Настоящее изобретение относится к новым соединениям и их фармацевтически приемлемым солям, которые применяются в лечении неопластических заболеваний или пролиферативных расстройств, к фармацевтической композиции, содержащей указанное соединение, и к способу получения этих соединений.
Хронический лимфоцитарный лейкоз (ХЛЛ) является наиболее распространенной формой лейкоза у взрослых в западных странах. Заболевание имеет очень гетерогенный характер, демонстрируя у некоторых пациентов крайне медленное развитие, в то время как у других пациентов быстро переходит в поздние стадии и требует немедленного лечения. (Cramer, P. and Hallek, M. (2011), "Prognostic factors in chronic lymphocytic leukemia-what do we need to know?", Nat Rev Clin Oncol 8: 38-47). В последнее десятилетие, несмотря на значительное улучшение терапевтических стратегий, при использовании традиционной химиоиммунотерапии, ХЛЛ остается неизлечимым заболеванием. Разработка новых методов лечения остается важной задачей.
Было показано, что нестероидные противовоспалительные средства (НПВС) могут использоваться не только в лечении боли, воспаления и лихорадки, но также обладают значительным противоопухолевым эффектом (Thun et al. (2002), "Nonsteroidal anti inflammatory drugs as anticancer agents: mechanistic, pharmacologic, and clinical issues", J. Natl Cancer Inst 94: 252-266; Shiff, S. J. and Rigas, B. (1999), "Aspirin for cancer", Nat Med 5: 1348-1349).
Что касается большей части классических НПВП, применение которых в качестве противоопухолевого средства ограничивается, главным образом, побочными эффектами со стороны желудочно-кишечного тракта и сердечно-сосудистой системы при необходимых концентрациях (для обзора см. Ng, S.C. and Chan, F. K. (2010), "NSAID-induced gastrointestinal and cardiovascular injury", Curr Opin Gastroenterol 26: 611-617), то были проведены химические модификации. Эти модификации направлены на объединение традиционных НПВП с фосфолипидами, циклодекстринами или химическими фрагментами, которые высвобождают гастропротекторные медиаторы, такие как оксид азота (NO), посредством алифатического, ароматического или гетероциклического спейсера. (для обзора см. Abdel-Tawab, M. et al. (2009), "Nonsteroidal anti-inflammatory drugs: a critical review on current concepts applied to reduce gastrointestinal toxicity", Curr Med Chem 16: 2042-2063) и Burgaud, J. L. et al., (2002), "Nitric-oxide releasing molecules: a new class of drugs with several major indications", Curr Pharm Des 8: 201-213). Фармакокинетические и фармакологические свойства конечного вещества в значительной степени зависят от химической структуры спейсера. Ацетилсалициловая кислота-донор NO (NO-ASA) не может рассматриваться в качестве классического NO-НПВП. При этом ароматический спейсер связывает классическую молекулу ацетилсалициловой кислоты с NO-высвобождающим фрагментом (-ONO2) (Baron, J. A., (2003), "Epidemiology of non-steroidal anti-inflammatory drugs and cancer", Prog Exp Tumor Res 37: 1-24). Полагают, что при пероральном введении эстеразы быстро расщепляют NO-ASA до ASA и NO-высвобождающего фрагмента, связанного со спейсером. Фактическое высвобождение NO происходит в последующем метаболизме спейсер/NO-высвобождающего комплекса (Wallace, J. L. et al. (2002), "Potential cardioprotective actions of NO-releasing aspirin", Nat Rev Drug Discov 1: 375-382).
Razavi, R. et al. в Clinical Cancer Research 17 (2), январь 15, 2011, стр. 286-293, отмечают, что пара-NO-ASA индуцирует апоптоз в клетках ХЛЛ in vitro и может ингибировать рост опухоли in vivo. Кроме того, Gehrke, I. et al. в Therapeutic Advance Hematology (2011) 2 (5), стр. 279-289 отмечают, что противоопухолевый эффект NO-ASA в клетках ХЛЛ в значительной степени зависит от его позиционной изомерии, а именно, что пара-NO-ASA показывает намного больший эффект, чем мета- или орто-изомер.
В WO 2005/065361 описаны соединения и композиции для лечения пролиферативных заболеваний, в частности, злокачественной опухоли, путем ингибирования роста диспролиферативных клеток. В настоящей заявке описаны несколько типов ароматических соединений, где в числе других описаны NO-ASA и его производные. Кроме того, в WO 02/30866 описаны нитрат-производные ароматических соединений в качестве препаратов для лечения заболеваний, имеющих в основе воспалительный процесс, в частности, заболеваний желудочно-кишечного тракта. При этом вновь в числе других описаны изомеры NO-ASA в качестве эффективных соединений.
В документе WO 01/04082 описаны (нитроокси)фениловые эфиры производных салициловой кислоты и способы их получения.
Кроме того, в WO 2009/023631 описаны соединения для лечения заболеваний, связанных с воспалением, таких как злокачественная опухоль, нейродегенеративных и сердечно-сосудистых заболеваний, где указанные соединения включают сложные эфиры ароматических производных.
Ни в одном из опубликованных ранее документов, указанных выше, не описаны соединения, приведенные в настоящем документе, в частности, не описано, что указанные соединения могут быть использованы для лечения неопластических заболеваний пролиферативных расстройств.
Задачей настоящего изобретения была разработка соединений, действующих в качестве эффективного и селективного лекарственного средства для лечения неопластических заболеваний или пролиферативных заболеваний, в частности, соединений, которые селективно индуцируют апоптоз дегенерированных клеток, обеспечивая уменьшение побочных эффектов в живых организмах.
Эта задача решается, когда соединение, соответствующее формуле:
[формула A]
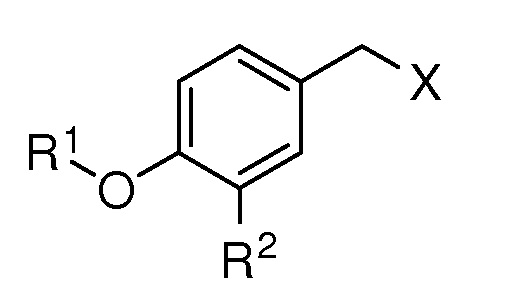
где R1 выбирают из
или [формула B],
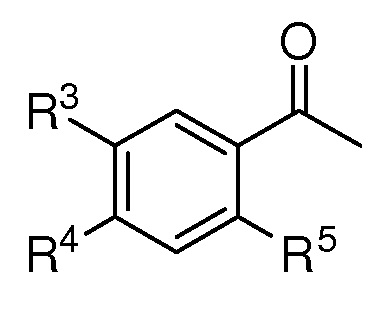
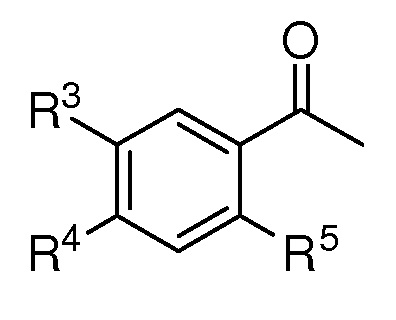
R2 представляет собой (С1-C5)алкил, (C1-C5)алкокси, (C2-C4)алкенил или алкинил, азидо(C1-C4)алкил или водород;
R3 представляет собой (С1-C5)алкил, (C1-C3)алкил с 1-3 галогеновыми заместителями, атом галогена или атом водорода;
R4 представляет собой (С1-C5)алкил, (C1-C5)алкокси или водород;
R5 представляет собой (С1-C5)алкил, (C1-C5)алкокси, ацетокси, атом галогена или атом водорода;
X представляет собой OTBS, гидрокси, формилокси, ацетокси, нитроокси, нитрооксиметил, или атом галогена;
при условии, что, если R1 представляет собой [формула B], R2, R3 и R5 представляют собой водород, и X представляет собой гидроксил, то R4 не представляет собой метокси;
или его фармацевтически приемлемая соль
используется в качестве лекарственного средства, в частности, соединение является подходящим для использования в лечении неопластического заболевания или пролиферативного расстройства. Хотя одно из соединений, соответствующее формуле, определенной выше, описано в документе WO 2001/021577 в качестве антагониста меланин-концентрирующего гормона, соединения по настоящему изобретению нигде не описаны в качестве потенциальных средств для лечения опухолевых заболеваний или (дис)пролиферативных расстройств.
Предпочтительные варианты осуществления включены в зависимые пункты формулы изобретения и описаны ниже.
В формуле (А) особый интерес представляет то, что остаток-OR1 и -CH2X связаны с бензольным кольцом в пара-конфигурации.
Настоящее изобретение относится также к указанному соединению для использования в лечении неопластического заболевания или (дис-)пролиферативного расстройства, где заболевание или расстройство представляет собой, предпочтительно, злокачественную опухоль. Более предпочтительно, злокачественную опухоль выбрают из группы, состоящей из злокачественной опухоли предстательной железы, поджелудочной железы, легких, кожи, молочной железы, мочевого пузыря, толстой кишки и крови, где особенно предпочтительно злокачественной опухолью является хронический лимфоцитарный лейкоз (ХЛЛ).
В соединениях по настоящему изобретению предпочтительно, чтобы путем связывания сложноэфирной группы остатка R1 образовывалось бензольное кольцо формулы А.
Соединения по настоящему изобретению оказывают эффект повышения апоптоза пролиферативных клеток. Не будучи связанными следующей теорией, предполагается, что указанный повышенный апоптоз дисфункциональных клеток обусловлен способностью соединений по настоящему изобретению образовывать нетипичные производные биологически активных соединений в клетках, как, например, производные нуклеотидной последовательности (ДНК, РНК), аминокислот, пептидов или белков, или соединений биологических путей или сигнальных путей. Эфирная группа соединений по настоящему изобретению может быть расщеплена посредством эстераз внутри клеток организмов/клеток с получением высоко реакционноспособных соединений, которые могут добавляться к биологическим соединениям, обычно присутствующим в клетке. Механизм получения указанных реакционноспособных соединений и образование производных биологических соединений для примера показан в виде общей схемы на фигуре 1. Присутствие полученных таким образом производных усиливает апоптоз клеток, содержащих указанные производные, и таким образом устраняет некоторое количество дисфункциональных клеток. Подробности указанного механизма, описанного в литературе, представлены на фигуре 2.
Соединения по настоящему изобретению обеспечивают повышенную селективность в отношении дисфункциональных клеток, в частности, злокачественных клеток. Селективность веществ исследовали in vitro с помощью анализа с использованием аннексина V/пропидийиодида (PI) (апоптоз/гибель клеток) с клетками первичного ХЛЛ и мононуклеарных клеток периферической крови (МКПК). Различия в чувствительности между клетками ХЛЛ и МКПК относительно соединения называют селективностью. Основной механизм селективности NO-ASA к клеткам злокачественной опухоли, как полагают, обусловлен ингибированием различных сигнальных путей, аналогично WNT или NF-каппаB путям, которые являются особенно важными для выживания клеток злокачественной опухоли.
Высокая селективность обычно указывает на невысокую вероятность побочных эффектов и, соответственно, является важным свойством современных химиотерапевтических средств. Фактическая токсичность и побочные эффекты препарата исследованы в следующих далее экспериментах на животных.
Настоящее изобретение дополнительно относится к фармацевтической композиции, содержащей, по меньшей мере, одно из соединений по настоящему изобретению или его фармацевтически приемлемую соль, предпочтительно, в смеси с одним или несколькими фармацевтически приемлемыми носителями.
Кроме того, настоящее изобретение относится к способам получения таких соединений.
"Фармацевтически приемлемая соль" относится к солям, которые сохраняют биологическую эффективность и свойства свободных оснований или свободных кислот, и которые не являются биологически или иным образом нежелательными, образованы с неорганическими кислотами, такими как соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и тому подобное, и органическими кислотами, такими как уксусная кислота, пропионовая кислота, гликолевая кислота, пировиноградная кислота, щавелевая кислота, яблочная кислота, малоновая кислота, янтарная кислота, малеиновая кислота, фумаровая кислота, винная кислота, лимонная кислота, бензойная кислота, коричная кислота, миндальная кислота, метансульфоновая кислота, этансульфоновая кислота, п-толуолсульфоновая кислота, салициловая кислота, аскорбиновая кислота и т.п., или с подходящими основаниями или солями, включая, но ими не ограничиваясь, например, соли алюминия, кальция, лития, магния, калия, натрия, цинка и диэтаноламина. Для обзора фармацевтически приемлемых солей смотри Berge et al., 66 J. PHARM. SCI. 1-19 (1977).
Термин "лечение", используемый в настоящем документе, охватывает любое лечение болезни у млекопитающего, в частности, человека, и включает:
(I) профилактику заболевания у субъекта, который может быть предрасположен к заболеванию, но еще не установлено, что имеет его;
(II) подавление заболевания, то есть прекращение его развития; или
(III) облегчение заболевания, то есть регресс заболевания.
Термин "неопластическое заболевание" или "(дис)пролиферативное расстройство", используемый в настоящем документе, охватывает болезненные состояния, показывающие формирование патологической массы ткани в результате неоплазии. Неоплазия представляет собой атипичную пролиферацию клеток. Перед неоплазией клетки обычно приобретают атипичный характер роста. Рост опухолевых клеток выходит за рамки нормального и не координируется клетками окружающей ее нормальной ткани. Этот превышающий норму рост сохраняется даже после прекращения стимулирования. Обычно это приводит к образованию узла или опухоли. Новообразование может быть доброкачественным, предзлокачественным и злокачественным (злокачественная опухоль). Пролиферативное заболевания или "дис"пролиферативное расстройство относится к дисфункции клеток, где скоординированная пролиферация (новый рост и развитие или биологических клеток) является дисрегулируемой, а продукция клеток и рост увеличиваются и превышают обычную клеточную скорость.
"Злокачественной опухолью" называют болезненное состояние, при котором неконтролируемый рост злокачественных клеток приводит к заметному увеличению массы клеток ткани, часто сопровождающийся вытеснением нормальной ткани. "Хронический лимфоцитарный лейкоз" является одним из видов лейкоза. Лейкозы представляют собой злокачественные опухоли белых кровяных клеток, где ХЛЛ поражает В-лимфоциты. В-клетки образуются в костном мозге, развиваются в лимфатических узлах и, в нормальном состоянии, борются с инфекциями, продуцируя антитела. При ХЛЛ B-клетки выходят из-под контроля и накапливаются в костном мозге и крови, где они вытесняют здоровые клетки крови.
Соединения по настоящему изобретению могут быть использованы в качестве лекарственного средства. Благодаря сродству соединений к злокачественным клеткам, соединения по настоящему изобретению являются пригодными для использования в лечении неопластических заболеваний или пролиферативных заболеваний. Кроме того, соединения оказывают воздействие на воспалительные заболевания. Предполагаемым основным эффектом соединений по настоящему изобретению является "маркирование" биологических клеточных молекул, как описано выше, приводя к апоптозу клеток, включающих маркированные соединения.
Соединения по настоящему изобретению демонстрируют хорошую селективность для клеток с патологической пролиферацией и, как полагают, обрабатываются эстеразой, обеспечивая активные компоненты, как показано на фиг.1 и 2.
В предпочтительном варианте осуществления настоящего изобретения соединения, которые могут быть использованы в качестве эффективного лекарственного средства, имеют следующий вид:
Соединение имеет формулу:
[формула A],
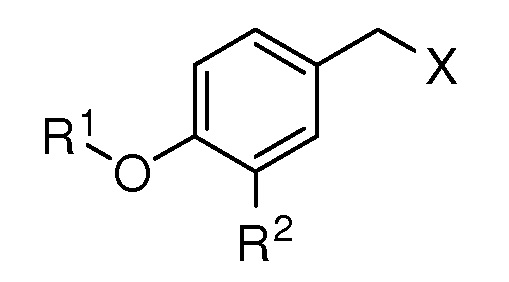
где R1 выбирают из
или [формула B],
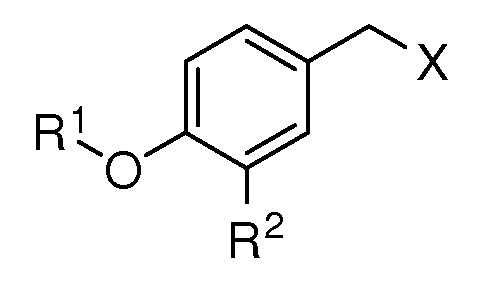
R2 представляет собой метокси, этинил, азидометил или водород;
R3 представляет собой метил, трифторметил, атом фтора или атом водорода;
R4 представляет собой метил, метокси или водород;
R5 представляет собой ацетокси, метокси, атом хлора или атом водорода;
X представляет собой OTBS, гидрокси, формилокси, нитроокси, нитрооксиметил или хлор; при условии, что, если R1 представляет собой [формула B], R2, R3 и R5 представляют собой водород, и X представляет собой гидроксил, то R4 не представляет собой метокси;
или его фармацевтически приемлемая соль;
в качестве лекарственного средства.
Некоторые соединения, представленные этой формулой, известны в данной области техники, однако они не описаны в качестве лекарственного средства. Тем не менее, большинство соединений, приведенных в настоящей заявке, являются новыми по сравнению с соединениями, известными из предшествующего уровня техники, которые, в частности, являются соединениями, соответствующими формуле:
[формула A]
где R1 выбирают из
или [формула B],
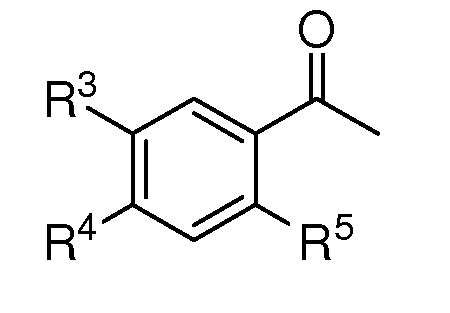
R2 представляет собой (С1-C5)алкил, (C1-C5)алкокси, (C2-C4)алкенил или алкинил, азидо(C1-C4)алкил или водород;
R3 представляет собой (С1-C5)алкил, (C1-C3)алкил с 1-3 галогеновыми заместителями, атом галогена или атом водорода;
R4 представляет собой (С1-C5)алкил, (C1-C5)алкокси или водород;
R5 представляет собой (С1-C5)алкил, (C1-C5)алкокси, ацетокси, атом галогена или атом водорода;
X представляет собой OTBS, гидрокси, формилокси, ацетокси, нитроокси, нитрооксиметил или атом галогена;
при условии, что, если R1 представляет собой [формула B], X представляет собой нитроокси и R5 представляет собой ацетокси, то, по крайней мере, один из R2-R4 не представляет собой водород; при условии, что, если R1 представляет собой [формула B], R3-R5 представляют собой водород, и X представляет собой гидроксил, то R2 не представляет собой водород и метокси; при условии, что, если R1 представляет собой [формула B], R2, R3 и R5 представляют собой водород, и X представляет собой гидроксил, то R4 не представляет собой метокси; при условии, что, если R1 представляет собой [формула B], R3-R5 представляют собой водород, и X представляет собой OTBS, то R2 не представляет собой метокси; и при условии, что, если R1 представляет собой метокси и X представляет собой нитроокси, то R2 не представляет собой водород.
В связи с этим предпочтительным является соединение, имеющее формулу (C),
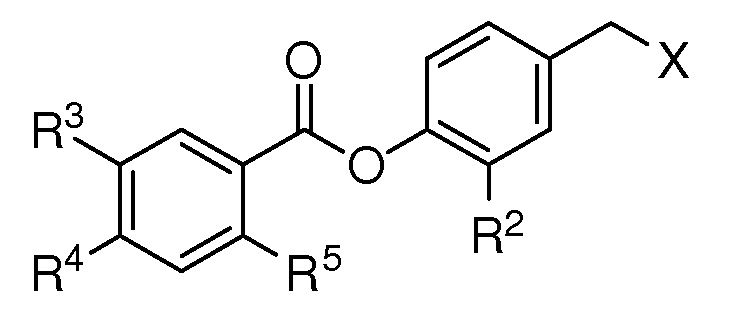
где
R2 представляет собой метокси, этинил, азидометил или водород;
R3 представляет собой метил, трифторметил, атом фтора или атом водорода;
R4 представляет собой метил, метокси или водород;
R5 представляет собой ацетокси, метокси, атом хлора или атом водорода;
X представляет собой OTBS, гидрокси, формилокси, нитроокси, нитрооксиметил или хлор;
при условии, что, если R1 представляет собой [формула B], X представляет собой нитроокси, и R5 представляет собой ацетокси, то, по крайней мере, один из R2-R4 не представляет собой водород; при условии, что, если R1 представляет собой [формула B], R3-R5 представляют собой водород, и X представляет собой гидроксил, то R2 не представляет собой водород и метокси; при условии, что, если R1 представляет собой [формула B], R2, R3 и R5 представляют собой водород, и X представляет собой гидроксил, то R4 не представляет собой метокси; при условии, что, если R1 представляет собой [формула B], R3-R5 представляют собой водород, и X представляет собой OTBS, то R2 не представляет собой метокси; и при условии, что, если R1 представляет собой метокси, и X представляет собой нитроокси, то R2 не представляет собой водород.
Из указанных выше соединений предпочтительными являются такие соединения, где Х представляет собой нитроокси или OTBS, R2 представляет собой водород, R3-R5, каждый, представляют собой водород, или R3 и R4 представляют собой метил, и R5 представляет собой ацетокси, и/или где R1 представляет собой [формула B], R2-R5, каждый, представляют собой водород, и Х выбирают из OTBS, гидроксила, нитроокси, нитрооксиметила, формилокси и хлора.
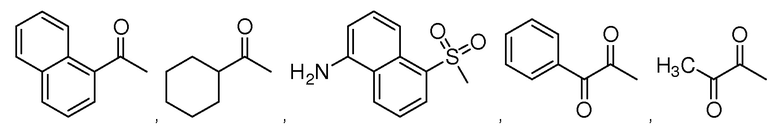
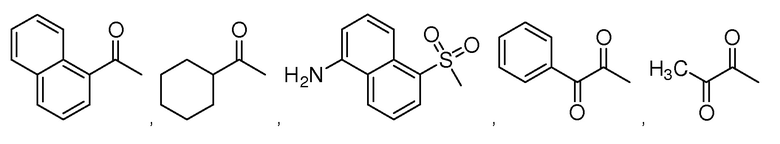
В особенно предпочтительном варианте осуществления настоящего изобретения соединение выбирают из группы, включающей 4-((нитроокси)метил)фенил 2-ацетокси-5-метилбензоат, 4-((нитроокси)метил)фенил 2-ацетокси-5-фторбензоат, 4-((нитроокси)метил)фенил 2-ацетокси-4-метилбензоат, 4-(((трет-бутилдиметилсилил)окси)метил)фенил 2-хлор-5-(трифторметил) бензоат, 4-(гидроксиметил)фенил 2-хлор-5-(трифторметил)бензоат, 4-((нитроокси)метил)фенил 2-хлор-5-(трифторметил)бензоат, 4-(((трет-бутилдиметилсилил)окси)метил)фенилбензоат, 4-((нитроокси)метил)фенилбензоат, 4-((формилокси)метил)фенилбензоат, 2-метокси-4-((нитроокси)метил)фенилбензоат, 4-(хлорметил)фенилбензоат, 4-((нитроокси)метил)фенил 1-нафтоат, 4-((нитроокси)метил)фенил циклогексан карбоксилат, 4-((нитроокси)метил)фенил 5-аминонафталин-1-сульфонат, 4-(2-(нитроокси)этил)фенилбензоат, 4-((нитроокси)метил)фенил 2-метоксибензоат, 4-((нитроокси)метил)фенил 4-метоксибензоат, 2-этинил-4-((нитроокси)метил)фенилбензоат, 2-(азидометил)-4-((нитроокси)метил)фенилбензоат, 4-((нитроокси)метил)фенил 2-оксо-2-фенилацетат и 4-((нитроокси)метил)фенил 2-оксопропаноат или их фармацевтически приемлемую соль.
В частности, предпочтительными соединениями по настоящему изобретению являются 4-((нитроокси)метил)фенил-2-ацетокси-5-метил бензоат, 4-((нитроокси)метил)фенил-2-ацетокси-4-метил бензоат, 4-((нитроокси)метил)фенилбензоат, 4-((хлор)метил)фенилбензоат, 4-((нитроокси)метил)фенил нафтоат, где 4-((нитроокси)метил)фенилбензоат и 4-((хлор)метил)фенилбензоат являются особенно предпочтительными. В частности, такие соединения предпочтительно обладают высокой эффективностью (низкая концентрация необходима для полезного эффекта, см. таблицу 1) и хорошей химической стабильностью.
Термин "алкил" означает прямую, разветвленную или циклическую алкильную группу с указанным числом атомов углерода. Примеры включают, но ими не ограничиваются, метил, этил, н-пропил, изо-пропил, н-бутил, изо-бутил, втор-бутил, трет-бутил, и пентил с прямой и разветвленной цепью и т.д., или соответствующие циклические алкилы. В любом случае, когда указан диапазон между двумя пределами, это означает, что описано любое значение или целое число в этом диапазоне. Например, "C1-C5" означает С1, С2, С3, С4 или С5, диапазон от "1-3" означает 1, 2 или 3, а диапазон между "0,1 и 1" означает, 0,1, 0,2, 0,3, 0,4, 0,5, 0,6, 0,7, 0,8, 0,9 или 1.
Термин "алкокси" означает алкильную группу, присоединенную через атом кислорода, как, например, метокси, этокси, пропокси, изо-пропокси, бутокси (н-бутокси, изо-бутокси, втор-бутокси, трет-бутокси) или пентокси и т.д., термин "алкенил" или "алкинил" означает алкильные группы, имеющие двойную или тройную связь в углеродной цепи.
Термин "гало" или "галоген" означает хлор, хлор, бром и йод.
Методы, используемые для синтеза новых соединений по настоящему изобретению, включают образование сложного эфира угольной или сульфоновой кислоты, и активированную алифатическую или ароматическую угольную или сульфоновую кислоту, подвергают взаимодействию с соединением формулы [D]:
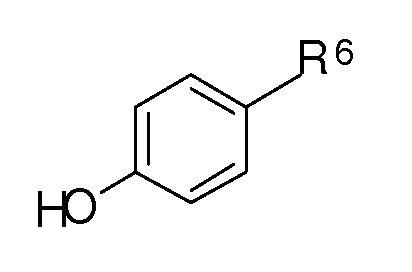
где R6 представляет собой метил-X или формил, X является таким, как определено выше.
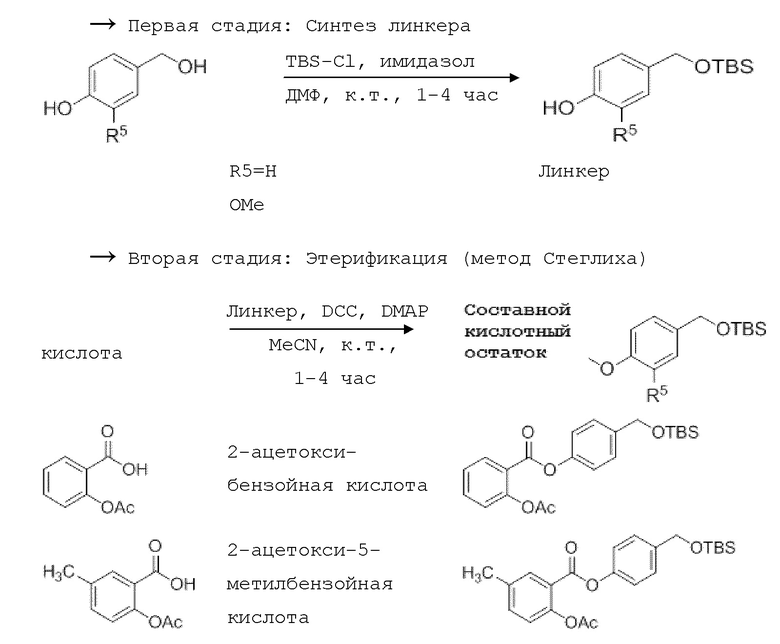
Общая схема I:
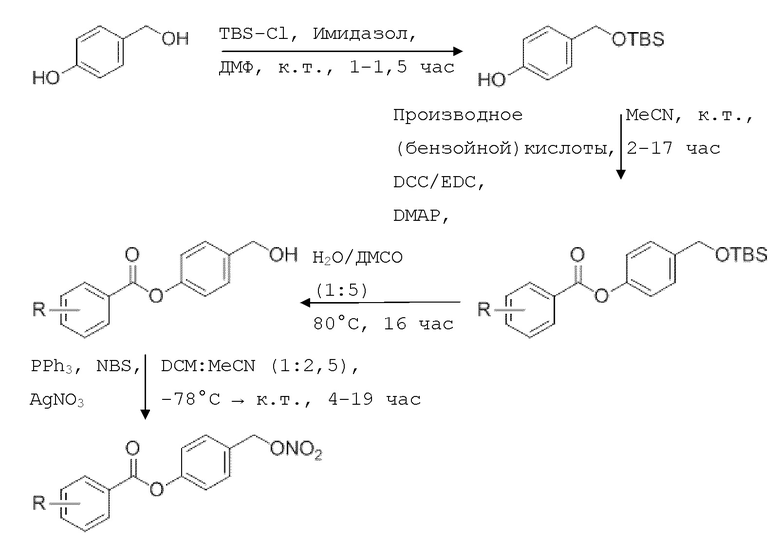
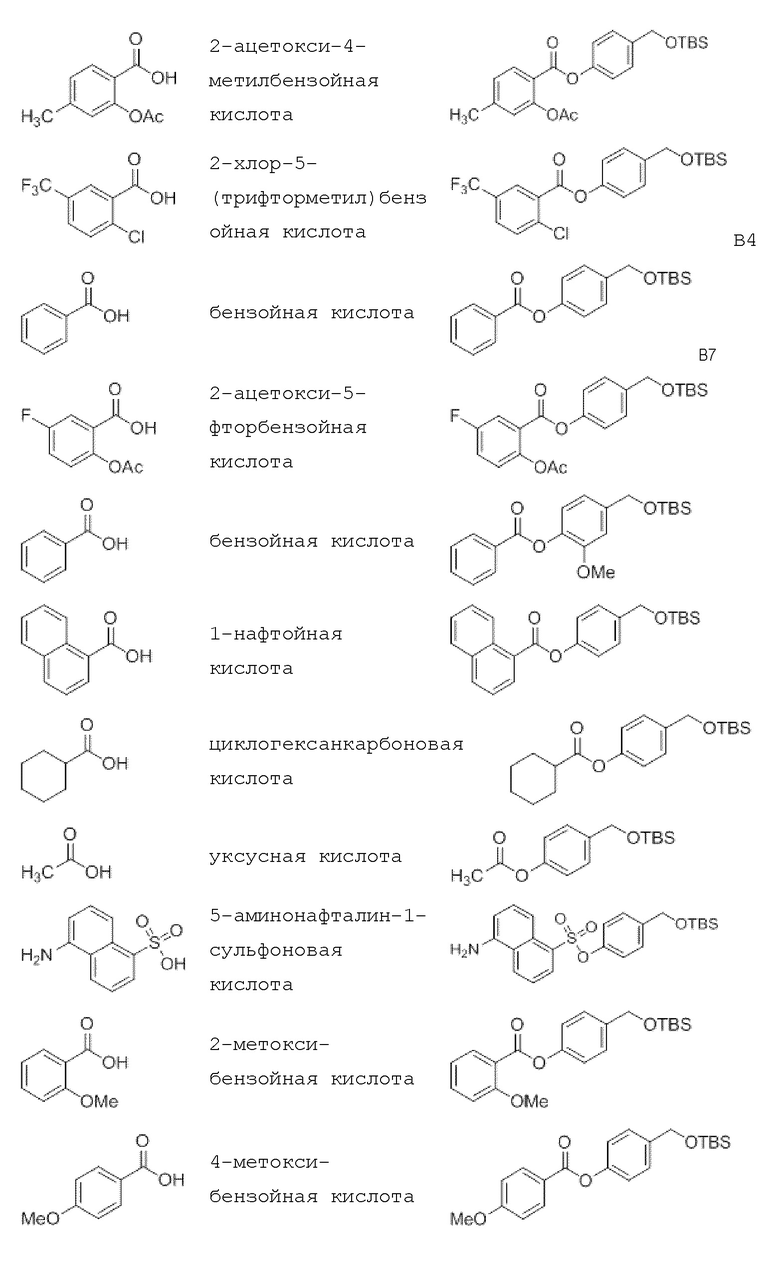
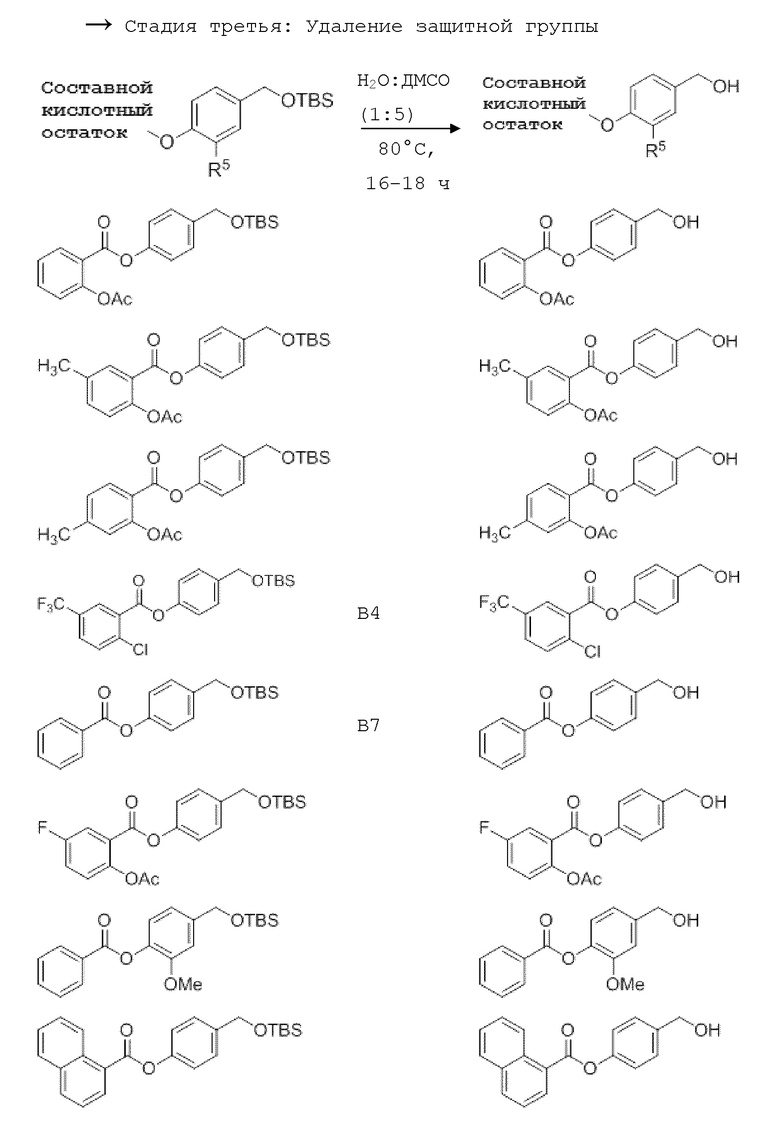
4-Гидроксибензил-трет-бутил триметилсилиловый (TBS) эфир получают путем обработки 4-гидроксибензилового спирта TBS-Cl и имидазолом. Производные бензойной кислоты, уксусную кислоту или производные кислот обычно этерифицируют методом, аналогичным методу Стеглиха (с ДКК/EDC и DMAP), с образованием OTBS-бензойной кислоты (OTBS-BA).
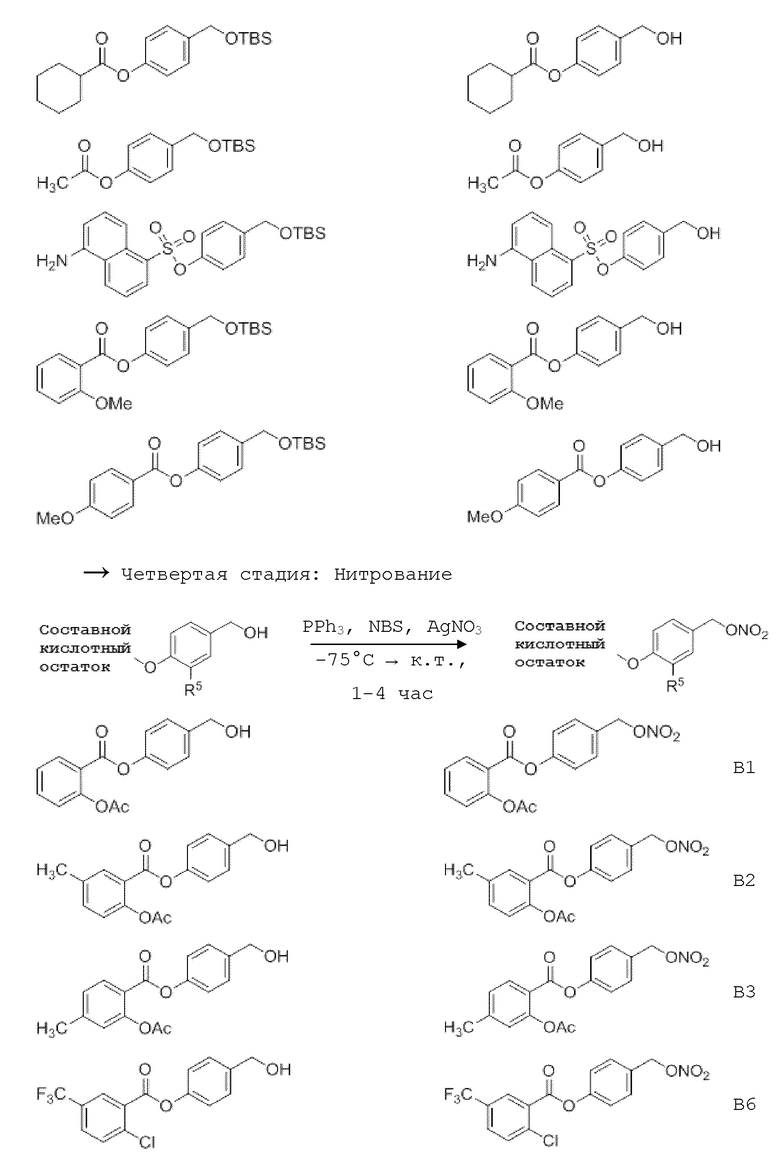
NO-Данзил (В16, см. таблицу 1 ниже) может быть синтезирован исходя из хлорида сульфоновой кислоты (данзил хлорид) с образованием сложного эфира сульфоновой кислоты. Следующими стадиями являются стадии, как описано выше (удаление защитных групп и, наконец, введение нитрата. Синтез этин-меченных соединений может начинаться с йод замещенной кислоты или линкер-структурного элемента. Этот субстрат может быть превращен в ацетиленовое соединение по реакции Соногашира с образованием с соответствующим аналогом сложного эфира впоследствии. После удаления защитной группы у обоих силиловых эфиров и последующего нитрования получают целевые молекулы. Все подробности этих процедур можно увидеть в примерах ниже.
Аббревиатуры, используемые в схемах настоящей заявки:
Общая схема II:
Для предпочтительных вариантов осуществления настоящего изобретения способы синтеза дополнительно показаны в примерах.
Кроме того, общие способы получения соединений настоящего типа также описаны в WO 2002/30866 и WO 2001/04082.
Соединения по настоящему изобретению являются эффективными в снижении дальнейшего развития опухоли или диспролиферативных клеток путем усиления апоптоза таких клеток. Благодаря селективности этих соединений побочные эффекты в живом организме уменьшаются и, следовательно, соединения являются пригодными в качестве фармацевтических средств.
Соответственно, соединения по настоящему изобретению используются для лечения неопластических заболеваний или (дис)пролиферативных расстройств. В частности, соединения по настоящему изобретению являются эффективными в лечении злокачественной опухоли. Злокачественная опухоль, которую можно эффективно лечить, представляет собой, например, злокачественную опухоль поджелудочной железы, легких, кожи, молочной железы, мочевого пузыря, толстой кишки и крови. В одном особенно предпочтительном варианте осуществления, злокачественная опухоль, которую подвергают лечению, представляет собой хронический лимфоцитарный лейкоз (ХЛЛ).
Селективность соединений для клеток, демонстрирующих пролиферативную дисфункцию (как в неоплазме или в пролиферативных расстройствах), может быть показана с помощью экспериментов in vitro, в которых способность соединений индуцировать апоптоз и/или гибель клеток или снижать пролиферацию в дисфункциональных клетках сравнивается с их воздействием на здоровые контрольные клетки.
Соединение, которое, как известно, является эффективным в лечении неопластических заболеваний, в частности, в лечении хронического лимфолейкоза (ХЛЛ), представляет собой 4-(нитроокси)метил фенил-2-ацетокси бензоат, известный как NO-ASA, смотри, например, Gehrke, I. et al. в "Therapeutic Advances in Hematology" (2011) 2(5), стр. 279-289. Таким образом, это соединение использовали в качестве эталона в исследованиях для анализа соединений по настоящему изобретению в отношении их эффективности, силы воздействия и эффектов на дисфункциональные клетки.
Экспериментальные данные показывают, что соединения по настоящему изобретению являются полезными в лечении неопластических заболеваний или (дис)пролиферативных расстройств благодаря повышенному апоптозу дисфункциональных клеток после добавления указанных соединений в in vitro анализе, описанном в примере 1. Результаты таких исследований показаны на фигуре 3 для соединений В1 (контрольный эталонный NO-ASA), В9, В12 и В13 (см. таблицу 1).
На фигуре 3 показана более высокая чувствительность клеток ХЛЛ относительно четырех препаратов при сравнении с МКПК. Препараты В1, В9, В12 и В13 являются, соответственно, селективными для клеток ХЛЛ. Релевантным для оценки селективности является соотношение ED50 для МКПК и клеток ХЛЛ (таблица 1).
В исследованиях, выполняемых с соединениями по настоящему изобретению, становится понятно, что соединения оказывают четкий эффект в отношении дисфункциональных клеток, причем некоторые из соединений были способны, в частности, эффективно усиливать клеточный апоптоз и, соответственно, ослаблять развитие клеток злокачественной опухоли.
В представленной ниже таблице 1 перечислены предпочтительные соединения, где соединения, показывающие самое низкое значение EC50 (эффективная концентрация 50%) в отношении клеток ХЛЛ, при этом оставаясь относительно нетоксичными для МКПК в анализе с аннексином V/PI, являются наиболее предпочтительными соединениями. Как видно из приведенной ниже таблицы, соединение, определяемое как "B9", показывает очень высокий эффект в анализе с использованием аннексина V и, соответственно, является наиболее предпочтительным соединением по настоящему изобретению. Кроме того, соединения "B9", "B12" и "B13" также являются предпочтительными благодаря их высокому эффекту в анализе с аннексином V/PI. Тем не менее, необходимо, в частности, отметить, что не только эффект в анализе с аннексином В/PI является существенным для предпочтения соединения, но также их стабильность, совместимость, развитие побочных эффектов и их селективность, и поэтому такие соединения, показывающие более высокое значение в анализе с аннексином V/PI по сравнению с NO-ASA, могут быть предпочтительными соединениями благодаря другим положительным эффектам.
Все соединения, описанные в настоящей заявке, и заявленные в прилагаемой формуле изобретения, можно использовать в качестве лекарственного средства, в частности, для лечения неопластического заболевания или (дис)пролиферативного расстройства. В частности, все эти соединения, а также NO-ASA являются эффективными лекарственными средствами для лечения злокачественной опухоли, где лечение ХЛЛ является особенно предпочтительным.
При применении соединений по настоящему изобретению в лечении указанных выше состояний, введение активного соединения и солей, описанных в настоящем документе, может быть осуществлено любым из приемлемых способов введения, включая пероральный, парентеральный и иной системный способ введения. Может быть использован любой фармацевтически приемлемый способ введения, включая твердые, полутвердые или жидкие лекарственные формы, такие как, например, таблетки, суппозитории, пилюли, капсулы, порошки, жидкости, суспензии или т.п., предпочтительно, в виде стандартных лекарственных форм, пригодных для однократного введения точных доз, или в лекарственных формах с пролонгированным или контролируемым высвобождением для длительного введения соединения с заранее определенной скоростью. Композиции типично включают обычный фармацевтический носитель или наполнитель и, по меньшей мере, одно из соединений по настоящему изобретению или их фармацевтически приемлемые соли и, кроме того, может включать другие лекарственные средства, фармацевтические агенты, носители, адъюванты и т.д.
Количество одного из вводимых производных по настоящему изобретению будет, безусловно, зависеть от субъекта, подвергаемого лечению, тяжести заболевания, способа введения и мнения лечащего врача. Однако эффективная доза для перорального, парентерального или иных системных способов введения находится в диапазоне 0,01-100 мг/кг/день, предпочтительно, 0,1-50 мг/кг/день. Для среднего человека массой 70 кг она составляет 0,7-7000 мг в день или, предпочтительно, 7-3500 мг/день.
Рядовой специалист в области лечения указанных заболеваний сможет без проведения лишних экспериментов и полагаясь на собственные знания и описание настоящей заявки определить терапевтически эффективное количество одного из соединений по изобретению для указанного заболевания.
В качестве твердых композиций могут быть использованы обычные нетоксичные твердые носители, включая, например, фармацевтические категории маннита, лактозы, целлюлозы, производных целлюлозы, натрий кроскармелозы, крахмала, стеарата магния, сахарина натрия, талька, глюкозы, сахарозы, карбоната магния и т.п. Активное соединение, как определено выше, может быть объединено в составе композиции в виде суппозиториев с использованием, например, полиалкиленгликолей, например, ПЭГ (полиэтиленгликоль) или производных ПЭГ, ацетилированных триглицеридов и т.п., в качестве носителя. Жидкие фармацевтически вводимые композиции могут, например, быть получены растворением, диспергированием и т.д. активного соединения, как определено выше, и необязательных фармацевтических адъювантов в носителе, таком как, например, вода, физиологический раствор, водный раствор декстрозы, глицерин, этанол и т.п., с образованием в результате раствора или суспензии. При желании, фармацевтическая композиция для введения может также содержать незначительные количества нетоксичных вспомогательных веществ, таких как смачивающие или эмульгирующие вещества, рН буферные вещества и т.п., например, ацетат натрия, сорбитан монолаурат, триэтаноламин ацетат натрия, олеат триэтаноламина и т.д. Вводимая композиция или препарат будет, в любом случае, содержать активное(ые) соединение(я) в количестве, эффективном для облегчения симптомов у субъекта, проходящего лечение.
Могут быть получены лекарственные формы или композиции, содержащие одно из соединений по настоящему изобретению в диапазоне от 0,25 до 95% по массе с наполнителем из нетоксичного носителя.
Для перорального введения фармацевтически приемлемую нетоксичную композицию получают путем включения любых обычно используемых вспомогательных веществ, таких как, например, маннит, лактоза, целлюлоза, производные целлюлозы, кроскармеллоза натрия, крахмал, стеарат магния, сахарин натрия, тальк, глюкоза, сахароза, карбонат магния и тому подобное. Такие композиции могут принимать форму растворов, суспензий, таблеток, пилюль, капсул, порошков, препаратов с замедленным высвобождением и тому подобное. Такие композиции могут содержать от 1 до 95% по массе одного из соединений по настоящему изобретению, более предпочтительно, от 2 до 50% по массе, наиболее предпочтительно, от 5 до 8% по массе.
Парентеральное введение обычно представляет собой инъекцию, либо подкожную, либо внутримышечную, либо внутривенную. Инъекционные препараты могут быть получены в виде обычных форм, либо в виде жидких растворов или суспензий, твердых форм, пригодных для растворения или суспендирования в жидкости перед инъекцией, либо в виде эмульсий. Подходящие вспомогательные вещества представляют собой, например, воду, физиологический раствор, декстрозу, глицерин, этанол или тому подобное. Кроме того, при желании, фармацевтические композиции для введения могут также содержать незначительные количества нетоксичных вспомогательных веществ, таких как смачивающие или эмульгирующие агенты, рН буферные вещества и тому подобное, такие как, например, ацетат натрия, сорбитанмонолаурат, триэтаноламинолеат, триэтаноламин ацетат натрия и т.д.
Трансдермальные или "импульсное" трансдермальное введение может осуществляться посредством кремов, гелей, дисперсий и тому подобное.
В недавно разработанном способе парентерального введения используют имплантацию системы с замедленным высвобождением или системы пролонгированного действия, посредством которой поддерживается постоянный уровень дозирования (смотри, например, US A3710795).
Выраженное в процентах количество активных соединений, содержащихся в указанных исходных композициях, значительно зависит от его конкретных свойств, а также от активности соединения и потребностей субъекта. Тем не менее, выраженные в процентах количества одного из предлагаемых в изобретении соединений, составляющие от 0,1 до 10% по массе в растворе, являются приемлемыми, и будут выше, если композиция представляет собой твердое вещество, которое впоследствии будет разведено до вышеуказанных процентных величин. Предпочтительно, композиция будет содержать от 0,2 до 2% по массе одного из соединений в растворе.
Предпочтительно, фармацевтическую композицию вводят в виде единичной стандартной лекарственной формы для непрерывного лечения или в виде единичной стандартной лекарственной формы, по желанию, когда конкретно требуется облегчение симптомов.
Фигуры
На фигуре 1 представлена достаточно общая схема предполагаемых механизмов действия фармацевтически активного вещества в пролиферативной клетке.
На фигуре 2 показан предполагаемый механизм, описанный в литературе, обеспечения фармацевтически активного средства, в частности, хинон метида (верхняя часть), или, в частности, NOx (нижняя часть), вызывающего апоптоз в клетке.
На фигуре 3 показано влияние соединений В1 (контрольное вещество NO-ASA), B9, B12 и B13 (смотри таблицу 1) на выживаемость первичных МКПК от здоровых доноров или клеток ХЛЛ. МКПК или клетки ХЛЛ (5*106 клеток/мл) инкубировали в течение 24 ч с различными соединениями в концентрациях от 0,01 до 100 мкM. Выживаемость клеток нормализовывали до контроля ДМСО [носитель]. См. пример 1.
На фигуре 4 показано ингибирование роста опухоли с использованием соединения B9 в ксенотрансплантатах ХЛЛ (смотри пример 2). Обработка B9 приводит к значительному ингибированию опухоли по сравнению с контролем носителем (р=0,015) через девять дней, с увеличением значения, до 19-го дня обработки (р=0,0003). Определяли значение IRmax 65% для B9 по сравнению с контролем носителем. *=P≤0,05, **=p≤0,01, ***=p≤0,001 вычисляли с помощью непарного двухстороннего теста Стьюдента, †=гибель, IR=степень ингибирования.
На фигуре 5 показано, что соединения В9 и В12 обладают превосходными цитотоксическими эффектами в отношении клеточных линий, несущих плохой прогноз (смотри пример 3). Несколько клеточных линий (n=5) обрабатывали различными концентрациями p-NO-ASA, B9, B12 и B13 в диапазоне от 0,01 мкM до 1000 мкM в течение 24 часов с последующим добавлением люминогенного реагента CellTiter-Glo®. Пара-NO-ASA, В9, В12 и В13 также значительно понижали содержание АТФ в клеточных линиях JVM-3, U2932 и EHEB, при этом пара-NO-ASA значительно менее эффективен в клеточных линиях MEC-1 и Granta-519. Для каждой клеточной линии порядок следования, слева направо, используемого соединения в столбчатом графике является следующим: p-NO-ASA, В9, В12, В13.
На фигуре 6 показано ингибирование роста клеток ХЛЛ с и без мутации TP53 посредством р-NO-ASA и производных B9 В12, В13 (смотри пример 4). Выделенные первичные клетки ХЛЛ обрабатывали различными соединениями при концентрации ЕС50 в течение 24 ч, и содержание АТР измеряли с помощью проточной цитометрии. Для каждого используемого соединения порядок, слева направо, средних концентраций EC50 в столбчатом графике является следующим: EC50 клеток ХЛЛ без мутации TP53, ЕС50 клеток ХЛЛ с мутацией TP53.
На фигуре 7 показано, что соединения B9, B12 и B13, демонстрируют превосходные цитотоксические эффекты в отношении линии клеток злокачественной опухоли толстой кишки SW480 по сравнению с р-NO-ASA. Клеточные линии (n=5) обрабатывали различными концентрациями p-NO-ASA, B9, B12 и B13 в диапазоне от 0,01 мкM до 100 мкM в течение 24 часов с последующим добавлением люминогенного реагента CellTiter-Glo® (смотри пример 5).
На фигуре 8 показано участие каспаза-опосредованного апоптоза в клетках ХЛЛ при обработке p-NO-ASA, B9, B12 и B13. Показаны типичные блоттинги трех независимых экспериментов. Необработанные и обработанные ДМСО (1%) клетки служили в качестве контроля. Бета-актин=(фигура 8А). Пара-NO-ASA и В9 индуцировали зависимое от концентрации увеличение активации каспаз-3/7 (фигура 8В).
На фигуре 9 показано зависимое от концентрации снижение активности NF-каппаB посредством В1 (р-NO-ASA), В9, В12 и В13 в анализах методом вестерн-блоттинга (смотри пример 7). Клетки ХЛЛ обрабатывали В1, В9, В12 и В13 (0,1 мкM, 1 мкM, 10 мкM) в течение 3 ч. Необработанные и обработанные ДМСО (1%) клетки служили в качестве контроля. GAPDH=контроль для нанесения.
Примеры
Пример 1: эффективные концентрации соединений в соответствии с изобретением:
Клетки первичного ХЛЛ или мононуклеарные клетки периферической крови здоровых доноров (5*106/мл) инкубировали в течение 24 ч с различными соединениями в соответствии с изобретением и NO-ASA в качестве контроля. Соединения добавляли в различных концентрациях, в частности, в концентрациях от 0,01 до 100 мкM. Выживаемость клеток оценивали посредством анализа с аннексином V/PI (набор можно приобрести, например, у компании Biotium Inc, США; или Phoenix Flow Systems, США), результаты нормализовывали к контролю ДМСО [носитель], и кривые доза-эффект вычисляли с использованием нелинейной регрессионной модели.
Эффективная концентрация 50% (EC50) различных производных NO-ASA. *=экстраполированный,/=не вычисляемый, н.и.=не исследовали
pNO-ASA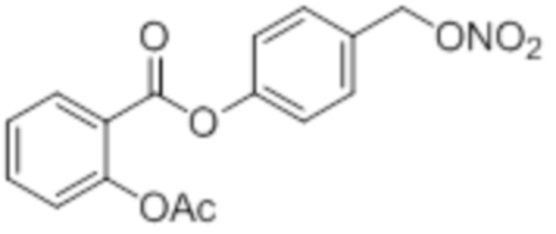
5Me-NO-ASA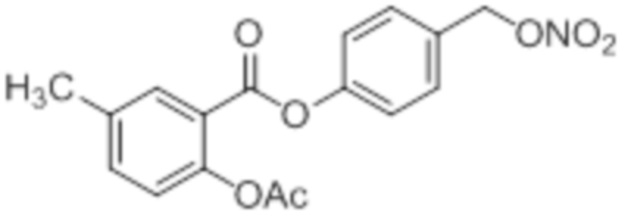
4Me-NO-ASA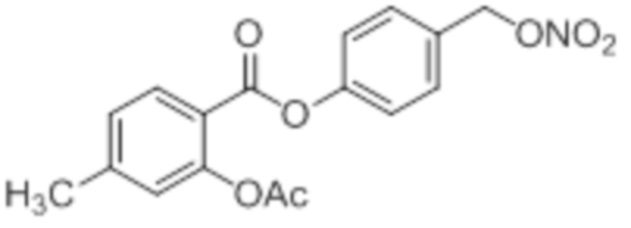
2Cl-5CF3-OTBS-BA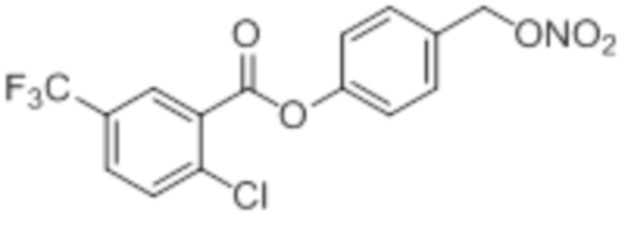
2Cl-5CF3-OH-BA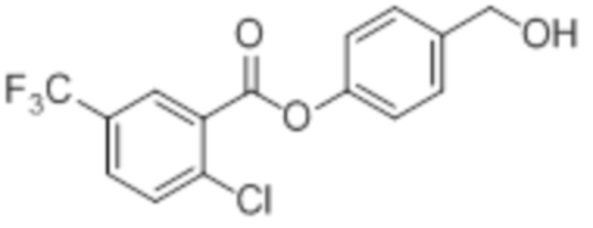
2Cl-5CF3-NO-BA
OTBS-BA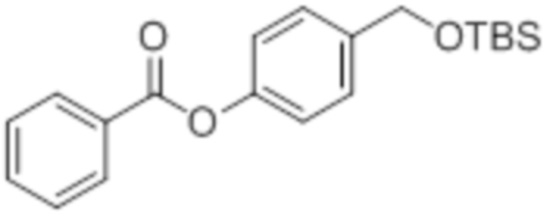
5F-NO-ASA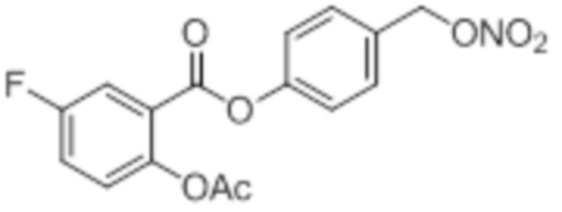
OH-BA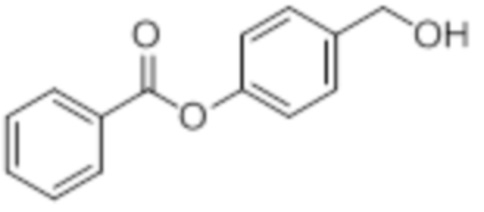
NO-BA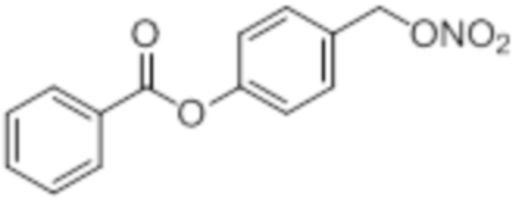
Форма-BA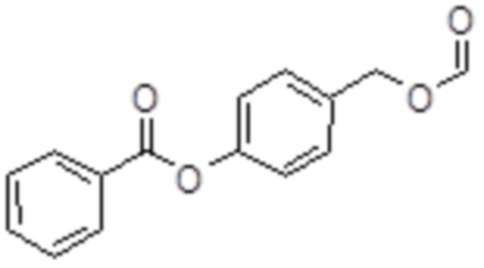
NO-OMe-BA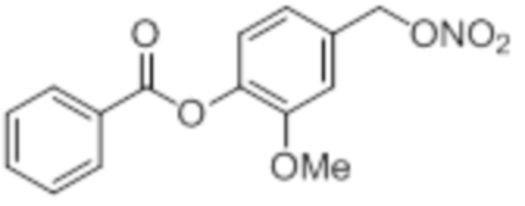
Cl-BA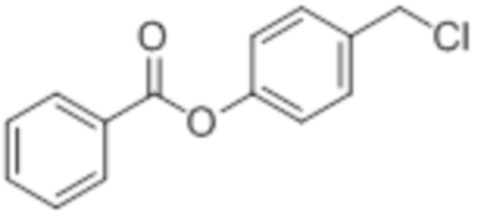
NO-Нафтил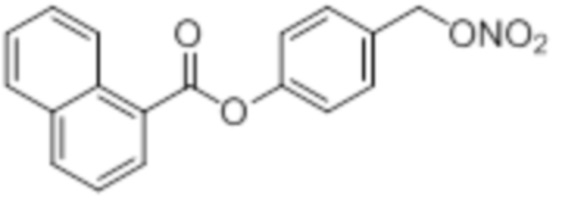
NO-cHex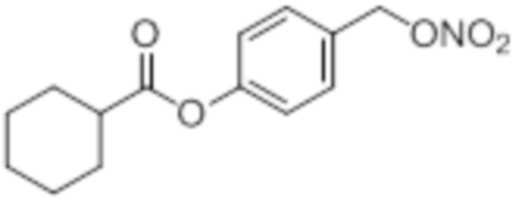
NO-AA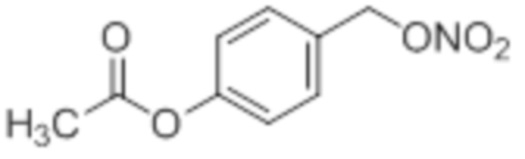
NO-Данзил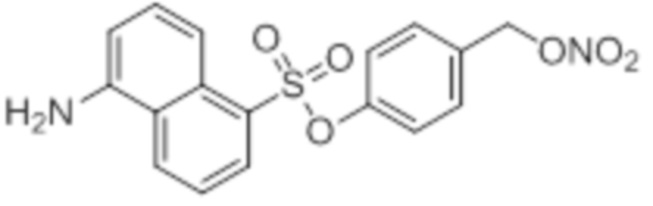
NO-Homo-BA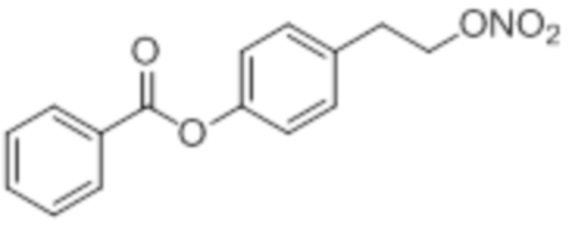
NO-2OMeBA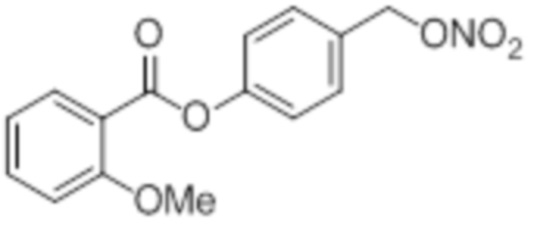
NO-4OMeBA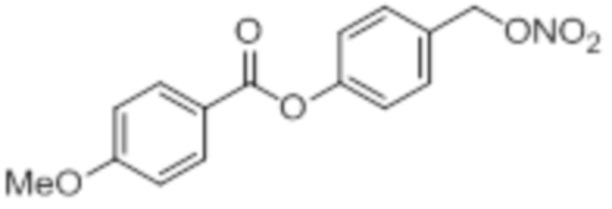
NO-2Этин-BA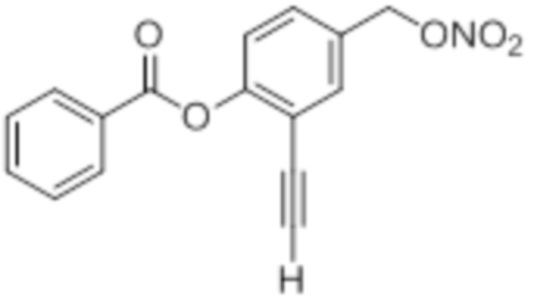
NO-2N3-BA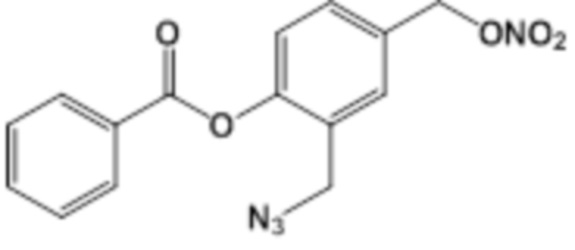
CO-NO-BA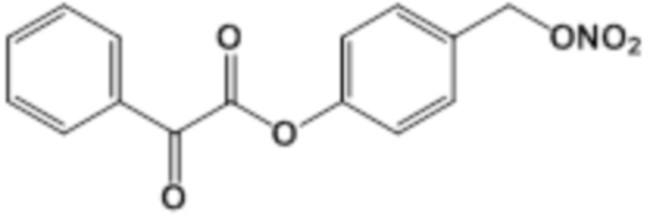
CO-NO-AA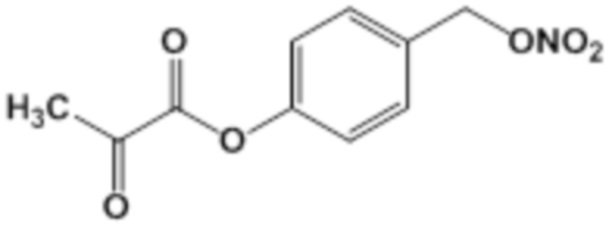
Пример 2
Благодаря своим предпочтительным характеристикам B9 был выбран для in vivo исследования на мышиной модели ксенотрансплантата ХЛЛ. Клетки JVM3 (клеточная линия хронического B-клеточного лейкоза человека) вводили подкожно в боковую область иммунокомпетентных мышей. Развивающуюся солидную опухоль обрабатывали внутрибрюшинными инъекциями в дозе 8 мг/кг соединения B9 или кунжутного масла (носитель) через день (см. фигуру 4).
Клетки JVM3 1*107 вводили подкожно бежевым мышам линии SCID (CB17.Cg-PrkdcscidLystbg-J/CRL). Опухоли измеряли через день с помощью циркуля, и рассчитывали объем опухоли V=(длина*(0,5*ширина2)). Мышей с опухолью более 50 мм3 обрабатывали через день либо кунжутным маслом (контроль носителем), либо 8 мг/кг В9, растворенном в кунжутном масле, с помощью внутрибрюшинных инъекций. Применяли критерии абортов, рекомендуемые GV-SOLAS для мышей с опухолью. С использованием непарного двухстороннего теста Стьюдента вычисляли р-значения.
На фигуре 4 показано значительное снижение роста опухоли при обработке В9. Ингибирования роста опухоли является высоко значимым после 11-го дня. Ингибирование скорости роста (IR), составляющее 65,33%, было самым высоким на 17-й день. Двух животных из контрольной группы пришлось умертвить, поскольку их опухоли превышали 15 мм в диаметре (критерии аборта). В процессе обработки носителем или B9 не наблюдались тяжелые побочные эффекты. Мыши реагировали на обработку незначительной ограниченной подвижностью в течение 15-30 мин, при этом потребление жидкости и кормление осуществлялось нормально. Снижения массы тела не наблюдалось. B9 значительно ослаблял рост опухоли в мышиной модели ксенотрансплантата (день 9: обработка B9=82,97 мм3).
Пример 3: in vitro эффективность производных NO-ASA в подгруппах ХЛЛ
Успех лечения ХЛЛ может зависеть от цитогенетических и молекулярных параметров, как, например, разрушение гена del13q или TP53. Таким образом, производные NO-ASA исследовали на клеточных линиях (хронической) B-клеточной лимфомы с различными генотипами и фенотипами (JVM3, EHEB, U2932, MEC-1, GRANTA-519). Клетки обрабатывали концентрациями от 0,01 до 1000 мкM в течение 24 ч с последующим добавлением люминогенного реагента CellTiter-Glo®.
р-NO-ASA был значительно менее эффективен против MEC-1 (ЕС50=53,44 мМ, р<0,001) и GRANTA-519 (ЕС50=22,21 мМ, р<0,001) по сравнению с В9 (MEC-1: EC5 0=6,62 мМ; Granta-519: EC50=2,28 мМ), В12 (MEC-1: EC50=3,24 мМ; GRANTA-519: EC50=0,68 мМ) и B13 (MEC-1: EC50=24,13 мМ; GRANTA-519: EC50=19,72 мМ). См. фигуру 5.
Пример 4:
Кроме того, производные B9, B12 и B13 анализировали в сравнении с пара-NO-ASA на клетках ХЛЛ, несущих мутацию TP53. Подгруппа пациентов с нарушением TP53 характеризуется в значительной степени неблагоприятным прогнозом. Клетки ХЛЛ пациентов с и без мутации ТР53 обрабатывали пятью различными концентрациями (0,01, 0,1, 1, 10, 100 мкM) пара-NO-ASA, B9, B12 и B13 в течение 24 ч.
На фигуре 6 представлены результаты анализа FACS указанных обработанных клеток, показывая, что все соединения, в частности B9 и B12, оказывают большой эффект в отношении клеток ХЛЛ без мутаций TP53. Кроме того, три соединения, В9, В12 и В13, были более эффективны в отношении ТР53-мутировавших клеток ХЛЛ по сравнению с пара-NO-ASA (B1). B9 представляло собой соединение указанной группы, показывающее наиболее существенный эффект в отношении клеток ХЛЛ с и без мутации TP53.
Пример 5:
В следующем эксперименте исследовали возможное терапевтическое окно для производных NO-ASA. Поэтому влияние наиболее эффективных производных на жизнеспособность клеток и индукцию апоптоза в нескольких линиях клеток злокачественной опухоли анализировали с помощью окрашивания аннексином. Линию клеток меланомы MelJuso, линию клеток карциномы толстой кишки SW480, линию клеток мелкоклеточного рака легкого HCC44, линию клеток аденокарциномы яичников COLO 704 и линию клеток острого миелоидного лейкоза SH2 обрабатывали р-NO-ASA и B9, B12 и B13 в диапазоне концентрации от 0,01 мкM до 100 мкM в течение 24 ч с последующим добавлением люминогенного реагента CellTiter-Glo®. Трое производных (В9, В12 и В13) показывали ясный цитотоксический эффект в отношении всех линий клеток злокачественной опухоли. На фигуре 7 показаны результаты в отношении указанных клеточных линий. р-NO-ASA, В9, В12 и В13 также значительно снижали содержание АТФ в клеточных линиях SW480, MelJuso, HCC44, SH2 и COLO704, в то время как p-NO-ASA был значительно меньше эффективен в линии SW480.
Результаты выживания, измеренного с помощью АТФ-анализа, дополнительно подчеркивают, что трое производных, В9, В12 и В13, демонстрируют терапевтическую активность в отношении различных неоплазий и твердых опухолей. В частности, В12 показывает токсические эффекты в отношении клеток злокачественной опухоли (SH2 EC50: 0,005 мкM, SW480 ЕС50: 129,5 мкM, MelJuso EC50: 0,54 мкM, HCC44 ЕС50: 1,05 мкM, COLO704 ЕС50: 2,77). Также результаты массива данных апоптоза показывают индукцию апоптоза в различных заболеваниях при концентрациях в диапазоне B9 1-9 мкM, B12 1-5 мкM и B13 7-57 мкM (смотрите таблицу ниже).
Таблица, сопровождающая пример 5. Обзор значений ЕС50 выживаемости клеток анализировали по содержанию АТФ и посредством исследования с аннексином V/PI. н.и.- не исследовали
EC50 [мкM] (n)
EC50 [мкM] (n)
EC50 [мкM] (n)
EC50 [мкM] (n)
EC50 [мкM] (n)
EC50 [мкM] (n)
Пример 6: Вовлечение каспаза-опосредованного апоптоза в клетках ХЛЛ при обработке p-NO-ASA, B9, B12 и B13.
Чтобы определить, является ли токсичность в отношении клеток ХЛЛ результатом каспаза-опосредованного апоптоза, анализировали расщепление PARP (поли (АДФ-рибоза)-полимераза 1) и XIAP (связанный с Х хромосомой ингибитор апоптоза) при помощи иммуноблота. Культивировали только клетки ХЛЛ с 1% ДМСО или р-NO-ASA, МЕТА-NO-ASA, B9, B12 и B13 при ЕС50 в течение 24 ч с последующим лизированием белка и вестерн-блоттингом с использованием антител для обнаружения прогностических апоптических белков (XIAP, PARP). Вещества для обработки при ЕС50 подвержены PARP расщеплению и явно понижают уровни антиапоптотических белков XIAP. Все исследуемые соединения индуцируют расщепление PARP и XIAP (фигура 8А). Кроме того, осуществляли анализ каспазы-3/7. Клетки ХЛЛ инкубировали с пара-NO-ASA и B9 при различных концентрациях в диапазоне от 0,01 мкM до 20 мкM в течение 6 ч с последующим добавлением люминогенного субстрата каспазы-3/7. Это указывало на снижение выживаемости клеток ХЛЛ при обработке p-NO-ASA и B9 в результате индукции каспаза-опосредованного апоптоза. Пара-NO-ASA и В9 также показали зависимую от концентрации активацию каспаз 3 и 7 (ЕС50 B9=0,23 мкM, 95% CI=0,11-0,49 мкM; EC50 р-NO-ASA=1,84 мкM, 95% CI=0,81-4,21 мкM) в исследовании специфической каспазы-3/7 (фигура 8В).
Пример 7: воздействие производных NO-ASA на основные внутриклеточные сигнальные пути ХЛЛ (NF-каппаB, WNT)
Сигнальный путь BCR играет важную патогенную роль в ХЛЛ и лимфомах, приводящий обычно к конститутивной активации NF-каппаB (в этом состоянии NF-каппаB фосфорилируется). Поэтому влияние производных на статус фосфорилирования NF-каппаB анализировали с помощью Вестерн-блоттинга. Клетки ХЛЛ обрабатывали 0,1 мкM, 1 мкM или 10 мкM каждого производного, соответственно, в течение 3 ч. Клетки ХЛЛ обрабатывали В1, В9, В12 и В13 (0,1 мкM, 1 мкM, 10 мкM) в течение 3 ч. Необработанные и обработанные ДМСО (1%) клетки служили в качестве контроля. GAPDH=контроль для нанесения.
Производные NO-ASA индуцируют зависимое от концентрации уменьшение фосфорилированного белка NF-каппаBp65 и, соответственно, репрессию сигнального пути NF-каппаB. В9, В12 и В13 индуцируют уменьшение концентрацией, составляющей только 10 мкM, при этом для индукции уменьшения белка NF-каппаB p65 была необходима двукратная концентрация p-NO-ASA (см. фигуру 9).
Пример 8: Способы синтеза
B1: pNO-ASA
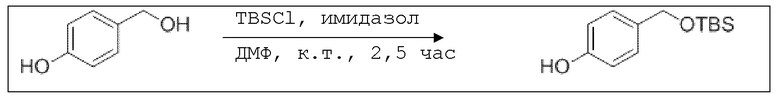
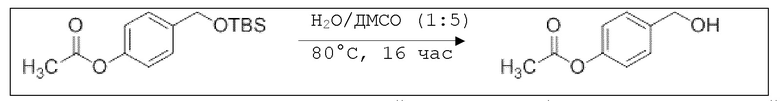
В 100 мл-овую трехгорлую колбу с инертной атмосферой добавляли 6,02 г (88,6 ммоль, 2,19 экв.) имидазола и 6,76 г (44,8 ммоль, 1,11 экв.) трет-бутил(хлор)диметилсилана. После двухкратного вакуумирования и заполнения аргоном, добавляли 40,0 мл сухого ДМФ и перемешивали в течение 10 минут при комнатной температуре. Потом добавляли 5,00 г (40,3 ммоль, 1,00 экв.) 4-(гидроксиметил)фенола. Перемешивание продолжали в течение 2,5 часов. Суспензию смешивали с 150 мл насыщенного солевого раствора и дважды экстрагировали 100 мл этилацетата. Растворитель удаляли при пониженном давлении, и сырой продукт очищали с помощью флэш-хроматографии на силикагеле (циклогексан/этилацетат=5:1) с получением указанного в заголовке соединения в виде бесцветного масла в количестве 6,78 г (28,5 ммоль, 71%).
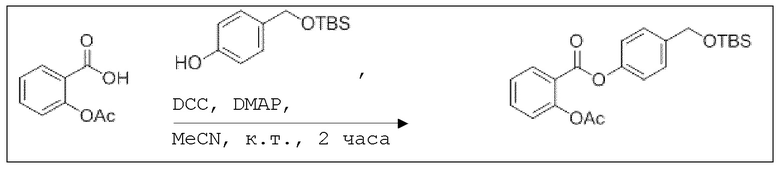
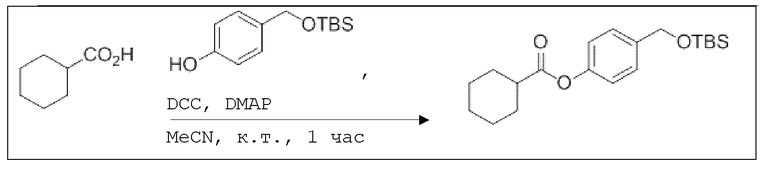
В колбе Шленка с инертной атмосферой 2,25 г (12,5 ммоль, 1,00 экв.) ацетилсалициловой кислоты растворяли в 45,0 мл ацетонитрила. Добавляли 2,98 г (12,5 ммоль, 1,00 экв.) 4-(((трет-бутилдиметилсилил)окси)метил)фенола, 153 мг (1,25 ммоль, 0,10 экв.) 4-(диметиламино)пиридина и 2,84 г (13,8 ммоль, 1,10 экв.) дициклогексилкарбодиимида. Через 2 часа растворитель удаляли при пониженном давлении, и сырой продукт очищали с помощью флэш-хроматографии на силикагеле (циклогексан/этилацетат=10:1) с получением указанного в заголовке соединения в виде бесцветного твердого вещества в количестве 3,14 г (7,85 ммоль, 63%).
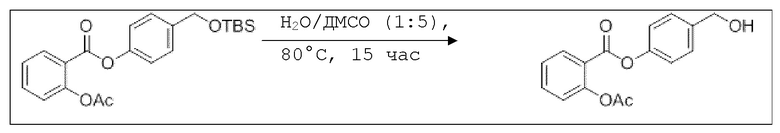
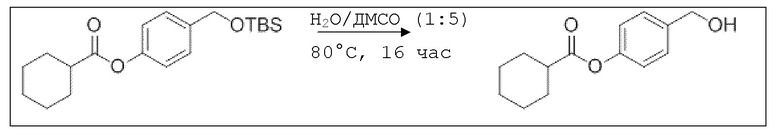
В 250 мл-овой трехгорлой колбе с инертной атмосферой растворяли 2,90 г (7,24 ммоль, 1,00 экв.) 4-(((трет-бутилдиметилсилил)окси)метил)фенил 2-ацетоксибензоата в 7,00 мл воды и 35,0 мл диметилсульфоксида. После перемешивания в течение 15 час при 80°C и охлаждения до комнатной температуры добавляли 60,0 мл воды. Смесь дважды экстрагировали 60,0 мл диэтилового эфира. Растворитель удаляли при пониженном давлении, и сырой продукт очищали с помощью флэш-хроматографии на силикагеле (циклогексан/этилацетат=1:1) с получением указанного в заголовке соединения в виде бесцветного твердого вещества в количестве 1,78 г (6,23 ммоль, 86%).

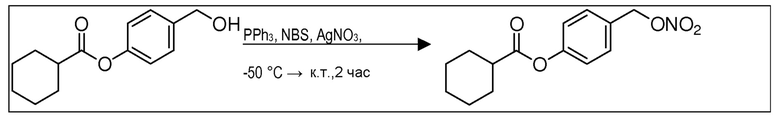
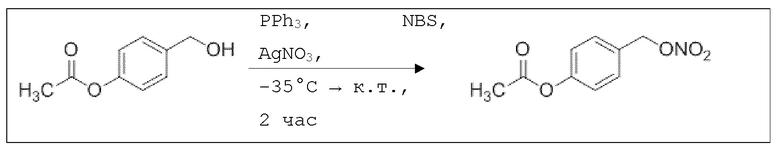
В 25,0 мл-овой колбе Шленка с инертной атмосферой растворяли 1,80 г (7,89 ммоль, 1,00 экв.) 4-(гидроксиметил)фенил 2-ацетоксибензоата и 2,07 г (7,89 ммоль, 1,00 экв.) трифенилфосфина в 8,00 мл ацетонитрила и 3,20 мл дихлорметана. Эту смесь охлаждали до -45°C и добавляли 1,40 г (7,89 ммоль, 1,00 экв.) N-бромсукцинимида. Охлаждение прекращали, при этом NBS медленно растворялся. Через 5 мин добавляли 2,01 г (11,84 ммоль, 1,50 экв.) нитрата серебра. Через 14 час перемешивания при комнатной температуре отфильтровывали осадок. Фильтрат удаляли из растворителя при пониженном давлении, и сырой продукт очищали с помощью флэш-хроматографии на силикагеле (циклогексан/этилацетат=10:1) с получением указанного в заголовке соединения в виде бесцветного твердого вещества в количестве 984 мг (2,97 ммоль, 57%).
B2: 5Me-NO-ASA
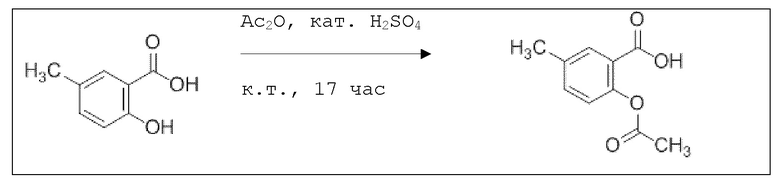
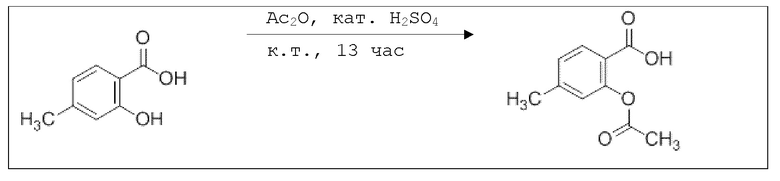
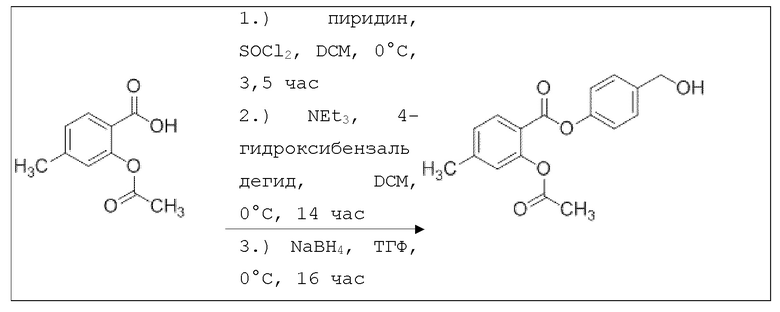
В 100 мл-вой трехгорлой круглодонной колбе с инертной атмосферой смешивали 5,00 г (32,9 ммоль, 1,00 экв.) 2-гидрокси-5-метилбензойной кислоты и 16,3 г (159 ммоль, 16,1 мл, 4,86 экв.) ангидрида уксусной кислоты. К этой суспензии добавляли каталитическое количество (6,44 мг (657 мкмоль, 3,50 мкл, 0,02 экв.)) концентрированной серной кислоты. Через 1 час добавляли 70,0 мл воды, и перемешивание продолжали в течение еще 17 ч. Осадок отфильтровывали, промывали 100 мл воды. Указанное в заголовке соединение получали в виде бесцветного твердого вещества в количестве 6,22 г (32,0 ммоль, 98%).
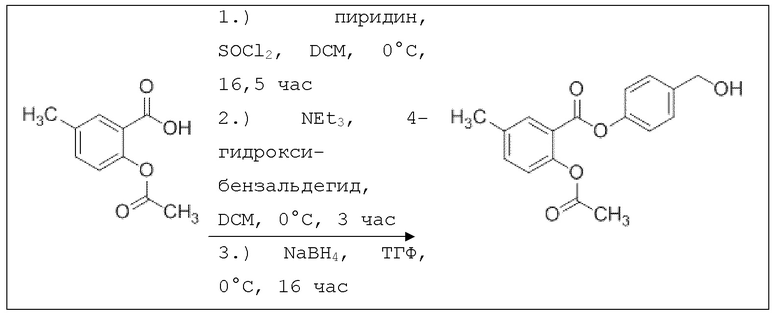
В 250 мл-вой трехгорлой круглодонной колбе с инертной атмосферой растворяли 5,00 г (25,8 ммоль, 1,00 экв.) 2-ацетокси-5-метилбензойной кислоты в 65,0 мл сухого DCM. После добавления 2,04 г (25,8 ммоль, 2,08 мл, 1,00 экв.) пиридина раствор охлаждали до 0°C. В течение 10 минут добавляли 4,60 г (38,7 ммоль, 2,81 мл, 1,50 экв.) тионилхлорида. Перемешивание продолжали в течение еще 16,5 час при 0°C, и потом растворитель удаляли. Масло абсорбировали 50,0 мл сухого DCM, и добавляли 3,14 г (31,0 ммоль, 4,29 мл, 1,20 экв.) триэтиламина. При 0°С добавляли 3,78 г (31,0 ммоль, 1,20 экв.) 4-гидроксибензальдегида. Полученный раствор перемешивали дополнительно в течение 3 час при 0°C. Смесь дважды промывали 50,0 мл воды и 30,0 мл насыщенного раствора гидрокарбоната натрия. После сушки над сульфатом магния растворитель удаляли при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии на силикагеле (циклогексан/этилацетат=2:1) с получением промежуточного продукта в виде бесцветного твердого вещества в количестве 4,16 г (14,0 ммоль, 54%). Этот промежуточный продукт обрабатывали 45,0 мл сухого ТГФ, охлаждали до 0°C и добавляли 491 мг (12,9 ммоль, 0,50 экв.) боргидрида натрия. После перемешивания в течение 16 час раствор промывали 45,0 мл насыщенного раствора хлорида аммония, сушили над сульфатом магния и растворитель удаляли при пониженном давлении. Сырой продукт очищали флэш-хроматографией на силикагеле (циклогексан/этилацетат=1:1) с получением указанного в заголовке соединения в виде бесцветного твердого вещества в количестве 1,91 г (6,37 мкмоль, 25%).
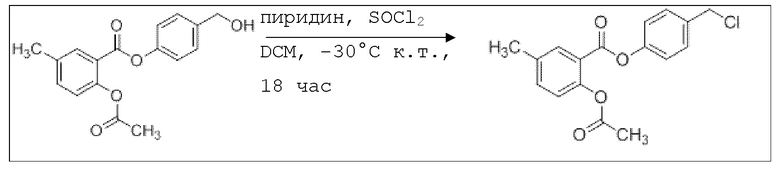
В 100 мл-вой трехгорлой круглодонной колбе с инертной атмосферой 800 мг (2,66 ммоль, 1,00 экв.) 4-(гидроксиметил)фенил 2-ацетокси-5-метилбензоата растворяли в 25,0 мл DCM, охлаждали до -30°C и в течение 1 мин добавляли 252 мг (3,19 ммоль, 283 мкл, 1,20 экв.) пиридина и 475 мг (3,99 ммоль, 283 мкл, 1,50 экв.) тионилхлорида. Перемешивание при -30°C продолжали в течение еще 45 минут и затем при комнатной температуре в течение 18 час. Раствор промывали 50,0 мл насыщенного солевого раствора и 25,0 мл воды. Органический слой сушили над сульфатом магния, и растворитель удаляли при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии на силикагеле (циклогексан/этилацетат=4:1) с получением указанного в заголовке соединения в виде бесцветного твердого вещества в количестве 543 мг (1,70 ммоль, 64%).
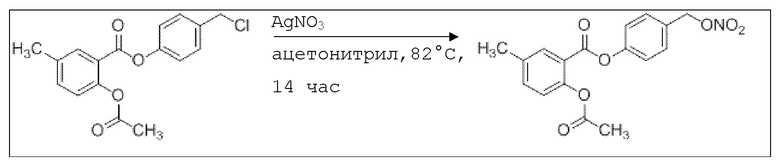
В 50,0 мл-овой трехгорлой круглодонной колбе с инертной атмосферой растворяли 450 мг (1,41 ммоль, 1,00 экв.) 4-(хлорметил)фенил 2-ацетокси-5-метилбензоата в 15,0 мл сухого ацетонитрила. После добавления 479 мг (2,82 ммоль, 2,00 экв.) нитрата серебра раствор нагревали в темноте с обратным холодильником в течение 14 час. Осадок отфильтровывали, и фильтрат сушили над сульфатом магния, и растворитель удаляли при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии на силикагеле (циклогексан/этилацетат=4:1) с получением указанного в заголовке соединения в виде твердого вещества светло-желтого цвета в количестве 437 мг (1,27 ммоль, 90%).
B3: 4Me-NO-ASA
В 250 мл-вой трехгорлой круглодонной колбе с инертной атмосферой смешивали 6,00 г (39,4 ммоль, 1,00 экв.) 2-гидрокси-4-метилбензойной кислоты и 13,1 г (159 ммоль, 12,1 мл, 3,26 экв.) ангидрида уксусной кислоты. К этой суспензии добавляли каталитическое количество (69,5 мг (990 мкмоль, 52,5 мкл, 0,03 экв.)) концентрированной серной кислоты. Через 1 час добавляли 83,7 мл воды, и перемешивание продолжали в течение еще 13 час. Осадок отфильтровывали, промывали 200 мл воды. Указанное в заголовке соединение получали в виде бесцветного твердого вещества в количестве 6,79 г (34,9 ммоль, 89%).
В 250 мл-вой трехгорлой круглодонной колбе с инертной атмосферой растворяли 5,00 г (25,8 ммоль, 1,00 экв.) 2-ацетокси-4-метилбензойной кислоты в 100,0 мл сухом DCM. После добавления 2,04 г (25,8 ммоль, 2,08 мл, 1,00 экв.) пиридина раствор охлаждали до 0°C. В течение 10 минут добавляли 4,60 г (38,7 ммоль, 2,81 мл, 1,50 экв.) тионилхлорида. Перемешивание продолжали в течение еще 3,5 час при 0°C, и потом растворитель удаляли. Масло обрабатывали 75,0 мл сухого DCM и добавляли 3,14 г (31,0 ммоль, 4,29 мл, 1,20 экв.) триэтиламина. При 0°C добавляли 3,78 г (31,0 ммоль, 1,20 экв.) 4-гидроксибензальдегида. Раствор перемешивали дополнительно в течение 14 час при 0°C. Смесь промывали 2×75,0 мл воды и 2×75,0 мл насыщенного раствора гидрокарбоната натрия. Затем сушили над сульфатом магния, и растворитель удаляли при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии на силикагеле (циклогексан/этилацетат=2:1) с получением промежуточного продукта в виде бесцветного твердого вещества в количестве 5,18 г (17,4 ммоль, 67%). Этот промежуточный продукт обрабатывали 50,0 мл сухого ТГФ, охлаждали до 0°C и добавляли 701 мг (18,4 ммоль, 0,72 экв.) боргидрида натрия.
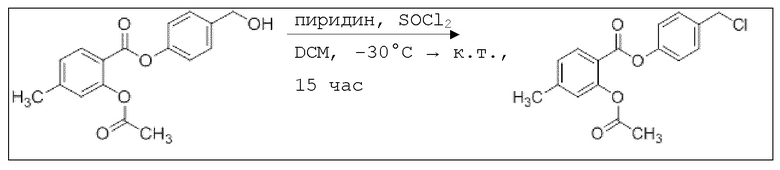
После перемешивания в течение 16 час раствор промывали 45,0 мл насыщенного раствора хлорида аммония, сушили над сульфатом магния, и растворитель удаляли при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии на силикагеле (циклогексан/этилацетат=2:1) с получением указанного в заголовке соединения в виде бесцветного твердого вещества в количестве 856 мг (2,85 ммоль, 11%).
В 50,0 мл-овой трехгорлой круглодонной колбе с инертной атмосферой растворяли 500 мг (1,67 ммоль, 1,00 экв.) 4-(гидроксиметил)фенил 2-ацетокси-4-метилбензоата в 25,0 мл DCM, охлаждали до -30°C и в течение 2 минут добавляли 158 мг (2,80 ммоль, 161 мкл, 1,20 экв.) пиридина и 297 мг (2,50 ммоль, 177 мкл, 1,50 экв.) тионилхлорида. Перемешивание продолжали при -30°C в течение еще 45 минут и затем при комнатной температуре в течение 15 час. Раствор промывали 50,0 мл насыщенного солевого раствора и 25,0 мл воды. Органический слой сушили над сульфатом магния, и растворитель удаляли при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии на силикагеле (циклогексан/этилацетат=10:1) с получением указанного в заголовке соединения в виде бесцветного твердого вещества в количестве 315 мг (988 мкмоль, 59%).
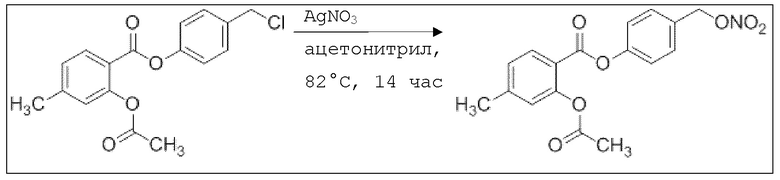
В 25,0 мл-овой трехгорлой круглодонной колбе с инертной атмосферой растворяли 200 мг (627 мкмоль, 1,00 экв.) 4-(хлорметил)фенил 2-ацетокси-4-метилбензоата в 7,00 мл сухого ацетонитрила. После добавления 213 мг (1,25 мкмоль, 2,00 экв.) нитрата серебра раствор нагревали в темноте с обратным холодильником в течение 14 час. Осадок отфильтровывали, и фильтрат сушили над сульфатом магния, и растворитель удаляли при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии на силикагеле (циклогексан/этилацетат=10:1) с получением указанного в заголовке соединения в виде твердого вещества в количестве 188 мг (544 мкмоль, 87%).
B4: 2Cl-5CF3-OTBS-BA
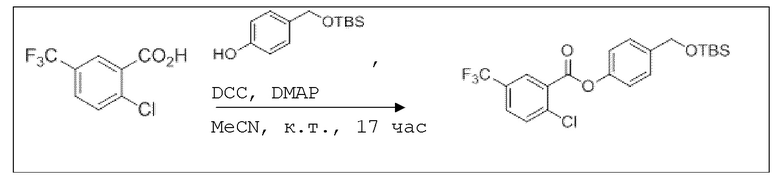
В 25,0 мл-овой колбе Шленка с инертной атмосферой растворяли 561 мг (2,50 ммоль, 1,00 экв.) 2-хлор-5-трифторметилбензойной кислоты в 10,0 мл ацетонитрила. Добавляли 596 мг (2,50 ммоль, 1,00 экв.) 4-(((трет-бутилдиметилсилил)окси)метил)фенола, 30,5 мг (250 мкмоль, 0,10 экв.) 4-(диметиламино)пиридина и 567 мг (2,75 ммоль, 1,10 экв.) дициклогексилкарбодиимида. Через 17 часов растворитель удаляли при пониженном давлении, и сырой продукт очищали с помощью флэш-хроматографии на силикагеле (циклогексан/этилацетат=20:1) с получением указанного в заголовке соединения в виде бесцветного твердого вещества в количестве 1,05 г (2,36 ммоль, 94%).
B5: 2Cl-5CF3-OH-BA
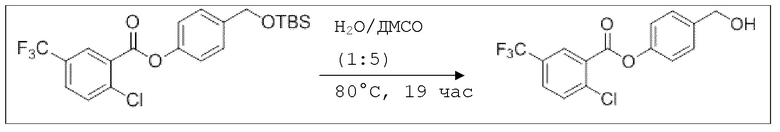
В 50 мл-овой трехгорлой колбе с инертной атмосферой растворяли 850 мг (1,91 ммоль, 1,00 экв.) 4-(((трет-бутилдиметилсилил)окси)метил)фенил 2-хлор-5-(трифторметил)бензоата в 2,00 мл воды и 10,0 мл диметилсульфоксида. После перемешивания в течение 19 час при 80°C и охлаждения до комнатной температуры добавляли 20,0 мл воды. Смесь дважды экстрагировали 20,0 мл диэтилового эфира. Растворитель удаляли при пониженном давлении, и сырой продукт очищали с помощью флэш-хроматографии на силикагеле (циклогексан/этилацетат=1:1) с получением указанного в заголовке соединения в виде бесцветного твердого вещества в количестве 619 мг (1,87 ммоль, 98%).
B6: 2Cl-5CF3-NO-BA

В 10,0 мл-овой колбе Шленка с инертной атмосферой растворяли 300 мг (910 мкмоль, 1,00 экв.) 4-(гидроксиметил)фенил 2-хлор-5-(трифторметил)бензоата и 238 мг (910 мкмоль, 1,00 экв.) трифенилфосфина в 1,00 мл ацетонитрила и 400 мкл дихлорметана. Эту смесь охлаждали до -45°C и добавляли 162 мг (910 мкмоль, 1,00 экв.) N-бромсукцинимида. Охлаждение прекращали, при этом NBS медленно растворялся. Через 5 мин добавляли 155 мг (1,37 ммоль, 1,50 экв.) нитрата серебра. Через 19 час перемешивания при комнатной температуре осадок отфильтровывали. Растворитель удаляли из фильтрата при пониженном давлении, и сырой продукт очищали с помощью флэш-хроматографии на силикагеле (циклогексан/этилацетат=2:1) с получением указанного в заголовке соединения в виде бесцветного твердого вещества в количестве 267 мг (711 мкмоль, 78%).
B7: OTBS-BA
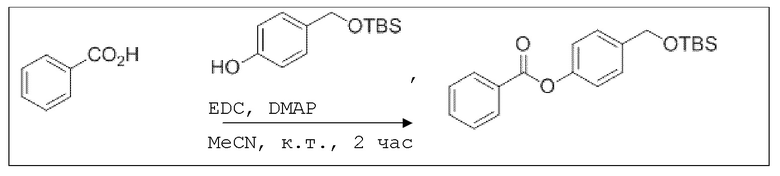
В 15,0 мл-овой колбе Шленка с инертной атмосферой растворяли 500 мг (4,09 ммоль, 1,00 экв.) бензойной кислоты в 10,0 мл ацетонитрила. Добавляли 975 мг (4,09 ммоль, 1,00 экв.) 4-(((трет-бутилдиметилсилил)окси)метил)фенола, 49,9 мг (409 мкмоль, 0,10 экв.) 4-(диметиламино)пиридина и 862 мг (4,50 ммоль, 1,10 экв.) EDC. Через 2 часа растворитель удаляли при пониженном давлении, и сырой продукт очищали с помощью флэш-хроматографии на силикагеле (циклогексан/этилацетат=10:1) с получением указанного в заголовке соединения в виде бесцветного твердого вещества в количестве 1,37 г (3,90 ммоль, 95%).
B7a: 5F-NO-ASA
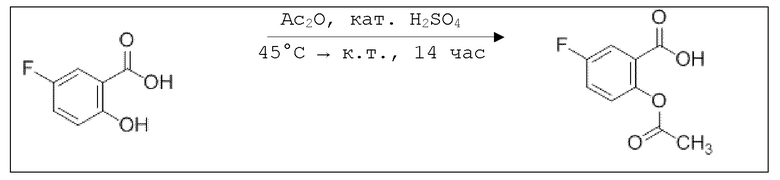
В 250-ой круглодонной колбе смешивали 5,00 г (32,0 ммоль, 1,00 экв.) 5-фтор-2-гидроксибензойной кислоты и 6,55 г (64,0 ммоль, 6,05 мл, 2,00 экв.) ангидрида уксусной кислоты. К этой суспензии добавляли каталитическое количество (6 капель) концентрированной серной кислоты при 35°C, после чего температура смеси повышалась до 45°C. Через 14 часов добавляли 67,0 мл воды. Осадок отфильтровывали, промывали 250 мл воды. Указанное в заголовке соединение получали в виде бесцветного твердого вещества в количестве 5,49 г (27,7 ммоль, 86%).
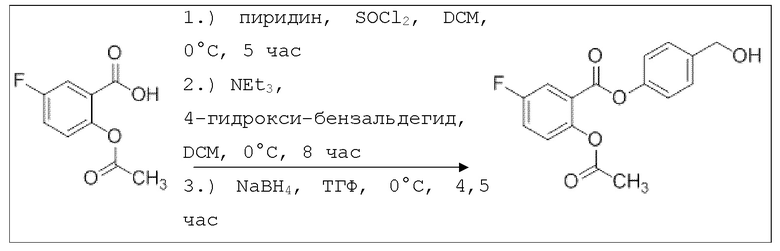
В 25,0 мл-овой трехгорлой круглодонной колбе с инертной атмосферой растворяли 872 мг (4,40 ммоль, 1,00 экв.) 2-ацетокси-5-фторбензойной кислоты в 11,2 мл сухого DCM. После добавления 872 мг (4,40 ммоль, 1,00 экв.) пиридина, раствор охлаждали до 0°C. В течение 15 минут добавляли 872 мг (4,40 ммоль, 1,00 экв.) тионилхлорида. Перемешивание продолжали в течение еще 5 час при 0°C, и потом растворитель удаляли. Масло поглощали с помощью 8,44 мл сухого DCM и добавляли 534 мг (5,28 ммоль, 732 мкл, 1,20 экв.) триэтиламина. При 0°C добавляли 537 мг (4,40 ммоль, 1,00 экв.) 4-гидроксибензальдегида. Раствор перемешивали дополнительно в течение 8 час при 0°C. Смесь дважды промывали 2×57,00 мл воды и 2×7,00 мл насыщенного раствора гидрокарбоната натрия. После сушки над сульфатом магния растворитель удаляли при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии на силикагеле (циклогексан/этилацетат=2:1) с получением промежуточного продукта в виде бесцветного твердого вещества в количестве 700 мг (2,32 ммоль, 53%). Этот промежуточный продукт обрабатывали 8,00 мл сухого ТГФ, охлаждали до 0°C и добавляли 88,6 мг (2,33 ммоль, 0,53 экв.) боргидрида натрия.
После перемешивания в течение 4,5 час раствор промывали 8,00 мл насыщенного раствора хлорида аммония, сушили над сульфатом магния, и растворитель удаляли при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии на силикагеле (циклогексан/этилацетат=1:1) с получением указанного в заголовке соединения в виде бесцветного твердого вещества в количестве 273 мг (897 мкмоль, 21%).
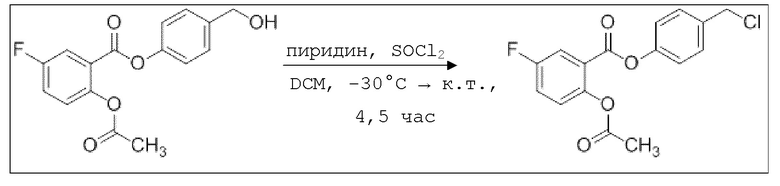
В 25,0 мл-овой трехгорлой круглодонной колбе с инертной атмосферой растворяли 350 мг (1,15 ммоль, 1,00 экв.) 4-(гидроксиметил)фенил 2-ацетокси-5-фторбензоата в 11,0 мл DCM, охлаждали до -30°C в течение 5 минут, затем добавляли 108 мг (1,37 ммоль, 111 мкл, 1,19 экв.) пиридина и 203 мг (1,68 ммоль, 121 мкл, 1,49 экв.) тионилхлорида. Перемешивание при -30°C продолжали в течение еще 45 минут и затем при комнатной температуре в течение 4,5 час. Раствор промывали 23,0 мл насыщенного солевого раствора и 11,0 мл воды. Органический слой сушили над сульфатом магния, и растворитель удаляли при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии на силикагеле (циклогексан/этилацетат=1:1) с получением указанного в заголовке соединения в виде бесцветного твердого вещества в количестве 156 мг (480 мкмоль, 42%).
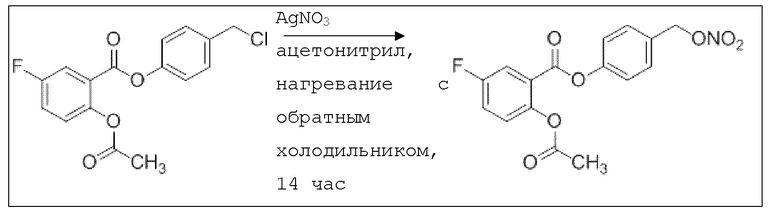
В 10,0 мл-овой трехгорлой круглодонной колбе с инертной атмосферой растворяли 85,0 мг (263 мкмоль, 1,00 экв.) 4-(хлорметил)фенил 2-ацетокси-5-фторбензоата в 3,00 мл сухого ацетонитрила. После добавления 88,3 мг (526 мкмоль, 2,00 экв.) нитрата серебра раствор нагревали в темноте с обратным холодильником в течение 14 час. Осадок отфильтровывали, фильтрат сушили над сульфатом магния, и растворитель удаляли при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии на силикагеле (циклогексан/этилацетат=4:1) с получением указанного в заголовке соединения в виде твердого вещества светло-желтого цвета в количестве 81,0 мг (232 мкмоль, 89%).
B8: OH-BA

В 50 мл-овой трехгорлой колбе с инертной атмосферой растворяли 1,03 г (3,00 ммоль, 1,00 экв.) 4-(((трет-бутилдиметилсилил)окси)метил)фенилбензоата в 3,00 мл воды и 15,0 мл диметилсульфоксида. После перемешивания в течение 16 час при 80°C и охлаждения до комнатной температуры добавляли 20,0 мл воды. Смесь дважды экстрагировали 40,0 мл диэтилового эфира. Растворитель удаляли при пониженном давлении, и сырой продукт очищали с помощью флэш-хроматографии на силикагеле (циклогексан/этилацетат=1:1) с получением указанного в заголовке соединения в виде бесцветного твердого вещества в количестве 682 мг (2,99 ммоль, 100%).
B9: NO-BA
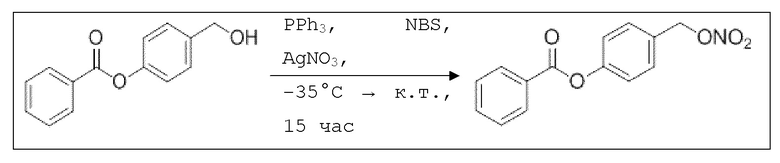
В 10,0 мл-овой колбе Шленка с инертной атмосферой растворяли 342 мг (1,50 ммоль, 1,00 экв.) 4-(гидроксиметил)фенилбензоата и 393 мг (1,50 ммоль, 1,00 экв.) трифенилфосфина в 1,50 мл ацетонитрила и 600 мкл дихлорметана. Раствор охлаждали до -45°C и добавляли 267 мг (1,50 ммоль, 1,00 экв.) N-бромсукцинимида. Охлаждение прекращали, при этом NBS медленно растворялся. Через 5 мин добавляли 382 мг (2,25 ммоль, 1,50 экв.) нитрата серебра. Через 15 час перемешивания при комнатной температуре осадок отфильтровывали. Фильтрат удаляли из растворителя при пониженном давлении, и сырой продукт очищали с помощью флэш-хроматографии на силикагеле (циклогексан/этилацетат=10:1) с получением указанного в заголовке соединения в виде бесцветного твердого вещества в количестве 355 мг (1,30 ммоль, 87%).
B10: Форма-BA
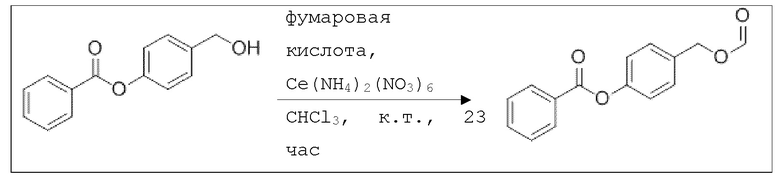
В 10,0 мл-ом круглодонной колбе растворяли 114 мг (500 мкмоль, 1,00 экв.) 4-(гидроксиметил)фенилбензоата, 22,9 мг (500 мкмоль, 18,8 мкл, 1,00 экв.) фумаровой кислоты и 27,4 мг (50,0 мкмоль, 0,10 экв.) церия аммония нитрата в 2,00 мл хлороформа. Раствор перемешивали при комнатной температуре в течение 23 час. Затем добавляли 10,0 мл охлажденной воды, и раствор дважды экстрагировали 10,0 мл MTBE. Сырой продукт очищали с помощью флэш-хроматографии на силикагеле (циклогексан/этилацетат=10:1) с получением указанного в заголовке соединения в виде бесцветного твердого вещества в количестве 113 мг (441 мкмоль, 88%).
B11: NO-OMe-BA
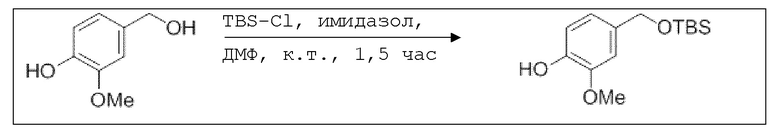
В 25,0 мл-ую трехгорлую колбу с инертной атмосферой помещали 899 мг (13,2 ммоль, 2,20 экв.) имидазола и 995 мг (6,60 ммоль, 1,10 экв.) трет-бутил(хлор)диметилсилана. После двухкратного вакуумирования и заполнения аргоном добавляли 7,00 мл сухого ДМФ и перемешивали в течение 10 минут при комнатной температуре. Затем добавляли 925 мг (6,00 ммоль, 1,00 экв.) 4-(гидроксиметил)-2-метоксифенола. Перемешивание продолжали в течение 1,5 часов. Суспензию смешивали с 20,0 мл насыщенного солевого раствора и дважды экстрагировали 20,0 мл этилацетата. Растворитель удаляли при пониженном давлении, и сырой продукт очищали с помощью флэш-хроматографии на силикагеле (циклогексан/этилацетат=10:1) с получением указанного в заголовке соединения в виде бесцветного масла в количестве 1,40 г (5,23 ммоль, 87%).
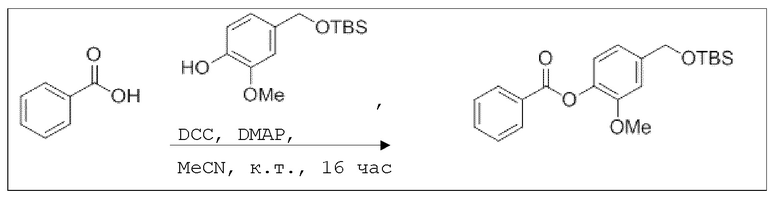
В 25,0 мл-овой колбе Шленка с инертной атмосферой растворяли 183 мг (1,50 ммоль, 1,00 экв.) бензойной кислоты в 7,0 мл ацетонитрила. Добавляли 403 мг (1,50 ммоль, 1,00 экв.) 4-((трет-бутилдиметилсилилоксиметил)-2-метокси)фенола, 18,0 мг (150 мкмоль, 0,10 экв.) 4-(диметиламино)пиридина, 340 мг (1,65 ммоль, 1,10 экв.) дициклогексилкарбодиимида. Через 16 часов растворитель удаляли при пониженном давлении, и сырой продукт очищали с помощью флэш-хроматографии на силикагеле (циклогексан/этилацетат=10:1) с получением указанного в заголовке соединения в виде бесцветного твердого вещества в количестве 543 мг (1,46 ммоль, 97%).
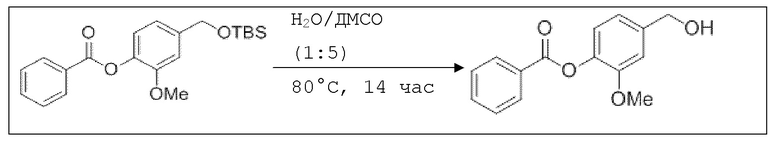
В 25,0 мл-овой колбе Шленка с инертной атмосферой растворяли 500 мг (1,34 ммоль, 1,00 экв.) 4-(((трет-бутилдиметилсилил)окси)метил)-2-метоксифенилбензоата в 1,50 мл воды и 7,50 мл диметилсульфоксида. После перемешивания в течение 14 час при 80°C и охлаждения до комнатной температуры добавляли 10,0 мл воды. Смесь дважды экстрагировали 10,0 мл диэтилового эфира. Растворитель удаляли при пониженном давлении, и сырой продукт очищали с помощью флэш-хроматографии на силикагеле (циклогексан/этилацетат=1:1) с получением указанного в заголовке соединения в виде бесцветного твердого вещества в количестве 344 мг (1,33 ммоль, 99%).
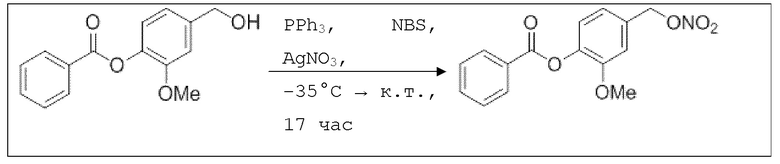
В 10,0 мл-овой колбе Шленка с инертной атмосферой растворяли 280 мг (1,08 ммоль, 1,00 экв.) 4-(гидроксиметил)-2-метоксифенилбензоата и 284 мг (1,08 ммоль, 1,00 экв.) трифенилфосфина в 1,08 мл ацетонитрила и 430 мкл дихлорметана. Раствор охлаждали до -45°C и добавляли 193 мг (1,08 ммоль, 1,00 экв.) N-бромсукцинимида. Охлаждение прекращали, при этом NBS медленно растворялся. Через 5 мин добавляли 280 мг (1,63 ммоль, 1,50 экв.) нитрата серебра. Через 17 час перемешивания при комнатной температуре осадок отфильтровывали. Растворитель удаляли из фильтрата при пониженном давлении, и сырой продукт очищали с помощью флэш-хроматографии на силикагеле (циклогексан/этилацетат=5:1) с получением указанного в заголовке соединения в виде бесцветного твердого вещества в количестве 322 мг (1,06 ммоль, 98%).
B12: Cl-BA
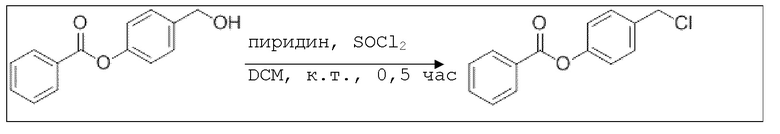
В 50,0 мл-овой колбе Шленка с инертной атмосферой растворяли 3,00 г (13,1 ммоль, 1,00 экв.) 4-гидроксиметилфенил)бензоата в 10,0 мл DCM, и охлаждали до -30°C. В течение 10 минут добавляли 321 мг (3,94 ммоль, 318 мкл, 1,19 экв.) пиридина и 2,35 г (3,94 ммоль, 1,43 мл, 1,49 экв.) тионилхлорида. После перемешивания при комнатной температуре в течение 0,5 часа добавляли 20,0 мл DCM и 20,0 мл воды к раствору, который затем промывали 20,0 мл насыщенного раствора карбоната натрия и 20,0 мл воды. Органический слой сушили над сульфатом магния, и растворитель удаляли при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии на силикагеле (циклогексан/этилацетат=5:1) с получением указанного в заголовке соединения в виде бесцветного твердого вещества в количестве 2,93 г (11,9 ммоль, 90%).
B13: NO-Нафтил
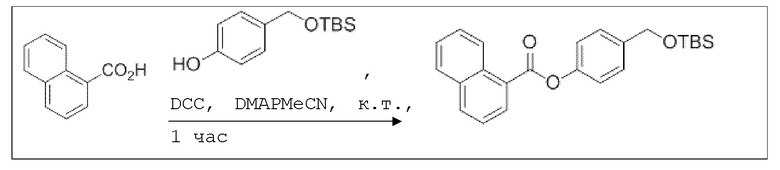
В колбе Шленка с инертной атмосферой растворяли 1,03 г (6,00 ммоль, 1,00 экв.) 1-нафтойной кислоты в 25,0 мл ацетонитрила. Добавляли 1,43 г (6,00 ммоль, 1,00 экв.) 4-(((трет-бутилдиметилсилил)окси)метил)фенола, 73,0 мг (600 мкмоль, 0,10 экв.) 4-(диметиламино)пиридина и 1,36 г (6,60 ммоль, 1,10 экв.) дициклогексилкарбодиимида. Через 1 час растворитель удаляли при пониженном давлении, и сырой продукт очищали с помощью флэш-хроматографии на силикагеле (циклогексан/этилацетат=10:1) с получением указанного в заголовке соединения в виде бесцветного твердого вещества в количестве 2,31 г (5,60 ммоль, 98%).
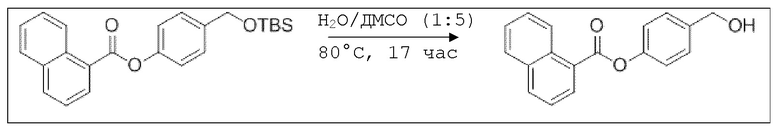
В колбе Шленка с инертной атмосферой растворяли 1,65 г (4,20 ммоль, 1,00 экв.) 4-(((трет-бутилдиметилсилил)окси)метил)фенил нафтоата в 6,50 мл воды и 32,5 мл диметилсульфоксида. После перемешивания в течение 17 час при 80°C и охлаждения до комнатной температуры добавляли 50,0 мл воды. Смесь дважды экстрагировали 50,0 мл диэтилового эфира. Растворитель удаляли при пониженном давлении, и сырой продукт очищали с помощью флэш-хроматографии на силикагеле (циклогексан/этилацетат=2:1) с получением указанного в заголовке соединения в виде бесцветного твердого вещества в количестве 1,13 г (4,05 ммоль, 96%).
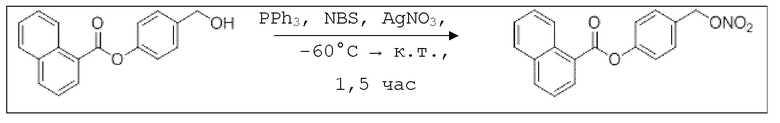
В 25,0 мл-овой колбе Шленка с инертной атмосферой растворяли 900 мг (3,23 ммоль, 1,00 экв.) 4-(гидроксиметил)фенил 1-нафтоата и 847 мг (3,23 ммоль, 1,00 экв.) трифенилфосфина в 3,50 мл ацетонитрила и 1,40 мл дихлорметана. Раствор охлаждали до -60°C и добавляли 575 мг (3,23 ммоль, 1,00 экв.) N-бромсукцинимида. Охлаждение прекращали, при этом NBS медленно растворялся. Через 15 мин добавляли 823 мг (4,85 ммоль, 1,50 экв.) нитрата серебра. Через 1,5 час перемешивания при комнатной температуре осадок отфильтровывали. Растворитель удаляли из фильтрата при пониженном давлении, и сырой продукт очищали с помощью флэш-хроматографии на силикагеле (циклогексан/этилацетат=5:1) с получением указанного в заголовке соединения в виде бесцветного твердого вещества в количестве 987 мг (3,05 ммоль, 94%).
B14: NO-cHex
В 25,0 мл-овой колбе Шленка с инертной атмосферой растворяли 269 мг (2,10 ммоль, 1,00 экв.) циклогексанкарбоновой кислоты в 10,0 мл ацетонитрила. Добавляли 500 мг (2,10 ммоль, 1,00 экв.) 4-(((трет-бутилдиметилсилил)окси)метил)фенола, 26,0 мг (210 мкмоль, 0,10 экв.) 4-(диметиламино)пиридина и 476 мг (2,31 ммоль, 1,10 экв.) дициклогексилкарбодиимида. Через 1 час растворитель удаляли при пониженном давлении, и сырой продукт очищали с помощью флэш-хроматографии на силикагеле (циклогексан/этилацетат=10:1) с получением указанного в заголовке соединения в виде бесцветного твердого вещества в количестве 723 мг (2,07 ммоль, 99%).
В 25,0 мл-овой колбе Шленка с инертной атмосферой растворяли 500 мг (1,44 ммоль, 1,00 экв.) 4-(((трет-бутилдиметилсилил)окси)метил)фенил циклогексан карбоксилата в 1,50 мл воды и 7,05 мл диметилсульфоксида. После перемешивания в течение 16 час при 80°C и охлаждения до комнатной температуры добавляли 10,0 мл воды. Смесь дважды экстрагировали 10,0 мл диэтилового эфира. Растворитель удаляли при пониженном давлении, и сырой продукт очищали с помощью флэш-хроматографии на силикагеле (циклогексан/этилацетат=1:1) с получением указанного в заголовке соединения в виде бесцветного твердого вещества в количестве 308 мг (1,32 ммоль, 92%).
4-(Гидроксиметил)фенил циклогексан карбоксилат и 224 мг (854 мкмоль, 1,00 экв.) трифенилфосфина растворяли в 2,50 мл ацетонитрила и 1,00 мл дихлорметана. Раствор охлаждали до -50°C и добавляли 152 мг (854 мкмоль, 1,00 экв.) N-бромсукцинимида. Охлаждение прекращали, при этом NBS медленно растворялся. Через 5 мин добавляли 218 мг (1,28 ммоль, 1,50 экв.) нитрата серебра. Через 2 часа перемешивания при комнатной температуре осадок отфильтровывали. Растворитель удаляли из фильтрата при пониженном давлении, и сырой продукт очищали с помощью флэш-хроматографии на силикагеле (циклогексан/этилацетат=10:1) с получением указанного в заголовке соединения в виде бесцветного твердого вещества в количестве 216 мг (773 мкмоль, 91%).
B15: NO-AA
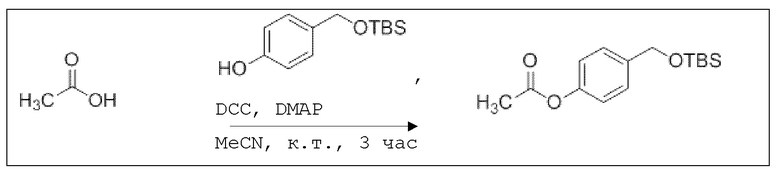
В 25,0 мл-овой колбе Шленка с инертной атмосферой растворяли 120 мкл (2,10 ммоль, 1,00 экв.) уксусной кислоты в 10,0 мл ацетонитрила. Добавляли 500 мг (2,10 ммоль, 1,00 экв.) 4-(((трет-бутилдиметилсилил)окси)метил)фенола, 26,0 мг (210 мкмоль, 0,10 экв.) 4-(диметиламино)пиридина и 476 мг (2,31 ммоль, 1,10 экв.) дициклогексилкарбодиимида. Через 3 часа растворитель удаляли при пониженном давлении, и сырой продукт очищали с помощью флэш-хроматографии на силикагеле (циклогексан/этилацетат=10:1) с получением указанного в заголовке соединения в виде бесцветного твердого вещества в количестве 531 мг (1,89 ммоль, 90%).
В 25,0 мл-овой колбе Шленка с инертной атмосферой растворяли 400 мг (1,43 ммоль, 1,00 экв.) 4-(((трет-бутилдиметилсилил)окси)метил)фенилацетата в 1,50 мл воды и 7,05 мл диметилсульфоксида. После перемешивания в течение 16 час при 80°C и охлаждения до комнатной температуры добавляли 10,0 мл воды. Смесь дважды экстрагировали с помощью 10,0 мл диэтилового эфира. Растворитель удаляли при пониженном давлении, и сырой продукт очищали с помощью флэш-хроматографии на силикагеле (циклогексан/этилацетат=1:1) с получением указанного в заголовке соединения в виде бесцветного твердого вещества в количестве 222 мг (1,34 ммоль, 94%).
В 25,0 мл-овой колбе Шленка с инертной атмосферой растворяли 100 мг (602 мкмоль, 1,00 экв.) 4-(гидроксиметил)фенилацетата и 158 мг (602 мкмоль, 1,00 экв.) трифенилфосфина в 2,50 мл ацетонитрила и 1,00 мл дихлорметана. Раствор охлаждали до -35°C и добавляли 152 мг (854 мкмоль, 1,00 экв.) N-бромсукцинимида. Охлаждение прекращали, при этом NBS медленно растворялся. Через 5 мин добавляли 218 мг (1,28 ммоль, 1,50 экв.) нитрата серебра. Через 2 час перемешивания при комнатной температуре осадок отфильтровывали. Растворитель удаляли из фильтрата при пониженном давлении, и сырой продукт очищали с помощью флэш-хроматографии на силикагеле (циклогексан/этилацетат=5:1) с получением указанного в заголовке соединения в виде бесцветного твердого вещества в количестве 105 мг (497 мкмоль, 83%).
B16: NO-Данзил
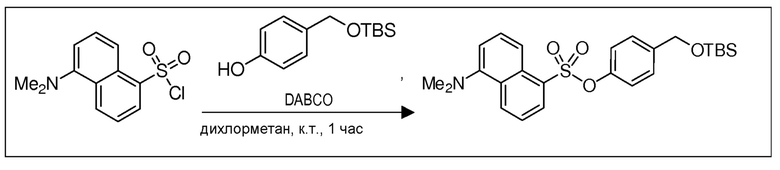
В 10,0 мл-овой колбе Шленка с инертной атмосферой растворяли 150 мг (556 мкмоль, 1,00 экв.) данзилхлорида и 133 мг (556 мкмоль, 1,00 экв.) 4-(((трет-бутилдиметилсилил)окси)метил)фенола в 2,00 мл дихлорметана. К этому раствору добавляли 75,0 мг (667 мкмоль, 1,20 экв.) DABCO. Через 1 час перемешивания при комнатной температуре растворитель удаляли при пониженном давлении, и сырой продукт очищали с помощью флэш-хроматографии на силикагеле (циклогексан/этилацетат=10:1) с получением указанного в заголовке соединения в виде масла в количестве 239 мг (507 мкмоль, 91%).
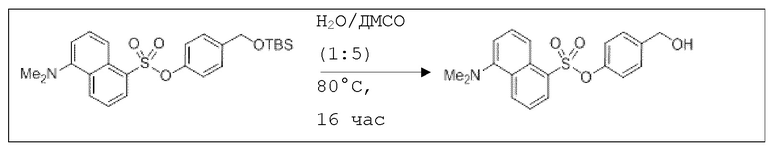
В 10,0 мл-овой колбе Шленка с инертной атмосферой растворяли 200 мг (424 мкмоль, 1,00 экв.) 4-(((трет-бутилдиметилсилил)окси)метил)фенил 5-(диметиламино)нафталин-1-сульфоната в 500 мкл воды и 2,05 мл диметилсульфоксида. После перемешивания в течение 14 час при 80°C и охлаждения до комнатной температуры добавляли 5,00 мл воды. Смесь дважды экстрагировали 5,00 мл диэтилового эфира. Растворитель удаляли при пониженном давлении, и сырой продукт очищали с помощью флэш-хроматографии на силикагеле (циклогексан/этилацетат=1:1) с получением указанного в заголовке соединения в виде бесцветного твердого вещества в количестве 135 мг (378 мкмоль, 89%).
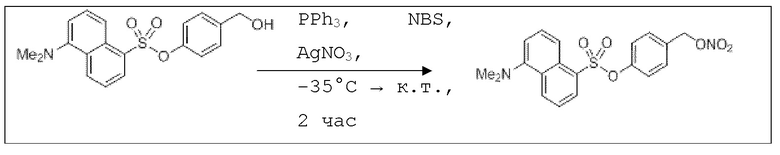
В 10,0 мл-овой колбе Шленка с инертной атмосферой растворяли 100 мг (280 мкмоль, 1,00 экв.) 4-(гидроксиметил)фенил 5-(диметиламино)нафталин-1-сульфоната и 73,0 мг (280 мкмоль, 1,00 экв.) трифенилфосфина в 1,00 мл ацетонитрила и 400 мкл дихлорметана. Эту смесь охлаждали до -35°C и добавляли 50,0 мг (280 мкмоль, 1,00 экв.) N-бромсукцинимида. Охлаждение прекращали, при этом NBS медленно растворялся. Через 5 мин добавляли 71,0 мг (420 мкмоль, 1,50 экв.) нитрата серебра. Через 2 час перемешивания при комнатной температуре осадок отфильтровывали. Растворитель удаляли из фильтрата при пониженном давлении, и сырой продукт очищали с помощью флэш-хроматографии (циклогексан/этилацетат=5:1) с получением указанного в заголовке соединения в виде масла желтого цвета в количестве 88,0 мг (219 мкмоль, 78%).
B17: NO-Homo-BA

В 50,0 мл-ую колбу Шленка с инертной атмосферой помещали 2,16 г (31,7 ммоль, 2,19 экв.) имидазола и 2,42 г (16,1 ммоль, 1,11 экв.) трет-бутил(хлор)диметилсилана. После двухкратного вакуумирования и заполнения аргоном добавляли 15,0 мл (14,3 g, 195 ммоль, 13,5 экв.) сухого ДМФ и перемешивали в течение 5 минут при комнатной температуре. Затем добавляли 2,00 г (14,5 ммоль, 1,00 экв.) 4-(2-гидроксиэтил)фенола. Перемешивание продолжали в течение 2 час. Суспензию смешивали с 70,0 мл насыщенного солевого раствора и дважды экстрагировали 50,0 мл этилацетата. Растворитель удаляли при пониженном давлении, и сырой продукт очищали с помощью флэш-хроматографии на силикагеле (циклогексан/этилацетат=5:1) с получением указанного в заголовке соединения в виде бесцветного твердого вещества в количестве 2,87 г (14,5 ммоль, 79%).
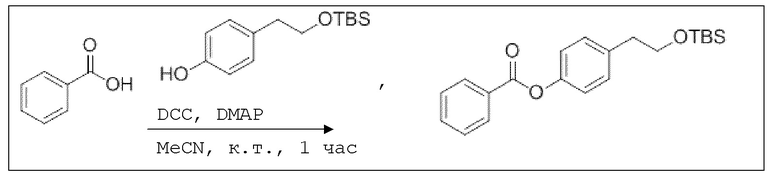
В 50,0 мл-овой колбе Шленка с инертной атмосферой растворяли 512 мг (4,19 ммоль, 1,00 экв.) бензойной кислоты в 20,0 мл ацетонитрила. Добавляли 1,06 г (4,19 ммоль, 1,00 экв.) 4-(2-((трет-бутилдиметилсилил)окси)этил)фенола, 51,0 мг (419 мкмоль, 0,10 экв.) 4-(диметиламино)пиридина и 952 мг (4,61 ммоль, 1,10 экв.) дициклогексилкарбодиимида. Через 1 час растворитель удаляли при пониженном давлении, и сырой продукт очищали с помощью флэш-хроматографии на силикагеле (циклогексан/этилацетат=10:1) с получением указанного в заголовке соединения в виде бесцветного твердого вещества в количестве 1,45 г (4,11 ммоль, 98%).
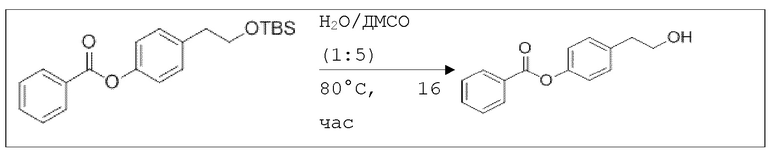
В 50 мл-овой колбе Шленка с инертной атмосферой растворяли 1,00 г (2,80 ммоль, 1,00 экв.) 4-(2-((трет-бутилдиметилсилил)окси)этил)фенилбензоата в 3,00 мл воды и 15,0 мл диметилсульфоксида. После перемешивания в течение 16 час при 80°C и охлаждения до комнатной температуры добавляли 20,0 мл воды. Смесь дважды экстрагировали 20,0 мл диэтилового эфира. Растворитель удаляли при пониженном давлении, и сырой продукт очищали с помощью флэш-хроматографии на силикагеле (циклогексан/этилацетат=2:1) с получением указанного в заголовке соединения в виде бесцветного твердого вещества в количестве 657 мг (2,71 ммоль, 97%).
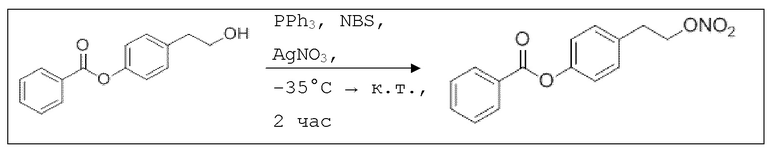
В 25,0 мл-овой колбе Шленка с инертной атмосферой растворяли 450 мг (1,86 ммоль, 1,00 экв.) 4-(2-гидроксиэтил)фенилбензоата и 487 мг (1,86 ммоль, 1,00 экв.) трифенилфосфина в 5,00 мл ацетонитрила и 2,00 мл дихлорметана. Раствор охлаждали до -35°C и добавляли 331 мг (1,86 мкмоль, 1,00 экв.) N-бромсукцинимида. Охлаждение прекращали, при этом NBS медленно растворялся. Через 5 мин добавляли 473 мг (2,79 ммоль, 1,50 экв.) нитрата серебра. Через 2 час перемешивания при комнатной температуре осадок отфильтровывали. Фильтрат удаляли из растворителя при пониженном давлении, и сырой продукт очищали с помощью флэш-хроматографии на силикагеле (циклогексан/этилацетат=5:1) с получением указанного в заголовке соединения в виде бесцветного твердого вещества в количестве 446 мг (1,55 ммоль, 84%).
B18: NO-2OmeBA
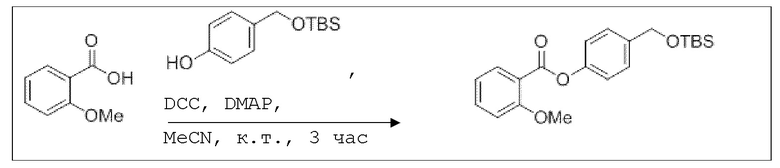
В 250 мл-овой колбе Шленка с инертной атмосферой растворяли 3,00 г (19,7 ммоль, 1,00 экв.) 2-метокси-бензойной кислоты в 60,0 мл ацетонитрила. Добавляли 4,70 г (19,7 ммоль, 1,00 экв.) 4-(((трет-бутилдиметилсилил)окси)метил)фенола, 241 мг (1,97 ммоль, 0,1 экв.) 4-(диметиламино)пиридина и 4,48 г (21,7 ммоль, 1,1 экв.) дициклогексилкарбодиимида. Через 3 часа растворитель удаляли при пониженном давлении, и сырой продукт очищали с помощью флэш-хроматографии на силикагеле (циклогексан/этилацетат=10:1) с получением указанного в заголовке соединения в виде бесцветного твердого вещества в количестве 6,56 г (17,6 ммоль, 89%).
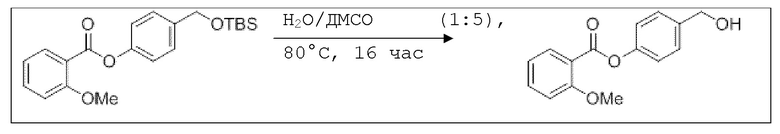
В 250 мл-овой трехгорлой колбе с инертной атмосферой растворяли 5,00 г (13,4 ммоль, 1,00 экв.) (трет-бутилдиметилсилил)окси)метилфенил)эфира 2-метоксибензойной кислоты в 15,00 мл воды и 75,0 мл диметилсульфоксид. После перемешивания в течение 16 час при 80°C и охлаждения до комнатной температуры добавляли 100,0 мл воды. Смесь дважды экстрагировали с помощью 100,0 мл диэтилового эфира. Растворитель удаляли при пониженном давлении, и сырой продукт очищали с помощью флэш-хроматографии на силикагеле (циклогексан/этилацетат=1:1) с получением указанного в заголовке соединения в виде бесцветного твердого вещества в количестве 3,28 г (12,7 ммоль, 95%).
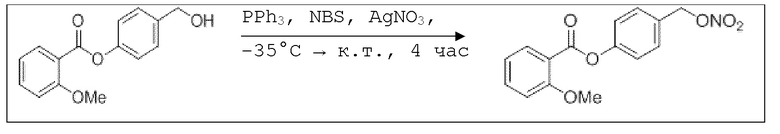
В 25,0 мл-овой колбе Шленка с инертной атмосферой растворяли 1,00 г (3,87 ммоль, 1,00 экв.) 4-(гидроксиметил)фенил 2-метоксибензоата и 1,02 г (3,87 ммоль, 1,00 экв.) трифенилфосфина в 10,0 мл ацетонитрила и 4,00 мл дихлорметана. Раствор охлаждали до -45°C и добавляли 689 мг (3,87 ммоль, 1,00 экв.) N-бромсукцинимида. Охлаждение прекращали, при этом NBS медленно растворялся. Через 5 мин добавляли 987 мг (5,81 ммоль, 1,50 экв.) нитрата серебра. Через 4 часа перемешивания при комнатной температуре осадок отфильтровывали. Растворитель удаляли из фильтрата при пониженном давлении, и сырой продукт очищали с помощью флэш-хроматографии на силикагеле (циклогексан/этилацетат=10:1) с получением указанного в заголовке соединения в виде бесцветного твердого вещества в количестве 889 мг (2,93 ммоль, 76%).
B19: NO-4OmeBA
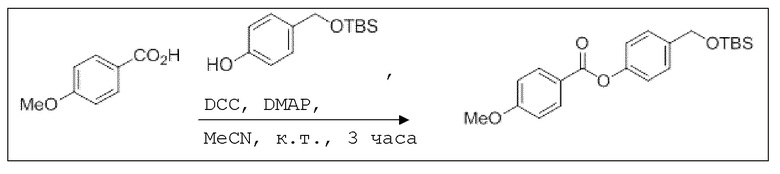
В 250 мл-овой колбе Шленка с инертной атмосферой растворяли 3,00 г (19,7 ммоль, 1,00 экв.) 2-метокси-бензойной кислоты в 60,0 мл ацетонитрила. Добавляли 4,70 г (19,7 ммоль, 1,00 экв.) 4-(((трет-бутилдиметилсилил)окси)метил)фенола, 241 мг (1,97 ммоль, 0,1 экв.) 4-(диметиламино)пиридина и 4,48 г (21,7 ммоль, 1,1 экв.) дициклогексилкарбодиимида. Через 3 часа растворитель удаляли при пониженном давлении, и сырой продукт очищали с помощью флэш-хроматографии на силикагеле (циклогексан/этилацетат=10:1) с получением указанного в заголовке соединения в виде бесцветного твердого вещества в количестве 6,60 г (17,7 ммоль, 90%).
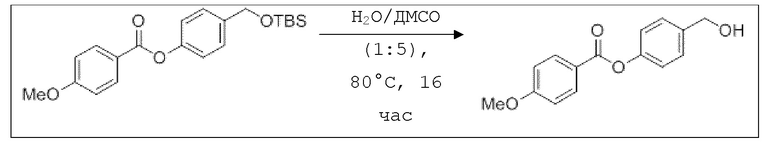
В 250 мл-овой трехгорлой колбе с инертной атмосферой растворяли 5,00 г (13,4 ммоль, 1,00 экв.) 4-(((трет-бутилдиметилсилил)окси)метил)фенил 4-метоксибензоата в 15,00 мл воды и 75,0 мл диметилсульфоксида. После перемешивания в течение 16 час при 80°C и охлаждения до комнатной температуры добавляли 100,0 мл воды. Смесь дважды экстрагировали 100,0 мл диэтилового эфира. Растворитель удаляли при пониженном давлении, и сырой продукт очищали с помощью флэш-хроматографии на силикагеле (циклогексан/этилацетат=1:1) с получением указанного в заголовке соединения в виде бесцветного твердого вещества в количестве 3,42 г (13,3 ммоль, 99%).
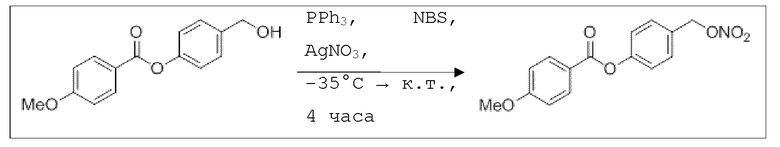
В 25,0 мл-овой колбе Шленка с инертной атмосферой растворяли 3,00 г (11,6 ммоль, 1,00 экв. 4 (гидроксиметил)фенил 4-метоксибензоата и 3,05 г (11,6 ммоль, 1,00 экв.) трифенилфосфина в 10,0 мл ацетонитрила и 4,00 мл дихлорметана. Раствор охлаждали до -45°C и добавляли 2,06 г (11,6 ммоль, 1,00 экв.) N-бромсукцинимида. Охлаждение прекращали, при этом NBS медленно растворялся. Через 5 мин добавляли 2,96 г (17,4 ммоль, 1,50 экв.) нитрата серебра. Через 4 часа перемешивания при комнатной температуре осадок отфильтровывали. Фильтрат удаляли из растворителя при пониженном давлении, и сырой продукт очищали с помощью флэш-хроматографии (циклогексан/этилацетат=2:1) с получением указанного в заголовке соединения в виде бесцветного твердого вещества в количестве 2,45 г (8,07 ммоль, 70%).
B20: NO-2Этин-BA
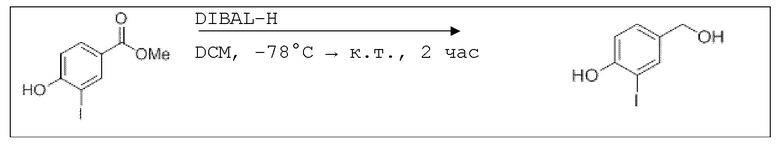
В 500 мл-овой колбе Шленка с инертной атмосферой растворяли 2,00 г (7,19 ммоль, 1,00 экв.) метил 4-гидрокси-3-йодбензоата в 200 мл дихлорметана и охлаждали до -78°C. Затем добавляли 22,9 мл (25,2 ммоль, 1,1M, 3,50 экв.) DIBAL-H. Через 0,5 часа охлаждение прекращали, и перемешивание продолжали в течение еще 2 часов. Смесь обрабатывали добавлением 200 мл воды и 30,0 мл уксусной кислоты и экстрагировали 2×200 мл дихлорметана. Растворитель удаляли из объединенных органических слоев при пониженном давлении, и сырой продукт очищали с помощью флэш-хроматографии на силикагеле (циклогексан/этилацетат=1:1) с получением указанного в заголовке соединения в виде бесцветного твердого вещества в количестве 1,74 г (6,96 ммоль, 98%).
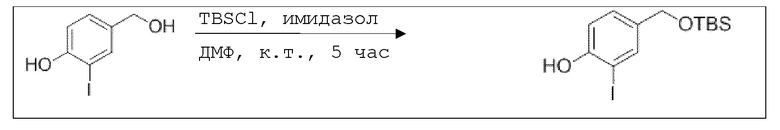
В 10,0 мл-ую колбу Шленка с инертной атмосферой помещали 408 мг (6,00 ммоль, 1,50 экв.) имидазола и 603 мг (4,00 ммоль, 1,00 экв.) трет-бутил(хлор)диметилсилана. После двухкратного вакуумирования и заполнения аргоном добавляли 4,0 мл сухого ДМФ и перемешивали в течение 5 минут при комнатной температуре. Затем добавляли 1,00 г (4,00 ммоль, 1,00 экв.) 4-(гидроксиметил)-2-йодфенола. Перемешивание продолжали в течение 5 час. Суспензию смешивали с 10 мл насыщенного солевого раствора и дважды экстрагировали 10 мл этилацетата. Растворитель удаляли при пониженном давлении, и сырой продукт очищали с помощью флэш-хроматографии на силикагеле (циклогексан/этилацетат=5:1) с получением указанного в заголовке соединения в виде бесцветного масла в количестве 213 мг (852 мкмоль, 21%).
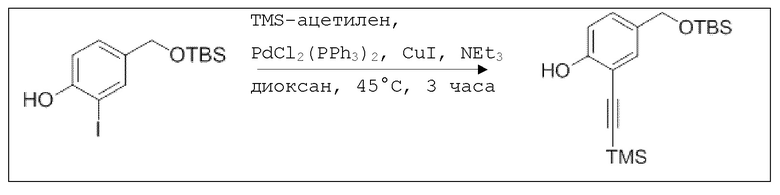
В 25,0 мл-овой трехгорлой колбе с инертной атмосферой растворяли 1,20 г (3,29 ммоль, 1,00 экв.) 4-(((трет-бутилдиметилсилил)окси)метил)-2-йодфенола в 15,0 мл 1,4-диоксана. К этому раствору добавляли 1,83 мл (13,2 ммоль, 4,00 экв.) триэтиламина, 599 мкл (4,28 ммоль, 1,30 экв.) триметилсилилацетилена, 23,0 мг (33,0 мкмоль, 0,01 экв.) бис(трифенилфосфин)палладий(II) дихлорида и 13,0 мг (66,0 мкмоль, 0,02 экв.) иодида меди(I). Смесь нагревали до 45°C. Через 3 часа добавляли 30,0 мл диэтилового эфира и 30,0 мл 0,1 н соляной кислоты. Органический слой промывали 30,0 мл насыщенного раствора гидрокарбоната натрия. Растворитель удаляли при пониженном давлении, и сырой продукт очищали с помощью флэш-хроматографии на силикагеле (циклогексан/этилацетат=20:1) с получением указанного в заголовке соединения в виде масла желтого цвета в количестве 21,09 г (3,27 ммоль, 99%).
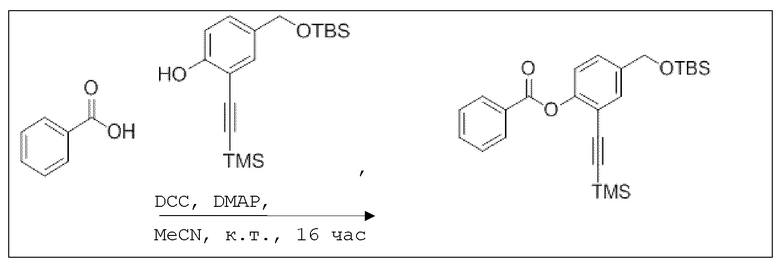
В 50,0 мл-овой трехгорлой колбе с инертной атмосферой растворяли 400 мг (1,20 ммоль, 1,00 экв.) бензойной кислоты в 4,00 мл ацетонитрила. Добавляли 400 мг (1,20 ммоль, 1,00 экв.) 4-(((трет-бутилдиметилсилил)окси)метил)-2-((триметилсилил)этинил)фенола, 15,0 мг (120 мкмоль, 0,10 экв.) 4-(диметиламино)пиридина и 271 мг (1,32 ммоль, 1,10 экв.) дициклогексилкарбодиимида. Через 1 час растворитель удаляли при пониженном давлении, и сырой продукт очищали с помощью флэш-хроматографии на силикагеле (циклогексан/этилацетат=20:1) с получением указанного в заголовке соединения в виде бесцветного масла в количестве 520 мг (1,19 ммоль, 99%).
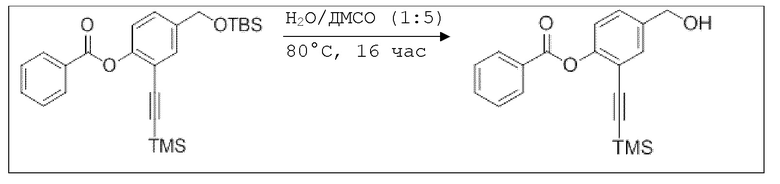
В 500 мл-ой круглодонной колбе с инертной атмосферой растворяли 970 мг (2,21 ммоль, 1,00 экв.) 4-(((трет-бутилдиметилсилил)окси)метил)-2-((триметилсилил)этинил)фенилбензоата в 3,00 мл воды и 15,0 мл диметилсульфоксида. После перемешивания в течение 16 час при 80°C и охлаждения до комнатной температуры добавляли 20,0 мл воды. Смесь дважды экстрагировали 20,0 мл диэтилового эфира. Растворитель удаляли при пониженном давлении, и сырой продукт очищали с помощью флэш-хроматографии на силикагеле (циклогексан/этилацетат=5:1) с получением указанного в заголовке соединения в виде бесцветного масла в количестве 656 мг (2,02 ммоль, 91%).

В 10,0 мл-овой колбе Шленка с инертной атмосферой растворяли 50,0 мг (154 мкмоль, 1,00 экв.) 4-(гидроксиметил)-2-((триметилсилил)этинил)фенилбензоата и 40,0 мг (154 мкмоль, 1,00 экв.) трифенилфосфина в 1,50 мл ацетонитрила и 600 мкл дихлорметана. Смесь охлаждали до -78°C и добавляли 27,0 мг (154 мкмоль, 1,00 экв.) N-бромсукцинимида. Охлаждение прекращали, при этом NBS медленно растворялся. Через 5 мин добавляли 9,00 мг (231 мкмоль, 1,50 экв.) нитрата серебра. Через 2,5 час перемешивания при комнатной температуре осадок отфильтровывали. Растворитель удаляли из фильтрата при пониженном давлении. Неочищенный продукт обрабатывали 293 мкл воды и 1,19 мл ацетона. К этому раствору добавляли 2,76 мг (16,0 мкмоль, 0,1 экв.) нитрата серебра. Через 72 час перемешивания при комнатной температуре добавляли 15,0 мл насыщенного солевого раствора. Смесь экстрагировали 2×15,0 мл дихлорметана. Растворитель удаляли из экстракта при пониженном давлении, и сырой продукт очищали с помощью флэш-хроматографии на силикагеле (циклогексан/этилацетат=5:1) с получением указанного в заголовке соединения в виде твердого вещества в количестве 20,0 мг (67,0 мкмоль, 41%) за две стадии.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ ТИРОЗИНКИНАЗЫ БРУТОНА | 2013 |
|
RU2619465C2 |
| Ингибитор CDK4/6 | 2017 |
|
RU2747311C2 |
| НИЗКОМОЛЕКУЛЯРНЫЕ КОНЪЮГАТЫ ДЛЯ ВНУТРИКЛЕТОЧНОЙ ДОСТАВКИ БИОЛОГИЧЕСКИ АКТИВНЫХ СОЕДИНЕНИЙ | 2011 |
|
RU2629957C2 |
| НИЗКОМОЛЕКУЛЯРНЫЕ КОНЪЮГАТЫ ДЛЯ ВНУТРИКЛЕТОЧНОЙ ДОСТАВКИ НУКЛЕИНОВЫХ КИСЛОТ | 2011 |
|
RU2582235C2 |
| 2,2,2-ТРИФТОРЭТИЛ-ТИАДИАЗИНЫ | 2015 |
|
RU2692102C2 |
| ПРОИЗВОДНОЕ РЕЗОРЦИНА В КАЧЕСТВЕ ИНГИБИТОРА HSP90 | 2016 |
|
RU2697703C2 |
| ПИРАЗОЛПИРИМИДИНОВОЕ ПРОИЗВОДНОЕ И ЕГО ПРИМЕНЕНИЕ | 2016 |
|
RU2735522C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ KIT- И PDGFRA-ОПОСРЕДОВАННЫХ ЗАБОЛЕВАНИЙ | 2020 |
|
RU2817354C2 |
| ЗАМЕЩЁННЫЕ ИНДОЛЬНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ПОНИЖАЮЩИХ РЕГУЛЯТОРОВ РЕЦЕПТОРОВ ЭСТРОГЕНА | 2017 |
|
RU2722441C2 |
| ПРОИЗВОДНЫЕ ПИРРОЛИДИНА В КАЧЕСТВЕ АГОНИСТОВ PPAR | 2017 |
|
RU2711991C1 |
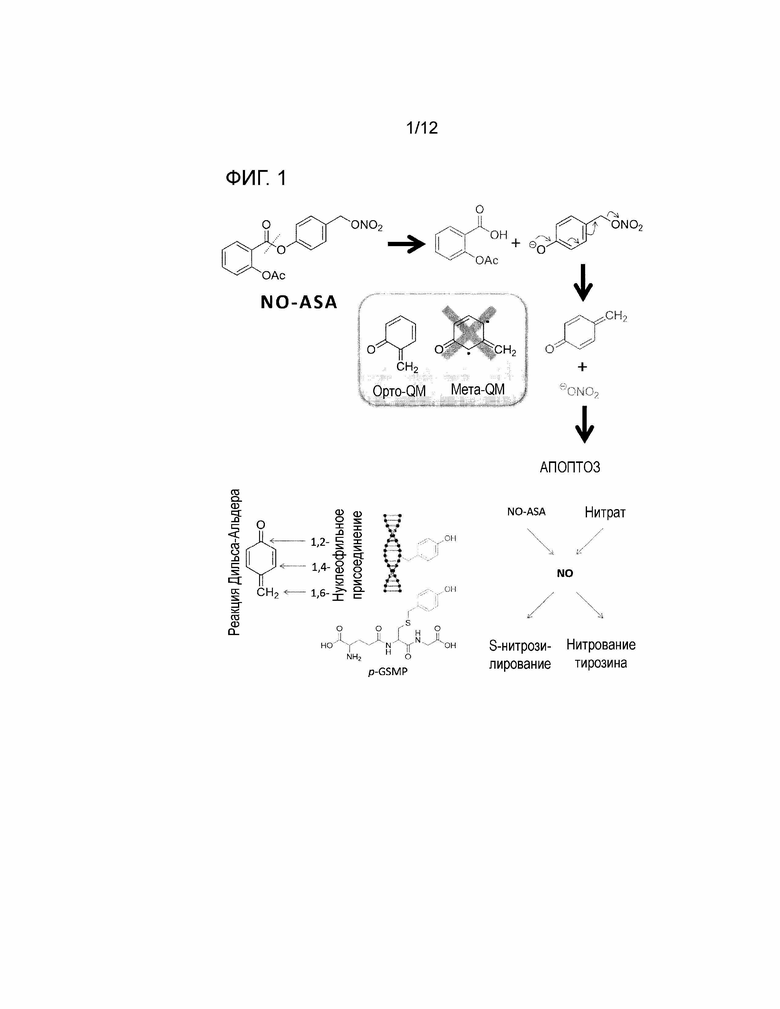
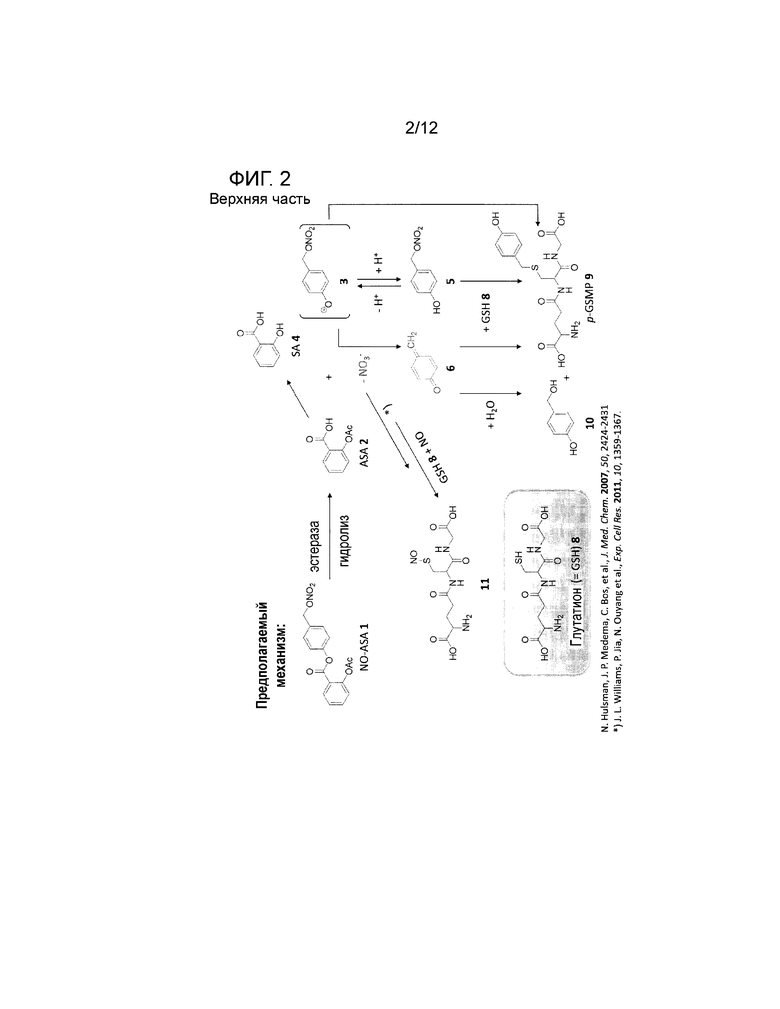
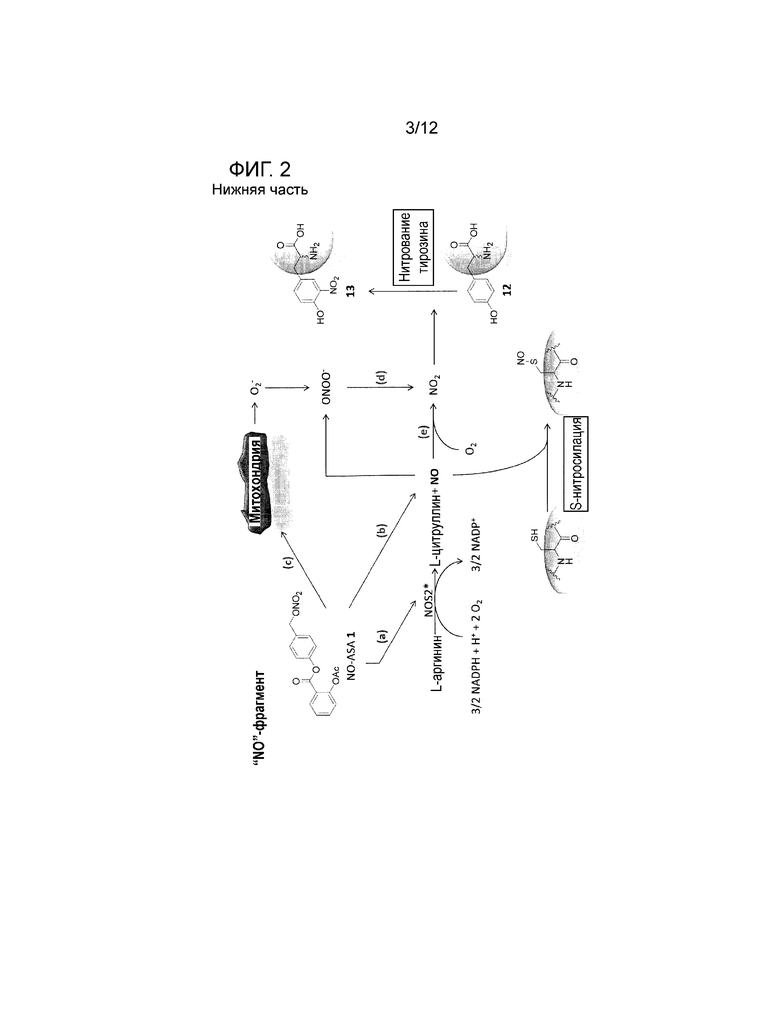
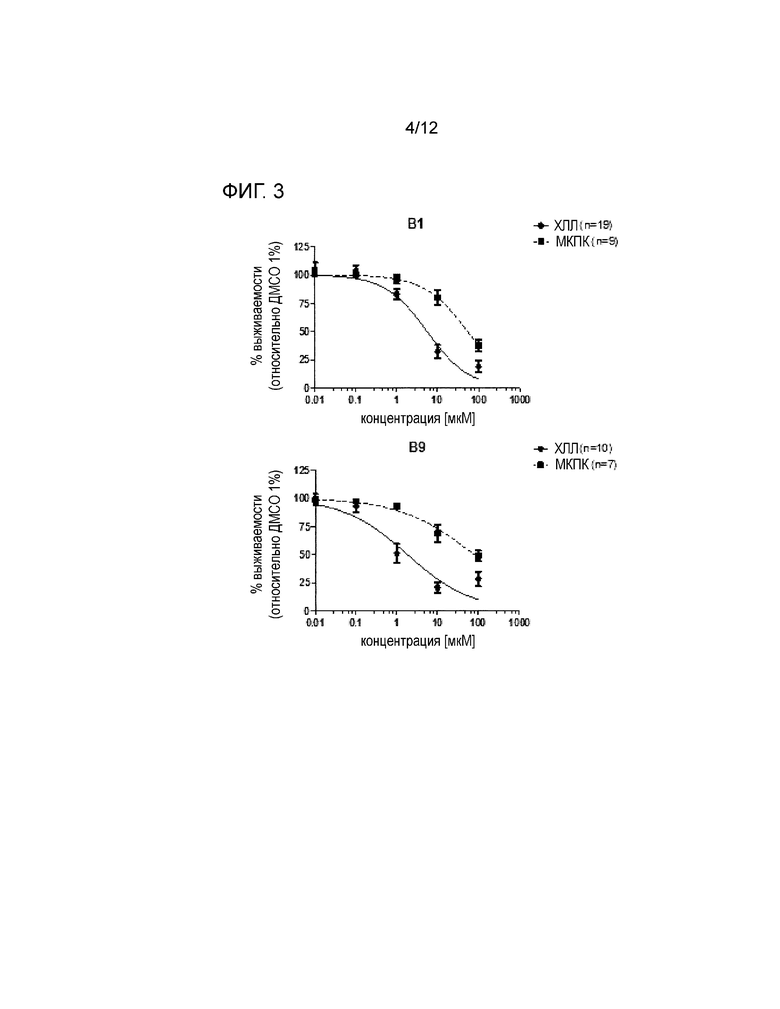
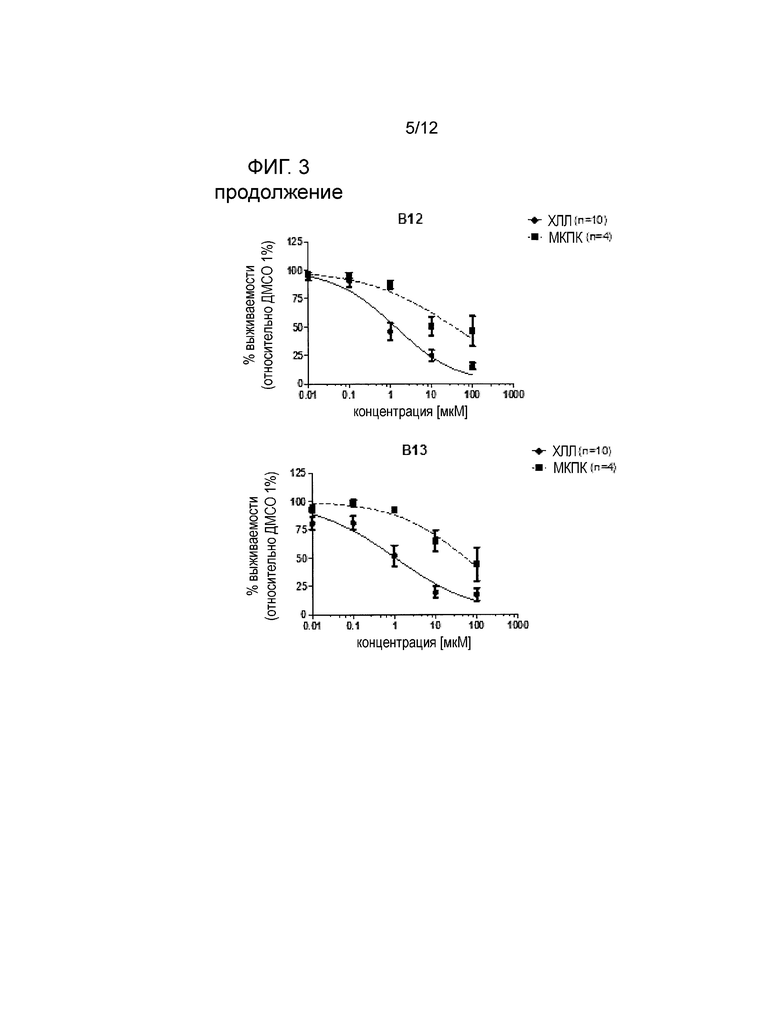
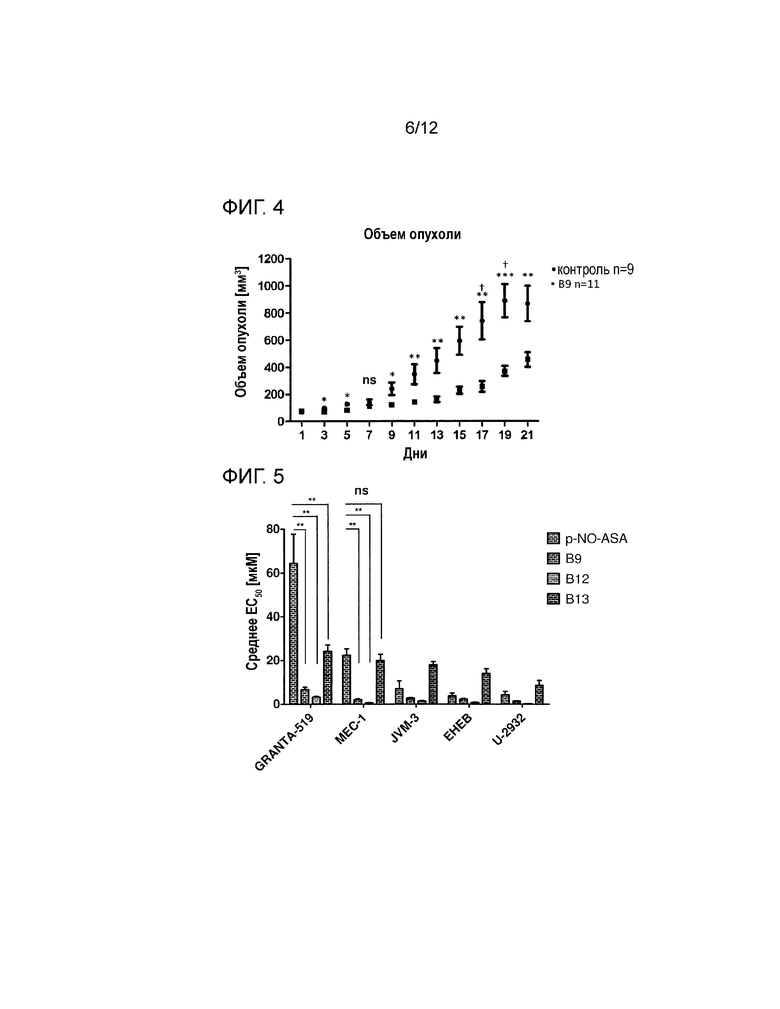
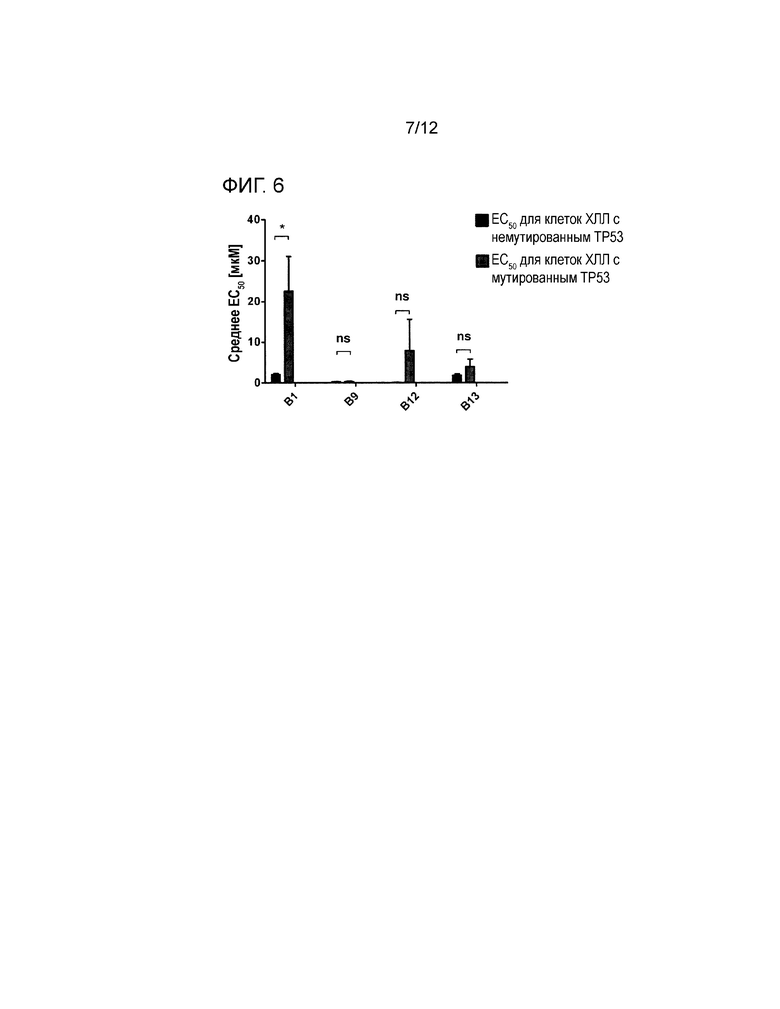
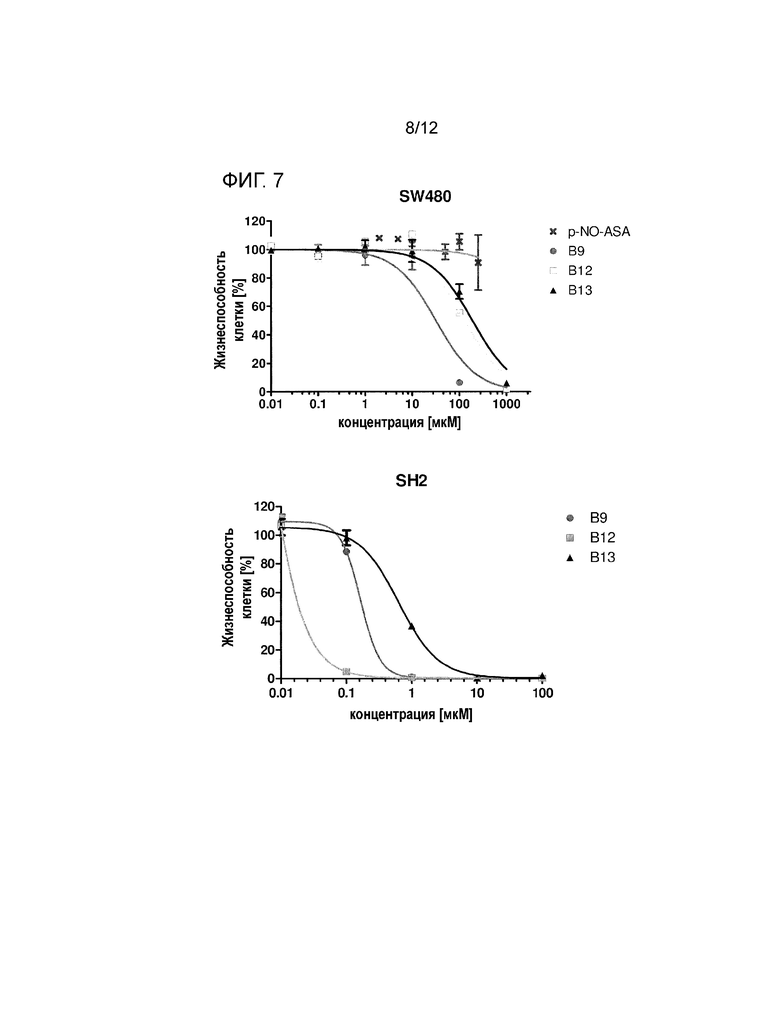
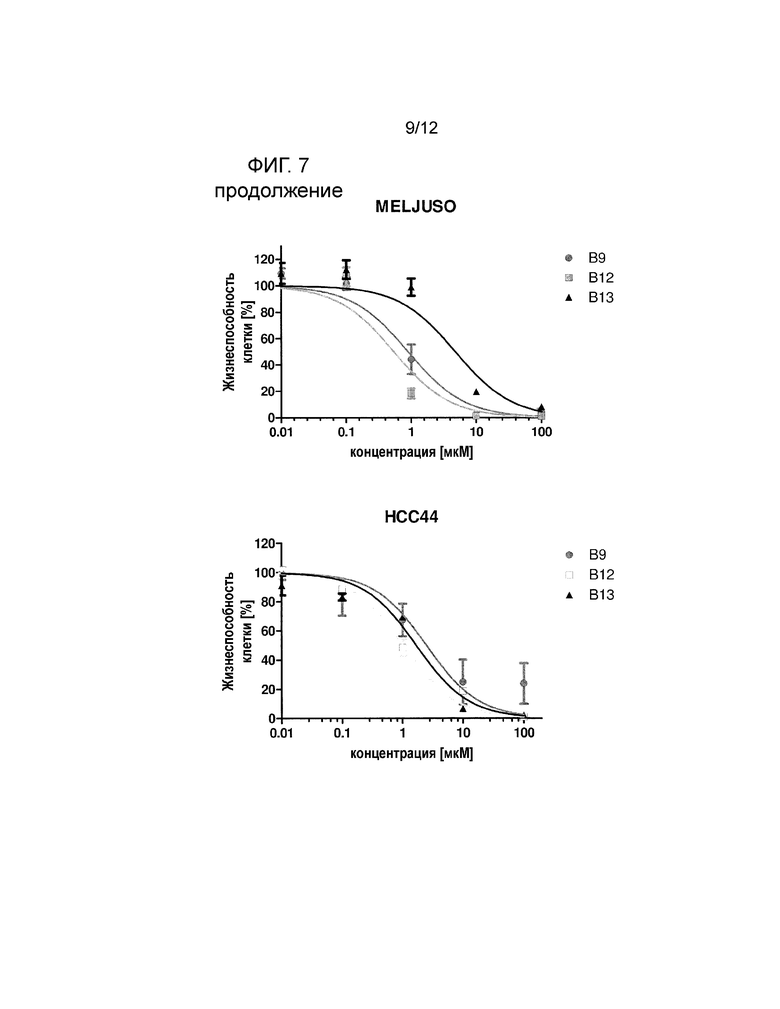
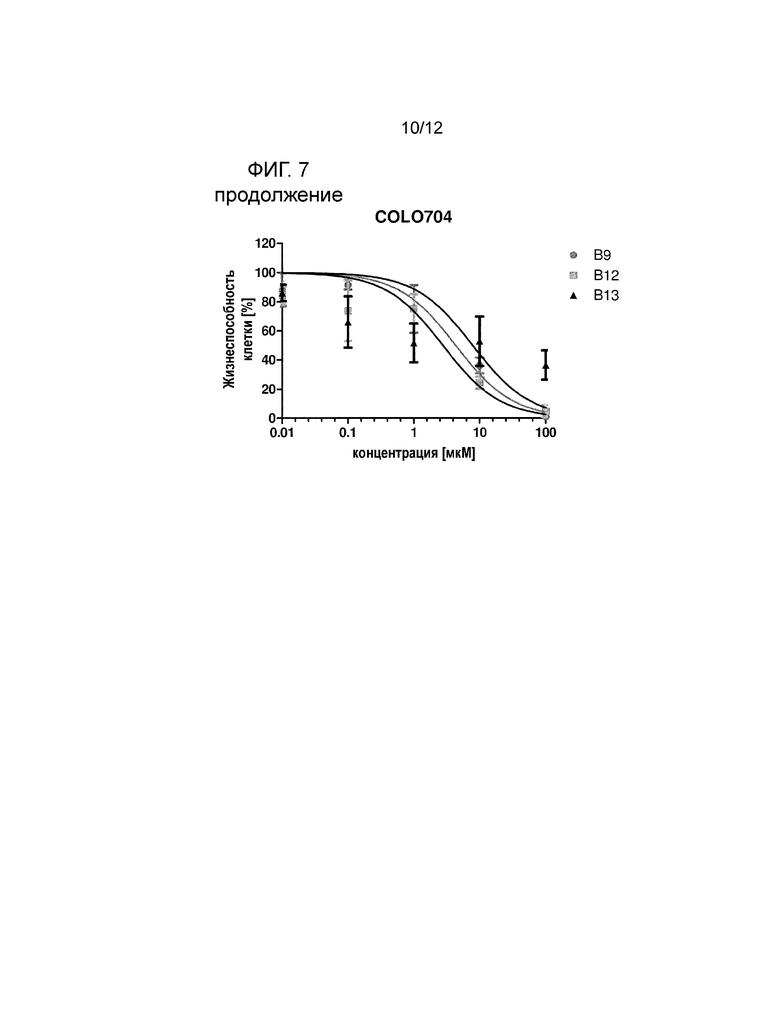
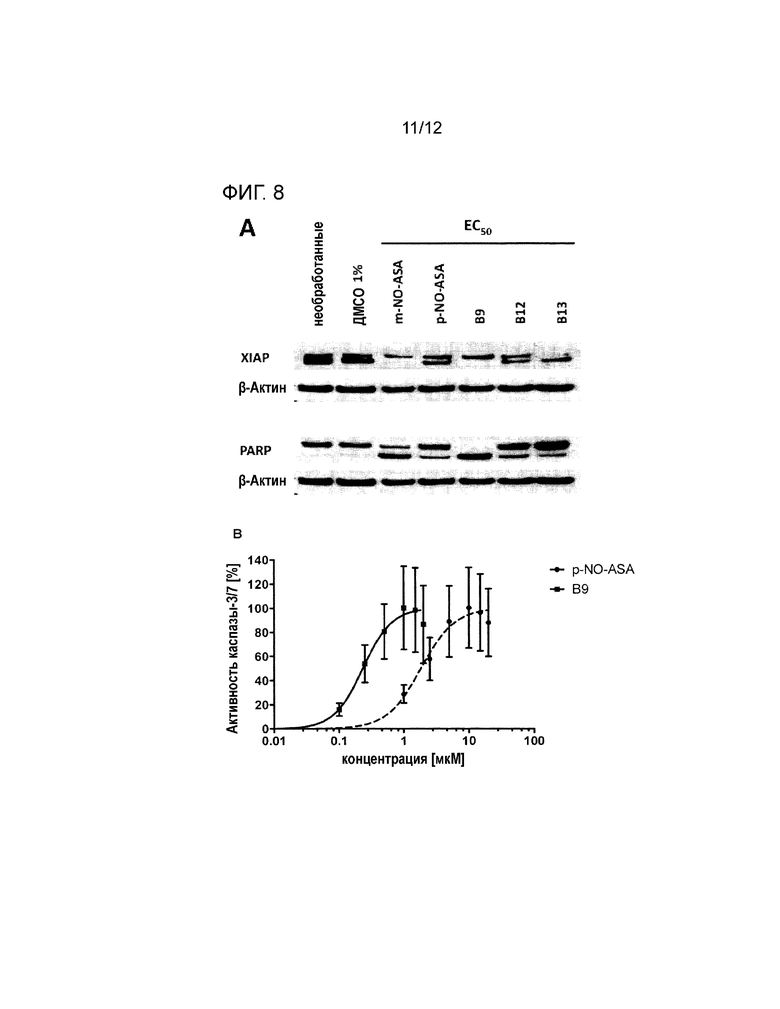
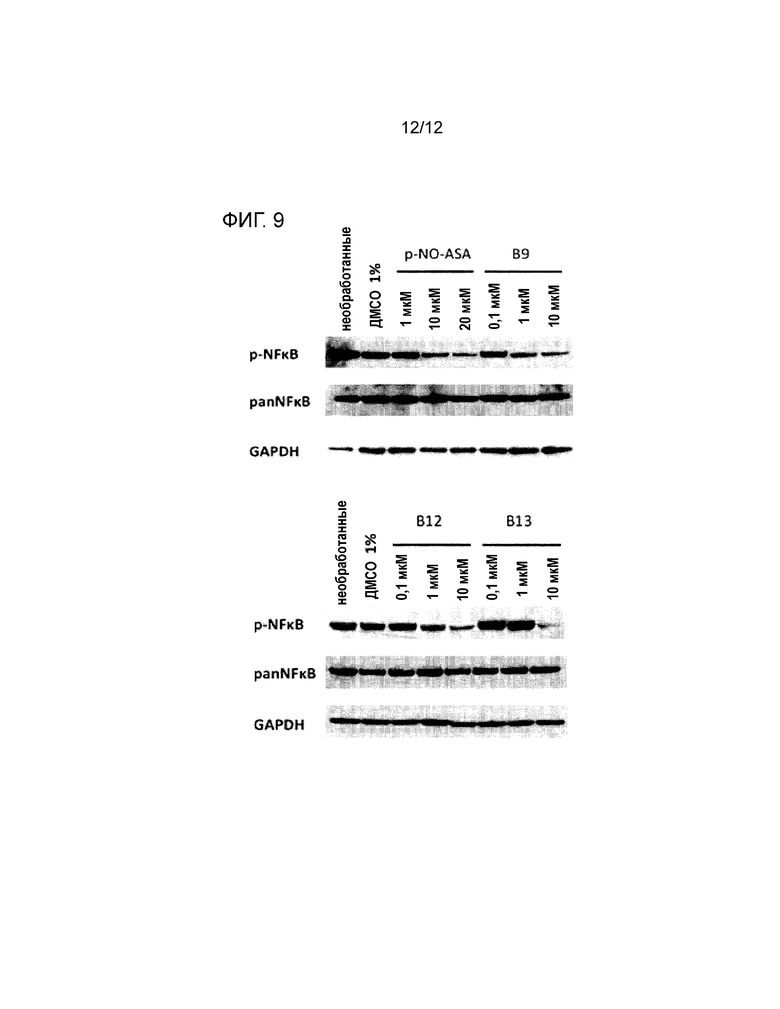
Настоящее изобретение относится к соединениям и их фармацевтически приемлемым солям, которые применяются в лечении неопластических заболеваний или расстройств, фармацевтической композиции, содержащей такое соединение, и способу получения этого соединения. Лекарственное средство для лечения неопластического заболевания или (дис)пролиферативного расстройства, содержащее соединение, выбранное из группы, состоящей из: 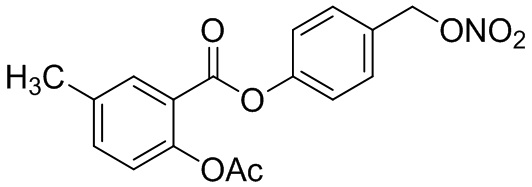 ,
, 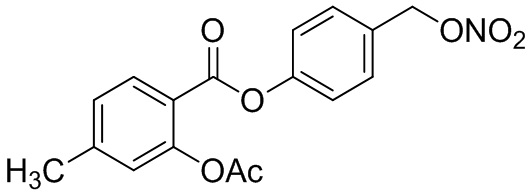 ,
, 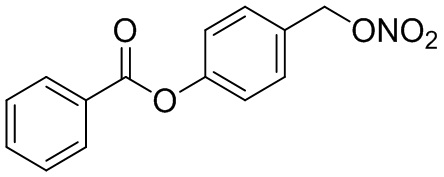 ,
, 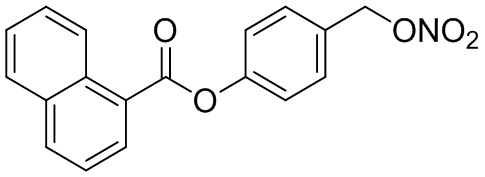 ,
, 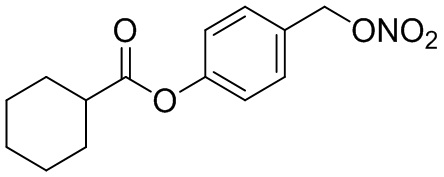 . Лекарственное средство для лечения злокачественной опухоли, содержащее соединение
. Лекарственное средство для лечения злокачественной опухоли, содержащее соединение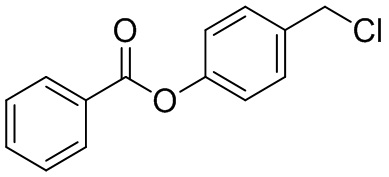 . 7 н. и 7 з.п. ф-лы, 9 ил., 2 табл., 8 пр.
. 7 н. и 7 з.п. ф-лы, 9 ил., 2 табл., 8 пр.
1. Лекарственное средство для лечения неопластического заболевания или (дис)пролиферативного расстройства, содержащее соединение, выбранное из группы, состоящей из:
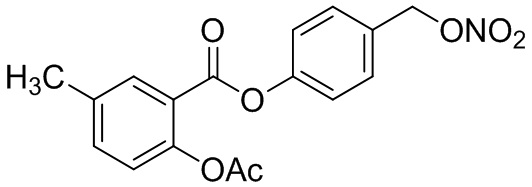 ,
, 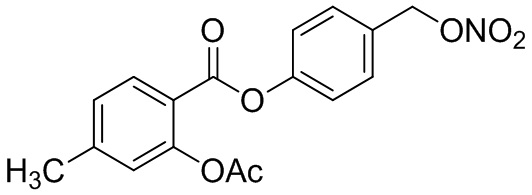 ,
, 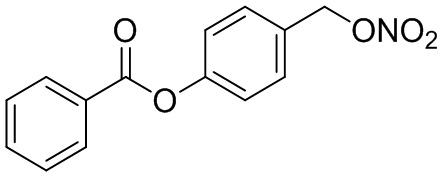 ,
, 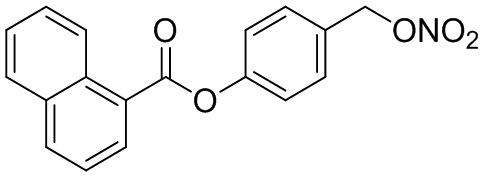 ,
, 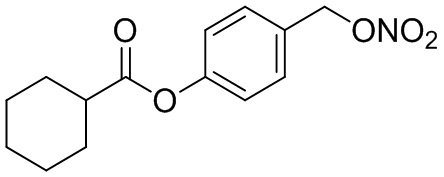 ,
,
или его фармацевтически приемлемую соль.
2. Лекарственное средство для лечения злокачественной опухоли, содержащее соединение
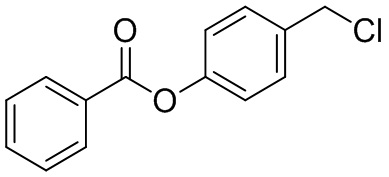 .
.
3. Лекарственное средство по п. 2, где злокачественную опухоль выбирают из группы, состоящей из злокачественной опухоли простаты, поджелудочной железы, легких, кожи, молочной железы, мочевого пузыря, толстой кишки и крови.
4. Лекарственное средство по п. 2, где злокачественная опухоль представляет собой злокачественную опухоль яичников.
5. Лекарственное средство по п. 2, где злокачественная опухоль представляет собой хронический лимфоцитарный лейкоз.
6. Применение соединения, выбранного из группы, состоящей из:
, , , , ,
или его фармацевтически приемлемой соли,
для лечения неопластического заболевания или (дис)пролиферативного расстройства.
7. Применение по п. 6, где заболевание или расстройство представляет собой злокачественную опухоль.
8. Применение соединения
для лечения злокачественной опухоли.
9. Применение по любому из пп. 6-8, где злокачественную опухоль выбирают из группы, состоящей из злокачественной опухоли простаты, поджелудочной железы, легких, кожи, молочной железы, мочевого пузыря, толстой кишки и крови.
10. Применение по любому из пп. 6-8, где злокачественная опухоль представляет собой злокачественную опухоль яичников.
11. Применение по любому из пп. 6-8, где злокачественная опухоль представляет собой хронический лимфоцитарный лейкоз.
12. Фармацевтическая композиция для лечения неопластического заболевания или (дис)пролиферативного расстройства, содержащая эффективное количество соединения, выбранного из группы, состоящей из
, , , , ,
или его фармацевтически приемлемую соль, в смеси, по крайней мере, с одним фармацевтически приемлемым носителем.
13. Фармацевтическая композиция для лечения злокачественной опухоли, содержащая эффективное количество соединения или его фармацевтически приемлемую соль, в смеси, по крайней мере, с одним фармацевтически приемлемым носителем.
14. Способ получения соединения, выбранного из группы, состоящей из
, , , , ,
включающий образование сложного эфира карбоновой или сульфоновой кислоты и взаимодействие активированной алифатической или ароматической карбоновой или сульфоновой кислоты с соединением в соответствии с формулой [формула D]:
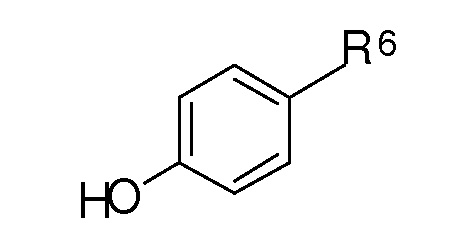 ,
,
где R6 представляет собой метил-X или формил, X представляет собой нитроокси.
| R | |||
| RAZAVI ET AL | |||
| "Nitric oxide-donating acetylsalicylic acid induces apoptosis in chronic lymphocytic leukaemia cells and shows strong antitumour efficacy in vivo", CLINICAL CANCER RESEARCH, vol | |||
| Печь для сжигания твердых и жидких нечистот | 1920 |
|
SU17A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| WO 2005065361 A2, 21.02.2005 | |||
| N | |||
| HULSMAN ET AL.: "Chemical insights in the concept of hybrid | |||

