Изобретение относится к химической промышленности, а именно, к области производства гетерогенных катализаторов процессов жидкофазного окисления органических соединений (в том числе - производных фенолов) и может быть применено на предприятиях различных отраслей промышленности для проведения реакций окисления, а также для каталитической очистки сточных вод от токсичных органических загрязнителей.
Известен гетерогенный катализатор окисления неорганических и/или органических соединений на полимерном носителе (RU 2255805, B01J 23/70, B01J 23/94, B01D 53/86, 10.07.2005), содержащий активный компонент на полимерном носителе - полиэтилене или полипропилене, или полистироле, или другом полимере, который в качестве активного компонента содержит оксиды и/или гидроксиды и/или шпинели металлов переменной валентности, и, дополнительно, модифицирующую добавку, в качестве которой используют органические основания и/или гетерополикислоты, и/или углеродсодержащий материал при следующем содержании компонентов катализатора, массовая доля в %: активный компонент 15-50; модифицирующая добавка 0,5-20; носитель остальное.
Однако его недостаточно высокая операционная стабильность из-за непрочного связывания активного компонента с поверхностью носителя, а также высокое содержание активного компонента обуславливает высокую стоимость катализатора и его низкую эффективность.
Известен также гетерогенный катализатор окисления органических соединений (RU 2288033; B01J 31/02, B01J 31/08, C12N 9/08, C02F 1/72; 15.11.2005), включающий полимерный носитель, модифицирующую добавку, активный компонент и сшивающий агент, при этом в качестве сшивающего агента используется глутаровый альдегид или карбодиимид, в качестве полимерного носителя катализатор содержит ионообменную смолу, в качестве модифицирующей добавки - альгинат натрия или хитозан, а в качестве активного компонента - экстракт корня хрена или редьки, при следующем соотношении компонентов в масс. %: активный компонент - 1-2%; модифицирующая добавка - 0,1-1%; сшивающий агент - 3-6%; носитель - 90-95%.
Однако недостаточно прочная сшивка активного компонента с поверхностью твердого носителя приводит к существенному снижению активности и стабильности катализатора в реакциях окисления органических соединений. Кроме того, активный компонент распределен только на поверхности носителя, не проникая в его поры, что существенно снижает эффективность катализатора.
Наиболее близким к предлагаемому катализатору является гетерогенный катализатор жидкофазного окисления органических соединений (RU 2626964, B01J 31/02, B01J 21/06, C12N 9/08, C12N 11/14, C02F 1/72, 14.11.2016), содержащий носитель, глутаровый диальдегид в качестве сшивающего агента и экстракт корня хрена (ArmoraciaRusticana) в качестве активного компонента, при этом в качестве носителя используют диоксид титана, модифицированный последовательно 0,095÷0,105 н. раствором соляной кислоты, 0,195÷0,205%-ным раствором хитозана в 0,0045÷0,0055 М растворе соляной кислоты и 4,95÷5,05%-ным раствором аминопропилтриэтоксисилана в 95,5÷96,5%-ном этаноле при следующем соотношении компонентов, % масс.: диоксид титана - 45÷55; хитозан - 7,5÷12,5; аминопропилтриэтоксисилан - 17,5÷22,5; сшивающий агент (глутаровыйдиальдегид) - 7,5÷12,5; активный компонент (экстракт корня хрена) - 7,5÷12,5.
Однако такой катализатор обладает сложной многокомпонентной структурой, вследствие чего активность его очень нестабильна и подвержена влиянию различных факторов реакционной среды, что в большинстве случаев приводит к существенному снижению эффективности катализатора.
Известен способ получения нерастворимых конъюгатов ферментов с производными полистирола (US 3860486; C07G 7/02; 14.01.1975), включающий обработку полимерного носителя на основе модифицированного полистирола водным раствором пероксидазы хрена в присутствии бикарбоната натрия при 0-4°С. Продолжительность обработки 20 ч.
Однако в данном способе необходимо проводить длительную обработку носителя раствором пероксидазы и проводить процесс иммобилизации при низких температурах.
Известен также способ получения иммобилизованной пероксидазы (SU 742434; C07G 7/02,C07G 7/022; 10.11.1978) путем обработки органического полимерного носителя раствором, содержащим фермент - пероксидазу, отличающийся тем, что, с целью упрощения процесса, в качестве носителя используют альгинатное волокно, в качестве раствора, содержащего пероксидазу культуральный фильтрат гриба Cerrerid, а обработку им носителя осуществляют при 18-22°С в течение 10-30 мин.
Однако материал носителя имеет низкую механическую, химическую и микробиологическую стойкость и активность иммобилизованной пероксидазы низкая.
Наиболее близким к предлагаемому способу является способ получения иммобилизованной пероксидазы (RU 2005784; C12N 11/14, C12N 11/10; 06.05.1990) предусматривающий взаимодействие ферментсодержащего раствора с модифицированным носителем, при этом с целью повышения удельной активности фермента, в качестве носителя используют аэросил, модифицированный 2-3% раствором декстрана и окисленный перхлоратом натрия для получения альдегидных групп на поверхности носителя.
Однако недостаточная стабилизация активного компонента на поверхности носителя из-за отсутствия химического взаимодействия между аэросилом и декстраном в этом способе существенно ухудшает каталитическую активность и стабильность катализатора.
Технической проблемой, решаемой при создании изобретения, является разработка высокоактивного, стабильного и селективного гетерогенного катализатора для многократного использования в реакции жидкофазного окисления органических соединений перекисью водорода, легко отделяемого от реакционной среды.
Техническим результатом изобретения является повышение активности, селективности, операционной стабильности гетерогенного катализатора в реакции жидкофазного окисления органических соединений перекисью водорода и его способности к отделению от реакционной среды за счет использования твердого носителя с большой площадью поверхности, высокореакционноспособными аминогруппами на поверхности и магнитными свойствами.
Решение поставленной проблемы и заявленный технический результат достигаются за счет того, что гетерогенный катализатор жидкофазного окисления органических соединений содержит носитель, модифицированный 3-аминопропилтриэтоксисиланом, глутаровый диальдегид в качестве сшивающего агента и пероксидазу корня хрена в качестве активного компонента при этом носителем являются магнитные наночастицы Fe3O4, модифицированные SiO2, при следующем соотношении компонентов в % масс.:
- Fe3O4 - 34,2÷34,6;
- SiO2 - 41,0÷41,4;
- 3-аминопропилтриэтоксисилан - 18,3÷18,8;
- глутаровый диальдегид - 3,8÷4,0;
- пероксидаза хрена - 1,9÷2,0.
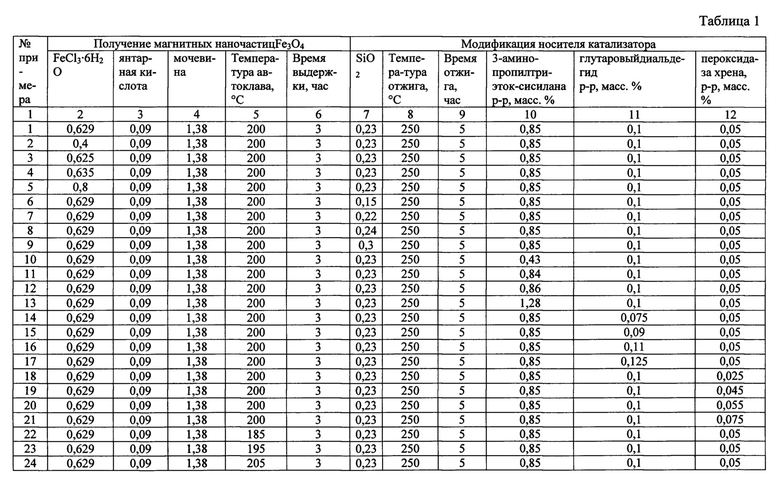
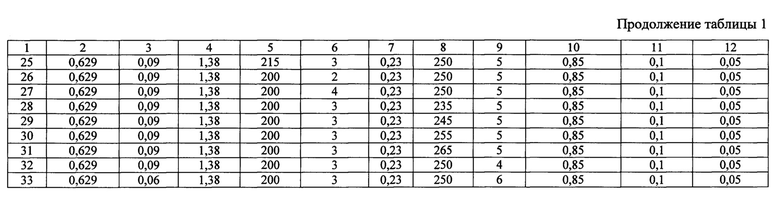
Способ получения гетерогенного катализатора жидкофазного окисления органических соединений включает взаимодействие фермент содержащего раствора с носителем, модифицированным для получения альдегидных групп на его поверхности, при этом для повышения удельной активности и стабильности фермента, в качестве носителя используют магнитные наночастицы Fe3O4, для получения которых растворяют 0,625÷0,635 г FeCl3, 0,09 г янтарной кислоты, 1,38 г мочевины в 30 мл этиленгликоля при постоянном перемешивании магнитной мешалкой в течение 30±1 минут, выдерживают смесь в автоклаве при температуре 200±5°С в течение 3±0,1 часов, охлаждают до 20±1°С, отделяют наночастицы с помощью постоянного магнита, промывают их этанолом и высушивают под вакуумом при 60±2°С в течение 6±0,1 часов, модификация носителя включает смешивание его с 10 мл этанола, добавление 0,22÷0,24 г SiO2, после чего смесь перемешивают в течение 12±0,5 часов, высушивают при комнатной температуре под вакуумом до постоянной массы, добавляют 25 капель этиленгликоля, нагревают в трубчатой печи в атмосфере аргона до 250±5°С со скоростью нагрева 2±0,1°С в секунду, отжигают в течение 5±0,1 часов при 250±5°С, охлаждают, измельчают пестиком в ступке, промывают ацетоном с использованием магнитной сепарации с постоянным магнитом, высушивают при комнатной температуре до постоянной массы, после чего полученный порошок суспендируют в 0,84÷0,86%(масс.) растворе 3-аминопропилтриэтоксисилана в 10% уксусной кислоте, добавляют к смеси7 мл глицерина, выдерживают при 90±1°С в течение 5±0,1 часов, промывают дистиллированной водой и метанолом, сушат в вакуумной печи в течение 12±0,5 часов при температуре 90±1°С, затем к полученному порошку добавляют 20 мл 0,09÷0,11% (масс.) раствора глутарового диальдегида в фосфатном буферном растворе (рН=7,0), перемешивают смесь в течение 12±0,5 часов, промывают порошок дистиллированной водой, добавляют 20 мл 0,045÷0,055%(масс.) раствора пероксидазы хрена в фосфатном буферном растворе (рН=7,0), перемешивают в течение 1±0,05 часа, промывают дистиллированной водой и высушивают при комнатной температуре до постоянной массы.
Предлагаемый катализатор обладает следующими преимуществами:
- более высокой активностью в реакции окисления органических субстратов в присутствии пероксида водорода;
- более высокой операционной стабильностью при многократном использовании;
- удобством полного отделения от реакционной среды практически без потерь с помощью постоянного магнита.
Включение в катализатор каждого из компонентов является обязательным и ни один из них нельзя исключить из состава катализатора, а также изменить их количественное соотношение, так как это приведет к существенному снижению активности и стабильности катализатора в реакции окисления органических соединений.
Магнитные наночастицы Fe3O4 необходимы для создания твердой основы для катализатора и придания ему магнитных свойств, что способствует легкому отделению катализатора от реакционной среды с помощью постоянного магнита.
Оксид кремния SiO2 необходим для создания твердой основы для катализатора, обеспечения присутствия на поверхности носителя гидроксидных-групп ОН-, способных к взаимодействию с другими функциональными группами, и увеличения площади поверхности носителя, что существенно увеличивает количество присоединенных к поверхности носителя молекул фермента, что существенно повышает активность и стабильность катализатора.
Аминопропилтриэтоксисилан необходим для образования прочной связи между компонентами катализатора за счет образования связи между гидроксильными группами на поверхности носителя и кремнием, входящим в состав аминопропилтриэтоксисилана, и для появления на поверхности носителя аминогрупп с высокой реакционной способностью.
Сшивающий агент - глутаровый диальдегид - необходим для образования азометиновой связи (оснований Шиффа) между аминогруппами на поверхности носителя и аминогруппами пероксидазы хрена, что способствует прочному закреплению фермента на поверхности носителя.
Активный компонент - фермент пероксидаза хрена - катализирует реакцию окисления органических соединений в присутствии пероксида водорода.
Относительное массовое содержание компонентов катализатора выбрано экспериментально.
Уменьшение содержания SiO2, 3-аминопропилтриэтоксисилана и глутарового диальдегида меньше представленных значений существенно снижает активность катализатора, так как значительно снижается количество пероксидазы хрена, ковалентно связанной с поверхностью носителя и фермент легко смывается с нее в процессе промывки. Увеличение содержания SiO2 больше указанного значения приводит к чрезмерному увеличению поверхности катализатора, что приводит к диффузионным ограничениям и существенно снижает активность катализатора из-за затруднения удаления субстрата из реакционной среды и отравления фермента.
Увеличение содержания 3-аминопропилтриэтоксисилана и глутарового альдегида выше указанных значений нецелесообразно, так как, по результатам экспериментов, не приводит к существенному увеличению активности и стабильности катализатора. Содержание активного компонента ниже представленного значения значительно снижает активность катализатора в реакции окисления органических субстратов, а увеличение его содержания нецелесообразно, так как, по результатам экспериментов, не приводит к существенному увеличению активности катализатора в реакции окисления органических субстратов.
Добавление янтарной кислоты и мочевины к FeCl3⋅6H2O необходимо для восстановления ионов железа и образования магнетита Fe3O4 (FeO⋅Fe2O3), который обладает магнитными свойствами. Восстановление проводится при температуре 200±5°С в течение 3±0,1 часов для наиболее полного восстановления ионов железа. Температура и время проведения реакции выбраны экспериментально. Уменьшение времени и температуры ниже указанных значений приводит к недовосстановлению ионов железа и существенному снижению магнитных свойств порошка, что существенно увеличивает потери катализатора в ходе реакции, а дальнейшее увеличение температуры и времени выше указанных значений не приводит к улучшению магнитных свойств, но нецелесообразно из-за существенного увеличения энергозатрат.
Промывка Fe3O4 этанолом необходима для удаления из порошка остатков неспецифически связанных компонентов.
Смешивание Fe3O4 с дальнейшим высушиванием и отжигом необходимо для формирования равномерной структуры SiO2 вокруг магнитных наночастиц, что необходимо для увеличения площади поверхности катализатора и сохранения его магнитных свойств.
Температура (250±5°С) и время (5±0,1 часов) отжига выбраны экспериментально. При уменьшении температуры и времени отжига ниже указанных значений уменьшается прочность и равномерность структуры носителя, что отрицательно сказывается на активности и стабильности катализатора. Дальнейшее увеличение температуры и времени отжига нецелесообразно, так как оно приводит к необратимым изменениям и разрушению структуры носителя, что существенно снижает активность и стабильность катализатора, а также требует дополнительных энергозатрат.
Промывка смеси Fe3O4 и SiO2 необходима для удаления из порошка остатков неспецифически связанных компонентов.
Температура (90±1°С) и время (5±0,1 часов) выдерживания носителя в растворе 3-аминопропилтриэтоксисилана выбраны экспериментально. При уменьшении температуры и времени выдерживания ниже указанных значений взаимодействие 3-аминопропилтриэтоксисилана с носителем проходит не полностью, что существенно снижает количество свободных функциональных аминогрупп на поверхности носителя, и, соответственно, снижает активность и стабильность получаемого катализатора. Дальнейшее увеличение температуры и времени выдерживания выше указанных значений нецелесообразно, так как оно не приводит существенному улучшению активности и стабильности катализатора, при этом требует дополнительных энергозатрат.
Время выдерживания модифицированного носителя в растворе глутарового диальдегида - 12±0,5 часов - выбрано экспериментально. При уменьшении времени выдерживания ниже указанного значения взаимодействие глутарового диальдегида с аминогруппами носителя проходит не полностью, что существенно снижает количество свободных функциональных альдегидных групп на поверхности носителя, и, соответственно, снижает активность и стабильность получаемого катализатора. Дальнейшее увеличение времени выдерживания выше указанного значения нецелесообразно, так как оно не приводит существенному улучшению активности и стабильности катализатора.
Время выдерживания модифицированного носителя в растворе пероксидазы хрена - 1±0,05 час - выбрано экспериментально. При уменьшении времени выдерживания ниже указанного значения взаимодействие аминогрупп пероксидазы хрена с альдегидными группами модифицированного носителя проходит не полностью, что существенно снижает количество прочно закрепленного на поверхности модифицированного носителя фермента, и, соответственно, снижает активность и стабильность получаемого катализатора. Дальнейшее увеличение времени выдерживания выше указанного значения нецелесообразно, так как оно не приводит существенному улучшению активности и стабильности катализатора.
Основными свойствами гетерогенных катализаторов процессов жидкофазного окисления органических соединений являются активность катализатора (ед/г), коэффициент иммобилизации (%) и сохранение начальной активности после 10 циклов использования в реакции окисления 2,2'-азино-бис-(3-этилбензтиозолин-6-сульфокислоты) (АБТС).
Активность катализатора определяли в реакции окисления 2,2'-азино-бис-(3-этилбензтиозолин-6-сульфокислоты) (АБТС). Для исследования активности катализатора в термостатируемую стеклянную ячейку с возвратно-поступательным качанием (300 мин-1) вносили 0,5 г катализатора, 25 мл фосфатного буферного раствора (рН=7,0), 2 мл раствора 2,2'-азино-бис-(3-этилбензтиозолин-6-сульфокислоты) (2 ммоль/л) и 2 мл раствора пероксида водорода (2,2 ммоль/л), через определенные промежутки времени отбирали пробы и измеряли оптическую плотность раствора при 440 нм (раствор сравнения - дистиллированная вода). Из полученных значений оптической плотности была рассчитана активность катализатора в единицах активности (ммоль субстрата в минуту) на 1 г катализатора, коэффициент иммобилизации, равный отношению активности синтезированного катализатора к активности исходного ферментативного экстракта (10 мг пероксидазы хрена в 20 мл фосфатного буферного раствора с рН=7,0), выраженный в процентах, а также процент сохранения начальной активности после 10 циклов использования в реакции окисления АБТС.
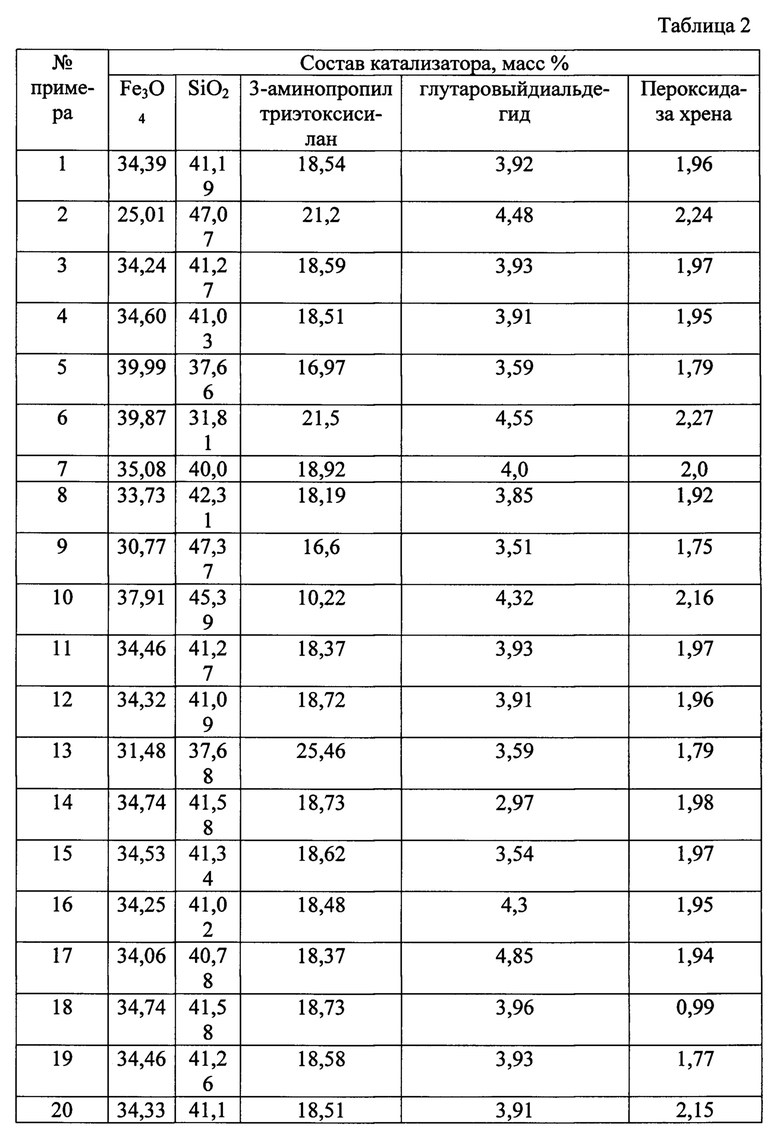
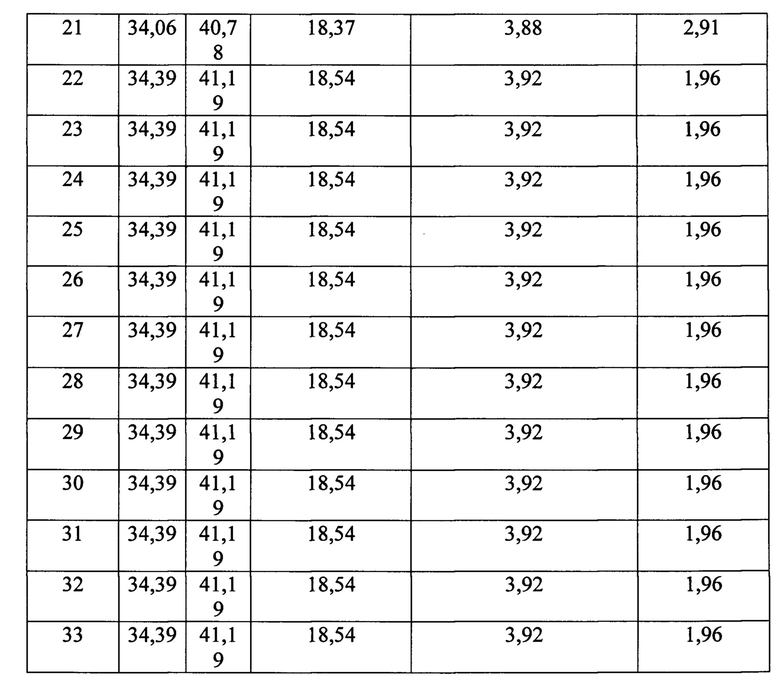
Сущность изобретения иллюстрируется следующими примерами и таблицами. Способ получения гетерогенного катализатора описывается примерами 1-33, таблицами 1 и 2. В таблице 2 представлен состав гетерогенных катализаторов процессов жидкофазного окисления органических соединений, получаемых в результате примеров 1-33.
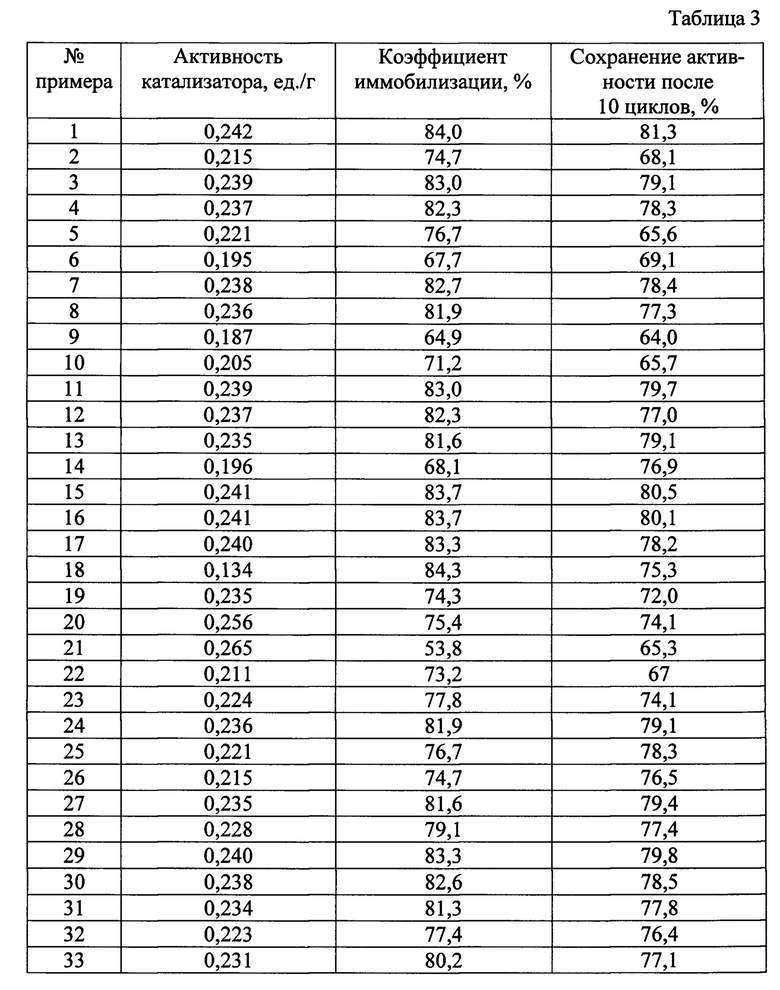
Свойства получаемых гетерогенных катализаторов процессов жидкофазного окисления органических соединений согласно примерам 1-33 представлены в таблице 3.
Пример 1
В химическом стакане в 30 мл этиленгликоля были растворены 0,629 г FeCl3⋅6H2O, 0,09 г янтарной кислоты, 1,38 г мочевины при постоянном перемешивании магнитной мешалкой в течение 30 минут. Полученный раствор переносился в тефлоновый стакан объемом 50 мл и выдерживался в автоклаве при температуре 200°С в течение 3 часов, после чего смесь охлаждалась до 20°С. Затем с помощью магнита отделялся черный осадок магнетита Fe3O4(FeO⋅Fe2O3), который промывался несколько раз этанолом и высушивался под вакуумом при температуре 60°С в течение 6 часов.
Высушенный осадок смешивали с 10 мл этанола и добавили 0,23 г SiO2, после чего перемешивали смесь в течение 12 часов. Далее смесь высушивали при комнатной температуре под вакуумом до постоянной массы. К высушенному порошку добавили 25 капель этиленгликоля, после чего его поместили в фарфоровую чашку, которую затем нагрели в кварцевой трубке в трубчатой печи в атмосфере аргона до 250°С со скоростью нагрева 2°С в секунду. Далее порошок отжигали в течение 5 часов при 250°С. После отжига порошок охлаждали и измельчали с помощью пестика в фарфоровой ступке. Измельченный порошок промывали 5 раз ацетоном с использованием магнитной сепарации с постоянным магнитом, а затем сушили при комнатной температуре до постоянной массы.
Порошок суспендировали в 11,6 мл 0,85% (масс.) раствора 3-аминопропилтриэтоксисилана (к 10 мл дистиллированной воды добавили 1,5 мл ледяной уксусной кислоты, после чего к ним добавляли 0,1 г 3-аминопропилтриэтоксисилана). Затем к смеси добавили 7 мл глицерина и выдерживали при 90°С в течение 5 часов. Затем смесь 3 раза промывали водой и 5 раз метанолом и сушили в вакуумной печи в течение 12 часов при температуре 90°С.
К полученному порошку добавили 20 мл 0,1% (масс.) раствора глутарового диальдегида (0,08 мл 25% (масс.) раствора глутарового диальдегида смешали с 20 мл фосфатного буферного раствора с рН=7,0) и перемешивали в течение 12 часов. После этого порошок промывали дистиллированной водой и добавляли 20 мл 0,05% (масс.) раствора пероксидазы хрена в фосфатном буферном растворе с рН=7,0 и перемешивали в течение 1 часа. Далее полученный катализатор промывали дистиллированной водой и высушивали при комнатной температуре до постоянной массы.
В результате сформировали катализатор со следующим соотношением компонентов, % масс.:
- Fe3O4 - 34,39%;
- SiO2 - 41,19%;
- 3-аминопропилтриэтоксисилан - 18,54%;
- глутаровый диальдегид - 3,92%;
- пероксидаза хрена -1,96%.
Соотношение компонентов в катализаторе представлены в таблице 2. Свойства полученного катализатора представлены в таблице 3.
Пример 2
Аналогичен примеру 1, однако использовали 0,4 г FeCl3⋅6H2O. Основные параметры способа получения представлены в таблице 1. Соотношение компонентов в катализаторе представлены в таблице 2. Свойства полученного катализатора представлены в таблице 3.
Пример 3
Аналогичен примеру 1, однако использовали 0,625 г FeCl3⋅6H2O. Основные параметры способа получения представлены в таблице 1. Соотношение компонентов в катализаторе представлены в таблице 2. Свойства полученного катализатора представлены в таблице 3.
Пример 4
Аналогичен примеру 1, однако использовали 0,635 г FeCl3⋅6Н2О.Основные параметры способа получения представлены в таблице 1. Соотношение компонентов в катализаторе представлены в таблице 2. Свойства полученного катализатора представлены в таблице 3.
Пример 5
Аналогичен примеру 1, однако использовали 0,8 г FeCl3⋅6Н2О. Основные параметры способа получения представлены в таблице 1. Соотношение компонентов в катализаторе представлены в таблице 2. Свойства полученного катализатора представлены в таблице 3.
Из результатов, сведенных в таблице 2 и таблице 3 по примерам 1-5 видно, что оптимальной активностью и стабильностью обладает катализатор, полученный при использовании 0,629 г FeCl3⋅6H2O.
Пример 6
Аналогичен примеру 1, однако использовали 0,15 г SiO2. Основные параметры способа получения представлены в таблице 1. Соотношение компонентов в катализаторе представлены в таблице 2. Свойства полученного катализатора представлены в таблице 3.
Пример 7
Аналогичен примеру 1, однако использовали 0,22 г SiO2. Основные параметры способа получения представлены в таблице 1. Соотношение компонентов в катализаторе представлены в таблице 2. Свойства полученного катализатора представлены в таблице 3.
Пример 8
Аналогичен примеру 1, однако использовали 0,24 г SiO2. Основные параметры способа получения представлены в таблице 1. Соотношение компонентов в катализаторе представлены в таблице 2. Свойства полученного катализатора представлены в таблице 3.
Пример 9
Аналогичен примеру 1, однако использовали 0,3 г SiO2. Основные параметры способа получения представлены в таблице 1. Соотношение компонентов в катализаторе представлены в таблице 2. Свойства полученного катализатора представлены в таблице 3.
Из результатов, сведенных в таблице 2 и таблице 3 по примерам 1, 6-9 видно, что оптимальной активностью и стабильностью обладает катализатор, полученный при использовании 0,23 г SiO2.
Пример 10
Аналогичен примеру 1, однако использовали 0,43% (масс.) раствор 3-аминопропилтриэтоксисилана. Основные параметры способа получения представлены в таблице 1. Соотношение компонентов в катализаторе представлены в таблице 2. Свойства полученного катализатора представлены в таблице 3.
Пример 11
Аналогичен примеру 1, однако использовали 0,84% (масс.) раствор 3-аминопропилтриэтоксисилана. Основные параметры способа получения представлены в таблице 1. Соотношение компонентов в катализаторе представлены в таблице 2. Свойства полученного катализатора представлены в таблице 3.
Пример 12
Аналогичен примеру 1, однако использовали 0,86% (масс.) раствор 3-аминопропилтриэтоксисилана. Основные параметры способа получения представлены в таблице 1. Соотношение компонентов в катализаторе представлены в таблице 2. Свойства полученного катализатора представлены в таблице 3.
Пример 13
Аналогичен примеру 1, однако использовали 1,28% (масс.) раствор 3-аминопропилтриэтоксисилана. Основные параметры способа получения представлены в таблице 1. Соотношение компонентов в катализаторе представлены в таблице 2. Свойства полученного катализатора представлены в таблице 3.
Из результатов, сведенных в таблице 2 и таблице 3 по примерам 1, 10-13 видно, что оптимальной активностью и стабильностью обладает катализатор, полученный при использовании 0,85% (масс.) раствора 3-аминопропилтриэтоксисилана.
Пример 14
Аналогичен примеру 1, однако использовали 0,075% (масс.) раствор глутарового диальдегида. Основные параметры способа получения представлены в таблице 1. Соотношение компонентов в катализаторе представлены в таблице 2. Свойства полученного катализатора представлены в таблице 3.
Пример 15
Аналогичен примеру 1, однако использовали 0,09% (масс.) раствор глутарового диальдегида. Основные параметры способа получения представлены в таблице 1. Соотношение компонентов в катализаторе представлены в таблице 2. Свойства полученного катализатора представлены в таблице 3.
Пример 16
Аналогичен примеру 1, однако использовали 0,11% (масс.) раствор глутарового диальдегида. Основные параметры способа получения представлены в таблице 1. Соотношение компонентов в катализаторе представлены в таблице 2. Свойства полученного катализатора представлены в таблице 3.
Пример 17
Аналогичен примеру 1, однако использовали 0,125% (масс.) раствор глутарового диальдегида. Основные параметры способа получения представлены в таблице 1. Соотношение компонентов в катализаторе представлены в таблице 2. Свойства полученного катализатора представлены в таблице 3.
Из результатов, сведенных в таблице 2 и таблице 3 по примерам 1, 14-17 видно, что оптимальной активностью и стабильностью обладает катализатор, полученный при использовании 0,1%(масс.) раствора глутарового диальдегида.
Пример 18
Аналогичен примеру 1, однако использовали 0,025% (масс.)раствор пероксидазы хрена. Основные параметры способа получения представлены в таблице 1. Соотношение компонентов в катализаторе представлены в таблице 2. Свойства полученного катализатора представлены в таблице 3.
Пример 19
Аналогичен примеру 1, однако использовали 0,045% (масс.) раствор пероксидазы хрена. Основные параметры способа получения представлены в таблице 1. Соотношение компонентов в катализаторе представлены в таблице 2. Свойства полученного катализатора представлены в таблице 3.
Пример 20
Аналогичен примеру 1, однако использовали 0,055% (масс.) раствор пероксидазы хрена. Основные параметры способа получения представлены в таблице 1. Соотношение компонентов в катализаторе представлены в таблице 2. Свойства полученного катализатора представлены в таблице 3.
Пример 21
Аналогичен примеру 1, однако использовали 0,075% (масс.) раствор пероксидазы хрена. Основные параметры способа получения представлены в таблице 1. Соотношение компонентов в катализаторе представлены в таблице 2. Свойства полученного катализатора представлены в таблице 3.
Из результатов, сведенных в таблице 2 и таблице 3 по примерам 1, 18-21 видно, что оптимальной активностью и стабильностью обладает катализатор, полученный при использовании 0,05% (масс.) раствора пероксидазы хрена.
Пример 22
Аналогичен примеру 1, однако смесь FeCl3⋅6H2O, янтарной кислоты и мочевины выдерживалась в автоклаве при температуре 185°С.Основные параметры способа получения представлены в таблице 1. Соотношение компонентов в катализаторе представлены в таблице 2. Свойства полученного катализатора представлены в таблице 3.
Пример 23
Аналогичен примеру 1, однако смесь FeCl3⋅6H2O, янтарной кислоты и мочевины выдерживалась в автоклаве при температуре 195°С. Основные параметры способа получения представлены в таблице 1. Соотношение компонентов в катализаторе представлены в таблице 2. Свойства полученного катализатора представлены в таблице 3.
Пример 24
Аналогичен примеру 1, однако смесь FeCl3⋅6H2O, янтарной кислоты и мочевины выдерживалась в автоклаве при температуре 205°С. Основные параметры способа получения представлены в таблице 1. Соотношение компонентов в катализаторе представлены в таблице 2. Свойства полученного катализатора представлены в таблице 3.
Пример 25
Аналогичен примеру 1, однако смесь FeCl3⋅6H2O, янтарной кислоты и мочевины выдерживалась в автоклаве при температуре 215°С. Основные параметры способа получения представлены в таблице 1. Соотношение компонентов в катализаторе представлены в таблице 2. Свойства полученного катализатора представлены в таблице 3.
Из результатов, сведенных в таблице 2 и таблице 3 по примерам 1, 22-25 видно, что оптимальной активностью и стабильностью обладает катализатор, полученный при выдерживании в автоклаве при температуре 200°С.
Пример 26
Аналогичен примеру 1, однако смесь FeCl3⋅6H2O, янтарной кислоты и мочевины выдерживалась в автоклаве в течение 2 часов. Основные параметры способа получения представлены в таблице 1. Соотношение компонентов в катализаторе представлены в таблице 2. Свойства полученного катализатора представлены в таблице 3.
Пример 27
Аналогичен примеру 1, однако смесь FeCl3⋅6H2O, янтарной кислоты и мочевины выдерживалась в автоклаве в течение 4 часов. Основные параметры способа получения представлены в таблице 1. Соотношение компонентов в катализаторе представлены в таблице 2. Свойства полученного катализатора представлены в таблице 3.
Из результатов, сведенных в таблице 2 и таблице 3 по примерам 1, 26-27 видно, что оптимальной активностью и стабильностью обладает катализатор, полученный при выдерживании в автоклаве в течение 3 часов.
Пример 28
Аналогичен примеру 1, однако отжиг смеси Fe3O4 и SiO2 проводился при температуре 235°С. Основные параметры способа получения представлены в таблице 1. Соотношение компонентов в катализаторе представлены в таблице 2. Свойства полученного катализатора представлены в таблице 3.
Пример 29
Аналогичен примеру 1, однако отжиг смеси Fe3O4 и SiO2 проводился при температуре 245°С. Основные параметры способа получения представлены в таблице 1. Соотношение компонентов в катализаторе представлены в таблице 2. Свойства полученного катализатора представлены в таблице 3.
Пример 30
Аналогичен примеру 1, однако отжиг смеси Fe3O4 и SiO2 проводился при температуре 255°С. Основные параметры способа получения представлены в таблице 1. Соотношение компонентов в катализаторе представлены в таблице 2. Свойства полученного катализатора представлены в таблице 3.
Пример 31
Аналогичен примеру 1, однако отжиг смеси Fe3O4 и SiO2 проводился при температуре 265°С. Основные параметры способа получения представлены в таблице 1. Соотношение компонентов в катализаторе представлены в таблице 2. Свойства полученного катализатора представлены в таблице 3.
Из результатов, сведенных в таблице 2 и таблице 3 по примерам 1, 28-31 видно, что оптимальной активностью и стабильностью обладает катализатор, полученный при отжиге смеси Fe3O4 и SiO2 при температуре 250°С.
Пример 32
Аналогичен примеру 1, однако отжиг смеси Fe3O4 и SiO2 проводился в течение 4 часов. Основные параметры способа получения представлены в таблице 1. Соотношение компонентов в катализаторе представлены в таблице 2. Свойства полученного катализатора представлены в таблице 3.
Пример 33
Аналогичен примеру 1, однако отжиг смеси Fe3O4 и SiO2 проводился в течение 6 часов. Основные параметры способа получения представлены в таблице 1. Соотношение компонентов в катализаторе представлены в таблице 2. Свойства полученного катализатора представлены в таблице 3.
Из результатов, сведенных в таблице 2 и таблице 3 по примерам 1, 32-33 видно, что оптимальной активностью и стабильностью обладает катализатор, полученный при отжиге смеси Fe3O4 и SiO2 в течение 5 часов.
Таким образом, по результатам экспериментов было определено оптимальное соотношение компонентов гетерогенного катализатора для реакций жидкофазного окисления органических соединений, % масс.:
- Fe3O4 - 34,2÷34,6;
- SiO2 - 41,0÷41,4;
- 3-аминопропилтриэтоксисилан - 18,3÷18,8;
- глутаровый диальдегид - 3,8÷4,0;
- пероксидаза хрена - 1,9÷2,0.
Предложенный способ позволяет получать гетерогенный катализатор для реакций жидкофазного окисления органических соединений перекисью водорода с повышенной активностью, селективностью, операционной стабильностью, который обладает способностью к отделению от реакционной среды за счет использования твердого носителя с большой площадью поверхности, высоко реакционноспособными аминогруппами на поверхности и магнитными свойствами.
Полученные результаты свидетельствуют о том, что применение катализатора на основе пероксидазы хрена, иммобилизованной на наночастицах Fe3O4, модифицированных SiO2 и 3-аминопропилтриэтоксисиланом, с использованием сшивающего агента глутарового диальдегида является перспективной возможностью для создания эффективных катализаторов окисления органических субстратов.
| название | год | авторы | номер документа |
|---|---|---|---|
| Магнитоотделяемый катализатор окисления органических соединений и способ его получения | 2023 |
|
RU2807591C1 |
| Магнитоотделяемый катализатор окисления органических соединений и способ его получения | 2024 |
|
RU2832335C1 |
| Гетерогенный катализатор жидкофазного окисления органических соединений | 2016 |
|
RU2626964C1 |
| ГЕТЕРОГЕННЫЙ КАТАЛИЗАТОР ОКИСЛЕНИЯ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ | 2005 |
|
RU2288033C1 |
| СПОСОБ ПОЛУЧЕНИЯ ГЕТЕРОГЕННОГО КАТАЛИЗАТОРА СИНТЕЗА УГЛЕВОДОРОДОВ ИЗ МЕТАНОЛА | 2015 |
|
RU2597269C1 |
| СПОСОБ ОКИСЛЕНИЯ ФЕНОЛЬНЫХ СОЕДИНЕНИЙ | 2006 |
|
RU2294321C1 |
| Катализатор синтеза Фишера-Тропша и способ его получения | 2024 |
|
RU2839626C1 |
| СИНТЕЗ МНОГОФУНКЦИОНАЛЬНОГО САМОНАСТРАИВАЮЩЕГОСЯ КАТАЛИЗАТОРА ОКИСЛИТЕЛЬНОГО КРЕКИНГА ОРГАНИЧЕСКОГО СЫРЬЯ И ЕГО ПРИМЕНЕНИЕ | 2010 |
|
RU2425715C1 |
| КАТАЛИЗАТОР ДЛЯ ОКИСЛЕНИЯ СЕРНИСТЫХ СОЕДИНЕНИЙ | 1994 |
|
RU2067496C1 |
| КАТАЛИЗАТОР СИНТЕЗА ФИШЕРА-ТРОПША И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2020 |
|
RU2745214C1 |
Изобретение относится к химической промышленности, а именно к области производства гетерогенных катализаторов процессов жидкофазного окисления органических соединений (в том числе производных фенолов), и может быть применено на предприятиях различных отраслей промышленности для проведения реакций окисления, а также для каталитической очистки сточных вод от токсичных органических загрязнителей. Гетерогенный катализатор жидкофазного окисления органических соединений содержит носитель, модифицированный 3-аминопропилтриэтоксисиланом, глутаровый диальдегид в качестве сшивающего агента и пероксидазу корня хрена в качестве активного компонента, в котором носителем являются магнитные наночастицы Fe3O4, модифицированные SiO2, при следующем соотношении компонентов, % мас.: Fe3O4 - 34,2÷34,6; SiO2 - 41,0÷41,4; 3-аминопропилтриэтоксисилан - 18,3÷18,8; глутаровый диальдегид - 3,8÷4,0; пероксидаза хрена - 1,9÷2,0. Способ получения гетерогенного катализатора жидкофазного окисления органических соединений включает взаимодействие фермент содержащего раствора с модифицированным для получения альдегидных групп на поверхности носителем, при этом в качестве носителя используют магнитные наночастицы Fe3O4. Модификация носителя включает смешивание его с SiO2, суспендирование полученного порошка в растворе 3-аминопропилтриэтоксисилана, добавление к смеси раствора глутарового диальдегида, раствора пероксидазы хрена, перемешивание, промывку дистиллированной водой и высушивание при комнатной температуре до постоянной массы. Техническим результатом изобретения является повышение активности, селективности, операционной стабильности гетерогенного катализатора в реакции жидкофазного окисления органических соединений перекисью водорода и его способности к отделению от реакционной среды за счет использования твердого носителя с большой площадью поверхности, высокореакционноспособными аминогруппами на поверхности и магнитными свойствами. 2 н.п. ф-лы, 3 табл., 33 пр.
1. Гетерогенный катализатор жидкофазного окисления органических соединений, содержащий носитель, модифицированный 3-аминопропилтриэтоксисиланом, глутаровый диальдегид в качестве сшивающего агента и пероксидазу корня хрена в качестве активного компонента, отличающийся тем, что носителем являются магнитные наночастицы Fe3O4, модифицированные SiO2, при следующем соотношении компонентов, % мас.:
Fe3O4 - 34,2÷34,6;
SiO2 - 41,0÷41,4;
3-аминопропилтриэтоксисилан - 18,3÷18,8;
глутаровый диальдегид - 3,8÷4,0;
пероксидаза хрена - 1,9÷2,0.
2. Способ получения гетерогенного катализатора по п.1 жидкофазного окисления органических соединений, включающий взаимодействие фермент содержащего раствора с модифицированным для получения альдегидных групп на поверхности носителем, отличающийся тем, что в качестве носителя используют магнитные наночастицы Fe3O4, для получения которых растворяют 0,625÷0,635 г FeCl3, 0,09 г янтарной кислоты, 1,38 г мочевины в 30 мл этиленгликоля при постоянном перемешивании магнитной мешалкой в течение 30±1 минут, выдерживают смесь в автоклаве при температуре 200±5°С в течение 3±0,1 часов, охлаждают до 20±1°С, отделяют наночастицы с помощью постоянного магнита, промывают их этанолом и высушивают под вакуумом при 60±2°С в течение 6±0,1 часов, модификация носителя включает смешивание его с 10 мл этанола, добавление 0,22÷0,24 г SiO2, после чего смесь перемешивают в течение 12±0,5 часов, высушивают при комнатной температуре под вакуумом до постоянной массы, добавляют 25 капель этиленгликоля, нагревают в трубчатой печи в атмосфере аргона до 250±5°С со скоростью нагрева 2±0,1°С в секунду, отжигают в течение 5±0,1 часов при 250±5°С, охлаждают, измельчают пестиком в ступке, промывают ацетоном с использованием магнитной сепарации с постоянным магнитом, высушивают при комнатной температуре до постоянной массы, после чего полученный порошок суспендируют в 0,84÷0,86% (мас.) растворе 3-аминопропилтриэтоксисилана в 10%-ной уксусной кислоте, добавляют к смеси 7 мл глицерина, выдерживают при температуре 90±1°С в течение 5±0,1 часов, промывают дистиллированной водой и метанолом, сушат в вакуумной печи при температуре 90±1°С в течение 12±0,5 часов, затем к полученному порошку добавляют 20 мл 0,9÷1,1% (мас.) раствора глутарового диальдегида в фосфатном буферном растворе (рН=7,0), перемешивают смесь в течение 12±0,5 часов, промывают порошок дистиллированной водой, добавляют 20 мл 0,045÷0,055% (мас.) раствора пероксидазы хрена в фосфатном буферном растворе (рН=7,0), перемешивают в течение 1±0,05 часа, промывают дистиллированной водой и высушивают при комнатной температуре до постоянной массы.
| Гетерогенный катализатор жидкофазного окисления органических соединений | 2016 |
|
RU2626964C1 |
| СПОСОБ ПОЛУЧЕНИЯ ИММОБИЛИЗОВАННОЙ ПЕРОКСИДАЗЫ | 1990 |
|
RU2005784C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПОВЕРХНОСТНО-МОДИФИЦИРОВАННЫХ НАНОЧАСТИЦ ДЛЯ ИММОБИЛИЗАЦИИ БИОЛОГИЧЕСКИХ ВЕЩЕСТВ | 2010 |
|
RU2425879C1 |
| CN 110438116 A, 12.11.2019 | |||
| CN 106434620 A, 22.02.2017 | |||
| О.В | |||
| Матвеева и др | |||
| "Влияние способа иммобилизации пероксидазы на активность биокатализатора в процессе окисления триметилфенола", Катализ в промышленности, номер 1, 2015, стр | |||
| Деревянный торцевой шкив | 1922 |
|
SU70A1 |
| Чан Т.Х | |||
| и др | |||
| "Магнетит с | |||

