворителями, главным образом, в качестве агентов маскировки вкуса, вкусовых добавок и ароматизаторов, антиоксидантов, стабилизаторов, модификаторов текстуры и вязкости и солюбилизаторов.
Суспензии, в дополнение к соединению по настоящему изобретению или комбинации, могут дополнительно включать носители, такие как суспендирующие агенты, например, этоксилированные изостеариловые спирты, сложные эфиры полиоксиэтилена и сорбитола или сорбитана, микрокристаллическая целлюлоза, метагидроксид алюминия, бентонит, агар-агар и трагакант или смеси этих веществ и тому подобное.
Композиции для ректального или вагинального введения предпочтительно включают суппозитории, которые могут быть приготовлены путем смешивания соединения по настоящему изобретению или комбинации с подходящими нераздражающими наполнителями или носителями, такими как масло какао, полиэтиленгликоль или воск для суппозиториев, которые являются твердыми при обычной комнатной температуре, но жидкими при температуре тела и, следовательно, тают в прямой кишке или полости влагалища, тем самым высвобождая активный компонент(ы).
Дозированные формы для местного применения соединений по настоящему изобретению или комбинаций включают мази, кремы, лосьоны, порошки и спреи. Лекарственные средства смешивают с фармацевтически приемлемым эксципиентом, разбавителем или носителем и любыми консервантами, буферами или пропеллентами, которые могут потребоваться.
Многие из соединений по настоящему изобретению плохо растворимы в воде, например, менее чем приблизительно 1 мкг/мл. Следовательно, жидкие композиции в солюбилизирующих неводных растворителях, такие как триглицеридные масла со средней длиной цепи, обсуждаемые выше, являются предпочтительной дозированной формой для этих соединений.
Твердые аморфные дисперсии, включая дисперсии, полученные способом распылительной сушки, также являются предпочтительной дозированной формой для плохо растворимых соединений по изобретению. Под "твердой аморфной дисперсией" подразумевается твердое вещество, в котором по меньшей мере часть плохо растворимого соединения находится в аморфной форме и диспергирована в водорастворимом полимере. Под "аморфной" подразумевается, что плохо растворимое соединение не является кристаллическим. Под "кристаллическим" подразумевается, что соединение проявляет дальний порядок в трех измерениях по меньшей мере 100 повторяющихся единиц в каждом измерении. Таким образом, подразумевается, что термин аморфный включает в себя не только вещество, которое по существу не имеет порядка, но также вещество, которое может иметь некоторую небольшую степень упорядоченности, но порядок имеет менее трех измерений и/или только на короткие расстояния. Аморфное вещество может быть охарактеризовано методами, известными в данной области техники, такими как порошковая рентгеновская дифракционная (PXRD) кристаллография, ЯМР в твердом состоянии или термические методы, такие как дифференциальная сканирующая калориметрия (ДСК).
Предпочтительно по меньшей мере основная часть (то есть по меньшей мере приблизительно 60% масс.) плохо растворимого соединения в твердой аморфной дисперсии является аморфной. Соединение может существовать в твердой аморфной дисперсии в относительно чистых аморфных доменах или областях, в виде твердого раствора соединения, гомогенно распределенного по всему полимеру, или любой комбинации этих состояний или тех состояний, которые находятся между ними. Предпочтительно твердая аморфная дисперсия является по существу гомогенной, так что аморфное соединение диспергируется как можно более однородно по всему полимеру. В контексте настоящего описания термин "по существу гомогенный" означает, что доля соединения, которое присутствует в относительно чистых аморфных доменах или областях в твердой аморфной дисперсии, является относительно небольшой, порядка менее 20% масс. и предпочтительно менее 10% масс. от общего количества лекарственного средства.
Водорастворимые полимеры, подходящие для использования в твердых аморфных дисперсиях, должны быть инертными в том смысле, что они не вступают в химическую реакцию с плохо растворимым соединением неблагоприятным образом, являются фармацевтически приемлемыми и имеют по меньшей мере некоторую растворимость в водном растворе при физиологически релевантных pH (например, 1-8). Полимер может быть нейтральным или ионизируемым и должен иметь растворимость в воде по меньшей мере 0,1 мг/мл по меньшей мере в части диапазона pH 1-8.
Водорастворимые полимеры, подходящие для использования с настоящим изобретением, могут быть целлюлозными или нецеллюлозными. Полимеры могут быть нейтральными или ионизируемыми в водном растворе. Из них ионизируемые и целлюлозные полимеры являются предпочтительными, причем ионизируемые целлюлозные полимеры являются более предпочтительными.
Типичные водорастворимые полимеры включают ацетат сукцинат гидроксипропилметилцеллюлозы (HPMCAS), гидроксипропилметилцеллюлозу (HPMC), фталат гидроксипропилметилцеллюлозы (HPMCP), карбоксиметилэтилцеллюлозу (CMEC), фталат ацетат целлюлозы (CAP), тримеллитат ацетат целлюлозы (CAT), поливинилпирролидон (PVP), гидроксипропилцеллюлозу (HPC), метилцеллюлозу (MC), блок-сополимеры этиленоксида и пропиленоксида (PEO/PPO, также известные как полоксамеры) и их смеси. Особенно предпочтительные полимеры включают HPMCAS, HPMC, HPMCP, CMEC, CAP, CAT, PVP, полоксамеры и их смеси. Наиболее предпочтительным является HPMCAS. См. публикацию европейской заявки на патент № 0 901 786 А2, раскрытие которой включено в настоящий документ посредством ссылки.
Твердые аморфные дисперсии могут быть получены в соответствии с любым способом образования твердых аморфных дисперсий, в результате которого по меньшей мере основная часть (по меньшей мере 60%) плохо растворимого соединения находится в аморфном состоянии. Такие способы включают механические, термические способы и способы из растворителя. Типичные механические способы включают измельчение и экструзию; способы плавления, включая способы высокотемпературного плавления, модифицированного растворителем плавления и застывания расплава; и способы из растворителя, включая осаждение в нерастворителе, распылительное покрытие и распылительную сушку. См., например, следующие патенты США, соответствующие раскрытия которых включены в настоящее описание посредством ссылки: №№ 5,456,923 и 5,939,099, которые описывают образование дисперсий посредством способов экструзии; №№ 5,340,591 и 4,673,564, которые описывают образование дисперсий посредством способов измельчения; и №№ 5,707,646 и 4,894,235, которые описывают образование дисперсий посредством способов, основанных на застывании расплава. В предпочтительном способе твердую аморфную дисперсию образуют распылительной сушкой, как раскрыто в публикации европейской патентной заявки № 0 901 786 А2. В этом способе соединение и полимер растворяют в растворителе, таком как ацетон или метанол, и растворитель затем быстро удаляют из раствора путем распылительной сушки с образованием твердой аморфной дисперсии. Твердые аморфные дисперсии могут быть получены так, чтобы они содержали приблизительно до 99% масс. соединения, например, 1% масс., 5% масс., 10% масс., 25% масс., 50% масс., 75% масс., 95% масс. или 98% масс., как желательно.
Твердую дисперсию можно использовать в качестве дозированной формы саму по себе или она может служить продуктом промышленного использования (MUP) при получении других дозированных форм, таких как капсулы, таблетки, растворы или суспензии. Примером водной суспензии является водная суспензия, полученная распылительной сушкой дисперсии 1:1 (масс./масс.) соединения/HPMCAS-HF, содержащей 2,5 мг/мл соединения в 2% полисорбате-80. Твердые дисперсии для применения в таблетках или капсулах обычно смешивают с другими эксципиентами или адъювантами, обычно присутствующими в таких дозированных формах. Например, примерный наполнитель для капсул содержит полученную распылительной сушкой дисперсию 2:1 (масс./масс.) соединения/HPMCAS-MF (60%), лактозу (легкотекучую) (15%), микрокристаллическую целлюлозу (например, Авицел®-102) (15,8%), крахмал натрия (7%), лаурилсульфат натрия (2%) и стеарат магния (1%).
Доступны полимеры HPMCAS низкого, среднего и высокого сорта как Аквоат®-LF, Аквоат®-MF и Аквоат®-HF соответственно от Shin-Etsu Chemical Co., LTD, Токио, Япония. Более высокие сорта MF и HF обычно являются предпочтительными.
Следующие абзацы описывают типичные составы, дозировки и т.д., полезные для животных, не являющихся человеком. Введение соединений по настоящему изобретению и комбинаций соединений по настоящему изобретению со средствами против ожирения может осуществляться перорально или не перорально.
Количество соединения по настоящему изобретению или комбинации соединения по настоящему изобретению с другим средством против ожирения вводят так, чтобы получить эффективную дозу. Обычно суточная доза, которую вводят животному перорально, составляет от приблизительно 0,01 до приблизительно 1000 мг/кг массы тела, например, от приблизительно 0,01 до приблизительно 300 мг/кг или от приблизительно 0,01 до приблизительно 100 мг/кг, или от приблизительно 0,01 до приблизительно 50 мг/кг массы тела, или от приблизительно 0,01 до приблизительно 25 мг/кг, или от приблизительно 0,01 до приблизительно 10 мг/кг, или от приблизительно 0,01 до приблизительно 5 мг/кг.
Для удобства соединение по настоящему изобретению (или комбинация) может быть внесено в питьевую воду, чтобы терапевтическая доза соединения принималась с ежедневным водоснабжением. Соединение может быть введено непосредственно в питьевую воду, предпочтительно в форме жидкого, водорастворимого концентрата (такого, как водный раствор водорастворимой соли).
Для удобства соединение по настоящему изобретению (или комбинацию) можно также добавлять прямо в корм как таковое или в форме кормовой добавки для животного, также называемой премиксом или концентратом. Для включения агента в корм более обычно используют премикс или концентрат соединения в эксципиенте, разбавителе или носителе. Подходящими эксципиентами, разбавителями или носителями являются жидкость или твердое вещество, по желанию, такие как вода, различные виды муки, такие как люцерновая мука, соевая мука, жмых от переработки хлопковых семян, кормовая мука из жмыха льняного семени, мука из кукурузных початков и кукурузная мука, кормовая патока, мочевина, костная мука и минеральные смеси, такие как те, что обычно используют в кормах для домашней птицы. Особенно эффективным эксципиентом, разбавителем или носителем является сам корм для соответствующего животного, а именно небольшая часть такого корма. Носитель способствует равномерному распределению соединения в готовом корме, к которому подмешивают премикс. Предпочтительно соединение тщательно подмешивают в премикс и затем в корм. При этом соединение можно диспергировать или растворить в подходящем масляном носителе, таком как соевое масло, кукурузное масло, хлопковое масло и тому подобное, или в летучем органическом растворителе и затем смешать с носителем. Понятно, что доли соединения в концентрате могут варьироваться в широких пределах, поскольку количество соединения в готовом корме может быть доведено до желаемого уровня смешиванием соответствующей доли премикса с кормом.
Производитель корма может смешивать сильнодействующие концентраты с белковым носителем, таким как соевый шрот и другие виды муки, которые описаны выше, с получением концентрированных добавок, подходящих для прямого кормления животных. В таких случаях животным позволяют потреблять обычный рацион. Альтернативно, такие концентрированные добавки можно добавлять прямо в корм, чтобы получить сбалансированный питательный готовый корм, содержащий терапевтически эффективный уровень соединения по настоящему изобретению. Смеси тщательно перемешивают стандартными способами, например в двухкорпусном смесителе, для обеспечения однородности.
Если добавку используют в качестве поверхностной обработки корма, это также способствует обеспечению равномерного распределения соединения через поверхность обработанного корма.
Питьевую воду и корм, эффективные для увеличения отложения постного мяса и для улучшения соотношения постного мяса и жира, обычно готовят смешиванием соединения по настоящему изобретению с достаточным количеством корма для животных для получения от приблизительно 10-3 до приблизительно 500 м.д. соединения в корме или воде.
Предпочтительный содержащий лекарственное средство корм для свиней, крупного рогатого скота, овец и коз обычно содержит от приблизительно 1 до приблизительно 400 граммов соединения по настоящему изобретению (или комбинации) на тонну корма, причем оптимальное количество для этих животных обычно составляет от приблизительно 50 до приблизительно 300 граммов на тонну корма.
Предпочтительные корма для домашней птицы и домашних животных обычно содержат от приблизительно 1 до приблизительно 400 граммов и предпочтительно от приблизительно 10 до приблизительно 400 граммов соединения по настоящему изобретению (или комбинации) на тонну корма.
Для парентерального введения животным соединения по настоящему изобретению (или комбинация) могут быть приготовлены в форме пасты или пеллета и вводиться в виде имплантата, обычно под кожу головы или уха животного, у которого необходимо увеличить отложение постного мяса и улучшить соотношение постного мяса к жиру.
Пастообразные составы могут быть получены путем диспергирования лекарственного средства в фармацевтически приемлемом масле, таком как арахисовое масло, кунжутное масло, кукурузное масло или тому подобное.
Пеллеты, содержащие эффективное количество соединения по настоящему изобретению, фармацевтической композиции или комбинации, могут быть получены путем смешивания соединения по настоящему изобретению или комбинации с разбавителем, таким как карбовакс, карнаубский воск и тому подобное, и смазывающее вещество, такое как стеарат магния или кальция, может быть добавлено для улучшения процесса пеллетирования.
Разумеется, для достижения желаемого уровня дозы, которая должна обеспечить желаемое увеличение отложения постного мяса и улучшение соотношение постного мяса и жира, животному можно вводить больше чем один пеллет. Более того, имплантаты также можно устанавливать периодически в течение периода лечения животного для того, чтобы поддерживать надлежащий уровень лекарственного средства в организме животного.
Настоящее изобретение имеет несколько выгодных ветеринарных отличий. Для владельца домашнего животного или ветеринара, который желает увеличить постность и/или убрать нежелательный жир у домашних животных, настоящее изобретение предоставляет способ, посредством которого это можно осуществить. Для тех, кто разводит домашнюю птицу, коров и свиней, применение способа по настоящему изобретению дает более постных животных, которые задают более высокие цены реализации в мясной промышленности.
ПРИМЕРЫ
Если не указано иное, исходные вещества обычно доступны из коммерческих источников, таких как Aldrich Chemicals Co. (Милуоки, Висконсин), Lancaster Synthesis, Inc. (Виндхам, Нью-Гэмпшир), Acros Organics (Фэр-Лон, Нью-Джерси), Maybridge Chemical Company, Ltd. (Корнуолл, Англия) и Tyger Scientific (Принстон, Нью-Джерси). Использовали некоторые общеупотребительные аббревиатуры и сокращения, которые могут включать: AcOH (уксусная кислота), DBU (1,8-диазабицикло[5.4.0]ундец-7-ен), CDI (1,1'-карбонилдиимидазол), ДХМ (дихлорметан), ДЭА (диэтиламин), ДИПЭА (N,N-диизопропилэтиламин), ДМАП (4-диметиламинопиридин), ДМФА (N,N'-диметилформамид), ДМСО (диметилсульфоксид), EDCI (N-(3-диметиламинопропил)-N'-этилкарбодиимид), Et2O (диэтиловый эфир), EtOAc (этилацетат), EtOH (этанол), HATU (2-(1H-7-азабензотриазол-1-ил)-1,1,3,3-тетраметилуронийгексафторфосфат метанаминия), HBTU (O-бензотриазол-1-ил-N,N,N',N'-тетраметилуронийгексафторфосфат), HOBT (1-гидроксибензотриазол), ИПС (изопропиловый спирт), KHMDS (гексаметилдисилазан калия), MeOH (метанол), MTBE (трет-бутилметиловый эфир), NaBH(OAc)3 (триацетоксиборгидрид натрия), NaHMDS (гексаметилдисилазан натрия), NMP (N-метилпирролидон), SEM ([2-(триметилсилил)этокси]метил), ТЭА (триэтиламин), ТФУК (трифторуксусная кислота), ТГФ (тетрагидрофуран) и T3P (ангдидрид пропанфосфоновой кислоты).
Реакции проводили на воздухе или, когда использовали чувствительные к кислороду или влаге реагенты или промежуточные соединения, в инертной атмосфере (азот или аргон). При необходимости реакционные аппараты сушили в динамическом вакууме, используя тепловую пушку, и использовали безводные растворители (продукты Sure-Seal™ от Aldrich Chemical Company, Милуоки, Висконсин или DriSolv™ от EMD Chemicals, Гибстаун, Нью-Джерси). Коммерческие растворители и реагенты использовали без дополнительной очистки. Когда указано, реакции нагревали микроволновым излучением с использованием микроволнового реактора Biotage Initiator или Personal Chemistry Emrys Optimizer. Ход реакции контролировали с использованием тонкослойной хроматографии (ТСХ), жидкостной хроматографии-масс-спектрометрии (ЖХМС), высокоэффективной жидкостной хроматографии (ВЭЖХ) и/или газовой хроматографии-масс-спектрометрии (ГХМС). ТСХ выполняли на пластинах, предварительно покрытых силикагелем, с индикатором флуоресценции (длина волны возбуждения 254 нм) и визуализировали в УФ-свете и/или с помощью I2, KMnO4, CoCl2, фосфомолибденовой кислоты и/или красителей на основе молибдата аммония-церия. Данные ЖХМС получали на приборе Agilent серии 1100 с автосэмплером Leap Technologies, колонками Gemini C18, градиентами MeCN/вода и модификаторами либо ТФУК, муравьиная кислота, либо гидроксид аммония. Элюент колонки анализировали с использованием масс-спектрометра Waters ZQ, сканирующего в режиме как положительных, так и отрицательных ионов от 100 до 1200 Да. Другие подобные приборы также использовали. Данные ВЭЖХ получали на приборе Agilent серии 1100 с использованием колонок Gemini или XBridge C18, градиентов MeCN/вода и модификаторов либо ТФУК, либо гидроксида аммония. Данные ГХМС получали с использованием печи Hewlett Packard 6890 с инжектором HP 6890, колонки HP-1 (12 м × 0,2 мм × 0,33 мкм) и газа-носителя гелия. Образец анализировали на масс-селективном детекторе HP 5973, сканирующем от 50 до 550 Да, с использованием электронной ионизации. Очистки выполняли с помощью среднеэффективной жидкостной хроматографии (СЭЖХ) с использованием приборов Isco CombiFlash Companion, AnaLogix IntelliFlash 280, Biotage SP1 или Biotage Isolera One и предварительно упакованных картриджей с силикагелем Isco RediSep или Biotage Snap. Хиральные очистки выполняли с помощью хиральной сверхкритической флюидной хроматографии (СФХ) с использованием приборов Berger или Thar; колонок ChiralPAK-AD, -AS, -IC, Chiralcel-OD или -OJ; и смесей CO2 с MeOH, EtOH, изоPrOH или MeCN, в чистом виде или модифицированных с использованием ТФУК или изоPrNH2. УФ-детекцию использовали для запуска сбора фракций.
Данные масс-спектрометрии описывают из анализа ЖХМС. Масс-спектрометрию (МС) выполняли посредством источников химической ионизации при атмосферном давлении (ХИАД), ионизации электроспреем (ИЭС), ионизации электронным ударом (ЭУ) или ионизации электронным рассеянием (ЭР). Химические сдвиги протонной ядерно-магнитной спектроскопии (1Н ЯМР) даны в миллионных долях в сторону слабого поля от тетраметилсилана и регистрировали на спектрометрах Varian 300, 400, 500 или 600 МГц. Химические сдвиги выражены в миллионных долях (м.д.,δ) по отношению к остаточным пикам дейтерированного растворителя. Форму пиков описывают следующим образом: с, синглет; д, дублет; т, триплет; кв, квартет; квин, квинтет; м, мультиплет; уш.с, уширенный синглет; каж., кажущийся. Аналитические данные СФХ получали на аналитическом приборе Berger, как описано выше. Данные оптического вращения получали на поляриметре PerkinElmer модель 343 с использованием ячейки 1 дм. Хроматографию на силикагеле проводили в основном с использованием систем Biotage или ISCO среднего давления с использованием колонок, предварительно упакованных различными коммерческими поставщиками, включая Biotage и ISCO. Микроанализы выполняли с помощью Quantitative Technologies Inc., и они находились в пределах 0,4% от расчетных значений.
Если не указано иное, химические реакции проводили при комнатной температуре (приблизительно 23 градуса по Цельсию).
Соединения и промежуточные соединения, описанные ниже, называли с использованием соглашения о присвоении имен, предоставленного ChemBioDraw Ultra, версия 12.0 (CambridgeSoft Corp., Кембридж, Массачусетс). Соглашение о присвоении имен, предоставленное ChemBioDraw Ultra, версия 12.0, хорошо известно специалистам в данной области техники, и считается, что соглашение о присвоении имен, предоставленное ChemBioDraw Ultra, версия 12.0, в целом соответствует рекомендациям ИЮПАК (Международного союза теоретической и прикладной химии) по номенклатуре органической химии и правилам присвоения индекса CAS. Если не указано иное, все реагенты получали коммерческим путем без дополнительной очистки или получали с использованием способов, известных в литературе.
Термины "концентрировали", "выпаривали" и "концентрировали в вакууме" относятся к удалению растворителя при пониженном давлении на роторном испарителе с температурой бани менее 60°C. Аббревиатуры "мин" и "ч" обозначают "минуты" и "часы", соответственно. Термин "ТСХ" относится к тонкослойной хроматографии, "комнатная температура или температура окружающей среды" означает температуру от 18 до 25°C, "ГХМС" относится к газовой хроматографии-масс-спектрометрии, "ЖХМС" относится к жидкостной хроматографии-масс-спектрометрии, "СВЭЖХ" относится к сверхэффективной жидкостной хроматографии, и "ЖХВД" относится к жидкостной хроматографии высокого давления, "СФХ" относится к сверхкритической флюидной хроматографии.
Гидрирование может быть проведено в шейкере Парра в атмосфере газообразного водорода под давлением или в аппарате для гидрирования в потоке H-Cube от Thales-nano при полном наполнении водородом и скорости потока от 1 до 2 мл/мин при определенной температуре.
Время удерживания ВЭЖХ, СВЭЖХ, ЖХМС, ГХМС и СФХ измеряли с использованием способов, указанных в процедурах.
Получение промежуточных соединений и примеров
Общая методика A: Добавить диизопропилэтиламин (3,0 экв.) к раствору промежуточного соединения 1 или промежуточного соединения 2 (1 экв.) в диметилформамиде или тетрагидрофуране (0,15 М). HATU (1,0 экв.) и соответствующий амин (2,0 экв.) добавляли последовательно во флакон. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь концентрировали и очищали препаративной ВЭЖХ, получая указанный продукт, если не указано иное.
Общая методика B: Добавить оксалилхлорид (2,0 экв., 2М в дихлорметане) к суспензии промежуточного соединения 1 или промежуточного соединения 2 (1,0 экв.) в дихлорметане (0,1М). К суспензии добавляли диметилформамид (10 мкл), и реакционную смесь перемешивали при комнатной температуре в течение 30 минут. За это время суспензия превращалась в раствор. Соответствующий амин (1,0 экв.) и диизопропилэтиламин (2,3 экв.) в дихлорметане (0,5 мл) добавляли к реакционной смеси и перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь разбавляли дихлорметаном (2 мл) и последовательно промывали 1н гидроксидом натрия (100 мкл) и рассолом (100 мкл). Органический слой сушили над сульфатом магния, фильтровали, и растворитель выпаривали при пониженном давлении. Полученный остаток очищали препаративной ВЭЖХ, получая указанный продукт, если не указано иное.
Промежуточное соединение 1: 2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)пиримидин-5-карбоновая кислота
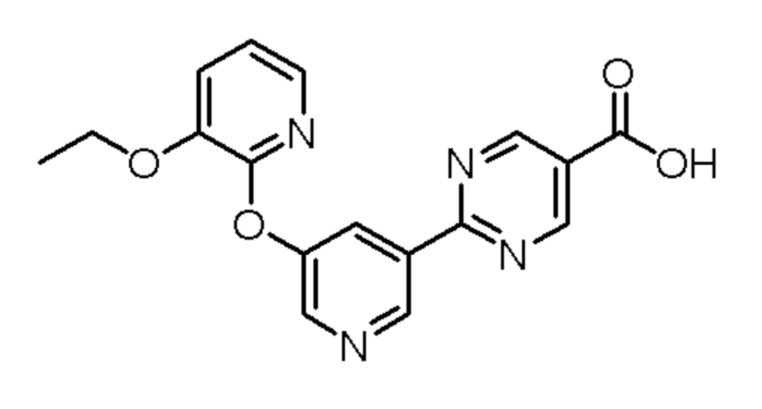
Стадия 1: 3-Этоксипиридин
Карбонат цезия (12 моль, 1,5 экв.) и йодистый этил (9,7 моль, 1,2 экв.) добавляли к раствору 3-гидроксипиридина (8,10 моль, 1,0 экв.) в ацетоне (12 л) при 15°C. Реакционную смесь перемешивали при комнатной температуре в течение 24 часов. Реакционную смесь фильтровали, и органический слой концентрировали, получая сырой продукт. Добавляли этилацетат (20 л) и промывали водой (3 × 5 л). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали, получая 3-этоксипиридин (620 г, 62%) в виде масла. 1H ЯМР (400 МГц, CDCl3) δ 1,44 (т, 3H), 4,07 (кв, 2H), 7,15-7,23 (м, 2H), 8,20 (дд, 1H), 8,30 (д, 1H).
Стадия 2: 3-Этоксипиридин-1-оксид
м-Хлоропероксибензойную кислоту (6,5 моль, 1,3 экв.) добавляли к раствору 3-этоксипиридина (5,0 моль, 1,0 экв.) в дихлорметане (12 л) при 10°C. Реакционную смесь перемешивали при комнатной температуре в течение 24 часов. Добавляли тиосульфат натрия (4 кг, в 5 л воды). Реакционную смесь перемешивали при 15°C в течение 2 часов. Добавляли еще одну порцию тиосульфата натрия (1,5 кг, в 5 л воды). Реакционную смесь перемешивали при 15°C в течение 1 часа. Смесь экстрагировали дихлорметаном (16 × 10 л). Объединенные органические слои концентрировали, получая сырой продукт. Сырой продукт очищали колоночной хроматографией на силикагеле (дихлорметан:метанол; 100:1-10:1), получая указанное в заголовке соединение (680 г, 97%) в виде коричневого масла. Затем его очищали растиранием с петролейным эфиром (4 л) при комнатной температуре в течение 24 часов, получая 3-этоксипиридин-1-оксид (580 г, 83%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 1,41 (т, 3H), 4,02 (кв, 2H), 6,84 (дд, 1H), 7,12 (дд, 1H), 7,85 (д, 1H), 7,91-7,95 (м, 1H).
Стадия 3: 2-((5-Бромпиридин-3-ил)окси)-3-этоксипиридин
Эту реакцию проводили пятью параллельными партиями.
Диизопропилэтиламин (2,69 моль, 3,7 экв.) и бромтрипирролидинофосфония гексафторфосфат (0,93 моль, 1,3 экв.) добавляли к перемешиваемому раствору 3-этоксипиридин-1-оксида (0,72 моль, 1,0 экв.) и 3-бром-5-гидроксипиридина (0,72 моль, 1,0 экв.) в тетрагидрофуране (2500 мл) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 2 дней, затем отдельные партии объединяли в одну партию. Полученную суспензию концентрировали досуха и растворяли в дихлорметане (25 л). Органический слой промывали 1н гидроксидом натрия (15 л), водой (3 × 20 л) и рассолом (20 л). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали, получая масло. Неочищенное масло очищали колоночной хроматографией на силикагеле (петролейный эфир:этилацетат; 10:1-1:1), получая сырой продукт в виде коричневого твердого вещества. Это твердое вещество растирали с метил-трет-бутиловым эфиром:петролейным эфиром (1:10; 11 л), получая 2-((5-бромпиридин-3-ил)окси)-3-этоксипиридин (730 г, 69%) в виде желтоватого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 1,49 (т, 3H), 4,16 (кв, 2H), 7,04 (дд, 1H), 7,25 (дд, 1H), 7,68-7,73 (м, 2H), 8,44 (д, 1H), 8,49 (д, 1H). МС (ЭС+) 297,1 (M+H).
Стадия 4: Этил 2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)пиримидин-5-карбоксилат
Раствор 2-((5-бромпиридин-3-ил)окси)-3-этоксипиридина (300 ммоль, 1,0 экв.) в тетрагидрофуране (1,3 л) дегазировали азотом в течение 30 минут. Турбо-Гриньяр (390 ммоль, 1,3 экв., 1,3 М в тетрагидрофуране) добавляли при комнатной температуре со скоростью, чтобы поддерживать внутреннюю температуру ниже 30°C. Реакционной смеси давали остыть до комнатной температуры и перемешивали в течение 3 часов. Реакционную смесь охлаждали до 10°C и добавляли хлорид цинка (390 ммоль, 1,3 экв., 1,9 М в 2-метилтетрагидрофуране) со скоростью, чтобы поддерживать температуру ниже 15°C. Полученную суспензию нагревали до комнатной температуры до полного растворения осадка и затем обратно охлаждали до 10°C. Этил 2-хлорпиримидин-5-карбоксилат (360 ммоль, 1,2 экв.) и дихлор[бис(2-(дифенилфосфино)фенил)эфир]палладий (II) (6,00 ммоль, 0,02 экв.) добавляли в виде твердых веществ. Полученную суспензию дегазировали азотом в течение 30 минут, затем нагревали до 50°C в течение 16 часов. Реакционную смесь обрабатывали в водных условиях, затем последовательно обрабатывали динатриевой солью этилендиаминтетрауксусной кислоты, функционализированным серой силикагелем и углем для удаления металлических примесей. Сырое соединение перекристаллизовывали из метанола (450 мл), получая этил 2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)пиримидин-5-карбоксилат (77 г, 70%) в виде бледно-желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 1,44 (т, 3H), 1,50 (т, 3H), 4,19 (кв, 2H), 4,46 (кв, 2H), 7,00-7,04 (м, 1H), 7,25 (с, 1H), 7,71 (д, 1H), 8,59 (с, 1H), 8,66 (д, 1H), 9,32 (с, 2H), 9,55 (с, 1H).
Стадия 5: 2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)пиримидин-5-карбоновая кислота
Гидроксид натрия (307 ммоль, 1,5 экв., 4М водный) и метанол (50 мл) добавляли к суспензии 2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)пиримидин-5-карбоксилата (205 ммоль, 1,0 экв) в тетрагидрофуране (300 мл). Полученный раствор перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь разбавляли водой (400 мл) и экстрагировали смесью 2:1 диэтиловый эфир:гептаны (2 × 300 мл). Водный слой подкисляли до pH 4 с помощью 4 М соляной кислоты. Полученную суспензию перемешивали при комнатной температуре в течение 1 часа. Твердое вещество отфильтровывали, промывали водой и сушили, получая 2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)пиримидин-5-карбоновую кислоту (69 г, 100%) в виде бледно-желтого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 1,37 (т, 3H), 4,18 (кв, 2H), 7,19 (дд, 1H), 7,58 (дд, 1H), 7,70 (дд, 1H), 8,35-8,40 (м, 1H), 8,66 (д, 1H), 9,33 (с, 2H), 9,41 (д, 1H), 13,9 (уш. с, 1H).
Промежуточное соединение 2: 3-{5-[(3-Этоксипиридин-2-ил)окси]пиридин-3-ил}-1,2,4-триазин-6-карбоновая кислота
Стадия 1. 6-Бром-1,2,4-триазин-3-амин
К смеси 3-амино-1,2,4-триазина (104 ммоль, 1,0 экв.) в ацетонитриле (120 мл) добавляли воду (120 мл) и перемешивали при комнатной температуре до образования коричневого раствора. Смесь охлаждали до 0°C, порциями обрабатывали N-бромсукцинимидом (109 ммоль, 1,05 экв.) и перемешивали при 0°C в течение 20 мин. После нагревания до комнатной температуры смесь разбавляли этилацетатом (350 мл) и охлаждали до 0°C. К смеси добавляли карбонат натрия (12 г) и перемешивали в течение 10 минут. Два слоя разделяли, и водную фазу экстрагировали этилацетатом (200 мл). Объединенные органические слои промывали водным бикарбонатом натрия, сушили над сульфатом натрия, фильтровали и концентрировали, получая 6-бром-1,2,4-триазин-3-амин (10,5 г, 58%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, CD3OD) 8,32 (с, 1Н).
Стадия 2. Этил 3-амино-1,2,4-триазин-6-карбоксилат
В двух отдельных партиях ацетат палладия (0,87 ммоль, 0,05 экв.) добавляли к раствору 6-бром-1,2,4-триазин-3-амина (17 ммоль, 1,0 экв.), триэтиламина (35 ммоль, 2,0 экв.) и ксантофоса (1,40 ммоль, 0,08 экв.) в этаноле (60 мл). Смесь дегазировали с помощью монооксида углерода и перемешивали при 85°C в атмосфере монооксида углерода (16 фунтов/кв. дюйм) в течение 16 часов. Охлажденную реакционную смесь разбавляли этилацетатом (60 мл), фильтровали через слой целита и концентрировали. Сырые продукты из обеих партий объединяли и очищали с использованием колоночной хроматографии (этилацетат/петролейный эфир=3:7) с получением этил 3-амино-1,2,4-триазин-6-карбоксилата (2,5 г, 88%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 1,49 (т, 3H), 4,50 (кв, 2H), 8,79 (с, 1H).
Стадия 3. Этил 3-хлор-1,2,4-триазин-6-карбоксилат
трет-Бутилнитрит (4,5 ммоль, 1,5 экв.) добавляли к раствору этил 3-амино-1,2,4-триазин-6-карбоксилата (3,0 ммоль, 1,0 экв.) и хлорида меди (II) (3,6 ммоль, 1,2 экв.) в ацетонитриле (15 мл) по каплям. Полученную смесь нагревали при 60°C в течение 1 часа. Реакционную смесь охлаждали до комнатной температуры и обрабатывали холодной соляной кислотой (10 мл, 1н). Смесь экстрагировали этилацетатом (3 × 30 мл), и объединенные органические слои сушили над сульфатом натрия, фильтровали и упаривали. Сырой продукт очищали с использованием колоночной хроматографии, элюируя смесью этилацетат/петролейный эфир (от 5:95 до 1:1), с получением этил 3-хлор-1,2,4-триазин-6-карбоксилата (300 мг, 54%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 1,50 (т, 3H), 4,58 (кв, 2H), 9,11 (с, 1H).
Стадия 4. 3-Этокси-2-{[5-(трибутилстаннанил)пиридин-3-ил]окси}пиридин
Тетракис(трифенилфосфин)палладий (0) (0,68 ммоль, 0,10 экв.) добавляли к раствору 2-[(5-бромпиридин-3-ил)окси]-3-этоксипиридина (6,8 ммоль, 1,0 экв.) и гексабутилдистаннана (7,5 ммоль, 1,1 экв.) в диоксане (40 мл) в атмосфере азота. Реакционную смесь нагревали до 110°C и перемешивали при этой температуре в течение 16 часов. Смесь гасили водным фторидом калия и перемешивали в течение 1 часа. Полученную суспензию фильтровали через слой целита, и фильтрат экстрагировали этилацетатом (3 × 60 мл). Объединенные органические слои промывали рассолом, сушили над сульфатом натрия, фильтровали и концентрировали. Сырой продукт очищали с помощью колоночной хроматографии (этилацетат/петролейный эфир=от 0:100 до 1:4), получая 3-этокси-2-{[5-(трибутилстаннанил)пиридин-3-ил]окси}пиридин (1,6 г, 47%) в виде желтого масла. 1H ЯМР (400 МГц, CDCl3) δ 0,88 (т, 9H), 1,06-1,11 (м, 6H), 1,32 (с, 6H), 1,47-1,58 (м, 9H), 4,17 (кв, 2H), 6,99 (дд, 1H), 7,22 (дд, 1H), 7,57 (дд, 1H), 7,70 (дд, 1H), 8,37-8,40 (м, 2H).
Стадия 5. Этил 3-{5-[(3-этоксипиридин-2-ил)окси]пиридин-3-ил}-1,2,4-триазин-6-карбоксилат
Тетракис(трифенилфосфин)палладий (0) (0,05 ммоль, 0,05 экв.) добавляли к смеси 3-этокси-2-{[5-(трибутилстаннанил)пиридин-3-ил]окси}пиридина (0,99 ммоль, 1,0 экв.) и этил 3-хлор-1,2,4-триазин-6-карбоксилата (0,99 ммоль, 1,0 экв) в диоксане (8 мл). Флакон дегазировали для удаления кислорода, осторожно барботируя газообразный азот в течение 2 минут. Затем флакон перемешивали при 115°C в течение 30 мин при микроволновом облучении. Реакционную смесь охлаждали до комнатной температуры, обрабатывали водным фторидом калия и перемешивали в течение 1 часа. Суспензию фильтровали через слой целита, и фильтрат экстрагировали этилацетатом (3 × 30 мл). Объединенные органические слои промывали рассолом, сушили над сульфатом натрия и концентрировали. Сырое вещество очищали с помощью колоночной хроматографии (этилацетат/петролейный эфир=от 1:4 до 1:1), получая этил 3-{5-[(3-этоксипиридин-2-ил)окси]пиридин-3-ил}-1,2,4-триазин-6-карбоксилат (150 мг, 41%) в виде желтого масла. МС (ЭС+) 368,0 (М+Н).
Стадия 6. 3-{5-[(3-этоксипиридин-2-ил)окси]пиридин-3-ил}-1,2,4-триазин-6-карбоновая кислота
Гидроксид натрия (1,0 ммоль, 20 экв., 2М) добавляли к раствору этил 3-{5-[(3-этоксипиридин-2-ил)окси]пиридин-3-ил}-1,2,4-триазин-6-карбоксилата (0,053 ммоль, 1,0 экв) в метаноле (1 мл) при комнатной температуре. Раствор перемешивали в течение 1 часа. Смесь концентрировали для удаления метанола, разбавляли водой и экстрагировали дихлорметаном (2 × 15 мл). Водный слой подкисляли до pH 5 с помощью 2н соляной кислоты и экстрагировали этилацетатом (3 × 10 мл). Объединенные органические слои промывали рассолом (20 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали досуха, получая продукт (15 мг, 83%) в виде светло-желтого твердого вещества. МС (ЭС+) 339,9 (М+Н).
Промежуточное соединение 3: 2-{5-[(3-этоксипиразин-2-ил)окси]пиридин-3-ил}пиримидин-5-карбоновая кислота
Стадия 1. 2-[(5-Бромпиридин-3-ил)окси]-3-этоксипиразин
5-Бромпиридин-3-ол (32 ммоль, 1,0 экв.) и карбонат цезия (39 ммоль, 1,3 экв.) добавляли к раствору 2-хлор-3-этоксипиразина (32 ммоль, 1,0 экв.) в N-метил-2-пирролидоне (250 мл). Реакционную смесь перемешивали при 150°C в течение 1 часа. Охлажденную реакционную смесь выливали в воду (300 мл) и экстрагировали этилацетатом (3 × 250 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали. Сырой продукт очищали с использованием колоночной хроматографии, элюируя смесью петролейный эфир/этилацетат (от 0% до 100%), с получением указанного в заголовке соединения (5,0 г, 54%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 1,50 (т, 3H), 4,54 (кв, 2H), 7,59 (д, 1H), 7,77 (т, 1H), 7,83 (д, 1H), 8,49 (д, 1H), 8,56 (д, 1H).
Стадия 2. Этил 2-{5-[(3-этоксипиразин-2-ил)окси]пиридин-3-ил}пиримидин-5-карбоксилат
Комплекс [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия (II) с дихлорметаном (124 мг, 0,05 экв.) добавляли к суспензии 2-((5-бромпиридин-3-ил)окси)-3-этоксипиразина (3,4 ммоль, 1,0 экв.), бис(пинаколато)диборона (4,1 ммоль, 1,2 экв.) и ацетата калия (13 ммоль, 4,0 экв.) в диоксане (5 мл). Реакционную смесь продували азотом и перемешивали при 100°C в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры и гасили водой (50 мл). Смесь экстрагировали этилацетатом (25 мл) и промывали рассолом (3 × 50 мл). Органический слой концентрировали с получением 2-этокси-3-((5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-3-ил)окси)пиразина (1,4 г, 110%) в виде черного масла, которое использовали непосредственно на следующей стадии.
Этил 2-хлорпиримидин-5-карбоксилат (930 мг, 1,2 экв.), карбонат калия (1,1 г, 2,0 экв.) и комплекс [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия (II) с дихлорметаном (150 мг, 0,05 экв.) добавляли к раствору 2-этокси-3-((5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-3-ил)окси)пиразина (1,4 г, 4,1 ммоль) в диоксане (5 мл). Черную суспензию продували азотом и перемешивали при 80°C в течение 16 часов. Реакционную смесь охлаждали до комнатной температуры и гасили водой (50 мл). Смесь экстрагировали этилацетатом (25 мл), промывали рассолом (3 × 50 мл) и сушили до черного остатка. Сырой продукт очищали флэш-хроматографией (этилацетат в петролейном эфире), получая этил 2-{5-[(3-этоксипиразин-2-ил)окси]пиридин-3-ил}пиримидин-5-карбоксилат (700 мг, 46%) в виде желтого твердого вещества. МС (ЭС+) 367,9 (М+Н).
Стадия 4. 2-{5-[(3-Этоксипиразин-2-ил)окси]пиридин-3-ил}пиримидин-5-карбоновая кислота
Гидроксид натрия (6,8 ммоль, 5,0 экв., 2М) добавляли к этил 2-{5-[(3-этоксипиразин-2-ил)окси]пиридин-3-ил}пиримидин-5-карбоксилату (1,4 ммоль, 1,0 экв.) в этаноле (5 мл). Реакционную смесь перемешивали при 30°C в течение 16 часов. Метил-трет-бутиловый эфир (30 мл) добавляли к реакционной смеси, и полученное твердое вещество отфильтровывали и сушили с получением 2-{5-[(3-этоксипиразин-2-ил)окси]пиридин-3-ил}пиримидин-5-карбоновой кислоты (500 мг, 101%) в виде натриевой соли. 1H ЯМР (300 МГц, ДМСО-d6) δ 1,42 (т, 3H), 4,48 (кв, 2H), 7,67 (с, 1H), 7,93 (с, 1H), 8,54 (с, 1H), 8,74 (с, 1H), 9,33 (с, 2H), 9,45 (с, 1H).
Промежуточное соединение 4: 2-(5-((3-(2-Фторэтокси)пиридин-2-ил)окси)пиридин-3-ил)пиримидин-5-карбоновая кислота

Стадия 1: 3-(2-Фторэтокси)пиридин
Карбонат калия (1,5 г, 2,0 экв.) добавляли к раствору 3-гидроксипиридина (500 мг, 1 экв.) и 1-фтор-2-йодэтана (920 мг, 1,0 экв.) в диметилформамиде (10 мл). Суспензию перемешивали при 30°C в течение 16 часов. Реакционную смесь разбавляли 10% метанолом в дихлорметане (90 мл) и промывали водой (20 мл). Органический слой промывали рассолом, сушили над сульфатом натрия и концентрировали. Сырое вещество очищали флэш-хроматографией (градиент: 0-4,5% метанол в дихлорметане) с получением 3-(2-фторэтокси)пиридина (500 мг, 67%) в виде коричневого масла. 1H ЯМР (400 МГц, CDCl3) δ 4,19-4,33 (м, 2H), 4,67-4,74 (м, 1H), 4,79-4,86 (м, 1H), 7,22 (дд, 2H), 8,24 (т, 1H), 8,34 (т, 1H).
Стадия 2: 3-(2-Фторэтокси)пиридин-1-оксид
м-Хлоропероксибензойную кислоту (2,49 г, 1,2 экв.) добавляли к раствору 3-(2-фторэтокси)пиридина (1,7 г, 1,0 экв.) в дихлорметане (30 мл). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь очищали непосредственно флэш-хроматографией (градиент: 0-5% метанола в дихлорметане) с получением 3-(2-фторэтокси)пиридин-1-оксида (1,2 г, 63%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 4,16-4,22 (м, 1H), 4,23-4,29 (м, 1H), 4,66-4,73 (м, 1H), 4,77-4,84 (м, 1H), 6,90 (дд, 1H), 7,17 (дд, 1H), 7,87-7,94 (м, 1H), 7,99 (т, 1H).
Стадия 3: 2-((5-Бромпиридин-3-ил)окси)-3-(2-фторэтокси)пиридин
Диизопропилэтиламин (5,2 мл, 3,8 экв.) добавляли к раствору 3-(2-фторэтокси)пиридин-1-оксида (1,2 г, 1,0 экв.), 3-бром-5-гидроксипиридина (1,3 г, 1,0 экв.) и бромтрипирролидинофосфония гексафторфосфата (4,6 г, 1,3 экв.) в тетрагидрофуране (25 мл) при 13°C. Смесь перемешивали при комнатной температуре в течение 16 часов. Реакцию гасили водой (20 мл) и экстрагировали этилацетатом (50 мл). Объединенные органические слои промывали насыщенным хлоридом аммония (3 × 20 мл) и рассолом (100 мл), сушили над сульфатом натрия и концентрировали. Сырое вещество очищали флэш-хроматографией (градиент: 0-70% этилацетата в петролейном эфире), получая 2-((5-бромпиридин-3-ил)окси)-3-(2-фторэтокси)пиридин (1,9 г, 81%) в виде желтого масла. 1H ЯМР (400 МГц, CDCl3) δ 4,28-4,33 (м, 1H), 4,34-4,41 (м, 2H), 4,70-4,78 (м, 1H), 4,82-4,82 (м, 1H), 7,05 (дд, 1H), 7,31 (дт, 1H), 7,69-7,73 (м, 1H), 7,74-7,80 (м, 2H), 8,44 (дд, 1H), 8,48-8,52 (м, 2H).
Стадия 4: 2-(5-((3-(2-Фторэтокси)пиридин-2-ил)окси)пиридин-3-ил)пиримидин-5-карбоновая кислота
Бис(пинаколато)диборон (580 мг, 1,2 экв.), ацетат калия (560 мг, 3,0 экв.) и комплекс [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия (II) с дихлорметаном (70 мг, 0,05 экв.) добавляли к раствору 2-((5-бромпиридин-3-ил)окси)-3-(2-фторэтокси)пиридина (600 мг, 1,0 экв.) в диоксане (10 мл) при комнатной температуре. Реакционную смесь перемешивали при 100°C в течение 16 часов. Реакционную смесь разбавляли этилацетатом (60 мл), промывали водой (20 мл) и рассолом, затем концентрировали до остатка. Остаток разбавляли диоксаном (15 мл) и водой (5 мл). Этил 2-хлорпиримидин-5-карбоксилат (310 мг, 1,0 экв.), комплекс [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия (II) с дихлорметаном (61 мг, 0,05 экв.) и карбонат калия (460 мг, 2,0 экв.) добавляли к реакционной смеси, и полученную суспензию перемешивали при 80°C в течение 16 часов. Суспензию фильтровали и затем распределяли между этилацетатом (30 мл) и водой (50 мл). Водный слой экстрагировали этилацетатом (2 × 30 мл). Объединенные органические слои промывали рассолом (50 мл), сушили над сульфатом натрия, фильтровали и концентрировали, получая сырое вещество, которое очищали препаративной ТСХ (5% метанола в дихлорметане) с получением 2-(5-((3-(2-фторэтокси)пиридин-2-ил)окси)пиридин-3-ил)пиримидин-5-карбоновой кислоты (80 мг, 13%). МС (ЭС+) 357,0 (М+Н).
Пример 1: (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамид

Оксалилхлорид (13,8 мл, 160 ммоль, 1,2 экв.) и диметилформамид (0,510 мл, 6,65 ммоль, 0,05 экв.) добавляли к суспензии 2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)пиримидин-5-карбоновой кислоты (45,0 г, 133 ммоль, 1,0 экв.) в дихлорметане (500 мл). Суспензию перемешивали в течение 2 часов, в то время как достигали раствор. Реакционную смесь концентрировали, получая сырой хлорангидрид в виде красного твердого вещества. Раствор (S)-тетрагидрофуран-3-амина (12,2 г, 140 ммоль, 1,05 экв.) и диизопропилэтиламина (51,0 мл, 293 ммоль, 2,2 экв.) в тетрагидрофуране (100 мл) добавляли по каплям к раствору сырого хлорангидрида в дихлорметане (200 мл) при 0°C. Реакционной смеси давали нагреться до комнатной температуры и перемешивали в течение 16 часов. Добавляли воду (1,0 л) и этилацетат (600 мл), и органический слой отделяли, промывали насыщенным бикарбонатом натрия, сушили над сульфатом магния и фильтровали. Фильтрат обрабатывали активированным углем (20 г) и перемешивали при 65°C в течение 20 минут. Суспензию фильтровали теплой, и фильтрат концентрировали до бледно-желтого твердого вещества, которое перекристаллизовывали из метанола в этилацетате (1:4, 1 л) с получением (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамида (43,5 г, 81%) в виде бесцветного твердого вещества. Указанное в заголовке соединение объединяли с предыдущими порциями (108,7 г, 266,8 ммоль), приготовленными таким же образом, и суспендировали с использованием этилацетата (1,0 л) при 80°C в течение 4 часов. Суспензии давали остыть до комнатной температуры и перемешивали в течение 4 дней. Твердое вещество отфильтровывали, промывали этилацетатом (3 × 200 мл) и сушили в высоком вакууме при 50°C в течение 24 часов, получая (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамид (100,5 г, 92%) в виде бесцветного твердого вещества. 1H ЯМР (300 МГц, ДМСО-d6) δ 1,38 (т, 3H), 1,89-1,98 (м, 1H), 2,15-2,26 (м, 1H), 3,65 (дд, 1H), 3,70-3,78 (м, 1H), 3,85-3,92 (м, 2H), 4,18 (кв, 2H), 4,46-4,55 (м, 1H), 7,18 (дд, 1H), 7,58 (дд, 1H), 7,69 (дд, 1H), 8,37 (дд, 1H), 8,64 (д, 1H), 8,95 (д, 1H), 9,28 (с, 2H), 9,39 (д, 1H). МС (ЭС+) 408,4 (М+Н). Температура плавления 177,5°C. Элементный анализ для C21H21N5O4: рассчитано, C, 61,91; H, 5,20; N, 17,19; найдено C 61,86; H 5,18; N, 17,30.
Твердую форму по этой методике охарактеризовывали с помощью порошковой рентгеновской дифракции (PXRD) и обозначали как Форма 1.
Альтернативное получение ( S )-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)- N -(тетрагидрофуран-3-ил)пиримидин-5-карбоксамида (Пример 1)
В реактор объемом 100 мл загружали ацетонитрил (35 мл), 2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)пиримидин-5-карбоновую кислоту (5,0 г, 15 ммоль) и (S)-тетрагидрофуран-3-амина гидрохлорид (2,2 г, 18 ммоль, 1,2 экв.). Диизопропилэтиламин (18 мл, 103 ммоль, 7,0 экв.) загружали, поддерживая температуру от 20°C до 30°C. Раствор ангидрида пропанфосфоновой кислоты (T3P) в ацетонитриле (21 мл, 30 ммоль, 2,0 экв.) загружали со скоростью, которая поддерживала температуру ниже 45°C. Реактор нагревали до 40±5°C в течение 1 часа, затем отбирали пробы для завершения реакции. Реакционную смесь охлаждали до температуры от 20°C до 25°C, и добавляли тетрагидрофуран (25 мл). Раствор бикарбоната натрия (0,5 М, 40 мл) загружали, и смесь перемешивали в течение 1 часа. pH проверяли и измеряли на уровне 8,5. Добавляли этилацетат (40 мл), и смесь перемешивали в течение 15 минут. Смесь отстаивали, и фазы разделялись. Водный слой переносили в делительную воронку и снова экстрагировали этилацетатом (100 мл). Органические фазы объединяли и промывали водой (40 мл). Органический слой переносили порциями в реактор объемом 100 мл и концентрировали в вакууме до небольшого объема. Добавляли метилэтилкетон (100 мл), и смесь концентрировали до конечного объема приблизительно 60 мл. Вакуум удаляли, и суспензию нагревали до температуры кипения с обратным холодильником и выдерживали до тех пор, пока твердые вещества не были смыты со стенок реактора. Суспензию охлаждали до 15°C в течение 2 часов и измельчали в течение ночи. Твердые вещества отделяли фильтрованием, дважды промывали реактор и осадок метилэтилкетоном (по 10 мл каждый). Твердые вещества сушили в вакуумной печи при 50°C с получением 4,86 г (81%) желаемого продукта. Твердую форму по этой методике охарактеризовывали с помощью порошковой рентгеновской дифракции (PXRD) и обозначали как Форма 2.
Преобразование Формы 2 в Форму 1
В 100 мл реактор загружали форму 2 (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамида (Примера 1) (10,0 г, 24,6 ммоль, 1,00 экв.), метилэтилкетон (8,8 мл/г, 88,0 мл) и воду (1,2 мл/г, 12,0 мл). Реактор нагревали до 50°C в течение 30 минут. Готовый раствор появился приблизительно при 44°C. Реактор охлаждали до 40°C в течение 30 минут, затем загружали затравочную форму 1 (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамида (Примера 1) (0,050 г, 0,123 ммоль, 0,0050 экв.). После внесения затравки мутную взвесь перемешивали в течение 1 часа, затем охлаждали до 5°C в течение 2 часов и затем перемешивали при 5°C в течение 12 часов. Контрольный образец в процессе извлекали и охарактеризовывали анализом PXRD для подтверждения того, что твердые вещества представляли собой форму 1. Суспензию фильтровали, и реактор и осадок промывали метилэтилкетоном (2,5 мл/г, 25 мл), охлажденным до 0°C. Твердые вещества сушили в вакуумной печи при 50°C с получением 8,15 г (81,5%) желаемого продукта. Дифрактограммы PXRD желаемого продукта соответствовали Форме 1.
Порошковая рентгеновская дифракция:
Анализ порошковый рентгеновской дифракцией проводили с использованием дифрактометра Bruker AXS D8 Advance, оборудованного источником излучения Cu (средняя длина волны Kα составляет 1,54056 Å), снабженного двойным индуктором, использующим зеркало Гебеля. Дифрагированное излучение детектировали детектором PSD-Lynx Eye. Как первый, так и второй оборудованы щелями Соллера 2.5. Напряжение и силу тока рентгеновской трубки устанавливали на 40 кВ и 40 мА, соответственно. Данные собирали с помощью гониометра Theta-Theta в режиме сканирования зажатой пары от 3,0 до 40,0 градусов 2-Тета с 1000 шагами, используя скорость сканирования 6 секунд на шаг. Образцы готовили путем размещения в силиконовом держателе для образцов с низким уровнем фона (C79298A3244B261). Данные собирали с использованием программного обеспечения Bruker DIFFRAC Plus. Анализ выполнен программным обеспечением EVA diffract plus.
Файл данных PXRD обрабатывали до выявления пиков. Используя алгоритм поиска пиков в программном обеспечении EVA, пики выбирали с пороговым значением 5 и значением ширины 0,2. Выходные данные автоматических присваиваний проверяли визуально, чтобы убедиться в их достоверности, и при необходимости вносили коррективы. Обычно выбирали пики с относительной интенсивностью≥3%. Пики, которые не были разрешены или соответствовали шуму, также отбрасывали. Типичная ошибка, связанная с положением пика из PXRD, установленная в ФСША (USP), находится в пределах +/- 0,2° (USP-941).
Таблица 1: Ключевые пики PXRD для характеристики кристаллического вещества примера 1
Фигура 1 представляет собой характеристическую рентгеновскую порошковую дифрактограмму, демонстрирующую кристаллическую форму 1 примера 1 (вертикальная ось: интенсивность (CPS); горизонтальная ось: два тета (градусы)).
Фигура 2 представляет собой характеристическую рентгеновскую порошковую дифрактограмму, демонстрирующую кристаллическую форму 2 примера 1 (вертикальная ось: интенсивность (CPS); горизонтальная ось: два тета (градусы)).
Пример 2: (R)-2-(5-((3-Этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамид
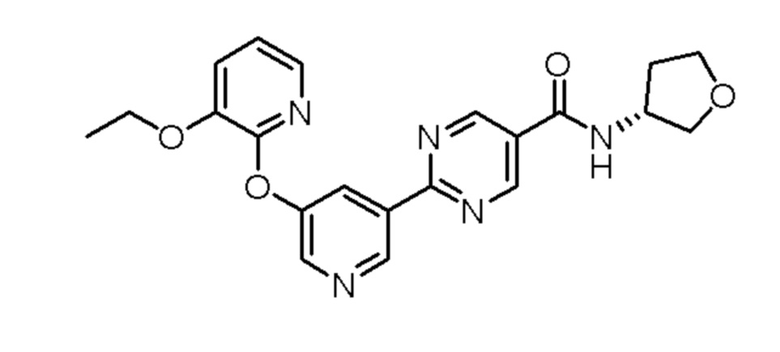
Указанное в заголовке соединение получали, используя общий метод A, с использованием промежуточного соединения 1 (0,31 ммоль, 1,0 экв.) и толуолсульфонатной соли (R)-(+)-тетрагидро-3-фуриламина (124 мг, 1,5 экв.). Сырой продукт очищали флэш-хроматографией с использованием этилацетата в гептанах с получением (R)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамида (91 мг, 70%). 1H ЯМР (400 МГц, ДМСО-d6) δ 1,38 (т, 3H), 1,89-1,98 (м, 1H), 2,15-2,28 (м, 1H), 3,5 (дд, 1H), 3,70-3,78 (м, 1H), 3,85-3,92 (м, 2H), 4,19 (кв, 2H), 4,46-4,55 (м, 1H), 7,19 (дд, 1H), 7,58 (дд, 1H), 7,69 (дд, 1H), 8,37 (дд, 1H), 8,64 (д, 1H), 8,96 (д, 1H), 9,28 (с, 2H), 9,39 (д, 1H). МС (ЭС+) 408,3 (М+Н).
Примеры 3.1-3.7: Примеры в таблице 2 получали по общей методике A с использованием соответствующих исходных веществ и анализировали с помощью методов, описанных ниже. Используемые переменная R3, промежуточные соединения (в которых варьируется R1, D1 и/или D2) и аналитический метод указаны в таблице 2.

Аналитические методы:
Метод A: Xbridge C18, 2,1 × 50 мм, 5 мкм, 40°C, подвижная фаза A 0,0375% ТФУК в воде, подвижная фаза B 0,01875% ТФУК в ацетонитриле, градиент: 0,00 мин 1% B, 0,60 мин 5% B, 4,00 мин 100% B, 0,8 мл/мин, ИАД-ЭС+.
Метод B: Xbridge C18, 2,1 × 50 мм, 5 мкм, 40°C, подвижная фаза A 0,05% NH4OH в воде, подвижная фаза B 100% ацетонитрил, градиент 0,00 мин 5% B, 3,40 мин 100% B, 0,8 мл/мин, ИАД-ЭС+.
Метод C: Waters Atlantis dC18 4,6 × 50, 5 мкм, подвижная фаза A: 0,05% ТФУК в воде (об./об.); Подвижная фаза B: 0,05% ТФУК в ацетонитриле (об./об.), Градиент: 95,0% H2O/5,0% ацетонитрила линейный до 5% H2O/95% ацетонитрила за 4,0 мин, удерживание на 5% H2O/95% ацетонитрила до 5,0 мин. Расход: 2 мл/мин
Метод D: Waters XBridge C18 4,6 × 50, 5 мкм, подвижная фаза A: 0,03% NH4OH в воде (об./об.); Подвижная фаза B: 0,03% NH4OH в ацетонитриле (об./об.), Градиент: 95,0% H2O/5,0% ацетонитрила линейный до 5% H2O/95% ацетонитрила за 4,0 мин, удерживание на 5% H2O/95% ацетонитрила до 5,0 мин. Расход: 2 мл/мин.
Метод E: Xtimate C18 5 × 30 мм, 3 мкм
Подвижная фаза A: 0,1% ТФУК в воде, Подвижная фаза B: ацетонитрил, Градиент: 0,00 мин 1% B, 1 мин 5% B, 5 мин 100% B, 8 мин 1% B. Скорость потока: 1,2 мл/мин
Метод F: ЖХМС E(4-302) XBridge C18 2,1 * 50 мм, 5 мкм
Подвижная фаза: 1,0% ацетонитрила в воде (0,1% муравьиной кислоты) до 5% ацетонитрила в воде (0,1% муравьиной кислоты) в течение 0,6 мин; затем от 5,0% ацетонитрила в воде (0,1% муравьиной кислоты) до 100% ацетонитрила (0,1% муравьиной кислоты) за 3,4 минуты; затем обратно до 1,0% ацетонитрила в воде (0,1% муравьиной кислоты) вплоть до 4,3 мин и удерживают 0,7 мин. Скорость потока: 0,8 мл/мин
Метод G: Xbridge C18, 2,0 × 50 мм, 5 мкм, 40°C, подвижная фаза A 10 мМ NH4HCO3 в воде, подвижная фаза B 100% ацетонитрил, градиент от 1,0% B до 5% B за 0,6 мин, 100% B за 3,4 минуты; затем обратно до 1,0% за 0,3 мин. Скорость потока: 0,8 мл/мин
Таблица 2
(ЭС+) (M+H)
Метод C
Метод D
Метод E
Метод F
Метод C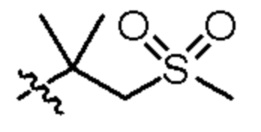
Метод F
Метод F
Пример 4: N-(2-цианопропан-2-ил)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)пиримидин-5-карбоксамид
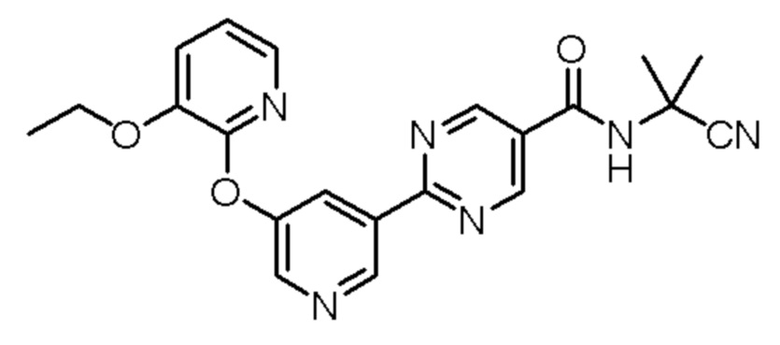
Указанное в заголовке соединение получали в соответствии с общей методикой B с использованием промежуточного соединения 1 (1,0 г, 1,0 экв.) и 2-амино-2-метилпропионитрила (436 мг, 1,1 экв.). Сырой продукт очищали флэш-хроматографией (50-100% этилацетата в гептанах) и обрабатывали углем. Полученный остаток перекристаллизовывали из этилацетата (8 мл), получая N-(2-цианопропан-2-ил)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)пиримидин-5-карбоксамид (1,1 г, 92%) в виде бесцветного твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 1,38 (т, 3H), 1,74 (с, 6H), 4,18 (кв, 2H), 7,18 (дд, 1H), 7,57 (дд, 1H), 7,69 (дд, 1H), 8,38 (дд, 1H), 8,66 (д, 1H), 9,18 (уш. с, 1H), 9,30 (с, 2H), 9,40 (д, 1H). МС (ЭС+) 405,3 (М+Н).
Пример 5: 2-(5-((3-Этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(1-гидрокси-2-метилпропан-2-ил)пиримидин-5-карбоксамид
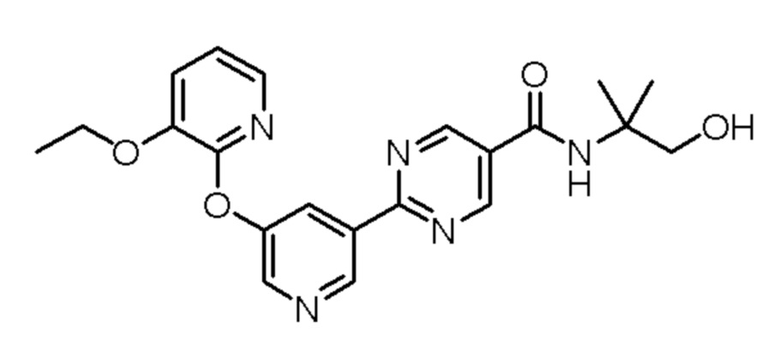
Указанное в заголовке соединение получали в соответствии с общей методикой A с использованием промежуточного соединения 1 (260 мг, 1,0 экв.) и 2-амино-2-метилпропан-1-ола (103 мг, 1,5 экв.). Сырой продукт очищали флэш-хроматографией (50-100% этилацетата в гептанах) и перекристаллизовывали из смеси этилацетат:метанол с получением 2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(1-гидрокси-2-метилпропан-2-ил)пиримидин-5-карбоксамида (236 мг, 75%) в виде бесцветного твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 1,33 (с, 6H), 1,38 (т, 3H), 3,55 (д, 2H), 4,18 (кв, 2H), 4,83 (т, 1H), 7,18 (дд, Гц, 1H), 7,58 (д, 1H), 7,70 (д, 1H), 8,04 (уш. с, 1H), 8,34-8,37 (м, 1H), 8,64 (д, 1H), 9,22 (с, 2H), 9,39 (д, 1H). МС (ЭС+) 410,3 (М+Н).
Таблица 3 включает профиль печеночного клиренса (Clint, app в HLM) примера 5, который продемонстрировал значительное улучшение по сравнению с примером 19.21 в WO2015140658. Этот значительно уменьшенный клиренс намного больше, чем можно было бы предсказать, учитывая разницу в структурах и значения IC50 DGAT2 в Примере 5 и Примере 19.21 в WO2015140658.
Таблица 3. Потенциал DGAT2 и метаболический клиренс в HLM
в HLM
[мкл/мин/мг]
Пример 19.21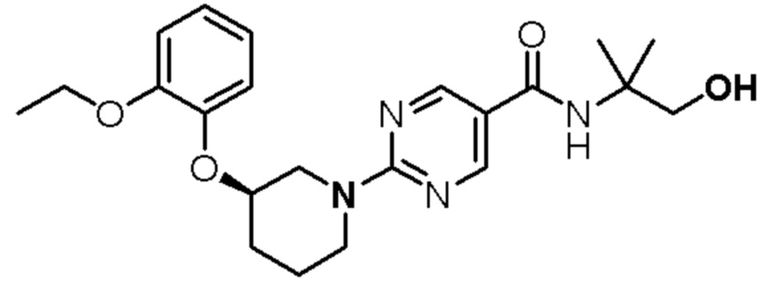
Примеры 6a и 6b: (R)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(3-(гидроксиметил)тетрагидрофуран-3-ил)пиримидин-5-карбоксамид и (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(3-(гидроксиметил)тетрагидрофуран-3-ил)пиримидин-5-карбоксамид

Указанные в заголовке соединения получали в соответствии с общей методикой A с использованием промежуточного соединения 1 (840 мг, 1 экв.) и (3-аминотетрагидрофуран-3-ил)метанола (320 мг, 1,1 экв.). Сырой продукт пропускали через слой силикагеля с получением рацемата в виде твердого вещества желтого цвета, которое разделяли путем очистки хиральной СФХ: Chiral Tech AD-H 250 мм × 4,6 мм 5 мкм; Изократический 70% A: диоксид углерода; Подвижная фаза 30% B: 0,2% изопропиламина в изопропаноле (об./об.). Расход: 60 мл/мин; противодавление=120 бар. Первый пик для элюирования с колонки представляет собой Пример 6a, а второй пик представляет собой Пример 6b.
Энантиомер 1: время удерживания СФХ=9,31 минут (330 мг). Это соединение дополнительно очищали колоночной хроматографией (метанол в дихлорметане), затем перекристаллизовывали из метанола в этилацетате с получением энантиомера 6a (218 мг, 20%) в виде бесцветного твердого вещества. 1H ЯМР (400 МГц, CD3OD) δ 1,40 (т, 3H), 2,23 (дт, 1H), 2,29-2,37 (м, 1H), 3,82-3,91 (м, 2H), 3,92-3,95 (м, 2H), 3,96-4,04 (м, 2H), 4,17 (кв, 2H), 7,19 (дд, 1H), 7,52 (дд, 1H), 7,71 (дд, 1H), 8,49 (дд, 1H), 8,52 (д, 1H), 9,22 (с, 2H), 9,41 (д, 1H). МС (ЭС+) 438,3 (М+Н).
Энантиомер 2: время удерживания СФХ=9,59 минут (300 мг). Это соединение дополнительно очищали с помощью колоночной хроматографии (метанол в дихлорметане), затем перекристаллизовывали из метанола в этилацетате с получением энантиомера 6b (205 мг, 19%) в виде бесцветного твердого вещества. 1H ЯМР (400 МГц, CD3OD) δ 1,40 (т, 3H), 2,23 (дт, 1H), 2,29-2,37 (м, 1H), 3,82-3,91 (м, 2H), 3,92-3,95 (м, 2H), 3,96-4,04 (м, 2H), 4,17 (кв, 2H), 7,19 (дд, 1H), 7,52 (дд, 1H), 7,71 (дд, 1H), 8,49 (дд, 1H), 8,52 (д, 1H), 9,22 (с, 2H), 9,41 (д, 1H). МС (ЭС+) 438,3 (М+Н).
Пример 7: 3-{5-[(3-Этоксипиридин-2-ил)окси]пиридин-3-ил}-N-(1-гидрокси-2-метилпропан-2-ил)-1,2,4-триазин-6-карбоксамид

Указанное в заголовке соединение получали в соответствии с общей методикой A, используя промежуточное соединение 2 (15 мг, 0,04 ммоль) и 2-амино-2-метил-1-пропанол (4,73 мг, 0,053 ммоль). Сырой продукт очищали препаративной обращено-фазной ВЭЖХ с получением указанного в заголовке соединения (2,8 мг, 16%) в виде светло-желтого твердого вещества. 1H ЯМР (400 МГц, CD3OD) δ 1,40 (т, 3H), 1,47 (с, 6H), 3,70 (с, 2H), 4,16 (кв, 2H), 7,20 (дд, 1H), 7,53 (дд, 1H), 7,71 (дд, 1H), 8,56-8,58 (м, 1H), 8,61 (д, 1H), 9,32 (с, 1H), 9,48 (д, 1H). МС (ЭС+) 411,0 (М+Н).
Пример 8: (S)-2-(5-((3-Этокси-5-фторпиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамид

Стадия 1: 3-этокси-5-фторпиридин-1-оксид
Карбонат цезия (21,6 г, 3,0 экв.) добавляли к раствору 5-фтор-3-пиридинола (25 г, 1,0 экв.) и йодистого этила (3,8 г, 1,1 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Смесь фильтровали и концентрировали, получая 3-этокси-5-фторпиридин (3,1 г, 100%) в виде желтого масла, которое использовали без дополнительной очистки. м-Хлорпероксибензойную кислоту (5,7 г, 1,5 экв.) добавляли к раствору 3-этокси-5-фторпиридина (3,1 г, 1,0 экв.) в дихлорметане (50 мл). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь очищали непосредственно флэш-хроматографией (градиент: 0-5% метанола в дихлорметане) с получением 3-этокси-5-фторпиридин-1-оксида (3,30 г, 95%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CD3OD) δ 1,40 (т, 3H), 4,14 (кв, 2H), 7,20 (дт, 1H), 7,96-7,98 (м, 1H), 8,04-8,08 (м, 1H).
Стадия 2: 2-((5-Бромпиридин-3-ил)окси)-3-этокси-5-фторпиридин
Диизопропилэтиламин (3,08 г, 3,8 экв.) добавляли к раствору 3-этокси-5-фторпиридин-1-оксида (1,0 г, 1,0 экв.) и бромтрипирролидинофосфония гексафторфосфата (3,86 г, 1,3 экв.) в тетрагидрофуране (60 мл) при 0°C. Смесь перемешивали при комнатной температуре в течение 16 часов. Реакцию гасили водой (150 мл) и экстрагировали этилацетатом (3 × 50 мл). Объединенные органические слои промывали рассолом (100 мл), сушили над сульфатом натрия и концентрировали. Сырое вещество очищали флэш-хроматографией (градиент: 4-24% этилацетата в петролейном эфире), получая 2-((5-бромпиридин-3-ил)окси)-3-этокси-5-фторпиридин (200 мг, 10%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 1,48 (т, 3H), 4,12 (кв, 2H), 7,04 (дд, 1H), 7,56-7,59 (м, 1H), 7,66-7,67 (м, 1H), 8,41-8,42 (м, 1H), 8,48-8,50 (м, 1H).
Стадия 3: Этил 2-(5-((3-этокси-5-фторпиридин-2-ил)окси)пиридин-3-ил)пиримидин-5-карбоксилат
Бис(пинаколато)диборон (243 мг, 1,2 экв.), ацетат калия (235 мг, 3,0 экв.) и комплекс [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия (II) с дихлорметаном (29 мг, 0,05 экв.) добавляли к раствору 2-((5-бромпиридин-3-ил)окси)-3-этокси-5-фторпиридина (250 мг, 1,0 экв.) в диоксане (5 мл) при комнатной температуре. Реакционную смесь перемешивали при 100°C в течение 2 часов и затем охлаждали до комнатной температуры. Реакционную смесь разбавляли этилацетатом (60 мл) и фильтровали через целит. Фильтрат концентрировали до остатка, затем разбавляли диоксаном (5 мл) и водой (1 мл). Этил 2-хлорпиримидин-5-карбоксилат (164 мг, 1,1 экв.), комплекс [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия (II) с дихлорметаном (18 мг, 0,03 экв.) и карбонат калия (166 мг, 1,5 экв.) добавляли к реакционной смеси, и полученную суспензию перемешивали при 80°C в течение 2 часов, затем оставляли стоять при комнатной температуре в течение 4 дней. Реакционную смесь распределяли между этилацетатом (30 мл) и водой (50 мл) и экстрагировали этилацетатом (2 × 30 мл). Органический слой отделяли, промывали рассолом (50 мл), сушили над сульфатом натрия, фильтровали и концентрировали, чтобы получить сырое вещество, которое очищали препаративной ТСХ (5:1 петролейный эфир:этилацетат) с получением этил 2-(5-((3-этокси-5-фторпиридин-2-ил)окси)пиридин-3-ил)пиримидин-5-карбоксилата (50 мг, 16%). МС (ЭС+) 385,0 (М+Н).
Стадия 4: 2-(5-((3-Этокси-5-фторпиридин-2-ил)окси)пиридин-3-ил)пиримидин-5-карбоновая кислота
Гидроксид натрия (0,20 мл, 3,0 экв., 2М) добавляли к этил 2-(5-((3-этокси-5-фторпиридин-2-ил)окси)пиридин-3-ил)пиримидин-5-карбоксилату (50 мг, 1,0 экв.) в этаноле (10 мл). Реакционную смесь перемешивали при 25°C в течение 16 часов. Раствор разбавляли водой (50 мл) и экстрагировали этилацетатом (3 × 30 мл). Водный слой подкисляли соляной кислотой (2н) до pH 3. Раствор экстрагировали этилацетатом (15 мл), сушили над сульфатом натрия и концентрировали, получая 2-(5-((3-этокси-5-фторпиридин-2-ил)окси)пиридин-3-ил)пиримидин-5-карбоновую кислоту (25 мг, 54%) в виде желтого твердого вещества. МС (ЭС+) 357,0 (М+Н).
Стадия 5: (S)-2-(5-((3-Этокси-5-фторпиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамид
Указанное в заголовке соединение получали в соответствии с общей методикой A, используя 2-(5-((3-этокси-5-фторпиридин-2-ил)окси)пиридин-3-ил)пиримидин-5-карбоновую кислоту (25 мг, 1,0 экв.) и (S)-тетрагидрофуран-3-амин (18,3 мг, 3,0 экв.). Сырое вещество очищали препаративной ВЭЖХ, получая (S)-2-(5-((3-этокси-5-фторпиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамид (20 мг, 67%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 1,38 (т, 3H), 1,87-2,00 (м, 1H), 2,14-2,28 (м, 1H), 3,65 (дд, 1H), 3,70-3,79 (м, 1H), 3,83-3,94 (м, 2H), 4,21 (кв, 2H), 4,46-4,56 (м, 1H), 7,65-7,74 (м, 2H), 8,37 (дд, 1H), 8,65 (д, 1H), 8,98 (д, 1H), 9,29 (с, 2H), 9,40 (д, 1H). МС (ЭС+) 425,9 (М+Н).
ФАРМАКОЛОГИЧЕСКИЕ ДАННЫЕ
Следующие протоколы, конечно, могут варьироваться специалистами в данной области техники.
Генерация конструкции человеческой DGAT2 (hDGAT2)
Конструкцию для hDGAT2 генерировали с помощью N-концевой метки FLAG (октапептид с аминокислотной последовательностью AspTyrLysAspAspAspAspLys). Для FLAG-меченой конструкции hDGAT2 кДНК для hDGAT2 специально синтезировали в Genscript и клонировали в вектор pFastBac1 (Invitrogen) путем использования рестрикционных ферментов BamHI/XhoI для генерации FLAG-меченой на N-конце конструкции pFastBac1-FLAG-hDGAT2 (аминокислоты 1-388). Конструкцию подтверждали секвенированием в обоих направлениях.
Экспрессия DGAT2 и получение мембранной фракции DGAT2
Рекомбинантный бакуловирус для FLAG-меченого hDGAT2 генерировали в клетках насекомых SF9 с использованием системы экспрессии бакуловируса Bac-to-Bac (Invitrogen) в соответствии с протоколом производителя. Для экспрессии hDGAT2 клетки SF9 (20 л), выращенные в среде Sf900II, инфицировали бакуловирусом hDGAT2 при множественности заражения 1 в пластиковом мешке для биореактора Wave Bioreactor System 20/50P (GE Healthcare). После 40 часов заражения клетки собирали центрифугированием при 5000 × g. Клеточные осадки промывали путем ресуспендирования в фосфатно-солевом буфере (ФСБ) и собирали центрифугированием при 5000 × g. Клеточную суспензию быстро замораживали в жидком N2 и хранили при -80°C до необходимости. Все приведенные ниже операции проводили при 4°C, если не указано иное. Клетки ресуспендировали в буфере для лизиса (50 мМ Трис-HCl, pH 8,0, 250 мМ сахарозы), содержащем 1 мМ этилендиаминтетрауксусной кислоты (EDTA) и полную смесь ингибиторов протеаз (Roche Diagnostics) в соотношении 3 мл буфера на 1 г клеточной суспензии. Клетки лизировали с помощью гомогенизатора Доунса. Клеточный дебрис удаляли центрифугированием при 1000 × g в течение 20 минут и супернатант центрифугировали при 100000 × g в течение 1 часа. Полученный осадок трижды промывали, заполняя ультрацентрифужные пробирки до верха ледяным ФСБ перед декантацией. Промытый осадок ресуспендировали при осторожном перемешивании в течение 1 часа в буфере для лизиса, содержащем 8 мМ 3-[(3-холамидопропил)диметиламмонио]-1-пропансульфоната (CHAPS) в соотношении 1 мл буфера на 1 г исходной клеточной суспензии, и снова центрифугировали при 100 000 × g в течение 1 часа. Полученный супернатант разделяли на аликвоты, быстро замораживали в жидком N2 и хранили при -80°C до использования.
Анализ DGAT2 in vitro и определение значений IC50 для ингибиторов DGAT2
Для определения значений IC50 реакции проводили в 384-луночных белых полимерных планшетах (Perkin Elmer) в общем объеме 20 мкл. К 1 мкл соединений, растворенных в 100% ДМСО и нанесенных на дно каждой лунки, добавляли 5 мкл 0,04% бычьего сывороточного альбумина (БСА) (без жирных кислот, Sigma Aldrich) и смесь инкубировали при комнатной температуре в течение 15 минут. Мембранные фракции hDGAT2 разбавляли в 100 мМ Hepes-NaOH, pH 7,4, 20 мМ MgCl2, содержащем 200 нМ метиларахидонилфторфосфоната (Cayman Chemical; высушивали из исходного раствора этилацетата в атмосфере газа аргона и растворяли в ДМСО в виде 5 мМ исходного раствора). 10 мкл этого рабочего раствора фермента добавляли в планшеты, и инкубирование продолжали в течение 2 часов при комнатной температуре. Реакции DGAT2 инициировали добавлением 4 мкл субстрата, содержащего 30 мкМ [1-14C]деканоил-КоА (синтезированного по заказу Perkin Elmer, 50 мКи/ммоль) и 125 мкМ 1,2-дидеканоил-sn-глицерина (Avanti Polar Lipids), растворенного в 12,5% ацетоне. Реакционные смеси инкубировали при комнатной температуре в течение 40 мин и реакции останавливали добавлением 5 мкл 1% H3PO4. После добавления 45 мкл MicroScint-E (Perkin-Elmer) планшеты герметизировали крышками Top Seal-A (Perkin-Elmer), и фазовое разделение субстратов и продуктов достигали с помощью орбитального шейкера для микропланшетов HT-91100 (Big Bear Automation, Санта-Клара, Калифорния). Планшеты центрифугировали при 2000 × g в течение 1 минуты в центрифуге Allegra 6R (Beckman Coulter) и затем снова герметизировали свежими крышками перед считыванием в сцинтилляционном счетчике 1450 Microbeta Wallac Trilux (Perkin Elmer). Активность DGAT2 измеряли путем количественного определения образовавшегося продукта [14C]тридеканоилглицерина в верхней органической фазе.
Фоновую активность, полученную с использованием 50 мкМ (R)-1-(2-((S)-1-(4-хлор-1H-пиразол-1-ил)этил)-3H-имидазо[4,5-b]пиридин-5-ил)пиперидин-3-ил)(пирролидин-1-ил)метанона (WO 2013150416, пример 196-A) для полного ингибирования DGAT2, вычитали из всех реакций. Ингибиторы тестировали в одиннадцати различных концентрациях, чтобы получить значения IC50 для каждого соединения. Используемые одиннадцать концентраций ингибитора обычно включали 50, 15,8, 5, 1,58, 0,50, 0,16, 0,05, 0,016, 0,005, 0,0016 и 0,0005 мкМ. Данные представляли в виде процента ингибирования в зависимости от концентрации ингибитора, и они соответствуют уравнению y=100/[1+(x/IC50)z], где IC50 представляет собой концентрацию ингибитора при 50% ингибировании, а z представляет собой угловой коэффициент Хилла (наклон кривой в точке перегиба).
В таблице 4 ниже приведены значения IC50 примеров для ингибирования DGAT2 в соответствии с вышеописанным анализом. Результаты представлены в виде средних геометрических значений IC50.
Таблица 4. Значения IC50 примеров для ингибирования DGAT2
Определение значений IC50 для ингибиторов DGAT2 в гепатоцитах человека
Для оценки действия ингибиторов DGAT2 в клеточных условиях криосохраненные гепатоциты человека (серия NON и EBS, Celsis, Чикаго, Иллинойс) размораживали и высевали на покрытые коллагеном типа I планшеты в соответствии с инструкциями производителя. После 24-часового периода восстановления в ночное время клетки покрывали средой, содержащей 250 мкг/мл Матригеля (BD Biosciences, Сан-Хосе, Калифорния). На следующий день среду аспирировали и заменяли бессывороточной средой Williams Media E (Life Technologies, Гранд-Айленд, Нью-Йорк), содержащей 400 мкМ додеканоата натрия (Sigma-Aldrich, Сент-Луис, Миссури). Сорок пять минут спустя селективный ингибитор DGAT1 (пример 3, WO2009016462, приготовленный в виде 100× исходные растворы в 25% ДМСО, 75% среды Williams' Media E) добавляли во все лунки в конечной концентрации (3 мкМ), которая полностью подавляла эндогенную активность DGAT1. Затем добавляли ингибиторы DGAT2 до желаемой конечной концентрации. После 15-минутной предварительной инкубации в каждую лунку добавляли 0,2 мкКи [1,3-14C]-глицерина (American Radio Chemicals, Сент-Луис, Миссури) с последующей 3-часовой инкубацией. В этот момент среду удаляли, клетки промывали один раз с помощью ФСБ и затем лизировали в изопропиловом спирте:тетрагидрофуране (9:1) перед центрифугированием при 3000 об/мин в течение 5 минут. Меченые радиоактивной меткой липиды разделяли с использованием системы 2 растворителей с помощью тонкослойной хроматографии с растворителем 1, состоящим из этилацетата:изопропилового спирта:хлороформа:метанола:0,25% хлорида калия в воде (100:100:100:40,2:36,1, об./об./об./об.), и растворителем 2, состоящим из гексана:диэтилового эфира:уксусной кислоты (70:27:3, об./об./об.). Пластины ТСХ хроматографировали до прохождения фронтом растворителя 1 одной трети от высоты пластины, пластины высушивали в атмосфере азота и затем хроматографировали до прохождения фронтом растворителя до верха пластины. После разделения меченые радиоактивной меткой липиды визуализировали с использованием системы Molecular Dynamics' PhosphorImager. Полумаксимальные ингибирующие концентрации (значения IC50) определяли с использованием GraphPad Prism (GraphPad Software, Inc., Ла-Холья, Калифорния) с использованием функции Хилла с фиксированной базовой линией=0 (контроль растворителем) и угловым коэффициентом Хилла=1.
В этих условиях в Примере 1 среднее геометрическое значение IC50 составило 2,8 нМ (N=10).
In Vivo эффекты ингибиторов DGAT2 на уровень триглицеридов в плазме и печени
Крысиную модель западной диеты использовали для оценки долгосрочных эффектов лечения ингибиторами DGAT2 на выработку триглицеридов в плазме и содержание триглицеридов в печени. Самцов крыс Sprague-Dawley содержали в стандартных лабораторных условиях с 12-часовым световым и 12-часовым темновым циклом (свет включался в 06:00). За две недели до начала исследования животным назначали диету с высоким содержанием жиров и холестерина (D12079b, Research Diets, Нью-Брансуик, Нью-Джерси). Эта диета обеспечивает ~ 43% килокалорий из углеводов и ~ 41% килокалорий из жиров. Ингибиторы DGAT2 вводили перорально в виде раствора (дозируемый объем 10 мл/кг) в 0,5% HPMCAS-HF и 0,015% SLS в деионизированной воде, pH 8,5 (метилцеллюлозу и бутилированный гидрокситолуол получали от Sigma Aldrich, Сент-Луис, Миссури). Животные, которых лечили инертным веществом, получали водный раствор 0,5% HPMCAS-HF и 0,015% SLS в деионизированной воде, pH 8,5 в чистом виде. Ингибиторы DGAT2 вводили перорально два раза в день в течение 7 дней в 08:00 и 16:00 в дозах 1, 3, 10, 30 и 90 мг/кг. На 8-й день всем животным ничего не давали в 06:00, в 10:00 вводили инертное вещество или ингибиторы DGAT2 и умерщвляли через 2 часа после введения дозы. Крыс умерщвляли асфиксией углекислым газом и собирали кровь через боковую хвостовую вену. Уровни ТГ в плазме определяли с использованием химического анализатора Roche Hitachi в соответствии с инструкциями производителя (Roche Diagnostics Corporation, Индианаполис, Индиана), а данные анализировали с использованием GraphPad Prism (GraphPad Software, Inc., Ла-Холья, Калифорния). Набор образцов печени для определения содержания триглицеридов в печени отделяли хирургически во время умерщвления, немедленно замораживали в жидком азоте и хранили при -80°C до анализа. Для оценки уровней триглицеридов в ткани срез печени, завернутый в алюминиевую фольгу, измельчали молотком на алюминиевом термостате в бане с жидким азотом. При измельчении ткани печени образуется гомогенный порошок. Буфер для гомогенизации, Трис pH 7,4, 98,9 миллилитров 0,9% NaCl и 100 микролитров Тритона Х 100, перемешивали на перемешивающей плите в течение 10 минут перед использованием. Вес образцов приблизительно 100 мг гомогенной ткани печени взвешивали и помещали в пробирку Lysing Matrix D (MP Biomedicals, Кат. № 6913-100) с 1 мл буфера для гомогенизации. Все образцы затем помещали в FastPrep FP120 (MP Biomedicals, Кат. № 6001-120) на 2 минуты или до тех пор, пока ткань не была должным образом гомогенизирована. Все образцы затем центрифугировали в течение 30 секунд при 10000 g, чтобы очистить от пены после гомогенизации. 50 микролитров образца переносили в стерильный планшет для смешивания с 450 микролитрами физиологического раствора с фосфатным буфером Дульбекко (DPBS) для создания разведения 1:10. После повторного суспендирования нового образца все образцы переносили в пробирки для отбора проб для клинического анализатора Siemens Advia XPT. Анализ триглицеридов проводили по поглощающей способности и выражали в миллиграммах на децилитр. Затем триглицериды нормализовали на грамм ткани в Microsoft Excel.
На фигурах 3 и 4 суммированы эффекты перорального введения с использованием примера 1 на уровни триглицеридов в плазме и печени у крыс Sprague Dawley, получавших западную диету, в соответствии с описанными выше способами. Данные представляют собой среднее значение±стандартное отклонение от 8 животных. Разницу между средними значениями в группе относительно инертного вещества определяли посредством однофакторного дисперсионного анализа ANOVA с последующим тестом множественного сравнения Даннетта ** p <0,01, **** p <0,0001.
В настоящей заявке приведены ссылки на различные публикации. Раскрытия этих публикаций во всей их полноте включены в настоящую заявку посредством ссылки для любых целей.
Специалистам в данной области техники будет очевидно, что различные модификации и изменения могут быть сделаны в настоящем изобретении без отклонения от объема или сущности изобретения. Другие варианты осуществления изобретения будут очевидны для специалистов в данной области техники из рассмотрения описания и практики осуществления настоящего изобретения, раскрытого в настоящем документе. Предполагается, что описание и примеры будут рассматриваться только как примерные, с истинным объемом и сущностью изобретения, указанными в следующей формуле изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОМБИНАЦИИ ДЛЯ ЛЕЧЕНИЯ НАСГ/НАЖБП И СВЯЗАННЫХ С НИМИ ЗАБОЛЕВАНИЙ | 2019 |
|
RU2776369C1 |
| НОВОЕ БИАРИЛЬНОЕ ПРОИЗВОДНОЕ, ПРИМЕНЯЕМОЕ В КАЧЕСТВЕ ИНГИБИТОРА ДИАЦИЛГЛИЦЕРОЛ-АЦИЛТРАНСФЕРАЗЫ 2, И ЕГО ПРИМЕНЕНИЕ | 2021 |
|
RU2808433C1 |
| НОВОЕ АМИНОАРИЛЬНОЕ ПРОИЗВОДНОЕ, ПРИГОДНОЕ В КАЧЕСТВЕ ИНГИБИТОРА ДИАЦИЛГЛИЦЕРОЛАЦИЛТРАНСФЕРАЗЫ 2, И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2799819C1 |
| НОВОЕ АМИДНОЕ ПРОИЗВОДНОЕ, ИСПОЛЬЗУЕМОЕ В КАЧЕСТВЕ ИНГИБИТОРА ДИАЦИЛГЛИЦЕРОЛ O-АЦИЛТРАНСФЕРАЗЫ 2, И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2810064C1 |
| ИНГИБИРУЮЩИЕ JAK СОЕДИНЕНИЯ НА ОСНОВЕ ПИРАЗОЛОПИРИМИДИНА И СПОСОБЫ | 2010 |
|
RU2567238C2 |
| ИНГИБИРУЮЩИЕ JAK СОЕДИНЕНИЯ НА ОСНОВЕ ПИРАЗОЛОПИРИМИДИНА И СПОСОБЫ | 2010 |
|
RU2675857C2 |
| ПИРАЗОЛОПИРИМИДИНОВЫЕ СОЕДИНЕНИЯ-ИНГИБИТОРЫ JAK И СПОСОБЫ | 2009 |
|
RU2539568C2 |
| СОЕДИНЕНИЯ СО СПИРО- И АРОМАТИЧЕСКИМИ КОЛЬЦАМИ И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2781100C1 |
| ХИНОЛИНИЛ-СОДЕРЖАЩЕЕ СОЕДИНЕНИЕ И ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРИМЕНЕНИЕ | 2020 |
|
RU2803116C2 |
| НОВЫЕ СОЕДИНЕНИЯ И ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ ВОСПАЛИТЕЛЬНЫХ ЗАБОЛЕВАНИЙ | 2018 |
|
RU2785126C2 |

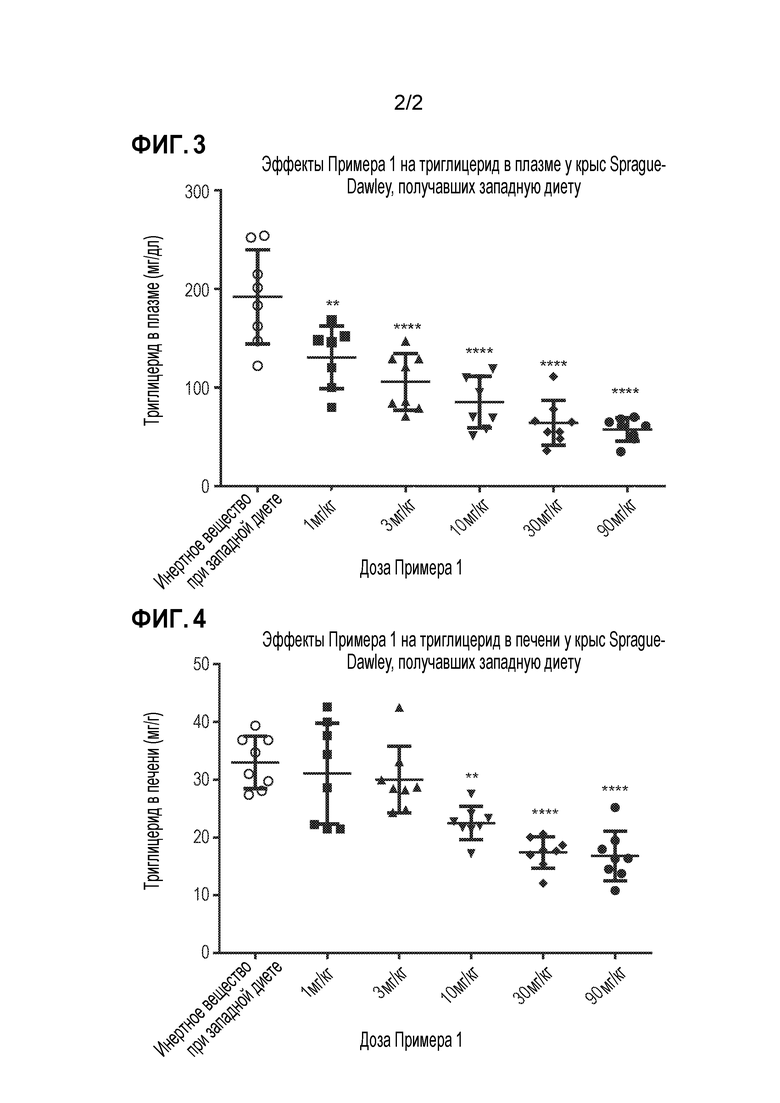
Изобретение относится к новому соединению формулы I и его фармацевтически приемлемой соли. Соединения ингибируют активность диацилглицерол-ацилтрансферазы 2 (DGAT2) и могут найти применение для лечения неалкогольной жировой дистрофии, неалкогольного стеатогепатита, гиперлипидемии, диабета и др. заболеваний, связанных с активностью DGAT2. В формуле (I)

(I)
D1 представляет собой СН; и D2 представляет собой N или СН; R1 представляет собой Н или (С1-С2)алкил, необязательно замещенный одним или двумя заместителями, каждый из которых независимо выбран из фтора; R2 представляет собой Н или фтор;
R3 представляет собой  ,
,  или
или  ; R4 представляет собой H, циано или (C1-C4)алкил, необязательно замещенный одним заместителем, каждый из которых независимо выбран из -OH и -S(O)2R6; R5 представляет собой Н или -OH; и R6 представляет собой (C1-C4)алкил. Предпочтительным соединением является соединение
; R4 представляет собой H, циано или (C1-C4)алкил, необязательно замещенный одним заместителем, каждый из которых независимо выбран из -OH и -S(O)2R6; R5 представляет собой Н или -OH; и R6 представляет собой (C1-C4)алкил. Предпочтительным соединением является соединение
 (I’)
(I’)
формулы (I’). Указанное соединение может быть получено в виде кристаллической формы I, имеющей показатели пиков порошковой рентгенограммы (PXRD) при излучении CuKα при угле 2-тета (°) 5,3, 7,7, 15,4, или в виде кристаллической формы II, имеющей показатели пиков порошковой рентгенограммы (PXRD) при излучении CuKα при угле 2-тета (°) 6,5, 9,3, 13,6. 6 н. и 8 з.п. ф-лы, 4 ил., 4 табл., 8 пр.
1. Соединение формулы (I)

(I)
где
D1 представляет собой СН; и D2 представляет собой N или СН;
R1 представляет собой Н или (С1-С2)алкил, необязательно замещенный одним или двумя заместителями, каждый из которых независимо выбран из фтора;
R2 представляет собой Н или фтор;
R3 представляет собой  ,
, 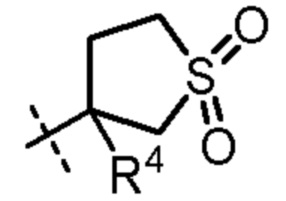 или
или 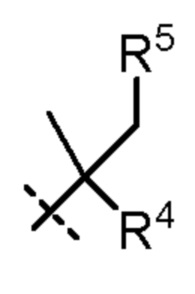 ;
;
R4 представляет собой H, циано или (C1-C4)алкил, необязательно замещенный одним заместителем, каждый из которых независимо выбран из -OH и -S(O)2R6;
R5 представляет собой Н или -OH; и
R6 представляет собой (C1-C4)алкил; или его фармацевтически приемлемая соль.
2. Соединение по п. 1, имеющее формулу (Ia)
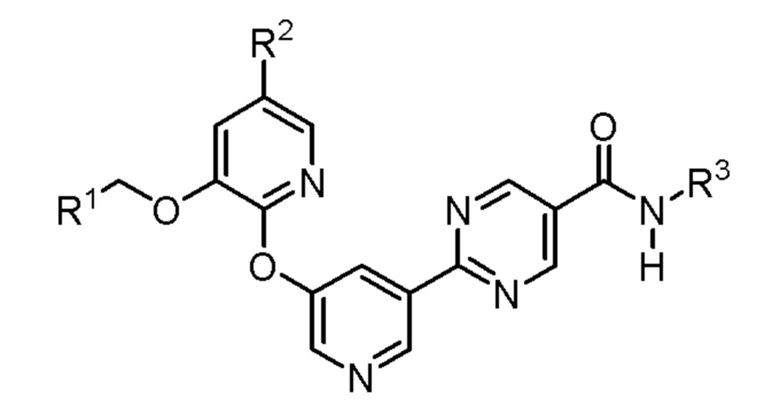
(Ia)
где R1-R3 имеют значения, указанные в п. 1,
или его фармацевтически приемлемая соль.
3. Соединение по п. 1 или его фармацевтически приемлемая соль, где R3 представляет собой  .
.
4. Соединение по п. 1 или его фармацевтически приемлемая соль, где R3 представляет собой  .
.
5. Соединение по п. 3 или его фармацевтически приемлемая соль, где R1 представляет собой метил.
6. Соединение по п. 4 или его фармацевтически приемлемая соль, где R1 представляет собой метил.
7. Соединение по п. 6 или его фармацевтически приемлемая соль, где R4 представляет собой H, -CH2OH или циано.
8. Соединение:
(S)-2-(5-((3-этокси-5-фторпиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамид;
N-(2-цианопропан-2-ил)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)пиримидин-5-карбоксамид;
2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(3-метил-1,1-диоксидотетрагидротиофен-3-ил)пиримидин-5-карбоксамид;
2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(1-гидрокси-2-метилпропан-2-ил)пиримидин-5-карбоксамид;
(S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамид;
(S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(3-(гидроксиметил)тетрагидрофуран-3-ил)пиримидин-5-карбоксамид;
(R)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(3-(гидроксиметил)тетрагидрофуран-3-ил)пиримидин-5-карбоксамид;
2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(2-метил-1-(метилсульфонил)пропан-2-ил)пиримидин-5-карбоксамид;
(S)-2-(5-((3-(2-фторэтокси)пиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамид;
3-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(1-гидрокси-2-метилпропан-2-ил)-1,2,4-триазин-6-карбоксамид;
N-(1,3-дигидрокси-2-метилпропан-2-ил)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)пиримидин-5-карбоксамид;
(S)-3-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)-1,2,4-триазин-6-карбоксамид;
N-(1,1-диоксидотетрагидротиофен-3-ил)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)пиримидин-5-карбоксамид;
(R)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамид; или
2-(5-((3-этоксипиразин-2-ил)окси)пиридин-3-ил)-N-(1-гидрокси-2-метилпропан-2-ил)пиримидин-5-карбоксамид;
или его фармацевтически приемлемая соль.
9. Соединение:
(R)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(3-(гидроксиметил)тетрагидрофуран-3-ил)пиримидин-5-карбоксамид; (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамид; или
(S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(3-(гидроксиметил)тетрагидрофуран-3-ил)пиримидин-5-карбоксамид, или его фармацевтически приемлемая соль.
10. Соединение, имеющее структуру:

или его фармацевтически приемлемая соль.
11. Фармацевтическая композиция, обладающая свойствами ингибирования активности диацилглицерол-ацилтрансферазы 2 (DGAT2), содержащая соединение по п. 1 или фармацевтически приемлемую соль указанного соединения, присутствующую в терапевтически эффективном количестве, в смеси по меньшей мере с одним фармацевтически приемлемым наполнителем.
12. Соединение, имеющее структуру:
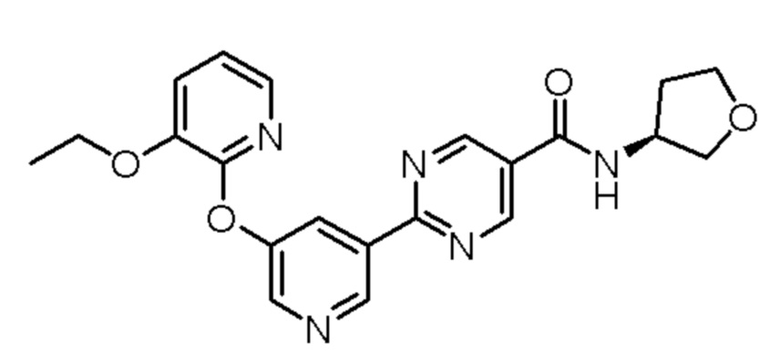
в виде кристаллической формы I, имеющей показатели пиков порошковой рентгенограммы (PXRD) при излучении CuKα при угле 2-тета (°) 5,3, 7,7, 15,4, или в виде кристаллической формы II, имеющей показатели пиков порошковой рентгенограммы (PXRD) при излучении CuKα при угле 2-тета (°) 6,5, 9,3, 13,6.
13. Кристаллическая форма I по п. 1, имеющая показатели пиков порошковой рентгенограммы, как показано на фиг. 1.
14. Кристаллическая форма II по п. 1, имеющая показатели пиков порошковой рентгенограммы, как показано на фиг. 2.
| US 20150259323 A1, 17.09.2015 & EA 201600589 А1, 31.03.2017 | |||
| WO 2014145025 A2, 18.09.2014 & RU 2015143717 А3, 28.03.2018 | |||
| RU 2014124953 A, 27.01.2016. |

