В настоящем изобретении предложены фармацевтические композиции факторов свертывания крови для подкожного введения.
Как правило, факторы свертывания крови предложены в виде фармацевтических композиций для внутривенного введения. Композиции основаны на применении активного белка, часто конъюгированного с полимером, таким как полиэтиленгликоль (ПЭГ) для увеличения времени полувыведения из кровяного русла. Следовательно, внутривенное введение конъюгированных с ПЭГ (ПЭГилированных) факторов свертывания крови в качестве медикаментозных средств общепризнанно и широко распространено. Также известны липосомальные составы очищенных (т.е., неконъюгированных и не содержащих модификации) факторов свертывания крови, таких как фактор VIII и фактор IX, см., например, WO 95/04524.
Фармацевтические композиции, содержащие фактор VIII и липосомы, модифицированные наличием полиэтиленгликоля, описаны в источнике WO 99/55306, согласно которому, фактор свертывания крови не инкапсулирован в липосому. Однако указанные составы подготовлены для внутривенного введения. В источнике WO 2004/091723 описаны дополнительные составы, содержащие другие белки, при этом указанные белки включают факторы свертывания крови. Сообщается, что указанные белки нековалентно связываются с липосомами за счет взаимодействия с полиэтиленгликолем, присутствующим на поверхности липосом. Однако составы с факторами свертывания крови, полученные в соответствии с примерами, приводимыми в указанном документе, также предназначены для внутривенного введения.
Другие примеры составов с факторами свертывания крови - фактором VIII и фактором VIIa, присутствующими в конъюгированном с ПЭГ состоянии, приведены в источниках WO 2011/135307 и WO 2011/135308, соответственно, где фактически полученные составы предназначались для внутривенного введения. В источнике WO 2013/156488 также описана лекарственная форма модифицированных терапевтических агентов, включающих факторы свертывания крови, такие как фактор VIII (FVIII) и фактор VIIa (FVIIa), для подкожного введения.
Было показано, что фактор VIII способен связываться с ПЭГилированными липосомами, т.е., указанный фактор свертывания крови не инкапсулирован внутри липосомы (Baru et al Thromb. Haemost., 93, pages 1061-1068, (2005)). Однако составы с FVIII получали только в виде составов для внутривенного введения.
В дальнейших исследованиях Peng et al in The AAPS Journal, 14(1), стр. 25-42 (2011), раскрывается альтернативный подход, основанный на FVIII, инкапсулированном в липосомы, которые впоследствии ПЭГилируются путем пассивного добавления ПЭГ к липосомам после получения. В одном эксперименте, проведенном Peng et al, липосомальный состав вводили подкожно (п/к) для изучения иммуногенности, но предложений по терапевтическому применению такого введения не было. В работе Peng et al также есть конкретная ссылка на статью Baru et al (2005), и утверждение, что при способе согласно Baru et al «фактор FVIII подвергался действию компонентов плазмы, таких как протеазы и IgG (иммуноглобулин G)». Липосомы, полученные в соответствии со способом Baru et аl (2005), содержащие рекомбинантный фактор VIII, вводили субъектам внутривенно (Spira et al Blood, 108 (12), стр. 3668-3673 (2012)).
Современные методологии получения составов с факторами свертывания крови для введения подразумевают внутривенный путь введения. Это проблематично, поскольку пациент неизбежно получает большую разовую дозу активного агента в несколько моментов времени, что приводит к неравномерному терапевтическому уровню агента в крови пациента.
Следовательно, существует потребность в фармацевтической композиции фактора свертывания крови, которая может обеспечить безопасное и эффективное дозирование.
Согласно настоящему изобретению предложена фармацевтическая композиция для подкожного введения, содержащая фактор свертывания крови и коллоидную частицу, содержащую приблизительно 0,5-20 мольных процентов амфифильного липида, в виде производного с биосовместимым гидрофильным полимером, причем указанный фактор свертывания крови не инкапсулирован в указанную коллоидную частицу.
Указанные коллоидные частицы могут быть по существу нейтральными и полимер может по существу не нести суммарного (результирующего) заряда. Коллоидные частицы могут иметь средний диаметр частиц от приблизительно 0,03 до приблизительно 0,4 микрона (мкм), например, средний диаметр частиц приблизительно 0,1 микрон (мкм). Средний диаметр частиц в данном диапазоне может увеличивать время пребывания частицы в кровяном русле in vivo и препятствовать ее всасыванию ретикуло-эндотелиальной системой (РЭС).
Фактор свертывания крови может быть выбран из группы, состоящей из фактора VIII, фактора VIIa, фактора VII, фактора IX, фактора X, фактора Ха, фактора XI, фактора V,
фактора XII, фактора XIII, фактора фон Виллебранда (vWF), протромбина или белка С и/или их. фрагментов. При приготовлении фармацевтической композиции фактор свертывания крови можно применять в лиофилизированной форме.
В случае, когда указанная композиция содержит фрагмент фактора свертывания крови, фактор может, соответственно, представлять собой активный фрагмент фактора, причем такой фрагмент сохраняет биологическую активность или по существу такую же биологическую активность, что и нативный фактор свертывания крови. Например, одним таким активным фрагментом является процессированная по В-домену последовательность фактора VIII, показанная на Фигуре 1. Другие фрагменты включают фрагмент фактора VII, показанный на Фигуре 2, фрагмент В-цепи тромбина, показанный на Фигуре 3. Фрагмент фактора XII, показанный на Фигуре 4, и D'D3-домены vWF.
Также возможно, что указанная композиция содержит и нативный фактор свертывания крови, и его фрагмент.
Фармацевтическая композиция согласно настоящему изобретению может дополнительно содержать другое терапевтически активное соединение или молекулу, например, противовоспалительный препарат, анальгетик или антибиотик, или другой фармацевтически активный агент, который может способствовать или усиливать активность фактора VIIa, фактора VII, фактора VIII, фактора IX, фактора X, фактора Ха, фактора XI, фактора V, фактора XIII, фактора фон Виллебранда (vWF), протромбина или белка С, или их фрагмента, например, другого фактора свертывания крови.
Термины фактор VIIa (FVIIa) и фактор VII (FVII) также применяются взаимозаменяемо, если контекст не указывает на другое. FVIII применяют в качестве сокращения для фактора VIII, FIX применяют в качестве сокращения для фактора IX, и т.д. для всех факторов свертывания крови, описываемых в настоящей заявке с соответствующими поправками.
Фактор свертывания (коагуляции) крови может быть получен из любого подходящего источника и может быть рекомбинантным белком, полученным по технологии рекомбинантных ДНК с применением молекулярно-биологических способов, или синтезирован химически, или полученным трансгенным способом в молоке млекопитающих, или указанный фактор может быть выделен из природного источника (например, очищен из плазмы крови). Соответственно, указанный фактор является фактором свертывания крови млекопитающих, например, фактор свертывания крови человека. Фактор свертывания крови может также называться фактором свертывающей системы крови.
Как обсуждалось выше, все факторы свертывания крови характеризуются помимо прочего свойством адгезии к поверхности. Это свойство является необходимым для функционирования каскада свертывания, для которого требуется, чтобы ферменты и кофакторы прикреплялись к другим участникам каскада, к поверхности тромбоцитов и к ткани в очаге повреждения. Действительно, особенно важно, чтобы сгусток крови оставался в очаге поражения и не смещался, вызывая опасный тромбоз. Данное свойство представляет проблему для получения составов лекарственных препаратов, поскольку факторы свертывания крови, такие как VIIa, VIII и IX, будут чрезмерно прикрепляться к любым стеклянным и пластиковым поверхностям. На практике степень прикрепления ослабляют применением полисорбата (например, Tween® 80) в избытке.
Коллоидные частицы согласно настоящему изобретению обычно находятся в форме липидных везикул или липосом, которые хорошо известны в данной области техники. Упоминания коллоидных частиц в настоящем описании включают липосомы и липидные везикулы, если контекст не указывает на другое.
В коллоидных частицах указанным амфифильным липидом может быть фосфолипид из природных или синтетических источников. Указанный амфифильный липид может содержать приблизительно от 0,5 до приблизительно 20 мольных процентов (%) частиц, например, приблизительно от 1 до 20%, или приблизительно от 1 до 6%, или приблизительно 3%.
Подходящие примеры таких амфифильных липидов включают фосфатидилэтаноламин (ФЭ), карбамат-сшитый незаряженный липополимер или аминопропандиол дистеароил (ДС) или их смесь. Подходящим примером фосфатидилэтаноламина (ФЭ) может быть 1,2-дистеароил-sn-глицеро-3-фосфоэтаноламин (ДСФЭ). Назначение биосовместимого гидрофильного полимера состоит в стерической стабилизации малых моноламеллярных везикул (ММВ), за счет чего предотвращается слияние везикул in vitro, и везикулы могут избегать всасывания в РЭС in vivo.
Коллоидные частицы могут дополнительно содержать второй амфифильный липид, полученный либо из природных, либо из синтетических источников. Вторым амфифильным липидом может быть фосфатидилхолин (ФХ). Подходящим примером фосфатидилхолина (ФХ) может быть пальмитоил-олеоил-фосфатидилхолин (ПОФХ).
Согласно такому варианту реализации фармацевтическая композиция может состоять из коллоидных частиц, которые содержат пальмитоил-олеоил-фосфатидилхолин (ПОФХ) и 1,2-дистеароил-sn-глицеро-3-фосфоэтаноламин (ДСПЭ) в соотношении ПОФХ:ДСПЭ от 85-99:15-1. В некоторых случаях соотношение ПОФХ:ДСПЭ может быть в диапазоне 90-99:10-1. Согласно одному варианту реализации соотношение ПОФХ:ДСПЭ может быть 97:3.
Согласно альтернативному варианту реализации фармацевтическая композиция согласно настоящему изобретению может дополнительно содержать холестерин.
Биосовместимый полимер может иметь молекулярную массу от приблизительно 500 до приблизительно 5000 Дальтон, например, приблизительно 2000 Дальтон.
Биосовместимый гидрофильный полимер, применяемый согласно настоящему изобретению, может быть выбран из группы, состоящей из полиалкилэфиров, полимолочных кислот и полигликолевых кислот. Биосовместимый гидрофильный полимер может быть полиэтиленгликолем (ПЭГ). Полиэтиленгликоль, применяемый в составе композиций согласно настоящему изобретению, может иметь молекулярную массу от приблизительно 500 до приблизительно 5000 Дальтон, например, он может иметь молекулярную массу приблизительно 1000, 2000 или 3000 Дальтон. Согласно одному варианту реализации молекулярная масса ПЭГ может составлять 2000 Дальтон. Полиэтиленгликоль может быть разветвленным или неразветвленным.
Примером подходящего амфифильного липида может быть 1,2-дистеарол-sn-глицеро-3-фосфоэтаноламин-N-[поли-(этиленгликоль)]. Если ПЭГ имеет молекулярную массу 2000 Дальтон, производное амфифильного липида может быть описано как 1,2-дистеарол-sn-глицеро-3-фосфоэтаноламин-N-[поли-(этиленгликоль)-2000] (ДСПЭ-ПЭГ 2000).
Фармацевтическая композиция может содержать любое подходящее вспомогательное вещество, буфер и/или адъювант. Примеры таких вспомогательных веществ, буферов и/или адъювантов включают фосфатно-солевой буфер (ФСБ), фосфат калия, фосфат натрия и/или цитрат натрия. Другие биологические буферы могут включать PIPES, MOPS и др.
Подходящие значения рН для фармацевтической композиции включают любые общепринятые значения рН для введения in vivo, такие как, например, рН 5,0 - рН 9,0, применимо от рН 6,8 до рН 7,2, или рН 7,0.
Авторы настоящего изобретения неожиданно показали, что составы с факторами свертывания крови в ассоциации с коллоидными частицами (липосомы), полученными путем соединения с биосовместимым полимером, могут успешно вводиться подкожно и достигать терапевтически эффективной дозы для фактора свертывания крови у субъекта, страдающего гемофилией. Соответственно, указанным биосовместимым полимером является полиэтиленгликоль.
Согласно примерам настоящего изобретения ПЭГ включают в липосомы во время образования везикул, перед соединением с фактором свертывания крови. Предполагается, что специфические аминокислотные последовательности в факторах свертывания крови могут связываться нековалентно функциональными группами карбамата на молекулах ПЭГ на наружной стороне липосомы.
Хотя в работе Peng et al (2011) есть упоминание о введении липосомального FVIII мышам подкожно (п/к), очевидно, что данный путь введения применяли только для оценки относительной иммуногенности и не рассматривали как перспективный вариант для лечения. Чтобы подчеркнуть это, авторы явно утверждают в начале страницы 41, что «FVIII-PI/ПЭГ вводился внутривенно, т.е. применяемым в клинике путем введения FVIII.» Иными словами, Peng et аl не раскрывают и даже не предлагают подкожное введение в качестве перспективного варианта лечения. Кроме того, авторы работы Peng et al (2011) также утверждают на стр. 40, что их подход «явно отличается» от подхода Baru et al (2005). Самые последние публикации в данной области, следовательно, представляют собой взаимоисключающие и разные альтернативы настоящему изобретению. Как обсуждалось выше, фактор(ы) свертывания крови не инкапсулированы в липосому, так что при желании можно применять липосомы меньшего размера, которые обладают большим временем полувыведения in vivo, поскольку они не удаляются ретикуло-эндотелиальной системой (РЭС). Активность факторов свертывания крови в составе лекарственных форм не ухудшается, как показано в разделе «Примеры», и in vitro, и сразу после инъекции in vivo обнаруживается полная активность.
Факторы свертывания крови взаимодействуют нековалентно с цепями полимеров на наружной поверхности липосом, и для активации цепей полимеров никакой химической реакции не проводится, в отличие от композиции, раскрываемой в ЕР-А-0689428. Природа взаимодействия между фактором свертывания крови и липосомой, полученной путем соединения с биосовместимым гидрофильным полимером, может подразумевать нековалентный механизм, такой как ионные взаимодействия, гидрофобные взаимодействия, водородные связи и вандервальсовы связи (Arakawa, Т. and Timasheff, S.N., Biochemistry 24: 6756-6762 (1985); Lee, J.C. and Lee, L.L.Y., J. Biol. Chem. 226: 625-631 (1981)). Примером такого полимера служит полиэтиленгликоль (ПЭГ).
Для получения образующих везикулы липидов, полученных путем соединения с гидрофильными полимерами, можно применять целый ряд известных реакций конденсации. Например, полимер (такой как ПЭГ), можно преобразовать в липид, такой как фосфатидилэтаноламин (ПЭ) при помощи цианурхлоридной группы. В качестве альтернативы кеппированный ПЭГ можно активировать при помощи связующего реагента карбонилдиимидазола, чтобы получить активированное имидазольное соединение. Карбамат-сшитое соединение можно получить путем реакции между терминальной гидроксильной группой МПЭГ (метокси-ПЭГ) с p-нитрофенилкарбнатом. Затем данный продукт реагировал с 1-амино-2,3-пропандеолом с образованием промежуточного карбамата. Гидроксильные группы диола ацетилируют с образованием конечного продукта. Аналогичный синтез с применением глицерина вместо 1-амино-2,3-пропандиола можно применять для получения карбонат-сшитого продукта, как описано в WO 01/05873. Другие реакции хорошо известны и описаны, например, в US 5013556.
Коллоидные частицы (липосомы) можно классифицировать по разным параметрам. Например, когда в качестве параметров применяют размер и число ламелл (структурные параметры), можно выделить три основных типа липосом: мультиламеллярные везикулы (МЛВ), малые одноламеллярные везикулы (MOB) и крупные одноламеллярные везикулы (КОВ).
МЛВ представляют собой разновидность, которая образуется спонтанно при гидратации высушенных фосфолипидов выше температуры их перехода из геля в жидкий кристалл (Tm). Размер МЛВ неоднороден, и их структура напоминает луковую кожуру из переменных, концентрических слоев жидкости и липидов.
MOB образуются из МЛВ при обработке ультразвуком или другими способами, такими как экструзия, гомогенизация при высоком давлении или смешивание с большой величиной сдвига, и являются однослойными. Они представляют собой самую маленькую разновидность с высоким отношением поверхность-объем, и, следовательно, имеют наименьший объем захвата водного пространства на массу липида.
Третий тип липосом - КОВ - обладает наибольшим водным компартментом и одним (одноламеллярные) или только несколькими (олиголамеллярные) липидными слоями. Более подробная информация раскрывается у D. Lichtenberg and Y. Barenholz, в ''Liposomes: Preparation, Characterization, and Preservation, in Methods of Biochemical Analysis'', Vol. 33, pp. 337-462 (1988).
В настоящей заявке термин «загрузка» относится к любой форме взаимодействия биополимерных веществ, которые требуется загрузить, например, к такому взаимодействию, как инкапсулирование, адгезия (к внутренней или наружной стенке везикулы) или запечатывание в стенку с экструзией или без нее биополимерных веществ.
В настоящей заявке, как указывалось выше, термин «липосома» относится к коллоидным частицам и должен включать в себя все сферы или везикулы из любых амфифильных соединений, которые могут спонтанно или неспонтанно образовывать везикулы, например, фосфолипиды, где по меньшей мере одна ацильная группа заменена на комплекс эфира фосфорной кислоты. Липосомы могут находиться в любом физическом состоянии от стекловидного состояния до жидкого кристалла. Подходит большинство триацилглицеридов, и наиболее распространенные фосфолипиды, подходящие для применения в настоящем изобретении, включают лектины (также называются фосфатидилхолины (ФХ)), которые представляют собой смеси диглицеридов стеариновой, пальметиновой и олеиновой кислот, сшитых с холиновым эфиром фосфорной кислоты. Лектины обнаруживаются у всех животных и растений, например, в яйцах, соевых бобах и в тканях животных (головной мозг, сердце и т.п.), и также могут быт получены путем синтеза. Источник фосфолипида или способ его синтеза не критичны, можно применять любой существующий в природе или синтетический фосфатид.
Примеры специфических фосфатидов включают L-а-(дистеарол) лектин, L-а-(дипальмитоил) лектин, L-a-фосфатидную кислоту, L-а-(дилауроил)-фосфатидную кислоту, L-а-(димиристоил) фосфатидную кислоту, L-а-(диолеил) фосфатидную кислоту, DL-а-(ди-пальмитоил) фосфатидную кислоту, L-а-(дистеароил) фосфатидную кислоту и разные типы L-a-фосфатидилхолинов, получаемых из головного мозга, печени, сердца, яичного желтка, соевых бобов и т.п., или синтезируемых, и их соли. Другие подходящие модификации включают контролируемое перекисное окисление жирных ацильных кросс-линкеров жирных ацильных остатков в фосфатидилхолинах (ФХ) и цвиттер-ионных амфифильных соединениях, которые образуют мицеллы сами или при их смешивании с ФХ, такими как алкильные аналоги ФХ.
Фосфолипиды могут варьировать по своей чистоте, а также могут быть гидрогенизированным эфиром, полным или частичным. Гидрогенизация снижает уровень нежелательного перекисного окисления, а также модифицирует и контролирует температуру фазового перехода гель-жидкость/кристалл (Тm), которая влияет на упаковку и текучесть.
Липосомы могут изготавливаться в соответствии с требованиями любого специфического резервуара, содержащего разные биологические жидкости, сохранять свою стабильность без агрегации или хроматографического разделения, и сохранять хорошую дисперсность и суспендирование в жидкости для инъекций. Текучесть in situ изменяется в зависимости от состава, температуры, солености, присутствия двухвалентных ионов и белков. Липосомы можно применять с другими растворителями или поверхностно-активными веществами или без них.
Как правило, подходящие липиды могут содержать состав ацильных цепей, который является характерным, по меньшей мере в том, что касается температуры перехода (Тm) компонентов с ацильными цепями в ФХ яйца или соевых бобов, т.е. одна цепь насыщенная и одна ненасыщенная, или обе цепи ненасыщенные. Однако не исключается возможность применения двух насыщенных цепей.
Указанные липосомы могут содержать другие липидные компоненты, при условии, что они не приводят к нестабильности и/или агрегации и/или хроматографическому разделению. Это может быть определено в ходе стандартных экспериментов.
Биосовместимый гидрофильный полимер можно физически присоединять к поверхности липосомы, или можно ввести в мембрану липосомы. Следовательно, полимер может быть ковалентно связан с липосомой.
Известен и доступен целый ряд способов получения модифицированных липосом, которые являются одноламеллярными или мультиламеллярными (см. Lichtenberg and Barenholz, (1988)):
1. Тонкую пленку фосфолипида гидратируют водной средой, а затем механически встряхивают и/или обрабатывают ультразвуком и/или подвергают экструзии через соответствующий фильтр;
2. Растворение фосфолипида в подходящем органическом растворителе, смешивание с водной средой и последующее удаление растворителя;
3. Применение газа выше его критической точки (т.е., фреонов и других газов, таких как СО2 или смеси СО2 и других газообразных углеводородов) или
4. Получение смешанных мицелл из липида и детергента, последующее снижение концентрации детергентов до уровня ниже его критической концентрации, при которой образуются мицеллы.
В целом такие способы приводят к образованию липосом с неоднородными размерами приблизительно от 0,02 до 10 мкм или больше. Поскольку липосомы, которые относительно малы и имеют хорошо определенные размеры, предпочтительны для применения согласно настоящему изобретению, для уменьшения размера и неоднородности по размеру суспензий липосом можно применять второй этап обработки, именуемый «уменьшение размеров липосом».
Суспензию липосом можно отсортировать по размеру, чтобы достичь селективного распределения по размеру везикул с разбросом по размерам менее чем приблизительно 5 мкм, например, <0,4 мкм. Согласно одному варианту реализации коллоидные частицы обладают средним диаметром частиц приблизительно от 0,03 до 0,4 микрон (мкм), применимо приблизительно 0,1 микрон (мкм).
Липосомы в данном диапазоне можно легко стерилизовать посредством фильтрации через соответствующий фильтр. Малые везикулы также демонстрируют меньшую склонность к агрегации при хранении, что сокращает потенциально серьезные проблемы с блокированием или закупоркой, когда липосомы вводят внутривенно или подкожно. Кроме того, липосомы, которые прошли сортировку по размеру с целью его уменьшения до субмикронного диапазона, демонстрируют более однородное распределение.
Для уменьшения размеров и неоднородности по размеру липосом в духе настоящего изобретения существует несколько технологий. Облучение ультразвуком суспензии липосом либо в стандартной ванне, либо с ультразвуковым зондом приводит к прогрессивному снижению размеров с образованием малых одноламеллярных везикул (MOB) размером от 0,02 до 0,08 мкм.
Другим способом является гомогенизация, и она основана на сдвиге энергии для фрагментации крупных липосом с образованием малых. При типичной процедуре гомогенизации суспензия липосом многократно пропускается через стандартный эмульсионный гомогенизатор до достижения избранного размера липосом, обычно приблизительно от 0,1 до 0,5 мкм. При обоих способах распределение по размерам частиц можно отслеживать при помощи традиционного определения размера частиц лазерным лучом.
Экструзия липосом через поликарбонатный фильтр с малым диаметром пор или эквивалентную мембрану также представляет собой эффективный способ уменьшения размеров липосом до относительно четкого распределения по размерам, которое в среднем составляет диапазон приблизительно от 0,02 до 5 мкм, в зависимости от диаметра пор мембраны.
Обычно суспензию циклически пропускают через одну или две расположенных друг над другом мембраны несколько раз, пока не будет достигнуто желаемое распределение липосом по размеру. Липосомы можно продавливать через мембраны с последовательно уменьшающимся диаметром пор для достижения постепенного уменьшения размера липосом.
Другие способы, которые существуют для получения суспензий липосом с размером частиц меньше избранного предела, менее 1 мкм, включают центрифугирование и хроматографию молекулярных сит. Указанные два способа, соответственно, подразумевают предпочтительное удаление крупных липосом, а не превращение крупных частиц в мелкие. Выход липосом соответственно уменьшается.
Суспензию липосом с улучшенными размерами можно легко стерилизовать путем пропускания через стерилизующую мембрану, обладающую размером дискриминации частиц приблизительно 0,4 мкм, традиционно объемный мембранный фильтр с порами 0,45 мкм. Указанные липосомы стабильны в лиофилизированной форме и могут быть быстро восстановлены перед применением путем поглощения воды.
Подходящие липиды для образования липосом описаны выше. Подходящие примеры включают без ограничения такие фосфолипиды, как димиристоилфосфатидилхолин (ДМФХ) и/или димиристоилфосфатидилдиглицерол (ДМФГ), фосфолипиды, полученные из яйца и соевых бобов, которые получают после частичной или полной очистки, напрямую или после частичной или полной дегидрогенизации.
Следующие четыре способа перечислены в заявке WO 95/4524 и в целом применимы для получения коллоидных частиц (липосом), применяемых согласно настоящему изобретению.
Способ А
- a) смешивание амфифильных веществ, таких как липиды, подходящих для образования везикул в смешивающихся с водой органических растворителях.
- b) удаление растворителя в присутствии твердой подложки, в качестве альтернативы, высушенные амфифильные вещества или их смеси можно применять в любой форме (порошок, гранулы и т.д.) напрямую,
- c) переведение продукта, полученного на этапе (b) в раствор биополимерных веществ в физиологически совместимом растворе
- d) добавление органического растворителя, обладающего солюбилизирующими или диспергирующими свойствами, а также
- е) высушивание фракции, полученной на этапе (d) при условиях, при которых сохраняется функция биополимерных веществ.
В соответствии с этапом (а) Способа А амфифильные вещества, подходящие для образования везикул, упоминаемые выше, смешивают в смешивающемся с водой органическом растворителе. Указанным смешивающимся с водой органическим растворителем может быть полярный протон-содержащий растворитель, такой как фторированные углеводороды, хлорированные углеводороды и т.п.
На этапе (b) способа согласно настоящему изобретению растворитель удаляют в присутствии твердой подложки. Указанной твердой подложкой может быть инертный органический или неорганический материал, обладающий гранулоподобной структурой. Материалом неорганической подложки может быть стекло, а органическим материалом может быть Тефлон™ или другие аналогичные полимеры.
Этап (с) Способа А согласно настоящему изобретению направлен на перевод продукта, полученного на этапе (b), в раствор веществ, которые должны икапсулироваться в физиологически совместимом растворе.
Указанный физиологически совместимый раствор может быть эквивалентен раствору хлорида натрия приблизительно до 1,5 по массе. Также возможно применять другие соли при условии, что они физиологически совместимы, например, такие как криопротектор, например, сахара и/или аминокислоты. Например, в качестве криопротектора можно применять лактозу, сахарозу или трегалозу.
Необязательно, между этапами (а) и (b) может быть предложен этап инактивации вирусов, стерилизации, депирогенизации, фильтрации фракции или подобного продукта из этапа (а). Это может иметь то преимущество, что раствор становится фармацевтически приемлемым на ранней стадии получения.
Этап (b) Способа А представляет собой добавление органического растворителя, обладающего солюбилизирующими или диспергирующими свойствами. Указанный органический растворитель может быть органическим полярным протон-содержащим растворителем, смешиваемым с водой. Также можно применять низшие алифатические спирты, содержащие от 1 до 5 атомов углерода в алкильной цепи, такие как третичный бутанол (трет-бутанол). Количество полярного протон-содержащего растворителя, смешиваемого с водой, в значительной степени зависит от того, насколько он препятствует функции вещества, которое требуется загрузить в липосомы. Например, если требуется загрузить белок, устанавливается верхний предел количества растворителя, при котором нарушается активность белка. Он может сильно варьировать в зависимости от природы вещества, которое требуется загружать. Например, если фактор свертывания крови включает фактор IX, то количество трет-бутанола составляет приблизительно 30%, а приемлемое количество фактора IX менее 10% от трет-бутанола (фактор VIII значительно более чувствителен к влиянию трет-бутанола). Процент трет-бутанола в данных примерах указан как процент по объему, рассчитанный для конечной концентрации. Необязательно после этапа (d) можно проводить инактивацию вирусов, стерилизацию и/или разделение фракции, полученной после этапа (d). Этап (е) согласно настоящему изобретению представляет собой сушку фракции, полученной на этапе (d) при условиях, при которых сохраняется функция вещества, которое требуется загружать. Одним из способов сушки является лиофилизация. Лиофилизацию можно проводить в присутствии криопротектора, например, лактозы или других сахаридов или аминокислот. В качестве альтернативы можно применять испарение или сушку распылением.
Сухой остаток затем перед применением может поглощаться водной средой. После поглощения твердого вещества оно образует дисперсию соответствующих липосом. Водная среда может содержать раствор хлорида натрия, и образующуюся дисперсию можно необязательно пропускать через соответствующий фильтр, чтобы при необходимости уменьшить размер липосом. Соответственно, липосомы могут иметь размер 0,02-5 мкм, например, в диапазоне <0,4 мкм.
Липосомы, которые можно получить при помощи способа А, демонстрируют высокую загрузку факторов свертывания крови.
Фармацевтические композиции согласно настоящему изобретению могут быть также промежуточным продуктом, получаемым при выделении любой из фракций из этапа (с) или (d) способа А. Соответственно, состав согласно настоящему изобретению также содержит водную дисперсию, получаемую после поглощения продукта, полученного на этапе (е) способа А водой в форме дисперсии (липосомы в водной среде).
В качестве альтернативы, фармацевтические композиции согласно настоящему изобретению также можно получить посредством следующих способов, которые называются способами В, С, D и Е.
Способ В
Данный способ включает также этапы (а), (b) и (с) способа А. Однако этап (d) и (е) способа А пропущены.
Способ С
В способе С этап (d) способа А заменен на цикл замораживания-размораживания, который должен повторяться по меньшей мере два раза. Данный этап хорошо известен на предыдущем уровне техники для получения липосом.
Способ D
В способе D исключено применение любого осмотического компонента. В способе D подразумеваются этапы получения везикул, смешивания и по существу бессолевой раствор веществ, которые требуется загружать, и совместное высушивание фракций, полученных таким образом.
Способ Е
Способ Е более простой, чем способы А - D, описанные выше. Для него требуется растворение соединений, применяемых при получении липосом (липидные антиоксиданты и др.), в полярном протон-содержащем растворителе, смешиваемом с водой, таком как трет-бутанол. Затем указанный раствор смешивают с водным раствором или дисперсией, содержащими фактор свертывания крови. Смешивание проводят при оптимальном соотношении объемов, необходимом для сохранения биологической и фармакологической активности агента.
Затем указанную смесь лиофилизируют в присутствии или отсутствии криопротектора. Перед применением указанного состава липосом требуется регидратация. Указанные липосомы мультиламеллярны, уменьшения их размера можно достичь при помощи одного из способов, описанных в WO 95/04524.
Изобретение также включает способы лечения заболеваний, связанных с нарушением свертывания крови (например, гемофилии), или травмы у субъекта, включающие подкожное введение фармацевтической композиции или дозы, описанных в настоящей заявке, субъекту, нуждающемуся в таком введении. Такие способы могут включать способ лечения заболеваний, связанных с нарушением свертывания крови, или травмы у субъекта, причем у указанного антитела вырабатываются антитела (т.е. ингибиторы) к фактору свертывания крови.
Заболевания, связанные с нарушением свертывания крови, могут характеризоваться утратой функции фактора свертывания крови или генерированием аутоантител. Примеры заболеваний, связанных с нарушением свертывания крови, включают гемофилию, такую как гемофилия А и гемофилия В.
Следовательно, настоящее изобретение распространяется на фармацевтическую композицию, определение которой дано выше, для применения при лечении заболеваний, связанных с нарушением свертывания крови (например, гемофилии) или травмы. Такие фармацевтические композиции для применения при лечении заболеваний, связанных с нарушением свертывания крови, или травмы можно применять, когда у пациента вырабатываются антитела на указанный фактор свертывания крови. Области применения согласно настоящему изобретению в соответствии с настоящим аспектом также включают применение фактора свертывания крови при производстве лекарственного препарата, который определен выше, для применения при лечении заболеваний, связанных с нарушением свертывания крови, или травмы.
Фактор VIIa можно применять при лечении эпизодов кровотечения при гемофилии А или В, или при лечении пациентов, у которых вырабатываются ингибиторные антитела к факторам FVIII или IX, соответственно. Фактор VIII можно применять при лечении эпизодов кровотечения у пациентов с гемофилией А, а фактор IX можно применять при лечении пациентов с гемофилией В.
В настоящей заявке термин «лечение» включает любой режим, который может принести пользу человеку или млекопитающему, отличному от человека. Лечение «млекопитающего, отличного от человека» распространяется на лечение домашних млекопитающих, включая лошадей и животных-компаньонов (например, кошек и собак), и сельскохозяйственных животных, включая членов семейств овечьих, козлиных, свинообразных, бычьих и лошадиных. Лечение может быть применительно к любому существующему состоянию или расстройству, или может быть профилактическим (превентивное воздействие). Лечение может проводиться в отношении врожденного или приобретенного заболевания. Лечение может проводиться в отношении острого или хронического заболевания.
Уровень активности каскада свертывания крови можно измерить при помощи любого подходящего анализа, например, по времени свертывания цельной крови (WBCT) или по активированному частичному тромбопластиновому времени (АЧТВ).
При анализе времени свертывания цельной крови (WBCT) измеряют время, затрачиваемое на образование сгустка в цельной крови в экспериментальной среде, обычно в стеклянной пробирке или чашке.
При анализе активированного частичного тромбопластинового времени (АЧТВ) измеряют параметр части пути свертывания крови. Он аномально большой при гемофилии и при внутривенном введении гепарина. Для оценки АЧТВ требуется отобрать несколько миллилитров крови из вены. АЧТВ является показателем для одной части системы свертывания, называемой «внутренним путем». Значение АЧТВ представляет собой время в секундах для специфического процесса свертывания, который происходит в лабораторной пробе. Данный результат всегда сравнивают с «контрольной» пробой нормальной крови. Если в исследуемой пробе время больше, чем в контрольной пробе, это указывает на снижение функции свертывания во внутреннем пути. Обычная медикаментозная терапия как правило направлена на достижение АЧТВ в диапазоне от 45 до 70 секунд, но значение можно также выражать как отношение исследуемой пробы к нормальной, например, 1,5-кратное увеличение по сравнению с нормой. Высокое значение АЧТВ в отсутствии терапии гепарином может быть связано с гемофилией, что может потребовать дальнейшего исследования.
Также согласно изобретению предложен набор из частей, включающих фармацевтическую композицию согласно настоящему изобретению и вспомогательные элементы для введения, включая раствор для инъекций для подкожного введения, причем указанный набор соответственно содержит инструкции по его применению.
Следовательно, согласно настоящему изобретению соответственно может быть предложена лекарственная форма фармацевтической композиции согласно настоящему изобретению. Такие лекарственные формы могут быть предложены в подходящих контейнерах или флаконах, содержащих соответствующую дозу для пациента.
Фармацевтические композиции для подкожного введения или лекарственные формы согласно настоящему изобретению можно вводить одни или в сочетании с другими соединениями, такими как терапевтические соединения или молекулы, например, противовоспалительные препараты, анальгетики или антибиотики, или другие фармацевтически активные агенты, которые могут способствовать или усиливать активность фактора VIIa, фактора VII, фактора VIII, фактора IX, фактора X, фактора Ха, фактора XI, фактора V, фактора XIII, фактора фон Виллебранда (vWF), протромбина или белка С, или их фрагмента, например, другого фактора свертывания крови. Такое введение с другими соединениями может быть одновременным, раздельным или последовательным. Компоненты могут быть получены в форме набора, который может содержать надлежащие инструкции.
Фармацевтические композиции согласно настоящему изобретению позволяют усовершенствовать лечение заболеваний, когда фактор свертывания крови вводят для лечения пациенту, страдающему заболеванием, связанным с нарушением свертывания крови, или с травмой.
Согласно одному варианту реализации настоящего изобретения предложена фармацевтическая композиция для подкожного введения, содержащая фактор свертывания крови и коллоидную частицу, содержащую приблизительно 1-20 мольных процентов амфифильного липида, модифицированного путем соединения с биосовмемтимым гидрофильным полимером, фармацевтически приемлемый буфер, скорректированный до физиологический рН, подходящий для подкожного введения, причем указанный фактор свертывания крови не инкапсулирован в указанную коллоидную частицу.
Специалисты в данной области техники должны понимать, что доза лекарственного препарата согласно настоящему изобретению зависит от концентрации эффективных биополимерных веществ, а также от их эффективности.
Пациенту можно вводить дозу до 2000 мг/липидные липосомы на кг массы тела, причем активные факторы в липосомы загружают с эффективностью более 50% от полной активности, применяемой при приготовлении загружаемых липосом.
Соответственно, согласно другому аспекту настоящего изобретения объем состава для доставки пациенту может быть не более 2 мл. Соответственно, объем доставки может составлять 5 мкл, 10 мкл, 25 мкл, 50 мкл, 100 мкл, 250 мкл, 500 мкл, 750 мкл или 1 мл. Согласно альтернативным вариантам реализации объем состава для доставки может быть не более 1,5 мл, 2 мл, 2,5 мл, 3,0 мл или 3,5 мл.
Важно отметить, что настоящее изобретение позволяет доставлять более высокую концентрацию активного агента в виде одной подкожной инъекции более безопасно, чем при внутривенной инъекции, поскольку она не попадает прямо в кровяное русло пациента. Это особенно важно, когда речь идет о факторах свертывания, поскольку высокая концентрация факторов свертывания крови, вводимая внутривенно может приводить к нежелательному и опасному образованию тромбов у указанного пациента.
Подкожная доставка позволяет добиться стабильного поступления активного агента в кровяное русло через лимфатическую систему, благодаря чему удается избежать эффекта опасных уровней активного агента, доставленных прямо в кровеносную систему. Следовательно, поскольку концентрация доставки агента в кровное русло регулируется лимфатической системой пациента, в составе дозы для подкожного введения может доставляться более высокая концентрация, что позволяет применять меньшие объемы, чем традиционно применяют при внутривенной доставке.
Составы согласно настоящему изобретению можно вводить по меньшей мере один раз в день, по меньшей мере два раза в день, приблизительно один раз в неделю, приблизительно два раза в неделю, приблизительно один раз в две недели или приблизительно один раз в месяц.
Для определенных терапевтических веществ будет достаточно режима дозирования один раз в день, тогда как для других более адекватным и желательным может быть режим с более частым дозированием, причем количество, доставляемое в составе каждой дозы можно уменьшить относительно стандартной внутривенной дозы. Так, например, состав согласно настоящему изобретению можно вводить один раз в день, два раза в день (или при необходимости чаще).
Настоящее изобретение позволяет предотвратить быстрое нарастание и последующее падение (т.е. «пиолообразную зависимость») концентрации терапевтического агента в крови. Согласно настоящему изобретению предложена более равномерная, предсказуемая концентрация агента в крови пациента в течение более длительного периода времени, чем традиционно наблюдается для стандартных лекарственных форм того же агента, когда он быстро доставляется внутривенно.
Дополнительная польза от настоящего изобретения состоит в том, что оно позволяет вводить подкожно более высокую дозу агента, чем можно безопасно вводить внутривенно. Это приводит к возможности более длительного полезного терапевтического действия, чем то, которое можно в обычных условиях и безопасно достичь при более высоком дозировании или более частом введении внутривенным путем. Например, в случае факторов свертывания крови, поскольку указанные продукты доставляются через грудной лимфатический проток в подключичную вену, указанный способ позволяет вводить большее количество продукта одномоментно в виде однократной дозы подкожно, чем можно вводить одномоментно в вену. Доставка болюсов с высокой дозой в вену может вызвать нежелательное явление тромбоза.
Дополнительное преимущество настоящего изобретения состоит в том, что оно позволяет повторно вводить терапевтический агент с интервалами, которые позволяют поддерживать концентрацию агента в крови на постоянном уровне, что дает длительное равномерное и предсказуемое терапевтическое действие без необходимости ждать возможности ввести повторную дозу, когда концентрация в крови упадет до терапевтически незначимых уровней. В традиционной практике внутривенное повторное введение факторов свертывания крови с немедленным достижением Сmax и началом действия откладывается до того, пока по оценкам уровень препарата не упадет до уровня, при котором появление Сmax от новой инъекции не достигнет потенциально тромбогенного уровня (т.е., уменьшение риска нежелательного явления), но это означает, что у пациента достигается «нездоровый» диапазон уровней агента в кровотоке. Иными словами, последующие дозы агента обычно не вводят пациенту, пока «здоровый уровень», или терапевтически эффективный уровень агента еще присутствует в кровяном русле пациента. Однако настоящее изобретение позволяет повторно вводить агент, пока уровень агента все еще находится в терапевтически эффективном диапазоне. Таким образом, изобретение обеспечивает более последовательный терапевтический уровень белка в кровяном русле, который идеальнее для профилактики. Благодаря непрерывной доставке агента в кровяное русло через грудной проток удается избежать проблемы повышения уровня агента в кровяном русле до нежелательно высокого значения.
Согласно настоящему изобретению предложен состав для подкожного введения субъекту, который позволяет субъекту получать лекарственную форму фактора свертывания крови, достаточную для поддержания времени свертывания цельной крови у указанного субъекта на уровне не более 20 минут, иными словами, для введения не более чем одной дозы в месяц. Также предложен состав с фактором свертывания крови для подкожного введения не чаще, чем один раз в месяц, причем указанная лекарственная форма демонстрирует Сmax по меньшей мере 10% и не более 90% от эквивалентной лекарственной формы сравнения, когда ту вводят внутривенно, для применения при лечении расстройств свертывания крови. Соответственно, Сmax составляет от 20 до 80%, или от 30 до 70%, или от 40 до 60%.
Выражение «не более» означает, что лекарственную форму можно вводить чаще, чем указанный период времени, но не обязательно делать это; действие от подкожного введения такой лекарственной формы означает, что эффекты наблюдаются на протяжении указанного периода времени. Однако из-за низкой и постоянной Сmax более частое дозирование можно проводить без нежелательных эффектов у пациента.
Соответственно, указанной лекарственной формы фактора свертывания крови может быть достаточно для поддержания времени свертывания цельной крови у указанного субъекта на уровне менее 15 минут, или, соответственно, менее 12 минут. Согласно одному варианту реализации указанная лекарственная форма фактора свертывания крови является лекарственной формой для введения по меньшей мере один раз в неделю, или по меньшей мере один раз в месяц, по меньшей мере один раз в две недели, по меньшей мере один раз в полнедели.
Также согласно настоящему изобретению предложена лекарственная форма, для которой доза фактора свертывания крови составляет от 1 до 1000 МЕ/кг, или от 5 до 500 МЕ/кг, или от 100 до 250 МЕ/кг, или от 25 до 50 МЕ/кг, или от 5 до 50 МЕ/кг.
Лекарственная форма согласно настоящему изобретению, содержащая фактор свертывания крови, позволяет менее часто вводить данную лекарственную форму, но этого все еще достаточно для поддержания времени свертывания цельной крови у указанного субъекта на уровне не более 20 минут, или не более 15 минут, или не более 10 минут. Согласно одному варианту реализации указанной лекарственной формы достаточно для поддержания времени свертывания цельной крови у указанного субъекта на уровне не более 12 минут. Лекарственная форма может подразумевать введение не чаще одного раза в две недели, не чаще одного раза в неделю, не чаще двух раз в неделю, не чаще одного раза в три дня, не чаще одного раза в 2 дня, не чаще одного раза в день или чаще или реже.
Важно отметить, что одним из преимущество настоящего изобретения является то, что лекарственную форму, в которой агентом является фактор свертывания крови, не требуется вводить пациенту чаще, чем с указанными интервалами, чтобы поддерживать время свертывания цельной крови в нормальном диапазоне, но ее может потребоваться вводить чаще, чтобы помочь обеспечить «устойчивое состояние», сходное с тем, которое достигается для формы с контролируемым высвобождением. Специалисты в данной области техники считают «нормальным» время свертывания цельной крови от 10 до 12 минут, и значение меньше 15 минут считается нормальным у людей без гемофилии. Когда время свертывания цельной крови превышает 20 минут, это считается за пределами нормального диапазона. Значения от 15 до 20 минут рассматривают как указывающие на то, что хотя кровотечение под контролем, но это не норма.
Согласно другому варианту реализации указанную лекарственную форму вводят реже, чем можно было бы предсказать по времени полувыведения из плазмы после болюсной внутривенной инъекции. Например, болюсная инъекция модифицированного фактора XI может требоваться один раз в неделю, тогда как тот же агент, вводимый подкожно согласно настоящему изобретению, может требоваться только раз в десять дней, или реже.
Согласно дополнительному аспекту настоящего изобретения предложена лекарственная форма фармацевтической композиции с дозой фактора свертывания крови 25-50 МЕ/кг для подкожного введения с той же или меньшей частотой, с которой указанный фактор свертывания вводят внутривенно.
Составы согласно настоящему изобретению, следовательно, позволяют поддерживать нормальный показатель гемостаза до семи дней, причем нормальный показатель определяют как время свертывания цельной крови (WBCT) менее 15 минут, соответственно, приблизительно 12 минут или менее.
Составы согласно специфическим вариантам реализации настоящего изобретения, в которых состав содержит фактор свертывания крови, могут содержать дозу агента от 25 до 50 МЕ/кг. Согласно некоторым вариантам реализации доза может составлять 25, 30, 35, 40, 45 или 50 МЕ/кг. Указанная доза может составлять от 25 МЕ/кг до 30 МЕ/кг, от 35 МЕ/кг до 40 МЕ/кг, или от 40 МЕ/кг до 50 МЕ/кг.
Составы согласно специфическим вариантам реализации настоящего изобретения, в которых состав содержит фактор свертывания крови, могут в качестве альтернативы содержать дозу агента от 5 до 50 МЕ/кг. Согласно некоторым вариантам реализации доза может составлять 5, 10, 15, 20, 25, 30, 35, 40, 45 или 50 МЕ/кг. Указанная доза может составлять от 5 МЕ/кг до 10 МЕ/кг, от 25 МЕ/кг до 30 МЕ/кг, от 35 МЕ/кг до 40 МЕ/кг, или от 40 МЕ/кг до 50 МЕ/кг.
Согласно одному варианту реализации, когда лекарственную форму получают с дозой 150 МЕ/кг, указанный состав может подходить для введения один раз в две недели субъекту, нуждающемуся в таком введении. Соответственно, указанный состав может быть предназначен для введения не чаще одного раза в две недели. В качестве альтернативы лекарственная форма может быть получена с дозой 100 МЕ/кг.
Согласно одному варианту реализации настоящего изобретения состав согласно настоящему изобретению, содержащий фактор свертывания крови, может приводить к нормализации гемостаза, которая сохраняется по меньшей мере полнедели.
Лекарственные формы согласно настоящему изобретению, когда их вводят подкожно, приводят к тому, что для достижения такой же терапевтической конечной точки требуется меньшее количество модифицированного фактора свертывания крови, таким образом, предложены более безопасные продукты для субъектов, нуждающихся в таком лечении. Согласно одному варианту реализации для достижения нормального гемостаза в течение по меньшей мере одной недели у субъекта достаточно половины скорректированной дозы модифицированного фактора свертывания крови, в частности, когда указанным фактором свертывания является фактор VIIa или фактор VIII. Подходящим показателем нормального гемостаза является время свертывания цельной крови (WBCT) приблизительно 12 минут, как было описано выше.
Составы согласно настоящему изобретению могут соответственно содержать менее половины скорректированного по дозе эффективного количества препарата сравнения, предназначенного для внутривенного введения, содержащей тот же модифицированный фактор свертывания, чтобы достичь того же терапевтического действия.
Следовательно, согласно настоящему изобретению также предложена лекарственная форма модифицированного фактора свертывания крови для подкожного введения, причем указанная лекарственная форма содержит 50% скорректированного по дозе количества, необходимого при внутривенном введении, чтобы достигнуть такой же продолжительности эффективного действия.
Состав, подходящий для подкожного введения, можно соответственно получить в виде водного или по существу водного раствора. Указанный состав может содержать такие дополнительные соли, консерванты и стабилизаторы и/или вспомогательные вещества, или при необходимости адъюванты. Лекарственные формы согласно настоящему изобретению могут быть предложены в виде безводных порошков, готовых к приготовлению непосредственно перед применением в подходящей водной среде.
Соответственно, такие лекарственные формы можно готовить в форме водных буферных составов. Подходящие буферные растворы могут включать без ограничений аминокислоты (например, гистидин), соли неорганических кислот и щелочных металлов или щелочноземельных металлов (например, соли натрия, соли магния, соли калия, соли лития или соли кальция - например, хлорид натрия, фосфат натрия или цитрат натрия). Могут присутствовать другие компоненты, такие как детергенты или эмульгаторы (например, Tween 80® или любая форма Tween®), и стабилизаторы (например, бензамидин или производное бензамидина). Также могут присутствовать вспомогательные вещества, такие как сахара (например, сахароза). Подходящие значения для рН включают физиологические рН, например, рН 6,8-7,4, или рН 7,0. В таких основах для введения можно получать жидкие лекарственные формы, готовые к употреблению.
Согласно одному конкретному варианту реализации настоящего изобретения предложена фармацевтическая композиция для подкожного введения со следующим составом:
- 50 мМ цитрата натрия
- pH 7,0
- 100 мМ фосфолипидов - мольное соотношение пальмитоил-олеил-фосфатидилхолина (ПОФХ) и 1,2-дистеароил-sn-глицеро-3-фосфоэтаноламин-N-[поли-(этиленгликоль)-2000] (ДСПЭ-ПЭГ 2000) в мольном отношении 97:3.
- Лиофилизированный rFVIII (Helixate NexGen)
Ниже настоящее изобретение описано посредством ссылок на конкретные примеры, которые приведены только в целях иллюстрации, но не накладывают никаких ограничений на изобретение.
Пример 1: Синтез липосом
Смешанные липиды получали из пальмитоил-олеил-фосфатидилхолина (ПОФХ) и 1,2-дистеароил-sn-глицеро-3-фосфоэтаноламин-N-[поли-(этиленгликоля)-2000], модифицированного с применением ПЭГ-2000 (ПЭГ с молекулярной массой 2000 Дальтон) (ДСПЭ-ПЭГ 2000), следующим образом:
Молекулярная масса ПОФХ: 760,08 г/моль
Молекулярная масса ДСПЭ-2kПЭГ: 2789,5 г/моль
Конечный препарат имеет концентрацию фосфолипидов 100 мМ. 15% смесь (масса/объем) липидов получали при мольном отношении ПОФХ:ДСПЭ-2kПЭГ97:3. Взвешивали и смешивали следующие количества веществ:
2,04 г ПОФХ
0,232 г ДСПЭ-2kПЭГ
14,9 мл трет-бутанола (плавится при 35°С на водяной бане), все вещества помещали во флакон компании Шотт объемом 100 мл.
Смесь держали при 35°С в водяной бане и периодически перемешивали до тех пор, пока все твердые вещества не растворятся/перейдут в дисперсию. Конечный материал представлял собой прозрачную бесцветную смесь. Смесь замораживали при -80°С в течение ночи.
Манипуляции проводили в вытяжном шкафу, чтобы содержимое после применения могло очистится от высушенного/конденсированного растворителя. Лиофильную сушилку Christ Alpha 1-2 LD и вакуумный насос прогревали в течение 20 минут, и замороженную смесь липид/растворитель вынимали из холодильника с температурой -80°С и высушивали в течение ночи.
Высушенные липиды доставали из сушилки следующим утром. Они выглядели как сухой кристаллический сгусток. Для дальнейшей работы требовался раствор липидов 100 мМ. Количество присутствующего липида рассчитывали как приблизительно 82 мкмоль ДСПЭ-2kПЭГ и 2,69 ммоль ПОФХ; то есть, приблизительно 2,77 ммоль липидов. Таким образом, требовалось 27,7 мл растворителя. К сухим липидам добавляли 27,7 мл 50 мМ буфера на основе цитрата натрия, и полученную смесь перемешивали и нагревали приблизительно до 35°С. Приблизительно через 120 минут получали белую эмульсию без явных крупных включений твердого вещества. Ее направляли на экструзию, как описано ниже.
Бак для фильтрации под давлением Sartorius 47 мм из нержавеющей стали монтировали и оборачивали кожухом водяного охлаждения (обернутые трубки, питаемые через термоциркулятор), в котором поддерживали температуру 35°С. Бак дополняли поликарбонатной трековой мембраной (подробное описание ниже), покрытой предварительным фильтром из стекловолокна (Whatman GF/D). Эмульсию выливали в бак и нагнетали под действием газообразного азота под давлением 4 бар, а фильтрат собирали в пробирки на 50 мл. Продолжительность каждой экструзии фиксировали и регистрировали.
Последовательность фильтрации была следующая: 0,8 мкм, 0,4 мкм, 0,2 мкм, 0,2 мкм, 0,1 мкм и 0,1 мкм (т.е., отдельное пропускание через более грубые фильтры и два пропускания через более тонкие фильтры 0,2 и 0,1 мкм), а фильтрат между фазами нагревали снова до 35°С. Липосомы подвергали экструзии, данные о ней представлены в таблице ниже:
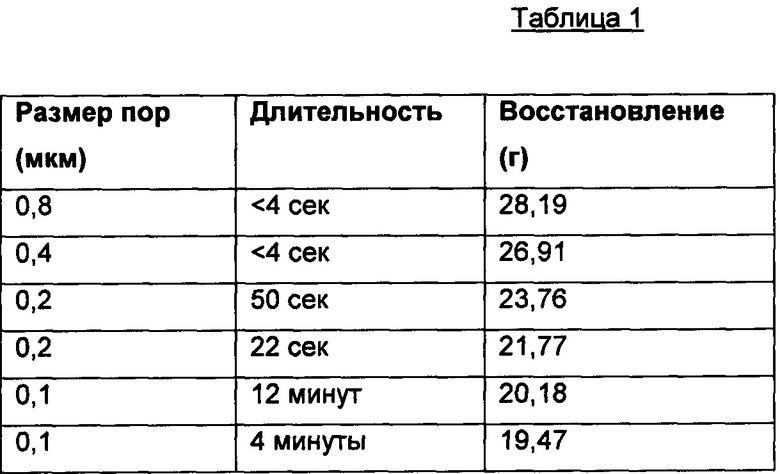
Полученные при экструзии липиды хранили при +5°С. 15 мл «экструдированных липосом» отбирали из места хранения в охлажденном состоянии и распределяли в стерильной пробирке на 50 мл в боксе микробиологической безопасности. Размер экструдированных липосом анализировали при помощи фотонно-корреляционного спектрометра ALV5000. Определенный средний радиус составил 75,4±0,86 нм, а средняя ширина пика 22,21±3,86 нм, что дает средний диаметр 150,080 нм, и коэффициент полидисперсности 0,087.
Пример 2
Фармакокинетика/Фармакодинамика рекомбинантного Фактора VIII человека, восстановленного в ПЭГилированных липосомах, у собак с гемофилией А после подкожного введения
Собаке с гемофилией А (обозначена как собака под номером «1») подкожно вводили дозы ПЭГилированных липосом, ассоциированных с фактором VIII (PEGLip FVIII п/к), как описано ниже:
Задачи данного исследования включали определение ФК и ФД полноразмерного rFVIII, востановленного в ПЭГилированных липосомах, у собаки с гемофилией А при его подкожном введении (п/к).
Полноразмерный rFVIII
В качестве исследуемого вещества применяли лиофилизированный, полноразмерный rFVIII (Helixate NexGen, партия 270LR8WB).
Получение ПЭГилированных липосом
ПЭГиилированные липосомы в цитратном буфере получали в соответствии с примером 1, описанном выше, по способу Baru et al. (2005). Состав липосом был следующим: 50 мМ цирта натрия, рН 7,0, 100 мМ фосфолипидов с мольным соотношением 97:3 пальмитоил-олеил-фосфатидилхолина (ПОФХ) и 1,2-дистеароил-sn-глицеро-3-фосфоэтаноламин-N-[поли-(этиленгликоля)-2000] (ДСПЭ-ПЭГ 2000).
Экспериментальное животное - собака - была из колонии собак с гемофилией А, живущей в Медицинской школе Университете Алабамы. Все собаки страдают врожденной тяжелой гемофилией А. Исследуемое животное было массой 16,4 кг, и ранее ему не вводили белков человека.
Перед введением препарата состояние собаки оценивали для подтверждения нормального состояния здоровья, включая биохимию крови, биохимию сыворотки крови, профиль фибриногена, белки-производные фибриногена (FDS), тромбиновое время и анализ мочи.
Дизайн данного исследования включал исследование применимости с однократным п/к введением одному животному.
Полноразмерный, рекомбинантный FVIII человека (Helixate NexGen, 2000 ME) восстанавливали в 13,3 мл растворителя из ПЭГилированных липосом. Восстановленный FVIII аккуратно перемешивали при комнатной температуре в течение 5-10 мин, чтобы белок мог абсорбироваться в липосомы перед применением. После восстановления суспензия обладала активностью FVIII 150 МЕ/мл.
Исследуемому животному п/к вводили 100 МЕ/кг. Расчет объема лекарственного препарата для введения проводили по следующей формуле:
Объемная доза (мл)=(а×b)/с
Где: а - целевая доза (100 МЕ/кг)
b - масса собаки (кг)
с - активность rFVIII (150 МЕ/мл)
После введения за исследуемым животным наблюдали, оценивая клинические признаки. Проводили скрининг на непредвиденную токсичность, который включал полный клинический анализ крови и исследование биохимии сыворотки через 48 ч и 5 дней после введения. Для оценки повышенного риска тромбоза оценивали уровень фибриногена, FDP и тромбиновое время (ТВ).
Пробы крови (5 мл) у собаки после п/к введения отбирали в следующие моменты времени после введения:
Перед введением и через 0,5, 2, 4, 8, 12, 24, 36, 48, 60, 72, 84, 96, 108 и 120 часов после введения.
Для оценки свертывания цельной крови и активированного времени свертывания применяли цельную кровь (без цитрата; 1 мл). Остающиеся пробы по 4 мл крови переносили в пробирки, содержащие 0,109М антикоагулнта тринатриевого цитрата (9:1 объем/объем) на льду.
В цельной крови с цитратом проводили такие оценки, как активированное частичное тромбопластиновое время (АЧТВ), активированное время свертывания (ACT) и тромбоэластограмма (ТЭГ).
Плазму получали путем центрифугирования оставшейся крови с цитратом, и полученные пробы плазмы хранили в аликвотах приблизительно по 100 мкл при -80°С.
Анализы
(i) Цельная кровь без цитрата: анализ свертывания цельной крови
Пробы крови делили между 2 вакуумными пробирками, (2×0,5 мл) и тщательно наблюдали путем периодического и разумного выравнивания пробирки до тех пор, пока не становилось видно, что сгусток препятствует потоку в полностью горизонтальном положении. Качество сгустка наблюдали путем удерживания пробирки в полностью перевернутом положении. Время свертывания цельной крови регистрировали как среднее полное время с момента отбора крови до визуального наблюдения сгустка крови для обеих проб, и отмечали качество сгустка в перевернутом положении.
(ii) Цельная кровь с цитратом: тромбоэластограмма (ТЭГ)
ТЭГ оценивали в рекальцифицированной цельной крови с цитратом с применением анализатора гемостаза - тромбоэластрографа модель 5000 (Haemoscope Corporation) в соответствии с инструкциями производителя. Если кратко, 1 мл цельной крови с цитратом помещали в коммерческий флакон (Teg®Hemostasis System Kaolin, Haemonetics), содержащий каолин. Смешивание проводили путем аккуратного 5-кратного переворачивания флаконов с каолином. Булавки и чашки помещали в ТЭГ-анализатор в соответствии со стандартной процедурой, рекомендованной производителем. Каждую стандартную чашку для ТЭГ помещали в предварительно нагретый до 37°С держатель прибора и заполняли 20 мкл хлорида кальция (0,2 М). Затем 340 мкл активированной каолином цельной крови с цитратом добавляли до общего объема 360 мкл.
(iii) Активированное время свертывания (ACT) и активированное частичное тромбопластиновое время (АЧТВ)
Анализ ACT и АЧТВ проводили при помощи анализатора свертывания Haemachron Jr (International Technidyne Corps.) в соответствии с инструкциями производителя.
(iv) Плазма: Анализ активности FVIII (хроматографический)
Активность FVIII в плазме определяли при помощи анализа Coatest Assay (Dia Pharma, West Chester, ОН). Пробы плазмы разводили в соотношении 1:20 - 1:80 растворителем для проб и оценивали в соответствии с инструкциями производителя. Стандартные кривые строили на основании нормальной эталонный плазмы для оценки гемостаза (american diagnostica inc, Stamford, CT) и очищенного белка ПЭГ-FVIII.
(v) Плазма: FVIII ELISA
Концентрацию антигена FVIII в пробах плазмы определяли посредством ELISA (твердофазного иммуноферментного анализа) с применением набора Visulize FVIII antigen от компании «Affmity Biologicals» (Анкастер, Онтарио, Канада) в соответствии с инструкциями производителя.
(vi) Плазма: Иммуногенность
Проводили Бетезда-анализ для разведений 1:4, 1:10 и 1:20 исследуемой плазмы в плазме человека, дефицитной по FVIII. Равные объемы разведенной исследуемой плазмы и нормальной эталонной плазмы человека инкубировали при 37°С в течение 2 часов, и титр Бетезда определяли при помощи анализа АЧТВ и стандартной кривой для стандарта нормальной плазмы человека, как описано выше.
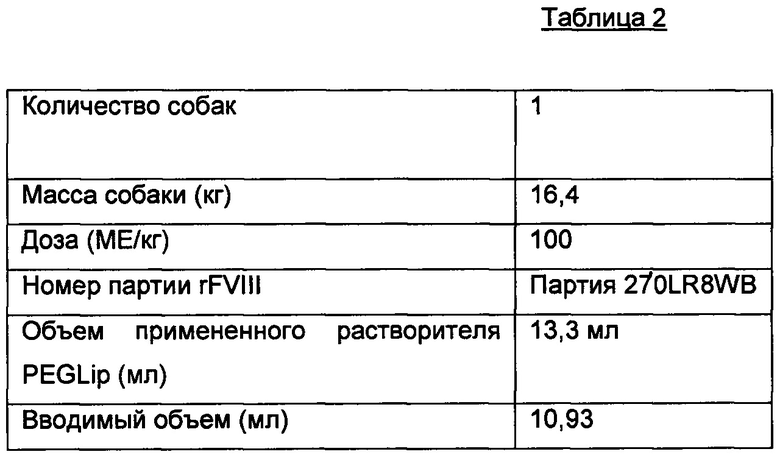
Результаты исследования показаны в Таблице 3.
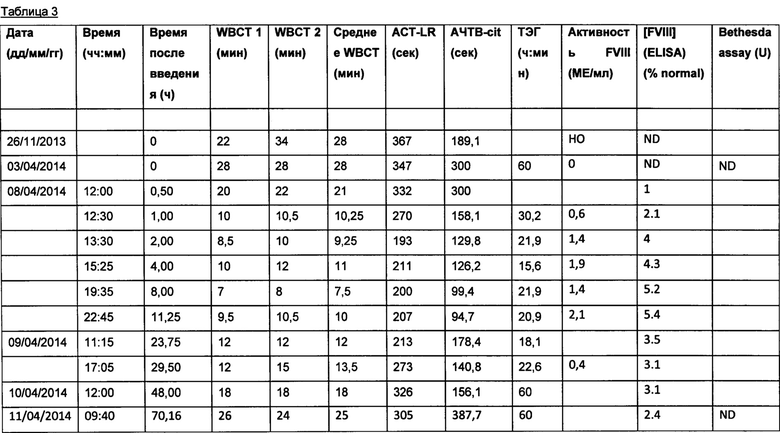
НО = Не определяется
--->
Перечень последовательностей
<110> Cantab Biopharmaceuticals Patents Limited
<120> ФАРМАЦЕВТИЧЕСКИЕ СОСТАВЫ ПЕГИЛИРОВАННЫХ ЛИПОСОМ И ФАКТОРОВ СВЕРТЫВАНИЯ КРОВИ
<130> P60726WO
<140> PCT/EP2015/073003
<141> 2015-10-06
<150> GB 1417589.7
<151> 2014-10-06
<160> 4
<170> PatentIn version 3.3
<210> 1
<211> 1438
<212> PRT
<213> Homo sapiens
<400> 1
Ala Thr Arg Arg Tyr Tyr Leu Gly Ala Val Glu Leu Ser Trp Asp Tyr
1 5 10 15
Met Gln Ser Asp Leu Gly Glu Leu Pro Val Asp Ala Arg Phe Pro Pro
20 25 30
Arg Val Pro Lys Ser Phe Pro Phe Asn Thr Ser Val Val Tyr Lys Lys
35 40 45
Thr Leu Phe Val Glu Phe Thr Asp His Leu Phe Asn Ile Ala Lys Pro
50 55 60
Arg Pro Pro Trp Met Gly Leu Leu Gly Pro Thr Ile Gln Ala Glu Val
65 70 75 80
Tyr Asp Thr Val Val Ile Thr Leu Lys Asn Met Ala Ser His Pro Val
85 90 95
Ser Leu His Ala Val Gly Val Ser Tyr Trp Lys Ala Ser Glu Gly Ala
100 105 110
Glu Tyr Asp Asp Gln Thr Ser Gln Arg Glu Lys Glu Asp Asp Lys Val
115 120 125
Phe Pro Gly Gly Ser His Thr Tyr Val Trp Gln Val Leu Lys Glu Asn
130 135 140
Gly Pro Met Ala Ser Asp Pro Leu Cys Leu Thr Tyr Ser Tyr Leu Ser
145 150 155 160
His Val Asp Leu Val Lys Asp Leu Asn Ser Gly Leu Ile Gly Ala Leu
165 170 175
Leu Val Cys Arg Glu Gly Ser Leu Ala Lys Glu Lys Thr Gln Thr Leu
180 185 190
His Lys Phe Ile Leu Leu Phe Ala Val Phe Asp Glu Gly Lys Ser Trp
195 200 205
His Ser Glu Thr Lys Asn Ser Leu Met Gln Asp Arg Asp Ala Ala Ser
210 215 220
Ala Arg Ala Trp Pro Lys Met His Thr Val Asn Gly Tyr Val Asn Arg
225 230 235 240
Ser Leu Pro Gly Leu Ile Gly Cys His Arg Lys Ser Val Tyr Trp His
245 250 255
Val Ile Gly Met Gly Thr Thr Pro Glu Val His Ser Ile Phe Leu Glu
260 265 270
Gly His Thr Phe Leu Val Arg Asn His Arg Gln Ala Ser Leu Glu Ile
275 280 285
Ser Pro Ile Thr Phe Leu Thr Ala Gln Thr Leu Leu Met Asp Leu Gly
290 295 300
Gln Phe Leu Leu Phe Cys His Ile Ser Ser His Gln His Asp Gly Met
305 310 315 320
Glu Ala Tyr Val Lys Val Asp Ser Cys Pro Glu Glu Pro Gln Leu Arg
325 330 335
Met Lys Asn Asn Glu Glu Ala Glu Asp Tyr Asp Asp Asp Leu Thr Asp
340 345 350
Ser Glu Met Asp Val Val Arg Phe Asp Asp Asp Asn Ser Pro Ser Phe
355 360 365
Ile Gln Ile Arg Ser Val Ala Lys Lys His Pro Lys Thr Trp Val His
370 375 380
Tyr Ile Ala Ala Glu Glu Glu Asp Trp Asp Tyr Ala Pro Leu Val Leu
385 390 395 400
Ala Pro Asp Asp Arg Ser Tyr Lys Ser Gln Tyr Leu Asn Asn Gly Pro
405 410 415
Gln Arg Ile Gly Arg Lys Tyr Lys Lys Val Arg Phe Met Ala Tyr Thr
420 425 430
Asp Glu Thr Phe Lys Thr Arg Glu Ala Ile Gln His Glu Ser Gly Ile
435 440 445
Leu Gly Pro Leu Leu Tyr Gly Glu Val Gly Asp Thr Leu Leu Ile Ile
450 455 460
Phe Lys Asn Gln Ala Ser Arg Pro Tyr Asn Ile Tyr Pro His Gly Ile
465 470 475 480
Thr Asp Val Arg Pro Leu Tyr Ser Arg Arg Leu Pro Lys Gly Val Lys
485 490 495
His Leu Lys Asp Phe Pro Ile Leu Pro Gly Glu Ile Phe Lys Tyr Lys
500 505 510
Trp Thr Val Thr Val Glu Asp Gly Pro Thr Lys Ser Asp Pro Arg Cys
515 520 525
Leu Thr Arg Tyr Tyr Ser Ser Phe Val Asn Met Glu Arg Asp Leu Ala
530 535 540
Ser Gly Leu Ile Gly Pro Leu Leu Ile Cys Tyr Lys Glu Ser Val Asp
545 550 555 560
Gln Arg Gly Asn Gln Ile Met Ser Asp Lys Arg Asn Val Ile Leu Phe
565 570 575
Ser Val Phe Asp Glu Asn Arg Ser Trp Tyr Leu Thr Glu Asn Ile Gln
580 585 590
Arg Phe Leu Pro Asn Pro Ala Gly Val Gln Leu Glu Asp Pro Glu Phe
595 600 605
Gln Ala Ser Asn Ile Met His Ser Ile Asn Gly Tyr Val Phe Asp Ser
610 615 620
Leu Gln Leu Ser Val Cys Leu His Glu Val Ala Tyr Trp Tyr Ile Leu
625 630 635 640
Ser Ile Gly Ala Gln Thr Asp Phe Leu Ser Val Phe Phe Ser Gly Tyr
645 650 655
Thr Phe Lys His Lys Met Val Tyr Glu Asp Thr Leu Thr Leu Phe Pro
660 665 670
Phe Ser Gly Glu Thr Val Phe Met Ser Met Glu Asn Pro Gly Leu Trp
675 680 685
Ile Leu Gly Cys His Asn Ser Asp Phe Arg Asn Arg Gly Met Thr Ala
690 695 700
Leu Leu Lys Val Ser Ser Cys Asp Lys Asn Thr Gly Asp Tyr Tyr Glu
705 710 715 720
Asp Ser Tyr Glu Asp Ile Ser Ala Tyr Leu Leu Ser Lys Asn Asn Ala
725 730 735
Ile Glu Pro Arg Ser Phe Ser Gln Asn Pro Pro Val Leu Lys Arg His
740 745 750
Gln Arg Glu Ile Thr Arg Thr Thr Leu Gln Ser Asp Gln Glu Glu Ile
755 760 765
Asp Tyr Asp Asp Thr Ile Ser Val Glu Met Lys Lys Glu Asp Phe Asp
770 775 780
Ile Tyr Asp Glu Asp Glu Asn Gln Ser Pro Arg Ser Phe Gln Lys Lys
785 790 795 800
Thr Arg His Tyr Phe Ile Ala Ala Val Glu Arg Leu Trp Asp Tyr Gly
805 810 815
Met Ser Ser Ser Pro His Val Leu Arg Asn Arg Ala Gln Ser Gly Ser
820 825 830
Val Pro Gln Phe Lys Lys Val Val Phe Gln Glu Phe Thr Asp Gly Ser
835 840 845
Phe Thr Gln Pro Leu Tyr Arg Gly Glu Leu Asn Glu His Leu Gly Leu
850 855 860
Leu Gly Pro Tyr Ile Arg Ala Glu Val Glu Asp Asn Ile Met Val Thr
865 870 875 880
Phe Arg Asn Gln Ala Ser Arg Pro Tyr Ser Phe Tyr Ser Ser Leu Ile
885 890 895
Ser Tyr Glu Glu Asp Gln Arg Gln Gly Ala Glu Pro Arg Lys Asn Phe
900 905 910
Val Lys Pro Asn Glu Thr Lys Thr Tyr Phe Trp Lys Val Gln His His
915 920 925
Met Ala Pro Thr Lys Asp Glu Phe Asp Cys Lys Ala Trp Ala Tyr Phe
930 935 940
Ser Asp Val Asp Leu Glu Lys Asp Val His Ser Gly Leu Ile Gly Pro
945 950 955 960
Leu Leu Val Cys His Thr Asn Thr Leu Asn Pro Ala His Gly Arg Gln
965 970 975
Val Thr Val Gln Glu Phe Ala Leu Phe Phe Thr Ile Phe Asp Glu Thr
980 985 990
Lys Ser Trp Tyr Phe Thr Glu Asn Met Glu Arg Asn Cys Arg Ala Pro
995 1000 1005
Cys Asn Ile Gln Met Glu Asp Pro Thr Phe Lys Glu Asn Tyr Arg
1010 1015 1020
Phe His Ala Ile Asn Gly Tyr Ile Met Asp Thr Leu Pro Gly Leu
1025 1030 1035
Val Met Ala Gln Asp Gln Arg Ile Arg Trp Tyr Leu Leu Ser Met
1040 1045 1050
Gly Ser Asn Glu Asn Ile His Ser Ile His Phe Ser Gly His Val
1055 1060 1065
Phe Thr Val Arg Lys Lys Glu Glu Tyr Lys Met Ala Leu Tyr Asn
1070 1075 1080
Leu Tyr Pro Gly Val Phe Glu Thr Val Glu Met Leu Pro Ser Lys
1085 1090 1095
Ala Gly Ile Trp Arg Val Glu Cys Leu Ile Gly Glu His Leu His
1100 1105 1110
Ala Gly Met Ser Thr Leu Phe Leu Val Tyr Ser Asn Lys Cys Gln
1115 1120 1125
Thr Pro Leu Gly Met Ala Ser Gly His Ile Arg Asp Phe Gln Ile
1130 1135 1140
Thr Ala Ser Gly Gln Tyr Gly Gln Trp Ala Pro Lys Leu Ala Arg
1145 1150 1155
Leu His Tyr Ser Gly Ser Ile Asn Ala Trp Ser Thr Lys Glu Pro
1160 1165 1170
Phe Ser Trp Ile Lys Val Asp Leu Leu Ala Pro Met Ile Ile His
1175 1180 1185
Gly Ile Lys Thr Gln Gly Ala Arg Gln Lys Phe Ser Ser Leu Tyr
1190 1195 1200
Ile Ser Gln Phe Ile Ile Met Tyr Ser Leu Asp Gly Lys Lys Trp
1205 1210 1215
Gln Thr Tyr Arg Gly Asn Ser Thr Gly Thr Leu Met Val Phe Phe
1220 1225 1230
Gly Asn Val Asp Ser Ser Gly Ile Lys His Asn Ile Phe Asn Pro
1235 1240 1245
Pro Ile Ile Ala Arg Tyr Ile Arg Leu His Pro Thr His Tyr Ser
1250 1255 1260
Ile Arg Ser Thr Leu Arg Met Glu Leu Met Gly Cys Asp Leu Asn
1265 1270 1275
Ser Cys Ser Met Pro Leu Gly Met Glu Ser Lys Ala Ile Ser Asp
1280 1285 1290
Ala Gln Ile Thr Ala Ser Ser Tyr Phe Thr Asn Met Phe Ala Thr
1295 1300 1305
Trp Ser Pro Ser Lys Ala Arg Leu His Leu Gln Gly Arg Ser Asn
1310 1315 1320
Ala Trp Arg Pro Gln Val Asn Asn Pro Lys Glu Trp Leu Gln Val
1325 1330 1335
Asp Phe Gln Lys Thr Met Lys Val Thr Gly Val Thr Thr Gln Gly
1340 1345 1350
Val Lys Ser Leu Leu Thr Ser Met Tyr Val Lys Glu Phe Leu Ile
1355 1360 1365
Ser Ser Ser Gln Asp Gly His Gln Trp Thr Leu Phe Phe Gln Asn
1370 1375 1380
Gly Lys Val Lys Val Phe Gln Gly Asn Gln Asp Ser Phe Thr Pro
1385 1390 1395
Val Val Asn Ser Leu Asp Pro Pro Leu Leu Thr Arg Tyr Leu Arg
1400 1405 1410
Ile His Pro Gln Ser Trp Val His Gln Ile Ala Leu Arg Met Glu
1415 1420 1425
Val Leu Gly Cys Glu Ala Gln Asp Leu Tyr
1430 1435
<210> 2
<211> 110
<212> PRT
<213> Homo sapiens
<400> 2
Ser Tyr Ser Asp Gly Asp Gln Cys Ala Ser Ser Pro Cys Gln Asn Gly
1 5 10 15
Gly Ser Cys Lys Asp Gln Leu Gln Ser Tyr Ile Cys Phe Cys Leu Pro
20 25 30
Ala Phe Glu Gly Arg Asn Cys Glu Thr His Lys Asp Asp Gln Leu Ile
35 40 45
Cys Val Asn Glu Asn Gly Gly Cys Glu Gln Tyr Cys Ser Asp His Thr
50 55 60
Gly Thr Lys Arg Ser Cys Arg Cys His Glu Gly Tyr Ser Leu Leu Ala
65 70 75 80
Asp Gly Val Ser Cys Thr Pro Thr Val Glu Tyr Pro Cys Gly Lys Ile
85 90 95
Pro Ile Leu Glu Lys Arg Asn Ala Ser Lys Pro Gln Gly Arg
100 105 110
<210> 3
<211> 17
<212> PRT
<213> Homo sapiens
<400> 3
Lys Pro Val Ala Phe Ser Asp Tyr Ile His Pro Val Cys Leu Pro Asp
1 5 10 15
Arg
<210> 4
<211> 110
<212> PRT
<213> Homo sapiens
<400> 4
Gly His Gln Phe Glu Gly Ala Glu Glu Tyr Ala Ser Phe Leu Gln Glu
1 5 10 15
Ala Gln Val Pro Phe Leu Ser Leu Glu Arg Cys Ser Ala Pro Asp Val
20 25 30
His Gly Ser Ser Ile Leu Pro Gly Met Leu Cys Ala Gly Phe Leu Glu
35 40 45
Gly Gly Thr Asp Ala Cys Gln Gly Asp Ser Gly Gly Pro Leu Val Cys
50 55 60
Glu Asp Gln Ala Ala Glu Arg Arg Leu Thr Leu Gln Gly Ile Ile Ser
65 70 75 80
Trp Gly Ser Gly Cys Gly Asp Arg Asn Lys Pro Gly Val Tyr Thr Asp
85 90 95
Val Ala Tyr Tyr Leu Ala Trp Ile Arg Glu His Thr Val Ser
100 105 110
<---
| название | год | авторы | номер документа |
|---|---|---|---|
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПРЕДНАЗНАЧЕННАЯ ДЛЯ ПРИМЕНЕНИЯ ДЛЯ ПРЕДУПРЕЖДЕНИЯ И/ИЛИ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЯ, КОТОРОЕ РАЗВИВАЕТСЯ ИЛИ ПРОГРЕССИРУЕТ ВСЛЕДСТВИЕ СНИЖЕНИЯ ИЛИ УТРАТЫ АКТИВНОСТИ ФАКТОРА СВЕРТЫВАНИЯ КРОВИ VIII И/ИЛИ АКТИВИРОВАННОГО ФАКТОРА СВЕРТЫВАНИЯ КРОВИ VIII | 2015 |
|
RU2721910C2 |
| УЛУЧШЕННЫЙ БЕЛОК СЛИЯНИЯ FVIII И ЕГО ПРИМЕНЕНИЕ | 2019 |
|
RU2789085C2 |
| Выделенная нуклеиновая кислота, которая кодирует слитый белок на основе FVIII-BDD и гетерологичного сигнального пептида, и ее применение | 2022 |
|
RU2818229C2 |
| Кодон-оптимизированная нуклеиновая кислота, которая кодирует белок фактора свёртывания крови IX, и ее применение | 2021 |
|
RU2831751C2 |
| Антитела против ингибитора пути тканевого фактора и их применения | 2016 |
|
RU2815681C1 |
| ВЫСОКОГЛИКОЗИЛИРОВАННЫЙ СЛИТЫЙ БЕЛОК НА ОСНОВЕ ФАКТОРА СВЕРТЫВАНИЯ КРОВИ ЧЕЛОВЕКА VIII, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2016 |
|
RU2722374C1 |
| УЛУЧШЕННЫЙ СЛИТЫЙ БЕЛОК ФАКТОРА IX И ЕГО КОНЪЮГАТ И ИХ ПРИМЕНЕНИЕ | 2020 |
|
RU2794350C1 |
| КОМПОЗИЦИИ И СПОСОБЫ ДЛЯ ПОВЫШЕНИЯ ИЛИ УСИЛЕНИЯ ТРАНСДУКЦИИ ВЕКТОРОВ ДЛЯ ГЕННОЙ ТЕРАПИИ И ДЛЯ УДАЛЕНИЯ ИЛИ СНИЖЕНИЯ ИММУНОГЛОБУЛИНОВ | 2021 |
|
RU2841237C1 |
| ГЛИКОЗИЛИРОВАННЫЕ СЛИТЫЕ БЕЛКИ VWF С УЛУЧШЕННОЙ ФАРМАКОКИНЕТИКОЙ | 2017 |
|
RU2782212C2 |
| АНТИТЕЛА ПРОТИВ ФАКТОРА СВЕРТЫВАНИЯ XI | 2017 |
|
RU2757314C2 |


Группа изобретений относится к области фармацевтики и раскрывает фармацевтическую композицию для подкожного введения, содержащую фактор свертывания крови, а также способ получения фармацевтической композиции. Композиция характеризуется тем, что содержит коллоидную частицу, содержащую приблизительно от 0,5 до 20 мольных процентов амфифильного липида, модифицированного биосовместимым гидрофильным полимером, причем указанный фактор свертывания крови не инкапсулирован в указанную коллоидную частицу. 2 н. и 21 з.п. ф-лы, 3 табл., 2 пр., 4 ил.
1. Применение фармацевтической композиции для лечения гемофилии А, при этом указанная фармацевтическая композиция содержит фактор свертывания крови, который представляет собой фактор VIII, и коллоидную частицу, содержащую от 0,5 до 20 мольных процентов амфифильного липида, модифицированного биосовместимым гидрофильным полимером, при этом указанный фактор свертывания не инкапсулирован указанной коллоидной частицей, и причем указанная фармацевтическая композиция предназначена для подкожного введения, при этом повторное подкожное введение указанной фармацевтической композиции осуществляют до того, как концентрация указанной фармацевтической композиции в крови снизится до терапевтически незначимых уровней.
2. Применение по п. 1, отличающееся тем, что указанная коллоидная частица по существу нейтральна и указанный полимер по существу не несет суммарного заряда.
3. Применение по п. 1, отличающееся тем, что указанная коллоидная частица обладает средним диаметром частиц от приблизительно 0,03 до приблизительно 0,4 микрон (мкм).
4. Применение по п. 3, отличающееся тем, что указанная коллоидная частица обладает средним диаметром частиц приблизительно 0,1 микрон (мкм).
5. Применение по любому из пп. 1-4, отличающееся тем, что указанный амфифильный липид представляет собой фосфолипид из природного или синтетического источника.
6. Применение по п. 5, отличающееся тем, что указанный амфифильный липид представляет собой фосфатидилэтаноламин (ФЭ).
7. Применение по любому из пп. 1-4, отличающееся тем, что указанный амфифильный липид представляет собой карбамат-сшитый незаряженный липополимер.
8. Применение по п. 7, отличающееся тем, что указанный амфифильный липид представляет собой аминопропандиол-дистеароил (ДС).
9. Применение по п. 1, отличающееся тем, что указанная коллоидная частица дополнительно содержит второй амфифильный липид, полученный из природного или синтетического источника.
10. Применение по п. 9, отличающееся тем, что указанный второй амфифильный липид представляет собой фосфатидилхолин.
11. Применение по п. 10, отличающееся тем, что указанная коллоидная частица содержит пальмитоил-олеоил-фосфатидилхолин (ПОФХ) и 1,2-дистеароил-sn-глицеро-3-фосфоэтаноламин (ДСПЭ) в соотношении (ПОФХ:ДСПЭ) 85 - 99:15 - 1.
12. Применение по п. 11, отличающееся тем, что указанное соотношение ПОФХ:ДСПЭ составляет 90 – 99:10 - 1.
13. Применение по п. 12, отличающееся тем, что указанное соотношение ПОФХ:ДСПЭ составляет 97:3.
14. Применение по п. 9, отличающееся тем, что указанная композиция дополнена холестерином.
15. Применение по любому из пп. 1-14, отличающееся тем, что указанный биосовместимый гидрофильный полимер выбран из группы, состоящей из полиалкилэфиров, полимолочных кислот и полигликолевых кислот.
16. Применение по п. 15, отличающееся тем, что указанный биосовместимый гидрофильный полимер представляет собой полиэтиленгликоль.
17. Применение по п. 16, отличающееся тем, что указанный полиэтиленгликоль обладает молекулярной массой от приблизительно 500 до приблизительно 5000 Дальтон.
18. Применение по п. 17, отличающееся тем, что указанный полиэтиленгликоль обладает молекулярной массой приблизительно 2000 Дальтон.
19. Применение по любому из пп. 16-18, отличающееся тем, что указанный модифицированный амфифильный липид представляет собой 1,2-дистеароил-sn-глицеро-3-фосфоэтаноламин-N-[поли-(этиленгликоль)].
20. Применение по любому из пп. 16-18, отличающееся тем, что указанный модифицированный амфифильный липид представляет собой 1,2-дистеароил-sn-глицеро-3-фосфоэтаноламин-N-[поли-(этиленгликоль)-2000] (ДСПЭ-ПЭГ 2000).
21. Применение по любому из пп. 1-20, отличающееся тем, что указанная композиция дополнительно содержит другое терапевтически активное соединение, выбранное из группы, содержащей противовоспалительный препарат, анальгетик, антибиотик или другой фармацевтически активный агент, который может способствовать или усиливать активность фактора свертывания крови.
22. Применение по п. 1, отличающееся тем, что у указанного пациента вырабатываются антитела против указанного фактора свертывания.
23. Способ лечения гемофилии А у пациента, при этом указанный способ включает подкожное введение фармацевтической композиции, содержащей фактор свертывания крови, который представляет собой фактор VIII, и коллоидную частицу, содержащую от 0,5 до 20 мольных процентов амфифильного липида, модифицированного биосовместимым гидрофильным полимером, при этом указанный фактор свертывания не инкапсулирован указанной коллоидной частицей, при этом повторное подкожное введение указанной фармацевтической композиции в эффективном количестве осуществляют до того, как концентрация указанной фармацевтической композиции в крови снизится до терапевтически незначимых уровней.
| WO 2004091723, 28.10.2004 | |||
| WO 2007002886, 04.01.2007 | |||
| BARU M | |||
| ET AL., Factor VIII efficient and specific non-covalent binding to PEGylated liposomes enables prolongation of its circulation time and haemostatic efficacy, Thrombosis and haemostasis, schattauer gmbh, (20050601), vol | |||
| Домовый номерной фонарь, служащий одновременно для указания названия улицы и номера дома и для освещения прилежащего участка улицы | 1917 |
|
SU93A1 |
| PENG A | |||
| ET AL., PEGylation of a Factor | |||

