ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Изобретение относится к новым фармацевтическим композициям, включающим (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамид или его фармацевтически приемлемую соль и, в этой же или в отдельной композиции, 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойную кислоту или ее фармацевтически приемлемую соль, для лечения неалкогольной жировой болезни печени (НАЖБП) и связанных с ней заболеваний, например неалкогольного стеатогепатита (НАСГ) и метаболических заболеваний.
УРОВЕНЬ ТЕХНИКИ ИЗОБРЕТЕНИЯ
Неалкогольный стеатогепатит (НАСГ) представляет собой клиническую и гистологическую разновидность неалкогольной жировой болезни печени (НАЖБП, определяемая наличием >5% жирового гепатоза), которая связана с повышенной смертностью, вызванной различными причинами, циррозом печени и заболеванием печени конечной стадии, повышенной смертностью, вызванной сердечно-сосудистыми осложнениями, и увеличением частоты онкологических осложнений как связанных, так и не связанных с печенью (Sanyal et al., Hepatology 2015;61(4):1392-1405). НАЖБП является печеночным проявлением метаболического синдрома и представляет собой спектр состояний печени, включая стеатоз (жировую дегенерацию печени), НАСГ, фиброз, цирроз и, в конечном счете, гепатоцеллюлярную карциному. НАЖБП и НАСГ считаются основными жировыми заболеваниями печени, поскольку среди пациентов с этими заболеваниями наибольшую долю составляют пациенты с повышенным уровнем липидов печени. Тяжесть НАЖБП/НАСГ определяет наличие липидов, инфильтрата воспалительных клеток, баллонной дистрофии клеток печени и распространенность фиброза. В настоящее время варианты лечения ограничиваются лечением сопутствующих состояний (EASL-EASD-EASO Clinical Practice Guidelines, J. Hepatol. 2016;64(6):1388-1402).
Предполагается, что изменения в липидном метаболизме способствуют молекулярному патогенезу НАЖБП и НАСГ. Стеатоз является необходимым, но не достаточным компонентом патогенеза НАСГ (Day C., and James O., Hepatology. 1998; 27(6):1463-6). Соответственно, многочисленные исследования показали, что тяжесть стеатоза определяет риск сопутствующего стеатогепатита, а также риск развития цирроза печени (Sorensen et al., Lancet. 1984; 2(8397): 241-4; Wanless I. and Lentz J., Hepatology 1990;12(5): 1106-10; Reeves H., et al. J. Hepatol. 1996; 25(5): 677-83). Стеатоз печени является следствием дисбаланса между продуцированием/поглощением TG печенью и клиренсом/выведением их из печени (Cohen J.C., et al., Science. 2011; 332(6037):1519-1523). Предполагается, что уменьшение стеатоза, метаболического фактора, лежащего в основе развития НАЖБП/НАСГ, будет приводить к последующим улучшениям при воспалении и фиброзе печени.
Ацетил-КoA-карбоксилаза (AКК) и диацилглицерол-ацилтрансфераза-2 (DGAT2) представляют собой два ключевых фермента, регулирующих метаболизм липидов. АКК в процессе первичного липогенеза (de novo lipogenesis - DNL) катализируют его жизненно важную и ограничивающую скорость стадию (Saggerson D., Annu. Rev. Nutr. 2008; 28:253-72.). Кроме того, АКК также регулирует митохондриальное бета-окисление жирных кислот через аллостерическую регуляцию фермента - картинин-пальмитоилтрансферазы 1 (carnitine palmitoyltransferase 1 - CPT1) (Saggerson, 2008; Waite M, and Wakil S.J. J. Biol. Chem. 1962;237:2750-2757.). Полученные данные также позволяют сделать предположение о том, что подавление DNL с помощью ингибирования АКК может непосредственно снижать воспаление посредством торможения образования воспалительного интерлейкина-17 (interleukin-17 - IL-17), секретирующего T-клетки линии T-хелперов (Th17 клетки), и способствовать развитию противовоспалительных FoxP3(+) регуляторных T-клеток (Treg) (Berod L., et al. Nat. Med. 2014; 20(11): 1327-33).
Предполагается, что ингибирование активности АКК оказывает полезное для пациентов с НАСГ действие посредством по меньшей мере двух независимых механизмов. Как отмечалось выше, у пациентов с НАЖБП наблюдается заметное повышение уровня печеночного DNL, и предполагается, что нормализация этого повышенного выделения через фармакологическое игибирование печеночного АКК снижает стеатоз. Кроме того, действие ингибиторов АКК, вызывающих повышение окисления жирных кислот, может также способствовать снижению содержания жира в печени. В соответствии с этим было показано, что ингибиторы АКК ингибируют DNL. См. Griffith D.A., et al. J. Med. Chem. 2014;57(24):10512-10526; Kim C.W., et al. Cell Metab. 2017;26, 394-406; Stiede K., et al. Hepatology. 2017;66(2):324-334; Lawitz E.J., et al. Clin Gastroenterol Hepatol. 2018 (https://doi.org/10.1016/j.cgh.2018.04.042). Кроме того, ожидается, что ингибирование DNL в IL-17, секретирующем T-клетки, подавляет воспаление печени ограничением образования воспалительных Th17 клеток (Berod et al., 2014) - путь метаболизма, который может быть важен в патогенезе НАСГ (Rau M., et al. J. Immunol. 2016;196(1):97-105) и благоприятен для развития противовоспалительных Treg-клеток. Кроме того, ингибирование АКК может снижать активацию звездчатых клеток и фиброз (Ross et al., 2019).
Триглицериды или триацилглицерины (TG) представляют собой основную форму накопления (хранения) энергии у млекопитающих. TG образуются посредством последовательной этерификации глицерина тремя жирными кислотами с различной длиной цепи и различной степенью насыщения (Coleman, R. A., and Mashek, D.G. 2011. Chem. Rev. 111: 6359-6386). TG, синтезированные в кишечнике или печени, собираются в хиломикроны или липопротеин очень низкой плотности (very low-density lipoprotein - VLDL), соответственно, и экспортируются в периферические ткани, где они гидролизуются липопротеинлипазой (lipoprotein lipase - LPL) до составляющих их жирных кислот и глицерина. Полученные неэтерифицированные жирные кислоты (non-esterified fatty acids - NEFA) могут далее метаболизироваться для получения энергии или снова подвергаться этерификации и поступать на хранение.
В нормальных физиологических условиях энергетически плотный TG остается секвестрированным в различных жировых депо до тех пор, пока не возникнет потребность в его высвобождении, после чего он гидролизуется до глицерина и свободных жирных кислот, которые затем высвобождаются в кровоток. Этот процесс жестко регулируется противоположно направленными действиями инсулина и гормонов, таких как катеоламины, которые способствуют отложению и мобилизации запасов TG в различных физиологических условиях. После приема пищи инсулин ингибирует липолиз, сдерживая таким образом высвобождение энергии в форме NEFA и обеспечивая соответствующее хранение пищевых липидов в жировых депо. Однако у пациентов с диабетом 2 типа способность инсулина подавлять липолиз повышается, и поток NEFA из адипоцитов неадекватно увеличивается. Это, в свою очередь, приводит к повышенной доставке липида к тканям, таким как мышца и печень. При отсутствии потребности в энергии TG и другие липидные метаболиты, такие как диацилглицерин (diacylglycerol - DAG), могут накапливаться и вызывать потерю чувствительности к инсулину (Erion, D.M., and Shulman, G.I. 2010. Nat Med 16: 400-402). Резистентность к инсулину в мышцах характеризуется пониженным поглощением глюкозы и депонированием гликогена, в то время как в печени потеря инсулиновых сигнальных путей приводит к нарушению регуляции выработки глюкозы и избыточному продуцированию TG-обогащенных VLDL, что является признаком диабета 2 типа (Choi, S.H., and Ginsberg, H.N. 2011. Trends Endocrinol. Metab. 22: 353-363). Считается, что повышенная секреция TG-обогащенных VLDL, так называемых VLDL1 частиц, стимулирует выработку малых плотных частиц липопротеинов низкой плотности (small dense low-density lipoprotein - sdLDL), проатерогенной субфракции LDL, которая связана с повышенным риском ишемической болезни сердца (St-Pierre, A.C. et al. 2005. Arterioscler. Thromb. Vasc. Biol. 25: 553-559).
У млекопитающих были охарактеризованы два фермента диацилглицерол-ацилтрансферазы (DGAT) - (DGAT1 и DGAT2). Хотя эти ферменты катализируют одну и ту же ферментативную реакцию, их соответствующие аминокислотные последовательности не являются родственными и относятся к разным семействам генов. Мыши с нарушением гена, кодирующего DGAT1, резистентны к ожирению, вызванному питанием, и проявляют повышенное потребление энергии и повышенную активность (Smith, S. J. et. al., 2000. Nat Genet 25: 87-90). Dgat1-/- мыши демонстрируют нерегулируемое постабсорбтивное (postaborpative) высвобождение хиломикронов и накапливают липиды в энтероцитах (Buhman, K.K. et.al. 2002. J. Biol. Chem. 277: 25474-25479). Предполагается, что метаболически благоприятный фенотип, наблюдаемый у этих мышей, обусловлен потерей экспрессии DGAT1 в кишечнике (Lee, B., et.al. 2010. J. Lipid Res. 51: 1770-1780). Важно отметить, что, несмотря на нарушение лактации у самок мышей Dgat1 -/-, эти животные сохраняют способность синтезировать TG, что указывает на существование дополнительных DGAT ферментов. Это наблюдение и выделение второго DGAT из гриба Mortierella rammaniana привело к идентификации и характеристическому описанию DGAT2 (Yen, C.L. et al. 2008. J. Lipid Res. 49: 2283-2301).
DGAT2 с высоким уровнем экспрессируется в печени и жировой ткани и в отличие от DGAT1 проявляет исключительную субстратную специфичность в отношении DAG (Yen, C.L., 2008). Делеция гена DGAT2 у грызунов приводит к нарушению внутриутробного развития, тяжелой липемии, нарушению барьерной функции кожи и ранней послеродовой смерти (Stone, S.J. et.al. 2004. J. Biol. Chem. 279: 11767-11776). Вследствие летальности, вызванной потерей DGAT2, большая часть сведений о физиологической роли DGAT2 получена из исследований, проведенных с антисмысловыми олигонуклеотидами (antisense oligonucleotide - ASO) в моделях метаболического заболевания грызунов. В этих условиях ингибирование печеночной DGAT2 приводит к улучшению профиля плазменных липопротеинов (снижение общего холестерина и TG) и к снижению липидной нагрузки печени, что сопровождается улучшением чувствительности к инсулину и контролем уровня глюкозы в организме (Liu, Y. et.al. 2008. Biochim. Biophys. Acta 1781: 97-104; Choi, C.S. et al. 2007. J. Biol. Chem. 282: 22678-22688; Yu, X.X. et al. 2005. Hepatology 42: 362-371). Хотя молекулярные механизмы, лежащие в основе этих наблюдений, полностью не изучены, ясно, что подавление DGAT2 приводит к подавлению экспрессии нескольких генов, кодирующих белки, участвующие в липогенезе, включая белки, связывающие стеролрегулирующие элементы 1c (SREBP1c) и стеароил-КоA-десатуразу 1 (stearoyl CoA-desaturase 1 - SCD1) (Choi, 2007; Yu, 2005). Параллельно индуцируются окислительные пути метаболизма, о чем свидетельствует повышенная экспрессия таких генов, как карнитинпальмитоилтрансфераза 1 (carnitine palmitoyl transferase 1 - CPT1) (Choi, 2007). Чистым результатом этих изменений является снижение уровней печеночных DAG и TG липидов, что, в свою очередь, приводит к улучшению чуствительности печени к инсулину. Кроме того, ингибирование DGAT2 подавляет печеночную секрецию c VLDL TG и снижение уровней циркулирующего холестерина. Наконец, уровни аполипопротеина В (apolipoprotein B - APOB) были понижены, возможно, благодаря снижению поступления TG для липидирования вновь синтезированного APOB белка (Liu, 2008; Yu, 2005). Полезное влияние ингибирования DGAT2 как на гликемический контроль, так и на профиль холестерина в плазме позволяет предположить, что эту мишень можно использовать при лечении метаболического заболевания (Choi, 2007). Кроме того, тот факт, что подавление активности DGAT2 приводит к снижению накопления липидов в печени, позволяет предположить, что ингибиторы этого фермента могут быть полезны при лечении НАСГ.
С учетом вышеизложенного, существует потребность в лекарственных средствах, например лекарственных средствах для перорального введения, содержащих комбинацию (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамида (ингибитор DGAT2) или его фармацевтически приемлемой соли и в той же или в отдельной композиции 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойной кислоты (ингибитор АККI) или ее фармацевтически приемлемой соли. Конкретные комбинации, описанные в настоящем документе, удовлетворяют существующую потребность.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к фармацевтической композиции, включающей (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамид или его фармацевтически приемлемую соль в комбинации по меньшей мере с 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойной кислотой или ее фармацевтически приемлемой солью, оба соединения - в терапевтичеси эффективных количествах, в смеси с фармацевтически приемлемым эксципиентом.
Настоящее изобретение также относится к фармацевтической композиции, включающей (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамид или его фармацевтически приемлемую соль и 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойную кислоту или ее фармацевтически приемлемую соль в смеси с фармацевтически приемлемым эксципиентом.
Настоящее изобретение относится также к способу лечения заболевания или состояния, выбранного из жировой инфильтрации печени, неалкогольной жировой болезни печени, неалкогольного стеатогепатита, неалкогольного стеатогепатита с фиброзом печени, неалкогольного стеатогепатита с циррозом печени и неалкогольного стеатогепатита с циррозом печени и с гепатоцеллюлярной карциномой или с метаболическим заболеванием, причем указанный способ включает введение пациенту, нуждающемуся в таком лечении, терапевтически эффективного количества (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамида или его фармацевтически приемлемой соли в комбинации с терапевтически эффективным количеством 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойной кислоты или ее фармацевтически приемлемой соли.
Настоящее изобретение относится также к способу снижения по меньшей мере на один пункт в балльных системах оценки тяжести неалкогольной жировой болезни печени или неалкогольного стеатогепатита, снижения уровня сывороточных маркеров активности неалкогольного стеатогепатита, снижения активности заболевания неалкогольного стеатогепатита или снижения медицинских последствий неалкогольного стеатогепатита у людей, включающему стадию введения пациенту, нуждающемуся в таком снижении, терапевтически эффективного количества (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамида или его фармацевтически приемлемой соли в комбинации по меньшей мере с терапевтически эффективным количеством 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойной кислоты или ее фармацевтически приемлемой соли.
Настоящее изобретение относится также к способу лечения жировой инфильтрации печени, неалкогольной жировой болезни печени, неалкогольного стеатогепатита, неалкогольного стеатогепатита с фиброзом печени, неалкогольного стеатогепатита с циррозом печени или неалкогольного стеатогепатита с циррозом печени и с гепатоцеллюлярной карциномой у людей, включающему стадию введения человеку, нуждающемуся в таком лечении, терапевтически эффективного количества первой и второй композиций и, необязательно, третьей композиции, где
i. первая композиция включает (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамид или его фармацевтически приемлемую соль и фармацевтически приемлемый эксципиент;
ii. вторая композиция включает 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойную кислоту или ее фармацевтически приемлемую соль и фармацевтически приемлемый эксципиент; и
iii. третья композиция включает фармацевтическое средство, выбранное из группы, состоящей из противовоспалительного средства, противодиабетического средства, противофиброзного средства, антистеатотического средства, холестерин/липид-модулирующего средства и противодиабетического средства, и фармацевтически приемлемый эксципиент.
Настоящее изобретение относится также к способу лечения жировой инфильтрации печени, неалкогольной жировой болезни печени, неалкогольного стеатогепатита, неалкогольного стеатогепатита с фиброзом печени, неалкогольного стеатогепатита с циррозом печени или неалкогольного стеатогепатита с циррозом печени и с гепатоцеллюлярной карциномой у людей, включающему стадию введения человеку, нуждающемуся в таком лечении, терапевтически эффективного количества двух отдельных фармацевтических композиций, в том числе
i. первой композиции, которая включает (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамид или его фармацевтически приемлемую соль в терапевтически эффективном количестве в смеси с фармацевтически приемлемым эксципиентом;
ii. второй композиции, которая включает 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойную кислоту или ее фармацевтически приемлемую соль в терапевтически эффективном количестве в смеси с фармацевтически приемлемым эксципиентом; и, необязательно,
iii. третьей композиции, включающей по меньшей мере одно дополнительное фармацевтическое средство, выбранное из группы, состоящей из агониста GLP-1R, ингибитора KHK или агониста FXR, и фармацевтически приемлемый эксципиент.
Изобретение также относится к способу лечения сердечной недостаточности, застойной сердечной недостаточности, ишемической болезни сердца, заболевания периферических сосудов, реноваскулярного заболевания, легочной гипертензии, васкулита, острых коронарных синдромов и модификации сердечно-сосудистого риска, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамида или его фармацевтически приемлемой соли в комбинации по меньшей мере с терапевтически эффективным количеством 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойной кислоты или ее фармацевтически приемлемой соли.
Изобретение также относится к способу лечения ожирения, диабета I типа, сахарного диабета II типа, идиопатического диабета I типа (типа Ib), латентного автоиммунного сахарного диабета взрослых (latent autoimmune diabetes in adults - LADA), юношеского диабета (early-onset diabetes - EOD) 2 типа, юношеского атипичного диабета (youth-onset atypical diabetes - YOAD), юношеского диабета взрослого типа (maturity onset diabetes of the young - MODY), диабета, связанного с недостаточностью питания, гестационного диабета, ишемической болезни сердца, ишемического инсульта, рестеноза после ангиопластики, заболевания периферических кровеносных сосудов, перемежающейся хромоты, инфаркта миокарда, дислипидемии, постпрандиальной липемии, состояний нарушенной толерантности к глюкозе (impaired glucose tolerance - IGT), состояний нарушения уровня глюкозы в плазме крови натощак, метаболического ацидоза, кетоза, артрита, диабетической ретинопатии, дегенерации желтого пятна, катаракты, диабетической нефропатии, гломерулосклероза, хронической почечной недостаточности, диабетической нейропатии, метаболического синдрома, синдрома X, гипергликемии, гиперинсулинемии, гипертриглицеридемии, инсулинорезистентности, нарушения метаболизма глюкозы, нарушений кожи и соединительной ткани, изъязвлений стопы и язвенного колита, эндотелиальной дисфункции и нарушения эластичности сосудов, гипераполипопротеинемии В и болезни мочи с запахом кленового сиропа, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамида или его фармацевтически приемлемой соли в комбинации по меньшей мере с терапевтически эффективным количеством 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойной кислоты или ее фармацевтически приемлемой соли.
Изобретение также относится к способу лечения гепатоцеллюлярной карциномы, светлоклеточного рака почки плоскоклеточного рака органов головы и шеи, колоректальной аденокарциномы, мезотелиомы, аденокарциномы желудка, адренокортикальной карциномы, папиллярно-клеточного рака почки, карциномы шейки матки и эндоцервикальной карциномы, уротелиальной карциномы мочевого пузыря, аденокарциномы легкого, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамида или его фармацевтически приемлемой соли в комбинации по меньшей мере с терапевтически эффективным количеством 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойной кислоты или ее фармацевтически приемлемой соли.
Следует иметь в виду, что приведенное выше общее описание и представленное далее подробное описание являются только иллюстративными и пояснительными и не ограничивают заявленное изобретение.
КРАТКОЕ ОПИСАНИЕ ФИГУР
Фигура 1 представляет собой характеристическую порошковую рентгеновскую дифрактограмму кристаллической формы 1 соединения DGAT2i примера 1 (вертикальная ось: интенсивность (число импульсов в секунду); горизонтальная ось: два тета (градусы)).
Фигура 2 представляет собой характеристическую порошковую рентгеновскую дифрактограмму кристаллической формы 2 соединения DGAT2i примера 1 (вертикальная ось: интенсивность (число импульсов в секунду); горизонтальная ось: два тета (градусы)).
Фигура 3 представляет собой иллюстративную порошковую рентгеновскую дифрактограмму (ПРДГ) формы 1 соединения A, полученную на дифрактометре Bruker AXS D4 Endeavor, снабженном Cu источником излучения.
Фигура 4 представляет собой иллюстративный рамановский спектр (спектр комбинационного рассеяния) формы 1 соединения A, полученный с использованием вспомогательного устройства Nicolet NXR FT-Raman, присоединенного к стенду FT-IR.
Фигура 5 представляет собой иллюстративный 13C ЯМР спектр твердого тела (13C ssЯМР) формы 1 соединения A, полученный с использованием зонда Bruker-BioSpin CPMAS, помещенного в ЯМР спектрометре Bruker-BioSpin Avance III 500 МHz (1H частота).
Фигура 6 представляет собой иллюстративную ПРДГ формы 2 соединения A, полученную на дифрактометре Bruker AXS D4 Endeavor, снабженном медным источником излучения.
Фигура 7 представляет собой иллюстративный рамановский спектр формы 2 соединения A, полученный с использованием вспомогательного устройства Nicolet NXR FT-Raman, присоединенного к стенду FT-IR.
Фигура 8 представляет собой иллюстративный 13C ssЯМР спектр формы 2 соединения A, полученный на зонде Bruker-BioSpin CPMAS, расположенном в ЯМР спектрометре Bruker-BioSpin Avance III 500 МHz (1H частота).
На фигуре 9 представлена иллюстративная монокристаллическая структура формы 2 соединения A.
На фигуре 10 показано влияние перорального введения соединения А и соединения D отдельно в качестве монотерапии и в комбинации на уровни содержания триглицеридов в плазме крови в состоянии «после еды» у крыс линии Sprague Dawley, получавших питание в соответствии с западной диетой.
На фигуре 11 показано влияние перорального введения соединения А и соединения D отдельно в качестве монотерапии и в комбинации на уровни содержания триглицеридов в плазме крови в состоянии «натощак» у крыс линии Sprague Dawley, получавших питание в соответствии с западной диетой.
На фигуре 12 показано влияние введения соединения А и соединения D отдельно в качестве монотерапии и в комбинации на ядерную локализацию SREBP-1 у крыс, получавших питание в соответствии с западной диетой.
На фигуре 13 показано влияние введения соединения А и соединения D отдельно в качестве монотерапии и в комбинации на экспрессию липогенных генов, в частности ацетил-КоА-карбоксилазы (АКК1), в печени крыс, получавших питание в соответствии с западной диетой.
На фигуре 14 показано влияние введения соединения А и соединения D отдельно в качестве монотерапии и в комбинации на экспрессию липогенных генов, в частности синтазы жирных кислот (fatty acid synthase - FASN), в печени крыс, получавших питание в соответствии с западной диетой.
На фигуре 15 показано влияние введения соединения А и соединения D отдельно в качестве монотерапии и в комбинации на экспрессию липогенных генов, в частности стеароил-КoA-десатуразы (SCD1), в печени крыс, получавших питание в соответствии с западной диетой.
На фигуре 16 показано влияние введения соединения А и соединения D отдельно в качестве монотерапии и в комбинации на экспрессию липогенных генов, в частности белка 1с, связывающего стеролрегулирующие элементы (SREBP-1c), в печени крыс, получавших питание в соответствии с западной диетой.
На фигуре 17 показано влияние введения соединения А и соединения D отдельно в качестве монотерапии и в комбинации на экспрессию липогенных генов, в частности пропротеинконвертазы 9-го субтилизин/кексинового типа (proprotein convertase subtilisin/kexin type 9 - PCSK9), в печени крыс, получавших питание в соответствии с западной диетой.
На фигуре 18 показано влияние перорального введения соединения А и соединения D отдельно в качестве монотерапии и в комбинации на уровни содержания триглицеридов в печени крыс линии Sprague Dawley, получавших питание в соответствии с западной диетой.
На фигуре 19 показано влияние перорального введения соединения А и соединения D отдельно в качестве монотерапии и в комбинации на эластичность печени, маркер воспаления и фиброза печени, самцов крыс линии Wistar-Hann, получавших питание в соответствии с диетой с низким содержанием холина и высоким содержанием жиров (choline deficient and high fat diet - CDAHFD).
На фигуре 20 показано влияние перорального введения соединения А и соединения D отдельно в качестве монотерапии и в комбинации на иммуногистохимию альфа-гладкомышечного актина (alpha smooth actin - αSMA), маркера активации миофибробластов и фиброгенеза, в печени самцов крыс линии Wistar Hann, получавших питание в соответствии с CDAHFD.
На фигуре 21 показано влияние перорального введения соединения А и соединения D отдельно в качестве монотерапии и в комбинации на окрашивание пикросириусом красным (Picrosirius red) печени самцов крыс линии Wistar Hann, получавших питание в соответствии с CDAHFD.
На фигуре 22 показано влияние перорального введения соединения А и соединения D отдельно в качестве монотерапии и в комбинации на генную экспрессию альфа-гладкомышечного актина (αSMA) в печени самцов крыс линии Wistar Hann, получавших питание в соответствии с CDAHFD.
На фигуре 23 показано влияние перорального введения соединения А и соединения D отдельно в качестве монотерапии и в комбинации на генную экспрессию коллагена 1A1 в печени самцов крыс линии Wistar Hann, получавших питани в соответствии с CDAHFD.
На фигуре 24 показано влияние перорального введения соединения А и соединения D отдельно в качестве монотерапии и в комбинации на окрашивание связывающей ионизированный кальций адаптерной молекулы 1 у самцов крыс линии Wistar Hann, получавших питание в соответствии с СDAHFD.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Изобретение можно легче понять, обратившись к представленному далее подробному описанию типичных примеров вариантов обуществления изобретения и включенных в него примеров.
Следует иметь в виду, что настоящее изобретение не ограничивается конкретными способами синтеза, которые, разумеется, могут изменяться. Следует также иметь в виду, что изпользуемая здесь терминология предназначена только для описания конкретных вариантов осуществления, но не для их ограничения. В данном описании и в прилагаемой формуле изобретения будет упоминаться ряд терминов, значение которых определено далее.
Использование в настоящем описании единственного числа означает и множественное число. Термин «другой», когда используется в настоящем описании, может означать по меньшей мере второй или больший по счету.
Термин «примерно» является относительным термином, обозначающим приближение в интервале плюс или минус 10% от номинального значения, который означает в одном варианте осуществления до плюс или минус 5%, в другом варианте осуществления - до плюс или минус 2%. Для области настоящего изобретения этот уровень аппроксимации является подходящим, если конкретно не указано, что величине требуется более узкий диапазон значений.
Термин «соединения», когда используется в настоящем описании, включает любое фармацевтически приемлемое производное или вариант, включая конформационные изомеры (например, цис- и транс-изомеры) и все оптические изомеры (например, энантиомеры и диастереомеры), рацемические, диастереомерные и другие смеси таких изомеров, а также сольваты, гидраты, изоморфы, полиморфы, таутомеры, сложные эфиры, солевые формы и пролекарства. Термин «пролекарство» относится к соединениям, которые являются предшественниками лекарственных средств и после введения высвобождают лекарственное средство in vivo в результате некоторого химического или физиологического процесса (например, пролекарство при обеспечении физиологического значения pH или под действие ферментов превращается в желаемую лекарственную форму). Типичные примеры пролекарств при расщеплении высвобождают соответствующую свободную кислоту, и такие гидролизуемые остатки, образующие сложные эфиры соединений по настоящему изобретению, включают, но не ограничиваются только ими, карбоксильный фрагмент, в котором свободный водород замещен (C1-C4)алкилом, (C2-C7)алканоилоксиметил, 1-(алканоилокси)этил, содержащий от 4 до 9 атомов углерода, 1-метил-1-(алканоилокси)этил, содержащий от 5 до 10 атомов углерода, алкоксикарбонилоксиметил, содержащий от 3 до 6 атомов углерода, 1-(алкоксикарбонилокси)этил, содержащий от 4 до 7 атомов углерода, 1-метил-1-(алкоксикарбонилокси)этил, содержащий от 5 до 8 атомов углерода, N-(алкоксикарбонил)аминометил, содержащий от 3 до 9 атомов углерода, 1-(N-(алкоксикарбонил(амино)этил, содержащий от 4 до 10 атомов углерода, 3-фталидил, 4-кротонолактонил, гамма-бутиролактон-4-ил, ди-N, N-(C1-C2)алкиламино(C2-C3)алкил (такой как β-диметиламиноэтил), карбамоил-(C1-C2)алкил, N, N-ди(C1-C2)алкилкарбамоил-(C1-C2)алкил и пиперидино-, пирролидино- или морфолино(C2-C3)алкил.
Стрелка-указатель « » или волнистая линия «
» или волнистая линия « », когда используются в настоящем описании, означают точку присоединения заместителя к другой группе.
», когда используются в настоящем описании, означают точку присоединения заместителя к другой группе.
Термин «пациент» относится к теплокровным животным, таким как, например, морские свинки, мыши, крысы, песчанки, кошки, кролики, собаки, крупный рогатый скот, козы, овцы, лошади, обезьяны, шимпанзе и люди. Термин «млекопитающее» означает «пациент».
Термин «фармацевтически приемлемый» означает, что вещество или композиция должны быть химически и/или токскологически совместимыми с другими ингредиентами, входящими в препарат и/или с млекопитающим, которое подлежит лечению ими.
Термины, относящиеся к введению фармацевтических средств, когда используются в настоящем описании, имеют следующие значения: «QD» означает один раз в день, «BID» означает два раза в день.
Выражения «инертный по отношению к реакции растворитель» и «инертный растворитель», когда используются в настоящем описании, относятся к растворителю или смеси растворителей, которые не взаимодействую с исходными веществами, реагентами, промежуточными соединениями или продуктами реакции и не оказывают неблагоприятного влияния на выход целевого продукта.
Термин «селективность» или «селективный», когда используется в настоящем описании, относится к большему эффекту соединения в первом анализе по сравнению с эффектом этого же соединения во втором анализе. Например, для соединений, «селективных в кишечнике», первый анализ проводится для периода полураспада соединения в кишечнике, а второй анализ - для периода полураспада соединения в печени.
Термин «терапевтически эффективное количество» означает количество 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойной кислоты (соединение A) в комбинации с количеством (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамида (соединение D) и, необязательно, в комбинации с количеством другого(их) соединения(ий), которое лечит конкретное заболевание, состояние или расстройство, описанное в настоящем изобретении.
4-(4-(1-Изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойная кислота является селективным ингибитором АКК и получена в форме свободной кислоты в примере 9 Патента США № 8859577, который представляет собой Национальную публикацию США Международной Заявки на патент № PCT/IB2011/054119, и содержание обоих указанных документов во всей полноте введено в настоящую заявку в виде ссылок для всех целей. Кристаллические формы соединения описаны в Международной Заявке на патент № PCT/IB2018/058966, опубликованной как WO 2019/102311 31 мая 2019 года.
(S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамид является ингибитором DGAT2 и описано в качестве соединения примера 1 в Публикации Заявки на патент США № 2018-0051012A1, содержание которой введено в настоящий документ в виде ссылки для всех целей.
Термин «лечащий», «лечить» или «лечение, когда используется в настоящем описании, означает превентивное, то есть профилактическое, лечение, паллиативное лечение, то есть ослабление, облегчение или замедление развития у пациента заболевания (или состояния) или любого повреждения тканей, связанного с заболеванием (или состоянием); и обращение развития, когда не только заболевание облегчается заболевание (или состояние) пациента, но и улучшается состояние тканей, поврежденных в связи с заболеванием (или состоянием) пациента, по сравнению с их состоянием в начале лечения. Это последнее может иметь место, когда наблюдается, но без ограничения, один или нескольких из следующих факторов: демонстрация устранения НАСГ и/или улучшения оценки фиброза, исходя из биопсии печени; снижение развития болезни до цирроза, гепатоцеллюлярной карциномы и/или других конечных результатов, связанных с печенью; снижение или улучшения показателя уровня сывороточных или визуализирующих маркеров активности неалкогольного стеатогепатита; снижение или улучшение активности неалкогольного стеатогепатита; или уменьшение медицинских последствий неалкогольного стеатогепатита.
Оказалось, что введение ингибитора АКК может оказывать воздействие, полезное для снижения содержания TG в печени и, возможно, другие полезные эффекты при лечении НАСГ. Сообщалось, что повышение уровней циркулирующих TG является механистическим следствием инигибрования АКК в печени (Kim et al., 2017), хотя дозы ингибиторов АКК, которые только частично ингибируют DNL, могут не вызывать повышения уровней циркулирующих TG (Bergman et al., (2018) J. of Hepatology, Volume 68, S582). В WO2016/112305 описываются способы лечения, стабилизации или снижения тяжести или развития неалкогольной жировой болезни печени с использованием одного ингибитора АКК или с одним или несколькими дополнительными терапевтическими средствами.
Было установлено, что введение 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойной кислоты, необязательно в форме фармацевтически приемлемой соли, может приводить к повышению уровней TG в крови (обычно измеряемых в плазме) крыс линии Sprague Dawley, получавших питание в соответствии с западной диетой, как наблюдалось у пациентов-людей.
Соединения по настоящему изобретению могут содержать асимметричные или хиральные центры и, следовательно, существовать в различных стереоизомерных формах. Если не указано иное, подразумевается, что все стереоизомерные формы соединений по настоящему изобретению, а также их смеси, включая рацемические смеси, составляют часть изобретения. Кроме того, изобретение охватывает все геометрические и позиционные изомеры. Например, если соединение по настоящему изобретению включает двойную связь или конденсированный цикл, цис- и трансформы, а также их смеси включены в область настоящего изобретения.
Хиральные соединения по настоящему изобретению (и их хиральные предшественники) могут быть получены в энантиомерно-обогащенной форме с использованием хроматографии, обычно жидкостной хроматографии высокого давления (ЖХВД) или сверхкритической жидкостной хроматографии (СЖХ), на смоле с асимметричной стациональной фазой и с подвижной фазой, содержащей углеводород, обычно гептан или гексан, содержащий от 0 до 50% изопропанола, обычно от 2 до 20%, и от 0 до 5% алкиламина, обычно 0,1% диэтиламина (DEA) или изопропиламина. Энантиомерно-обогащенную смесь получают концентрированием элюента.
Смеси диастереомеров могут быть разделены на отдельные диастереоизомеры с использованием различий их физико-химических свойств методами, хорошо известными специалистам в данной области, такими как хроматография и/или фракционная кристаллизация. Энантиомеры могут быть разделены превращением энантиомерной смеси в диастереомерную смесь взаимодействием с подходящим оптически активным соединением (например, хиральным вспомогательным веществом, таким как хиральный спирт или хлорангидрид кислоты Мошера), разделением диастереоизомеров и превращением (например, гидролизом) разделенных диастереоизомеров в соответствующие чистые энантиомеры. Энантиомеры также могут быть разделены с помощью хиральной колонки для ВЭЖХ. Альтернативно, конкретные стереоизомеры могут быть синтезированы с использованием оптически активного исходного материала, асимметрическим синтезом с использованием оптически активных реагентов, субстратов, катализаторов или растворителей или превращением одного стереоизомера в другой с помощью асимметричного преобразования.
Когда соединения по настоящему изобретению содержат два или более стереогенных центров и абсолютная или относительная стереохимия указана в названии, обозначения R и S относятся, соответственно, к каждому стереогенному центру в порядке возрастания номеров (1, 2, 3 и т.д.) в соответствии с общепринятыми схемами нумерации IUPAC для каждой молекулы. Когда соединения по настоящему изобретению содержат один или несколько стереогенных центров и в названии или структуре не указана стереохимия, то подразумевается, что название или структура охватывает все формы соединения, включая рацемическую форму.
Возможно также, что промежуточные соединения и соединения по настоящему изобретению могут существовать в различных таутомерных формах, тогда все такие формы включены в область настоящего изобретения. Термин «таутомер» или «таутомерная форма» относится к структурным изомерам с различными энергиями и низких энергетическим барьером их взаимного превращения. Например, взаимные превращения протонных таутомеров (известных также как прототропные таутомеры) включают взаимные превращения через миграцию протона, такие как кето-енольная и имин-енаминная изомеризации.
Взаимные превращения таутомеров валентности включают взаимные превращения посредством перераспределения некоторых из связывающих электронов.
В объем заявленных соединений по настоящему изобретению включены все стереоизомеры, геометрические изомеры и таутомерные формы соединений по настоящему изобретению, включая соединения, проявляющие изомерию нескольких типов, и смеси изомеров одного или нескольких типов. В объем настоящего изобретения включены также кислотно-аддитивные или основные соли, в которых противоион является оптически активным, например D-лактат или L-лизин или их рацетамы, например DL-тартрат или DL-аргинин.
Изобретение включает все фармацевтически приемлемые изотопно-меченые соединения по настоящему изобретению, в которых один или несколько атомов замещены на атомы с таким же атомным номером, но с атомной массой или массовым числом, отличным от атомной массы или массового числа, которые обычно встречают в природе.
Примеры изотопов, подходящих для включения в соединения по настоящему изобретению, включают изотопы водорода, такие как 2H и 3H, углерода, такие как 11C, 13C и 14C, хлора, такие как 36Cl, фтора, такие как 18F, йода, такие как 123I, 124I и 125I, азота, такие как 13N и 15N, кислорода, такие как 15O, 17O и 18O, фосфора, такие как 32P, и серы, такие как 35S.
Некоторые изотопно-меченые соединения по настоящему изобретению, например соединения, содержащие радиоактивный изотоп, могут применяться в исследованиях распределения лекарственных средств и/или субстратов в тканях. Радиоактивные изотомы трития, т.е. 3H, и углерода-14, т.е. 14C, особенно полезны для этой цели ввиду простоты их введения и готовых средств обнаружения.
Замещение более тяжелыми изотопами, такими как дейтерий, т.е. 2H, может приводить к некоторым терапевтическим преимуществам, которые являются результатом большей метаболической стабильности, например увеличению периода полувыведения in vivo или снижению необходимой дозы, и, следовательно, может быть предпочтительным в некоторых случаях.
Замещение изотопами, испускающими позитроны, такими как 11C, 18F, 15O и 13N, может применяться в исследованиях позитронно-эмиссионной томографии (ПЭТ) для определения степени занятости рецепторов субстратами.
Изотопно-меченые соединения по настоящему изобретению вообще могут быть получены стандартными способами, известными специалистам в данной области техники, или способами, аналогичным описанным в прилагаемых примерах и примерах получения с использованием подходящих изотопно-меченых реагентов вместо немеченых реагентов, применяемых ранее.
Соединения по настоящему изобретению могут быть выделены и использоваться сами по себе или, когда это возможно, в форме их фармацевтически приемлемых солей. Термин «соли» относится к неорганическим и органическим солям соединения по настоящему изобретению. Эти соли могут быть получены in situ во время окончательного выделения и очистки соединения или с помощью отдельной обработки соединения подходящей органической или неорганической кислотой или органическим или неорганическим основанием с выделением образованной таким образом соли. Кислоты, которые используются для получения фармацевтически приемлемых кислотно-аддитивных солей упомянутых выше основных соединений по настоящему изобретению, представляют собой кислоты, которые образуют нетоксичные кислотно-аддитивные соли (т.е. соли, содержащие фармакологически приемлемые анионы, такие как гидрохлорид, гидробромид, гидройодид, нитрат, сульфат, бисульфат, фосфат, кислый фосфат, ацетат, лактат, цитрат, кислый цитрат, тартрат, битартрат, сукцинат, малеат, фумарат, глюконат, сахарат, бензоат, метансульфонат, этансульфонат, бензолсульфонат, нафтилат, мезилат, глюкогептонат, лактобионат, лаурилсульфонат, гексафторфосфат, бензолсульфонат, тозилат, формиат, трифторацетат, оксалат, безилат, пальмитат, памоат, малонат, стеарат, лаурат, малат, борат, п-толуолсульфонат и памоат (т.е. 1,1'-метилен-бис-(2-гидрокси-3-нафтоат)).
Изобретение также относится к основно-аддитивным солям соединений по настоящему изобретению. Химические основания, которые могут использоваться в качестве реагентов для получения фармацевтически приемлемых основных солей соединений по настоящему изобретению, кислотных по природе, представляют собой основания, которые образую нетоксичные основные соли с такими соединениями. Такие нетоксичные основные соли включают, но не ограничиваются ими, соли, полученные из фармакологически приемлемых катионов, таких как катионы щелочных металлов (например, лития, калия и натрия) и катионы щелочно-земельных металлов (например, кальция и магния), соли аммония или растворимые в воде амино-аддитивные соли, такие как N-метилглюкамин (меглюмин), тетраметиламмоний, тетраэтиламмоний, метиламин, диметиламин, триметиламин, триэтиламин, этиламин и низший алканоламмоний и другие основные соли фармакологически приемлемых органических аминов (см., например, Berge, et al. J. Pharm. Sci. 66, 1-19 (1977).
Некоторые соединения по настоящему изобретению могут существовать в нескольких кристаллических формах (обычно называемых «полиморфы»). Полиморфы могут быть получены кристаллизацией в различных условиях, например с использованием различных растворителей или различных смесей растворителей перекристаллизации; кристаллизацией при различных температурах; и/или при различных способах охлаждения - от очень быстрого до очень медленного охлаждения в процессе кристаллизации. Полиморфы также могут быть получены нагревом или плавлением соединения по настоящему изобретению с последующим постепенным или быстрым охлаждением. Наличие полиморфов может быть определено с помощью ЯМР спектроскопии твердого тела, ИК спектроскопии, дифференциальной сканирующей каллориметрии, порошковой рентгеновской дифрации или другими такими методами.
В одном варианте осуществления настоящее изобретение относится к фармацевтической композиции, включающей (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамид или его фармацевтически приемлемую соль в комбинации по меньшей мере с 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойной кислотой или ее фармацевтически приемлемой солью, каждое из указанных соединений - в терапевтически эффективном количестве, в смеси с фармацевтически приемлемым эксципиентом.
В другом варианте осуществления настоящего изобретения, композиция дополнительно включает по меньшей мере одно дополнительное фармацевтическое средство, выбранное из группы, состоящей из противовоспалительного средства, противодиабетического средства и холестерин/липид-модулирующего средства.
В другом варианте осуществления настоящего изобретения композиция включает также по меньшей мере одно дополнительное фармацевтическое средство, выбранное из группы, состоящей из [(1R,5S,6R)-3-{2-[(2S)-2-метилазетидин-1-ил]-6-(трифторметил)пиримидин-4-ил}-3-азабицикло[3.1.0]гекс-6-ил]уксусной кислоты; 2-[(1R,3R,5S)-3-({5-циклопропил-3-[2-(трифторметокси)фенил]-1,2-оксазол-4-ил}метокси)-8-азабицикло[3.2.1]октан-8-ил]-4-фтор-1,3-бензотиазол-6-карбоновой кислоты; или 2-[(4-{6-[(4-циано-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновой кислоты, или его фармацевтически приемлемую соль.
В другом варианте осуществления настоящего изобретения фармацевтическая композиция содержит (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамид в форме твердого кристаллического вещества структуры:
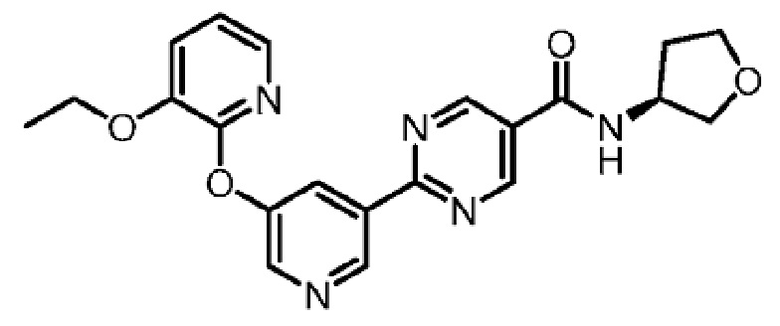
или его фармацевтически приемлемую соль.
В другом варианте осуществления настоящего изобретения порошковая рентгеновская дифрактограмма указанного твердого кристаллического вещества содержит пики при значениях угла 2-тета (CuKα излучение, длина волны 1,54056 Å) 5,3 ± 0,2, 7,7 ± 0,2 и 15,4 ± 0,2.
В другом варианте осуществления настоящего изобретения порошковая рентгеновская дифрактограмма указанного твердого кристаллического вещества содержит пики при значениях угла 2-тета (CuKα излучение, длина волны 1,54056Å) 6,5 ± 0,2, 9,3 ± 0,2 и 13,6 ± 0,2.
В еще одном варианте осуществления настоящего изобретения фармацевтическая композиция содержит 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойную кислоту или ее фармацевтически приемлемую соль в форме твердого кристаллического вещества структуры:
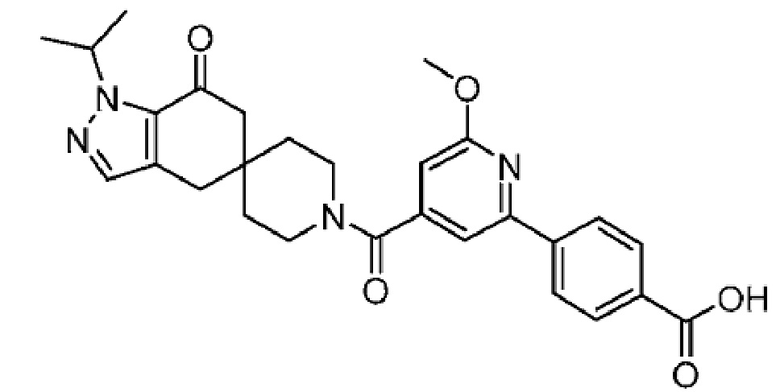
или ее фармацевтически приемлемую соль.
В еще одном варианте осуществления настоящего изобретения твердое кристаллическое вещество представляет собой 2-амино-2-(гидроксиметил)пропан-1,3-диольную соль 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойной кислоты.
В еще одном варианте осуществления настоящего изобретения фармацевтическая композиция включает также по меньшей мере одно дополнительное фармацевтическое средство, выбранное из группы, состоящей из [(1R,5S,6R)-3-{2-[(2S)-2-метилазетидин-1-ил]-6-(трифторметил)пиримидин-4-ил}-3-азабицикло[3.1.0]гекс-6-ил]уксусной кислоты; 2-[(1R,3R,5S)-3-({5-циклопропил-3-[2-(трифторметокси)фенил]-1,2-оксазол-4-ил}метокси)-8-азабицикло[3.2.1]октан-8-ил]-4-фтор-1,3-бензотиазол-6-карбоновой кислоты; или 2-[(4-{6-[(4-циано-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновой кислоты, или его фармацевтически приемлемую соль.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения заболевания или состояния, выбранного из жировой инфильтрации печени, неалкогольной жировой болезни печени, неалкогольного стеатогепатита, неалкогольного стеатогепатита с фиброзом печени, неалкогольного стеатогепатита с циррозом печени и неалкогольного стеатогепатита с циррозом печени и с гепатоцеллюлярной карциномой или с метаболическим заболеванием, причем указанный способ включает введение пациенту, нуждающемуся в таком лечении, терапевтически эффективного количества (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамида или его фармацевтически приемлемой соли в комбинации по меньшей мере с терапевтически эффективным количеством 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойной кислоты или ее фармацевтически приемлемой соли.
В еще одном варианте осуществления настоящего изобретения заболевание или состояние представляет собой жировую инфильтрацию печени. В другом варианте осуществления настоящего изобретения заболевание или состояние представляет собой неалкогольную жировую болезнь печени. В еще одном варианте осуществления настоящего изобретения заболевание или состояние представляет собой неалкогольный стеатогепатит. В еще одном варианте осуществления настоящего изобретения заболевание или состояние представляет собой неалкогольный стеатогепатит с фиброзом печени. В еще одном варианте осуществления настоящего изобретения заболевание или состояние представляет собой неалкогольный стеатогепатит с циррозом печени. В еще одном варианте осуществления настоящего изобретения заболевание или состояние представляет собой неалкогольный стеатогепатит с циррозом печени и с гепатоцеллюлярной карциномой. В еще одном варианте осуществления настоящего изобретения заболевание или состояние представляет собой неалкогольный стеатогепатит с циррозом печени и с метаболическим заболеванием.
В еще одном варианте осуществления настоящего изобретения способ включает по меньшей мере одно другое фармацевтическое средство, где указанное фармацевтическое средство выбрано из группы, состоящей из ингибитора ацетил-КоА-карбоксилазы (АКК), ингибитора диацилглицерол-О-ацилтрансферазы 1 (DGAT-1), ингибиторов моноацилглицерол-О-ацилтрансферазы, ингибитора фосфодиэстеразы-10 (phosphodiesterase 10 - PDE-10), активатора AMPK, сульфонилмочевины, меглитинида, ингибитора α-амилазы, ингибитора α-глюкозидгидролазы, ингибитора α-глюкозидазы, агониста PPARγ, агониста PPAR α/γ, бигуанида, модулятора глюкагоноподобного пептида 1 (glucagon-like peptide 1 - GLP-1), лираглутида, албиглутида, эксенатида, албиглутида, ликсисенатида, дулаглутида, семаглутида, ингибитора протеинтирозинфосфатазы-1В (protein tyrosine phosphatase-1B - PTP-1B), активатора SIRT-1, ингибитора дипептидилпептидазы IV (DPP-IV), стимулятора секреции инсулина, ингибитора окисления жирных кислот, A2 антагониста, ингибитора с-Jun N-терминальной киназы (JNK), активаторов глюкокиназы (GKa), инсулина, инсулиноподобного средства, ингибитора гликогенфосфорилазы, агониста рецептора VPAC2t, ингибиторов SGLT2, модулятора рецептора глюкагона, модуляторы GPR119, производных и аналогов FGF21, модуляторов рецептора TGR5, модуляторов рецептора GPBAR1, агонистов GPR40, модуляторов GPR120, активаторов рецепторов никотиновой кислоты высокого сродства (HM74A), ингибиторов SGLT1, ингибиторов или модуляторов ферментов карнитинпальмитоилтрансферазы, ингибиторов фруктозо-1,6-дифосфатазы, ингибиторов альдозоредуктазы, ингибиторов минералокортикоидного рецептора, ингибиторов TORC2, ингибиторов CCR2 и/или CCR5, ингибиторов изоформ PKC (например, PKCα, PKCβ, PKCγ), ингибиторов синтетазы жирных кислот, ингибиторов серинпальмитоилтрансферазы, модуляторов GPR81, GPR39, GPR43, GPR41, GPR105, Kv1.3, ретинолсвязывающего белка-4, глюкокортикоидного рецептора, рецепторов соматостатина, ингибиторов или модуляторов PDHK2 или PDHK4, ингибиторов MAP4K4, модуляторов семейства IL1, включая IL1beta, ингибиторов гидроксиметилглутарил-кофермент А-редуктазы (ГМГ-КoA редуктазы), ингибиторов скваленсинтетазы, фибратов, секвестрантов желчных кислот, ингибиторов ACAT, ингибиторов MTP, ингибиторов липооксигеназы, ингибиторов абсорбции холестерина, модуляторов PCSK9, ингибиторов транспортного белка холестериновых эфиров и модуляторов RXRalpha.
В еще одном варианте осуществления настоящего изобретения способ включает по меньшей мере одно другое фармацевтическое средство, выбранное из группы, состоящей из цистеамина или его фармацевтически приемлемой соли, цистамина или его фармацевтически приемлемой соли, антиоксидантного соединения, лецитина, комплекса витамина В, препаратов желчных солей, антагонистов рецептора каннабиноидов 1 (Cannabinoid-1 - CB1), обратных антагонистов рецептора каннабиноидов 1 (CB1), регуляторов активности рецепторов, активируемых пролифератором пероксисом, производного бензотиазепина или бензотиепина, конструкта антисмысловой РНК для ингибирования протеинтирозинфосфатазы PTPRU, пипередина, связанного через гетероатом, и его производных, производного азациклопентана, способного ингибировать стеароил-коэнзим-альфа-дельта-9-десатуразу, ациламидного соединения, обладающего активностью стимулятора секреции или индуктора активности адипонектина, четвертичного аммониевого соединения, ацетата глатирамера, пентатраксиновых белков, ингибитора ГМГ-КoA редуктазы, н-ацетилцистеина, соединения изофлавона, макролидного антибиотика, ингибитора галектина, антитела или любой их комбинации.
В еще одном варианте осуществления настоящего изобретения способ включает по меньшей одно другое фармацевтическое средство, где указанное фармацевтическое средство выбрано из группы, состоящей из [(1R,5S,6R)-3-{2-[(2S)-2-метилазетидин-1-ил]-6-(трифторметил)пиримидин-4-ил}-3-азабицикло[3.1.0]гекс-6-ил]уксусной кислоты; 2-[(1R,3R,5S)-3-({5-циклопропил-3-[2-(трифторметокси)фенил]-1,2-оксазол-4-ил}метокси)-8-азабицикло[3.2.1]октан-8-ил]-4-фтор-1,3-бензотиазол-6-карбоновой кислоты; или 2-[(4-{6-[(4-циано-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновой кислоты, или его фармацевтически приемлемую соль.
В другом варианте осуществления настоящее изобретение относится также к способу снижения по меньшей мере на один пункт в балльных системах оценки тяжести неалкогольной жировой болезни печени или неалкогольного стеатогепатита, снижения уровня сывороточных маркеров активности неалкогольного стеатогепатита, снижения активности заболевания неалкогольного стеатогепатита или снижения медицинских последствий неалкогольного стеатогепатита у людей, включающему стадию введения пациенту, нуждающемуся в таком снижении, терапевтически эффективного количества (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамида или его фармацевтически приемлемой соли в комбинации по меньшей мере с терапевтически эффективным количеством 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойной кислоты или ее фармацевтически приемлемой соли.
В еще одном варианте осуществления настоящего изобретения способ включает по меньшей мере одно другое фармацевтическое средство, где указанное фармацевтическое средство выбрано из группы, состоящей из [(1R,5S,6R)-3-{2-[(2S)-2-метилазетидин-1-ил]-6-(трифторметил)пиримидин-4-ил}-3-азабицикло[3.1.0]гекс-6-ил]уксусной кислоты; 2-[(1R,3R,5S)-3-({5-циклопропил-3-[2-(трифторметокси)фенил]-1,2-оксазол-4-ил}метокси)-8-азабицикло[3.2.1]октан-8-ил]-4-фтор-1,3-бензотиазол-6-карбоновой кислоты; или 2-[(4-{6-[(4-циано-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновой кислоты, или его фармацевтически приемлемую соль.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения жировой инфильтрации печени, неалкогольной жировой болезни печени, неалкогольного стеатогепатита, неалкогольного стеатогепатита с фиброзом печени, неалкогольного стеатогепатита с циррозом печени или неалкогольного стеатогепатита с циррозом печени и с гепатоцеллюлярной карциномой у людей, включающему стадию введения человеку, нуждающемуся в таком лечении, терапевтически эффективного количества двух отдельных фармацевтических композиций, в том числе
i. первой композиции, которая включает (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамид или его фармацевтически приемлемую соль в терапевтически эффективном количестве в смеси с фармацевтически приемлемым эксципиентом;
ii. второй композиции, которая включает 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойную кислоту или ее фармацевтически приемлемую соль в терапевтически эффективном количестве в смеси с фармацевтически приемлемым эксципиентом; и необязательно,
iii. третьей композиции, включающей по меньшей мере одно фармацевтическое средство, выбранное из группы, состоящей из противовоспалительного средства, противодиабетического средства, противофиброзного средства, антистеатотического средства, холестерин/липид-модулирующего средства и противодиабетического средства, и фармацевтически приемлемый эксципиент.
В еще одном варианте осуществления настоящего изобретения указанная первая композиция и указанная вторая композиция вводятся одновременно. В другом варианте осуществления настоящего изобретения композиция включает первую композицию, вторую композицию и третью композицию.
В еще одном варианте осуществления настоящего изобретения способ включает третью композицию, в которой по меньшей мере одно другое фармацевтическое средство выбрано из группы, состоящей из [(1R,5S,6R)-3-{2-[(2S)-2-метилазетидин-1-ил]-6-(трифторметил)пиримидин-4-ил}-3-азабицикло[3.1.0]гекс-6-ил]уксусной кислоты; 2-[(1R,3R,5S)-3-({5-циклопропил-3-[2-(трифторметокси)фенил]-1,2-оксазол-4-ил}метокси)-8-азабицикло[3.2.1]октан-8-ил]-4-фтор-1,3-бензотиазол-6-карбоновой кислоты; или 2-[(4-{6-[(4-циано-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновой кислоты или их фармацевтически приемлемых солей.
В еще одном варианте осуществления настоящего изобретения (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамид или его фармацевтически приемлемая соль в комбинации с 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойной кислотой или ее фармацевтически приемлемой солью присутствует в фармацевтической композиции в терапевтически эффективном количестве в смеси с фармацевтически приемлемым эксципиентом.
В еще одном варианте осуществления настоящего изобретения (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамид или его фармацевтически приемлемая соль в комбинации по меньшей мере с 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойной кислотой или ее фармацевтически приемлемой солью присутствуют в фармацевтической композиции в терапевтически эффективном количестве в смеси с фармацевтически приемлемым эксципиентом.
В еще одном варианте осуществления настоящего изобретения композиция включает также по меньшей мере одно дополнительное фармацевтическое средство, выбранное из группы, состоящей из агониста GLP-1R, ингибитора KHK, агониста FXR, противовоспалительного средства, противодиабетического средства, противофиброзного средства, антистеатотического средства и холестерин/липид-модулирующего средства.
В еще одном варианте осуществления настоящего изобретения способ лечения метаболического заболевания, состояния или расстройства или заболевания, состояния или расстройства, связанного с метаболизмом, включает стадию введения пациенту терапевтически эффективного количества (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамида или его фармацевтически приемлемой соли в комбинации с 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойной кислотой или ее фармацевтически приемлемой солью.
В еще одном варианте осуществления настоящего изобретения способ лечения состояния, выбранного из группы, состоящей из гиперлипидемии, диабета I типа, сахарного диабета II типа, идиопатического диабета I типа (типа Ib), латентного аутоиммунного диабета взрослых (LADA), юношеского диабета (EOD) 2 типа, юношеского атипичного диабета (YOAD), юношеского диабета взрослого типа (MODY), диабета, связанного с недостаточностью питания, гестационного диабета, ишемической болезни сердца, ишемического инсульта, рестеноза после ангиопластики, заболевания периферических кровеностных сосудов, меремежающейся хромоты, инфаркта миокарда (например, некроза и апоптоза), дислипидемии, постпрандиальной липемии, состояний нарушенной толерантности к глюкозе (IGT), состояний нарушения уровня глюкозы в плазме крови натощак, метаболического ацидоза, кетоза, артрита, ожирения, остеопороза, гипертензии, застойной сердечной недостаточности, гипертрофии левого желудочка, периферической артериальной болезни, диабетической ретинопатии, дегенерации желтого пятна, катаракты, диабетической нефропатии, гломерулосклероза, хронической почечной недостаточности, диабетической нейропатии, метаболического синдрома, синдрома X, предменструального синдрома, ишемической болезни сердца, стенокардии, тромбоза, атеросклероза, инфаркта миокарда, тразиторных ишемических атак, инсульта, васкулярного рестеноза, гипергликемии, гиперинсулинемии, гиперлипидемии, гипертриглицеридемии, инсулинорезистентности, нарушения метаболизма глюкозы, состояний нарушенной толерантности к глюкозе, состояний нарушения уровня глюкозы натощак, ожирения, эректильной дисфункции, нарушений кожи и соединительной ткани, изъязвлений стопы и язвенного колита, эндотелиальной дисфункции и нарушения эластичности сосудов, гипераполипопротеинемии В, болезни Альцгеймера, шизофрении, ухудшения когнитивной деятельности, воспалительного заболевания кишечника, язвенного колита, болезни Крона и синдрома раздраженного кишечника, неалкогольного стеатогепатита (НАСГ), неалкогольной жировой болезни печени (НАЖБП), включает введение терапевтически эффективного количества соединения А или его фармацевтически приемлемой соли и терапевтически эффективного количества соединения D или его фармацевтически приемлемой соли.
В еще одном варианте осуществления настоящего изобретения способ лечения метаболического заболевания, состояния или расстройства или заболевания, состояния или расстройства, связанного с метаболизмом, включает стадию введения пациенту, нуждающемуся в таком лечении, по меньшей мере двух отдельных фармацевтических композиций, в том числе
(i) первой композиции, которая включает (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамид или его фармацевтически приемлемую соль в терапевтически эффективном количестве в смеси с фармацевтически приемлемым эксципиентом;
(ii) второй композиции, которая включает 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойную кислоту или ее фармацевтически приемлемую соль в терапевтически эффективном количестве в смеси с фармацевтически приемлемым эксципиентом; и, необязательно,
(iii) третьей композиции, включающей по меньшей мере одно фармацевтическое средство, выбранное из группы, состоящей из агониста GLP-1R, ингибитора KHK, агониста FXR, противовоспалительного средства, противодиабетического средства, противофиброзного средства, антистеатотического средства, холестерин/липид-модулирующего средства и противодиабетического средства, и фармацевтически приемлемый эксципиент.
В еще одном варианте осуществления настоящего изобретения способ по настоящему изобретению осуществляется, когда указанная первая композиция, указанная вторая композиция и указанная третья композиция вводятся одновременно.
В еще одном варианте осуществления настоящего изобретения способ по настоящему изобретению осуществляется, когда указанная первая композиция, указанная вторая композиция и указанная третья композиция вводятся последовательно в любом порядке.
В одном варианте осуществления настоящего изобретения, когда вводятся три фармацевтических средства, первое фармацевтическое средство и второе фармацевтическое средство вводятся одновременно, а третье фармацевтическое средство вводится последовательно. В другом варианте осуществления настоящего изобретения три отдельных фармацевтических средства вводятся последовательно в любом порядке.
В одном варианте осуществления настоящего изобретения, когда вводятся три фармацевтических средства, третье фармацевтическое средство включает агонист GLP-1R. Агонист GLP-1R - 2-[(4-{6-[(4-циано-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновая кислота или ее фармацевтически приемлемая соль [такая как ее 2-амино-2-(гидроксиметил)пропан-1,3-диольная соль, также известная как ее трис-соль], а также другие агонисты GLP-1R и способы получения этих соединений описаны в Патенте США № 10208019, содержание которого введено в настоящее описание в виде ссылки для всех целей.
В некоторых вариантах осуществления настоящего изобретения агонист GLP-1R выбран из группы, состоящей из лираглутида, албиглутида, эксенатида, албиглутида, ликсисенатида, дулаглутида, семаглутида, HM15211, LY3298176, Medi-0382, NN-9924, TTP-054, TTP-273, эфпенгленатида, агонистов, описанных в WO2018109607, и DIAST-X2.
GLP-1 представляет собой инкретиновый гормон длиной 30 аминокислот, секретируемый L-клетками кишечника в ответ на прием пищи. Было показано, что GLP-1 стимулирует секрецию инсулина физиологическим и глюкозозависимым образом, снижает секрецию глюкагона, ингибирует опорожнение желудка, снижает аппетит и стимулирует пролиферацию бета-клеток. В неклинических экспериментах GLP-1 способствует продолжающейся компетентности бета-клеток, стимулируя транскрипцию генов, важных для глюкозозависимой секреции инсулина, и стимулируя неогенез бета-клеток (Meier, et al. Biodrugs. 2003; 17 (2): 93-102).
У здорового человека GLP-1 играет важную роль в регуляции уровней глюкозы в крови после приема пищи, стимулируя глюкозозависимую секрецию инсулина поджелудочной железой, что приводит к увеличению всасывания глюкозы на периферии. GLP-1 также подавляет секрецию глюкагона, что приводит к снижению выработки глюкозы в печени. Кроме того, GLP-1 задерживает опорожнение желудка и замедляет перистальтику тонкой кишки, задерживая всасывание пищи. У людей с СД2 нормальное повышение уровня GLP-1 после приема пищи отсутствует или снижается (Vilsboll T., et al. Diabetes. 2001. 50; 609-613).
Holst (Physiol. Rev. 2007, 87, 1409) и Meier (Nat. Rev. Endocrinol. 2012, 8, 728) сообщают, что агонисты рецептора GLP-1, такие как GLP-1, лираглутид и эксендин-4, обладают тремя основными фармакологическими активностями по улучшению гликемического контроля у пациентов, таких как пациенты с СД2, за счет снижения уровней содержания глюкозы натощак и постпрандиальной глюкозы (FPG и PPG): (i) повышенная глюкозозависимая секреция инсулина (улучшенные первая и вторая фазы), (ii) активность подавления глюкагона в состояниях гипергликемии, (iii) замедление скорости опорожнения желудка, приводящее к замедленному всасыванию глюкозы, полученной из пищи.
В другом варианте осуществления настоящего изобретения, когда вводятся три фармацевтических средства, третье средство включает ингибитор KHK.
В некоторых вариантах осуществления настоящего изобретения ингибитор KHK представляет собой [(1R,5S,6R)-3-{2-[(2S)-2-метилазетидин-1-ил]-6-(трифторметил)пиримидин-4-ил}-3-азабицикло[3.1.0]гекс-6-ил]уксусную кислоту или ее фармацевтически приемлемые соли. [(1R,5S,6R)-3-{2-[(2S)-2-метилазетидин-1-ил]-6-(трифторметил)пиримидин-4-ил}-3-азабицикло[3.1.0]гекс-6-ил]уксусная кислота (включая ее кристаллическую форму свободной кислоты) представляет собой ингибитор кетогексокиназы и описана в примере 4 Патента США № 9809579, содержание которого во всей полноте включено в настоящий документ в виде ссылки для всех целей.
В некоторых вариантах осуществления настоящего изобретения ингибитор KHK представляет собой кристаллическую форму свободной [(1R,5S,6R)-3-{2-[(2S)-2-метилазетидин-1-ил]-6-(трифторметил)пиримидин-4-ил}-3-азабицикло[3.1.0]гекс-6-ил]уксусной кислоты.
Кетогексокиназа (KHK) является ключевым ферментом в метаболизме фруктозы и катализирует превращение фруктозы во фруктозо-1-фосфат (F1P). KHK экспрессируется в виде двух альтернативных сплайс-вариантов мРНК, обозначаемых как KHKa и KHKc, образующихся в результате альтернативного сплайсинга третьего эксона. Сродство и способность к фосфорилированию фруктозы у KHKc значительно выше, чем у KHKa, о чем свидетельствует гораздо более низкий Km (Ishimoto, Lanaspa et al., PNAS 109, 4320-4325, 2012). Хотя KHKa экспрессируется повсеместно, экспрессия KHKc является наивысшей в печени, почках и кишечнике, основных участках метаболизма фруктозы в организме (Diggle C.P., et al. (2009) J. Histochem. Cytochem. 57:763-774; Ishimoto, Lanaspa, et al., PNAS 109, 4320-4325, 2012). Описана также у людей мутация фруктозы с потерей функции без каких-либо побочных эффектов, за исключением появления фруктозы в моче после приема сахара, указанная мутация названа эссенциальной фруктозурией (OMIM #229800).
Более тяжелым состоянием, связанным с метаболизмом фруктозы, является наследственная непереносимость глюкозы (Hereditary Fructose Intolerance - HFI, OMIM #229600), которая обусловлена дефектами альдолазы B (GENE: ALDOB), фермента, ответственного за расщепление F1P и расположенного непосредственно ниже стадии KHK в метаболизме (Bouteldja N., et. al, J. Inherit. Metab. Dis. 2010 Apr;33(2):105-12; Tolan, D.R., Hum. Mutat. 1995;6(3):210-8; http://www.omim.org/entry/229600). Это редкое заболевание, которым страдает примерно 1 из 20000 человек, и указанные мутации приводят к накоплению F1P, деплеции ATP и повышению уровня мочевой кислоты, что наряду с другими нарушениями метаболизма приводит к гипогликемии, гиперурикемии и лактодозу. HFI нарушает способность организма метаболизировать фруктозу, поступающую с пищей, вызывая проявление острых симптом, таких как рвота, тяжелая гипогликемия, диарея и абдоминальное расстройство, приводящих к длительным нарушениям роста, повреждению печени и почек и потенциальной смерти (Ali M. et al., J. Med. Genet. 1998 May:35(5):353-65). Пациенты обычно страдают в течение первых лет жизни до постановки диагноза, и единственным курсом лечения является исключение фруктозы из рациона питания. Лечение усложняется наличием этого макроэлемента в большинстве продуктов питания. Помимо физических симптомов многие пациенты чувствуют эмоциональную и социальную изолированность как следствие их необычной диеты и постоянно следят за соблюдением строгих диетических ограничений (HFI-INFO Discussion Board, http://hfiinfo.proboards.com. Accessed 14 December 2015). Даже когда они кажутся бессимптомными, у некоторых пациентов развиваются НАЖБП и заболевание почек, что подчеркивает неадекватность добровольного ограничения в питании как единственного варианта лечения и высокую потребность в методах медицинского лечения этого состояния.
При гипергликемических состояниях эндогенное продуцирование глюкозы происходит через полиольный путь метаболизма, посредством которого глюкоза превращается во фруктозу с сорбитом в качестве промежуточного продукта. Активность этого пути повышается при гипергликемии. В этих исследованиях авторы продемонстрировали, что KHK-нулевые мыши были защищены от индуцированного глюкозой повышения массы тела, инсулинорезистентности и стеатоза печени, и высказали предположение о том, что в состояниях гипергликемии эндогенно продуцируемая фруктоза может способствовать развитию инсулинорезистентности и стеатозу печени (Lanaspa, M.A., et al., Nature Comm. 4, 2434, 2013). Поэтому ожидается, что ингибирование KHK полезно при многих заболеваниях, в которых задействована эндогенная или поступившая с пищей фруктоза или оба эти вида фруктозы.
В другом варианте осуществления настоящего изобретения, когда вводятся три фармацевтических средства, третье фармацевтическое средство включает агонист FXR. В некоторых вариантах осуществления настоящего изобретения агонист FXR выбран из группы, состоящей из тропифексора (2-[(1R,3R,5S)-3-({5-циклопропил-3-[2-(трифторметокси)фенил]-1,2-оксазол-4-ил}метокси)-8-азабицикло[3.2.1]октан-8-ил]-4-фтор-1,3-бензотиазол-6-карбоновой кислоты) («тропифексор»); цилофексора (GS-9674); обетихолевой кислоты; LY2562175; Met409; TERN-101; и EDP-305 и их фармацевтически приемлемых солей. Агонист FXR тропифексор или его фармацевтически приемлемая соль описаны, например, в примере 1-1B Патента США № 9150568, содержание которого во всей полноте включено в настоящее описание в виде ссылки для всех целей. Химическое название тропифексора - 2-[(1R,3R,5S)-3-({5-циклопропил-3-[2-(трифторметокси)фенил]-1,2-оксазол-4-ил}метокси)-8-азабицикло[3.2.1]октан-8-ил]-4-фтор-1,3-бензотиазол-6-карбоновая кислота.
Фарнезоидный Х-рецептор (farnesoid X receptor - FXR) является представителем надсемейства ядерных гормонов и экспрессируется главным образом в печи, почках и кишечнике (см., например, Seol et al. (1995) Mol. Endocrinol. 9:72-85; и Forman et al. (1995) Cell 81:687-693). Он функционирует как гетеродимер с ретиноидным X рецептором (retinoid X receptor - RXR) и связывается с элементами отклика в промоторах генов-мишеней для регулирования генной транскрипции. Гетеродимер FXR-RXR связывается с наивысшим сродством с элементом отклика на инвертированный повтор-1 (inverted repeat-1 - IR-1), в котором консенсусные гексамеры, связывающиеся с рецептором, разделены одним нуклеотидом. FXR является частью взаимосвязанного процесса, в котором FXR активируется желчными кислотами (конечными продуктами метаболезма холестерина) (см., например, Makishima et al. (1999) Science 284: 1362-1365, Parks et al. (1999) Science 284:1365-1368, Wang et al. (1999) Mol. Cell. 3:543-553), которые служат для ингибирования катаболизма холестерина (см. также Urizar et al. (2000) J. Biol. Chem. 275:39313-39317).
FXR является ключевым регулятором гомеостаза холестерина, синтеза триглицеридов и липогенеза (Crawley, Expert Opinion Ther. Patents (2010), 20(8): 1047-1057). В дополнение к лечению дислипидемии было описано множестсво показаний для FXR, включая лечение заболеваний печени, диабета, заболеваний, связанных с витамином D, побочных эффектов, вызванных лекарственными средствами, и гепатита (Crawley, см. выше). Несмотря на успехи в разработке новых агонистов FXR, сохраняются значительные возможности для улучшения.
В некоторых других вариантах осуществления, когда вводятся три фармацевтических средства, третье фармацевтическое средство включает ингибитор SGLT2, метформин, аналоги инкретина, модулятор рецептора инкретина, ингибитор DPP-4 или агонист PPAR.
В некоторых других вариантах осуществления, когда вводятся три фармацевтических средства, третье фармацевтическое средство представляет собой противодиабетическое средство, выбранное из из метформина, ситаглиптина или эртуглифлозина.
В некоторых других вариантах осуществления, когда вводятся три фармацевтических средства, третье фармацевтическое средство представляет собой средство от сердечной недостаточности, выбранное из ингибитора ACE, блокатора ангиотензиновых рецепторов, блокатора кальциевых каналов или вазодилататора.
Настоящее изобретение дополнительно включает наборы, которые подходят для применения при осуществлении способов лечения, описанных выше. В одном варианте осуществления настоящего изобретения набор содержит первую лекарственную форму, включающую одно или несколько лекарственных средств (соединений), или их фармацевтически приемлемые соли по настоящему изобретению и контейнер для лекарственной формы, в количествах, достаточных для осуществления способов по настоящему изобретению.
В другом варианте осуществления настоящего изобретения набор содержит первую лекарственную форму, включающую одно из фармацевтических средств (соединений) или его фармацевтически приемлемую соль по настоящему изобретению, и контейнер для первой лекарственной формы, и вторую лекарственную форму, включающую другое фармацевтическое средство (соединение) или его фармацевтически приемлемую соль по настоящему изобретению, и контейнер для второй лекарственной формы, где обе лекарственные формы присутствуют в количествах, достаточных для осуществления способов по настоящему изобретению.
В другом варианте осуществления настоящего изобретения набор содержит первую лекарственную форму, включающую одно из фармацевтический средств (соединений) или его фармацевтически приемлемую соль по настоящему изобретению, и контейнер для первой лекарственной формы, вторую лекарственную форму, включающую другое фармацевтическое средство (соединение) или его фармацевтически приемлемую соль по настоящему изобретению, и контейнер для второй лекарственной формы и третью лекарственную форму, включающую еще одно фармацевтическое средство (соединение) или его фармацевтически приемлемую соль по настоящему изобретению, и контейнер для третьей лекарственной формы, где все три лекарственные формы присутствуют в количествах, достаточных для осуществления способов по настоящему изобретению.
В некоторых вариантах осуществления настоящее изобретение относится к набору, который включает первую лекарственную форму, включающую (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамид или его фармацевтически приемлемую соль, контейнер для первой лекарственной формы и вторую лекарственную форму, включающую 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойную кислоту или ее фармацевтически приемлемую соль, и контейнер для второй лекарственной формы, где обе лекарственные формы присутствуют в количествах, достаточных для осуществления способов по настоящему изобретению.
В другом варианте осуществления настоящее изобретение относится к набору, который включает первую лекарственную форму, включающую (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамид или его фармацевтически приемлемую соль, контейнер для первой лекарственной формы, вторую лекарственную форму, включающую 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойную кислоту или ее фармацевтически приемлемую соль, контейнер для второй лекарственной формы и третью лекарственную форму, включающую другое лекарственное средство (соединение) или его фармацевтически приемлемую соль, и контейнер для третьей лекарственной формы, где все три лекарственные формы присутствуют в количествах, достаточных для осуществления способов по настоящему изобретению.
В некоторых вариантах осуществления лекарственная(ые) форма(ы) набора могут вводиться одновременно или хронологически отсроченно, то есть в разные моменты времени и с равными или разными временными интервалами для любой из лекарственных форм набора.
В любом из описанных выше наборов предоставлены средства для отдельного хранения каждой лекарственной формы, такие как контейнер, разделенный флакон или разделенный пакет из фольги. Примером такого набора является известная блистерная упаковка, используемая для расфасовки таблеток, капсул и т.п. Такой набор особенно подходит для введения разных лекарственных форм, например лекарственных форм для перорального и парентерального введения, для введения отдельных лекарственных средств (соединений) с различными интервалами дозирования или для титрования отдельного лекарственного средства (соединения) относительно другого. Для соблюдения режима терапии набор может включать указания по введению и может быть предоставлен с так называемой памяткой.
Настоящее изобретение также включает различных другие наборы, применяемые для упаковки, распределения и введения фармацевтических средств (соединений) по настоящему изобретению. В еще одном варианте осуществления настоящее изобретение относится к фармацевтической композиции, включающей 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойную кислоту или ее фармацевтически приемлемую соль, ингибитор ГМГ-КoA редуктазы и фармацевтически приемлемый эксципиент.
В некоторых вариантах осуществления настоящее изобретение относится к фармацевтической композиции, включающей 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойную кислоту или ее фармацевтически приемлемую соль, ингибитор ГМГ-КoA редуктазы, выбранный из группы, состоящей из правастатина, питавастатина, ловастатина, аторвастатина, симвастатина, флувастатина, итавастатина, нисвастатина, нисбастатина, росувастатина, атавастатина и висастатина, и фармацевтически приемлемый эксципиент.
В некоторых вариантах осуществления настоящее изобретение относится к фармацевтической композиции, включающей 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойную кислоту или ее фармацевтически приемлемую соль, аторвастатин и фармацевтически приемлемый эксципиент.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения заболевания или состояния, выбранного из жировой инфильтрации печени, неалкогольной жировой болезни печени, неалкогольного стеатогепатита, неалкогольного стеатогепатита с фиброзом печени, неалкогольного стеатогепатита с циррозом печени и неалкогольного стеатогепатита с циррозом печени и с гепатоцеллюлярной карциномой или с метаболическим заболеванием, причем указанный способ включает введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойной кислоты или ее фармацевтически приемлемой соли и ингибитора ГМГ-КoA редуктазы. В некоторых вариантах осуществления настоящего изобретения ингибитор ГМГ-КoA редуктазы представляет собой аторвастатин.
В еще одном варианте осуществления настоящее изобретение относится к способу снижения по меньшей мере на один пункт в балльных системах оценки тяжести неалкогольной жировой болезни печени или неалкогольного стеатогепатита, снижения уровня сывороточных маркеров активности неалкогольного стеатогепатита, снижения активности заболевания неалкогольного стеатогепатита или снижения медицинских последствий неалкогольного стеатогепатита у людей, включающему стадию введения пациенту, нуждающемуся в таком снижении, терапевтически эффективного количества 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойной кислоты или ее фармацевтически приемлемой соли и ингибитора ГМГ-КoA редуктазы. В некоторых вариантах осуществления настоящего изобретения ингибитор ГМГ-КoA редуктазы представляет собой аторвастатин.
В еще одном варианте осуществления настоящее изобретение относится к фармацевтической композиции, включающей 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойную кислоту или ее фармацевтически приемлемую соль, фибратное средство и фармацевтически приемлемый эксципиент.
В некоторых вариантах осуществления настоящее изобретение относится к фармацевтической композиции, включающей 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойную кислоту или ее фармацевтически приемлемую соль, фибратное средство, выбранное из группы, состоящей из фиброзила, фенофибрата и клофибрата, и фармацевтически приемлемый эксципиент.
В некоторых вариантах осуществления настоящее изобретение относится к фармацевтической композиции, включающей 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойную кислоту или ее фармацевтически приемлемую соль, фенофибрат и фармацевтически приемлемый эксципиент.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения заболевания или состояния, выбранного из жировой инфильтрации печени, неалкогольной жировой болезни печени, неалкогольного стеатогепатита, неалкогольного стеатогепатита с фиброзом печени, неалкогольного стеатогепатита с циррозом печени и неалкогольного стеатогепатита с циррозом печени и с гепатоцеллюлярной карциномой или с метаболическим заболеванием, причем указанный способ включает введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойной кислоты или ее фармацевтически приемлемой соли и фибратного средства. В некоторых вариантах осуществления настоящего изобретения фибратное средство представляет собой фенофибрат.
В дополнительном варианте осуществления настоящее изобретение относится к способу снижения по меньшей мере на один пункт в балльных системах оценки тяжести неалкогольной жировой болезни печени или неалкогольного стеатогепатита, снижения уровня сывороточных маркеров активности неалкогольного стеатогепатита, снижения активности заболевания неалкогольного стеатогепатита или снижения медицинских последствий неалкогольного стеатогепатита у людей, включающему стадию введения пациенту, нуждающемуся в таком снижении, терапевтически эффективного количества 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойной кислоты или ее фармацевтически приемлемой соли и фибратного средства. В некоторых вариантах осуществления настоящего изобретения фибратное средство представляет собой фенофибрат.
В еще одном варианте осуществления настоящее изобретение относится к фармацевтической композиции, включающей 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойную кислоту или ее фармацевтически приемлемую соль, секвестрант желчных кислот и фармацевтически приемлемый эксципиент.
В некоторых вариантах осуществления настоящее изобретение относится к фармацевтической композиции, включающей 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойную кислоту или ее фармацевтически приемлемую соль, секвестрант желчных кислот, выбранный из группы, состоящей из квестрана, колестипола и колесевелама, и фармацевтически приемлемый эксципиент.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения заболевания или состояния, выбранного из жировой инфильтрации печени, неалкогольной жировой болезни печени, неалкогольного стеатогепатита, неалкогольного стеатогепатита с фиброзом печени, неалкогольного стеатогепатита с циррозом печени и неалкогольного стеатогепатита с циррозом печени и с гепатоцеллюлярной карциномой или с метаболическим заболеванием, причем указанный способ включает введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойной кислоты или его фармацевтически приемлемой соли и секвестранта желчных кислот.
В еще одном варианте осуществления настоящее изобретение относится к способу снижения по меньшей мере на один пункт в балльных системах оценки тяжести неалкогольной жировой болезни печени или неалкогольного стеатогепатита, снижения уровня сывороточных маркеров активности неалкогольного стеатогепатита, снижения активности заболевания неалкогольного стеатогепатита или снижения медицинских последствий неалкогольного стеатогепатита у людей, включающему стадию введения пациенту, нуждающемуся в таком снижении, терапевтически эффективного количества 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойной кислоты или ее фармацевтически приемлемой соли и секвестранта желчных кислот.
В еще одном варианте осуществления настоящее изобретение относится к фармацевтической композиции, включающей 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойную кислоту или ее фармацевтически приемлемую соль, ингибитор абсорбции холестерина и фармацевтически приемлемый эксципиент. В некоторых вариантах осуществления настоящего изобретения ингибитор абсорбции холестерина представляет собой эзетимиб.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения заболевания или состояния, выбранного из жировой инфильтрации печени, неалкогольной жировой болезни печени, неалкогольного стеатогепатита, неалкогольного стеатогепатита с фиброзом печени, неалкогольного стеатогепатита с циррозом печени и неалкогольного стеатогепатита с циррозом печени и с гепатоцеллюлярной карциномой или с метаболическим заболеванием, причем указанный способ включает введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойной кислоты или ее фармацевтически приемлемой соли и ингибитора абсорбции холестерина.
В еще одном варианте осуществления настоящее изобретение относится к способу снижения по меньшей мере на один пункт в системах оценки тяжести неалкогольной жировой болезни печени или неалкогольного стеатогепатита, снижения уровня сывороточных маркеров активности неалкогольного стеатогепатита, снижения активности заболевания неалкогольного стеатогепатита или снижения медицинских последствий неалкогольного стеатогепатита у людей, включающему стадию введения пациенту, нуждающемуся в таком снижении, терапевтически эффективного количества 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойной кислоты или ее фармацевтически приемлемой соли и ингибитора абсорбции холестерина.
В еще одном варианте осуществления настоящее изобретение относится к фармацевтической композиции, включающей 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойную кислоту или ее фармацевтически приемлемую соль, фармацевтическое средство никотиновой кислоты и фармацевтически приемлемый эксципиент.
В некоторых вариантах осуществления настоящее изобретение относится к фармацевтической композиции, включающей 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойную кислоту или ее фармацевтически приемлемую соль, фармацевтическое средство никотиновой кислоты, выбранное из группы, состоящей из ниацина, ниакора и сло-ниацина, и фармацевтически приемлемый эксципиент.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения заболевания или состояния, выбранного из жировой инфильтрации печени, неалкогольной жировой болезни печени, неалкогольного стеатогепатита, неалкогольного стеатогепатита с фиброзом печени, неалкогольного стеатогепатита с циррозом печени и неалкогольного стеатогепатита с циррозом печени и с гепатоцеллюлярной карциномой или с метаболическим заболеванием, причем указанный способ включает введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойной кислоты или ее фармацевтически приемлемой соли и фармацевтического средства никотиновой кислоты.
В еще одном варианте осуществления настоящее изобретение относится к способу снижения по меньшей мере на один пункт в балльных системах оценки тяжести неалкогольной жировой болезни печени или неалкогольного стеатогепатита, снижения уровня сывороточных маркеров активности неалкогольного стеатогепатита, снижения активности заболевания неалкогольного стеатогепатита или снижения медицинских последствий неалкогольного стеатогепатита у людей, включающему стадию введения пациенту, нуждающемуся в таком снижении, терапевтически эффективного количества 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойной кислоты или ее фармацевтически приемлемой соли и фармацевтического средства никотиновой кислоты.
В еще одном варианте осуществления настоящее изобретение относится к фармацевтической композиции, включающей 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойную кислоту или ее фармацевтически приемлемую соль, модулятор PCSK9 и фармацевтически приемлемый эксципиент.
В некоторых вариантах осуществления настоящее изобретение относится к фармацевтической композиции, включающей 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойную кислоту или ее фармацевтически приемлемую соль, модулятор PCSK9, выбранный из группы, состоящей из алирокумаба и эволукумаба, и фармацевтически приемлемый эксципиент.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения заболевания или состояния, выбранного из жировой инфильтрации печени, неалкогольной жировой болезни печени, неалкогольного стеатогепатита, неалкогольного стеатогепатита с фиброзом печени, неалкогольного стеатогепатита с циррозом печени и неалкогольного стеатогепатита с циррозом печени и с гепатоцеллюлярной карциномой или с метаболическим заболеванием, причем указанный способ включает введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойной кислоты или ее фармацевтически приемлемой соли и модулятора PCSK9.
В еще одном варианте осуществления настоящее изобретение относится к способу снижения по меньшей мере на один пункт в балльных системах оценки тяжести неалкогольной жировой болезни печени или неалкогольного стеатогепатита, снижения уровня сывороточных маркеров активности неалкогольного стеатогепатита, снижения активности заболевания неалкогольного стеатогепатита или снижения медицинских последствий неалкогольного стеатогепатита у людей, включающему стадию введения пациенту, нуждающемуся в таком снижении, терапевтически эффективного количества 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойной кислоты или ее фармацевтически приемлемой соли и модулятора PCSK9.
В любом из предыдущих вариантов осуществления 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойная кислота представляет собой твердое кристаллическое вещество. В некоторых вариантах осуществления настоящего изобретения твердое кристаллическое вещество представляет собой 2-амино-2-(гидроксиметил)пропан-1,3-диольную соль 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойной кислоты.
В еще одном варианте осуществления настоящее изобретение относится к фармацевтической композиции, включающей (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамид или его фармацевтически приемлемую соль, ингибитор ГМГ-КoA редуктазы и фармацевтически приемлемый эксципиент.
В некоторых вариантах осуществления настоящее изобретение относится к фармацевтической композиции, включающей (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамид или его фармацевтически приемлемую соль, ингибитор ГМГ-КoA редуктазы, выбранный из группы, состоящей из правастатина, питавастатина, ловастатина, аторвастатина, симвастатина, флувастатина, итавастатина, нисвастатина, нисбастатина, росувастатина, атавастатина и висастатина, и фармацевтически приемлемый эксципиент.
В некоторых вариантах осуществления настоящее изобретение относится к фармацевтической композиции, включающей (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамид или его фармацевтически приемлемую соль, аторвастатин и фармацевтически приемлемый эксципиент.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения заболевания или состояния, выбранного из жировой инфильтрации печени, неалкогольной жировой болезни печени, неалкогольного стеатогепатита, неалкогольного стеатогепатита с фиброзом печени, неалкогольного стеатогепатита с циррозом печени и неалкогольного стеатогепатита с циррозом печени и с гепатоцеллюлярной карциномой или с метаболическим заболеванием, причем указанный способ включает введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамида или его фармацевтически приемлемой соли и ингибитора ГМГ-КoA редуктазы. В некоторых вариантах осуществления настоящего изобретения ингибитор ГМГ-КoA редуктазы представляет собой аторвастатин.
В еще одном варианте осуществления настоящее изобретение относится к способу снижения по меньшей мере на один пункт в балльных системах оценки тяжести неалкогольной жировой болезни печени или неалкогольного стеатогепатита, снижения уровня сывороточных маркеров активности неалкогольного стеатогепатита, снижения активности заболевания неалкогольного стеатогепатита или снижения медицинских последствий неалкогольного стеатогепатита у людей, включающему стадию введения пациенту, нуждающемуся в таком снижении, терапевтически эффективного количества (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамида или его фармацевтически приемлемой соли и ингибитора ГМГ-КoA редуктазы. В некоторых вариантах осуществления настоящего изобретения ингибитор ГМГ-КoA редуктазы представляет собой аторвастатин.
В еще одном варианте осуществления настоящее изобретение относится к фармацевтической композиции, включающей (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамид или его фармацевтически приемлемую соль, фибратное средство и фармацевтически приемлемый эксципиент.
В некоторых вариантах осуществления настоящее изобретение относится к фармацевтической композиции, включающей (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамид или его фармацевтически приемлемую соль, фибратное средство, выбранное из группы, состоящей из гемфиброзила, фенофибрата и клофибрата, и фармацевтически приемлемый эксципиент.
В некоторых вариантах осуществления настоящее изобретение относится к фармацевтической композиции, включающей (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамид или его фармацевтически приемлемую соль, фенофибрат и фармацевтически приемлемый эксципиент.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения заболевания или состояния, выбранного из жировой инфильтрации печени, неалкогольной жировой болезни печени, неалкогольного стеатогепатита, неалкогольного стеатогепатита с фиброзом печени, неалкогольного стеатогепатита с циррозом печени и неалкогольного стеатогепатита с циррозом печени и с гепатоцеллюлярной карциномой или с метаболическим заболеванием, причем указанный способ включает введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамида или его фармацевтически приемлемой соли и фибратного средства. В некоторых вариантах осуществления настоящего изобретения фибратное средство представляет собой фенофибрат.
В еще одном варианте осуществления настоящее изобретение относится к способу снижения по меньшей мере на один пункт в балльных системах оценки тяжести неалкогольной жировой болезни печени или неалкогольного стеатогепатита, снижения уровня сывороточных маркеров активности неалкогольного стеатогепатита, снижения активности заболевания неалкогольного стеатогепатита или снижения медицинских последствий неалкогольного стеатогепатита у людей, включающему стадию введения пациенту, нуждающемуся в таком снижении, терапевтически эффективного количества (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамида или его фармацевтически приемлемой соли и фибратного средства. В некоторых вариантах осуществления настоящего изобретения фибратное средство представляет собой фенофибрат.
В еще одном варианте осуществления настоящее изобретение относится к фармацевтической композиции, включающей (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамид или его фармацевтически приемлемую соль, секвестрант желчных кислот и фармацевтически приемлемый эксципиент.
В некоторых вариантах осуществления настоящее изобретение относится к фармацевтической композиции, включающей (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамид или его фармацевтически приемлемую соль, секвестрант желчных кислот, выбранный из группы, состоящей из квестрана, колестипола и колесевелама, и фармацевтически приемлемый эксципиент.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения заболевания или состояния, выбранного из жировой инфильтрации печени, неалкогольной жировой болезни печени, неалкогольного стеатогепатита, неалкогольного стеатогепатита с фиброзом печени, неалкогольного стеатогепатита с циррозом печени и неалкогольного стеатогепатита с циррозом печени и с гепатоцеллюлярной карциномой или с метаболическим заболеванием, причем указанный способ включает введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамида или его фармацевтически приемлемой соли и секвестранта желчных кислот.
В еще одном варианте осуществления настоящее изобретение относится к способу снижения по меньшей мере на один пункт в балльных системах оценки тяжести неалкогольной жировой болезни печени или неалкогольного стеатогепатита, снижения уровня сывороточных маркеров активности неалкогольного стеатогепатита, снижения активности заболевания неалкогольного стеатогепатита или снижения медицинских последствий неалкогольного стеатогепатита у людей, включающему стадию введения человеку, нуждающемуся в таком снижении, терапевтически эффективного количества (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамида или его фармацевтически приемлемой соли и секвестранта желчных кислот.
В еще одном варианте осуществления настоящее изобретение относится к фармацевтической композиции, включающей (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамид или его фармацевтически приемлемую соль, ингибитор абсорбции холестерина и фармацевтически приемлемый эксципиент. В некоторых вариантах осуществления настоящего изобретения ингибитор абсорбции холестерина представляет собой эзетимиб.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения заболевания или состояния, выбранного из жировой инфильтрации печени, неалкогольной жировой болезни печени, неалкогольного стеатогепатита, неалкогольного стеатогепатита с фиброзом печени, неалкогольного стеатогепатита с циррозом печени и неалкогольного стеатогепатита с циррозом печени и с гепатоцеллюлярной карциномой или с метаболическим заболеванием, причем указанный способ включает введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамида или его фармацевтически приемлемой соли и ингибитора абсорбции холестерина.
В еще одном варианте осуществления настоящее изобретение относится к способу снижения по меньшей мере на один пункт в балльных системах оценки тяжести неалкогольной жировой болезни печени или неалкогольного стеатогепатита, снижения уровня сывороточных маркеров активности неалкогольного стеатогепатита, снижения активности заболевания неалкогольного стеатогепатита или снижения медицинских последствий неалкогольного стеатогепатита у людей, включающему стадию введения пациенту, нуждающемуся в таком снижении, терапевтически эффективного количества (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамида или его фармацевтически приемлемой соли и ингибитора абсорбции холестерина.
В еще одном варианте осуществления настоящее изобретение относится к фармацевтической композиции, включающей (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамид или его фармацевтически приемлемую соль, фармацевтическое средство никотиновой кислоты и фармацевтически приемлемый эксципиент.
В некоторых вариантах осуществления настоящее изобретение относится к фармацевтической композиции, включающей (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамид или его фармацевтически приемлемую соль, фармацевтическое средство никотиновой кислоты, выбранное из группы, состоящей из ниацина, ниакора и сло-ниацина, и фармацевтически приемлемый эксципиент.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения заболевания или состояния, выбранного из жировой инфильтрации печени, неалкогольной жировой болезни печени, неалкогольного стеатогепатита, неалкогольного стеатогепатита с фиброзом печени, неалкогольного стеатогепатита с циррозом печени и неалкогольного стеатогепатита с циррозом печени и с гепатоцеллюлярной карциномой или с метаболическим заболеванием, причем указанный способ включает введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамида или его фармацевтически приемлемой соли и фармацевтического средства никотиновой кислоты.
В еще одном варианте осуществления настоящее изобретение относится к способу снижения по меньшей мере на один пункт в балльных системах оценки тяжести неалкогольной жировой болезни печени или неалкогольного стеатогепатита, снижения уровня сывороточных маркеров активности неалкогольного стеатогепатита, снижения активности заболевания неалкогольного стеатогепатита или снижения медицинских последствий неалкогольного стеатогепатита у людей, включающему стадию введения пациенту, нуждающемуся в таком снижении, терапевтически эффективного количества (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамида или его фармацевтически приемлемой соли и фармацевтического средства никотиновой кислоты.
В еще одном варианте осуществления настоящее изобретение относится к фармацевтической композиции, включающей (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамид или его фармацевтически приемлемую соль, модулятор PCSK9 и фармацевтически приемлемый эксципиент.
В некоторых вариантах осуществления настоящее изобретение относится к фармацевтической композиции, включающей (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамид или его фармацевтически приемлемую соль, модулятор PCSK9, выбранный из группы, состоящей из алирокумаба и эволукумаба, и фармацевтически приемлемый эксципиент.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения заболевания или состояния, выбранного из жировой инфильтрации печени, неалкогольной жировой болезни печени, неалкогольного стеатогепатита, неалкогольного стеатогепатита с фиброзом печени, неалкогольного стеатогепатита с циррозом печени и неалкогольного стеатогепатита с циррозом печени и с гепатоцеллюлярной карциномой или с метаболическим заболеванием, причем указанный способ включает введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамида или его фармацевтически приемлемой соли и модулятора PCSK9.
В еще одном варианте осуществления настоящее изобретение относится к способу снижения по меньшей мере на один пункт в балльных системах оценки тяжести неалкогольной жировой болезни печени или неалкогольного стеатогепатита, снижения уровня сывороточных маркеров активности неалкогольного стеатогепатита, снижения активности заболевания неалкогольного стеатогепатита или снижения медицинских последствий неалкогольного стеатогепатита у людей, включающему стадию введения пациенту, нуждающемуся в таком снижении, терапевтически эффективного количества (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамида или его фармацевтически приемлемой соли и модулятора PCSK9.
В любом из предыдущих вариантов осуществления настоящего изобретения (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамид представляет собой твердое кристаллическое вещество. В некоторых вариантах осуществления настоящего изобретения порошковая рентгеновская дифрактограмма указанного твердого кристаллического вещества включает пики при значениях угла 2-тета (CuKα излучение, длина волны 1,54056 Å) 5,3±0,2, 7,7±0,2 и 15,4±0,2. В некоторых других вариантах осуществления порошковая рентгеновская дифрактограмма указанного твердого кристаллического вещества включает пики при значениях угла 2-тета (CuKα излучение, длина волны 1,54056Å) 6,5±0,2, 9,3±0,2 и 13,6±0,2.
Соединения по настоящему изобретению могут быть синтезированы способами синтеза, которые включают процессы, аналогичные хорошо известным в данной области техники, в частности описанными в настоящем документе. Исходные вещества обычно доступны от коммерческих источников, таких как Aldrich Chemicals (Milwaukee, WI) или получают с использованием способов, хорошо известных специалистам в данной области техники (например, способами, описанными в публикации Louis F. Fieser and Mary Fieser, Reagents for Organic Synthesis, v. 1-19, Wiley, New York (1967-1999 ed.), или Beilsteins Handbuch der organischen Chemie, 4, Aufl. ed. Springer-Verlag, Berlin, включая приложения (также доступные через on line базу данных Beilstein)). Получение (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамида представлено в примере 1 Заявки на патент США 2018-0051012A1, содержание которой введено в настоящее описание в виде ссылки для всех целей. Получение 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойной кислоты описано в примере 9 Патента США № 8859577, содержание которого введено в настоящее описание в виде ссылки для всех целей. Получение [(1R,5S,6R)-3-{2-[(2S)-2-метилазетидин-1-ил]-6-(трифторметил)пиримидин-4-ил}-3-азабицикло[3.1.0]гекс-6-ил]уксусной кислоты (включая ее кристаллическую форму свободной кислоты) описано в примере 4 Патента США № 9809579. Получение агонистов GLP-1R описано в Патенте США № 10208019.
ФАРМАЦЕВТИЧЕСКИЕ СРЕДСТВА ДЛЯ КОМБИНИРОВАННОГО ВВЕДЕНИЯ
Соединения по настоящему изобретению могут вводиться отдельно или совместно в качестве отдельных фармацевтических средств, в комбинации фиксированной дозы или в комбинации с одним или несколькими дополнительными терапевтическими средствами. Термин «введение в комбинации» или «комбинированная терапия» означает, что соединение A и соединение D вводятся вместе в виде двух отдельных терапевтических средств или в комбинации с одним или несколькими дополнительными терапевтическими средствами, вводимыми одновременно, млекопитающему, подлежащему лечению. При введении в комбинации каждый компонент может вводиться в то же самое время или последовательно в любом порядке в разные моменты времени. Следовательно, каждый компонент может вводиться отдельно, но в достаточно близкие моменты времени, чтобы обеспечить желаемый терапевтический эффект. Таким образом, способы лечения, описанные в настоящем изобретении, включают применение комбинированных средств для введения трех или более средств в комбинации.
Комбинированные средства вводятся млекопитающему в терапевтически эффективном количестве. Термин «терапевтически эффективное количество» означает количество соединений по настоящему изобретению, которое при введении млекопитающему отдельно или в комбинации с дополнительным терапевтическим средством эффективно для лечения желаемого заболевания/состояния, например НАСГ.
Предпочтительными средствами для лечения неалкогольного стеатогепатита (НАСГ) и/или неалкогольной жировой болезни печени (НАЖБП) (т.е. анти-НАСГ и анти-НАЖБП средствами) являются ингибитор ацетил-КоА-карбоксилазы (АКК), ингибитор кетогексогиназы (KHK), агонист рецептора GLP-1, агонист FXR, антагонист CB1, ингибитор ASK1, ингибитор CCR2 и/или CCR5, ингибитор PNPLA3, ингибитор гидроксистероид-17-β-дегидрогеназы (HSD17B13), ингибитор DGAT1, аналог FGF21, аналог FGF19, ингибитор SGLT2, агонист PPAR, активатор AMPK, ингибитор SCD1 или ингибитор MPO. Традиционно упоминаемая заявка на патент PCT/IB2017/057577, поданная 12/01/2017, относится к агонисту рецептора GLP-1. Более предпочтительными являются агонист FXR, ингибитор регулирующей апоптотические сигналы киназы 1 (apoptosis signal-regulating kinase 1 - ASK1), агонист PPAR, агонист рецептора GLP-1, ингибитор SGLT, ингибитор АКК и ингибитор KHK.
Принимая во внимание активность соединений по настоящему изобретению в отношении НАСГ/НАЖБП, они могут вводиться совместно с другими лекарственными средствами для лечения неалкогольного стеатогепатита (НАСГ) и/или неалкогольной жировой болезни печени (НАЖБП) и связанных с ними заболеваний/состояний, такими как орлистат (Orlistat), производные тиазолидиндиона (TZD) и другие инсулин-сенсибилизирующие лекарственные средства, аналоги FGF21, метформин, этиловые эфиры омега-3-кислот (например, Lovaza), фибраты, ингибиторы гидроксиметилглутарил-кофермент-А-редуктазы, эзетимиб, пробукол, уродезоксихолевая кислота, агонисты TGR5, агонисты FXR, витамин E, бетаин, пентоксифиллин, антагонисты CB1, карнитин, N-ацетилцистеин, восстановленный глутатион, лоркасерин, комбинация налтрексона с бупроприоном, ингибиторы SGLT2 (включая дапаглифлозин, канаглифлозин, эмпаглифлозин, тофоглифлозин, эртуглифлозин, ASP-1941, THR1474, TS-071, ISIS388626 и LX4211, а также ингибиторы SGLT2, раскрытые в WO2010023594), фентермин, топирамат, агонисты рецептора GLP-1, агонисты рецептора GIP, двойные агонисты рецепторов GLP-1/глюкагона (т.е. OPK88003, MEDI0382, JNJ-64565111, NN9277, BI 456906), двойные агонисты рецепторов GLP-1/GIP (т.е. тирзепатид (LY3298176), NN9423), блокаторы ангиотензиновых рецепторов, ингибитор ацетил-КоА-карбоксилазы (АКК), ингибитор BCKDK, ингибитор кетогексокиназы (KHK), ингибиторы ASK1, ингибиторы киназы дегидрогеназы альфа-кетокислот с разветвленной цепью (branched-chain alpha keto acid dehydrogenase kinase - BCBK), ингибиторы CCR2 и/или CCR5, ингибиторы PNPLA3, ингибиторы DGAT1, аналог FGF21, аналоги FGF19, агонисты PPAR, агонисты FXR, активаторы AMPK, ингибиторы SCD1 или ингибиторы MPO.
Типичные примеры ингибиторов АКК включают 4-(4-[(1-изопропил-7-оксо-1,4,6,7-тетрагидро-1'H-спиро[индазол-5,4'-пиперидин]-1'-ил)карбонил]-6-метоксипиридин-2-ил)бензойную кислоту и фирсокостат (GS-0976) и их фармацевтически приемлемые соли.
Типичные примеры ингибиторов DGAT2 включают (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамид;
2-(5-((3-этокси-5-фторпиридин-2-ил)окси)пиридин-3-ил)-N-((3R,4S)-4-фторпиперидин-3-ил)пиримидин-5-карбоксамид;
2-(5-((3-этокси-5-фторпиридин-2-ил)окси)пиридин-3-ил)-N-((3S,5S)-5-фторпиперидин-3-ил)пиримидин-5-карбоксамид;
2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-((3R,4S)-4-фторпиперидин-3-ил)пиримидин-5-карбоксамид;
2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-((3R,4R)-4-фторпиперидин-3-ил)пиримидин-5-карбоксамид;
2-(5-((3-этокси-5-фторпиридин-2-ил)окси)пиридин-3-ил)-N-((3R,4R)-4-фторпиперидин-3-ил)пиримидин-5-карбоксамид; и
2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-((3S,5S)-5-фторпиперидин-3-ил)пиримидин-5-карбоксамид.
Примеры подходящих противодиабетических средств включают, например, инсулины, метформин, ингибиторы DPPIV, агонисты, аналоги и миметики рецептора GLP-1, ингибиторы SGLT1 и SGLT2. Подходящие противодиабетические средства включают ингибиторы ацетил-КоА-карбоксилазы (АКК), например описанные в WO2009144554, WO2003072197, WO2009144555 и WO2008065508, ингибиторы диацилглицерол-O-ациaтрансферазы 1 (DGAT-1), например описанные в WO09016462 или WO2010086820, AZD7687 или LCQ908, ингибиторы моноацилглицерол-О-ацилтрансферазы, ингибитор фосфодиэстеразы-10 (PDE-10), активатор AMPK, сульфонилмочевины (например, ацетогексамид, хлорпропамид, диабинес, глибенкламид, глипизид, глибурид, глимепирид, гликлазид, глипентид, гликвидон, глисоламид, толазамид и толбутамид), меглитинид, ингибитор α-амилазы (например, тендамистат, трестатин и AL-3688), ингибиторы α-глюкозидгидролазы (например, акарбозу), ингибитор α-глюкозидазы (например, адипозин, камиглибозу, эмиглитат, миглитол, воглибозу, прадимицин-Q и салбостатин), агонист PPARγ (например, балаглитазон, циглитазон, дарглитазон, энглитазон, исаглитазон, пиоглитазон и росиглитазон), агонист PPAR α/γ (например, CLX-0940, GW-1536, GW-1929, GW-2433, KRP-297, L-796449, LR-90, MK-0767 и SB-219994), бигуанид (например, метформин), модулятор глюкагон-подобного пептида 1 (glucagon-like peptide 1 - GLP-1), такой как агонист (например, эксендин-3 и эксендин-4), лираглутид, албаглутид, эксенатид (Byetta®), албиглутид, ликсисенатид, дулаглутид, семаглутид, NN-9924, TTP-054, ингибитор протеинтирозинфосфатазы-1B (protein tyrosine phosphatase-1B - PTP-1B) (например, тродусквемин, гиртиосаловый экстракт (hyrtiosal extract) и соединения, описанные в публикации Zhang, S., et al., Drug Discovery Today, 12(9/10), 373-381 (2007)), активатор SIRT-1 (например, ресвератрол, GSK2245840 или GSK184072), ингибитор дипептидилпептидазы-IV (dipeptidyl peptidase IV - DPP-IV) (например, описанный в WO2005116014, ситаглиптин, вилдаглиптин, алоглиптин, дутоглиптин, линаглиптин и саксаглиптин), стимулятор секреции инсулина, ингибитор окисления жирных кислот, A2 антагонист, ингибитор с-Jun N-терминальной киназы (JNK), активаторы глюкокиназы (GKa), такие как активаторы, описанные в WO2010103437, WO2010103438, WO2010013161, WO2007122482, TTP-399, TTP-355, TTP-547, AZD1656, ARRY403, MK-0599, TAK-329, AZD5658 или GKM-001, инсулин, инсулиноподобное средство, ингибитор гликогенфосфорилазы (например GSK1362885), агонист рецептора VPAC2t, ингибиторы SGLT2, например, описанные в публикации E.C. Chao et al. Nature Reviews Drug Discovery 9, 551-559 (July 2010) включая дапаглифлозин, канаглифлозин, эмпаглифлозин, тофоглифлозин (CSG452), эртуглифлозин, ASP-1941, THR1474, TS-071, ISIS388626 и LX4211, а также соединения, раскрытые в WO2010023594, модулятор рецептора глюкагона, например, описанный в публикации Demong, D.E. et al. Annual Reports in Medicinal Chemistry 2008, 43, 119-137, модуляторы GPR119, в частности агонисты, например описанные в WO2010140092, WO2010128425, WO2010128414, WO2010106457, а также в публикации Jones, R.M. et al., Medicinal Chemistry 2009, 44, 149-170 (например, MBX-2982, GSK1292263, APD597 и PSN821), производные и аналоги FGF21, например описанные в публикации Kharitonenkov, A. et al., Current Opinion in Investigational Drugs 2009, 10(4)359-364, модуляторы рецептора TGR5 (называемого также GPBAR1), в частности агонисты, например описанные в Zhong, M., Current Topics in Medicinal Chemistry, 2010, 10(4), 386-396, и INT777, агонисты GPR40, например описанные в публикации Medina, J.C., Annual Reports in Medicinal Chemistry, 2008, 43, 75-85, включая, но без ограничения только ими, TAK-875, модуляторы GPR120, в частности агонисты, активаторы рецепторов никотиновой кислоты высокого сродства (HM74A) и ингибиторы SGLT1, такие как GSK1614235. Дополнительный репрезентативный перечень противодиабетических средств, которые могут комбинироваться с соединениями по настоящему изобретению, можно найти, например, в WO2011005611 (страница 28 строка 35 - страницы 30 строка 19). Предпочтительными противодиабетическими средствами являются метформин и ингибиторы DPP-IV (например, ситаглиптин, вилдаглиптин, алоглиптин, дутоглиптин, линаглиптин и саксаглиптин). Другие противодиабетические средства могут включать ингибиторы или модуляторы ферментов карнитинпальмитоилтрансферазы, ингибиторы фруктозо-1,6-дифосфатазы, ингибиторы альдозоредуктазы, ингибиторы минералокортикоидного рецептора, ингибиторы TORC2, ингибиторы CCR2 и/или CCR5, ингибиторы изоформ PKC (например, PKCα, PKCβ, PKCγ), ингибиторы синтетазы жирных кислот, ингибиторы серинпальмитоилтрансферазы, модуляторы GPR81, GPR39, GPR43, GPR41, GPR105, Kv1.3, ретинолсвязывающий белок, глюкокортикоидный рецептор, рецепторы соматостатина (например, SSTR1, SSTR2, SSTR3 и SSTR5), ингибиторы или модуляторы PDHK2 или PDHK4, ингибиторы MAP4K4, модуляторы семейства IL1, включая IL1beta, модуляторы RXRalpha. Кроме того, подходящие противодиабетические средства включают механизмы, описанные в публикации Carpino, P.A., Goodwin, B. Expert Opin. Ther. Pat, 2010, 20(12), 1627-51.
Подходящие средства против ожирения включают ингибиторы 11β-гидроксистероиддегидрогеназы-1 (11β-hydroxy steroid dehydrogenase-1-11β-HSD type 1), ингибитор стеароил-КоА-десатуразы-1 (stearoyl-CoA desaturase-1 - SCD-1), агонисты MCR-4, агонисты холецистокинина-А (cholecystokinin-A - CCK-A), ингибиторы обратного захвата моноаминов (такие как сибутрамин), симпатомиметические средства, β3-адренергические агонисты, агонисты допаминовых рецепторов (такие как бромокриптин), аналоги меланоцитостимулирующего гормона, агонисты 5HT2c, антагонисты меланиноконцентрирующего гормона, лептин (OB белок), аналоги лептина, агонисты лептина, антагонисты галанина, ингибиторы липазы (такие как тетрагидролипстатин, т.е. орлистат), аноректические средства (такие как агонисты бомбезина), антагонисты нейропептида Y (neuropeptide-Y - NPY) (например, антагонисты NPY Y5), PYY3-36 (включая его аналоги), тиромиметики, дегидростерон или его аналоги, агонисты или антагонисты глюкокортикоидов, антагонисты арексиновых рецепторов, агонисты рецептора глюкагоноподобного пептида-1, циллиарные нейротропные факторы (такие как Axokine™, доступный от Regeneron Pharmaceuticals, Inc., Tarrytown, NY и Procter & Gamble Company, Cincinnati, OH), ингибиторы агути-связанного белка (agouti-related protein - AGRP), антагонисты грелина, антагонисты или обратные агонисты гистамина 3, агонисты нейромедина U, ингибиторы MTP/ApoB (например, селективные для кишечника ингибиторы MTP, такие как дирлотапид), опиоидный антагонист, антагонисты орексиновых рецепторов, комбинацию налтрексона с бупроприоном и т.п.
Предпочтительные средства против ожирения для применения в комбинации по настоящему изобретению включают селективные для кишечника ингибиторы MTP (например, дирлотапид, митратапид и имплитапид, R56918 (CAS № 403987) и CAS № 913541-47-6), агонисты CCKa (например, N-бензил-2-[4-(1H-индол-3-илметил)-5-оксо-1-фенил-4,5-дигидро-2,3,6,10b-тетраазабензо[e]азулен-6-ил]-N-изопропилацетамид, описанный в Публикации PCT № WO 2005/116034 или Публикации США № 2005-0267100 A1), агонисты 5HT2c (например, лоркасерин), агонисты MCR4 (например, соединения, описанные в Патенте США № 6818658), ингибитор липазы (например, Cetilistat), PYY3-36 (где «PYY3-36», когда используется в настоящем описании, включает его аналоги, такие как пегилированный PYY3-36, например описанные в Публикации США 2006/0178501), опиодные антагонисты (например, налтрексон), комбинацию налтрексона с бупроприоном, олеоил-эстрон (CAS № 180003-17-2), обинепитид (™30338), прамлинтид (Symlin®), тезофензин (NS2330), лептин, лираглутид, бромокриптин, орлистат, эксенатид (Byetta®), AOD-9604 (CAS № 221231-10-3), фентермин, топирамат (торговое название: Qsymia) и сибутрамин. Предпочтительно, соединения по настоящему изобретению и средства комбинированной терапии вводятся в сочетании с физическими упражнениями и разумной диетой.
Соединения по настоящему изобретению могут использоваться в комбинации с холестерин-модулирующими средствами (включая средства, понижающие холестерин), такими как ингибитор липазы, ингибитор ГМГ-КoA редуктазы, ингибитор ГМГ-КoА синтазы, ингибитор генной экспрессии ГМГ-КoA редуктазы, ингибитор генной экспрессии ГМГ-КoА синтазы, ингибитор секреции MTP/Apo B, ингибитор CETP, ингибитор абсорбции желчных кислот, ингибитор абсорбции холестерина, ингибитор синтеза холестерина, ингибитор скваленсинтетазы, ингибитор скваленэпоксидазы, ингибитор скваленциклазы, комбинированный ингибитор скваленэпоксидазы/скваленциклазы, фибрат, ниацин, ионно-обменная смола, антиоксидант, ингибитор ACAT или секвестрант желчных кислот или такое средство, как мипомерсен.
Примеры подходящих холестерин/липид-понижающих средств и терапевтических средств липидного профиля включают ингибиторы ГМГ-КoА редуктазы (например, правастатин, питавастатин, ловастатин, аторвастатин, симвастатин, флувастатин, NK-104 (известный также как итавастатин, нисвастатин или нисбастатин) и ZD-4522 (известный также как росувастатин, атавастатин или висастатин); ингибиторы скваленсинтетазы; фибраты (например, фиброзил, фенофибрат, клофибрат); секвестранты желчных кислот (такие как квестран, колестипол, колесевелам); ингибиторы ACAT; ингибиторы MTP; ингибиторы липооксигеназы; ингибиторы абсорбции холестерина (например, эзетимиб); фармацевтические средства никотиновой кислоты (например, ниацин, ниакор, сло-ниацин); омега-3-жирные кислоты; и ингибиторы транспортного белка холестериновых эфиров. Другие противоатеросклеротические средства включают модуляторы PCSK9 (например, алирокумаб и эволокумаб).
В другом варианте осуществления настоящего изобретения соединения по настоящему изобретению могут вводиться совместно с фармацевтическими средствами для лечения неалкогольного стеатогепатита (НАСГ) и/или неалкогольной жировой болезни печени (НАЖБП), такими как орлистат, TZD и другие инсулин-сенсибилизирующие средства, аналоги FGF21, метформин, сложные эфиры омега-3-кислот (например, ловаза), фибраты, ингибиторы ГМГ-КоA-редуктазы, эзитимиб, пробукол, уродезоксихолевая кислота, агонисты TGR5, агонисты FXR, витамин E, бетаин, пентоксифиллин, антагонисты CB1, карнитин, N-ацетилцистеин, восстановленный глутатион, лоркасерин, комбинация налтрексона с бупроприоном, ингибиторы SGLT2, фентермин, топирамат, аналоги инкретина (GLP и GIP) и блокаторы ангиотензиновых рецепторов.
В другом варианте осуществления настоящего изобретения дополнительное фармацевтическое средство выбрано из группы, состоящей из цистеамина или его фармацевтически приемлемой соли, цистамина или его фармацевтически приемлемой соли, антиоксиданта, лецитина, комплекса витамина В, препаратов желчных солей, антагонистов рецептора каннабиноида 1 (CB1), обратных агонистов рецептора каннабиноида 1 (CB1), регуляторов активности рецепторов, активируемых пролифератором пероксисом, производного бензотиазепина или бензотиепина, конструкта антисмысловой РНК для ингибирования протеинтирозинфосфатазы PTPRU, пипередина, связанного через гетероатом, и его производных, производного азациклопентана, способного ингибировать стеароил-коэнзим-альфа-дельта-9-десатуразу, ациламидного соединения, обладающего активностью стимулятора секреции или индуктора активности адипонектина, четвертичного аммониевого соединения, ацетата глатирамера, пентатраксиновых белков, ингибитора ГМГ-КoА редуктазы, н-ацетилцистеина, соединения изофлавона, макролидного антибиотика, ингибитора галектина, антитела или любой их комбинации.
Дополнительные терапевтические средства включают антикоагулянты или средства, ингибирующие коагуляцию, антитромбоцитарных или тромбоцит-ингибирующих средств, ингибиторы тромбина, тромболитические или фибринолитические средства, противоаритмические средства, гипотензивные средства, блокаторы кальциевых каналов (L-типа и T-типа), сердечные гликозиды, диуретические средства, агонисты минералокортикоидного рецептора, NO-обеспечивающие средства, такие как органические нитраты, NO-промотирующие средства, такие как ингибиторы фосфодиэстеразы, холестерин/липид-понижающие средства, терапевтические средства липидного профиля, противодиабетические средства, антидепрессанты, противовоспалительные средства (стероидные и нестероидные), противоостепорозные средства, гормон-заместительные терапевтические средства, пероральные контрацептивы, средства против ожирения, анксиолетические средства, антипролиферативные средства, противоопухолевые средства, противоязвенное средства и средства против гастроэзофагеального рефлюксного заболевания, гормон роста и/или средства, усиливающие секрецию соматотропного гормона, тироидные миметики (включая антагонисты рецепторов тироидного гормона), противоинфекционные средства, противовирусные средства, бактерицидные и фунгицидные средства.
Средства, применяемые в условиях интенсивной терапии, включают, например, добутамин, допамин, эпинефрин, нитроглицерин, нитропруссид и т.д.
Средства для комбинированного применения, подходящие для лечения васкулита, включают, например, азатиоприн, циклофосфамид, микофенолят, мофетил, ритуксимаб и т.д.
В другом варианте осуществления настоящее изобретение относится к комбинации, в которой третье фармацевтическое средство представляет собой по меньшей мере одно фармацевтическое средство, выбранное из ингибитора фактора Xa, противокоагулянтного средства, антитромбоцитарного средства, средства ингибирования тромбина, тромболитического средства и фибринолитического средства. Типичные примеры ингибиторов фактора Xa включают апиксабан и ривароксабан. Примеры подходящих антикоагулянтов для применения в комбинации с соединениями по настоящему изобретению включают гепарины (например, нефракционированные и низкомолекулярные гепарины, такие как эноксапарин и далтепарин).
В другом предпочтительном варианте осуществления третье фармацевтическое средство представляет собой по меньшей мере одно средство, выбранное из варфарина, дабигатрана, нефракционированного гепарина, низкомолекулярного гепарина, синтетического пентасахарида, гирудина, аргатробана, аспирина, ибупрофена, напроксена, сулиндака, индометацина, мефенамата, дроксикама, диклофенака, сульфинпиразона, пироксикама, тиклопидина, клопидогрела, тирофибана, эптифибатида, абциксимаба, мелагатрана, дисульфатогирудина, активатора тканевого плазминогена, модифицированного активатора тканевого плазминогена, анистреплазы, урокиназы и стрептокиназы.
Предпочтительное третье фармацевтическое средство представляет собой по меньшей мере одно антитромбоцитарное средство. Особенно предпочтительными антитромбоцитарными средствами являются аспирин и клопидогрел.
Термин «антитромбоцитарные средства» (или «тромбоцит-ингибирующие средства»), когда используется в настоящем описании, означает средства, которые ингибируют функцию тромбоцитов, например ингибированием агрегации, адгезии или гранулярной секреции тромбоцитов. Указанные средства включают, но не ограничиваются ими, различные известные нестероидные противовоспалительные лекарственные средства (non-steroidal anti-inflammatory drugs - NSAID), такие как аспирин, ибупрофен, напроксен, сулиндак, индометацин, мефенамат, дроксикам, диклофенак, сульфинпиразон, пироксикам и их фармацевтически приемлемые соли и пролекарства. Среди NSAIDS предпочтительными являются аспирин (ацетилсалициловая кислота или ASA) и ингибиторы ЦОГ-2, такие как целебрекс или пироксикам. Другие подходящие средства ингибирования тромбоцитов включают антагонисты IIb/IIIa (например, тирофибан, эптифибатид и абциксимаб), антагонисты рецептора тромбоксана-A2 (например, ифетробан), ингибиторы тромбоксан-A2-синтетазы, ингибиторы PDE-III (например, плетал, дипиридамол) и их фармацевтически приемлемые соли и пролекарства.
Подразумевается, что термин «антитромбоцитарные средства» (или «тромбоцит-ингибирующие средства»), когда используется в настоящем описании, включает антагонисты рецепторов ADP (аденозиндифосфата), предпочтительно антагонисты пуринергических рецепторов P2Y1 и P2Y12, более предпочтительно P2Y12. Предпочтительные антагонисты рецепторов P2Y12 включают тикагрелор, прасугрел, тиклопидин и клопидогрел и их фармацевтически приемлемые соли и пролекарства. Клопидогрел является еще более предпочтительным средством. Тиклопидин и клопидогрел также являются предпочтительными соединениями, поскольку известно, что при применении они мягко воздействуют на желудочно-кишечный тракт.
Термин «ингибиторы тромбина» (или «антитромбиновые средства»), когда используется в настоящем изобретении, означает ингибиторы тромбина серинпротиазы. С помощью ингибирования тромбина происходит нарушение различных процессов, опосредуемых тромбином, таких как опосредуемая тромбином активация тромбоцитов (то есть, например, агрегация тромбоцитов и/или гранулярная секреция ингибитора-1 активатора плазминогена и/или серотонина) и/или образование фибрина. Специалисту в данной области техники известны ингибиторы тромбина, и подразумевается, что эти ингибиторы предназначены для применения в комбинации с соединениями по настоящему изобретению. Такие ингибиторы включают, но не ограничиваются только ими, производные бороаргинина, боропептиды, дабигатран, гепарины, гирудин, аргатробан и мелагатран, включая их фармацевтически приемлемые соли и пролекарства. Производные бороаргинина и боропептиды включают N-ацетильные и пептидные производные бороновой кислоты, такие как производные C-концевой альфа-аминобороновой кислоты лизина, орнитина, аргинина, гомоаргинина и их соответствующие изотиоурониевые аналоги. Термин «гирудин», когда используется в настоящем описании, включает подходящие производные или аналоги гирудина, называемые в описании гирулогами, такие как дисульфатогирудин. Термин «тромболитики» или «фибринолитические средства» (или «тромболитики» или «фибринолитики»), когда используется в настоящем описании, означает фармацевтические средства, которые лизируют сгустки крови (тромбы). Такие средства включают тканевый активатор плазминогена (природный или рекомбинантный) и его модифицированные формы, анистреплазу, урокиназу, стрептокиназу, тенектеплазу (TNK), ланотеплазу (nPA), ингибиторы фактора VIIa, ингибиторы PAI-1 (т.е. инактиваторы ингибиторов тканевого активатора плазминогена), ингибиторы альфа-2-антиплазмина и анизоилированный комплекс активатора плазминогена и стрептокиназы, включая их фармацевтически приемлемые соли и пролекарства. Термин «анистреплаза», когда используется в настоящем описании, относится к анизоилированному комплексу активатора плазминогена и стрептокиназы, как описано, например, в публикации EP 028489, содржание которой включено в настоящее описание в виде ссылки. Термин «урокиназа», когда используется в настоящем описании, означает как двухцепочечную, так и одноцепочечную урокиназу, причем последняя также упоминается в настоящем описании как проурокиназа.
Примеры подходящих противоаритмических средств включают фармацевтические средства I класса (такие как пропафенон); фармацевтические средства II класса (такие как метопролол, атенолол, карведилол и пропранолол); фармацевтические средства III класса (такие как соталол, дофетилид, амиодарон, азимилид и ибутилид); фармацевтические средства IV класса (такие как дилтиазем и верапамил); активаторы калиевых (K+) каналов, такие как ингибиторы IAch и ингибиторы IKur (например, описанные в WO01/40231).
Соединения по настоящему изобретению могут совместно вводиться со средствами против сердечной недостаточности, такими как ингибиторы ACE (например, каптоприл, эналаприл, фосиноприл, лизиноприл, периндоприл, квинаприл, рамиприл, трандолаприл), блокаторы рецепторов ангиотензина II (например, кандесартан, лозартан, валсартан), ингибиторы ангиотензиновых рецепторов наприлизина (сакубитрил/валсартан), блокатор If каналов ивабрадин, бета-адренергические блокаторы (например, бисопролол, метопролола сукцинат, карведилол), антагонист альдестерона (например, спиронолактон, эплеренон), гидралазин и изосорбида динитрат, диуретические средства (например, фуросемид, буметанид, торсемид, хлортиазид, амилорид, гидрохлортиазид, индапамид, метолазон, триамтерен) или дигоксин.
Соединения по настоящему изобретению могут использоваться в комбинаци с гипотензивными средствами, и такая гипотензивная активность легко определяется специалистами в данной области технии в соответствии со стандартными анализами (например, измерением кровяного давления). Примеры подходящих гипотензивных средств включают альфа-адреноблокаторы; бета-адреноблокаторы; блокаторы кальциевых каналов (например, дилтиазем, верапамил, нифедипин и амлодипин); вазодилататоры (например, гидралазин), диуретические средства (например, хлортиазид, гидрохлортиазид, флуметиазид, гидрофлуметиазид, бендрофлуметиазид, метилхлортиазид, трихлорметиазид, политиазид, бензтиазид, этакриновую кислоту, трикринафен, хлорталидон, торасемид, фуросемид, индапамид, метозолон (metozolone), мусолимин, буметанид, триамтерен, амилорид, спиронолактор); ингибиторы ренина; ингибиторы ACE (например, каптоприл, зофеноприл, фосиноприл, эналаприл, кераноприл, цилазоприл, делаприл, пентоприл, периндоприл, квинаприл, рамиприл, трандолаприл, лисиноприл); антагонисты рецептора AT-1 (например, лозартан, ирбесартан, валсартан); ингибиторы рецепторов ангиотензина/неприлизина (сакубитрил/валсартан); блокаторы бета-адренергических рецепторов (например, бисопролол, метапролола сукцинат, карведилол); антагонисты рецептора ET (например, ситаксентан, антрасентан и соединения, описанные в Патентах США № 5612359 и 6043265); двойные антагонисты ET/AII (например, соединения, описанные в WO 00/01389); ингибиторы нейтральной эндопептидазы (neutral endopeptidase - NEP); ингибиторы вазопептидаз (двойные ингибиторы NEP-ACE) (например, гемопатрилат и нитраты). Типичным антиангинальным средством является ивабрадин.
Примеры подходящих блокаторов кальциевых каналов (L-типа или T-типа) включают дилтиазем, варапамил, нифедипин, амлодипин и мибефрадил.
Примеры подходящих сердечных гликозидов включают дигиталис и уабаин.
В одном варианте осуществления настоящего изобретения, соединения по настоящему изобретению могут совместно вводиться с одним или несколькими диуретическими средствами. Примеры подходящих диуретических средств включают (a) петлевые диуретики, такие как фуросемид (например, LASIX™), торсемид (например, DEMADEX™), беметанид (например, BUMEX™) и этакриновая кислота (например, EDECRIN™); (b) тиазидные диуретики, такие как хлортиазид (например, DIURIL™, ESIDRIX™ или HYDRODIURIL™), гидрохлортиазид (например, MICROZIDE™ или ORETIC™), бензтиазид, гидрофлуметиазид (например, SALURON™), бендрофлуметиазид, метихлортиазид, политиазид, трихлорметиазид и индапамид (например, LOZOL™); (c) фталимидиновые диуретики, такие как хлортиалидон (например, HYGROTON™) и метолазон (например, ZAROXOLYN™); (d) хиназолиновые диуретики, такие как хинеразон; и (e) калийсберегающие диуретики, такие как триамтерен (например, DYRENIUM™) и амилорид (например, MIDAMOR™ или MODURETIC™).
В другом варианте осуществления настоящего изобретения соединения по настоящему изобретению могут вводиться совместно с петлевым диуретиком. В еще одном варианте осуществления петлевой диуретик выбран из фуросемида и торсемида. В еще одном варианте осуществления соединения по настоящему изобретению могут вводиться совместно с фуросемидом. В еще одном варианте осуществления соединения по настоящему изобретению могут совместно вводиться с торсемидом, который необязательно может быть представлен в форме с контролируемым или модифицированным высвобождением действующего вещества.
В другом варианте осуществления настоящего изобретения, соединения по настоящему изобретению могут совместно вводиться с тиазидным диуретиком. В еще одном варианте осуществления тиазидный диуретик выбран из группы, состоящей из хлортиазида и гидрохлортиазида. В еще одном варианте осуществления соединения по настоящему изобретению могут совместно вводиться с хлортиазидом. В еще одном варианте осуществления соединения по настоящему изобретению могут совместно вводиться с гидрохлортиазидом.
В другом варианте осуществления настоящего изобретения, соединения по настоящему изобретению могут совместно вводиться с фталимидиновым диуретиком. В еще одном варианте осуществления фталимидиновый диуретик представляет собой хлорталидон. Примеры подходящих антагонистов минералокортикоидных рецепторов включают спиронолактон и эплеренон. Примеры подходящих ингибиторов фосфодиэстеразы включают ингибиторы PDE II (такие как цилостазол); и ингибиторы PDE V (такие как силденафил).
Специалистам в данной области техники понятно, что соединения по настоящему изобретению могут использоваться в сочетании с другим сердечно-сосудистым или цереброваскулярным лечением, включая PCI, стентирование, стенты с лекарственным покрытием, лечение стволовыми клетками и изделия медицинского назначения, такие как имплантируемые кардиостимуляторы, дефибрилляторы или сердечная ресинхронизирующая терапия.
В частности, при использовании в виде лекарственной формы стандартной дозы существует возможность химического взаимодействия между объединенными активными ингредиентами. По этой причине, когда первое терапевтическое средство и второе терапенвтическое средство объединяются в одну лекарственную форму стандартной дозы, препарат получают так, что, хотя активные ингредиенты объединяются в лекарственную форму стандартной дозы, физический контакт между активными ингредиентами минимизируется (то снижается). Например, один активный ингредиент может покрываться энтеросолюбильным покрытием. С помощью энтеросолюбильного покрытия одного из активных ингредиентов можно не только минимизировать контакт между объединенными активными ингредиентами, но и контролировать высвобождение одного из этих компонентов в желудочно-кишечном тракте, так что один их этих компонентов высвобождается не в желудке, а в кишечнике. Один из активных ингредиентов также может быть покрыт материалом, который замедленно высвобождает активный ингредиент по всему желудочно-кишечному тракту, а также служит для снижения до минимума физического контакта между объединенными активными ингредиентами. Кроме того, компонент с замедленным высвобождением действующего вещества может дополнительно покрываться энтеросолюмибьным покрытием, так что высвобождение этого компонента происходит только в кишечнике. Еще один подход предполагает получение комбинированного продукта, в котором один компонент покрыт полимером замедленного и/или энтеросолюбильного высвобождения действующего вещества, а другой компонент также покрыт полимером, таким как гидроксипропилметилцеллюлоза (ГПМЦ) низкой вязкости или другими подходящими материалами, которые известны в данной области техники, для дополнительного разделения активных компонентов. Полимерное покрытие служит для образования дополнительного барьера, препятствующего взаимодействию с другим компонентом.
Эти, а также другие способы минимизации контакта между компонентами комбинированных продуктов по настоящему изобретению, вводимых в одной лекарственной форме или вводимых в отдельных формах, но одновременно одним и тем же способом введения, будут понятны квалифицированным специалистам данной области техники после ознакомления с настоящим описанием.
В лечении комбинированной терапией соединения по настоящему изобретения и другие лекарственные средства вводят млекопитающим (например, людям, самцам или самкам) стандартными методами.
Доза каждого терапевтического средства, например соединения A, соединения D и любого дополнительного терапевтического средства, обычно зависит от ряда факторов, включая состояние здоровья субъекта, подлежащего лечению, объем необходимого лечения, природу и вид сопутствующего лечения, если таковое имеется, а также частоту вводимого лечения и характер желаемого эффекта. Обычно доза каждого терапевтического средства находится интервале от примерно 0,001 мг до примерно 100 мг на килограмм массы тела индивидуума в день, предпочтительно от примерно 0,1 мг до примерно 10 мг на килограмм массы тела индивидуума в день. Однако может также потребоваться некоторое изменение в общем интервале доз в зависимости от возраста и массы тела субъекта, подлежащего лечению, предполагаемого способа введения, конкретного вводимого средства против ожирения и т.п. Определение интервалов доз и оптимальных доз для конкретного пациента также находится в компетенции специалистов в данной области техники, располагающего преимуществами настоящего изобретения.
В соответствии со способами лечения по настоящему изобретению соединение по настоящему изобретению или комбинации соединения по настоящему изобретению и по меньшей мере одного дополнительного фармацевтического средства (называемая в описание «комбинация») вводится субъекту, нуждающемуся в таком лечении, предпочтительно в форме фармацевтической композиции. В аспекте комбинации по настоящему изобретению соединение по настоящему изобретению и по меньшей мере одно другое фармацевтическое средство (например, другое средство против ожирения) могут вводиться раздельно или в фармацевтической композиции, включающей оба активных ингредиента. Обычно такое введение предпочтительно является пероральным.
Когда комбинация соединения по настоящему изобретению и по меньшей мере одно другое фармацевтическое средство вводятся совместно, такое введение может быть последовательным по времени или одновременным. Одновременное введение лекарственных комбинаций является предпочтительным. При последовательном введении соединение по настоящему изобретению и дополнительное фармацевтическое средство могут вводиться в любом порядке. Обычно такое введение предпочтительно является пероральным. Особенно предпочтительным является пероральное и одновременное введение. Когда соединение по настоящему изобретению и дополнительное фармацевтическое средство вводятся последовательно, их введение может осуществляться одинаковыми или разными способами.
В соответствии со способами по настоящему изобретению соединение по настоящему изобретению или комбинация предпочтительно вводится в форме фармацевтической композиции. Соответственно, соединение по настоящему изобретению или комбинация может вводиться пациенту отдельно или вместе в любой обычной лекарственной форме для перорального, ректального, трансдермального, парентерального (например, внутривенного, внутримышечного или подкожного), интрацистернального, интравагинального, интраперитонеального, местного (например, в форме порошка, мази, крема, спрея или лосьона), буккального или назального введения (например, спрей, капли или препарат для ингаляции).
Соединения по настоящему изобретению или комбинации могут вводиться сами по себе, но обычно будут вводиться в смеси с одним или несколькими подходящими фармацевтическими эксципиентами, адъювантами, разбавителями или носителями, известными в данной области техники и выбранными с учетом предполагаемого способа введения и стандартной фармацевтической практики. Соединение по настоящему изобретению или комбинация могут вводиться в препарат для получения лекарственных форм с немедленным, отсроченным, модифицированным, замедленным, импульсным или контролируемым высвобождением действующего вещества в зависимости от желаемого способа введения и специфичности профиля высвобождения, соизмеримых с терапевтическими потребностями.
Фармацевтическая композиция включает соединение по настоящему изобретению или комбинацию в количестве обычно в интервале от примерно 1% до примерно 75%, 80%, 85%, 90% или даже 95% (по массе) композиции, обычно в интервале от примерно 1%, 2% или 3% до примерно 50%, 60% или 70%, чаще в интервале от примерно 1%, 2% или 3% до менее 50%, например примерно 25%, 30% или 35%.
Способы получения различных фармацевтических композиций с конкретным количеством активного соединения известны специалистам в данной области техники (см., например, Remington: The Practice of Pharmacy, Lippincott Williams и Wilkins, Baltimore Md. 20.sup.th ed. 2000).
Композиции, подходящие для парентеральной инъекции, обычно включают фармацевтически приемлемые стерильные водные или неводные растворы, дисперсии, суспензии или эмульсии, а также стерильные порошки для разведения с получением стерильных растворов или дисперсий для инъекций. Примеры подходящих водных и неводных носителей или разбавителей (включая растворители и несущие среды для лекарственных веществ) включают воду, этанол, многоатомные спирты (пропиленгликоль, полиэтиленгликоль, глицерин и т.п.), их подходящие смеси, триглицериды, включая растительные масла, такие как оливковое масло, и съедобные органические сложные эфиры, такие как этилолеат. Предпочтительным носителем является Miglyol® (торговая марка) - сложный эфир каприловой/каприновой кислоты с пропиленгликолем (например, Miglyol.R™. 812, Miglyol.R™. 829, Miglyol.R™. 840), доступный от Condea Vista Co., Cranford, N.J. Необходимую текучесть можно поддерживать, например, с помощью применения покрытия, такого как лецитин, обеспечения требуемого размера частиц в случае дисперсии и с помощью поверхностно-активных веществ.
Эти композиции для парентеральной инъекции также могут содержать эксципиенты, такие как консерванты, смачивающие агенты, эмульгаторы и дисперсанты. Предотвращение заражения композиций микроорганизмами может осуществляться с помощью различных бактерицидных и фунгицидных срдств, например парабенов, хлорбутанола, фенола, сорбиновой кислоты и т.п. Может быть желательно применение изотонических средств, например сахаров, хлорида натрия и т.п. Пролонгированная абсорбция вводимых инъекцией фармацевтических композиций может быть достигнута применением добавок, способных замедлять абсорбцию, например моностеарата алюминия и желатина.
Твердые лекарственные формы для перорального введения включают капсулы, таблетки, жевательные таблетки, пастилки, пилюли, порошки и препараты, состоящие из множества частиц (гранулы). В таких твердых лекарственных формах соединение по настоящему изобретению или комбинация смешивается по меньшей мере с одним инертным эксципиентом, разбавителем или носителем. Подходящие эксципиенты, разбавители или носители включают такие материалы, как цитрат натрия или дикальцийфосфат и/или (a) один или несколько наполнителей или разбавителей (например, микрокристаллическую целлюлозу (доступную как Avicel.™. от FMC Corp.), крахмалы, лактоза, сахарозу, маннит, кремниевую кислоту, ксилит, сорбит, декстрозу, гидрофосфат кальция, декстрин, альфа-циклодекстрин, бета-циклодекстрин, полиэтиленгликоль, жирные кислоты со средней длиной цепи, оксид титана, оксид магния, оксид алюминия и т.п.); (b) одно или несколько связующих веществ (например, карбоксиметилцеллюлозу, метилцеллюлозу, гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу, желатин, гуммиарабик, этилцеллюлозу, поливиловый спирт, пуллулан, прежелатинированный крахмал, агар, трагакант, альгинаты, желатин, поливинилпирролидон, сахарозу, гуммиарабик и т.п.); (c) один или несколько увлажнителей (например, глицерин и т.п.); (d) один или несколько дезинтегрирующих веществ (например, агар-агар, карбонат кальция, картофельный крахмал или крахмал из тапиоки, алльгиновая кислота, некоторые комплексные силикаты, карбонат натрия, лаурилсульфат натрия, натрийкрахмалгликолят (доступный как Explotab™ от Edward Mendell Co.), поперечно сшитый поливинилпирролидон, кросскармеллоза натрия A-типа (доступная как Ac-di-sol.™.), полиакрилин калия (polyacrilin potassium) (ионно-обменная смола) и т.п.); (e) один или несколько замедлителей растворения (например, парафин и т.п.); (f) один или несколько ускорителей абсорбции (например, четвертичные аммониевые соединения и т.п.); (g) один или несколько смачивающихся агентов (например, цетиловый спирт, глицеринмоностеарат и т.п.); (h) один или несколько адсорбентов (например, каолин, бентонит и т.п.); и/или (i) одно или несколько смазывающих веществ (например, тальк, стеарат кальция, стеарат магния, стеариновая кислота, полиоксилстеарат, цетанол, тальк, гидрированное касторовое масло, сложные эфиры сахарозы и жирных кислот, диметилполисилоксан, микрокристаллический воск, желтый пчелиный воск, белый пчелиный воск, твердые полиэтиленгликоли, лаурилсульфат натрия и т.п.). В случае капсул и таблеток лекарственные формы могут также включать буферные агенты.
Твердые композиции аналогичного типа могут также использоваться в качестве наполнителей в мягкие или твердые желатиновые капсулы с использованием таких эксципиентов как лактоза, молочный сахар, а также высокомолекулярные полиэтиленгликоли и т.п.
Твердые лекарственные формы, такие как таблетки, драже, капсулы и гранулы, могут быть получены с покрытиями и оболочками, такими как энтеросолюбильные покрытия и др., хорошо известными в данной области техники. Они также могут содержать опалесцирующие компоненты и могут быть представлены в композиции, которые высвобождают соединение по настоящему изобретению и/или дополнительное фармацевтическое средство замедленным образом. Примерами встраиваемых композиций, которые могут использоваться, являются полимерные вещества и воски. Лекарственное средство также может быть представлено в микроинкапсулированной форме, если это подходит, с одним или несколькими указанными выше эксципиентами.
В таблетках активный ингредиент будет обычно составлять менее 50% (по массе) препарата, например менее примерно 10%, например 5% или 2,5% по массе. Большую часть такого препарата составляют наполнители, разбавители, дезинтеграторы, смазочные материалы и, возможно, вкусовые добавки. Композиция этих вспомогательных веществ хорошо известна в данной области. Часто наполнители/разбавители содержат смеси двух или более следующих компонентов: микрокристаллическая целлюлоза, маннит, лактоза (все виды), крахмал и дикальцийфосфат. Смеси наполнитель/разбавитель обычно составляют менее 98% препарата, предпочтительно менее 95%, например 93,5%. Предпочтительные дезинтегрирующие вещества включают Ac-di-sol.™., Explotab.™., крахмал и лаурилсульфат натрия. Дезинтегрирующее вещество, когда присутствует, обычно составляет менее 10% препарата или менее 5%, например примерно 3%. Предпочтительным смазывающим веществом является стеарат магния. Смазывающее вещество, когда присутствует, обычно составляет менее 5% препарата или менее 3%, например примерно 1%.
Таблетки могут производиться стандартными способами таблетирования, например прямым прессованием либо влажной или сухой грануляцией или грануляцией из расплава и экструзией. Ядра таблеток могут быть однослойными или многослойными и могут быть покрыты соответствующими покрытиями, известными в данной области техники.
Жидкие лекарственные формы для перорального введения включают фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы и эликсиры. Помимо соединения по настоящему изобретению или комбинации жидкая лекарственная форма может содержать инертные разбавители, обычно используемые в данной области техники, такие как вода или другие растворители, солюбилизаторы и эмульгаторы, например этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, диметилформамид, масла (например, хлопковое масло, арахисовое масло, масло зародышей кукурузы, оливковое масло, касторовое масло, кунжутное масло и др.), Miglyole™ (доступный от CONDEA Vista Co., Cranford, N.J.), глицерин, тетрагидрофуриловый спирт, полиэтиленгликоли и сложные эфиры жирных кислот сорбитана или смеси этих веществ и т.п.
Помимо таких инертных разбавителей композиция может также включать такие эксципиенты как смачивающие агенты, эмульгирующие и суспендирующие агенты, подсластители, вкусовые добавки и отдушки.
Пероральные жидкие формы соединений по настоящему изобретению или их комбинации включают растворы, в которых активное соединение является полностью растворенным. Примеры растворителей включают все известные фармацевтические растворители, подходящие для перорального введения, в частности растворители, в которых соединения изобретения проявляют хорошую растворимость, например полиэтиленгликоль, полипропиленгликоль, пищевые масла и системы на основе глицерина и глицеридов. Системы на основе глицерина и глицеридов могут включать, например, следующие фирменные продукты (и соответствующие джнериковые препараты): Captex™ 355EP (глицерилтрикаприлат/капрат, от Abitec, Columbus Ohio), Crodamol™ GTC/C (среднецепочечный триглицерид, от Croda, Cowick Hall, UK) или Labrafac™ CC (среднецепочечный триглицерид, от Gattefosse), Captex™ 500P (глицерилтриацетат, т.е. триацетин, от Abitec), Capmul ™ MCM (среднецепочечные моно- и диглицериды, от Abitec), Migyol™ 812 (каприловый/каприновый триглицерид, от Condea, Cranford N.J.), Migyol™ 829 (каприловый/каприновый/янтарный триглицерид, от Condea), Migyol™ 840 (пропиленгликольдикаприлат/дикапрат, от Condea), Labrafil™ M1944CS (олеоилмакрогол-6-глицериды, от Gattefosse), Peceol™ (глицерилмоноолеат, от Gattefosse) и Maisine™ 35-1 (глицерилмоноолеат, от Gattefosse). Особый интерес представляют среднецепочечные (примерно от С8 до С10) триглицеридные масла. Эти растворители часто составляют преобладающую часть композиции, т.е. более примерно 50%, обычно более примерно 80%, например примерно 95% или 99%. Адъюванты и добавки также могут быть включены в состав растворителей, главным образом в качестве маскирующих вкус веществ, улучшителей вкуса и вкусовых добавок, антиоксидантов, стабилизаторов, модификаторов текстуры и вязкости и солюбилизаторов.
Суспензии помимо соединения по настоящему изобретению или комбинации могут дополнительно содержать носители, такие как суспендирующие агенты, например, этоксилированные изостеариловые спирты, полиоксиэтиленсорбитол и сложные эфиры сорбита, микрокристаллическая целлюлоза, метагидроксид алюминия, бентонит, агар-агар и трагакант или смеси этих веществ и т.п.
Композиции для ректального или вагинального введения предпочтительно включают суппозитории, которые могут быть получены смешиванием соединения по настоящему изобретению или комбинации с подходящими, не вызывающими раздражения эксципиентами или носителями, такими как масло какао, полиэтиленгликоль или воск для суппозиторий, которые являются твердыми при обычной комнатной температуре, но жидкими при температуре тела и, следовательно, плавятся в прямой кишке или вагинальной полости, высвобождая таким образом активный(е) компонент(ы).
Лекарственные формы для местного введения соединений по настоящему изобретению или комбинаций включают мази, кремы, лосьоны, порошки и спреи. Лекарственные средства смешиваются с фармацевтически приемлемым наполнителем, разбавителем или носителем, а также с любыми консервантами, буферными агентами или пропеллентами, которые могут потребоваться.
Если соединения являются плохорастворимыми в воде, например растворимость составляет менее 1 мкг/мл, то для этих соединений предпочтительной лекарственной формой являются жидкие композиции в солюбилизирующих неводных растворителях, таких как рассмотренные выше среднецепочечные триглицеридные масла.
Твердые аморфные дисперсии, в том числе дисперсии, полученные в процессе распылительной сушки, также являются предпочтительной лекарственной формой для плохорастворимых соединений по настоящему изобретению. Термин «твердая аморфная дисперсия» означает твердый материал, в котором по меньшей мере часть плохорастворимого соединения находится в аморфной форме и диспергирована в растворимом в воде полимере. Термин «аморфный» означает то, что плохорастворимое соединение не является кристаллическим. Термин «кристаллический» означает, что соединение проявляет дальний порядок в трех измерениях по меньшей мере 100 повторяющихся единиц в каждом измерении. Таким образом, подразумевается, что термин «аморфный» включает не только материала, который по существу не имеет порядка, но и материал, который может иметь некоторую небольшую степень порядка, но порядок находится менее чем в трех измерениях и/или только на коротких расстояниях. Аморфный материал может быть характеризоваться с использованием методов, известных в данной области техники, такими как порошковая рентгеновская дифракционная кристаллография (ПРДК), ЯМР твердого вещества или термические методы, такие как дифференциальная сканирующая калориметрия (ДСК).
Предпочтительно, по меньшей мере основная часть (т.е. по меньшей мере примерно 60% масс.) плохорастворимого соединения в твердой аморфной дисперсии является аморфной. Соединение может существовать внутри твердой аморфной дисперсии в относительно чистых аморфных доменах или областях, в виде твердого раствора соединения, гомогенно распределенного по всему полимеру, или в любой комбинации этих состояний или состояний, которые занимают промежуточное положение между ними. Предпочтительно, твердая аморфная дисперсия является по существу гомогенной, чтобы аморфное соединение было диспергировано гомогенно, насколько это возможно, по всему полимеру. Термин «по существу гомогенный», когда используется в настоящем описании, означает, что доля соединения, которая присутствует в относительно чистых аморфных доменах или областях внутри твердой аморфной дисперсии, относительно мала, составляет менее 20% масс., предпочтительно менее 10% масс., общего количества лекарственного средства.
Растворимые в воде полимеры, подходящие для использования в твердых аморфных дисперсиях, должны быть инертными, то есть не вступать в химическую реакцию с плохорастворимым соединением неблагоприятным образом, быть фармацевтически приемлемыми и обладать по меньшей мере некоторой растворимостью в водном растворе при физиологически значимых рН (например, 1-8). Полимер может быть нейтральным или ионизируемым, и его растворимость в воде должна составлять минимум 0,1 мг/мл по меньшей мере при значениях рН в интервале 1-8.
Растворимые в воде полимеры, подходящие для использования в настоящем изобретении, могут быть целлюлозными или нецеллюлозными. Полимеры могут быть нейтральными или ионизируемыми в водном растворе. Из них предпочтительными являются ионизируемые и целлюлозные полимеры, причем более предпочтительными являются ионизируемые целлюлозные полимеры.
Типичные растворимые в воде полимеры включают сукцинат ацетата гидроксипропилметилцеллюлозы (hydroxypropyl methyl cellulose acetate succinate - HPMCAS), гидроксипропилметилцеллюлозу (hydroxypropyl methyl cellulose - HPMC), фталат гидроксипропилметилцеллюлозы (hydroxypropyl methyl cellulose phthalate - HPMCP), карбоксиметилэтилцеллюлозу (carboxy methyl ethyl cellulose - CMEC), фталат ацетата целлюлозы (cellulose acetate phthalate - CAP), тримеллит ацетата целлюлозы (cellulose acetate trimellitate - CAT), поливинилпирролидон (polyvinylpyrrolidone - PVP), гидроксипропилцеллюлозу (hydroxypropyl cellulose - HPC), метилцеллюлозу (methyl cellulose - MC), блок-сополимеры оксида этилена и оксида пропилена (PEO/PPO, также известные как полоксамеры) и их смеси. Особенно предпочтительными полимерами являются HPMCA, HPMC, HPMCP, CMEC, CAP, CAT, PVP, полоксамеры и их смеси. Наиболее предпочтительны HPMCA (см. Публикацию Европейской патентной заявки № 0901786 А2, содержание которой включено в настоящее описание в виде ссылки).
Твердые аморфные дисперсии могут быть получены в соответствии с любым способом получения твердых аморфных дисперсий, в результате которого по меньшей мере большая часть (по меньшей мере 60%) плохорастворимого соединения находится в аморфном состоянии. К таким способам относятся механические и термические способы, а также способы с применением растворителей. Типичные примеры механических способов включают измельчение и экструзию; к термическим относятся способы получения расплава, включая высокотемпературное плавление, плавление, модифицированное растворителем, способы с использованием застывания расплава; и способы с применением растворителей включают осаждение нерастворителем, нанесение покрытия опрыскиванием и распылительную сушку (см., например, следующие Патенты США № 5456923 и 5939099, в которых описывается получение дисперсий с помощью процессов экструзии; № 5340591 и 4673564, в которых описывается получение дисперсий с помощью измельчения; и № 5707646 и 4894235, в которых описывается получение дисперсий с помощью застывания расплава; содержание всех указанных патентов включено в настоящее описание в виде ссылок). В предпочтительном способе твердую аморфную дисперсию получают распылительной сушкой, как описано в публикации Европейской Заявки на патент № 0901786 А2. В этом способе соединение и полимер растворяют в растворителе, таком как ацетон или метанол, а затем растворитель быстро удаляют из раствора распылительной сушкой с образованием твердой аморфной дисперсии. Твердые аморфные дисперсии могут быть получены так, чтобы при желании они содержали до 99% масс. соединения, например 1% масс., 5% масс., 10% масс., 25% масс., 50% масс., 75% масс., 95% масс. или 98% масс.
Твердая дисперсия может использоваться сама по себе в качестве лекарственной формы или в качестве изделия для промышленного применения (manufacturing-use-product - MUP) при получении других лекарственных форм, таких как капсулы, таблетки, растворы или суспензии. Примером водной суспензии является водная суспензия 1:1 (масс./масс.) соединение/HPMCASHF, высушенная распылением дисперсии, содержащей 2,5 мг/мл соединения в 2% полисорбате-80. Твердые дисперсии для использования в таблетке или капсуле обычно смешиваются с другими вспомогательными веществами или адъювантами, традиционно использующимися в таких лекарственных формах. Например, типичный наполнитель для капсул содержит смесь 2:1 (масс./масс.) соединение/дисперсия HPMCAS-MF, высушенная распылением (60%), лактозу (высокотекучести) (15%), микрокристаллическую целлюлозу (например, Avicel(R0-102) (15,8%), натриевый крахмал (7%), лаурилсульфат натрия (2%) и стеарат магния (1%).
HPMCAS полимеры доступны в качестве изделий низкого, среднего и высокого сортов, таких как Aqoa(R)-LF, Aqoat(R)-MF и Aqoat(R)-HF, соответственно, от Shin-Etsu Chemical Co., LTD, Токио, Япония. Обычно предпочтительными являются продукты более высоких сортов - MF и HF.
Далее описываются типичные примеры препаратов, доз и т.д., применимые для животных кроме человека. Введение соединения А или его фармацевтически приемлемой соли в комбинации с соединением D или его фармацевтически приемлемой солью в качестве двух фармацевтических средств или в комбинации с еще одним фармацевтическим средством может осуществляться перорально или не перорально.
Количество соединения А или его фармацевтически приемлемой соли с соединением D или его фармацевтически приемлемой солью вместе или в комбинации с другим фармацевтическим средством вводят таким образом, чтобы была получена эффективная доза. Как правило, суточная доза, которая вводится животному перорально, составляет от 0,01 до 1000 мг/кг массы тела, например от 0,01 до 300 мг/кг, от 0,01 до 100 мг/кг, от 0,01 до 50 мг/кг массы тела, от 0,01 до 25 мг/кг, от 0,01 до 10 мг/кг или от 0,01 до 5 мг/кг. Суточная доза вводимого соединения А может составлять 2 мг, 3 мг, 5 мг, 10 мг, 15 мг, 20 мг, 25 мг, 30 мг или 50 мг. Суточная доза может быть разделена на несколько доз, например вводимых два раза в день с 12-часовым интервалом введения. Например, в некоторых случаях суточная доза соединения А может вводиться по 15 мг раз в 12 часов. Суточная доза вводимого соединения D может составлять 50 мг, 100 мг, 200 мг или 300 мг. Суточная доза может быть разделена на несколько доз, например вводиться два раза в день с интервалом в 12 часов. Например, в некоторых случаях суточная доза соединения D может вводиться введением 300 мг раз в 12 часов.
Обычно соединение по настоящему изобретению (или комбинацию) можно вводить в питьевой воде, чтобы терапевтическая доза соединения принималась с ежедневным потреблением воды. Соединение может быть непосредственно дозировано в питьевую воду, предпочтительно в виде жидкого растворимого в воде концентрата (например, водного раствора растворимой в воде соли).
Обычно соединение по настоящему изобретению (или комбинацию) также может добавляться непосредственно в корм или в виде кормовой добавки для животных, также называемой премиксом или концентратом. Премикс или концентрат соединения в эксципиенте, разбавителе или носителе чаще всего используется для включения фармацевтического средства в корм. Подходящие эксципиенты, разбавители или носители являются жидкими или твердыми, по желанию, такими как вода, различные виды муки, например люцерновая мука, соевая мука, хлопковая мука, льняная мука, мука кукурузных початков и кукурузная мука, меласса, мочевина, костная мука, и минеральные смеси, которые обычно используются в кормах для домашней птицы. Особенно эффективным эксципиентом, разбавителем или носителем является непосредственно соответствующий корм для животных, то есть небольшая часть такого корма. Носитель способствует равномерному распределению смеси в готовом корме, с которым смешивается премикс. Предпочтительно, смесь тщательно смешивается с премиксом, а затем с кормом. Для этого соединение может быть диспергировано или растворено в подходящем маслянистом носителе, таком как соевое масло, кукурузное масло, хлопковое масло и т.п., или в летучем органическом растворителе, а затем смешано с носителем. Следует иметь в виду, что пропорции соединения в концентрате могут значительно изменяться, поскольку количество соединения в готовом корме можно регулировать посредством смешивания в соответствующей пропорции премикса с кормом для получения нужного содержания соединения.
Высокоэффективные концентраты могут быть смешаны производителем корма с белковым носителем, таким как соевый шрот и другие шроты, как описано выше, для получения концентрированных добавок, которые подходят для непосредственного кормления животных. В таких случаях животные могут принимать обычный рацион. В качестве альтернативы такие концентрированные добавки могут добавляться непосредственно в корм для получения сбалансированного по питательным веществам готового корма, содержащего терапевтически эффективный уровень соединения по изобретению. Смеси тщательно смешиваются стандартными способами, например в двухшнековом блендере, для обеспечения однородности.
Если добавка вводится как верхняя пропитка в корм, она также способствует равномерному распределению соединения по верхней части пропитанного ею корма.
Питьевую воду и корм, применяемые для наращивания постного мяса и для улучшения соотношения постного мяса и жира, обычно готовят путем смешивания соединения по настоящему изобретению с достаточным количеством корма для животных, чтобы обеспечить от примерно 10-3 до примерно 500 м.д. соединения в корме или воде.
Предпочтительный лечебный корм для свиней, крупного рогатого скота, овец и коз обычно содержит от 1 до 400 граммов соединения по настоящему изобретению (или комбинации) на тонну корма, причем оптимальное количество для этих животных обычно составляет от 50 до 300 граммов на тонну корма.
Предпочтительные корма для домашней птицы и домашних животных обычно содержат от 1 до 400 граммов, предпочтительно от 10 до 400 граммов соединения по настоящему изобретению (или комбинации) на тонну корма.
Обычно для парентерального введения животным соединения по настоящему изобретению (или их комбинации) могут быть приготовлены в виде пасты или гранул и введены в виде имплантата под кожу головы или уха животного, которому требуется увеличение наращивания постного мяса и улучшение соотношения постного мяса и жира.
Препараты в форме пасты могут быть получены диспергированием лекарственного средства в фармацевтически приемлемом масле, таком как арахисовое масло, кунжутное масло, кукурузное масло или т.п.
Пеллеты, содержащие терапевтически эффективное количество соединения А или его фармацевтически приемлемой соли в комбинации с соединением D или его фармацевтически приемлемой солью, фармацевтическая композиция или комбинация могут быть получены посредством смешивания соединения А или его фармацевтически приемлемой соли с соединением D или его фармацевтически приемлемой солью и разбавителем, таким как карбовакс, карнубский воск и т.п., и для улучшения процесса гранулирования может быть добавлено смазывающее вещество, такое как стеарат магния или кальция.
Разумеется, понятно, что животному можно вводить более одного пеллета для достижения желаемой дозы, которая обеспечит наращивание постного мяса и улучшение соотношения постного мяса и жира. Кроме того, имплантаты также могут вводиться периодически во время лечения животных, чтобы поддерживать надлежащий уровень содержания препарата в организме животного.
Изобретение обладает несколькими полезными для ветеринарии отличительными признаками. Для владельца домашнего животного или ветеринара, который хочет добиться поджарости домашнего животного и/или избавить домашнее животное от нежелательного жира, настоящее изобретение предоставляет средства, с помощью которых это может буть достигнуто. Для производителей мяса птицы, говядины и свинины использование способа по изобретению приводит к получению более постного мяса животных, которое должно поступать в продажу по более высоким отпускным ценам.
ПРИМЕРЫ
Если не указано иное, исходные материалы обычно доступны из коммерческих источников, таких как Aldrich Chemicals Co. (Milwaukee, WI), Lancaster Synthesis, Inc. (Windham, NH), Acros Organics (Fairlawn, NJ), Maybridge Chemical Company, Ltd. (Cornwall, England) и Tyger Scientific (Princeton, NJ). В описании используются некоторые стандартные аббревиатуры и сокращения, такие как AcOH (уксусная кислота), DBU (1,8-диазабицикло[5.4.0]ундец-7-ен), CDI (1,1'-карбонилдиимидазол), ДХМ (дихлорметан), DEA (диэтиламин), DIPEA (N, N-диизопропилэтиламин), DMAP (4-диметиламинопиридин), ДМФА (N, N'-диметилформамид), ДМСО (диметилсульфоксид), EDCI (N-(3-диметиламинопропил)-N'-этилкарбодиимид), Et2O (диэтиловый эфир), EtOAc (этилацетат), EtOH (этанол), Г или г (грамм), HATU (гексафторфосфатметанаминий 2-(1H-7-азабензотриазол-1-ил)-1,1,3,3-тетраметилурония), HBTU (гексафторфосфат O-бензотриазол-1-ил-N, N,N',N'-тетраметилурония), HOBT (1-гидроксибензотриазол), Час. или час. (час), IPA (изопропиловый спирт), KHMDS (гексаметилдисилазан калия), MeOH (метанол), Л или л (литр), мл (миллилитр) MTBE (трет-бутилметиловый эфир), мг (миллиграмм), NaBH(OAc)3 (триацетоксиборогидрид натрия), NaHMDS (гексаметилдисилазан натрия), NMP (N-метилпирролидон), RH (относительная влажность), КТ или кт (комнатная температура, которая равна температуре окружающей среды (примерно от 20 до 25°C)), SEM ([2-(триметилсилил)этокси]метил), TEA (триэтиламин), ТФУК (трифторуксусная кислота), ТГФ (тетрагидрофуран) и T3P (ангидрид пропанфосфорной кислоты).
Спектры 1H ядерного магнитного резонанса (ЯМР) во всех случаях соответствуют предполагаемым структурам. Характеристические химические сдвиги (δ) приводятся в миллионных долях (м.д.) относительно остаточного протонного сигнала в дейтерированном растворителе (CHCl3 при 7,27 м.д.; CD2HOD при 3,31 м.д.) и описаны с использованием стандартных аббревиатур для обозначения основных пиков; например, с - синглет; д - дублет; т - триплет; кв - квартет; м - мультиплет; уш. - уширенный.
ssЯМР означает ЯМР спектр вещества в твердом состоянии.
ПРДГ означает порошковую рентгеновскую дифракцию.
Термин «по существу одинаковые», когда используется для описания порошковых рентгеновских дифрактограмм, означает дифрактограммы, включающие пики при углах дифракции, значения которых находятся в пределах стандартного отклонения +/- 0,2° 2Θ.
Термин «по существу чистая», когда используется в отношении конкретной кристаллической формы, означает, что указанная кристаллическая форма включает менее 10%, предпочтительно менее 5%, предпочтительно менее 3%, предпочтительно менее 1% по массе любой другой физической формы соединения A или соединения D.
Реакции обычно проводят на воздухе или, когда используются чувствительные к кислороду или влаге реагенты или промежуточные продукты, в инертной атмосфере (в атмосфере азота или аргона). При необходимости реакторы сушат в динамическом вакууме с использованием теплового пистолета и безводных растворителей (изделия Sure-Seal™ от Aldrich Chemical Company, изделия Milwaukee, Wisconsin или DriSolv™ от EMD Chemicals, Gibbstown, NJ). Коммерческие растворители и реагенты используют без дополнительной очистки. Когда указано, нагрев реакционный смесей проводят с использованием микроволнового излучения от Biotage Initiator или Personal Chemistry Emrys Optimizer. Ход реакций контролируют с использованием тонкослойной хроматографии (ТСХ), жидкостной хроматографии-масс-спектрометрии (ЖХМС), высокоэффективной жидкостных хроматографии (ВЭЖХ) и/или газовой хроматографии-масс-спектрометрии (ГХМС). ТСХ проводят на пластинах, предварительно покрытых силикагелем, с индикатором флуоресценции (с возбуждающей длиной волны 254 нм) и визуализацией под УФ светом и/или с помощью I2, KMnO4, CoCl2, фосфорномолибденовой кислоты и/или молибдата церия-аммония. ЖХМС данные получают на аппарате Agilent 1100 Series с автоматическим пробоотборником Leap Technologies, колонками Gemini C18 и с использованием градиентов MeCN/вода и в качестве модификаторов ТФУК, муравьиной кислоты или гидроксида аммония. Элюент колонки анализируют с использованием масс-спектрометра Waters ZQ, сканирующего в режимах как положительных, так и отрицательных ионов от 100 до 1200 Да. Используют также другие аналогичные инструменты. Данные ВЭЖХ получают на аппарате Agilent 1100 Series с использованием колонок Gemini или XBridge C18, градиентов MeCN/вода и в качестве модификаторов ТФУК или гидроксида аммония. ГХМС данные получают с использованием сушильной камеры Hewlett Packard 6890 с инжектором HP 6890, колонкой HP-1 (12 м × 0,2 мм × 0,33 мкм) и гелием в качестве газа-носителя. Образец анализируют на масс-селективном детекторе HP 5973, сканирующем от 50 до 550 Да, с использованием электронной ионизации. Очистку проводят с помощью жидкостной хроматографии средней производительности (medium performance liquid chromatography - MPLC) с использованием аппаратов Isco CombiFlash Companion, AnaLogix IntelliFlash 280, Biotage SP1 или Biotage Isolera One instruments, предварительно оснащенных картриджами диоксида кремния Isco RediSep или Biotage Snap. Хиральную очистку выполняют с помощью хиральной сверхкритической жидкостной хроматографии (сверхкритическая жидкостная хроматография - СЖХ) с использованием аппаратов Berger или Thar; колонок ChiralPAK-AD, -AS, -IC, Chiralcel-OD или -OJ; и смесей CO2 с MeOH, EtOH, iPrOH или MeCN или модифицированных с использованием ТФУК или iPrNH2. УФ детектирование используют для инициирования сбора фракций.
Данные масс-спектрометрии записывают с использованием ЖХМС анализа. Масс-спектрометрический (МС) анализ выполняют с помощью химической ионизации при атмосферном давлении (a™ospheric pressure chemical ionization - APCI), ионизации электрораспылением (electrospray ionization - ESI), ионизации электронным ударом (electron impact ionization - EI) или электронного рассеяния (electron scatter - ES). Химические сдвиги спектроскопии протонного ядерного магнитного резонанса (1H ЯМР) в сторону слабого поля относительно тетраметилсилана представляют в миллионных долях и записывают на спектрометрах Varian с частотой 300, 400, 500, или 600 МГц. Химические сдвиги выражают в миллионных долях (м.д., δ) относительно остаточных пиков дейтерированного растворителя. Формы пиков описывают следующими аббревиатурами: с - синглет; д - дублет; т - триплет; кв - квартет; квин - квинтет; м - мультиплет; уш. с - уширенный синглет; предп. - предполагаемый. Аналитические данные СЖХ получают с помощью аналитического инструмента Berger, как описано выше. Данные оптического вращения получают на поляриметре PerkinElmer model 343 с использованием кюветы 1 дм. Хроматографию на силикагеле выполняют главным образом с использованием систем среднего давления Biotage или ISCO и колонок, предварительно заполненных различными коммерческими поставщиками, включая Biotage и ISCO. Микроанализы выполняют Quantitative Technologies Inc., и полученные значения отличаются от расчетных значений не более чем на 0,4%.
Если не указано иное, химические реакции проводят при комнатной температуре (примерно 23 градуса Цельсия).
Названия соединений и промежуточных продуктов, описанных ниже, составляют с использованием способа, предоставленного ChemBioDraw Ultra, Version 12.0 (CambridgeSoft Corp., Cambridge, Massachusetts). Способ получения названий, предоставленный ChemBioDraw Ultra, Version 12.0, хорошо известен специалистам данной области техники, и считается, что способ получения названий, предоставленный ChemBioDraw Ultra, Version 12.0, соответствует рекомендациям IUPAC (International Union for Pure и Applied Chemistry), относящимся к номенклатуре органической химии, и правилам CAS Index. Если не указано иное, все реагенты получают от коммерческих поставщиков и используют без дополнительной очистки или синтезируют с использованием методов, описанных в технической литературе.
Термины «концентрируют, «выпаривают» и «концентрируют в вакууме» относятся к удалению растворителя при пониженном давлении на роторном испарителе при температуре бани менее 60°C. Аббревиатуры «мин.» и «час.» используются для обозначения «минут» и «часов», соответственно. Аббревиатура «ТСХ» означает тонкослойную хроматографию, термин «комнатная температура или температура окружающей среды» означает температуру в интервале от 18 до 25°C, аббревиатура «ГХМС» означает газовую хроматографию-масс-спектрометрию, аббревиатура «ЖХМС» означает жидкостную хроматографию-масс-спектрометрию, аббревиатура «СВЭЖХ» означает сверхэффективную жидкостную хроматографию, аббревиатура «ЖХВД» относится к жидкостной хроматографии высокого давления, аббревиатура «СЖХ» означает сверхкритическую жидкостную хроматографию.
Гидрирование может проводиться в смесительном реакторе Парра под давлением газообразного водорода или в аппарате гидрирования в потоке Thales-nano H-Cube при заполнении водородом и скорости потока 1-2 мл/мин. при указанной температуре.
Время удерживания ВЭЖХ, СВЭЖХ, ЖХМС, ГХМС и СЖХ измеряют с пользованием методов, указанных в методиках.
Получение промежуточных соединений и соединений примеров
Пример 1. (DGAT2i-соединение/соединение D): (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамид
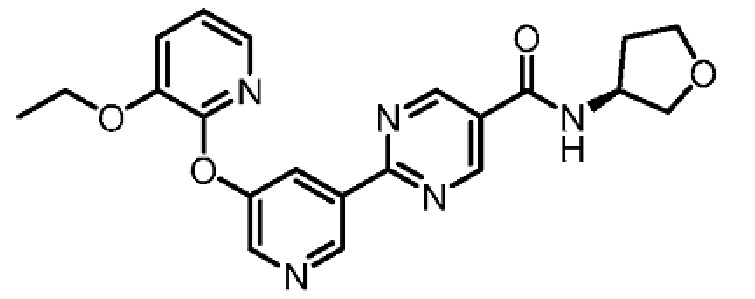
Стадия 1: 3-этоксипиридин
Карбонат цезия (12 моль, 1,5 эквив.) и этилйодид (9,7 моль, 1,2 эквив.) добавляют к раствору 3-гидроксипиридина (8,10 моль, 1,0 эквив.) в ацетоне (12 л) при 15°C. Реакционную смесь перемешивают при комнатной температуре в течение 24 часов. Реакционную смесь фильтруют и органический слой концентрируют с получением сырого продукта. Добавляют этилацетат (20 л) и промывают смесь водой (3 × 5 л). Органический слой сушат над сульфатом натрия, фильтруют и концентрируют с получением 3-этоксипиридина (620 г, 62%) в виде масла. 1H ЯМР (400 МГц, CDCl3) δ 1,44 (т, 3H), 4,07 (кв, 2H), 7,15-7,23 (м, 2H), 8,20 (дд, 1H), 8,30 (д, 1H).
Стадия 2: 3-этоксипиридин-1-оксид
м-Хлорпероксибензойную кислоту (6,5 моль, 1,3 эквив.) добавляют к раствору 3-этоксипиридина (5,0 моль, 1,0 эквив.) в дихлорметанe (12 л) при 10°C. Реакционную смесь перемешивают при комнатной температуре в течение 24 часов. Добавляют тиосульфат натрия (4 кг в 5 л воды). Реакционную смесь перемешивают при 15°C в течение 2 часов. Добавляют другую порцию тиосульфата натрия (1,5 кг, в 5 л воды). Реакционную смесь перемешивают при 15°C в течение 1 часа. Смесь экстрагируют дихлорметаном (16 × 10 л). Объединенные органические слои концентрируют с получением сырого продукта. Сырой продукт очищают хроматографией (силикагель, дихлорметан:метанол; 100:1-10:1) с получением указанного в заголовке соединения (680 г, 97%) в виде масла коричневого цвета. Продукт дополнительно очищают растиранием с петролейным эфиром (4 л) при комнатной температуре в течение 24 часов с получением 3-этоксипиридин-1-оксида (580 г, 83%) в виде твердого вещества желтого цвета. 1H ЯМР (400 МГц, CDCl3) δ 1,41 (т, 3H), 4,02 (кв, 2H), 6,84 (дд, 1H), 7,12 (дд, 1H), 7,85 (д, 1H), 7,91-7,95 (м, 1H).
Стадия 3: 2-((5-бромпиридин-3-ил)окси)-3-этоксипиридин
Данную реакцию проводят с использованием пяти параллельных загрузок.
Диизопропилэтиламин (2,69 моль, 3,7 эквив.) и гексафторфосфат бромтрипирролидинофосфония (0,93 моль, 1,3 эквив.) при перемешивании добавляют к раствору 3-этоксипиридин-1-оксида (0,72 моль, 1,0 эквив.) и 3-бром-5-гидроксипиридина (0,72 моль, 1,0 эквив.) в тетрагидрофуране (2500 мл) при комнатной температуре. Реакционную смесь перемешивают при комнатной температуре в течение 2 дней, затем отдельные загрузки объединяют в единую массу. Полученную суспензию концентрируют досуха и остаток растворяют в дихлорметанe (25 л). Органический слой промывают 1N гидроксидом натрия (15 л), водой (3 × 20 л) и насыщенным раствором соли (20 л). Органический слой сушат над сульфатом натрия, фильтруют и концентрируют с получением масла. Сырое масло очищают колоночной хроматографией (силикагель, петролейный эфир:этилацетат; 10:1-1:1) с получением сырого продукта в виде твердого вещества коричневого цвета. Полученный твердый продукт растирают со смесью метил-трет-бутиловый эфир:петролейный эфир (1:10; 11 л) с получением 2-((5-бромпиридин-3-ил)окси)-3-этоксипиридина (730 г, 69%) в виде твердого вещества желтоватого цвета. 1H ЯМР (400 МГц, CDCl3) δ 1,49 (т, 3H), 4,16 (кв, 2H), 7,04 (дд, 1H), 7,25 (дд, 1H), 7,68-7,73 (м, 2H), 8,44 (д, 1H), 8,49 (д, 1H). МС (ES+) 297,1 (M+H).
Стадия 4: этил-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)пиримидин-5-карбоксилaт
Раствор 2-((5-бромпиридин-3-ил)окси)-3-этоксипиридина (300 ммоль, 1,0 эквив.) в тетрагидрофуране (1,3 л) дегазируют азотом в течение 30 минут. Turbo Grignard (390 ммоль, 1,3 эквив., 1,3 M в тетрагидрофуране) добавляют при комнатной температуре со скоростью, при которой температура смеси остается ниже 30°C. Реакционной смеси дают возможность охладиться до комнатной температуры и перемешивают смесь в течение 3 часов. Реакционную смесь охлаждают до 10°C и добавляют к смеси хлорид цинка (390 ммоль, 1,3 эквив., 1,9 M в 2-метилтетрагидрофуране) со скоростью, при которой температура смеси остается ниже 15°C. Полученную суспензию нагревают до комнатной температуры для полного растворения осадка и затем снова охлаждают до 10°C. К смеси добавляют этил-2-хлорпиримидин-5-карбоксилaт (360 ммоль, 1,2 эквив.) и дихлор[бис(2-(дифенилфосфино)фенил)эфир]палладий (II) (6,00 ммоль, 0,02 эквив.) в виде твердых веществ. Полученную суспензию дегазируют азотом в течение 30 минут, затем нагревают до 50°C и выдерживают при указанной температуре в течение 16 часов. Реакционную смесь обрабатывают в водных условиях, затем нагревают с последовательным добавлением натриевой соли этилендиаминтетрауксусной кислоты, сульфида кремния (thiosilica) и угля для удаления металлических примесей. Сырой продукт перекристаллизовывают из метанола (450 мл) с получением этил-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)пиримидин-5-карбоксилата (77 г, 70%) в виде твердого вещества бледно-желтого цвета. 1H ЯМР (400 МГц, CDCl3) δ 1,44 (т, 3H), 1,50 (т, 3H), 4,19 (кв, 2H), 4,46 (кв, 2H), 7,00-7,04 (м, 1H), 7,25 (с, 1H), 7,71 (д, 1H), 8,59 (с, 1H), 8,66 (д, 1H), 9,32 (с, 2H), 9,55 (с, 1H),
Стадия 5: 2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)пиримидин-5-карбоновая кислота (промежуточное соединение 1)
Гидроксид натрия (307 ммоль, 1,5 эквив., 4M водный раствор) и метанол (50 мл) добавляют к суспензии 2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)пиримидин-5-карбоксилата (205 ммоль, 1,0 эквив.) в тетрагидрофуране (300 мл). Полученный раствор перемешивают при комнатной температуре в течение 3 часов. Реакционную смесь разбавляют водой (400 мл) и экстрагируют смесью 2:1 диэтиловый эфир:гептаны (2 × 300 мл). Водный слой подкисляют до pH 4 добавлением 4M соляной кислоты. Полученную суспензию перемешивают при комнатной температуре в течение 1 часа. Твердый продукт собирают фильтрацией, промывают водой и сушат с получением 2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)пиримидин-5-карбоновой кислоты (69 г, 100%) в виде твердого вещества бледно-желтого цвета. 1H ЯМР (400 МГц, ДМСО-d6) δ 1,37 (т, 3H), 4,18 (кв, 2H), 7,19 (дд, 1H), 7,58 (дд, 1H), 7,70 (дд, 1H), 8,35-8,40 (м, 1H), 8,66 (д, 1H), 9,33 (с, 2H), 9,41 (д, 1H), 13,9 (уш. с, 1H).
Стадия 6: (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамид (пример 1 (DGAT2i-соединение))
Оксалилхлорид (13,8 мл, 160 ммоль, 1,2 эквив.) и диметилформамид (0,510 мл, 6,65 ммоль, 0,05 эквив.) добавляют к суспензии 2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)пиримидин-5-карбоновой кислоты (45,0 г, 133 ммоль, 1,0 эквив.) в дихлорметанe (500 мл). Суспензию перемешивают смесь в течение 2 часов с получением раствора. Реакционную смесь концентрируют с получением сырого хлорангидрида в виде твердого вещества красного цвета. Раствор (S)-тетрагидрофуран-3-амина (12,2 г, 140 ммоль, 1,05 эквив.) и диизопропилэтиламина (51,0 мл, 293 ммоль, 2,2 эквив.) в тетрагидрофуране (100 мл) по каплям добавляют к раствору сырого хлорангидрида в дихлорметанe (200 мл) при 0°C. Реакционной смеси дают возможность нагреться до комнатной температуры и перемешивают смесь в течение 16 часов. К смеси добавляют воду (1,0 л) и этилацетат (600 мл), органический слой отделяют, промывают насыщенным раствором гидрокарбоната натрия, сушат над сульфатом магния и фильтруют. Фильтрат обрабатывают активированным углем (20 г) и перемешивают при 65°C в течение 20 минут. Теплую суспензию фильтруют и фильтрат концентрируют с получением твердого остатка бледно-желтого цвета, который перекристаллизовывают из смеси метанола и этилацетата (1:4, 1 л) с получением (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамида (43,5 г, 81%) в виде твердого бесцветного вещества. Указанное в заголовке соединение объединяют с продуктами (108,7 г, 266,8 ммоль), полученными таким же образом, суспендируют в этилацетате (1,0 л) при 80°C и выдерживают в указанных условиях в течение 4 часов. Суспензии дают возможность охладиться до комнатной температуры и перемешивают смесь в течение 4 дней. Твердый продукт собирают фильтрацией, промывают этилацетатом (3 × 200 мл) и сушат в высоком вакууме при 50°С в течение 24 часов с получением (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамида (100,5 г, 92%) в виде твердого бесцветного вещества. 1H ЯМР (300 МГц, ДМСО-d6) δ 1,38 (т, 3H), 1,89-1,98 (м, 1H), 2,15-2,26 (м, 1H), 3,65 (дд, 1H), 3,70-3,78 (м, 1H), 3,85-3,92 (м, 2H), 4,18 (кв, 2H), 4,46-4,55 (м, 1H), 7,18 (дд, 1H), 7,58 (дд, 1H), 7,69 (дд, 1H), 8,37 (дд, 1H), 8,64 (д, 1H), 8,95 (д, 1H), 9,28 (с, 2H), 9,39 (д, 1H). МС (ES+) 408,4 (M+H). Температура плавления 177,5°C. Элементный анализ для C21H21N5O4: вычислено C 61,91; H 5,20; N 17,19; найдено C 61,86; H 5,18; N 17,30.
Твердое вещество, полученное с помощью данной методики, характеризуют с помощью порошковой дифракционной рентгенографии (ПРДГ) и определяют как форму 1 соединения D.
Альтернативная стадия 6 получения (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамида (соединение D примера 1)
В реактор объемом 100 мл загружают ацетонитрил (35 мл), 2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)пиримидин-5-карбоновую кислоту (5,0 г, 15 ммоль) и гидрохлорид (S)-тетрагидрофуран-3-амина (2,2 г, 18 ммоль, 1,2 эквив.). Добавляют диизопропилэтиламин (18 мл, 103 ммоль, 7,0 эквив.), поддерживая температуру в интервале от 20°C до 30°C. Раствор ангидрида пропанфосфорной кислоты (T3P) в ацетонитриле (21 мл, 30 ммоль, 2,0 эквив.) загружают со скоростью, при которой температура смеси остается ниже 45°C. Реактор нагревают до 40±5°С и выдерживают при указанной температуре в течение 1 часа, затем отбирают образец реакционной смеси для контроля хода реакции. Реакционную смесь охлаждают до температуры в интервале от 20°C до 25°C и добавляют тетрагидрофуран (25 мл). Загружают раствор бикарбоната натрия (0,5 M, 40 мл) и смесь перемешивают в течение 1 часа. Проверяют значение pH, которое составляет 8,5. Добавляют этилацетат (40 мл) и перемешивают смесь в течение 15 минут. Смесь оставляют и фазы разделяют. Водный слой переносят в делительную воронку и снова экстрагируют этилацетатом (100 мл). Органические фазы объединяют и промывают смесь водой (40 мл). Органический слой переносят в реактор объемом 100 мл порциями и концентрируют в вакууме до малого объема. Добавляют метилэтилкетон (100 мл) и смесь концентрируют до конечного объема приблизительно 60 мл. Вакуум удаляют, суспензию кипятят с обратным холодильником до тех пор, пока твердые частицы не смываются со стенок реактора. Суспензию охлаждают до 15°C в течение 2 часов и гранулируют в течение ночи. Твердое вещество выделяют фильтрацией, промывают реактор и осадок дважды метилэтилкетоном (10 мл каждый). Твердые вещества сушат в вакуумной печи при 50°C с получением 4,86 г (81%) целевого продукта. Твердый продукт, полученный в помощью данной методики, характеризуют с помощью порошковой рентгеновской дифрактометрии и обозначают ее как форму 2 соединения D.
Превращение формы 2 соединения D в форму 1 соединения D
В реактор объемом 100 мл загружают форму 2 (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамида (пример 1) (10,0 г, 24,6 ммоль, 1,00 эквив.), метилэтилкетон (8,8 мл/г, 88,0 мл) и воду (1,2 мл/г, 12,0 мл). Реактор нагревают до 50°C в течение 30 минут. Полное растворение наблюдают при температуре примерно 44°C. Реактор охлаждают до 40°C в течение 30 минут, затем добавляют затравку формы 1 (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамида (соединение D примера 1) (0,050 г, 0,123 ммоль, 0,0050 эквив.). После засевания мутную суспензию перемешивают в течение 1 часа, после чего охлаждают до 5°C в течение 2 часов и затем перемешивают при 5°С в течение 12 часов. В процессе перемешивания отбирают контрольный образец и анализируют его с использованием ПРДГ для подтверждения того, что твердые частицы представляют собой форму 1 соединения D. Суспензию фильтруют, реактор и осадок на фильтре промывают охлажденным до 0°C метилэтилкетоном (2,5 мл/г, 25 мл). Твердый продукт сушат в вакуумной печи при 50°C с получением 8,15 г (81,5%) целевого продукта. ПРДГ целевого продукта соответствует форме 1 соединения D.
Порошковая рентгеновская дифрактограмма (ПРДГ)
Анализ методом порошковой рентгеновской дифрактометрии проводят с использованием дифрактометра Bruker AXS D8 Advance с медным источником излучения (Kα средняя длина волны 1,54056Å), снабженного основным двойным зеркалом Гобеля. Дифрагированное излучение регистрируют с помощью детектора PSD-Lynx Eye. Первичное и вторичное устройства снабжены 2,5 щелями Соллера. Напряжение и силу тока рентгеновской трубки устанавливают на уровне 40 кВ и 40 мA, соответственно. Данные собирают в гониометре Theta-Theta при закрытом двойном сканировании от 3,0 до 40,0 градусов 2-тета со 1000 шагов и скоростью сканирования 6 секунд на шаг. Образцы помещают в кремниевый держатель образца с низким фоном (C79298A3244B261). Данные собирают с использованием программного обеспечения Bruker DIFFRAC Plus. Анализ данных выполняют с помощью программного обеспечения EVA DIFFRACT Plus.
Файл данных ПРДГ не обрабатывают до поиска пиков. Используя алгоритм поиска пиков программного обеспечения EVA, выбирают пики с пороговым значением 5 и шириной 0,2. Автоматизированный вывод значений визуально проверяют на предмет достоверности и при необходимости регулируют вручную. Обычно выбирают пики с относительной интенсивностью ≥3%. Пики, которые не были выделены и которые соответствуют шумовым помехам, также не принимают во внимание. Типичная ошибка, связанная с положением пика в ПРДГ, заявленным USP, находится в пределах +/- 0,2° (USP-941).
Таблица 1. Основные пики на ПРДГ, характеризующие кристаллические соединения примера 1 (соединения D)
На фигуре 1 представлена характеристическая порошковая рентгеновская дифрактограмма кристаллической формы 1 соединения примера 1 (соединение D) (вертикальная ось: интенсивность (импульсы в секунду); горизонтальная ось: угол два тета (градусы).
На фигуре 2 представлена характеристическая порошковая рентгеновская дифрактограмма кристаллической формы 2 соединения примера 1 (соединение D) (вертикальная ось: интенсивность (импульсы в секунду); горизонтальная ось: угол два тета (градусы)).
Пример 2. Получение 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойной кислоты (соединение A (АККi-соединение))
Следует отметить, что при получении соединения А в некотрых способах, описанных в настоящем документе, может потребоваться защита удаленной функциональности (например, группы первичного амина, вторичного амина, карбоксильной группы в предшественниках формулы I). Потребность в такой защите будет изменяться в зависимости от природы удаленной функциональности и условий способов получения. Необходимость такой защиты легко определяется специалистом в данной области техники. Применение таких методов введения/удаления защиты также входит в компетенцию специалиста в данной области техники. Общее описание защитных групп и способов их применения см. в монографии T.W. Greene, Protective Groups in Organic Synthesis, John Wiley & Sons, New York, 1991. Кроме того, настоящее изобретение не ограничивается конкретными методами синтеза, представленными в описании, которые могут изменяться.
Промежуточное соединение A1: гидрохлоридная соль 1-изопропил-4,6-дигидроспиро[индазол-5,4'-пиперидин]-7(1H)-она
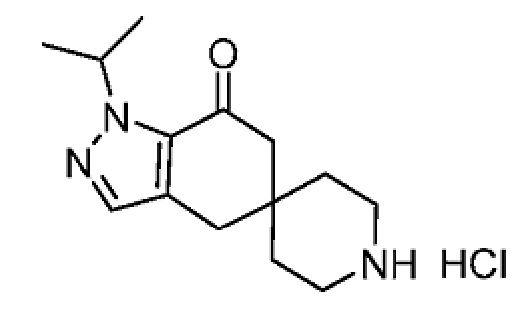
Стадия 1: трет-бутил-9-оксо-3-азаспиро[5.5]ундец-7-ен-3-карбоксилат
В сухой реактор загружают трет-бутил-4-формилпиперидин-1-карбоксилат (108 кг), циклогексан (1080 л) и пирролидин(64,8 кг) при 25-30°С. Смесь перемешивают в течение 5-10 минут и затем кипятят с обратным холодильником в течение 12-16 часов, собирая воду с использованием ловушки Дина-Старка. Реакционную смесь охлаждают до 50-60°С и при указанной температуре применяют вакуум для отгонки избытка пирролидина и циклогексана. Реакционную смесь охлаждают до 25-30°С и последовательно добавляют циклогексан (648 л) и метилвинилкетон (49,63 кг). Смесь перемешивают в течение 12-16 часов, затем фильтруют и фильтрат загружают в чистый сухой реактор. Раствор охлаждают до 10-15°С, затем медленно добавляют раствор уксусной кислоты (54,75 кг) в воде (54 л), поддерживая температуру ниже 15°С. После добавления смесь нагревают до 25-30°С и перемешивают в течение 12-16 часов. Слои разделяют и водный слой экстрагируют этилацетатом (324 л). Объединенные органические слои промывают раствором бикарбоната натрия (32,34 кг) в воде (324 л), затем сушат над сульфатом натрия. Твердые вещества промывают этилацетатом (54 л) и объединенные фильтраты концентрируют при пониженном давлении при температуре ниже 40°С. В реактор загружают н-гептан (216 л) и растворитель отгоняют досуха при пониженном давлении при температуре ниже 40°С. Смесь охлаждают до 25-30°С и в реактор загружают н-гептан (216 л). Смесь перемешивают в течение 1-2 часов после образование твердого осадка. Твердый продукт затем собирают фильтрацией, промывают н-гептаном (54 л) и сушат при 40-50°С в течение 10-12 часов с получением целевого продукта (90,1 кг, выход 67%).
Стадия 2: (E)-трет-бутил-10-((диметиламино)метилен)-9-оксо-3-азаспиро[5.5]ундец-7-ен-3-карбоксилат
В чистый сухой реактор загружают трет-бутил-9-оксо-3-азаспиро[5.5]ундец-7-ен-3-карбоксилат (50 кг), N, N-диметилформамид (500 л) и N, N-диметилформамид-диметилацеталь (135 кг) при 25-30°С в атмосфере азота. Реакционную смесь перемешивают в течение 5-10 минут, затем нагревают до 120-130°С и выдерживают при указанной температуре в течение 20 часов. Смесь затем охлаждают до 50-60°С и отгоняют растворитель в высоком вакууме при температуре ниже 60°С. Смесь ксилолов (200 л) загружают при температуре ниже 45°С и растворитель отгоняют в высоком вакууме при температуре ниже 60°С. Эту операцию повторяют с другой загрузкой ксилолов (200 л). Затем в реактор загружают толуол (200 л) и отгоняют растворитель в высоком вакууме при температуре ниже 60°С. Операцию повторяют со второй загрузкой толуола (200 л). Затем при температуре ниже 30°С загружают метил-трет-бутиловый эфир (100 л) и растворитель отгоняют в высоком вакууме при температуре ниже 40°С. Смесь охлаждают до 15-20°С и загружают метил-трет-бутиловый эфир (100 л) при температуре ниже 20°С. Смесь перемешивают в течение 20-30 минут, твердый продукт собирают фильтрацией, промывают метил-трет-бутиловым эфиром (50 л) и сушат без применения вакууме при 50-55°С в течение 10 часов с получением целевого соединения (52,1 кг, выход 87%). 1H ЯМР (400 МГц, CDCl3) δ м.д. 7,48 (с, 1H), 6,57 (д, J=9,97 Гц, 1H), 5,99 (д, J=10,16 Гц, 1H), 3,32-3,51 (м, 4H), 3,06 (с, 6H), 2,72 (с, 2H), 1,57-1,66 (м, 2H), 1,41-1,53 (м, 11H).
Стадия 3: трет-бутил-1-изопропил-1,4-дигидроспиро[индазол-5,4'-пиперидин]-1'-карбоксилат
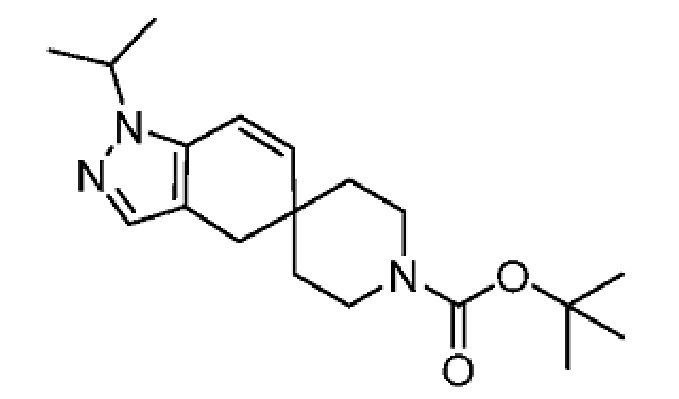
В чистый сухой реактор загружают (E)-трет-бутил-10-((диметиламино)метилен)-9-оксо-3-азаспиро[5.5]ундец-7-ен-3-карбоксилат (80 кг), толуол (704 л) и триметиламин (16 л) при 25-30°С. Реакционную смесь нагревают до 70-80°С и в течение 4-5 часов добавляют раствор гидрохлоридной соли изопропилгидразина в метаноле (1,25 эквив., 141 кг). Реакционную смесь затем перемешивают в течение 8-10 часов при 70-80°С и охлаждают до 15-25°С. Затем медленно добавляют раствор лимонной кислоты (48 кг) в воде (480 л), поддерживая внутреннюю температуру ниже 25°С. Этилацетат (208 л) добавляют и перемешивают смесь в течение 10 минут. Слои разделяют, органический слой последовательно промывают раствором лимонной кислоты (48 кг) в воде (480 л) и затем только водой (320 л). Объединенные водные слои экстрагируют этилацетатом (320 л). Объединенные органические слои сушат над сульфатом натрия (8 кг) и растворители упаривают досуха при пониженном давлении при температуре ниже 40°С. В реактор загружают дихлорметан (240 л) и смесь перемешивают при 25-30°С до получения прозрачного раствора. Последовательно загружают активированный уголь (1,84 кг), силикат магния (1,84 кг) и силикагель (32 кг, 100-200 мееш) при 25-30°С и гетерогенную смесь перемешивают в течение 1 часа. Суспензию затем фильтруют на слое Hyflow, полученном смешением Hyflow supercell (8 кг) и дихлорметана (40 л). Осадок на фильтре промывают дихлорметаном (три раза по 120 л). Объединенные фильтраты снова загружают в реактор и растворитель выпаривают при пониженном давлении при температуре ниже 40°C. н-Гептан (160 л) загружают в реактор и отгоняют при пониженном давлении при температуре ниже 40°C. н-Гептан (200 л) загружают в реактор и охлаждают смесь до 0-5°C. После перемешивания в течение 12-15 часов твердый продукт собирают фильтрацией при 0°C, промывают охлажденным (0-5°C) н-гетаном (160 л) и сушат в вакууме при 40-50°C с получением указанного в заголовке соединения (82,4 кг, 75%). 1H ЯМР (400 МГц, CDCl3) δ м.д. 7,25 (с, 1H), 6,42 (дд, J=10,05, 0,49 Гц, 1H) 5,84 (д, J=9,95 Гц, 1H), 4,42-4,52 (м, 1H), 3,36-3,53 (м, 4H), 2,62 (с, 2H) 1,56-1,68 (м, 2H) 1,45-1,55 (м, 17H).
Стадия 4: гидрохлоридная соль 1-изопропил-4,6-дигидроспиро[индазол-5,4'-пиперидин]-7(1H)-она
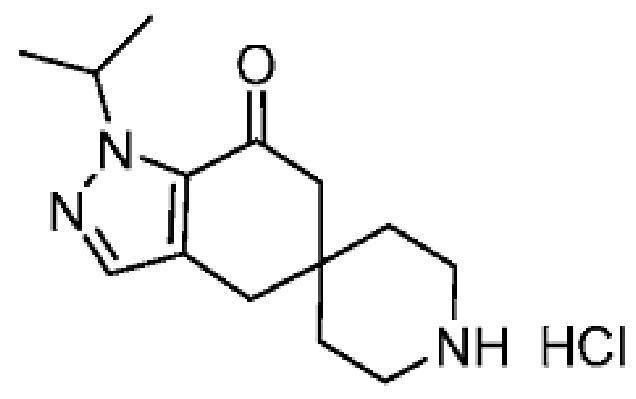
В чистый сухой реактор при 25-30°С загружают трет-бутил-1-изопропил-1,4-дигидроспиро[индазол-5,4'-пиперидин]-1'-карбоксилат (60 кг) и метанол (600 л). К смеси пятью порциями в течение 30-40 минут добавляют N-бромсукцинимид (32,4 кг) при 25-30°С и перемешивание продолжают в течение 30-60 минут. К смеси медленно добавляют раствор пентагидрата тиосульфата натрия (5,4 кг) в воде (102 л), поддерживая внутреннюю температуру смеси ниже 30°С. Смесь перемешивают в течение 20-30 минут, затем растворитель выпаривают при пониженном давлении при температуре ниже 45°С. Остаток охлаждают до 25-30°С и в реактор загружают 2-метилтетрагидрофуран (420 л) и воду (90 л). Смесь перемешивают в течение 15-20 минут, затем слои разделяют и водный слой дополнительно экстрагируют 2-метилтетрагидрофураном (120 л). Объединенный органические экстракты обрабатывают в течение 15-20 минут при 25-30°С раствором гидроксида натрия (4,8 кг) в воде (120 л). Слои разделяют, органический слой промывают водой (120 л), раствором хлорида натрия (12 кг) в воде (120 л) и затем сушат над сульфатом натрия (6 кг). Смесь фильтруют, осадок на фильтре промывают 2-метилтетрагидрофураном (30 л) и объединенный фильтрат снова загружают в реактор. Растворитель полностью отгоняют при температуре ниже 45°С при пониженном давлении и остаток снова растворяют в тетрагидрофуране (201 л). В другой чистый сухой реактор при 25-30°С загружают трет-бутоксид калия (60,6 кг) и тетрагидрофуран (360 л). К полученной смеси медленно добавляют раствор остатка в тетрагидрофуране, поддерживая температуру ниже 30°С. Реакционную смесь нагревают до 60-65°С и выдерживают при указанной температуре в течение 1-2 часов. После завершения реакции смесь охлаждают до 0-10°С и медленно гасят раствором соляной кислоты (1 N, 196 л), поддерживая внутреннюю температуру ниже 10°С. Реакционной смеси дают возможность нагреться до 25-30°С и добавляют этилацетат (798 л). После перемешивания в течение 15-20 минут слои разделяют и водный слой дополнительно экстрагируют эилацетатом (160 л). Объединенные органические слои промывают водой (160 л), сушат над сульфатом натрия (8 кг), фильтруют и осадок на фильтре промывают этилацетатом (300 л). Растворители полностью отгоняют при пониженном давлении при температуре ниже 45°С, в реактор загружают этилацетат (540 л) при 25-30°С и затем метанол (156 л). Смесь охлаждают до 0-5°С и при указанной температуре медленно добавляют ацетилхлорид (79,8 кг), поддерживая температуру в указанной области значений. После этого смеси дают возможность нагреться до 20-25°С и смесь перемешивают при указанной температуре в течение 4-5 часов. Полученную суспензию фильтруют, твердый продукт промывают этилацетатом (120 л), затем сушат при 40-45°С в течение 8-10 часов, получая сырой продукт (33,5 кг, 65%).
Конечную стадию очистки проводят солюбилизацией полученного твердого продукта (56,8 кг) в метаноле (454,4 л) в чистом сухом реакторе при 25-30°С. Раствор перемешивают в течение 30-45 минут, затем через картриджный фильтр (0,2 микрона) загружают в чистый сухой реактор при 25-30°С. Метанол отгоняют при пониженном давлении при температуре ниже 50°С до тех пор, пока не останется ~1 об. растворителя. Реакционную смесь охлаждают до 25-30°С и через картриджный фильтр (0,2 микрон) загружают свежий ацетонитрил (113,6 л). Растворители отгоняют при пониженном давлении при температуре ниже 50°С до тех пор, пока не останется ~1 об. растворителя. Реакционную смесь охлаждают до 25-30°С и загружают в резактор через картриджный фильтр (0,2 микрон) свежий ацетонитрил (190 л). Смесь нагревают до 65-70°С и перемешивают в течение 45 минут, затем охлаждают до 25-30°С и перемешивают в течение 1 часа. Полученную суспензию фильтруют, осадок на фильтре промывают охлажденным (15°С) ацетонитрилом (56,8 л). Твердый продукт сушат при пониженном давлении при 40-50°С в течение 8 часов с получением промежуточного соединения A1 (36,4 кг, 64%). 1H ЯМР (400 МГц, CD3OD) δ м.д. 7,43 (с, 1H), 5,32-5,42 (м, 1H), 3,15-3,25 (м, 4H), 2,89 (с, 2H), 2,64 (с, 2H), 1,69-1,90 (м, 4H), 1,37-1,45 (м, 6H); ESI [M+H]+ = 248.
Промежуточное соединение A2: 2-(4-(трет-бутоксикарбонил)фенил)-6-метоксиизоникотиновая кислота
В чистый сухой реактор загружают 2,6-дихлоризоникотиновую кислоту (30 кг) и метанол (120 л) при 20-25°С. Суспензию перемешивают в течение 5 минут и затем нагревают до 65°С (температура образования флегмы). К смеси через капельную воронку в течение по меньшей мере 4 часов медленно добавляют раствор метоксида натрия в метаноле (30%, 87,2 кг). Воронку промывают метанолом (15 л) и перемешивают смесь при 65°С в течение по меньшей мере 15 часов. Смесь охлаждают до 45°С и растворители отгоняют при пониженном давлении до конечного объема ~90 л. В реактор добавляют раствор бикарбоната калия (28,2 кг) и карбоната калия (21,6 кг) в воде (180 л) при 40-45°С. Реактор, содержащий водный раствор, промывают водой (21 л) и промывную жидкость загружают в реакционную смесь. Смесь отгоняют при пониженном давлении при температуре ниже 80°С до конечного объема ~240 л, затем охлаждают до 20-25°С.
В другой чистый сухой реактор загружают трет-бутил-4-(4,4,5,5-тетраметил-1,3,2-диоксоборолан-2-ил)бензоат (52,3 кг) и диоксан (340 кг) и перемешивают смесь при 2-25°С до полного растворения. Содержимое первого реактора затем нагревают до 40°С до полного растворения и переносят раствор в этот новый реактор. Реакционную смесь охлаждают до 20-25°С и выполняют операцию деоксигенации, циклически создавая вакуум и заполняя азотом. Смесь охлаждают до 0-10°С, в реактор загружают ацетат палладия (0,65 кг) и затем трифенилфосфин (2,46 кг) в потоке азота. Смесь нагревают до 20-25°С и снова выполняют операцию деоксигенации, циклически повторяя создание вакуума и заполнение реактора азотом. Смесь нагревают до 80°С и выдерживают при указанной температуре в течение по меньшей мере 18 часов. Смесь охлаждают до 20-25°С, затем последовательно добавляют в реактор метил-трет-бутиловый эфир (133,2 кг) и воду (30 л). Слои разделяют, водный слой разбавляют водой (110 л) и затем экстрагируют метил-трет-бутиловым эфиром (110 л). Объединенные органические экстракты промывают раствором лимонной кислоты (52 кг) в воде (84 л) и слои разделяют. Водный слой дополнительно экстрагируют метил-трет-бутиловым эфиром (88,8 кг), органические слои объединяют, затем три раза промывают каждый раз третьей частью раствора хлорида натрия (43 кг) в воде (80 л). После конечного разделения слоев органический слой фильтруют через палл-фильтр, содержащий угольный картридж, и осадок на фильтре промывают метил-трет-бутиловым эфиром (11,2 кг). Фильтрат отгоняют при пониженном давлении при температуре ниже 50°С до объема ~90 л и затем совместно отгоняют с гептаном (120 л) при температуре ниже 50°С и объема ~120 л. Смесь охлаждают до 20-25°С в течение 1 часа, затем перемешивают при указанной температуре в течение дополнительного 1 часа. Суспензию фильтруют, осадок на фильтре промывают гептаном (3 × 18 л) и затем ацетонитрилом (3 × 18 л). Полученное влажное твердое вещество сушат в вакууме и в потоке азота при температуре ниже 45°С в течение по меньшей мере 15 часов с получением промежуточного соединения A2 (44,6 кг, выход 87%). 1H ЯМР (400 МГц, CDCl3) δ м.д. 8,13 (с, 2H), 8,09 (с, 2H), 7,97 (д, J=1,17 Гц, 1H), 7,34 (д, J=0,98 Гц, 1H), 4,08 (с, 3H), 1,61 (с, 9H); ESI [M+H]+ = 330.
Промежуточное соединение А3: трет-бутил-4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензоат
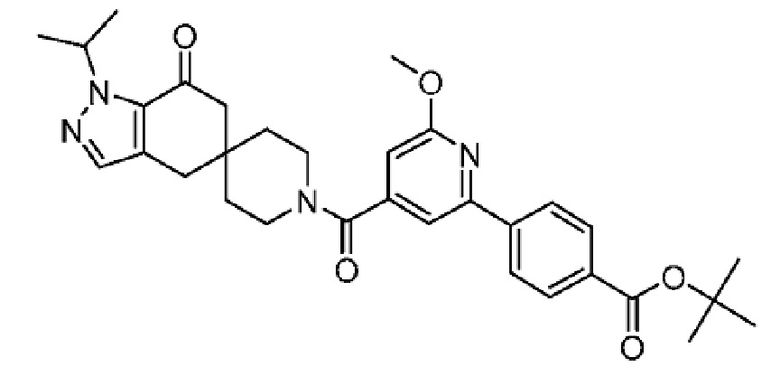
В круглодонную колбу загружают 2-(4-(трет-бутоксикарбонил)фенил)-6-метоксиизоникотиновую кислоту (промежуточное соединение A2, 15,2 г, 46,2 ммоль) и этилацетат (140 мл). К смеси одной порцией добавляют 1,1'-карбонилдиимидазол (8,98 г, 55,4 ммоль) и перемешивают смесь в течение 1 часа при комнатной температуре. К смеси добавляют гидрохлорид 1-изопропил-4,6-дигидроспиро[индазол-5,4'-пиперидин]-7(1H)-она (промежуточное соединение A1, 14,8 г, 52,2 ммоль) и затем N, N-диизопропилэтиламин (9,1 мл, 52,2 мл) и перемешивают реакционную смесь в течение 18 часов при комнатной температуре. Добавляют водную 2 M HCl (40 мл) с последующим добавлением 1 M гидросульфата калия (40 мл) и 50 мл гептана. Полученную смесь перемешивают в течение 1 часа при комнатной температуре. Смесь переносят в делительную воронку. Органическую фазу отделяют, промывают последовательно водой (20 мл), насыщенным раствором бикарбоната натрия (30 мл), водой (20 мл) и насыщенным раствором соли (20 мл), сушат над 20 г сульфата магния и 10 г силикагеля, фильтруют и концентрируют в вакууме. В процессе концентрирования начинает образовываться твердый продукт. Остаток смешивают с 40 мл этилацетата при 80°С и медленно по каплям добавляют гептан (120 мл). Смесь перемешивают при 80°С в течение 1 часа, затем медленно охлаждают до комнатной температуры с перемешиванием в течение 1 часа и перемешивают смесь в течение 18 часов при комнатной температуре. Твердое вещество собирают фильтрацией, промывают водой и смесью этилацетат-гептан (1:3) и сушат в вакууме при 50°С в течение 18 часов с получением промежуточного соединения A3 (19,64 г, выход 76%).
Альтернативное получение промежуточного соединения A3
В чистый сухой реактор загружают ацетонитрил (219 кг) и 2-(4-(трет-бутоксикарбонил)фенил)-6-метоксиизоникотиновую кислоту (промежуточное соединение A2, 34,8 кг) при 20-25°С. Смесь перемешивают в течение 5 минут и тремя последовательными порциями добавляют 1,1-карбодиимидазол (18,9 кг). Суспензию дополнительно перемешивают при 20-25°C в течение по меньшей мере 1 часа, затем добавляют гидрохлоридную соль 1-изопропил-4,6-дигидроспиро[индазол-5,4'-пиперидин]-7(1H)-она (промежуточное соединение A1, 33,0 кг) с последующим добавлением N, N-диизопропилэтиламина (20,5 кг) через насос. Насос реагента, а также стенки реактора ополаскивают ацетонитрилом (13,7 кг) и смесь перемешивают при 20-25°С в течение по меньшей мере 2 часов. После этого в смесь вносят затравку кристаллизации трет-бутил-4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензоата (промежуточное соединение A3, 209 г) и перемешивают смесь в течение по меньшей мере 30 минут. После подтверждения начала кристаллизации в течение 1 часа добавляют раствор моногидрата лимонной кислоты (58,5 кг) в воде (257 л). Полученную суспензию дополнительно перемешивают при 20-25°С в течение по меньшей мере 2 часов, затем фильтруют и осадок на фильтре промывают смесью ацетонитрила (68,4 кг) и воды (87 л). Промывную жидкость используют также для ополаскивания реактора. Твердый продукт сушат при пониженном давлении при температуре ниже 55°С с получение промежуточного соединения A3 (43,44 кг, выход 73%).
Соединение A (в форме свободной кислоты): 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойная кислота
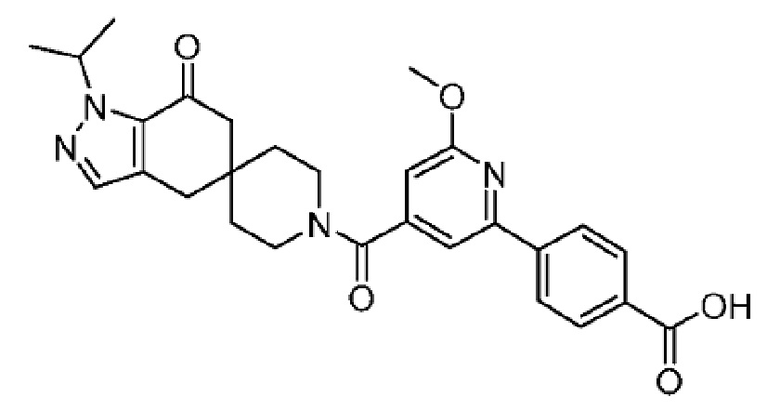
В круглодонную колбу загружают трет-бутил-4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензоат (3,7 г, 6,6 ммоль) и толуол (25 мл). К смеси при перемешивании по каплям добавляют 85% фосфорную кислоту (3,0 мл), реакционную смесь нагревают до 60°С и выдерживают при указанной температуре в течение 4 часов. Образуется бесцветная густая смола. Реакционную смесь охлаждают до комнатной температуры и добавляют воду. Образуется белый осадок. Органический толуольный слой утилизируют, оставляя водный слой и твердый продукт. Добавляют этилацетат (60 мл) и 4N раствор NaOH для достижения значения pH ~7. Слои разделяют и водный слой экстрагируют этилацетатом (50 мл). Объединенные этилацетатные органические слои сушат над сульфатом натрия, фильтруют и концентрируют в вакууме с получением продукта в виде твердого белого вещества. Продукт растворяют в этилацетате (80 мл) при 50°С и медленно добавляют гептан (90 мл). Негрев удаляют, смесь охлаждают до комнатной температуры и перемешивают в течение 16 часов. Образованный твердый осадок собирают фильтрацией, промывают маточной жидкостью и сушат с получением указанного в заголовке соединения (соединении A в форме свободной кислоты, 2,15 г, выход 65%) в виде твердого белого вещества.
Альтернативное получение соединения A (в форме свободной кислоты)
В чистый сухой реактор загружают ацетонитрил (130,4 кг) и трет-бутил-4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензоат (промежуточное соединение A3, 20,72 кг) при 20-25°С. Смесь перемешивают в течение 5 минут, затем загружают п-толуолсульфокислоту (8,5 кг) с осторожной продувкой азотом. Реакционную смесь нагревают до 70°С и выдерживают при указанной температуре в течение по меньшей мере 6,5 часов. После этого смесь охлаждают до 40°С, вносят затравку кристаллизации соединения A (104 г) и медленно в течение по меньшей мере 1 часа добавляют воду (83 л). Смесь дополнительно перемешивают при 40°С в течение по меньшей мере 4 часов, затем охлаждают до 20-25°С в течение 2 часов. Смесь перемешивают в течение по меньшей мере 2 часов, затем фильтруют и промывают осадок на фильтре раствором ацетонитрила (33 кг) в воде (41 л). Эту промывную жидкость используют также для ополаскивания реактора. Полученный твердый продукт сушат при пониженном давлении при температуре ниже 55°С с получением соединения A (16,5 кг, выход 89%).
Получение формы 1 соединения А: безводная моно-трис-соль соединения А
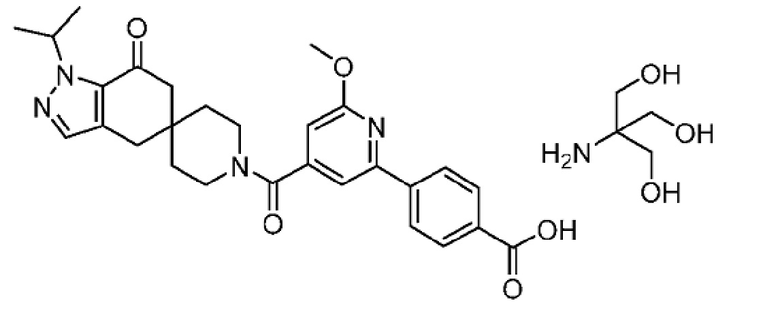
В колбу загружают 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойную кислоту (151 мг, 0,300 ммоль) и 3 мл этанола. Смесь нагревают до 80°С в течение 5 минут для растворения твердого вещества и затем охлаждают до комнатной температуры. Добавляют трис(гидроксиметил)аминометан (39 мг, 0,32 ммоль) и смесь перемешивают в течение ночи при комнатной температуре. К смеси по каплям добавляют гептан (2,25 мл) с получением суспензии, которую нагревают до 50°С с получением прозрачного раствора. Смесь охлаждают до комнатной температуры в течение ночи при перемешивании. Образуется твердый белый осадок, и смесь перемешивают в течение дополнительных 3 дней. Осадок собирают фильтрацией и сушат в вакуумной печи при 50°С в течение ночи с получением формы 1 (151 мг, 0,242 ммоль, выход 81%).
Альтернативное получение формы 1 соединения А: безводная моно-трис соль соединения А
В чистый сухой реактор загружают этанол (83 л) с последующим добавлением соединения А (9,43 кг) и трис (2,55 кг), поддерживая температуру смеси на уровне 20-25°С. Стенки реактора ополаскивают этанолом (2 л), полученную смесь нагревают до 65-70°С, поддерживая указанную температуру в течение по меньшей мере 30 минут до полного растворения твердых веществ, затем охлаждают до 45-50°С. Теплую смесь фильтруют через 10 мкм пропиленовый фильтр in-line и промывают реактор и фильтр этанолом (9 л). н-Гептан (24 л) загружают в теплый раствор через этот же in-line фильтр и вносят в смесь затравку кристаллизации безводной трис-соли 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойной кислоты (100 г) в этаноле (0,5 л) при 45-50°С. Указанную температуру поддерживают в течение по меньшей мере 2 часов перед охлаждением до 20-25°С в течение по меньшей мере 2 часов. Смесь перемешивают в течение по меньшей мере 5 дней. Затем суспензию фильтруют и осадок на фильтре промывают смесью этанола (13 л) и н-гептана (6 л). Твердый продукт сушат при пониженном давлении при температуре ниже 45°С в течение по меньшей мере 12 часов с получением соединения примера 1 (11,7 кг, 77%).
Получение формы 2 соединения А: тригидрат моно-трис-соли соединения А
Форму 2 соединения А получают из формы 1 соединения А. В реактор EasyMax объемом 50 мл загружают форму 1 соединения А (1,7214 г, 2,760 ммоль), изопропанол (16,50 мл, 215,8 ммоль) и воду (688 мкл, 38,190 ммоль). Смесь перемешивают (300 об./мин.) в течение примерно 72 часов при температуре рубашки реактора 25°С. Реакционную смесь нагревают до 40°С в течение 15 минут, выдерживают при указанной температуре в течение примерно 24 часов, охлаждая один раз до 20°С для отбора образца для тестирования. ПРДГ показывает наличие смеси форм; поэтому добавляют дополнительное количество воды (688 мкл, 38,190 ммоль). Скорость перемешивания увеличивают до 400 об./мин., суспензию перемешивают в течение 6 часов и затем охлаждают до 15°С. Твердый продукт собирают фильтрацией на фильтре 60 мл/40 M и промывают смесью изопропанол/вода (96/4). Полученный продукт представляет собой форму 2 соединения А, что подтверждает ПРДГ.
Альтернативное получение формы 2 соединения А: тригидрат моно-трис соли соединения А
В чистый сухой реактор загружают изопропанол (60,4 кг) и добавляют соединение А (16,68 кг) и трис (4,42 кг), поддерживая температуру на уровне 20-25°С. Смесь перемешивают в течение 5 минут, затем добавляют воду (6,7 кг) и нагревают суспензию до 55°С. Новый чистый раствор загружают через in-line 10 мкм полипропиленовый фильтр в предварительно нагретый чистый сухой реактор (50-55°С). После этого в раствор вносят затравку кристаллизации моно-трис-соли соединения А в форме тригидрата (167 г). После проверки того, что затравка кристаллизации сохраняется, смесь охлаждают до 15°С в течение по меньшей мере 2 часов, затем выдерживают 15°С в течение по меньшей мере 16 часов. Суспензию фильтруют и осадок на фильтре промывают охлажденным изопропанолом (13,1 кг). Твердый продукт затем сушат при пониженном давлении при температуре ниже 25°С с получением формы 2 соединения А (22,1 кг, выход 98%) в качестве единственного продукта.
Форма 1 соединения А является безводной и термодинамически стабильной ниже активности воды на примерно 0,2 (20% RH) при температуре окружающей среды. ПРДГ формы 1 соединения А по существу является такой же, как ПРДГ, представленная на фигуре 3. Характеристические пики формы 1 соединения А на ПРДГ наблюдаются при следующих углах 2-тета (2Θ±0,2°): 9,6, 10,7 и 11,3. Расположение пиков и их интенсивность в ПРДГ, представленной на фигуре 3, приведены в таблице 2.
Таблица 2. Расположение пиков в ПРДГ формы 1 соединения A и их относительные интенсивности
± 0,2°
± 0,2°
± 0,2°
Рамановский спектр формы 1 соединения А по существу является таким же, как рамановский спектр, представленный на фигуре 4. На рамановском спектре формы 1 соединения А наблюдаются характеристические пиковые сдвиги, выраженные в см-1, при 568, 698, 989, 1218, 1511, 1561 и 1615±2 см-1. Положения пиков (± 2 см-1) формы 1 соединения А и их нормализованная интенсивность (W=слабая, M=средняя, S=сильная) спектра, представленного на фигуре 4, приведены в таблице 3.
Таблица 3. Расположение пиков в рамановском спектре формы 1 соединения А и их нормализованная интенсивность
(см-1)
(см-1)
(см-1)
13C ssЯМР спектр формы 1 соединения А по существу является таким же, как 13C ssЯМР спектр, представленный на фигуре 5. Характеристические химические сдвиги 13C ssЯМР спектра формы 1 соединения А, выраженные в м.д., наблюдаются при 22,9 м.д., 146,2 м.д., 157,9 м.д., 161,9 м.д. и 172,9 м.д.±0,2 м.д. Химические сдвиги (± 0,2 м.д.) в 13C ssЯМР спектре формы 1 соединения А, представленном на фигуре 5, приведены в таблице 4.
Таблица 4. 13C химические сдвиги формы 1 соединения А и их интенсивность
Форма 2 соединения А представляет собой тригидрат и является термодинамически стабильной выше активности воды на примерно 0,2 при температуре окружающей среды и 20% RH. ПРДГ формы 2 соединения А является такой же, как ПРДГ, представленная на фигуре 6. Характеристически пики формы 2 соединения А на ПРДГ наблюдаются при следующих углах 2-тета (2Θ±0,2°): 8,4, 9,0, 10,5, 15,0 и 24,7. Расположение пиков и их интенсивность в ПРДГ, представленной на фигуре 6, приведены в таблице 5.
Таблица 5. Расположение пиков в ПРДГ формы 2 соединения A и их относительная интенсивность
± 0,2°
± 0,2°
± 0,2°
Рамановский спектр формы 2 соединения А по существу является таким же, как рамановский спектр, представленный на фигуре 7. На рамановском спектре формы 2 соединения А наблюдаются характеристические пиковые сдвиги, выраженные в см-1, при 562, 692, 984, 1225, 1507, 1557 и 1610±2 см-1. Положения пиков (± 2 см-1) формы 2 соединения А и их нормализованная интенсивность (W=слабая, M=средняя, S=сильная) спектра, представленного на фигуре 7, приведены в таблице 6.
Таблица 6. Расположение пиков в рамановском спектре формы 2 соединения А и их нормализованная интенсивность
(см-1)
(см-1)
(см-1)
13C ssЯМР спектр формы 2 соединения А по существу является таким же, как 13C ssЯМР спектр, представленный на фигуре 8. Характеристические химические сдвиги спектра 13C ssЯМР формы 2 соединения А, выраженные в м.д., наблюдаются при 19,2 м.д., 149,5 м.д., 155,6 м.д., 163,8 м.д. и 188,3 м.д.±0,2 м.д. Химические сдвиги (± 0,2 м.д.) 13C ssЯМР спектра формы 2 соединения А, представленного на фигуре 8, приведены в таблице 7.
Таблица 7. 13C химические сдвиги и их интенсивность для формы 2 соединения A
На основании представленного в настоящем документе описания специалисту в данной области техники будет понятно, что форма 1 и форма 2 соединения А могут быть однозначно идентифицироваться несколькими разными пиками в спектрах или их контурами в различных комбинациях. Ниже представлены типичные примеры комбинаций характеристических пиков, которые могут применяться для раздельной идентификации формы 1 и формы 2 соединения А, представленные комбинации не следует рассматривать как ограничивающие другие комбинации параметров пиков, описанных в настоящем документе.
Данные, подтверждающие присутствие трех молекул воды в форме 2 соединения А, получают с использованием дифрактометра Bruker D8 Venture при комнатной температуре (см. фигуру 9). Структуру определяют с помощью внутреннего фазирования с использованием программного обеспечения SHELX в моноклинной пространственной группе (Monoclinic class space group) P21/c (Version 5.1, Bruker AXS, 1997). Структуру затем уточняют полноматричным методом наименьших квадратов. Все атомы, которые не являются атомами водорода, определяют и уточняют с использованием параметров анизотропного смещения.
Атомы водорода, расположенные на атомах азота и кислорода, определяют из разностной карты Фурье и уточняют с ограничением расстояний. Остальные атомы водорода помещают в расчетные положения, позволяя им находиться на их атомах-носителях.
Итоговый R-индекс составляет 7,2%. Конечная разность Фурье не выявляет отсутствующей или неправильно определенной электронной плотности.
В таблице 8 представлены данные, относящиеся к форме 2 соединения А:
Таблица 8
b=13,2753(7) Å
c=14,6480(8) Å
β=92,451(3)°
α=90°
Кристаллическая 2-амино-2-(гидроксиметил)пропан-1,3-диольная соль 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойной кислоты. Эту кристаллическую соль обычно называют трис-солью соединения А.
Кристаллическая трис-соль соединения А, где соотношение 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойной кислоты и соли составляет 1:1.
Кристаллическая трис-соль соединения А, где кристаллическая соль является безводной кристаллической солью.
Безводная кристаллическая трис-соль соединения А, где ПРДГ указанной безводной кристаллической соли включает пики при углах дифракции 2Θ 9,6°, 10,7° и 11,3°±0,2°.
Безводная кристаллическая трис-соль соединения А, где рамановский спектр указанной безводной кристаллической соли включает сдвиги пиков при 1511 см-1, 1561 см-1 и 1615 см-1±2 см-1.
Безводная кристаллическая трис-соль соединения А, где 13C ssЯМР спектр указанной безводной кристаллической соли включает химические сдвиги при 22,9 м.д., 146,2 м.д. и 161,9 м.д.±0,2 м.д.
Безводная кристаллическая трис-соль соединения А, где аналитические параметры указанной безводной кристаллической соли выбраны из группы, состоящей из рамановского спектра, включающего сдвиги пиков при 1511 см-1 и 1615 см-1±2 см-1, и 13C ssЯМР спектра, включающего по меньшей мере один химический сдвиг при 22,9 м.д., 146,2 м.д. или 161,9 м.д.±0,2 м.д.
Безводная кристаллическая трис-соль соединения А, где указанная безводная кристаллическая соль является по существу чистой.
Кристаллическая трис-соль соединения А, где кристаллическая соль представляет собой тригидратную кристаллическую соль.
Тригидратная кристаллическая трис-соль соединения А, где ПРДГ указанной тригидратной кристаллической соли включает пики при углах дифракции 2Θ 8,4°, 9,0° и 10,5°±0,2°.
Тригидратная кристаллическая трис-соль соединения А, где рамановский спект указанной тригидратной кристаллической соли включает сдвиги пиков при 1507 см-1, 1557 см-1 и 1610 см-1±2 см-1.
Тригидратная кристаллическая трис-соль соединения А, где 13C ssЯМР спектр указанной тригидратной кристаллической соли включает химические сдвиги при 19,2, 149,5 и 163,8 м.д.±0,2 м.д.
Тригидратная кристаллическая трис-соль соединения А, где аналитический параметр указанной тригидратной кристаллической соли выбран из группы, состоящей из
ПРДГ, включающей пики при углах дифракции 2Θ 8,4° и 9,0°±0,2°,
рамановского спектра, включающего сдвиги пиков при 1557 см-1 и 1610 см-1±2 см-1, и
13C ssЯМР спектра, включающего по меньшей мере один химический сдвиг при 19,2 м.д., 149,5 м.д. или 163,8 м.д.±0,2 м.д.
Тригидратная кристаллическая трис-соль соединения А, где аналитический параметр указанной тригидратной кристаллической соли выбран из группы, состоящей из ПРДГ, включающей пики при углах дифракции 2Θ 8,4° и 9,0°±0,2°, и рамановского спектра, включающего по меньшей мере один сдвиг пика при 1507 см-1, 1557 см-1 или 1610 см-1±2 см-1.
Тригидратная кристаллическая трис-соль соединения А, где аналитический параметр указанной тригидратной кристаллической соли выбран из группы, состоящей из ПРДГ, включающей пики при углах дифракции 2Θ 8,4° и 9,0°±0,2°, и 13C ssЯМР спектра, включающего по меньшей мере один химический сдвиг при 19,2 м.д., 149,5 м.д. или 163,8 м.д.±0,2 м.д.
ФАРМАКОЛОГИЧЕСКИЕ ДАННЫЕ
Представленные далее протоколы, разумеется, могут изменяться специалистами в данной области техники.
Рандомизированное исследование с 8 параллельными группами и использованием в качестве контроля разбавителя проводят на самцах крас линии Sprague-Dawley (Charles River (Boston, MA)) для определения уровней TG в крови и в печени. Для 96 крыс (с массой тела ~200 г) используют стандартные условия содержания; крыс размещают в клетки по две особи и содержат при обращенном цикле дня и ночи 12:12 часов (свет выключают в 8 часов утра). После получения крыс рандомизируют на группы, получающие стандартный корм для крыс или корм в соответствии с западной диетой, и дозовые группы и перед исследованием устанавливают 14-дневный вводный период, в течение которого крысы получают стандартный корм для крыс или корм в соответствии с западной диетой. Стандартный корм для лабораторных грызунов, 5053, получают от LabDiet (PMI, St Louis, Missouri). Корм, соответствующий западной диете, D12079Bi, получают от Research Diets (New Brunswick, NJ).
Для этих исследований приготавливают разбавитель (носитель), содержащий 0,5% (масс./об.) метилцеллюлозы (MЦ, Sigma Aldrich; 274429) в деионизированной воде. Исходные растворы соединения А (полученного из трис-соли) или соединения D приготавливают из соответствующего соединения с получением концентрации, при которой 10 мл раствора обеспечивают нужную дозу в мг/кг, где средняя массы используемых крыс составляет примерно 200 г.
Начиная с 1 дня исследования крысам вводят перорально (10 мл/кг) разбавитель (контроль) (0,5% MЦ (% масс./об.) в деионизированной воде), низкие или высокие дозы соединения А (1 мг/кг или 10 мг/кг QD, соответственно), низкие или высокие дозы соединения D (5 мг/кг или 30 мг/кг BID, соответственно), либо совместно вводят низкие дозы (соединение А при 1 мг/кг QD и сoединение D при 5 мг/кг BID) или высокие дозы (соединение А при 10 мг/кг QD и соединение D при 30 мг/кг BID) соединения А и соединения D..
Образцы для анализа плазмы в состоянии «после еды». Кровь для определения концентраций TG в состоянии «после еды» собирают через 2 часа после введения дозы (2 часа в темном цикле) из боковой хвостовой вены, переносят в BD пробирки микроконтейнера, покрытые двукалиевой солью этилендиаминтетрауксусной кислоты (K2EDTA) (PN365974) и центрифугируют при 4°С. Образцы полученной плазмы затем анализируют на клиническом анализаторе Siemens Chemistry XPT (Malven, PA) с использованием реагентов для анализа триглицеридов Siemen triglycerides_2 (ref 10335892).
Образцы для анализа плазмы крови в состоянии «натощак». Кровь для определения концентраций TG в плазме крови в состоянии «натощак» собирают после 4-часового голодания через 2 часа после введения дозы (2 часа в темном цикле) из боковой хвостовой вены, переносят в BD пробирки микроконтейнера, покрытые двукалиевой солью этилендиаминтетрауксусной кислоты (K2EDTA) (PN365974) и центрифугируют при 4°С. Образцы полученной плазмы затем анализируют на клиническом анализаторе Siemens Chemistry XPT (Malven, PA) с использованием реагентов для анализа триглицеридов Siemen triglycerides_2 (ref 10335892).
В последний день исследования (день 28) крыс умерщвляют для сбора ткани после 4 часового голодания, через 2 часа после введения дозы: через два часа после введения дозы кровь для определения компонентов плазмы собирают через боковую хвостовую вену, а затем животных умерщвляют аспирацией в СО2. Кровь переносят в BD пробирки микроконтейнера, покрытые двукалиевой солью этилендиаминтетрауксусной кислоты (K2EDTA) (PN365974), центрифугируют при 4°С, плазму переносят в 96-луночный микротитровальный планшет и хранят при -20°С. Печень быстро извлекают, замораживают в зажиме Волленберга, предварительно охлажденном в жидком N2, отдельно завернутом в алюминиевую фольгу, и хранят при -80°С.
Измельчение ткани: замороженную печень быстро измельчают на алюминиевом блоке, охлажденном в жидком N2, гарантируя таким образом, что ткань останется замороженной во время измельчения. Измельченные ткани переносят в конические пробирки из полипропилена объемом 7 мл и хранят при -80°С до проведения анализа.
Экстракция печеночного триглицерида: приблизительно от 50 до 100 мг измельченной ткани добавляют в матричную D пробирку для лизиса (MP Bio) объемом 2 мл, содержащую 800 мкл охлажденной льдом смеси (1:1) CHCl3:MeOH. Образцы сразу экстрагируют при 4°С с использованием Qiagen Tissue Lyser II (Qiagen Cat № 85300) в течение 4 минут при 30 Гц. После этого гомогенат переносят в стеклянные пробирки (13×100 мм) и помещают на лед. Пробирки для лизиса затем промывают 800 мкл смеси (1:1) CHCl3:MeOH, встряхивают в течение 30 секунд и переносят экстракт в стеклянные пробирки (13 × 100 мм). Во все стеклянные пробирки, помещенные на лед, добавляют 2,4 мл 100% CHCl3 для доведения соотношения CHCl3:MeOH до 4:2. После этого образцы выдерживают в охлаждающей камере при -20°С. На следующий день добавляют 1,75 мл 1M KCl H2O с получением соотношения CHCl3:MeOH:H2O 4:2:1,75. Образцы встрахивают в течение 30 секунд и центрифугируют (1500 об./мин. × 15 мин.) при 4°С. После центрифугирования органическую фазу переносят в свежую пробирку для экстракции (13 × 100 мм), сушат при 37°С в атмосфере N2 и снова суспендируют в 750 мкл CHCl3. Картриджи для твердофазной экстракции (ТФЭ) с аминопропилом (Waters Cat № 054560, 6 мл, 500 мг) смачивают и промывают 5 мл гексана. После промывки образцы экстракта объемом 200 мкл в CHCl3 наносят на картридж и удаляют вакуумом без сушки колонки. Нейтральные липиды затем элюируют 5 мл смеси 2:1 CHCl3:изопропанол/50 мкM бутилированный гидрокситолуол. Затем образцы сушат при 37°С в атмосфере N2 и снова суспендируют в 1,75 мл смеси 98:2 изооктан:изопропанол. Образцы фильтруют через фильтр-шприц (0,2 мкM) перед впрыском в ВЭЖХ колонку Cyanopropy (размер частиц 3,5 мкM - 4,6 × 150 мм, колонка Agilent Zorbax Eclipse XBD-CN). Метод: введение впрыскиванием 4 мкл при продолжительности анализа 27 минут с использованием растворителя А (1000:1:2, изооктан:изопропанол:уксусная кислота) и растворителя B (50:50 изопропанол:метил-трет-бутиловый эфир). В 0-3 минуты состав растворителя: 100% растворителя A. Минуты 3-8: состав растворителя изменяется от 100% растворителя A до 95% растворителя A и 5% растворителя B. Минуты 8-18: состав растворителя меняется до соотношения 50:50. Минуты 18-19: состав растворителя снова изменяется до 100% растворителя A, и данный состав сохраняется в течение 19-27 минут.
Фракции ядер и мембран получают из части измельченных образцов печени, которые объединяют на одну группу лечения, ультрацентрифугированием с использованием стандартных методов. Образцы ядерного экстракта и мембранных фракций анализируют вестерн-блоттингом для SREBP1. В качестве маркера для фракции мембран используют вестерт-блоты для калнексина, в качестве маркера общей загрузки образца - актин, и в качестве маркера для фракции ядер используют гистон 2B. Уровни содержания SREBP1 ядер количественно определяют с использованием относительных единиц и нормализуют по гистону 2B для контроля потерь образца в процессе ядерного фракционирования и загрузки геля.
Другую часть измельченной печени обрабатывают и анализируют на экспрессию липогенных генов. Линейные разрушаемые зонды (taqman probes) крыс в отношении АКК1, FASN, SCD1, PCSK9 и SREBP-1c оценивают с использованием Actb в качестве гена «домашнего хозяйства» на кПЦР.
Введение (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамида (соединение D) или его фармацевтически приемлемой соли в комбинации с 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойной кислотой (соединение А) или ее фармацевтически приемлемой солью приводит к значительному снижению уровней содержания TG в плазме (фигуры 10, 11) и печени (фигура 18) относительно содержания TG в плазме и печени при введении соединения А в качестве монотерапии.
Кормление в соответствии с западной диетой приводит к повышению содержания TG в плазме крыс в состоянии «натощак» в 2,2 раза по сравнению с крысами, получающими стандартный корм (фигура 10). Пероральное введение в низкой дозе (1 мг/кг QD) или в высокой дозе (10 мг/кг QD) только соединения А (монотерапия) приводит к повышению содержания TG в плазме в 1,7 раза и 1,3 раза, соответственно, по сравнению с крысами, получающими корм в соответствии с западной диетой, которым вводят разбавитель. И наоборот, пероральное введение только соединения D (монотерапия) в низкой (5 мг/кг BID) или высокой (30 мг/кг BID) дозе снижает содержание TG в плазме в состоянии после еды на 55% и 63%, соответственно, относительно крыс, которых получающих корм в соответствии с западной диетой, которым вводят разбавитель. Совместное введение соединения А и соединения D приводит к полной блокаде опосредованного соединением А повышения содержания TG в плазме в состоянии после еды. Пероральное совместное введение соединения А и соединения D в низких или в высоких дозах каждого приводит к снижению содержания TG в плазме в состоянии после еды на 37% и 64%, соответственно, по сравнению с крысами, которых кормят в соответствии с западной диетой и которым вводят разбавитель.
Кормление в соответствии с западной диетой приводит к повышению содержания TG в плазме в состоянии «натощак» в 1,6 раза по сравнению с крысами, которых кормят стандартным кормом (фигура 11). Пероральное введение в низкой и высокой дозах соединения А в качестве монотерапия приводит к повышению содержания TG в плазме в состоянии «натощак» в 2,4 и 1,7 раз, соответственно, по сравнению с крысами, получающими корм в соответствии с западной диетой, которым вводят разбавитель. И наоборот, пероральное введение низкой и высокой доз соединения D в качестве монотерапии приводит к снижению содержания TG в плазме в состоянии «натощак» на 20% и 35%, соответственно, по сравнению с крысами, получающими корм в соответствии с западной диетой, которым вводят разбавитель. Пероральное совместное введение соединения А и соединения D как в низкой, так и в высокой дозе, полностью подавляет опосредуемое соединением А повышение содержания TG в плазме в состоянии «натощак», наблюдаемое при введении тольо соединения А. Уровни содержания TG в плазме крови в состоянии «натощак» как для группы низкой дозы (109 мг/дл), так и для группы высокой дозы (81 мг/дл) совместного введения соединения А и соединения D аналогичны уровням содержания TG у крыс, получающих корм в соответствии с западной диетой, которым вводят разбавитель (96 мг/дл).
Внутриядерную локализацию SREBP-1 сравнивают в образцах крыс, получающих корм в соответствии с западной диетой, которым вводят разбавитель, отдельно высокую дозу соединения А в качестве монотерапии, отдельно высокую дозу соединения D в качестве монотерапии или совместно вводят в высоких дозах соединение А и соединение D (фигура 12). Относительно крыс, получающих корм в соответствии с западной диетой, которым вводят разбавитель, введение соединения А приводит к повышению ядерной локализации SREBP-1, что указывает на повышенную активацию SREBP-1. Введение соединение D приводит, наоборот, к пониженной ядерной локализации SREBP-1 и активации SREBP-1. Совместное введение соединения А и соединения D блокирует опосредуемое соединением А повышение ядерной локализации SREBP-1, приводя к 50% снижению по сравнению с использованием в качестве монотерапии только соединения А.
Среди крыс, которым вводят разбавитель, крысы, получающие корм в соответствии с западной диетой, проявляют повышенную экспрессию липогенных генов по сравнению с крысами, получающими стандартный корм: АКК1 (фигура 13), FASN (фигура 14), SCD1 (фигура 15) и SREBP1 (фигура 16), но не PCSK9 (фигура 17), содержание которого ниже у крыс, получающих корм в соответствии с западной диетой. По сравнению с введением разбавителя животным, получающим корм в соответствии с западной диетой, введение соединения А имеет тенденцию дополнительного повышения экспрессии АКК1, FASN (только высокая доза соединения А), SCD, но не PCSK9 и SREBP1. И наоборот, введение соединение D снижает экспрессию всех липогенных генов. Совместное введение соединения А и соединения D приводит к уровням экспрессии, которые сравнимы или ниже уровней, наблюдаемых у крыс, получающих корм в соответствии с западной диетой, которым вводят разбавителью.
По сравнению с крысами, которые получают стандартный корм и которым вводят разбавитель, крысы, которые получают корм в соответствии с западной диетой и которым вводят разбавитель, показывают повышение аккумуляции гепатических триглицеридов примерно в 2,7 раз (фигура 18). Пероральное введение низкой дозы и высокой дозы соединения А приводит к снижению стеатоза соответственно на 36% и 53% сравнению с крысами, которые получают корм в соответствии с западной диетой и которым вводят разбавитель. Аналогично, пероральное введение низкой дозы и высокой дозы соединения D приводит к снижению стеатоза на 25% и 30%, соответственно, по сравнению с крысами, которые получают корм в соответствии с западной диетой и которым вводят разбавитель. Пероральное совместное введение низкой или высокой дозы соединения А и соединения D снижает стеатоз на 50% и 73%, соответственно, по сравнению с крысами, которые получают корм в соответствии с западной диетой и которым вводят разбавитель. Пероральное введение комбинации соединения А и соединения D в низких дозах или высоких дозах приводит к большему снижению стеатоза, чем при отдельном введении соединения А или соединения D в качестве монотерапии в таких же дозах.
Рандомизированное исследование с 5 параллельными группами и использованием в качестве контроля разбавителя проводят на самцах крыс линии Wistar-Han rats (Charles River (Boston, MA)), получающих корм с низким содержание холина и высоким содержанием жиров (choline-deficient and high fat diet - CDAHFD) (Research diets; A16092003) для идентификации различий в улучшении маркеров воспаления и фиброза печени при отдельном введении соединения А или соединения D в качестве монотерапии и при их введении в комбинации. Для 60 крыс используют стандартные условия содержания (~200 г); крыс размещают по две особи в клетку и содержат при обращенном цикле дня и ночи 12:12-часов (свет выключают в 8 часов утра). Крыс кормят в соответствии с диетой с низким содержанием холина и высоким содержанием жиров (CDAHFD), начиная за 6 недель до начала исследования. Крысы, рандомизированные на 4 группы дозирования (n=12/группа), получают два раза в день разбавитель, соединение А (5 мг/кг) в качестве монотерапии, соединение D (30 мг/кг) в качестве монотерапии или комбинацию соединения А (5 мг/кг) и соединения D (30 мг/кг) в течение 6 недель. В качестве контрольной группы используют животных (n=12), получающих стандартное питание в процессе исследования, которым дважды в сутки вводят разбавитель. Образцы крови собирают перед началом введения соединений и через 3 и 6 недель после начала введения соединений для оценки циркулирующих маркеров. Сдвиговолновые эластографические измерения (Aixplorer Ultimate imager, Supersoinc imagine) проводят в -3 неделю, 0 неделю (перед введением 1-й дозы), в 3 неделю и 6 неделю для оценки развития воспаления и фиброза во времени. Гистологию оценивают после 6-недельного введения лекарственного средства, что соответствует 12 неделям кормления животных в соответствии с CDAHFD. Результаты представляют в виде средней величины по животным на каждую дозовую группу.
После 12-недельного кормления в соответствии с CDAHFD животных умерщвляют асфиксией в CO2. Собирают правую боковую, среднюю и левую боковую доли печени. Из левой боковой, правой средней и правой боковой долей печени каждого животного берут срезы, фиксируют их в формалине и перерабатывают в парафиновые блоки. Один срез левой боковой доли на животное криконсервируют при оптимальных температурах среза (OCT). Остаток печени от каждого животного замораживают и быстро измельчают на алюминиевом блоке, охлажденном в жидком N2, гарантируя таким образом, что ткань останется замороженной во время измельчения. Измельченные ткани переносят и хранят при -80°С до проведения анализа. Часть образца измельченной печени каждого животного обрабатывают и анализируют на маркеры генной экспрессии фиброгенеза. Линейные разрушаемые зонды (taqman probes) крыс в отношении SMA и COL1A1 оценивают с использованием Actb в качестве гена «домашнего хозяйства» на кПЦР.
С помощью качественной гистологической оценки сертифицированного ветеринара-патолога и количественной гистоморфометрии оценивают следующие конечные показатели: активация звездчатых клеток печени и дифференциация в миофибробласты с помощью иммуногистохмии αSMA (immunohistochemistry - IHC); коллаген как коррелят фиброза с помощью красителя Picrosirius Red. Изображения анализируют с помощью программного обеспечения Visiopharm software. Приложения Visiopharm с пороговыми параметрами применяют равномерно для идентификации срезов тканей и количественной оценки мишеней на каждой IHC (DAB (3,3'-диаминобензидин) положительной) или гистохимически окрашенных слайдах как процент площади: интересующая окрашенная площадь/общая ROI ткани - площадь пустого пространства) × 100%. Для анализа данных, полученных в этом исследовании, используют непараметрическую статистику. Групповые значения записывают как среднее значение +/- стандартная ошибка среднего значения.
По сравнению с контрольными животными, которые получают стандартный корм и которым вводят разбавитель, животные, получающие CDAHFD, которым вводят разбавитель, показывают значительное повышение жесткости печени (определяемую с использованием сдвиговолновой эластографии (shearwave elastography - SWE) в пилопаскалях (кПа)) в течение исследования, что указывает на прогрессирующие воспаление и фиброз печени (фигура 19). Отдельное введение соединения А или соединения D в качестве монотерапии снижает жесткость печени, позволяя сделать предположение о снижении воспаления печени и/или фиброза. Совместное введение соединения А и соединения D приводит к большему снижению жесткости печени, чем отдельное введение каждого соединения в качестве монотерапии (фигура 19).
По сравнению с контрольными животными, которые получают стандартный корм и которым вводят разбавитель, животные, которые получают CDAHFD и которым вводят разбавитель, показывают значительное увеличение окрашивания печени альфа-гладкомышечным актином (αSMA), указывающего на фибробластную активацию и фиброгенез (фигура 20). Отдельное введение соединения А или соединения D в качестве монотерапии снижает αSMA окрашивание на 41% и 23%, соответственно, позволяя сделать предположение о снижении активации печеночных миофибробластов и фиброгенеза. Совместное введение соединения А и соединения D приводит к большему снижения αSMA окрашивания, чем введение каждого соединения отдельно при применении в качестве монотерапии, снижая окрашивание на 72% (фигура 20).
По сравнению с контрольными животными, которые получают стандартный корм и которым вводят разбавитель, животные, получающие CDAHFD, которым вводят разбавитель, показывают значительное увеличение окрашивания красителем Picosrius red (PSR), указывающее на отложение коллагена и фиброз (фигура 21). Отдельное введение соединения А или соединения D в качестве монотерапии снижает PSR окрашивание на 26% и 20%, соответственно, позволяя сделать предположение о снижении отложения коллагена и фиброза. Совместное введение соединения А и соединения D приводит к большему снижению PSR окрашивания, чем отдельное введение каждого соединения в качестве монотерапии, снижая окрашивание на 56% (фигура 21).
По сравнению с контрольными животными, которые получают стандартный корм и которым вводят разбавитель, животные, которые получают CDAHFD и которым вводят разбавитель, показывают значительное увеличение окрашивания адаптерной молекулы, связывающей ионизированный кальций 1 (Iba1), указывающее на активацию печеночных макрофагов (фигура 24). Введение соединения А в качестве монотерапии снижает Iba1 окрашивание на 15%, позволяя сделать предположение о снижении воспалительного тонуса печени. Хотя введение D в качестве монотерапии не изменяет Iba1 окрашивания, совместное введение A и D дает большее снижение Iba1 окрашивания, чем отдельное введение соединения A в качестве монотерапии, снижая окрашивание на 33% (фигура 24).
По сравнению с контрольными животными, которые получают стандартный корм и которым вводят разбавитель, животные, которые получают CDAHFD и которым вводят разбавитель, показывают значительное повышение генной экспрессии альфа-гладкомышечного актина (α-alpha smooth muscle actin - αSMA) (фигура 22) и A1A (COL1A1) (фигура 23), что указывает на активацию миофибробластов и фиброгенез. Отдельное введение соединения А или соединения D в качестве монотерапии снижает генную экспрессию гепатического αSMA и COL1A1, позволяя сделать предположение о снижении активации печеночных миофибробластов и фиброгенеза. Совместное введение соединения А и соединения D приводит к большему снижению генной экспрессии печеночного αSMA (фигура 22) и COL1A1 (фигура 23), чем при введении каждого соединения отдельно в качестве монотерапии.
В настоящем описании приводятся ссылки на различные публикации. Содержание этих публикаций во всей полноте включается в настоящее описании в виде ссылки для всех целей.
Специалистам в данной области техники будет понятно, что в изобретение могут быть внесены различные модификации и вариации, не выходящие за рамки объема или сущности настоящего изобретения. Другие варианты осуществления будут очевидны специалистам в данной области техники из рассмотрения описания и вариантов осуществления изобретения, раскрытых в настоящем описании. Предполагается, что описание и примеры следует рассматривать только как иллюстрацию изобретения, истинный объем и сущность которого представлены в приведенной далее формуле изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ ДИАЦИЛГЛИЦЕРОЛ-АЦИЛТРАНСФЕРАЗЫ 2 | 2017 |
|
RU2719589C1 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ПРЕДУПРЕЖДЕНИЯ ИЛИ ЛЕЧЕНИЯ НЕАЛКОГОЛЬНОГО СТЕАТОГЕПАТИТА | 2021 |
|
RU2803733C1 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ПРЕДУПРЕЖДЕНИЯ ИЛИ ЛЕЧЕНИЯ НЕАЛКОГОЛЬНОЙ ЖИРОВОЙ ИНФИЛЬТРАЦИИ ПЕЧЕНИ, СОДЕРЖАЩАЯ ЛИГАНД GPR119 В КАЧЕСТВЕ АКТИВНОГО ИНГРЕДИЕНТА | 2019 |
|
RU2768943C1 |
| НОВЫЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ, СОДЕРЖАЩИЙ ИХ | 2019 |
|
RU2793138C2 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ОКСИДОЛА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ЛИГАНДОВ РЕЦЕПТОРА ВАЗОПРЕССИНА | 2007 |
|
RU2461556C2 |
| АНТАГОНИСТЫ ПЕПТИДНОГО РЕЦЕПТОРА, СВЯЗАННОГО С ГЕНОМ КАЛЬЦИТОНИНА | 2003 |
|
RU2341526C2 |
| N1/N2-ЛАКТАМНЫЕ ИНГИБИТОРЫ АЦЕТИЛ-КоА-КАРБОКСИЛАЗ | 2011 |
|
RU2540337C2 |
| ПРОИЗВОДНЫЕ ПИРИДИНОНА И ПИРИДАЗИНОНА В КАЧЕСТВЕ ИНГИБИТОРОВ ПОЛИ(ADP-РИБОЗА) ПОЛИМЕРАЗЫ (PARP) | 2007 |
|
RU2472782C2 |
| КОМБИНАЦИИ ИНГИБИТОРОВ FGFR4 И СЕКВЕСТРАНТОВ ЖЕЛЧНЫХ КИСЛОТ | 2017 |
|
RU2742033C2 |
| ТРИЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2020 |
|
RU2833354C1 |
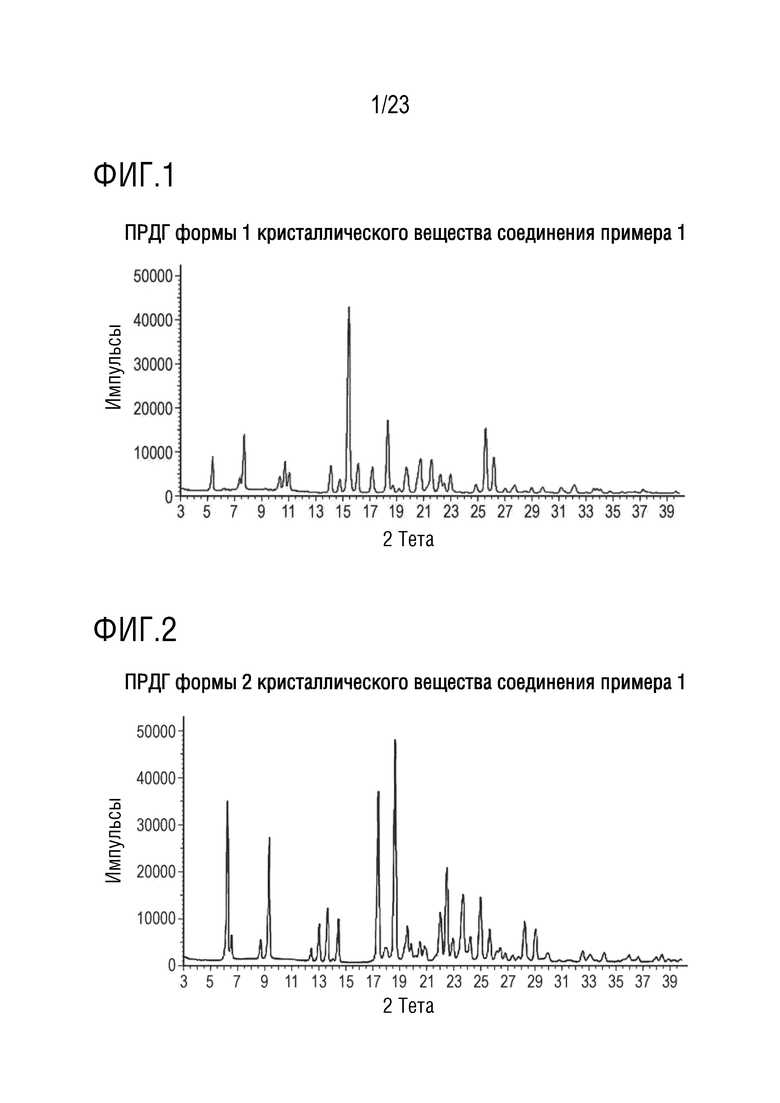
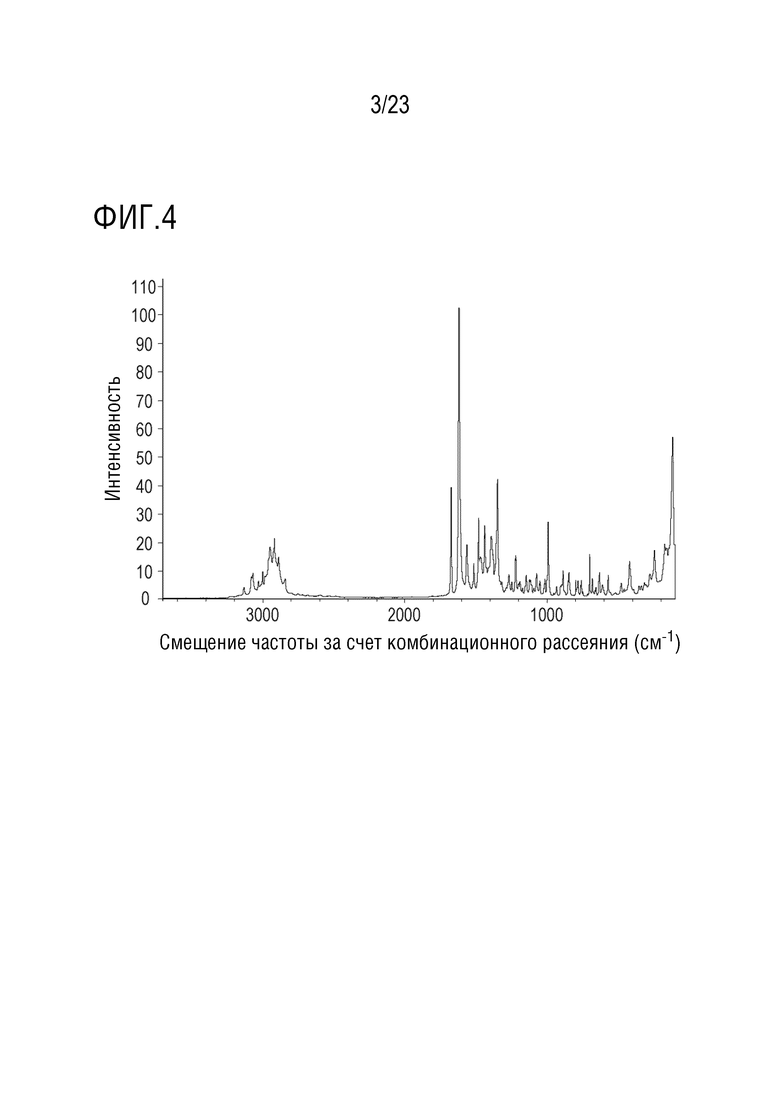
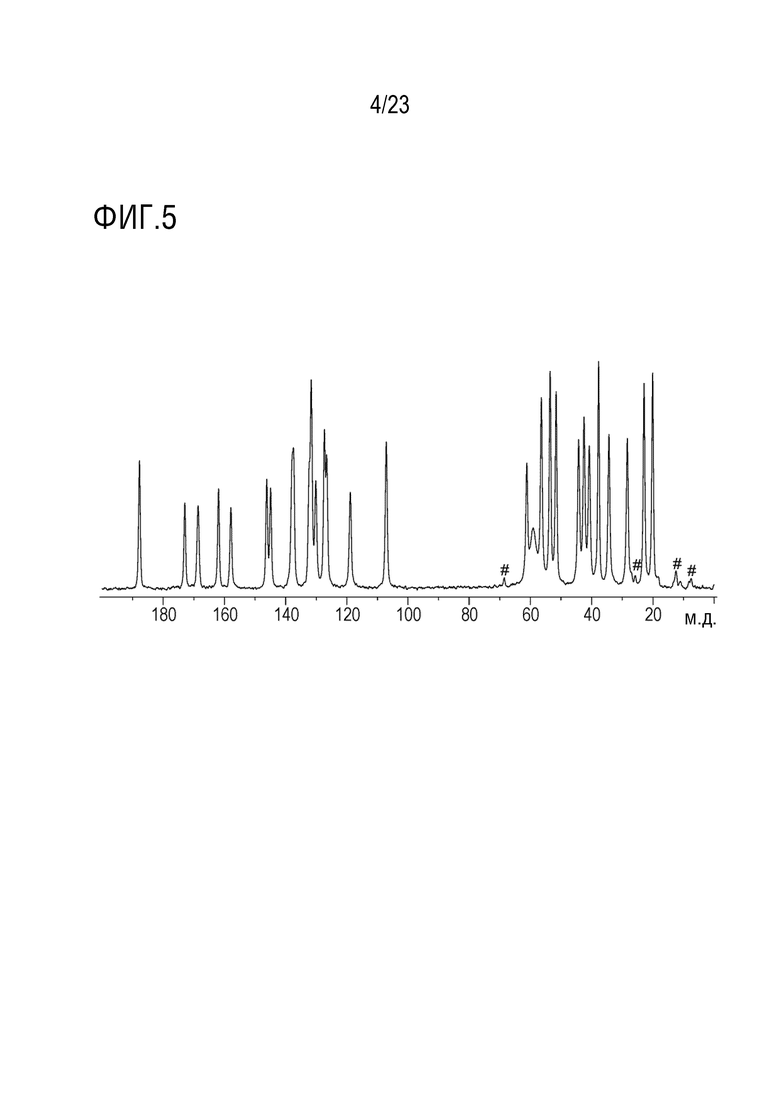
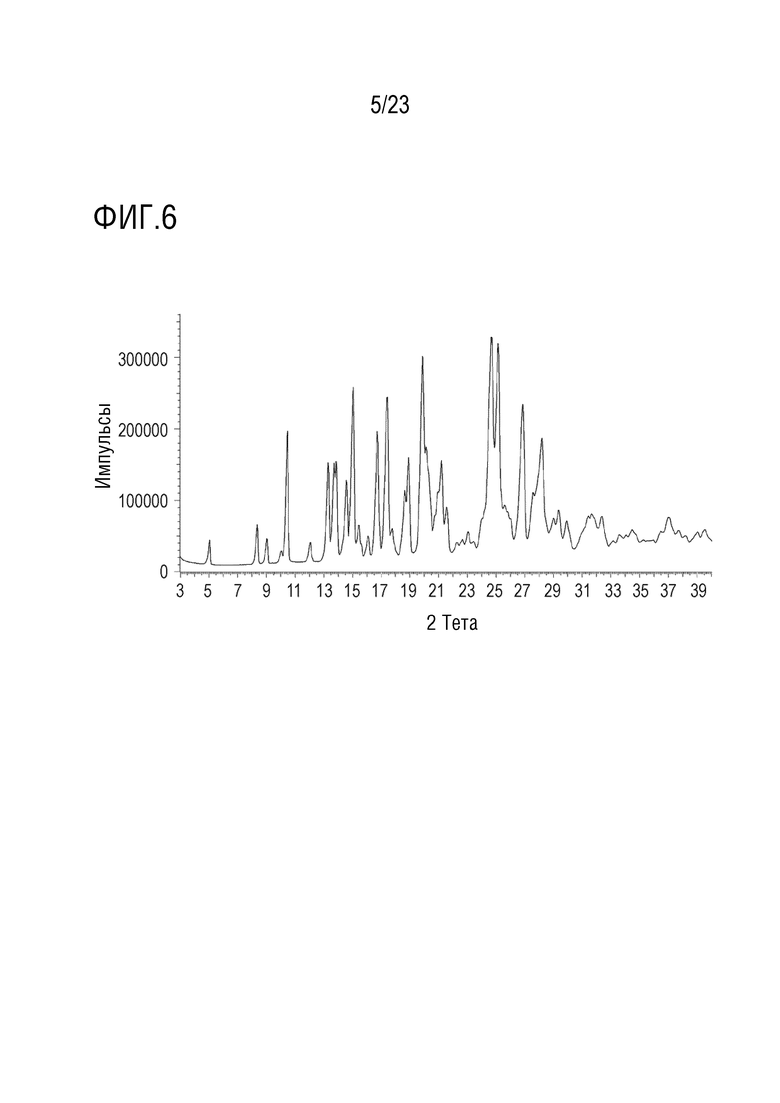
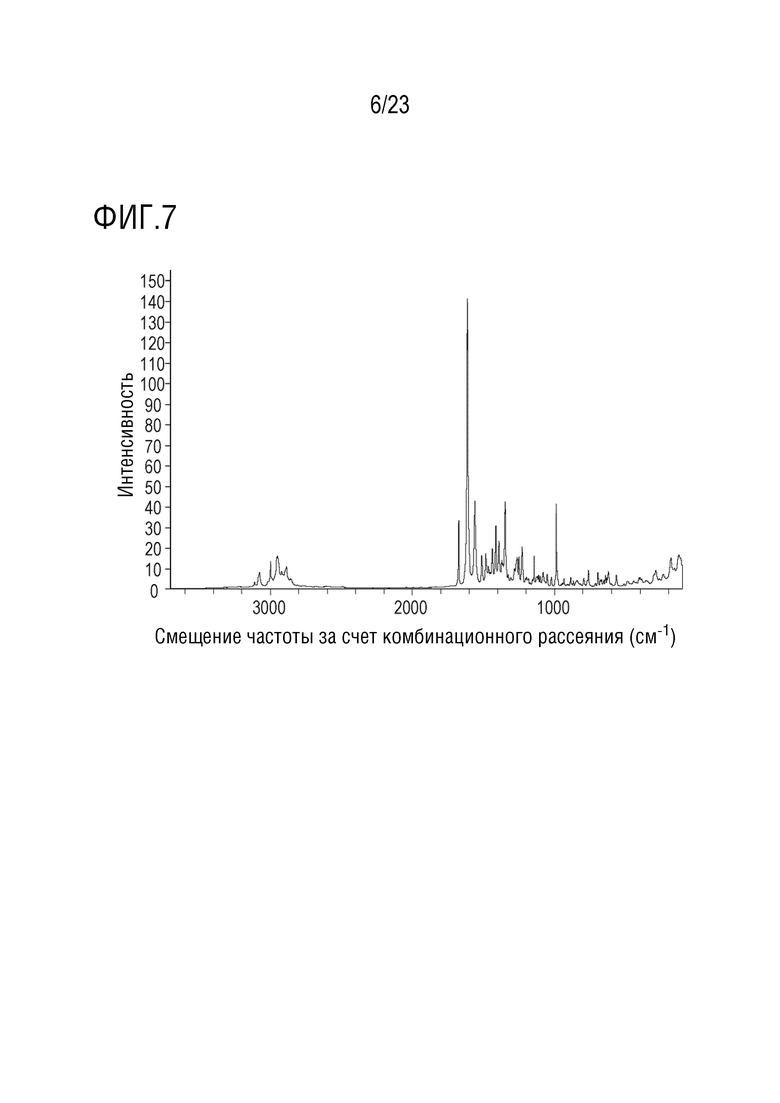
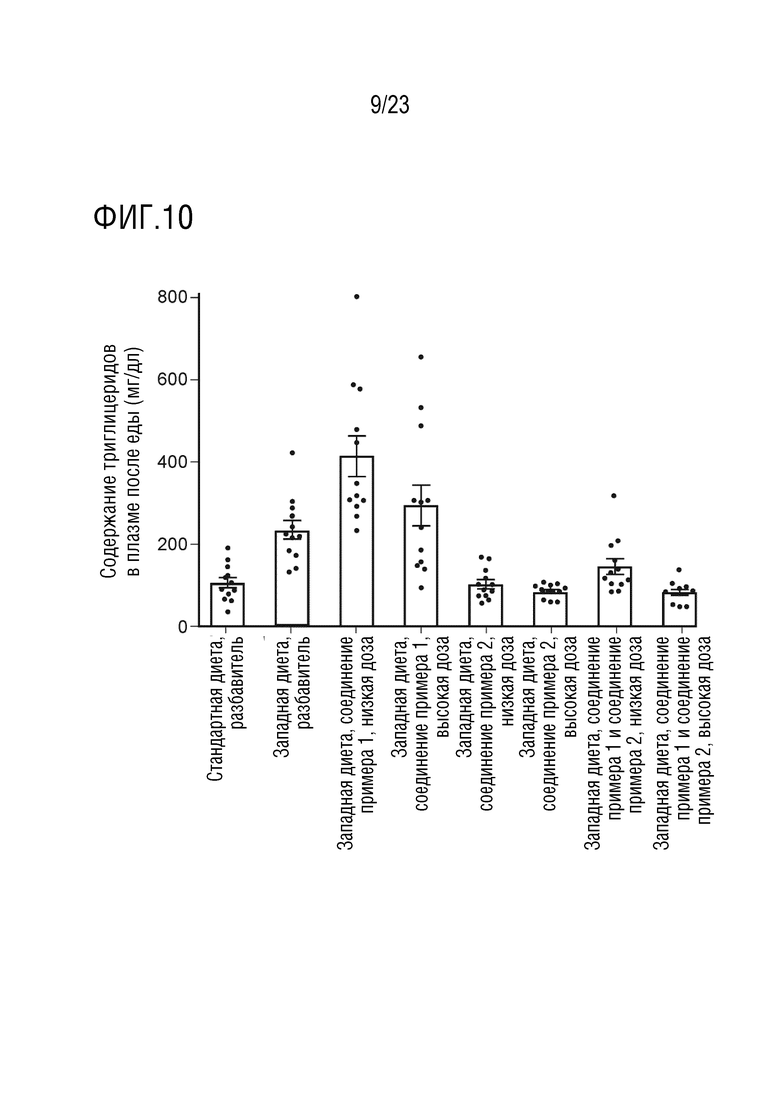
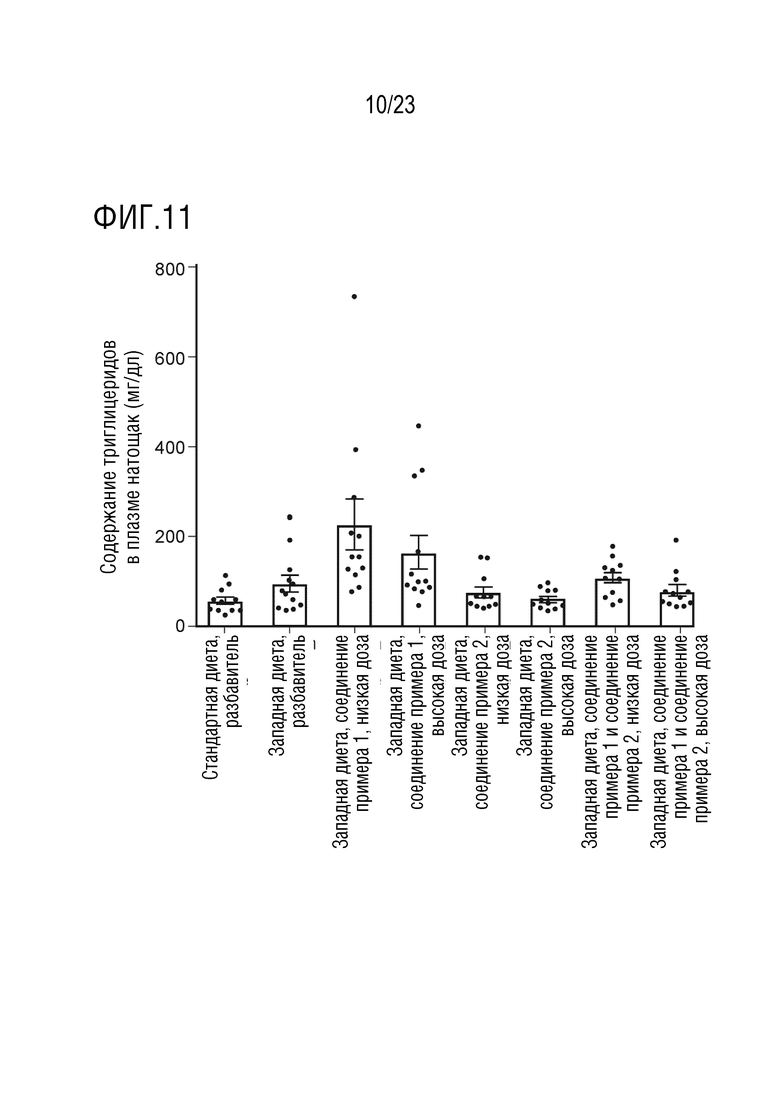
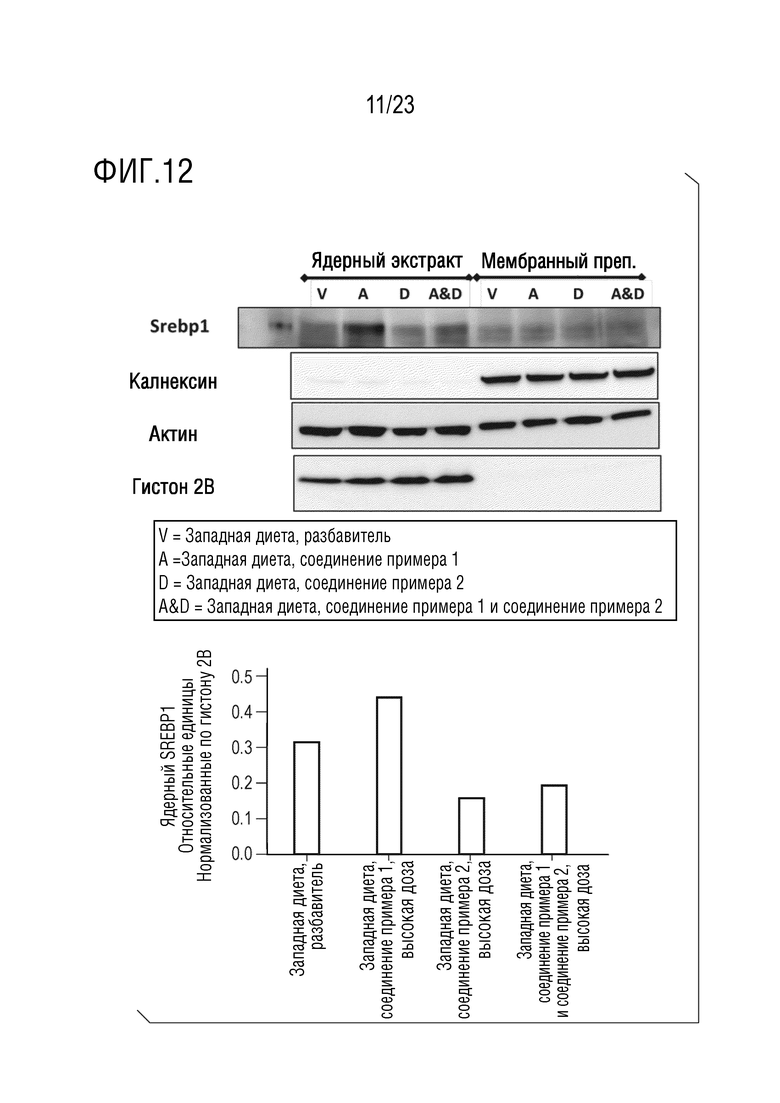
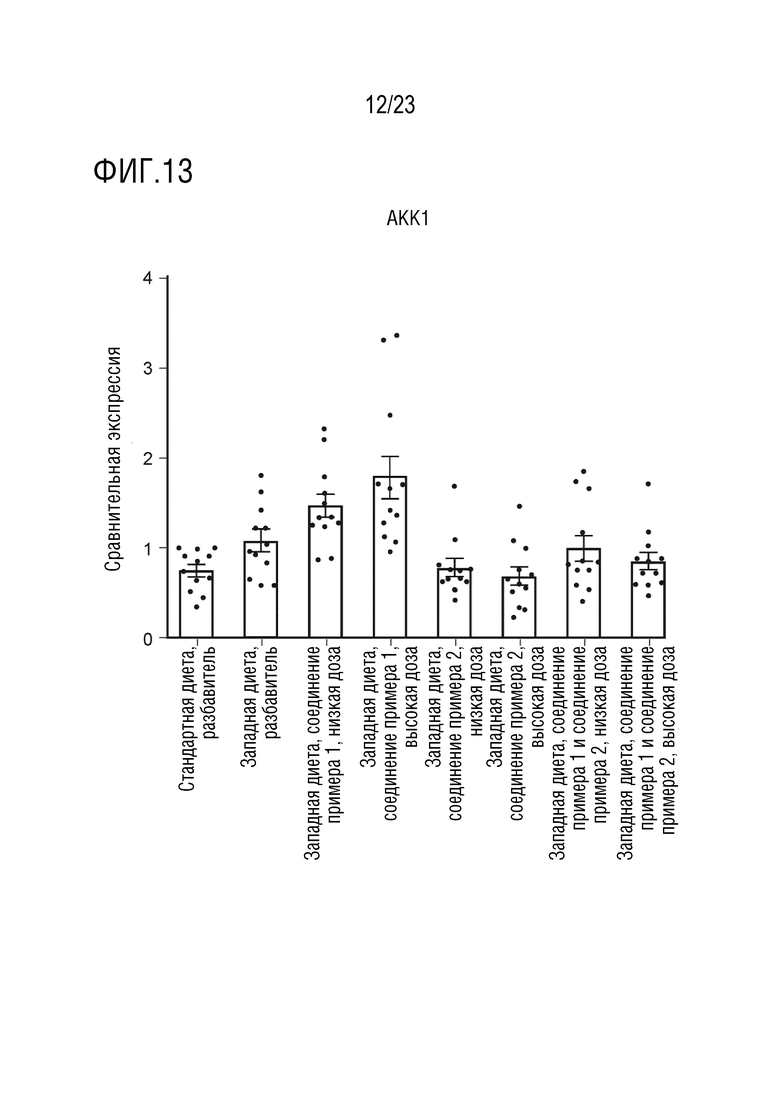
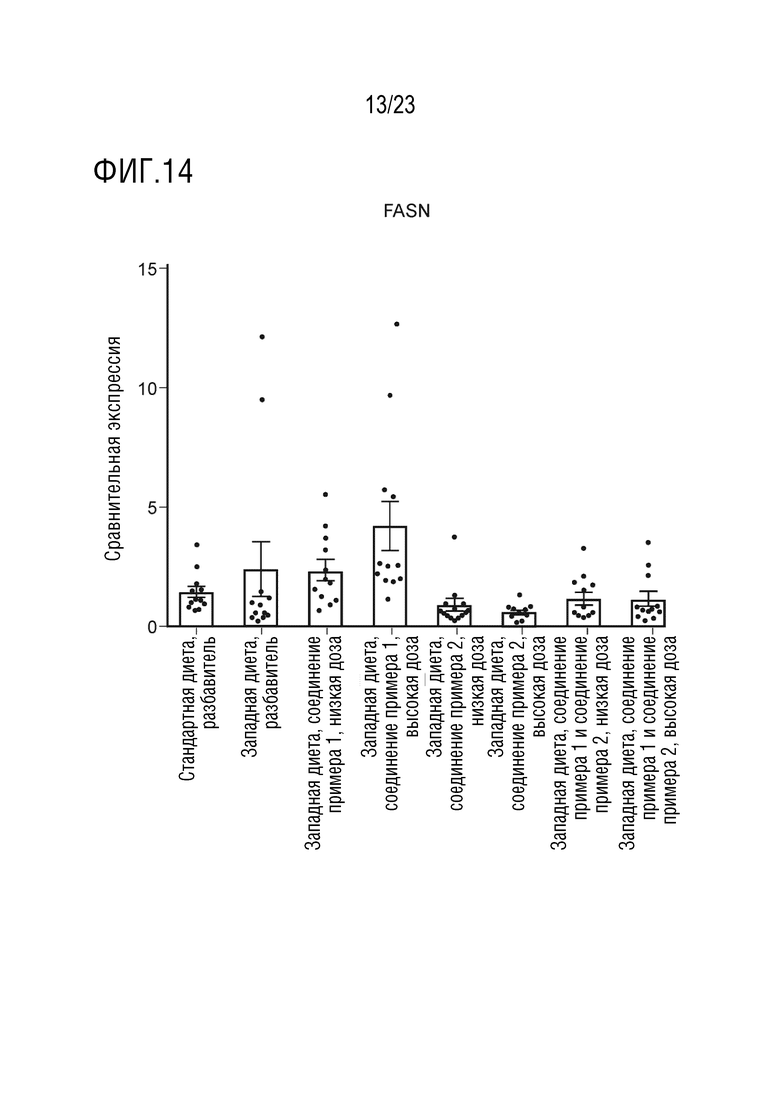
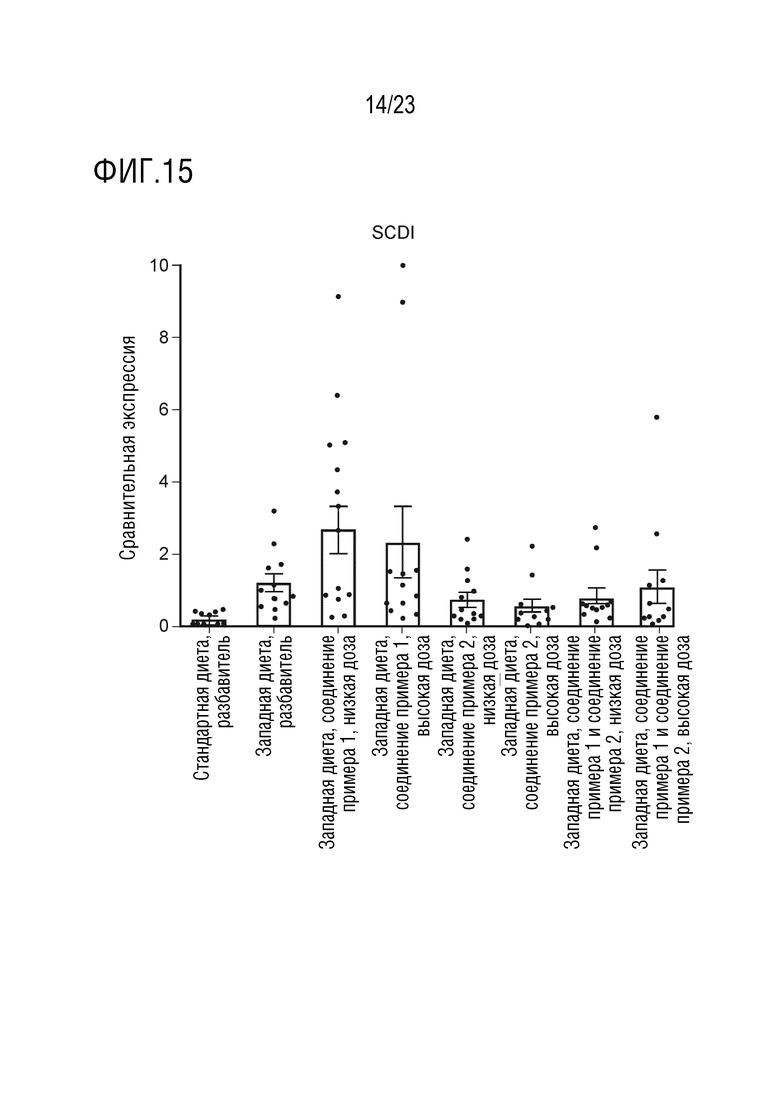
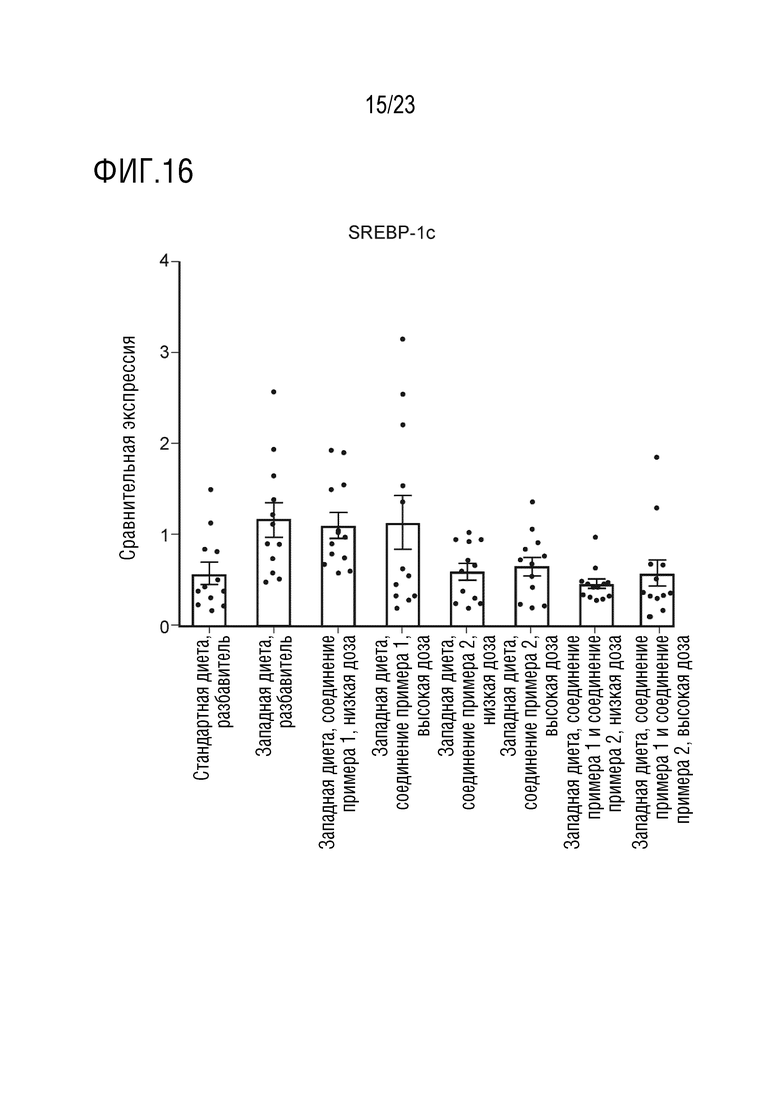
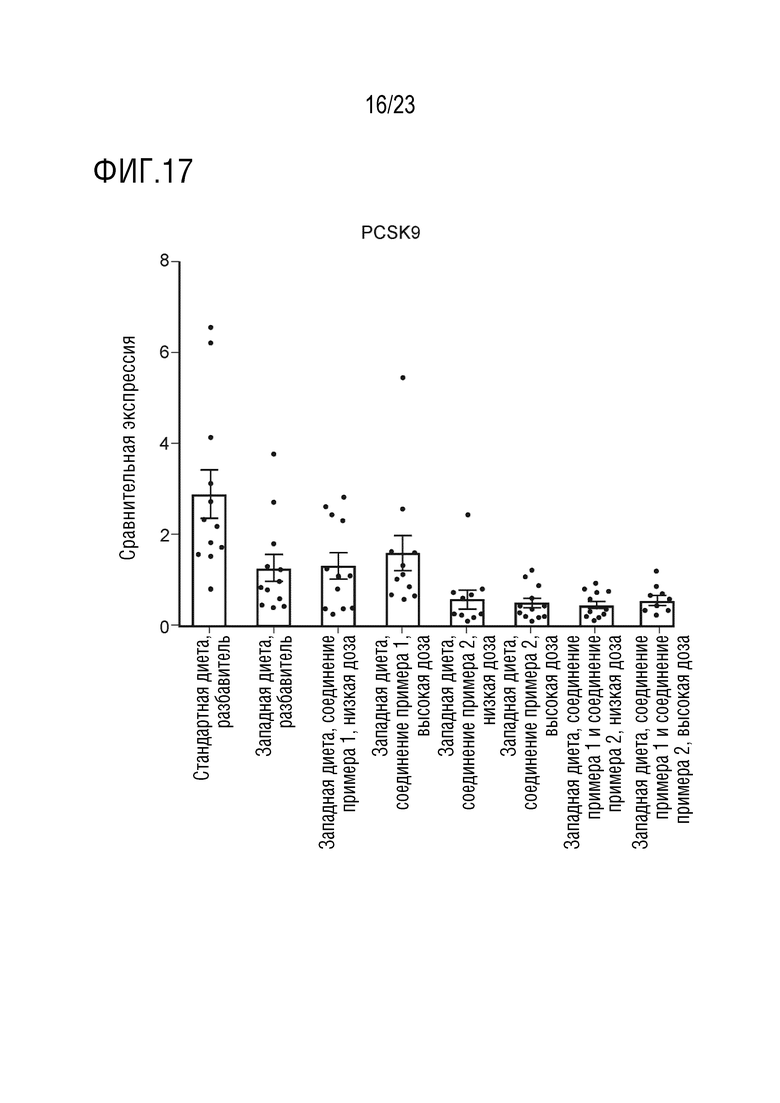
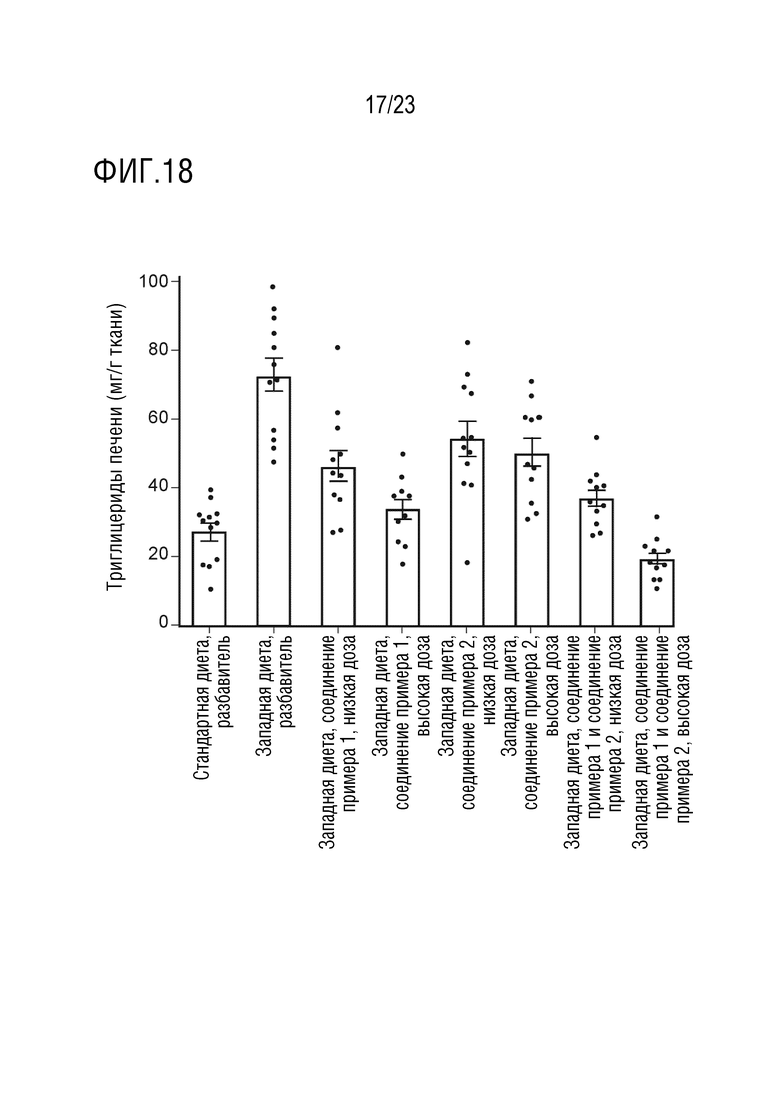
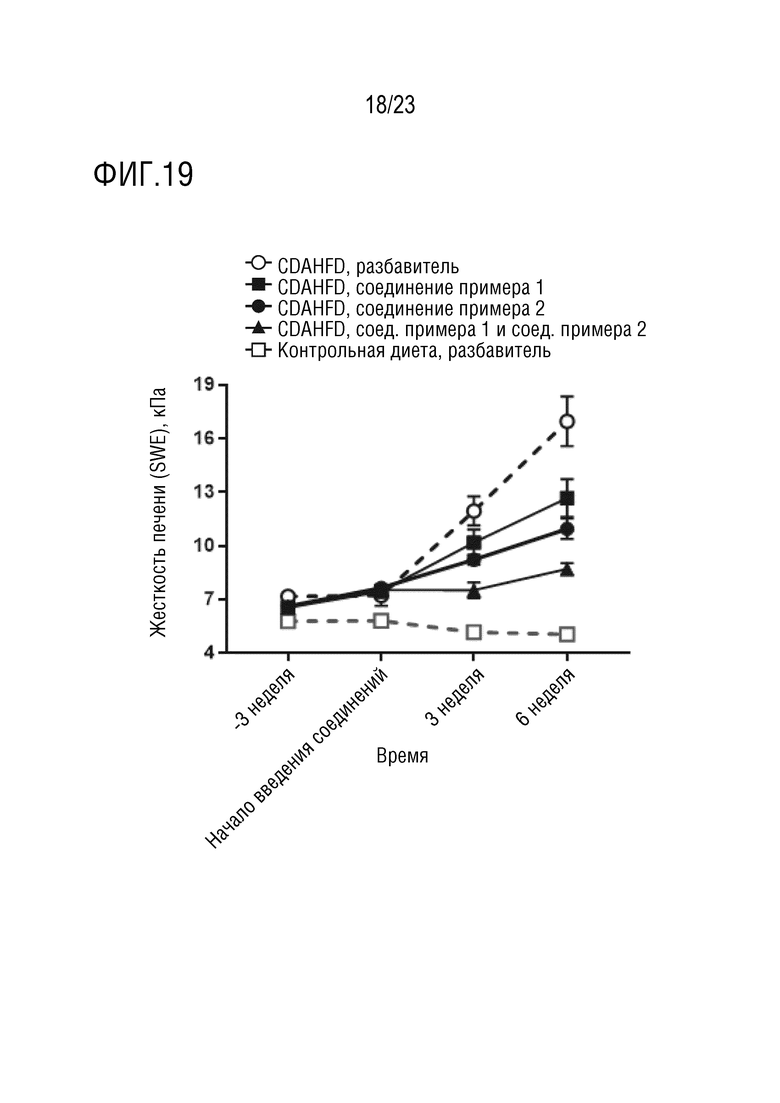
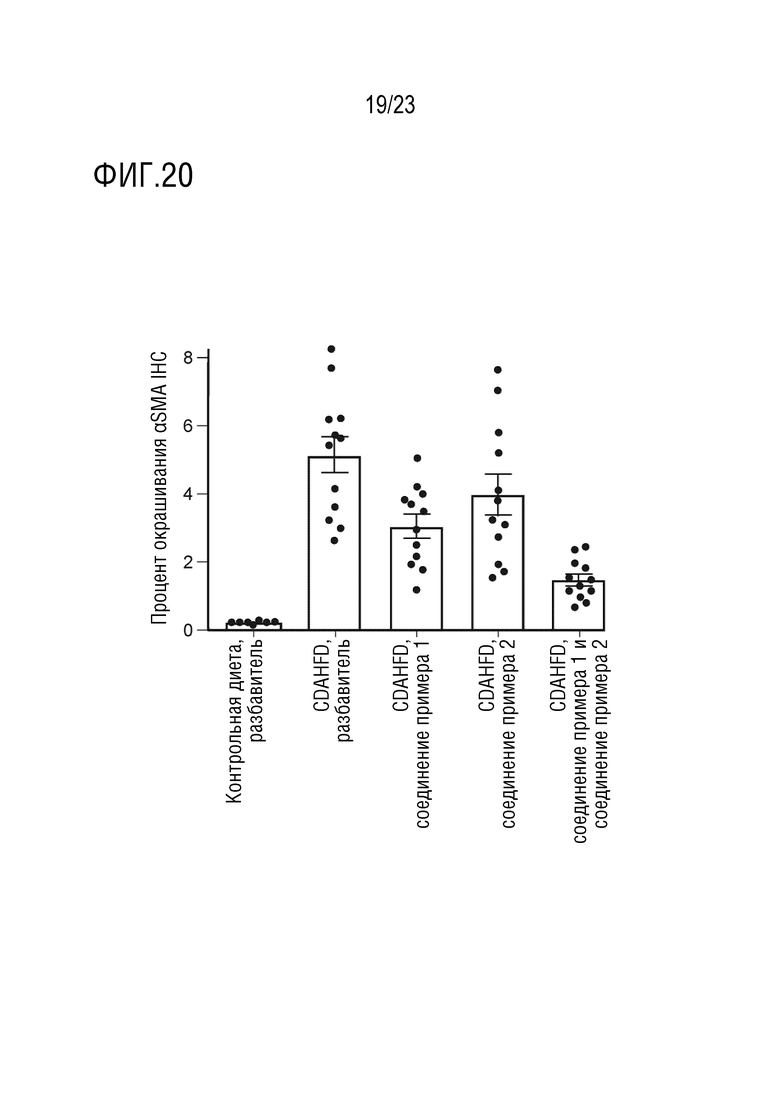
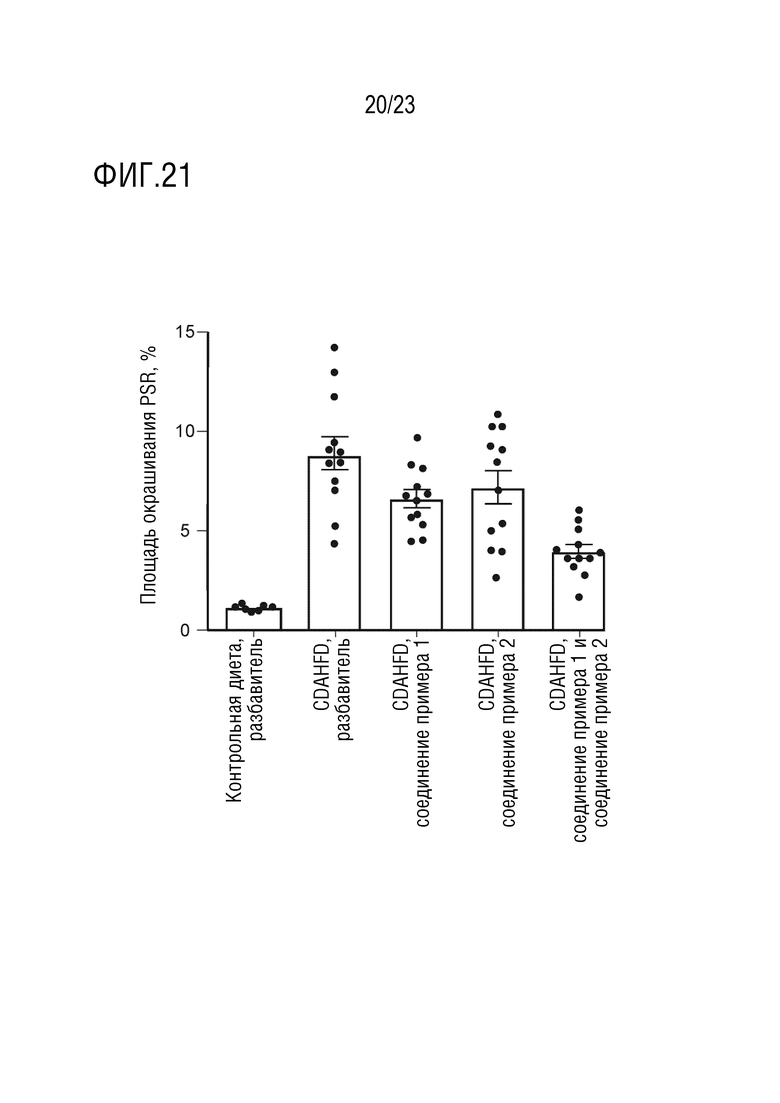
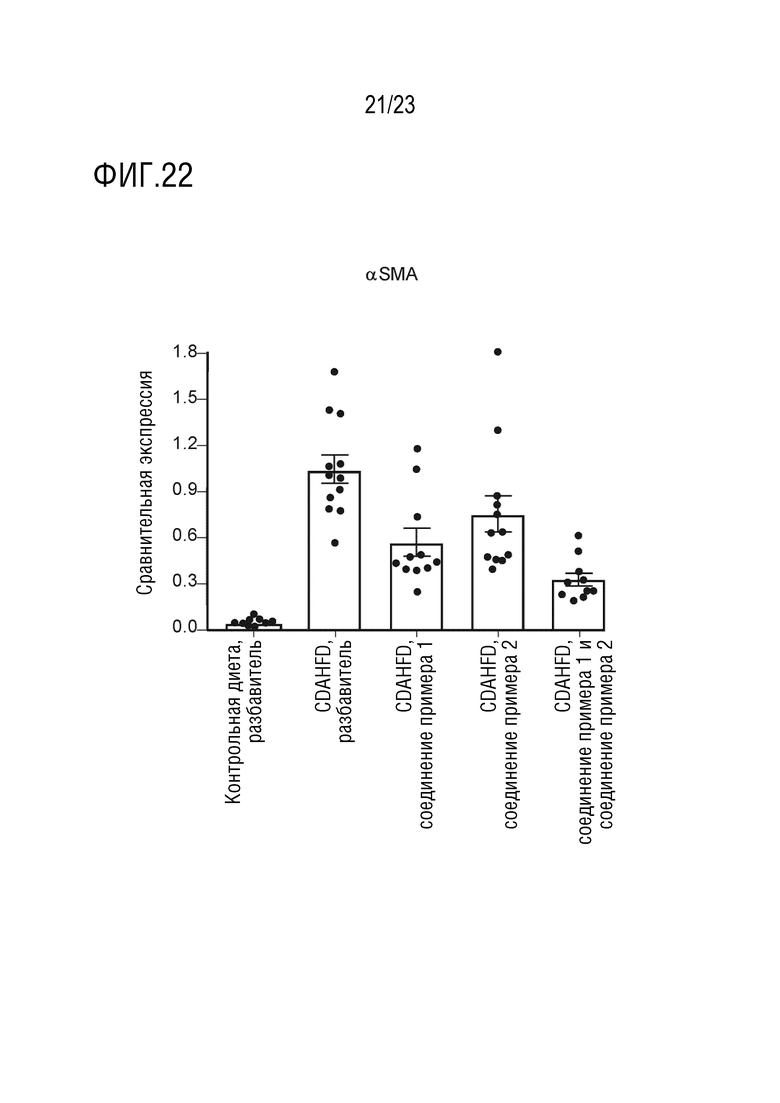
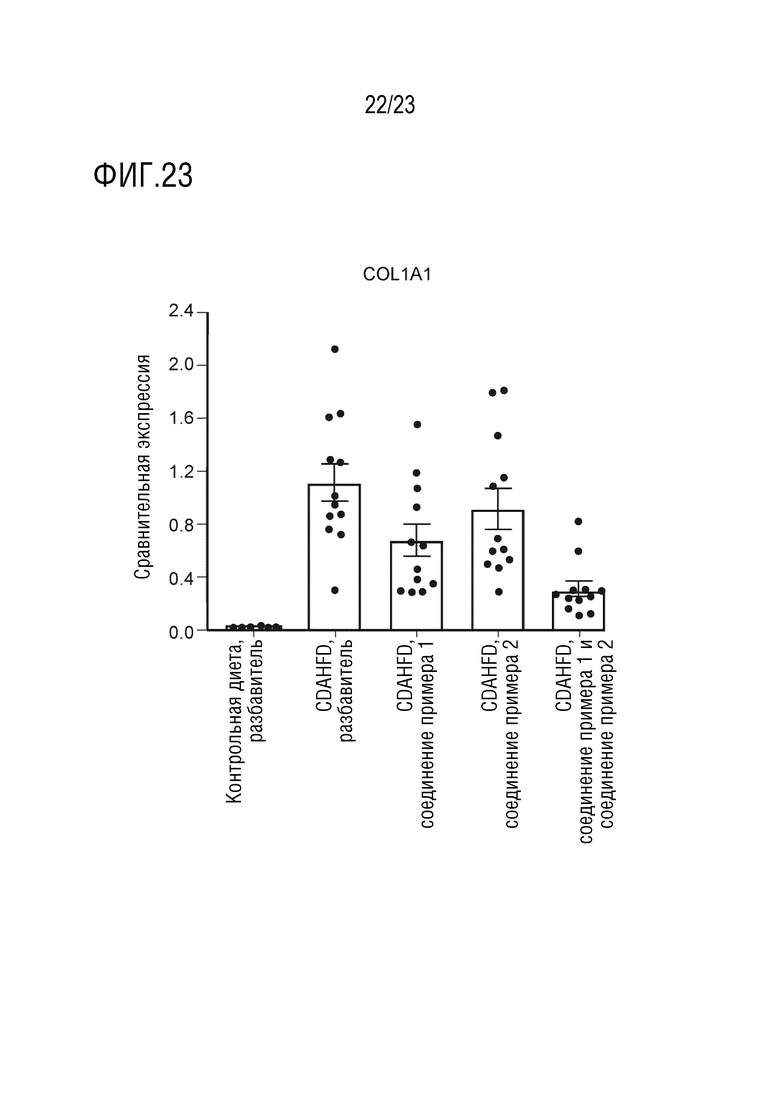
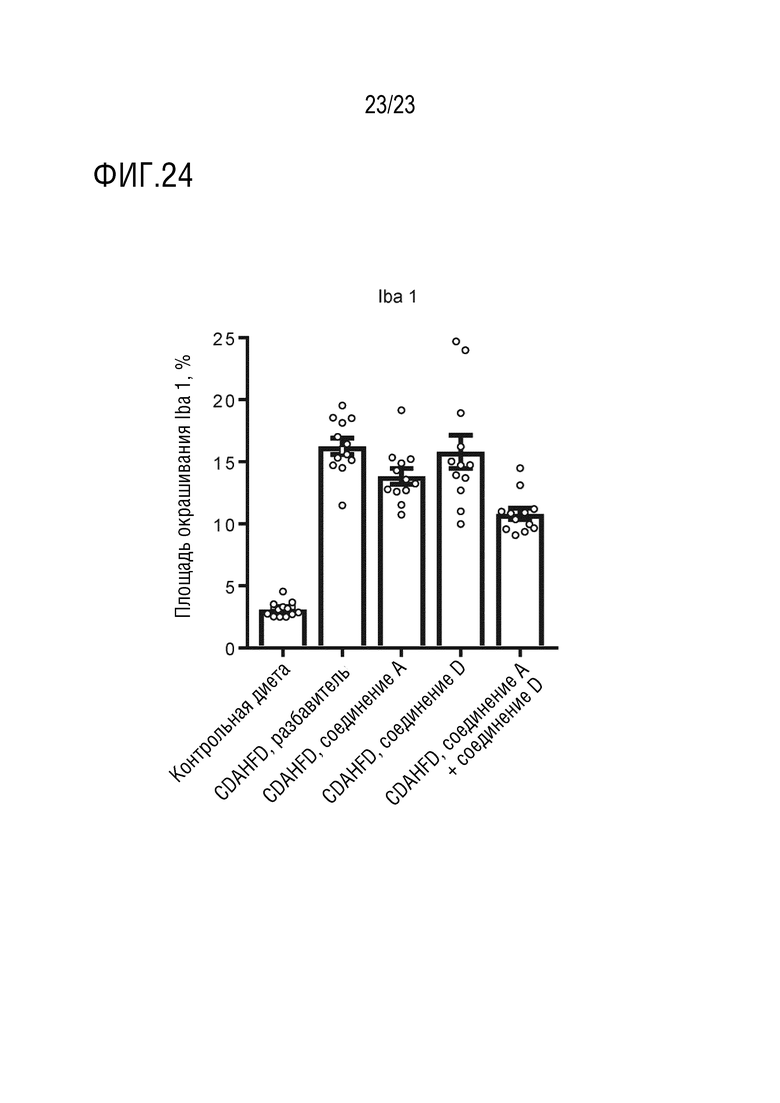
Группа изобретений относится к области медицины и фармацевтики и может быть использована для лечения заболевания или состояния, выбранного из жирной печени, неалкогольной жировой болезни печени, неалкогольного стеатогепатита, неалкогольного стеатогепатита с фиброзом печени, неалкогольного стеатогепатита с циррозом печени и неалкогольного стеатогепатита с циррозом печени и с гепатоцеллюлярной карциномой или с метаболическим заболеванием. Предложена фармацевтическая композиция для лечения указанных заболеваний, включающая (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамид или его фармацевтически приемлемую соль в комбинации по меньшей мере с 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойной кислотой или ее фармацевтически приемлемой солью, каждое соединение в терапевтически эффективном количестве, в смеси с фармацевтически приемлемым эксципиентом. Группа изобретений касается также способов лечения указанных заболеваний, включающих введение терапевтически эффективного количества указанных соединений. Обеспечивается значительное снижение уровня триглицеридов в плазме и печени. 6 н. и 19 з.п. ф-лы, 24 ил., 8 табл., 2 пр.
1. Фармацевтическая композиция для лечения заболевания или состояния, выбранного из жирной печени, неалкогольной жировой болезни печени, неалкогольного стеатогепатита, неалкогольного стеатогепатита с фиброзом печени, неалкогольного стеатогепатита с циррозом печени и неалкогольного стеатогепатита с циррозом печени и с гепатоцеллюлярной карциномой или с метаболическим заболеванием, включающая (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамид или его фармацевтически приемлемую соль в комбинации по меньшей мере с 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойной кислотой или ее фармацевтически приемлемой солью, каждое соединение в терапевтически эффективном количестве, в смеси с фармацевтически приемлемым эксципиентом.
2. Фармацевтическая композиция для лечения заболевания или состояния, выбранного из жирной печени, неалкогольной жировой болезни печени, неалкогольного стеатогепатита, неалкогольного стеатогепатита с фиброзом печени, неалкогольного стеатогепатита с циррозом печени и неалкогольного стеатогепатита с циррозом печени и с гепатоцеллюлярной карциномой или с метаболическим заболеванием, включающая (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамид или его фармацевтически приемлемую соль, 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойную кислоту или ее фармацевтически приемлемую соль и фармацевтически приемлемый эксципиент.
3. Композиция по п. 2, кроме того включающая по меньшей мере одно дополнительное фармацевтическое средство, выбранное из группы, состоящей из противовоспалительного средства, противодиабетического средства и холестерин/липид-модулирующего средства.
4. Способ лечения заболевания или состояния, выбранного из жирной печени, неалкогольной жировой болезни печени, неалкогольного стеатогепатита, неалкогольного стеатогепатита с фиброзом печени, неалкогольного стеатогепатита с циррозом печени и неалкогольного стеатогепатита с циррозом печени и с гепатоцеллюлярной карциномой или с метаболическим заболеванием, причем указанный способ включает введение пациенту, нуждающемуся в таком лечении, терапевтически эффективного количества (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамида или его фармацевтически приемлемой соли в комбинации по меньшей мере с терапевтически эффективным количеством 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойной кислоты или ее фармацевтически приемлемой соли.
5. Способ лечения заболевания или состояния, выбранного из жирной печени, неалкогольной жировой болезни печени, неалкогольного стеатогепатита, неалкогольного стеатогепатита с фиброзом печени, неалкогольного стеатогепатита с циррозом печени и неалкогольного стеатогепатита с циррозом печени и с гепатоцеллюлярной карциномой или с метаболическим заболеванием, причем указанный способ включает введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамида или его фармацевтически приемлемой соли и 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойной кислоты или ее фармацевтически приемлемой соли.
6. Способ по п. 5, в котором заболевание или состояние представляет собой жирную печень.
7. Способ по п. 5, в котором заболевание или состояние представляет собой неалкогольную жировую болезнь печени.
8. Способ по п. 5, в котором заболевание или состояние представляет собой неалкогольный стеатогепатит.
9. Способ по п. 5, в котором заболевание или состояние представляет собой неалкогольный стеатогепатит с фиброзом печени.
10. Способ по п. 5, в котором заболевание или состояние представляет собой неалкогольный стеатогепатит с циррозом печени.
11. Способ по п. 10, в котором заболевание или состояние представляет собой неалкогольный стеатогепатит с циррозом печени и с гепатоцеллюлярной карциномой.
12. Способ по п. 5, который включает дополнительное введение терапевтически эффективного количества по меньшей мере одного другого фармацевтического средства, выбранного из группы, состоящей из ингибитора ацетил-КоА-карбоксилазы (АКК), ингибитора диацилглицерол-О-ацилтрансферазы 1 (DGAT-1), ингибиторов моноацилглицерол-О-ацилтрансферазы, ингибитора фосфодиэстеразы-10 (PDE-10), активатора AMPK, сульфонилмочевины, меглитинида, ингибитора α-амилазы, ингибитора α-глюкозидгидролазы, ингибитора α-глюкозидазы, агониста PPARγ, агониста PPAR α/γ, бигуанида, модулятора глюкагоноподобного пептида (GLP-1), лираглутида, албиглутида, эксенатида, албиглутида, ликсисенатида, дулаглутида, семаглутида, ингибитора протеинтирозинфосфатазы-1В (РТР-1В), активатора SIRT-1, ингибитора дипептидилпептидазы IV (DPP-IV), стимулятора секреции инсулина, ингибитора окисления жирных кислот, антагониста A2, ингибитора с-Jun N-терминальной киназы (JNK), активаторов глюкокиназы (GKa), инсулина, инсулиноподобного средства, ингибитора гликогенфосфорилазы, агониста рецептора VPAC2t, ингибиторов SGLT2, модуляторов рецептора глюкагона, модуляторов GPR119, производных и аналогов FGF21, модуляторов рецептора TGR5, модуляторов рецептора GPBAR1, агонистов GPR40, модуляторов GPR120, активаторов рецепторов никотиновой кислоты высокого сродства (HM74A), ингибиторов SGLT1, ингибиторов или модуляторов ферментов карнитинпальмитоилтрансферазы, ингибиторов фруктозо-1,6-дифосфатазы, ингибиторов альдозоредуктазы, ингибиторов минералокортикоидного рецептора, ингибиторов TORC2, ингибиторов CCR2 и/или CCR5, ингибиторов PKCα, ингибиторов PKCβ, ингибиторов PKCγ, ингибиторов синтетазы жирных кислот, ингибиторов серинпальмитоилтрансферазы, модуляторов GPR81, GPR39, GPR43, GPR41, GPR105, Kv1.3, ретинолсвязывающего белка-4, глюкокортикоидного рецептора, рецепторов соматостатина, ингибиторов или модуляторов PDHK2 или PDHK4, ингибиторов MAP4K4, модуляторов IL1beta, ингибиторов ГМГ-КoA редуктазы, ингибиторов скваленсинтетазы, фибратов, секвестрантов желчных кислот, ингибиторов ACAT, ингибиторов MTP, ингибиторов липооксигеназы, ингибиторов абсорбции холестерина, модуляторов PCSK9, ингибиторов транспортного белка холестериновых эфиров и модуляторов RXRalpha.
13. Способ лечения жирной печени, неалкогольной жировой болезни печени, неалкогольного стеатогепатита, неалкогольного стеатогепатита с фиброзом печени, неалкогольного стеатогепатита с циррозом печени или неалкогольного стеатогепатита с циррозом печени и с гепатоцеллюлярной карциномой у людей, включающий стадию введения человеку, нуждающемуся в таком лечении, терапевтически эффективного количества первого средства, второго средства и, необязательно, третьего средства, где:
i. первое средство включает (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамид или его фармацевтически приемлемую соль;
ii. второе средство включает 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойную кислоту или ее фармацевтически приемлемую соль; и
iii. третье средство включает фармацевтическое средство, выбранное из группы, состоящей из противовоспалительного средства, противодиабетического средства, противофиброзного средства, антистеатотического средства, холестерин/липид-модулирующего средства и противодиабетического средства.
14. Способ по п. 5, в котором (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамид представляет собой твердое кристаллическое вещество структуры:
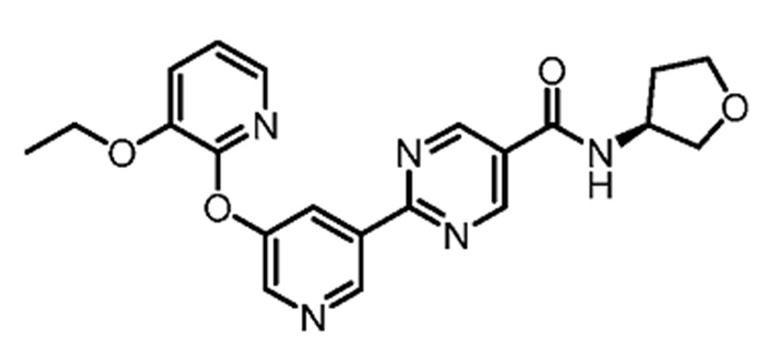
или его фармацевтически приемлемую соль.
15. Способ по п. 14, в котором порошковая рентгеновская дифрактограмма твердого кристаллического вещества включает пики при значениях угла 2-тета (CuKα излучение, длина волны 1,54056 Å) 5,3 ± 0,2, 7,7 ± 0,2 и 15,4 ± 0,2.
16. Способ по п. 14, в котором порошковая рентгеновская дифрактограмма твердого кристаллического вещества включает пики при значениях угла 2-тета (CuKα излучение, длина волны 1,54056 Å) 6,5 ± 0,2, 9,3 ± 0,2 и 13,6 ± 0,2.
17. Способ по п. 5, в котором 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойная кислота представляет собой твердое кристаллическое вещество структуры:
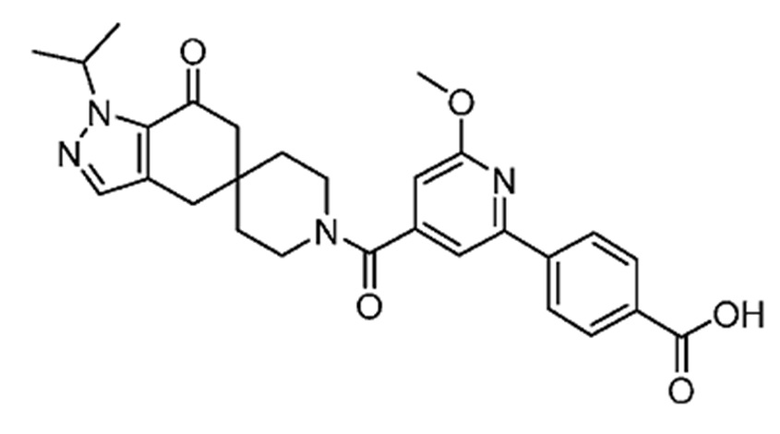
или его фармацевтически приемлемую соль.
18. Способ по п. 17, в котором твердое кристаллическое вещество представляет собой 2-амино-2-(гидроксиметил)пропан-1,3-диольную соль 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойной кислоты.
19. Фармацевтическая композиция по п. 2, в которой (S)-2-(5-((3-этоксипиридин-2-ил)окси)пиридин-3-ил)-N-(тетрагидрофуран-3-ил)пиримидин-5-карбоксамид представляет собой твердое кристаллическое вещество структуры:
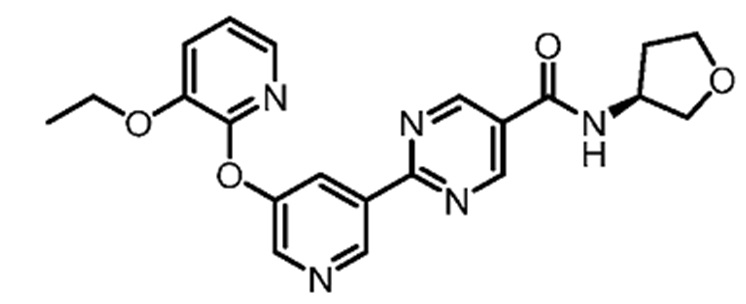
или его фармацевтически приемлемую соль.
20. Фармацевтическая композиция по п. 19, где порошковая рентгеновская дифрактограмма указанного твердого кристаллического вещества включает пики при значениях угла 2-тета (CuKα излучение, длина волны 1,54056 Å) 5,3 ± 0,2, 7,7 ± 0,2 и 15,4 ± 0,2.
21. Фармацевтическая композиция по п. 19, где порошковая рентгеновская дифрактограмма указанного твердого кристаллического вещества включает пики при значениях угла 2-тета (CuKα излучение, длина волны 1,54056 Å) 6,5 ± 0,2, 9,3 ± 0,2 и 13,6 ± 0,2.
22. Фармацевтическая композиция по п. 2, в которой 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойная кислота представляет собой твердое кристаллическое вещество структуры:
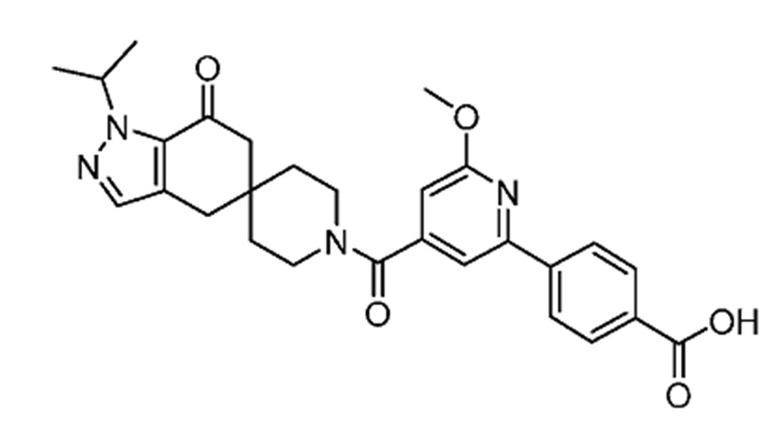
или ее фармацевтически приемлемую соль.
23. Фармацевтическая композиция по п. 22, в которой твердое кристаллическое вещество представляет собой 2-амино-2-(гидроксиметил)пропан-1,3-диольную соль 4-(4-(1-изопропил-7-оксо-1,4,6,7-тетрагидроспиро[индазол-5,4'-пиперидин]-1'-карбонил)-6-метоксипиридин-2-ил)бензойной кислоты.
24. Фармацевтическая композиция по п. 22, в которой по меньшей мере одно дополнительное фармацевтическое средство выбрано из группы, состоящей из [(1R,5S,6R)-3-{2-[(2S)-2-метилазетидин-1-ил]-6-(трифторметил)пиримидин-4-ил}-3-азабицикло[3.1.0]гекс-6-ил]уксусной кислоты; 2-[(1R,3R,5S)-3-({5-циклопропил-3-[2-(трифторметокси)фенил]-1,2-оксазол-4-ил}метокси)-8-азабицикло[3.2.1]октан-8-ил]-4-фтор-1,3-бензотиазол-6-карбоновой кислоты; или 2-[(4-{6-[(4-циано-2-фторбензил)окси]пиридин-2-ил}пиперидин-1-ил)метил]-1-[(2S)-оксетан-2-илметил]-1H-бензимидазол-6-карбоновой кислоты или его фармацевтически приемлемой соли.
25. Способ лечения заболевания или состояния, выбранного из жирной печени, неалкогольной жировой болезни печени, неалкогольного стеатогепатита, неалкогольного стеатогепатита с фиброзом печени, неалкогольного стеатогепатита с циррозом печени и неалкогольного стеатогепатита с циррозом печени и с гепатоцеллюлярной карциномой или с метаболическим заболеванием, причем указанный способ включает введение человеку, нуждающемуся в таком лечении, композиции по п. 2.
| US 20180051012 A1, 22.02.2018 | |||
| WO 2012042433 A1, 05.04.2012 | |||
| WO 2018109607 A1, 21.06.2018 | |||
| WO 2016112305 A1, 14.07.2016 | |||
| DANIEL BLANCO-ANIA ET AL | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Автомобиль-сани, движущиеся на полозьях посредством устанавливающихся по высоте колес с шинами | 1924 |
|
SU2017A1 |

